 C. David Pauza
C. David Pauza Bhawna Poonia
Bhawna Poonia Haishan Li
Haishan Li Cristiana Cairo
Cristiana Cairo Suchita Chaudhry
Suchita Chaudhry- Institute of Human Virology and Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, USA
Human immunodeficiency virus (HIV) type 1 dysregulates γδ T cells as part of an immune evasion mechanism. Nearly three decades of research defined the effects of HIV on γδ T cells and how this impacts disease. With highly effective antiretroviral therapy providing virus suppression and longer survival, we expected a return to normal for γδ T cells. This is not the case. Even in patients with CD4 T cell reconstitution, normal γδ T cell levels and function are not recovered. The durable damage to Vδ2 T cells is paralleled by defects in NK, CD8 T cells, and dendritic cells. Whether these consequences of HIV stem from similar or distinct mechanisms are not known and effective means for recovering the full range of cellular immunity have not been discovered. These unanswered questions receive too little attention in the overall program of efforts to cure HIV this disease. Approved drugs capable of increasing Vδ2 T cell function are being tested in clinical trials for cancer and hold promise for restoring normal function in patients with HIV disease. The impetus for conducting clinical trials will come from understanding the significance of γδ T cells in HIV disease and what might be gained from targeted immunotherapy. This review traces the history and current progress of AIDS-related research on γδ T cells. We emphasize the damage to γδ T cells that persists despite effective virus suppression. These chronic immune deficits may be linked to the comorbidities of AIDS (cancer, cardiovascular disease, metabolic disease, and others) and will hinder efforts to eradicate HIV by cytotoxic T or NK cell killing. Here, we focus on one subset of T cells that may be critical in the pathogenesis of HIV and an attractive target for new immune-based therapies.
Original Studies on HIV and γδ T Cells
Human immunodeficiency virus (HIV) is an aggressive, lymphotropic virus known for CD4 depletion and immune suppression. In addition to killing CD4 T cells, HIV affects several other lymphocyte subsets (1) and impairs both acquired and innate immunity (2). We focus on HIV damage to γδ T cells and how this is related to acute or chronic disease. The two major subsets of human γδ T cells (designated Vδ1 or Vδ2) are both altered after HIV infection. Early reports that Vδ1 T cells were increased (3) and that the normal ratio of Vδ2:Vδ1 cells was inverted (4) identified an important and reproducible effect of HIV on these CD4-negative T cells. The increased levels of Vδ1 cells suggested they may be involved in antiviral immunity (5) and parallels were drawn between the expansion of Vδ1 cells and similar increases in CD8+ T cells (5) that contribute to the inverted CD4:CD8 T cell ratio (6). Non-human primate studies showed that Vδ1 cell expansion is an indirect consequence of viral infection and reflects increased translocation of stimulatory bacterial products across the gut epithelium (7). The Vδ1 cells may have antiviral functions through killing of infected cells using the NKp30 (8) or NKG2C recognition receptors (9). Killing of uninfected CD4 T cells by Vδ1 cells may also be a mechanism for HIV disease (10). Whether Vδ1 T cells accelerate or slow HIV progression has not been resolved.
In contrast to Vδ1 cells, the Vδ2 subset is uniformly depleted in HIV disease with the greatest declines seen among patients with high viremia (11). These early studies [before the introduction of combination antiretroviral therapy (ART)] involved patients who were untreated or received single drug therapy where viremia was reduced 10–50-fold but never suppressed fully. The lowest levels of Vδ2 cells were found in patients with opportunistic infections or CD4 T cells < 200 cell/mm3 of blood (12, 13). These individuals frequently had no detectable cells bearing the phosphoantigen-responsive Vγ9 chain (14). Importantly, decreases in Vδ2 cells and inversion of the Vδ2:Vδ1 cell ratio were early events in HIV disease that occurred while CD4 cell counts and their CD4:CD8 T cell ratio were still in the normal ranges.
Molecular analysis of γ and δ T cell receptor (TCR) chain usage showed that HIV-driven expansion of Vδ1 cells did not select for specific Vδ chain rearrangements or individual Vγ chains (15). Sequencing studies to describe the TCR repertoire confirmed that the population of Vδ1 chains was similar in donors with or without HIV infection and that HIV-driven Vδ1 cell expansion was not similar to antigen selection in αβ T cells (16) but more likely was a polyclonal or super-antigen response. In contrast to the case for Vδ1 cells, there was strong evidence for selective Vδ2 cell depletion based on TCR structure. Flow cytometry studies documented the specific loss of Vγ9 (also called Vγ2) expressing cells, a chain more frequently associated with Vδ2 than Vδ1 in healthy controls. Finding that the relative proportion of Vγ9 expressing cells was decreased in HIV patients, being lower in both blood and bronchoalveolar lavage specimens despite having increased Vδ1 cells (17), encouraged a closer look at the Vγ9Vδ2 subset. The importance of Vγ9Vδ2 T cell depletion and the relationship between depletion and TCR became more clear as other groups defined antigens for these unusual cells.
Finding the Antigens for Vδ2 T Cells
Major activities of Vγ9Vδ2 T cells include a strong response to Mycobacterium-infected human PBMC, even though these cells do not react to purified mycobacterial 65-kd heat shock protein (18). The Vγ9Vδ2 T cells respond to Mycobacteria-pulsed cells or antigens found on the human MOLT-4 lymphoma cell line (19), which was confusing in the context of the model for MHC-restriction that explained αβ T cell recognition of peptide antigens. Recognition of either Mycobacterium-pulsed cells or lymphoma cell lines required a specific rearrangement between the variable γ9 (Vγ9) and joining P (JP) segments; constant region 1 (C1) was incorporated by mRNA splicing to form Vγ9JPC1 chains that combine with a Vδ2DJC chains to form functional receptors (20, 21). In an alternate nomenclature, the V-J rearrangement is designated Vγ2Jγ1.2; the nomenclature is consistent for δ chains. This combination of specifically rearranged Vγ9 with Vδ2 chains endowed the capacity for recognizing mycobacterial antigens (22, 23). Efforts to characterize stimulatory molecules showed first they were phosphorylated (24) and later that they were prenyl pyrophosphate intermediates of sterol synthesis in mammalian cells (25). A variety of natural and non-natural molecules were tested for Vγ9Vδ2 T cell stimulation to define the essential antigenic structure (26) within a group of chemicals now known as phosphoantigens. The natural abundance of mammalian phosphoantigens, being made in every living cell, in addition to structurally similar but often more potent compounds produced by bacteria, protists, plants, or fungi, serves to inundate human physiology with stimulators of Vγ9Vδ2 T cells. These compounds select for and amplify the Vγ9JPVδ2 cell subset during early life as was described in a remarkable paper from Michael Brenner’s group (27).
In children, a fetal Vγ9 chain repertoire is replaced slowly with one dominated by the Vγ9JγP rearrangement. Under continuous positive selection, the Vγ9Vδ2 cell count rises and phosphoantigen-responsive cells are increasingly found in blood as central or effector memory types with declining proportions of naïve cells. Healthy adults maintain a diverse but highly redundant repertoire such that 1 in 40 circulating memory T cells is a phosphoantigen-responsive Vγ9Vδ2 T cell. Clearly, the Vγ9JPVδ2 cell dominates T cell memory in healthy adults. Baseline levels of Vγ9Vδ2 T cells differ by two to fourfold between white European-origin or Asian-origin (high) and African-origin (low) peoples but repertoire complexity is similar among these groups and in vitro responses to phosphoantigen are also similar (28). Positive selection and amplification of Vγ9JPVδ2 T cells is ubiquitous in man and present in most non-human primate species studied so far, but is not present in lower mammals including rodents that lack both a gamma chain gene similar to Vγ9 and butyrophilin 3A1 that is also required for phosphoantigen responses (29–34).
Specific Destruction of Antigen-Specific Vδ2 T Cells in HIV Disease
Two important papers in 1996 and 1997 helped to bridge HIV studies with the emerging understanding of phosphoantigens and their importance to γδ T cell biology. Gougeon’s group confirmed earlier studies on Vδ2 cell depletion in HIV patients and reported a disease-associated “functional anergy” measured by lack of proliferation or cytokine responses after stimulation with mycobacterial antigens (35). These authors studied the junctional diversity of Vγ9Vδ2 TCR chains expressed in HIV+ individuals and reported that the Vδ2 cell chain repertoire remained diverse. They also noted there were no differences in spontaneous apoptosis between HIV patients or uninfected control donors after in vitro phosphoantigen stimulation. A second group led by Malkovsky confirmed the functional anergy in Vδ2 T cells from HIV patients by documenting decreased responses to phosphoantigen or to the prototypical cell target Daudi B cell (36). Both groups noted that Vδ2 T cells were reduced but not eliminated in HIV disease, and were substantially deficient in their response to phosphoantigen due to anergy that may have resulted from inappropriate activation in vivo. A smaller study of HIV+ individuals noted differences from controls regarding γδ T cell responses to Salmonella typhimurium or Candida albicans, and reported that Vδ2 cell responses to Mycobacteria remained intact only in patients with >500 CD4 T cells/mm3 (37). Further, Vδ2 cells were depleted from blood but increased in liver from both HIV patients and HIV-negative patients who had disseminated Mycobacterium avium complex (38). Vδ1 cells were increased in tissue sites among HIV patients, notably liver (39) or bone marrow (40). The pattern of changes among γδ T cells for both Vδ2 and Vδ1 cells was a distinguishing feature of HIV disease.
Milestone Achievements from Early Studies on γδ T Cells in HIV Disease
By 1997, there was a basic understanding of HIV infection and its impact on γδ T cells. Four major concepts had emerged: (1) Inversion of the Vδ2:Vδ1 cell ratio was an early event, occurring prior to inversion of the CD4:CD8 T cell ratio. (2) Vδ1 cells are increased in patients with HIV. (3) The Vδ2 cell depletion was accompanied by decreased responsiveness to phosphoantigens or tumor cells. (4) Loss of Vδ2 cells was greatest in patients with low CD4+ T cells, high viremia, opportunistic infections and late stage disease (AIDS). Consequently, HIV-mediated changes in γδ T cells appear to be part of the mechanism for evading antiviral immunity and establishing persistent infection with chronic disease. Persistent infection is essential for viruses like HIV that are transmitted with relatively low efficiency and require direct person-to-person contact. These studies highlighted the need to understand mechanisms for γδ T cell dysregulation, define impacts of these changes on immunity to HIV and look more broadly at “unintended consequences” of the viral immune evasion strategy.
Mechanisms for Dysregulating γδ T Cells
Model studies in non-human primates have helped to explain some of the γδ T cell changes during disease. Because rodents lack the TCR sequences needed for phosphoantigen recognition, studies on Vγ9Vδ2 T cells have been restricted to human beings and non-human primates. A recent genome mining study revealed that functional genes for Vγ9Vδ2 and butyrophilin 3A1 were actually present in a few other placental mammals including alpaca, sloth, bottlenose dolphin, killer whale, horse, and armadillo (41). Most of these species are unfamiliar experimental systems with the exception of armadillo that has been used for research on Mycobacterium leprae, but we can expect humans and non-human primate models to dominate this field for the foreseeable future.
When peripheral blood Vγ9Vδ2 T cells were isolated from uninfected rhesus monkeys (naïve to viral antigens) they were directly cytotoxic for SIV-infected target cells (42). There were rapid increases in blood Vγ9Vδ2 T cell counts and higher expression of activation markers within a few days after SIV infection of rhesus monkeys even though these cells were already showing decreased proliferation responds in vitro (43). The Vγ9Vδ2 T cell expansion seen early after SIV infection of macaques was brief and was followed by a rapid decline in cell count and function. This animal model recapitulates the decline in Vγ9Vδ2 T cells and inversion of the Vδ2:Vδ1 cell ratio. Similarly, Vγ9Vδ2 cell lines or clones from HIV-negative donors recognized and killed HIV-infected cells (44). It was also known that healthy Vγ9Vδ2 cells produced large amounts of interferon-γ, tumor necrosis factor-α, and chemokines RANTES or MIP-1β (45) that were associated with antiviral immunity. Normally, the circulating Vγ9Vδ2 cells (mostly memory phenotype) have preformed cytoplasmic vesicles containing RANTES that are released immediately upon phosphoantigen stimulation or after contact with target cells (46). The release of RANTES and other chemokines suppressed HIV replication in vitro by blocking co-receptors for virus entry (47, 48). The CCR5 receptor is highly expressed on Vδ2 cells (49) meaning RANTES release would attract even more Vγ9Vδ2 cells able to release additional chemokine that would block HIV entry, kill already infected cells through direct cytotoxicity, or mediate antibody-dependent cellular cytotoxicity through cell surface Fc γ receptors (50). Circulating Vγ9Vδ2 cells also activate innate and acquired immunity through the release of pro-inflammatory IFN-γ and TNF-α or regulatory cytokines (51).
While it seemed clear that γδ T cell dysregulation is part of HIV immune evasion, the precise impact on pathogen immunity was less clear. Little was known about how Vδ1 expansion or Vδ2 T cell depletion affect normal immunity and whether these are important aspects of viral immune suppression linked to progressing disease. Brenchley’s group (7) reported that Vδ1 cell expansion in SIV-infected rhesus monkeys was related to pathologic changes in the intestinal epithelium that increased bacterial translocation causing higher levels of bacterial products in circulation that would stimulate Vδ1 T cells. The Vδ1 cells were also increased by influenza vaccination in HIV patients but only if the vaccine contained MF-59 adjuvant (52). Thus, Vδ1 cells are responsive to stimulation in HIV patients and expansion could be linked to bacterial translocation or reflect the normal responses to Candida albicans (53) and other common intercurrent infections in HIV patients.
Like Vδ1, phosphoantigen-responsive Vδ2 cells increase soon after infection as was documented in the SIV infection of macaques (43) but then are depleted and often extinguished. When cloned Vδ2 T cells are stimulated by anti-CD3 monoclonal antibody plus IL-2 they frequently die due to apoptosis, yet anti-CD3 stimulation plus feeder cells in the absence of exogenous IL-2 leads to proliferation (54). Thus, stimulation conditions impact outcome. It remains difficult to extrapolate in vitro conditions into explanations for in vivo outcomes and the mechanism for HIV-mediated depletion are still unclear. Laboratories working with Vδ2 T cells, including our groups, know that phosphoantigen plus IL-2 stimulation of human or macaque PBMC results in rapid cell death followed by outgrowth of a surviving population that peaks around 10–14 days later. The repertoire of Vγ9JPC1 chains is essentially unchanged in the expanded subset compared to fresh cells (55), despite the fact that stimulation indices for individual clones vary 10–100-fold (56). Surprisingly, there were no significant differences in apoptosis after in vitro stimulation comparing Vδ2 cells from HIV-infected patients or uninfected controls (35) but the preferential loss of phosphoantigen responses in HIV disease still argues for a direct link between antigen specificity and depletion, apparently a different type of activation-induced cell death.
Gene expression array studies provided a surprising insight into infection and γδ T cell dysregulation that may impact Vδ2 cells. During HIV infection, mRNA for enzymes in the cholesterol biosynthesis pathway is upregulated (57, 58). Higher production of cholesterol is needed to meet the demands for viral membrane synthesis and more of the biosynthetic intermediates are required including prenyl pyrophosphates (phosphoantigens). With more phosphoantigen present, it is reasonable to expect greater activation of Vγ9Vδ2 T cells and this may explain the rapid decreases during acute infection when viremia is highest and the frequency of infected cells is also highest compared to other stages of disease.
We also know (49, 59) that a large proportion of activated γδ T cells express chemokine receptors including CCR5 and CXCR4 that are major co-receptors for HIV (Figure 1). These receptors bind sequences in the V3 loop of envelope glycoprotein gp120 and are important for viral entry. Both Vδ2 and Vδ1 cells express α4β7 integrin that also binds the V2 loop of gp120 (60, 61). CCR5 expression was only seen in the Vδ2 subset where it was estimated to be present at >50,000 molecules per cell surface (62), or roughly 10-fold higher surface density compared to activated αβ CD4 T cells. Treating expanded Vδ2 cells with gp120 lead to induction/activation of caspases followed by apoptosis. Inhibitor studies confirmed that signaling through CCR5 and its associated p38 MAP kinase, was necessary for this cell death pathway (62). We also detected gp120 binding to Vδ1 cells that could be blocked by ligands for α4β7 but was not affected by the CCR5 antagonistic drug Maraviroc. Flow cytometry showed that Vδ1 cells have no detectable surface CCR5 (63). The presence of high density CCR5 on Vδ2 T cells and its absence on Vδ1 T cells may explain specific cell killing of Vδ2 cells by gp120 that is similar to the pattern seen during HIV infection. Since high density CCR5 expression occurs on phosphoantigen-stimulated cells, this mechanism might account for the connection between antigen specificity and cell loss.
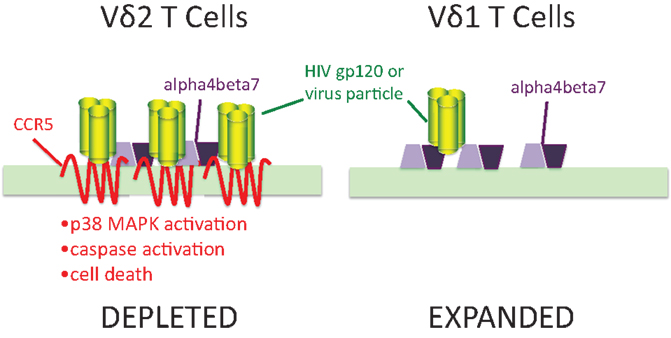
Figure 1. Human immunodeficiency virus envelopes gp120 signaling through CCR5 is a mechanism for specific depletion of Vδ2 T cells. Vδ2 T cells express high levels of two binding molecules for HIV gp120: chemokine receptor CCR5 and integrin α4β7. In Vδ1 cells, gp120 (green trimers) bind to α4β7 (purple polygons) but do not signal caspase activation or cause cell death. In Vδ2 T cells, the presence of 7-transmembrane CCR5 (red) binds gp120 and signals through its associated p38 MAP kinase to increase caspase activity and cell death. Adapted from Ref. (62,63).
Several other models have been proposed to explain the γδ T cell defect in HIV disease. We first reported that Vγ9Vδ2 T cells activated in vitro were permissive for HIV infection (36) but the number of productively infected cells in patient samples seems to be very low. However, it was reported that human herpes virus 6 elevates CD4 on several cell types including γδ T cells (64) and could impact susceptibility of these cells in vivo. Both direct infection of intrathymic γδ T cell precursors (65) and inhibition of thymic development by HIV-infected γδ T cells (66) were proposed as models for specific or general T cell depletion. However, it is important to note that Vδ1 cells are not depleted during HIV disease as would be expected for a mechanism acting at the level of thymopoiesis. In addition, the normal human blood repertoire includes Vγ9Vδ2 cells that use both the JP and other rearrangements even though we emphasize Vγ9JP because of its responses to phosphoantigens. Sequencing studies showed that Vγ9Vδ2 cells using J segments other than JP, remained at normal levels and appeared to be unchanged during HIV disease (14). Further, the rapid depletion of Vγ9Vδ2 T cells before the onset of immunodeficiency and reactivation of pathogens like HHV-6, argues this is not a major mechanism for depletion. The impact of direct infection, despite infrequent CD4 expression or negative effects on thymopoiesis may contribute to Vγ9Vδ2 cell depletion but are not likely to be the major mechanisms.
Based on studies of former plasma donors in southern China who were infected at the same time and with similar strains of HIV, we know that Vδ2 levels correlate with viremia (hence levels of gp120) but not with CD4 T cell count (67). This adds support to a model where viral proteins are directly responsible for Vδ2 cell depletion as was proposed earlier (11). During natural history studies of HIV disease it will be important to identify patients being treated with CCR5 antagonists including Maraviroc, who may have increased Vδ2 T cells compared to patients treated with regimens that do not include this drug class.
In order to understand better the dynamics of Vδ2 T cell depletion and the potential for immune reconstitution during therapy, extensive use was made of TCR repertoire analysis (Figure 2). The focus has been on the Vγ9 chain that is most tightly linked to phosphoantigen responsiveness due to the predominant Vγ9JP rearrangement (also called Vγ2Jγ1.2). Indeed, sequencing studies on the γ chain repertoire established the specificity of HIV-mediated deletion in the Vδ2 cell population (12) and subsequent work [reviewed in Ref. (68)] showed this TCR-specific defect is common to all HIV patients regardless of virus type, geographic distribution, race, or ethnicity. In the era before effective antiretroviral therapy, repertoire sequencing studies showed that patients with CD4 < 200 cells/mm3 had no detectable Vγ9JP cells in PBMC (14). When these patients initiated combination antiretroviral therapy and achieved virus suppression, we did not observe rapid rebound of Vδ2 cells in blood despite increases in CD4 cell count during 2–2.5 years of treatment. The lack of rapid Vδ2 cell recovery argues against the idea that depletion in peripheral blood is due to increased tissue compartmentalization, which is an important mechanism for controlling CD4 T cell count early in disease (69). A subsequent study of patients with longer intervals of antiretroviral therapy provided some initial evidence that Vγ9JP cells were reconstituted during virus suppression but the recovery rates were slow (13). More recently, studies with patients after >7 years of antiretroviral therapy revealed extensive reconstitution of the TCR cell repertoire (based on Vγ9 chain sequencing) and a population of Vγ9JP chains having nearly the complexity found in healthy, HIV-negative controls. Considering that some of the treated patients had nadir CD4 counts below 100 cells/mm3 and were expected to have no circulating Vγ9JP cells, reconstitution of this population was a surprising result (70).
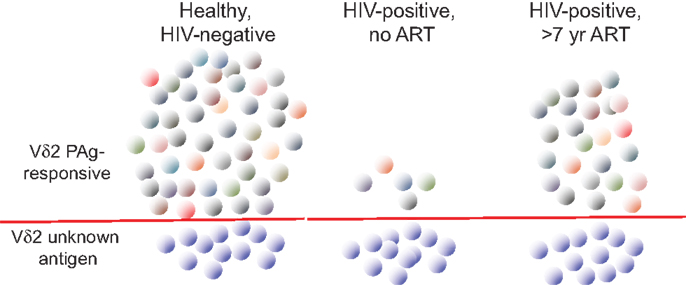
Figure 2. The impact of HIV infection on circulating Vγ9Vδ2 T cells. The characteristics of circulating Vγ9Vδ2 T cells (abbreviated Vδ2) are summarized for healthy, HIV-uninfected, and HIV-infected patients without antiretroviral therapy (ART) or after >7 years of ART. Individual cell clones are designated by colored symbols. Below the red line, we show the population of Vγ9Vδ2 T cells that do not respond to phosphoantigen (PAg) and remain at roughly equal numbers irrespective of infection or treatment. Without ART, HIV infection drives down the number and TCR complexity of PAg-responsive Vγ9Vδ2 (above the red line). After prolonged ART, TCR complexity recovers in the PAg-responding population (indicated by different colors) but the numbers and functions remain below uninfected controls. Adapted from Ref. (50, 70,74).
The γδ T cell repertoire is defined by analyzing the number and frequency of clonotypes (predicted peptide sequences in the CDR3 or V-J region of the gamma chain) or nucleotypes (nucleotide sequences for these peptides) within a sample of the total population of TCR sequences in a blood or tissue specimen. These sequences are classified as public (identical amino acid sequences present in unrelated donors) or private (unique to one individual in our population of donors). For healthy, HIV-negative adults most of the clonotypes are public and many of the nucleotypes are also public indicating a high degree of similarity among unrelated individuals. This pattern is consistent with the use of monomorphic antigen-presenting molecules and is probably biased by nucleotide sequences in the germline regions of Vγ9 and JP that provide alternate routes to expressing the “germline” Vγ9JP rearrangement (70) using the mechanism of convergent recombination (71–73). In long-term treated HIV patients, there were clear differences between uninfected controls or treated patients who reconstituted the Vγ9 repertoire as measured by clonotype and nucleotype abundance (70). These differences prove that the repertoire was reconstituted by new cell synthesis and not by regrowth of a cell population that survived an initial HIV attack. Further, the reconstituted TCR repertoire included many sequences that should respond to phosphoantigen. Despite having a reconstituted repertoire and TCR against phosphoantigen, Vδ2 cells in these patients remained at levels well below matched, HIV-negative controls and had greater proportions of naïve cells indicating a lack of phosphoantigen responses and impaired positive selection in vivo. Patients also had lower expression of the CD56 cell surface marker (74) that is associated with cytotoxic effector function (75). Vδ2 cells from these long-term treated patients could be activated by potent in vitro stimulation whereupon they regained Fc receptor expression (CD16) and effector function in antibody-dependent cellular cytotoxicity (50). Despite reconstitution of the Vδ2 T cell population by new cell synthesis during prolonged ART, this subset does not recover to normal levels or function after treatment intervals of years to decades. The majority of Vδ2 cells found in treated HIV patients were generated and selected in an environment almost devoid of HIV due to effective therapy. The nature of this long-term defect is an obstacle to immune reconstitution and might contribute to chronic comorbid diseases in HIV patients.
A very small fraction of HIV patients actually have normal Vδ2 cell levels and function; these are elite controllers or natural virus suppressors defined as HIV patients with undetectable viremia except for occasional blips and no history of antiretroviral therapy (76). Among natural virus suppressor patients (approximately 0.5% of all persons with HIV infection), Vδ2 cell levels are equivalent to age, gender, and race-matched uninfected controls but there are significant differences in the Vγ9 chain repertoire (77). These repertoire differences reflect an early impact of HIV before the time when virus replication was controlled by host immunity and in this way, are similar to what we observed in treated patients where viremia was suppressed by chemotherapy. This also tells us that normal function of Vγ9Vδ2 T cells can return in HIV-infected patients but as yet, we have not uncovered the critical mechanisms for recovery that might help us to find new therapeutic targets.
Milestone Achievements from the Middle Period of Studying γδ T Cells in HIV Disease
Nearly three decades of research produced many insights into HIV and γδ T cells. Studying HIV disease proved that Vγ9JPVδ2 cell depletion explained the defective response to phosphoantigen. Mechanisms were described for specific loss of Vγ9JPVδ2 cells that account for the relationship between depletion and phosphoantigen responsiveness. The findings showing that therapy reconstituted the Vγ9 repertoire but did not restore normal cell counts or function, were both encouraging and cautionary. Changes in γδ T cells are clearly related to HIV immune evasion but the long-term defects pose substantial challenges to the clinical management of HIV and efforts to reduce comorbid diseases.
Problem of Durable γδ T Cell Damage Despite Chemotherapeutic Suppression of HIV
Natural history studies of HIV infection documented a brief, violent interval of acute infection characterized by extraordinarily high viremia and dramatic changes in immune cell populations, which continued until a stable viral set-point (78) was established. We prefer the term “immune set-point” that better describes the balance between immune system destruction and chronic infection that allows HIV to persist in an individual or in the human population. The immune set-point includes damage to Vδ2 T cell function that goes beyond what is seen in acute, adult malaria (79) for example, where there was a more limited transient loss of Vγ9Vδ2 T cells.
Both viral and immunological set-points designate the phase of disease when acute HIV infection gives way to a stable, persistent infection with slower progression and more time for virus to spread by sexual contact. In some virus examples, the acute to persistent transition reflects changes in viral gene regulation but in HIV disease the most important mechanisms are viral evasion of host immunity. Once persistence is established, chronic disease progression causes death around 11 years later in untreated HIV disease, as declining CD4 T cell counts and collapsing innate immunity increase the incidence and severity of lethal opportunistic infections, cancer, cardiovascular disease, and other terminal conditions. Evolutionary selection of HIV created a pathogen capable of spreading in the human population by relatively inefficient sexual transmission even when the median life expectancy after infection was only 11 years for adults and shorter for infected infants or elderly persons. Now, in the era of effective ART and much longer survival times, we are seeing a new scenario that was not part of the selection pressures driving natural evolution of HIV. Therapy with effective virus suppression repairs some of the earlier immune damage and likely reduces transmission rates (80) but key cell types including γδ T cells fail to recover normal numbers, phenotypes, or function, even after prolonged treatment that also fails to eradicate the virus. These enduring defects and their impacts on durable virus persistence or comorbidities of HIV are unanticipated – or better said – unselected consequences of HIV biology that are continuing decades after a viral immune evasion mechanism that caused damage to host immunity during the interval of acute infection.
Why these defects linger is a critical and unsolved question. We know that low numbers of viral RNA remain in plasma even during successful antiretroviral therapy (81) but these levels are around 105 vRNA copies per ml of plasma lower that what is found during acute viremia. Accordingly, when the γ chain repertoire is reconstituted by new cell synthesis (70), we are dealing with cells that were birthed in an environment nearly devoid of HIV replication. Reconstitution is indeed slow as was predicted several years ago (13), but the return of function lags behind the gain of TCR repertoire complexity and as of now, there is no reliable estimate for the kinetics of functional recovery.
The durable impact of HIV on γδ T cells impedes their contributions to key immune effector and regulatory mechanisms. Relating the list of possible γδ T cell functions to the known outcomes of HIV disease is a challenging undertaking and will trigger much debate. Our list of relevant Vδ2 T cell functions (Figure 3) includes control of antiviral immunity (47), tumor or infected cell killing (82, 83), antigen presentation (84), B helper T cell function (85), and costimulation of NK cells (86, 87) although alternate lists with additional functions can be imagined. In this review, we focus on the impact of losing Vδ2 T cell costimulation and why this loss can be an important underlying factor in HIV-associated comorbid diseases that are causing death and disability in persons despite effective viral suppression by ART.

Figure 3. Some of the known functions for human Vδ2 T cells that are related to HIV infection. Naïve Vδ2 T cells can be stimulated to produce Type 1 cytokines/chemokines for antiviral immunity (47) or display potent lytic effect function against tumor cells (82, 83) or infected cells (47). Vδ2 T cells also provide and receive costimulation from both NK cells (86) and dendritic cells (87). They display some similarity to T follicular helper cells required for B cell responses (85) and can function as antigen-presenting cells (84).
The consequences of prolonged depression in Vδ2 T cell function may appear in unexpected comorbidities of HIV disease. For example, psoriasis affects 1–3% of patients with HIV (88), which seems paradoxical since this is a T cell-mediated autoimmune disorder (89). Normally, psoriasis is associated with influx of blood Vγ9Vδ2 T cells into the inflamed skin that may be a mechanism for resolving the acute condition. In HIV disease, we can imagine that the absence of Vγ9Vδ2 T cells or their chronically poor responses in patients treated with antiretroviral therapy will reduce their beneficial effect and increase the severity or duration of psoriasis in these patients. The Vγ9Vδ2 T cells are intimately related to both NK and dendritic cells through interactions that control cell activation and inflammation. Psoriasis is an example where the loss of Vγ9Vδ2 function removes a key protective mechanism.
Cell:cell interactions (cross-talk) involving γδ T cells were implicated in the activation of dendritic cells (90), enhancement of antibody production by γδ T cells with T follicular helper cell activity (85) and antigen presentation or cross presentation (84). An additional and more specific example of cross-talk involved NK tumor cytotoxicity, which was activated by Vδ2 T cells (86). The critical role for Vδ2 T cells in NK tumor cytotoxicity was mapped to expression of the 4-1BB ligand (CD137L). Phosphoantigen stimulation of human PBMC expanded the Vδ2 subset and upregulated both 4-1BB (costimulatory receptor) and NKG2D (activating receptor that recognizes stressed-self antigens) on the NK cells (Figure 4). A murine cell line expressing 4-1BBL could be substituted for Vδ2+ T cells indicating that this costimulatory ligand was the major signal for Vδ2 T cell enhancement of NK tumor cytotoxicity (86). This surprising result linked the anti-tumor effector activity of NK cells to the presence of phosphoantigens and activation of Vδ2 T cells.
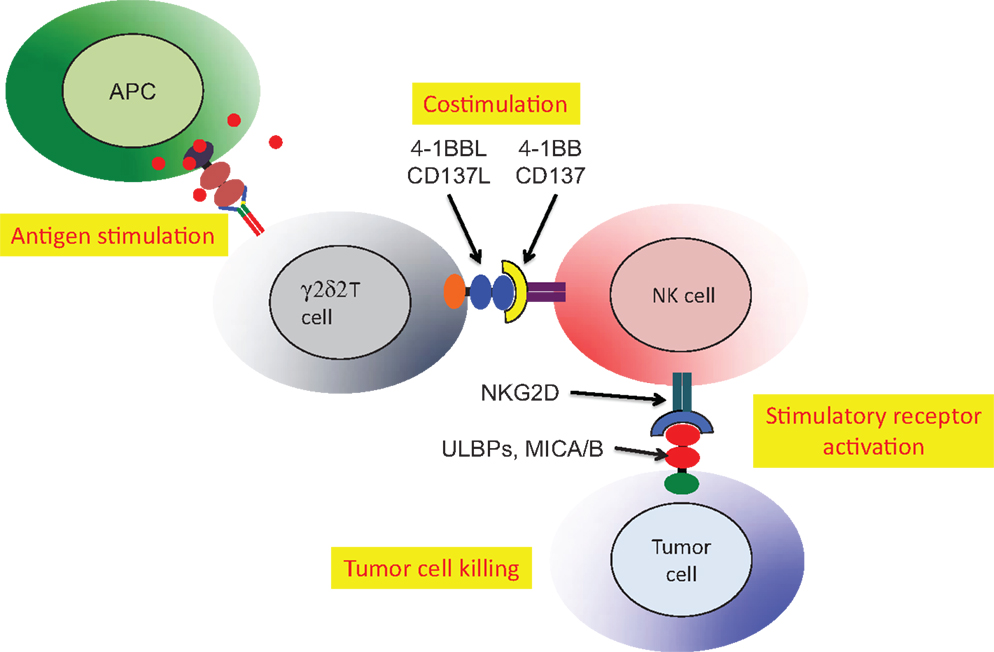
Figure 4. Stimulated Vδ2 T cells co-stimulate NK to increase tumor cell cytolysis. Activated Vδ2 T cells express 4-1BBL that binds 4-1BB on NK cells. The NKG2D receptor on NK is upregulated allowing stronger recognition of tumor cells expressing self-stress ligands ULBP or MIC A/B (86). HIV-mediated destruction of Vδ2 T cells will cripple the important mechanism for NK costimulation and reduce natural tumor surveillance.
A second example of Vδ2 T cell costimulation of NK is enhancement of effector activity capable of eliminating immature or mature, antigen-presenting dendritic cells (Figure 5). Again, phosphoantigen-expanded Vδ2 T cells delivered a costimulatory signal to NK that increased the killing of autologous dendritic cells. Along with increased production of Type 1 cytokines and chemokines, the increased levels of ICOS on Vδ2 T cells binding to ICOSL on NK cells was related to improving cytotoxic killing of dendritic cells (87). The Vδ2 T cells are reciprocally activated by the cross-talk with NK and may also be effectors for dendritic cell killing, but these experiments are complicated by the use of anti-TCR or anti-CD3 antibodies to purify Vδ2 cells. When antibodies are used for positive selection, Vδ2 T cells are super-activated and the effects of costimulation are obscured. These studies on costimulation support an earlier model for NK:γδ:DC interactions that focused on γδ T cell cytokines that regulate NK and DC function (91). In this example, we see a key role for antigen-specific Vδ2 T cells in costimulation of NK cells that will then gain the capacity for killing activated dendritic cells and help to control chronic inflammation.
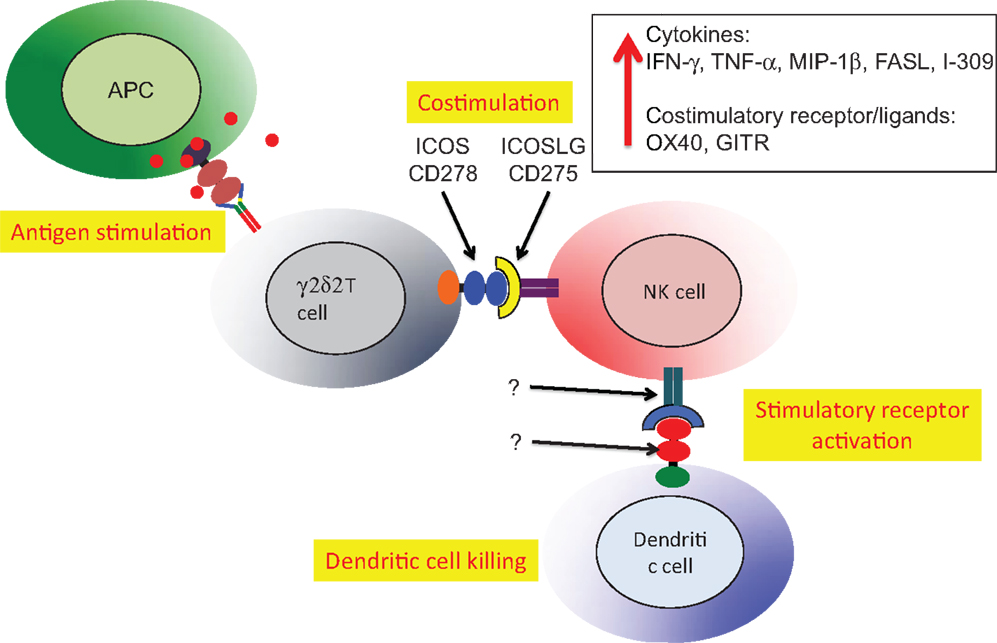
Figure 5. Vδ2 costimulation of NK cells increases lytic effector activity against autologous dendritic cells. The Vδ2 and NK cells interact through ICOS/ICOSL interactions resulting in upregulation of cytokine expression, increased mRNA for several other costimulatory receptors and increase lytic effector activity against dendritic cells (87). The relevant receptor(s) on NK and recognition molecules on dendritic cells have not been identified. Similar to the example in Figure 4, HIV-mediated depletion of Vδ2 is expected to reduce the capacity for NK killing of dendritic cells that will accumulate and promote chronic immune activation/inflammation that is a hallmark of HIV disease.
When Vδ2 T cell costimulation of NK cells is lost we might expect increased cancer risk among HIV patients, despite effective viral suppression by therapy. In HIV clinics of the University of Maryland, Baltimore, more than 15% of all HIV patients will have a cancer diagnosis and in 85% of those cases, cancer will be the cause of death (92). The great variety of cancers that increase in HIV patients suggests that a common mechanism of tumor control is missing or ineffective. Decreased tumor surveillance by NK cells may contribute to this comorbidity of HIV-associated cancer and the loss of Vδ2 T cell costimulation could be a key mechanism for the loss of effector function.
Vδ2 T cell depletion, loss of costimulation, and the consequent reduction in dendritic cell killing may have important effects on several HIV-associated comorbidities. Chronic immune activation with inflammation is a hallmark of HIV disease and a danger to patients even after virus replication is suppressed. The lingering defect in Vδ2 T cells and the linked failure to co-stimulate NK for dendritic cell killing, likely increases the risk for chronic activation/inflammation as potent antigen-presenting cells accumulate in the absence of normal control mechanisms. Indeed, Fauci’s group noted the lack of potent dendritic cell editing by NK in HIV-infected and treated patients (93) and defects in plasmacytoid dendritic cell–NK cross-talk in HIV patient specimens that reflected poor activation of NK during innate immune responses (94). Vδ2 T cells are also involved in reciprocal interactions with dendritic cells that affect activation or maturation of both cell types (90). Consequently, dysfunction of Vδ2 cells will affect NK and dendritic cell interactions as all three cell types are interdependent. When all three cell types are normal, there is a balance between cell activation and cell killing that modulates inflammation. When one piece is missing, in this case Vδ2 T cells, the control over inflammation is lost.
The examples for defective Vδ2 T cells in HIV-infected and treated patients have important parallels in the NK literature. There are many reports about the phenotype of circulating NK cells in patients where HIV is controlled by antiretroviral therapy. In these patients, NK have lower density of NKp46, NKp30, and NKp44 receptors (95) and often appear as CD56−/CD16+ cells lacking cytotoxic effector activity (96). Because HIV infection is associated with decreased NKG2D (activating) and increased NKG2A (inhibiting) receptor expression (97), even the increased levels of ULBP target ligands failed to trigger cytolysis of HIV-infected target cells. The earliest mark of NK dysfunction seems to be the decreased expression of Siglec-7 (98). When the lack of Siglec-7 is combined with lower expression of CD56 (99), we are beginning to define the phenotype of defective NK cells that is common among HIV patients. Whether these defects could be corrected by costimulation has not yet been tested. Clearly, there is a convergence of functional defects among Vδ2, NK, and dendritic cells in HIV and potent antiretroviral therapy does not restore the normal cell interactions or function of this regulatory triangle. It is worthwhile to look for defects in multiple mechanisms of immunity including those highlighted in Figure 3.
With highly effective antiretroviral therapy and increasing interest in eradicating HIV, it is important to look at the future of research on γδ T cells. Surely, Vδ2 T cells provide an attractive target for immunotherapy, probably using aminobisphosphonate compounds plus IL-2 or IL-15 to increase cell levels and functions. Patterned on a number of ongoing clinical studies in cancer (100–105), aminobisphosphonate drugs may be useful for correcting the HIV-associated Vδ2 T cell defect. A pilot study showed this approach was safe for HIV patients (106) but these studies have not been repeated or expanded to include clinical and immunological endpoints such as NK or dendritic cell phenotype and function. The lack of progress may be due to insufficient justification for the outcome of bisphosphonate/IL-2 therapy, since viremia can be suppressed more easily with once-a-day antiretroviral agents.
Plea for Innovative Clinical Trials Targeting γδ T Cells in HIV Disease
Clinical research on therapeutic restoration of γδ T cells in HIV disease will require innovative clinical trial strategies. It may be difficult to launch therapeutic interventions such as aminobisphosphonate plus IL-2 for 2 or 3 months, then wait 20 years or more to accumulate significant data on changing rates of pulmonary arterial hypertension (found in <5% of HIV patients) or other comorbidities. While each of the comorbid conditions affects only a portion of the HIV population, their combined impact is substantial but clinical trials rarely lump disparate conditions into single endpoints. We might overcome this problem by using immunological endpoints that are related to chronic disease mechanisms. For example, direct immunotherapy targeting γδ T cells might be tested as a way to activate Vδ2 T cells that would normalize NK cell phenotype and function. Recovering of normal properties in two distinct lymphocyte subsets may be sufficient to document a clinical benefit, even though neither change is likely to result in short term changes to the incidence, prevalence, or severity of comorbid diseases.
Summary
The Vγ9Vδ2 T cells in HIV-infected individuals contain the historical record of disease, documenting the intensity of initial depletion and reconstitution during therapy by their abundance, activity, and sequences of their TCR. They are barometers for current disease status, reflecting the capacity for resisting opportunistic infections, natural tumor surveillance, the control of immune activation/inflammation, and likely several other conditions critical to the clinical management of HIV disease. In addition, reconstitution of the TCR repertoire may be a measure for treatment success and help to identify patients with the highest potential response to immunotherapies targeted at Vδ2 T cell activation. Their compact TCR repertoire and extraordinarily high proportion of public Vγ9 chains facilitates comparisons among individuals or groups of patients. When combined with the sophisticated knowledge about cell surface marker expression including costimulatory receptors/ligands, and their patterns of cytokine/chemokine expression, these cells become critical tools for the immunology of infectious diseases. Adding to the interest is the availability of approved drugs that activate Vγ9Vδ2 T cells in vivo with high specificity, and a rich pipeline of compounds under development that may do even better. The missing link is a commitment to small interventional clinical trials testing the capacity for γδ T cell-targeted immunotherapies to alter the profile of immunity and recover normal control in diseases like HIV/AIDS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the many participants in research protocols around the world for donating specimens needed to study gamma delta T cells and HIV disease. We also thank our many colleagues, friends, and laboratory members present or past who contributed ideas and criticism that have shaped our views. We are particularly grateful to Miroslav Malkovsky for first encouraging us to study γδ T cells in HIV disease and to Maria S. Salvato for editing an early version of this review and providing invaluable insights into many viral disease models. This work was funded in part by Public Health Service grants CA142458, AI096949 and AI102680 (C. David Pauza), AI110229 (Bhawna Poonia), AI104702 (Cristiana Cairo), and support to Haishan Li from the Creative and Novel Ideas in HIV Research Program through a supplement to the University of California at San Francisco (UCSF) Center For AIDS Research P30AI027763.
References
1. Moir S, Fauci AS. B-cell exhaustion in HIV infection: the role of immune activation. Curr Opin HIV AIDS (2014) 9:472–7. doi:10.1097/COH.0000000000000092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Kottilil S, Jackson JO, Reitano KN, O’shea MA, Roby G, Lloyd M, et al. Innate immunity in HIV infection: enhanced susceptibility to CD95-mediated natural killer cell death and turnover induced by HIV viremia. J Acquir Immune Defic Syndr (2007) 46:151–9. doi:10.1097/QAI.0b013e3180dc9909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Autran B, Triebel F, Katlama C, Rozenbaum W, Hercend T, Debre P. T cell receptor gamma/delta+ lymphocyte subsets during HIV infection. Clin Exp Immunol (1989) 75:206–10.
4. De Paoli P, Gennari D, Martelli P, Basaglia G, Crovatto M, Battistin S, et al. A subset of gamma delta lymphocytes is increased during HIV-1 infection. Clin Exp Immunol (1991) 83:187–91. doi:10.1111/j.1365-2249.1991.tb05612.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Margolick JB, Scott ER, Odaka N, Saah AJ. Flow cytometric analysis of gamma delta T cells and natural killer cells in HIV-1 infection. Clin Immunol Immunopathol (1991) 58:126–38. doi:10.1016/0090-1229(91)90154-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. De Maria A, Ferrazin A, Ferrini S, Ciccone E, Terragna A, Moretta L. Selective increase of a subset of T cell receptor gamma delta T lymphocytes in the peripheral blood of patients with human immunodeficiency virus type 1 infection. J Infect Dis (1992) 165:917–9. doi:10.1093/infdis/165.5.917
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Harris LD, Klatt NR, Vinton C, Briant JA, Tabb B, Ladell K, et al. Mechanisms underlying gammadelta T-cell subset perturbations in SIV-infected Asian rhesus macaques. Blood (2010) 116:4148–57. doi:10.1182/blood-2010-05-283549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Hudspeth K, Fogli M, Correia DV, Mikulak J, Roberto A, Della Bella S, et al. Engagement of NKp30 on Vdelta1 T cells induces the production of CCL3, CCL4, and CCL5 and suppresses HIV-1 replication. Blood (2012) 119:4013–6. doi:10.1182/blood-2011-11-390153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Fausther-Bovendo H, Wauquier N, Cherfils-Vicini J, Cremer I, Debre P, Vieillard V. NKG2C is a major triggering receptor involved in the V[delta]1 T cell-mediated cytotoxicity against HIV-infected CD4 T cells. AIDS (2008) 22:217–26. doi:10.1097/QAD.0b013e3282f46e7c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Sindhu ST, Ahmad R, Morisset R, Ahmad A, Menezes J. Peripheral blood cytotoxic gammadelta T lymphocytes from patients with human immunodeficiency virus type 1 infection and AIDS lyse uninfected CD4+ T cells, and their cytocidal potential correlates with viral load. J Virol (2003) 77:1848–55. doi:10.1128/JVI.77.3.1848-1855.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Hermier F, Comby E, Delaunay A, Petitjean J, Favennec L, Bazin C, et al. Decreased blood TcR gamma delta+ lymphocytes in AIDS and p24-antigenemic HIV-1-infected patients. Clin Immunol Immunopathol (1993) 69:248–50. doi:10.1006/clin.1993.1176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Enders PJ, Yin C, Martini F, Evans PS, Propp N, Poccia F, et al. HIV-mediated gammadelta T cell depletion is specific for Vgamma2+ cells expressing the Jgamma1.2 segment. AIDS Res Hum Retroviruses (2003) 19:21–9. doi:10.1089/08892220360473934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Bordon J, Evans PS, Propp N, Davis CE Jr, Redfield RR, Pauza CD. Association between longer duration of HIV-suppressive therapy and partial recovery of the V gamma 2 T cell receptor repertoire. J Infect Dis (2004) 189:1482–6. doi:10.1086/382961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Hebbeler AM, Propp N, Cairo C, Li H, Cummings JS, Jacobson LP, et al. Failure to restore the Vgamma2-Jgamma1.2 repertoire in HIV-infected men receiving highly active antiretroviral therapy (HAART). Clin Immunol (2008) 128:349–57. doi:10.1016/j.clim.2008.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Boullier S, Cochet M, Poccia F, Gougeon ML. CDR3-independent gamma delta V delta 1+ T cell expansion in the peripheral blood of HIV-infected persons. J Immunol (1995) 154:1418–31.
16. Hinz T, Wesch D, Friese K, Reckziegel A, Arden B, Kabelitz D. T cell receptor gamma delta repertoire in HIV-1-infected individuals. Eur J Immunol (1994) 24:3044–9. doi:10.1002/eji.1830241219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Agostini C, Zambello R, Trentin L, Cerutti A, Bulian P, Crivellaro C, et al. Gamma delta T cell receptor subsets in the lung of patients with HIV-1 infection. Cell Immunol (1994) 153:194–205. doi:10.1006/cimm.1994.1017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Kabelitz D, Bender A, Schondelmaier S, Schoel B, Kaufmann SH. A large fraction of human peripheral blood gamma/delta + T cells is activated by Mycobacterium tuberculosis but not by its 65-kD heat shock protein. J Exp Med (1990) 171:667–79. doi:10.1084/jem.171.3.667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. De Libero G, Casorati G, Giachino C, Carbonara C, Migone N, Matzinger P, et al. Selection by two powerful antigens may account for the presence of the major population of human peripheral gamma/delta T cells. J Exp Med (1991) 173:1311–22. doi:10.1084/jem.173.6.1311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Davodeau F, Peyrat MA, Hallet MM, Gaschet J, Houde I, Vivien R, et al. Close correlation between Daudi and mycobacterial antigen recognition by human gamma delta T cells and expression of V9JPC1 gamma/V2DJC delta-encoded T cell receptors. J Immunol (1993) 151:1214–23.
21. Davodeau F, Peyrat MA, Hallet MM, Houde I, Vie H, Bonneville M. Peripheral selection of antigen receptor junctional features in a major human gamma delta subset. Eur J Immunol (1993) 23:804–8. doi:10.1002/eji.1830230405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Constant P, Davodeau F, Peyrat MA, Poquet Y, Puzo G, Bonneville M, et al. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science (1994) 264:267–70. doi:10.1126/science.8146660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Schoel B, Sprenger S, Kaufmann SH. Phosphate is essential for stimulation of V gamma 9V delta 2 T lymphocytes by mycobacterial low molecular weight ligand. Eur J Immunol (1994) 24:1886–92. doi:10.1002/eji.1830240826
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Burk MR, Mori L, De Libero G. Human V gamma 9-V delta 2 cells are stimulated in a cross-reactive fashion by a variety of phosphorylated metabolites. Eur J Immunol (1995) 25:2052–8. doi:10.1002/eji.1830250737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Morita CT, Beckman EM, Bukowski JF, Tanaka Y, Band H, Bloom BR, et al. Direct presentation of nonpeptide prenyl pyrophosphate antigens to human gamma delta T cells. Immunity (1995) 3:495–507. doi:10.1016/1074-7613(95)90178-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature (1995) 375:155–8. doi:10.1038/375155a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Parker CM, Groh V, Band H, Porcelli SA, Morita C, Fabbi M, et al. Evidence for extrathymic changes in the T cell receptor gamma/delta repertoire. J Exp Med (1990) 171:1597–612. doi:10.1084/jem.171.5.1597
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Cairo C, Armstrong CL, Cummings JS, Deetz CO, Tan M, Lu C, et al. Impact of age, gender, and race on circulating gammadelta T cells. Hum Immunol (2010) 71:968–75. doi:10.1016/j.humimm.2010.06.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gammadelta T-cell subset. Blood (2012) 120:2269–79. doi:10.1182/blood-2012-05-430470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Palakodeti A, Sandstrom A, Sundaresan L, Harly C, Nedellec S, Olive D, et al. The molecular basis for modulation of human Vgamma9Vdelta2 T cell responses by CD277/butyrophilin-3 (BTN3A)-specific antibodies. J Biol Chem (2012) 287:32780–90. doi:10.1074/jbc.M112.384354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Vavassori S, Kumar A, Wan GS, Ramanjaneyulu GS, Cavallari M, El Daker S, et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human gammadelta T cells. Nat Immunol (2013) 14:908–16. doi:10.1038/ni.2665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Wang H, Henry O, Distefano MD, Wang YC, Raikkonen J, Monkkonen J, et al. Butyrophilin 3A1 plays an essential role in prenyl pyrophosphate stimulation of human Vgamma2Vdelta2 T cells. J Immunol (2013) 191:1029–42. doi:10.4049/jimmunol.1300658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Decaup E, Duault C, Bezombes C, Poupot M, Savina A, Olive D, et al. Phosphoantigens and butyrophilin 3A1 induce similar intracellular activation signaling in human TCRVgamma9+ gammadelta T lymphocytes. Immunol Lett (2014) 161:133–7. doi:10.1016/j.imlet.2014.05.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Sandstrom A, Peigne CM, Leger A, Crooks JE, Konczak F, Gesnel MC, et al. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vgamma9Vdelta2 T cells. Immunity (2014) 40:490–500. doi:10.1016/j.immuni.2014.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Poccia F, Boullier S, Lecoeur H, Cochet M, Poquet Y, Colizzi V, et al. Peripheral V gamma 9/V delta 2 T cell deletion and anergy to nonpeptidic mycobacterial antigens in asymptomatic HIV-1-infected persons. J Immunol (1996) 157:449–61.
36. Wallace M, Scharko AM, Pauza CD, Fisch P, Imaoka K, Kawabata S, et al. Functional gamma delta T-lymphocyte defect associated with human immunodeficiency virus infections. Mol Med (1997) 3:60–71.
37. Chervenak KA, Lederman MM, Boom WH. Bacterial antigen activation of Vdelta1 and Vdelta2 gammadelta T cells of persons infected with human immunodeficiency virus type 1. J Infect Dis (1997) 175:429–33. doi:10.1093/infdis/175.2.429
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Grottrup-Wolfers E, Grunewald T, Strzelecki R, Pohle HD, Ruf B. Selective expansion of gammadelta T cells among liver-derived lymphocytes of AIDS patients with disseminated Mycobacterium avium complex infection. Clin Immunol Immunopathol (1997) 85:151–7. doi:10.1006/clin.1997.4410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Agrati C, D’offizi G, Narciso P, Selva C, Pucillo LP, Ippolito G, et al. Gammadelta T cell activation by chronic HIV infection may contribute to intrahepatic vdelta1 compartmentalization and hepatitis C virus disease progression independent of highly active antiretroviral therapy. AIDS Res Hum Retroviruses (2001) 17:1357–63. doi:10.1089/08892220152596614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Rossol R, Dobmeyer JM, Dobmeyer TS, Klein SA, Rossol S, Wesch D, et al. Increase in Vdelta1+ gammadelta T cells in the peripheral blood and bone marrow as a selective feature of HIV-1 but not other virus infections. Br J Haematol (1998) 100:728–34. doi:10.1046/j.1365-2141.1998.00630.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Karunakaran MM, Gobel TW, Starick L, Walter L, Herrmann T. Vgamma9 and Vdelta2 T cell antigen receptor genes and butyrophilin 3 (BTN3) emerged with placental mammals and are concomitantly preserved in selected species like alpaca (Vicugna pacos). Immunogenetics (2014) 66:243–54. doi:10.1007/s00251-014-0763-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Wallace M, Gan YH, Pauza CD, Malkovsky M. Antiviral activity of primate gamma delta T lymphocytes isolated by magnetic cell sorting. J Med Primatol (1994) 23:131–5. doi:10.1111/j.1600-0684.1994.tb00113.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Gan YH, Pauza CD, Malkovsky M. Gamma delta T cells in rhesus monkeys and their response to simian immunodeficiency virus (SIV) infection. Clin Exp Immunol (1995) 102:251–5. doi:10.1111/j.1365-2249.1995.tb03773.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Wallace M, Bartz SR, Chang WL, Mackenzie DA, Pauza CD, Malkovsky M. Gamma delta T lymphocyte responses to HIV. Clin Exp Immunol (1996) 103:177–84. doi:10.1046/j.1365-2249.1996.d01-625.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Boismenu R, Feng L, Xia YY, Chang JC, Havran WL. Chemokine expression by intraepithelial gamma delta T cells. Implications for the recruitment of inflammatory cells to damaged epithelia. J Immunol (1996) 157:985–92.
46. Tikhonov I, Deetz CO, Paca R, Berg S, Lukyanenko V, Lim JK, et al. Human Vgamma2Vdelta2 T cells contain cytoplasmic RANTES. Int Immunol (2006) 18:1243–51. doi:10.1093/intimm/dxl055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Poccia F, Battistini L, Cipriani B, Mancino G, Martini F, Gougeon ML, et al. Phosphoantigen-reactive Vgamma9Vdelta2 T lymphocytes suppress in vitro human immunodeficiency virus type 1 replication by cell-released antiviral factors including CC chemokines. J Infect Dis (1999) 180:858–61. doi:10.1086/314925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Lehner T, Mitchell E, Bergmeier L, Singh M, Spallek R, Cranage M, et al. The role of gammadelta T cells in generating antiviral factors and beta-chemokines in protection against mucosal simian immunodeficiency virus infection. Eur J Immunol (2000) 30:2245–56. doi:10.1002/1521-4141(2000)30:8<2245::AID-IMMU2245>3.0.CO;2-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Glatzel A, Wesch D, Schiemann F, Brandt E, Janssen O, Kabelitz D. Patterns of chemokine receptor expression on peripheral blood gamma delta T lymphocytes: strong expression of CCR5 is a selective feature of V delta 2/V gamma 9 gamma delta T cells. J Immunol (2002) 168:4920–9. doi:10.4049/jimmunol.168.10.4920
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Poonia B, Pauza CD. Gamma delta T cells from HIV+ donors can be expanded in vitro by zoledronate/interleukin-2 to become cytotoxic effectors for antibody-dependent cellular cytotoxicity. Cytotherapy (2012) 14:173–81. doi:10.3109/14653249.2011.623693
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Biswas P, Ferrarini M, Mantelli B, Fortis C, Poli G, Lazzarin A, et al. Double-edged effect of Vgamma9/Vdelta2 T lymphocytes on viral expression in an in vitro model of HIV-1/mycobacteria co-infection. Eur J Immunol (2003) 33:252–63. doi:10.1002/immu.200390028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Fenoglio D, Zocchi MR, Parodi A, Durando P, Gabutitp G, Gasparini R, et al. MF-59 adjuvant influence on the functions of gammadelta T cells in HIV-1+ adults immunized with influenza seasonal vaccine. J Prev Med Hyg (2011) 52:137–41.
53. Fenoglio D, Poggi A, Catellani S, Battaglia F, Ferrera A, Setti M, et al. Vdelta1 T lymphocytes producing IFN-gamma and IL-17 are expanded in HIV-1-infected patients and respond to Candida albicans. Blood (2009) 113:6611–8. doi:10.1182/blood-2009-01-198028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Kabelitz D, Pechhold K, Bender A, Wesselborg S, Wesch D, Friese K, et al. Activation and activation-driven death of human gamma/delta T cells. Immunol Rev (1991) 120:71–88. doi:10.1111/j.1600-065X.1991.tb00588.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Evans PS, Enders PJ, Yin C, Ruckwardt TJ, Malkovsky M, Pauza CD. In vitro stimulation with a non-peptidic alkylphosphate expands cells expressing Vgamma2-Jgamma1.2/Vdelta2 T-cell receptors. Immunology (2001) 104:19–27. doi:10.1046/j.1365-2567.2001.01282.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Hebbeler AM, Cairo C, Cummings JS, Pauza CD. Individual Vgamma2-Jgamma1.2+ T cells respond to both isopentenyl pyrophosphate and Daudi cell stimulation: generating tumor effectors with low molecular weight phosphoantigens. Cancer Immunol Immunother (2007) 56:819–29. doi:10.1007/s00262-006-0235-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. van ‘t Wout AB, Lehrman GK, Mikheeva SA, O’keeffe GC, Katze MG, Bumgarner RE, et al. Cellular gene expression upon human immunodeficiency virus type 1 infection of CD4(+)-T-cell lines. J Virol (2003) 77:1392–402. doi:10.1128/JVI.77.2.1392-1402.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. van ‘t Wout AB, Swain JV, Schindler M, Rao U, Pathmajeyan MS, Mullins JI, et al. Nef induces multiple genes involved in cholesterol synthesis and uptake in human immunodeficiency virus type 1-infected T cells. J Virol (2005) 79:10053–8. doi:10.1128/JVI.79.15.10053-10058.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Imlach S, Leen C, Bell JE, Simmonds P. Phenotypic analysis of peripheral blood gammadelta T lymphocytes and their targeting by human immunodeficiency virus type 1 in vivo. Virology (2003) 305:415–27. doi:10.1006/viro.2002.1759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Arthos J, Cicala C, Martinelli E, Macleod K, Van Ryk D, Wei D, et al. HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nat Immunol (2008) 9:301–9. doi:10.1038/ni1566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Cicala C, Martinelli E, Mcnally JP, Goode DJ, Gopaul R, Hiatt J, et al. The integrin alpha4beta7 forms a complex with cell-surface CD4 and defines a T-cell subset that is highly susceptible to infection by HIV-1. Proc Natl Acad Sci U S A (2009) 106:20877–82. doi:10.1073/pnas.0911796106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Li H, Pauza CD. HIV envelope-mediated, CCR5/alpha4beta7-dependent killing of CD4-negative gammadelta T cells which are lost during progression to AIDS. Blood (2011) 118:5824–31. doi:10.1182/blood-2011-05-356535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Li H, Pauza CD. The alpha4beta7 integrin binds HIV envelope but does not mediate bystander killing of gammadelta T cells. Blood (2012) 120:698–9. doi:10.1182/blood-2012-03-420117
64. Biswas P, Mantelli B, Sica A, Malnati M, Panzeri C, Saccani A, et al. Expression of CD4 on human peripheral blood neutrophils. Blood (2003) 101:4452–6. doi:10.1182/blood-2002-10-3056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Schnittman SM, Denning SM, Greenhouse JJ, Justement JS, Baseler M, Kurtzberg J, et al. Evidence for susceptibility of intrathymic T-cell precursors and their progeny carrying T-cell antigen receptor phenotypes TCR alpha beta + and TCR gamma delta + to human immunodeficiency virus infection: a mechanism for CD4+ (T4) lymphocyte depletion. Proc Natl Acad Sci U S A (1990) 87:7727–31. doi:10.1073/pnas.87.19.7727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Gurney KB, Yang OO, Wilson SB, Uittenbogaart CH. TCR gamma delta+ and CD161+ thymocytes express HIV-1 in the SCID-hu mouse, potentially contributing to immune dysfunction in HIV infection. J Immunol (2002) 169:5338–46. doi:10.4049/jimmunol.169.9.5338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Li H, Peng H, Ma P, Ruan Y, Su B, Ding X, et al. Association between Vgamma2Vdelta2 T cells and disease progression after infection with closely related strains of HIV in China. Clin Infect Dis (2008) 46:1466–72. doi:10.1086/587107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Li H, Chaudhry S, Poonia B, Shao Y, Pauza CD. Depletion and dysfunction of Vgamma2Vdelta2 T cells in HIV disease: mechanisms, impacts and therapeutic implications. Cell Mol Immunol (2013) 10:42–9. doi:10.1038/cmi.2012.50
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Schenkel AR, Uno H, Pauza CD. Asymptomatic simian immunodeficiency virus infection decreases blood CD4(+) T cells by accumulating recirculating lymphocytes in the lymphoid tissues. J Virol (1999) 73:601–7.
70. Chaudhry S, Cairo C, Venturi V, Pauza CD. The gammadelta T-cell receptor repertoire is reconstituted in HIV patients after prolonged antiretroviral therapy. AIDS (2013) 27:1557–62. doi:10.1097/QAD.0b013e3283611888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Quigley MF, Greenaway HY, Venturi V, Lindsay R, Quinn KM, Seder RA, et al. Convergent recombination shapes the clonotypic landscape of the naive T-cell repertoire. Proc Natl Acad Sci U S A (2010) 107:19414–9. doi:10.1073/pnas.1010586107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Venturi V, Quigley MF, Greenaway HY, Ng PC, Ende ZS, Mcintosh T, et al. A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J Immunol (2011) 186:4285–94. doi:10.4049/jimmunol.1003898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Greenaway HY, Ng B, Price DA, Douek DC, Davenport MP, Venturi V. NKT and MAIT invariant TCRalpha sequences can be produced efficiently by VJ gene recombination. Immunobiology (2013) 218:213–24. doi:10.1016/j.imbio.2012.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Boudova S, Li H, Sajadi MM, Redfield RR, Pauza CD. Impact of persistent HIV replication on CD4 negative Vgamma2Vdelta2 T cells. J Infect Dis (2012) 205:1448–55. doi:10.1093/infdis/jis217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Urban EM, Li H, Armstrong C, Focaccetti C, Cairo C, Pauza CD. Control of CD56 expression and tumor cell cytotoxicity in human Vgamma2Vdelta2 T cells. BMC Immunol (2009) 10:50. doi:10.1186/1471-2172-10-50
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Sajadi MM, Heredia A, Le N, Constantine NT, Redfield RR. HIV-1 natural viral suppressors: control of viral replication in the absence of therapy. AIDS (2007) 21:517–9. doi:10.1097/QAD.0b013e328013d9eb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Riedel DJ, Sajadi MM, Armstrong CL, Cummings JS, Cairo C, Redfield RR, et al. Natural viral suppressors of HIV-1 have a unique capacity to maintain gammadelta T cells. AIDS (2009) 23:1955–64. doi:10.1097/QAD.0b013e32832ff1ff
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Maldarelli F, Palmer S, King MS, Wiegand A, Polis MA, Mican J, et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog (2007) 3:e46. doi:10.1371/journal.ppat.0030046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Martini F, Paglia MG, Montesano C, Enders PJ, Gentile M, Pauza CD, et al. V gamma 9V delta 2 T-cell anergy and complementarity-determining region 3-specific depletion during paroxysm of nonendemic malaria infection. Infect Immun (2003) 71:2945–9. doi:10.1128/IAI.71.5.2945-2949.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Quinn TC, Wawer MJ, Sewankambo N, Serwadda D, Li C, Wabwire-Mangen F, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N Engl J Med (2000) 342:921–9. doi:10.1056/NEJM200003303421303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Ripamonti D, Hill A, Lauthouwers E, Van Delft Y, Moecklinghoff C. Time to HIV-1 RNA suppression below 5 copies/ml during first-line protease inhibitor-based antiretroviral treatment - any impact of residual viremia on treatment success? AIDS Rev (2013) 15:230–6.
82. Fisch P, Malkovsky M, Braakman E, Sturm E, Bolhuis RL, Prieve A, et al. Gamma/delta T cell clones and natural killer cell clones mediate distinct patterns of non-major histocompatibility complex-restricted cytolysis. J Exp Med (1990) 171:1567–79. doi:10.1084/jem.171.5.1567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Fisch P, Kovats S, Fundim N, Sturm E, Braakman E, Demars R, et al. Function and specificity of human V gamma 9/V delta 2 T lymphocytes. Curr Top Microbiol Immunol (1991) 173:179–82.
84. Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human gammadelta T Cells. Science (2005) 309:264–8. doi:10.1126/science.1110267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Caccamo N, Todaro M, La Manna MP, Sireci G, Stassi G, Dieli F. IL-21 regulates the differentiation of a human gammadelta T cell subset equipped with B cell helper activity. PLoS One (2012) 7:e41940. doi:10.1371/journal.pone.0041940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Maniar A, Zhang X, Lin W, Gastman BR, Pauza CD, Strome SE, et al. Human gammadelta T lymphocytes induce robust NK cell-mediated antitumor cytotoxicity through CD137 engagement. Blood (2010) 116:1726–33. doi:10.1182/blood-2009-07-234211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Cairo C, Surendran N, Harris KM, Mazan-Mamczarz K, Sakoda Y, Diaz-Mendez F, et al. Vgamma2Vdelta2 T cell costimulation increases NK cell killing of monocyte-derived dendritic cells. Immunology (2014). doi:10.1111/imm.12386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Patel RV, Weinberg JM. Psoriasis in the patient with human immunodeficiency virus, part 1: review of pathogenesis. Cutis (2008) 82:117–22.
89. Morar N, Willis-Owen SA, Maurer T, Bunker CB. HIV-associated psoriasis: pathogenesis, clinical features, and management. Lancet Infect Dis (2010) 10:470–8. doi:10.1016/S1473-3099(10)70101-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Scotet E, Nedellec S, Devilder MC, Allain S, Bonneville M. Bridging innate and adaptive immunity through gammadelta T-dendritic cell crosstalk. Front Biosci (2008) 13:6872–85. doi:10.2741/3195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Nussbaumer O, Gruenbacher G, Gander H, Thurnher M. DC-like cell-dependent activation of human natural killer cells by the bisphosphonate zoledronic acid is regulated by gammadelta T lymphocytes. Blood (2011) 118:2743–51. doi:10.1182/blood-2011-01-328526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Riedel DJ, Mwangi EI, Fantry LE, Alexander C, Hossain MB, Pauza CD, et al. High cancer-related mortality in an urban, predominantly African-American, HIV-infected population. AIDS (2013) 27:1109–17. doi:10.1097/QAD.0b013e32835dc068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Mavilio D, Lombardo G, Kinter A, Fogli M, La Sala A, Ortolano S, et al. Characterization of the defective interaction between a subset of natural killer cells and dendritic cells in HIV-1 infection. J Exp Med (2006) 203:2339–50. doi:10.1084/jem.20060894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Reitano KN, Kottilil S, Gille CM, Zhang X, Yan M, O’shea MA, et al. Defective plasmacytoid dendritic cell-NK cell cross-talk in HIV infection. AIDS Res Hum Retroviruses (2009) 25:1029–37. doi:10.1089/aid.2008.0311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. De Maria A, Fogli M, Costa P, Murdaca G, Puppo F, Mavilio D, et al. The impaired NK cell cytolytic function in viremic HIV-1 infection is associated with a reduced surface expression of natural cytotoxicity receptors (NKp46, NKp30 and NKp44). Eur J Immunol (2003) 33:2410–8. doi:10.1002/eji.200324141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Mavilio D, Lombardo G, Benjamin J, Kim D, Follman D, Marcenaro E, et al. Characterization of CD56-/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc Natl Acad Sci U S A (2005) 102:2886–91. doi:10.1073/pnas.0409872102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Zhang R, Xu J, Hong K, Yuan L, Peng H, Tang H, et al. Increased NKG2A found in cytotoxic natural killer subset in HIV-1 patients with advanced clinical status. AIDS (2007) 21(Suppl 8):S9–17. doi:10.1097/01.aids.0000304691.32014.19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Brunetta E, Fogli M, Varchetta S, Bozzo L, Hudspeth KL, Marcenaro E, et al. The decreased expression of Siglec-7 represents an early marker of dysfunctional natural killer-cell subsets associated with high levels of HIV-1 viremia. Blood (2009) 114:3822–30. doi:10.1182/blood-2009-06-226332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Brunetta E, Hudspeth KL, Mavilio D. Pathologic natural killer cell subset redistribution in HIV-1 infection: new insights in pathophysiology and clinical outcomes. J Leukoc Biol (2010) 88:1119–30. doi:10.1189/jlb.0410225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Caccamo N, Meraviglia S, Scarpa F, La Mendola C, Santini D, Bonanno CT, et al. Aminobisphosphonate-activated gammadelta T cells in immunotherapy of cancer: doubts no more. Expert Opin Biol Ther (2008) 8:875–83. doi:10.1517/14712598.8.7.875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Dieli F, Caccamo N, Meraviglia S. Advances in immunotherapy of castration-resistant prostate cancer: bisphosphonates, phosphoantigens and more. Curr Opin Investig Drugs (2008) 9:1089–94.
102. Caccamo N, Dieli F, Meraviglia S, Guggino G, Salerno A. Gammadelta T cell modulation in anticancer treatment. Curr Cancer Drug Targets (2010) 10:27–36. doi:10.2174/156800910790980188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Meraviglia S, Eberl M, Vermijlen D, Todaro M, Buccheri S, Cicero G, et al. In vivo manipulation of Vgamma9Vdelta2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin Exp Immunol (2010) 161:290–7. doi:10.1111/j.1365-2249.2010.04167.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Kalyan S, Wesch D, Kabelitz D. Aminobisphosphonates and toll-like receptor ligands: recruiting Vgamma9Vdelta2 T cells for the treatment of hematologic malignancy. Curr Med Chem (2011) 18:5206–16. doi:10.2174/092986711798184280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Braza MS, Klein B. Anti-tumour immunotherapy with Vgamma9Vdelta2 T lymphocytes: from the bench to the bedside. Br J Haematol (2013) 160:123–32. doi:10.1111/bjh.12090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Poccia F, Gioia C, Martini F, Sacchi A, Piacentini P, Tempestilli M, et al. Zoledronic acid and interleukin-2 treatment improves immunocompetence in HIV-infected persons by activating Vgamma9Vdelta2 T cells. AIDS (2009) 23:555–65. doi:10.1097/QAD.0b013e3283244619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: gamma delta T cell, HIV, Vdelta1 gamma delta T cells, Vgamma9Vdelta2 T cells, immunotherapy
Citation: Pauza CD, Poonia B, Li H, Cairo C and Chaudhry S (2015) γδ T cells in HIV disease: past, present, and future. Front. Immunol. 5:687. doi: 10.3389/fimmu.2014.00687
Received: 17 October 2014; Accepted: 22 December 2014;
Published online: 30 January 2015.
Edited by:
Dieter Kabelitz, Christian-Albrechts University Kiel, GermanyReviewed by:
Christian Schönbach, Nazarbayev University, KazakhstanAlessandro Poggi, IRCCS AOU San Martino IST, Italy
Copyright: © 2015 Pauza, Poonia, Li, Cairo and Chaudhry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. David Pauza, Institute of Human Virology and Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, USA e-mail:Y2RwYXV6YUBpaHYudW1hcnlsYW5kLmVkdQ==