 Andrea Gras Navarro
Andrea Gras Navarro Andreas T. Björklund
Andreas T. Björklund Martha Chekenya
Martha Chekenya- 1Department of Biomedicine, University of Bergen, Bergen, Norway
- 2Karolinska University Hospital, Hematology Center and Karolinska Institute, Stockholm, Sweden
Natural killer (NK) cells are innate lymphoid cells that hold tremendous potential for effective immunotherapy for a broad range of cancers. Due to the mode of NK cell killing, requiring one-to-one target engagement and site-directed release of cytolytic granules, the therapeutic potential of NK cells has been most extensively explored in hematological malignancies. However, their ability to precisely kill antibody coated cells, cancer stem cells, and genotoxically altered cells, while maintaining tolerance to healthy cells makes them appealing therapeutic effectors for all cancer forms, including metastases. Due to their release of pro-inflammatory cytokines, NK cells may potently reverse the anti-inflammatory tumor microenvironment (TME) and augment adaptive immune responses by promoting differentiation, activation, and/or recruitment of accessory immune cells to sites of malignancy. Nevertheless, integrated and coordinated mechanisms of subversion of NK cell activity against the tumor and its microenvironment exist. Although our understanding of the receptor ligand interactions that regulate NK cell functionality has evolved remarkably, the diversity of ligands and receptors is complex, as is their mechanistic foundations in regulating NK cell function. In this article, we review the literature and highlight how the TME manipulates the NK cell phenotypes, genotypes, and tropism to evade tumor recognition and elimination. We discuss counter strategies that may be adopted to augment the efficacy of NK cell anti-tumor surveillance, the clinical trials that have been undertaken so far in solid malignancies, critically weighing the challenges and opportunities with this approach.
Introduction
Cancer is a generic group of over 100 diseases unified by fundamental characteristics acquired during their clonal evolution (1), including tumor-promoting inflammation and escape from immune surveillance (2, 3). Increased cancer incidence is associated with immunodeficiency (4, 5) and sustained immunosuppression (6–8). This supports the immunological surveillance hypothesis that postulates that as well as conferring protection from infectious pathogens, the immune system also guards against cancer (9). A prospective study of a large cohort of Japanese inhabitants followed up for 11 years uncovered an association of low natural cytotoxicity of peripheral blood natural killer (NK) cells with approximately 40% increased cancer risk compared to individuals with high cytotoxicity activity (10). This is reinforced by findings of decreased NK-cell activity among relatives of patients diagnosed with familial melanoma (11), implicating NK cells in tumor surveillance. Novel immunotherapies have recently demonstrated unprecedented success in selected cancer types (12, 13). Heightened interest in applicability of NK cells for cancer immunotherapy has been evoked by improved methodologies for their purification, molecular and phenotypic characterization. This progress is a result of a deeper understanding emerging from 40 years of scrutiny of their roles in anti-cancer immunity. In this article, we discuss the current knowledge on how NK cells recognize and kill cancer cells while sparing the normal healthy cells, how the cancer and its associated cells exploit this biology. We discuss the strategies that may be employed to derive effective medicine, the promise and challenges of these approaches for solid malignancies.
NK Phenotypes and Natural Cytotoxicity Against Solid Tumors
Natural killer cells comprise 10–15% of peripheral blood lymphocytes and classically display a half-life of approximately 7–10 days in circulation (14–16). However, some subpopulations generated by cytokine stimulation or viral infections may display prolonged persistence (17–19). NK cells arise from lineage restricted progenitor cells derived from CD34+ common lymphoid progenitors through a process regulated by basic leucine zipper transcription factors, E4-promoter binding protein 4 (E4pb4), Ets1 (20, 21), and the T-box transcription factors, T-bet, Eomes, as well as ID2 (inhibitor of DNA binding). In their absence, fewer numbers of NK cells develop (22–25) and exhibit functional alterations (26). Terminally differentiated NK cells lack phenotypic markers of B and T lymphocytes and are distinguished further by their greater size and cytoplasmic granularity. They express CD56 neural cell adhesion molecule and are thus phenotypically categorized as CD3-CD19-CD56+ innate lymphoid cells (27–29).
Functionally, NK cells can lyse directly virus-infected or transformed cells without prior sensitization (28, 30), mediated by natural cytotoxicity receptors (NCRs) NKp30 (CD337), NKp44 (CD336), and NKp46 (CD335) that belong to the Immunoglobulin superfamily (31). NKp30 is constitutively expressed and recognizes B7-H6 tumor antigens (expressed on leukemia, lymphomas, carcinomas, and melanomas) (32). NKp44 is expressed only on activated human NK cells and binds viral hemagglutinin (HA) and HA-neuraminidase (HN) as well as tumor-associated ligands (33). NKp46 is expressed on both resting and activated NK cells in mice and men and can inhibit growth of tumor metastases in mice (34, 35), although its ligands have yet to be identified. The NK group 2D (NKG2D, CD314) is a lectin-like type 2 transmembrane homodimeric receptor that is constitutively expressed on all NK cells (36). It transduces activating signals upon binding to its ligands major histocompatibility complex (MHC) class 1-related chains A and B (MICA and MICB) and viral UL16 binding proteins (ULBPs). These stress induced-ligands are expressed on tumor, but not healthy cells, as a result of heat shock, viral infection, or genotoxic stress by DNA damage (37). NKG2D ligation induces phosphorylation of YINM motifs on DAP10, allowing recruitment and activation of the p85 subunit of phosphatidylinositol-3-kinase (PI3K) and growth factor receptor-bound protein 2 (GRB2) to trigger NK cytotoxicity (38). We recently demonstrated that NKG2D was highly expressed by glioblastoma (GBM) infiltrating NK cells in situ (39). Antibody blockade of NKG2D rescued approximately 50% stress ligand-bearing GBM but not K562 chronic myelogenous leukemia (AML) cells, from lysis by donor NK cells in vitro (40). This emphasizes the importance of activation signaling via NKG2D for NK cell cytotoxicity. Indeed, proteolytic cleavage of NKG2D ligands by ADAM 10 and 17 proteases (a disintegrin and metalloproteinase) sheds soluble ligands into serum to circumvent cytotoxicity via NKG2D receptor (41, 42), and is a common aberration in cancer (43). Soluble MICA/B and ULBPs have been detected in sera of patients with diverse solid malignancies (44), where soluble ULBP2 distinguished early stage pancreatic adenocarcinoma from healthy subjects. Elevated ULBP2 could identify melanoma patients at risk for disease progression and was prognostic in patients with early stage B-cell chronic lymphocytic leukemia (45–47). Conversely, others demonstrated that hypoxia induced microRNAs miR-20a, miR-93, and miR-106b downregulated NKG2D ligands on GBM cells as a mechanism of immunological escape (48). Genome wide association studies also identified a MICA-A5.1 allelic variant with a frameshift mutation that results in a truncated protein that is released as a membrane-anchored molecule in exosomes in human papilloma virus induced cervical cancer in a Swedish cohort (49, 50). Another MICA variant, rs23596542, was identified in hepatitis C virus induced hepatocellular carcinomas (HCC) from a Japanese population (51). Both cleaved MICA and exosomal MICA-A5.1 result in high serum levels of soluble MICA that interacts with NKG2D and prevents its interaction with membrane bound ligands. Recently, the GBM derived metabolite, lactate dehydrogenase isoform 5 (LDH5), was demonstrated to upregulate the NKG2D ligands MICA/B and ULBPs on monocytes from healthy individuals in vitro and on circulating macrophages from patient derived breast, prostate, and HCC as a further means to subvert NK cell surveillance (52). This would lead to NKG2D receptor downregulation through internalization, degradation, and/or desensitization (53). Ultimately, diminished NK cytotoxicity ensues due to chronic exposure to ligand expressing cells, consistent with the discontinuity theory of immunity (54). A caveat to interpreting causality of soluble ligands in patient sera to attenuated NKG2D receptor levels is the presence of transforming growth factor β (TGFβ) that also diminishes NKG2D, as reported in GBM (55). Another emerging concept coined split anergy proposes that NK cell-monocyte/macrophage cross-talk in vitro results in anergic NK cells that are not cytotoxic but secrete cytokines that enhance differentiation of cancer stem cells (CSCs) (56). CSCs are minor subpopulations within the tumor capable of self-renewal by asymmetrical cell division to maintain the tumor’s cellular heterogeneity (57). CSCs are resistant to conventional anti-cancer therapy (57, 58) and are proposed to drive malignant progression. Differentiated cells are thought to be more resistant to NK lysis (59, 60), but more responsive to the standard treatment. Thus, NK-cell/macrophage crosstalk may halt malignant progression by directly killing and/or differentiating the CSCs (56). Although largely observed in vitro, split anergy may have clinical implications for the impact of NK-cell/macrophage crosstalk in tumor surveillance in vivo. Nevertheless, natural cytotoxicity is a tumor-dependent killing mechanism that varies in potency because although NCRs and NKG2D are constitutively expressed on NK cells in most individuals, different tumors variably express tumor-associated antigens and NKG2D ligands.
NK Subsets
On the basis of the density of CD56, NK cells can be further subclassified as CD56bright subsets (61) that also express high levels of l-selectin (CD62L), an adhesion molecule that mediates interaction with the vascular endothelium (62). CD56bright NK cells account for only 10% of circulating peripheral blood NK cells but represent a dominant phenotype in secondary lymphoid tissues. They constitutively express high affinity heterotrimeric interleukin (IL)-2Rαβγ receptors, maintain long telomeres, and proliferate in response to low concentrations of IL-2 (63, 64). Unlike T cells, NK cells do not secrete IL-2 (65) but CD56bright subsets are denoted by secretion of other cytokines, including interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), IL-5, IL-10, and IL-13. Notably, IFN-γ promotes T-helper type 1- (Th1) responses that enhance effector functions of both NK cells and cytotoxic T lymphocytes (CTLs) (66, 67), up-regulate class I and class II MHC (68) on antigen presenting cells, as well as costimulatory molecules on macrophages (67). CD56bright subsets express CC-chemokine receptors 7 (CCR7), CCR5, and CXCR3 (69) that allow their preferential recruitment to tumor and inflamed tissues (70, 71). CD56bright NK cells also secrete granulocyte colony-stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF) (67, 72, 73) that promote homing to secondary lymphoid organs through endothelial venules (74).
In contrast, the majority (approximately 90%) of NK cells in steady state peripheral blood are CD56dim, and are considered to differentiate from the CD56bright CD94/NKG2A+ subsets through transitional loss of CD94/NKG2A and CD62L. Acquisition of CD57 and killer immunoglobulin like receptors (KIRs) denotes their terminal differentiation to CD56dim CD57+CD62L-CD94/NKG2A- KIR+ mature NK cells. The CD56dim subsets are more cytotoxic against target cells compared to the CD56bright subsets and proliferate in response to IL-2 and IL-15 stimulation through signaling via their heterodimeric IL-2Rβγ/IL-15Rα. CD56dim CD57+ subsets exhibit replicative senescence as indicated by shortened telomeres and diminished proliferation in response to stimulation with cytokine combinations ex vivo (75, 76). CD56dim subsets secrete low IFN-γ, even after activation with IL-2, or combination IL-15/IL-21. They lack CCR7 but do express CXCR1, CXCR2, and low density CXCR3, as well as CX3C chemokine receptors 1 (CX3CR1high). This traditional designation of CD56dim as “potent killers” and CD56bright subsets as “cytokine producers” might be oversimplified, as both subsets can perform either function when appropriately stimulated (77). NK cells dynamically adjust their phenotypes in response to the changing cytokine concentrations, ligand density, and cell types present in their microenvironment. Thus, it is debated whether the phenotypic subsets represent distinct maturation stages that are also functionally independent subpopulations, regardless of age, diurnal fluctuations, and microenvironments in diseases states, such as cancer (78). If subset characteristics change dynamically depending on their microenvironment, challenges for selecting a priori suitable subsets for anti-cancer therapy will be inevitable. All human NK cell subsets express a range of other adhesion molecules, including CD2, CD44, VLA-5 α chain (CD49e), lymphocyte function associated antigen (LFA-1), and intracellular adhesion molecule-1 (ICAM-1). Their density vary and thus impact trafficking patterns to various tissues and ability to form functional immunological synapses during immune responses (71). Both CD56bright and CD56dim NK cell subsets secrete chemokines such as monocyte chemotactic protein-1 (MCP-1, CCL2), macrophage inflammatory protein 1-α (MIP1-α, CCL3), macrophage inflammatory protein 1-β (MIP1-β, CCL4), and regulated on activation normal T cell expressed and secreted (RANTES, CCL5) (72). These chemokines further recruit, activate, and enhance antigen presentation of other immune cells at sites of inflammation, required for full-fledged immune responses (79, 80).
Trafficking to the Solid Tumor Microenvironment
The microenvironment of solid tumors poses a formidable challenge to NK cell efficacy due to chronic immune suppressive signals that select for tumor cells with altered immunogenicity, while simultaneously hindering both infiltration and activation of NK cells at tumor nests (81). The tumor secretes TGFβ (82, 83) vascular endothelial growth factor (VEGF), prostaglandin E2 (PGE2), and IL-10 that suppress T cell proliferation and cytotoxic responses (84) (Figure 1). These factors also downregulate class I MHC, skew dendritic cells toward an immature, tolerogenic, immature phenotype (85). TGFβ and PGE2 generate a highly heterogeneous population of myeloid derived suppressor cells (MDSCs) that comprise granulocytes, macrophages, and dendritic cells arrested at various differentiation stages (86). In response to hypoxia in the tumor microenvironment (TME), MDSCs upregulate arginase activity and inducible nitric oxide synthase (iNOS) (Figure 1) that inhibit T cells through NO signaling (87). This further depletes intracellular l-arginine and enhances production of reactive oxygen species (ROS) that diminish immune surveillance through shedding of NKG2D ligands (87). In addition, MDSCs secrete IL-10 and through membrane bound TGFβ, polarize tumor associated macrophages (TAMs) toward an M2- like anti-inflammatory phenotype (86) (Figure 1). A seminal study demonstrated that a subpopulation of mouse GR-1+CD11b+F4/80+ mononuclear MDSCs expressing the NKG2D ligand RAE-1 did not suppress, but in fact activated NK cells to produce IFN-γ and abolished tumor growth (88). Depletion of this population fueled tumor growth confirming their protective role against cancer. Although this study defines functional plasticity within the highly heterogeneous MDSC pool, the study was performed in a NK cell sensitive, relatively homogeneous mouse tumor. The findings may not easily translate to biochemically complex microenvironments of heterogeneous solid tumors in humans. The function of the RAE-1 expressing GR-1+CD11b+F4/80+ does not easily reconcile findings of the NK tolerizing role of NKG2D ligands on TAMs (39, 52). Others reported that coculture with Gr-1+CD11b+ MDSCs inhibited cytotoxicity of IL-2 activated NK cells against Yac-1 cells and that this was mediated via membrane bound TGFβ and signaling via signal transducer and activator of transcription-5 (STAT5) (89). Thus, polyclonal MDSCs may exhibit functional plasticity to induce anti-tumor or pro-tumor effects depending on their subset phenotype, tumor type, or microenvironment encountered (Figure 1).
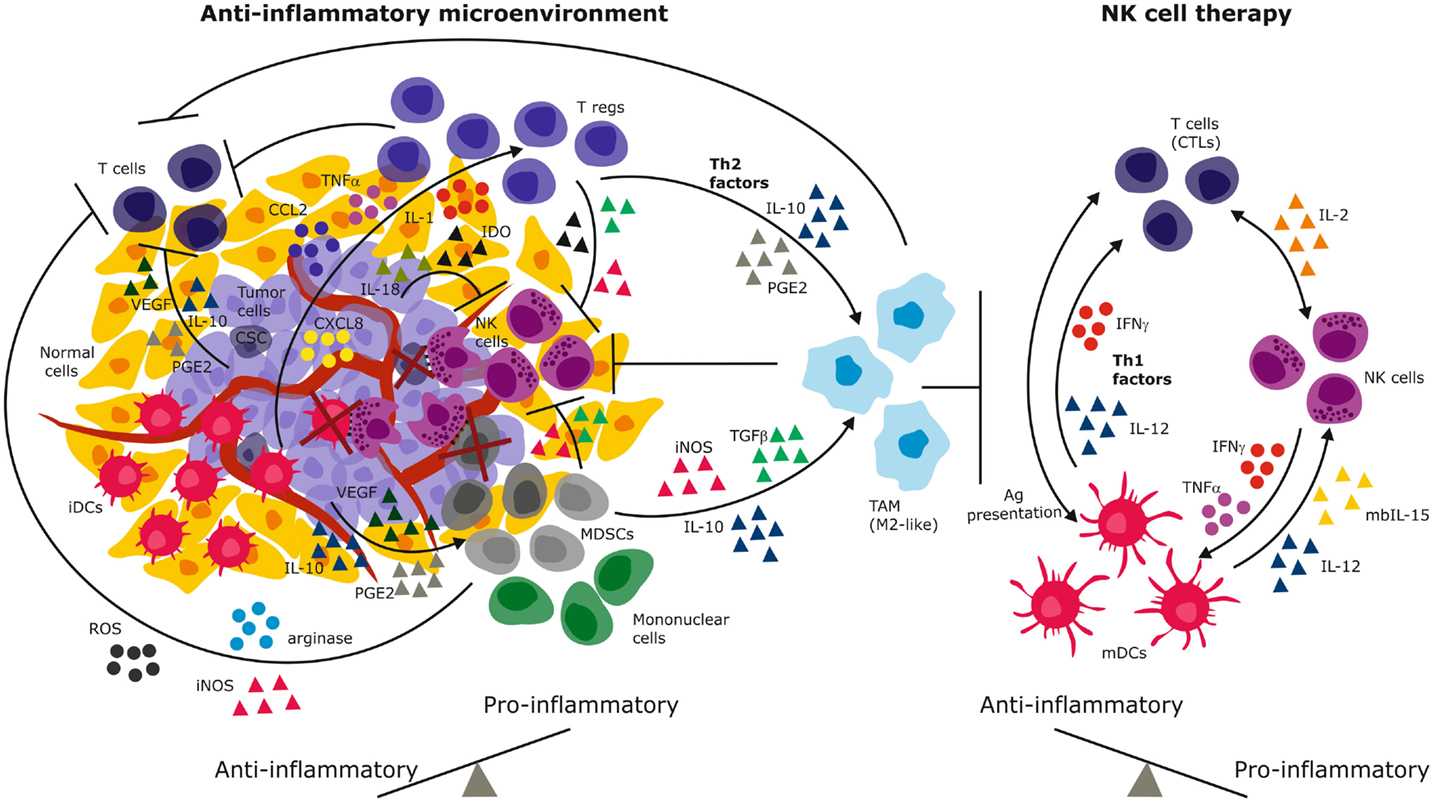
Figure 1. Immune suppressive mechanisms of the solid tumor microenvironment. Tumor-derived immunosuppressive mechanisms include secreted cytokines and chemokines that recruit and sustain immature and suppressive immune cells. These in turn secrete Th2 factors that propagate the cycle to create a chronic anti-inflammatory environment that promotes and predominates malignant progression (left). NK cell therapy has the potential to reverse the anti-inflammation to engender dominant pro-inflammatory signaling through secretion of IFN-γ, Th1 cytokines, and activation of both innate and adaptive immune cells (right). By elimination of tumor cells and immature suppressive cells (e.g., iDCs, MDSCs, cancer stem cells), NK cells have the potential to reverse the anti-inflammatory signals and eliminate tumor progression. iDCs, immature dendritic cells; mDCs, mature dendritic cells; MDSCs, myeloid derived suppressor cells; PGE2, prostaglandin E-2; IDO, indoleamine-2,3-dioxygenase; VEGF, vascular endothelial growth factor; TGFβ, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha; IFN-γ, interferon-gamma; IL, interleukin; iNOS, inducible nitric oxide synthase; ROS, reactive oxygen species; CTLs, cytotoxic T lymphocytes (CTLs); Tregs, regulatory T lymphocytes; CSC, cancer stem cells.
Although CSCs represent a minor population in the TME, they too contribute to immune suppression (90, 91) through STAT3 (92), a signaling hub utilized by diverse cytokines and tumor infiltrating immune cells to markedly suppress their functionality (93). The transcriptional activity of nuclear factor-kappa beta (NFκB) also regulates the function of CSCs and immune effectors at the tumor site (94). Differentiated oral epithelial tumors exhibit higher NFκB activity and are resistant to NK-mediated cytotoxicity compared to their CSC counterparts (59). Inhibition of NFκB in differentiated carcinoma cells, or in non-transformed keratinocytes, increased sensitivity to NK cell lysis (95), implicating NFκB survival signaling in resistance to NK lysis. Indeed, CSCs from oral squamous carcinomas and colorectal cancers with diminished NFκB levels are more sensitive to NK lysis compared to their differentiated counterparts (59, 60, 96). In contrast, NK cells preferentially lyse GBM CSCs despite constitutively activated STAT3 and NFκB signaling (97, 98). Instead, it was reported that low class I MHC and high PVR and Nectin-2 expression levels underlie their sensitivity to NK cells (97, 98). This discrepancy may reflect diversity in oncogenic signaling pathways of CSCs from different tissues.
The tumor secretes chemokines CXCL8, CCL2, IL-1, and TNF-α that recruits macrophages and suppressive CD4+CD25+CD127lowFOXP3+ Tregs to the tumor site (Figure 1). Tregs have been demonstrated to inhibit NK cells from gastrointestinal stromal tumors (GIST) by competing for IL-2 availability and secrete IL-4, indoleamine-2,3-dioxygenase (IDO). Tregs express membrane bound TGFβ that inhibits NKG2D and NKp30 by cleaving their ligands off malignant cells (99, 100). Tumor-derived IDO promotes production of the immunosuppressive tryptophan catabolite l-kynurenine that interferes with IL-2 induced expression of NKp46 and NKG2D receptors that are required for target cell recognition and killing (101). In contrast to CD4+CD25+CD127lowFOXP3+ Tregs, we recently identified CD8+CD28–Foxp3+ Tregs that were induced in the GBM microenvironment (39). These Tregs might tolerize tumor infiltrating antigen presenting cells through IL-10 induced immunoglobulin-like transcripts (ILT)-2, ILT3, ILT4, and decreased expression of CD40, CD80, and CD86 costimulatory molecules (39). Decreased Tregs have been associated with improved NK cell activity in GIST, melanoma, and GBM after treatment with Imatinib, and DC exosomes (102) or vaccination (103), underscoring the clinical impact of these suppressive cells. Melanoma and colorectal tumor associated fibroblasts could inhibit NK cell activity through cell–cell contact in vitro (104, 105) and through secretion of PGE2 or IDO that abrogated IL-2 induced NKp44, DNAM-1, and NKp30 upregulation (106). Thus, targeting IDO in cervical cancer increased NK cell accumulation at the tumor site and inhibited growth (101).
Indeed, increased infiltration of NK cells into tumor lesions of diverse histological types, such as GBM (107), solid metastases (108) lung, gastric, colorectal, as well as head and neck cancers, have been associated with good prognosis (70, 109–112). In gastric cancer, increased NK cell infiltration correlated with reduced invasion, reduced lymph nodes metastases, and improved outcome. A caveat to these early studies, however, was the utilization of single staining immunohistochemistry against CD56 or CD57 markers that are also expressed by CD8+ T cells (107) and some tumor types (113). Moreover, CD57 is only expressed on a subset of CD56dim mature NK cells (75, 76). Thus, the degree of NK cell infiltration in tumors is difficult to ascertain from these studies, as both overestimation and under representation of the figures is possible. Staining for NKp46 that is expressed by all NK cells revealed that renal cell carcinomas (RCC) and GIST had substantial NK cell infiltration (114, 115). However, NK cells infiltrating the RCC were not cytotoxic and upregulated CD94/NKG2A inhibitory receptor. NK cells infiltrating the GIST tumors ostensibly expressed the immunosuppressive NKp30c isoform whose surface levels were also prognostic in these patients (114, 115). Other studies reported scarce infiltration of HCC and RCC by NK cells. The NK cells were functionally anergic as indicated by diminished NKp30, DNAM-1 expression, reduced degranulation or stayed in the stroma, and did not contact the tumor cells (104). Extracellular matrix and tumor cell derived gangliosides (sialic acid glycoproteins), GM2, and GM3 have been demonstrated to diminish NK cell cytotoxicity through contact inhibition to targets, and abrogation of IL-2 dependent proliferation (116). Furthermore, pre-incubation of NK cells with GD3 and GM3, or serum from cancer patients containing shed GD3 prior to addition of target cells, strongly inhibited NK cell lysis in a dose dependent manner (117, 118). Again, the effects of serum could be explained through other mechanisms, such as TGFβ. GD3 and various gangliosides are abundantly secreted into the TME of patient GBMs in situ (119, 120), potentially providing similar immunosuppressive mechanisms of escape from NK cell lysis. Precise mechanisms regulating NK cell recruitment to tissues are poorly delineated but may depend on both tumor histological type and chemokine profiles. Therefore, several experimental therapies have attempted to augment endogenous NK cell trafficking and activity at tumor nests. CD27high NK cells in mice are orthologs of human CD56bright cells, and in both species, these NK subsets express CXCR3 and are attracted to CXCL9 and CXCL10 chemokines at tumor sites (121, 122). CD56dim NK cells, however, express CX3CR1 and have been detected in breast, lung, and colorectal cancer biopsies. In the latter, the NK cells remained in the stroma and were not in direct contact with the malignant cells despite elevated levels of chemokines typically expressed by both CD56dim and CD56bright subpopulations (123). This might indicate insufficiency of chemokines alone in determining the impact of tumor infiltrating NK cells. Remarkably, the chemoattractant chemerin has been shown to favor infiltration of melanoma by NK cells, reduced MDSCs, and immunosuppressive plasmocytoid DCs (81, 124). Whole genome expression datasets confirmed that chimerin was prognostic of improved outcome in melanoma patients and expression is lost during malignant progression of several solid tumors. In B16 transplantable mouse melanoma, chimerin overexpression or exogenous injection in the context of its chemokine-like receptor 1 diminished tumor growth (124). It is not known whether CD56dim and CD56bright subsets are differentially recruited to the tumor because of their different chemokine receptor profiles. It is possible that CD56bright subsets might be increased in the TME as a consequence of their enhanced proliferative capacity and or propensity for surviving oxidative stress conditions (125) that typify the TME. Both CD56dim and CD56bright subsets may express CD16, a low affinity receptor (FcγRIII) that binds the constant Fc chain of antibody (126), inducing NK cell activation and killing of antibody coated cells through antibody-dependent cellular cytotoxicity (ADCC). Human breast cancer infiltrating CD56bright NK cells expressing low levels of CD16 but heightened expression of activation markers NKp44, CD25, CD69, and NKG2D were reported (127). Similarly, we reported that CD56dim CD16- NK cells that infiltrate patient GBM lesions expressed NKG2D (39). Loss of CD16 on tumor infiltrating NK cells is one mechanism proposed for induction of split anergy (60, 128). Tumor infiltrating NK cells purified by negative selection from ascites fluid from ovarian cancer patients produced less IFN-γ and IL-4, but more IL-10 compared to corresponding peripheral blood NK cells (129). Although the CD56dim CD16- subsets might indicate potential cytotoxicity, their diminished proliferative potential may result in low intra-tumor effector-target ratios, thus reducing their killing efficiency. Attenuated cytokine production and inability to execute ADCC of antibody-coated cells due to lack of CD16 might render their cytotoxicity unsustainable in hostile solid TMEs. Collectively, these studies indicate a consistent characteristic in solid cancers of loss of NK cell-tumor engagement, cleavage of stress ligands, loss of NKG2D receptor and or CD16, thus diminishing tumor recognition, cytotoxicity, and ability to execute ADCC of antibody-coated targets.
Antibody-Dependent Cellular Cytotoxicity
Indeed, the induction of ADCC may represent the underlying mechanism for the unprecedented success of monoclonal antibodies to enhance tumor cell recognition by the immune system, exemplified by Rituximab against CD20 in lymphoma (130) and Ipilumimab targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4) in metastatic melanoma (12). IgG1 is the most frequently used isotype for humanized antibodies due to serum stability and high affinity for CD16 on NK cells, or CD32 on neutrophils and monocytes/macrophages (FcγRII). The homozygous FcγRIII-158Val and FcγRII-131His allelic polymorphisms result in proteins with higher affinity for IgG1, IgG3, and IgG4. Patients with these polymorphisms demonstrated greater NK cell–mediated ADCC (131–133). ADCC is executed by the lytic granules, perforin, and granzymes A/B, whereas the release of cytokines and chemokines leads to inhibition of cell proliferation and angiogenesis (134, 135). Thus, as well as exerting efficacy through immune checkpoint inhibition, ipilumimab also triggers ADCC and TNF-α mediated killing by NK cells (134). Nivolumimab, an IgG4 antibody against another immune checkpoint molecule, programed death-1 (PD-1), has been combined with Ipilumimab to harness synergism in blocking immune checkpoints and evoke therapeutic benefits greater than either treatment alone (13). Since tumor cells secrete IL-18 that upregulates PD-1 on NK cells, anti-IL-18 neutralizing mAb in combination with Nivolumimab may circumvent tolerization of NK cells. Strong evidence for the anti-cancer efficacy of ADCC was obtained with Rituximab in patients with various lymphoid malignancies of B-cell origin, including follicular and aggressive large B-cell non-Hodgkin’s lymphoma (130). Trastuzumab/Herceptin for treatment of patients with metastatic breast (136) and early stage gastric carcinomas (137), humanized anti-GD2 mAb in melanoma, neuroblastoma, osteosarcoma, and soft tissue sarcoma patients (138–141) induce potent ADCC. As well as allelic polymorphisms in FcγRIII, high antigen expression is required for improved efficacy. A humanized CD16-CD33 BiKE (bispecific killer engager) antibody was demonstrated to strongly activate NK cells against CD33+ AML blasts when combined with pre-treatment with an ADAM17 small molecule inhibitor that prevented shedding of CD16 in vitro (142). Cetuximab, a chimeric human/mouse antibody targeting epidermal growth factor receptors (EGFR) for treatment of GBM, advanced non-small cell lung cancer (NSCLC), ovarian cancer, and colorectal cancer, induced NK cell mediated ADCC, and extended overall survival by 3 months in head and neck cancer patients (97, 143, 144). EGFR variant III (EGFRvIII) is a tumor specific mutant epitope that is expressed on one-third of GBMs and mediates an aggressive phenotype. Unarmed mAbs against the EGFRvIII showed promise in GBM preclinical studies, but limited efficacy in patients (145). The latter was likely the result of limited penetration of the tumor bed, regrowth of heterogeneous tumor due to antigen loss variants, and or resistance due to redundancy in oncogenic signaling pathways (146, 147). A promising GBM associated antigen is the cell surface chondroitin sulfate proteoglycan NG2/CSPG4 that is also expressed on various cancer forms, including melanomas, leukemia, breast, and sarcomas, but not in normal differentiated cells in the corresponding tissues (73, 148–150). Elevated NG2/CSPG4 enhances tumorigenicity, angiogenesis and migration, as well as conferring resistance to chemotherapy and radiotherapy via distinct mechanisms (119, 151–157). We demonstrated that NG2/CSPG4 is highly expressed by approximately 20–30% of treatment resistant cells and angiogenic vessels in the TME of 50% of all patient GBMs (119, 151–156). Elevated NG2/CSPG4 expression was an independent prognostic biomarker for poor survival. We showed that combination treatment of adoptively transferred NK cells with mAb9.2.27 against NG2/CSPG4 in preclinical models of GBM induced synergistic therapeutic effects through TNF-α, a IFN-γ release, diminished IL-10, IL-6, and IL-1α. Combination NK cells + mAb9.2.27 induced potent ADCC mediated by Fcγ-IIR on microglia/macrophages that resulted in prolonged survival (158, 159). mAb9.2.27 could not induce ADCC by NK cells proper, likely because of its IgG2a isotype, known to engage weakly the Fcγ-RIII on NK cells. However, improved chimeric antigen receptor conjugated or humanized bispecific antibody constructs against this antigen have recently been tested in pre-clinical studies with promising results (160, 161). Natural cytotoxicity and ADCC are not the only mechanisms NK cells use to eliminate malignant cells.
NK-Tumor Cells’ KIR-HLA Interactions: “Missing Self”, “Induced Self” and Tolerance
Natural killer cells express activating and inhibitory killer immunoglobulin-like receptor (KIR) genes encoded on chromosome 19q13.4 that are inherited and segregate separately from their ligands. These class 1 human leukocyte antigen (HLA)-A, -B, and -C are allelic variants that are encoded on chromosome 6p21.3. Mutations in the β2-microglobulin gene and loss of heterozygosity on chromosome 6 that harbors the class I and II HLA are common aberrations in cancer (162, 163). They invariably lead to downregulated expression of HLA molecules. Although the current review focuses on NK cells, in heterogeneous tissues KIRs are also expressed on CD8+ T cell subsets, albeit as a unique repertoire of single dominant KIR that impacts their function (164, 165). Through ligation of their KIRs to cognate HLA ligands on autologous cells, NK cells become educated to distinguish healthy self-cells (Figure 2A) from non-self cells that lack or possess poorly recognized polymorphic HLA ligands [deemed “missing self” (166); Figure 2B]. These malignant cells exhibiting HLA loss are thus recognized as missing self by educated NK cells, triggering their cytotoxicity (166, 167) (Figure 2B).
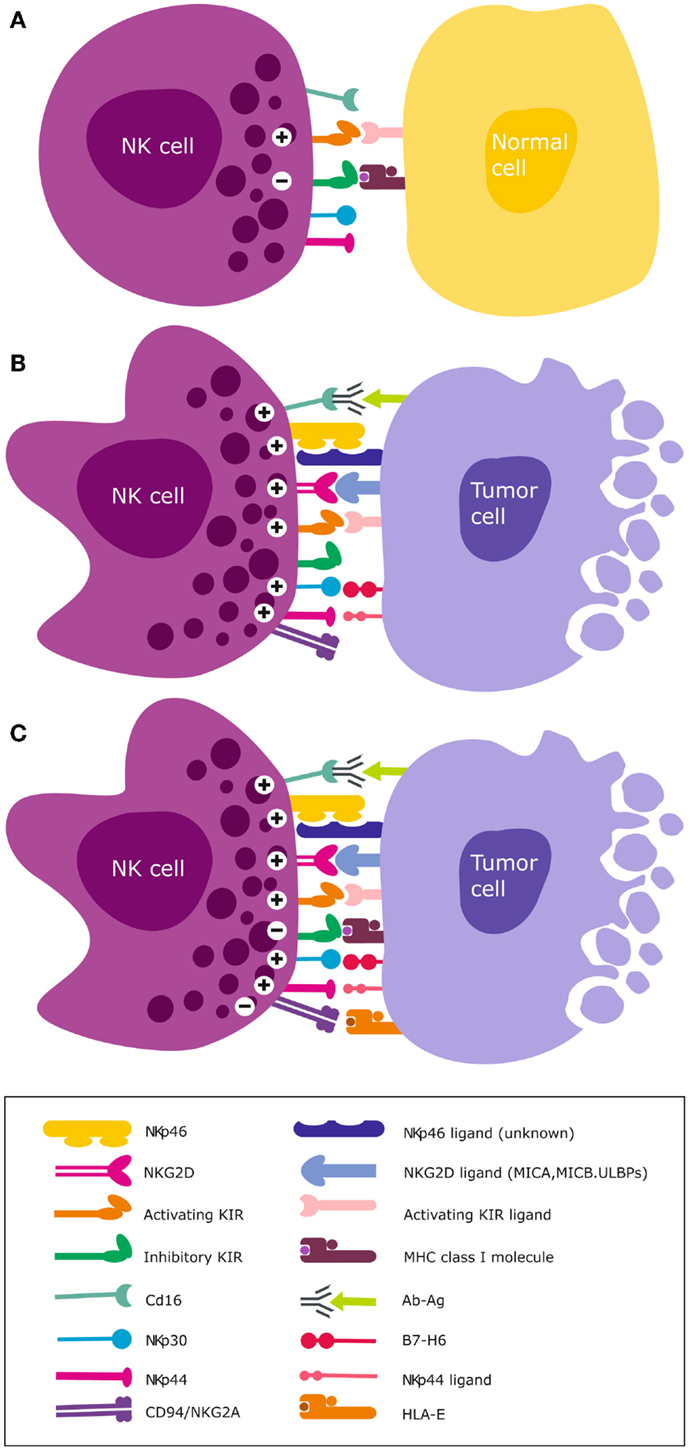
Figure 2. NK cell activation and target recognition. (A) Tolerance to self. NK cells express inhibitory and activating receptors on the surface and when interaction with cognate ligands for both activating and inhibitory receptors is balanced, NK cells remain tolerant and unable to kill the target cell e.g., normal unstressed self-cells that express class I HLA ligands for inhibitory receptors. (B) Missing self. A malignantly transformed cell may downregulate class I HLA but concomitantly express stress induced ligands that are recognized by NK cell activating receptors. A dominant activating signal is transduced that outweighs the inhibitory signals, resulting in NK cell activation and subsequent lysis of the target cell. (C) Altered self. Some cancer types express normal levels of class I HLA but concomitantly over-express stress-induced ligands e.g., GBM. Only when the activation signal transduced by ligation of stress ligands to cognate activating receptors overcomes the inhibitory signal, will NK cells become activated to lyse the target cell.
Educated NK cells can also distinguish healthy self-cells from altered-self cells that may express appropriate HLA ligands but concomitantly express stress-induced ligands (Figure 2C). Inhibitory KIRs are essential for this exquisite distinction of self from non-self cells and their main HLA ligand specificities are HLA-C. We recently demonstrated that although GBMs express high levels of class I HLA (39, 40), these malignant brain tumors may nevertheless become targets for NK cells due to the “altered self” mechanism (67, 72, 168) (Figure 2C). This is because the tumor cells concomitantly overexpress stress-induced ligands that are recognized by activating NKG2D receptors (39, 40, 169). Although GBM cells proper express low to negligent non-classical HLA-E, antigen presenting cells that constitute upto 50% of cells of the TME highly express HLA-E and may contribute to further tolerization of tumor infiltrating KIR-NKG2A+ NK cells (39, 40). Ligation of the self-HLA ligands to cognate inhibitory KIR results in phosphorylation of the ITIMs by Src family protein tyrosine kinases (PTKs). This creates docking sites for protein tyrosine phosphatases SHP-1 and SHP-2 (170) that inhibit NK cell activation by dephosphorylating proteins involved in downstream activation signaling. This inhibitory signaling maintains NK cells tolerant to normal self-cells. Ligation of activating KIRs transduces activating signals through phosphorylation of DAP12 or FcεRI-γ adaptor proteins containing immunoreceptor tyrosine-based activation motif (ITAM) by the Src PTKs (171). Downstream NK cell cytotoxicity pathways converge on the PI3K/Akt and mitogen-activated protein kinase (MAPK) pathways. The ensuing activating signal may override the inhibitory signals and ultimately, the overall threshold strength of activating or competing inhibitory signals determines whether the NK cell will be triggered for cytotoxicity (172, 173), cytokine release, or tolerance against encountered target cells.
However, only 5% of individuals possess all KIR genes in their genome and different individuals display highly variable KIR repertoires with up to 30,000 distinct NK cell subsets in peripheral blood (174). It is estimated that 10–20% autologous NK cells express inhibitory KIRs that do not recognize HLA-A, HLA-Bw4, or HLA-C self-alleles. These NK cells are considered “uneducated” and remain functionally tolerant against self-cells (175, 176). This is likely due to constitutively expressed CD94/NKG2A receptor that ligates non-classical HLA-E alleles to maintain tolerance, in complementation with KIRs to prevent NK cell autoreactivity (177, 178). This interpretation may be debated since uneducated cells should not express CD94/NKG2A since by definition this receptor provides NK cell education. However, there might be a qualitative difference in KIR-inhibition and NKG2A inhibition (179). NKG2A induces stronger cytokine responses and less degranulation than KIRs do. The NKG2A-inhibition may be easier to overcome and may be more important in microenvironments where HLA-expression is low. The ligand HLA-E is more commonly expressed by hematopoietic tumors but is less abundantly expressed by solid tumors, in particular GBM.
Other studies, however, demonstrated that NK cells that lack KIR for self-HLA, or lack self-HLA to cognate KIRs may instead be educated through CD94/NKG2A/HLA-E interactions. Functional activity manifests in transplant cases where donor NK cells express low frequencies of KIR or during the early phases after haploidentical hematopoietic stem cell transplantation (HSCT) (180, 181) when the donor NK cells’ KIRs have yet to reconstitute to pre-transplant levels. However, this might not be a general mechanism as it was also demonstrated that tolerance is maintained after HLA-matched sibling transplantation (182). Discrepancies between these two clinical studies may be due to emergence of conditions that influence endogenous cytokine levels. These include graft versus host disease (GvHD), bacterial infections or reactivation of human cytomegalovirus (CMV), as well as use of immunosuppressive drugs. Likewise, tolerance of uneducated NK cells may be abrogated by exogenously administered cytokines (179). These NK cells were subsequently shown to execute graft versus malignancy effects and improve survival after HSCT for AML/myelodysplastic syndrome (AML/MDS) (183). In an elegant study, neuroblastoma patients possessing one or more inhibitory KIRs missing self-HLA ligands (NS-KIRs) exhibited superior overall survival and progression free survival when treated with the anti-GD2 mAb 3F8 compared to patients possessing all KIRs in the presence of self-HLA (S-KIRs) following chemotherapy or autologous stem cell transplantation (141). This effect was attributed to the uneducated NK cells, even though in vitro experiments demonstrated that both educated and uneducated NK subpopulations were activated for ADCC by 3F8mAb. The difference between the in vitro and in vivo effects was attributed to increased IFN-γ release after ADCC that upregulated expression of self-HLA ligands and subsequently tolerized the educated NK cells expressing S-KIRs. It is also possible that other cellular subsets may have contributed to the inhibition signal of the effector cells in patients but not in vitro, such as γδT cells that also express CD94/NKG2A (184). In fact the interpretation can be further refined when considering that NK responses are not of “all or nothing” binary function. NK cell responses are functionally fine-tuned by the varying combinations of particular inhibitory KIRs with distinct affinities to cognate HLA ligands in different individuals formulated in the “rheostat” model (185, 186). However, the contribution of activating receptors has also been proposed (187). Furthermore, uneducated NK cells edit and eliminate immature DCs (iDCs) that express low levels HLA-E through contact dependent ligation of NKp30 (188), and thus enhance the adaptive response by potentiation of antigen presentation by mature DCs (Figure 1). Thus, uneducated NK cells play a significant role in shaping NK cell mediated tumor surveillance and treatment response.
Clinical Basis of “Missing Self” Mechanism
The missing-self recognition (166, 189) has been exploited to the greatest extent clinically in the context of HSCT for treatment of leukemia (189). When HLA-matched related or unrelated donors possess NK cell subsets that express KIRs with specificity for particular class I HLA ligands that are missing in the recipient’s cancer, a receptor-ligand mismatch (190) between the donor’s inhibitory KIR and recipient’s HLA arises (KIR-HLA mismatch) (Figure 2B) (in allogeneic, transplant setting). This mechanism has been shown to induce potent NK cell alloreactivity and to mediate graft versus malignancy effect but not treatment limiting GvHD in some leukemia types (189, 191). Since NK cells are the first lymphocyte subset to recover following HSCT, it is likely that they mediate the early graft versus malignancy effects (192). However, the benefit of KIR-HLA mismatch in unrelated donor, allogeneic HSCT has been debated. Several contradicting studies reported increased transplant related mortality (193), reduced overall survival and progression-free-survival (194) due to infections, resulting in higher relapse rates. Discrepancies such as age and disease stage of the patient at the time of allograft, differences in extent of T cell depletion through, either antithymocyte globuline (ATG) use, myeloablative regimen, or source and number of CD34+ stem cells, may account for the conflicting benefits of KIR-HLA mismatch reported (191, 194). The KIR-HLA interaction models used to determine NK-mediated graft-versus-leukemia (GVL) may also generate different results [i.e., HLA-ligand mismatch (189); KIR mismatch (195); receptor-ligand mismatch (196) or missing ligand (183)]. Recently KIR allelic polymorphisms have been reported to impact the effect of KIR-HLA ligand interactions, for example, the 25 alleles of KIR2DL1 have been shown to display variable inhibitory strengths and duration of expression at the surface after interaction with HLA-ligands. Thus, donor grafts containing KIR alleles with appropriate polymorphism motifs are associated clinically with fewer relapses, less transplant related mortality, and improved survival (197) regardless of primary disease, T cell depletion, or donor type. Increased GVL can be demonstrated under some transplantation conditions pointing to the possibility that KIR-HLA interactions are important in tumor control.
Activating KIRs and Haplotypes for Predicting NK Cell Potency Against Cancer
A study with mixed hematological malignancies reported that in the context of KIR-HLA ligand mismatch, donor haplotype B with high activating KIR genes (4–8 versus 1–3) was associated with poor disease-free survival (194), consistent with the disarming theory (198). However, they also analyzed impact of single KIR genes on relapse and revealed increased risk for AML, MDS, and CML patients transplanted with donors possessing KIR3DS1, KIR2DS1, and KIR2DS5. The latter two also display high linkage disequilibrium with KIR2DS3. Other studies demonstrated improved relapse-free and overall survival for AML patients with haplotype B (199), with the strongest prediction in the centromeric B region (Cen B) (200). Recently, haplotype B with telomeric A/B genotype, KIR2DL5, and KIR2DL1 in the presence of HLA-C2 strongly predicted post induction minimal residual disease with 100% sensitivity and 80% specificity in a cohort of 244 childhood lymphoblastic leukemia (201). We recently reported that NK cells isolated from healthy donors with haplotype B, KIR2DS2, and centromeric A/B or B/B, telomeric A/A or A/B genotype were the most potent against GBM cells in vitro and in vivo in mice compared to donors with KIR2DS4 (haplotype A, centromeric A/A, telomeric A/A) and KIR2DS2-/KIR2DS4- negative donors (centromeric A/A, telomeric A/A, A/B, or B/B) (40). The KIR2DS2+ NK cell subsets constitutively elevated CD69 and CD16 activation markers that remained elevated when in contact with GBM target cells. The KIR2DS2+ NK cell subsets preferentially degranulated granzyme A from elevated LAMP-1+ cytolytic lysosomes (40). Others reported that KIR2DS1+ NK cells from HLA-C1 donors efficiently lysed target cells with HLA-C2 ligand in vitro (202), although this was possibly mediated through KIR-HLA mismatch. Another study with fewer patients demonstrated contradicting results of worse outcome (203). The presence of KIR3DS1 was reported to decrease relapse rates in Bw4+ recipients (204), diminish risk for acute GvHD, and increase overall survival (205, 206). These effects were potentiated in the presence of two copies of KIR3DS1 gene. From these cumulative studies, it appears both inhibitory and activating KIRs are important for anti-tumor responses, although qualitative differences in the composite KIRs defining the haplotypes may be better predictive of treatment response.
Ultimately, cytotoxicity requires that the NK cell contacts its target cell and form a functional immunological synapse (207, 208) for optimal site directed secretion of perforin and granzymes A/B proteases (Figure 3). Intuitively, treatment response is also determined by the sensitivity of the cancer cells to the particular mode of death induced. Granzyme B induces rapid, intrinsic apoptosis mediated through cleavage of BH3-only pro-apoptotic protein Bid, cytochrome c, and downstream effector caspases 3 and 8 (209). Activated NK cells also express Fas Ligand [CD95L, (196), TNF-α, and TNF-related apoptosis inducing ligand (TRAIL)], and ligation of Fas (CD95) or TNF-α to death receptors leads to apoptosis of the receptor-bearing cell (67) (Figure 3). Although GBMs express the DR4, DR5 TNFR death domains, and CD95, they are inherently resistant to TRAIL and FasL. This is possibly due to deregulated downstream cascades involving BCL-2 and X-linked inhibitor of apoptosis proteins (XIAP) (147). Granzyme B also induces cytochrome c release by cleavage and inactivation of the anti-apoptotic BCL-2-family member Mcl-1 and substrates such as poly (ADP-ribose) polymerase (PARP), DNA-dependent protein kinase (DNA-PK), ICAD, and lamin B to generate double DNA-strand breaks. However, this signaling cascade is deregulated in solid cancers due to frequent p53 mutations, for example, in GBMs where p53 is mutated in 40% of cases (210). This results in upregulation of the pro-survival proteins, BCL-2 and BCL-XL, but downregulation of Bax, rendering them refractory to apoptosis (147). In contrast, granzyme A induced non-apoptotic cell death through release of ROS (211, 212), cleavage of nuclear lamins and histone H1 that are required for nuclear envelope stabilization and maintenance of chromatin structure (212, 213) to facilitate activity of DNAses. We recently demonstrated that potent alloreactive and highly activated KIR2DS2+ NK cell subsets killed apoptosis resistant GBM cells by preferentially degranulating granzyme A (40). Multiple mechanisms of inducing cell death are necessary to ensure the elimination of apoptosis-resistant cells (Figure 3).
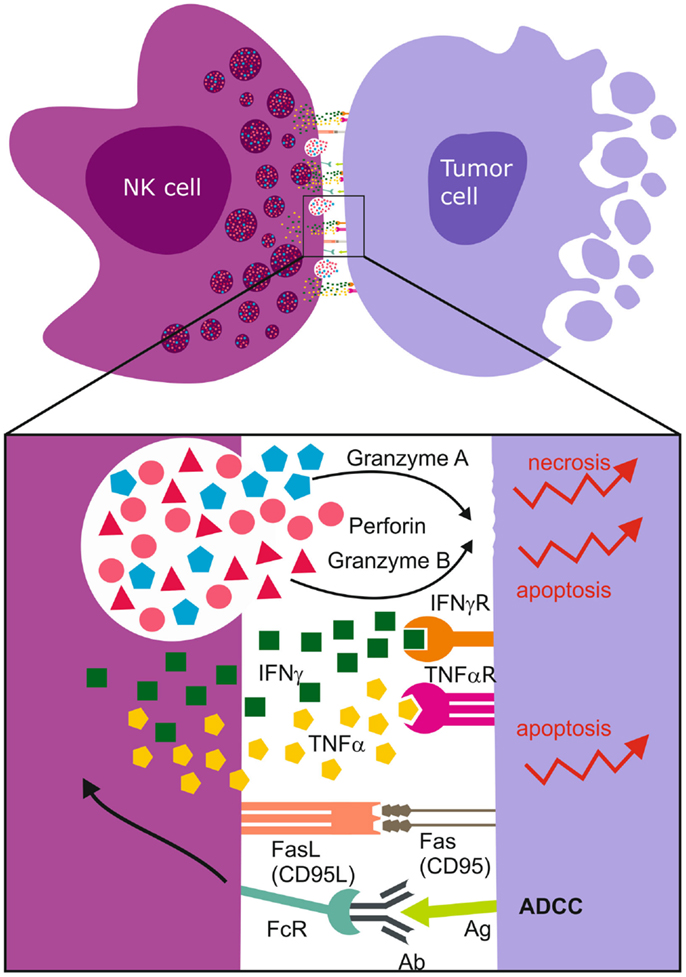
Figure 3. Immunological synapse and NK cell mediated cell death. Upon synapsing with target cells, NK cells release proteolytic enzymes, granzyme A and B, as well as perforins that induce necrotic death. The ligation of Fas Ligand (FasL) to Fas/CD95 receptor together with the binding of TNF-α to death receptors induce apoptosis of the tumor cell. NK cell FcγRIII (CD16) recognizes antibody Fc constant domains resulting in cytokine secretion and NK cell mediated antibody dependent cellular cytotoxicity (ADCC). The most abundantly secreted cytokines are TNF-α and IFN-γ.
Clinical Trials Exploiting NK Cell Activity Against Cancer
Autologous NK Cell Transfer
To circumvent the poor tropism into the tumor of high numbers of patients’ own NK cells, the latter can be expanded to high yields, activated ex vivo, and re-infused into patients’ circulation or tumor. This approach has the advantage of decreased risk for transplant related infections, graft rejection, or GvHD, lack of requirement for immunosuppression and histocompatibility matching. However, patients’ own educated NK cells might be inhibited to efficiently kill the tumor by self-HLA and the hostile immunosuppressiveTME. Autologous lymphokine-activated killer cells (LAK: polyclonal fractions of NK and T cells) were activated ex vivo in IL-2 and re-infused into melanoma and renal cell carcinoma patients, followed by adjuvant high dose IL-2 (214). The treatment was partially effective, although limited by low NK cell composition of the LAK product and severe toxicity due to IL-2 induced capillary leak syndrome. Furthermore, the adoptively transferred cells failed to persist in vivo. Although follow-up Phase II studies demonstrated that daily low dose (1.75 × 106 IU/m2/day) subcutaneous IL-2 was better tolerated in relapsed non-Hodgkin’s lymphoma and breast cancer patients and expanded cytotoxic T cells, no improvement in patient outcomes was reported. Instead, the IL-2 treatment also expanded Tregs (217). One trial using recurrent malignant glioma patients’ own expanded and enriched NK cell fractions placed them in an Ommaya reservoir (215, 216, 218) in the resection cavity. The treatment was supplemented with 100 IU/kg IL-2 at implantation, followed by low dose IFN-β (6 × 106 IU/week). The treatment was tolerated and partial radiological responses were reported (215). Attempts to circumvent the underlying tolerance administered a humanized IgG4 antibody IPH2101 that blocks KIR2DL1, KIR2DL2, and KIR2DL3 inhibitory signaling. However, as IPH2101 also binds KIR2DS1 and KIR2DS2, it may block crucial dominant activation signals provided by potent KIR2DS1, KIR2DS2 NK cell subsets (40). Indeed, IPH2101 has been investigated in early phase clinical trials for multiple myeloma and AML, and although few adverse events and toxicity were reported in multiple myeloma, long lasting objective responses were also not registered in the patients (219, 220), except for transient NK cell activation and cytotoxicity ex vivo. The next generation hinge-stabilized IPH2102 was recently shown to synergize with Lenalidomide to increase NK cytotoxicity of myeloma patients with FcγRIII and FcγRII polymorphisms treated with Daratumumab against CD38 (221). While IPH2102 blocked KIR-inhibitory signaling on the NK cells, Lenalidomide activated them to increase production of TNF-α, IFN-γ, and granzyme B. The tumor can also be sensitized to autologous NK cells by coadministering proteosome inhibitors (e.g., Bortezomib), doxorubicin, or histone deacetylase inhibitors that upregulate stress ligands for NKG2D receptors, induce pro-inflammatory cytokines or death receptors that bind TNF-α. Although Bortezomib was effective in preclinical models mediated by upregulated death receptors (TRAILR1-DR4, TNFR1, DR3 and DR6) as well as stress ligands on the tumor cells, it was not effective in AML and RCC patients (99, 222). The efficacy of Bortezomib might be contingent upon the inherent susceptibility of the tumor cell to undergo TRAIL and FASL dependent apoptosis. The dose, duration, and sequence of administration of agents when combining with NK cells requires fine-tuning as unexpected effects on NK cell function and or tumor sensitization may occur (223). During NK cell therapy, concurrent treatment with azacytidine and sorafenib, which impair PI3K and ERK phosphotylation might also hamper NK cell activity and be counter productive in augmenting tumor cell kill by the immunological mechanisms (224, 225). Lenalidomide was shown to enhance Rituximab induced ADCC against non-Hodgkin lymphoma and B-cell chronic lymphocytic leukemia through enhanced CD4+ T cell dependent IL-2 release, resulting in augmented NK cell activation, granzyme B, and FasL release (226). NK cells could also be stimulated to enhanced activation, proliferation, and persistence with anti-CD137 agonistic antibodies.
Allogeneic NK Cell Transfer
Due to challenges posed by tolerance to self-cells, infusions of allogeneic NK cells derived from related or HLA-matched unrelated donors are increasingly pursued for adoptive cellular therapies. The cells can be obtained from donor bone marrow progenitors, umbilical cord, or peripheral blood leukapheresis product. Subsequently, T lymphocytes and B cells are depleted with anti-CD3 or anti-CD19 microbeads, respectively. This is essential to prevent GvHD and lymphoproliferative disorders (227) induced by transfer of T cells and Epstein Barr virus (EBV) transformed B cells, respectively. To prevent GvHD, the final T cell dose should be <3 × 105 cells/kg or below 0.1% total lymphocyte count (99, 228). 1 × 108 purified allogeneic NK cells have been safely transfused to patients without major toxicity, although a threshold of maximum tolerated doses have yet to be established. Initial studies demonstrated that absence of recipient lymphoid and myeloid cell depletion prior to donor cell infusion resulted in diminished NK cell persistence in vivo and reduced therapeutic efficacy. Thus, an immune suppressive “preparative” regimen became imperative prior to donor cell infusions. In a non-transplant treatment of AML patients, investigators compared low dose non-myeloablative fludarabine (25 mg/m2/day) for 5 days or high cyclophosphomide (60 mg/m2/day) for 2 days chased by fludarabine (25 mg/m2/day) for 5 days (Hi-Cy/Flu) prior to donor NK cell infusions and adjuvant IL-2 (1.75 million U/m2) for 14 days. Only patients receiving Hi-Cy/Flu regimen had in vivo detectable NK cells in peripheral blood 2 weeks post treatment based on PCR donor-recipient chimerism assay and measurable circulating IL-15 levels (99). Thus, depletion of the lymphoid and myeloid cells that also utilize and compete for IL-15 resulted in elevated serum levels of IL-15 and IL-7, allowing for homeostatic proliferation and expansion of the NK and CD8T cells in vivo. Objective therapeutic responses were correlated with the prolonged presence of donor NK cells in the recipient. The infused NK cells were tolerated and did not induce GvHD and notably, 25% high risk, poor prognosis AML patients exhibited complete responses (229). This regimen is widely used with minor modifications for trials in leukemia, non-Hodgkin’s lymphoma, breast, ovarian, and NSCLCs. Allogeneic NK cells combined with 2 weeks’ adjuvant IL-2 were administered in a phase II trial in 20 patients with solid tumors (14 ovarian, 6 breast cancer) following the low dose, non-myeloablative preparative regimen (230). Very few peripheral blood NK cells were detected after 1 week in the majority of patients and the expanded cells were mostly Tregs (99) due to the IL-2 treatment. Although proven safe in these settings, a major bottleneck to clinical efficacy remains consistent in vivo NK cell expansion and abrogation of the tumor induced immunosuppressive mechanisms.
Methods for Expanding NK Cells in High Numbers
Cytokines
Depending on whether a lymphodepletive regimen is given, typical NK cell infusions may vary from 8–20 × 106 to 1 × 109 cells/kg. In unconditioned patients, high NK cells doses are required to achieve therapeutic effects, thus high throughput expansion methods are required. Moreover, since NK cells constitute only 10–15% of peripheral blood lymphocytes, and freshly isolated cells are in resting state and not optimally cytotoxic, activation by cytokines in culture is additionally required. Initially, cytokine cocktails were administered in vivo but they evoked disappointing efficacy in clinical studies largely due to toxicity, or activation-induced cell death (AICD), or exhaustion (231) where the effector cells succumbed to apoptosis upon contact with vascular endothelium. Recently, cytokine therapy in preclinical studies demonstrated that IL-12, IL-18, and the genetically engineered “superkine” H9 reverted the functional anergy of NK cells that was induced by class I MHC deficient tumors and prolonged animal survival (232). NK cell stimulation with IL-12 and IL-18 elevated expression of T-cell immunoglobulin and mucin domain-containing (Tim)-3 receptor (233), previously associated with T cell exhaustion (234). However, in this study, TIM-3 was conversely proposed to denote NK cell activation and maturation, and only weakly suppressed NK cell cytotoxicity (233). “Memory” NK cells with enhanced proliferation and IFN-γ production could be generated by pre-activation with a cytokine cocktail including IL-12, IL-18, and IL-15 followed by a 3-week recovery period prior to re-stimulation with IL-12 + IL-15, or IL-12 + IL-18, or K562 (235). IL-2 increases NK cytotoxicity in vitro by increasing density of activation receptors (236), and sustains NK survival in vivo but off-target effects associated with vascular leak syndrome induced by stimulation of IL-2R on endothelial cells are problematic. The H9 superkine that signals independently of the IL-2Rα chain (237) may redress the above shortcoming and derive therapeutic benefits in the clinic. Combination IL-2 and IL-15 markedly prolongs NK cell survival. Uniquely, IL-15 inhibits IL-2 dependent AICD, (238) and decreases expression of PD-1. IL-15 promotes NK cell survival and expansion, especially when presented to NK cells in trans as a membrane bound complex with IL-15Rα by DCs and monocytes. Recently, a first in human trial investigated safety and maximum tolerated dose of a recombinant human (rh) IL-15 in patients with metastatic malignancy (239). Higher doses of IL-15 resulted in rapid redistribution of NK and CD8T cells from peripheral blood within 48 h followed by hyper-proliferation, where NK cells expanded upto 10-fold of baseline. The cells were highly activated and intensely released cytokines, IL-8, IL-10, TNF-α, IL-1β, IL-6, and IFN-γ, resulting in radiologically visible anti-tumor effects in some patients. However, the treatment was associated with dose limiting severe adverse events and the authors concluded the MTD for IL-15 in this format was 0.3 mg/kg/day (239). In other studies, CD34+ progenitor cells from umbilical cord blood (UCB) cells could be differentiated to NK cells in medium supplemented with low-dose GM-CSF, G-CSF, IL-6, and a high-dose cytokine cocktail consisting of IL-7, stem cell factor (SCF), IL-15, and IL-2. However, the cells displayed low CD16 and KIR expression at the end of the culture process (240, 241). Alternatively, CD34+ cells from adult peripheral blood could be differentiated in the presence of SCF, FLT3 ligand, IL-7, and hydrocortisone, followed by IL-7, IL-15, and hydrocortisone. No major toxicity was recorded when using these cells (240). These methods typically produce small-scale NK expansions in the range 10- to 20-fold that might not be sufficient to treat patients in trials.
Feeder Cells
Coculture of CD56+ purified NK cells with irradiated feeder cells has been demonstrated to expand and activate NK cells from T and B cell depleted peripheral blood of patients 800- to 1000-fold in a short time scale of 2 weeks (236). This NK cell product upregulated expression of NKG2D, NKG2C, secreted FasL, IFN-γ, IL-2, TRAIL, Granzymes A/B, and was highly cytotoxic against targets. The EBV transformed lymohoblastoid (EBV-LCL) feeder cells were irradiated with 100 Gy and cocultured in 500 IU/ml IL-2 at 20:1 ratio with 2 × 108 purified peripheral blood NK cells obtained from 15 l leukapheresis product. Clinical grade NK cells expanded upto 3 × 1010 within a 3-week period (236). However, attempts to further expand beyond this failed, and it is not clear if this is due to AICD or replicative senescence due to shortened telomeres. Importantly, this study also demonstrated that previously expanded and frozen NK cells, when subsequently thawed, require further activation in culture with IL-2 to maintain their cytotoxicity although their viability is substantially compromised. Others have utilized feeder cell-free, automated bioreactor systems to expand highly cytotoxic cells from polyclonal PBMCs with 77-fold yield in 21 days. However, these were impure and consisted mostly T cells (CD3+CD56-) and NKT cells (CD56+CD3+) (242). This product had very low NK cells, average 38% (range 10–80%), and bears the treatment limiting risk of inducing GvHD. However, an autologous setting may be more permissive of T cell contamination, although they may differentiate into Tregs. Further manipulation to deplete the T cells prior to infusion in the patient would be required and is potentially costly (243). The K562 CML cells have been engineered to express membrane bound IL-15 gene fused to CD8α transmembrane domain as well as membrane bound 4-1BBL (mb15-41BBL) and shown to induce nearly 22-fold NK expansion within 1 week culture in 10 IU/ml IL-2 (244). In other studies, K562-based artificial antigen presenting cells were modified to express membrane bound IL-15 or IL-21 (245). Upon comparison, MbIL-21 expanded cells were superior to mbIL-15 expanded NK cells in that they exhibited an activated phenotype (denoted by elevated CD160 expression, (246), proliferated better, and elongated their telomeres (postulated due to STAT3 dependent signaling). They retained expression of KIR, CD16, produced cytokines, and were potent mediators of cellular cytotoxicity. Depending on the class I HLA ligands expressed on the surface of the feeder cells, it may be possible to selectively expand NK cells with particular KIR repertoires selected for potent efficacy (247). Since the alloreactive fraction in the polyclonal NK cell pool varies widely from 0 to 62% among potential donors (177), not all individuals are expected to be potent donors. Improving our understanding of molecular KIR/HLA combinations that predict best therapeutic benefit is imperative in next generation NK cell therapy. We recently demonstrated that donor NK cells with KIR2DS2 immunogenotype exhibited potent efficacy against GBM targets (40), even though they represented only 36% of the total NK cell pool. It is highly compelling to investigate whether selective expansion of these highly activated KIR2DS2 NK cell fractions would yield greater clinical benefits for GBM patients than using bulk NK cells.
Conclusion and Perspectives
Our deeper understanding of NK cell biology and function in anti-cancer immunity, the increasingly precise methodologies for their characterization, purification, and expansion have all heightened our interest in exploring the applicability of NK cells for cancer immunotherapy. However, good manufacturing practice approved protocols for generating high quantities of pure, activated NK cells for repeated infusion into patients remain complicated, expensive, and are still somewhat experimental. Likewise, appropriate conditioning regimens to prevent graft rejection, that can be used for a wide range of cancer types, the right choice of exogenous cytokines to safely sustain homeostatic NK cell expansion, activation, and survival in vivo are all issues still under investigation. Nevertheless, concerted research efforts are underway to refine and renew the protocols to establish NK cells as efficacious effectors against malignancy. It is intriguing that the multitude of ways to enhance NK cytotoxicity seems to impact fundamental regulatory mechanisms such as receptor-ligand mismatch, expression of activating receptors, increasing receptor affinity, augmentation of ADCC. All the same, we do not yet know how to reap maximal benefit of this expertise in the clinic. It may be because the clearest evidence accrued so far is most often seen in larger cohort studies. Nevertheless, the salient question is what will it take to make NK cell therapy standard care for GBM and other solid cancers? Due to the poor tumor infiltration and limited passage across the blood brain barrier (39, 248), NK cell therapy in the brain might be best suited as a haplotype-matched, intralesional combination NK cell therapy with mAbs. Exploiting NK cell subsets with best predictive KIR/HLA combinations might be advantageous, since autologous NK cells previously demonstrated limited efficacy. For this to be effective, tumor reduction prior to NK therapy is an imperative in order to give the NK cells a task they can manage. The cells could be injected into and around the resection cavity and administered as an adjuvant therapy post standard treatment that combines debulking surgery, concomitant Temozolomide chemotherapy, and conformal ionizing radiation (249). This would exploit the period of a relative immune compromise (250) to promote homeostatic proliferation of grafted cells and prevent graft rejection. A phase I dose escalation toxicity study would be required as the brain may present unique toxicity and necessitates careful trial design. Strategies to consider might be single treatment with high numbers (effector: target ratios) of maximally activated cells or lower numbers and repeated doses to promote in vivo persistence in the recipient. We recently demonstrated in preclinical models that large, single NK cell doses were efficient and tolerated whereas repeated high doses were poorly tolerated in mice (40). If these results were translatable to humans, in a patient phase I trial, smaller dose escalations with maximally alloreactive subsets may be better tolerated, not least more feasible with regards to ex vivo NK cell expansions. Moreover, we demonstrated using multiparametric physiological magnetic resonance imaging (MRI) to identify predictive biomarkers, that 2 × 106 intralesional NK cells were more effective compared to 1 × 106 cells, even when these were combined with mAb9.2.27 in nude rats (158, 251). Ultimately, NK cells in solid tumors should be combined with mAbs against abundant tumor specific antigens to enhance their efficacy via ADCC and overcome inherent immunosuppression in the tumor bed (158). Immunologic surrogates that predict efficacy in patients could be IFN-γ, as this has been previously correlated with anti-tumor responses and overall survival (103).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to The Norwegian Research Council (FRIMEDBIO grant 230691) for supporting our research and thank Mireia Gras Navarro for assistance with illustrations.
References
1. Nowell PC. The clonal evolution of tumor cell populations. Science (1976) 194(4260):23–8. doi:10.1126/science.959840
2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi:10.1016/S0092-8674(00)81683-9
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
4. Roder JC, Haliotis T, Klein M, Korec S, Jett JR, Ortaldo J, et al. A new immunodeficiency disorder in humans involving NK cells. Nature (1980) 284(5756):553–5. doi:10.1038/284553a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Sullivan JL, Byron KS, Brewster FE, Purtilo DT. Deficient natural killer cell activity in x-linked lymphoproliferative syndrome. Science (1980) 210(4469):543–5. doi:10.1126/science.6158759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Penn I. Development of cancer as a complication of clinical transplantation. Transplant Proc (1977) 9(1):1121–7.
7. Corthay A. Does the immune system naturally protect against cancer? Front Immunol (2014) 5:197. doi:10.3389/fimmu.2014.00197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Engels EA, Pfeiffer RM, Fraumeni JF Jr, Kasiske BL, Israni AK, Snyder JJ, et al. Spectrum of cancer risk among US solid organ transplant recipients. JAMA (2011) 306(17):1891–901. doi:10.1001/jama.2011.1592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet (2000) 356(9244):1795–9. doi:10.1016/S0140-6736(00)03231-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Hersey P, Edwards A, Honeyman M, McCarthy WH. Low natural-killer-cell activity in familial melanoma patients and their relatives. Br J Cancer (1979) 40(1):113–22. doi:10.1038/bjc.1979.147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
13. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol (2004) 22:405–29. doi:10.1146/annurev.immunol.22.012703.104711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Zhang Y, Wallace DL, de Lara CM, Ghattas H, Asquith B, Worth A, et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology (2007) 121(2):258–65. doi:10.1111/j.1365-2567.2007.02573.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Moretta A, Bottino C, Mingari MC, Biassoni R, Moretta L. What is a natural killer cell? Nat Immunol (2002) 3(1):6–8. doi:10.1038/ni0102-6
17. Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A (2009) 106(6):1915–9. doi:10.1073/pnas.0813192106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104(12):3664–71. doi:10.1182/blood-2004-05-2058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Guma M, Budt M, Saez A, Brckalo T, Hengel H, Angulo A, et al. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood (2006) 107(9):3624–31. doi:10.1182/blood-2005-09-3682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Barton K, Muthusamy N, Fischer C, Ting CN, Walunas TL, Lanier LL, et al. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity (1998) 9(4):555–63. doi:10.1016/S1074-7613(00)80638-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Gascoyne DM, Long E, Veiga-Fernandes H, de Boer J, Williams O, Seddon B, et al. The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol (2009) 10(10):1118–24. doi:10.1038/ni.1787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Kallies A, Carotta S, Huntington ND, Bernard NJ, Tarlinton DM, Smyth MJ, et al. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood (2011) 117(6):1869–79. doi:10.1182/blood-2010-08-303123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Lacorazza HD, Miyazaki Y, Di Cristofano A, Deblasio A, Hedvat C, Zhang J, et al. The ETS protein MEF plays a critical role in perforin gene expression and the development of natural killer and NK-T cells. Immunity (2002) 17(4):437–49. doi:10.1016/S1074-7613(02)00422-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Ohno S, Sato T, Kohu K, Takeda K, Okumura K, Satake M, et al. Runx proteins are involved in regulation of CD122, Ly49 family and IFN-gamma expression during NK cell differentiation. Int Immunol (2008) 20(1):71–9. doi:10.1093/intimm/dxm120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Samson SI, Richard O, Tavian M, Ranson T, Vosshenrich CA, Colucci F, et al. GATA-3 promotes maturation, IFN-gamma production, and liver-specific homing of NK cells. Immunity (2003) 19(5):701–11. doi:10.1016/S1074-7613(03)00294-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Kaisho T, Tsutsui H, Tanaka T, Tsujimura T, Takeda K, Kawai T, et al. Impairment of natural killer cytotoxic activity and interferon gamma production in CCAAT/enhancer binding protein gamma-deficient mice. J Exp Med (1999) 190(11):1573–82. doi:10.1084/jem.190.11.1573
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol (2012) 30:647–75. doi:10.1146/annurev-immunol-020711-075053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5(2):112–7. doi:10.1002/eji.1830050208
29. Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity (1997) 7(4):493–504. doi:10.1016/S1074-7613(00)80371-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer (1975) 16(2):216–29. doi:10.1002/ijc.2910160204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol (2001) 19:197–223. doi:10.1146/annurev.immunol.19.1.197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med (2009) 206(7):1495–503. doi:10.1084/jem.20090681
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol (2013) 34(4):182–91. doi:10.1016/j.it.2013.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Pessino A, Sivori S, Bottino C, Malaspina A, Morelli L, Moretta L, et al. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J Exp Med (1998) 188(5):953–60. doi:10.1084/jem.188.5.953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, Enk J, et al. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol (2012) 188(6):2509–15. doi:10.4049/jimmunol.1102461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol (2003) 3(10):781–90. doi:10.1038/nri1199
37. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A (1996) 93(22):12445–50. doi:10.1073/pnas.93.22.12445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science (1999) 285(5428):730–2. doi:10.1126/science.285.5428.730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol (2013) 264(1–2):71–83. doi:10.1016/j.jneuroim.2013.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Gras Navarro A, Kmiecik J, Leiss L, Zelkowski M, Engelsen A, Bruserud O, et al. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J Immunol (2014) 193(12):6192–206. doi:10.4049/jimmunol.1400859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res (2008) 68(15):6368–76. doi:10.1158/0008-5472.CAN-07-6768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. White JM. ADAMs: modulators of cell-cell and cell-matrix interactions. Curr Opin Cell Biol (2003) 15(5):598–606. doi:10.1016/j.ceb.2003.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Chitadze G, Bhat J, Lettau M, Janssen O, Kabelitz D. Generation of soluble NKG2D ligands: proteolytic cleavage, exosome secretion and functional implications. Scand J Immunol (2013) 78(2):120–9. doi:10.1111/sji.12072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169(8):4098–102. doi:10.4049/jimmunol.169.8.4098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Chang YT, Wu CC, Shyr YM, Chen TC, Hwang TL, Yeh TS, et al. Secretome-based identification of ULBP2 as a novel serum marker for pancreatic cancer detection. PLoS One (2011) 6(5):e20029. doi:10.1371/journal.pone.0020029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Nuckel H, Switala M, Sellmann L, Horn PA, Durig J, Duhrsen U, et al. The prognostic significance of soluble NKG2D ligands in B-cell chronic lymphocytic leukemia. Leukemia (2010) 24(6):1152–9. doi:10.1038/leu.2010.74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Paschen A, Sucker A, Hill B, Moll I, Zapatka M, Nguyen XD, et al. Differential clinical significance of individual NKG2D ligands in melanoma: soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin Cancer Res (2009) 15(16):5208–15. doi:10.1158/1078-0432.CCR-09-0886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Codo P, Weller M, Meister G, Szabo E, Steinle A, Wolter M, et al. MicroRNA-mediated down-regulation of NKG2D ligands contributes to glioma immune escape. Oncotarget (2014) 5(17):7651–62.
49. Chen D, Juko-Pecirep I, Hammer J, Ivansson E, Enroth S, Gustavsson I, et al. Genome-wide association study of susceptibility loci for cervical cancer. J Natl Cancer Inst (2013) 105(9):624–33. doi:10.1093/jnci/djt051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Chen D, Gyllensten U. MICA polymorphism: biology and importance in cancer. Carcinogenesis (2014) 35(12):2633–42. doi:10.1093/carcin/bgu215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lo PH, Urabe Y, Kumar V, Tanikawa C, Koike K, Kato N, et al. Identification of a functional variant in the MICA promoter which regulates MICA expression and increases HCV-related hepatocellular carcinoma risk. PLoS One (2013) 8(4):e61279. doi:10.1371/journal.pone.0061279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci U S A (2014) 111(35):12823–8. doi:10.1073/pnas.1413933111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature (2007) 447(7143):482–6. doi:10.1038/nature05768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Pradeu T, Jaeger S, Vivier E. The speed of change: towards a discontinuity theory of immunity? Nat Rev Immunol (2013) 13(10):764–9. doi:10.1038/nri3521
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol (2004) 172(12):7335–40. doi:10.4049/jimmunol.172.12.7335
56. Tseng HC, Bui V, Man YG, Cacalano N, Jewett A. Induction of split anergy conditions natural killer cells to promote differentiation of stem cells through cell-cell contact and secreted factors. Front Immunol (2014) 5:269. doi:10.3389/fimmu.2014.00269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Sakariassen PO, Immervoll H, Chekenya M. Cancer stem cells as mediators of treatment resistance in brain tumors: status and controversies. Neoplasia (2007) 9(11):882–92. doi:10.1593/neo.07658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature (2012) 488(7412):522–6. doi:10.1038/nature11287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Tseng HC, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A, et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS One (2010) 5(7):e11590. doi:10.1371/journal.pone.0011590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Tseng HC, Cacalano N, Jewett A. Split anergized natural killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget (2015).
61. Lanier LL, Testi R, Bindl J, Phillips JH. Identity of Leu-19 (CD56) leukocyte differentiation antigen and neural cell adhesion molecule. J Exp Med (1989) 169(6):2233–8. doi:10.1084/jem.169.6.2233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Frey M, Packianathan NB, Fehniger TA, Ross ME, Wang WC, Stewart CC, et al. Differential expression and function of l-selectin on CD56bright and CD56dim natural killer cell subsets. J Immunol (1998) 161(1):400–8.
63. Caligiuri MA, Murray C, Robertson MJ, Wang E, Cochran K, Cameron C, et al. Selective modulation of human natural killer cells in vivo after prolonged infusion of low dose recombinant interleukin 2. J Clin Invest (1993) 91(1):123–32. doi:10.1172/JCI116161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Romagnani C, Juelke K, Falco M, Morandi B, D’Agostino A, Costa R, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol (2007) 178(8):4947–55. doi:10.4049/jimmunol.178.8.4947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Smith KA. Interleukin-2. Curr Opin Immunol (1992) 4(3):271–6. doi:10.1016/0952-7915(92)90076-Q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol (2004) 5(12):1260–5. doi:10.1038/ni1138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer (2002) 2(11):850–61. doi:10.1038/nrc928
68. Reddy PG, Graham GM, Datta S, Guarini L, Moulton TA, Jiang HP, et al. Effect of recombinant fibroblast interferon and recombinant immune interferon on growth and the antigenic phenotype of multidrug-resistant human glioblastoma multiforme cells. J Natl Cancer Inst (1991) 83(18):1307–15. doi:10.1093/jnci/83.18.1307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Campbell JJ, Qin S, Unutmaz D, Soler D, Murphy KE, Hodge MR, et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol (2001) 166(11):6477–82. doi:10.4049/jimmunol.166.11.6477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Stojanovic A, Cerwenka A. Natural killer cells and solid tumors. J Innate Immun (2011) 3(4):355–64. doi:10.1159/000325465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol (2001) 22(11):633–40. doi:10.1016/S1471-4906(01)02060-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331(6013):44–9. doi:10.1126/science.1198687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Smith FO, Rauch C, Williams DE, March CJ, Arthur D, Hilden J, et al. The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood (1996) 87(3):1123–33.
74. Campbell JJ, Hedrick J, Zlotnik A, Siani MA, Thompson DA, Butcher EC. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science (1998) 279(5349):381–4. doi:10.1126/science.279.5349.381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Bjorkstrom NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood (2010) 116(19):3853–64. doi:10.1182/blood-2010-04-281675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Lopez-Verges S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood (2010) 116(19):3865–74. doi:10.1182/blood-2010-04-282301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood (2010) 115(11):2167–76. doi:10.1182/blood-2009-08-238469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Lanier LL. Of snowflakes and natural killer cell subsets. Nat Biotechnol (2014) 32(2):140–2. doi:10.1038/nbt.2810
79. Mocikat R, Braumuller H, Gumy A, Egeter O, Ziegler H, Reusch U, et al. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity (2003) 19(4):561–9. doi:10.1016/S1074-7613(03)00264-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Adam C, King S, Allgeier T, Braumuller H, Luking C, Mysliwietz J, et al. DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction. Blood (2005) 106(1):338–44. doi:10.1182/blood-2004-09-3775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol (2014) 44(6):1582–92. doi:10.1002/eji.201344272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Bodmer S, Huber D, Heid I, Fontana A. Human glioblastoma cell derived transforming growth factor-beta 2: evidence for secretion of both high and low molecular weight biologically active forms. J Neuroimmunol (1991) 34(1):33–42. doi:10.1016/0165-5728(91)90096-P
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Elliott RL, Blobe GC. Role of transforming growth factor beta in human cancer. J Clin Oncol (2005) 23(9):2078–93. doi:10.1200/JCO.2005.02.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Roszman T, Elliott L, Brooks W. Modulation of T-cell function by gliomas. Immunol Today (1991) 12(10):370–4. doi:10.1016/0167-5699(91)90068-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol (2006) 6(10):715–27. doi:10.1038/nri1936
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi:10.1038/nri3175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H, et al. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res (2008) 68(12):4746–53. doi:10.1158/0008-5472.CAN-08-0054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood (2008) 112(10):4080–9. doi:10.1182/blood-2008-03-143776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182(1):240–9. doi:10.4049/jimmunol.182.1.240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res (2010) 16(3):800–13. doi:10.1158/1078-0432.CCR-09-2730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Schatton T, Schutte U, Frank NY, Zhan Q, Hoerning A, Robles SC, et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res (2010) 70(2):697–708. doi:10.1158/0008-5472.CAN-09-1592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther (2010) 9(1):67–78. doi:10.1158/1535-7163.MCT-09-0734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med (2005) 11(12):1314–21. doi:10.1038/nm1325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM, et al. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J Biol Chem (2013) 288(36):26167–76. doi:10.1074/jbc.M113.477950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Jewett A, Cacalano NA, Teruel A, Romero M, Rashedi M, Wang M, et al. Inhibition of nuclear factor kappa B (NFkappaB) activity in oral tumor cells prevents depletion of NK cells and increases their functional activation. Cancer Immunol Immunother (2006) 55(9):1052–63. doi:10.1007/s00262-005-0093-7
96. Tallerico R, Todaro M, Di Franco S, Maccalli C, Garofalo C, Sottile R, et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol (2013) 190(5):2381–90. doi:10.4049/jimmunol.1201542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Avril T, Vauleon E, Hamlat A, Saikali S, Etcheverry A, Delmas C, et al. Human glioblastoma stem-like cells are more sensitive to allogeneic NK and T cell-mediated killing compared with serum-cultured glioblastoma cells. Brain Pathol (2012) 22(2):159–74. doi:10.1111/j.1750-3639.2011.00515.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol (2009) 182(6):3530–9. doi:10.4049/jimmunol.0802845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Knorr DA, Bachanova V, Verneris MR, Miller JS. Clinical utility of natural killer cells in cancer therapy and transplantation. Semin Immunol (2014) 26(2):161–72. doi:10.1016/j.smim.2014.02.002
100. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A (2003) 100(7):4120–5. doi:10.1073/pnas.0730640100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Sato N, Saga Y, Mizukami H, Wang D, Takahashi S, Nonaka H, et al. Downregulation of indoleamine-2,3-dioxygenase in cervical cancer cells suppresses tumor growth by promoting natural killer cell accumulation. Oncol Rep (2012) 28(5):1574–8. doi:10.3892/or.2012.1984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Ghiringhelli F, Menard C, Martin F, Zitvogel L. The role of regulatory T cells in the control of natural killer cells: relevance during tumor progression. Immunol Rev (2006) 214:229–38. doi:10.1111/j.1600-065X.2006.00445.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Pellegatta S, Eoli M, Frigerio S, Antozzi C, Bruzzone MG, Cantini G, et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology (2013) 2(3):e23401. doi:10.4161/onci.23401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett (2012) 318(2):154–61. doi:10.1016/j.canlet.2011.12.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Li T, Yi S, Liu W, Jia C, Wang G, Hua X, et al. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med Oncol (2013) 30(3):663. doi:10.1007/s12032-013-0663-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A (2009) 106(49):20847–52. doi:10.1073/pnas.0906481106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Kmiecik J, Zimmer J, Chekenya M. Natural killer cells in intracranial neoplasms: presence and therapeutic efficacy against brain tumours. J Neurooncol (2014) 116(1):1–9. doi:10.1007/s11060-013-1265-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Kondo E, Koda K, Takiguchi N, Oda K, Seike K, Ishizuka M, et al. Preoperative natural killer cell activity as a prognostic factor for distant metastasis following surgery for colon cancer. Dig Surg (2003) 20(5):445–51. doi:10.1159/000072714
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Hsia JY, Chen JT, Chen CY, Hsu CP, Miaw J, Huang YS, et al. Prognostic significance of intratumoral natural killer cells in primary resected esophageal squamous cell carcinoma. Chang Gung Med J (2005) 28(5):335–40.
110. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer (2000) 88(3):577–83. doi:10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Villegas FR, Coca S, Villarrubia VG, Jimenez R, Chillon MJ, Jareno J, et al. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer (2002) 35(1):23–8. doi:10.1016/S0169-5002(01)00292-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Plonquet A, Haioun C, Jais JP, Debard AL, Salles G, Bene MC, et al. Groupe d’etude des lymphomes de: peripheral blood natural killer cell count is associated with clinical outcome in patients with aaIPI 2-3 diffuse large B-cell lymphoma. Ann Oncol (2007) 18(7):1209–15. doi:10.1093/annonc/mdm110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Mechtersheimer G, Staudter M, Moller P. Expression of the natural killer cell-associated antigens CD56 and CD57 in human neural and striated muscle cells and in their tumors. Cancer Res (1991) 51(4):1300–7.
114. Schleypen JS, Von Geldern M, Weiss EH, Kotzias N, Rohrmann K, Schendel DJ, et al. Renal cell carcinoma-infiltrating natural killer cells express differential repertoires of activating and inhibitory receptors and are inhibited by specific HLA class I allotypes. Int J Cancer (2003) 106(6):905–12. doi:10.1002/ijc.11321
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, Lyonnet L, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med (2011) 17(6):700–7. doi:10.1038/nm.2366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Merritt WD, Bailey JM, Pluznik DH. Inhibition of interleukin-2-dependent cytotoxic T-lymphocyte growth by gangliosides. Cell Immunol (1984) 89(1):1–10. doi:10.1016/0008-8749(84)90191-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Prokazova NV, Dyatlovitskaya EV, Bergelson LD. Sialylated lactosylceramides. Possible inducers of non-specific immunosuppression and atherosclerotic lesions. Eur J Biochem (1988) 172(1):1–6. doi:10.1111/j.1432-1033.1988.tb13847.x
118. Prokazova NV, Orekhov AN, Mukhin DN, Mikhailenko IA, Kogtev LS, Sadovskaya VL, et al. The gangliosides of adult human aorta: intima, media and plaque. Eur J Biochem (1987) 167(2):349–52. doi:10.1111/j.1432-1033.1987.tb13343.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Chekenya M, Enger PO, Thorsen F, Tysnes BB, Al-Sarraj S, Read TA, et al. The glial precursor proteoglycan, NG2, is expressed on tumour neovasculature by vascular pericytes in human malignant brain tumours. Neuropathol Appl Neurobiol (2002) 28(5):367–80. doi:10.1046/j.1365-2990.2002.00412.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Wagener R, Rohn G, Schillinger G, Schroder R, Kobbe B, Ernestus RI. Ganglioside profiles in human gliomas: quantification by microbore high performance liquid chromatography and correlation to histomorphology and grading. Acta Neurochir (Wien) (1999) 141(12):1339–45. doi:10.1007/s007010050439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother (2014) 64(2):225–35. doi:10.1007/s00262-014-1629-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res (2008) 68(20):8437–45. doi:10.1158/0008-5472.CAN-08-1440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res (2011) 17(4):678–89. doi:10.1158/1078-0432.CCR-10-2173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Pachynski RK, Zabel BA, Kohrt HE, Tejeda NM, Monnier J, Swanson CD, et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J Exp Med (2012) 209(8):1427–35. doi:10.1084/jem.20112124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Harlin H, Hanson M, Johansson CC, Sakurai D, Poschke I, Norell H, et al. The CD16- CD56(bright) NK cell subset is resistant to reactive oxygen species produced by activated granulocytes and has higher antioxidative capacity than the CD16+ CD56(dim) subset. J Immunol (2007) 179(7):4513–9. doi:10.4049/jimmunol.179.7.4513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Lanier LL, Le AM, Phillips JH, Warner NL, Babcock GF. Subpopulations of human natural killer cells defined by expression of the Leu-7 (HNK-1) and Leu-11 (NK-15) antigens. J Immunol (1983) 131(4):1789–96.
127. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest (2011) 121(9):3609–22. doi:10.1172/JCI45816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Jewett A, Man YG, Tseng HC. Dual functions of natural killer cells in selection and differentiation of stem cells; role in regulation of inflammation and regeneration of tissues. J Cancer (2013) 4(1):12–24. doi:10.7150/jca.5519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Rabinowich H, Suminami Y, Reichert TE, Crowley-Nowick P, Bell M, Edwards R, et al. Expression of cytokine genes or proteins and signaling molecules in lymphocytes associated with human ovarian carcinoma. Int J Cancer (1996) 68(3):276–84. doi:10.1002/(SICI)1097-0215(19961104)68:3<276::AID-IJC2>3.3.CO;2-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol (1998) 16(8):2825–33.
131. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol (2003) 21(21):3940–7. doi:10.1200/JCO.2003.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood (2002) 99(3):754–8. doi:10.1182/blood.V99.3.754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Lehrnbecher T, Foster CB, Zhu S, Leitman SF, Goldin LR, Huppi K, et al. Variant genotypes of the low-affinity Fcgamma receptors in two control populations and a review of low-affinity Fcgamma receptor polymorphisms in control and disease populations. Blood (1999) 94(12):4220–32.
134. Laurent S, Queirolo P, Boero S, Salvi S, Piccioli P, Boccardo S, et al. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-alpha production. J Transl Med (2013) 11:108. doi:10.1186/1479-5876-11-108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Starnes T, Robertson MJ, Sledge G, Kelich S, Nakshatri H, Broxmeyer HE, et al. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J Immunol (2001) 167(8):4137–40. doi:10.4049/jimmunol.167.8.4137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Beano A, Signorino E, Evangelista A, Brusa D, Mistrangelo M, Polimeni MA, et al. Correlation between NK function and response to trastuzumab in metastatic breast cancer patients. J Transl Med (2008) 6:25. doi:10.1186/1479-5876-6-25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Kono K, Takahashi A, Ichihara F, Sugai H, Fujii H, Matsumoto Y. Impaired antibody-dependent cellular cytotoxicity mediated by herceptin in patients with gastric cancer. Cancer Res (2002) 62(20):5813–7.
138. Betting DJ, Yamada RE, Kafi K, Said J, van Rooijen N, Timmerman JM. Intratumoral but not systemic delivery of CpG oligodeoxynucleotide augments the efficacy of anti-CD20 monoclonal antibody therapy against B cell lymphoma. J Immunother (2009) 32(6):622–31. doi:10.1097/CJI.0b013e3181ab23f1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Parihar R, Nadella P, Lewis A, Jensen R, De Hoff C, Dierksheide JE, et al. A phase I study of interleukin 12 with trastuzumab in patients with human epidermal growth factor receptor-2-overexpressing malignancies: analysis of sustained interferon gamma production in a subset of patients. Clin Cancer Res (2004) 10(15):5027–37. doi:10.1158/1078-0432.CCR-04-0265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Yamane BH, Hank JA, Albertini MR, Sondel PM. The development of antibody-IL-2 based immunotherapy with hu14.18-IL2 (EMD-273063) in melanoma and neuroblastoma. Expert Opin Investig Drugs (2009) 18(7):991–1000. doi:10.1517/13543780903048911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Tarek N, Le Luduec JB, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest (2012) 122(9):3260–70. doi:10.1172/JCI62749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res (2013) 19(14):3844–55. doi:10.1158/1078-0432.CCR-13-0505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Baumann M, Krause M. Targeting the epidermal growth factor receptor in radiotherapy: radiobiological mechanisms, preclinical and clinical results. Radiother Oncol (2004) 72(3):257–66. doi:10.1016/j.radonc.2004.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med (2008) 359(11):1116–27. doi:10.1056/NEJMoa0802656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med (2008) 359(5):492–507. doi:10.1056/NEJMra0708126
146. Congdon KL, Gedeon PC, Suryadevara CM, Caruso HG, Cooper LJ, Heimberger AB, et al. Epidermal growth factor receptor and variant III targeted immunotherapy. Neuro Oncol (2014) 16(Suppl 8):viii20–5. doi:10.1093/neuonc/nou236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: opportunities for targeted therapeutics. Mol Cancer (2010) 9:135. doi:10.1186/1476-4598-9-135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Behm FG, Smith FO, Raimondi SC, Pui CH, Bernstein ID. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood (1996) 87(3):1134–9.
149. Li Y, Madigan MC, Lai K, Conway RM, Billson FA, Crouch R, et al. Human uveal melanoma expresses NG2 immunoreactivity. Br J Ophthalmol (2003) 87(5):629–32. doi:10.1136/bjo.87.5.629
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Wang X, Osada T, Wang Y, Yu L, Sakakura K, Katayama A, et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst (2010) 102(19):1496–512. doi:10.1093/jnci/djq343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Brekke C, Lundervold A, Enger PO, Brekken C, Stalsett E, Pedersen TB, et al. NG2 expression regulates vascular morphology and function in human brain tumours. Neuroimage (2006) 29(3):965–76. doi:10.1016/j.neuroimage.2005.08.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Chekenya M, Hjelstuen M, Enger PO, Thorsen F, Jacob AL, Probst B, et al. NG2 proteoglycan promotes angiogenesis-dependent tumor growth in CNS by sequestering angiostatin. FASEB J (2002) 16(6):586–8.
153. Chekenya M, Krakstad C, Svendsen A, Netland IA, Staalesen V, Tysnes BB, et al. The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene (2008) 27(39):5182–94. doi:10.1038/onc.2008.157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Chekenya M, Pilkington GJ. NG2 precursor cells in neoplasia: functional, histogenesis and therapeutic implications for malignant brain tumours. J Neurocytol (2002) 31(6–7):507–21. doi:10.1023/A:1025795715377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Svendsen A, Verhoeff JJ, Immervoll H, Brogger JC, Kmiecik J, Poli A, et al. Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol (2011) 122(4):495–510. doi:10.1007/s00401-011-0867-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Wang J, Svendsen A, Kmiecik J, Immervoll H, Skaftnesmo KO, Planaguma J, et al. Targeting the NG2/CSPG4 proteoglycan retards tumour growth and angiogenesis in preclinical models of GBM and melanoma. PLoS One (2011) 6(7):e23062. doi:10.1371/journal.pone.0023062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Al-Mayhani MT, Grenfell R, Narita M, Piccirillo S, Kenney-Herbert E, Fawcett JW, et al. NG2 expression in glioblastoma identifies an actively proliferating population with an aggressive molecular signature. Neuro Oncol (2011) 13(8):830–45. doi:10.1093/neuonc/nor088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Poli A, Wang J, Domingues O, Planaguma J, Yan T, Rygh CB, et al. Targeting glioblastoma with NK cells and mAb against NG2/CSPG4 prolongs animal survival. Oncotarget (2013) 4(9):1527–46.
159. Kmiecik J, Gras Navarro A, Poli A, Planaguma JP, Zimmer J, Chekenya M. Combining NK cells and mAb9.2.27 to combat NG2-dependent and anti-inflammatory signals in glioblastoma. Oncoimmunology (2014) 3(1):e27185. doi:10.4161/onci.27185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Geldres C, Savoldo B, Hoyos V, Caruana I, Zhang M, Yvon E, et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res (2014) 20(4):962–71. doi:10.1158/1078-0432.CCR-13-2218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Torisu-Itakura H, Schoellhammer HF, Sim MS, Irie RF, Hausmann S, Raum T, et al. Redirected lysis of human melanoma cells by a MCSP/CD3-bispecific BiTE antibody that engages patient-derived T cells. J Immunother (2011) 34(8):597–605. doi:10.1097/CJI.0b013e3182307fd8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. D’Urso CM, Wang ZG, Cao Y, Tatake R, Zeff RA, Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in B2m gene expression. J Clin Invest (1991) 87(1):284–92. doi:10.1172/JCI114984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Jimenez P, Canton J, Collado A, Cabrera T, Serrano A, Real LM, et al. Chromosome loss is the most frequent mechanism contributing to HLA haplotype loss in human tumors. Int J Cancer (1999) 83(1):91–7. doi:10.1002/(SICI)1097-0215(19990924)83:1<91::AID-IJC17>3.3.CO;2-W
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Uhrberg M, Valiante NM, Young NT, Lanier LL, Phillips JH, Parham P. The repertoire of killer cell Ig-like receptor and CD94:NKG2A receptors in T cells: clones sharing identical alpha beta TCR rearrangement express highly diverse killer cell Ig-like receptor patterns. J Immunol (2001) 166(6):3923–32. doi:10.4049/jimmunol.166.6.3923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Bjorkstrom NK, Beziat V, Cichocki F, Liu LL, Levine J, Larsson S, et al. CD8 T cells express randomly selected KIRs with distinct specificities compared with NK cells. Blood (2012) 120(17):3455–65. doi:10.1182/blood-2012-03-416867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today (1990) 11(7):237–44. doi:10.1016/0167-5699(90)90097-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319(6055):675–8. doi:10.1038/319675a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Ljunggren HG, Malmberg KJ. Prospects for the use of NK cells in immunotherapy of human cancer. Nat Rev Immunol (2007) 7(5):329–39. doi:10.1038/nri2073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285(5428):727–9. doi:10.1126/science.285.5428.727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Vivier E, Nunes JA, Vely F. Natural killer cell signaling pathways. Science (2004) 306(5701):1517–9. doi:10.1126/science.1103478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Chiesa S, Tomasello E, Vivier E, Vely F. Coordination of activating and inhibitory signals in natural killer cells. Mol Immunol (2005) 42(4):477–84. doi:10.1016/j.molimm.2005.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Kohler K, Xiong S, Brzostek J, Mehrabi M, Eissmann P, Harrison A, et al. Matched sizes of activating and inhibitory receptor/ligand pairs are required for optimal signal integration by human natural killer cells. PLoS One (2010) 5(11):e15374. doi:10.1371/journal.pone.0015374
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Lanier LL. NK cell recognition. Annu Rev Immunol (2005) 23:225–74. doi:10.1146/annurev.immunol.23.021704.115526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med (2013) 5(208):208ra145. doi:10.1126/scitranslmed.3006702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Anfossi N, Andre P, Guia S, Falk CS, Roetynck S, Stewart CA, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity (2006) 25(2):331–42. doi:10.1016/j.immuni.2006.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature (2005) 436(7051):709–13. doi:10.1038/nature03847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Fauriat C, Andersson S, Bjorklund AT, Carlsten M, Schaffer M, Bjorkstrom NK, et al. Estimation of the size of the alloreactive NK cell repertoire: studies in individuals homozygous for the group A KIR haplotype. J Immunol (2008) 181(9):6010–9. doi:10.4049/jimmunol.181.9.6010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Valiante NM, Uhrberg M, Shilling HG, Lienert-Weidenbach K, Arnett KL, D’Andrea A, et al. Functionally and structurally distinct NK cell receptor repertoires in the peripheral blood of two human donors. Immunity (1997) 7(6):739–51. doi:10.1016/S1074-7613(00)80393-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. NK cell education after allogeneic transplantation: dissociation between recovery of cytokine-producing and cytotoxic functions. Blood (2011) 118(10):2784–92. doi:10.1182/blood-2011-04-347070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Andersson S, Fauriat C, Malmberg JA, Ljunggren HG, Malmberg KJ. KIR acquisition probabilities are independent of self-HLA class I ligands and increase with cellular KIR expression. Blood (2009) 114(1):95–104. doi:10.1182/blood-2008-10-184549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Bjorklund AT, Schaffer M, Fauriat C, Ringden O, Remberger M, Hammarstedt C, et al. NK cells expressing inhibitory KIR for non-self-ligands remain tolerant in HLA-matched sibling stem cell transplantation. Blood (2010) 115(13):2686–94. doi:10.1182/blood-2009-07-229740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Yu J, Venstrom JM, Liu XR, Pring J, Hasan RS, O’Reilly RJ, et al. Breaking tolerance to self, circulating natural killer cells expressing inhibitory KIR for non-self HLA exhibit effector function after T cell-depleted allogeneic hematopoietic cell transplantation. Blood (2009) 113(16):3875–84. doi:10.1182/blood-2008-09-177055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Hsu KC, Keever-Taylor CA, Wilton A, Pinto C, Heller G, Arkun K, et al. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood (2005) 105(12):4878–84. doi:10.1182/blood-2004-12-4825
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Angelini DF, Borsellino G, Poupot M, Diamantini A, Poupot R, Bernardi G, et al. FcgammaRIII discriminates between 2 subsets of Vgamma9Vdelta2 effector cells with different responses and activation pathways. Blood (2004) 104(6):1801–7. doi:10.1182/blood-2004-01-0331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Joncker NT, Fernandez NC, Treiner E, Vivier E, Raulet DH. NK cell responsiveness is tuned commensurate with the number of inhibitory receptors for self-MHC class I: the rheostat model. J Immunol (2009) 182(8):4572–80. doi:10.4049/jimmunol.0803900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Joncker NT, Shifrin N, Delebecque F, Raulet DH. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J Exp Med (2010) 207(10):2065–72. doi:10.1084/jem.20100570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Fauriat C, Ivarsson MA, Ljunggren HG, Malmberg KJ, Michaelsson J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood (2010) 115(6):1166–74. doi:10.1182/blood-2009-09-245746
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Van Elssen CH, Oth T, Germeraad WT, Bos GM, Vanderlocht J. Natural killer cells: the secret weapon in dendritic cell vaccination strategies. Clin Cancer Res (2014) 20(5):1095–103. doi:10.1158/1078-0432.CCR-13-2302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295(5562):2097–100. doi:10.1126/science.1068440
190. Leung W. Infusions of allogeneic natural killer cells as cancer therapy. Clin Cancer Res (2014) 20(13):3390–400. doi:10.1158/1078-0432.CCR-13-1766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood (2003) 102(3):814–9. doi:10.1182/blood-2003-01-0091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Savani BN, Mielke S, Adams S, Uribe M, Rezvani K, Yong AS, et al. Rapid natural killer cell recovery determines outcome after T-cell-depleted HLA-identical stem cell transplantation in patients with myeloid leukemias but not with acute lymphoblastic leukemia. Leukemia (2007) 21(10):2145–52. doi:10.1038/sj.leu.2404892
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Schaffer M, Malmberg KJ, Ringden O, Ljunggren HG, Remberger M. Increased infection-related mortality in KIR-ligand-mismatched unrelated allogeneic hematopoietic stem-cell transplantation. Transplantation (2004) 78(7):1081–5. doi:10.1097/01.TP.0000137103.19717.86
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Kroger N, Binder T, Zabelina T, Wolschke C, Schieder H, Renges H, et al. Low number of donor activating killer immunoglobulin-like receptors (KIR) genes but not KIR-ligand mismatch prevents relapse and improves disease-free survival in leukemia patients after in vivo T-cell depleted unrelated stem cell transplantation. Transplantation (2006) 82(8):1024–30. doi:10.1097/01.tp.0000235859.24513.43
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Gagne K, Brizard G, Gueglio B, Milpied N, Herry P, Bonneville F, et al. Relevance of KIR gene polymorphisms in bone marrow transplantation outcome. Hum Immunol (2002) 63(4):271–80. doi:10.1016/S0198-8859(02)00373-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Leung W, Iyengar R, Turner V, Lang P, Bader P, Conn P, et al. Determinants of antileukemia effects of allogeneic NK cells. J Immunol (2004) 172(1):644–50. doi:10.4049/jimmunol.172.1.644
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Bari R, Rujkijyanont P, Sullivan E, Kang G, Turner V, Gan K, et al. Effect of donor KIR2DL1 allelic polymorphism on the outcome of pediatric allogeneic hematopoietic stem-cell transplantation. J Clin Oncol (2013) 31(30):3782–90. doi:10.1200/JCO.2012.47.4007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol (2006) 6(7):520–31. doi:10.1038/nri1863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT, et al. Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood (2009) 113(3):726–32. doi:10.1182/blood-2008-07-171926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Marsh SG, et al. Donor killer cell Ig-like receptor B haplotypes, recipient HLA-C1, and HLA-C mismatch enhance the clinical benefit of unrelated transplantation for acute myelogenous leukemia. J Immunol (2014) 192(10):4592–600. doi:10.4049/jimmunol.1302517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Sullivan EM, Jeha S, Kang G, Cheng C, Rooney B, Holladay M, et al. NK cell genotype and phenotype at diagnosis of acute lymphoblastic leukemia correlate to post-induction residual disease. Clin Cancer Res (2014) 20(23):5986–94. doi:10.1158/1078-0432.CCR-14-0479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Chewning JH, Gudme CN, Hsu KC, Selvakumar A, Dupont B. KIR2DS1-positive NK cells mediate alloresponse against the C2 HLA-KIR ligand group in vitro. J Immunol (2007) 179(2):854–68. doi:10.4049/jimmunol.179.2.854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Giebel S, Nowak I, Dziaczkowska J, Czerw T, Wojnar J, Krawczyk-Kulis M, et al. Activating killer immunoglobulin-like receptor incompatibilities enhance graft-versus-host disease and affect survival after allogeneic hematopoietic stem cell transplantation. Eur J Haematol (2009) 83(4):343–56. doi:10.1111/j.1600-0609.2009.01280.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Gagne K, Busson M, Bignon JD, Balere-Appert ML, Loiseau P, Dormoy A, et al. Donor KIR3DL1/3DS1 gene and recipient Bw4 KIR ligand as prognostic markers for outcome in unrelated hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2009) 15(11):1366–75. doi:10.1016/j.bbmt.2009.06.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Venstrom JM, Gooley TA, Spellman S, Pring J, Malkki M, Dupont B, et al. Donor activating KIR3DS1 is associated with decreased acute GVHD in unrelated allogeneic hematopoietic stem cell transplantation. Blood (2010) 115(15):3162–5. doi:10.1182/blood-2009-08-236943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med (2012) 367(9):805–16. doi:10.1056/NEJMoa1200503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science (1999) 285(5425):221–7. doi:10.1126/science.285.5425.221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature (1998) 395(6697):82–6. doi:10.1038/25764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Trapani JA, Sutton VR. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol (2003) 15(5):533–43. doi:10.1016/S0952-7915(03)00107-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455(7216):1061–8. doi:10.1038/nature07385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Bots M, Medema JP. Granzymes at a glance. J Cell Sci (2006) 119(Pt 24):5011–4. doi:10.1242/jcs.03239
212. Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, et al. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell (2006) 23(1):133–42. doi:10.1016/j.molcel.2006.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Lieberman J, Fan Z. Nuclear war: the granzyme A-bomb. Curr Opin Immunol (2003) 15(5):553–9. doi:10.1016/S0952-7915(03)00108-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med (1985) 313(23):1485–92. doi:10.1056/NEJM198512053132327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Ishikawa E, Tsuboi K, Saijo K, Harada H, Takano S, Nose T, et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res (2004) 24(3b):1861–71.
216. Ishikawa E, Takano S, Ohno T, Tsuboi K. Adoptive cell transfer therapy for malignant gliomas. Adv Exp Med Biol (2012) 746:109–20. doi:10.1007/978-1-4614-3146-6_9
217. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant (2003) 32(2):177–86. doi:10.1038/sj.bmt.1704086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. Ommaya AK. Subcutaneous reservoir and pump for sterile access to ventricular cerebrospinal fluid. Lancet (1963) 2(7315):983–4. doi:10.1016/S0140-6736(63)90681-0
219. Benson DM Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120(22):4324–33. doi:10.1182/blood-2012-06-438028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120(22):4317–23. doi:10.1182/blood-2012-06-437558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Nijhof IS, Lammerts van Bueren JJ, van Kessel B, Andre P, Morel Y, Lokhorst HM, et al. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica (2015) 100(2):263–8. doi:10.3324/haematol.2014.117531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J Cancer (2011) 2:383–5. doi:10.7150/jca.2.383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
223. Hallett WH, Ames E, Motarjemi M, Barao I, Shanker A, Tamang DL, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol (2008) 180(1):163–70. doi:10.4049/jimmunol.180.1.163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Krusch M, Salih J, Schlicke M, Baessler T, Kampa KM, Mayer F, et al. The kinase inhibitors sunitinib and sorafenib differentially affect NK cell antitumor reactivity in vitro. J Immunol (2009) 183(12):8286–94. doi:10.4049/jimmunol.0902404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
225. Schmiedel BJ, Arelin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int J Cancer (2011) 128(12):2911–22. doi:10.1002/ijc.25635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res (2008) 14(14):4650–7. doi:10.1158/1078-0432.CCR-07-4405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Skeate R, Singh C, Cooley S, Geller M, Northouse J, Welbig J, et al. Hemolytic anemia due to passenger lymphocyte syndrome in solid malignancy patients treated with allogeneic natural killer cell products. Transfusion (2013) 53(2):419–23. doi:10.1111/j.1537-2995.2012.03942.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118(12):3273–9. doi:10.1182/blood-2011-01-329508
229. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105(8):3051–7. doi:10.1182/blood-2004-07-2974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
230. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13(1):98–107. doi:10.3109/14653249.2010.515582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
231. Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest (2012) 122(4):1271–82. doi:10.1172/JCI59806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Ardolino M, Azimi CS, Iannello A, Trevino TN, Horan L, Zhang L, et al. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J Clin Invest (2014) 124(11):4781–94. doi:10.1172/JCI74337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood (2012) 119(16):3734–43. doi:10.1182/blood-2011-11-392951
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med (2008) 205(12):2763–79. doi:10.1084/jem.20081398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
235. Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature (2009) 457(7229):557–61. doi:10.1038/nature07665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
236. Berg M, Lundqvist A, McCoy P Jr, Samsel L, Fan Y, Tawab A, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy (2009) 11(3):341–55. doi:10.1080/14653240902807034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
237. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature (2012) 484(7395):529–33. doi:10.1038/nature10975
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
238. Waldmann TA, Lugli E, Roederer M, Perera LP, Smedley JV, Macallister RP, et al. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood (2011) 117(18):4787–95. doi:10.1182/blood-2010-10-311456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
239. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33(1):74–82. doi:10.1200/JCO.2014.57.3329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
240. Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One (2011) 6(6):e20740. doi:10.1371/journal.pone.0020740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
241. Spanholtz J, Tordoir M, Eissens D, Preijers F, van der Meer A, Joosten I, et al. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS One (2010) 5(2):e9221. doi:10.1371/journal.pone.0009221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
242. Sutlu T, Stellan B, Gilljam M, Quezada HC, Nahi H, Gahrton G, et al. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy (2010) 12(8):1044–55. doi:10.3109/14653249.2010.504770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
243. Berg M, Childs R. Ex-vivo expansion of NK cells: what is the priority – high yield or high purity? Cytotherapy (2010) 12(8):969–70. doi:10.3109/14653249.2010.536216
244. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood (2005) 106(1):376–83. doi:10.1182/blood-2004-12-4797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
245. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One (2012) 7(1):e30264. doi:10.1371/journal.pone.0030264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
246. Le Bouteiller P, Tabiasco J, Polgar B, Kozma N, Giustiniani J, Siewiera J, et al. CD160: a unique activating NK cell receptor. Immunol Lett (2011) 138(2):93–6. doi:10.1016/j.imlet.2011.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
247. Rose MJ, Brooks AG, Stewart LA, Nguyen TH, Schwarer AP. Killer Ig-like receptor ligand mismatch directs NK cell expansion in vitro. J Immunol (2009) 183(7):4502–8. doi:10.4049/jimmunol.0803323
248. Poli A, Kmiecik J, Domingues O, Hentges F, Blery M, Chekenya M, et al. NK cells in central nervous system disorders. J Immunol (2013) 190(11):5355–62. doi:10.4049/jimmunol.1203401
249. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi:10.1056/NEJMoa043330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
250. Fadul CE, Fisher JL, Gui J, Hampton TH, Cote AL, Ernstoff MS. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol (2011) 13(4):393–400. doi:10.1093/neuonc/noq204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
251. Rygh CB, Wang J, Thuen M, Gras Navarro A, Huuse EM, Thorsen F, et al. Dynamic contrast enhanced MRI detects early response to adoptive NK cellular immunotherapy targeting the NG2 proteoglycan in a rat model of glioblastoma. PLoS One (2014) 9(9):e108414. doi:10.1371/journal.pone.0108414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: NK-cell subsets, KIR-HLA interactions, tumor microenvironment, cancer stem cells, clinical application
Citation: Gras Navarro A, Björklund AT and Chekenya M (2015) Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front. Immunol. 6:202. doi: 10.3389/fimmu.2015.00202
Received: 20 February 2015; Accepted: 14 April 2015;
Published: 29 April 2015
Edited by:
Anahid Jewett, UCLA School of Dentistry and Medicine, USAReviewed by:
Viktor Umansky, German Cancer Research Center (DKFZ), GermanyJennifer Wu, Medical University of South Carolina, USA
Copyright: © 2015 Gras Navarro, Björklund and Chekenya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martha Chekenya, Brain Tumour Immunology and Therapy Group, Department of Biomedicine, University of Bergen, Jonas Lies vei 91, Bergen 5009, Norway,bWFydGhhLmNoZWtlbnlhQGJpb21lZC51aWIubm8=