 Nicholas M. de la Rua
Nicholas M. de la Rua Derrick R. Samuelson
Derrick R. Samuelson Tysheena P. Charles
Tysheena P. Charles David A. Welsh
David A. Welsh Judd E. Shellito
Judd E. Shellito- Section of Pulmonary/Critical Care and Allergy/Immunology, Department of Medicine, Louisiana State University Health Sciences Center, New Orleans, LA, USA
Pneumocystis pneumonia is a major cause of morbidity and mortality among immunocompromised patients, especially in the context of HIV/AIDS. In the murine model of Pneumocystis pneumonia, CD4+ T-cells are required for clearance of a primary infection of Pneumocystis, but not the memory recall response. We hypothesized that the memory recall response in the absence of CD4+ T-cells is mediated by a robust memory humoral response, CD8+ T-cells, and IgG-mediated phagocytosis by alveolar macrophages. To investigate the role of CD8+ T-cells and alveolar macrophages in the immune memory response to Pneumocystis, mice previously challenged with Pneumocystis were depleted of CD8+ T-cells or alveolar macrophages prior to re-infection. Mice depleted of CD4+ T-cells prior to secondary challenge cleared Pneumocystis infection within 48 h identical to immunocompetent mice during a secondary memory recall response. However, loss of CD8+ T-cells or macrophages prior to the memory recall response significantly impaired Pneumocystis clearance. Specifically, mice depleted of CD8+ T-cells or alveolar macrophages had significantly higher fungal burden in the lungs. Furthermore, loss of alveolar macrophages significantly skewed the lung CD8+ T-cell response toward a terminally differentiated effector memory population and increased the percentage of IFN-γ+ CD8+ T-cells. Finally, Pneumocystis-infected animals produced significantly more bone marrow plasma cells and Pneumocystis-specific IgG significantly increased macrophage-mediated killing of Pneumocystis in vitro. These data suggest that secondary immune memory responses to Pneumocystis are mediated, in part, by CD8+ T-cells, alveolar macrophages, and the production of Pneumocystis-specific IgG.
Introduction
Pneumonia due to the opportunistic human fungal pathogen Pneumocystis jirovecii is an AIDS-defining illness, and there is a direct inverse relationship between CD4+ T-cell counts in the blood and the risk for infection (1). Pneumocystis is also a major cause of mortality in patients whose CD4+ T-cell number or function is significantly depressed due to malignancy, chemotherapy, or other immunosuppression (1, 2). Animal models of immunodeficiency demonstrate that the loss of CD4+ T-cells renders mammals susceptible to Pneumocystis lung infection (2). Additionally, CD4+ T-cells from Pneumocystis-infected mice are able to mediate clearance of Pneumocystis infection upon adoptive transfer into Rag1(−/−) mice (3). As such, CD4+ T-cells have been a primary focus in the study of host defense against this pathogen.
However, it has also been shown that adoptive transfer of hyperimmune serum mediates effective passive immunity to Pneumocystis in the absence of T cells (4). Further, Gigliotti and colleagues have shown that immunocompetent mice immunized against Pneumocystis by intratracheal inoculations with Pneumocystis are protected from subsequent lung infection following depletion of CD4+ T cells with anti-CD4 monoclonal antibodies, which suggests that CD4+ T-cells are not required for the memory recall response (5). In addition, these investigators demonstrated that antibodies are responsible for the observed protection against P. carinii in the absence of CD4+ T cells (5). These results suggest that CD4+ T-cells are not required for secondary immune recall to Pneumocystis infection. These results also suggest that more focus should be given to immune cells other than CD4+ T-cells in host defense against Pneumocystis. This is highlighted by the fact that exposure to Pneumocystis is ubiquitous in humans and that the memory response to infection is often called into play in the context of CD4+ T-cell depletion (HIV infection).
In this study, we evaluated the cellular immune memory recall response to murine Pneumocystis infection in the absence of CD4+ T-cells. Specifically, we depleted CD8+ T-cells or alveolar macrophages prior to a secondary infection and evaluated the host’s memory response to Pneumocystis infection. Mice depleted of CD4+ T-cells prior to secondary challenge cleared Pneumocystis infection within 48 h identical to immunocompetent mice during a secondary memory recall response. However, loss of CD8+ T-cells or macrophages prior to the memory recall response significantly impaired Pneumocystis clearance. Specifically, mice depleted of CD8+ T-cells or alveolar macrophages had significantly higher fungal burden in the lungs, and loss of alveolar macrophages significantly increased the percentage of IFN-γ+ CD8+ T-cells. Finally, Pneumocystis-infected animals generated significantly more bone marrow plasma cells, and Pneumocystis-specific IgG purified from sera of previously infected mice significantly increased macrophage-mediated killing of Pneumocystis in vitro. These data suggest that secondary immune memory responses to Pneumocystis are mediated, in part, by CD8+ T-cells and alveolar macrophages, in addition to the production of Pneumocystis-specific IgG.
Materials and Methods
Immune Memory Model
Pneumocystis murina was propagated in B10;B6 Rag2/IL2rg double knockout mice from Taconic (Model 4111F; Hudson, NY, USA). Inocula and naive lung homogenates were prepared as previously described (2), and recipient mice were infected, as previously described (6). C57Bl/6 female mice (6–8 weeks) were either intratracheally challenged with 2 × 105 cysts or naive lung homogenate. Mice were depleted of CD4+ T-cells, CD8+ T-cells, or macrophages at the indicated time points prior to infection by intraperitoneal (i.p.) injection with 100 μg of anti-CD4 mAb (hybridoma GK 1.5; Taconic), 100 μg of anti-CD8 mAb (hybridoma 58.6.72; National Cell Culture Center), or intratracheal administration (i.t.) of 100 μL of clodronate liposomes (http://clodronateliposomes.com; Netherlands). Control animals were given IgG isotype control via i.p. injection or PBS containing empty liposomes via i.t. inoculation. Depletions were maintained by dosing animals every 6 days, which is sufficient to maintain significant cellular depletion (data not shown).
Quantitation of Pneumocystis-Specific IgG
Whole blood was either collected from tail bleeds or cardiac puncture, and serum was obtained via centrifugation of whole blood in BD serum separator tubes at 1,500 × g for 10 min at 4°C. Serum was stored at −20°C. Pneumocystis whole cell lysate was used to quantify the IgG humoral response. In brief, Pneumocystis-infected lungs from B10;B6 Rag2/IL2rg double knockout mice were homogenized over a 70-μm filter and washed with chilled magnesium-free PBS supplemented with 5% glutathione. The filtrate was centrifuged at 300 × g for 10 min, and the pellet was resuspended in approximately 1 mL of residual supernatant. The resuspended pellet was then pipetted on top of 30 mL of 1:2 Centricoll (density ~1.40; Sigma C-0580) diluted in PBS. The preparation was centrifuged for 15 min at 275 × g. Pneumocystis organisms were collected from the PBS–Centricoll interphase and were washed with PBS containing 5% glutathione. The pellet was then sonicated using a water bath sonicator. Total protein was quantified using Nanodrop spectrophotometer. Total Pneumocystis-specific IgG was quantified using Mouse IgG Total ELISA Ready-SET-Go Kit (Affymetrix eBiosciences). Pneumocystis whole cell lysate was coated on a Thermo Scientific Nunc 96-well flat bottom plates at 5 μg/mL in 50 μL of 0.1M Na2CO3/0.1M NaHCO3 (pH 9.5) per well. The plates were incubated overnight at 4°C. Wells were aspirated and washed twice with PBS 0.05% Tween20. Plates were blocked according to the manufacture’s protocol. Serum was diluted 1:500 and allowed to incubate in the presence of anti-mouse IgG HRP-conjugated detection antibodies for 3 h at room temperature with shaking at 400 rpm. Plates were aspirated and washed four times. Plates were developed with TMB solution for 15 min at room temperature; the reactions were stopped via the addition of 2 N H2SO4. Optical density (OD) was recorded at 450 nm with a BioTek ELX800 plate reader. Values are reported as the mean OD450 ± SEM with the value of the control wells (BSA coated) subtracted.
Quantitation of Pneumocystis Fungal Burden
The right lung was placed in 1 mL of Trizol and briefly stored at −20°C. RNA was extracted from each lung following the manufacturer’s protocol. RNA was quantified using a Nanodrop spectrophotometer. cDNA was synthesized from 1 μg of Lung RNA using BioRad Reverse Transcription Supermix for RT-qPCR following the manufacturer’s protocol. Pneumocystis fungal burden was assayed via amplifying a 166-bp fragment of the small ribosomal rRNA (ssRNA) from the mitochondrial genome, as described previously (6, 7).
Flow Cytometric Analysis of T-Cells, B-Cells, and Macrophages in the Lung
Left lung tissue was homogenized over a 70-μm filter with 10 mL of homogenization buffer that comprised RPMI 1640 10% FBS, 1 mg/mL collagenase type 1 (Worthington Biochemical, Lakewood, NJ, USA), and 30 μg/mL DNAse I (Roche Diagnostics, Indianapolis, IN, USA). Lung homogenates were incubated at 37°C with shaking at 200 rpm for 30 min. All cell suspensions were centrifuged at 300 × g for 10 min at 4°C. Cells were resuspended in 5 mL of chilled 1× RBC lysis buffer (Biolegend, San Diego, CA, USA) for 4 min on ice. RBC lysis reactions were diluted with 25-mL chilled PBS. Lymphocytes were enumerated using a hemocytometer and trypan blue exclusion. Prior to all extracellular staining, cells were treated with TruStain fcX (Biolegend) to block Fc receptors. To conduct extracellular phenotype studies, 1 × 106 cells were stained with various combinations of the following fluorochrome-conjugated Abs specific for murine CD3, CD4, CD8, CD19, CD28, CD45, CD45RA, CD197 (CCR7), CD11b, CD11c, and Zombie NIR (Biolegend) suspended in MACS buffer (Miltenyi Biotec, San Diego, CA, USA) at the manufacturer’s recommended concentrations for 30 min at 4°C.
Pan T-cell, B-cells, and macrophages in the lung were assayed by gating singlet events (FSH vs. FSA), lymphocyte size gate (FSC vs. SSC), and selecting viable CD45+ events indicated by a CD45 expression and lack of uptake of Zombie NIR, intracellular uptake of this dye indicates cell death. T-cells were enumerated as a percent of CD45+ live events by gating on CD3+, CD4+, and CD8a+ expression. B-cells were enumerated as a percentage of CD45+ live events by gating on CD45+CD19+ expression. Alveolar macrophages were enumerated as a percentage of CD45+ live events by gating on CD11c+CD11b− events. These data provided evidence of successful depletion and of cellular response to secondary infection with Pneumocystis.
In order to assess the CD8+ T-cell response to secondary infection with Pneumocystis, four extracellular phenotypes for CD8+ T-cells were defined as naive (CD45+CD3+CD4−CD8+CD45RA+CD28+), central memory (CD45+CD3+CD4−CD8+CD45RA−CD197+), effector memory (CD45+CD3+CD4−CD8+CD45RA−CD197−), and terminally differentiated effector memory re-expressing CD45RA (CD45+CD3+CD4−CD8+CD45RA+CD28−) cells.
For intracellular staining (ICS) experiments, cells were resuspended in BD GolgiPlug and incubated for 5 h. Following GolgiPlug treatment, cells were stained with TruStain fcX (Biolegends) for 5 min prior to extracellular staining with CD45, CD3, CD4, and CD8 antibodies. Cells were then washed with FACS buffer, and resuspended in Cytofix/Cytoperm (Fixation/Permeablization Kit; BD Biosciences) and incubated for 20 min, as per the manufacturer’s instructions. Cells were then resuspended in 1× Permwash and incubated for 5 min at room temperature. After washing, cells were incubated for 30 min at 4°C with fluorescently labeled anti-mouse IFN-γ, IL-4, IL-17, or GM-CSF antibodies (Biolegend, San Diego, CA, USA), diluted in Permwash at appropriate concentrations. Wells were then washed with FACS buffer and fixed with PBS + 1% formalin. For all experiments, cells were acquired using LSRII flow cytometer (BD Biosciences, San Jose, CA, USA). All analyses of FACS data were performed using FlowJo software Version 9.4 (Tree Star; Ashland, OR, USA). Intracellular phenotyping is presented as a percentage of CD45+, CD3+CD4+, or CD45+CD3+CD8+ events.
Isolation and Culture of Bone Marrow Plasma Cells to Assess Their Generation after Infection and Potential Secretion of Pneumocystis-Specific IgG
In order to assess whether infection with Pneumocystis generates Pneumocystis-specific bone marrow plasma cells (BMPCs), BMPCs were enumerated using flow cytometry and cultured. BMPCs (CD45R−CD138+) are long-lived cells that secrete neutralizing antibody after exposure to antigen. As terminally differentiated B-cells of the bone marrow compartment, they do not survive or secrete antibody in vitro without the support of mesenchymal stem cells (MSCs). Mouse MSCs were isolated from the femur and the tibia of uninfected animals, as described previously (8). Specifically, the epiphysis from each bone was removed and the bone marrow from the diaphysis was flushed with a 26-gage needle with a syringe filled with culture media, making sure that the bone was flushed completely. Bones were then cut into small chips ~1–3 mm in size and treated with DMEM containing 20% FBS and collagenase II (1 mg/mL) for 1 h at 37°C with shaking at 200 rpm. The bone chips were then resuspended in 10 mL of DMEM containing 20% FBS and 0.1% penicillin–streptomycin for 5 days in a T-25 flask and monitored for mesenchymal cell migration from bone chips and growth onto the flask. After sufficient migration from bone chips (~5–7 days), cells were passed into a T75 flask and maintained in culture for the rest of the experiment.
In order to quantify total BMPCs, mouse BMPCs were enriched from total bone marrow cells via the mouse CD138+ Plasma Cell Isolation Kit (Miltenyi Biotec). Briefly, cells were resuspended in MACS buffer and incubated with 100 μL of CD138 Microbeads per 108 total cells for 15 min at 4–8°C. Cells were then washed and resuspended in 500 μL of MACS buffer. Plasma cells were then obtained by placing a MS column (Miltenyi Biotec) in the magnetic field of an octoMACs separator (Miltenyi Biotec) and applying the labeled cells to the column and washing two times with MACS buffer. Plasma cells were collected by removing the column from the magnetic field and eluting with MACs buffer. BMPCs were further purified via flow cytometric sorting. Briefly, purified BMPCs were incubated Fc block prior to staining with anti-mouse CD138 and anti-mouse/human CD45R/B220 for 30 min at 4°C. Cells were then washed and resuspended in MACS buffer for flow sorting. Enriched BMPCs (CD138+/CD45R−) were sorted directly into culture media and immediately cultured. Magnetically and flow cytometric-sorted CD138+ BMPCs were immediately cocultured with 5 × 103 MSCs in 96-well plates in DMEM containing 20% FBS and 0.1% pen–strep. After 72 h, supernatant was collected from wells containing MSCs, BMPCs, or cocultured BMPCs and MSCs, and Pneumocystis-specific IgG was quantified as described above.
Purification of Pneumocystis-Specific Mouse IgG
Sera from animals previously infected with Pneumocystis or sera from uninfected animals were pooled and desalted using the Zeba desalting columns (Life Technologies), as per the manufacturer’s instructions. Desalted sera were then used to purify IgG using the Melon Gel Purification Kit (Life Technologies Corporation, Grand Island, NY, USA) as per the manufacturer’s instructions. Purified IgG was then quantified using a Nanodrop spectrophotometer and used for in vitro macrophage killing assays.
Macrophage-Mediated Pneumocystis Killing Assay
MH-S alveolar macrophages were acquired from ATTC (ATCC® CRL-2019™). MH-S cells were maintained in complete media (RPMI 1640 containing 0.05-mM 2-mercaptoethanol and 10% FBS) and were passed every 2–3 days throughout the experiment. Macrophage-mediated Pneumocystis killing assays were performed by plating 100,000 Pneumocystis cysts and 100,000 alveolar macrophages (1:1 ratio) per well in a 96-well flat bottom plate. Each experimental group was run in sextuplet. Purified immune IgG or naive IgG was then added to the wells at the following concentrations (0.002, 0.004, and 0.008 μg), and cultures were incubated for 24 h. Control wells contained cysts + macrophages without the addition of IgG. Following incubation, 250 μL of Trizol was added to each well and incubated for 5 min and repeated one more time. RNA extractions and Pneumocystis enumeration were then performed as described above.
Statistical Analysis
Results are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism 5 (La Jolla, CA, USA), and statistical significance was measured at P ≤ 0.05. The non-parametric Kruskal–Wallis one-way analysis of variance (ANOVA), followed by post hoc Dunn’s multiple comparisons of the means, was used for all in vivo assays.
Results
Immune Memory Recall Does Not Require CD4+ T-Cells
To evaluate the immune memory recall responses to Pneumocystis pneumonia, we first investigated the requirement of CD4+ T-cells during secondary immune responses, as well as establishment of cellular and humoral immune memory. C57BL/6 mice were grouped into the following categories: uninfected animals (designated none), immune-intact animals (isotype), and three groups of mice depleted of CD4+ T cells. CD4 depletion was maintained: (a) prior to primary infection and continuously throughout the experiment (cont.), (b) only prior to a primary infection (1°), or (c) only prior to a secondary infection (2°). Mice were infected intratracheally with live P. murina (~2 × 105 cysts in 100 μL of PBS) and were allowed to clear the infection naturally (isotype, 1° and 2°) or were given Bactrim (trimethoprim and sulfamethoxazole) ad libitum in the water (cont.), following 4 weeks of persistent infection. All mice had no detectable P. murina in lung tissue by qPCR prior to re-infection (not shown). Animals were then re-infected with P. murina (~2 × 105 cysts in 100 μL of PBS) and pathogen clearance, P. murina-specific IgG, and lung immune responses were evaluated.
Figure 1A describes the experimental design used in this study. P. murina-specific IgG in the serum was assayed by ELISA (Figure 1B). Levels of P. murina in the lung following secondary infection at the following time points: 2 days post-lung infection (Figure 1C), 4 days post-lung infection (Figure 1D), and 6 days post-lung infection (Figure 1E) were assessed via qPCR. We also assessed the levels of CD4+ T cells (Figure 1F), CD8+ T cells (Figure 1G), and CD19+ B cells (Figure 1H) in the lungs using flow cytometry. The representative gating strategy to determine the number of CD4+ T cells, CD8+ T cells, and CD19+ B cells is shown in Figure 1I. We found that CD4+ T-cells are required to generate Pneumocystis-specific IgG, but are not required for IgG recall responses during re-infection (Figure 1B). Secondary immune memory responses do not require CD4+ T-cells, as the levels of P. murina in the lung did not significantly differ from immune-intact animals (Figures 1C–E). However, CD4+ T-cells are required for generation of immune memory, as mice depleted of CD4+ T-cells prior to a primary infection or continuously depleted are unable to mount an effective memory immune response (Figures 1C–E). In fact, mice continuously depleted of CD4+ T-cells do not generate any immune memory, as indicated by fungal burden and IgG levels identical to CD4-depleted animals experiencing P. murina infection for the first time. In addition, evaluation of the immune cell populations in the lungs following secondary infection showed a significant increase in the number of CD8+ T-cells and CD19+ B-cells in animals depleted of CD4+ T-cells prior to a secondary infection (Figures 1G,H). Levels of CD8+ T-cells and CD19+ B-cells in mice depleted of CD4+ T-cells prior to a secondary infection were similar to those observed in immune-intact animals, suggesting that CD8+ T-cells and CD19+ B-cells may be important for a maximal secondary immune response. These results further indicate that secondary immune memory responses do not require CD4+ T-cells, but that the development of protective memory does require CD4+ T-cells during a primary infection. We then went on to investigate other cellular populations that could be involved in secondary immune memory responses to Pneumocystis.
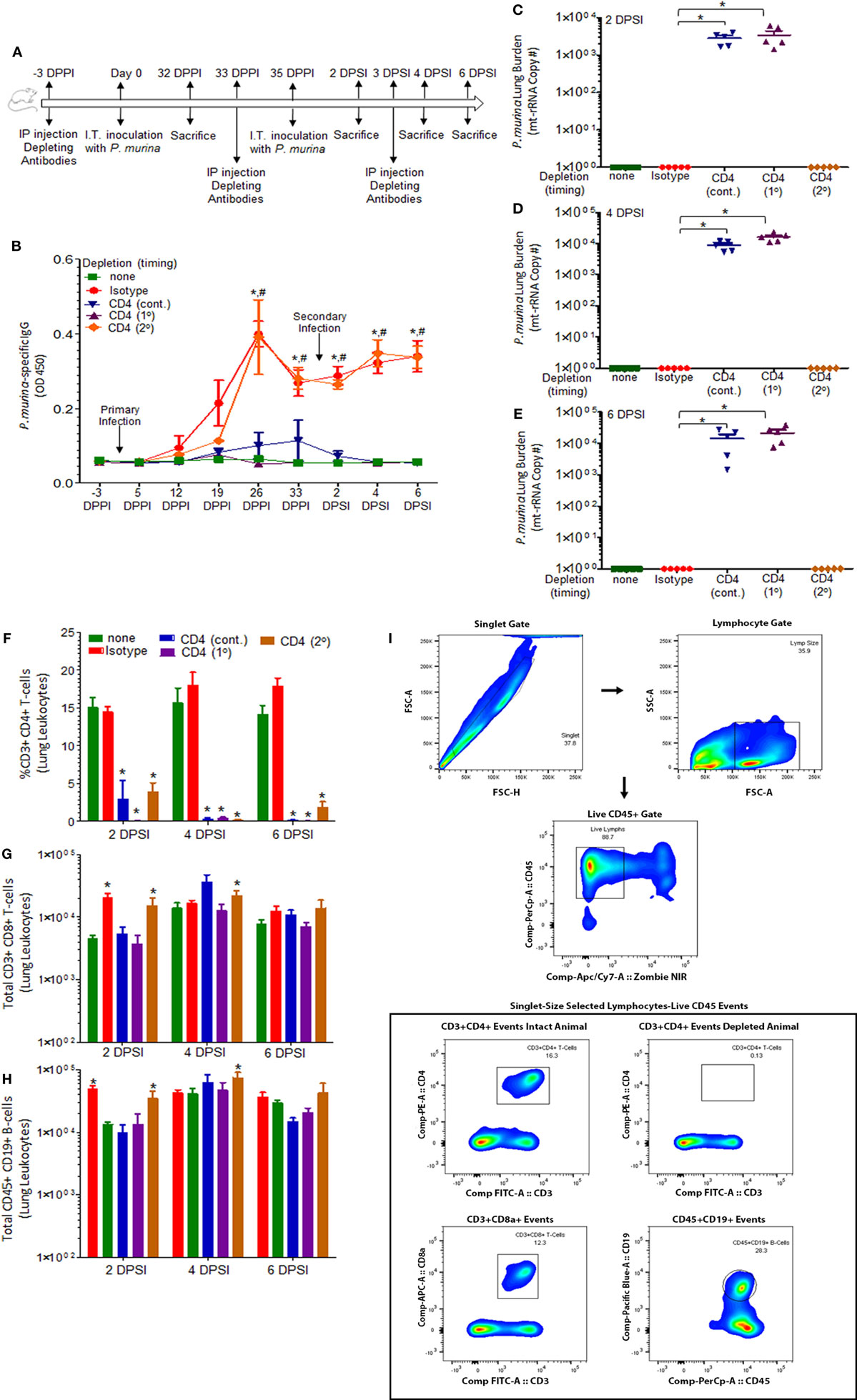
Figure 1. Immune memory recall does not require CD4+ T-cells. (A) Schematic outline of the experimental protocol used in this study. C57BL/6 mice were infected intratracheally with live P. murina (~2 × 105 cysts in 100 μL of PBS). Three groups of mice were depleted of CD4+ T cells every 6 days with CD4+ T-cell depletion occurring continuously throughout the experiment (cont.) or prior to primary infection (1°) or prior to secondary infection (2°). (B) P. murina-specific IgG in the serum was assayed by ELISA. Levels of P. murina in the lung following secondary infection at the following time points: (C) 2 days post-lung infection, (D) 4 days post-lung infection, and (E) 6 days post-lung infection via qPCR. Lung immunological responses: (F) CD4+ T cells, (G) CD8+ T cells, and (H) CD19+ B cells were quantified via flow cytometry. (I) Representative gating strategy used for flow cytometry analysis. Dots and bars represent mean ± SEM, N = 5 at each time points, * indicates P < 0.05, when comparing isotype to sham-infected animals and # indicates P < 0.05, when comparing CD4 (2°) to sham-infected animals, for each of the indicated time point, by Kruskal–Wallis ANOVA followed by Dunn’s posttest. DPPI, days post-primary infection; DPSI, days post-secondary infection.
Maximal Host Defense against Secondary Infection Requires CD8+ T-Cells and Alveolar Macrophages
Mice were infected with P. murina as described above, and groups of mice were depleted of CD4+ T cells, CD8+ T-cells, CD4+/CD8+ T-cells, macrophages, and CD4+ T-cells/macrophages every 6 days with depletion occurring prior to secondary infection (2°). Levels of P. murina in the lung following secondary infection at the following time points: 2 days post-lung infection (Figure 2A), 4 days post-lung infection (Figure 2B), and 6 days post-lung infection (Figure 2C) were determined via qPCR. Additionally, the levels of lung CD4+ T cells (Figure 2D), CD8+ T cells (Figure 2D), and CD11c+CD11b− macrophages (Figure 2D) were quantified via flow cytometry. P. murina-specific IgG in the serum was also assessed (Figure 2E). The representative gating strategy to determine the number of CD4+ T cells, CD8+ T cells, and CD11c+CD11b− macrophages is shown in Figure 2F. Flow cytometric analysis demonstrated that all targeted immune cell populations in each group were significantly depleted prior to re-infection (Figures 2D,F). Further, loss of CD4+ T cells, CD8+ T-cells, macrophages, CD4+ and CD8+ T cells, or CD4+ T cells and macrophages did not significantly affect the levels of Pneumocystis-specific IgG after re-challenge, suggesting that the humoral response is still intact (Figure 2E). However, mice depleted of CD8+ T-cells prior to secondary infection with P. murina had a significant increase in lung burden compared to either CD4-depleted or immune-intact animals at 2 and 4 days post-infection (DPI) (Figures 2A,B), despite similar amounts of Pneumocystis-specific IgG when compared to immune-intact animals. Similarly, mice depleted of macrophages exhibited a significantly increased level of P. murina in the lungs compared to control mice at 2 and 4 DPI (Figures 2A,B). However, mice depleted of CD8+ T-cells or macrophages cleared lung infection at 6 DPI (Figure 2C), suggesting that the cellular recall response is not essential for secondary host defense against Pneumocystis infection. These data also suggest that the immune memory response to Pneumocystis is, in part, mediated by CD8+ T-cells and macrophages, but that CD4+ T-cells play little role. We then sought to examine the cellular phenotypes associated with CD4+ T-cells and CD8+ T-cells during secondary immune responses to Pneumocystis.
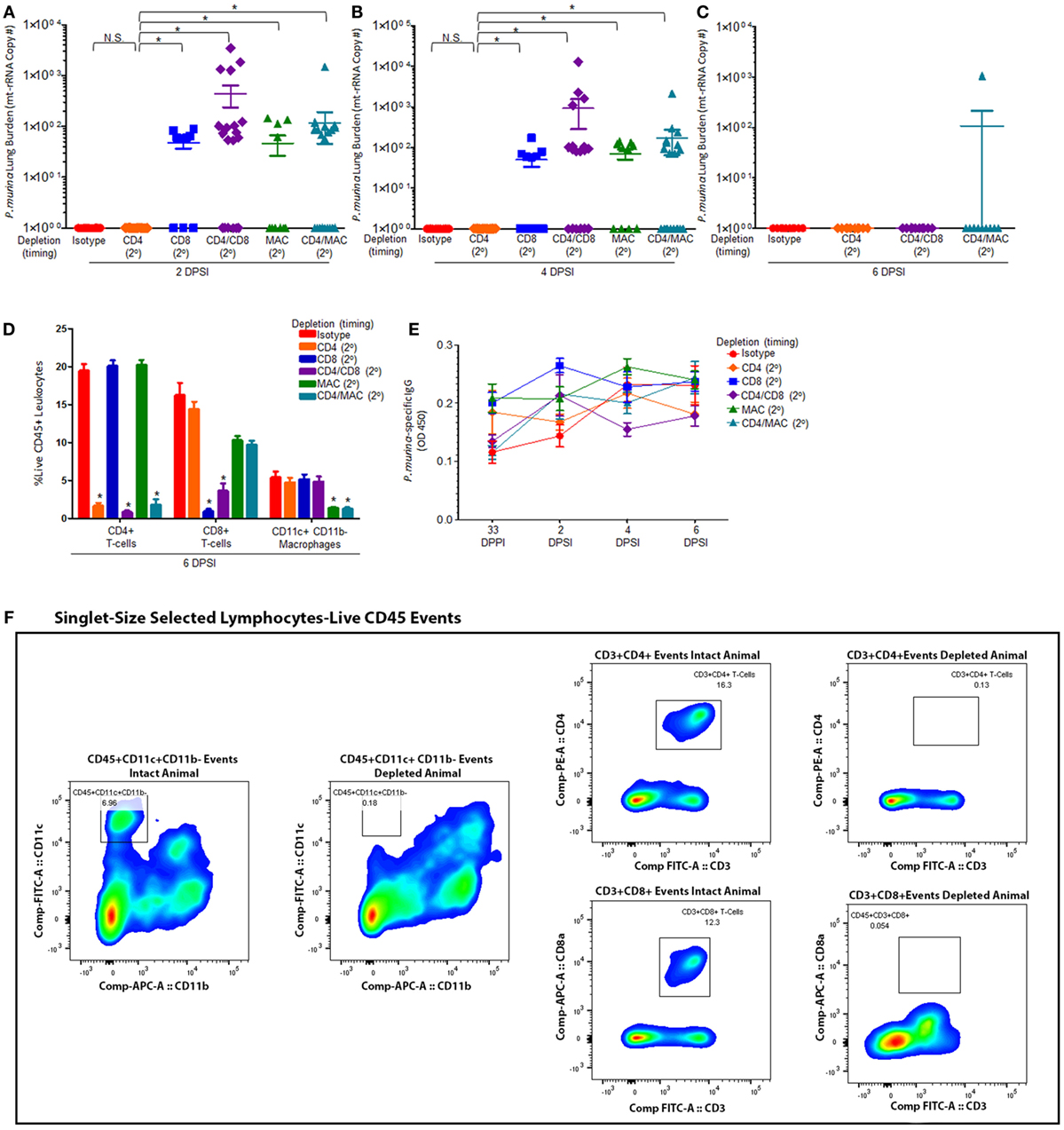
Figure 2. Maximal immune memory recall requires CD8+ T-cells and alveolar macrophages. C57BL/6 mice were infected intratracheally with live P. murina (~2 × 105 cysts in 100 μL of PBS), and groups of mice were depleted of CD4+ T cells, CD8+ T-cells, CD4+ and CD8+ T-cells, macrophages, or CD4+ T-cells and macrophages every 6 days with depletion occurring prior to secondary infection (2°). Levels of P. murina in the lung following secondary infection at the following time points: (A) 2 days post-lung infection, (B) 4 days post-lung infection, and (C) 6 days post-lung infection was assessed via qPCR. Lung immunological responses: (D) CD4+ T cells, CD8+ T cells, and CD11c+CD11b− macrophages were quantified via flow cytometry. (E) P. murina-specific IgG, in the serum was assayed by ELISA. (F) Representative gating strategy used for flow cytometry analysis. Dots and bars represent mean ± SEM, N = 10–15 at each time points, * indicates P < 0.05, for the indicated comparisons for each of the indicated time point, by Kruskal–Wallis ANOVA followed by Dunn’s posttest. DPPI, days post-primary infection; DPSI, days post-secondary infection.
Loss of CD8+ T-Cells or Alveolar Macrophages Delays IFN-γ Production by CD4+ T-Cells in Secondary Immune Responses to Pneumocystis
Mice were infected with P. murina as described above, and groups of mice were depleted of CD8+ T-cells and macrophages every 6 days with depletion occurring prior to secondary infection (2°). Lung CD4+ T-cell subsets were then examined at 2 and 4 days post-secondary infection: GM-CSF+ CD4+ T cells (Figures 3A,E, respectively), IL-4+ CD4+ T cells (Figures 3B,F, respectively), IFN-γ+ CD4+ T cells (Figures 3C,G, respectively), and IL-17+ CD4+ T cells (Figures 3D,H, respectively). The gating strategy used to determine the various CD4+ T-cell phenotypes are depicted in Figure 3I. The percentage of GM-CSF+, IL-17+, or IL-4+ CD4+ T cells in the lung 2 or 4 DPI in immune-intact animals did not significantly differ from CD8-depleted animals or macrophage-depleted mice. Conversely, loss of CD8+ T-cells or macrophages significantly decreased the percentage of IFN-γ+ CD4+ T cells compared to control animals 2 DPI. However, by 4 DPI, there was a significant increase in the percentage of IFN-γ+ CD4+ T cells compared to control animals.
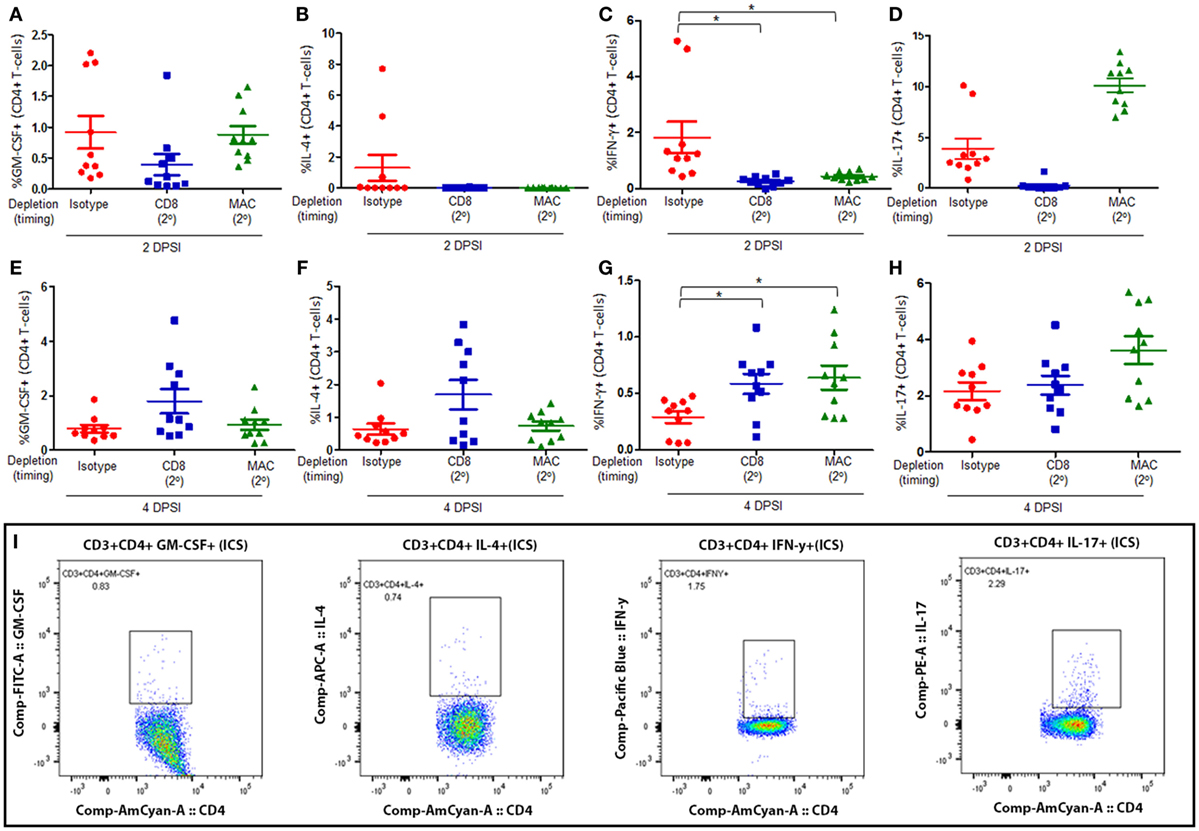
Figure 3. Loss of CD8+ T-cells or alveolar macrophages delays IFN-γ production by CD4+ T-cells in secondary immune responses to Pneumocystis. C57BL/6 mice were infected intratracheally with live P. murina, and groups of mice were depleted of CD8+ T-cells and macrophages every 6 days with depletion occurring prior to secondary infection (2°). Lung CD4+ T-cell subsets: GM-CSF+CD4+ T cells at 2 and 4 days post-secondary infection [(A,E), respectively], IL-4+ CD4+ T cells at 2 and 4 DPI [(B,F), respectively], IFN-γ+CD4+ T cells at 2 and 4 DPI [(C,G), respectively], and IL-17+CD4+ T cells at 2 and 4 DPI [(D,H), respectively], were quantified via flow cytometry. (I) Representative gating strategy used for flow cytometry analysis. Dots and bars represent mean ± SEM, N = 10 at each time points, * indicates P < 0.05, for the indicated comparisons for each of the indicated time point, by Kruskal–Wallis ANOVA followed by Dunn’s posttest. DPSI, days post-secondary infection.
Maximal CD8+ T-Cell Response Requires Alveolar Macrophages but Not CD4+ T-Cells
We then assessed the composition and quantity of various lung CD8+ T-cell subsets. We first examined the levels of naive CD8+ T (Figures 4A,E), central memory CD8+ T (Figures 4B,F), effector memory CD8+ T cells (Figures 4C,G), and terminally differentiated effector memory (TEMRA) CD8+ T cells (Figures 4D,H) in mice depleted of CD4+ T-cells, alveolar macrophages, or CD4+ T-cells and alveolar macrophages prior to a secondary infection at 2 and 4 days post-secondary infection. The gating strategy used to determine the various CD8+ T-cell phenotypes are depicted in Figure 4I. We defined the CD8+ T-cell subsets as naive (CD45+CD3+CD4−CD8+CD45RA+CD28+), central memory (CD45+CD3+CD4−CD8+CD45RA−CD197+), effector memory (CD45+CD3+CD4−CD8+CD45RA−CD197−), and TEMRA (CD45+CD3+CD4−CD8+CD45RA+CD28−). Interestingly, loss of CD4+ T-cells did not significantly affect the percentage of any of the CD8+ T-cell subsets, suggesting that CD8+ T-cell immune memory remains active despite loss of CD4+ T-cells. However, mice depleted of alveolar macrophages prior to a secondary infection exhibited a significant decrease in the percentage of effector memory CD8+ T-cells, as well as a significant increase in the percentage of TEMRA CD8+ T cells at both 2 and 4 DPI. We also found that mice depleted of macrophages and CD4+ T-cells prior to a secondary infection exhibited a significant decrease in the amount of naive CD8+ T-cells at 2 DPI, and this trend remained at 4 DPI. The percentage of central memory CD8+ T cells in the lung 2 DPI in immune-intact animals did not significantly differ from CD4-depleted animals or macrophage-depleted mice. However, by 4 DPI, the percentage of central memory CD8+ T cells significantly decreased in macrophage-depleted mice. These results suggest that loss of CD4+ T-cells does not affect the immune memory response of CD8+ T-cells, but that loss of macrophages or loss of both macrophages and CD4+ T-cells drives the CD8+ T-cell response toward a terminally differentiated effector memory phenotype.
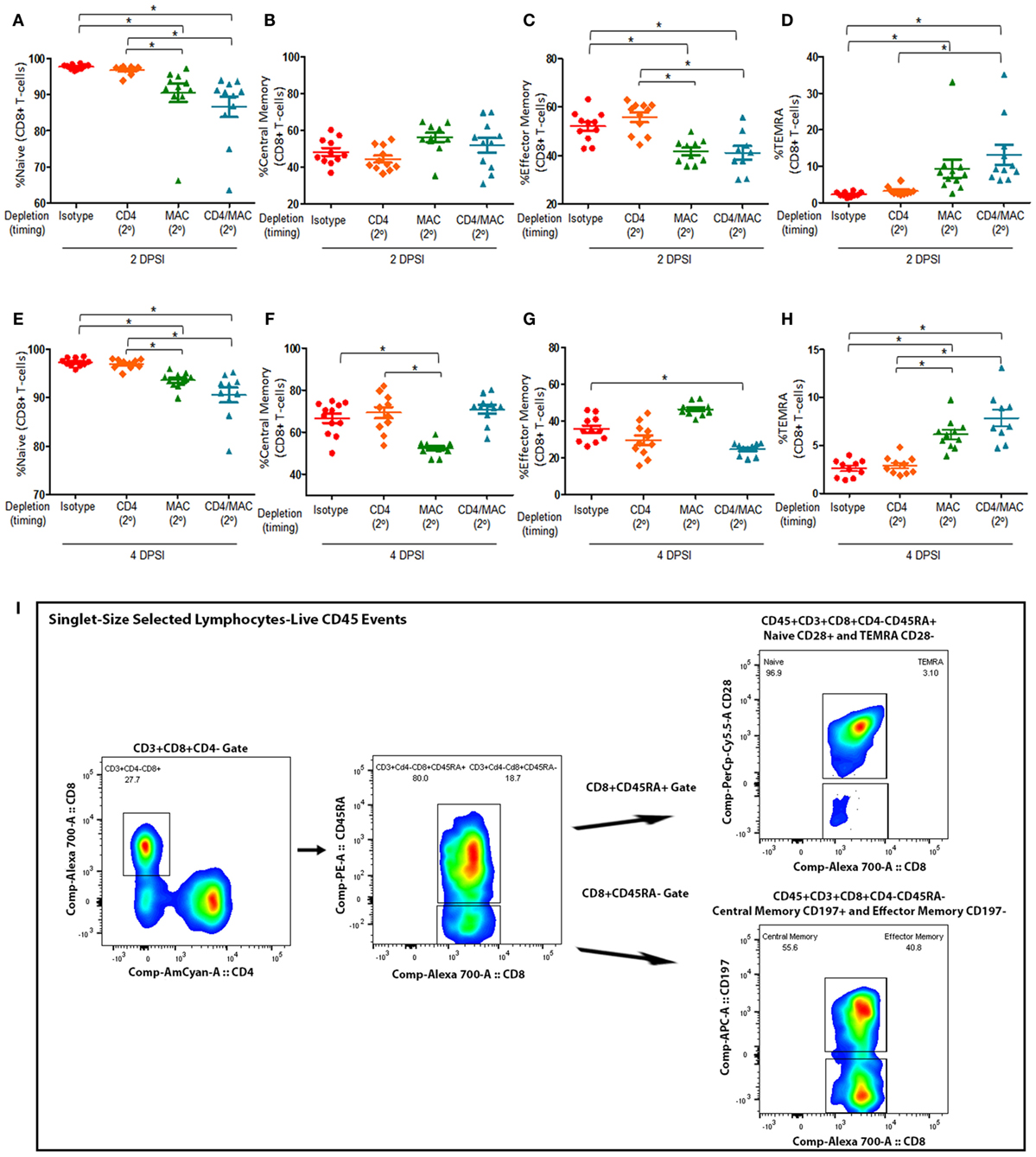
Figure 4. Maximal CD8+ T-cell response require alveolar macrophages but not CD4+ T-cells. Mice were infected intratracheally with P. murina, and groups of mice were depleted of CD4+ T-cells, macrophages, or CD4+ T-cells and macrophages every 6 days with depletion occurring prior to secondary infection (2°). Lung CD8+ T-cell subsets: naive CD8+ T cells at 2 and 4 days post-secondary infection [(A,E), respectively], central memory CD8+ T cells at 2 and 4 DPI [(B,F), respectively], effector memory CD8+ T cells at 2 and 4 DPI [(C,G), respectively], and TEMRA CD8+ T cells at 2 and 4 DPI [(D,H), respectively] were quantified via flow cytometry. (I) Representative gating strategy used for flow cytometry analysis. Dots and bars represent mean ± SEM, N = 10 at each time points, * indicates P < 0.05, for the indicated comparisons for each of the indicated time point, by Kruskal–Wallis ANOVA followed by Dunn’s posttest. DPSI, days post-secondary infection.
Loss of Alveolar Macrophages Increases IFN-γ Production by CD8+ T-Cells in Secondary Immune Responses to Pneumocystis
Mice were infected with P. murina as described above, and groups of mice were depleted of CD4+ T-cells and macrophages every 6 days with depletion occurring prior to secondary infection (2°). Lung CD8+ T-cell subsets were then examined at 2 and 4 days post-secondary infection: GM-CSF+ CD8+ T cells (Figures 5A,E, respectively), IL-4+ CD8+ T cells (Figures 5B,F, respectively), IFN-γ+ CD8+ T cells (Figures 5C,G, respectively), and IL-17+ CD8+ T cells (Figures 5D,H, respectively). The gating strategy used to determine the various CD8+ T-cell phenotypes are depicted in Figure 5I. The percentage of GM-CSF+, IL-17+, or IL-4+ CD8+ T cells in the lung 2 or 4 DPI in immune-intact animals did not significantly differ from CD4-depleted animals or macrophage-depleted mice. However, loss of macrophages significantly increased the percentage of IFN-γ+ CD8+ T cells compared to control animals 4 DPI. These results suggest that loss of macrophages significantly increases the production of IFN-γ by CD8+ T-cells.
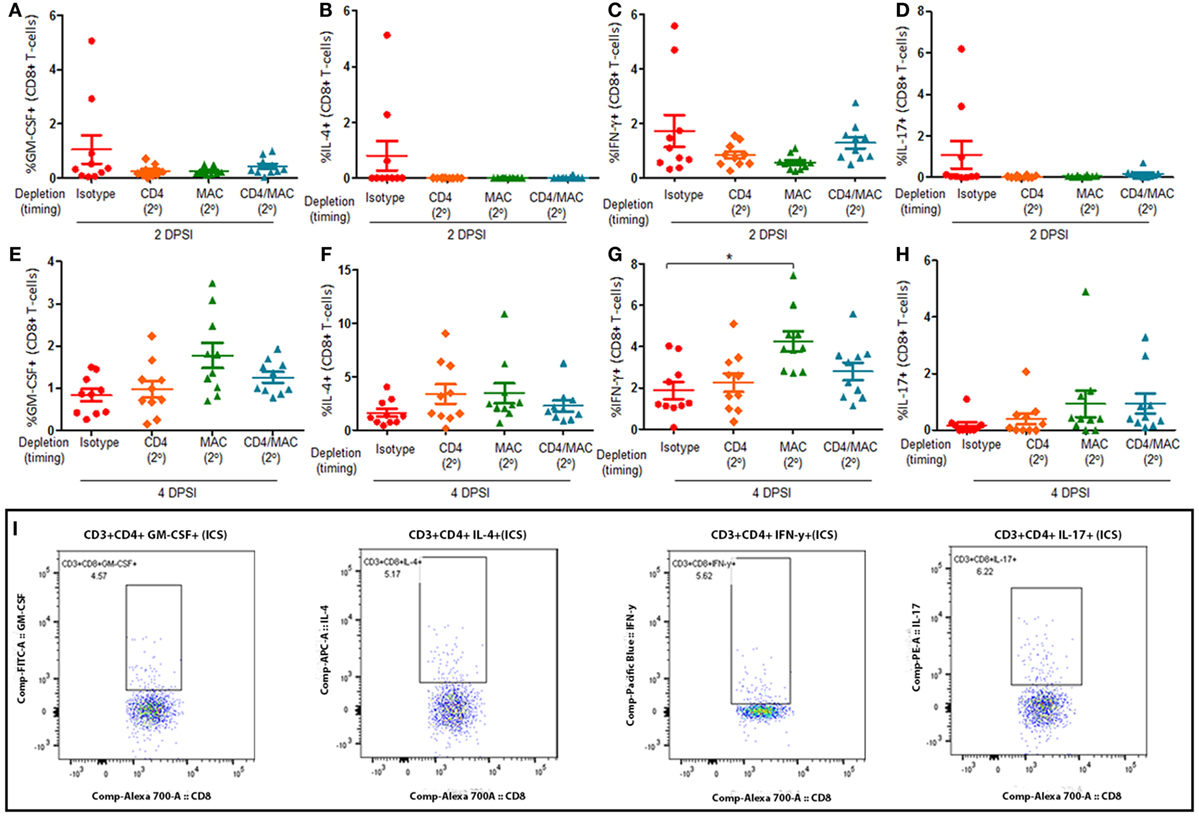
Figure 5. Loss of alveolar macrophages increases IFN-γ production by CD8+ T-cells in secondary immune responses to Pneumocystis. C57BL/6 mice were infected with P. murina, and depleted of CD4+ T-cells, macrophages, or CD4+ T-cells and macrophages every 6 days with depletion occurring prior to secondary infection (2°). Lung CD8+ T-cell subsets: GM-CSF+ CD8+ T cells at 2 and 4 days post-secondary infection [(A,E), respectively], IL-4+ CD8+ T cells at 2 and 4 DPI [(B,F), respectively], IFN-γ+ CD8+ T cells at 2 and 4 DPI [(C,G), respectively], and IL-17+ CD8+ T cells at 2 and 4 DPI [(D,H), respectively] were quantified via flow cytometry. (I) Representative gating strategy used for flow cytometry analysis. Dots and bars represent mean ± SEM, N = 10 at each time points, * indicates P < 0.05, for the indicated comparisons for each of the indicated time point, by Kruskal–Wallis ANOVA followed by Dunn’s posttest. DPSI, days post-secondary infection.
Secondary Immune Responses Significantly Increase the Number of P. murina-Specific Bone Marrow Plasma Cells and P. murina-Specific IgG Enhances Macrophage Killing
We next wanted to assess if infection with Pneumocystis generates long-lived bone marrow plasma cells and if Pneumocystis-specific antibodies enhance macrophage-mediated killing. Mice were infected with P. murina and depleted of CD4+ T-cells every 6 days with depletion occurring prior to secondary infection (2°), as described previously. The gating strategy used to flow sort BMPCs is described in Figure 6A. We first examined the absolute number of bone marrow plasma cells in P. murina-infected animals and found that there was a significant increase in the amount of obtainable BMPCs relative to sham-infected controls (Figure 6B). Additionally, loss of CD4+ T-cells prior to secondary infection did not significantly affect the number of BMPCs. To confirm that the BMPCs isolated from infected mice secreted Pneumocystis-specific IgG, BMPCs were grown in coculture with MSCs, and the levels of P. murina-specific IgG in the supernatant were determined. We found that the bone marrow plasma cells isolated from infected animals secreted high levels of P. murina-specific IgG (Figure 6C). A representative micrograph of bone marrow plasma cells isolated from infected animals is shown in Figure 6C and 1. We also found that the levels of P. murina-specific IgG in the serum was significantly increased in infected animals and was correlated with the increase in BMPCs (Figure 6D). Finally, we assessed the capacity of P. murina-specific IgG to enhance in vitro killing of P. murina by macrophages. We found that IgG from infected animals significantly increased the killing of P. murina by macrophages, in a dose-dependent manner (Figure 6E).
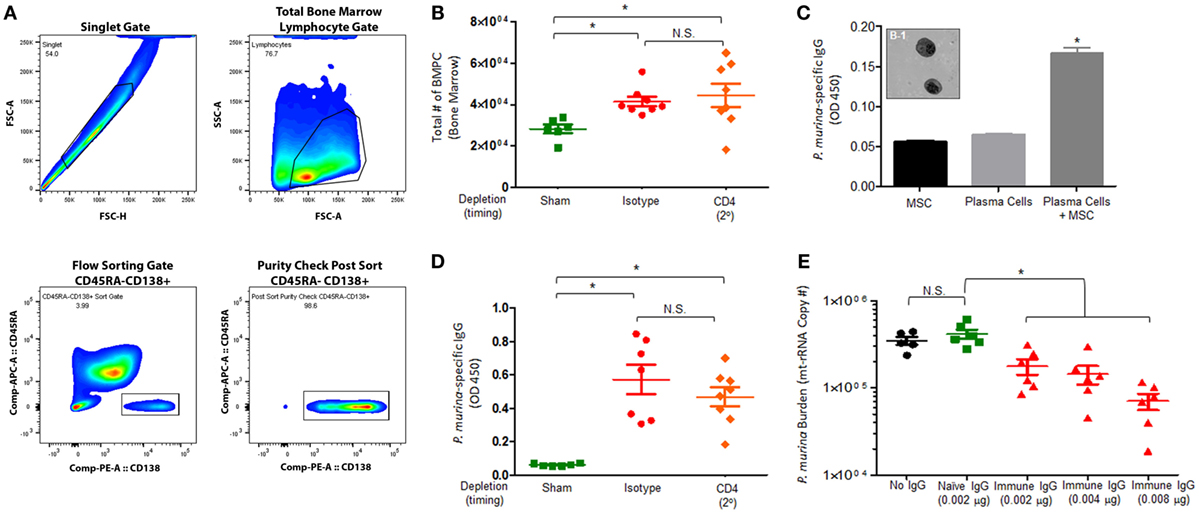
Figure 6. Pneumocystis pneumonia significantly increases the level of P. murina-specific bone marrow plasma cells and P. murina-specific IgG enhances macrophage killing. C57BL/6 mice were infected with P. murina and depleted of CD4+ T-cells every 6 days with depletion occurring prior to secondary infection (2°). (A) Representative gating strategy used for flow cytometry analysis. (B) The absolute number of bone marrow plasma cell significantly increases in P. murina infected animals. (C) Bone marrow plasma cells isolated from infected animals secreted high levels of P. murina-specific IgG. (C-1) Representative micrograph of bone marrow plasma cells isolated from infected animals. (D) P. murina-specific IgG in the serum was assayed by ELISA. (E) P. murina-specific IgG enhanced in vitro killing of P. murina by macrophages in a dose-dependent manner. Dots and bars represent mean ± SEM, N = 10 at each time points, * indicates P < 0.05, for the indicated comparisons for each of the indicated time point, by Kruskal–Wallis ANOVA followed by Dunn’s posttest.
Discussion
Exposure to Pneumocystis is ubiquitous in humans and occurs at an early age, around 2 years of life (2, 9). However, Pneumocystis pneumonia is a major cause of morbidity and mortality among immunocompromised patients, especially in the context of HIV/AIDS. This is surprising as the majority of adults have detectable levels of Pneumocystis-specific antibodies at the time of infection (10). These data suggest that immunological memory against Pneumocystis in humans may be insufficient or suboptimal, that other cellular populations (non-CD4+ T-cells) may be required for maximal secondary immune responses, or that HIV infection disrupts immune memory responses. Despite the fact that nearly all humans are exposed to Pneumocystis, there remains a paucity of data investigating the immunological memory and secondary immune response against Pneumocystis, especially in the context of CD4+ T-cell depletion. Therefore, we sought to investigate immunological memory against Pneumocystis in the context of CD4+ T-cell depletion.
Our current experiments demonstrate that secondary immune responses to Pneumocystis do not require CD4+ T-cells, which is consistent with the previous reports (3, 11, 12). However, we further went on to investigate other cellular populations that could be involved in secondary immune memory responses by investigating the role of CD8+ T-cells and macrophages during the immune memory recall response to Pneumocystis. We found that loss of CD8+ T-cells or macrophages alone or in the context of CD4+ T-cell depletion significantly impaired the ability of the host to clear Pneumocystis infection. We also evaluated the cellular phenotypes associated with CD4+ T-cells and CD8+ T-cells during secondary immune responses, in the context of CD4+ T-cell depletion, CD8+ T-cell depletion, and alveolar macrophage depletion. Loss of CD8+ T-cells or macrophages delayed the production of IFN-γ by CD4+ T-cells; however, by 4 DPI, the production is increased, which is likely due to increased fungal burden and the host trying to mediate a more robust immune response. While CD4+ T-cells are absolutely critical for resolution of Pneumocystis through the coordination of host inflammatory responses by recruitment and activation of effector cells, which ultimately lead to the elimination of the organism during a primary infection, loss of CD4+ T-cells does not render a previously exposed animal susceptible to re-infection with Pneumocystis (1).
Additionally, loss of CD4+ T-cells does not affect the immune memory response of CD8+ T-cells, but loss of macrophages or loss of both macrophages and CD4+ T-cells drives the CD8+ T-cell response toward terminally differentiated effector memory phenotype. Loss of macrophages also significantly increased the production of IFN-γ by CD8+ T-cells, which is likely due to increased fungal burden. Several studies have examined the role of CD8+ T-cells during primary infection, yet the role of CD8+ T-cells, particularly in the setting of CD4+ T-cell deficiency, remains controversial. Specifically, Gigliotti et al. demonstrated that there was no difference in organism burden in CD4+ T-cell depleted vs. CD4+ and CD8+ T-cell-depleted animals, indicating that CD8+ T cells are not involved in fungal clearance (13). However, other studies have shown that CD8+ T cells can be protective against Pneumocystis infection, though this is dependent on their cytotoxic phenotype, which is defined by high levels of endogenous IFN-γ production (14). In fact, animal studies have demonstrated that delivery of IFN-γ via aerosol or by an adenoviral-mediated gene transfer results in clearance of Pneumocystis in the absence of CD4+ T cells, and this was associated with an increase in recruited IFN-γ-producing CD8+ T cells (15). Further, McAllister et al. showed that IFN-γ stimulated CD8+ T cells from Pneumocystis-infected CD4-depleted mice enhanced macrophage-mediated in vitro killing (16). These studies suggest that the role of CD8+ T-cells during a primary infection is to enhance macrophage-mediated killing through the production of IFN-γ, which is consistent with our data assessing the role of CD8+ T-cells in secondary immune responses. More precisely, we observe loss of macrophages, but not CD4+ T-cells, significantly increases the production of IFN-γ by CD8+ T-cells and drives CD8+ T-cells toward a terminally differentiated effector memory phenotype [a cellular phenotype characterized by high levels of IFN-γ production (17)], all of which suggest that CD8+ T-cells upregulate IFN-γ responses to combat Pneumocystis infection during a secondary immune response. While we cannot fully explain these results, we believe that the increased fungal burden seen in macrophage-depleted mice results in increased activation of the remaining immune effector cells in the lung, including IFN-γ producing CD8+ T-cells.
Finally, we found that Pneumocystis-specific IgG from infected animals significantly increases the killing of P. murina by macrophages, in a dose-dependent manner. Alveolar macrophages are critical in optimal host defense against Pneumocystis as they have ability to directly kill both trophozoites and cysts (18, 19). Several studies have shown an inverse correlation between macrophage numbers and severity of Pneumocystis infection (20, 21). Specifically, animal studies demonstrate that chemical depletion of alveolar macrophages in normal rats, followed by Pneumocystis challenge results in significantly higher fungal load in the lungs compared with non-depleted mice (22). However, little data exist examining the role of macrophages during secondary immune responses to Pneumocystis. Macrophages utilize several mechanisms to kill Pneumocystis (i.e., nitric oxide release and opsonization) (23). More precisely, alveolar macrophages primed by IFN-γ produce a nitrosative burst that is toxic to Pneumocystis (23). Additionally, Roths and Sidman found that anti-Pneumocystis serum was effective at reducing the number of organisms in all stages of Pneumocystis infection in SCID mice (24). Taken together, these studies suggest that macrophage-mediated killing of Pneumocystis is mediated, in part, by IFN-γ stimulation and opsonization via Pneumocystis-specific IgG, which is consistent with our data that demonstrate that Pneumocystis-specific IgG from infected animals significantly increases the killing of P. murina by macrophages, and that loss of macrophages significantly increases the production of IFN-γ by CD8+ T-cells.
The host immune memory response during Pneumocystis infection involves a complex interaction between CD4+ T-cells, CD8+ T-cells, alveolar macrophages, and soluble factors that together facilitate clearance of the infection. Our data suggest that the immune memory response to Pneumocystis is, in part, mediated by CD8+ T-cells, macrophages, and Pneumocystis-specific IgG, but that CD4+ T-cells play little role. In addition, infection with Pneumocystis significantly increases the levels of Pneumocystis-specific long-lived BMPCs, and numbers of BMPCs are not significantly affected by loss of CD4+ T-cells. Taken together, these results suggest that investigation of immune cell populations other than CD4+ T-cells during immune responses to Pneumocystis in the HIV+ population may be extremely valuable.
Ethics Statement
The study was approved by the LSUHSC Institutional Animal Care and Use Committee.
Author Contributions
Conception and design: DS, TC, NR, DW, and JS; analysis and interpretation: DS, TC, NR, JS, and DW; drafting the manuscript: DS and NR.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the members of the Section of Pulmonary/Critical Care and Allergy/Immunology for advice and discussions. In addition, the authors thank Connie Porretta for expert flow cytometry assistance. This work was supported by The National Institutes of Health (NIH) Public Health Service grant # P01-HL076100, the NIH grant # P60-AA009803 and # R24-AA019661, and by National Institute of General Medical Sciences grant # UG54-GM104940.
References
1. Beck JM, Warnock ML, Kaltreider HB, Shellito JE. Host defenses against Pneumocystis carinii in mice selectively depleted of CD4+ lymphocytes. Chest (1993) 103(2 Suppl):116s–8s. doi:10.1378/chest.103.2.116S
2. Shellito J, Suzara VV, Blumenfeld W, Beck JM, Steger HJ, Ermak TH. A new model of Pneumocystis carinii infection in mice selectively depleted of helper T lymphocytes. J Clin Invest (1990) 85(5):1686–93. doi:10.1172/JCI114621
3. Opata MM, Hollifield ML, Lund FE, Randall TD, Dunn R, Garvy BA, et al. B lymphocytes are required during the early priming of CD4+ T cells for clearance of Pneumocystis infection in mice. J Immunol (2015) 195(2):611–20. doi:10.4049/jimmunol.1500112
4. Roths JB, Sidman CL. Both immunity and hyperresponsiveness to Pneumocystis carinii result from transfer of CD4+ but not CD8+ T cells into severe combined immunodeficiency mice. J Clin Invest (1992) 90(2):673–8. doi:10.1172/JCI115910
5. Harmsen AG, Chen W, Gigliotti F. Active immunity to Pneumocystis carinii reinfection in T-cell-depleted mice. Infect Immun (1995) 63(7):2391–5.
6. Kelly MN, Zheng M, Ruan S, Kolls J, D’Souza A, Shellito JE. Memory CD4+ T cells are required for optimal NK cell effector functions against the opportunistic fungal pathogen Pneumocystis murina. J Immunol (2013) 190(1):285–95. doi:10.4049/jimmunol.1200861
7. Samuelson DR, de la Rua NM, Charles TP, Ruan S, Taylor CM, Blanchard EE, et al. Oral immunization of mice with live Pneumocystis murina protects against Pneumocystis pneumonia. J Immunol (2016) 196(6):2655–65. doi:10.4049/jimmunol.1502004
8. Zhu H, Guo ZK, Jiang XX, Li H, Wang XY, Yao HY, et al. A protocol for isolation and culture of mesenchymal stem cells from mouse compact bone. Nat Protoc (2010) 5(3):550–60. doi:10.1038/nprot.2009.238
9. Vargas SL, Hughes WT, Santolaya ME, Ulloa AV, Ponce CA, Cabrera CE, et al. Search for primary infection by Pneumocystis carinii in a cohort of normal, healthy infants. Clin Infect Dis (2001) 32(6):855–61. doi:10.1086/319340
10. Gingo MR, Lucht L, Daly KR, Djawe K, Palella FJ, Abraham AG, et al. Serologic responses to Pneumocystis proteins in human immunodeficiency virus patients with and without Pneumocystis jirovecii pneumonia. J Acquir Immune Defic Syndr (2011) 57(3):190–6. doi:10.1097/QAI.0b013e3182167516
11. Zheng M, Ramsay AJ, Robichaux MB, Norris KA, Kliment C, Crowe C, et al. CD4+ T cell-independent DNA vaccination against opportunistic infections. J Clin Invest (2005) 115(12):3536–44. doi:10.1172/JCI26306
12. Wells J, Haidaris CG, Wright TW, Gigliotti F. Active immunization against Pneumocystis carinii with a recombinant P. carinii antigen. Infect Immun (2006) 74(4):2446–8. doi:10.1128/IAI.74.4.2446-2448.2006
13. Gigliotti F, Crow EL, Bhagwat SP, Wright TW. Sensitized CD8+ T cells fail to control organism burden but accelerate the onset of lung injury during Pneumocystis carinii pneumonia. Infect Immun (2006) 74(11):6310–6. doi:10.1128/IAI.00668-06
14. Li L, Sad S, Kagi D, Mosmann TR. CD8Tc1 and Tc2 cells secrete distinct cytokine patterns in vitro and in vivo but induce similar inflammatory reactions. J Immunol (1997) 158(9):4152–61.
15. Kolls JK, Habetz S, Shean MK, Vazquez C, Brown JA, Lei D, et al. IFN-gamma and CD8+ T cells restore host defenses against Pneumocystis carinii in mice depleted of CD4+ T cells. J Immunol (1999) 162(5):2890–4.
16. McAllister F, Steele C, Zheng M, Young E, Shellito JE, Marrero L, et al. T cytotoxic-1 CD8+ T cells are effector cells against Pneumocystis in mice. J Immunol (2004) 172(2):1132–8. doi:10.4049/jimmunol.172.2.1132
17. Barrera L, Mendoza F, Zuniga J, Estrada A, Zamora AC, Melendro EI, et al. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med (2008) 177(1):44–55. doi:10.1164/rccm.200701-093OC
18. Ezekowitz RA, Williams DJ, Koziel H, Armstrong MY, Warner A, Richards FF, et al. Uptake of Pneumocystis carinii mediated by the macrophage mannose receptor. Nature (1991) 351(6322):155–8. doi:10.1038/351155a0
19. Fraser IP, Takahashi K, Koziel H, Fardin B, Harmsen A, Ezekowitz RA. Pneumocystis carinii enhances soluble mannose receptor production by macrophages. Microbes Infect (2000) 2(11):1305–10. doi:10.1016/S1286-4579(00)01283-1
20. Fleury J, Escudier E, Pocholle MJ, Carre C, Bernaudin JF. Cell population obtained by bronchoalveolar lavage in Pneumocystis carinii pneumonitis. Acta Cytol (1985) 29(5):721–6.
21. Young JA, Stone JW, McGonigle RJ, Adu D, Michael J. Diagnosing Pneumocystis carinii pneumonia by cytological examination of bronchoalveolar lavage fluid: report of 15 cases. J Clin Pathol (1986) 39(9):945–9. doi:10.1136/jcp.39.9.945
22. Limper AH, Hoyte JS, Standing JE. The role of alveolar macrophages in Pneumocystis carinii degradation and clearance from the lung. J Clin Invest (1997) 99(9):2110–7. doi:10.1172/JCI119384
23. Shellito JE, Kolls JK, Olariu R, Beck JM. Nitric oxide and host defense against Pneumocystis carinii infection in a mouse model. J Infect Dis (1996) 173(2):432–9. doi:10.1093/infdis/173.2.432
Keywords: immune memory, pneumonia, Pneumocystis, CD8+ T-cells, macrophages, BMPCs
Citation: de la Rua NM, Samuelson DR, Charles TP, Welsh DA and Shellito JE (2016) CD4+ T-Cell-Independent Secondary Immune Responses to Pneumocystis Pneumonia. Front. Immunol. 7:178. doi: 10.3389/fimmu.2016.00178
Received: 19 January 2016; Accepted: 25 April 2016;
Published: 11 May 2016
Edited by:
Pushpa Pandiyan, Case Western Reserve University, USACopyright: © 2016 de la Rua, Samuelson, Charles, Welsh and Shellito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Judd E. Shellito, anNoZWxsQGxzdWhzYy5lZHU=
†Nicholas M. de la Rua and Derrick R. Samuelson contributed equally to this work.