 Spyridon Makris
Spyridon Makris Michelle Paulsen
Michelle Paulsen Cecilia Johansson
Cecilia Johansson- Section of Respiratory Infections, National Heart and Lung Institute, Imperial College London, London, UK
Immune responses to lung infections must be tightly regulated in order to permit pathogen eradication while maintaining organ function. Exuberant or dysregulated inflammation can impair gas exchange and underlies many instances of lung disease. An important driver of inflammation in the lung is the interferon (IFN) response. Type I IFNs are antiviral cytokines that induce a large range of proteins that impair viral replication in infected cells. This cell-intrinsic action plays a crucial role in protecting the lungs from spread of respiratory viruses. However, type I IFNs have also recently been found to be central to the initiation of lung inflammatory responses, by inducing recruitment and activation of immune cells. This helps control virus burden but can cause detrimental immunopathology and contribute to disease severity. Furthermore, there is now increasing evidence that type I IFNs are not only induced after viral infections but also after infection with bacteria and fungi. The pro-inflammatory function of type I IFNs in the lung opens up the possibility of immune modulation directed against this antiviral cytokine family. In this review, the initiation and signaling of type I IFNs as well as their role in driving and maintaining lung inflammation will be discussed.
Lung Inflammation
Mucosal surfaces such as those found in the intestine and the lungs are the most common targets for invading pathogens. The surface area of the human lung is approximately 70 m2, and its main function is gas exchange (1). The lung is constantly in direct contact with the environment and the cells of the lung need to be able to tolerate non-harmful stimuli but react appropriately to harmful pathogens. When lung cells respond to invading pathogens, the regulation of inflammation is particularly important since the lung comprises delicate structure crucial for conducting gas exchange.
Interferons (IFNs), discovered in the 1950s, represent a family of cytokines, which induce robust antiviral and immunomodulatory responses to interfere with virus replication and spread (2–4). IFNs can be classified into three main subclasses: type I, II, and III. Type I IFNs consist of several IFN-α isoforms (13 in human and 14 in mice), IFN-β, IFN-ɛ, IFN-κ, and IFN-ω. In contrast, type II IFNs include only one member, IFN-γ. The most recently discovered group of IFNs are the type III IFNs, including IFN-λ1 (IL-29; non-functional pseudogene in mice), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and the very recently described human IFN-λ4 (5–7). This review will focus on the role of type I IFNs in the lung, however, many of the effects induced by type I IFNs will also be mediated by the other types of IFNs.
Sensing of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) results in production of many cytokines and chemokines including the type I IFNs. These cytokines have a whole array of functions. First, they elicit an antiviral state in infected and neighboring, uninfected cells. Second, type I IFNs modulate the immune response by promoting antigen presentation, cytokine production, dendritic cell (DC) and natural killer (NK) cell activity, and macrophage function. Third, they enhance the adaptive immune response by manipulating T cell effector function and antibody production. Overall, the antiviral and immune stimulatory potential of type I IFNs is required for the effective clearance of acute viral infections (2–4, 8). Furthermore, the impact of type I IFNs on the inflammatory response during other types of infections is also starting to be appreciated.
Induction of Type I IFNs
Multiple cell types including leukocytes and structural cells can detect PAMPs, in various cellular compartments and in all tissues. Recognition of PAMPs by the PRRs initiates an intracellular signaling cascade that causes the translocation of transcription factors to the nucleus initiating innate immune gene expression. The PRRs that can couple pathogen detection to type I IFN induction are toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG)-I-like receptors (RLRs), and cGAS/cGAMP/stimulator of IFN genes [stimulator of interferon genes (STING); Figure 1].
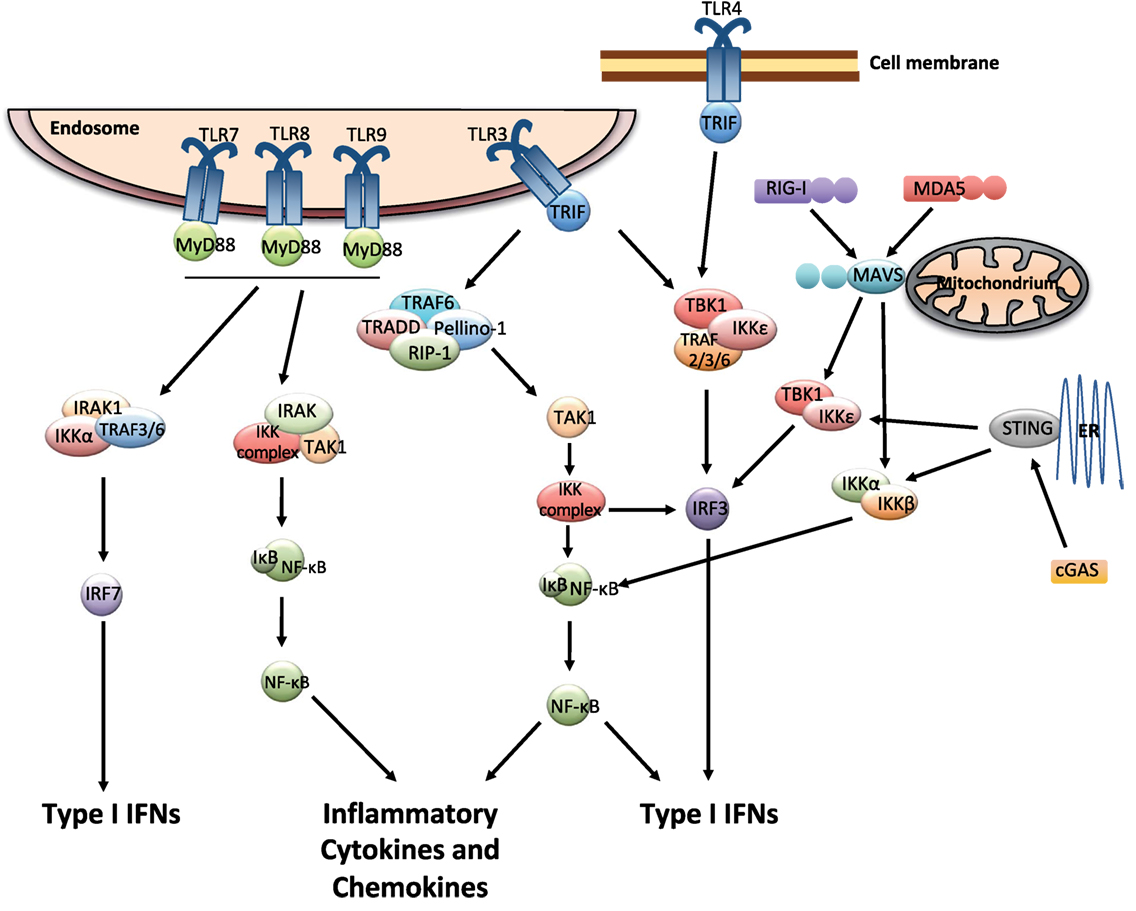
Figure 1. Pattern recognition receptor signaling that leads to the induction of type I interferons (IFNs). The endosomally expressed TLR-3, -7, -8, and -9, cell surface expressed TLR4, the RLRs [retinoic acid-inducible gene I (RIG)-I and MDA-5], and cGAMP synthase (cGAS) can couple pathogen detection to type I IFN induction. TLR3 and TLR4 signal via TRIF, which occurs through inhibitor of kappa-B (IκB) kinases (IKKs), tumor necrosis factor (TNF) receptor-associated factor (TRAF) family associated NF-κB activator (TANK)-binding kinase-1 (TBK1), and IKK-ɛ. This causes the activation of IRF3, which in turn induces the expression of type I IFNs. The activation of TLR3 can also induce the production of inflammatory mediators via TRIF by activating a complex formed by TRAF-6, TNF receptor type I DEATH domain-associated protein (TRADD), Pellino-1, and the receptor-interacting kinase (RIP)-1. This causes the activation of NF-κB pathway, which is mediated by the IKK complex and transforming growth factor beta activated kinase (TAK)-1. TLR7, 8, and 9 use MyD88 for downstream signaling and can activate IRF and NF-κB pathways. RIG-I and MDA5 signal through the adaptor molecule mitochondrial antiviral signaling protein (MAVS). cGAS signals via the adaptor protein stimulator of interferon genes (STING). MAVS and STING further recruit signaling molecules (involving the IKK complex, TBK1, and several TRAF proteins) and lead to the activation of NF-κB and IRF3, resulting in gene expression of various antiviral cytokines including type I IFNs (9–13).
The endosomally expressed TLRs that induce type I IFN gene expression are TLR-3, -7, -8, and -9, each of which detects different forms of nucleic acids (14). TLR3 recognizes double-stranded (ds) RNA which initiates the TRIF-dependent signaling cascade (15). TLR-7/8 and -9 recognize GU-rich single-stranded (ss) RNA and unmethylated CpG DNA, respectively (9), and they require MyD88 for their signaling. Finally, one cell surface expressed TLR, TLR4, which recognize lipopolysaccharides (LPS), and respiratory syncytial virus (RSV) F protein (16), can induce type I IFNs via TRIF-mediated signaling (15) (Figure 1).
The three receptors in the RLRs family, RIG-I, melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) detect viral RNA in the cytosol (10, 11). Due to their localization within the cytoplasm, these receptors are constantly exposed to host RNA, and it is therefore important to distinguish it from that of viral origin. For this reason, the receptors can detect features that are common in many viral genomes and viral replication intermediates but absent from the host (12). RIG-I specifically binds to short uncapped 5′-triphosphate (5′-ppp) and 5′-diphosphate (5′-pp)-bearing base-paired RNA molecules, an RNA motif known to be present in some viral genomes but not in host RNA (17–19). MDA5 binds long stretches of base-paired RNA, which, again, are absent from uninfected cells but often produced as a consequence of viral replication (11). Structurally, RIG-I and MDA5 share many similarities as they both have caspases activation and recruitment domains (CARDs) essential for the downstream signaling (10, 11). LGP2 possesses the RNA-binding domain but it lacks the CARD domains and is therefore not involved in direct signaling. Instead, a role for LGP2 in assisting MDA5-mediated signaling has been suggested (20). For both RIG-I and MDA5, binding of agonists allows downstream signaling through the adaptor molecule mitochondrial antiviral signaling protein (MAVS), leading to the activation of NF-κB and IRF3 and subsequent induction of gene expression of various antiviral mediators including type I IFNs (11, 21, 22) (Figure 1).
The first DNA sensor described was DNA-dependent activator of IFN-regulatory factors (DAI), which was shown to induce type I IFNs during in vitro infections of fibroblast with either herpes simplex virus 1 or cytomegalovirus (CMV). However, the in vivo role for DAI remains unclear (23). Another pathway of recognizing infections with DNA viruses is via RNA polymerase III (PolIII), which transforms DNA into a 5′-ppp RNA, the ligand for RIG-I, yet the physiological role of PolIII remains elusive (24, 25). Other proteins belonging to the PYHIN (IFI16; IFN gamma-inducible protein 16, and AIM2; absent in melanoma 2) or DExD/H-box helicase (DDX) protein families were suggested likely DNA sensors, but their specific role in vivo is unclear and is being investigated [(26); for an extensive review about DNA sensing, see Ref. (24)]. The discovery of the adaptor protein STING identified a pathway crucial for the recognition of foreign dsDNA in vivo. Located at the endoplasmic reticulum, STING activates signaling via NF-κB and IRF3, resulting in the production of type I IFNs (27) (Figure 1). Recently, guanosine-monophosphate adenosine-monophosphate (cyclic GMP-AMP or cGAMP) synthase (cGAS) was identified as a DNA sensor leading to the downstream activation of STING. cGAS binds dsDNA and catalyzes the synthesis of the second messenger cGAMP from ATP and GTP, which binds to STING and activates the signaling cascade leading to type I IFN production (28–30). Interestingly, cGAMP can be transferred through gap junctions from infected cell to neighboring cells and thereby spread antiviral immunity (31).
Different cells of the lung will respond differently to infections depending on both the tropism of the pathogen (which cells that are infected) and which PRRs that are triggered. Lung epithelial cells are the first and most abundant cell type that will interact with the pathogens, and they have the ability to induce IFN-β production especially after influenza virus infection (32). In addition, plasmacytoid dendritic cells (pDCs) have a constitutive expression of IRF-7, which make them able to respond rapidly to TLR7 ligation and induce type I IFNs (33). This is especially important during influenza virus infection (34). However, during RSV infection, it is the alveolar macrophages (AMs) that are the major source of type I IFNs and they use MAVS-dependent PRRs for sensing the virus (35). Thus, in the lung as well as on other sites, many different pathogens can be recognized by several sets of PRRs expressed on the cell surface, in endosomes, or in the cytosol by different cell types. In combination, this will result in the induction of type I IFNs and an efficient antiviral response.
Signaling through the Type I IFN Receptor
Type I IFNs bind to the heterodimeric transmembrane IFN-α/β receptor (IFNAR), which is expressed on all nucleated cells and composed of the two subunits: IFNAR1 and IFNAR2 (36) (Figure 2). Via signaling through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, the induction of several hundreds of interferon stimulated genes (ISGs) is initiated (36–40) (Figure 2). These ISGs interfere with various stages of the viral cycle and change the state of the infected and neighboring cells. Type I IFN signaling also drive the immune response to a number of pathogens by, for example, enhancing the production of inflammatory mediators, cell communication, and the induction of apoptosis in infected cells (see below). It is interesting that the type I IFN receptor has so many ligands, and it is still unclear if all type I IFNs bind the receptor in the same way or if binding of certain IFNs results in a functional difference. There is evidence that different IFNs bind to different anchor points resulting in variations to the binding affinities and conformational change of the IFNAR1 (41). In addition, IFN-β can ligate only the subunit IFNAR1 and signal independently of JAK–STAT pathways (42). Type I IFN production and signaling are tightly regulated by a positive feedback loop, with early IFNs (in the mouse IFN-β and IFN-α4) stimulating the expression of the ISG IRF7 and other important signaling molecules (43). This regulates the expression of all IFN-α isotypes (44) resulting in enhanced signaling through the type I IFN receptor (Figure 2). While the positive feedback loop is important for enhancing the production and effect of type I IFNs, equally important are the negative regulators that are required to restore cellular homeostasis. The type I IFN response is tightly controlled by a series of mechanism that are dependent on cell-intrinsic factors, ISG-mediated proteins, and miRNA. The activity of STAT proteins can be regulated by protein inhibitors of activated STAT (PIAS) and the suppressor of cytokine proteins (SOCS). Another important ISG for the regulation of type I IFN signaling is the ubiquitin carbol-terminal hydrolase protein, USP18 (40, 45).
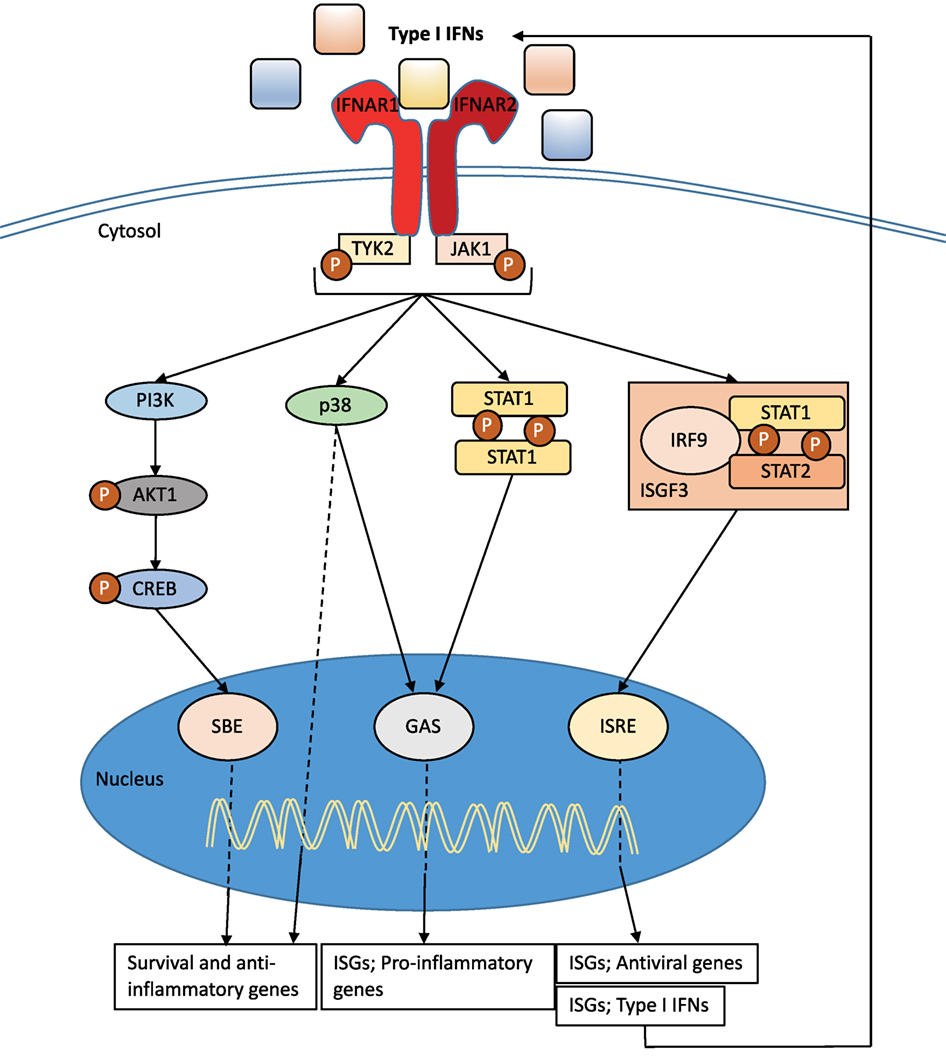
Figure 2. Type I interferon (IFN) signaling. Type I IFNs bind to the heterodimeric transmembrane IFN-α/β receptor (IFNAR), which is composed of the two subunits: IFNAR1 and IFNAR2. The c-termini of IFNAR1 and IFNAR2 are associated with the tyrosine kinase 2 (TYK2) and Janus kinase 1 (JAK1), respectively, and activation of the receptor transduces the phosphorylation of JAK1 and TYK2 by tyrosine phosphorylation. This initiates a signaling cascade composed of proteins of the signal transducer and activator of transcription (STAT) family. The STAT1 and STAT2 proteins are activated upon JAK1 phosphorylation, dimerize and together with IRF9, form the ISG factor 3 (ISGF3) complex. This complex translocates to the nucleus and binds to IFN-stimulated response elements (ISREs) in interferon-stimulated genes (ISGs) promoters to initiate gene transcription. Signaling through IFNAR can also occur independent of IRF9 recruitment through STAT1 homodimers that can bind to IFN-γ-activated sites (GAS) in ISG promoters. Both pathways initiate transcription that promotes the induction of a range of pro-inflammatory mediators and enhance the antiviral state. The JAK–TYK signaling pathway can also promote signaling pathways independent of STAT signaling. One such pathway includes MAPKs, which are important for signals regulating important cellular functions such as gene transcription, post-transcription, apoptosis, and cell-cycle progression. Specifically, the p38 signaling cascade after IFN-stimulation drives transcription of genes that are important for inducing the antiviral effects of type I IFNs and are regulated by ISREs and GAS. Further to MAPK, the type I IFN receptor signaling can also activate the phosphoinositide 3-kinase (PI3K) signaling pathway. The phosphorylation of PI3K causes the activation of the RAC-α serine/threonine-protein kinase (AKT1)/cAMP responsive-element-binding protein that can bind smad binding elements (SBE). This signaling pathway is believed to be important for transcription of genes controlling cellular survival and inflammatory (36, 38–40).
Type I IFN responses are difficult to study in humans since by the time patients with severe lower respiratory tract infections are likely to be admitted to hospital several days have passed since the initial infection, at which time the production of type I IFNs is declining and not easily detectable. However, it is interesting that complete type I IFN deficiency has not been described in humans but mutations in STAT1, tyrosine kinase 2, or NEMO are associated with poor control of viral infections (46, 47). Recently, a mutation in IRF7, leading to impaired type I and type III IFN responses, was shown to result in severe influenza infection in one case (48). In addition, severe RSV disease has been associated with polymorphisms in several innate immune response genes, in particular many that control the type I IFN system (49, 50). Also, several ISGs have been associated with severe influenza infection in mouse and man, for example, IFITM3 and MX1 (51–54). Thus, most data suggest an important role for type I IFNs during respiratory infections.
Type I IFNs Driving Lung Inflammation
The type I IFN signaling is especially important for the control of viral infections. However, in many diseases, the usefulness of type I IFNs has been debated (55). Like all immune responses, a balance is required and launching a response is as important as dampening it. As type I IFNs can lead to both cellular recruitment and activation, an imbalance of the type I IFN response can influence the cellular responses to either result in immunosuppression or immunopathology.
Type I IFNs have multiple effects in the lung. They have been shown to result in the production of chemokines such as CCL2, a monocyte chemoattractant, and CXCL10, important for the recruitment of monocytes/macrophages, T cells, NK cells, and DCs, therefore directly influencing inflammation in the lung (35, 56–58). The type I IFNs also drive a multitude of events in DCs including the differentiation of precursors, increased antigen presentation and cross-presentation, expression of costimulatory molecules, and promoting chemokine secretion and migration (8, 59–66). Interestingly, different subsets of DCs have a different degree of sensitivity to type I IFN signaling, which determines the susceptibility to influenza virus infection and thereby how much antigen they can present (67). Type I IFNs are also important for the activation of macrophages and NK cells (8, 68). Also, CD4+ and CD8+ T cells are directly affected by type I IFNs during various infections and the effect of type I IFNs can either be stimulatory or inhibitory by stimulating proliferation, differentiation, survival as well as inducing anti-proliferative and pro-apoptotic responses (63, 69). Type I IFN receptor signaling in T cells is also important for cytokine secretion, cytotoxicity, and memory formation (63). This varied effect of type I IFNs on T cells is partly dependent on the different STATs induced by type I IFNs where for example STAT1 is pro-inflammatory, pro-apoptotic, and anti-proliferative, while STAT4 promotes proliferation and cell survival (63). The different effects are dependent on when and where the T cells get the signal via the type I IFN receptor and will therefore be dependent on the site of infection, timing, magnitude, and source of type I IFN responses. B cells are also influenced by type I IFNs, and type I IFN receptor signaling on both B and T cells is important for antibody production (70, 71). Interestingly, it has been suggested that during LCMV infection, type I IFN receptor signaling in CD8+ T cells increase their killing of B cells and therefore decrease the production of neutralizing antibodies (72). If this is also occurring during respiratory infections remain to be elucidated.
In the absence of type I IFNs, there is often less inflammation in the lung. Interestingly, intranasal administration of IFN-α alone can stimulate the expression of pro-inflammatory cytokines (56, 73). Also, IFN-α can potentiate the pro-inflammatory responses after intranasal LPS challenge (56). How this occurs considering most cytokines and chemokines are induced by NF-κβ and not IFNAR signaling is unknown. One possibility is the fact that type I IFNs increase the expression of many PRRs or related signaling molecules, which can enhance signaling via other pathways for example those involved in NF-κB activation (74). Nevertheless, all cellular components involved in the mutual influence of NF-κB and type I IFN pathways are not fully investigated and might depend on the cellular context. It is therefore a likely scenario that type I IFNs play a role in NF-κB-induced expression of pro-inflammatory cytokines and chemokines.
Lung Viral Infections
Type I IFNs are rapidly produced during all lung viral infections and contribute to the initial control of viral replication before effective innate and adaptive cellular responses are generated to clear the virus. Signaling via the IFNAR activates a cascade of ISGs that directly interfere with viral replication and viral spread. These include proteins that inhibit virus entry (e.g., MX1, IFITM proteins, TRIM proteins), modulate membrane lipids to prevent viral release (e.g., Viperin, Tetherin), induce apoptosis of infected cells [e.g., protein kinase R (PKR)], regulate transcriptional (e.g., Viperin) and posttranscriptional (e.g., OAS/RNaseL, PKR) mechanisms, and posttranslational events (e.g., ISG15) (38, 40). In addition to the ISG-mediated effects, type I IFNs modulate cell viability and function (e.g., apoptosis, inhibiting cell death, differentiation, migration, proliferation) to support antiviral defense (75). The amplification of the primary signal by type I IFNs is achieved by upregulation of many PRR molecules and associated signaling molecules like TLR3, RIG-I, MDA5, MAVS, MyD88, IRF3, and IRF7, which are themselves ISGs and therefore amplify the type I IFN response to viral infections [see above and Ref. (76, 77)]. Without a functioning IFNAR loop the detection of accumulating viral RNA and the further downstream processing of the signal is compromised in infected cells as they lack the feedback-mediated boosted expression of the viral RNA sensors. This imbalance will eventually promote viral replication and spread early during infection and also influence the degree of inflammation. For example, in a severe acute respiratory syndrome-CoV infection, type I IFN induction is delayed resulting in an overwhelming viral burden (78). Interestingly, many viruses have virulence factors that antagonize type I IFN responses [reviewed elsewhere (79–81)], indicating the importance of type I IFN responses during viral infection. It is also important to note that, especially during influenza virus infection, type III IFNs (IFN-λ) are highly induced at the same time as type I IFNs (82, 83). Interestingly, the receptor for IFN-λ is mostly expressed by epithelial cells and therefore these cytokines have a more restricted effect directed to intrinsic antiviral mechanisms (73). The effects of IFN-λ have recently been reviewed (5).
There are also age-related effects of type I IFN production in response to respiratory infections as both cells from infants and neonatal mice show a reduced type I IFN production and ISG induction after RSV exposure (84–86). Furthermore, monocytes from elderly have a diminished type I IFN response after exposure to influenza virus (53). Interestingly, these age groups are also very vulnerable to respiratory infections.
In addition to the cell-intrinsic responses, type I IFNs are known to enhance immune responses especially by activating and recruiting immune cells. During RSV infection, type I IFNs produced by AMs, induced the production of CCL2 and other chemoattractants crucial for monocyte extravasation into the lung during RSV infection (35). These recruited monocytes are important for controlling the virus (35). Interestingly, the expression of pro-inflammatory cytokines and chemokines was significantly reduced in RSV, influenza virus, and human metapneumovirus-infected Ifnar1−/− mice (56, 87, 88). This highlights again a synergizing effect of NF-κB and type I IFN pathways in inducing the optimal secretion of cytokines and chemokines. However, NF-κB translocation into the nucleus can be detected as early as 0.5 h post RSV inoculation without the necessity of viral replication (89). This might provide an explanation for the early induction of some of the measured cytokines such as IFN-β, IL-6, TNF-α, or IL-12 in the lungs of RSV-infected Ifnar1−/− mice, but perhaps sustained NF-κB activation may be in part type I IFN dependent. In contrast to Ifnar1−/− mice, wt mice have the ability to further enhance this “first signal” through the IFNAR-driven autocrine and paracrine amplification loop to maximize the responses. Moreover, the lack of responsiveness to viruses in Ifnar1−/− mice could also suppress the recruitment of additional cells to the site of infection, which could otherwise contribute to local cytokine secretion. These usually specifically recruited cells are most likely of non-polymorphonuclear origin, since neutrophil recruitment was comparable or increased in RSV or influenza virus infected Ifnar1−/− mice compared to wt mice (56, 90). Furthermore, during influenza or Sendai virus infection type I IFNs can act as messengers from the lung to the bone marrow where they instruct leukocytes to activate an antiviral transcriptional program, resulting in an increased antiviral state of these cells before they migrate to the lung (91).
Additional to T cell expansion, type I IFNs have been shown to promote T effector cell function due to the dependence on IFNAR mediated STAT1 signaling for the cytolytic activity of memory CD8+ T cells during recall responses with respiratory viruses (92). In contrast, after influenza virus infection type I IFNs can induce the secretion of IL-10 from CD8+ T cells (93) and the expression of programmed cell death ligand 1 (PDL-1) on epithelial cells (94). Furthermore, blocking of PDL-1 enhanced CD8+ T cells function and viral clearance (94). Thus, type I IFNs are important both to induce T cell effector functions and also to induce anti-inflammatory mechanisms that can suppress the T cell response.
Despite the essential role for type I IFNs in activating the immune response to successfully combat viral infection and to guarantee survival of the organism, they can have deleterious effects on the host and cause acute immunopathology. High type I IFN production during influenza virus infection mediated by recruited inflammatory monocytes and pDCs cause the upregulation of TNF-related apoptosis-inducing ligand (TRAIL) (8, 95). This “death” ligand binds to the death receptor 5 on epithelial cells, the expression of which is also upregulated by type I IFNs. Thus, the TRAIL-expressing inflammatory monocytes cause the death of the epithelial cells, which in turn increases morbidity or death of the mice (83, 96). Therefore, the potential of type I IFNs to drive and amplify pro-inflammatory responses (56, 83) could, if type I IFNs are produced excessively or for an extended time, cause the increased morbidity and mortality during lung viral infections (78, 83, 96). Thus, the timing and magnitude of type I IFN responses are crucial to obtain an efficient cell-intrinsic response and a balanced cell-extrinsic response that together lead to the clearance of the virus without causing detrimental immunopathology.
Lung Bacterial Infections
Since type I IFNs have an important role in preventing replication and spread during viral infections their role is overall beneficial. During lung bacterial infections, type I IFNs are induced but the role of these cytokines is unclear and their ability to drive inflammation might in these cases be more detrimental. For example, mice deficient in IFNAR1 or TLR9 showed an improved clearance of Staphylococcus aureus (97). Also, a lung infection model of Chlamydia trachomatis mouse pneumonitis (Chlamydia muridarum) showed that deficiency in the IFNAR1 resulted in less bacterial burden and bodyweight loss, and milder pathological changes (98). Mycobacterium tuberculosis (MTb) infection of TB-susceptible Ifnar1−/− mice showed enhanced protection from death, lower bacterial burden in the lungs, and decreased degree of lung inflammation compared to wt mice (99). In addition, an interferon signature is evident in patients with active MTb disease (100), and mouse studies have shown that the type I IFN responses during MTb infection is tightly regulated by IL-1 and PGE2 (101). More contradictory data are presented for Streptococcus pneumoniae infection, where an invasive strain of S. pneumoniae induces type I IFNs in the lungs and blocking the IFNAR decreased the systemic bacteremia (102). In contrast, another study showed that S. pneumoniae infection of Ifnar1−/− mice or mice treated with an antibody against the type I IFN receptor displayed enhanced bacterial spread and increase bacteremia (103). In addition, if mice were given rIFN-β, this reduced the bacteremia after intranasal S. pneumoniae infection (103). Thus, most data suggest that type I IFNs induced during lung bacterial infection are part of the inflammatory response and might be important to initiate immune responses to the infection. However, since these cytokines lead to the recruitment and activation of immune cells, this can enhance inflammation and result in bacterial dissemination and spread.
Interestingly, bacterial secondary infections are common after a severe lung viral infection and the host is more susceptible to infection by Escherichia coli, Pseudomonas aeruginosa, S. pneumoniae, or S. aureus after influenza virus infection (104–106). This increased susceptibility is probably due to many factors such as a lower activation threshold of lung cells, inhibition of important signaling pathways and cytokines induction and exhaustion of immune cells resulting in non-appropriate immune responses elicited to the new pathogen. Noteworthy, this increased susceptibility cease to exist as soon as the virus-induced type I IFNs are decreasing (104) and IFNAR1 deficient mice can effectively clear a secondary S. pneumoniae infection (106). Furthermore, the induction of type I IFNs during influenza virus infection attenuates chemokines important for neutrophil recruitment, which can promote secondary bacterial pneumonia (107). A possible mechanism for this is the type I IFN-dependent upregulation of the methyltransferase Setdb2, which can repress chemokines such as CXCL1 at the chromatin level (108). Also, influenza virus infection before or during MTb infection increases the severity of the MTb infection via type I IFN signaling (109). Interestingly, type I IFNs have been shown to inhibit inflammasome activation and IL-1 responses and increase IL-10 production (110). Altogether, this suggests that type I elicited during a lung viral infection makes it more possible for a subsequent bacterial infection to establish.
Lung Fungal Infections
The role of type I IFNs during fungal infections has also been investigated using mice deficient in the IFNAR1. During a lung Pneumocystis infection, a decreased pro-inflammatory response was detected and even if the pathogen burden was the same, the clearance was delayed in Ifnar1−/− mice and this resulted in an exacerbated Th2 response and fibrosis (111). The lack of type I IFN signaling during Cryptococcus neoformans infection has been shown both to result in a decreased pathogen burden (112) and increased pathogen burden with increased death (113). However, both studies showed higher Th2 cytokine levels in the Ifnar1−/− mice after C. neoformans infection (112, 113). Thus, type I IFN responses during lung fungal infection are also part of driving the inflammatory response but it is still unclear in which magnitude or which mechanisms that are used.
Summary
There are obvious benefits of type I IFNs during a lung viral infection as these cytokines have a vital cell-intrinsic antiviral effect limiting viral replication. However, the cell-extrinsic effects of type I IFNs are important during all lung infections as type I IFNs directly drive lung inflammation, most likely by amplifying primary signals initiated by other stimuli. When considering to use type I IFN as a potential antiviral agent, the immune-modulating effects during lung infections needs to be considered as the type I IFN response has to be tightly regulated so that a balance of beneficial (initiation of inflammation) and detrimental (immunopathology) effects is achieved and gas exchange is not impaired.
Author Contributions
CJ conceptualized the scope of the review article. CJ, SM, and MP wrote the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors want to thank the Medical Research Council (Grant G0800311), the Rosetrees Trust (M370), and the National Heart and Lung Institute Foundation (registered charity number 1048073) for research support. The authors also want to thank Caetano Reis e Sousa for critically reading the review.
References
1. Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol (2014) 16:36–44. doi:10.1038/ni.3052
2. Durbin RK, Kotenko SV, Durbin JE. Interferon induction and function at the mucosal surface. Immunol Rev (2013) 255:25–39. doi:10.1111/imr.12101
3. González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12:125–35. doi:10.1038/nri3133
4. Swiecki M, Colonna M. Type I interferons: diversity of sources, production pathways and effects on immune responses. Curr Opin Virol (2011) 1:463–75. doi:10.1016/j.coviro.2011.10.026
5. Wack A, Terczyńska-Dyla E, Hartmann R. Guarding the frontiers: the biology of type III interferons. Nat Immunol (2015) 16:802–9. doi:10.1038/ni.3212
6. Egli A, Santer DM, O’Shea D, Barakat K, Syedbasha M, Vollmer M, et al. IL-28B is a key regulator of B- and T-cell vaccine responses against influenza. PLoS Pathog (2014) 10:e1004556. doi:10.1371/journal.ppat.1004556
7. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet (2013) 45:164–71. doi:10.1038/ng.2521
8. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi:10.1038/nri3787
9. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi:10.1016/j.immuni.2011.05.006
10. Loo Y-M, Gale M. Immune signaling by RIG-I-like receptors. Immunity (2011) 34:680–92. doi:10.1016/j.immuni.2011.05.003
11. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity (2013) 38:855–69. doi:10.1016/j.immuni.2013.05.007
12. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol (2015) 33:257–90. doi:10.1146/annurev-immunol-032414-112240
13. Gürtler C, Bowie AG. Innate immune detection of microbial nucleic acids. Trends Microbiol (2013) 21:413–20. doi:10.1016/j.tim.2013.04.004
14. O’Neill LAJ, Golenbock D, Bowie AG. The history of toll-like receptors – redefining innate immunity. Nat Rev Immunol (2013) 13:453–60. doi:10.1038/nri3446
15. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med (2003) 198:1043–55. doi:10.1084/jem.20031023
16. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol (2000) 1:398–401. doi:10.1038/80833
17. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature (2014) 514:372–5. doi:10.1038/nature13590
18. Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science (2006) 314:994–7. doi:10.1126/science.1132505
19. Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science (2006) 314:997–1001. doi:10.1126/science.1132998
20. Bruns AM, Leser GP, Lamb RA, Horvath CM. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol Cell (2014) 55:771–81. doi:10.1016/j.molcel.2014.07.003
21. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347(6227):aaa2630. doi:10.1126/science.aaa2630
22. Wu B, Hur S. How RIG-I like receptors activate MAVS. Curr Opin Virol (2015) 12:91–8. doi:10.1016/j.coviro.2015.04.004
23. Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature (2008) 451:725–9. doi:10.1038/nature06537
24. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity (2013) 38:870–80. doi:10.1016/j.immuni.2013.05.004
25. Chiu Y-H, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell (2009) 138:576–91. doi:10.1016/j.cell.2009.06.015
26. Schattgen SA, Gao G, Kurt-Jones EA, Fitzgerald KA. Cutting edge: DNA in the lung microenvironment during influenza virus infection tempers inflammation by engaging the DNA sensor AIM2. J Immunol (2016) 196:29–33. doi:10.4049/jimmunol.1501048
27. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal (2012) 5:ra20. doi:10.1126/scisignal.2002521
28. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi:10.1126/science.1232458
29. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498:380–4. doi:10.1038/nature12306
30. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339:826–30. doi:10.1126/science.1229963
31. Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature (2013) 503:530–4. doi:10.1038/nature12640
32. Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog (2013) 9:e1003773. doi:10.1371/journal.ppat.1003773
33. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11:373–84. doi:10.1038/ni.1863
34. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol (2014) 14:315–28. doi:10.1038/nri3665
35. Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med (2015) 212:699–714. doi:10.1084/jem.20140825
36. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi:10.1038/nri1604
37. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature (2011) 472:481–5. doi:10.1038/nature09907
38. Schoggins JW. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol (2014) 6:40–6. doi:10.1016/j.coviro.2014.03.006
39. Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi:10.1016/j.immuni.2012.03.013
40. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi:10.1146/annurev-immunol-032713-120231
41. Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell (2011) 146:621–32. doi:10.1016/j.cell.2011.06.048
42. De Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff S-J, et al. Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat Immunol (2013) 14:901–7. doi:10.1038/ni.2667
43. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434:772–7. doi:10.1038/nature03464
44. Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J (1998) 17:6660–9. doi:10.1093/emboj/17.22.6660
45. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2013) 14:36–49. doi:10.1038/nri3581
46. Sancho-Shimizu V, Pérez de Diego R, Jouanguy E, Zhang S-Y, Casanova J-L. Inborn errors of anti-viral interferon immunity in humans. Curr Opin Virol (2011) 1:487–96. doi:10.1016/j.coviro.2011.10.016
47. Zhang S-Y, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev (2008) 226:29–40. doi:10.1111/j.1600-065X.2008.00698.x
48. Ciancanelli MJ, Huang SXL, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science (2015) 348:448–53. doi:10.1126/science.aaa1578
49. Janssen R, Bont L, Siezen CLE, Hodemaekers HM, Ermers MJ, Doornbos G, et al. Genetic susceptibility to respiratory syncytial virus bronchiolitis is predominantly associated with innate immune genes. J Infect Dis (2007) 196:826–34. doi:10.1086/520886
50. Siezen CLE, Bont L, Hodemaekers HM, Ermers MJ, Doornbos G, Van’t Slot R, et al. Genetic susceptibility to respiratory syncytial virus bronchiolitis in preterm children is associated with airway remodeling genes and innate immune genes. Pediatr Infect Dis J (2009) 28:333–5. doi:10.1097/INF.0b013e31818e2aa9
51. Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature (2012) 484:519–23. doi:10.1038/nature10921
52. Zhang Y-H, Zhao Y, Li N, Peng Y-C, Giannoulatou E, Jin R-H, et al. Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat Commun (2013) 4:1418. doi:10.1038/ncomms2433
53. Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science (2016) 352:463–6. doi:10.1126/science.aaf3926
54. Ciancanelli MJ, Abel L, Zhang S-Y, Casanova J-L. Host genetics of severe influenza: from mouse Mx1 to human IRF7. Curr Opin Immunol (2016) 38:109–20. doi:10.1016/j.coi.2015.12.002
55. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207:2053–63. doi:10.1084/jem.20101664
56. Goritzka M, Durant LR, Pereira C, Salek-Ardakani S, Openshaw PJM, Johansson C. Interferon-α/β receptor signaling amplifies early pro-inflammatory cytokine production in the lung during respiratory syncytial virus infection. J Virol (2014) 88:6128–36. doi:10.1128/JVI.00333-14
57. Lehmann MH, Torres-Domínguez LE, Price PJR, Brandmüller C, Kirschning CJ, Sutter G. CCL2 expression is mediated by type I IFN receptor and recruits NK and T cells to the lung during MVA infection. J Leukoc Biol (2016) 99:1057–64. doi:10.1189/jlb.4MA0815-376RR
58. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25:373–81. doi:10.1016/j.immuni.2006.08.007
59. Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol (2001) 166:2961–9. doi:10.4049/jimmunol.166.5.2961
60. Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol (2003) 4:1009–15. doi:10.1038/ni978
61. Rouzaut A, Garasa S, Teijeira A, González I, Martinez-Forero I, Suarez N, et al. Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to IFN-α. Eur J Immunol (2010) 40:3054–63. doi:10.1002/eji.201040523
62. Spadaro F, Lapenta C, Donati S, Abalsamo L, Barnaba V, Belardelli F, et al. IFN-α enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood (2012) 119:1407–17. doi:10.1182/blood-2011-06-363564
63. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol (2015) 15:231–42. doi:10.1038/nri3806
64. Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O’Garra A, Vicari A, et al. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med (2005) 201:1157–67. doi:10.1084/jem.20041930
65. Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med (2009) 206:1589–602. doi:10.1084/jem.20090247
66. Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, et al. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A (2003) 100:10872–7. doi:10.1073/pnas.1934678100
67. Moltedo B, Li W, Yount JS, Moran TM. Unique type I interferon responses determine the functional fate of migratory lung dendritic cells during influenza virus infection. PLoS Pathog (2011) 7:e1002345. doi:10.1371/journal.ppat.1002345
68. Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res (2011) 17:2619–27. doi:10.1158/1078-0432.CCR-10-1114
69. Tough DF. Modulation of T-cell function by type I interferon. Immunol Cell Biol (2012) 90:492–7. doi:10.1038/icb.2012.7
70. Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol (2006) 176:2074–8. doi:10.4049/jimmunol.176.4.2074
71. Chang WLW, Coro ES, Rau FC, Xiao Y, Erle DJ, Baumgarth N. Influenza virus infection causes global respiratory tract B cell response modulation via innate immune signals. J Immunol (2007) 178:1457–67. doi:10.4049/jimmunol.178.3.1457
72. Moseman EA, Wu T, la Torre de JC, Schwartzberg PL, McGavern DB. Type I interferon suppresses virus-specific B cell responses by modulating CD8+ T cell differentiation. Sci Immunol (2016) 1(4). doi:10.1126/sciimmunol.aah3565
73. Davidson S, McCabe TM, Crotta S, Gad HH, Hessel EM, Beinke S, et al. IFNλ is a potent anti-influenza therapeutic without the inflammatory side effects of IFNα treatment. EMBO Mol Med (2016) 8:1099–112. doi:10.15252/emmm.201606413
74. Pfeffer LM. The role of nuclear factor κB in the interferon response. J Interferon Cytokine Res (2011) 31:553–9. doi:10.1089/jir.2011.0028
75. Mattei F, Schiavoni G, Tough DF. Regulation of immune cell homeostasis by type I interferons. Cytokine Growth Factor Rev (2010) 21:227–36. doi:10.1016/j.cytogfr.2010.05.002
76. Haller O, Kochs G, Weber F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology (2006) 344:119–30. doi:10.1016/j.virol.2005.09.024
77. Jaitin DA, Schreiber G. Upregulation of a small subset of genes drives type I interferon-induced antiviral memory. J Interferon Cytokine Res (2007) 27:653–64. doi:10.1089/jir.2006.0162
78. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe (2016) 19:181–93. doi:10.1016/j.chom.2016.01.007
79. Wang BX, Fish EN. The yin and yang of viruses and interferons. Trends Immunol (2012) 33:190–7. doi:10.1016/j.it.2012.01.004
80. Schulz KS, Mossman KL. Viral evasion strategies in type I IFN signaling – a summary of recent developments. Front Immunol (2016) 7:459. doi:10.3389/fimmu.2016.00498
81. Coccia EM, Battistini A. Early IFN type I response: learning from microbial evasion strategies. Semin Immunol (2015) 27:85–101. doi:10.1016/j.smim.2015.03.005
82. Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS, Bakaletz LO, et al. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J Virol (2007) 81:9790–800. doi:10.1128/JVI.00530-07
83. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon αβ in acute influenza infection. Nat Commun (2014) 5:3864. doi:10.1038/ncomms4864
84. Cormier SA, Shrestha B, Saravia J, Lee GI, Shen L, DeVincenzo JP, et al. Limited type I interferons and plasmacytoid dendritic cells during neonatal respiratory syncytial virus infection permit immunopathogenesis upon reinfection. J Virol (2014) 88:9350–60. doi:10.1128/JVI.00818-14
85. Remot A, Descamps D, Jouneau L, Laubreton D, Dubuquoy C, Bouet S, et al. Flt3 ligand improves the innate response to respiratory syncytial virus and limits lung disease upon RSV re-exposure in neonate mice. Eur J Immunol (2015) 46:874–84. doi:10.1002/eji.201545929
86. Marr N, Wang TI, Kam SHY, Hu YS, Sharma AA, Lam A, et al. Attenuation of respiratory syncytial virus-induced and RIG-I-dependent type I IFN responses in human neonates and very young children. J Immunol (2014) 192:948–57. doi:10.4049/jimmunol.1302007
87. Hastings AK, Erickson JJ, Schuster JE, Boyd KL, Tollefson SJ, Johnson M, et al. Role of type I interferon signaling in human metapneumovirus pathogenesis and control of viral replication. J Virol (2015) 89:4405–20. doi:10.1128/JVI.03275-14
88. Duerr CU, McCarthy CDA, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol (2016) 17:65–75. doi:10.1038/ni.3308
89. Haeberle HA, Takizawa R, Casola A, Brasier AR, Dieterich H-J, van Rooijen N, et al. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J Infect Dis (2002) 186:1199–206. doi:10.1086/344644
90. Stifter SA, Bhattacharyya N, Pillay R, Flórido M, Triccas JA, Britton WJ, et al. Functional interplay between type I and II interferons is essential to limit influenza A virus-induced tissue inflammation. PLoS Pathog (2016) 12:e1005378. doi:10.1371/journal.ppat.1005378
91. Hermesh T, Moltedo B, Moran TM, López CB. Antiviral instruction of bone marrow leukocytes during respiratory viral infections. Cell Host Microbe (2010) 7:343–53. doi:10.1016/j.chom.2010.04.006
92. Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, Woodland DL. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity (2010) 33:96–105. doi:10.1016/j.immuni.2010.06.016
93. Jiang L, Yao S, Huang S, Wright J, Braciale TJ, Sun J. Type I IFN signaling facilitates the development of IL-10-producing effector CD8(+) T cells during murine influenza virus infection. Eur J Immunol (2016) 46(12):2778–88. doi:10.1002/eji.201646548
94. McNally B, Ye F, Willette M, Flaño E. Local blockade of epithelial PDL-1 in the airways enhances T cell function and viral clearance during influenza virus infection. J Virol (2013) 87:12916–24. doi:10.1128/JVI.02423-13
95. Chaperot L, Blum A, Manches O, Lui G, Angel J, Molens J-P, et al. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J Immunol (2006) 176:248–55. doi:10.4049/jimmunol.176.1.248
96. Herold S, Steinmueller M, Wulffen von W, Cakarova L, Pinto R, Pleschka S, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med (2008) 205:3065–77. doi:10.1084/jem.20080201
97. Parker D, Prince A. Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J Immunol (2012) 189:4040–6. doi:10.4049/jimmunol.1201055
98. Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol (2008) 181:2092–102. doi:10.4049/jimmunol.181.3.2092
99. Dorhoi A, Yeremeev V, Nouailles G, Weiner J III, Jörg S, Heinemann E, et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur J Immunol (2014) 44:2380–93. doi:10.1002/eji.201344219
100. Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 466:973–7. doi:10.1038/nature09247
101. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 522:99–103. doi:10.1038/nature13489
102. Hughes CE, Harvey RM, Plumptre CD, Paton JC. Development of primary invasive pneumococcal disease caused by serotype 1 pneumococci is driven by early increased type I interferon response in the lung. Infect Immun (2014) 82:3919–26. doi:10.1128/IAI.02067-14
103. LeMessurier KS, Häcker H, Chi L, Tuomanen E, Redecke V. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog (2013) 9:e1003727. doi:10.1371/journal.ppat.1003727
104. Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, et al. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol (2015) 309:L158–67. doi:10.1152/ajplung.00338.2014
105. Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep (2015) 16:1203–18. doi:10.15252/embr.201540473
106. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γd T cells. J Virol (2012) 86:12304–12. doi:10.1128/JVI.01269-12
107. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest (2009) 119:1910–20. doi:10.1172/JCI35412
108. Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminathan S, Bosnjak B, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol (2015) 16:67–74. doi:10.1038/ni.3046
109. Redford PS, Mayer-Barber KD, McNab FW, Stavropoulos E, Wack A, Sher A, et al. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J Infect Dis (2014) 209:270–4. doi:10.1093/infdis/jit424
110. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34:213–23. doi:10.1016/j.immuni.2011.02.006
111. Meissner NN, Swain S, Tighe M, Harmsen A, Harmsen A. Role of type I IFNs in pulmonary complications of Pneumocystis murina infection. J Immunol (2005) 174:5462–71. doi:10.4049/jimmunol.174.9.5462
112. Sato K, Yamamoto H, Nomura T, Matsumoto I, Miyasaka T, Zong T, et al. Cryptococcus neoformans infection in mice lacking type I interferon signaling leads to increased fungal clearance and IL-4-dependent mucin production in the lungs. PLoS One (2015) 10:e0138291. doi:10.1371/journal.pone.0138291
Keywords: lung, infection, inflammation, type I interferons, pattern recognition receptors
Citation: Makris S, Paulsen M and Johansson C (2017) Type I Interferons as Regulators of Lung Inflammation. Front. Immunol. 8:259. doi: 10.3389/fimmu.2017.00259
Received: 21 December 2016; Accepted: 21 February 2017;
Published: 10 March 2017
Edited by:
Claudia U. Duerr, McGill University, CanadaReviewed by:
Sabine Riffault, Centre de Recherches de Jouy-en-Josas (INRA), FranceAndreas Wack, Francis Crick Institute, UK
Copyright: © 2017 Makris, Paulsen and Johansson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Johansson, Yy5qb2hhbnNzb25AaW1wZXJpYWwuYWMudWs=
†Present address: Michelle Paulsen, Department of Translational Pulmonology, Translational Lung Research Center Heidelberg (TLRC), German Center for Lung Research (DZL), University of Heidelberg, Germany