 Junyang Yu
Junyang Yu Yuzhang Wu*
Yuzhang Wu* Jingxue Wang
Jingxue Wang- Institute of Immunology, Third Military Medical University, Chongqing, China
NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome activation and effects during ribonucleic acid (RNA) viral infection are the focus of a wide range of research currently. Both the pathogen-associated molecule pattern derived from virions and intracellular stress molecules involved in the process of viral infection lead to activation of the NLRP3 inflammasome, which in turn triggers inflammatory responses for antiviral defense and tissue healing. However, aberrant activation of the NLRP3 inflammasome can instead support viral pathogenesis and promote disease progression. Here, we summarize and expound upon the recent literature describing the molecular mechanisms underlying the activation and effects of the NLRP3 inflammasome in RNA viral infection to highlight how it provides protection against RNA viral infection.
Introduction
The ribonucleic acid (RNA) virus has single-stranded (ss)RNA or double-stranded (ds)RNA as its genetic material. Infections with RNA viruses are responsible for a variety of diseases are significant threats to public health, including influenza, hepatitis, viral encephalitis, and autoimmune deficiency syndrome, to name a few of the most prominent. Host antiviral defense mechanisms are critical for clearing RNA viral infections and controlling the manifested diseases. The initiation of antiviral defense in response to RNA viruses involves interferon (IFN) response and inflammasome activation. The RNA viral infection usually leads to activation of the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome, which is the intracellular multiprotein complex formed upon sensing pathogen-associated molecule patterns and/or damage-associated molecule patterns (1). The inflammasome response is critical for the innate immune response’s ability to initiate innate and adaptive immunity pathways during RNA viral infections (2).
The NLRP3 inflammasome has been extensively investigated. Its major components include pro-caspase-1 and apoptosis-associated speck-like protein containing a CARD (ASC), in addition to the NLRP3 molecules. Upon sensing activating signals, the NLRP3 oligomerize to generate a caspase-1-activating scaffold, resulting in autocleavage, and activation of pro-caspase-1. The activated caspase-1 then shears pro-interleukin (IL)-1β and pro-IL-18 into IL-1β and IL-18, leading to production of their functional forms and release from cells to mediate protective (or sometimes detrimental) inflammatory responses. The activated caspase-1, however, can also promote caspase-1-dependent pyroptosis, a type of inflammatory programmed cell death; although, the consequence of pyroptosis induced by the NLRP3 inflammasome remains to be fully elucidated in RNA viral infection (3).
Multiple lines of evidence have shown that the NLRP3 inflammasome can be activated by various viruses, including type A Influenza virus (IAV), human immunodeficiency virus-type 1 (HIV-1), vesicular stomatitis virus (VSV), respiratory syncytial virus (RSV), enterovirus 71 (EV71), hepatitis C virus (HCV), and SARS coronavirus (SARS-CoV), as detailed in Table 1. This review will focus on recent advances in our knowledge of the activation mechanisms and functions of the NLRP3 inflammasome in response to such RNA viral infections, discussing the related protective, and/or detrimental mechanisms that may represent manipulable targets for prevention and/or treatment.
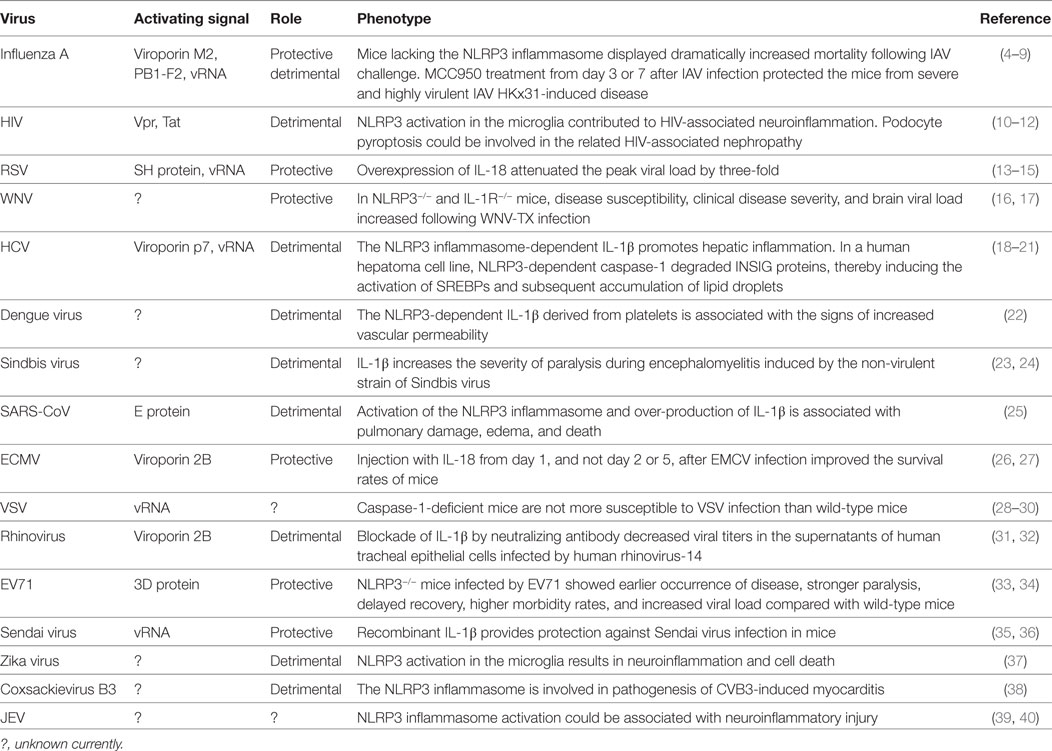
Table 1. Activating signals for and roles of the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome in ribonucleic acid (RNA) viral infections.
Priming Mechanisms of the NLRP3 Inflammasome in RNA Viral Infections
NACHT, LRR, and PYD domains-containing protein 3 inflammasome activation requires two types of signals. Signal 1 primes the NLRP3 inflammasome and signal 2 initiates the sequence of its assembly, activation of the pro-caspase-1, and cleavage of pro-IL-1β and pro-IL-18.
During RNA viral infections, signal 1 is usually derived from the signals evoked upon pathogen recognition by toll-like receptors (TLRs) (6) and retinoic acid-inducible gene-I (RIG-I)-like receptors (41). Recent research has shown that signal 1 triggering of the NLRP3 inflammasome involves transcription-dependent and post-translation-dependent priming. The transcription-dependent priming is executed by activation of nuclear factor (NF)-κB and IFN-regulatory factor transcription factors, and subsequent up-regulation of transcription of genes encoding the major inflammasome components through the classical TLR signaling pathway: the MyD88-mediated and TRIF-mediated pathway. The specific role of TLR7 was shown by the reduced pro-IL-1β transcription in bone marrow-derived macrophages from TLR7−/− mice in response to 24 h of IAV infection (6). Another in vivo study of the IAV infection model provided evidence that commensal bacteria lipopolysaccharide can provide priming signal 1 (42). Investigations of VSV infection showed that RIG-I promotes the synthesis of pro-IL-1β through engagement by the viral 5′-triphosphate RNA (3pRNA). Moreover, bone marrow-derived cells lacking RIG-I, as well as the downstream molecules MAVS and CARD9, showed reduced synthesis of pro-IL-1β but normal caspase-1 p10 subunits when stimulated with VSV and 3pRNA (41), suggesting that the RIG-I/MAVS/CARD9 signaling pathway provides the signal 1 in VSV infection.
Post-translation-dependent priming occurs upon the simultaneous engagement of TLRs and Nod-like receptors with their ligands, even causing rapid activation of the NLRP3 inflammasome (within 15 min) (43, 44). Viral RNA (vRNA) molecule-induced NLRP3 inflammasome activation also comes about by post-translational priming, but involving the RIP1/caspase 8/RIP3 signaling pathway (29, 45), which may ultimately result in the deubiquitination of NLRP3 protein by BRCC3 (46, 47). RIP3-deficient mice showed reduced caspase-1 activation and IL-1β production following infection with VSV, Sendai virus, or IAV (29). Bone marrow-derived macrophages and mice lacking RIP1 both displayed substantial reduction in caspase-1 cleavage and levels of IL-1β and IL-18 following challenge with IAV (8, 29). In various RNA viral infection studies, the RIP1/RIP3-mediated signaling pathway has been demonstrated to provide the endogenous signal 2 for the NLRP3 inflammasome, being derived from reactive oxygen species (ROS) production by mitochondrial fission initiated by the RIP1/RIP3/DRP pathway (29, 45, 48).
Viral Proteins Involved in NLRP3 Inflammasome Activation Induced by RNA Virus
Signal 2 is responsive to a wide range of stimuli [i.e., NLRP3 agonists, such as adenosine triphosphate (ATP), silica, alum, and ultraviolet radiation]. However, the spectrum of different structures represented by these varied and distinctive agonists indicates that NLRP3 does not simply bind directly to each but instead relies on a common intracellular signal transmission pathway, triggered by the agonists themselves, for activation of the NLRP3 inflammasome. To date, five major signal mechanisms have been proposed as those responsible for or contributing to the NLRP3 inflammasome activation (1, 49). In RNA viral infections, in particular, viroporins and some viral functional proteins act as the stimuli for signal 2 (as shown in Table 1).
Viroporins are a group of virus-encoded proteins that enhance permeability of host cell membrane (through interactions with the lipid bilayer) to promote release of viral particles from cells (50), making them a critical component of virus replication and virion release in the host system. The viroporin effect on permeability also modifies the cell’s ability to regulate ion passage, disrupting ion homeostasis (51). Several viroporins, such as the IAV M2 protein (6), RSV SH protein (15), HCV p7 protein (21), SARS-CoV E protein (25), EMCV 2B protein (27), and rhinovirus 2B protein (32), have been reported to activate the NLRP3 inflammasome by disturbing intracellular ionic concentrations, particularly through potassium efflux, calcium flux, and pH alteration. In the viroporin-induced NLRP3 activation itself, the host-encoded NEK7 protein may contribute to the NLRP3 inflammasome assembly and activation via formation of the NLRP3-NEK7 complex (52).
Among the other viral proteins involved in NLRP3 inflammasome activation are IAV PB1-F2 (a small protein encoded by an alternate + 1 open reading frame in the viral PB1 gene) (7) and EV71 3D protein (an RNA-dependent RNA polymerase) (34). PB1-F2 activates the NLRP3 inflammasome through mitochondrial ROS production, which could be a major mechanism of pathogenic inflammation induced by highly pathogenic IAV strains (53). The ZBP1 protein can interact with RIP3 upon sensing the viral protein PB1, and contributes to NLRP3 inflammasome activation in IAV but not in VSV infection through post-translational regulation (8). In contrast, the EV71 3D protein can directly bind to NLRP3 and ASC, and promotes NLRP3 inflammasome assembly and activation (34).
vRNA-Associated Mechanisms Contributing to NLRP3 Inflammasome Activation
Plasma vRNA, another common agonist for NLRP3, may bind with NLRP3 and act to subsequently activate it. Although NLRP3 contains a nucleotide-binding domain (54), RNA-sensing molecules have been reported to participate in the process by which vRNA activates the NLRP3 inflammasome; these include the DExD/H-box helicase (DHX) family members (14, 41, 55, 56) and the 2′,5′-oligoadenylate synthetase (OAS, or 2-5A)/RNase L system (30). However, some debate exists as to the exact role of DHX33, in particular, in VSV-induced NLRP3 inflammasome activation (29).
The DHX family members contain some domains required for ATP binding, hydrolysis, and nucleic acid binding and unwinding, which usually acts as a cytosolic RNA sensor and triggers type I IFN response. DHX33, DDX19A, and DDX58 have been reported to regulate NLRP3 inflammasome activation. DHX33 was shown to combine directly with vRNA to bind to NLRP3 through the DEAD domain of DHX33 and the NACHT domain of NLRP3, forming a DHX33/NLRP3/ASC inflammasome complex to promote NLRP3 inflammasome activation (14). In that same study, down-regulation of DHX33 expression led to a substantial reduction in cleavage of caspase-1 and secretion of IL-18 and IL-1β from the human monocyte cell line, THP-1, upon stimulation with viral dsRNA purified from reovirus, and produced by RSV during the replication process (14). DHX33 has also been demonstrated to contribute to NLRP3 inflammasome activation and IL-1β secretion during VSV and IAV infections (30), the process of which was found to be dependent upon the OAS/RNase L system induced by IFN. OASs recognize cytosolic viral dsRNA and subsequently trigger synthesis of 2–5A in the presence of three ATP molecules. Then, RNase L is activated by 2–5A and cleaves intracellular vRNA. These RNase L-mediated RNA cleavage products will directly interact with DHX33 and induce a DHX33/MAVS/NLRP3 complex to trigger assembly of the NLRP3 inflammasome.
Another study demonstrated that DDX19A binds porcine reproductive and respiration syndrome virus (PRRSV) genomic RNA through its ATP-binding domain and C-terminal domain and directly interacts with the NLRP3 inflammasome in highly pathogenic-PRRSV-infected primary porcine alveolar macrophages, dependent on the DDX19A ATP-binding domain, and the NLRP3 NACHT and LRR domains. Knockdown of DDX19A expression led to reductions in both IL-1β secretion and virus titers in the PRRSV-infected porcine alveolar macrophages (56). Thus, DDX19A appears to be required for both antiviral defense and NLRP3 inflammasome activation. RIG-I (DDX58) is a key intracellular sensor for cytosol ssRNA and provides the transcriptional priming signal for NLRP3 inflammasome during infections with VSV (41) and IAV (55), upon recognition of the vRNA. However, in primary respiratory epithelial cells, IAV infection promotes RIG-I binding with ASC and caspase-1, implying that RIG-I contributes to inflammasome assembly (55).
Effects Induced by the NLRP3 Inflammasome During RNA Viral Infections
During the process of RNA viral infection, NLRP3 inflammasome activation contributes to either enhancing antiviral defense and tissue healing, or inflammatory pathological injury, which depends mainly on the time point and extent of NLRP3 activation. The appropriate and early phase activation of the NLRP3 inflammasome in some RNA viral infection systems, such those of IAV, encephalomyocarditis virus, and West Nile virus (WNV) (shown in Table 1), usually provide protection, thereby decreasing mortality and viral load, as shown in virus-infected mouse models (4, 9, 16, 57). In rhesus monkeys, neither NLRP3 inflammasome activation nor IFN response can be established successfully within 24 h following challenge with the simian immunodeficiency virus, which may be the key reason underlying the observation of viral dissemination occurring rapidly from the inoculation site to a distal site (within the first day of infection) (58). Reciprocally, when using HIV-1-specific broadly neutralizing antibodies, PGT121 is capable of inducing innate immune responses such as NLRP3 inflammasome activation in vRNA-positive tissues within 24 h; in addition, viral replication is reduced in distal tissues significantly (59). These findings imply the early NLRP3 inflammasome activation as well as IFN response play critical roles in establishment of effective antiviral defense. Similarly, the administration of MC9550 (a specific NLRP3 inhibitor) on days 1, 3, and 5 but not days 3–5 following IAV challenge also resulted in accelerated weight loss and mortality in IAV-infected mice (9). At 2 weeks after challenge with IAV, the mortality rates among NLRP3−/−, caspase-1−/− (4, 5), and IL-1RI−/− (57) mice are significantly higher (by 60–80%) than for the wild-type counterparts. Thus, proper NLRP3 inflammasome activation in the early phase of RNA viral infection protects the infected host. In contrast, if activated at a later phase or over-activated, the NLRP3 inflammasome response to RNA virus results in inflammatory injury.
Production of IL-1β and IL-18, and pyroptosis are the critical and fundamental reactions directly induced by activated NLRP3 inflammasome (Figure 1). IL-1β and IL-18 serve to activate myriad downstream cell responses, and orchestrate innate and adaptive immunity through MyD88/IRAK4/TRAF6-mediated NF-κB signaling and the JNK/p38 mitogen-activated protein kinase pathways (60–63), which may represent key events for the NLRP3 inflammasome-dependent antiviral defense. Pyroptosis is a pro-inflammatory caspase-dependent programed cell death (64). NLRP3 inflammasome-dependent pyroptosis has been described in HCV-infected hepatoma cells (65), Dengue virus (DV)-infected monocytes (66), and HIV-infected podocytes (10). The data to date suggest that pyroptosis in viral infection leads to host injury and pathogenesis.
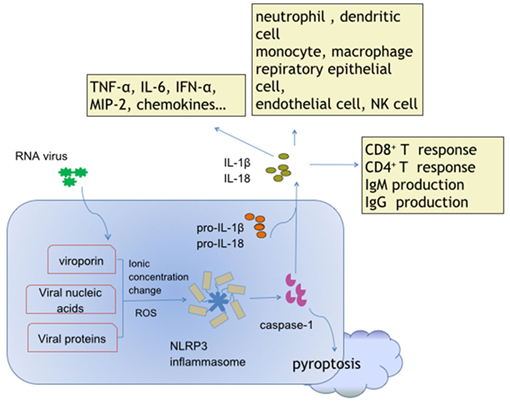
Figure 1. Activation and effects of NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome in response to ribonucleic acid (RNA) virus infections. Viroporin, viral nucleic acids, and some virus-encoded functional proteins are able to activate the NLRP3 inflammasome. Viroporin-mediated ion concentration change, virus replication-produced reactive oxygen species, and even viral protein direct binding promote assembly of the NLRP3 inflammasome. The cytokines interleukin (IL)-1 and IL-18 contain extensive downstream effector molecules and effector cells, which ultimately orchestrate innate and adaptive immunity, leading to antiviral defense, and/or inflammatory injury during RNA virus infections.
In mice, IAV-induced NLRP3 inflammasome activation initiates protective inflammation by recruitment of monocytes and neutrophils to the lung (as noted in the bronchoalveolar lavage fluid), which occurs within 3–7 days of the IAV infection, and the subsequent promotion of secretion of various cytokines, such as tumor necrosis factor (TNF)-α, IFN-α, and MIP-2α (4, 5). Furthermore, the impaired IL-1 response by IL-1RI deficiency results in impaired production of immunoglobulin M, reduced recruitment of CD4+ T cells in the bronchoalveolar lavage fluid (57), and impaired priming of CD8+ T cells in the lung and draining mesenteric lymph nodes through the intervention of CD11bloCD103+ dendritic cell migration and maturation (67). In a recent study, IL-18 was confirmed to be required for IFN-γ production in CD161+Vα7.2+ mucosal-associated invariant T cells, which has been observed clinically to be increased in recovered but not succumbed patients hospitalized with avian H7N9 influenza pneumonia and which implies a protective role in influenza virus infection (68). In WNV infected mice, the IL-1β produced by CD45+CD11b+ infiltrating T cells promotes the WNV-specific CD8+ T cell entry to the central neuronal system via regulation of CXCL12-mediated T cell adhesion, eventually controlling the viral infection (16, 17). In BV-2 mouse microglia cells infected by Japanese encephalitis virus, the NLRP3 inflammasome induces production of IL-1β and IL-18 rapidly (within 3 h of exposure) and of TNF-α, CCL2, and IL-6 later (within 6 h after exposure) (40); the findings suggest that the NLRP3-dependent protective inflammatory response is a very early phase innate immune response against RNA viral infection. In addition to initiating protective inflammation to regulate and orchestrate innate and adaptive immunity, the NLRP3 inflammasome also promotes tissue healing and reduces respiratory epithelial necrosis, which contributes to enhancing host disease tolerance (5, 69). Thus, the NLRP3 inflammasome provides protective roles through enhancing host tolerance capacity and orchestrating innate immunity and adaptive immunity during RNA viral infections.
Aberrant NLRP3 inflammasome activation usually results in progression of viral pathogenesis and the manifested disease in host, such as the robust NLRP3 activation induced by PB1-F2 during the IAV infection (7). The major pathogenic mechanism associated with PB1-F2-induced aberrant NLRP3 inflammasome activation involves excessive neutrophil influx (7, 53). The NLRP3 activation induced by HCV promotes chronic intrahepatic inflammation in macrophages (18) and interrupts cellular lipid metabolism (20) in HCV-infected hepatoma cells, both of which contribute to chronic liver injury and liver disease progression. In HIV-infected microglia, NLRP3 inflammasome activation is involved in the occurrence of chronic neuroinflammation (12). In addition, NLRP3-dependent pyroptosis during RNA viral infection also supports the pathogenesis of virus infection diseases. HIV-induced podocyte pyroptosis could be associated with HIV-associated nephropathy (10). NLRP3 inflammasome-mediated pyroptosis is also observed in DV-infected monocytes (66) and in HCV-infected and bystander hepatoma cells (65), and could be associated with viral pathogenesis (Table 1).
Negative Regulation of NLRP3 Inflammasome by RNA Virus
Unlike RNA viruses, the DNA viruses, which have a large DNA genome, can encode viral homologs of some complex intracellular inflammasome-regulating molecules to regulate inflammasome activity; an example of this is the pyrin-only proteins and BCL-2 encoded by poxvirus (70, 71). The RNA viruses, in contrast, have evolved some simple yet effective strategies to inhibit activation of the NLRP3 inflammasome and evade host immunity. Specifically, the proteases 3C and 2A of EV71 are able to cleave NLRP3 at the G493-L494 or Q225-G226 junction (34). Furthermore, the protease 3C of EV71 is capable of degrading gasdermin D, a protein critical to the induction of pyroptosis (72), and host failure to inhibit EV71 replication (73). The NS1 protein of influenza virus can bind with NLRP3 directly, thereby inhibiting assembly of the NLRP3-ASC-caspase-1 complex and secretion of IL-1β; this inhibition is dependent on RNA and the TRIM-25 domain of the NS1 protein (74). The measles virus V protein also targets NLRP3, probably serving to interfere with the NLRP3 inflammasome assembly and ultimately facilitating escape from the host immune response (75). Finally, the non-structural protein 11 encoded by PRRSV also antagonizes the NLRP3 inflammasome effect in the macrophages, through decreasing expression of pro-IL-1β (76).
Concluding Remarks
NACHT, LRR, and PYD domains-containing protein 3 inflammasome activation in response to RNA virus is a fundamental innate immune response as well as an IFN response, which usually contributes to antivirus defense, although aberrant and inappropriate NLRP3 inflammasome response still results in the immunopathology and exaggeration of infectious disease, especially in some persistent virus infections. The activation itself is induced by different signals and probably occurs in different phases of the viral infection, triggering inflammatory response (appropriate/protective or inappropriate/detrimental), and affecting outcome of the viral infection substantially. Thus, the proper regulation of NLRP3 inflammasome activation with specific NLRP3 inhibitors may have therapeutic benefit in controlling virus-related diseases and in clearance of the infecting virus.
Author Contributions
JY collecting literatures and drafting manuscript. JW design of the work, collecting literatures, drafting, and revising manuscript. YW discussing literatures, design of the work, and revising manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from the National Natural Science Foundation of China (grant no. 31670899 and 31070796) and the Natural Science Foundation of Chongqing (grant no. CSTC, 2011BB5029).
References
1. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi:10.1016/j.cell.2010.01.040
2. Lupfer C, Malik A, Kanneganti TD. Inflammasome control of viral infection. Curr Opin Virol (2015) 12:38–46. doi:10.1016/j.coviro.2015.02.007
3. Danthi P. Viruses and the diversity of cell death. Annu Rev Virol (2016) 3(1):533–53. doi:10.1146/annurev-virology-110615-042435
4. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity (2009) 30(4):556–65. doi:10.1016/j.immuni.2009.02.005
5. Thomas PG, Dash P, Aldridge JR Jr, Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity (2009) 30(4):566–75. doi:10.1016/j.immuni.2009.02.006
6. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol (2010) 11(5):404–10. doi:10.1038/ni.1861
7. McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, Stutz A, et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog (2013) 9(5):e1003392. doi:10.1371/journal.ppat.1003392
8. Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol (2016) 1(2). doi:10.1126/sciimmunol.aag2045
9. Tate MD, Ong JD, Dowling JK, McAuley JL, Robertson AB, Latz E, et al. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci Rep (2016) 6:27912. doi:10.1038/srep27912
10. Haque S, Lan X, Wen H, Lederman R, Chawla A, Attia M, et al. HIV promotes NLRP3 inflammasome complex activation in murine HIV-associated nephropathy. Am J Pathol (2016) 186(2):347–58. doi:10.1016/j.ajpath.2015.10.002
11. Chivero ET, Guo ML, Periyasamy P, Liao K, Callen SE, Buch S. HIV-1 Tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci (2017) 37(13):3599–609. doi:10.1523/jneurosci.3045-16.2017
12. Mamik MK, Hui E, Branton WG, McKenzie BA, Chisholm J, Cohen EA, et al. HIV-1 viral protein R activates NLRP3 inflammasome in microglia: implications for HIV-1 associated neuroinflammation. J Neuroimmune Pharmacol (2017) 12(2):233–48. doi:10.1007/s11481-016-9708-3
13. Harker JA, Godlee A, Wahlsten JL, Lee DC, Thorne LG, Sawant D, et al. Interleukin 18 coexpression during respiratory syncytial virus infection results in enhanced disease mediated by natural killer cells. J Virol (2010) 84(8):4073–82. doi:10.1128/jvi.02014-09
14. Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity (2013) 39(1):123–35. doi:10.1016/j.immuni.2013.07.001
15. Triantafilou K, Kar S, Vakakis E, Kotecha S, Triantafilou M. Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to signal inflammasome activation. Thorax (2013) 68(1):66–75. doi:10.1136/thoraxjnl-2012-202182
16. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog (2012) 8(11):e1003039. doi:10.1371/journal.ppat.1003039
17. Durrant DM, Daniels BP, Klein RS. IL-1R1 signaling regulates CXCL12-mediated T cell localization and fate within the central nervous system during West Nile Virus encephalitis. J Immunol (2014) 193(8):4095–106. doi:10.4049/jimmunol.1401192
18. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog (2013) 9(4):e1003330. doi:10.1371/journal.ppat.1003330
19. Chen W, Xu Y, Li H, Tao W, Xiang Y, Huang B, et al. HCV genomic RNA activates the NLRP3 inflammasome in human myeloid cells. PLoS One (2014) 9(1):e84953. doi:10.1371/journal.pone.0084953
20. McRae S, Iqbal J, Sarkar-Dutta M, Lane S, Nagaraj A, Ali N, et al. The hepatitis C virus-induced NLRP3 inflammasome activates the sterol regulatory element-binding protein (SREBP) and regulates lipid metabolism. J Biol Chem (2016) 291(7):3254–67. doi:10.1074/jbc.M115.694059
21. Farag NS, Breitinger U, El-Azizi M, Breitinger HG. The p7 viroporin of the hepatitis C virus contributes to liver inflammation by stimulating production of Interleukin-1beta. Biochim Biophys Acta (2017) 1863(3):712–20. doi:10.1016/j.bbadis.2016.12.006
22. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood (2013) 122(20):3405–14. doi:10.1182/blood-2013-05-504449
23. Liang XH, Goldman JE, Jiang HH, Levine B. Resistance of interleukin-1beta-deficient mice to fatal Sindbis virus encephalitis. J Virol (1999) 73(3):2563–7.
24. Prow NA, Irani DN. The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J Neurochem (2008) 105(4):1276–86. doi:10.1111/j.1471-4159.2008.05230.x
25. Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Castano-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology (2015) 485:330–9. doi:10.1016/j.virol.2015.08.010
26. Kanda T, Tanaka T, Sekiguchi K, Seta Y, Kurimoto M, Wilson McManus JE, et al. Effect of interleukin-18 on viral myocarditis: enhancement of interferon-gamma and natural killer cell activity. J Mol Cell Cardiol (2000) 32(12):2163–71. doi:10.1006/jmcc.2000.1242
27. Ito M, Yanagi Y, Ichinohe T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog (2012) 8(8):e1002857. doi:10.1371/journal.ppat.1002857
28. Rajan JV, Rodriguez D, Miao EA, Aderem A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol (2011) 85(9):4167–72. doi:10.1128/jvi.01687-10
29. Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, et al. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol (2014) 15(12):1126–33. doi:10.1038/ni.3015
30. Chakrabarti A, Banerjee S, Franchi L, Loo YM, Gale M Jr, Nunez G, et al. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe (2015) 17(4):466–77. doi:10.1016/j.chom.2015.02.010
31. Terajima M, Yamaya M, Sekizawa K, Okinaga S, Suzuki T, Yamada N, et al. Rhinovirus infection of primary cultures of human tracheal epithelium: role of ICAM-1 and IL-1beta. Am J Physiol (1997) 273(4 Pt 1):L749–59.
32. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol (2013) 49(6):923–34. doi:10.1165/rcmb.2013-0032OC
33. Wang H, Lei X, Xiao X, Yang C, Lu W, Huang Z, et al. Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep (2015) 12(1):42–8. doi:10.1016/j.celrep.2015.05.047
34. Wang W, Xiao F, Wan P, Pan P, Zhang Y, Liu F, et al. EV71 3D protein binds with NLRP3 and enhances the assembly of inflammasome complex. PLoS Pathog (2017) 13(1):e1006123. doi:10.1371/journal.ppat.1006123
35. Azuma I, Ishihara C, Iida J, Yoo YC, Yoshimatsu K, Arikawa J. Stimulation of host-defense mechanism with synthetic adjuvants and recombinant cytokines against viral infection in mice. Adv Exp Med Biol (1992) 319:253–63. doi:10.1007/978-1-4615-3434-1_26
36. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem (2006) 281(48):36560–8. doi:10.1074/jbc.M607594200
37. Tricarico PM, Caracciolo I, Crovella S, D’Agaro P. Zika virus induces inflammasome activation in the glial cell line U87-MG. Biochem Biophys Res Commun (2017) 492(4):597–602. doi:10.1016/j.bbrc.2017.01.158
38. Wang Y, Gao B, Xiong S. Involvement of NLRP3 inflammasome in CVB3-induced viral myocarditis. Am J Physiol Heart Circ Physiol (2014) 307(10):H1438–47. doi:10.1152/ajpheart.00441.2014
39. Mishra MK, Kumawat KL, Basu A. Japanese encephalitis virus differentially modulates the induction of multiple pro-inflammatory mediators in human astrocytoma and astroglioma cell-lines. Cell Biol Int (2008) 32(12):1506–13. doi:10.1016/j.cellbi.2008.08.020
40. Kaushik DK, Gupta M, Kumawat KL, Basu A. NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS One (2012) 7(2):e32270. doi:10.1371/journal.pone.0032270
41. Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol (2010) 11(1):63–9. doi:10.1038/ni.1824
42. Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A (2011) 108(13):5354–9. doi:10.1073/pnas.1019378108
43. Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol (2013) 191(8):3995–9. doi:10.4049/jimmunol.1301681
44. Lin KM, Hu W, Troutman TD, Jennings M, Brewer T, Li X, et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A (2014) 111(2):775–80. doi:10.1073/pnas.1320294111
45. Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun (2015) 6:7515. doi:10.1038/ncomms8515
46. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem (2012) 287(43):36617–22. doi:10.1074/jbc.M112.407130
47. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell (2013) 49(2):331–8. doi:10.1016/j.molcel.2012.11.009
48. Rayamajhi M, Miao EA. The RIP1-RIP3 complex initiates mitochondrial fission to fuel NLRP3. Nat Immunol (2014) 15(12):1100–2. doi:10.1038/ni.3030
49. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev (2015) 265(1):6–21. doi:10.1111/imr.12296
50. Gonzalez ME, Carrasco L. Viroporins. FEBS Lett (2003) 552(1):28–34. doi:10.1016/S0014-5793(03)00780-4
51. Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol (2012) 10(8):563–74. doi:10.1038/nrmicro2820
52. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–7. doi:10.1038/nature16959
53. Pinar A, Dowling JK, Bitto NJ, Robertson AB, Latz E, Stewart CR, et al. PB1-F2 derived from avian influenza a virus H7N9 induces inflammation via activation of the NLRP3 inflammasome. J Biol Chem (2016) 292(3):826–36. doi:10.1074/jbc.M116.756379
54. Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A (2007) 104(19):8041–6. doi:10.1073/pnas.0611496104
55. Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, et al. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog (2013) 9(4):e1003256. doi:10.1371/journal.ppat.1003256
56. Li J, Hu L, Liu Y, Huang L, Mu Y, Cai X, et al. DDX19A senses viral RNA and mediates NLRP3-dependent inflammasome activation. J Immunol (2015) 195(12):5732–49. doi:10.4049/jimmunol.1501606
57. Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol (2005) 79(10):6441–8. doi:10.1128/jvi.79.10.6441-6448.2005
58. Barouch DH, Ghneim K, Bosche WJ, Li Y, Berkemeier B, Hull M, et al. Rapid inflammasome activation following mucosal SIV infection of rhesus monkeys. Cell (2016) 165(3):656–67. doi:10.1016/j.cell.2016.03.021
59. Liu J, Ghneim K, Sok D, Bosche WJ, Li Y, Chipriano E, et al. Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science (2016) 353(6303):1045–9. doi:10.1126/science.aag0491
60. Akira S. The role of IL-18 in innate immunity. Curr Opin Immunol (2000) 12(1):59–63. doi:10.1016/S0952-7915(99)00051-5
61. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol (2010) 10(2):89–102. doi:10.1038/nri2691
62. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal (2010) 3(105):cm1. doi:10.1126/scisignal.3105cm1
63. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity (2013) 39(6):1003–18. doi:10.1016/j.immuni.2013.11.010
64. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev (2017) 277(1):61–75. doi:10.1111/imr.12534
65. Kofahi HM, Taylor NG, Hirasawa K, Grant MD, Russell RS. Hepatitis C virus infection of cultured human hepatoma cells causes apoptosis and pyroptosis in both infected and bystander cells. Sci Rep (2016) 6:37433. doi:10.1038/srep37433
66. Tan TY, Chu JJ. Dengue virus-infected human monocytes trigger late activation of caspase-1, which mediates pro-inflammatory IL-1beta secretion and pyroptosis. J Gen Virol (2013) 94(Pt 10):2215–20. doi:10.1099/vir.0.055277-0
67. Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol (2013) 14(3):246–53. doi:10.1038/ni.2514
68. Loh L, Wang Z, Sant S, Koutsakos M, Jegaskanda S, Corbett AJ, et al. Human mucosal-associated invariant T cells contribute to antiviral influenza immunity via IL-18-dependent activation. Proc Natl Acad Sci U S A (2016) 113(36):10133–8. doi:10.1073/pnas.1610750113
69. Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science (2016) 352(6284):463–6. doi:10.1126/science.aaf3926
70. Le HT, Harton JA. Pyrin- and CARD-only proteins as regulators of NLR functions. Front Immunol (2013) 4:275. doi:10.3389/fimmu.2013.00275
71. Shrivastava G, Leon-Juarez M, Garcia-Cordero J, Meza-Sanchez DE, Cedillo-Barron L. Inflammasomes and its importance in viral infections. Immunol Res (2016) 64(5–6):1101–17. doi:10.1007/s12026-016-8873-z
72. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi:10.1038/nature15514
73. Lei X, Zhang Z, Xiao X, Qi J, He B, Wang J. Enterovirus 71 inhibits pyroptosis through cleavage of gasdermin D. J Virol (2017) 91(18). doi:10.1128/jvi.01069-17
74. Moriyama M, Chen IY, Kawaguchi A, Koshiba T, Nagata K, Takeyama H, et al. The RNA- and TRIM25-binding domains of influenza virus NS1 protein are essential for suppression of NLRP3 inflammasome-mediated interleukin-1beta secretion. J Virol (2016) 90(8):4105–14. doi:10.1128/jvi.00120-16
75. Komune N, Ichinohe T, Ito M, Yanagi Y. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1beta secretion. J Virol (2011) 85(24):13019–26. doi:10.1128/jvi.05942-11
76. Wang C, Shi X, Zhang X, Wang A, Wang L, Chen J, et al. The endoribonuclease activity essential for the nonstructural protein 11 of porcine reproductive and respiratory syndrome virus to inhibit NLRP3 inflammasome-mediated IL-1beta induction. DNA Cell Biol (2015) 34(12):728–35. doi:10.1089/dna.2015.2929
Keywords: ribonucleic acid virus, NACHT, LRR, and PYD domains-containing protein 3 inflammasome, activation, effects, viral infection
Citation: Yu J, Wu Y and Wang J (2017) Activation and Role of NACHT, LRR, and PYD Domains-Containing Protein 3 Inflammasome in RNA Viral Infection. Front. Immunol. 8:1420. doi: 10.3389/fimmu.2017.01420
Received: 19 August 2017; Accepted: 12 October 2017;
Published: 31 October 2017
Edited by:
Hao Shen, University of Pennsylvania, United StatesReviewed by:
Etienne Meunier, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceHaitao Guo, University of North Carolina at Chapel Hill, United States
Copyright: © 2017 Yu, Wu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuzhang Wu, d3V5dXpoYW5nQHlhaG9vLmNvbQ==;
Jingxue Wang, amluZ3h1ZS53YW5nQGhvdG1haWwuY29t