 Shui-Xing Yu†
Shui-Xing Yu† Wei Chen†Zhen-Zhen Liu
Wei Chen†Zhen-Zhen Liu Feng-Hua Zhou
Feng-Hua Zhou Shi-Qing YanGui-Qiu HuXiao-Xia QinJie ZhangKe MaChong-Tao Du
Shi-Qing YanGui-Qiu HuXiao-Xia QinJie ZhangKe MaChong-Tao Du Jing-Min Gu
Jing-Min Gu Xu-Ming DengWen-Yu Han
Xu-Ming DengWen-Yu Han Yong-Jun Yang*
Yong-Jun Yang*
- Key Laboratory of Zoonosis, Ministry of Education, College of Veterinary Medicine, Jilin University, Changchun, China
The intestinal mucosal barrier is critical for host defense against pathogens infection. Here, we demonstrate that the mixed lineage kinase-like protein (MLKL), a necroptosis effector, promotes intestinal epithelial barrier function by enhancing inflammasome activation. MLKL−/− mice were more susceptible to Salmonella infection compared with wild-type counterparts, with higher mortality rates, increased body weight loss, exacerbated intestinal inflammation, more bacterial colonization, and severe epithelial barrier disruption. MLKL deficiency promoted early epithelial colonization of Salmonella prior to developing apparent intestinal pathology. Active MLKL was predominantly expressed in crypt epithelial cells, and experiments using bone marrow chimeras found that the protective effects of MLKL were dependent on its expression in non-hematopoietic cells. Intestinal mucosa of MLKL−/− mice had impaired caspase-1 and gasdermin D cleavages and decreased interleukin (IL)-18 release. Moreover, administration of exogenous recombinant IL-18 rescued the phenotype of increased bacterial colonization in MLKL−/− mice. Thus, our results uncover the role of MLKL in enhancing inflammasome activation in intestinal epithelial cells to inhibit early bacterial colonization.
Introduction
The gastrointestinal (GI) tract of mammals is colonized by tens of trillions of microorganisms, which are primarily composed of bacteria, viruses, fungi, parasites, and archaea, and is constantly exposed to a wide array of these microbial antigens, requiring the mucosal immune system to induce tolerance to the commensal microbes while still mounting potent responses to pathogens (1, 2). The Salmonella enterica serovar typhimurium (Salmonella) is among the main causes of bacterial GI infections in humans and animals. According to the statistics, an estimated 20 million cases and 200,000 deaths occur due to typhoidal Salmonella annually (3). When Salmonella first enters the host it initially propagates inside the GI tract and overcomes colonization resistance provided by the gut microbiota and the innate immune system (4–6). Subsequently, to gain access to the host, it must breach the intestinal epithelial barrier (7, 8) and then translocate and/or replicate outside the gut, such as the mesenteric lymph nodes (MLNs), spleen, and liver, causing a severe inflammation of the intestinal mucosal epithelium, resulting gastroenteritis in humans, and typhoid-like systemic illness in mice (9).
As the forefront of defense against pathogens, the intestinal epithelial cells (IECs) and mononuclear phagocytes through several types of pattern recognition receptors sense both microbes and host cells that are damaged by infection, resulting in the initiation of immune responses to orchestrate a protective inflammatory response against dangerous microbes. Several families of innate receptors expressed by IECs are involved in recognition of bacterial moieties, such as Toll-like receptors (10), NOD-like receptors (11), and HIN-200 families (12, 13), which in turn help to prevent bacterial invaders and disease progression. Thus, it is not surprising that IEC-mediated innate immune signaling pathways serve a pivotal role in maintaining intestinal homeostasis.
The mixed lineage kinase-like protein (MLKL), a member of the pseudokinase family, was originally identified as an essential necroptosis effector that operates downstream of the receptor interacting protein kinase (RIPK) 1 and RIPK3 (14, 15). The activated MLKL multimerizes and translocates to the cell membrane and exposes the killer N-terminal four-helix bundle domain that results in disruption of the plasma membrane (16, 17). Since MLKL direct membrane permeabilization or pore-forming capacity was disclosed, research on MLKL has predominantly focused on its role in cell death. Evidence has shown that MLKL-mediated necroptosis participates in many inflammatory diseases (15, 18, 19) and is sufficient to protect the host against pathogens infection (20–22). However, much less is known about the biological implications of MLKL in gut including the possibility that it might elicit mucosal immunity in response to defense pathogens invasion.
In the current study, we showed that MLKL plays a critical role in intestinal defense against Salmonella infection and MLKL-mediated inflammasome activation in the epithelial compartment limits early bacterial mucosal colonization.
Materials and Methods
Mice
MLKL−/− mice (C57BL/6 background, a gift from Dr. Jia-Huai Han, Xiamen University, China) (14) were backcrossed to C57BL/6 background for eight generations. C57BL/6 (WT) and MLKL−/− were housed in a pathogen-free facility and have no obvious GI disorders symptoms with normal diet. The animal studies were conducted according to the experimental practices and standards approved by the Animal Welfare and Research Ethics Committee at Jilin University (No. 20150601).
Salmonella Infections
Six- to eight-week-old sex-matched mice were used in this study. Salmonella strain SL1344 (gift from Dr. Xiang-Chao Cheng, Henan University of Science and Technology, Luoyang, China) was grown overnight at 37°C in LB broth supplemented with streptomycin (4 µg/ml) and was subsequently transferred to a high osmotic medium (a LB broth containing high concentration of sodium chloride) and incubated for 4–6 h until culture density of OD 600 reaches ~0.6. The Salmonella-induced colitis model was established as described previously (8). Briefly, following the administration of 20 mg of streptomycin per mouse, mice were orally inoculated with 5 × 107 CFU of Salmonella, and 6 or 48 h p.i. Log CFU per organ was determined. For the survival study, mice were orally challenged with 1 × 108 CFU of Salmonella.
Evaluation of Intestinal Inflammation
Tissue samples of cecum were fixed in buffered formalin solution (4%) and embedded in paraffin. Sections (5 µm thick) were then stained with hematoxylin and eosin (H&E). Salmonella-induced colitis was assessed in a blinded manner using an amended version of a previously described scoring criteria (23, 24).
In brief, tissue pathology scores were determined as follows: submucosal edema (scores 0 to 3), goblet cell depletion (scores 0 to 3), epithelial integrity (scores 0 to 3), and polymorphonuclear leukocyte (PMN) infiltration (scores 0 to 4).
Immunohistochemistry
Cecum tissue sections (5 µm) were deparaffinized and rehydrated using xylene and ethanol, respectively. Following antigen retrieval in citrate buffer (10 mM, pH = 6), samples were blocked in normal goat serum (5%) or donkey serum (5%). For immunohistochemistry, sections were stained with mucin 2 (Santa Cruz), claudin 3 (Abcam), p-MLKL (Abcam), Ly-6G/Ly-6c (BioLegend), and F4/80 (BioLegend) antibodies. Subsequently, specific staining was detected using the UltraSensitive S-P Kit and DAB Detection Kit (Maixin-Bio, China) according to the manufacturer’s directions. For immunofluorescence, tissue sections were stained with rabbit-anti-PCNA (Santa Cruz) and Alexa Fluor® 488-conjugated anti-rabbit IgG (Invitrogen). Epithelial cell apoptosis was analyzed by TUNEL staining using a commercial kit (KeyGEN Biotech). DAPI (1 µg/ml) was used to stain nuclei.
Cytokine and Chemokine Measurements
To measure the cytokine and chemokine amounts in cecum tissue, a part of cecum was homogenized mechanically in cold PBS (at a ratio of 4 ml per gram tissue) containing 1% Triton X-100 and complete protease inhibitor cocktail (Sigma-Aldrich). The mouse IL-18 ELISA kit was purchased from Arigo Biolaboratories. The other ELISA kits were purchased from R&D Systems. Cytokines and chemokines in cecum tissue were measured by ELISA according to the manufacturer’s instructions.
Real-time PCR
RNA was isolated using TRI reagent (Sigma-Aldrich) and converted into cDNA. Subsequently, Real-time PCR assays were performed using SYBR Green (Roche) on ABI Prism 7500 sequence detection system (Applied Biosystems). Gene expression levels were calculated using the 2−ΔCt method. The following primer sequences were used: GAPDH sense 5′-CACCCCAGCAAGGACACTGAGCAAG -3′, antisense 5′-GGGGGTCTGGGATGGAAATTGTGAG-3′. Occludin sense 5′-CAGCCTTCTGC TTCATCG-3′, antisense 5′-GTCGGGTTCACTCCCATTA-3′. ZO-1 sense 5′-GACCTTGAGCAGCC -GTCATA-3′, antisense 5′-CCGTAGGCGATGGTCATAGTT-3′. Claudin3 sense 5′-CCTAGGAACT -GTCCAAGCCG-3′, antisense 5′-CCCGTTTCATGGTTTGCCTG-3′.
Immunoblotting
The ceca of mice were harvested and then homogenized in lysis buffer solution [1% Triton X-100, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1 mM Na3VO4] supplemented with complete protease inhibitor cocktail (Sigma-Aldrich). Total cell lysates were separated by SDS-PAGE and transferred to PVDF membrane. The membranes were incubated with primary antibodies against iNOS (Abcam), Cox-2 (Abcam), Claudin3 (Abcam), Caspase-1 (Santa Cruz), ASC (Santa Cruz), gasdermin D (GSDMD) (Santa Cruz), and GAPDH (Proteintech), and proteins were detected with appropriate secondary anti-rabbit or anti-mouse antibody (Santa Cruz) conjugated to horseradish and then were visualized by enhanced chemiluminescence detection reagent (Millipore).
In Vivo Intestinal Permeability
An in vivo permeability assay was performed using FITC-dextran as described previously (25, 26). Streptomycin-pretreated WT and MLKL−/− mice were orally administrated with 5 × 107 CFU of Salmonella for 48 h. Mice were gavaged with FITC-dextran (Sigma-Aldrich) at a dose of 600 mg/kg body weight 4 h before harvest. The serum fluorescence intensity of the FITC-dextran was determined using a microplate fluorometer (Infinite M200 PRO, Tecan) with an excitation wavelength of 490 nm and an emission wavelength of 530 nm.
Bone Marrow Chimeras
Six- to eight-week-old mice were lethally irradiated with 10 Gy of γ radiation at a rate of 1.5 Gy/min in a 137Cs irradiator. Within 24 h of irradiation, mice received an intravenous injection of 8 × 106 bone marrow cells harvested from the femurs and tibias of WT or MLKL−/− mice. The following chimeras were generated (donor bone marrow > irradiated recipient): WT > WT, WT > MLKL−/−, MLKL−/− > MLKL−/−, MLKL−/− > WT. Mice were allowed to recover at least 7 weeks to reconstitute the hematopoietic compartment before being used for the experiments. Efficient reconstitution by donor bone marrow cells was confirmed by PCR for the MLKL gene in splenocytes isolated from bone marrow chimeric mice (1, 27).
Administration of Recombinant IL-18
MLKL−/− mice (seven mice per group) were injected daily i.p. with PBS alone or with recombinant IL-18 (MBL International) at a dose of 1.0 µg per mouse in 100 µl PBS on days 0, 1, and 2. WT mice were injected daily i.p. with PBS alone.
Statistical Analysis
Date are represented as mean ± SEM. Differences between mean values of normally distributed data were assessed with one-way ANOVA (Dunnett’s t-test) and two-tailed Student’s t-test. Log-rank test was used for statistical analysis of animal mortality. *p < 0.05 and **p < 0.01 compared with control group. Statistical analysis was performed using Prism (GraphPad Software, La Jolla, CA, USA).
Results
MLKL Plays a Critical Role in Restricting Salmonella Colitis
To determine the biological role of MLKL in Salmonella infection in vivo, streptomycin-pretreated WT and MLKL−/− mice were orally administrated with 1 × 108 CFU of Salmonella enterica subsp. enterica serovar Typhimurium SL1344 (Salmonella strain SL1344), and the mortality of mice were monitored over 15 days. On day 8 p.i., we observed that all of MLKL−/− mice had died, whereas almost 66.7% of WT mice remained alive, and the entire cohort of WT mice succumbed to Salmonella infection within 15 days (Figure 1A). The experiment was repeated with a lower infective dose of Salmonella (5 × 107 CFU) to study the phenotype of MLKL−/− mice under milder conditions. In accordance with the high level of mortality, MLKL−/− mice also tended to lose more body weight and cecal weight compared to WT mice (Figures 1B–D). Simultaneously, these clinical assessments were validated by the histologic analysis of cecums. MLKL−/− mice had severe intestinal damage and exacerbated intestinal inflammation, demonstrated by intense submucosal edema, goblet cell loss, massive PMN infiltration, and complete destruction of epithelial integrity (Figures 1E,F).
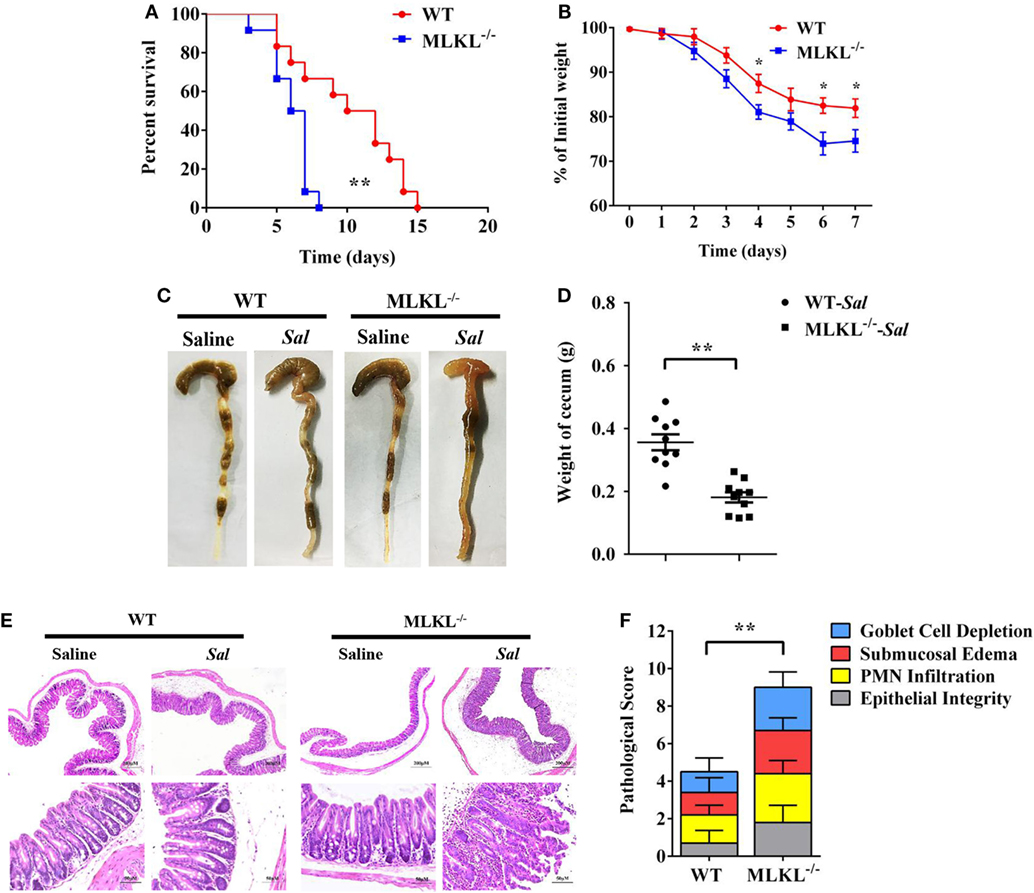
Figure 1. MLKL is sufficient to protect against intestinal salmonella infection. Streptomycin-pretreated WT and MLKL−/− mice were orally infected with Salmonella. (A) Survival (1 × 108 CFU, n = 12 each group). (B) Body weight loss (5 × 107 CFU, n = 12 each group). (C,D) Representative gross appearance and weight of cecum (5 × 107 CFU, n = 10 each group, at 48 h p.i.). (E,F) Representative H&E staining of cecum tissue and pathological score (5 × 107 CFU, n = 10 each group, at 48 h p.i.). All data are shown as mean ± SEM. Student’s t-test was performed. Log-rank test was used for statistical analysis of animal mortality. Statistical significance is indicated by *p < 0.05, **p < 0.01.
To further determine the pathologic changes in protein levels, the secreted amounts of cytokines and chemokines in cecum tissue homogenates were examined. At 48 h p.i., amounts of inflammatory cytokines (TNF-α, IL-6, IL-1β, IFN-γ, and IL-12) and chemokines (KC, CCL2, and CXCL10) were higher in the cecum tissues of MLKL−/− mice than in those of WT mice (Figures 2A–H), although CCL5 secretion was comparable between both genotypes (Figure 2I). In addition, the expression of Cox-2 and iNOS, two major inflammatory mediators implicated in colorectal inflammation and cancer (28), were enhanced in MLKL−/− mice (Figure 2J). Strikingly, neutrophil and macrophage accumulation were also elevated in MLKL−/− mice following Salmonella challenge (Figures 2K,L). Conclusively, these results suggested that MLKL plays an indispensable role for protection against Salmonella-induced colitis.
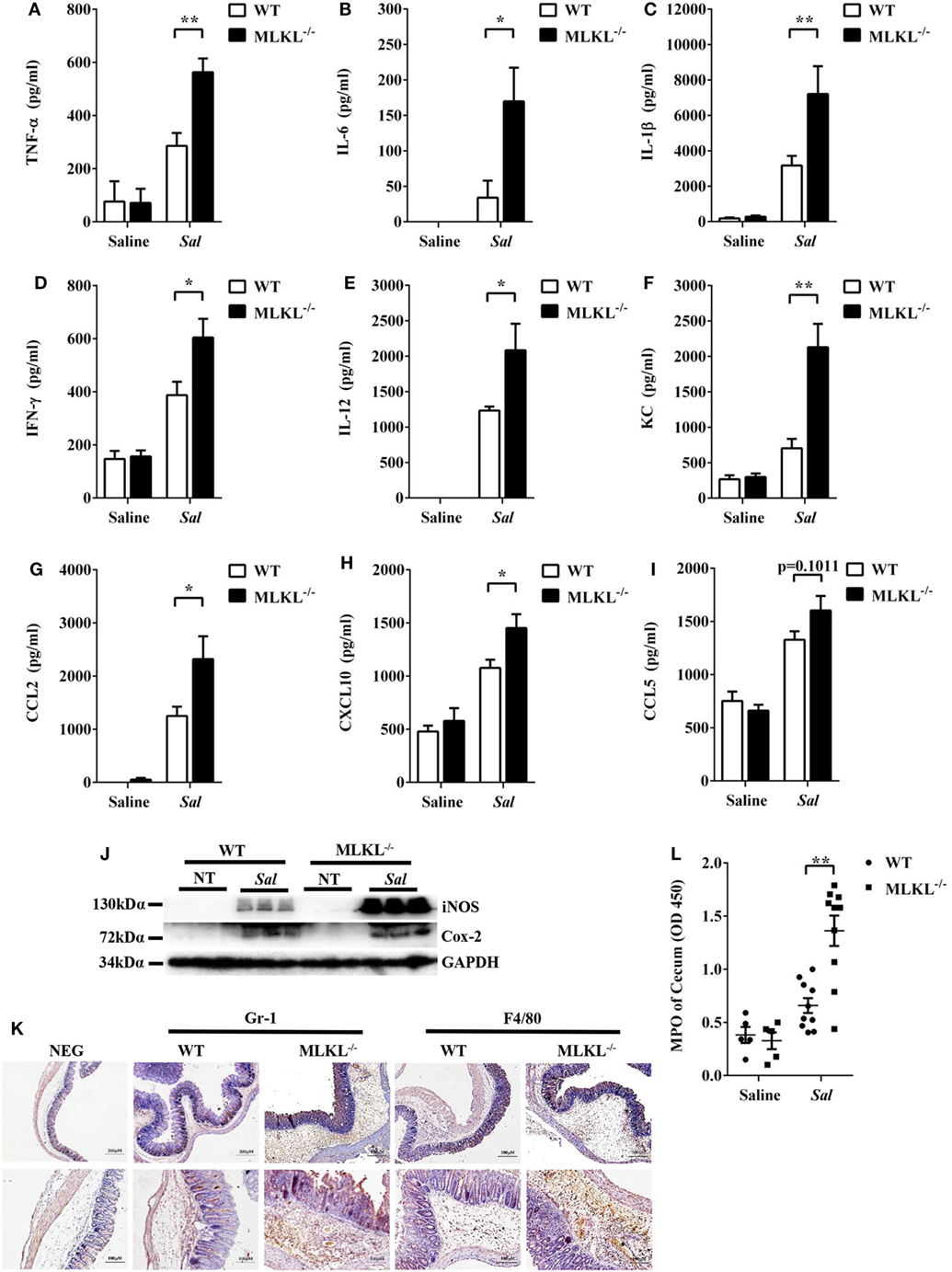
Figure 2. MLKL deficiency leads to enhanced pro-inflammatory mediator production following Salmonella intestinal infection. Streptomycin-pretreated WT and MLKL−/− mice were orally infected with Salmonella (5 × 107 CFU, n = 10 each group) for 48 h. The homogenate supernatant of cecum was detected for concentrations of indicated cytokines and chemokines by ELISA. (A) TNF-α, (B) IL-6, (C) IL-1 β, (D) IFN-γ, (E) IL-12, (F) KC, (G) CCL2, (H) CXCL10, (I) CCL5. (J) Representative cecum tissue lysate was analyzed for iNOS and Cox-2 by western blotting. GAPDH was used as a loading control. (K) Representative immunohistochemical staining of Gr-1 (a neutrophil marker) and F4/80 (a macrophagocyte marker) were performed in the cecal sections. (L) The homogenate supernatant of cecum was also used to determine activity of MPO (a neutrophil marker). All data are shown as mean ± SEM. Student’s t-test was performed. Statistical significance is indicated by *p < 0.05, **p < 0.01.
MLKL Deficiency Results in Increased Tissue Salmonella Numbers
Based on the above results, we speculated that increased pathology in MLKL−/− mice during Salmonella infection might link to increased colonization levels. Whereas recovery of Salmonella from feces of MLKL−/− mice did not differ from that from feces of WT mice, MLKL−/− mice harbored significantly elevated loads of Salmonella in the cecum and the MLN (Figures 3A–C), indicating that MLKL plays a protective role against intestinal translocation of Salmonella. Moreover, the Salmonella CFU were strongly increased in the liver and spleen of MLKL−/− mice compared to WT mice (Figures 3D,E), indicating that MLKL−/− mice are more susceptible to dissemination of Salmonella. Thus, these results suggested that MLKL-mediated protection against mucosal Salmonella infection may depend on the limitation of initial intestinal invasion.
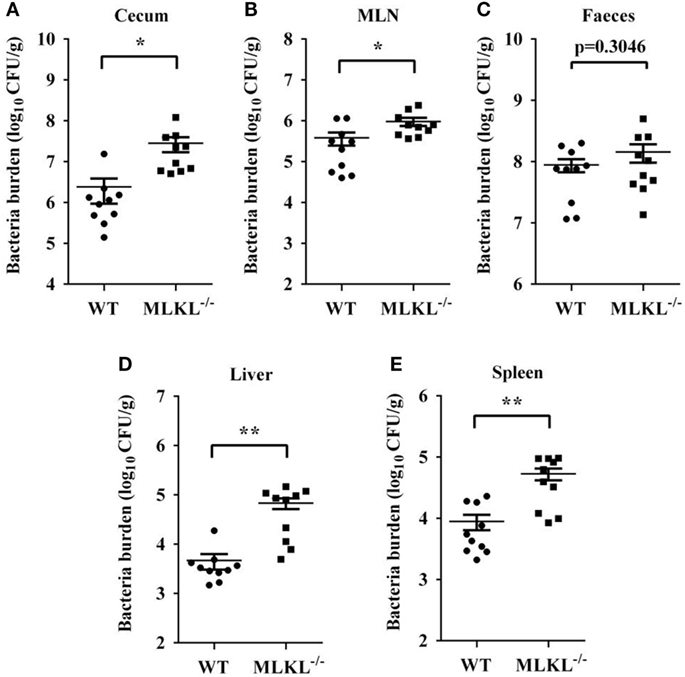
Figure 3. MLKL restricts Salmonella invasion and dissemination. Streptomycin-pretreated WT and MLKL−/− mice were orally infected with Salmonella (5 × 107 CFU, n = 10 each group) for 48 h. Bacterial load in the cecum (A), mesenteric lymph node (MLN) (B), feces (C), liver (D), and spleen (E) were detected. All data are shown as mean ± SEM. Student’s t-test was performed. Statistical significance is indicated by *p < 0.05, **p < 0.01.
MLKL Alleviates the Disruption of Intestinal Mucosal Barrier Integrity
Next, we sought the cause of the heightened bacteria load seen in Salmonella-infected MLKL−/− mice. The intestinal epithelia barrier, primarily made up of the mucus gel layer covering the epithelium, IECs, and tight junctions, can effectively prevent enteric bacterial pathogens from penetrating the intestinal mucosa and invading into deep tissues (29, 30). We first investigate whether MLKL deficiency affects mucin secretion and glycosylation patterns. At 48 h p.i., cecal tissues were collected and stained them with alcian blue as well as periodic acid–Schiff’s reagent (AB-PAS staining). Indisputably, AB-PAS stains acidic carbohydrates blue and neutral carbohydrates pink or magenta, while tissues containing both acidic and neutral mucins are stained dark blue or purple. In WT mice, the amount of mucoprotein expression was relatively larger in Salmonella-infected mice than that of uninfected mice. Compared with WT mice, the AB-PAS staining mucins were dramatically reduced in MLKL−/− mice after infection (Figure 4A). Mucin 2, the major component of the mucus layer, also trended to decrease in MLKL−/− mice following Salmonella infection (Figure 4B). Thus, these data imply that mucosal infection leads to deregulation of the expression of mucoproteins in MLKL−/− mice, potentially as a pathogenic mechanism in Salmonella colitis.
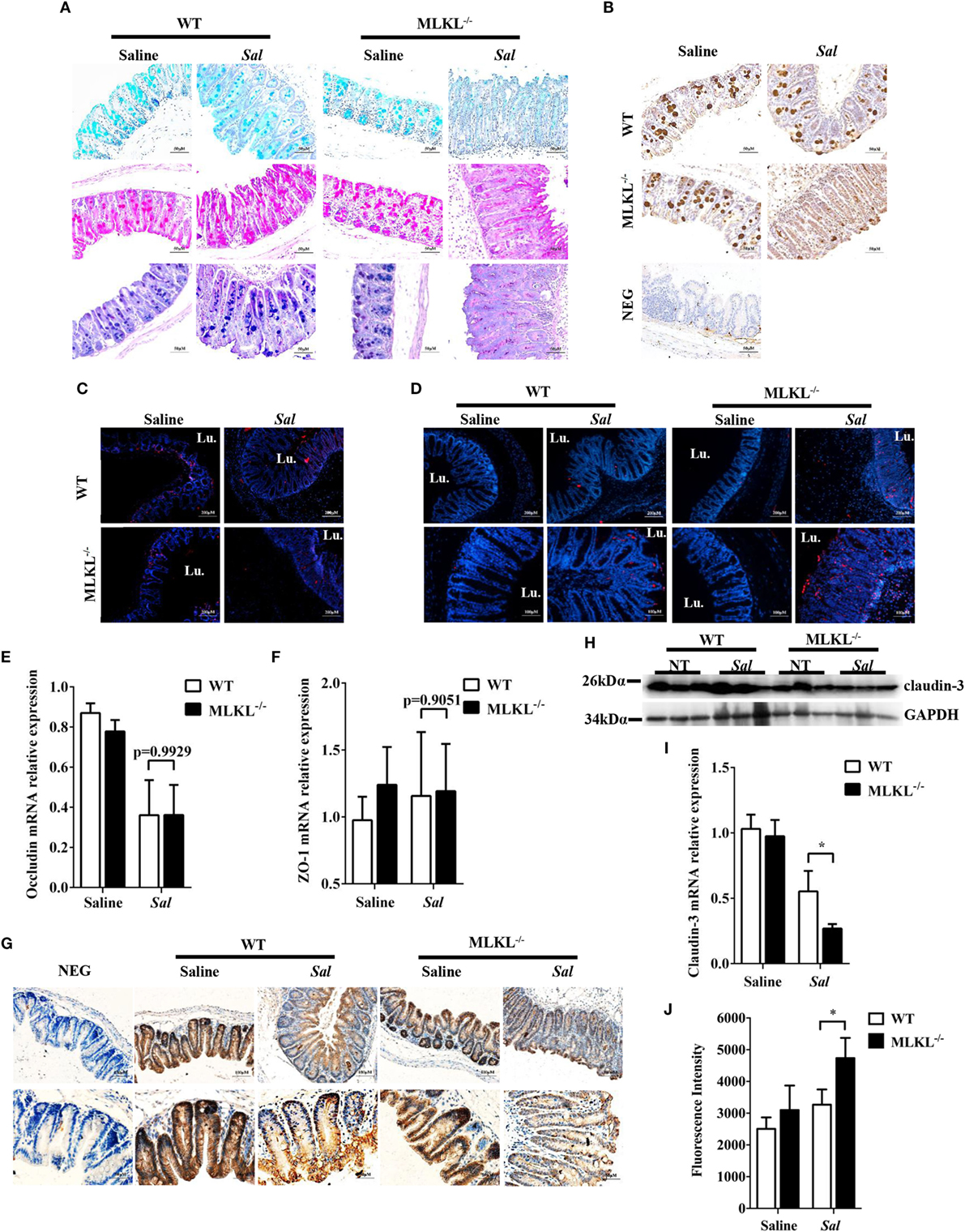
Figure 4. MLKL deficiency leads to severe disruption of intestinal mucosal barrier following Salmonella intestinal infection. Streptomycin-pretreated WT and MLKL−/− mice were orally infected with Salmonella (5 × 107 CFU) (n = 10 each group) for 48 h. (A) AB-PAS staining. (B) Mucin 2 (IHC) stained brown. (C,D) PCNA and TUNEL staining of proliferating cells and apoptotic cells in the cecum tissue, respectively. The cecum tissue mRNA was examined for Occludin (E) and ZO-1 (F) and claudin 3 (I) by QRT-PCR. The data were normalized to GAPDH expression and are showed as the fold increase in mRNA. (G) Claudin-3 (IHC) stained brown. (H) The cecum tissue lysate was analyzed for claudin-3 by western blotting. GAPDH was used as a loading control. (J) The intestinal permeability was measured using FITC-dextran as described in Section “Materials and Methods” (n = 5–7 each group). All data are shown as mean ± SEM. Student’s t-test was performed. Statistical significance is indicated by *p < 0.05, **p < 0.01.
Subsequently, we examined the impact of MLKL deficiency on tissue homeostasis by examining the epithelial cell proliferation and cell death. Although there was no obvious difference in the PCNA-positive cells between WT and MLKL−/− epithelial crypts (Figure 4C), TUNEL staining of histological sections of cecum showed that MLKL−/− mice had greatly increased numbers of TUNEL-positive cells in the epithelium and lamina propria than in those of WT mice (Figure 4D), indicating that MLKL−/− mice have a defect in maintaining enterocyte proliferative/apoptotic homeostasis following mucosal Salmonella challenge.
As intercellular junctions are crucial for epithelial barrier function, we wondered if Salmonella infection also induced any changes in tight junctions between WT mice and MLKL−/− mice. The tight junction, composed of claudin, occludin, and zonula occludens (ZO)-1, has been shown previously to protect against Salmonella infection (8). Despite there were no difference in the expression levels of occludin and ZO-1 (Figures 4E,F), the expression level of claudin-3, a major sealing junctional component for early noninvasive detection of intestinal tight junction integrity loss (31), was significant lower in MLKL−/− mice than in WT mice (Figures 4G–I). Subsequently, to further determine whether MLKL protection against Salmonella was associated with preservation of intestinal barrier function, paracellular intestinal permeability was assessed by oral administration of the fluorescent tracer FITC-dextran. As expected, fluorescence intensity of serum showed that MLKL−/− mice had the highest levels of FITC-dextran in the blood than in those of WT mice (Figure 4J), suggesting that MLKL−/− mice exhibit a disruption of barrier function. Conclusively, these results indicated that MLKL deficiency results in increased intestinal barrier damage following mucosal Salmonella infection.
MLKL Potentially Limits Early Intestinal Bacterial Colonization
The exaggerated loss of the intestinal mucosal integrity in MLKL−/− mice on 48 h p.i. may be the result of exacerbated inflammation or increased bacteria localization. Therefore, to further investigate the mechanisms for MLKL mediates protection, we analyzed the initial interplay between bacteria and the host at 6 h after gavage with 5 × 107 CFU Salmonella, since the cecal lumen was fully colonized by 2–6 h p.i (32). The clinical and histologic assessments of the infected MLKL−/− and WT mice showed no obvious change compared with the controls (Figures 5A–C). Simultaneously, mucins secretion in MLKL−/− mice was not different from that in WT mice before and after infection (Figures 5D,E). Subsequently, we enumerated Salmonella in feces, cecum, MLN, liver, and spleen. Bacteria loads in feces of both WT and MLKL−/− mice were comparable at 6 h p.i. (p = 0.4954, Figure 5F), like those at 48 h p.i. Although no viable bacteria were detected in the liver and spleen of mice at this early stage of infection, we noted that a 0.5–1 Log increase in bacterial numbers was detected in cecum (p = 0.0742) and MLN tissues (p = 0.1456) of MLKL−/− mice compared with WT mice (Figures 5G,H). These data suggest that the potential capacity of limiting early intestinal colonization underlies the protection role of MLKL, and the compromised colonization defense of MLKL−/− mice leads to severe intestinal inflammation and barrier integrity disruption.
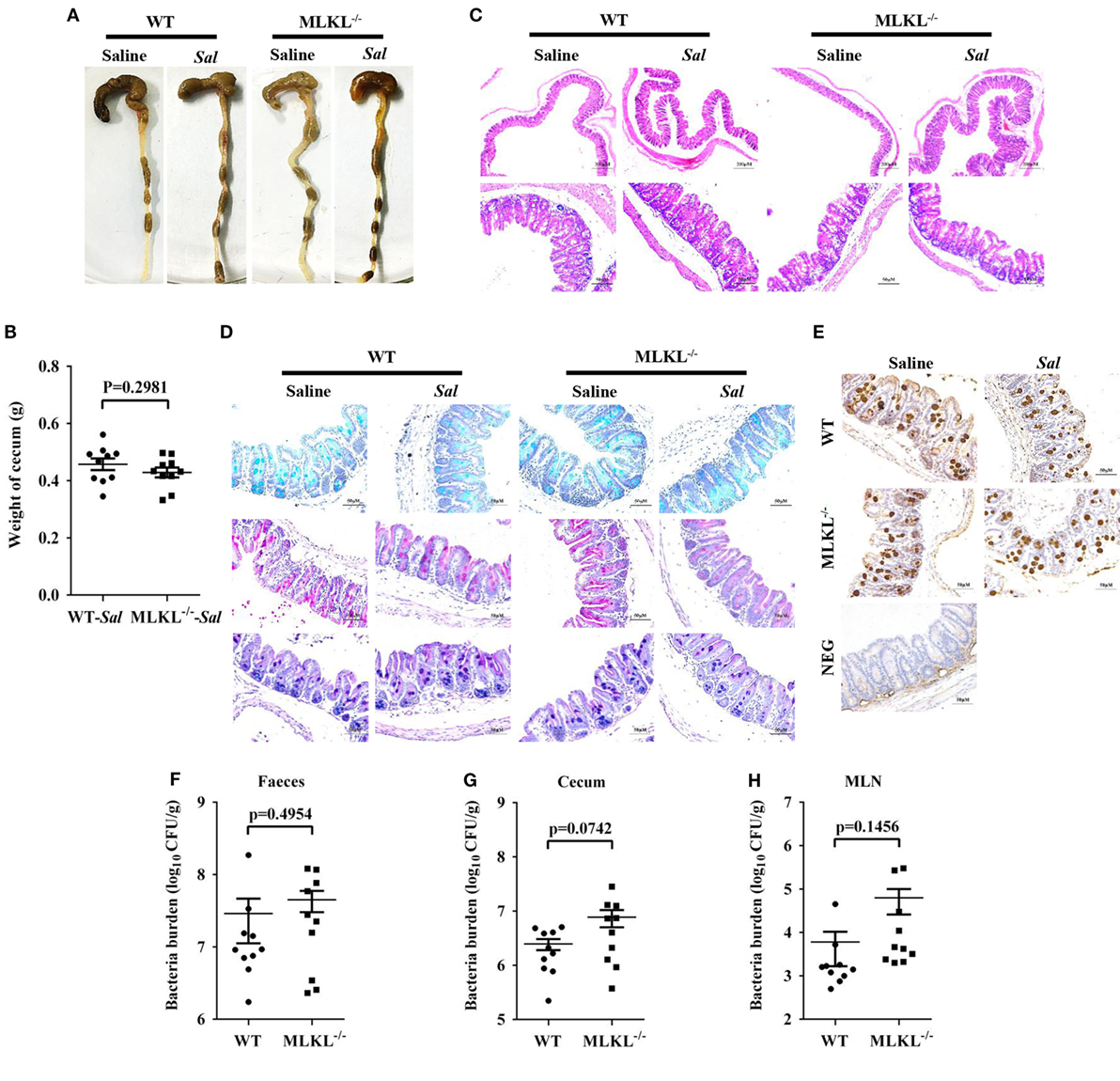
Figure 5. MLKL effectively restricts the acute Salmonella mucosal infection. Streptomycin-pretreated WT and MLKL−/− mice were orally infected with Salmonella (5 × 107 CFU) (n = 10 each group) for 6 h. (A,B) Gross appearance and weight of cecum. (C) Representative H&E staining of cecum tissue. (D) AB-PAS staining. (E) Mucin 2 (IHC) stained brown. (F–H) Bacterial load in the feces, cecum, and MLN. All data are shown as mean ± SEM. Student’s t-test was performed. Statistical significance is indicated by *p < 0.05.
MLKL Expression in Non-Hematopoietic Cells Mediates Protection against Infection
To further characterize the role of MLKL in defense against intestinal colonization with Salmonella, the expression of phosphorylated MLKL (p-MLKL) in the cecal tissues of infected WT mice was investigated by immunohistochemical staining. Our results showed that p-MLKL was located in inflammatory cells which infiltrated to the submucosa, but predominantly expressed in crypt epithelial cells (Figure 6A). To determine in which cell populations MLKL expression mediated protective effects against Salmonella infection, we generated bone marrow chimeras. Thus, WT or MLKL−/− recipient mice were lethally irradiated and injected with bone marrow cells from WT or MLKL−/− donors. After hematopoietic reconstitution, these mice were infected with Salmonella. Similar to complete MLKL-deficient chimeras (MLKL−/− donor > MLKL−/− recipient), chimeras selectively deficient in MLKL expression in the non-hematopoietic cells (WT > MLKL−/−) were susceptible to oral Salmonella infection, as seen by severe histopathology. Conversely, chimeras in which MLKL was selective deficient in hematopoietic cells (MLKL−/− > WT) were resistant to oral Salmonella infection, as seen by minimal histopathology identical to that observed in control chimeras (WT > WT) (Figures 6B–E). The same pattern was observed with respect to bacterial localization, with chimeras deficient in MLKL in the non-hematopoietic cells (MLKL−/− > MLKL−/− and WT > MLKL−/−) having higher bacterial burden in liver, spleen, MLN, cecum, and feces compared to chimeras with a WT non-hematopoietic compartment (WT > WT and MLKL−/− > WT) (Figures 6F–J). Collectively, these data suggest that MLKL expression in epithelial cell mediates host defense against Salmonella mucosal invasion.
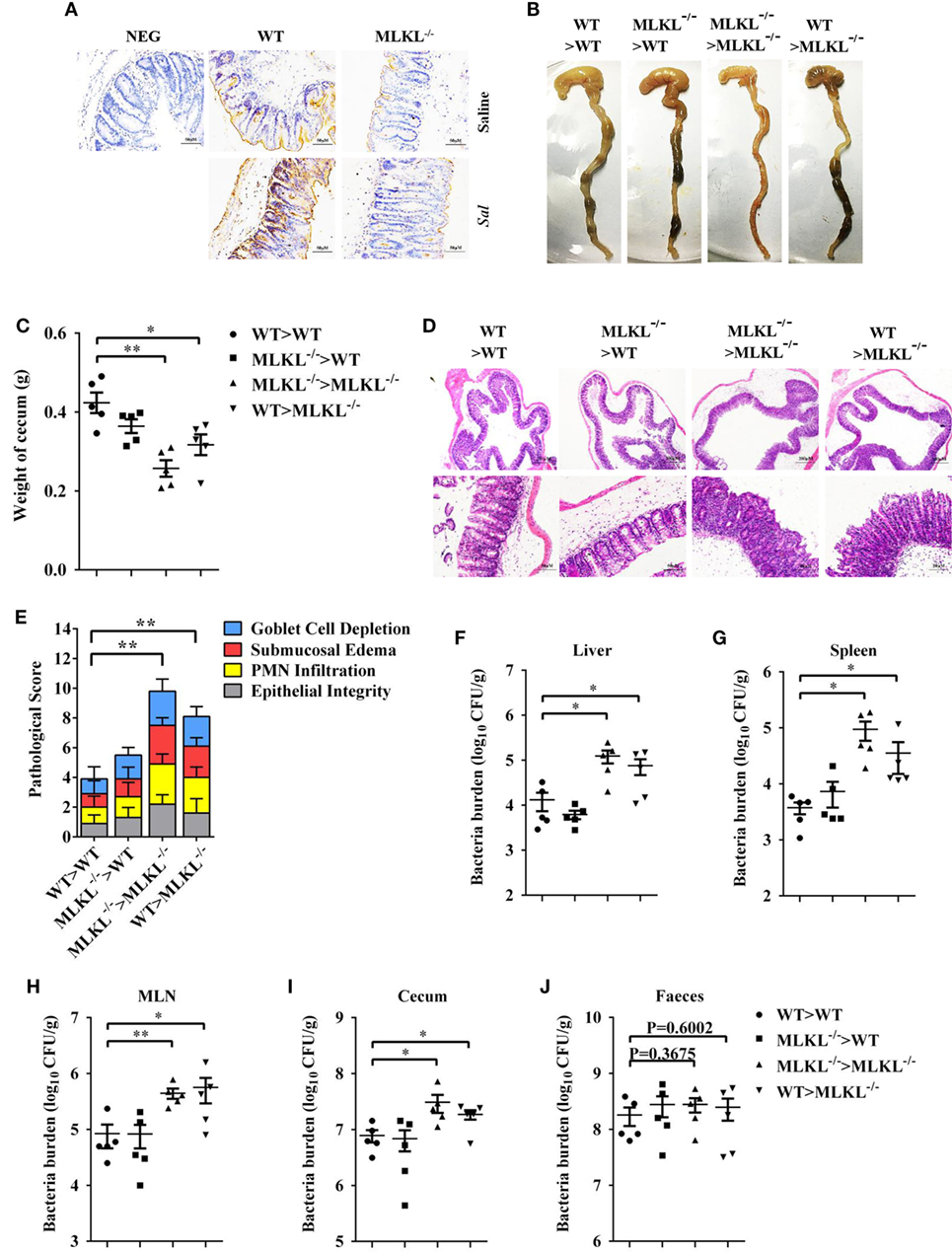
Figure 6. MLKL expression in non-hematopoietic cells contributes to protection during Salmonella infection. Streptomycin-pretreated WT and MLKL−/− mice were orally infected with Salmonella (5 × 107 CFU, n = 10 each group) for 48 h. (A) Representative cecum sections. Phosphorylated MLKL (p-MLKL IHC) stained brown. (B–J) MLKL−/− and WT bone marrow chimeric mice were generated as described in Section “Materials and Methods.” Streptomycin-pretreated mice were infected orally with Salmonella (5 × 107 CFU, n = 5 each group). (B,C) Gross appearance and weight of cecum. (D,E) Representative H&E staining of cecum tissue and pathological score, and bacteria numbers in livers (F), spleens (G), MLN (H), cecum (I), and feces (J) were determined. All data are shown as mean ± SEM. Student’s t-test was performed. Statistical significance is indicated by *p < 0.05, **p < 0.01.
MLKL-Mediated Inflammasome Activation Limits Bacterial Cololization
Inflammasome activation and the release of the pro-inflammatory cytokine interleukin (IL)-18 in the epithelial compartment contribute to repair and restitution of ulcerated epithelium (33, 34) and host protection against mucosal bacterial infections (1, 35, 36). To determine whether there is a difference in inflammasome activation that can explain the higher susceptibility of MLKL−/− mice to Salmonella invasion, we first evaluated caspase-1, the best characterized protein to date, which is cleaved and activated upon recruitment to the multiprotein complex inflammasome (37). Our results revealed that caspase-1 cleavage markedly decreased in the ceca of infected MLKL−/− mice relative to the levels observed in the ceca of infected WT mice, whereas the expression of ASC did not appear significantly different on 6 or 48 h p.i. (Figures 7A,B). Specially, we found that GSDMD (38, 39), an executor of pyroptosis and inflammasome-dependent cytokines release, also prominently reduced in the ceca of infected MLKL−/− mice (Figures 7A,B). Importantly, the amount of inflammasome-dependent IL-18 was relatively lower on 6 h p.i. (p = 0.1302) and significantly inhibited on 48 h p.i. in the cecum tissues of MLKL−/− mice than in those of WT mice (Figures 7C,D). These results demonstrate that MLKL deficiency potently inhibits inflammasome activation during mucosal Salmonella infection. Subsequently, to determine whether MLKL-mediated inflammasome activation is implicated in protection against Salmonella infection, we treated MLKL−/− mice with recombinant IL-18 (rIL-18) before Salmonella infection and examined whether exogenous IL-18 administration could rescue the defect in pathogen control. We found that injection of recombinant IL-18 strongly reduced the Salmonella burdens in MLKL−/− mice, as indicated by the levels of viable bacteria in liver, spleen, MLN, cecum and feces of MLKL−/− mice, and which were returned to almost WT mice levels (Figures 7E–I). In conclusion, these data establish that MLKL mediates protection against Salmonella epithelial localization through promoting inflammasome activation.
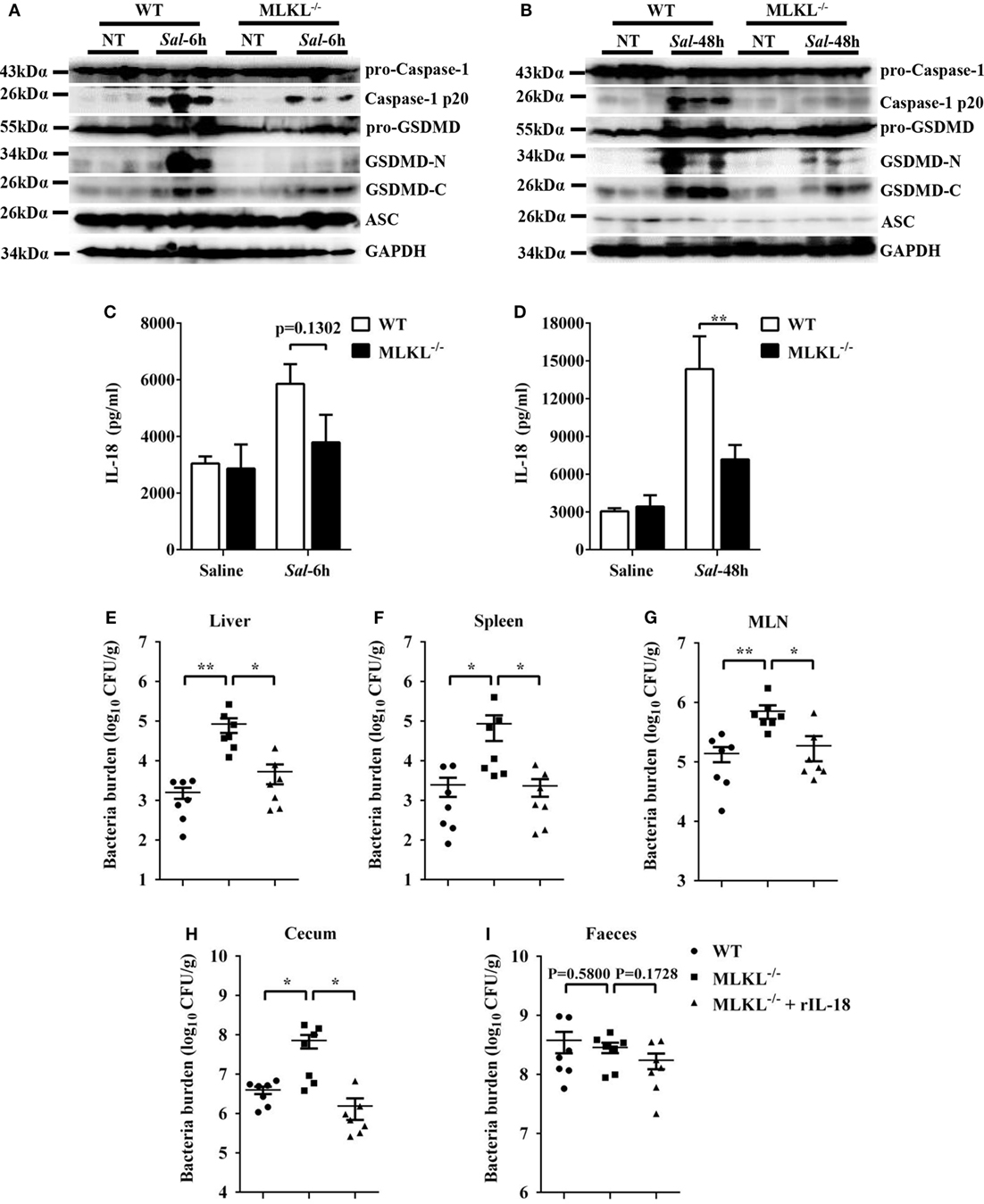
Figure 7. Inflammasome signaling downstream of MLKL confers protection against Salmonella infection. Streptomycin-pretreated WT and MLKL−/− mice were orally infected with Salmonella (5 × 107 CFU, n = 10 each group) for 6 or 48 h. (A,B) The cecum tissue lysate was analyzed for caspase-1, GSDMD, and ASC by western blotting. GAPDH was used as a loading control. (C,D) The homogenate supernatant of the cecum tissue was analyzed for the presence of IL-18 protein using ELISA. For one group of MLKL−/− mice, 1.0 µg recombinant IL-18 was injected intraperitoneally daily starting the day prior to Salmonella challenge (5 × 107 CFU, n = 7 each group, at 48 h p.i.). Bacterial burdens in the liver (E), spleen (F), MLN (G), cecum (H), and feces (I) were determined, respectively. All data are shown as mean ± SEM. Student’s t-test was performed. Statistical significance is indicated by *p < 0.05, **p < 0.01.
Discussion
As a key component of necroptosis signaling pathway, MLKL is the most known terminal protein in this kinase cascade (14, 40). Although MLKL-mediated necroptosis is involved in host defense to microbial infection (41), the biological implications of MLKL in bacterially triggered colitis have not been characterized. Using a colitis model, we found that MLKL−/− mice were more sensitive to Salmonella-induced colitis, as indicated by increased mortality, body weight loss, and intestinal-associated pathologic changes including cecal weight loss, goblet cells loss, submucosal edema, PMN infiltration, complete destruction of epithelial integrity, and pro-inflammatory mediator production. After gavage administration, Salmonella initially breaches the intestinal epithelium and then disseminates from the GI tract to colonize systemic sites, ultimately resulting in typhoid-like systemic illness (5, 9, 42). MLKL deficiency resulted in increased bacterial burdens in both intestinal and systemic sites, highlighting the key role for the MLKL in constraining Salmonella initial intestinal invasion and subsequent systemic dissemination.
On account of Salmonella is primarily a food-borne pathogen, and the primary and essential step in Salmonella infection is to cross the intestinal epithelial barrier (9). To better define how MLKL prevents Salmonella pathogenesis, it is worthwhile to investigate whether intestinal barrier function is impaired in MLKL−/− mice. The intestinal mucus provides the first line of defense for the host against various infectious agent and effectively prevents enteric pathogens from reaching and damaging the epithelium (43). Our present studies recall earlier studies with mucosal Salmonella infection, the level of mucins synthesis in WT mice was relatively increased during infection, potentially promoting host defense by removing bacteria from the mucosal surface. However, mucin expressions were dramatically reduced in MLKL−/− mice following Salmonella infection. Especially, mucin 2, a predominant component of the intestinal mucus layer, was also materially reduced in MLKL−/− mice.
Aside from the mucins secretion, there are other factors that determine host susceptibility to an enteric bacterial pathogen. Tight junction dysfunction has been linked to a variety of local and systemic diseases (44), such as Crohn’s disease (45), leaky diarrhea (46), inflammatory bowel diseases, and inflammatory bowel disease associated colorectal cancer (47). Importantly, Salmonella can cause tight junction disruption in infected epithelial cells, leading to increased epithelial permeability (8, 29, 48). Thus, we hypothesize that the susceptibility of the MLKL−/− mice to Salmonella reflects not only their impaired mucins secretion but also the exaggerated tight junction dysfunction. As expected, following infection, the expression of claudin 3 was dramatically reduced in MLKL−/− epithelium. Simultaneously, in vivo permeabilization assay using FITC-dextran showed that fluorescence intensity of serum was significantly increased in MLKL−/− mice, suggesting that intestinal barrier function is more readily disrupted in the absence of MLKL signaling.
Cell turnover is maintained by a balance between the rate of cell proliferation and apoptosis and has been offered to account for maintenance of the epithelial barrier (49, 50). Furthermore, previous reports have shown that pathologically induced epithelial cell apoptosis was associated with increased epithelial permeability (51, 52). Similarly, despite we observed that there were no significant difference in PCNA-positive cells between WT and MLKL−/− mice, MLKL−/− mice presented with notably increased TUNEL-positive cells during Salmonella infection, indicating that activation of the MLKL signal may induce a compensatory tissue homeostatic response in order to preserve the integrity of the epithelial layer during Salmonella infection.
Although these data suggest MLKL promoting intestinal mucosal barrier integrity following Salmonella challenge, we cannot exclude the possibility that the disrupted gut mucosal barrier in MLKL−/− mice on 48 h p.i. is a consequence rather than the cause of increased bacteria localization. Therefore, to further determine whether the fragile intestinal barrier is a fatal defect in MLKL−/− mice, we analyzed the initial interplay between Salmonella and the host’s gut mucosa. At 6 h p.i., intestinal pathological features of infected MLKL−/− and WT mice were not obvious. The synthesis or secretion of mucins before and after acute infection was not affected in the absence of MLKL. Exhilaratingly, we observed higher Salmonella burdens MLKL−/− epithelium at the acute stage of infection. There results suggest that MLKL potentially limits early epithelial colonization of Salmonella to prevent subsequent intestinal barrier disruption and bacterial systemic dissemination.
The question arose as to how MLKL limits early intestinal colonization of Salmonella. Using immunohistochemistry of cecum samples, we observed that active MLKL was primarily located in the crypt epithelial cells. Moreover, MLKL expression in non-hematopoietic cells had a critical role in protection during Salmonella infection, as evidenced by WT and MLKL−/− chimeric mice. Thus, MLKL expression in IECs mediates protection against early bacterial colonization. Since the enterocyte inflammasome activation emerges as an important aspect of epithelial defense against microbial infiltration (32, 36, 53). Thus, we examined whether the inflammasome modulates MLKL-mediated intestinal protection. Strikingly, we found that Salmonella-induced inflammasome activation was notably inhibited in MLKL−/− mice compared to WT mice. Importantly, inflammasome dependent IL-18 production in MLKL−/− epithelium was lower compared with the control, and systemic administration of recombinant IL-18 strongly increased the protective effects of Salmonella-infected MLKL−/− mice reflected by reduced bacteria colonization. This is consistent with previous data showing that IL-18 is associated with repair and restitution of ulcerated epithelium and contribute to host protection against mucosal bacterial infections (35, 54, 55). IL-1β, another inflammasome-mediated cytokine, increased in the cecum tissues of infected MLKL−/− mice. Intestinal levels of IL-1β correlate well with severity of intestinal inflammation and NLRC4 inflammasome deficiency still results in IL-1β production, suggesting other signaling rather than NLRC4 inflammasome plays dominant role in IL-1β production (56). IL-1α/β and IL-18 are not required for epithelial restriction of early Salmonella infection (18 h p.i.), and IL-18 contributes to host protection at later stages (36 h p.i.) (32). Consistent with this finding, we observed rIL-18 restricts Salmonella translocation at 48 h p.i. However, MLKL−/− epithelium had a modest higher bacterial burden at the acute stage of infection (6 h p.i.); it is probable that pyroptosis rather than IL-18 contributes to MLKL-mediated protection against early Salmonella epithelial colonization.
Recent studies have shown that necroptotic stimuli-mediated MLKL signaling can promote NLRP3 inflammasome activation in macrophages (17, 57). NLRP3 inflammasome is required for host control of mucosal pathogen Citrobacter rodentium (54), and Salmonella can also induce NLRP3 inflammasome activation in macrophages (58, 59). However, neither ASC nor NLRP3 is essential for host defense against Salmonella intestinal infection (60). To date, NLRP1, NLRP3, NLRC4, AIM2, and pyrin are well established to assemble a canonical inflammasome complex. Of these, actually only NLRC4 inflammasome has been shown to mediate mucosal protection against Salmonella infection (32, 56, 61). Epithelium-intrinsic NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa via activation of caspase-1 and -8 (32, 61). Therefore, we suspect that non-hematopoietic MLKL-mediated host protection may primarily rely on NLRC4 inflammasome activation, which need extensive study.
Ethics Statement
All animal studies were conducted according to experimental practices and standards approved by the Animal Welfare and Research Ethics Committee at Jilin University (No. 20150601).
Author Contributions
SXY, WC, and YJY designed experiments. SXY, WC, ZZL, FHZ, SQY, GQH, XXQ, JZ, KM, CTD, and JMG performed the experiments and analyzed the data. SXY wrote the manuscript. XMD, YWH, and YJY revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by National Key Research and Development Program of China (No. 2017YFD0501000) and National Natural Science Foundation of China (No. 31372410).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00119/full#supplementary-material.
Abbreviations
MLKL, mixed lineage kinase-like protein; GSDMD, gasdermin D; Salmonella, Salmonella enterica serovar typhimurium; MLNs, mesenteric lymph nodes; IECs, intestinal epithelial cells; PRR, pattern recognition receptors; TLRs, toll-like receptors; RIPK1, receptor interacting protein kinase 1; RIPK3, receptor interacting protein kinase 3; CFU, colony-forming unit; H&E, hematoxylin and eosin; PMN, polymorphonuclear leukocyte; ZO-1, zonula occludens-1; rIL-18, recombinant IL-18.
References
1. Nordlander S, Pott J, Maloy KJ. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol (2014) 7:775–85. doi:10.1038/mi.2013.95
2. Yang JY, Kim MS, Kim E, Cheon JH, Lee YS, Kim Y, et al. Enteric viruses ameliorate gut inflammation via toll-like receptor 3 and toll-like receptor 7-mediated interferon-beta production. Immunity (2016) 44:889–900. doi:10.1016/j.immuni.2016.03.009
3. Boyle EC, Bishop JL, Grassl GA, Finlay BB. Salmonella: from pathogenesis to therapeutics. J Bacteriol (2007) 189:1489–95. doi:10.1128/JB.01730-06
4. Lawley TD, Walker AW. Intestinal colonization resistance. Immunology (2013) 138:1–11. doi:10.1111/j.1365-2567.2012.03616.x
5. Broz P, Ohlson MB, Monack DM. Innate immune response to Salmonella typhimurium, a model enteric pathogen. Gut Microbes (2012) 3:62–70. doi:10.4161/gmic.19141
6. Ahmer BM, Gunn JS. Interaction of Salmonella spp. with the intestinal microbiota. Front Microbiol (2011) 2:101. doi:10.3389/fmicb.2011.00101
7. Corr SC, Palsson-McDermott EM, Grishina I, Barry SP, Aviello G, Bernard NJ, et al. MyD88 adaptor-like (Mal) functions in the epithelial barrier and contributes to intestinal integrity via protein kinase C. Mucosal Immunol (2014) 7:57–67. doi:10.1038/mi.2013.24
8. Hu GQ, Song PX, Li N, Chen W, Lei QQ, Yu SX, et al. AIM2 contributes to the maintenance of intestinal integrity via Akt and protects against Salmonella mucosal infection. Mucosal Immunol (2016) 9:1330–9. doi:10.1038/mi.2015.142
9. Schultz BM, Paduro CA, Salazar GA, Salazar-Echegarai FJ, Sebastián VP, Riedel CA, et al. A potential role of Salmonella infection in the onset of inflammatory bowel diseases. Front Immunol (2017) 8:191. doi:10.3389/fimmu.2017.00191
10. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol (2010) 10:131–44. doi:10.1038/nri2707
11. Opipari A, Franchi L. Role of inflammasomes in intestinal inflammation and crohn’s disease. Inflamm Bowel Dis (2015) 21:173–81. doi:10.1097/MIB.0000000000000230
12. Man SM, Zhu Q, Zhu L, Liu Z, Karki R, Malik A, et al. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell (2015) 162:45–58. doi:10.1016/j.cell.2015.06.001
13. Swamy M, Jamora C, Havran W, Hayday A. Epithelial decision makers: in search of the ‘epimmunome’. Nat Immunol (2010) 11:656–65. doi:10.1038/ni.1905
14. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res (2013) 23:994–1006. doi:10.1038/cr.2013.91
15. Yu SX, Zhou FH, Chen W, Jiang GM, Du CT, Hu GQ, et al. Decidual stromal cell necroptosis contributes to polyinosinic-polycytidylic acid-triggered abnormal murine pregnancy. Front Immunol (2017) 8:916. doi:10.3389/fimmu.2017.00916
16. Murphy JM, Vince JE. Post-translational control of RIPK3 and MLKL mediated necroptotic cell death. F1000Res (2015) 4:F1000 Faculty Rev-1297. doi:10.12688/f1000research.7046.1
17. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A (2017) 114:E961–9. doi:10.1073/pnas.1613305114
18. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol (2017) 18:127–36. doi:10.1038/nrm.2016.149
19. Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol (2014) 35:14–23. doi:10.1016/j.semcdb.2014.07.013
20. Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe (2010) 8:44–54. doi:10.1016/j.chom.2010.06.007
21. Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, et al. Cell death and infection: a double-edged sword for host and pathogen survival. J Cell Biol (2011) 195:931–42. doi:10.1083/jcb.201108081
22. Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe (2016) 20:13–24. doi:10.1016/j.chom.2016.05.011
23. Barthel M, Hapfelmeier S, Quintanilla-Martínez L, Kremer M, Rohde M, Hogardt M, et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun (2003) 71:2839–58. doi:10.1128/IAI.71.5.2839-2858.2003
24. Lei QQ, Hu GQ, Chen W, Yu SX, Qi S, Du CT, et al. RCAN1 deficiency protects against Salmonella intestinal infection by modulating JNK activation. Mol Immunol (2016) 77:26–33. doi:10.1016/j.molimm.2016.07.009
25. Oppong GO, Rapsinski GJ, Newman TN, Nishimori JH, Biesecker SG, Tükel Ç. Epithelial cells augment barrier function via activation of the toll-like receptor 2/phosphatidylinositol 3-kinase pathway upon recognition of Salmonella enterica serovar typhimurium curli fibrils in the gut. Infect Immun (2013) 81:478–86. doi:10.1128/IAI.00453-12
26. Wu X, Sun L, Zha W, Studer E, Gurley E, Chen L, et al. HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Gastroenterology (2010) 138:197–209. doi:10.1053/j.gastro.2009.08.054
27. Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity (2012) 36:276–87. doi:10.1016/j.immuni.2011.12.011
28. Zhu Y, Zhu M, Lance P. iNOS signaling interacts with COX-2 pathway in colonic fibroblasts. Exp Cell Res (2012) 318:2116–27. doi:10.1016/j.yexcr.2012.05.027
29. Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A, et al. The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar typhimurium colitis. Infect Immun (2013) 81:3672–83. doi:10.1128/IAI.00854-13
30. Yan L, Yang C, Tang J. Disruption of the intestinal mucosal barrier in Candida albicans infections. Microbiol Res (2013) 168:389–95. doi:10.1016/j.micres.2013.02.008
31. Thuijls G, Derikx JP, de Haan JJ, Grootjans J, de Bruïne A, Masclee AA, et al. Urine-based detection of intestinal tight junction loss. J Clin Gastroenterol (2010) 44:e14–9. doi:10.1097/MCG.0b013e31819f5652
32. Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe (2014) 16:237–48. doi:10.1016/j.chom.2014.07.001
33. Reuter BK, Pizarro TT. Commentary: the role of the IL-18 system and other members of the IL-1R/TLR superfamily in innate mucosal immunity and the pathogenesis of inflammatory bowel disease: friend or foe? Eur J Immunol (2004) 34:2347–55. doi:10.1002/eji.200425351
34. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32:379–91. doi:10.1016/j.immuni.2010.03.003
35. Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, et al. Host-protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell (2016) 167(444–456):e414. doi:10.1016/j.cell.2016.08.076
36. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe (2014) 16:249–56. doi:10.1016/j.chom.2014.07.002
37. Yu SX, Du CT, Chen W, Lei QQ, Li N, Qi S, et al. Genipin inhibits NLRP3 and NLRC4 inflammasome activation via autophagy suppression. Sci Rep (2015) 5:17935. doi:10.1038/srep17935
38. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25:1285–98. doi:10.1038/cr.2015.139
39. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi:10.1038/nature18629
40. Petrie EJ, Hildebrand JM, Murphy JM. Insane in the membrane: a structural perspective of MLKL function in necroptosis. Immunol Cell Biol (2017) 95:152–9. doi:10.1038/icb.2016.125
41. Pearson JS, Murphy JM. Down the rabbit hole: is necroptosis truly an innate response to infection? Cell Microbiol (2017) 19:e12750. doi:10.1111/cmi.12750
42. Santos RL, Zhang S, Tsolis RM, Kingsley RA, Adams LG, Bäumler AJ. Animal models of Salmonella infections: enteritis versus typhoid fever. Microbes Infect (2001) 3:1335–44. doi:10.1016/S1286-4579(01)01495-2
43. Cornick S, Tawiah A, Chadee K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers (2015) 3:e982426. doi:10.4161/21688370.2014.982426
44. Buckley A, Turner JR. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harb Perspect Biol (2018) 10:a029314. doi:10.1101/cshperspect.a029314
45. Qian XX, Peng JC, Xu AT, Zhao D, Qiao YQ, Wang TR, et al. Noncoding transcribed ultraconserved region (T-UCR) uc.261 participates in intestinal mucosa barrier damage in crohn’s disease. Inflamm Bowel Dis (2016) 22:2840–52. doi:10.1097/MIB.0000000000000945
46. Barmeyer C, Fromm M, Schulzke JD. Active and passive involvement of claudins in the pathophysiology of intestinal inflammatory diseases. Pflugers Arch (2017) 469:15–26. doi:10.1007/s00424-016-1914-6
47. Landy J, Ronde E, English N, Clark SK, Hart AL, Knight SC, et al. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J Gastroenterol (2016) 22:3117–26. doi:10.3748/wjg.v22.i11.3117
48. Boyle EC, Brown NF, Finlay BB. Salmonella enterica serovar typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell Microbiol (2006) 8:1946–57. doi:10.1111/j.1462-5822.2006.00762.x
49. Edelblum KL, Yan F, Yamaoka T, Polk DB. Regulation of apoptosis during homeostasis and disease in the intestinal epithelium. Inflamm Bowel Dis (2006) 12:413–24. doi:10.1097/01.MIB.0000217334.30689.3e
50. Negroni A, Cucchiara S, Stronati L. Apoptosis, necrosis, and necroptosis in the gut and intestinal homeostasis. Mediators Inflamm (2015) 2015:250762. doi:10.1155/2015/250762
51. Yasuda T, Takeyama Y, Ueda T, Shinzeki M, Sawa H, Nakajima T, et al. Breakdown of intestinal mucosa via accelerated apoptosis increases intestinal permeability in experimental severe acute pancreatitis. J Surg Res (2006) 135:18–26. doi:10.1016/j.jss.2006.02.050
52. Chaïbi C, Cotte-Laffitte J, Sandré C, Esclatine A, Servin AL, Quéro AM, et al. Rotavirus induces apoptosis in fully differentiated human intestinal Caco-2 cells. Virology (2005) 332:480–90. doi:10.1016/j.virol.2004.11.039
53. Broz P. Getting rid of the bad apple: inflammasome-induced extrusion of Salmonella-infected enterocytes. Cell Host Microbe (2014) 16:153–5. doi:10.1016/j.chom.2014.07.010
54. Song-Zhao GX, Srinivasan N, Pott J, Baban D, Frankel G, Maloy KJ. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol (2014) 7:763–74. doi:10.1038/mi.2013.94
55. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev (2017) 277:61–75. doi:10.1111/imr.12534
56. Carvalho FA, Nalbantoglu I, Aitken JD, Uchiyama R, Su Y, Doho GH, et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol (2012) 5:288–98. doi:10.1038/mi.2012.8
57. Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of gasdermin-D. J Immunol (2017) 198:2156–64. doi:10.4049/jimmunol.1601757
58. Pereira M, Tourlomousis P, Wright J, Monie TP, Bryant CE. CARD9 negatively regulates NLRP3-induced IL-1beta production on Salmonella infection of macrophages. Nat Commun (2016) 7:12874. doi:10.1038/ncomms12874
59. Diamond CE, Leong KWK, Vacca M, Rivers-Auty J, Brough D, Mortellaro A. Salmonella typhimurium-induced IL-1 release from primary human monocytes requires NLRP3 and can occur in the absence of pyroptosis. Sci Rep (2017) 7:6861. doi:10.1038/s41598-017-07081-3
60. De Jong HK, Koh GC, van Lieshout MH, Roelofs JJ, van Dissel JT, van der Poll T, et al. Limited role for ASC and NLRP3 during in vivo Salmonella typhimurium infection. BMC Immunol (2014) 15:30. doi:10.1186/s12865-014-0030-7
Keywords: mixed lineage kinase-like protein, inflammasome, gasdermin D, Salmonella, colitis
Citation: Yu SX, Chen W, Liu ZZ, Zhou FH, Yan SQ, Hu GQ, Qin XX, Zhang J, Ma K, Du CT, Gu JM, Deng XM, Han WY and Yang YJ (2018) Non-Hematopoietic MLKL Protects Against Salmonella Mucosal Infection by Enhancing Inflammasome Activation. Front. Immunol. 9:119. doi: 10.3389/fimmu.2018.00119
Received: 20 November 2017; Accepted: 15 January 2018;
Published: 02 February 2018
Edited by:
Eric Cox, Ghent University, BelgiumReviewed by:
Nobuhiko Kamada, University of Michigan Health System, United StatesJuan Li, Rockefeller University, United States
Copyright: © 2018 Yu, Chen, Liu, Zhou, Yan, Hu, Qin, Zhang, Ma, Du, Gu, Deng, Han and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Jun Yang, eW91bmdqdW5lQGpsdS5lZHUuY24=
†These authors contributed equally to this work.