 Yotam Lior1†
Yotam Lior1† Mariana Zaretsky2†
Mariana Zaretsky2† David E. Ochayon1†
David E. Ochayon1† Diana Lotysh2Boris M. Baranovski1
Diana Lotysh2Boris M. Baranovski1 Ronen Schuster1Ofer Guttman1
Ronen Schuster1Ofer Guttman1 Amir Aharoni2Eli C. Lewis1*
Amir Aharoni2Eli C. Lewis1*
- 1Department of Clinical Biochemistry and Pharmacology, Faculty of Health Sciences, Ben-Gurion University of the Negev, Be’er Sheva, Israel
- 2Department of Life Sciences, Ben-Gurion University of the Negev and National Institute for Biotechnology, Be’er Sheva, Israel
Introduction: Human α1-antitrypsin (hAAT) is a 394-amino acid long anti-inflammatory, neutrophil elastase inhibitor, which binds elastase via a sequence-specific molecular protrusion (reactive center loop, RCL; positions 357–366). hAAT formulations that lack protease inhibition were shown to maintain their anti-inflammatory activities, suggesting that some attributes of the molecule may reside in extra-RCL segments. Here, we compare the protease-inhibitory and anti-inflammatory profiles of an extra-RCL mutation (cys232pro) and two intra-RCL mutations (pro357cys, pro357ala), to naïve [wild-type (WT)] recombinant hAAT, in vitro, and in vivo.
Methods: His-tag recombinant point-mutated hAAT constructs were expressed in HEK-293F cells. Purified proteins were evaluated for elastase inhibition, and their anti-inflammatory activities were assessed using several cell-types: RAW264.7 cells, mouse bone marrow-derived macrophages, and primary peritoneal macrophages. The pharmacokinetics of the recombinant variants and their effect on LPS-induced peritonitis were determined in vivo.
Results: Compared to WT and to RCL-mutated hAAT variants, cys232pro exhibited superior anti-inflammatory activities, as well as a longer circulating half-life, despite all three mutated forms of hAAT lacking anti-elastase activity. TNFα expression and its proteolytic membranal shedding were differently affected by the variants; specifically, cys232pro and pro357cys altered supernatant and serum TNFα dynamics without suppressing transcription or shedding.
Conclusion: Our data suggest that the anti-inflammatory profile of hAAT extends beyond direct RCL regions. Such regions might be relevant for the elaboration of hAAT formulations, as well as hAAT-based drugs, with enhanced anti-inflammatory attributes.
Introduction
Human α1-antitrypsin (hAAT) is a 52-kDa, 394-amino acid long serum glycoprotein, a member of the serine protease inhibitor superfamily. The molecule is secreted primarily by hepatocytes to the circulation, in both steady state and acute-phase responses (1–3). Additionally, hAAT is produced by lung epithelia, intestinal paneth cells, and M2-like macrophages (4). Several mutations in the gene coding for hAAT have been known to result in significantly low circulating levels of hAAT, a rare genetic condition termed α1-antitrypsin deficiency (AATD). The Z variant (Glu342Lys) is the most common variant of AATD, followed by the S variant (Glu264Val) (5). AATD is most commonly associated with early-onset non-smoker lung emphysema as well as liver cirrhosis, vasculitis, and bacterial pneumonia (5–8). Being a one-gene disease, several research teams have been making advances in the field of hAAT gene-therapy, primarily via an adenoviral backbone construct (clinical trials NCT01054339, NCT02168686, NCT00377416, and NCT00430768) (9, 10), the only standard of care for AATD at present involves life-long weekly infusions of affinity-purified human plasma-derived AAT, aimed at restoring circulating hAAT levels (7, 11).
While globular in structure, hAAT has a reactive center loop (RCL, positions 357–366) that protrudes from its surface, and that acts as a sequence-specific bait for serine-proteases (2, 12), among which are neutrophil elastase, cathepsin G, and proteinase-3 (13, 14). RCL cleavage leads to the covalent attachment of the targeted protease to hAAT, followed by a conformational change and the removal of the hAAT:protease complex from the circulation (14).
Interestingly, proteases outside the serine-protease family are also inhibited by hAAT, albeit to a lesser extent. These include metalloproteases [e.g., MMP-9 (15–17), ADAMTS-4 (18), and cysteine-proteases (e.g., caspase-3) (19, 20)], suggesting that some functions of the molecule may extend beyond the specificity conferred by the primary sequence of the RCL. As such, it has been proposed that the globular surface of hAAT may contain significant functional attributes. Indeed, it has been established that hAAT directly binds IL-8 (21, 22), as well as to polymeric immunoglobulin receptor (23), gp96 (24), HSP70 (25), oxidized cholesterol within lipid rafts and serum lipids (26–29), HDL particles (30–32), and LRP1 receptor (33). Furthermore, certain activities that are attributed to hAAT appear to be reproducible in formulations that lack elastase inhibition as in the case of recombinant Fc-hAAT and truncated hAAT (26, 34–36).
The breadth of anti-inflammatory and immunomodulatory functions of hAAT has gained increased recognition in the past decade. hAAT promotes production of anti-inflammatory cytokines, such as IL-10 (37) and IL-1 receptor antagonist (IL-1Ra) (38), and inhibits the release of pro-inflammatory cytokines and chemokines, such as IL-6 and TNFα (3, 39–43). In the context of allograft transplantation, hAAT modifies dendritic cell responses (37, 44) and B lymphocyte activities (45), reduces the levels of inducible co-stimulatory molecules, e.g., CD40 and CD86, and promotes regulatory T cell expansion (4, 41, 46). Of particular interest, hAAT reduces soluble TNFα levels (42, 43) and interferes with TNFα-dependent responses. Inducible membrane-associated TNFα appears to accumulate on the surface of hAAT-treated leukocytes (47), even though TNFα cleavage requires ADAM metallopeptidase domain 17 (ADAM17/TACE) (43), which is outside the repertoire of hAAT protease inhibition.
Mutations within the RCL usually alter hAAT protease-inhibiting specificity or total protease-inhibiting capacity, as reported with regards to a mutation in which proline is substituted with cysteine within the RCL region (pro357cys) (48). However, little is known regarding the effect of such mutations in as far as the anti-inflammatory properties of hAAT are concerned. Furthermore, only a few studies explored non-AATD-causing mutations outside the RCL in terms of anti-proteases and anti-inflammatory effects.
In the present study, we revisited a previously described intra-RCL mutation (pro357cys) known to lack anti-protease activities (48). To better understand the effects of this intra-RCL mutation, we compared its anti-inflammatory attributes to those of wild-type (WT) hAAT as well as to those of novel intra-RCL (pro357ala) and extra-RCL (cys232pro) hAAT variants. Indeed, the functions of hAAT evaluated hereby, appear to extend beyond protease-inhibition and include both in vitro and in vivo anti-inflammatory activities.
Materials and Methods
Plasmid Constructs
Human AAT EST clone was purchased from Open Biosystems (GE Healthcare, Chicago, IL, USA) and amplified by PCR using FW 5′-GATCACCG-GTGAATTCGATATCTCGAGCACCATGGTTATGCCGTCTTCTGTCTCGTGGGGCATCC-3′ and RE 5′-GCTGGGCAAGGTGGGCACTCCACAGATCTCTACTA-GTGATGGTGATGATGATGATGATGTTTTTGGGTGGGATTCACCAC-3′ primers. A His-tag sequence was added to the C terminal. Specific mutations for the replacement of C232 and P357 were inserted by assembly PCR using the primers: FW-AAT-C232P TTTAGGCATGTTTAACATC-CAGCACCCCAAGAAGCTGTCCAGCTGGGTGCTGCTG and RE-asm-AAT GTGCTGGATGTTAAACATGCCTAAACG for C232P, FW-asm-PtoC-AAT TTAGAGGCCATATGCATGTCTATCCCCCCCGAGG and RE-asm-PtoC-AAT CCTCGGGGGGGATAGACATGCATATGGCCTCTAA for P357C, and FW-asm-PtoA-AAT GTTTTTAGAGGCCATAGCCATGTCTATCCCCCCCGAG and RE-asm-PtoA-AAT CTCGGGGGGGATAGACATGGCTATGGCCTCTAAAAAC for P357A. Sequences were cloned into pFUSE plasmid (Invivogen, San Diego, CA, USA) using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA, USA), according to manufacturer’s instructions. Naïve human AAT signal peptide was used in protein expression. Plasmids were replicated in E. coli (HIT Competent Cells-DH5α, Real Biotech Corporation, Banqiao city, Taiwan) and purified using Wizard® Plus SV Minipreps DNA Purification Systems (Promega, Fitchburg, WI, USA), according to manufacturer’s instructions.
Recombinant Protein Production and Purification
HEK-293F cells (CRL-1573, ATCC, Manassas, WV, USA) were cultured in FreeStyle 293 expression medium (Invitrogen, Carlsbad, CA, USA) in 8% CO2 shaking incubator. Cells were transfected using GeneTran™ transfection reagent (Biomega, San Diego, CA, USA) according to manufacturer’s instructions. Six days post-transfection, supernatants were collected and secreted hAAT was purified using Ni beads (Calbiochem, Merck Millipore, Darmstadt, Germany) by standard protocol. After protein purification, samples were assessed for purity and molecular weight on a 10% polyacrylamide gel stained with coomassie brilliant blue; commercial clinical-grade serum-purified hAAT (Glassia, Kamada, Ness-Ziona, Israel) was used as reference. Protein concentrations were determined using micro-volume spectrophotometer (Nanodrop, ThemoFisher Scientific, Waltham, MA, USA) and Bradford Protein Assay (Bio-Rad Laboratories, Rishon-LeZion, Israel).
Neutrophil Elastase Activity Assay
Neutrophil elastase activity was determined in acellular conditions using a designated kit (Sigma-Aldrich, Lois, MO, USA), according to manufacturer’s instruction (final elastase concentration per well: 0.39 μM). rhAAT variants were pre-incubated with the commercial enzyme prior to kinetic evaluation of color-producing substrate processing.
Mice
C57BL/6 mice (6–8 weeks old males and females from Harlan Laboratories Ltd., Jerusalem, Israel) were used for all experiments. The study was approved by the Ben-Gurion University of the Negev Animal Care and Use Committee.
Production of Bone Marrow-Derived Macrophages (BMDM)
The tibia and femur of C57BL/6 mice were surgically removed and thoroughly flushed through a 70-μM sterile nylon cell strainer (Falcon; BD Biosciences Discovery Labware, San Jose, CA, USA) with PBS (Biological Industries, Beit Ha’emek, Israel). Cells were resuspended and cultured in 10 ml complete RPMI 1640 (containing 10% fetal bovine serum, 50 U/ml streptomycin/penicillin, 50 μg/ml l-glutamine, all from Biological Industries), 50 μM β2-mercaptoethanol (Sigma-Aldrich, Rehovot, Israel) and 20 ng/ml recombinant Granulocyte Macrophage Colony-Stimulating Factor (rGM-CSF, PeproTech, Rocky Hill, NJ, USA). Medium containing rGM-CSF was added on day 3 and on day 6. Cell populations were confirmed as being >95% CD11b+ after 9 days of incubation with rGM-CSF by flow cytometry.
Thioglycolate-Elicited Primary Peritoneal Cells
C57BL/6 mice were injected with thioglycolate (3% v/v, Sigma-Aldrich; i.p., 1.5 ml per mouse). Five days later, peritoneal lavage was performed with cold PBS. Recovered cell suspensions were filtered through a 70-μM sterile nylon strainer. Cells were then resuspended in complete RPMI 1640. Cell cultures were routinely verified to be >95% CD11b+/F4-80+ cells by flow cytometry.
Cell Activation Assays and Flow Cytometry
Peritoneal macrophages and BMDMs, as indicated, were seeded at 2–3 × 105 cells per well in 300 μl complete RPMI 1640. Recombinant hAAT variants were added at indicated concentrations for overnight incubation. Cells were then carefully washed with PBS and medium was replaced with the same concentrations of rhAAT variants, as well as LPS (Sigma-Aldrich) at indicated concentrations. Twenty-four hours later, supernatants were collected and analyzed for IL-6 and TNFα concentrations using specific ELISA (Biolegend, San Diego, CA, USA).
Cells were gently removed with a rubber policeman and suspended in FACS buffer (PBS containing 1% BSA from Biological Industries, 0.1% sodium azide and 2 mM EDTA, both from Sigma-Aldrich). Blocking was performed at room temperature for 20 min using anti-CD16/32 antibody (Biolegend). Staining was performed at 4°C for an additional 20 min using the following anti-mouse antibodies: anti-CD40-FITC (3/2.3), anti-CD86-PE (GL-1), anti-TNFα-APC (MP6-XT22), anti-CD11b-Pacific blue (M1/70), all from Biolegend, and anti-F4/80-PerCP-Cy5.5 (BM8.1) (Merc, Temecula, CA, USA). Fluorescent readout was determined using BD Canto II and data were analyzed by FLOWJO 10.0.8r1 software (Flowjo, LLC Data Analysis Software, Ashland, OR, USA). After exclusion of cellular debris and duplicated cells, F4-80+/CD11b+ population was selected and surface expression levels of CD40 and CD86 were assessed and compared between samples.
In Vivo LPS-Induced Peritonitis
Mice were pretreated with 100 μl of PBS or rhAAT variants (50 μg per mouse i.p., n = 20 per experiment) for 3 h, then treated with 1 mg/kg LPS (i.p.). Blood samples (20 μl) were collected from the tail vein at 1.5, 3, and 24 h later, and serum was separated by centrifuge; sera were analyzed for TNFα concentrations using specific ELISA (R&D Systems).
Real-Time Quantitative PCR
RAW264.7 cells (TIB-71, ATCC) were seeded at 5 × 105 cells per well in 500 μl complete RPMI 1640. Cells were carefully washed and medium replaced with identical concentrations of rhAAT variants and LPS at indicated concentrations. Total RNA was purified at 1, 3, and 6 h poststimulation using total RNA purification kit (Norgen, Thorold, ON, Canada), according to manufacturer’s instructions. Sample concentrations were normalized to RNA content using micro-volume spectrophotometer (Nanodrop) and then reverse-transcribed with qScript cDNA synthesis kit (Quanta Biosciences, Gaithersburg, MD, USA), according to manufacturer’s instructions. cDNA amplification was performed and gene transcription was analyzed by qPCR (StepOnePlus real-time PCR system, ThemoFisher Scientific) using the following primers: 18S FW 5′-TCAACACAGGGATCGGACAACACA-3′ RE 5′-GCCTTGGATCAAGTTCACAGGCAA-3′; TNFα FW 5′-CCCACGTCGTAGCAAACCAC-3′ RE 5′-CCCTTGAAGAGAACCTGGGAG-3′.
Pharmacokinetics Study
rhAAT variants were introduced into mice (50 μg/mouse, i.v.). Blood samples (40 μl) were collected from the tail vein and circulating serum hAAT levels were determined at 1, 12, and 24 h from injections using species-specific hAAT ELISA (ICL Lab, Portland, OR, USA). T0.5 and distribution volume were calculated using PKsolver add-in for Microsoft Excel (49).
Statistical Analysis
Two-tailed Mann–Whitney test was used to assess differences between selected experimental conditions. Results are expressed as mean ± SEM, p ≤ 0.05 was considered significant. All statistical analyses were performed using GraphPad Prism version 6.01.
Results
Recombinant hAAT Variants
Mutations at amino acid positions 357 (inside the RCL) and 232 (outside the RCL) were generated, as illustrated in Figures 1A,B; for this, HEK-T293F cells were transfected with respective plasmid constructs and allowed to release His-tag WT recombinant hAAT (WT-rhAAT) and its mutated variants (C232P, P357C and P357A). hAAT variants were then affinity-purified, and their size confirmed to be consistent with serum-purified commercially available clinical-grade human AAT (Figure 1C). According to neutrophil elastase inhibition assays (Figure 1D), WT-rhAAT inhibition profile appears consistent with that of serum-purified hAAT, requiring concentrations in the range of micrograms (in contrast to the later experimental 200 ng/ml concentration range, arrow). The variants C232P (CP), P357C (PC), and P357A (PA) failed to inhibit neutrophil elastase at all tested concentrations (not shown); expectedly, inhibition of ADAM17 in an acellular inhibition assay was negative for all formulations of rhAAT, including WT-rhAAT (not shown).
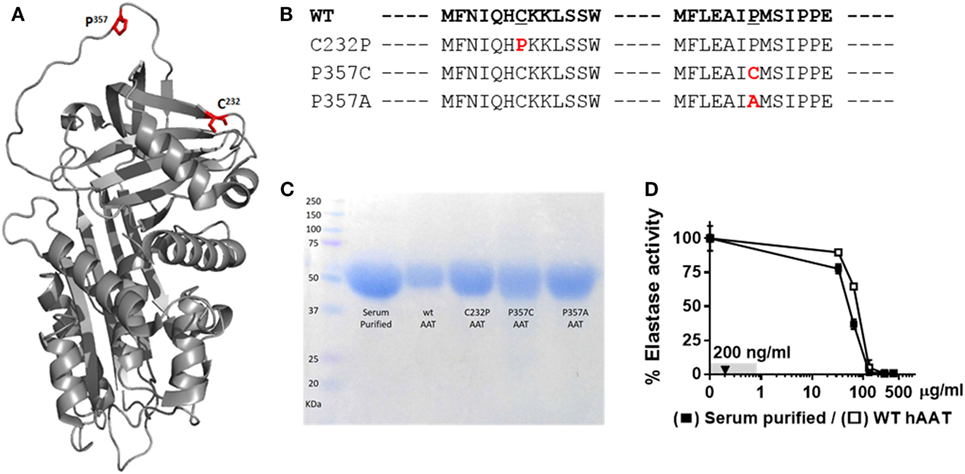
Figure 1. α1-Antitrypsin. (A) Computerized 3D model of AAT. Red, mutagenesis specific loci, C232 and P357. (B) FASTA sequence of WT-hAAT and three generated mutations. Red, mutation sites. (C) Representative Commassie brilliant blue blot of purified rhAAT variants and serum-purified human α1-antitrypsin (hAAT). (D) Inhibitory potency of WT-hAAT and serum-purified hAAT over neutrophil elastase (0.39 μM). Gray, concentrations range used in this study. Triangle, concentration used in anti-inflammatory studies. Data representative of three independent experimental repeats.
Anti-Inflammatory Attributes of rhAAT Variants at Below Protease-Inhibitory Concentrations
The response of primary murine BMDMs to LPS was tested in the presence of hAAT variants. As shown in Figure 2, the cellular response to LPS included inducible IL-6 and TNFα release (Figure 2A), and increased expression levels of surface CD40 and CD86 (Figure 2B). WT-rhAAT pretreatment at 200 ng/ml resulted in a significant reduction of inducible IL-6 level (31.5% from LPS alone). While PC and PA pretreatment at the same concentration failed to achieve a statistically significant reduction in IL-6 (7 and 12.5%, respectively), CP achieved a significant inhibition of inducible IL-6 levels at concentrations as low as 50 ng/ml (Figure 2A, arrowhead). Inducible TNFα supernatant levels displayed a different pattern to that of inducible IL-6. WT-rhAAT pretreatment reached a significant decrease at 100 ng/ml, while CP, PC and PA caused a comparable decline at 50 ng/ml (24.9, 30.4, 25.9, and 14.1%, respectively).
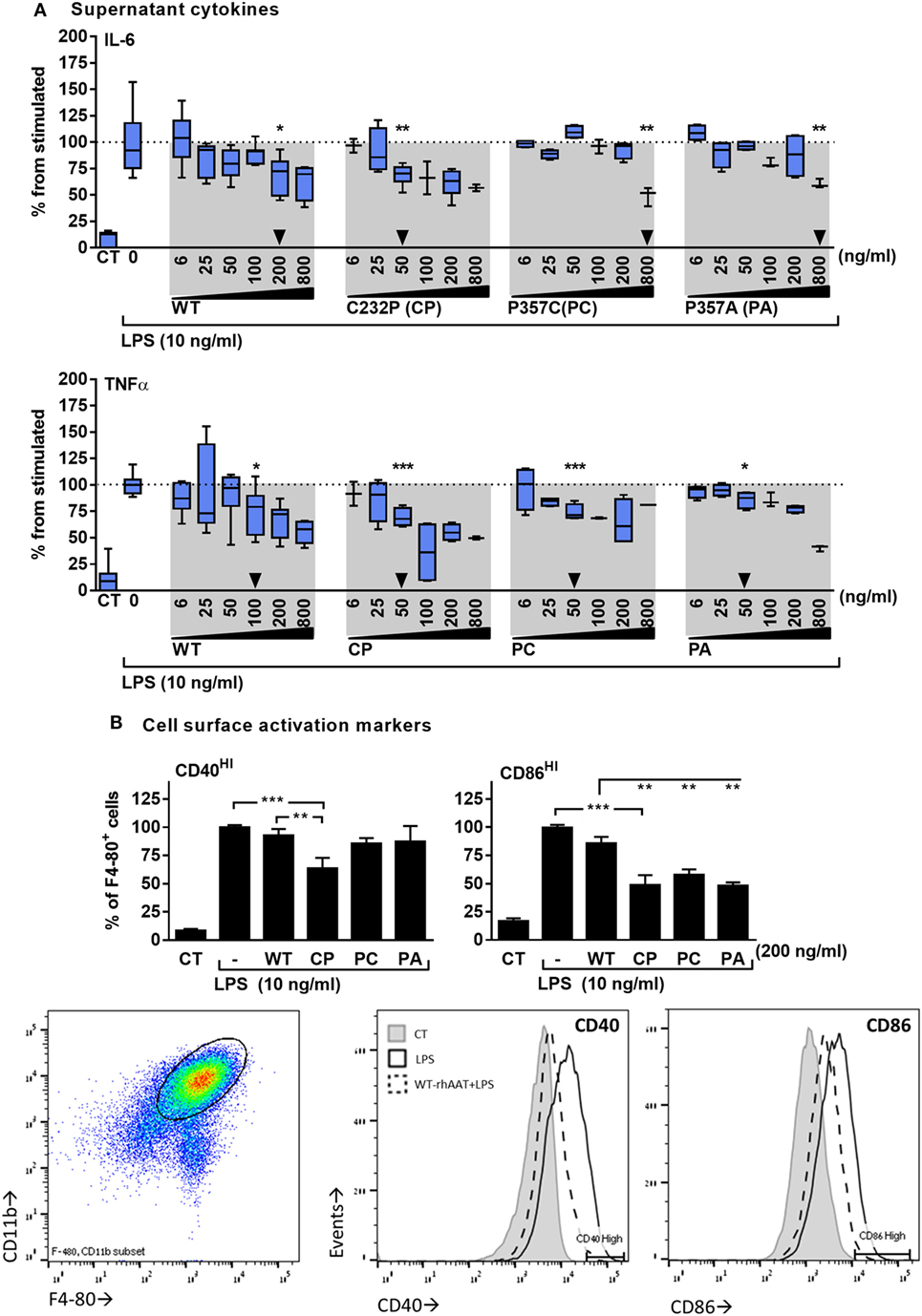
Figure 2. rhAAT anti-inflammatory potency variations on leukocyte LPS-responses. (A) Cytokine release, bone marrow-derived macrophages (BMDM) (2 × 105 per well) overnight incubation with complete medium containing indicated doses of rhAAT followed by PBS wash and re-incubation with complete medium containing LPS (10 ng/ml, 24 h). Supernatants analysis for IL-6 and TNFα concentrations by specific ELISA. Triangle, first statistically significant difference from stimulated sample. (B) Membranal activation markers, BMDM (5 × 105 per well) overnight incubation with complete medium containing 200 ng/ml rhAAT, followed by LPS addition (10 ng/ml, 24 h). Flow cytometric analysis for CD40HI and CD86HI. Gate, CD11b+/F4-80+ cells. Data representative of four to five independent experimental repeats. Mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
Based on these observations, the concentration of 200 ng/ml was used for evaluating the effect of rhAAT variants on CD40 and CD86 surface expression (Figure 2B). As shown, changes in CD40 and CD86 displayed a pattern similar to that of released inflammatory cytokines: at 200 ng/ml, WT-rhAAT was ineffective in reducing CD40HI or CD86HI cell population proportions, while CP pretreatment resulted in significant reduction in CD40HI and CD86HI cell populations (36 and 51%, respectively). CD86 was responsive to PC and PA, exhibiting a reduction of 42 and 51%, respectively, as opposed to CD40 (14 and 13%, respectively).
In the absence of LPS, WT-rhAAT did not elevate IL-6 and TNFα supernatant levels nor the expression of CD40 and CD86 compared to non-exposed cells (data not shown).
In vivo, the effect of rhAAT on leukocyte responses to LPS was evaluated in a peritoneal LPS-induced sterile inflammatory model. In this model, activated infiltrating monocytes are readily depicted upon peritoneal lavage. Here, monocytes were characterized by staining for F4-80 and CD11b and then further tested for the proportion of co-stimulatory activation. As shown in Figure 3A, animals pretreated with WT-rhAAT exhibited a 36% reduction in CD11b+ F4-80+ cell population size compared to the LPS group (set at 100%). While pretreatment with CP or PA led to a 21 and 29% reduction in elicited CD11b+ F4-80+ cell population, respectively, pretreatment with PC was ineffective in altering cell subtype ratio. The degree of activation of CD11b+ F4-80+ cells (Figure 3B) depicted a rise in LPS-induced CD40HI cells to 24% of CD11b+ F4-80+ cells, while pretreatment with WT-rhAAT resulted in a reduction of the CD40HI population to 13% of CD11b+ F4-80+ cells, similar to the 12% observed by PC pretreatment. In contrast, pretreatment with CP resulted in a greater decline in the proportion of CD40HI cells to 5% of the LPS group, while PA was ineffective in altering the inducible profile of CD40HI on cells. Compared to control untreated animals, CD86HI cell population size was unaffected by in vivo LPS stimulation. Nonetheless, significant reductions in CD86HI cell population size were observed under pretreatment with WT, CP, PC and PA rhAAT (31, 29, 34, and 34%, respectively).
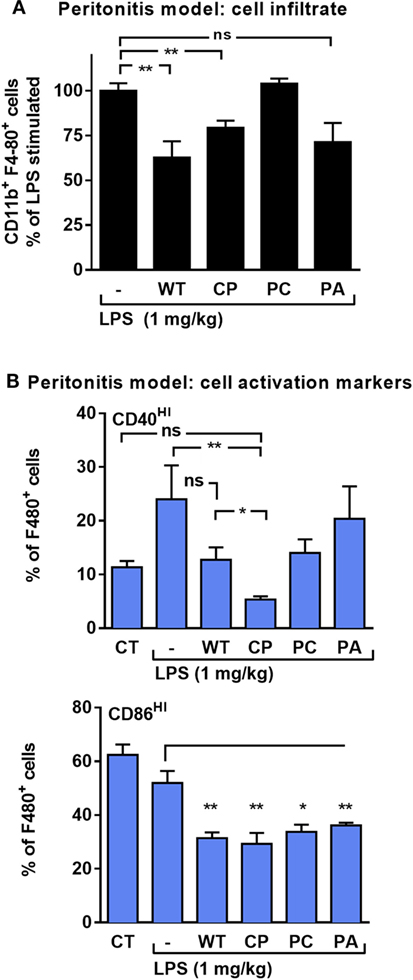
Figure 3. rhAAT anti-inflammatory potency variations in sterile peritonitis in vivo models. C57BL/6 mice (n = 5 per group) injected with rhAAT (50 μg per mouse) i.p. and 3 h afterward, LPS (1 mg/kg). Peritoneal lavage performed 24 h post LPS injection. Flow cytometric analysis for (A) CD11b+ F4-80+, % of LPS-stimulated (B) CD40HI and CD86HI, Gate, CD11b+ F4-80+. CT, PBS injection. Mean ± SEM, *p < 0.05, **p < 0.01 compared to LPS-stimulated group.
Unique Pharmacokinetics of the CP Variant
Half-life and distribution volume for each rhAAT variant were calculated based on time-dependent circulating hAAT concentrations, as determined in mice injected with each rhAAT variant (50 μg, i.v.). As shown in Figure 4, the kinetics of the circulating recombinant forms appears uniform between WT, PC, and PA. However, the levels of circulating CP were 7.13 ± 0.08-fold lower than in WT-rhAAT at as early as 1 h after injection (Figure 4A). Accordingly, its distribution volume was calculated to be 9.5 ± 3.0-fold greater than that of WT-rhAAT (Figure 4B), and its half-life significantly extended (Figure 4C).
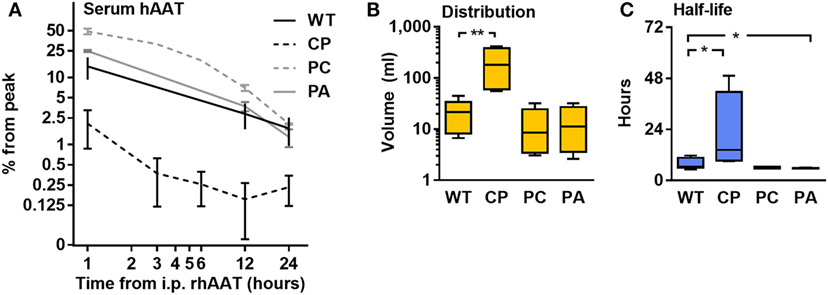
Figure 4. Pharmacokinetics. C57BL/6 mice (n = 5 per group) were injected with rhAAT i.v. human α1-antitrypsin (hAAT) concentration analysis by species-specific ELISA from serum samples (1, 12, 24 h). (A) hAAT serum concentrations, Mean ± SEM. (B) Calculated distribution volume. (C) Calculated half-life time. Data representative of three independent experimental repeats., *p < 0.05, **p < 0.01 compared to wild-type (WT).
TNFα Expression, Production, and Release
LPS-stimulated RAW264.7 cells were pretreated with rhAAT variants and several aspects of TNFα expression were determined (Figure 5A transcription, Figure 5B membrane-associated, Figures 5C and 5D soluble form in supernatant and serum in vivo, respectively). As shown, LPS had induced a spike in relative TNFα transcript levels (Figure 5A, shaded); accordingly, LPS-treated cultured primary peritoneal cells displayed a rise in TNFα release (Figure 5C). Animals injected with LPS exhibited a time-dependent rise in serum TNFα levels (Figure 5D, shaded). Membrane-associated TNFα levels (Figure 5B) displayed no significant change upon LPS stimulation, agreeing with the anticipated dynamic of ADAM17-dependent cleavage of membrane-associated TNFα during inflammatory conditions. Unexpectedly, pretreatment with WT-rhAAT resulted in a significant rise in relative TNFα transcript levels 1-h poststimulation (1.53 ± 0.12-fold from LPS alone, Figure 5A), coupled with a decline in serum TNFα levels 3 h poststimulation (1.5 ± 0.02-fold lower than LPS alone, Figure 5D).
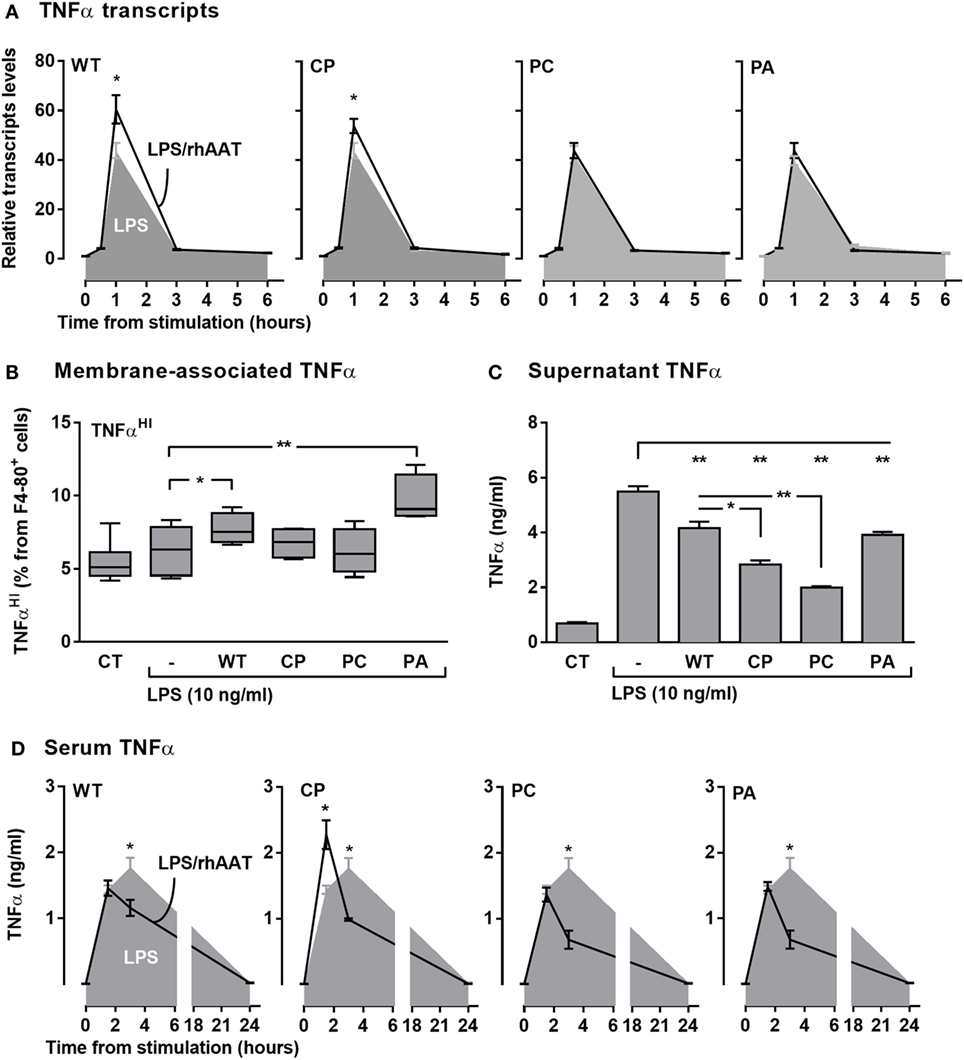
Figure 5. rhAAT effect potency variation on TNFα transcription, expression, and release. (A) TNFα transcription, Raw 264.7 cells (0.5 × 106 per well) overnight incubation with complete medium containing 200 ng/ml rhAAT, followed by LPS addition (10 ng/ml, 24 h), nucleic acids extracted (0.5, 1, 3, 6 h) and TNFα transcription assessed by qPCR. Results presented as fold from control. (B,C) Peritoneal macrophages (3 × 105 per well) overnight incubation with complete medium containing 200 ng/ml rhAAT, followed by LPS addition (10 ng/ml, 24 h). (B) Flow cytometric analysis for membrane-associated TNFα. Gate, CD11b+. (C) Supernatant TNFα analysis by specific ELISA. (D) In vivo sterile peritonitis model serum TNFα levels. C57BL/6 mice (n = 5 per group) injected with PBS or rhAAT (50 μg/mouse) i.p. and 3 h afterward, LPS (1 mg/kg). TNFα serum analysis by ELISA (1.5, 3, and 24 h). CT, non-stimulated cells. Mean ± SEM. Data representative of two independent experimental repeats. *p < 0.05, **p < 0.01 Compared to LPS-stimulated group.
Interestingly, while the three rhAAT variants displayed no significant effect on LPS-induced TNFα transcription levels (Figure 5A), pretreatment with CP resulted in a significant earlier narrow spike in serum TNFα levels (Figure 5D). PC displayed a pattern of inhibition similar to that of WT-rhAAT, and PA did not exert a significant effect on LPS-stimulated TNFα transcript levels nor on serum levels. In vitro (Figure 5C), the levels of released TNFα levels were consistent with in vivo findings in that treatment with WT, CP, PC, and PA caused a decline in soluble TNFα concentrations, most effectively by CP and PC variants.
Expression of TNFα without the emergence of its soluble form could be caused by inhibition of ADAM17 activity (43); such a process is expected to result in elevated levels of membrane-associated non-cleaved TNFα. As shown in Figure 5B, membrane-associated TNFα levels were evaluated after pretreatment with each of the rhAAT variants. Pretreatment with WT-rhAAT resulted in a significant increase in membrane-associated TNFα, corresponding to 24% lower soluble TNFα levels under the same conditions (Figure 5C). Membrane-associated and soluble TNFα levels responded differentially to the various variants; while the membranous effect of CP and PC seemed to be minimal, soluble TNFα levels were reduced by 48 and 63%, respectively. Interestingly, PA was the sole variant which resulted in a significant rise in membrane-associated TNFα levels. However, this rise was coupled with only a 29% decrease in soluble TNFα level, similar to the change observed under WT-rhAAT pretreatment. The overall effect of CP pretreatment, compared to WT-rhAAT pretreatment, appears to involve a minor shift in transcript levels and rapidly declining soluble TNFα levels with only a minimal change in membrane-associated TNFα levels; at the same time, the first evidence of circulating TNFα in vivo is pushed up to the 1-h region, temporarily exhibiting serum TNFα levels higher than those encountered in LPS treatment alone.
Discussion
Recent years have witnessed an expansion of potential clinical applications for hAAT treatment beyond that of straightforward augmentation therapy for genetic hAAT deficiency; these include type 1 diabetes (37, 50–52), allogeneic and xenogeneic transplants (44–46, 53, 54), graft-versus-host disease (53, 55, 56), acute myocardial infarction (57–59), inflammatory bowel disease (52, 60), rheumatoid arthritis (61–63), multiple sclerosis (41), and osteoporosis (64, 65). Collectively, these represent an extension of earlier preclinical studies that portray hAAT as possessing anti-inflammatory and immunoregulatory properties (3, 4, 26, 35).
Unexpectedly, a major part of the anti-inflammatory and immunoregulatory properties of hAAT were shown to be independent of protease inhibition (34–36). Evidence for several molecular binding partners is presently on the rise, supporting the possibility that the globular surface of hAAT is relevant to its anti-inflammatory functions. It is, therefore, timely that the functionality of molecular attributes outside the protease inhibitory region of hAAT be investigated. Such an effort coincides with three decades of attempts to generate straightforward clinical-grade recombinant WT-hAAT for AATD augmentation therapy (36, 66–76). The relevance of non-RCL hAAT segments has been addressed in the past primarily in the context of aberrant hAAT aggregation in AATD (14); little attention is drawn to any specific functional significance to non-RCL segments in the molecule. Thus, we suggest that the development of recombinant hAAT may benefit from a better understanding of its non-RCL-related biology, as it may host biologically important anti-inflammatory and immunomodulatory activities.
This study examines the structure–function relationships of hAAT using recombinant proteins. While recombinant proteins are known to vary from their biological counterparts in multiple aspects, e.g., posttranslational modifications, it is important to note that native hAAT varies between individuals, particularly with regards to their glycosylation patterns (1, 66, 67, 77). Moreover, factory release criteria for clinical-grade hAAT do not require structural homogenicity, introducing a large number of isoforms to experiments that utilize clinical-grade hAAT. Thus, the use of recombinant protein technology in the present experimental design facilitates an evaluation of molecularly uniform sequence-modified species of hAAT.
In the present study, we revisit a previously reported intra-RCL mutation (P357C); this mutation has been established as lacking anti-elastase activity, but was never evaluated for anti-inflammatory or immunomodulatory functions. Here, the functionality of a recombinant hAAT that bares this mutation is compared to an amino acid switch at the same position (P357A). Per our findings, both variants were compared to recombinant hAAT formulation that bare a mutation outside the RCL: cysteine 232 was replaced with proline (C232P).
As expected, disruption of the primary sequence of the RCL indeed nullified elastase inhibition. Interestingly, the mutation outside the RCL, C232P, also resulted in lack of elastase inhibitory capacity. Based on structural prediction of naïve hAAT, it is noted that position 232 is relatively proximal to the RCL and thus may partake in the conformational changes associated with protease binding. Nevertheless, it represents a variant of hAAT that, like PC and PA, fails to inhibit elastase and is thus of interest for studying elastase-independent anti-inflammatory and immunomodulatory molecular aspects of hAAT.
Similar to plasma-derived hAAT, the anti-inflammatory qualities of recombinant WT-hAAT have been established in other studies (26, 34, 36, 68). However, in the present study, variations surfaced in the anti-inflammatory potency between recombinant variants of hAAT, most notably in the case of the extra-RCL mutant, CP. In this mutated recombinant form of hAAT, greater anti-inflammatory effects were consistently observed. For example, pretreatment of cells with CP-hAAT resulted in reduced supernatant levels of both TNFα and IL-6 at lower concentrations than those required by recombinant WT-hAAT, and in a concentration-dependent manner. Additionally, at a uniform concentration of 200 ng/ml of recombinant formulations of hAAT, CP-hAAT was more potent than the other formulations with regards to changes in cell activation markers, in vitro. In vivo, CP-hAAT treatment resulted in LPS-stimulated animals displaying 2.4-fold lower proportions of CD40HI cells, than in the LPS-stimulated WT-hAAT-treated group.
While some mutations may result in reduced half-life time due to unexpected binding partners, as well as changes in distribution volume and altered susceptibility to proteolytic attacks, the study of hAAT variant pharmacokinetics indicates that WT, PC, and PA share similar properties. In contrast, CP presented with a larger distribution volume and an extended half-life. While these results increase our confidence that the novel variants share other pharmacokinetic qualities to WT, the possibility of unique binding partners should be addressed in future studies.
In regard to CP increased distribution volume and half-life, since these distinctions appear to accompany this variant’s enhanced anti-inflammatory capacity, it is suggested that its physical properties render it functionally unique. It has been shown that exogenously administered hAAT is readily detected on the surface of activated immune cells (78), coinciding with observations by Subramaniyam et al., in which hAAT localizes on membrane lipid rafts (29). This property is in agreement with evidence for direct binding of oxidized cholesterol (31) and fatty acids (79) by hAAT, and suggests that hAAT membranal presence might serve as a platform for several of its functional attributes (80–82). Considering the abrupt structural change that a proline is predicted to exert at position 232, it is possible that the structural properties of CP allow it to better adhere to cell membranes, accounting for the rapid decline in serum levels of its soluble form on the one hand, and its significantly prolonged half-life on the other. Given that CD40 and CD86 signal transduction requires surface di- or -trimerization events, and that TNFα release is dependent on an intact surface protease, it is possible that increased membranal presence of CP might disrupt these processes. Specific studies are required in order to confirm this hypothesis.
Particular attention was relegated to TNFα in the present study. TNFα is one of the most consistent responders to hAAT treatment across inflammatory models (4, 83). Given that soluble TNFα levels are the result of transcription, expression, and proteolytic shedding processes, we sought to investigate the mechanisms by which hAAT treatment facilitates a reduction in the inducible levels of soluble TNFα. In the present study, the effect of each of the rhAAT variants over TNFα transcript levels, as well as membrane-associated TNFα and soluble supernatant/serum levels, were assessed. Our findings show that, as expected, inducible serum TNFα levels are reduced under WT-rhAAT treatment in vivo, consistent with other studies (84, 85). However, the effect of the variants varied: PA treatment resulted in minimal changes compared to the non-treated group, while both CP and PC treatments resulted in a sharp decline in TNFα levels. Intriguingly, CP treatment resulted in an isolated spike in TNFα levels at the 90-min time-point. This latter finding is in accordance with a study by Janciauskiene et al., in which a brief treatment of human monocytes with hAAT in vitro, results in a short-lived elevation in TNFα, IL-1β and IL-8 levels (86), an overt inflammatory response. While pretreatment of peritoneal macrophages with any of the rhAAT variants indeed resulted in reduced soluble TNFα levels within 24 h, only two variants, WT and PA, resulted in a detectable change in inducible membrane-associated TNFα levels. Considering that IL-6 secretion is ADAM17-independent, it is interesting to note that exposure to increasing WT or CP concentrations, but not PC or PA, resulted in similar IL-6 and TNFα supernatant concentrations, hinting at a more elaborate anti-inflammatory mechanism than the otherwise anticipated anti-proteolytic aspect of hAAT. Regarding intracellular expression of TNFα and its transport to the cellular membrane, an unexpected outcome was observed; pretreatment of cells with either WT-rhAAT or any of the other three hAAT variants not only did not reduce TNFα transcripts levels, but, in the cases of WT-rhAAT and CP treated cells, TNFα mRNA transcript levels increased. Collectively, the findings suggest that the effect of hAAT on the production and shedding of TNFα from leukocytes may involve multiple structural domains on hAAT, altogether irrespective of the RCL, affecting both intra- and extracellular elements in TNFα levels. In support of this possibility, antithrombin III, which shares structural homology to hAAT outside the RCL, has been shown to produce several overlapping outcomes to some of those obtained using hAAT (87, 88), including reduced LPS-stimulated TNFα release in monocytes (89).
While the majority of structural studies on hAAT focus on AATD-related aspects of the molecule, such as the occurrence of naturally occurring mutations, their potential to aggregate and their anti-proteolytic qualities, our data suggest that the non-RCL sections on the generous surface of hAAT may be of relevance to novel modifications for the purpose of enhanced functionality. Future potentiated variants of hAAT with longer half-life may be pharmaceutically attractive in as far as the renowned low patient compliance to repeated i.v. infusions, potentially offering future recombinant regimen that achieves the desired outcomes of hAAT at several-fold lower concentrations of the molecule. As such, one of the major limitation of introducing large volumes of hAAT may be lifted, promoting the exploration of subcutaneous hAAT treatment in humans.
Further studies are required in order to fully explore the immunological effects of the described structural variations of hAAT, including their potential clinical applicability for employing specific enhanced qualities of hAAT, such as half-life or anti-inflammatory potency. The outcomes hereby, once further investigated and developed, may be translated into the design of context- and disease-oriented therapeutic hAAT variants.
Ethics Statement
All experiments involving animals conducted in this study were approved by the Ben-Gurion University of the Negev Animal Care and Use Committee.
Author Contributions
YL: protein design, in vitro and in vivo immunological experiments design, execution and analysis, and writing. MZ: protein design and fabrication and purification. DO: protein design, in vitro and in vivo immunological experiments design, execution, and analysis. DL: protein fabrication and purification. BB: in vitro and in vivo immunological experiments execution. RS: in vitro and in vivo immunological experiments execution. AA: protein design and fabrication and purification, and writing. EL: mentoring, experimental design and analysis, and writing.
Conflict of Interest Statement
All authors declare no personal, professional, or financial conflict of interest present at the conduction of this work.
Acknowledgments
The authors thank Dr. Sofiya Kolusheva from the Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, for her instrumental advice regarding structure analysis. The authors also wish to thank Mrs. Valeria Frishman for her invaluable technical support.
Funding
This research was funded by Ben-Gurion University of the Negev.
References
1. Kolarich D, Weber A, Turecek PL, Schwarz HP, Altmann F. Comprehensive glyco-proteomic analysis of human alpha1-antitrypsin and its charge isoforms. Proteomics (2006) 6(11):3369–80. doi:10.1002/pmic.200500751
2. Kalsheker N. Alpha 1-antitrypsin: structure, function and molecular biology of the gene. Biosci Rep (1989) 9(2):129–38. doi:10.1007/BF01115992
3. Hunt JM, Tuder R. Alpha 1 anti-trypsin: one protein, many functions. Curr Mol Med (2012) 12(7):827–35. doi:10.2174/156652412801318755
4. Guttman O, Baranovski BM, Schuster R, Kaner Z, Freixo-Lima GS, Bahar N, et al. Acute-phase protein alpha1-anti-trypsin: diverting injurious innate and adaptive immune responses from non-authentic threats. Clin Exp Immunol (2015) 179(2):161–72. doi:10.1111/cei.12476
5. Townsend SA, Edgar RG, Ellis PR, Kantas D, Newsome PN, Turner AM. Systematic review: the natural history of alpha-1 antitrypsin deficiency, and associated liver disease. Aliment Pharmacol Ther (2018) 47(7):877–85. doi:10.1111/apt.14537
6. Chapman KR, Chorostowska-Wynimko J, Koczulla AR, Ferrarotti I, McElvaney NG. Alpha 1 antitrypsin to treat lung disease in alpha 1 antitrypsin deficiency: recent developments and clinical implications. Int J Chron Obstruct Pulmon Dis (2018) 13:419–32. doi:10.2147/COPD.S149429
7. Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med (2009) 360(26):2749–57. doi:10.1056/NEJMcp0900449
8. Cowden DI, Fisher GE, Weeks RL. A pilot study comparing the purity, functionality and isoform composition of alpha-1-proteinase inhibitor (human) products. Curr Med Res Opin (2005) 21(6):877–83. doi:10.1185/030079905X46395
9. Chiuchiolo MJ, Crystal RG. Gene therapy for alpha-1 antitrypsin deficiency lung disease. Ann Am Thorac Soc (2016) 13(Suppl 4):S352–69. doi:10.1513/AnnalsATS.201506-344KV
10. Mueller C, Flotte TR. Gene-based therapy for alpha-1 antitrypsin deficiency. COPD (2013) 10(Suppl 1):44–9. doi:10.3109/15412555.2013.764978
11. Hubbard RC, Crystal RG. Alpha-1-antitrypsin augmentation therapy for alpha-1-antitrypsin deficiency. Am J Med (1988) 84(6A):52–62. doi:10.1016/0002-9343(88)90159-3
12. Kang UB, Baek JH, Ryu SH, Kim J, Yu MH, Lee C. Kinetic mechanism of protease inhibition by alpha1-antitrypsin. Biochem Biophys Res Commun (2004) 323(2):409–15. doi:10.1016/j.bbrc.2004.08.105
13. Duranton J, Bieth JG. Inhibition of proteinase 3 by [alpha]1-antitrypsin in vitro predicts very fast inhibition in vivo. Am J Respir Cell Mol Biol (2003) 29(1):57–61. doi:10.1165/rcmb.2002-0258OC
14. Krishnan B, Gierasch LM. Dynamic local unfolding in the serpin alpha-1 antitrypsin provides a mechanism for loop insertion and polymerization. Nat Struct Mol Biol (2011) 18(2):222–6. doi:10.1038/nsmb.1976
15. Koepke J, Dresel M, Schmid S, Greulich T, Beutel B, Schmeck B, et al. Therapy with plasma purified alpha1-antitrypsin (Prolastin(R)) induces time-dependent changes in plasma levels of MMP-9 and MPO. PLoS One (2015) 10(1):e0117497. doi:10.1371/journal.pone.0117497
16. Han YP, Yan C, Garner WL. Proteolytic activation of matrix metalloproteinase-9 in skin wound healing is inhibited by alpha-1-antichymotrypsin. J Invest Dermatol (2008) 128(9):2334–42. doi:10.1038/jid.2008.77
17. Churg A, Dai J, Zay K, Karsan A, Hendricks R, Yee C, et al. Alpha-1-antitrypsin and a broad spectrum metalloprotease inhibitor, RS113456, have similar acute anti-inflammatory effects. Lab Invest (2001) 81(8):1119–31. doi:10.1038/labinvest.3780324
18. Yoshida K, Suzuki Y, Saito A, Fukuda K, Hamanishi C, Munakata H. Aggrecanase-1 (ADAMTS-4) interacts with alpha1-antitrypsin. Biochim Biophys Acta (2005) 1725(2):152–9. doi:10.1016/j.bbagen.2005.06.009
19. Gold M, Koczulla AR, Mengel D, Koepke J, Dodel R, Dontcheva G, et al. Reduction of glutamate-induced excitotoxicity in murine primary neurons involving calpain inhibition. J Neurol Sci (2015) 359(1–2):356–62. doi:10.1016/j.jns.2015.11.016
20. Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, et al. alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol (2006) 169(4):1155–66. doi:10.2353/ajpath.2006.060058
21. Tamura K, Takashima H, Fumoto K, Kajihara T, Uchino S, Ishihara O, et al. Possible role of alpha1-antitrypsin in endometriosis-like grafts from a mouse model of endometriosis. Reprod Sci (2015) 22(9):1088–97. doi:10.1177/1933719115570901
22. Bergin DA, Reeves EP, Meleady P, Henry M, McElvaney OJ, Carroll TP, et al. alpha-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest (2010) 120(12):4236–50. doi:10.1172/JCI41196
23. Eckman EA, Mallender WD, Szegletes T, Silski CL, Schreiber JR, Davis PB, et al. In vitro transport of active alpha(1)-antitrypsin to the apical surface of epithelia by targeting the polymeric immunoglobulin receptor. Am J Respir Cell Mol Biol (1999) 21(2):246–52. doi:10.1165/ajrcmb.21.2.3687
24. Ochayon DE, Mizrahi M, Shahaf G, Baranovski BM, Lewis EC. Human alpha1-antitrypsin binds to heat-shock protein gp96 and protects from endogenous gp96-mediated injury in vivo. Front Immunol (2013) 4:320. doi:10.3389/fimmu.2013.00320
25. Finotti P, Pagetta A. A heat shock protein70 fusion protein with alpha1-antitrypsin in plasma of type 1 diabetic subjects. Biochem Biophys Res Commun (2004) 315(2):297–305. doi:10.1016/j.bbrc.2004.01.058
26. Janciauskiene S, Welte T. Well-known and less well-known functions of alpha-1 antitrypsin. Its role in chronic obstructive pulmonary disease and other disease developments. Ann Am Thorac Soc (2016) 13(Suppl 4):S280–8. doi:10.1513/AnnalsATS.201507-468KV
27. Mashiba S, Wada Y, Takeya M, Sugiyama A, Hamakubo T, Nakamura A, et al. In vivo complex formation of oxidized alpha(1)-antitrypsin and LDL. Arterioscler Thromb Vasc Biol (2001) 21(11):1801–8. doi:10.1161/hq1101.098232
28. Talmud PJ, Martin S, Steiner G, Flavell DM, Whitehouse DB, Nagl S, et al. Progression of atherosclerosis is associated with variation in the alpha1-antitrypsin gene. Arterioscler Thromb Vasc Biol (2003) 23(4):644–9. doi:10.1161/01.ATV.0000065196.61663.8D
29. Subramaniyam D, Zhou H, Liang M, Welte T, Mahadeva R, Janciauskiene S. Cholesterol rich lipid raft microdomains are gateway for acute phase protein, SERPINA1. Int J Biochem Cell Biol (2010) 42(9):1562–70. doi:10.1016/j.biocel.2010.06.009
30. Gordon SM, McKenzie B, Kemeh G, Sampson M, Perl S, Young NS, et al. Rosuvastatin alters the proteome of high density lipoproteins: generation of alpha-1-antitrypsin enriched particles with anti-inflammatory properties. Mol Cell Proteomics (2015) 14(12):3247–57. doi:10.1074/mcp.M115.054031
31. Kotani K, Yamada T, Taniguchi N. The association between adiponectin, HDL-cholesterol and alpha1-antitrypsin-LDL in female subjects without metabolic syndrome. Lipids Health Dis (2010) 9:147. doi:10.1186/1476-511X-9-147
32. Moreno JA, Ortega-Gomez A, Rubio-Navarro A, Louedec L, Ho-Tin-Noe B, Caligiuri G, et al. High-density lipoproteins potentiate alpha1-antitrypsin therapy in elastase-induced pulmonary emphysema. Am J Respir Cell Mol Biol (2014) 51(4):536–49. doi:10.1165/rcmb.2013-0103OC
33. Strickland DK, Muratoglu SC, Antalis TM. Serpin-enzyme receptors LDL receptor-related protein 1. Methods Enzymol (2011) 499:17–31. doi:10.1016/B978-0-12-386471-0.00002-X
34. Jonigk D, Al-Omari M, Maegel L, Muller M, Izykowski N, Hong J, et al. Anti-inflammatory and immunomodulatory properties of alpha1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci U S A (2013) 110(37):15007–12. doi:10.1073/pnas.1309648110
35. Lior Y, Geyra A, Lewis EC. Therapeutic compositions and uses of alpha1-antitrypsin: a patent review (2012–2015). Expert Opin Ther Pat (2016) 26(5):581–9. doi:10.1517/13543776.2016.1165210
36. Lee S, Lee Y, Hong K, Hong J, Bae S, Choi J, et al. Effect of recombinant alpha1-antitrypsin Fc-fused (AAT-Fc)protein on the inhibition of inflammatory cytokine production and streptozotocin-induced diabetes. Mol Med (2013) 19:65–71. doi:10.2119/molmed.2012.00308
37. Ozeri E, Mizrahi M, Shahaf G, Lewis EC. alpha-1 antitrypsin promotes semimature, IL-10-producing and readily migrating tolerogenic dendritic cells. J Immunol (2012) 189(1):146–53. doi:10.4049/jimmunol.1101340
38. Abecassis A, Schuster R, Shahaf G, Ozeri E, Green R, Ochayon DE, et al. alpha1-antitrypsin increases interleukin-1 receptor antagonist production during pancreatic islet graft transplantation. Cell Mol Immunol (2014) 11(4):377–86. doi:10.1038/cmi.2014.17
39. Bergin DA, Reeves EP, Hurley K, Wolfe R, Jameel R, Fitzgerald S, et al. The circulating proteinase inhibitor alpha-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med (2014) 6(217):217ra1. doi:10.1126/scitranslmed.3007116
40. Geraghty P, Eden E, Pillai M, Campos M, McElvaney NG, Foronjy RF. alpha1-antitrypsin activates protein phosphatase 2A to counter lung inflammatory responses. Am J Respir Crit Care Med (2014) 190(11):1229–42. doi:10.1164/rccm.201405-0872OC
41. Subramanian S, Shahaf G, Ozeri E, Miller LM, Vandenbark AA, Lewis EC, et al. Sustained expression of circulating human alpha-1 antitrypsin reduces inflammation, increases CD4+FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab Brain Dis (2011) 26(2):107–13. doi:10.1007/s11011-011-9239-9
42. Lockett AD, Kimani S, Ddungu G, Wrenger S, Tuder RM, Janciauskiene SM, et al. alpha(1)-antitrypsin modulates lung endothelial cell inflammatory responses to TNF-alpha. Am J Respir Cell Mol Biol (2013) 49(1):143–50. doi:10.1165/rcmb.2012-0515OC
43. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature (1997) 385(6618):729–33. doi:10.1038/385729a0
44. Chen G, Li J, Chen L, Lai X, Qiu J. alpha1-antitrypsin-primed tolerogenic dendritic cells prolong allograft kidney transplants survival in rats. Int Immunopharmacol (2016) 31:216–21. doi:10.1016/j.intimp.2015.12.038
45. Mizrahi M, Cal P, Rosenthal M, Ochayon D, Shahaf G, Kaner Z, et al. Human alpha1-antitrypsin modifies B-lymphocyte responses during allograft transplantation. Immunology (2013) 140(3):362–73. doi:10.1111/imm.12149
46. Ashkenazi E, Baranovski BM, Shahaf G, Lewis EC. Pancreatic islet xenograft survival in mice is extended by a combination of alpha-1-antitrypsin and single-dose anti-CD4/CD8 therapy. PLoS One (2013) 8(5):e63625. doi:10.1371/journal.pone.0063625
47. Lewis EC, Shapiro L, Bowers OJ, Dinarello CA. Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci U S A (2005) 102(34):12153–8. doi:10.1073/pnas.0505579102
48. Stephens AW, Thalley BS, Hirs CH. Antithrombin-III Denver, a reactive site variant. J Biol Chem (1987) 262(3):1044–8.
49. Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed (2010) 99(3):306–14. doi:10.1016/j.cmpb.2010.01.007
50. ClinicalTrials.gov. Study of the Safety and Efficacy of Intravenous Alpha-1 Antitrypsin in Type 1 Diabetes Mellitus. Bethesda, MD: National Library of Medicine (US) (2016). Available from: http://clinicaltrials.gov/ct/show/NCT00287391?order=1 (Accessed: March 31, 2018).
51. Fleixo-Lima G, Ventura H, Medini M, Bar L, Strauss P, Lewis EC. Mechanistic evidence in support of alpha1-antitrypsin as a therapeutic approach for type 1 diabetes. J Diabetes Sci Technol (2014) 8(6):1193–203. doi:10.1177/1932296814547096
52. Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, et al. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta-cell autoimmunity in patients with type 1 diabetes. Diabetes (2014) 63(12):4239–48. doi:10.2337/db14-0480
53. Goldstein S, Reddy P. Tolerance without toxicity? Alpha1-antitrypsin as a novel alternative to immunosuppression. Expert Rev Clin Immunol (2012) 8(5):397–9. doi:10.1586/eci.12.33
54. Bellacen K, Kalay N, Ozeri E, Shahaf G, Lewis EC. Revascularization of pancreatic islet allografts is enhanced by alpha-1-antitrypsin under anti-inflammatory conditions. Cell Transplant (2013) 22(11):2119–33. doi:10.3727/096368912X657701
55. Marcondes AM, Karoopongse E, Lesnikova M, Margineantu D, Welte T, Dinarello CA, et al. alpha-1-Antitrypsin (AAT)-modified donor cells suppress GVHD but enhance the GVL effect: a role for mitochondrial bioenergetics. Blood (2014) 124(18):2881–91. doi:10.1182/blood-2014-04-570440
56. Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, Nieves E, et al. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci U S A (2012) 109(2):564–9. doi:10.1073/pnas.1117665109
57. Gilutz H, Siegel Y, Paran E, Cristal N, Quastel MR. Alpha 1-antitrypsin in acute myocardial infarction. Br Heart J (1983) 49(1):26–9. doi:10.1136/hrt.49.1.26
58. Toldo S, Seropian IM, Mezzaroma E, Van Tassell BW, Salloum FN, Lewis EC, et al. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury. J Mol Cell Cardiol (2011) 51(2):244–51. doi:10.1016/j.yjmcc.2011.05.003
59. Abbate A, Van Tassell BW, Christopher S, Abouzaki NA, Sonnino C, Oddi C, et al. Effects of prolastin C (plasma-derived alpha-1 antitrypsin) on the acute inflammatory response in patients with ST-segment elevation myocardial infarction (from the VCU-alpha 1-RT pilot study). Am J Cardiol (2015) 115(1):8–12. doi:10.1016/j.amjcard.2014.09.043
60. Yang P, Tremaine WJ, Meyer RL, Prakash UB. Alpha1-antitrypsin deficiency and inflammatory bowel diseases. Mayo Clin Proc (2000) 75(5):450–5. doi:10.1016/S0025-6196(11)64212-2
61. Grimstein C, Choi YK, Wasserfall CH, Satoh M, Atkinson MA, Brantly ML, et al. Alpha-1 antitrypsin protein and gene therapies decrease autoimmunity and delay arthritis development in mouse model. J Transl Med (2011) 9:21. doi:10.1186/1479-5876-9-21
62. Abboud RT, Chalmers A, Gofton JP, Richter AM, Enarson DA. Relationship between severity of rheumatoid arthritis and serum alpha 1-antitrypsin. J Rheumatol (1991) 18(10):1490–5.
63. Cox DW, Huber O. Association of severe rheumatoid arthritis with heterozygosity for alpha 1-antitrypsin deficiency. Clin Genet (1980) 17(2):153–60. doi:10.1111/j.1399-0004.1980.tb00125.x
64. Akbar MA, Lu Y, Elshikha AS, Chen MJ, Yuan Y, Whitley EM, et al. Transplantation of adipose tissue-derived mesenchymal stem cell (ATMSC) expressing alpha-1 antitrypsin reduces bone loss in ovariectomized osteoporosis mice. Hum Gene Ther (2017) 28(2):179–89. doi:10.1089/hum.2016.069
65. Akbar MA, Nardo D, Chen MJ, Elshikha AS, Ahamed R, Elsayed EM, et al. Alpha-1 antitrypsin inhibits RANKL-induced osteoclast formation and functions. Mol Med (2017) 23:57–69. doi:10.2119/molmed.2016.00170
66. Chung HS, Kim JS, Lee SM, Park SJ. Additional N-glycosylation in the N-terminal region of recombinant human alpha-1 antitrypsin enhances the circulatory half-life in Sprague-Dawley rats. Glycoconj J (2016) 33(2):201–8. doi:10.1007/s10719-016-9657-3
67. Jaberie H, Naghibalhossaini F. Recombinant production of native human alpha-1-antitrypsin protein in the liver HepG2 cells. Biotechnol Lett (2016) 38(10):1683–90. doi:10.1007/s10529-016-2150-z
68. Karnaukhova E, Ophir Y, Golding B. Recombinant human alpha-1 proteinase inhibitor: towards therapeutic use. Amino Acids (2006) 30(4):317–32. doi:10.1007/s00726-005-0324-4
69. Bischoff R, Speck D, Lepage P, Delatre L, Ledoux C, Brown SW, et al. Purification and biochemical characterization of recombinant alpha 1-antitrypsin variants expressed in Escherichia coli. Biochemistry (1991) 30(14):3464–72. doi:10.1021/bi00228a017
70. Casolaro MA, Fells G, Wewers M, Pierce JE, Ogushi F, Hubbard R, et al. Augmentation of lung antineutrophil elastase capacity with recombinant human alpha-1-antitrypsin. J Appl Physiol (1987) 63(5):2015–23. doi:10.1152/jappl.1987.63.5.2015
71. Jha S, Agarwal S, Sanyal I, Jain GK, Amla DV. Differential subcellular targeting of recombinant human alpha(1)-proteinase inhibitor influences yield, biological activity and in planta stability of the protein in transgenic tomato plants. Plant Sci (2012) 196:53–66. doi:10.1016/j.plantsci.2012.07.004
72. Agarwal S, Singh R, Sanyal I, Amla DV. Expression of modified gene encoding functional human alpha-1-antitrypsin protein in transgenic tomato plants. Transgenic Res (2008) 17(5):881–96. doi:10.1007/s11248-008-9173-8
73. Nadai M, Bally J, Vitel M, Job C, Tissot G, Botterman J, et al. High-level expression of active human alpha1-antitrypsin in transgenic tobacco chloroplasts. Transgenic Res (2009) 18(2):173–83. doi:10.1007/s11248-008-9209-0
74. Carver A, Wright G, Cottom D, Cooper J, Dalrymple M, Temperley S, et al. Expression of human alpha 1 antitrypsin in transgenic sheep. Cytotechnology (1992) 9(1–3):77–84. doi:10.1007/BF02521734
75. Carver AS, Dalrymple MA, Wright G, Cottom DS, Reeves DB, Gibson YH, et al. Transgenic livestock as bioreactors: stable expression of human alpha-1-antitrypsin by a flock of sheep. Biotechnology (1993) 11(11):1263–70.
76. Archibald AL, McClenaghan M, Hornsey V, Simons JP, Clark AJ. High-level expression of biologically active human alpha 1-antitrypsin in the milk of transgenic mice. Proc Natl Acad Sci U S A (1990) 87(13):5178–82. doi:10.1073/pnas.87.13.5178
77. McCarthy C, Saldova R, Wormald MR, Rudd PM, McElvaney NG, Reeves EP. The role and importance of glycosylation of acute phase proteins with focus on alpha-1 antitrypsin in acute and chronic inflammatory conditions. J Proteome Res (2014) 13(7):3131–43. doi:10.1021/pr500146y
78. Baranovski BM, Ozeri E, Shahaf G, Ochayon DE, Schuster R, Bahar N, et al. Exploration of alpha1-antitrypsin treatment protocol for islet transplantation: dosing plan and route of administration. J Pharmacol Exp Ther (2016) 359:482–90. doi:10.1124/jpet.116.236067
79. Frenzel E, Wrenger S, Brugger B, Salipalli S, Immenschuh S, Aggarwal N, et al. alpha1-antitrypsin combines with plasma fatty acids and induces angiopoietin-like protein 4 expression. J Immunol (2015) 195(8):3605–16. doi:10.4049/jimmunol.1500740
80. Dykstra M, Cherukuri A, Sohn HW, Tzeng SJ, Pierce SK. Location is everything: lipid rafts and immune cell signaling. Annu Rev Immunol (2003) 21:457–81. doi:10.1146/annurev.immunol.21.120601.141021
81. Varshney P, Yadav V, Saini N. Lipid rafts in immune signalling: current progress and future perspective. Immunology (2016) 149(1):13–24. doi:10.1111/imm.12617
82. Katagiri YU, Kiyokawa N, Fujimoto J. A role for lipid rafts in immune cell signaling. Microbiol Immunol (2001) 45(1):1–8. doi:10.1111/j.1348-0421.2001.tb01259.x
83. Koulmanda M, Bhasin M, Fan Z, Hanidziar D, Goel N, Putheti P, et al. Alpha 1-antitrypsin reduces inflammation and enhances mouse pancreatic islet transplant survival. Proc Natl Acad Sci U S A (2012) 109(38):15443–8. doi:10.1073/pnas.1018366109
84. Nita I, Hollander C, Westin U, Janciauskiene SM. Prolastin, a pharmaceutical preparation of purified human alpha1-antitrypsin, blocks endotoxin-mediated cytokine release. Respir Res (2005) 6:12. doi:10.1186/1465-9921-6-12
85. Janciauskiene S, Larsson S, Larsson P, Virtala R, Jansson L, Stevens T. Inhibition of lipopolysaccharide-mediated human monocyte activation, in vitro, by alpha1-antitrypsin. Biochem Biophys Res Commun (2004) 321(3):592–600. doi:10.1016/j.bbrc.2004.06.123
86. Nita IM, Serapinas D, Janciauskiene SM. alpha1-Antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int J Biochem Cell Biol (2007) 39(6):1165–76. doi:10.1016/j.biocel.2007.02.017
87. Gmyr V, Bonner C, Moerman E, Tournoys A, Delalleau N, Quenon A, et al. Human recombinant antithrombin (ATryn((R))) administration improves survival and prevents intravascular coagulation after intraportal islet transplantation in a piglet model. Cell Transplant (2017) 26(2):309–17. doi:10.3727/096368916X693554
88. Wang J, Sun Z, Gou W, Adams DB, Cui W, Morgan KA, et al. alpha-1 antitrypsin enhances islet engraftment by suppression of instant blood-mediated inflammatory reaction. Diabetes (2017) 66(4):970–80. doi:10.2337/db16-1036
Keywords: α1-antitryspin, protein structure, reactive center loop, recombinant protein, pharmacokinetics, inflammation, anti-inflammatory
Citation: Lior Y, Zaretsky M, Ochayon DE, Lotysh D, Baranovski BM, Schuster R, Guttman O, Aharoni A and Lewis EC (2018) Point Mutation of a Non-Elastase-Binding Site in Human α1-Antitrypsin Alters Its Anti-Inflammatory Properties. Front. Immunol. 9:759. doi: 10.3389/fimmu.2018.00759
Received: 30 January 2018; Accepted: 27 March 2018;
Published: 01 May 2018
Edited by:
Janos G. Filep, Université de Montréal, CanadaReviewed by:
Angelo A. Manfredi, Vita-Salute San Raffaele University, ItalySusana Seixas, i3S, Instituto de Investigação e Inovação em Saúde, Portugal
Copyright: © 2018 Lior, Zaretsky, Ochayon, Lotysh, Baranovski, Schuster, Guttman, Aharoni and Lewis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eli C. Lewis, bGV3aXNAYmd1LmFjLmls
†These authors have contributed equally in this work.