 Aparna P. Sajja
Aparna P. Sajja Aditya A. Joshi
Aditya A. Joshi Heather L. Teague
Heather L. Teague Nehal N. Mehta
Nehal N. Mehta- National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, United States
Preclinical and clinical research provide strong evidence that chronic, systemic inflammation plays a key role in development and progression of atherosclerosis. Indeed, chronic inflammatory diseases, such as psoriasis, are associated with accelerated atherosclerosis and increased risk of cardiovascular events. Contemporary research has demonstrated plausible mechanistic links between immune cell dysfunction and cardiometabolic disease in psoriasis. In this review, we describe the role of potential common immunological mechanisms underlying both psoriasis and atherogenesis. We primarily discuss innate and adaptive immune cell subsets and their contributions to psoriatic disease and cardiovascular morbidity. Emerging efforts should focus on understanding the interplay among immune cells, adipose tissue, and various biomarkers of immune dysfunction to provide direction for future targeted therapy.
Introduction
Inflammation is the hallmark of atherosclerosis (1). Preclinical and clinical research provide strong evidence that chronic inflammation is critical to the process of atherogenesis. Chronic inflammatory diseases, such as psoriasis, are associated with accelerated atherosclerosis and increased risk of cardiovascular events (2–6). Atherosclerosis is increasingly recognized as an inflammatory process, thus similarities between atherosclerosis and chronic, systemic inflammatory diseases have become an emerging focus of interest. Almost 20% patients with coronary heart disease lack conventional risk factors (7), supporting the importance of evaluating residual inflammatory risk (8). Chronic inflammatory diseases such as psoriasis have been shown to add 6% additional risk (9, 10) to the Framingham Risk Score (8, 9) highlighting the need to understand the role of immunological processes in cardiovascular disease (CVD) for better risk stratification and treatment strategies.
Chronic Inflammation and Cardiovascular Co-Morbidities
Patients with chronic inflammatory diseases are predisposed to cardiometabolic diseases including obesity, hypertension, and dyslipidemia (11–16)—chronic inflammatory conditions common in the general population (17–19). Obesity, particularly visceral, is strongly associated with dysregulated expression of inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and IL-6, as well as adiponectin and leptin, contributing to metabolic derangement and insulin resistance (13, 18, 20). Atherogenic metabolic dyslipidemia is common in chronic inflammation. Abnormalities include impaired reverse cholesterol transport ability of the HDL, increased LDL particle number, and decreased LDL size (21–23). Animal and human models have demonstrated innate immunity as well as experimental in vivo induction of inflammation via bolus of an inflammatory cytokine such as TNF-α or IL-6, results in release of adipokines and generation of peripheral insulin resistance (24–27). Moreover, anti-inflammatory therapies such as aspirin, colchicine, and more recently canakinumab have been effective in CVD treatment, supporting the critical role of inflammation in CVD (28–32).
One of the most common co-morbid conditions associated with psoriasis is psoriatic arthritis (PsA). Epidemiological data indicate that almost one-third patients with psoriasis also have prevalent PsA (33). Similar to psoriasis, PsA is associated with increased prevalence of traditional cardiovascular risk factors, greater subclinical CVD assessed as vascular inflammation (VI) by 18-FDG PET/CT and ultrasound-guided carotid plaque assessment and intima-media thickness measurement, and elevated rates of major adverse cardiovascular events (MACEs) (34–38). Furthermore, like psoriasis, traditional risk factors do not fully capture the risk of CVD in PsA (39, 40).
Recently, there is growing focus on shared immunological links between atherosclerosis and several other autoimmune diseases such as systemic lupus erythematosus, inflammatory bowel disease, human immunodeficiency virus infection, rheumatoid arthritis, and psoriasis. These all carry an accelerated CVD risk, thought to be partly attributable to inflammation-driven endothelial dysfunction, lipoprotein derangement, and metabolic dysfunction stemming from chronic inflammation (41, 42). In order to speed understanding of inflammatory cardiometabolic dysfunction, psoriasis has been utilized as a human model (3) to understand the role of innate and adaptive immunity in subclinical CVD (43, 44). The clinical implications of understanding how the inflammatory processes in psoriasis contribute to cardiovascular morbidity are vast since approximately 3% of the US population has psoriasis. Furthermore, observational reports have suggested that anti-inflammatory therapies commonly used to treat psoriasis may associate with reduced cardiovascular risk (45, 46).
Potential Immunologic Links Between Psoriasis and CVD
Psoriasis Is Associated With Subclinical and Clinical Atherosclerosis
In the last decade, multiple studies have demonstrated an association between psoriasis and both subclinical and clinical atherosclerosis, such as VI by 18F-FDG PET/CT, coronary artery calcium and non-calcified coronary plaque burden by coronary computed tomography angiography (44, 47–51). Population-based studies provide evidence of early subclinical and clinical CVD in psoriasis (2, 4, 52, 53). Research into the concept of psoriatic march (54) has led to an understanding of common cellular and molecular level links between psoriasis and atherosclerosis (55).
Common Immune Cells Between Psoriasis and Atherosclerosis
T Cells
Studies in the last two decades have established psoriasis primarily as a T-cell-mediated disorder (56–60). While initial evidence implicated a predominant role of helper T cells type 1 (Th1) through downstream activation of macrophages, neutrophils, and CD8+ cytotoxic T lymphocytes (61), recent research shows the importance of the Th17 and other IL-17 producing cell types such as CD8+ T cells and γδ T cells (62). Although Th1 subtype is the most studied cell-type in psoriasis, different stages of this chronic inflammatory disease employ various cells of innate and adaptive immunity (62). All the subtypes of T cells involved in pathogenesis of psoriasis are also involved in atherosclerosis (63).
Th1 Cells—Helper T Cells Type 1
Activation of the innate immune system is the key event in beginning the inflammatory cascade in psoriasis. It primarily includes differentiation of T cells into Th1 cells catalyzed by IL-12 (62). Mechanistic studies in patients with psoriasis have suggested a preference of hematopoietic progenitors toward Th1 subtype (64). Th1 cells induce psoriatic inflammation by activating neutrophils, macrophages, and CD8+ cytotoxic T lymphocytes (61). Primary mediators of Th1 activity are interferon-gamma (IFN-γ), IL-2, and TNF-α, which act on keratinocytes and induce antimicrobial peptide production that subsequently continues the inflammatory cascade. Th1 cells are also critical to the process of atherosclerosis, a process thought to be primarily driven by IFN-γ, the hallmark cytokine of the Th1 response (65). In patients with unstable angina and acute coronary syndrome (ACS), Th1 cells were found to be elevated (66, 67). Furthermore, mechanistic studies have also established the role of IL-12 in the development and progression of early atherosclerotic plaques (68–70). In addition, IL-18, a Th1-promoting cytokine, has also been shown to have a role in atherosclerosis (71, 72). Finally, targeting Th1 differentiating transcription factor is shown to associate with reduced atherosclerotic plaques (73). An IL-12 stimulated activation of Th1 response with downstream release of pro-inflammatory cytokines is a common feature between psoriasis and atherosclerosis and is thought to contribute to subsequent endothelial dysfunction and T cell recruitment to the sites of atherosclerotic plaques (74). While the role of Th1 cells is profoundly studied, the function of Th2 cells remains a topic of controversy as multiple studies exist that support pro-atherosclerotic (75), atherosclerosis protective (76), and also null effect (77) of Th2 cells.
Th17 Cells—Helper T Cells Type 17
Th17 cells in psoriasis release different cytokines such as IL-17, IL-22, and TNF-α (78) and are also involved in macrophage-dependent and -independent stimulation of dendritic cells (DCs) to propagate the inflammatory response (79). They may be involved in increased production of angiogenic inflammatory mediators such as monocyte chemoattractant protein (MCP-1), nitric oxide, and vascular endothelial growth factor (80, 81). Similar to Th2 helper cells, there is conflicting data on the role of Th17 cells in atherosclerosis (82). Patients with ACS show increased Th17 cells and IL-17 compared with those with stable angina or non-cardiac chest pain (83, 84). There is mixed evidence from mechanistic models: with some mouse models supporting the pro-atherogenic role of Th17 and IL-17 (85–87), while others have found low IL-17 mRNA in atherosclerotic plaques and overall attenuated disease development with high prevalence of Th17 cells (88–90). We later discuss the emerging role of neutrophils in the IL-17 axis, a possible mechanistic link; however, further clinical and translational research is necessary to elucidate the differential roles of Th17 and neutrophils in this pathway.
Regulatory T Cells (Treg Cells)
Regulatory T cells are a subset of T lymphocytes with a primary function to inhibit T cell activation and proliferation, through both cell-contact-dependent and cell-contact-independent anti-inflammatory cytokine (mainly TGFβ and IL-10) driven mechanisms (91). Treg inhibitory function is distinctly impaired in psoriasis (92, 93), contributing to the chronic auto- inflammation in psoriasis. ACS patients are also known to have decreased levels of circulating Treg cells with reduced efficacy and increased apoptosis susceptibility (94–97). Treg cells play an anti-inflammatory role in atherosclerosis through endothelial cell modulation, plaque stabilization by decreasing macrophages and lipid content and increasing smooth muscle cell and collagen, inhibition of pro-inflammatory cytokines, and secretion of anti-inflammatory cytokines such as TGFβ, IL-10, and IL-35 (91). Identification of common targets to reverse Treg cell dysfunction or to augment their activity in psoriasis may represent treatment mechanisms for both psoriasis and atherosclerosis simultaneously.
Finally, there are several other T cell phenotypes that have been identified in psoriasis skin lesions, such as CD4+, CD8+ T cells, CD146+, and γδ T cells (98). However, their role in psoriasis and atherosclerosis need to be further explored. While the traditional paradigm of T cell lineages might predominate shared mechanistic links between psoriasis and atherosclerosis, there is significant heterogeneity and plasticity within the T cell subtypes. T cell predominance may change in context of subtype preponderance with the natural disease course, specifically, a switch from Th1 dominated profile in early initiation phase of psoriasis to a Th17 governed response in the chronic inflammatory phase with both involved in atherosclerosis progression (99).
Dendritic Cells
In psoriasis, DCs not only act as antigen presenters and cytokine producers but also play an important part of bridging the innate and adaptive immune systems in continuing the chronic inflammation inducing cascade (43, 79). While pDCs are important in initiation of psoriasis via type 1 IFN responses (62, 100), mDCs are key mediators for specific Th cell expansion via IL-12 and IL-23 secretion (79). While new evidence suggests a role for DCs in atherosclerotic plaque build-ups, plaque vulnerability through cholesterol metabolism and adaptive immune response modulation (101), their shared role in psoriasis and atherosclerosis needs further research.
Monocytes and Macrophages
Monocytes and macrophages are cellular hallmark of atherosclerosis (1) and are also involved in pathogenesis of psoriasis (102). While macrophages are traditionally subclassified as pro-inflammatory (M1) and anti-inflammatory (M2), they are known to be plastic and adapt to the surrounding milieu according to the underlying pathological state (103, 104). Furthermore, a preclinical in vivo and in vitro study demonstrated that chronic skin inflammation in psoriasis polarizes them toward the pro-atherosclerotic phenotype (99). These cells are involved in ACS, and their increased expression and activity is also present in vulnerable plaques (105). Novel evidence has recently suggested that a complex interplay involving neutrophil–macrophage cross-talk is crucial to the process of atherosclerosis and ACS (106–108). As these cells are involved throughout the process of atherosclerosis from plaque development to complications, such as ACS, and also play a significant role in psoriasis, further research may provide new avenues for treatment of both these conditions.
Neutrophils
Despite being the most abundant white blood cell in the circulation, neutrophils have received little attention in the pathophysiology of atherosclerosis and psoriasis. Recent mouse models and clinical trials have demonstrated the mechanistic role of neutrophils in psoriasis and atherosclerosis through the IL-17 driven keratinocyte hyper-proliferation, leading to chronic skin inflammation (109, 110). Psoriasis patients are known to have higher serum levels of IL-17 compared with healthy controls; however, the paradigm of Th17 as the predominant cellular source of IL-17 in psoriatic lesions is no longer fully valid (111). Recent studies have demonstrated that cells of the innate immune system, such as neutrophils, mast cells, γδ T cells, and innate lymphoid cells, are the main sources of IL-17 in psoriasis. Furthermore, despite controversies, IL-17 is shown to have a role in atherosclerosis in clinical and mouse model-based studies (83–85, 87).
Psoriasis increases neutrophil activation and release of neutrophil-associated proteins. Proteins associated with neutrophils such as S100A8/A9 may further provide a link between psoriasis and cardiometabolic diseases (100). S100A8/A9 (MRP8/14) is released by activated neutrophils and upregulated in psoriatic lesional skin (100, 112). We demonstrated its strong association with both skin disease severity and VI (100). Collectively, evidence suggests that neutrophils and their proteins may contribute to the early atherosclerotic milieu in psoriasis and independently predict endothelial dysfunction.
A novel subtype of neutrophils, the low-density granulocytes (LDGs), are moving to the forefront of research in psoriasis and CVD pathophysiology. LDGs are characterized by high pro-inflammatory activity, altered phagocytic function, elevated type I interferon production, and high abundance in atherosclerotic plaques and plasma of psoriasis patients (113). At the gene expression level, LDGs differ from their autologous normal-density granulocytes (NDGs) counterparts, as well as from healthy control neutrophils (114–116). LDGs also differ phenotypically from NDGs. Of these differences, the most compelling is their enhanced capacity to spontaneously form neutrophil extracellular traps (NETs). This novel defense mechanism termed NETosis goes beyond classical phagocytosis, where NETs are formed as a result of release of cytosolic granule proteins bound to nuclear material catalyzed by peptidylarginine deiminase 4 (117). Although NETs are beneficial in antimicrobial defense, they may act as a source of autoantigens and are implicated in the development of autoimmune diseases especially psoriasis, as well as other diseases including systemic lupus erythematosus, atherosclerosis, preeclampsia, acute lung injury, deep vein thrombosis, and cancer-associated thrombosis (118–121). Cholesterol crystals are shown to trigger NETosis, further potentiating atherosclerosis by macrophage priming, Th17 activation, and immune cell recruitment in plaques (108). NETs are also shown through immunochemical stains to directly induce endothelial dysfunction and plaque rupture in human carotid plaque sections (122). NETs may be involved in the initial injury of the endothelium during atherogenesis, with recent evidence demonstrating the presence of neutrophils and NETs at sites of plaque rupture and endothelial cell erosion in human carotid plaques, features which we hypothesized would be evident in early atherosclerosis in psoriasis.
Adipose Dysfunction in Psoriasis
Systemic inflammation associated with psoriasis also contributes to inflammation of the adipose tissue (20), harboring components of the innate immune system (Figure 1) (63, 123). The physiological distinction between visceral and subcutaneous adiposity has been considered an important determinant in assessing CVD risk. Visceral adiposity is highly metabolically active, and its dysregulation can alter the immune cell and adipokine profile, exacerbating endothelial dysfunction. Visceral adiposity is associated with subclinical CVD measured as VI by 18F-FDG PET/CT independent of cardiovascular risk factors in psoriasis (124). Furthermore, a decrease in visceral adiposity associated with an improvement of VI following 1 year of biologic anti-inflammatory therapy.
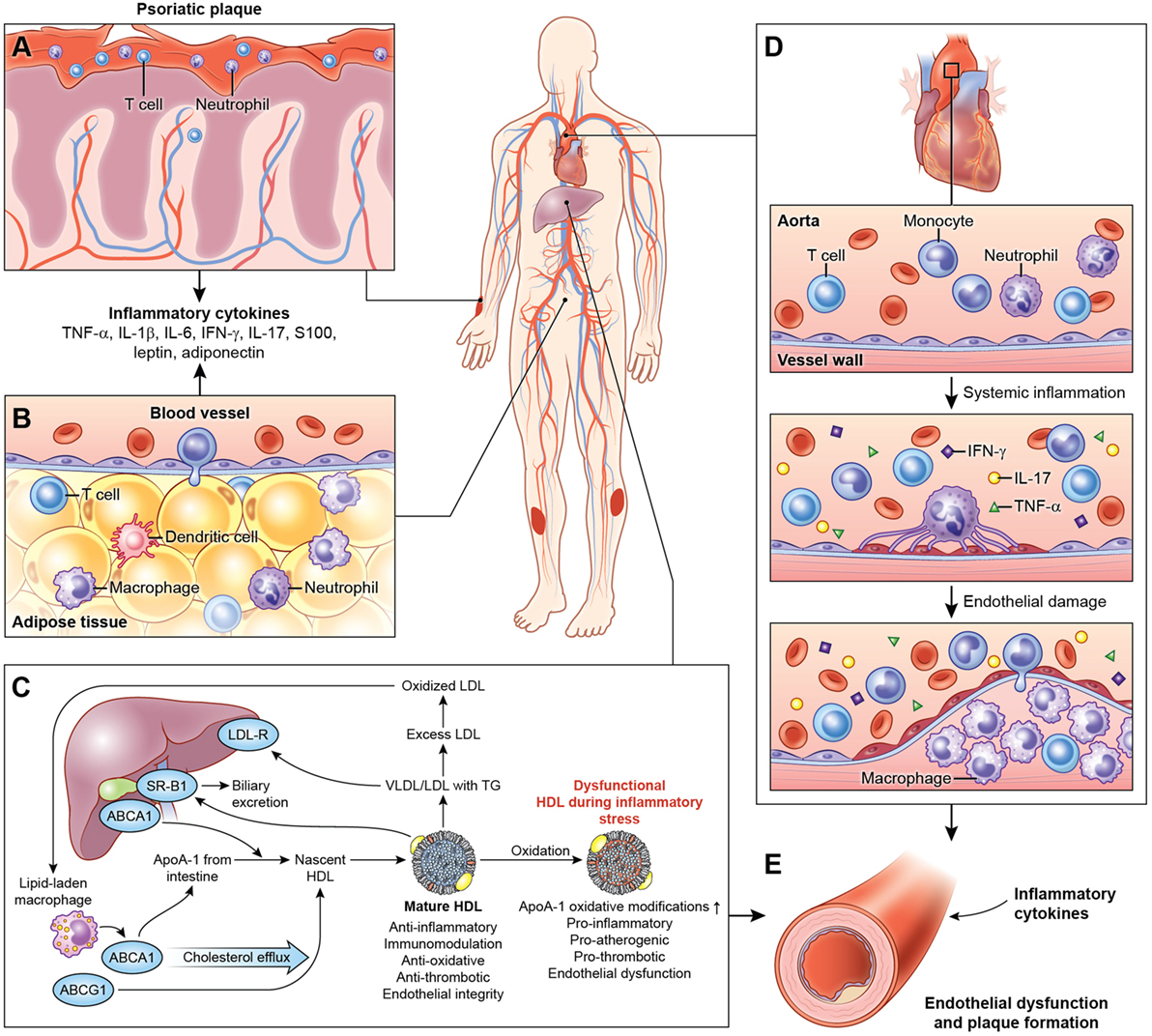
Figure 1. Systemic effects of chronic low-grade inflammation in psoriasis. (A) Psoriasis, both cutaneous and arthritic, is a low-grade chronic, systemic inflammatory disease associated with increased circulating pro-inflammatory cytokines. (B) Chronic inflammation in psoriasis is associated with adipose tissue dysfunction characterized by pro-inflammatory cytokines and adipokines associated with endothelial dysfunction. (C) Furthermore, psoriasis exhibits a deranged lipid profile and impaired HDL function, which in combination with chronic inflammation accelerate atherosclerotic vascular disease. (D) The vessel wall is infiltrated through a complex interplay of pro-inflammatory cellular components, cholesterol crystals, and various lipoproteins. Over the time, with build-up of the plaque, this atherosclerotic lesion poses a significant threat to blood flow and is prone to rupture, often accelerated by inflammation leading to myocardial infarction. (E) Thus, psoriasis and psoriatic arthritis upregulate T-cell, neutrophil chemotaxis, and keratinocyte activation and endothelial dysfunction leading to increased atherosclerosis in blood vessels. Abbreviations: TNF-α, tumor necrosis factor-alpha; IL, interleukin; IFN-γ, interferon-gamma.
Psoriatic adipose tissue contains immune cells that influence cardiometabolic disease (20). T cells, B cells, DCs, neutrophils, mast cells, and adipose tissue macrophages (ATM) may contribute to obesity and insulin resistance, while eosinophils and Treg may protect against insulin resistance. ATM represent unique functional subset in psoriasis that are predisposed toward pro-inflammatory cytokine expression and adipose dysfunction, extending beyond the M1/M2 macrophage paradigm (20, 125, 126).
While visceral abdominal adiposity is being increasingly studied, there is emerging research that a local type of visceral adipose tissue, known as perivascular adipose tissue (PVAT), which surrounds most blood vessels (coronary arteries, the aorta, and microcirculation of the mesentery), may contribute to cardiometabolic disease (127, 128). Its anatomic proximity to the vasculature has led to research investigating the mechanisms of dysfunctional PVAT driven immune-mediated cross-talk in endothelial and vascular function under inflammatory conditions (127, 128). Mechanistic studies have demonstrated significant adipokine and chemokine (MCP-1, IL-8) production by PVAT and its ability to stimulate chemotaxis, contributing to progression of atherosclerosis (129, 130). Multiple pathways have been identified through which adipokines are implicated in CVD development—from direct vascular effects on endothelial function and smooth muscle migration to immune cell migration into the vascular wall through a potential “outside-in” inflammatory cascade (127). Recent efforts have led to a novel approach to image the PVAT and showed that it is associated with coronary inflammation in a dynamic fashion (131), with potential for prospective risk stratification.
Leptin is shown to be elevated in patients with psoriasis, to correlate with psoriasis disease severity and with indices of subclinical atherosclerosis (132, 133). We have previously exhibited an association between enhanced leptin and resistin activity with attenuated adiponectin activity in innate immune activation (24). Increased leptin and resistin promote expression of pro-inflammatory cytokines including TNF-α, IL-2, IL-6, and MCP-1, all of which are prothrombotic and drive VI through monocyte migration and macrophage activation (134). Finally, adipokines may contribute to the effect of insulin on the vasculature by contributing to changes in capillary recruitment (127).
Peri- and epicardial fat tissue are additional sources of visceral fat deposition, and a rich source of inflammatory cytokines that are associated with both subclinical and clinical coronary heart disease (128). Epicardial fat tissue has been reported to be significantly increased in psoriasis patients and may represent an independent risk factor for atherosclerosis (135).
Biologic Therapies
The current generation of biologic agents target cytokines critical to the pathogenesis of psoriasis, including the three known major drivers: TNF-α, IL-23, and IL-17. The majority of most effective psoriasis treatments target the IL-23/Th17 pathway. These medications include the anti-IL-17 and anti-IL23p19 agents (Table 1). However, as novel therapies emerge, even today, anti-TNF agents remain the standard of care in general clinical practice (43, 136). While observational data in large payer-based or veterans association-based cohorts suggest a reduced risk for MACEs primarily with anti-TNF agents, no trials assessing direct cardiovascular effects of these medications in psoriasis patients exist to date (137–140). Although effective in treating psoriasis, interestingly, these therapies have been proven of no use in rheumatoid arthritis, another chronic inflammatory disease where the IL-23/Th17 axis plays an important role. The rationale behind these contradictory findings in two major inflammatory diseases currently remains unclear (141, 142).
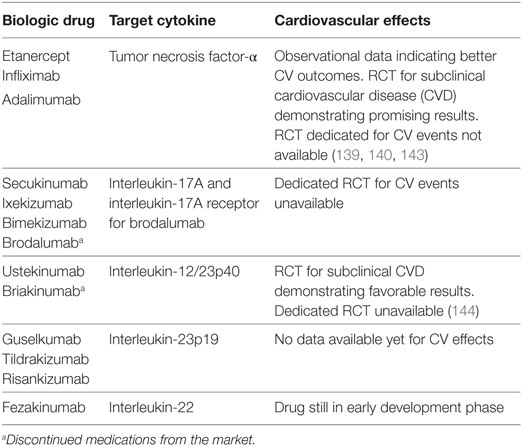
Table 1. Biologic treatment options to treat psoriasis.
Future Directions
Over the last decade, remarkable progress has been made for the treatment of moderate-to-severe psoriasis, especially with the advent of biologic therapies, which target specific cytokines, immune cells, and pathways. Moreover, the recent success of CANTOS (32) has demonstrated that inflammation reduction through direct IL-1β inhibition using a monoclonal antibody, canakinumab, in the absence of lipid lowering, can reduce CV event rates. As such, the emerging field of biologic treatments is exciting as it may provide therapeutic utility in psoriasis with added benefits of modulating CVD risk. Furthermore, completed and ongoing trials assessing the subclinical CVD in psoriasis have demonstrated promising findings (143, 144).
Finally, future research should focus on examination of complex inter-relationships between various conventional and non-conventional, inflammatory and non-inflammatory pathways to understand the heightened risk of CVD in disease conditions with underlying chronic inflammation.
Conclusion
Increasing evidence demonstrates an important role of immune dysfunction linking psoriasis to cardiometabolic diseases including atherosclerosis. Future efforts in patients with chronic inflammatory disease like psoriasis should focus on elucidating the complex interplay among immune cells, adipose tissue, and various biomarkers of immune dysfunction. The shared mechanistic links between psoriasis and atherosclerosis provide promising avenues in targeted treatment for both diseases, especially in light of the recent trial CANTOS (32), which demonstrated reduced incidence of recurrent cardiovascular events after treating residual inflammation in patients with known coronary artery disease.
Author Contributions
AS and NM conceived and designed research. AS, AJ, HT, AD, and NM contributed to both manuscript writing and critical review.
Conflict of Interest Statement
NM is a full-time US Government Employee and receives research grants to the NHLBI from AbbVie, Janssen, Celgene, and Novartis. All other authors have nothing to disclose.
Acknowledgments
We would like to acknowledge Ethan Tyler for his contribution to the figure.
Funding
NM’s Lab is supported by the National Heart, Lung, and Blood Institute Intramural Program (HL006193-02), National Institutes of Health.
References
1. Libby P, Ridker PM, Hansson GK, Leducq Transatlantic Network on Atherothrombosis. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol (2009) 54(23):2129–38. doi:10.1016/j.jacc.2009.09.009
2. Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. JAMA (2006) 296(14):1735–41. doi:10.1001/jama.296.14.1735
3. Harrington CL, Dey AK, Yunus R, Joshi AA, Mehta NN. Psoriasis as a human model of disease to study inflammatory atherogenesis. Am J Physiol Heart Circ Physiol (2017) 312(5):H867–73. doi:10.1152/ajpheart.00774.2016
4. Mehta NN, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Patients with severe psoriasis are at increased risk of cardiovascular mortality: cohort study using the General Practice Research Database. Eur Heart J (2010) 31(8):1000–6. doi:10.1093/eurheartj/ehp567
5. Brauchli YB, Jick SS, Miret M, Meier CR. Psoriasis and risk of incident myocardial infarction, stroke or transient ischaemic attack: an inception cohort study with a nested case-control analysis. Br J Dermatol (2009) 160(5):1048–56. doi:10.1111/j.1365-2133.2008.09020.x
6. Prodanovich S, Kirsner RS, Kravetz JD, Ma F, Martinez L, Federman DG. Association of psoriasis with coronary artery, cerebrovascular, and peripheral vascular diseases and mortality. Arch Dermatol (2009) 145(6):700–3. doi:10.1001/archdermatol.2009.94
7. Khot UN, Khot MB, Bajzer CT, Sapp SK, Ohman EM, Brener SJ, et al. Prevalence of conventional risk factors in patients with coronary heart disease. JAMA (2003) 290(7):898–904. doi:10.1001/jama.290.7.898
8. Ridker PM. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation (2003) 107(3):363–9. doi:10.1161/01.CIR.0000053730.47739.3C
9. Mehta NN, Krishnamoorthy P, Yu Y, Khan O, Raper A, Van Voorhees A, et al. The impact of psoriasis on 10-year Framingham risk. J Am Acad Dermatol (2012) 67(4):796–8. doi:10.1016/j.jaad.2012.05.016
10. Mehta NN, Yu Y, Pinnelas R, Krishnamoorthy P, Shin DB, Troxel AB, et al. Attributable risk estimate of severe psoriasis on major cardiovascular events. Am J Med (2011) 124(8):775.e1–6. doi:10.1016/j.amjmed.2011.03.028
11. Cohen AD, Dreiher J, Shapiro Y, Vidavsky L, Vardy DA, Davidovici B, et al. Psoriasis and diabetes: a population-based cross-sectional study. J Eur Acad Dermatol Venereol (2008) 22(5):585–9. doi:10.1111/j.1468-3083.2008.02636.x
12. Cohen AD, Sherf M, Vidavsky L, Vardy DA, Shapiro J, Meyerovitch J. Association between psoriasis and the metabolic syndrome. A cross-sectional study. Dermatology (2008) 216(2):152–5. doi:10.1159/000111512
13. Davidovici BB, Sattar N, Prinz J, Puig L, Emery P, Barker JN, et al. Psoriasis and systemic inflammatory diseases: potential mechanistic links between skin disease and co-morbid conditions. J Invest Dermatol (2010) 130(7):1785–96. doi:10.1038/jid.2010.103
14. Langan SM, Seminara NM, Shin DB, Troxel AB, Kimmel SE, Mehta NN, et al. Prevalence of metabolic syndrome in patients with psoriasis: a population-based study in the United Kingdom. J Invest Dermatol (2012) 132(3 Pt 1):556–62. doi:10.1038/jid.2011.365
15. Love TJ, Qureshi AA, Karlson EW, Gelfand JM, Choi HK. Prevalence of the metabolic syndrome in psoriasis: results from the National Health and Nutrition Examination Survey, 2003–2006. Arch Dermatol (2011) 147(4):419–24. doi:10.1001/archdermatol.2010.370
16. Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB, Gelfand JM. Prevalence of cardiovascular risk factors in patients with psoriasis. J Am Acad Dermatol (2006) 55(5):829–35. doi:10.1016/j.jaad.2006.08.040
17. Eckel RH, Krauss RM. American Heart Association call to action: obesity as a major risk factor for coronary heart disease. AHA Nutrition Committee. Circulation (1998) 97(21):2099–100. doi:10.1161/01.CIR.97.21.2099
18. Maresca F, Di Palma V, Bevilacqua M, Uccello G, Taglialatela V, Giaquinto A, et al. Adipokines, vascular wall, and cardiovascular disease: a focused overview of the role of adipokines in the pathophysiology of cardiovascular disease. Angiology (2015) 66(1):8–24. doi:10.1177/0003319713520463
19. Rao G, Powell-Wiley TM, Ancheta I, Hairston K, Kirley K, Lear SA, et al. Identification of obesity and cardiovascular risk in ethnically and racially diverse populations: a scientific statement from the American Heart Association. Circulation (2015) 132(5):457–72. doi:10.1161/CIR.0000000000000223
20. Rose S, Stansky E, Dagur PK, Samsel L, Weiner E, Jahanshad A, et al. Characterization of immune cells in psoriatic adipose tissue. J Transl Med (2014) 12:258. doi:10.1186/s12967-014-0258-2
21. McGillicuddy FC, de la Llera Moya M, Hinkle CC, Joshi MR, Chiquoine EH, Billheimer JT, et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation (2009) 119(8):1135–45. doi:10.1161/CIRCULATIONAHA.108.810721
22. Mehta NN, Li R, Krishnamoorthy P, Yu Y, Farver W, Rodrigues A, et al. Abnormal lipoprotein particles and cholesterol efflux capacity in patients with psoriasis. Atherosclerosis (2012) 224(1):218–21. doi:10.1016/j.atherosclerosis.2012.06.068
23. Rosenson RS, Brewer HB Jr, Ansell BJ, Barter P, Chapman MJ, Heinecke JW, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol (2016) 13(1):48–60. doi:10.1038/nrcardio.2015.124
24. Anderson PD, Mehta NN, Wolfe ML, Hinkle CC, Pruscino L, Comiskey LL, et al. Innate immunity modulates adipokines in humans. J Clin Endocrinol Metab (2007) 92(6):2272–9. doi:10.1210/jc.2006-2545
25. de la Llera Moya M, McGillicuddy FC, Hinkle CC, Byrne M, Joshi MR, Nguyen V, et al. Inflammation modulates human HDL composition and function in vivo. Atherosclerosis (2012) 222(2):390–4. doi:10.1016/j.atherosclerosis.2012.02.032
26. Ferguson JF, Shah RY, Shah R, Mehta NN, Rickels MR, Reilly MP. Activation of innate immunity modulates insulin sensitivity, glucose effectiveness and pancreatic beta-cell function in both African ancestry and European ancestry healthy humans. Metabolism (2015) 64(4):513–20. doi:10.1016/j.metabol.2014.12.007
27. Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, et al. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes (2010) 59(1):172–81. doi:10.2337/db09-0367
28. Nidorf M, Thompson PL. Effect of colchicine (0.5 mg twice daily) on high-sensitivity C-reactive protein independent of aspirin and atorvastatin in patients with stable coronary artery disease. Am J Cardiol (2007) 99(6):805–7. doi:10.1016/j.amjcard.2006.10.039
29. Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol (2013) 61(4):404–10. doi:10.1016/j.jacc.2012.10.027
30. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med (1997) 336(14):973–9. doi:10.1056/NEJM199704033361401
31. Vaidya K, Arnott C, Martinez GJ, Ng B, McCormack S, Sullivan DR, et al. Colchicine therapy and plaque stabilization in patients with acute coronary syndrome: a CT Coronary Angiography Study. JACC Cardiovasc Imag (2018) 11(2 Pt 2):305–16. doi:10.1016/j.jcmg.2017.08.013
32. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377(12):1119–31. doi:10.1056/NEJMoa1707914
33. Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med (2017) 376(10):957–70. doi:10.1056/NEJMra1505557
34. Armstrong AW, Gelfand JM, Boehncke WH, Armstrong EJ. Cardiovascular comorbidities of psoriasis and psoriatic arthritis: a report from the GRAPPA 2012 annual meeting. J Rheumatol (2013) 40(8):1434–7. doi:10.3899/jrheum.130457
35. Eder L, Jayakar J, Shanmugarajah S, Thavaneswaran A, Pereira D, Chandran V, et al. The burden of carotid artery plaques is higher in patients with psoriatic arthritis compared with those with psoriasis alone. Ann Rheum Dis (2013) 72(5):715–20. doi:10.1136/annrheumdis-2012-201497
36. Kolliker Frers RA, Cosentino V, Tau J, Kerzberg EM, Urdapilleta A, Chiocconi M, et al. Immune-mediated inflammation promotes subclinical atherosclerosis in recent-onset psoriatic arthritis patients without conventional cardiovascular risk factors. Front Immunol (2018) 9:139. doi:10.3389/fimmu.2018.00139
37. Ogdie A, Yu Y, Haynes K, Love TJ, Maliha S, Jiang Y, et al. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis (2015) 74(2):326–32. doi:10.1136/annrheumdis-2014-205675
38. Rose S, Dave J, Millo C, Naik HB, Siegel EL, Mehta NN. Psoriatic arthritis and sacroiliitis are associated with increased vascular inflammation by 18-fluorodeoxyglucose positron emission tomography computed tomography: baseline report from the psoriasis atherosclerosis and cardiometabolic disease initiative. Arthritis Res Ther (2014) 16(4):R161. doi:10.1186/ar4676
39. Eder L, Chandran V, Gladman DD. The Framingham Risk Score underestimates the extent of subclinical atherosclerosis in patients with psoriatic disease. Ann Rheum Dis (2014) 73(11):1990–6. doi:10.1136/annrheumdis-2013-203433
40. Shen J, Lam SH, Shang Q, Wong CK, Li EK, Wong P, et al. Underestimation of risk of carotid subclinical atherosclerosis by cardiovascular risk scores in patients with psoriatic arthritis. J Rheumatol (2018) 45(2):218–26. doi:10.3899/jrheum.170025
41. Teague H, Mehta NN. The link between inflammatory disorders and coronary heart disease: a look at recent studies and novel drugs in development. Curr Atheroscler Rep (2016) 18(1):3. doi:10.1007/s11883-015-0557-y
42. Teague HL, Ahlman MA, Alavi A, Wagner DD, Lichtman AH, Nahrendorf M, et al. Unraveling vascular inflammation: from immunology to imaging. J Am Coll Cardiol (2017) 70(11):1403–12. doi:10.1016/j.jacc.2017.07.750
43. Greb JE, Goldminz AM, Elder JT, Lebwohl MG, Gladman DD, Wu JJ, et al. Psoriasis. Nat Rev Dis Primers (2016) 2:16082. doi:10.1038/nrdp.2016.82
44. Shaharyar S, Warraich H, McEvoy JW, Oni E, Ali SS, Karim A, et al. Subclinical cardiovascular disease in plaque psoriasis: association or causal link? Atherosclerosis (2014) 232(1):72–8. doi:10.1016/j.atherosclerosis.2013.10.023
45. Boehncke S, Fichtlscherer S, Salgo R, Garbaraviciene J, Beschmann H, Diehl S, et al. Systemic therapy of plaque-type psoriasis ameliorates endothelial cell function: results of a prospective longitudinal pilot trial. Arch Dermatol Res (2011) 303(6):381–8. doi:10.1007/s00403-010-1108-6
46. Prodanovich S, Ma F, Taylor JR, Pezon C, Fasihi T, Kirsner RS. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol (2005) 52(2):262–7. doi:10.1016/j.jaad.2004.06.017
47. Dey AK, Joshi AA, Chaturvedi A, Lerman JB, Aberra TM, Rodante JA, et al. Association between skin and aortic vascular inflammation in patients with psoriasis: a case-cohort study using positron emission tomography/computed tomography. JAMA Cardiol (2017) 2(9):1013–8. doi:10.1001/jamacardio.2017.1213
48. Lerman JB, Joshi AA, Chaturvedi A, Aberra TM, Dey AK, Rodante JA, et al. Coronary plaque characterization in psoriasis reveals high-risk features that improve after treatment in a prospective observational study. Circulation (2017) 136(3):263–76. doi:10.1161/CIRCULATIONAHA.116.026859
49. Mehta NN, Yu Y, Saboury B, Foroughi N, Krishnamoorthy P, Raper A, et al. Systemic and vascular inflammation in patients with moderate to severe psoriasis as measured by [18F]-fluorodeoxyglucose positron emission tomography-computed tomography (FDG-PET/CT): a pilot study. Arch Dermatol (2011) 147(9):1031–9. doi:10.1001/archdermatol.2011.119
50. Hjuler KF, Bottcher M, Vestergaard C, Botker HE, Iversen L, Kragballe K. Association between changes in coronary artery disease progression and treatment with biologic agents for severe psoriasis. JAMA Dermatol (2016) 152(10):1114–21. doi:10.1001/jamadermatol.2016.1984
51. Mansouri B, Kivelevitch D, Natarajan B, Joshi AA, Ryan C, Benjegerdes K, et al. Comparison of coronary artery calcium scores between patients with psoriasis and type 2 diabetes. JAMA Dermatol (2016) 152(11):1244–53. doi:10.1001/jamadermatol.2016.2907
52. Gelfand JM, Dommasch ED, Shin DB, Azfar RS, Kurd SK, Wang X, et al. The risk of stroke in patients with psoriasis. J Invest Dermatol (2009) 129(10):2411–8. doi:10.1038/jid.2009.112
53. Gelfand JM, Troxel AB, Lewis JD, Kurd SK, Shin DB, Wang X, et al. The risk of mortality in patients with psoriasis: results from a population-based study. Arch Dermatol (2007) 143(12):1493–9. doi:10.1001/archderm.143.12.1493
54. Boehncke WH, Boehncke S, Tobin AM, Kirby B. The ‘psoriatic march’: a concept of how severe psoriasis may drive cardiovascular comorbidity. Exp Dermatol (2011) 20(4):303–7. doi:10.1111/j.1600-0625.2011.01261.x
55. Shlyankevich J, Mehta NN, Krueger JG, Strober B, Gudjonsson JE, Qureshi AA, et al. Accumulating evidence for the association and shared pathogenic mechanisms between psoriasis and cardiovascular-related comorbidities. Am J Med (2014) 127(12):1148–53. doi:10.1016/j.amjmed.2014.08.008
56. Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest (1999) 103(9):1243–52. doi:10.1172/JCI5857
57. Chamian F, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, Sullivan-Whalen M, et al. Alefacept (anti-CD2) causes a selective reduction in circulating effector memory T cells (Tem) and relative preservation of central memory T cells (Tcm) in psoriasis. J Transl Med (2007) 5:27. doi:10.1186/1479-5876-5-27
58. Chamian F, Lowes MA, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A (2005) 102(6):2075–80. doi:10.1073/pnas.0409569102
59. Gottlieb SL, Gilleaudeau P, Johnson R, Estes L, Woodworth TG, Gottlieb AB, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med (1995) 1(5):442–7. doi:10.1038/nm0595-442
60. Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, Nussbaum R, et al. Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci U S A (2005) 102(52):19057–62. doi:10.1073/pnas.0509736102
61. Nickoloff BJ. Skin innate immune system in psoriasis: friend or foe? J Clin Invest (1999) 104(9):1161–4. doi:10.1172/JCI8633
62. Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol (2014) 32:227–55. doi:10.1146/annurev-immunol-032713-120225
63. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol (2011) 12(3):204–12. doi:10.1038/ni.2001
64. Zhang K, Li X, Yin G, Liu Y, Tang X. Functional characterization of T cells differentiated in vitro from bone marrow-derived CD34 cells of psoriatic patients with family history. Exp Dermatol (2010) 19(8):e128–35. doi:10.1111/j.1600-0625.2009.01016.x
65. Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol (2006) 1:297–329. doi:10.1146/annurev.pathol.1.110304.100100
66. Liuzzo G, Vallejo AN, Kopecky SL, Frye RL, Holmes DR, Goronzy JJ, et al. Molecular fingerprint of interferon-gamma signaling in unstable angina. Circulation (2001) 103(11):1509–14. doi:10.1161/01.CIR.103.11.1509
67. Methe H, Kim JO, Kofler S, Weis M, Nabauer M, Koglin J. Expansion of circulating toll-like receptor 4-positive monocytes in patients with acute coronary syndrome. Circulation (2005) 111(20):2654–61. doi:10.1161/CIRCULATIONAHA.104.498865
68. Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol (2003) 163(3):1117–25. doi:10.1016/S0002-9440(10)63471-2
69. Hauer AD, Uyttenhove C, de Vos P, Stroobant V, Renauld JC, van Berkel TJ, et al. Blockade of interleukin-12 function by protein vaccination attenuates atherosclerosis. Circulation (2005) 112(7):1054–62. doi:10.1161/CIRCULATIONAHA.104.533463
70. Lee TS, Yen HC, Pan CC, Chau LY. The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol (1999) 19(3):734–42. doi:10.1161/01.ATV.19.3.734
71. Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, et al. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res (2003) 59(1):234–40. doi:10.1016/S0008-6363(03)00343-2
72. Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(-/-) mice through release of interferon-gamma. Circ Res (2002) 90(2):E34–8. doi:10.1161/hh0202.105292
73. Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci U S A (2005) 102(5):1596–601. doi:10.1073/pnas.0409015102
74. Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med (2013) 368(21):2004–13. doi:10.1056/NEJMra1216063
75. King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor-/- mice. Arterioscler Thromb Vasc Biol (2002) 22(3):456–61. doi:10.1161/hq0302.104905
76. Huber SA, Sakkinen P, David C, Newell MK, Tracy RP. T helper-cell phenotype regulates atherosclerosis in mice under conditions of mild hypercholesterolemia. Circulation (2001) 103(21):2610–6. doi:10.1161/01.CIR.103.21.2610
77. King VL, Cassis LA, Daugherty A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am J Pathol (2007) 171(6):2040–7. doi:10.2353/ajpath.2007.060857
78. Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol (2011) 131(3):677–87. doi:10.1038/jid.2010.340
79. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med (2009) 361(5):496–509. doi:10.1056/NEJMra0804595
80. Numasaki M, Lotze MT, Sasaki H. Interleukin-17 augments tumor necrosis factor-alpha-induced elaboration of proangiogenic factors from fibroblasts. Immunol Lett (2004) 93(1):39–43. doi:10.1016/j.imlet.2004.01.014
81. Takahashi H, Numasaki M, Lotze MT, Sasaki H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol Lett (2005) 98(2):189–93. doi:10.1016/j.imlet.2004.11.012
82. Liuzzo G, Trotta F, Pedicino D. Interleukin-17 in atherosclerosis and cardiovascular disease: the good, the bad, and the unknown. Eur Heart J (2013) 34(8):556–9. doi:10.1093/eurheartj/ehs399
83. Cheng X, Yu X, Ding YJ, Fu QQ, Xie JJ, Tang TT, et al. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol (2008) 127(1):89–97. doi:10.1016/j.clim.2008.01.009
84. Zheng Y, Wang Z, Deng L, Zhang G, Yuan X, Huang L, et al. Modulation of STAT3 and STAT5 activity rectifies the imbalance of Th17 and Treg cells in patients with acute coronary syndrome. Clin Immunol (2015) 157(1):65–77. doi:10.1016/j.clim.2014.12.012
85. Erbel C, Akhavanpoor M, Okuyucu D, Wangler S, Dietz A, Zhao L, et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J Immunol (2014) 193(9):4344–55. doi:10.4049/jimmunol.1400181
86. Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, et al. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol (2009) 183(12):8167–75. doi:10.4049/jimmunol.0901126
87. Smith E, Prasad KM, Butcher M, Dobrian A, Kolls JK, Ley K, et al. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation (2010) 121(15):1746–55. doi:10.1161/CIRCULATIONAHA.109.924886
88. de Boer OJ, van der Meer JJ, Teeling P, van der Loos CM, Idu MM, van Maldegem F, et al. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J Pathol (2010) 220(4):499–508. doi:10.1002/path.2667
89. Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation (2009) 119(10):1424–32. doi:10.1161/CIRCULATIONAHA.108.827618
90. Taleb S, Tedgui A, Mallat Z. IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles. Arterioscler Thromb Vasc Biol (2015) 35(2):258–64. doi:10.1161/ATVBAHA.114.303567
91. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol (2015) 13:167. doi:10.1038/nrcardio.2015.169
92. Kagen MH, McCormick TS, Cooper KD. Regulatory T cells in psoriasis. Ernst Schering Res Found Workshop (2006) (56):193–209.
93. Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, et al. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol (2005) 174(1):164–73. doi:10.4049/jimmunol.174.1.164
94. Han SF, Liu P, Zhang W, Bu L, Shen M, Li H, et al. The opposite-direction modulation of CD4+CD25+ Tregs and T helper 1 cells in acute coronary syndromes. Clin Immunol (2007) 124(1):90–7. doi:10.1016/j.clim.2007.03.546
95. Mor A, Luboshits G, Planer D, Keren G, George J. Altered status of CD4(+)CD25(+) regulatory T cells in patients with acute coronary syndromes. Eur Heart J (2006) 27(21):2530–7. doi:10.1093/eurheartj/ehl222
96. Wigren M, Bjorkbacka H, Andersson L, Ljungcrantz I, Fredrikson GN, Persson M, et al. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler Thromb Vasc Biol (2012) 32(8):2000–4. doi:10.1161/ATVBAHA.112.251579
97. Zhang WC, Wang J, Shu YW, Tang TT, Zhu ZF, Xia N, et al. Impaired thymic export and increased apoptosis account for regulatory T cell defects in patients with non-ST segment elevation acute coronary syndrome. J Biol Chem (2012) 287(41):34157–66. doi:10.1074/jbc.M112.382978
98. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol (2012) 9(4):302–9. doi:10.1038/cmi.2012.15
99. Baumer Y, Ng Q, Sanda GE, Dey AK, Teague HL, Sorokin AV, et al. Chronic skin inflammation accelerates macrophage cholesterol crystal formation and atherosclerosis. JCI Insight (2018) 3(1). doi:10.1172/jci.insight.97179
100. Naik HB, Natarajan B, Stansky E, Ahlman MA, Teague H, Salahuddin T, et al. Severity of psoriasis associates with aortic vascular inflammation detected by FDG PET/CT and neutrophil activation in a prospective observational study. Arterioscler Thromb Vasc Biol (2015) 35(12):2667–76. doi:10.1161/ATVBAHA.115.306460
101. Zernecke A. Dendritic cells in atherosclerosis: evidence in mice and humans. Arterioscler Thromb Vasc Biol (2015) 35(4):763–70. doi:10.1161/ATVBAHA.114.303566
102. Clark RA, Kupper TS. Misbehaving macrophages in the pathogenesis of psoriasis. J Clin Invest (2006) 116(8):2084–7. doi:10.1172/JCI29441
103. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol (2013) 13(10):709–21. doi:10.1038/nri3520
104. Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest (2017) 127(8):2904–15. doi:10.1172/JCI75005
105. Ghattas A, Griffiths HR, Devitt A, Lip GY, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: where are we now? J Am Coll Cardiol (2013) 62(17):1541–51. doi:10.1016/j.jacc.2013.07.043
106. Frodermann V, Nahrendorf M. Neutrophil-macrophage cross-talk in acute myocardial infarction. Eur Heart J (2017) 38(3):198–200. doi:10.1093/eurheartj/ehw085
107. Nahrendorf M, Swirski FK. Immunology. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science (2015) 349(6245):237–8. doi:10.1126/science.aac7801
108. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (2015) 349(6245):316–20. doi:10.1126/science.aaa8064
109. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol (2009) 129(9):2175–83. doi:10.1038/jid.2009.65
110. Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol (2009) 160(2):319–24. doi:10.1111/j.1365-2133.2008.08902.x
111. Keijsers RR, Joosten I, van Erp PE, Koenen HJ, van de Kerkhof PC. Cellular sources of IL-17 in psoriasis: a paradigm shift? Exp Dermatol (2014) 23(11):799–803. doi:10.1111/exd.12487
112. Kerkhoff C, Voss A, Scholzen TE, Averill MM, Zanker KS, Bornfeldt KE. Novel insights into the role of S100A8/A9 in skin biology. Exp Dermatol (2012) 21(11):822–6. doi:10.1111/j.1600-0625.2012.01571.x
113. Sanda GE, Belur AD, Teague HL, Mehta NN. Emerging associations between neutrophils, atherosclerosis, and psoriasis. Curr Atheroscler Rep (2017) 19(12):53. doi:10.1007/s11883-017-0692-8
114. Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol (2013) 35(4):455–63. doi:10.1007/s00281-013-0375-7
115. Nakou M, Knowlton N, Frank MB, Bertsias G, Osban J, Sandel CE, et al. Gene expression in systemic lupus erythematosus: bone marrow analysis differentiates active from inactive disease and reveals apoptosis and granulopoiesis signatures. Arthritis Rheum (2008) 58(11):3541–9. doi:10.1002/art.23961
116. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187(1):538–52. doi:10.4049/jimmunol.1100450
117. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi:10.1126/science.1092385
118. Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol (2017) 8:81. doi:10.3389/fimmu.2017.00081
119. Doring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res (2017) 120(4):736–43. doi:10.1161/CIRCRESAHA.116.309692
120. Qi H, Yang S, Zhang L. Neutrophil extracellular traps and endothelial dysfunction in atherosclerosis and thrombosis. Front Immunol (2017) 8:928. doi:10.3389/fimmu.2017.00928
121. Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M, et al. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol (2016) 36(10):2078–87. doi:10.1161/ATVBAHA.116.307634
122. Quillard T, Araujo HA, Franck G, Shvartz E, Sukhova G, Libby P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J (2015) 36(22):1394–404. doi:10.1093/eurheartj/ehv044
123. Shah A, Mehta N, Reilly MP. Adipose inflammation, insulin resistance, and cardiovascular disease. JPEN J Parenter Enteral Nutr (2008) 32(6):638–44. doi:10.1177/0148607108325251
124. Rivers JP, Powell-Wiley TM, Dey AK, Rodante JA, Chung JH, Joshi AA, et al. Visceral adiposity in psoriasis is associated with vascular inflammation by 18fluorodeoxyglucose positron-emission tomography/computed tomography beyond cardiometabolic disease risk factors in an observational cohort study. J Cardiovasc Imag (2017) 11(2 Pt 2):349–57. doi:10.1016/j.jcmg.2017.08.014
125. Bourlier V, Zakaroff-Girard A, Miranville A, De Barros S, Maumus M, Sengenes C, et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation (2008) 117(6):806–15. doi:10.1161/CIRCULATIONAHA.107.724096
126. Fjeldborg K, Pedersen SB, Moller HJ, Christiansen T, Bennetzen M, Richelsen B. Human adipose tissue macrophages are enhanced but changed to an anti-inflammatory profile in obesity. J Immunol Res (2014) 2014:309548. doi:10.1155/2014/309548
127. Britton KA, Fox CS. Perivascular adipose tissue and vascular disease. Clin Lipidol (2011) 6(1):79–91. doi:10.2217/clp.10.89
128. Fitzgibbons TP, Czech MP. Epicardial and perivascular adipose tissues and their influence on cardiovascular disease: basic mechanisms and clinical associations. J Am Heart Assoc (2014) 3(2):e000582. doi:10.1161/JAHA.113.000582
129. Henrichot E, Juge-Aubry CE, Pernin A, Pache JC, Velebit V, Dayer JM, et al. Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis? Arterioscler Thromb Vasc Biol (2005) 25(12):2594–9. doi:10.1161/01.ATV.0000188508.40052.35
130. Karastergiou K, Evans I, Ogston N, Miheisi N, Nair D, Kaski JC, et al. Epicardial adipokines in obesity and coronary artery disease induce atherogenic changes in monocytes and endothelial cells. Arterioscler Thromb Vasc Biol (2010) 30(7):1340–6. doi:10.1161/ATVBAHA.110.204719
131. Antonopoulos AS, Sanna F, Sabharwal N, Thomas S, Oikonomou EK, Herdman L, et al. Detecting human coronary inflammation by imaging perivascular fat. Sci Transl Med (2017) 9(398):eaal2658. doi:10.1126/scitranslmed.aal2658
132. Cerman AA, Bozkurt S, Sav A, Tulunay A, Elbasi MO, Ergun T. Serum leptin levels, skin leptin and leptin receptor expression in psoriasis. Br J Dermatol (2008) 159(4):820–6. doi:10.1111/j.1365-2133.2008.08742.x
133. Robati RM, Partovi-Kia M, Haghighatkhah HR, Younespour S, Abdollahimajd F. Increased serum leptin and resistin levels and increased carotid intima-media wall thickness in patients with psoriasis: is psoriasis associated with atherosclerosis? J Am Acad Dermatol (2014) 71(4):642–8. doi:10.1016/j.jaad.2014.06.006
134. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol (2011) 11(2):85–97. doi:10.1038/nri2921
135. Wang X, Guo Z, Zhu Z, Bao Y, Yang B. Epicardial fat tissue in patients with psoriasis: a systematic review and meta-analysis. Lipids Health Dis (2016) 15:103. doi:10.1186/s12944-016-0271-y
136. Mansouri B, Patel M, Menter A. Biological therapies for psoriasis. Expert Opin Biol Ther (2013) 13(12):1715–30. doi:10.1517/14712598.2013.853739
137. Rungapiromnan W, Yiu ZZN, Warren RB, Griffiths CEM, Ashcroft DM. Impact of biologic therapies on risk of major adverse cardiovascular events in patients with psoriasis: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol (2017) 176(4):890–901. doi:10.1111/bjd.14964
138. Wu JJ, Choi YM, Bebchuk JD. Risk of myocardial infarction in psoriasis patients: a retrospective cohort study. J Dermatolog Treat (2015) 26(3):230–4. doi:10.3109/09546634.2014.952609
139. Wu JJ, Joshi AA, Reddy SP, Batech M, Egeberg A, Ahlehoff O, et al. Anti-inflammatory therapy with tumor necrosis factor inhibitors is associated with reduced risk of major adverse cardiovascular events in psoriasis. J Eur Acad Dermatol Venereol (2018). doi:10.1111/jdv.14951
140. Wu JJ, Poon KY, Channual JC, Shen AY. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol (2012) 148(11):1244–50. doi:10.1001/archdermatol.2012.2502
141. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis – shaping the immunological landscape. Nat Rev Rheumatol (2016) 12(1):63–8. doi:10.1038/nrrheum.2015.171
142. Yago T, Nanke Y, Kawamoto M, Kobashigawa T, Yamanaka H, Kotake S. IL-23 and Th17 disease in inflammatory arthritis. J Clin Med (2017) 6(9):E81. doi:10.3390/jcm6090081
143. Gelfand JM, Joshi A, Shin D, Dey A, Torrigian D, Rader D, et al. A trial to determine the effect of psoriasis treatment (adalimumab, phototherapy, and placebo) on cardiometabolic disease: the vascular inflammation in psoriasis (VIP) trial. J Invest Dermatol (2017) 138(5):S67. doi:10.1016/j.jid.2018.03.400
Keywords: psoriasis, cardiovascular disease, inflammation, atherosclerosis, vascular inflammation, inflammatory cytokines
Citation: Sajja AP, Joshi AA, Teague HL, Dey AK and Mehta NN (2018) Potential Immunological Links Between Psoriasis and Cardiovascular Disease. Front. Immunol. 9:1234. doi: 10.3389/fimmu.2018.01234
Received: 27 March 2018; Accepted: 16 May 2018;
Published: 01 June 2018
Edited by:
Eva Reali, Istituto Ortopedico Galeazzi (IRCCS), ItalyReviewed by:
Pasquale Maffia, University of Glasgow, United KingdomFrancisco Capani, University of Buenos Aires, Argentina
Copyright: © 2018 Sajja, Joshi, Teague, Dey and Mehta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nehal N. Mehta, bmVoYWwubWVodGFAbmloLmdvdg==