 Isabelle Serr1,2
Isabelle Serr1,2 Carolin Daniel1,2*
Carolin Daniel1,2*
- 1Research Group Immune Tolerance in Diabetes, Institute for Diabetes Research, Helmholtz Diabetes Center at Helmholtz Zentrum München, Munich, Germany
- 2German Center for Diabetes Research (DZD), Munich, Germany
T follicular helper (TFH) cells are an integral part of humoral immunity by providing help to B cells to produce high-affinity antibodies. The TFH precursor compartment circulates in the blood and TFH cell dysregulation is implied in various autoimmune diseases including type 1 diabetes (T1D). Symptomatic T1D is preceded by a preclinical phase (indicated by the presence of islet autoantibodies) with a highly variable progression time to the symptomatic disease. This heterogeneity points toward differences in immune activation in children with a fast versus slow progressor phenotype. In the context of T1D, previous studies on TFH cells have mainly focused on the clinically active state of the disease. In this review article, we aim to specifically discuss recent insights on TFH cells in human islet autoimmunity before the onset of symptomatic T1D. Furthermore, we will highlight advances in the field of TFH differentiation and function during human islet autoimmunity. Specifically, we will focus on the regulation of TFH cells by microRNAs (miRNAs), as well as on the potential use of miRNAs as biomarkers to predict disease progression time and as future drug targets to interfere with autoimmune activation.
Introduction
T follicular helper (TFH) cells are a subset of CD4+ T cells characterized by the expression of the C-X-C chemokine receptor type 5 (CXCR5) (1–3) and their master transcription factor B-cell lymphoma 6 (BCL6) (4–6) as well as secretion of the cytokine interleukin-21 (IL-21) (7–9). The expression of CXCR5 together with a low expression of C-C chemokine receptor 7 (CCR7) allows these T cells to enter the B cell follicle in the secondary lymphoid organs (10, 11), where they take part in the germinal center reaction. Specifically, TFH cells interact with germinal center B cells to induce maturation, class switching, and the production of high-affinity antibodies and are therefore an integral part of humoral immunity (1–3).
Although their primary point of action is in the lymph nodes, studies have demonstrated that TFH cell precursors can be found in the blood circulation. These circulating TFH precursors are characterized by the expression of CXCR5, high expression of programmed cell death 1 and low expression of CCR7. Furthermore, circulating TFH precursors are clonally related and phenotypically similar to germinal center TFH cells and comprise a memory compartment that can be reactivated and expanded in response to immunization (12). Therefore, changes in the frequency and phenotype of circulating TFH precursors correlate with those of active TFH cells in the lymph nodes during infections (13). Since continuous stimulation of TFH cells with antigen, in the follicles provided by germinal center B cells, is important to maintain high levels of BCL6 (14), circulating TFH precursors display low or intermediate levels of BCL6 (13).
T follicular helper precursor cells can be subdivided into different subsets according to the effector cytokines they express in parallel to IL-21. Three TFH subsets can be distinguished based on their surface expression of CXCR3 and CCR6. Th1-like TFH cells are CXCR3+CCR6− and produce IFNγ, Th2-like TFH cells are CXCR3−CCR6− and produce IL-4, IL-5, and IL-13, and Th17-like TFH cells are CXCR3−CCR6+ and secrete IL-17A and IL-22 (15). Whereas Th2- and Th17-like TFH cells can induce naïve B cells to become plasma cells and produce antibodies, Th1-like TFH cells are suggested to lack this ability (15, 16). CXCR3+ TFH precursors were shown to correlate with effective vaccination responses by inducing antibody release from pre-existing memory B cells (16). However, also the memory B cell help by CXCR3+ TFH precursors is less efficient compared to that of their CXCR3− counterparts (13, 17). Th2- and Th17-like TFH cells do, however, impact differentially on the class switching of B cells, with Th2-like TFH cells promoting rather IgG and IgE responses and Th17-like TFH cells promoting IgG and IgA responses (15). A recent study on prostate cancer suggests that Th2- and Th17-like TFH cells also impact differentially on the subtype of IgG antibodies produced (18).
Because of their integral role in humoral immunity, TFH cells have been studied in depth in the context of vaccination. Their function of inducing high-affinity antibody responses additionally implies a role of TFH cells in the development and progression of autoimmune diseases that are characterized by the presence of autoantibodies.
One such autoimmune disease is type 1 diabetes (T1D). T1D is the most common metabolic disorder in children and its incidence is rising steadily, especially in young children (19). Impairments in immune tolerance mechanisms can lead to the destruction of the pancreatic insulin-producing β-cells and consequently a failure of blood glucose control, making life-long insulin replacement therapy necessary for patients with symptomatic T1D.
Symptomatic T1D is preceded by a presymptomatic phase (termed islet autoimmunity), characterized by the presence of autoantibodies against islet autoantigens (insulin, insulinoma antigen 2, glutamic acid decarboxylase, zinc transporter 8). The presence of multiple islet autoantibodies increases the life-long risk to develop the symptomatic disease to approximately 100% (20). The time taken for the progression from the development of the first autoantibodies (seroconversion) to the development of the symptomatic disease is, however, very heterogeneous and can range from months (fast progressors) to decades (slow progressors) (20). Accordingly, in our studies, we distinguish different stages of islet autoimmunity: recent onset of islet autoimmunity with islet autoantibodies for less than 5 years and long-term islet autoimmunity with islet autoantibodies for more than 10 years without progression to clinical overt T1D (21–23). However, the immunological mechanisms underlying these differences in disease progression remain poorly understood (24).
TFH Cells in Presymptomatic T1D
Alterations in the frequency or function of TFH precursor populations in the peripheral blood have been implicated in various autoimmune disorders, including systemic lupus erythematosus and T1D (25–27). Regarding T1D, Kenefeck et al. have demonstrated in a transgenic TCR model that the transfer of TFH cells can induce diabetes. Specifically, they transferred ovalbumin-specific CXCR5+ or CXCR5−CD4+ T cells into recipient mice expressing ovalbumin under the insulin promoter in the β-cells and observed a significant increase in diabetes incidence in mice receiving CXCR5+CD4+ T cells (28). Furthermore, Ferreira et al. observed increased IL-21 production by CD4+ T cells in T1D patients (29). These previous studies on TFH cells in T1D have focused on symptomatic T1D, which excludes conclusions regarding the involvement of TFH cells in the presymptomatic phase or the progression to clinical T1D. The development of multiple islet autoantibodies characterizes the onset of presymptomatic T1D. The important contribution of TFH cells to humoral immunity therefore implicates an involvement of these cells also in disease onset and progression. Accordingly, we found insulin-specific and polyclonal TFH precursor frequencies to be increased during recent onset of islet autoimmunity. This increase was, however, transient and in children with long-term islet autoimmunity without progression to symptomatic T1D, the TFH precursor frequency was similar to that observed in children without islet autoantibodies (22) (Figure 1A). This is in accordance with the observation that children with long-term islet autoimmunity tend to lose their first islet autoantibodies, most commonly insulin autoantibodies (30). Data from birth cohort studies highlight that proinsulin-specific CD4+ T cells of children who developed islet autoantibodies show a gene expression signature resembling TFH/TH17 cell responses already very early on in infancy, well before the development of islet autoantibodies (31). In a recent Finnish study, no alterations in circulating TFH precursors were observed in normoglycemic children with multiple islet autoantibodies (32) (Figure 1A). However, study participants were not discriminated according to the duration of islet autoantibody positivity. These seemingly divergent results highlight the heterogeneity of T1D and underline the necessity to more precisely discriminate the stages of islet autoimmunity and age of study participants.
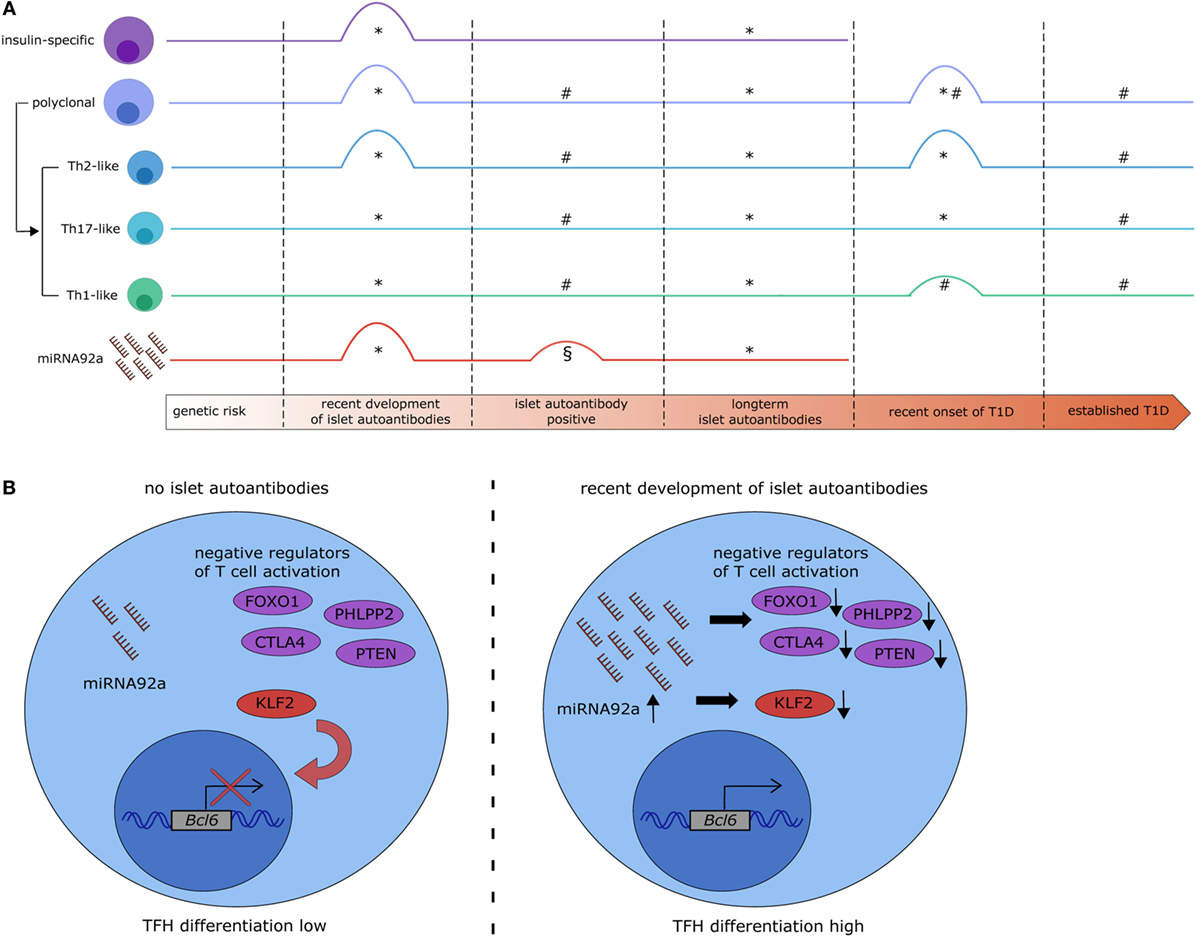
Figure 1. MicroRNA (miRNA)92a expression links alterations in T follicular helper (TFH) precursor frequencies with islet autoimmunity. (A) Overview of recent studies on the dynamics of circulating TFH precursor frequencies and miRNA92a abundance in islet autoimmunity. *Serr et al. (22), #Viisanen et al. (32), §Snowhite et al. (33). #§: islet autoantibody positive participants were not stratified based on the duration of islet autoantibody positivity. §: increase in miRNA92a was borderline significant and statistical significance was not reached after additional data processing. (B) Potential signaling mechanisms in CD4+ T cells targeted by miRNA92a. In states of no islet autoimmunity (left) miRNA92a is expressed at low levels, allowing for the expression of its targets. Targets of miRNA92a are among others negative regulators of T cell activation (e.g., FOXO1, PHLPP2, CTLA4, and PTEN) and negative regulators of TFH differentiation [e.g., krueppel-like factor 2 (KLF2)] which contributes to a reduced expression of the TFH transcription factor B-cell lymphoma 6 (BCL6) and reduced TFH differentiation. During recent onset of islet autoimmunity (right) the expression of miRNA92a is upregulated, leading to a decreased expression of its targets, increased expression of BCL6, and increased TFH differentiation.
Regarding the function of circulating TFH precursors, the analysis of Th1-, Th2-, and Th17-like TFH precursors is relevant, because of differences in their ability to provide B cell help and impact on antibody isotype production (15). Data regarding TFH precursor subsets in autoimmune diseases is limited; however, we reported an increase specifically in the Th2-like TFH subset in children with recent onset of islet autoimmunity and in children with newly diagnosed clinical T1D, whereas Th1- and Th17-like TFH cells were unaltered (22). Although Ig subtypes were not analyzed in our study, previous studies highlighted that Ig isotypes of islet autoantibodies and even IgG subtypes induced in the presymptomatic phase of the disease might influence the disease progression (34–36). Similarly, regarding autoimmune diseases other than T1D, a study by Le Coz et al. highlighted an increase of Th2-like TFH cells, accompanied by a decrease in Th1-like TFH cells in patients with systemic lupus erythematosus (37). In this study, Le Coz et al. demonstrate that IgE levels in the serum of lupus patients correlate with disease activity and are associated with high frequencies of Th2-like TFH cells (37).
Mechanisms of TFH Induction in Islet Autoimmunity
The TFH differentiation process is highly complex, involving several steps and factors (25–27). In 2013, two research groups demonstrated an important role of the microRNA17~92 (miRNA17~92) cluster, which is essential for normal TFH development and function in mice (38, 39). miRNAs are small, ~22 nucleotide long, non-coding RNAs which can complementarily bind their target mRNAs in the RNA-induced silencing complex and induce their translational silencing or degradation (40–42). miRNAs usually have a multitude of targets and induce rather modest regulation (43, 44), enabling them to regulate complex cellular states, such as T cell activation (45, 46) and making them suitable targets for immune modulating therapies.
The miRNA17~92 cluster transcribes six mature miRNAs (miRNA17, miRNA18a, miRNA19a, miRNA19b, miRNA20a, and miRNA92a). The relevance of these miRNAs in autoimmune diseases is highlighted by the fact that overexpression of the cluster leads to autoimmunity and autoantibody production in mice (47). Regarding the role of the cluster in murine TFH cell differentiation, miRNA17~92 regulates differentiation and migration of TFH cells together with Bcl6 by repressing TFH subset inappropriate genes like retinoid-related orphan receptor α (Rora) and by regulating signaling molecules important for TFH differentiation and function, such as inducible T cell costimulator (Icos) and phosphatidylinositol-3-kinase (PI3K)/protein kinase B signaling (38, 39). Accordingly, two validated targets of miRNA92a are the phosphatase and tensin homolog (Pten) and PH domain and leucine rich repeat protein phosphatase 2 (Phlpp2), both negative regulators of PI3K signaling (47, 48). In line with its role in TFH cell differentiation and function, additional confirmed targets for miRNA92a are other negative regulators of T cell activation, such as forkhead box protein O1 (Foxo1) and cytotoxic T-lymphocyte associated protein 4 (Ctla4) (47, 48).
In an miRNA profiling approach to investigate miRNAs in T cells that could be involved in human autoimmune activation, we identified miRNA92a to be significantly increased in CD4+ T cells from children with ongoing islet autoimmunity compared to healthy controls (22). Confirmation via RT-qPCR highlighted an increase in miRNA92a specifically in T cells from children with recent onset of islet autoimmunity and not in children with long-term islet autoimmunity (Figure 1A). Our analysis demonstrated furthermore that this increase in miRNA92a expression correlates with TFH precursor frequencies in the peripheral blood. Accordingly, the lowest expression of miRNA92a was found in T cells from children with long-term islet autoimmunity.
For the investigation of the role of miRNA92a in human TFH differentiation, in vitro TFH induction assays, relying on the stimulation of human naïve CD4+ T cells with anti-CD3 and anti-CD28 antibodies in the presence of memory B cells, were established. In line with a role of miRNA92a in TFH induction, human TFH induction was decreased in in vitro assays, when miRNA92a activity was blocked, whereas an miRNA92a mimic promoted TFH induction (Figure 1B). In assays with an miRNA92a mimic, negative regulators of T cell activation such as PTEN, PHLPP2, FOXO1, and CTLA4 that are confirmed targets of miRNA92a, were reduced in their expression (22) (Figure 1B). These findings are in line with previous studies, highlighting that TFH cell differentiation is largely dependent on low levels of FOXO1, maintained either by ICOS-PI3K signaling or by degradation via the E3 ubiquitin ligase ITCH (49, 50). miRNA92a mediated TFH induction likewise depends on PI3K signaling, since in vitro TFH induction with an miRNA92a mimic is blunted in the presence of a PI3K inhibitor, whereas it is increased when PTEN is inhibited (22). PTEN, as a negative regulator of PI3K signaling, is critically involved in the de novo induction of regulatory T cells (Tregs). Accordingly, in vitro Treg induction from naïve CD4+ T cells was found to be impaired in the presence of an miRNA92a mimic. Moreover, insulin-specific Treg frequencies are reduced in children with recent onset of islet autoimmunity, a disease state where miRNA92a abundance was shown to be significantly enhanced in T cells (21, 22) (Figure 2A).
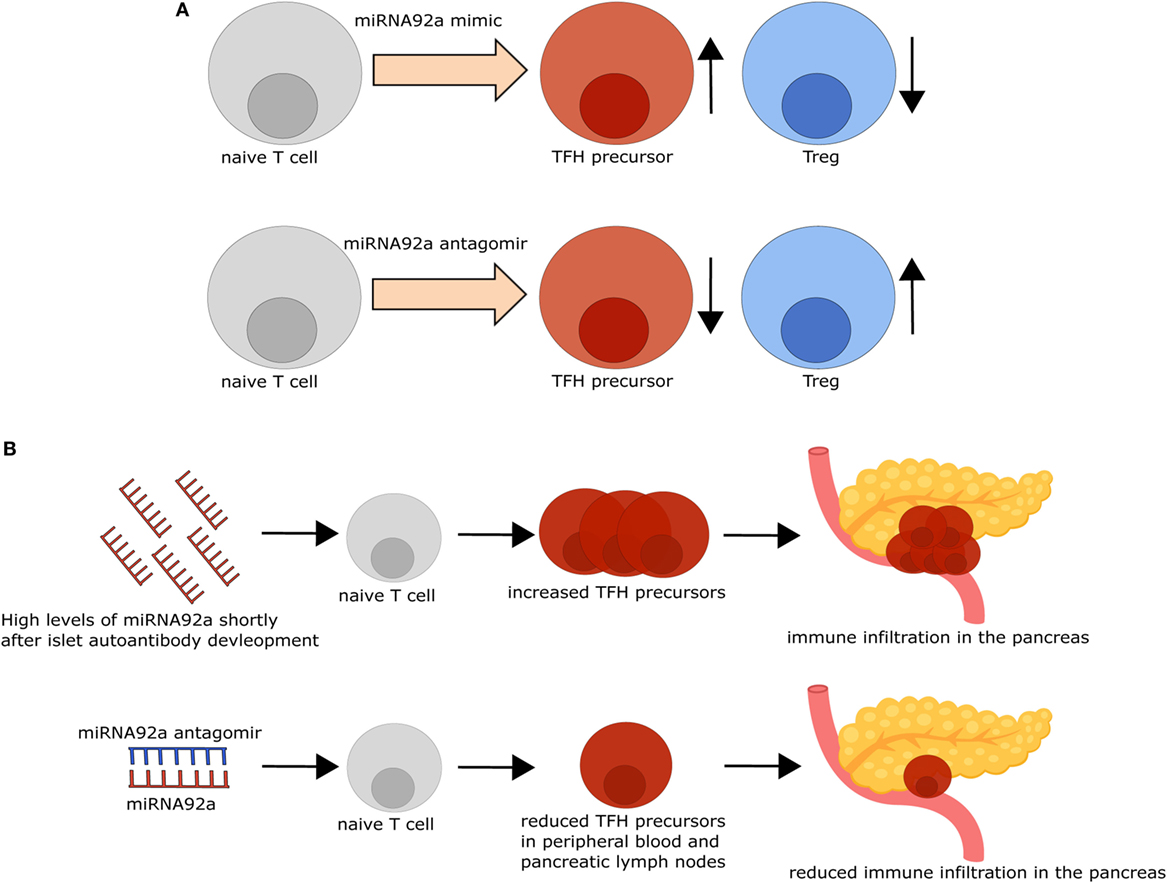
Figure 2. Modifying microRNA (miRNA)92a activity impacts T follicular helper (TFH) and regulatory T cell (Treg) induction in vitro and immune activation in vivo. (A) TFH induction in vitro is increased, while Treg induction in vitro is decreased in the presence of an miRNA92a mimic (upper row). By contrast, inhibition of miRNA92a, using an miRNA92a antagomir (lower row), results in decreased TFH induction and increased Treg induction in vitro. (B) Non-obese diabetic (NOD) mice with islet autoantibodies express increased amounts of miRNA92a and increased TFH precursor frequencies, accompanied by immune infiltration in the pancreas. Treatment of NOD mice with an miRNA92a antagomir reduces circulating TFH precursor frequencies and immune cell infiltration in the pancreas.
T follicular helper cell function is largely dependent on their ability to enter the B cell follicle in the lymph nodes. Therefore, molecules that regulate lymphocyte trafficking and homing are important mediators of TFH cell function. One example is krueppel-like factor 2 (KLF2). Lee et al. demonstrated that TFH differentiation is dependent on low levels of Klf2, since Klf2 induces the expression of sphingosine 1 phosphate receptor 1 (S1pr1), which opposes TFH induction (51). Furthermore, Klf2 was shown to inhibit Bcl6 expression by upregulating B-lymphocyte induced maturation protein 1 (51). Interestingly, our data suggest that KLF2 can be directly targeted by miRNA92a, since a target site blocker, that inhibits the binding of miRNA92a specifically to KLF2 abolishes in vitro TFH induction (22) (Figure 1B), thereby offering one additional mechanism of miRNA92a-mediated TFH differentiation.
miRNAs as Biomarkers in Islet Autoimmunity
The heterogeneous disease progression from the development of islet autoantibodies to the symptomatic disease necessitates the discovery of biomarkers that will enable a better prediction of the progression time to the clinically active disease. To that end, it remains to be determined, whether changes in miRNA92a expression can also be observed in the serum of children with recent development of islet autoantibodies, or whether the detection of these alterations is limited to the CD4+ T cell population. One recent study by Snowhite et al. aimed at identifying differentially expressed miRNAs in the serum of children with and without autoantibodies. miRNA92a was one of the identified miRNAs that was increased in children with autoantibodies, however, this increase was only borderline significant and not significant after further data processing (33) (Figure 1A). The autoantibody positive children investigated in this study were not stratified based on the duration of autoantibody positivity, which might account for this outcome. The study of longitudinal samples from children at risk of developing T1D will help to assess the usefulness of miRNA92a, TFH cell frequencies, and their respective subsets as biomarkers to predict the progression to clinically overt T1D. More specifically, the analysis of possible correlations between these markers and autoantibody titers or subtypes might be of interest. In this context, a correlation of miRNA92a expression in T cells with TFH precursor frequencies in the blood as well as a modest correlation with insulin autoantibody titers was reported (22). A more detailed analysis of TFH precursor subsets might be especially relevant, because of their divergent functions with respect to providing B cell help and impacting Ig subtype production. Moreover, given the negative impact of high miRNA92a levels on Treg induction, the analysis of Treg frequencies and possible inverse correlations with miRNA92a abundance or TFH subset frequencies can be envisioned. Together, these analyses could be useful to define TFH signature profiles that might serve as biomarkers for assessing T1D disease progression.
Targeting miRNAs to Interfere with Autoimmune Activation
miRNAs can function as promising novel potential drug targets, since they can be targeted by small, highly specific oligonucleotides. In this regard, clinical trials for the treatment of hepatitis C virus infections with an miRNA inhibitor have been successfully conducted (52). Targeting specific cell types, especially immune cells, with miRNA inhibitors is, however, challenging, because of the negative charge of the oligonucleotides which inhibits penetration of the cell membrane (53). Research efforts focus mainly on encapsulation techniques, and various nanoparticles were shown to mediate an efficient uptake of small RNAs by lymphocyte populations (54). Other techniques, targeting T cells more specifically, are, e.g., the use of a single chain CD7 antibody (scFvCD7) fused to an oligonucleotide-nona-arginine peptide (55).
The possibility of altering immune activation and regulation by targeting miRNAs was demonstrated in insulin autoantibody positive non-obese diabetic mice, the most commonly used mouse model for T1D. I.p. Application of an miRNA92a antagomir, optimized for in vivo use, decreased TFH frequencies and immune activation in the pancreas, accompanied by decreased insulitis scores and autoantibody titers (22). Furthermore, this decreased immune activation went along with increased frequencies of Tregs in treated animals, suggesting that, apart from reducing immune activation, inhibition of miRNA92a positively impacts on mediators of T cell tolerance (Figure 2B).
The restoration of immune tolerance mechanisms in autoimmune diseases is a long envisioned goal. Since Tregs are important mediators of T cell tolerance in the periphery and can be induced in an antigen-specific fashion, Treg induction could contribute to interfering with the progression of autoimmune activation in autoimmune diseases. This notion is supported by identified associations indicating high frequencies of insulin-specific Tregs accompanied by reduced numbers of insulin-specific TFH precursors in the peripheral blood of children with long-term islet autoimmunity without progression to clinically active T1D. During recent onset of islet autoimmunity, a significant decrease in insulin-specific Treg frequencies was observed accompanied by impaired in vitro Treg induction (23). Specifically, during this critical time frame we found an increased sensitivity to antigenic stimulation in naïve CD4+ T cells and reduced expression of negative regulators of T cell activation which can interfere with efficient Treg induction (23). Using miRNAs to tame T cell activation during ongoing islet autoimmunity might therefore open a window of opportunity for improving Treg induction potential in a setting, where the autoimmune process is already in progress. However, the effectiveness of inhibiting miRNA92a to interfere with autoimmune activation and progression to T1D requires long-term in vivo studies in animal models of T1D, which are missing so far.
Conclusion
Accumulating evidence points toward a role of TFH cells in the development of autoimmune diseases including T1D. During recent onset of islet autoimmunity, children display increased frequencies of TFH precursor cells, specifically Th2-like TFH precursors, whereas this increase is absent in children with long-term islet autoimmunity without overt T1D (22). The analysis of TFH cell frequencies or miRNAs involved in TFH development in longitudinal samples could therefore help to identify biomarkers in order to improve our ability to predict the progression time to clinically overt T1D, as well as to improve the stratification of respective disease groups. In addition, progress is made regarding the cell type-specific delivery of miRNA inhibitors or mimics. Since miRNAs regulate cellular states, rather than single targets, they compose a new, promising group of future drug targets. In this regard, miRNAs such as miRNA92a that regulate TFH differentiation and function might be targeted to limit immune activation in settings of autoimmunity such as T1D.
Author Contributions
IS wrote the manuscript and designed illustrations. CD conceptualized, wrote, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
CD is supported by a Research Group at Helmholtz Zentrum München, by the German Center for Diabetes Research (DZD), and through a membership in the CRC1054 of the Deutsche Forschungsgemeinschaft (B11).
References
1. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192(11):1545–52. doi:10.1084/jem.192.11.1545
2. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med (2000) 192(11):1553–62. doi:10.1084/jem.192.11.1553
3. Kim CH, Rott LS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J Exp Med (2001) 193(12):1373–81. doi:10.1084/jem.193.12.1373
4. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity (2008) 29(1):138–49. doi:10.1016/j.immuni.2008.05.009
5. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi:10.1126/science.1175870
6. Yu D, Batten M, Mackay CR, King C. Lineage specification and heterogeneity of T follicular helper cells. Curr Opin Immunol (2009) 21(6):619–25. doi:10.1016/j.coi.2009.09.013
7. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med (2010) 207(1):155–71. doi:10.1084/jem.20091706
8. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med (2010) 207(2):353–63. doi:10.1084/jem.20091738
9. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med (2010) 207(2):365–78. doi:10.1084/jem.20091777
10. Hardtke S, Ohl L, Förster R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood (2005) 106(6):1924–31. doi:10.1182/blood-2004-11-4494
11. Haynes NM, Allen CDC, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol (2007) 179(8):5099–108. doi:10.4049/jimmunol.179.8.5099
12. Heit A, Schmitz F, Gerdts S, Flach B, Moore MS, Perkins JA, et al. Vaccination establishes clonal relatives of germinal center T cells in the blood of humans. J Exp Med (2017) 214(7):2139–52. doi:10.1084/jem.20161794
13. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity (2013) 39(4):770–81. doi:10.1016/j.immuni.2013.09.007
14. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity (2013) 38(3):596–605. doi:10.1016/j.immuni.2012.11.020
15. Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity (2011) 34(1):108–21. doi:10.1016/j.immuni.2010.12.012
16. Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med (2013) 5(176):176ra132. doi:10.1126/scitranslmed.3005191
17. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39(4):758–69. doi:10.1016/j.immuni.2013.08.031
18. Tan J, Jin X, Zhao R, Wei X, Liu Y, Kong X. Beneficial effect of T follicular helper cells on antibody class switching of B cells in prostate cancer. Oncol Rep (2015) 33(3):1512–8. doi:10.3892/or.2014.3684
19. Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G, Group ES. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: a multicentre prospective registration study. Lancet (2009) 373(9680):2027–33. doi:10.1016/S0140-6736(09)60568-7
20. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion tomultiple islet autoantibodies and risk of progression to diabetes in children. JAMA (2013) 309(23):2473–9. doi:10.1001/jama.2013.6285
21. Serr I, Furst RW, Achenbach P, Scherm MG, Gokmen F, Haupt F, et al. Type 1 diabetes vaccine candidates promote human Foxp3(+)Treg induction in humanized mice. Nat Commun (2016) 7:10991. doi:10.1038/ncomms10991
22. Serr I, Fürst RW, Ott VB, Scherm MG, Nikolaev A, Gökmen F, et al. miRNA92a targets KLF2 and the phosphatase PTEN signaling to promote human T follicular helper precursors in T1D islet autoimmunity. Proc Natl Acad Sci U S A (2016) 113(43):E6659–68. doi:10.1073/pnas.1606646113
23. Serr I, Scherm MG, Zahm AM, Schug J, Flynn VK, Hippich M, et al. A miRNA181a/NFAT5 axis links impaired T cell tolerance induction with autoimmune type 1 diabetes. Sci Transl Med (2018) 10(422):eaag1782. doi:10.1126/scitranslmed.aag1782
24. Achenbach P, Hummel M, Thumer L, Boerschmann H, Hofelmann D, Ziegler AG. Characteristics of rapid vs slow progression to type 1 diabetes in multiple islet autoantibody-positive children. Diabetologia (2013) 56(7):1615–22. doi:10.1007/s00125-013-2896-y
25. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly – TFH cells in human health and disease. Nat Rev Immunol (2013) 13(6):412–26. doi:10.1038/nri3447
26. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi:10.1016/j.immuni.2014.10.004
27. Scherm MG, Ott VB, Daniel C. Follicular helper T cells in autoimmunity. Curr Diab Rep (2016) 16(8):75. doi:10.1007/s11892-016-0770-2
28. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest (2015) 125(1):292–303. doi:10.1172/JCI76238
29. Ferreira RC, Simons HZ, Thompson WS, Cutler AJ, Dopico XC, Smyth DJ, et al. IL-21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia (2015) 58(4):781–90. doi:10.1007/s00125-015-3509-8
30. Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity (2010) 32(4):468–78. doi:10.1016/j.immuni.2010.03.018
31. Heninger A-K, Eugster A, Kuehn D, Buettner F, Kuhn M, Lindner A, et al. A divergent population of autoantigen-responsive CD4+ T cells in infants prior to β cell autoimmunity. Sci Transl Med (2017) 9(378):eaaf8848. doi:10.1126/scitranslmed.aaf8848
32. Viisanen T, Ihantola E-L, Näntö-Salonen K, Hyöty H, Nurminen N, Selvenius J, et al. Circulating CXCR5+PD-1+ICOS+ follicular T helper cells are increased close to the diagnosis of type 1 diabetes in children with multiple autoantibodies. Diabetes (2016) 66(2):437–47. doi:10.2337/db16-0714
33. Snowhite IV, Allende G, Sosenko J, Pastori RL, Messinger Cayetano S, Pugliese A. Association of serum microRNAs with islet autoimmunity, disease progression and metabolic impairment in relatives at risk of type 1 diabetes. Diabetologia (2017) 60(8):1409–22. doi:10.1007/s00125-017-4294-3
34. Petersen JS, Kulmala P, Clausen JT, Knip M, Dyrberg T; The Childhood Diabetes in Finland Study Group. Progression to type 1 diabetes is associated with a change in the immunoglobulin isotype profile of autoantibodies to glutamic acid decarboxylase (GAD65). Clin Immunol (1999) 90(2):276–81. doi:10.1006/clim.1998.4641
35. Hoppu S, Ronkainen MS, Kimpimaki T, Simell S, Korhonen S, Ilonen J, et al. Insulin autoantibody isotypes during the prediabetic process in young children with increased genetic risk of type 1 diabetes. Pediatr Res (2004) 55(2):236–42. doi:10.1203/01.PDR.0000100905.41131.3F
36. Hoppu S, Härkönen T, Ronkainen MS, Simell S, Hekkala A, Toivonen A, et al. IA-2 antibody isotypes and epitope specificity during the prediabetic process in children with HLA-conferred susceptibility to type I diabetes. Clin Exp Immunol (2006) 144(1):59–66. doi:10.1111/j.1365-2249.2006.03033.x
37. Le Coz C, Joublin A, Pasquali J-L, Korganow A-S, Dumortier H, Monneaux F. Circulating TFH subset distribution is strongly affected in lupus patients with an active disease. PLoS One (2013) 8(9):e75319. doi:10.1371/journal.pone.0075319
38. Baumjohann D, Kageyama R, Clingan JM, Morar MM, Patel S, de Kouchkovsky D, et al. The microRNA cluster miR-17-92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat Immunol (2013) 14(8):840–8. doi:10.1038/ni.2642
39. Kang SG, Liu WH, Lu P, Jin HY, Lim HW, Shepherd J, et al. MicroRNAs of the miR-17-92 family are critical regulators of T(FH) differentiation. Nat Immunol (2013) 14(8):849–57. doi:10.1038/ni.2648
40. Olsen PH, Ambros V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol (1999) 216(2):671–80. doi:10.1006/dbio.1999.9523
41. Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian MicroRNA targets. Cell (2003) 115(7):787–98. doi:10.1016/S0092-8674(03)01018-3
42. Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell (2005) 122(4):553–63. doi:10.1016/j.cell.2005.07.031
43. Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature (2008) 455(7209):64–71. doi:10.1038/nature07242
44. Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature (2008) 455(7209):58–63. doi:10.1038/nature07228
45. Cobb BS, Nesterova TB, Thompson E, Hertweck A, O’Connor E, Godwin J, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med (2005) 201(9):1367–73. doi:10.1084/jem.20050572
46. Cobb BS, Hertweck A, Smith J, O’Connor E, Graf D, Cook T, et al. A role for Dicer in immune regulation. J Exp Med (2006) 203(11):2519–27. doi:10.1084/jem.20061692
47. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol (2008) 9(4):405–14. doi:10.1038/ni1575
48. Jiang P, Rao EY, Meng N, Zhao Y, Wang JJ. MicroRNA-17-92 significantly enhances radioresistance in human mantle cell lymphoma cells. Radiat Oncol (2010) 5(1):100. doi:10.1186/1748-717x-5-100
49. Xiao N, Eto D, Elly C, Peng G, Crotty S, Liu Y-C. The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat Immunol (2014) 15(7):657–66. doi:10.1038/ni.2912
50. Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai C-Y, Emslie E, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity (2015) 42(2):239–51. doi:10.1016/j.immuni.2015.01.017
51. Lee J-Y, Skon CN, Lee YJ, Oh S, Taylor JJ, Malhotra D, et al. The transcription factor KLF2 restrains CD4+ T follicular helper cell differentiation. Immunity (2015) 42(2):252–64. doi:10.1016/j.immuni.2015.01.013
52. Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting MicroRNA. N Engl J Med (2013) 368(18):1685–94. doi:10.1056/NEJMoa1209026
53. Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov (2014) 13(8):622–38. doi:10.1038/nrd4359
54. Peer D, Park EJ, Morishita Y, Carman CV, Shimaoka M. Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science (2008) 319(5863):627–30. doi:10.1126/science.1149859
Keywords: T follicular helper cells, islet autoimmunity, microRNA92a, krueppel-like factor 2, type 1 diabetes
Citation: Serr I and Daniel C (2018) Regulation of T Follicular Helper Cells in Islet Autoimmunity. Front. Immunol. 9:1729. doi: 10.3389/fimmu.2018.01729
Received: 04 May 2018; Accepted: 12 July 2018;
Published: 23 July 2018
Edited by:
Georgia Fousteri, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Lucy S. K. Walker, University College London, United KingdomJoanna Groom, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright: © 2018 Serr and Daniel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolin Daniel, Y2Fyb2xpbi5kYW5pZWxAaGVsbWhvbHR6LW11ZW5jaGVuLmRl