 Shuang Song
Shuang Song Patrick D. Matthias
Patrick D. Matthias- 1Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland
- 2Faculty of Sciences, University of Basel, Basel, Switzerland
Germinal centers (GCs) are essential structures of the humoral immune response, which form in the periphery in response to T cell dependent antigens. During the GC reaction, B cells undergo critical differentiation steps, which ultimately lead to the generation of antibodies with altered effector function and higher affinity for the selected antigen. Remarkably, many of the B cell tumors have their origin in the GCs; thus, understanding how the formation of these structures is regulated or deregulated is of high medical importance. This review gives an overview of the transcription factors that have been linked to the generation of GCs, and of their roles in the process.
Background to B Cell Development
B (and T) cells represent a unique model of cellular development, in which cells of multiple differentiation stages can be identified based on surface markers and readily isolated. Owing to these advantages, the lymphoid system has been used widely, beyond immunology, as a developmental paradigm in which the role of transcription factors (TFs) or signaling molecules can be tested experimentally. Several excellent reviews exist that describe in detail how B lymphocytes develop, what regulatory circuits are critical, or the details of GC development (1–11). We will therefore not discuss these aspects in detail, but will only give a high-level overview, and then focus this review on the transcriptional control of GCs formation.
B cells originate and develop in the bone marrow from hematopoietic stem cells (HSCs) that differentiate into progenitor stages of increasingly restricted potential. Once committed to the B lineage, B cell progenitors go through several successive stages, at which key events of their developmental fate take place. In particular, the PreB stage represents the phase during which immunoglobulin (Ig) genes, which code for the antibody molecules, rearrange their DNA segments in order to produce functional genes. The heavy chain rearranges first at the ProB stage, followed by the light chain at the small PreB-II stage. Immature B cells then express IgM at their surface and exit the bone marrow to enter the circulation and move to peripheral lymphoid organs such as the spleen or the lymph nodes. There, marginal zone (MZ) B cells play vital functions in T cell-independent humoral immune responses against blood-borne pathogens, follicular B cells can capture antigen presented by Follicular Dendritic Cells (FDCs) and present it to CD4+ follicular T helper cells (TFH) that are located around the B cell zone of the developing GC. This is the time during which critical signals, sent by the TFH cells, induce isotype switching (so-called class switching, which exchanges IgM for IgG) and expansion of B cell clones starts. These B cells are called centroblasts and form the dark zone (DZ) of the GC. After several rounds of proliferation, somatic hypermutation begins, a process by which the Ig DNA becomes mutated under the action of activation-induced cytidine deaminase (AID), leading to the generation of diverse clones expressing antibodies with different, potentially higher, affinity for antigen. From there, the B cells (centroblasts) move to the adjacent region called the light zone (LZ), where they express their antibody on the cell surface. GC B cells in the light zone are called centrocytes and are in a near apoptotic state. It is there that selection for the quality (affinity) of the antibody takes place: based on the affinity of the antibody for the antigen, the B cell can be eliminated or rescued and sent back to the dark zone as centroblast for an additional round of mutations, followed by renewed entry into the light zone and further antibody affinity testing. At some point in this dark zone–light zone selection cycle, the B cell expresses a high affinity antibody and can now exit the GC as a plasma cell that secretes high amounts of the antibody, or as a memory B cell that is ready to be reactivated upon future encounter with the antigen.
The rest of this review will put the emphasis on the transcriptional control of the formation and function of GCs, and highlight in particular TFs that are essential.
Transcription Factors Regulating GC Formation
GC Initiation
Initiation of the GC reaction involves activation of the B cell receptor (BCR) by antigen engagement, followed by interaction of these B cells with antigen presenting cells and TFH cells, which provide further activation signals (2, 3). Figure 1 summarizes the molecular networks regulating initiation and function of the germinal centers reaction.
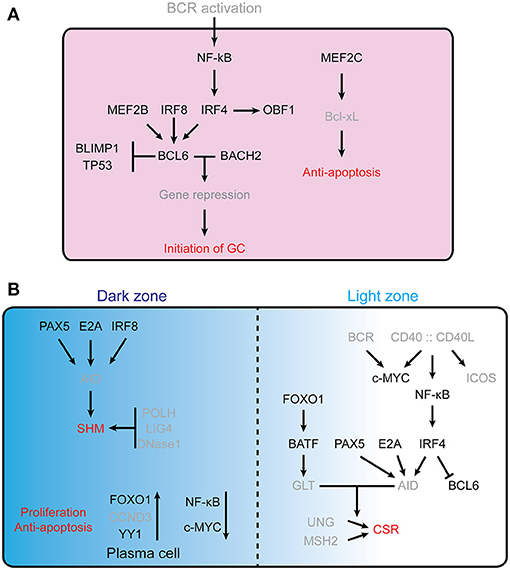
Figure 1. Transcription factors controling GC formation. (A) Initiation of the GC reaction in follicular B cells. For clarity, TFs are indicated in black, while other molecules (e.g., receptors, cytokines, etc…) are in gray. B cell lymphoma 6 (BCL6) is essential for the initiation of germinal center, MEF2B, IRF8, IRF4, BLIMP1, and TP53 are involved in regulating the expression of Bcl6. BCL6 and Bach2 cooperatively (12) repress gene expression and thus allow the establishment of the germinal center B cell program. MEF2C is required for B cell survival post-antigen stimulation by upregulating the Bcl2l1. (B) Schematic representation of the dark zone and light zone of the GC. The different TFs involved are indicated, as well as some of the processes regulated (SHM, Proliferation, anti-apoptosis, CSR), see text for further details. In the GC DZ, AID is a key enzyme for SHM; its expression is controlled by PAX5, E2A, and IRF8. POLH, LIG4, and DNaseI are required for SHM and are highly expressed in DZ B cells. FOXO1 is a key factor for maintaining the GC DZ B cell program, CCND3 is preferentially expressed in GC DZ B cells and YY1 is required for GC DZ B cell proliferation and survival. NF-κB signaling and c-Myc are not essential for GC DZ B cells. In the GC LZ, CD40 signaling stimulated NF-κB further stimulates IRF4 expression, which suppresses Bcl6 gene (13, 14). PAX5, E2A, and IRF4 are key factors in regulating AID level. BATF, a downstream target of FOXO1, regulates germline transcripts (GLTs) in centrocytes. GLT levels are highly correlated with accessibility of AID in CSR.
Transcription factors that are downstream of the BCR, such as the transcription coactivator OBF1 (a.k.a. OCA-B, or Bob1), a B cell-specific coactivator for the octamer transcription factors OCT1 and OCT2, are critical for GC formation (15–18). Mice deficient in Pou2f2 (encoding OCT2), Pou2af1 (encoding OBF1) or both showed complete lack of GCs (19). The underlying molecular mechanism is not clear yet, and the target genes of OBF1/OCT2 in the context of the germinal center reaction are largely unknown, although Spi-B which itself is required for GCs (20, 21) has been identified as a downstream target of OBF1 (22). Moreover, in CD4+ T cells OBF1 and OCT1/OCT2 directly bind to the promoter region of Bcl6 and activate its transcription, thereby promoting the development of TFH cells (23). The putative role of these factors in regulating Bcl6 expression in early GC B cells remains to be investigated.
BCL6 is a zinc finger TF that is essential for germinal center formation, as Bcl6-null mice completely lack GCs and affinity maturation (3, 24). During the early phase of the GC response, antigen stimulated B cells rely on TFH cells for differentiation into GC B cells, and interaction between TFH and B cells leads to the upregulation of BCL6 (25). Moreover, the upregulation of BCL6 leads to stabilized conjugation between B and TFH cells, creating a positive feedback loop that enhances the GC formation program (3, 25). Failure in BCL6 upregulation prevents B cells from entering GC clusters and impairs the upregulation of CXCR4, a chemokine receptor expressed on germinal center DZ B cells that is critical for the maintenance of GC structural integrity (25).
IRF4 is required at the early stage of GC formation. In transplantation experiments, Irf4−/− B cells fail to differentiate into GC B cells (26). Conditional knockout of Irf4 by CD19cre which deletes from early B cells onwards leads to impaired GC formation (26). In contrast, once GCs have formed or initiated, IRF4 is no longer needed, as conditional knockout by Cγ1cre which deletes in already formed GC cells has minimal effects on GC differentiation (27). These results suggest that IRF4 is required for the very early phase upon T-cell-dependent antigen stimulation. Additional evidence supporting this idea is the rapid upregulation of IRF4 following BCR stimulation (28). Moreover, IRF4 is involved in modulating the expression of BCL6 and OBF1, which both are key factors for GC initiation (3, 26). Taken together, IRF4 plays an important role in the early initiation phase of GC formation, possibly by regulating the induction of Bcl6 and Pou2af1.
IRF8 was reported to upregulate BCL6 and AID levels in GC B cells (29, 30), and it was shown to promote GC B cells survival by regulating the expression level of MDM2 (31). However, deletion of IRF8 in B cells did not affect GC formation (32). Moreover, IRF8 is involved in the regulation of the BCL6-related transcriptional program in GC cells by directly interacting with BCOR (B cell lymphoma 6 corepressor) and BCL6. In transactivation assays, IRF8 augments the transcription repressive activity of BCL6 (33).
MEF2C is required for the proliferation and survival of B cells upon antigen receptor stimulation by upregulating the expression level of Bcl2l1 (encoding the Bcl-xL protein) and several cell cycle related genes (34). Specific deletion of Mef2c in B cells leads to reduced proliferation and increased cell apoptosis upon anti-IgM stimulation. However, the responses are normal in the case of LPS, CD40, IL4, BAFF and RP105 stimulations. By histological examination, reduced number of GC follicules are observed in the spleens of Mef2Cfl/fl-CD19cre mice immunized with sheep red blood cells (SRBC) (34). MEF2B, another member of the MEF2 family, has been found to be mutated in ca. 11% of diffuse large B cell lymphoma (DLBCL), which are GC-derived tumors (35). MEF2B directly activates Bcl6 transcription by binding to the regulatory region 1 kb upstream of the Bcl6 gene transcription start site (35). Mutation of the MEF2B binding motif in the Bcl6 gene promoter abrogates Bcl6 transcription activity in cotransfection assays in 293T cells. Furthermore, knockdown of MEF2B protein by shRNAs leads to downregulation of BCL6 and upregulation of BCL6 target genes. These data suggest that MEF2B plays an important role in early GC formation by modulating Bcl6 expression (35, 36).
BATF is a transcription factor of the AP-1 family, which is involved in GC structure establishment and class switch recombination. Batf−/− mice failed to develop normal GC structures when immunized with SRBC, as characterized by a lack of CD95 or GL7 positive B cells (37). Batf-null TFH cells lack expression of the chemokine receptor CXCR5, which is essential for GC structure integrity. Additionally, the expression of Bcl6 and c-Maf, both of which are important factors for TFH cells development, is downregulated in absence of BATF (37).
c-MYC is another TF indispensable during the early phase of germinal center formation. Its expression is induced already 1–2 days after immunization (38) and it is required for GC maintenance, as conditional deletion of c-Myc by Cγ1cre leads to impaired GCs (39).
GC Development
The dark zone and the light zone of the GC are organized by the expression of the chemokine receptors CXCR4 and CXCR5, respectively (40). Thus, one can expect that TFs critical for CXCR4 and CXCR5 expression will be important for GCs.
GC Dark Zone
The germinal center DZ is characterized by an interconnected network of CXCL12 expressing reticular cells and compactly filled with rapidly proliferating centroblasts (41).
FOXO1 is highly expressed in human and mouse GC B cells, and its expression is largely specific to DZ B cells (with also some expression in naïve B cells) (42). Like in Cxcr4−/− mice, GCs from Foxo1fl/fl-Cγ1cre mice completely lack a DZ structure, while the differentiation of plasma cells is normal (42, 43). Foxo1-null GCs lack proper structural polarization and show an even distribution of the FDC network (42). FOXO1, together with BCL6, represses the expression of B lymphocyte induced maturation protein 1 (BLIMP1), a key factor promoting differentiation of GC B cells into plasma cells, which is encoded by the Prdm1 gene. By binding to the Prdm1 promoter region, FOXO1 and BCL6 maintain the germinal center DZ program (42).
Bcl6-null GC precursor B cells fail to upregulate the expression of CXCR4 (25), which is a crucial chemokine receptor for GC DZ B cells. c-MYC is required throughout the early and late initiation phases of GC formation, but is not expressed in the proliferating DZ B cells (3), where it is repressed by BCL6 (38).
YY1 is required for GC B cell proliferation and GC development at least partly by modulating cell apoptosis (44). Deletion of Yy1 specifically in GC B cells leads to a significant decrease in the number of DZ B cells, and elevated cell apoptosis (44).
Somatic hypermutation (SHM)
SHM generates a wide repertoire of affinities toward specific antigens, and mainly takes place in the DZ (45), although some extrafollicular SHM has been reported in transgenic mice deficient in the ability to establish GCs (46). AID, encoded by the Aicda gene, is the enzyme responsible for SHM and class switch recombination (47, 48). AID deaminates cytidines in DNA (49–54), followed by error-prone repair involving different DNA repair factors and ultimately leading to the introduction of somatic mutations (55). Thus, transcription factors which affect the expression of Aicda and DNA-damage tolerance related genes should be important for SHM. E proteins (56), PAX5 (57) and IRF8 (29) have been associated with positive regulation of Aicda transcription.
FOXO1 is involved in SHM by affecting the protein level of AID: Foxo1-null GC B cells show reduced level of AID enzyme, while mRNA level of Aicda is unchanged. Therefore, Foxo1-null GC B cells carry lower level of mutations in Ig locus than control cells (58).
Irf8 mRNA level peaks in centroblasts, and IRF8 regulates SHM by modulating the expression of Aicda and Bcl6: knockdown of IRF8 by siRNA leads to decreased transcription of Aicda and Bcl6 (29). By ChIP, IRF8 binds to the promoter regions of Aicda and Bcl6 in both human and mouse B cells. Furthermore, luciferase assays showed that IRF8 directly regulates the transcription of Aicda and Bcl6 in HeLa cells cotransfected with an IRF8 expression vector and a reporter containing promoter regions of Aicda or Bcl6 (29). Moreover, IRF8 promotes GC B cells survival by regulating the expression level of MDM2 in the case of DNA damage (31).
Aicda expression is significantly reduced in activated B cells in which the helix-loop-helix factor ID3 is ectopically expressed. The possible mechanism is that ID3 inhibits the DNA-binding activity of E-proteins which activate the expression of Aicda (56, 59).
Light Zone
Three crucial B-cell developmental processes take place in the GC light zone: (i) selection of B cells that produce high-affinity antibodies, (ii) CSR, and (iii) initiation of centrocytes differentiation into plasma cells or memory B cells (9).
After rapid expansion in the DZ, B cells migrate to the LZ where those carrying high affinity B cell receptor genes are selected. The BCR pathway plays a fundamental role in this process: BCR signaling leads to phosphorylated AKT, and activated AKT further phosphorylates FOXO1 which then relocates from the nucleus to the cytoplasm (60). CD40 stimulation leads to NF-κB-mediated upregulation of IRF4 (13), which in turn represses Bcl6 transcription (61). Together, these coordinated actions terminate the dark zone-associated transcriptional programme and allow establishment of the LZ transcriptome (13).
c-MYC is absent in most GC B cells, however, its expression is induced in high affinity BCR presenting GC B cells when receiving help from TFH cells, in a process that requires both BCR and CD40 signaling (60). In addition to the requirement of c-MYC activity during the initial stage of GC formation, c-MYC is needed for GC maintenance in the late GC response (38). With the help from TFH cells, c-MYC is transiently induced and upregulated in a small fraction of high affinity BCR expressing GC B cells within the LZ compartment. The Omomyc protein inhibits c-MYC function by antagonizing its DNA binding activity (62). Specific inhibition of c-MYC function by Doxycyclin-induced Omomyc expression in late GC B cells (10 days post-immunization by SRBC) leads to reduced GC size, indicating that c-MYC is required for GC maintenance once GCs are established (38).
Affinity maturation
FOXO1 is necessary for effective antibody affinity maturation: SHM frequency is comparable between WT and Foxo1-null GC B cells, but a severely decreased number of GC B cells harboring high affinity antibodies is observed in Foxo1-null GCs (42). Furthermore, Foxo1-null GC B cells have a lower level of cell surface BCR and are ineffective in activating TFH cells in the LZ; this leads to lower stimulation of TFH cells in the GC microenvironment and reduced production of IL-21, a cytokine that is vital for antibody affinity maturation (58). Thus, FOXO1 regulates antibody affinity maturation through both antigen presentation and TFH cell activation (42, 58).
c-MYC is transiently induced in LZ B cells after receiving help from TFH cells (38); selected GC B cells with induced c-MYC present high affinity BCR on the cell surface, and subsequently migrate into the DZ for the next iteration of proliferation and SHM (38, 39). However, in Foxo1-null GC B cells, the expression level of c-MYC is downregulated even under the help from TFH cells. BATF, which controls the expression of Aicda, is another TF downregulated in the absence of Foxo1 (58).
Class Switch Recombination
Like SHM, CSR also requires the expression of Aicda (47, 48). However, CSR depends on a different domain of the AID protein (63, 64). It is worthwhile mentioning that CSR already takes place before the GCs are formed, following B cell activation (65, 66). Much of the knowledge about CSR and the required factors originates from in vitro B cell activation experiments.
IRF4 was shown to regulate CSR in CD40 and IL4 stimulated B cells (27, 67). In the absence of IRF4, the Aicda expression level is decreased (67), and CSR is impaired (67). However, the expression of other genes important for CSR, such as Ung or Msh2 remains normal in Irf4−/− B cells. Therefore, the CSR defects in Irf4-null B cells seem to mainly reflect the impaired Aicda expression (67).
FOXO1 deficiency results in impaired class-switching: the compartment of Igg1-switched B cells in Foxo1-null GCs is heavily reduced, with accumulation of IgM+ GC B cells. Yet, the expression level of Aicda is similar between WT and Foxo1fl/fl-Cγ1cre GC B cells (42). In addition, Foxo1-null GC B cells display significantly lower expression of germline transcripts (GLTs) across the Ig locus. GLTs coincide with open chromatin and allow the exposure of switch regions to AID, which in turn induces single-strand DNA breaks through which class switch recombination is accomplished (68, 69). Thus, lower levels of GLTs correlate with reduced accessibility of AID toward the class switch regions. At the molecular level, FOXO1 possibly modulates GLT and post-switch transcripts by binding to I-mu, the 3′ IgH enhancer and a super-enhancer (70) in the Ig locus. Moreover, the transcription factor BATF, which is necessary for the expression of Ig GLTs and subsequent CSR (37), is downregulated upon FOXO1 depletion (42).
PAX5 binds to the promoter region of Aicda and activates its expression. Overexpression of PAX5 in a ProB cell line induces the expression Aicda, while ID2 has an antagonizing effect on this induction. Moreover, PAX5, E2A, and AID directly interact with each other and form a complex, which contributes to directing AID to the Igh locus for CSR (71). In addition, ID2 and ID3 negatively regulate CSR by repressing Aicda expression (56, 57).
BATF directly controls the expression of Aicda. By ChIP-seq and EMSA, BATF was shown to bind to the regulatory region of Aicda. In line with this, the expression level of Aicda is downregulated in Batf−/− mice (37). Consequently, production of isotype switched antibodies is almost completely missing, although IgM production upon T-cell-dependent or -independent antigen stimulation is still normal in Batf−/− mice (37). Moreover, Batf−/− mice display a reduction in GLTs from different isotypes, except those from μ-chain. Germline transcription initiated by switch region (I) region promoters, located upstream of the different constant heavy chain exons, is required for AID targeting and successful CSR (8, 54). Taken together, BATF regulates CSR by modulating the expression of Aicda and GLTs from the Ig locus.
TFs Controlling the Migration of Cells Between Dark and Light Zone
The LZ-to-DZ transition is mainly driven by high affinity antibody presentation on the surface of GC B cells and the subsequent help from TFH cells toward high affinity antibody LZ cells (58, 72–74).
Foxo1-null GC B cells showed reduced level of surface BCR and Igβ when compared to WT cells (58). Therefore, reduced antigen presentation on Foxo1−/− GC LZ B cells fails to effectively activate TFH cells, resulting in lower number of TFH cells and decreased production of IL-21, an important cytokine for GC B cell differentiation and affinity maturation promoted by TFH cells (75, 76). Moreover, Foxo1-null GC LZ B cells express less IL21R, further abrogating the ability to receive help from TFH cells (BCR and CD40 signal). Therefore, the LZ-to-DZ migration of selected cells is impaired in the absence of Foxo1 (58) and Foxo1-null GC B cells are trapped in the LZ compartment. Furthermore, Foxo1-null GC B cells showed lower proliferation rate in the LZ compartment, in spite of harboring a high affinity BCR, indicating defects in cyclic reentry mediated by help from TFH cells which is coupled with the LZ-to-DZ migration (58).
Interaction between high affinity BCR expressing LZ B cells and TFH cells leads to activation of c-MYC expression (60), which promotes cyclic reentry and LZ-to-DZ migration (58).
Differentiation: Memory/Plasma Cell Fate Decision
Figure 2 summarizes the molecular networks involved in plasma cell and memory B cell differentiation.
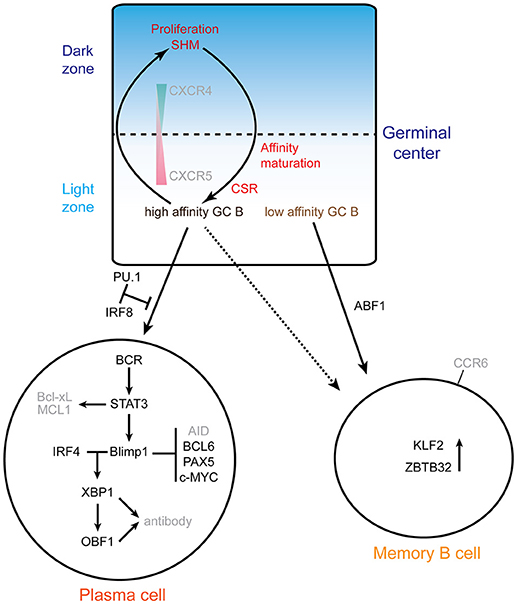
Figure 2. Exit from the GC: memory vs. plasma cells. The top scheme highlights the cycling of B cells between the dark zone and the light zone. The gradients of the chemokine receptors CXCR4 and 5 are indicated, as well as the processes involved. GC B cells proliferate and undergo somatic hypermutation (SHM) in the GC DZ, and then migrate to the light zone where the affinity of mutated BCRs is selected. B cells with high affinity BCR are selected to reenter the DZ for several rounds of SHM or can differentiate into plasma cells. Memory B cells are mainly originated from B cells with low affinity BCR. The alternative fates of B cells exiting the GC are depicted at the bottom: plasma cells or memory B cells. The main networks that have been identified are indicated. In the plasma cell differentiation pathway, PU.1 and IRF8 negatively regulate plasma cell differentiation. IRF4 and BLIMP1 form the central axis in establishing plasma cells. BLIMP1 further suppresses the expression of Aicda, Bcl6, Pax5 and c-Myc genes and finally terminates the GC program. BLIMP1 positively regulates Xbp1 expression, makes the cells ready for antibody production and secretion. ABF1 promotes memory B cell differentiation and inhibits plasma cell differentiation. ZBTB32 and KLF2 are highly expressed in memory B cells.
Transient and low expression of Irf4 leads to the expression of Bcl6 and Pou2af1 during the early phase of GC formation, while sustained and high level expression of Irf4 is required for plasma cell differentiation (26). Irf4 expression is induced to a high level in the fraction of LZ B cells which present high affinity antibodies (26, 28). BLIMP1 is a transcriptional repressor that is essential for plasma cell development: Prdm1-deficient mice cannot produce plasma cells and overexpression of BLIMP1 is sufficient to induce plasmablast differentiation (77, 78). Prdm1 is a downstream target of IRF4, and BLIMP1 can further increase the level of IRF4, thus reinforcing the plasma cell differentiation program in a feed forward loop (26, 79). Morever, BLIMP1 represses the expression of Aicda (79) and Abf-1 (80).
BLIMP1, together with IRF4, acts upstream of X-box binding protein 1 (XBP1), a transcription factor that is essential for upregulation of the secretory apparatus required for antibody production in plasma cells (27, 81, 82). PAX5 represses XBP1 and thus prevents plasma cell differentiation (83). Conversely, downregulation of Pax5 by BLIMP1 is a necessary step for induction of Xbp-1 and activation of the plasma cell program (81, 84). However, in the absence of IRF4, BLIMP1 alone is not sufficient for plasma cell differentiation. Moreover, BLIMP1 is required for post-transcriptional regulation of XBP1 mRNA, by which the active form XBP1 protein is generated (85). XBP1 directly regulates Pou2af1 expression in plasma cells (86), which might be important for IgG production, since OBF1 and OCT2 are required for normal IgG expression (87). Irf4-deficient (Cγ1cre) mice completely lack CD138+ (aka Syndecan1hi) plasma cells in spleen, peripheral blood and bone marrow (27). In addition to the important yet mechanistically unclear role of OBF1 in the early development of GC formation, this factor is required for antibody production (both unswitched and switched isotypes) in T-dependent antigen stimulation and normal antibody secreting cell differentiation, as the number of antibody-secreting Syndecan1hi cells is dramatically reduced in absence of OBF1 (88). Moreover, OBF1 is required for the induction of Prdm1 (88). Taken together, IRF4 and BLIMP1 function together to drive the transition from GC B cells to plasma cells by repressing the GC program and enhancing the plasma cell differentiation program.
In contrast, IRF8 together with PU.1 inhibit the GC B cell differentiation toward plasma cells, suggesting that the balance between IRF4 and IRF8 may be critical for the fate of B cells at this developmental transition (89). In addition, STAT3 regulates the differentiation of plasma cells possibly by promoting cell survival through activating the expression of pro-survival genes such as Bcl2l1 and Mcl1 (90, 91).
It is not resolved yet which transcription factor(s) play a major role in regulating the differentiation of memory B cells (92); however, recent evidence suggests that memory B cells originate from the low affinity compartment of the LZ (92, 93). Comparison of the transcriptome profiles from high- and low-affinity BCR expressing GC B cells in the LZ compartment showed that genes involved in DZ maintenance and cyclic reentry are downregulated in low-affinity fractions (with switched isotype), where Bach2 and Pax5 are upregulated (93). As indicated above, upregulation of these transcription factors blocks plasma cell differentiation (94–96).
BACH2 regulates LZ B cells to commit to the memory B cell differentiation path in a dosage dependent manner, as complete knockout or haploinsufficiency of Bach2 lead to lower memory B cell differentiation (93). Furthermore, TFH cell interaction and affinity maturation in LZ compartment are negatively correlated with Bach2 expression, thus confirming that memory B cells are generated from low affinity fraction in the LZ compartment (92, 93).
Activated B cell Factor 1 (ABF-1), a helix-loop-helix TF predominantly expressed in memory B cells, blocks the plasma cell differentiation program (80). An inducible ABF-1-ER mouse model demonstrated that induction of ABF-1 promotes GC formation and memory B cell differentiation (80, 97). ZBTB32 and KLF2 are two factors expressed in memory B cells and which inhibit the GC response (98). They have been associated with memory B cells (99), but further mechanistic studies are needed to understand their specific role. BCL6 is a known inhibitor of plasma cell differentiation which directly represses the expression of Prdm1 (100, 101). STAT5 directly modulates the expression level of Bcl6, which in turn directly represses Prdm1 expression, and thus promotes memory B cell differentiation.
Outlook and Open Questions
A central theme is that often multiple distinct TFs act in concert to promote, or repress, specific steps of GC development. Some factors, such as BCL6 or BLIMP1, are considered to be master regulators for GC or plasma cell development, respectively. Yet, many other factors have been shown to be important (FOXO1, IRF4) or sometimes essential (OBF1, OCT2). In most cases, however, additional mechanistic studies are required to precisely understand how these factors fit in the overall regulatory circuitry. Moreover, it is often not clear how the TFs perform their function in this biological paradigm: which co-activators or co-repressors are involved and what epigenetic regulators are required?
Finally, intensive investigations have been conducted to understand the interaction between high affinity BCR expressing GC B cells and TFH cells, yet little is known about how GC B cells are determined by transcriptional regulators to proceed with the LZ-to-DZ migration, or commit to the plasma cell differentiation cascade.
Author Contributions
SS wrote the manuscript and PM corrected the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Novartis Research Foundation. We thank all lab members and colleagues for useful discussions. This review is dedicated to our friend and colleague Antonius (Ton) Rolink, with whom we have had a very long-standing collaboration; it was always fun.
References
1. Matthias P, Rolink AG. Transcriptional networks in developing and mature B cells. Nat Rev Immunol. (2005) 5:497–508. doi: 10.1038/nri1633
2. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
3. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. (2015) 15:137–48. doi: 10.1038/nri3804
4. Mesin L, Ersching J, Victora GD. Germinal center B Cell Dynamics. Immunity (2016) 45:471–82. doi: 10.1016/j.immuni.2016.09.001
5. Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. (2005) 5:230–42. doi: 10.1038/nri1572
6. Wagner SD, Neuberger MS. Somatic hypermutation of immunoglobulin genes. Annu Rev Immunol. (1996) 14:441–57. doi: 10.1146/annurev.immunol.14.1.441
7. Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity (2007) 27:190–202. doi: 10.1016/j.immuni.2007.07.009
8. Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. (2008) 26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248
9. Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. (2008) 8:22–33. doi: 10.1038/nri2217
10. Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol. (2009) 9:767–77. doi: 10.1038/nri2656
11. Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. (2001) 19:595–621. doi: 10.1146/annurev.immunol.19.1.595
12. Huang C, Geng H, Boss I, Wang L, Melnick A. Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood (2014) 123:1012–20. doi: 10.1182/blood-2013-07-518605
13. Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. (2007) 12:280–92. doi: 10.1016/j.ccr.2007.08.011
14. Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, et al. Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J Exp Med. (2014) 211:2103–18. doi: 10.1084/jem.20132613
15. Kim U, Qin XF, Gong S, Stevens S, Luo Y, Nussenzweig MC, et al. The B-cell-specific transcription coactivator OCA-B-OBF-1-Bob-1 is essential for normal production of immunoglobulin isotypes. Nature (1996) 383:542–7.
16. Schubart DB, Rolink A, Kosco-Vilbois MH, Botteri F, Matthias P. B-cell-specific coactivator OBF-1/OCA-B/Bob1 required for immune response and germinal centre formation. Nature (1996) 383:538–42. doi: 10.1038/383538a0
17. Luo Y, Roeder RG. Cloning, functional characterization, and mechanism of action of the B-cell-specific transcriptional coactivator OCA-B. Mol Cell Biol. (1995) 15:4115–24. doi: 10.1128/MCB.15.8.4115
18. Strubin M, Newell JW, Matthias P. OBF-1, a novel B cell-specific coactivator that stimulates immunoglobulin promoter activity through association with octamer-binding proteins. Cell (1995) 80:497–506. doi: 10.1016/0092-8674(95)90500-6
19. Schubart K, Massa S, Schubart D, Corcoran LM, Rolink AG, Matthias P. B cell development and immunoglobulin gene transcription in the absence of Oct-2 and OBF-1. Nat Immunol. (2001) 2:69–74. doi: 10.1038/83190
20. Garrett-Sinha LA, Su GH, Rao S, Kabak S, Hao Z, Clark MR, et al. PU.1 and Spi-B are required for normal B cell receptor-mediated signal transduction. Immunity (1999) 10:399–408. doi: 10.1016/S1074-7613(00)80040-0
21. Su GH, Chen HM, Muthusamy N, Garrett-Sinha LA, Baunoch D, Tenen DG, et al. Defective B cell receptor-mediated responses in mice lacking the Ets protein, Spi-B. EMBO J. (1997) 16:7118–29. doi: 10.1093/emboj/16.23.7118
22. Bartholdy B, Du Roure C, Bordon A, Emslie D, Corcoran LM, Matthias P. The Ets factor Spi-B is a direct critical target of the coactivator OBF-1. Proc Natl Acad Sci USA. (2006) 103:11665–70. doi: 10.1073/pnas.0509430103
23. Stauss D, Brunner C, Berberich-Siebelt F, Hopken UE, Lipp M, Muller G. The transcriptional coactivator Bob1 promotes the development of follicular T helper cells via Bcl6. EMBO J. (2016) 35:881–98. doi: 10.15252/embj.201591459
24. Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. (2010) 11:114–20. doi: 10.1038/ni.1837
25. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity (2011) 34:961–72. doi: 10.1016/j.immuni.2011.03.025
26. Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity (2013) 38:918–29. doi: 10.1016/j.immuni.2013.04.009
27. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. (2006) 7:773–82. doi: 10.1038/ni1357
28. Sciammas R, Li Y, Warmflash A, Song Y, Dinner AR, Singh H. An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Mol Syst Biol. (2011) 7:495. doi: 10.1038/msb.2011.25
29. Lee CH, Melchers M, Wang H, Torrey TA, Slota R, Qi CF, et al. Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J Exp Med. (2006) 203:63–72. doi: 10.1084/jem.20051450
30. Qi CF, Li ZY, Raffeld M, Wang HS, Kovalchuk AL, Morse HC. Differential expression of IRF8 in subsets of macrophages and dendritic cells and effects of IRF8 deficiency on splenic B cell and macrophage compartments. Immunol Res. (2009) 45:62–74. doi: 10.1007/s12026-008-8032-2
31. Zhou JX, Lee CH, Qi CF, Wang H, Naghashfar Z, Abbasi S, et al. IFN regulatory factor 8 regulates MDM2 in germinal center B cells. J Immunol. (2009) 183:3188–94. doi: 10.4049/jimmunol.0803693
32. Feng J, Wang H, Shin DM, Masiuk M, Qi CF, Morse HC III. IFN regulatory factor 8 restricts the size of the marginal zone and follicular B cell pools. J Immunol. (2011) 186:1458–66. doi: 10.4049/jimmunol.1001950
33. Yoon J, Feng X, Kim YS, Shin DM, Hatzi K, Wang H, et al. Interferon regulatory factor 8 (IRF8) interacts with the B cell lymphoma 6 (BCL6) corepressor BCOR. J Biol Chem. (2014) 289:34250–7. doi: 10.1074/jbc.M114.571182
34. Wilker PR, Kohyama M, Sandau MM, Albring JC, Nakagawa O, Schwarz JJ, et al. Transcription factor Mef2c is required for B cell proliferation and survival after antigen receptor stimulation. Nat Immunol. (2008) 9:603–12. doi: 10.1038/ni.1609
35. Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. (2013) 14:1084–92. doi: 10.1038/ni.2688
36. Pon JR, Marra MA. MEF2 transcription factors: developmental regulators and emerging cancer genes. Oncotarget (2016) 7:2297–312. doi: 10.18632/oncotarget.6223
37. Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol. (2011) 12:536–43. doi: 10.1038/ni.2037
38. Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol. (2012) 13:1083–91. doi: 10.1038/ni.2428
39. Calado DP, Sasaki Y, Godinho SA, Pellerin A, Kochert K, Sleckman BP, et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol. (2012) 13:1092–100. doi: 10.1038/ni.2418
40. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. (2004) 5:943–52. doi: 10.1038/ni1100
41. Bannard O, Horton RM, Allen CD, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity (2013) 39:912–24. doi: 10.1016/j.immuni.2013.08.038
42. Dominguez-Sola D, Kung J, Holmes AB, Wells VA, Mo T, Basso K, et al. The FOXO1 transcription factor instructs the germinal center dark zone program. Immunity (2015) 43:1064–74. doi: 10.1016/j.immuni.2015.10.015
43. Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, et al. PI3 Kinase and FOXO1 transcription factor activity differentially control b cells in the germinal center light and dark zones. Immunity (2015) 43:1075–86. doi: 10.1016/j.immuni.2015.10.021
44. Trabucco SE, Gerstein RM, Zhang H. YY1 Regulates the germinal center reaction by inhibiting apoptosis. J Immunol. (2016) 197:1699–707. doi: 10.4049/jimmunol.1600721
45. Oropallo MA, Cerutti A. Germinal center reaction: antigen affinity and presentation explain it all. Trends Immunol. (2014) 35:287–9. doi: 10.1016/j.it.2014.06.001
46. Toyama H, Okada S, Hatano M, Takahashi Y, Takeda N, Ichii H, et al. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity (2002) 17:329–39. doi: 10.1016/S1074-7613(02)00387-4
47. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102:553–63. doi: 10.1016/S0092-8674(00)00078-7
48. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell (2000) 102:565–75. doi: 10.1016/S0092-8674(00)00079-9
49. Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature (2002) 418:99–103. doi: 10.1038/nature00862
50. Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci USA. (2003) 100:4102–7. doi: 10.1073/pnas.0730835100
51. Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature (2003) 422:726–30. doi: 10.1038/nature01574
52. Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature (2002) 419:43–8. doi: 10.1038/nature00981
53. Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med. (2003) 197:1291–6. doi: 10.1084/jem.20030481
54. Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. (2003) 4:452–6. doi: 10.1038/ni920
55. Chahwan R, Edelmann W, Scharff MD, Roa S. AIDing antibody diversity by error-prone mismatch repair. Semin Immunol. (2012) 24:293–300. doi: 10.1016/j.smim.2012.05.005
56. Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. (2003) 4:586–93. doi: 10.1038/ni923
57. Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, et al. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med. (2003) 198:1427–37. doi: 10.1084/jem.20030802
58. Inoue T, Shinnakasu R, Ise W, Kawai C, Egawa T, Kurosaki T. The transcription factor Foxo1 controls germinal center B cell proliferation in response to T cell help. J Exp Med. (2017) 214:1181–98. doi: 10.1084/jem.20161263
59. Quong MW, Harris DP, Swain SL, Murre C. E2A activity is induced during B-cell activation to promote immunoglobulin class switch recombination. EMBO J. (1999) 18:6307–18. doi: 10.1093/emboj/18.22.6307
60. Luo W, Weisel F, Shlomchik MJ. B cell receptor and CD40 signaling are rewired for synergistic induction of the c-Myc transcription factor in germinal center B cells. Immunity (2018) 48:313–26 e5. doi: 10.1016/j.immuni.2018.01.008
61. Allman D, Jain A, Dent A, Maile RR, Selvaggi T, Kehry MR, et al. BCL-6 expression during B-cell activation. Blood (1996) 87:5257–68.
62. Soucek L, Jucker R, Panacchia L, Ricordy R, Tato F, Nasi S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. (2002) 62:3507–10.
63. Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Molecular Cell (2003) 12:501–8. doi: 10.1016/S1097-2765(03)00309-5
64. Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, et al. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol. (2003) 4:843–8. doi: 10.1038/ni964
65. Chan TD, Gatto D, Wood K, Camidge T, Basten A, Brink R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J Immunol. (2009) 183:3139–49. doi: 10.4049/jimmunol.0901690
66. Zhang Y, Garcia-Ibanez L, Toellner KM. Regulation of germinal center B-cell differentiation. Immunol Rev. (2016) 270:8–19. doi: 10.1111/imr.12396
67. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity (2006) 25:225–36. doi: 10.1016/j.immuni.2006.07.009
68. Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. (2007) 157–214. doi: 10.1016/S0065-2776(06)94006-1
69. Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Adv Immunol. (2011) 110:1–26. doi: 10.1016/B978-0-12-387663-8.00005-3
70. Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell (2014) 159:1524–37. doi: 10.1016/j.cell.2014.11.013
71. Hauser J, Grundstrom C, Kumar R, Grundstrom T. Regulated localization of an AID complex with E2A, PAX5 and IRF4 at the Igh locus. Mol Immunol. (2016) 80:78–90. doi: 10.1016/j.molimm.2016.10.014
72. Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science (2007) 315:528–31. doi: 10.1126/science.1136736
73. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143:592–605. doi: 10.1016/j.cell.2010.10.032
74. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature (2015) 517:214–8. doi: 10.1038/nature13803
75. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. (2010) 207:353–63. doi: 10.1084/jem.20091738
76. Zotos D, Coquet JM, Zhang Y, Light A, D'Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. (2010) 207:365–78. doi: 10.1084/jem.20091777
77. Turner CA Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell (1994) 77:297–306. doi: 10.1016/0092-8674(94)90321-2
78. Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity (2003) 19:607–20. doi: 10.1016/S1074-7613(03)00267-X
79. Minnich M, Tagoh H, Bonelt P, Axelsson E, Fischer M, Cebolla B, et al. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol. (2016) 17:331–43. doi: 10.1038/ni.3349
80. Chiu YK, Lin IY, Su ST, Wang KH, Yang SY, Tsai DY, et al. Transcription factor ABF-1 suppresses plasma cell differentiation but facilitates memory B cell formation. J Immunol. (2014) 193:2207–17. doi: 10.4049/jimmunol.1400411
81. Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature (2001) 412:300–7. doi: 10.1038/35085509
82. Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity (2004) 21:81–93. doi: 10.1016/j.immuni.2004.06.010
83. Recaldin T, Fear DJ. Transcription factors regulating B cell fate in the germinal centre. Clin Exp Immunol. (2016) 183:65–75. doi: 10.1111/cei.12702
84. Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. (2002) 22:4771–80. doi: 10.1128/MCB.22.13.4771-4780.2002
85. Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, et al. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol. (2016) 17:323–30. doi: 10.1038/ni.3348
86. Shen Y, Hendershot LM. Identification of ERdj3 and OBF-1/BOB-1/OCA-B as direct targets of XBP-1 during plasma cell differentiation. J Immunol. (2007) 179:2969–78. doi: 10.4049/jimmunol.179.5.2969
87. Ren X, Siegel R, Kim U, Roeder RG. Direct interactions of OCA-B and TFII-I regulate immunoglobulin heavy-chain gene transcription by facilitating enhancer-promoter communication. Mol Cell (2011) 42:342–55. doi: 10.1016/j.molcel.2011.04.011
88. Corcoran LM, Hasbold J, Dietrich W, Hawkins E, Kallies A, Nutt SL, et al. Differential requirement for OBF-1 during antibody-secreting cell differentiation. J Exp Med. (2005) 201:1385–96. doi: 10.1084/jem.20042325
89. Carotta S, Willis SN, Hasbold J, Inouye M, Pang SH, Emslie D, et al. The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. J Exp Med. (2014) 211:2169–81. doi: 10.1084/jem.20140425
90. Fornek JL, Tygrett LT, Waldschmidt TJ, Poli V, Rickert RC, Kansas GS. Critical role for Stat3 in T-dependent terminal differentiation of IgG B cells. Blood (2006) 107:1085–91. doi: 10.1182/blood-2005-07-2871
91. Ding C, Chen X, Dascani P, Hu X, Bolli R, Zhang HG, et al. STAT3 signaling in B cells is critical for germinal center maintenance and contributes to the pathogenesis of murine models of lupus. J Immunol. (2016) 196:4477–86. doi: 10.4049/jimmunol.1502043
92. Suan D, Krautler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, et al. CCR6 Defines memory B cell precursors in mouse and human germinal centers, revealing light-zone location and predominant low antigen affinity. Immunity (2017) 47:1142–53.e4. doi: 10.1016/j.immuni.2017.11.022
93. Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, et al. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol. (2016) 17:861–9. doi: 10.1038/ni.3460
94. Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev. (2012) 247:172–83. doi: 10.1111/j.1600-065X.2012.01112.x
95. Igarashi K, Ochiai K, Muto A. Architecture and dynamics of the transcription factor network that regulates B-to-plasma cell differentiation. J Biochem. (2007) 141:783–9. doi: 10.1093/jb/mvm106
96. Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. (2007) 8:463–70. doi: 10.1038/ni1454
97. Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. (2014) 15:631–7. doi: 10.1038/ni.2914
98. Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, et al. Germinal-center development of memory B cells driven by IL-9 from follicular helper T cells. Nat Immunol. (2017) 18:921–30. doi: 10.1038/ni.3788
99. Bhattacharya D, Cheah MT, Franco CB, Hosen N, Pin CL, Sha WC, et al. Transcriptional profiling of antigen-dependent murine B cell differentiation and memory formation. J Immunol. (2007) 179:6808–19. doi: 10.4049/jimmunol.179.10.6808
100. Scheeren FA, Naspetti M, Diehl S, Schotte R, Nagasawa M, Wijnands E, et al. STAT5 regulates the self-renewal capacity and differentiation of human memory B cells and controls Bcl-6 expression. Nat Immunol. (2005) 6:303–13. doi: 10.1038/ni1172
Keywords: hematopoiesis, transcription factors, B cell development, germinal center (GCs), transcriptional regulation, germinal center development, germinal center maintenance, plasma cell and memory B cell differentiation
Citation: Song S and Matthias PD (2018) The Transcriptional Regulation of Germinal Center Formation. Front. Immunol. 9:2026. doi: 10.3389/fimmu.2018.02026
Received: 30 June 2018; Accepted: 16 August 2018;
Published: 05 September 2018.
Edited by:
Rhodri Ceredig, National University of Ireland Galway, IrelandReviewed by:
Antony Basten, Garvan Institute of Medical Research, AustraliaKai-Michael Toellner, University of Birmingham, United Kingdom
Copyright © 2018 Song and Matthias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick D. Matthias, cGF0cmljay5tYXR0aGlhc0BmbWkuY2g=