 Alireza Saeidi1,2
Alireza Saeidi1,2 Keivan Zandi3
Keivan Zandi3 Yi Ying Cheok1Hamidreza Saeidi4
Yi Ying Cheok1Hamidreza Saeidi4 Won Fen Wong1
Won Fen Wong1 Chalystha Yie Qin Lee1
Chalystha Yie Qin Lee1 Heng Choon Cheong1
Heng Choon Cheong1 Yean Kong Yong2,5
Yean Kong Yong2,5 Marie Larsson6
Marie Larsson6 Esaki Muthu Shankar7*
Esaki Muthu Shankar7*- 1Department of Medical Microbiology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
- 2Center of Excellence for Research in AIDS, University of Malaya, Kuala Lumpur, Malaysia
- 3Department of Pediatrics School of Medicine Emory University, Atlanta, GA, United States
- 4Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, University of Putra Malaysia, Selangor, Malaysia
- 5Laboratory Center, Xiamen University Malaysia, Sepang, Malaysia
- 6Division of Molecular Virology, Department of Clinical and Experimental Medicine, Linköping University, Linköping, Sweden
- 7Division of Infection Biology and Medical Microbiology, Department of Life Sciences, School of Life Sciences, Central University of Tamil Nadu, Thiruvarur, India
T-cell exhaustion is a phenomenon of dysfunction or physical elimination of antigen-specific T cells reported in human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV) infections as well as cancer. Exhaustion appears to be often restricted to CD8+ T cells responses in the literature, although CD4+ T cells have also been reported to be functionally exhausted in certain chronic infections. Although our understanding of the molecular mechanisms associated with the transcriptional regulation of T-cell exhaustion is advancing, it is imperative to also explore the central mechanisms that control the altered expression patterns. Targeting metabolic dysfunctions with mitochondrion-targeted antioxidants are also expected to improve the antiviral functions of exhausted virus-specific CD8+ T cells. In addition, it is crucial to consider the contributions of mitochondrial biogenesis on T-cell exhaustion and how mitochondrial metabolism of T cells could be targeted whilst treating chronic viral infections. Here, we review the current understanding of cardinal features of T-cell exhaustion in chronic infections, and have attempted to focus on recent discoveries, potential strategies to reverse exhaustion and reinvigorate optimal protective immune responses in the host.
Introduction
T cells play a key role in orchestrating pathogen-specific adaptive immune responses. During primary infection, naive T cells recognize antigenic peptides presented on major histocompatibility complex (MHC) molecules via their T-cell receptors (TCRs), leading to their activation and differentiation into effector T cells over the course of ~1–2 weeks (1, 2). This differentiation results in robust proliferation, transcriptional, epigenetic, and metabolic changes, as well as the acquisition of effector functions, altered tissue homing, and massive clonal expansion (1, 3). Following antigenic clearance, a vast majority of the effector T cells die by apoptosis. However, ~5–10% of the cells persist and differentiate into memory T cells such as central memory, effector memory, and tissue resident memory T cells (1, 3). Memory T cells are maintained after the effector phase and can rapidly execute their effector functions in response to reinfection/exposure to previously encountered antigens. The rapid effector function arises when the antigen is present transitory during an acute infection. Nonetheless, this programming of memory T cell differentiation is distinctly altered during chronic viral and bacterial infections, and also in chronic diseases such as cancer due to persistent antigenic exposure and/or inflammation (3, 4). When altered differentiation progresses, the immune response fails, and antigen-specific T cells progress to a state called T-cell exhaustion.
T-cell exhaustion was first defined in 1993 by Moskophidis and colleagues when they demonstrated impaired cytotoxic functions during viral persistence in murine models (5). T-cell exhaustion denotes the physical elimination of antigen-specific T cells, also observed in chronic lymphocytic choriomeningitis virus (LCMV) infection of mice (6, 7). T-cell exhaustion has also been reported in various human chronic viral infections such as human immunodeficiency virus (HIV), hepatitis B (HBV), and hepatitis C (HCV), as well as in cancer (4, 8, 9). Exhaustion has been mostly described for CD8+ T cells responses although CD4+ T cells have also been reported to be functionally unresponsive in several chronic infections (10, 11). Wherry et al. firstly described the molecular signature of CD8+ T-cell exhaustion during chronic viral infection (12). Later, advances in biomedical technologies in research including the utilization of MHC multimers that can recognize antigen-specific T cells without reliance on T cell functions as readout, as well as the progress in improved methods to evaluate the phenotypic and functional portfolios of single cells have enhanced our understanding of the complexities underlying the T-cell exhaustion phenomenon (10, 13).
The state of exhaustion is mainly characterized by sequential loss of T cell effector functions in a hierarchical manner where loss of interleukin (IL)-2 production is the earliest sign (14, 15). Subsequently, the production of tumor necrosis factor (TNF) and other cytokines is dramatically reduced. However, interferon (IFN)-γ, beta-chemokine production and perhaps cytotoxic activities, are more resilient to inactivation (16–19). Exhausted T cells also have altered proliferative abilities, sustained upregulation of a wide array of co-inhibitory receptors, unique transcriptional and epigenetic signatures, altered metabolic fitness, failure for transition to quiescence, and acquisition of antigen-independent memory T cell responsiveness (3, 4, 8, 20, 21). Notably, severely exhausted T cells appear to undergo apoptosis and become eliminated leading to marked decline in virus-specific T cells (5, 22, 23).
It is imperative to also understand that T-cell exhaustion should be clearly delineated from T-cell anergy and senescence. T-cell anergy is a state of non-responsiveness molecularly distinct from T-cell exhaustion, which is induced by excessive stimulation of TCR and either robust co-inhibitory molecule signaling or restricted presence of concomitant co-stimulation through CD28 (4, 24). On the other hand, senescent T cells are often described by increased expression of markers such as killer-cell lectin-like receptor G1 (KLRG1) and/or CD57 (25–27), which exhausted T cells have in low levels on their surfaces (12). The expression of PD-1 is also increased on exhausted T cells whereas senescent cells seldom express this marker (28). In this review, we will discuss our current understanding of cardinal features of T-cell exhaustion in chronic infections, while we will attempt to also focus on recent discoveries and potential strategies for reversing the state of exhaustion with a view to reinvigorate immune responses.
Immune Checkpoints With Therapeutic Potentials in T-Cell Exhaustion
In acute infections, co-inhibitory receptors function to dampen the magnitude of immune responses, which are in fact, down-regulated after pathogen clearance to achieve homeostasis, and development of memory T cells. However, this pattern diverges during chronic infections, where higher and sustained expression of co-inhibitory receptors is characteristic (3, 10). Co-inhibitory receptors vary in expression pattern, ligands, and signaling motifs and our understanding of the molecular mechanisms whereby they control T-cell exhaustion is seldom understood (29). However, the identification of their significance in the dysregulation of cellular immune responses in chronically infected hosts has provided newer avenues for designing therapeutic molecules to restore optimum immune responses (10).
PD-1 Plays a Pivotal Role in Regulating T-Cell Exhaustion
The dominant role of programmed death-1 (PD-1) in regulating T-cell exhaustion was first revealed following gene expression profiling of virus-specific CD8+ T cells during chronic LCMV infection (30). Exhausted T cells up-regulated PD-1 expression, and blockade of the PD-1 pathway promoted effector functions of virus-specific T cells and significantly reduced the viral load in the experimental animals. This result has been further substantiated in many other chronic infections in mice, non-human primates, and humans.
During HIV-1 infection, PD-1 expression on HIV-specific CD8+ T cells positively correlates with high viral load, impairment of CD8+ T-cell function, disease progression, and reduced CD4+ T-cell counts. Antiretroviral therapy (ART) can reduce the expression of PD-1 on virus-specific T cells in HIV-infected patients. Long-term non-progressors (LTNPs) have low expression of PD-1 on virus-specific T cells and these populations are more polyfunctional than T cells of progressors (10, 31, 32). In vitro studies describe that blocking the PD-1 pathway restores T-cell functions and improves pathogen control by enhancing the proliferation potentials of T cells and promoting cytokine production (31–33). Moreover, in vivo administration of anti-PD-L1 antibody increased both CD4+ and CD8+ T cells with the ability to inhibit viral replication, i.e., decreasing the plasma viral load, in mice chronically infected with HIV-1 (34). More recently, treatment with PD-1 inhibitory antibody during simian immunodeficiency virus (SIV) infection increased the frequencies and functional quality of SIV-specific CD8+ T cells detectable in the blood and gut, viral loads declined, and significantly improved the survival rates in infected macaques (35, 36). In addition to HIV, the dynamics and significance of the PD-1 pathway has been investigated in HBV and HCV infections (37–41). In chimpanzees chronically infected with HCV, a 100-fold suppression of viremia was observed in one of three animals treated with anti-PD-1 antibodies. Control of virus replication was associated with reinvigoration of HCV-specific CD4+ and CD8+ T cell responses (42). Interestingly, PD-1 expression noticeably increased on HCV-specific CD8+ T cells in the liver although the blocking of PD-1 had no enhancing effect on the functions of these cells (41). This explains that multiple factors must contribute and control the maintenance of T-cell exhaustion and also indicates that the severity of exhaustion is highly influenced by the location and levels of viral antigen and the compartmentalization of the virus-specific T cells (10).
Clinical trials have so far only evaluated single-dose regimens in chronically infected patients, due to considerations of potential toxicities of PD-1-targeted therapy in otherwise healthy individuals (29). Even though there was only a modest response rate for chronic HCV, among 20 patients receiving the highest anti-PD-1 dose, three showed remarkable reduction in viral RNA, and in 1 patient, HCV was undetectable for at least 1 year. Mild to moderate immune-related adverse events were reported in six of 54 patients, which were resolved without specific intervention (43). Single-dose PD-1-targeted therapy, i.e., anti-PD-L1, has been evaluated in HIV infected patients on clinically effective combination ART (cART). In this study, Gay et al. described an increase in HIV-specific CD8+ T cell responses in the blood in two of six patients, but without any effects on HIV viral load. This result could likely be attributed to the dosage of anti-PD-L1 antibodies used, which was 10-fold lower than dosages selected for activity in patients with cancer (44). These clinical trials suggest that there is potential to use PD-1-targeted therapy in some patients for overcoming chronic infections and that combination treatments should further be assessed (29).
Contribution of Other Co-Inhibitory Receptors for T-Cell Exhaustion
There are several co-inhibitory molecules other than PD-1, which are expressed on exhausted T cells. Exhausted T cells can co-express PD-1 together with cytotoxic T lymphocyte antigen-4 (CTLA-4), T cell immunoglobulin domain and mucin domain-containing protein 3 (TIM-3), 2B4 (CD244), lymphocyte activation gene 3 protein (LAG-3), CD160, and several others (45). The individual expression of PD-1 or other co-inhibitory receptors does not define a state of exhaustion rather a co-expression of multiple co-inhibitory receptors do. Interestingly, the indicated co-expression patterns are mechanistically related, as concurrent blockade of these multiple co-inhibitory receptors lead to synergistic reversal of exhaustion (3). Direct in vivo blockade of CTLA-4 during chronic viral infections such as LCMV, SIV, and HIV suggest that blockade of CTLA-4 fail to decrease the viral load or increase T cell functionalities (30, 46). In HCV infection, in vitro blockade of PD-1 alone failed to restore the functions of hepatic PD-1+ CTLA-4+ virus-specific CD8+ T cells although concurrent blockade of CTLA-4 and PD-1 reinvigorated HCV-specific CD8+ T cells in a CD4+ T cell–independent manner (41). Impressive results in controlling cancer has been demonstrated for combined PD-1 and CTLA-4 blockade in patients with melanoma, and drugs targeting at least three other immune checkpoints, i.e., LAG-3, TIM-3, and TIGIT, are now in clinical trials (29, 47). The investigation of co-inhibitory molecules in co-regulating T-cell exhaustion indicates that these pathways are non-redundant. It is also important to consider that the molecules constitute diverse structural families, which bind ligands with unique expression patterns and poses distinct intracellular signaling pathways. Hence, there is a potential to negate exhaustion through manipulation of the pathways where these molecules are involved (3).
Cytokines and T-Cell Exhaustion
Cytokines are molecules that facilitate communications, activation, differentiation and de-activation of immune cells. Involvement of both pro- and anti-inflammatory cytokines in T-cell functions has been extensively studied to understand the physiology of T cells under stressed conditions (48). Of these modulators is the immunosuppressive cytokine IL-10, which has been identified as a potential target to reinvigorate exhausted T cells. IL-10 has been reported to be positively associated with persistence of viral infections such as HCV, HIV, and Epstein-Barr virus (EBV), a possible strategy for viruses to evade host immune defenses (49, 50). In LCMV infection models, the blockade of IL-10 appears to inhibit viral persistence and enhances T-cell functions (51, 52). IL-10 blockade is also employed in HIV infection, whereby IL-10Rα blockade results in markedly increased secretion of IFN-γ by CD4+ T cells. However, combining IL-10 blockade with PD-1 blockade can only restore a restricted number of cytokines produced by T cells including IL-2 (53). On the contrary, in a mouse model of LCMV infection, combined anti-IL-10 and anti-PD-1 therapy synergistically enhanced anti-viral response of T cells (54). Despite the benefits of anti-IL-10 therapy, there are still disadvantages in modulating this otherwise important anti-inflammatory cytokine. For instance, IL-10 production and downstream signaling paves the way for regulating inflammation against gut microbiome (55). Taken together, with the observations of IL-10-mediated regulation of liver inflammation following LCMV infection, treatment with anti-IL-10 should be carefully calibrated to prevent undesirable side-effects (56). This concern also warrants a thorough revisit on the role of IL-10 blockade in immunotherapy (50).
Apart from IL-10, IL-2 also serves as a target for immunotherapy. Exogenous administration of IL-2 in vitro is able to abrogate PD-1 inhibitory signaling (57). Grint et al. demonstrated that IL-2 treatment successfully decreased the virus RNA levels in HCV in HCV/HIV co-infected patients (58). Importantly, IL-2 therapy is highly dependent on the state of infection and can give rise to opposing consequences, such as that previously described with the administration of IL-2/anti-IL-2 immune complexes, which results in expansion of regulatory T cells (Tregs) that impede virus clearance and anti-viral functions of T-cells (59).
Other cytokines serving as potential immunotherapeutic targets in reverting T-cell exhaustion include IL-7, IL-17, IL-21, IL-22, and many others, which are also regarded as key mediators during chronic infections (60–63). IL-21, for instance, improves the clinical outcome in SIV-infected macaques with reduction in the non-AIDS-associated morbidities when administered as supplements with probiotics and ART (64). Other combinations of ART with type-1 interferon (IFN-I) has also yielded positive results by reducing the viral loads and restoring CD8+ T-cell functions in humanized mice infected with HIV (65).
Cross-Talk of Exhausted T Cells With Other Immune Cells
The frequency of Tregs may be increased in HIV and HCV infections where Tregs likely limit the in vitro responses of effector T cells (66). The direct role of Tregs in exhaustion of CD8+ T cells as well as FOXP3–CD4+ T cells remains unclear. There is also a possibility for Tregs to play a role in exhaustion considering that Tregs are a source of IL-10, TGF-β and perhaps other suppressive cytokines, for instance IL-35 (66). Interestingly, recent reports in a chronic LCMV model described an interaction between Tregs and the PD-1 signaling pathway in regulating exhausted CD8+ T cells because simultaneous reduction of Tregs and blockade of PD-1 signaling pathway appears to have a robust synergistic effect on viral control and reversal of exhaustion (67). Furthermore, other immune cell types such as antigen-presenting cells (APCs), myeloid-derived suppressor cells (MDSCs) (68, 69) natural killer (NK) cells and even CD8+ regulatory populations (70–72) have been reported to directly or indirectly promote T-cell exhaustion during chronic infections.
Accumulating evidence indicates that functional impairment of DCs could be associated with exhaustion of T cell functions and progression of disease in HIV, HBV, HCV, and LCMV infections (70, 71, 73–75) although the molecular mechanism behind the impairment of T cell functions mediated via DCs during chronic infections still remain ambiguous. Recent studies showed that DCs promote T-cell exhaustion through signaling by inhibitory receptors such as PD-1 and CTLA-4 (75, 76). Indeed, PD-L1 is up-regulated on mDCs, although MHC molecules and co-stimulatory molecules such as B7-1, B7-2, and CD40 are down-regulated (74, 75, 77). Intriguingly, up-regulated PD-L1 appears to impair DC functions and associate with disease progression during HIV and HBV infections (74, 75, 77).
Transcriptomic Changes in T-Cell Exhaustion
Several recent studies demonstrated that exhausted T cells have a transcriptional profile remarkably different from their effector and memory counterparts. These differences include major alternations in the expression level of co-inhibitory and co-stimulatory molecules, transcription factors, signaling molecules, chemokine and cytokine receptors, and genes involved in metabolism (12, 20, 78). There are several transcription factors that play significance roles in T-cell exhaustion, including T-bet, EOMES, FOXO1, FOXP1, Blimp-1, NFAT, BATF, IRF4, and von Hippel–Lindau disease tumor suppressor (VHL) (16, 79–86). Intriguingly, the main transcription factors, which regulate establishment of T-cell exhaustion function in a specific manner. During acute infection, terminally differentiated CD8+ T cells express T-bet that has a functional role in the development of these cell subsets (87) and differentiation of Th1 cells. However, this transcription factor regulates the population of non-terminal progenitor cells within the exhausted T cell subsets during chronic infections (2, 81). It is described that EOMES favor the development of central memory T cells during acute infection by regulating quiescence and homeostatic turnover (88–90). On the other hand, it is reported that EOMES regulates the development of a terminally-differentiated subset of exhausted T cells during chronic infection (81). There are two phenotypically characterized subsets of exhausted T cells that are described by intermediate expression of inhibitory receptor PD-1 and high expression of the transcription factor T-bet and (PD-1IntT-betHi) or high Eomes and high PD-1 expression (EomesHiPD-1Hi). Although both the populations are required for control of chronic infection, the PD-1Int subset have been shown to contribute distinctly to clearance of pathogens upon PD-1 blockade (3, 81, 91).
In a study using toxoplasma encephalitis (TE)–susceptible model, the CD4 +T cells experienced a more pronounced exhaustion in comparison with CD8+ T cells. It has also been demonstrated that deletion of Blimp-1 from exhausted CD4+ T cells led to reversal of CD8+ T-cell exhaustion and improved pathogen control (92). The transcription factor interferon regulatory factor 9 (IRF9) has an integral role in the antiviral immune response and is considered as a component of IFN type I signaling pathway downstream of the IFN-I receptor (IFNAR) (93). Using LCMV acute infection model, it has been demonstrated that IRF9 limited early LCMV replication by controlling the expression of IFN-stimulated genes and IFN-I, and by regulating the levels of IRF7, a transcription factor necessary for IFN-I production. The study has also revealed that infection of IRF9- or IFNAR-deficient mice resulted in the loss of early restriction of viral replication and impaired anti-viral responses among dendritic cells, leading to CD8+ T-cell exhaustion and chronic infection (94). Our understanding of the transcriptional regulation of T-cell exhaustion is progressing, although it is important to elucidate the mechanisms controlling the altered pattern of gene expression. Moreover, there is also a necessity to identify a master lineage-specific transcription factor for exhausted T cells (63).
Signaling Pathways and T-Cell Exhaustion
T-cell exhaustion involves a tight control by intricate signaling pathways, either through active or passive suppression (Figure 1). The signaling pathways involved in the up- and down-streams of PD-1, a central player in rendering T-cell exhaustion, are as discussed herein.
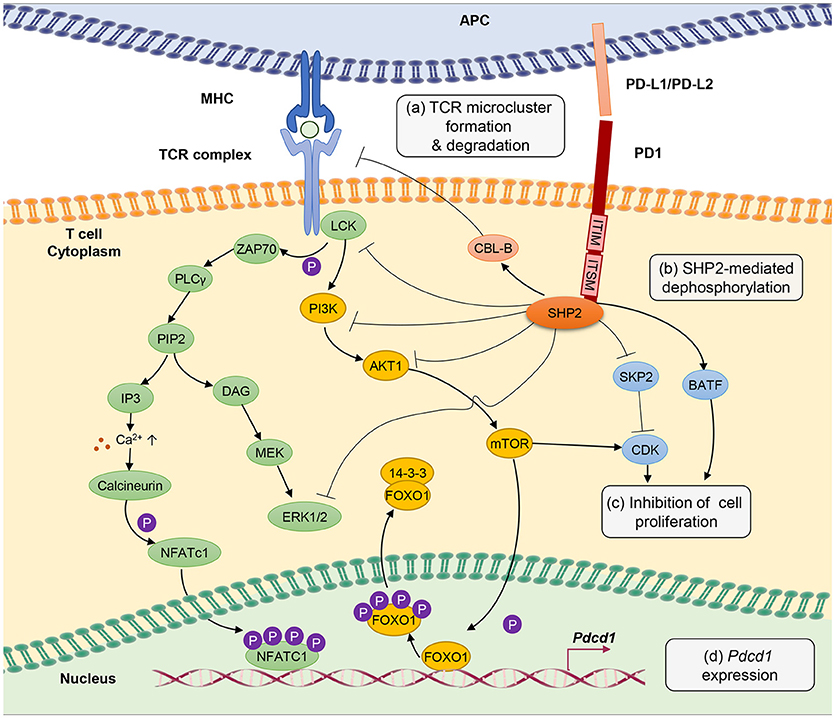
Figure 1. PD-1 signal inhibits T-cell receptor (TCR) signaling pathway through several different mechanisms. (a) PD-1 engagement with PD-L1 or PD-L2 ligands blocks TCR signal transmission by promoting microcluster formation and degradation of TCR. Accumulation of PD-1 within the synapse stabilizes the interaction between T cells and antigen presenting cells causing “immune paralysis” and cell motility arrest. PD-1 ligation also enhances TCR internalization and degradation by Casitas B-lymphoma (CBL)-B E3 ubiquitin ligase. (b) PD-1 signaling recruits SHP2 phosphatase to immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). SHP2 dephosphorylates ZAP70, ERK1/2 and suppresses the phosphatidylinositide 3 kinase (PI3K)/AKT/Mammalian Target of Rapamyc from (mTOR) pathways, thus inhibiting multiple T-cell activation pathways. (c) PD-1 suppresses T-cell proliferation by blocking the transcription of Skp1/Cullin/F-box protein ubiquitin ligase (SKP2), which controls cyclin-dependent kinases (Cdks) activation. A Basic leucine transcription factor, ATF-like (BATF) is also a downstream target of PD-1 signaling that causes repression of T-cell proliferation and cytokine secretion. Besides, by inhibiting TCR signaling, PD-1 blocks IL-2 production to limit T-cell proliferation. (d) PD-1 signaling promotes FOXO1 retention in nucleus and enables Pdcd1 gene transactivation. Nuclear factor of activated T cells (NFATc1) in the absence of AP-1 interaction promotes the expression of Pdcd1 (PD-1 encoding gene). The transcription of Pdcd1 is inhibited by AKT/mTOR signaling, which promotes phosphorylation of FOXO1 and 14-3-3 docking to sequester FOXO1 molecule from the nucleus into the cytoplasm. PD-1 signaling stops the process by targeting the AKT/mTOR pathway.
Chronic antigen stimulation alone is known to be adequate in conferring T-cell exhaustion and inducing PD-1 expression (3). PD-1 limits T-cell activation by attenuating TCR signaling, thus preventing immunopathology. PD-1 is a transmembrane molecule, and it's C-terminal domain at the cytoplasmic tail harbors an V/IxY225xxL/V immunoreceptor tyrosine-based inhibitory motif (ITIM) and an TxY248xxV/I immunoreceptor tyrosine-based switch motif (ITSM) inhibitory domains, which serve as binding sites for Src homology 2 domain-containing tyrosine phosphatase 2 (SHP2) (95–97).
Mutation of the ITSM tyrosine domain, which prevents SHP2 recruitment attenuates the ability of PD-1 to suppress T cell activation (96, 98). Engagement of PD-1 receptor with PD-L1 or PD-L2 recruits SHP2 phosphatase to its cytoplasmic domain, which functions to inhibit TCR signaling pathway by preventing ZAP70 phosphorylation and its association with CD3ζ at TCR complex (99). SHP2 also interferes with CD28 costimulatory signaling by blocking PKC-θ activation and dephosphorylates TCR signaling molecules including phospholipase C γ 1 (PLCγ1) and Extracellular signal-regulated protein kinases (ERK1/2) (100).
Besides, PD-1 antagonizes T-cell signaling by inhibiting phosphatidylinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway. The PI3K/AKT/mTOR pathway is well established in regulating cell survival, proliferation and metabolism. Recruitment of SHP2 to the ITIM and ITSM cytoplasmic domains of PD-1 inhibits PI3K thus further blocks AKT phosphorylation and mTOR pathway (101). It is important to note that reports from cancer studies suggest that PD-1 engagement with PD-L1 does not cause dephosphorylation, but leads to phosphorylation and activation of AKT and ERK, which increases chemo resistance of the cancer cells (102, 103), implying a difference in PD-1 signaling between chronic infection and cancer that requires further investigation. Through inhibiting these signaling molecules, PD-1 indirectly inhibits the production of cytokines including IL-2, hence interferes with T cell growth and function. Anti-PD-1 treatment is able to restore anti-viral T-cell signaling such as phosphor-JNK, phosphor-ZAP-70 and phosphor-ERK, besides cytokine production (104).
T-cell activation promotes clathrin-independent internalization TCRζ internalization as reported by Compeer et al. (105) but TCR unit is often directed to the recycling compartment. PD-1 ligation antagonizes TCR signaling by causing internalization and ubiquitin-mediated degradation by casitas B-lymphoma (Cbl)-b E3 ubiquitin ligase (106). Besides, results from single cell imaging suggest that engagement of PD-1 receptors form a microcluster consisting of PD-1 and TCR within the synapse (100). This PD-1-TCR microcluster formation interferes with the TCR signal and mediates T-cell suppression. In case of chronic infection, viral persistence causes the arrest of T cell motility in the splenic red pulp, as PD-1 engagement provides stability to the immunological synapse resulting in immune paralysis in which the unresponsive T cell cannot be released from the sites to engage other targets (104). However, T-cell motility arrest can be completely restored by therapeutic blockade of intravenous injection of antibodies against PD-1 and PD-L1 (104).
One of the hallmarks of T-cell exhaustion is impaired cell proliferation. PD-1 inhibits cell proliferation by attenuating TCR-mediated activation of IL-2 production (99). In addition, PD-1 blocks cell cycle progression through the G1 phase by suppressing multiple transcription of SKP2, downstream of PI3K-AKT and ERK pathways (107). SKP2 is a component of Skp1/Cullin/F-box protein ubiquitin ligase, which functions to degrade p27 (kip1) and inactivates cyclin-dependent kinases (Cdks). By interfering with TCR signaling pathway, PD-1 engagement also leads to changes in transcriptional program of T cells, as seen in the gene expression profiles from HIV-specific CD8+ T cells in HIV-infected individuals. Among the list of the regulated genes include basic leucine transcription factor, ATF-like (BATF), an AP-1 family of transcription factor, of which overexpression impairs T-cell proliferation and cytokine functions. In contrast, BATF silencing rescue HIV-specific T cells derived from individuals with chronic viremia (80).
Epigenetic Alternations of T-Cell Exhaustion
Several reports have described that the epigenetic landscape of a cell directly influences the transcriptional regulation of T-cell exhaustion. Therefore, decoding the language of epigenome specific to exhausted T cells appears to be one of the fundamental steps toward developing therapeutic strategies for overcoming T-cell exhaustion (108).
Currently, there is a paucity for information on global epigenetic landscape for exhausted T cells although recent studies of the Pdcd1 locus (which encodes PD-1) revealed important information (29). During acute LCMV infection, the regulatory region of the Pdcd1 locus is completely demethylated in exhausted CD8+ T cells compared to effector and memory T cells counterparts, and that reduction of virus titers is unlikely to affect methylation pattern as the Pdcd1 regulatory region remained unmethylated when virus titers decreased (109). Moreover, chromatin remodeling of CD8+ T cells from LCMV-infected mice revealed that diacetylated histone H3 was downregulated in total and virus-specific CD8+ T cells suggesting loss of epigenetically active genes (110). Intriguingly, in vitro treatment of exhausted CD8+ T cells with histone deacetylase (HDAC) inhibitors improved function of exhausted T cells in this recent study.
Effector T cells and memory T cells display a number of chromatin accessible regions (ChARs) within the Ifng locus which are absent in exhausted T cells. However, ChARs of the exhausted T cells are only moderately altered after treatment with PD-L1 inhibitor (111). Assessment of the epigenetic state during T-cell exhaustion in acute and chronic infections has identified a number of notable changes to the chromatin accessibility unique to exhausted T cells, which were marked by distinct regulatory sequences possessing characteristics of enhancer, such as enrichment at the intergenic and intronic regions, depletion of transcription start sites (TSS), as well as gene-distal regulatory elements (21, 111). These ChARs were grouped into modules, which upregulate adjacent genes, including Pdcd1. Of note, deletion of an −23.8 kb ChAR harboring transcription factors binding motifs of Sox3, Tbx21 (Encodes T-bet), and Rara (encodes for retinoic acid receptor, RAR) led to dramatic reduction in the expression of PD-1, illustrating its role as an enhancer in PD-1 regulation (21). These described observations, along with other studies (112, 113) indicate that continued antigen burden imparts a stable pattern of chromatin accessibility in exhausted T cells with functional consequences on transcription factors. Nevertheless, the molecular basis by which transcription factors exert their influence on cell fate remains unclear (3).
Exhausted CD8+ T cells reinvigorated by PD-L1 blockade have a distinct epigenetic profile in comparison with memory T cells (111). This epigenetic state is reported to be maintained after PD-1/PD-L1 blockade. Subsequent work involving transferring exhausted T cells from chronically infected mice with LCMV to mice with resolved acute infection showed sustained PD-1 expression as well demethylation of PD-1 promoter in the exhausted CD8+ T cells post transfer (114). Youngblood and colleagues investigated the capacity of HIV-specific CD8+ T cells to modify the PD-1 epigenetic program after reduction in viral load. They reported that PD-1 promotor region was unmethylated in the PD-1 hi virus-specific CD8+ T cells, whereas it remained methylated in donor-matched naive cells at acute and chronic stages of HIV infection. Interestingly, the transcriptional regulatory region sustained unmethylated in virus-specific T cells from individual with a viral load controlled by ART or from elite controllers (115).
Metabolic Programming in T-Cell Exhaustion
An increasing body of evidence indicates that sufficient nutrient supply and energy generation are main determinants of the T cell's ability to proliferate and mediate effector function (116, 117). Although alternations in the transcription program of T cells be linked to T-cell exhaustion, several reports have also suggested that metabolic deficiency and deregulation of nutrient sensing signaling pathways contribute to T-cell exhaustion (12, 118, 119).
Bengsch et al. have recently demonstrated that glycolytic and mitochondrial metabolism in early effector CD8+ T cells is inhibited by PD-1 signaling in chronic LCMV infection. They reported that PD-1 signals inhibit the expression of key metabolic regulator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) and interestingly overexpression of PGC1α corrected some metabolic alterations in developing exhausted T cells and improved effector function (120). Importantly, the authors of this study also reported that T-betHiPD-1Int cells have higher glucose uptake and a decrease in mitochondrial mass in comparison to EomesHiPD-1Hi cells and only PD-1Int cells could be rescued metabolically and functionally.
Naive and resting T cells make use of fatty acid oxidation (FAO) and the mitochondrial tricarboxylic acid (TCA) cycle to generate large amounts of ATP through oxidative phosphorylation (OXPHOS) (121, 122). Recent studies in a murine model revealed that mitochondrial activity was one of the requirements for the activation and sustenance of antigen-specific responses (123, 124). Upon activation, T cells switch their metabolism to high rates of glycolysis even in the presence of sufficient oxygen and this support proliferation and effector function via providing fast energy and metabolites (125).
HIV-specific T cells upregulated OXPHOS owing to an increased mitochondrial mass (126). However, it's not clear whether the observed increase was due to an augmented number of functional mitochondria or the emergence of massive non-functional mitochondria (127, 128). Schurich et al. recently described that the poorly functional PD-1hi T cell responses against HBV upregulate GLUT1, which is a constitutive glucose transporter (116). They also showed that Glut1hi HBV-specific T cells are dependent on glucose supplies, unlike the more functional CMV-specific T cells that could utilize OXPHOS in the absence of glucose. The exhausted HBV-specific T cells were unable to switch to OXPHOS and had also increased mitochondrial size and lower mitochondrial potential, all suggestive of mitochondrial dysfunction (116). Intriguingly, their defect in mitochondrial metabolism was rescued by the proinflammatory cytokine interleukin (IL)-12, which recovered the exhausted HBV-specific T cell effector function, increased their mitochondrial potential, and reduced their dependence on glycolysis.
The transcription factor mTOR is a key molecule sensing ATP and intracellular amino acids (129) that also regulates the fatty acid metabolism in memory T cells (122). The PD-1 signaling pathway also affects T-cell functions through metabolism. PD-1 signaling reduces AKT activation and thus suppress mTOR activity, switching T cell metabolism from glycolysis to FOA (Figure 2) (84, 130, 131). Declined mTOR activation in exhausted PD-1+ CD8+ T cells give rise to increased activity of the transcription factor forkhead box O1 (FoxO1), which sustains PD-1 expression and survival of exhausted CD8 T cells (84). A recent report indicated that IL-12 enhanced mTOR expression in antigen-stimulated CD8+ T cells, thereby modulating CD8+ T effector differentiation and metabolism. Furthermore, blockade of the mTOR signaling pathway via rapamycin inhibits IL-12-induced expression of T-bet and skews the CD8+ T cell response toward EOMES dependent memory development (132). Notably, T-bet has a significant role in sustaining the limited effector functional capacity of T cells in chronic infections (81). Moreover, T-bet expression is increased by IL-12 in exhausted T cells in chronic HBV infection and this enhance their functionality (133).
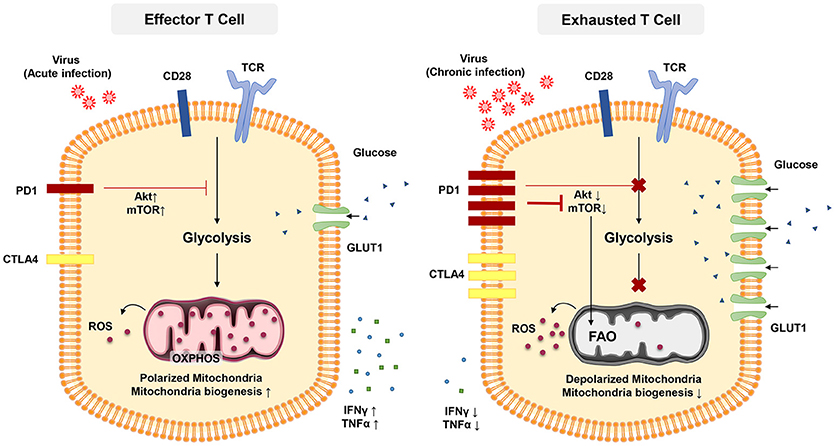
Figure 2. In acute infection, effector CD8 + T cells induce glycolysis after activation to sustain effector functions. Akt and mTOR promote glycolysis and support effector T cell functions. In chronic infection, exhausted T cells express inhibitory receptors such as PD-1 and CTL4. PD-1 signaling reduces AKT activation and thus suppress mTOR activity, switching T cell metabolism from glycolysis to FAO. These metabolic reprogramming may lead to mitochondrial depolarization, reduction of mitochondrial biogenesis and higher rate of ROS production which is associated with functional impairment in exhausted T cells.
Targeting metabolic dysfunctions with mitochondrion-targeted antioxidants are reported to remarkably improve the antiviral activity of exhausted HBV-specific CD8+ T cells suggesting the pivotal role for ROS in T-cell exhaustion (134). It is crucial to further investigate the contributions of mitochondrial biogenesis on T-cell exhaustion and how we can target mitochondrial metabolism of T cells when treating chronic viral infection (135).
Other Potential Strategies to Overcome T-Cell Exhaustion
Engineered T cells, such as T cells expressing chimeric antigen receptors (CARs), are another strategy to overcome exhaustion in cancer and chronic infections (136–138). Nevertheless, CAR-T cells also become exhausted and require immune checkpoint blockade so that they can restore their functionality (139, 140). Instead of targeting the PD-1/PD-L1 signaling pathway, PD-1 expression could be declined by gene editing approach, made possible by the CRISPR-Cas9 system. A recent report showed a decreased PD-1 expression on primary human cells, utilizing the CRISPR-Cas9 system (141), demonstrating a method to generate CAR-T cells with more resistance to exhaustion. Nonetheless, as up-regulation of inhibitory receptors represents a way for the immune system to restrict immunopathology triggered by prolonged exposure to antigen, a more fine-tuned and adjustable approach to avoid exhaustion could be preferable (142–150).
Recently a stem cell–like CD8+ T-cell subset was discovered among exhausted PD-1-expressing CD8+ T cells during chronic viral infection and this subset can expand in response to PD-1-targeted immunotherapy in contrast with terminally differentiated/exhausted PD-1-expressing CD8+ T cells (151). These two CD8+ T cell populations have distinct expression profiles of co-inhibitory receptors and co-stimulatory molecules, so describing how different immunotherapeutic interventions affect these two population is highly relevant for understanding the mechanistic basis of the efficacy of present and future immunotherapies that target exhausted T cells (29).
Conclusions
Our understanding of T-cell exhaustion is advancing at a rapid pace. However, it remains unclear as to what key transcription factors are involved in critical signaling pathways related to exhaustion and how these transcription factors are regulated by epigenomic alterations. Moreover, most of the studies of T-cell exhaustion have been focused on LCMV model and the critical changes in T cell phenotype and functional impairment of exhausted T cells utilizing human infected samples has been neglected due to lack of in vitro models. Finally, more detailed understanding of human anti-viral immunity is still critical to develop novel immunotherapies to reverse the state of exhaustion.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The work has also been funded by the Universiti Malaya Research Grant No. RP021A-13HTM to AS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. (2013) 13:309–20. doi: 10.1038/nri3442
2. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
3. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
5. Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature (1993) 362:758–61. doi: 10.1038/362758a0
6. Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. (1998) 188:2205–13. doi: 10.1084/jem.188.12.2205
7. Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliott T, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. (1998) 187:1383–93. doi: 10.1084/jem.187.9.1383
8. Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. (2014) 35:51–60. doi: 10.1016/j.it.2013.10.001
9. Tan A, Koh S, Bertoletti A. Immune response in hepatitis B virus infection. Cold Spring Harbor Perspect Med. (2015) 5:a021428. doi: 10.1101/cshperspect.a021428
10. Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology (2010) 129:474–81. doi: 10.1111/j.1365-2567.2010.03255.x
11. Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc Natl Acad Sci USA. (2010) 107:20453–8. doi: 10.1073/pnas.1008437107
12. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
13. Welsh RM. Assessing CD8 T cell number and dysfunction in the presence of antigen. J Exp Med. (2001) 193:F19–22. doi: 10.1084/jem.193.5.F19
14. Fuller MJ, Khanolkar A, Tebo AE, Zajac AJ. Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J Immunol. (2004) 172:4204–14. doi: 10.4049/jimmunol.172.7.4204
15. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. (2003) 77:4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003
16. Agnellini P, Wolint P, Rehr M, Cahenzli J, Karrer U, Oxenius A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc Natl Acad Sci USA. (2007) 104:4565–70. doi: 10.1073/pnas.0610335104
17. Zhou S, Ou R, Huang L, Price GE, Moskophidis D. Differential tissue-specific regulation of antiviral CD8+ T-cell immune responses during chronic viral infection. J Virol. (2004) 78:3578–600. doi: 10.1128/JVI.78.7.3578-3600.2004
18. Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, et al. A role for the transcriptional repressor Blimp-1 in CD8+ T cell exhaustion during chronic viral infection. Immunity (2009) 31:309–20. doi: 10.1016/j.immuni.2009.06.019
19. Mackerness KJ, Cox MA, Lilly LM, Weaver CT, Harrington LE, Zajac AJ. Pronounced virus-dependent activation drives exhaustion but sustains IFN-gamma transcript levels. J Immunol. (2010) 185:3643–51. doi: 10.4049/jimmunol.1000841
20. Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity (2012) 37:1130–44. doi: 10.1016/j.immuni.2012.08.021
21. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (2016) 354:1165–9. doi: 10.1126/science.aae0491
22. Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. (2004) 78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004
23. Williams MA, Bevan MJ. Effector and memory CTL differentiation. Ann Rev Immunol. (2007) 25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548
24. Kasakovski D, Xu L, Li Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J Hematol. Oncol. (2018) 11:91. doi: 10.1186/s13045-018-0629-x
25. Dolfi DV, Mansfield KD, Polley AM, Doyle SA, Freeman GJ, Pircher H, et al. Increased T-bet is associated with senescence of influenza virus-specific CD8 T cells in aged humans. J Leukocyte Biol. (2013) 93:825–36. doi: 10.1189/jlb.0912438
26. Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood (2003) 101:2711–20. doi: 10.1182/blood-2002-07-2103
27. Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. (2011) 11:289–95. doi: 10.1038/nri2959
28. Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, Harty JT, et al. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8+ T cell differentiation. Immunity (2010) 33:128–40. doi: 10.1016/j.immuni.2010.06.014
29. Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Ann Rev Med. (2018) 69:301–18. doi: 10.1146/annurev-med-012017-043208
30. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. doi: 10.1038/nature04444
31. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443:350–4. doi: 10.1038/nature05115
32. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. (2006) 12:1198–202. doi: 10.1038/nm1482
33. Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. (2006) 203:2281–92. doi: 10.1084/jem.20061496
34. Palmer BE, Neff CP, Lecureux J, Ehler A, Dsouza M, Remling-Mulder L, et al. In vivo blockade of the PD-1 receptor suppresses HIV-1 viral loads and improves CD4+ T cell levels in humanized mice. J Immunol. (2013) 190:211–9. doi: 10.4049/jimmunol.1201108
35. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature (2009) 458:206–10. doi: 10.1038/nature07662
36. Dyavar Shetty R, Velu V, Titanji K, Bosinger SE, Freeman GJ, Silvestri G, et al. PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J Clin Invest. (2012) 122:1712–6. doi: 10.1172/JCI60612
37. Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. (2007) 81:4215–25. doi: 10.1128/JVI.02844-06
38. Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. (2007) 81:2545–53. doi: 10.1128/JVI.02021-06
39. Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, et al. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. (2006) 80:11398–403. doi: 10.1128/JVI.01177-06
40. Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. (2007) 81:9249–58. doi: 10.1128/JVI.00409-07
41. Nakamoto N, Kaplan DE, Coleclough J, Li Y, Valiga ME, Kaminski M, et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology (2008) 134:1927–37, 37 e1–2. doi: 10.1053/j.gastro.2008.02.033
42. Fuller MJ, Callendret B, Zhu B, Freeman GJ, Hasselschwert DL, Satterfield W, et al. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc Natl Acad Sci USA. (2013) 110:15001–6. doi: 10.1073/pnas.1312772110
43. Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS ONE (2013) 8:e63818. doi: 10.1371/journal.pone.0063818
44. Gay CL, Bosch RJ, Ritz J, Hataye JM, Aga E, Tressler RL, et al. Clinical trial of the Anti-PD-L1 antibody BMS-936559 in HIV-1 infected participants on suppressive antiretroviral therapy. J Infect Dis. (2017) 215:1725–33. doi: 10.1093/infdis/jix191
45. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. (2009) 10:29–37. doi: 10.1038/ni.1679
46. Kaufmann DE, Walker BD. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J Immunol. (2009) 182:5891–7. doi: 10.4049/jimmunol.0803771
47. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Eng J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
48. Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell (1994) 76:241–51. doi: 10.1016/0092-8674(94)90332-8
49. Blackburn SD, Wherry EJ. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. (2007) 15:143–6. doi: 10.1016/j.tim.2007.02.006
50. Ni G, Wang T, Walton S, Zhu B, Chen S, Wu X, et al. Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol. (2015) 293:126–9. doi: 10.1016/j.cellimm.2014.12.012
51. Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. (2006) 203:2461–72. doi: 10.1084/jem.20061462
52. Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, Sela J, et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood (2009) 114:346–56. doi: 10.1182/blood-2008-12-191296
53. Porichis F, Hart MG, Zupkosky J, Barblu L, Kwon DS, McMullen A, et al. Differential impact of PD-1 and/or interleukin-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J Virol. (2014) 88:2508–18. doi: 10.1128/JVI.02034-13
54. Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci USA. (2008) 105:20428–33. doi: 10.1073/pnas.0811139106
55. Blander JM, Longman RS, Iliev ID, Sonnenberg GF, Artis D. Regulation of inflammation by microbiota interactions with the host. Nat Immunol. (2017) 18:851–60. doi: 10.1038/ni.3780
56. Gaddi PJ, Crane MJ, Kamanaka M, Flavell RA, Yap GS, Salazar-Mather TP. IL-10 mediated regulation of liver inflammation during acute murine cytomegalovirus infection. PLoS ONE (2012) 7:e42850. doi: 10.1371/journal.pone.0042850
57. Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al. PD-1:PD-L inhibitory pathway affects both CD4+ and CD8+ T cells and is overcome by IL-2. Eur J Immunol. (2002) 32:634–43. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9
58. Grint D, Tedaldi E, Peters L, Mocroft A, Edlin B, Gallien S, et al. Hepatitis C virus (HCV) RNA profiles among chronic HIV/HCV-coinfected individuals in ESPRIT; spontaneous HCV RNA clearance observed in nine individuals. HIV Med. (2017) 18:430–4. doi: 10.1111/hiv.12466
59. Schmitz I, Schneider C, Frohlich A, Frebel H, Christ D, Leonard WJ, et al. IL-21 restricts virus-driven Treg cell expansion in chronic LCMV infection. PLoS Pathog. (2013) 9:e1003362. doi: 10.1371/journal.ppat.1003362
60. Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov. (2014) 13:379–95. doi: 10.1038/nrd4296
61. Zhao J, Zhang Z, Luan Y, Zou Z, Sun Y, Li Y, et al. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology (2014) 59:1331–42. doi: 10.1002/hep.26916
62. Isailovic N, Daigo K, Mantovani A, Selmi C. Interleukin-17 and innate immunity in infections and chronic inflammation. J Autoimmun. (2015) 60:1–11. doi: 10.1016/j.jaut.2015.04.006
63. Kahan SM, Wherry EJ, Zajac AJ. T cell exhaustion during persistent viral infections. Virology (2015) 479–480:180–93. doi: 10.1016/j.virol.2014.12.033
64. Ortiz AM, Klase ZA, DiNapoli SR, Vujkovic-Cvijin I, Carmack K, Perkins MR, et al. IL-21 and probiotic therapy improve Th17 frequencies, microbial translocation, and microbiome in ARV-treated, SIV-infected macaques. Mucosal Immunol. (2016) 9:458–67. doi: 10.1038/mi.2015.75
65. Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J, et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J Clin Invest. (2017) 127:260–8. doi: 10.1172/JCI89488
66. Veiga-Parga T, Sehrawat S, Rouse BT. Role of regulatory T cells during virus infection. Immunol Rev. (2013) 255:182–96. doi: 10.1111/imr.12085
67. Penaloza-MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, West EE, et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med. (2014) 211:1905–18. doi: 10.1084/jem.20132577
68. Ng CT, Snell LM, Brooks DG, Oldstone MB. Networking at the level of host immunity: immune cell interactions during persistent viral infections. Cell Host Microbe (2013) 13:652–64. doi: 10.1016/j.chom.2013.05.014
69. Goh C, Narayanan S, Hahn YS. Myeloid-derived suppressor cells: the dark knight or the joker in viral infections? Immunol Rev. (2013) 255:210–21. doi: 10.1111/imr.12084
70. Joosten SA, van Meijgaarden KE, Savage ND, de Boer T, Triebel F, van der Wal A, et al. Identification of a human CD8+ regulatory T cell subset that mediates suppression through the chemokine CC chemokine ligand 4. Proc Natl Acad Sci USA. (2007) 104:8029–34. doi: 10.1073/pnas.0702257104
71. Holderried TA, Lang PA, Kim HJ, Cantor H. Genetic disruption of CD8+ Treg activity enhances the immune response to viral infection. Proc Natl Acad Sci USA. (2013) 110:21089–94. doi: 10.1073/pnas.1320999110
72. Waggoner SN, Kumar V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front Immunol. (2012) 3:377. doi: 10.3389/fimmu.2012.00377
73. Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, et al. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci USA. (2007) 104:15430–5. doi: 10.1073/pnas.0702579104
74. Schacker T. The role of secondary lymphatic tissue in immune deficiency of HIV infection. Aids (2008) 22 Suppl 3:S13–8. doi: 10.1097/01.aids.0000327511.76126.b5
75. Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J Clin Invest. (2004) 113:737–45. doi: 10.1172/JCI20243
76. Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science (2009) 324:1569–72. doi: 10.1126/science.1174182
77. Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science (2009) 324:1576–80. doi: 10.1126/science.1172815
78. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity (2014) 40:289–302. doi: 10.1016/j.immuni.2014.01.005
79. Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity (2015) 42:265–78. doi: 10.1016/j.immuni.2015.01.006
80. Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. (2010) 16:1147–51. doi: 10.1038/nm.2232
81. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science (2012) 338:1220–5. doi: 10.1126/science.1229620
82. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol. (2011) 12:663–71. doi: 10.1038/ni.2046
83. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat Immunol. (2013) 14:1173–82. doi: 10.1038/ni.2714
84. Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8+ T cells during chronic infection. Immunity (2014) 41:802–14. doi: 10.1016/j.immuni.2014.10.013
85. Stephen TL, Rutkowski MR, Allegrezza MJ, Perales-Puchalt A, Tesone AJ, Svoronos N, et al. Transforming growth factor beta-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression. Immunity (2014) 41:427–39. doi: 10.1016/j.immuni.2014.08.012
86. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor IRF4 promotes CD8+ T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity (2017) 47:1129–41 e5. doi: 10.1016/j.immuni.2017.11.021
87. Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, et al. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med. (2007) 204:2015–21. doi: 10.1084/jem.20070841
88. Paley MA, Gordon SM, Bikoff EK, Robertson EJ, Wherry EJ, Reiner SL. Technical advance: fluorescent reporter reveals insights into eomesodermin biology in cytotoxic lymphocytes. J Leukocyte Biol. (2013) 93:307–15. doi: 10.1189/jlb.0812400
89. Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, et al. Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol. (2010) 185:4988–92. doi: 10.4049/jimmunol.1002042
90. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity (2010) 33:229–40. doi: 10.1016/j.immuni.2010.08.002
91. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci USA. (2008) 105:15016–21. doi: 10.1073/pnas.0801497105
92. Hwang S, Cobb DA, Bhadra R, Youngblood B, Khan IA. Blimp-1-mediated CD4 T cell exhaustion causes CD8 T cell dysfunction during chronic toxoplasmosis. J Exp Med. (2016) 213:1799–818. doi: 10.1084/jem.20151995
93. Suprunenko T, Hofer MJ. The emerging role of interferon regulatory factor 9 in the antiviral host response and beyond. Cytokine Growth Fact Rev. (2016) 29:35–43. doi: 10.1016/j.cytogfr.2016.03.002
94. Huber M, Suprunenko T, Ashhurst T, Marbach F, Raifer H, Wolff S, et al. IRF9 Prevents CD8+ T cell exhaustion in an extrinsic manner during acute Lymphocytic choriomeningitis virus infection. J Virol. (2017) 91:e01219-17. doi: 10.1128/JVI.01219-17
95. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. (2009) 229:114–25. doi: 10.1111/j.1600-065X.2009.00767.x
96. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. (2004) 173:945–54. doi: 10.4049/jimmunol.173.2.945
97. Arasanz H, Gato-Canas M, Zuazo M, Ibanez-Vea M, Breckpot K, Kochan G, et al. PD1 signal transduction pathways in T cells. Oncotarget (2017) 8:51936–45. doi: 10.18632/oncotarget.17232
98. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA. (2001) 98:13866–71. doi: 10.1073/pnas.231486598
99. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. (2004) 574:37–41. doi: 10.1016/j.febslet.2004.07.083
100. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
101. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
102. Black M, Barsoum IB, Truesdell P, Cotechini T, Macdonald-Goodfellow SK, Petroff M, et al. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget (2016) 7:10557–67. doi: 10.18632/oncotarget.7235
103. Liu S, Chen S, Yuan W, Wang H, Chen K, Li D, et al. PD-1/PD-L1 interaction up-regulates MDR1/P-gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget (2017) 8:99901–12. doi: 10.18632/oncotarget.21914
104. Zinselmeyer BH, Heydari S, Sacristan C, Nayak D, Cammer M, Herz J, et al. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med. (2013) 210:757–74. doi: 10.1084/jem.20121416
105. Compeer EB, Kraus F, Ecker M, Redpath G, Amiezer M, Rother N, et al. A mobile endocytic network connects clathrin-independent receptor endocytosis to recycling and promotes T cell activation. Nat Commun. (2018) 9:1597. doi: 10.1038/s41467-018-04088-w
106. Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. (2011) 3:581–92. doi: 10.1002/emmm.201100165
107. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. (2012) 5:ra46. doi: 10.1126/scisignal.2002796
108. Wu J, Shi H. Unlocking the epigenetic code of T cell exhaustion. Trans Cancer Res. (2017) 6(Suppl. 2):S384–S7. doi: 10.21037/tcr.2017.03.02
109. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity (2011) 35:400–12. doi: 10.1016/j.immuni.2011.06.015
110. Zhang F, Zhou X, DiSpirito JR, Wang C, Wang Y, Shen H. Epigenetic manipulation restores functions of defective CD8(+) T cells from chronic viral infection. Mol Ther. (2014) 22:1698–706. doi: 10.1038/mt.2014.91
111. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (2016) 354:1160–5. doi: 10.1126/science.aaf2807
112. Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, et al. Dynamic changes in chromatin accessibility occur in CD8(+) T cells responding to viral infection. Immunity (2016) 45:1327–40. doi: 10.1016/j.immuni.2016.10.028
113. Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O'Brien S, et al. Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T cells. Immunity (2018) 48:1029–45 e5. doi: 10.1016/j.immuni.2018.04.026
114. Ahn E, Youngblood B, Lee J, Lee J, Sarkar S, Ahmed R. Demethylation of the PD-1 promoter is imprinted during the effector phase of CD8 T cell exhaustion. J Virol. (2016) 90:8934–46. doi: 10.1128/JVI.00798-16
115. Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, Austin JW, et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol. (2013) 191:540–4. doi: 10.4049/jimmunol.1203161
116. Schurich A, Pallett LJ, Jajbhay D, Wijngaarden J, Otano I, Gill US, et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep. (2016) 16:1243–52. doi: 10.1016/j.celrep.2016.06.078
117. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
118. West EE, Youngblood B, Tan WG, Jin HT, Araki K, Alexe G, et al. Tight regulation of memory CD8(+) T cells limits their effectiveness during sustained high viral load. Immunity (2011) 35:285–98. doi: 10.1016/j.immuni.2011.05.017
119. Assmann N, Finlay DK. Metabolic regulation of immune responses: therapeutic opportunities. J Clin Invest. (2016) 126:2031–9. doi: 10.1172/JCI83005
120. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity (2016) 45:358–73. doi: 10.1016/j.immuni.2016.07.008
121. Brand K, Leibold W, Luppa P, Schoerner C, Schulz A. Metabolic alterations associated with proliferation of mitogen-activated lymphocytes and of lymphoblastoid cell lines: evaluation of glucose and glutamine metabolism. Immunobiology (1986) 173:23–34. doi: 10.1016/S0171-2985(86)80086-9
122. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi: 10.1038/nature08097
123. Okoye I, Wang L, Pallmer K, Richter K, Ichimura T, Haas R, et al. T cell metabolism. The protein LEM promotes CD8(+) T cell immunity through effects on mitochondrial respiration. Science (2015) 348:995–1001. doi: 10.1126/science.aaa7516
124. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
125. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Ann Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
126. Petrovas C, Mueller YM, Dimitriou ID, Altork SR, Banerjee A, Sklar P, et al. Increased mitochondrial mass characterizes the survival defect of HIV-specific CD8(+) T cells. Blood (2007) 109:2505–13. doi: 10.1182/blood-2006-05-021626
127. Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, et al. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol. (2006) 209:468–80. doi: 10.1002/jcp.20753
128. Schurich A, Henson SM. The many unknowns concerning the bioenergetics of exhaustion and senescence during chronic viral infection. Front Immunol. (2014) 5:468. doi: 10.3389/fimmu.2014.00468
129. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Ann Rev Immunol. (2012) 30:39–68. doi: 10.1146/annurev-immunol-020711-075024
130. Patsoukis N, Bardhan K, Weaver J, Herbel C, Seth P, Li L, et al. The role of metabolic reprogramming in T cell fate and function. Curr Trends Immunol. (2016) 17:1–12.
131. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
132. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity (2010) 32:67–78. doi: 10.1016/j.immuni.2009.10.010
133. Schurich A, Pallett LJ, Lubowiecki M, Singh HD, Gill US, Kennedy PT, et al. The third signal cytokine IL-12 rescues the anti-viral function of exhausted HBV-specific CD8 T cells. PLoS Pathog. (2013) 9:e1003208. doi: 10.1371/journal.ppat.1003208
134. Fisicaro P, Barili V, Montanini B, Acerbi G, Ferracin M, Guerrieri F, et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med. (2017) 23:327–36. doi: 10.1038/nm.4275
135. Chao T, Wang H, Ho PC. Mitochondrial control and guidance of cellular activities of T cells. Front Immunol. (2017) 8:473. doi: 10.3389/fimmu.2017.00473
136. Feldman SA, Assadipour Y, Kriley I, Goff SL, Rosenberg SA. Adoptive cell therapy–tumor-infiltrating lymphocytes, T-cell receptors, and chimeric antigen receptors. Sem Oncol. (2015) 42:626–39. doi: 10.1053/j.seminoncol.2015.05.005
137. Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH. CAR T-cell therapy of solid tumors. Immunol Cell Biol. (2017) 95:356–63. doi: 10.1038/icb.2016.128
138. Krebs K, Bottinger N, Huang LR, Chmielewski M, Arzberger S, Gasteiger G, et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology (2013) 145:456–65. doi: 10.1053/j.gastro.2013.04.047
139. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
140. John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology (2013) 2:e26286. doi: 10.4161/onci.26286
141. Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci USA. (2015) 112:10437–42. doi: 10.1073/pnas.1512503112
142. Marcus B, Johanna T, Annika CK. Genetic footprints of T cell exhaustion. Trans Cancer Res. (2017) 5:S65–7. doi: 10.21037/tcr.2017.02.11
143. Barathan M, Mohamed R, Yong YK, Kannan M, Vadivelu J, Saeidi A, Larsson M, Shankar EM. Viral persistence and chronicity in hepatitis C virus infection: role of T-cell apoptosis, senescence and exhaustion. Cells (2018) 7:E165. doi: 10.3390/cells7100165
144. Yong YK, Saeidi A, Tan HY, Rosmawati M, Enström PF, Batran RA, Vasuki V, et al. Hyper-expression of PD-1 is associated with the levels of exhausted and dysfunctional phenotypes of circulating CD161++TCR iVα7.2+ mucosal-associated invariant T cells in chronic hepatitis B virus infection. Front Immunol. (2018) 9:472. doi: 10.3389/fimmu.2018.00472
145. See JX, Chandramathi S, Abdulla MA, Vadivelu J, Shankar EM. Persistent infection due to a small-colony variant of Burkholderia pseudomallei leads to PD-1 upregulation on circulating immune cells and mononuclear infiltration in viscera of experimental BALB/c mice. PLoS Negl Trop Dis. (2017) 11:e0005702. doi: 10.1371/journal.pntd.0005702
146. Ahmad F, Shankar EM, Yong YK, Tan HY, Ahrenstorf G, Jacobs R, et al. Negative checkpoint regulatory molecule 2B4 (CD244) upregulation is associated with invariant natural killer T cell alterations and human immunodeficiency virus disease progression. Front Immunol. (2017) 8:338. doi: 10.3389/fimmu.2017.00338
147. Barathan M, Mohamed R, Vadivelu J, Chang LY, Saeidi A, Yong YK, et al. Peripheral loss of CD8(+) CD161(++) TCRVα7·2(+) mucosal-associated invariant T cells in chronic hepatitis C virus-infected patients. Eur J Clin Invest. (2016) 46:170–80. doi: 10.1111/eci.12581
148. Saeidi A, Tien Tien VL, Al-Batran R, Al-Darraji HA, Tan HY, Yong YK, et al. Attrition of TCR Vα7.2+ CD161++ MAIT cells in HIV-tuberculosis co-infection is associated with elevated levels of PD-1 expression. PLoS ONE (2015) 10:e0124659. doi: 10.1371/journal.pone.0124659
149. Barathan M, Gopal K, Mohamed R, Ellegård R, Saeidi A, Vadivelu J, et al. Chronic hepatitis C virus infection triggers spontaneous differential expression of biosignatures associated with T cell exhaustion and apoptosis signaling in peripheral blood mononucleocytes. Apoptosis (2015) 20:466–80. doi: 10.1007/s10495-014-1084-y
150. Larsson M, Shankar EM, Che KF, Saeidi A, Ellegård R, Barathan M, et al. Molecular signatures of T-cell inhibition in HIV-1 infection. Retrovirology (2013) 10:31. doi: 10.1186/1742-4690-10-31
Keywords: T-cell exhaustion, PD-1, immunotherapy, rejuvenation, T-bet, metabolism, epigenetics
Citation: Saeidi A, Zandi K, Cheok YY, Saeidi H, Wong WF, Lee CYQ, Cheong HC, Yong YK, Larsson M and Shankar EM (2018) T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 9:2569. doi: 10.3389/fimmu.2018.02569
Received: 09 August 2018; Accepted: 18 October 2018;
Published: 09 November 2018.
Edited by:
Cristina Bonorino, Pontifícia Universidade Católica do Rio Grande do Sul, BrazilReviewed by:
Xing Chang, Shanghai Institutes for Biological Sciences (CAS), ChinaStefan Carl Wilhelm Meuer, Universität Heidelberg, Germany
Copyright © 2018 Saeidi, Zandi, Cheok, Saeidi, Wong, Lee, Cheong, Yong, Larsson and Shankar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esaki Muthu Shankar, c2hhbmthcmVtQGN1dG4uYWMuaW4=