 Jessica D. Gereige
Jessica D. Gereige Paul J. Maglione
Paul J. Maglione- Department of Pulmonary, Allergy, Sleep & Critical Care Medicine, Boston Medical Center, Boston University School of Medicine, Boston, MA, United States
Common variable immunodeficiency (CVID) is the most prevalent symptomatic primary immunodeficiency and comprises a group of disorders with similar antibody deficiency but a myriad of different etiologies, most of which remain undefined. The variable aspect of CVID refers to the approximately half of patients who develop non-infectious complications in addition to heightened susceptibility to infection. The pathogenesis of these complications is poorly understood and somewhat counterintuitive because these patients that are defined by their immune futility simultaneously have elevated propensity for autoimmune disease. There are numerous aspects of immune dysregulation associated with autoimmunity in CVID that have only begun to be studied. These findings include elevations of T helper type 1 and follicular helper T cells and B cells expressing low levels of CD21 as well as reciprocal decreases in regulatory T cells and isotype-switched memory B cells. Recently, advances in genomics have furthered our understanding of the fundamental biology underlying autoimmunity in CVID and led to precision therapeutic approaches. However, these genetic etiologies are also associated with clinical heterogeneity and incomplete penetrance, highlighting the fact that continued research efforts remain necessary to optimize treatment. Additional factors, such as commensal microbial dysbiosis, remain to be better elucidated. Thus, while recent advances in our understanding of CVID-associated autoimmunity have been exciting and substantial, these current scientific advances must now serve as building blocks for the next stages of discovery.
Introduction
The diagnosis common variable immunodeficiency (CVID) is used to denote a group of disorders that, together, account for more than 50% of symptomatic primary immune deficiencies (PID) (1) with an estimated incidence of 1:50,000–1:25,000 (2). The term common variable immunodeficiency was originally used to describe patients with a primary antibody deficiency who did not meet criteria for the more well-defined PIDs such as Burton's agammaglobulinemia (3). We have come to appreciate that CVID is best understood to constitute a group of hypogammaglobulinemic disorders with heterogeneous phenotypic presentations, rather than a single entity (4). The application of genomics has only furthered the evidence of heterogeneity within CVID.
Diagnostic criteria for CVID have evolved since it was first made a diagnosis of exclusion in 1971 by the World Health Organization. The International Consensus Document on CVID put forward in 2016 agreed on a definition requiring IgG levels two standard deviations below the age-appropriate reference as well as either low IgA or IgM levels, and poor antibody response to vaccination in an individual that is at least 4 years old with no secondary cause of hypogammaglobulinemia (1). The European Society of Immune Deficiencies (ESID) diagnostic criteria differ slightly from these criteria in that they require the presence of symptoms such as infections or autoimmune manifestations, in addition to the laboratory abnormalities cited above, to make the diagnosis of CVID (5). Notably, non-infectious presentations are an under-recognized feature of CVID and often the predominant clinical presentation, resulting in diagnostic delays of several years in many cases (6). While historically the clinical presentation of CVID was focused on the susceptibility to infections, the decision to include autoimmune and inflammatory conditions as primary clinical presentations reflects the heterogeneity of CVID and highlights the importance of recognizing these non-infectious entities as a feature of this immune deficiency.
Immunoglobulin replacement therapy is the standard of care for CVID. Since the widespread adoption of this treatment, mortality of patients with CVID has decreased from 30% in the early 1990s (7) to 15% in the early 2000s (8), in a cohort of 240 patients in the United Kingdom and 334 patients from the ESID registry, respectively, all followed for approximately two decades. The improved survival in CVID patients has been attributed to the reduction of infectious complications thanks to the widespread use of immunoglobulin replacement and improved anti-microbial therapies (9–12). While overall survival has improved, patients with CVID continue to have reduced survival compared to age-matched controls (13). When comparing mortality within CVID patients, subjects with at least one non-infectious complication had significantly higher mortality compared to patients with only infectious complications (13, 14). These non-infectious complications including autoimmune, gastrointestinal, pulmonary, lymphoproliferative, and malignant complications (1), are not ameliorated by immunoglobulin replacement therapy alone. The clear necessity to address non-infectious complications of CVID has led to attempts at categorizing these heterogeneous disorders into distinct phenotypes, with the hopes of elucidating endotypes. The ultimate goal of such stratification of CVID is to identify targeted treatments that will improve outcomes in CVID patients with non-infectious complications (14–16).
The first attempt at phenotyping CVID patients was done by Chapel et al. (14) by categorizing the various associated complications and looking at the independence of one from the other. They identified five distinct phenotypes: no complications, autoimmunity, polyclonal lymphocytic infiltration, enteropathy, and lymphoid malignancy (14). Studies that followed have confirmed categorization of CVID based upon the presence of complications, with certain features like autoimmunity, lymphocytic interstitial lung disease, and lymphoid hyperplasia typically occurring together (6, 17). Assuming that CVID endotypes present within these phenotypic clusters, the pathogenesis and genetic mechanisms underlying the disease may be related. Autoimmunity and immune deficiency have been shown to have genetic overlap and occur together beyond CVID (18–20), suggesting a common pathophysiologic mechanism underlying both forms of immune dysregulation. In this review, we focus on one aspect of immune dysregulation, the autoimmune manifestations of CVID, providing an overview of these complications as well as an update on research and treatment advances.
Overview of Autoimmune Disease in Common Variable Immunodeficiency
The initial clustering of CVID patients with autoimmunity included organ specific autoimmune disease (e.g., Grave's thyroiditis, insulin dependent diabetes mellitus), systemic autoimmune disease (e.g., rheumatoid arthritis, systemic lupus erythematosus), and autoimmune cytopenias (e.g., immune thrombocytopenia, autoimmune hemolytic anemia) (14). Further analysis on two other cohorts showed that within the autoimmune cluster, only autoimmune cytopenias had decreased survival and that organ-specific and systemic autoimmune disease showed no association with cytopenias or the other clinical phenotypes (16). This led to a revision of the clinical phenotypes with more emphasis being placed on autoimmune cytopenias as opposed to autoimmunity in general (16). Unbiased network clustering in a separate CVID cohort yielded similar phenotypes, with systemic and organ-specific autoimmune diseases clustering separately from autoimmune cytopenias (21). While this distinction likely carries implications regarding underlying pathophysiology, many studies continue to combine autoimmune cytopenias with other autoimmunity as they compare CVID patients. For this reason, we will make distinctions between autoimmune cytopenias and organ specific or systemic autoimmunity in this review.
Autoimmune diseases are one of the most common non-infectious complications, occurring in ~20–30% of patients with CVID, with autoimmune cytopenias being the most common (9, 22, 23). In a European cohort of 2700 CVID patients taken from the ESID registry, autoimmune cytopenias were found to be 700 times more prevalent in CVID patients compared to the general population (9). Among autoimmune cytopenias, autoimmune thrombocytopenia and autoimmune hemolytic anemia occur most frequently, either separately or concurrently as Evan's syndrome. Autoimmune neutropenia also occurs in CVID, although more rarely than thrombocytopenia or anemia (9, 24). Importantly, the diagnosis of cytopenia precedes that of CVID by several years in up to 60% of patients (22).
Autoimmune cytopenias are often associated with other non-infectious complications in CVID. Compared with other CVID patients, those with ILD are more likely to have had autoimmune cytopenias (6, 25). Conversely, CVID patients with autoimmune cytopenias had a higher frequency of CVID-associated non-infectious complications, including granulomatous and lymphoproliferative disease, as well as organ-specific autoimmune disease, but interestingly, not systemic autoimmunity (26). In a recent study of a 295 patient CVID cohort in the Czech Republic, immune thrombocytopenic purpura (ITP) was identified as a risk factor for malignancy, with over a 3-fold increase compared to those without ITP (27). Splenomegaly was also more common in patients with autoimmune cytopenias and was found to share some immunophenotypic characteristics, but the pathophysiology of this link is still not clearly understood (28).
Though autoimmune cytopenias are highly associated with CVID, other forms of autoimmunity have also been frequently reported. In a recent study of 870 CVID patients from the USIDNET registry, 5% were found to have rheumatologic disease, which accounted for 40% of the autoimmune complications of this cohort. Although the male-to-female ratio was almost equal for overall autoimmune complications, there was a clear female predominance for the rheumatologic manifestations (24). Several other studies have shown a similar female predominance for systemic autoimmune complications in CVID (29, 30). The most common rheumatologic manifestation reported in CVID is inflammatory arthritis (juvenile and adult), occurring in ~3% of patients (13, 24, 31, 32). Other rheumatologic manifestations include systemic lupus erythematosus, Sjogren's disease, Behcet's disease, and psoriasis (24, 32). Among organ specific autoimmune manifestations, hypothyroidism was the most prevalent at 3.5%, followed by alopecia areata and vitiligo at 2.7%, and type 1 diabetes at 1.6% in an ESID registry of 2700 CVID patients (9). Autoimmunity may also underlie gastrointestinal complications of CVID, including inflammatory bowel disease, autoimmune enteropathy, and autoimmune gastritis (33).
Immunophenotypic Markers Associated With Autoimmunity in CVID and Implications on Pathogenesis
T Cell Dysregulation in CVID Autoimmune Disease
While PIDs in general are associated with a higher risk of autoimmune manifestations, when comparing across all PIDs, the greatest risk of autoimmune cytopenia and rheumatologic disease was seen in two specific subsets of PID: CVID and PID with T-cell deficiencies (34). This finding highlights the importance of T cell dysfunction in CVID autoimmunity and demonstrates that pathogenesis of this immune disorder extends beyond defects of B cells exclusively. Indeed, CVID patients with autoimmunity have been found to have lower total T cells compared to those without autoimmunity (30). Such T cell deficiency is not necessarily profound, as a CD4+ T cell count <200 or significantly impaired lymphocyte proliferation may result in categorization as a combined immunodeficiency rather than CVID (35). When Chapel et al. classified CVID patients into specific phenotypes, they found that low proportions of CD8+ T cells were predictive of autoimmunity; in fact, in this cohort, each additional 10% increase in CD8+ T cells reduced the odds of autoimmunity by 18% (14). Later studies found that autoimmunity in CVID is associated with lower naïve CD8+ T cells specifically, with terminally differentiated CD8+ T cells actually increased, suggesting a hyperactivated T cell phenotype as the defining feature (36). While several studies have shown reduced CD4+ cells in CVID (29, 37), those with autoimmune cytopenias and organ specific autoimmunity had the most significantly reduced CD4+ T cells (36, 38, 39). Within CD4+ T cells, autoimmunity in CVID was particularly associated with reduced number of regulatory T cells (TR) compared to CVID patients without autoimmunity (39, 40). Expression of the canonical TR transcription factor forkhead box P3 (FOXP3) was reduced in CVID patients with autoimmunity compared to those without (39), and the suppressive activity of TR cells in CVID with autoimmunity was also reduced, with the degree of dysfunction correlating with the extent of FOXP3 downregulation (41). Other findings in CVID patients with autoimmunity include lower CCR7+ T cells, also considered to be key mediators in immune tolerance (42). Overall, T cell-mediated processes that help promote immune tolerance appear to break down in a subset of CVID patients, contributing to the development of autoimmunity.
In addition to loss of naïve and regulatory T cells, an increase in T helper type 1 (TH1) and T follicular helper (TFH) CD4+ T cells have been described in association with autoimmunity in CVID. TFH cells provide help to activate and diversify B cell responses within secondary and tertiary lymphoid tissues (43). TFH are elevated in CVID patients with autoimmunity, particularly those producing type 1 cytokines or otherwise known as TFH1 (44). While TFH provide most of their function within germinal centers, it is notable that their increase is associated with germinal center enlargement and disorganization in CVID patients with autoimmunity (45). This increased TFH development has been linked with greater IgA deficiency and resultant endotoxemia, presumably due to bacterial translocation from mucosal surfaces in the absence of IgA (46). Expansion of TH17 cells has also been associated with autoimmunity in patients with CVID (47, 48). Thus, it is clear that T cell dysregulation, particularly loss of regulatory subsets with concurrent increase in proinflammatory lymphocytes, is a fundamental feature of CVID patients with autoimmunity. Continued efforts toward improved understanding of this form of immune dysregulation will be vital to improving treatment of CVID associated autoimmune disease.
B Cell Dysfunction in CVID Autoimmune Disease
Low frequency of TR cells in CVID patients with autoimmunity is associated with expansion of a particular B cell type, CD21low B cell (37), linking T and B cell pathology in CVID. Early studies have shown that reduced switched memory B cell (CD19+CD27+IgD–) percentage correlates more strongly with autoimmunity in patients with CVID compared to serum IgG levels (49). Indeed, patients with reduced numbers of switched memory B cells (≤0.55% of B-cells) had greater than a 3-fold increase in their risk of autoimmune cytopenias and systemic autoimmune disease (50). Subsequent studies found increased proportions of CD21low B cells in patients with CVID (37, 51, 52), with clustering of these low levels in patients with autoimmunity (28, 53). Interestingly, non-CVID patients with autoimmune disease have also been found to have expansion of CD21low B cells (54). These cells were found to have preferential homing to peripheral tissues such as the synovial fluid of rheumatoid arthritis patients and the bronchoalveolar space of CVID patients (55). Further analysis found that CD21low B cells from both rheumatoid arthritis and CVID patients expressed germline autoreactive antibodies which recognized nuclear and cytoplasmic structures (56). Authors concluded that CD21low B cells are a “distinct, polyclonal, pre-activated, partially autoreactive, functionally attenuated B cell population with preferential enrichment in peripheral tissues” thus offering another possible mechanism of autoimmunity in patients with CVID (55).
Notably, CD21low B cells were found to produce significantly more IgM than naïve B cells after stimulation with CD40L, IL-2, and IL-10 (55). Along these lines, CVID patients with autoimmunity have been found to have higher levels of IgM compared with non-autoimmune phenotypes (31, 57). High levels of IgM have also been associated with autoimmunity in other PIDs, such as Wiskott-Aldrich syndrome (58). While this may be a marker for increased risk of autoimmune disease, and may be related to the aforementioned CD21low B cells, there may be a pathogenic role for IgM autoantibodies, as IgM may underlie the autoimmune cytopenias that are the predominant autoimmune manifestation of CVID (57).
B cell receptor diversity is diminished in CVID patients with autoimmunity (59). As a consequence of this diminished B cell repertoire, the presence of certain B cell receptors with autoreactive proclivity, such as those that express the VH4-34 heavy chain, may be more prominent in CVID patients as has been demonstrated in other forms of PID associated with autoimmunity (60, 61). Thus, B cell developmental defects that impair immunity may also contribute to the propensity for autoimmunity in some CVID patients.
B cell-activating factor (BAFF), a cytokine that promotes both maturation and survival of B cells, has long been linked with autoimmunity when its levels are elevated (62–64). BAFF also stimulates T cells via the BAFF receptor (BAFF-R) and skews the inflammatory response by augmenting TH1 cytokine production (65). Though BAFF levels were found to be elevated in CVID, an association with autoimmunity was not found (66). One possible explanation for this discrepancy is that the sample size of this cohort did not provide enough power to reach significance (only 17 of 77 patients had autoimmunity). In a separate study, BAFF levels were elevated in CVID patients with active interstitial lung disease, an inflammatory pulmonary disease linked with autoimmune cytopenias (17, 25). On the other hand, BAFF levels were not elevated in those with quiescent stable disease, suggesting that increases of this cytokine might be closely tied to disease activity (67) thus offering another possible explanation for the lack of association in the aforementioned cohort. While shedding of BAFF receptors has been postulated to regulate BAFF-driven inflammation, there was no relationship of serum levels of the BAFF receptor B cell maturation antigen (BCMA) and the development of autoimmunity in CVID (68, 69). Yet, elevated BAFF levels have been shown to inhibit negative selection of autoreactive B cells, in CVID autoimmunity as in other diseases, which apparently contributes to the increased autoimmunity seen in CVID patients with mutations in the BAFF receptor transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI) (70–72). Further studies are needed to refine our understanding of the complex relationship between BAFF and autoimmunity in CVID.
Other biomarkers of immune dysregulation have been linked with autoimmunity, as well as other non-infectious complications, in CVID. Importantly, an elevated TH1 signature has been found in the peripheral blood of these patients (73). This signature includes the prototypical TH1 cytokine interferon-γ (IFN-γ) and its downstream effects. This heightened TH1 signature in the blood is consistent with the heighted TH1 cellular response previously mentioned to be increased in CVID patients with autoimmune complications and elevated CD21low B cells. Elevated IFN-γ producing innate lymphoid cells have also been found to distinguish CVID patients with non-infectious complications (74). Thus, systemic immune dysregulation favoring TH1 cytokine production appears to be an important feature of CVID patients with non-infectious complications, yet the pathological basis of this skewed cytokine response remains unclear. Likewise, the therapeutic benefit of trying to neutralize this heightened TH1 response remains inadequately explored (Figure 1).
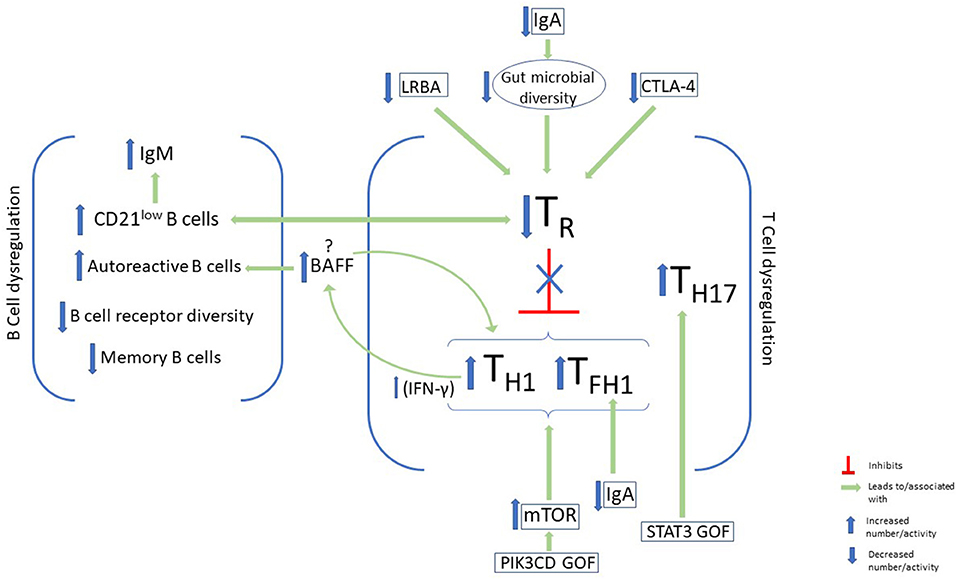
Figure 1. B-cell and T-cell dysregulation and immunophenotypic characteristics of autoimmunity in CVID. Some features of B-cell dysregulation in CVID patients with autoimmunity include elevated CD21low B cells and IgM and increased autoreactive B cells in the periphery. On the other hand, memory B-cells, and B-cell receptor diversity are decreased. Disturbance of T-cell homeostasis, with a decrease in T regulatory cells (TR) and a skew toward type 1 immunity, has been associated with autoimmunity in CVID. Factors that have been identified in CVID patients which lead to downregulation of TR cells include LRBA and CTLA-4 deficiency, as well as IgA deficiency. Upregulation of mTOR activity in GOF PIK3CD may also promote this immune dysregulation, while STAT3 GOF leads to expansion of TH17. BAFF augments TH1 cytokine production via its effects on the BAFF receptor on T cells, and promotes the survival of autoreactive antibodies via its receptor on B cells.
Genetics of CVID Autoimmunity
As CVID is a phenotypically heterogeneous disease, the expansive genetic landscape of these patients is perhaps unsurprising. While many CVID patients may have polygenic disease, recent advances in next generation sequencing (NGS) techniques have increased the discovery of monogenic forms of CVID to 15–30% of cases (75, 76) from 2–10% in 2016 (77). The majority of identified monogenic mutations encode proteins present in immune cells, which may reflect the nature of this immune disorder or a bias of the genomic analysis that is still nascent in its sophistication (77, 78). Out of the 12 monogenic mutations listed on the Online Mendelian Inheritance in Man (OMIM) database (79) in association with CVID, we will focus on those associated with autoimmunity. It is worth noting that while these mutations are associated with a clinical presentation that fits the diagnosis of CVID, many instances may not meet the full diagnostic criteria, in particular the extent of hypogammaglobulinemia typically needed. As these mutations were described in patients with CVID or CVID-like disorders and are likely to be encountered in a clinical evaluation of such patients, we include them in our discussion of CVID-associated autoimmunity even though there is an emerging trend to categorize them separately.
Defects in the TACI, a BAFF and APRIL (a proliferation inducing ligand) receptor encoded by the TNFRSF13B gene, is one of the first mutations to be linked to CVID (80). It is also among the most common genetic variants found, detected in up to 10% of CVID patients who can be either heterozygous or homozygous for the mutation (81). Heterozygous TACI mutations may be more appropriately defined as a risk factor for CVID, as some are not adequately rare to be considered monogenic etiologies and are frequently found in unaffected individuals (81). Notably, CVID patients heterozygous for the TNFRSF13B variant have a higher risk of developing autoantibody-mediated autoimmunity than those with homozygous mutations (82). It has been hypothesized that this difference may be due to the level of dysfunction in the TACI receptor: by regulating the function of several other receptors, TACI may be involved in central B cell tolerance and that reduced function results in loss of tolerance and resultant autoimmunity. By contrast, in homozygous individuals, the complete loss of TACI function results in the inability to maintain continuous autoantibody production that would otherwise result in autoimmunity (82).
LRBA (lipopolysaccharide-responsive beige-like anchor) and CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) deficiencies are two closely related protein deficiencies that were detected in patients with CVID and autoimmunity (83). While mutations in LRBA and CTLA4 have phenotypic variance thought to be due to incomplete penetrance and epigenetic changes, a common finding in these patients is hypogammaglobulinemia and early onset severe autoimmunity (77). CTLA-4 is an inhibitory T cell receptor that negatively regulates immunity by inhibiting excessive T cell activation and maintaining immune tolerance via its effect on TR cells (83). LRBA, on the other hand, is thought to play a role in CTLA-4 cell surface expression, hence the phenotypic similarities in the two deficiencies (84). Deficiencies in both these proteins thus cause excessive T cell activation and breakdown of immune tolerance, resulting in autoimmunity. They are both examples of how T cell-intrinsic genetic defects can lead to hypogammaglobulinemia, further highlighting how T cell dysfunction is key to the pathogenesis of at least some cases of CVID.
Gain-of-function (GOF) mutations in STAT3 have been identified in CVID as well as those with less profound antibody defects (75, 78). Patients with STAT3 GOF mutations also present with early-onset and quite severe manifestations of autoimmune disease (85, 86). One mechanism through which STAT3 is thought to lead to autoimmunity is by promoting the activation and expansion of autoimmunity-associated TH17 cells (47, 48). While a heightened TH1 response has been linked to CVID complications, features of these STAT3 GOF patients indicate that other forms of hyperactivated T cell responses, namely TH17, may also promote an autoimmune CVID phenotype. Additionally, increased STAT3 activation may impair B cell differentiation (87) leading to hypogammaglobulinemia and heightened autoreactivity found in association with CVID or more mild forms of hypogammaglobulinemia. Thus, STAT3 GOF may have both B cell-extrinsic and -intrinsic effects contributing to the immunological phenotype of affected patients.
Class IA phosphoinositide 3-kinases (PI3Ks) are heterodimeric lipid kinases that are involved in regulating cell growth, survival, and activity. Recently, a GOF mutation in the gene PIK3CD encoding PI3Kδ has been found in patients with CVID-like disease and autoimmunity. PI3Kδ is a PI3K subunit exclusively expressed in leukocytes. Patients heterozygous for this mutation are now said to have “activated PI3Kδ syndrome,” or APDS, of which ~200 patients have been described to date (88). Activated PI3Kδ syndrome is characterized by impaired T- and B-cell development and function, autoimmunity, and lymphoproliferation. One of the major downstream effectors of PI3K is mTOR, which regulates cell growth and survival and is critical for TH1 and TFH cell differentiation (89, 90). While effector cells proliferate, naïve, and central memory T-cell subsets remain metabolically quiescent, likely contributing to autoimmunity, lymphoproliferation, and immunodeficiency seen in this syndrome (91). As is the case with STAT3 GOF mutations, both B cell-intrinsic and -extrinsic effects have been described as PI3Kδ is expressed in B cells and other leukocytes alike.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a family of transcription factors that are crucial for B-cell maturation, survival, differentiation, class switching, as well as self-tolerance (92). It is also a fundamental transcription factor for cytokine production by innate immune cells as well as other vital cell signaling pathways that expand beyond the immune system (93). NF-κB1 and NF-κB2 deficiencies were first described in patients of CVID affected families who were found to carry autosomal dominant mutations in NFKB1 and NFKB2 genes, respectively (94, 95). Mutations affecting the inducible T-cell co-stimulator (ICOS) are closely related to NF-κB deficiencies since NF-κB are activated by ICOS receptors. Because ICOS activation is essential for terminal B cell differentiation and immune tolerance (96) both ICOS and NF-κB deficiencies result in CVID-like immunodeficiency syndromes and autoimmunity (77). NFKB1 mutation has also been linked with cytokine dysregulation, namely the elevation of type 1 cytokines that mirrors the immune profile characterizing the broader population of genetically-undefined CVID with non-infectious complications (97). Some with NFKB1 mutations have antibody deficiency without associated non-infectious complications (98). NFKB2 mutations lead to autoimmunity affecting the skin, hair and nails, such as alopecia and trachyonychia, and less frequently autoimmune cytopenias, and are characterized by pituitary hormone deficiencies (99) (Table 1).
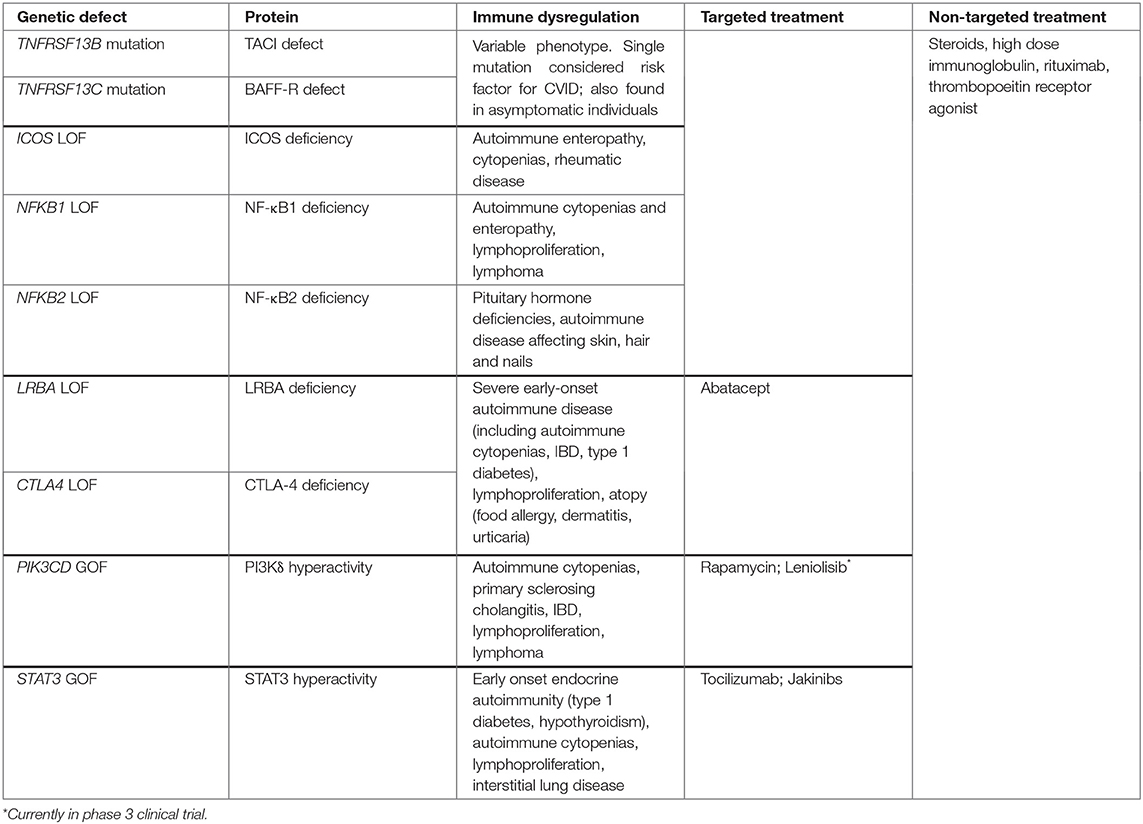
Table 1. Monogenic defects associated with autoimmunity and CVID.
Microbiome in CVID and Autoimmunity
As noted above, monogenic mutations represent a minority of CVID patients with CVID. The majority of cases may be polygenic, or perhaps better defined as simply multifactorial, with environmental, epigenetic, or other factors contributing. Over the past decade, the microbiome has been implicated in the manifestation of immune dysregulation (100), and has been explored in the pathogenesis of CVID complications (46).
It has been hypothesized that impaired immunity results in increased microbial translocation across the gut barrier. This in turn drives persistent systemic immune activation leading to the disruption of the immune homeostasis (46, 101, 102). While regular immune sampling and microbial translocation occur in healthy individuals (103), the increased frequency occurring in CVID patients with diminished barrier function may lead to both local and systemic inflammation and immune dysregulation. Lipopolysaccharides (LPS) and soluble CD14 and IL-2 are used as markers for endotoxemia since their presence in systemic circulation is indicative of increased gut microbial translocation (46). CVID patients have higher levels of LPS and soluble CD14 and IL-2 compared to healthy controls, and CVID patients with autoimmune complications have higher levels compared to CVID without complications (102).
Low IgA levels are thought to play an important role in microbial dysbiosis seen in CVID since IgA is directly associated with the gastrointestinal tract immunity. Patients with isolated IgA deficiency have been found to have lower frequencies of TR cells, especially in those patients with IgA deficiency and autoimmune disease (104). They have also been found to have reduced gut microbial diversity (105). Separately, a feedback loop has been described whereby healthy microbiota stimulate TR cells and leads to the formation of germinal centers and IgA, which in turn maintains a healthy microbiome (106). The disruption of this feedback loop by low IgA levels and an unhealthy microbiome leads to reduction in TR cells and subsequent immune dysregulation. Importantly, systemic IgG responses may be significant in preventing inflammation in those with IgA deficiency as a consequence of microbial dysbiosis (107). Such protective IgG responses may be significantly impaired in some CVID patients, thus predisposing to inflammation and, potentially, autoimmune disease.
Similarly to IgA deficient patients, CVID patients' gut microbial diversity was found to be reduced compared to healthy controls, with again, higher levels of soluble IL-2 and LPS detected in CVID patients with inflammatory and autoimmune complications (102). They also found an inverse correlation between T cell activation and gut microbial diversity, again suggesting that those with reduced microbial diversity have higher levels of T cell activation, and thus autoimmunity. While these findings offer insight into some drivers of immune dysregulation, further work is needed to unravel the specific mechanisms by which the microbiome affects the immune system, and how it is altered in patients with CVID, to best understand how it should be modulated or otherwise harnessed to treat complications of this immune deficiency.
Treatment of Autoimmunity in CVID
Classically, autoimmunity in CVID has been treated with broad immunosuppressive agents, including corticosteroids, methotrexate, and azathioprine among others, which place already immunodeficient patients at an even higher risk of infections. For genetically undefined CVID, the use of rituximab for autoimmune cytopenias in CVID is one of the most efficacious and safe treatments. Its use was first documented in 2004 (108), and its efficacy and safety have been well-established, especially for ITP (109). While ritixumab's efficacy with autoimmune cytopenias may be in part due to its B-cell depleting properties resulting in depletion of autoantibodies, it is thought that its success in CVID patients is also partially due to its effect on T cells (110), again highlighting the importance of T cell abnormalities in CVID. There is the documented potential risk of persistent B-cell lymphopenia after treatment with rituximab (111), but this risk is offset by the ongoing use of immunoglobulin replacement therapy. Other therapies include thrombopoietin-receptor agonists, such as romiplostim and eltrombopag which were approved by the FDA in 2008 for the treatment of cirrhosis-associated thrombocytopenia, have shown success in the treatment of thrombocytopenia in CVID and other immunodeficiencies (112, 113).
In recent years, thanks to the recent molecular and genetic findings in CVID, more targeted approaches have led to improved results through precision medicine therapy. In CTLA-4 deficiency, corticosteroids have been the most consistently used immunosuppressive agents for autoimmunity, while other steroid-sparing agents (such as mycophenolate mofetil, cyclosporine, rituximab, anti-TNF drugs) have had mixed results (114). Abatacept, a CTLA-4 immunoglobulin fusion protein, considered as CTLA-4 replacement precision therapy for these patients, has been used to treat autoimmune manifestations and shown promising results. Ten patients treated with abatacept, showed either complete resolution or partial response with regards to stabilization of their cytopenia and improvement in gastrointestinal symptoms, with no reports of adverse outcomes (114, 115). Since LRBA deficiency is related to CTLA-4 deficiency as described above, it is no surprise that abatacept has also shown similar results in LRBA-deficient patients (84). These precision therapy approaches exemplify the potential of harnessing genomics and fundamental biology to improve care of patients with PID.
STAT3 activation occurs downstream of IL-6 signaling, a cytokine implicated in autoimmune disease, such as rheumatoid arthritis (86). Thus, upon discovery of STAT3 GOF mutations, IL-6 emerged as a potential target for treatment. Tocilizumab, an IL-6 receptor antagonist, was trialed successfully in 2015 in a patient with STAT3 GOF mutation who had failed other treatments; their TH17 cells which were elevated pre-treatment, similarly to what has been observed in other patients, normalized after treatment with tocilizumab (85). More recently, jakinibs, inhibitors of Janus kinases (JAKs) which are also involved in the activation cascade of STAT3, have been used adjunctly with tocilizumab in six patients and have yielded more sustained results (116). The authors of this latest study suggest that the combination of IL-6 blockade and a jakinib may be the most effective treatment strategy for patients with STAT3 GOF mutations (116).
Targeted therapy in PI3Kδ mutations have focused on the mTOR pathway and its inhibitor, rapamycin. A cohort of 26 APDS patients from the ESID registry were treated with rapamycin which showed excellent effects on the lymphoproliferative aspect of the disease, but less promising results on autoimmune cytopenias and enteropathy (117). Directed targeted inhibition of PI3Kδ is being explored with the use of leniolisib: early results of the clinical trial published in 2017 showed improvement in lymphoproliferation as well as cytopenias (118). Nemiralisib, an inhaled PI3Kδ inhibitor, has thus far shown safety and tolerability in healthy, asthmatic, and COPD patients (91) but no data has been published on APDS patients as of the publication of this review. The field of CVID has grown by leaps and bounds in recent years, coupling genomic studies with precision therapeutic approaches that have significantly improved both the efficacy and tolerability of immunosuppressive treatment for non-infectious complications.
Conclusion
Autoimmunity in CVID is profoundly shaped by the nature of immune dysregulation that accompanies the immune deficiency in these patients. For many years CVID patients with autoimmunity have been set apart from other CVID patients on the basis of immunophenotypic characteristics, but recent advances in our understanding of genetic defects associated with CVID have shed light on the underlying pathophysiology of autoimmunity. This improved understanding has inspired treatment of autoimmunity with targeted therapies in patients who would otherwise be subjected to broad immunosuppression. Some monogenic defects have now been listed as separate immunodeficiency syndromes, such as APDS. Importantly, many of these monogenic defects have variable clinical presentation, attributed to incomplete penetrance or variable expressivity. Additional factors, such as microbial dysbiosis, may also contribute to the pathophysiology of the disease, leading to greater heterogeneity. Future research that focuses on the immune dysregulation caused by the alteration of the gut microbiota may lead to a completely new line of therapies for these patients, such as probiotics, fecal transplantation, or even dietary recommendations. Examination of large CVID cohorts in non-Western countries may shed further light on these alternative mechanisms that may shape disease manifestations, especially given profound dietary differences and possible changes in gut microbiota between globally diverse populations. Ethnic, racial, gender, and socioeconomic factors are likely to be important to explore within Western countries. As we continue to understand the mechanisms underlying the multifactorial physiology that underlies CVID disorders, we will move closer to elucidating the fundamental immune changes that can be targeted with precision therapies to optimize disease management.
Author Contributions
JG wrote the review with support from PM.
Funding
PM was supported by the National Institutes of Health grant K23 AI137183, a faculty development grant from the American Association of Allergy, Asthma and Immunology Foundation, and investigator-initiated grants from Horizon Pharma and Takeda.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, Teresa de la Morena M, et al. International Consensus Document (ICON): common variable immunodeficiency disorders HHS public access. J Allergy Clin Immunol Pr. (2016) 4:38–59. doi: 10.1016/j.jaip.2015.07.025
2. Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. (2012) 129:1425–6.e3. doi: 10.1016/j.jaci.2012.03.025
3. Cooper MD, Faulk WP, Fudenberg HH, Good RA, Hitzig W, Kunkel H, et al. Classification of primary immunodeficiencies. N Engl J Med. (1973) 288:966–7. doi: 10.1056/NEJM197305032881814
4. Notarangelo LD, Fischer A, Geha RS, Casanova J-L, Chapel H, Conley ME, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. (2009) 124:1161–78. doi: 10.1016/j.jaci.2009.10.013
5. Edgar D, Ehl S. ESID Registry - Working Definitions for Clinical Diagnosis of PID. (2016). p. 1–33.
6. Gathmann B, Mahlaoui N, Ceredih, Gérard L, Oksenhendler E, Warnatz K, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. (2014) 134:116–26.e11. doi: 10.1016/j.jaci.2013.12.1077
7. Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q J Med. (1993) 86:31–42.
8. Cunningham-Rundles C. How I treat common variable immune deficiency. Blood. (2010) 116:7–15. doi: 10.1182/blood-2010-01-254417
9. Odnoletkova I, Kindle G, Quinti I, Grimbacher B, Knerr V, Gathmann B, et al. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J Rare Dis. (2018) 13:201. doi: 10.1186/s13023-018-0941-0
10. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. (2007) 27:308–16. doi: 10.1007/s10875-007-9075-1
11. Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. (2010) 125:1354–60.e4. doi: 10.1016/j.jaci.2010.02.040
12. Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol. (2010) 137:21–30. doi: 10.1016/j.clim.2010.06.012
13. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
14. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
15. Jolles S. The variable in common variable immunodeficiency: a disease of complex phenotypes. J Allergy Clin Immunol Pract. (2013) 1:545–56. doi: 10.1016/j.jaip.2013.09.015
16. Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol. (2012) 130:1197–8.e9. doi: 10.1016/j.jaci.2012.05.046
17. Maglione PJ, Overbey JR, Radigan L, Bagiella E, Cunningham-Rundles C. Pulmonary radiologic findings in common variable immunodeficiency: Clinical and immunological correlations. Ann Allergy Asthma Immunol. (2014) 113:452–9. doi: 10.1016/j.anai.2014.04.024
18. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. (2014) 506:376–81. doi: 10.1038/nature12873
19. Li YR, Zhao SD, Li J, Bradfield JP, Mohebnasab M, Steel L, et al. Genetic sharing and heritability of paediatric age of onset autoimmune diseases. Nat Commun. (2015) 6:8442. doi: 10.1038/ncomms9442
20. Li J, Wei Z, Li YR, Maggadottir SM, Chang X, Desai A, et al. Understanding the genetic and epigenetic basis of common variable immunodeficiency disorder through omics approaches. Biochim Biophys Acta Gen Subj. (2016) 1860:2656–63. doi: 10.1016/j.bbagen.2016.06.014
21. Farmer JR, Ong MS, Barmettler S, Yonker LM, Fuleihan R, Sullivan KE, et al. Common variable immunodeficiency non-infectious disease endotypes redefined using unbiased network clustering in large electronic datasets. Front Immunol. (2018) 8:1740. doi: 10.3389/fimmu.2017.01740
22. Patuzzo G, Barbieri A, Tinazzi E, Veneri D, Argentino G, Moretta F, et al. Autoimmunity and infection in common variable immunodeficiency (CVID). Autoimmun Rev. (2016) 15:877–82. doi: 10.1016/j.autrev.2016.07.011
23. Song J, Lleo A, Yang GX, Zhang W, Bowlus CL, Gershwin ME, et al. Common variable immunodeficiency and liver involvement. Clin Rev Allergy Immunol. (2018) 55:340–51. doi: 10.1007/s12016-017-8638-z
24. Gutierrez MJ, Sullivan KE, Fuleihan R, Bingham CO. Phenotypic characterization of patients with rheumatologic manifestations of common variable immunodeficiency. Semin Arthritis Rheum. (2018) 48:318–26. doi: 10.1016/j.semarthrit.2018.02.013
25. Weinberger T, Fuleihan R, Cunningham-Rundles C, Maglione PJ. Factors beyond lack of antibody govern pulmonary complications in primary antibody deficiency. J Clin Immunol. (2019) 39:440–7. doi: 10.1007/s10875-019-00640-5
26. Feuille EJ, Anooshiravani N, Sullivan KE, Fuleihan RL, Cunningham-Rundles C. Autoimmune cytopenias and associated conditions in CVID: a report from the USIDNET registry. J Clin Immunol. (2018) 38:28–34. doi: 10.1007/s10875-017-0456-9
27. Kralickova P, Milota T, Litzman J, Malkusova I, Jilek D, Petanova J, et al. CVID-associated tumors: Czech nationwide study focused on epidemiology, immunology, and genetic background in a cohort of patients with CVID. Front Immunol. (2018)9:3135. doi: 10.3389/fimmu.2018.03135
28. Boileau J, Mouillot G, Gérard L, Carmagnat M, Rabian C, Oksenhendler E, et al. Autoimmunity in common variable immunodeficiency: correlation with lymphocyte phenotype in the French DEFI study. J Autoimmun. (2011) 36:25–32. doi: 10.1016/j.jaut.2010.10.002
29. Xiao X, Miao Q, Chang C, Gershwin ME, Ma X. Common variable immunodeficiency and autoimmunity–an inconvenient truth. Autoimmun Rev. (2014) 13:858–64. doi: 10.1016/j.autrev.2014.04.006
30. Azizi G, Kiaee F, Hedayat E, Yazdani R, Dolatshahi E, Alinia T, et al. Rheumatologic complications in a cohort of 227 patients with common variable immunodeficiency. Scand J Immunol. (2018) 87:1–9. doi: 10.1111/sji.12663
31. Abolhassani H, Amirkashani D, Parvaneh N, Mohammadinejad P, Gharib B, Shahinpour S, et al. Autoimmune phenotype in patients with common variable immunodeficiency. J Investig Allergol Clin Immunol. (2013) 23:323–9.
32. Maglione PJ. Autoimmune and lymphoproliferative complications of common variable immunodeficiency. Curr Allergy Asthma Rep. (2016) 16:1–10. doi: 10.1007/s11882-016-0597-6
33. Pikkarainen S, Martelius T, Ristimäki A, Siitonen S, Seppänen MRJ, Färkkilä M. A High prevalence of gastrointestinal manifestations in common variable immunodeficiency. Am J Gastroenterol. (2019) 114:648–55. doi: 10.14309/ajg.0000000000000140
34. Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, Adoue D, et al. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol. (2017) 140:1388–93.e8. doi: 10.1016/j.jaci.2016.12.978
35. Azizi G, Rezaei N, Kiaee F, Tavakolinia N, Yazdani R, Mirshafiey A, et al. T-cell abnormalities in common variable immunodeficiency. J Investig Allergol Clin Immunol. (2016) 26:233–43. doi: 10.18176/jiaci.0069
36. Bateman EAL, Ayers L, Sadler R, Lucas M, Roberts C, Woods A, et al. T cell phenotypes in patients with common variable immunodeficiency disorders: associations with clinical phenotypes in comparison with other groups with recurrent infections. Clin Exp Immunol. (2012) 170:202–11. doi: 10.1111/j.1365-2249.2012.04643.x
37. Arumugakani G, Wood PMD, Carter CRD. Frequency of Treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J Clin Immunol. (2010) 30:292–300. doi: 10.1007/s10875-009-9351-3
38. Mouillot G, Carmagnat M, Gérard L, Garnier J-L, Fieschi C, Vince N, et al. B-cell and T-cell phenotypes in CVID patients correlate with the clinical phenotype of the disease. J Clin Immunol. (2010) 30:746–55. doi: 10.1007/s10875-010-9424-3
39. Genre J, Errante PR, Kokron CM, Toledo-Barros M, Câmara NOS, Rizzo LV. Reduced frequency of CD4+CD25HIGHFOXP3+ cells and diminished FOXP3 expression in patients with common variable immunodeficiency: a link to autoimmunity? Clin Immunol. (2009) 132:215–21. doi: 10.1016/j.clim.2009.03.519
40. Arandi N, Mirshafiey A, Abolhassani H, Jeddi-Tehrani M, Edalat R, Sadeghi B, et al. Frequency and expression of inhibitory markers of CD4 + CD25 + FOXP3 + regulatory T cells in patients with common variable immunodeficiency. Scand J Immunol. (2013) 77:405–12. doi: 10.1111/sji.12040
41. Yu GP, Chiang D, Song SJ, Hoyte EG, Huang J, Vanishsarn C, et al. Regulatory T cell dysfunction in subjects with common variable immunodeficiency complicated by autoimmune disease. Clin Immunol. (2009) 131:240–53. doi: 10.1016/j.clim.2008.12.006
42. Kutukculer N, Azarsiz E, Aksu G, Karaca NE. CD4+CD25+Foxp3+ T regulatory cells, Th1 (CCR5, IL-2, IFN-γ) and Th2 (CCR4, IL-4, Il-13) type chemokine receptors and intracellular cytokines in children with common variable immunodeficiency. Int J Immunopathol Pharmacol. (2016) 29:241–51. doi: 10.1177/0394632015617064
43. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. (2019) 50:1132–48. doi: 10.1016/j.immuni.2019.04.011
44. Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The TH1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol. (2018) 141:730–40. doi: 10.1016/j.jaci.2017.04.041
45. Romberg N, Le Coz C, Glauzy S, Schickel JN, Trofa M, Nolan BE, et al. Patients with common variable immunodeficiency with autoimmune cytopenias exhibit hyperplastic yet inefficient germinal center responses. J Allergy Clin Immunol. (2019) 143:258–65. doi: 10.1016/j.jaci.2018.06.012
46. Berbers RM, Nierkens S, van Laar JM, Bogaert D, Leavis HL. Microbial dysbiosis in common variable immune deficiencies: evidence, causes, and consequences. Trends Immunol. 38:206–16. doi: 10.1016/j.it.2016.11.008
47. Yang X-P, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. (2011) 12:247–54. doi: 10.1038/ni.1995
48. Haapaniemi EM, Kaustio M, Rajala HLM, van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood. (2015) 125:639–48. doi: 10.1182/blood-2014-04-570101
49. Alachkar H, Taubenheim N, Haeney MR, Durandy A, Arkwright PD. Memory switched B cell percentage and not serum immunoglobulin concentration is associated with clinical complications in children and adults with specific antibody deficiency and common variable immunodeficiency. Clin Immunol. (2006) 120:310–8. doi: 10.1016/j.clim.2006.05.003
50. Sánchez-Ramón S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. (2008) 128:314–21. doi: 10.1016/j.clim.2008.02.013
51. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. (2008) 111:77–85. doi: 10.1182/blood-2007-06-091744
52. Warnatz K, Wehr C, Dräger R, Schmidt S, Eibel H, Schlesier M, et al. Expansion of CD19hiCD21lo/neg B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology. (2002) 206:502–13. doi: 10.1078/0171-2985-00198
53. Warnatz K, Denz A, Dräger R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cells (CD27+IgM−IgD−) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. (2002) 99:1544–51. doi: 10.1182/blood.V99.5.1544
54. Wehr C, Eibel H, Masilamani M, Illges H, Schlesier M, Peter H-H, et al. A new CD21low B cell population in the peripheral blood of patients with SLE. Clin Immunol. (2004) 113:161–71. doi: 10.1016/j.clim.2004.05.010
55. Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc Natl Acad Sci USA. (2009) 106:13451–6. doi: 10.1073/pnas.0901984106
56. Isnardi I, Ng Y-S, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. (2010) 115:5026–36. doi: 10.1182/blood-2009-09-243071
57. Picchianti Diamanti A, Rosado MM, Scarsella M, Ceccarelli S, Laganà B, D'Amelio R, et al. Increased serum IgM, immunodeficiency, and autoimmunity: a clinical series. Int J Immunopathol Pharmacol. (2015) 28:547–56. doi: 10.1177/0394632015600231
58. Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, et al. Autoimmunity in Wiskott-Aldrich Syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. (2003) 111:e622–7. doi: 10.1542/peds.111.5.e622
59. Roskin KM, Simchoni N, Liu Y, Lee JY, Seo K, Hoh RA, et al. IgH sequences in common variable immune deficiency reveal altered B cell development and selection. Sci Transl Med. (2015) 7:302ra135. doi: 10.1126/scitranslmed.aab1216
60. Kolhatkar NS, Brahmandam A, Thouvene CD, Becker-Herman S, Jacobs HM, Schwartz MA, et al. Altered BCR and TLR signals promote enhanced positive selection of autoreactive transitional B cells in Wiskott-Aldrich syndrome. J Exp Med. (2015) 212:1663–7. doi: 10.1084/jem.20150585
61. Schickel JN, Glauzy S, Ng YS, Chamberlain N, Massad C, Isnardi I, et al. Self-reactive VH4-34-expressing IgG B cells recognizecommensal bacteria. J Exp Med. (2017) 214:1991–2003. doi: 10.1084/jem.20160201
62. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. (2009) 9:491–502. doi: 10.1038/nri2572
63. Matsushita T, Hasegawa M, Yanaba K, Kodera M, Takehara K, Sato S. Elevated serum BAFF levels in patients with systemic sclerosis: enhanced BAFF signaling in systemic sclerosis B lymphocytes. Arthritis Rheum. (2006) 54:192–201. doi: 10.1002/art.21526
64. Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. (2002) 2:465–75. doi: 10.1038/nri844
65. Sutherland APR, Ng LG, Fletcher CA, Shum B, Newton RA, Grey ST, et al. BAFF augments certain Th1-associated inflammatory responses. J Immunol. (2005) 174:5537–44. doi: 10.4049/jimmunol.174.9.5537
66. Knight AK, Radigan L, Marron T, Langs A, Zhang L, Cunningham-Rundles C. High serum levels of BAFF, APRIL, and TACI in common variable immunodeficiency. Clin Immunol. (2007) 124:182–9. doi: 10.1016/j.clim.2007.04.012
67. Maglione PJ, Gyimesi G, Cols M, Radigan L, Ko HM, Weinberger T, et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI Insight. (2019) 4:122728. doi: 10.1172/jci.insight.122728
68. Maglione PJ, Ko HM, Tokuyama M, Gyimesi G, Soof C, Li M, et al. Serum B cell maturation antigen (BCMA) levels differentiate primary antibody deficiencies. J Allergy Clin Immunol Pract. (2019). doi: 10.1016/j.jaip.2019.08.012. [Epub ahead of print].
69. Meinl E, Thaler FS, Lichtenthaler SF. Shedding of BAFF/APRIL receptors controls B cells. Trends Immunol. (2018) 39:673–6. doi: 10.1016/j.it.2018.07.002
70. Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. (2008) 20:632–8. doi: 10.1016/j.coi.2008.09.001
71. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu H-B, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF Immunity. (2004) 20:441–53. doi: 10.1016/S1074-7613(04)00079-2
72. Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest. (2013) 123:4283–93. doi: 10.1172/JCI69854
73. Park J, Munagala I, Xu H, Blankenship D, Maffucci P, Chaussabel D, et al. Interferon signature in the blood in inflammatory common variable immune deficiency. PLoS ONE. (2013) 8:e74893. doi: 10.1371/journal.pone.0074893
74. Cols M, Rahman A, Maglione PJ, Garcia-Carmona Y, Simchoni N, Ko HBM, et al. Expansion of inflammatory innate lymphoid cells in patients with common variable immune deficiency. J Allergy Clin Immunol. (2016) 137:1206–15.e6. doi: 10.1016/j.jaci.2015.09.013
75. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova J-L, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol. (2016) 7:220. doi: 10.3389/fimmu.2016.00220
76. de Valles-Ibáñez G, Esteve-Solé A, Piquer M, Azucena González-Navarro E, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the genetics of common variable immunodeficiency: monogenetic model and beyond. Front Immunol. (2018) 9:636. doi: 10.3389/fimmu.2018.00636
77. Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. (2016) 53:575–90. doi: 10.1136/jmedgenet-2015-103690
78. Russell MA, Pigors M, Houssen ME, Manson A, Kelsell D, Longhurst H, et al. A novel de novo activating mutation in STAT3 identified in a patient with common variable immunodeficiency (CVID). Clin Immunol. (2018) 187:132–6. doi: 10.1016/j.clim.2017.11.007
79. Online Mendelian Inheritance in Man OMIM®. Baltimore, MD: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University. Available online at: https://omim.org/ (accessed August 20, 2019).
80. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. (2005) 37:829–34. doi: 10.1038/ng1601
81. Martinez-Gallo M, Radigan L, Almejún MB, Martínez-Pomar N, Matamoros N, Cunningham-Rundles C. TACI mutations and impaired B-cell function in subjects with CVID and healthy heterozygotes. J Allergy Clin Immunol. (2013) 131:468–76. doi: 10.1016/j.jaci.2012.10.029
82. Zhang Y, Li J, Zhang Y-M, Zhang X-M, Tao J. Effect of TACI signaling on humoral immunity and autoimmune diseases. J Immunol Res. (2015) 2015:1–12. doi: 10.1155/2015/247426
83. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. (2014) 20:1410–6. doi: 10.1038/nm.3746
84. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. (2015) 349:436–40. doi: 10.1126/science.aaa1663
85. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. (2015) 125:591–9. doi: 10.1182/blood-2014-09-602763
86. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. (2014) 46:812–4. doi: 10.1038/ng.3040
87. Clemente A, Pons J, Lanio N, Cunill V, Frontera G, Crespí C, et al. Increased STAT3 phosphorylation on CD27 + B-cells from common variable immunodeficiency disease patients. Clin Immunol. (2015) 161:77–88. doi: 10.1016/j.clim.2015.09.004
88. Preite S, Gomez-Rodriguez J, Cannons JL, Schwartzberg PL. T and B-cell signaling in activated PI3K delta syndrome: from immunodeficiency to autoimmunity. Immunol Rev. (2019) 291:154–73. doi: 10.1111/imr.12790
89. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
90. Thauland TJ, Pellerin L, Ohgami RS, Bacchetta R, Butte MJ. Case study: mechanism for increased follicular helper T cell development in activated PI3K Delta Syndrome. Front Immunol. (2019)10:753. doi: 10.3389/fimmu.2019.00753
91. Nunes-Santos CJ, Uzel G, Rosenzweig SD. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol. (2019) 143:1676–87. doi: 10.1016/j.jaci.2019.03.017
92. Scott O, Roifman CM. NF-κB pathway and the Goldilocks principle: lessons from human disorders of immunity and inflammation. J Allergy Clin Immunol. (2019) 143:1688–701. doi: 10.1016/j.jaci.2019.03.016
93. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
94. Fliegauf M, Bryant VL, Frede N, Slade C, Woon S-T, Lehnert K, et al. Haploinsufficiency of the NF-κB1 Subunit p50 in common variable immunodeficiency. Am J Hum Genet. (2015) 97:389–403. doi: 10.1016/j.ajhg.2015.07.008
95. Chen K, Coonrod EM, Kumánovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet. (2013) 93:812–24. doi: 10.1016/j.ajhg.2013.09.009
96. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. (2003) 4:261–8. doi: 10.1038/ni902
97. Dieli-Crimi R, Martínez-Gallo M, Franco-Jarava C, Antolin M, Blasco L, Paramonov I, et al. Th1-skewed profile and excessive production of proinflammatory cytokines in a NFKB1-deficient patient with CVID and severe gastrointestinal manifestations. Clin Immunol. (2018) 195:49–58. doi: 10.1016/j.clim.2018.07.015
98. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol. (2018) 142:1285–96. doi: 10.1016/j.jaci.2018.01.039
99. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol. (2019) 10:297. doi: 10.3389/fimmu.2019.00297
100. Jørgensen SF, Fevang B, Aukrust P. Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modifications. J Clin Immunol. (2019) 39:30–6. doi: 10.1007/s10875-018-0574-z
101. Perreau M, Vigano S, Bellanger F, Pellaton C, Buss G, Comte D, et al. Exhaustion of bacteria-specific CD4 T cells and microbial translocation in common variable immunodeficiency disorders. J Exp Med. (2014) 211:2033–45. doi: 10.1084/jem.20140039
102. Jørgensen SF, Trøseid M, Kummen M, Anmarkrud JA, Michelsen AE, Osnes LT, et al. Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation. Mucosal Immunol. (2016) 9:1455–65. doi: 10.1038/mi.2016.18
103. Laugerette F, Vors C, Géloën A, Chauvin M-A, Soulage C, Lambert-Porcheron S, et al. Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. J Nutr Biochem. (2011) 22:53–9. doi: 10.1016/j.jnutbio.2009.11.011
104. Soheili H, Abolhassani H, Arandi N, Khazaei HA, Shahinpour S, Hirbod-Mobarakeh A, et al. Evaluation of natural regulatory T cells in subjects with selective IgA deficiency: from senior idea to novel opportunities. Int Arch Allergy Immunol. (2013)160:208–14. doi: 10.1159/000339867
105. Jørgensen SF, Holm K, Macpherson ME, Storm-Larsen C, Kummen M, Fevang B, et al. Selective IgA deficiency in humans is associated with reduced gut microbial diversity. J Allergy Clin Immunol. (2019) 143:1969–71.e11. doi: 10.1016/j.jaci.2019.01.019
106. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, et al. Foxp3+ T cells regulate immunoglobulin A selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity. (2014) 41:152–65. doi: 10.1016/j.immuni.2014.05.016
107. Fadlallah J, Sterlin D, Fieschi C, Parizot C, Dorgham K, El Kafsi H, et al. Synergistic convergence of microbiota-specific systemic IgG and secretory IgA. J Allergy Clin Immunol. (2019) 143:1575–85.e4. doi: 10.1016/j.jaci.2018.09.036
108. Michel M, Chanet V, Galicier L, Ruivard M, Levy Y, Hermine O, et al. Autoimmune thrombocytopenic purpura and common variable immunodeficiency. Medicine. (2004) 83:254–63. doi: 10.1097/01.md.0000133624.65946.40
109. Gobert D, Bussel JB, Cunningham-Rundles C, Galicier L, Dechartres A, Berezne A, et al. Efficacy and safety of rituximab in common variable immunodeficiency-associated immune cytopenias: a retrospective multicentre study on 33 patients. Br J Haematol. (2011) 155:498–508. doi: 10.1111/j.1365-2141.2011.08880.x
110. Pecoraro A, Crescenzi L, Galdiero MR, Marone G, Rivellese F, Rossi FW, et al. Immunosuppressive therapy with rituximab in common variable immunodeficiency. Clin Mol Allergy. (2019)17:9. doi: 10.1186/s12948-019-0113-3
111. Kano G, Nakatani T, Yagi K, Sakamoto I, Imamura T. Complicated pathophysiology behind rituximab-induced persistent hypogammaglobulinemia. Immunol Lett. (2014) 159:76–8. doi: 10.1016/j.imlet.2013.10.005
112. Carrabba M, Barcellini W, Fabio G. Use of thrombopoietin-receptor agonist in CVID-associated immune thrombocytopenia. J Clin Immunol. (2016) 36:434–6. doi: 10.1007/s10875-016-0282-5
113. González-López TJ, Alvarez-Román MT, Pascual C, Sánchez-González B, Fernández-Fuentes F, Pérez-Rus G, et al. Use of eltrombopag for secondary immune thrombocytopenia in clinical practice. Br J Haematol. (2017) 178:959–70. doi: 10.1111/bjh.14788
114. Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol. (2017) 190:1–7. doi: 10.1111/cei.12997
115. Uzel G, Karanovic D, Su H, Rump A, Agharahimi A, Holland SM, et al. Management of cytopenias in CTLA4 haploinsufficiency using abatacept and sirolimus. Blood. (2018) 132(Suppl 1):2409. doi: 10.1182/blood-2018-99-120185
116. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol. (2018) 142:1665–9. doi: 10.1016/j.jaci.2018.07.020
117. Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ syndrome: The European Society for immunodeficiencies-activated phosphoinositide 3-kinase δ syndrome registry. Front Immunol. (2018) 9:543. doi: 10.3389/fimmu.2018.00338
Keywords: primary immunodeficieny, common variable immunodeficiency, autoimmunity, cytopenia, genetics, microbiome, precision therapy
Citation: Gereige JD and Maglione PJ (2019) Current Understanding and Recent Developments in Common Variable Immunodeficiency Associated Autoimmunity. Front. Immunol. 10:2753. doi: 10.3389/fimmu.2019.02753
Received: 13 September 2019; Accepted: 11 November 2019;
Published: 10 December 2019.
Edited by:
Isabella Quinti, Sapienza University of Rome, ItalyReviewed by:
David Andrew Fulcher, Australian National University, AustraliaFilomeen Haerynck, Ghent University, Belgium
Copyright © 2019 Gereige and Maglione. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica D. Gereige, amVzc2lrYWdAYnUuZWR1; Paul J. Maglione, cG1hZ2xpb25AYnUuZWR1