 Daniel R. Laucirica
Daniel R. Laucirica Luke W. Garratt
Luke W. Garratt Anthony Kicic
Anthony Kicic- 1Faculty of Health and Medical Sciences, University of Western Australia, Nedlands, WA, Australia
- 2Telethon Kids Institute, University of Western Australia, Nedlands, WA, Australia
- 3Department of Respiratory and Sleep Medicine, Perth Children's Hospital, Nedlands, WA, Australia
- 4School of Public Health, Curtin University, Bentley, WA, Australia
In response to recurrent infection in cystic fibrosis (CF), powerful innate immune signals trigger polymorphonuclear neutrophil recruitment into the airway lumen. Exaggerated neutrophil proteolytic activity results in sustained inflammation and scarring of the airways. Consequently, neutrophils and their secretions are reliable clinical biomarkers of lung disease progression. As neutrophils are required to clear infection and yet a direct cause of airway damage, modulating adverse neutrophil activity while preserving their pathogen fighting function remains a key area of CF research. The factors that drive their pathological behavior are still under investigation, especially in early disease when aberrant neutrophil behavior first becomes evident. Here we examine the latest findings of neutrophils in pediatric CF lung disease and proposed mechanisms of their pathogenicity. Highlighted in this review are current and emerging experimental methods for assessing CF mucosal immunity and human neutrophil function in the laboratory.
Introduction
Polymorphonuclear neutrophils are the most abundant immune cells in human blood and act as first responders to sites of infection. Their function is a key component of host defense against invading pathogens. In the autosomal recessive disorder cystic fibrosis (CF), persistent microbial colonization in the lungs induces abundant and continuous migration of neutrophils to the airways via powerful inflammatory signals of IL-6, IL-8, and leukotriene B4 (1). Recruited CF neutrophils secrete high levels of proteolytic compounds such as neutrophil elastase (NE), which damage airway tissue and highly correlate with disease severity (2, 3). Despite recognition of neutrophils in the progression of CF lung disease, mechanisms modulating their pathological role are not well-characterized. Past investigations have been hampered by a lack of widely available CF animal models and no suitable in vitro infection models that effectively incorporate multiple factors driving complex in vivo disease. New data from modern clinical studies are changing the view that neutrophils are a fixed population and are revealing a spectrum of functional phenotypes neutrophils employ to address the variety of pathogenic scenarios they encounter (4). Understanding neutrophil phenotypes and mechanisms in early CF disease, when the airway environment is less complex and more responsive to intervention, will require researchers to revisit or adapt many models of CF. With this review, we present new insights, challenges, and considerations for researchers studying neutrophils in early CF lung disease.
Pediatric CF Lung Disease
The clinical picture of early life cystic fibrosis has changed significantly since CF was first identified in the mid twentieth century, when patients rarely survived to the age of 10 (5). Improvements in diagnostics, from sweat tests to genetic testing, and wider screening of newborns by immunoreactive trypsinogen, has permitted earlier detection of CF and management of the disease. Increased antibacterial therapy, mucolytic and osmotic agents, and the advent of CFTR modulators have further increased the lifespan of many CF patients to beyond 40 years of age (6). Despite these advances, signs of airway inflammation and lung damage are still evident in CF from an early age. In 2005, a pioneering study assessing bronchoalveolar lavage fluid (BALF) after CF newborn screening demonstrated that infection in the first year of life is linked to early airway inflammation (7). Subsequent surveillance studies have now established that for most children, inflammation, altered microbiome, active neutrophil proteolytic function, and lung damage all become evident within the first 2 years of life (8–11), before children are old enough to be treated with CFTR modulators (12, 13). Lung function declines can be evident in infants and continue into childhood (14–16). However, early airway disease can occur in the absence of overt respiratory symptoms (17) or infection (18, 19). Computed Tomography (CT) screening has revealed that CF associated structural changes diagnosed in early life persist into childhood and adolescence. Permanent bronchial wall thickening, or bronchiectasis, is detectable in about 8.5% of pediatric CF patients in the first year of life, and this increases to 36% by 4 years (8). In addition to cytokine release, neutrophil influx into early CF airways may be supported by chemotactic fragments from the extracellular matrix (20, 21). Overall, CF lung damage and declines in function are linked to neutrophil counts and levels of neutrophil proteases, that are often a response to early incidence of infection. Understanding the pathology of early lung disease as it appears today will be key to maximizing long-term benefits from subsequent CFTR modulator therapies.
Neutrophils in CF Airways
Neutrophil Elastase and Serine Proteases
Early in vertebrate immunity, neutrophils evolved a granule system to separately store enzymes and antimicrobial factors safely until fused with a phagosome (22). Neutrophil elastase (NE) and other serine proteases are a central component of the neutrophil antimicrobial arsenal, stored in the primary granules that are the last granule to mobilize and are highly resistant to fusion with the outer membrane (23). Yet uninhibited NE activity can be detected in over 30% of BALF samples from young children with CF (11, 24). Activity of NE is considered one of the most significant biomarkers in CF lung disease, as activity significantly correlates with lung damage and functional declines at all stages of life with CF (25–28). Unregulated activity is destructive to airway epithelial cells and the lamina propria, and can impede microbial clearance through destruction of host immune factors (29). In vitro and in vivo studies have demonstrated how elevated NE activity induces epithelial senescence in CF airway cells (30, 31), prevents epithelial repair mechanisms (24), and is a key driver of airway inflammation and mucus production (25–28). Neutrophil Elastase and other serine proteases digest a variety of host proteins, suggesting multiple mechanisms that implicate these compounds in CF airway pathology. Along with neutrophil serine proteases cathepsin G and proteinase 3, NE directly interacts with cytokines, including IL-8 and IL-1α, increasing their potency (32–34). Counter-intuitively, serine proteases also degrade antimicrobial peptides (AMPs), including lactoferrin, midkine, and surfactant protein-A (SP-A) (35–37). In particular, NE has been shown to degrade pattern recognition receptors including toll-like receptor 4 (TLR4), reducing bacterial lipopolysaccharide (LPS) sensitivity and increasing inflammation (38). In addition to modulating mucosal immunity, serine proteases may promote airway epithelial dysfunction in CF. For example, NE cleaves E-cadherin, an important component of adherens junctions, compromising epithelial integrity (39). It also induces CFTR protein degradation by calpain activity in both in vitro epithelial cells and in vivo mouse models, resulting in impaired channel function as well as increasing sodium transport into cells through proteolytic activation of sodium ion channels (ENaC) (40–42).
Cysteine Proteases, Matrix Metalloproteinases, and Reactive Oxygen Species
In addition to NE and other enzymes found in primary granules, additional neutrophil derived compounds may contribute to CF airway pathology. Crucial for intracellular degradation of pathogens, secreted cysteine proteases have similar deleterious effects as their serine counterparts. Cathepsins B and S positively correlate with clinical markers of inflammation in pediatric CF airways, including NE, IL-8, and TNFα (43, 44). They selectively maintain neutrophil influx through activation of chemokines containing glutamic acid-leucine-arginine (ELR) motifs and inactivation of lymphocyte attracting non-ELR chemokines (45). Similar to serine proteases, cathepsins can compromise immunity through degradation of AMPs such as lactoferrin, LL-37, SP-A, and β-defensins (46–49). Cathepsins B and S are also implicated in airway mucus dehydration through induction of ENaC activity (50, 51). Matrix metalloproteinases (MMPs) are additional proteases implicated in CF associated with airway remodeling following lung injury. They can originate from any tissue, but neutrophil derived MMP-9 is particularly linked to airway damage, inflammation, and lung function decline in early CF (52, 53). Furthermore, MMP-9 sustains airway neutrophilia through potentiation of IL-8 and generation of proline-glycine-proline (PGP) matrikine fragments from breakdown of collagen (21, 54). Upon phagocytosis of pathogens, neutrophils produce large amounts of superoxide radicals for microbial killing. Broadly termed reactive oxygen species (ROS), neutrophils are among the most potent producers of these compounds (55). Oxidative stress as shown by elevated airway ROS is observed in chronic obstructive pulmonary disease (COPD) as well as CF (56–58). Increased ROS production results in destruction of antiproteases, which are crucial for protecting tissue from unregulated proteolysis (59, 60). In the context of CF, ROS may impede the function of antiproteases such as alpha-1-antitrypsin, an important NE inhibitor, prolonging airway neutrophil proteolytic activity (24, 61).
CFTR in Neutrophils
A central conundrum of CF is why proteolytic activity develops in such early, mild stages of CF lung inflammation. One obvious area of investigation has been whether neutrophil dysfunction in CF airways is exclusively influenced by factors in the lung environment or is also a consequence of inherent CFTR defects. Since the discovery of the CFTR gene, there have been studies suggesting CFTR protein is routinely expressed in cells of myeloid origin and has a role in microbial clearance within phagosomes (62–64). Hypochlorous acid (HOCl) is an important antimicrobial component of neutrophil phagosomes whose formation is proposed to be dependent on CFTR-mediated chloride ion transport (65). CFTR is reported to traffic to phagosomal membranes in peripheral blood neutrophils, with CFTR mutation resulting in defective phagosomal chlorination, affecting clearance of microbes such as P. aeruginosa (66–68). Contrasting findings have shown normal respiratory burst activity and production of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase components in CF blood neutrophils and no detectable CFTR protein in these cells (69). Additional evidence for the role of CFTR in neutrophils comes from a small number of studies showing restoration of CF neutrophil functions including CFTR phagosomal trafficking (67), leukocyte activation (70), and intracellular ion regulation (71) in response to CFTR modulator treatment. Still, further research is needed to clarify the presence and function of CFTR in neutrophils, and how defects in the gene influence the pathological activity of CF airway neutrophils. One consistent observation is a CFTR mutation dependent effect on in vivo neutrophil lifespan, with CF neutrophils displaying delayed apoptosis compared to non-CF neutrophils, possibly preventing resolution of neutrophilic inflammation (72–74). The most recent of these studies demonstrated a link between delayed apoptosis by CF neutrophils and propensity to form neutrophil extracellular traps (74).
Neutrophil Extracellular Traps
The identification of neutrophil extracellular traps (NETs), extracellular networks of DNA containing azurophilic granules, neutrophil elastase and other antimicrobial components, was a significant event in neutrophil biology (75). NET formation was initially viewed as a form of active cell death upon which nuclear and granular membranes were disintegrated, contents ejected and mixed in the cytoplasm, then released upon deterioration of the cell membrane (76). The process was later termed NETosis and proposed to be an alternative strategy used by neutrophils upon failing to clear infection via traditional phagocytosis. While NETs can trap and neutralize invading pathogens, the extent of their microbe killing abilities is debated (77, 78). A significant amount of research into NETosis has been undertaken, as recently reviewed by this journal (79). Multiple studies have described forms of NETosis that result in mitochondrial DNA release rather than nuclear DNA, or allow neutrophils to remain viable and motile after NET formation (80–83). The ability of NETs to harbor NE, the presence of NET derived DNA in CF sputum, and increased pathogen resistance in response to NETs, suggest NETosis is likely to play a role in CF lung disease (84, 85). Yet the question remains on how frequently NETosis occurs during early CF airway inflammation, prior to significant biofilm formation that reduces availability of bacteria to neutrophils.
Neutrophil Exocytosis
Perhaps the most intriguing hypothesis explaining early airway neutrophil proteolytic activity is that upon recruitment to CF airways, neutrophils reprogram toward an aberrant granular-releasing, immunoregulatory, and metabolically distinct (GRIM) phenotype that includes exocytosis of primary granules—as evidenced by high CD63 expression (86–88). The GRIM phenotype is specific to recruited neutrophils as peripheral blood neutrophils from CF patients exhibit a normal phenotype (89). However, when naïve neutrophils from either CF or non-CF donors are stimulated in an in vitro transmigration model of neutrophil recruitment by adult CF BALF or sputum, cells from both groups of donors undergo GRIM reprogramming (89). While factors such as tumor necrosis factor-alpha (TNF-α) can prime exocytosis of neutrophil azurophilic granules (90), Forrest and colleagues observed GRIM reprogramming only upon stimulation with ex vivo CF samples but not with exogenously added chemokines, suggesting a yet unidentified factor in CF airways is responsible for changes in neutrophil activity (89).
Most significantly, GRIM neutrophils were also found to have reduced bacterial killing capacity, which aligns with the apparent disconnect between NE release and inability to resolve infection in CF airways (89). More recent studies have reported how Staphylococcal superantigen-like protein 13 (SSL13) from Staphylococcus aureus, a common early CF pathogen, can induce neutrophil exocytosis (91) and whose production is evident in the CF microbiome (92). In an age related cohort of non-CF children admitted for acute respiratory distress syndrome (ARDS), neutrophil exocytosis and reduced bacterial killing was observed in individuals co-infected with virus and bacteria but not viral infection alone, suggesting that neutrophil exocytosis may be linked to responses against polymicrobial infection (93). This relationship with infection is yet to be established in early CF disease, however neutrophil exocytosis markers correlate positively with disease severity more so than free NE activity (11). Therefore, changes in airway neutrophil functional markers may be more reliable indicators of disease progression in children with CF and should be a focus of early CF lung disease research.
Modeling Infection And Inflammation
Characterizing the early mechanisms that trigger phenotype shifts in airway neutrophils may be key for preventing progressive lung disease. Clinical surveillance gives valuable insights into disease phenotypes in vivo, however, basic science is crucial for understanding the biology of CF lung disease and the role of the airway epithelium. Over the years, researchers have developed a variety of approaches for this purpose (Figure 1). The following is a summary of some of the more important, biologically relevant models currently in use to study infection and inflammation in CF airways.
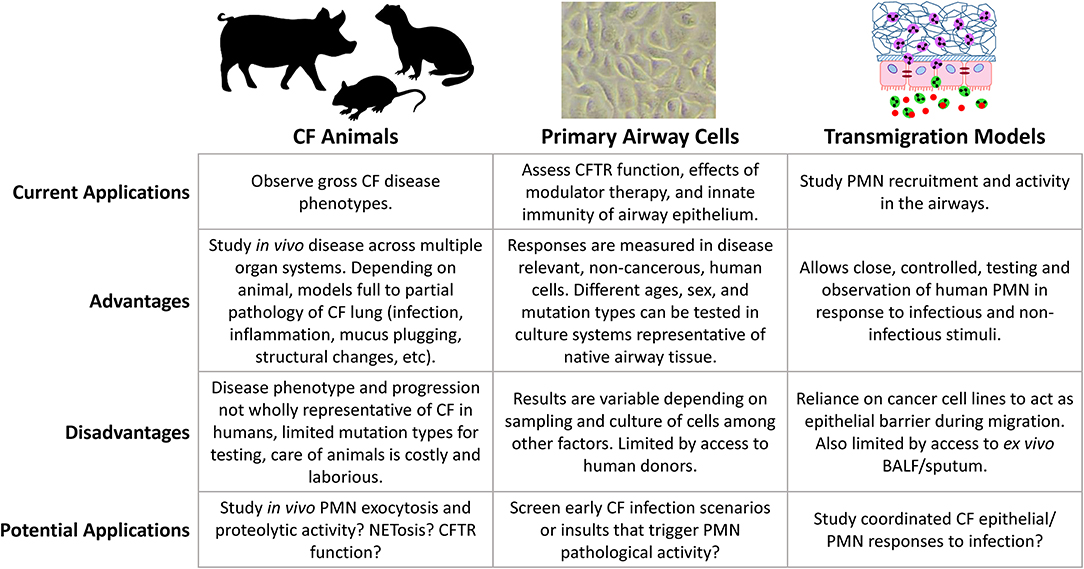
Figure 1. Comparison of model systems for studying CF lung disease.
Animal Models
While CFTR mutant and knockout mice were developed shortly after discovery of the CFTR gene, their use as animal models for CF lung disease is controversial as they lack a robust CF lung phenotype of spontaneous infection and disease (94–97). Despite extensive similarity, mouse immune cells can behave differently to human counterparts in their response to pathogens, for example, murine neutrophils are not activated by SSL13 (91). Mice also express toll-like receptor 11 (TLR11), a TLR not expressed in humans, that detects profilin and bacterial flagellin (98). Still, mouse models of induced airway infection have provided insights into CF airway inflammation and disease. Studies of acute Pseudomonas aeruginosa infection have observed poor growth, increased mortality, and reduced bacterial clearance in CF vs. wild-type mice (99, 100). Additionally, CF mice have exaggerated levels of murine inflammatory cytokines and airway neutrophilia in response to infection, as well as prolonged inflammation compared to wild-type mice (101, 102). Most of these studies have inoculated animals through intratracheal delivery of agarose beads embedded with bacteria, an unrealistic representation of how CF patients normally acquire these organisms. Of interest has been the observation that environmental acquisition of P. aeruginosa can be modeled in mice through inoculated drinking water, with CF mice more susceptible to chronic colonization via this route (103). Chronic exposure of CF mice to P. aeruginosa LPS also results in increased airway inflammation, neutrophilia and airway remodeling (104, 105). A common theme emerging from these studies, is that neutrophils and their products play a central role in CF lung pathology. With the development of Cre recombinase mice targeting the neutrophil-specific locus Ly6G (106), future studies utilizing this model will continue to play a very useful role in elucidating CF airway neutrophil biology.
The more physiologically relevant animal model for studying CF lung disease include CFTR disrupted pigs and ferrets, as they recapitulate the CF phenotype across all organ systems implicated in human disease (107). Both models were developed just over a decade ago using adenoviral vectors, generating CFTR full or partial knockout animals in both species via exon 10 disruption, as well as a ΔF508 pig (108–110). Pigs are suitable human disease models due to their analogous physiology, and in the case of respiratory disease, similar bronchial structure and distribution of submucosal glands (111). CF pigs have CFTR protein similar to that of humans (112). Neonatal CF pigs have little airway inflammation and normal levels of IL-8 and neutrophil counts in BAL compared to non-CF pigs (109). Neonatal CF pigs also have increased presence of microbes in the lungs as shown by culture from ex vivo tissue samples, and are less likely to have sterile BAL samples compared to non-CF pigs (113). In the months following birth, CF pigs develop signs of lung disease such as mucus accumulation, inflammation, infection, and airway remodeling (113). While lung disease progression in the CF pig model reflects progression in humans, there are obvious drawbacks of cost and time of pig husbandry and the need of adequate facilities and resources. Furthermore, virtually all CF pigs develop meconium ileus and require early surgical intervention; in contrast, the condition is present in only 20% of infants with CF (109, 113, 114).
Ferret CFTR protein length, amino acid sequence, and function is also similar to that of humans (115). Like CF humans, CF ferrets are prone to spontaneous airway infection; however, infection in these animals is far more severe, with CF ferrets requiring continuous antibiotic treatment immediately after birth to survive (116). Additionally, CF ferrets demonstrate abnormally high levels of lung inflammation from birth, and lung disease progresses rapidly upon cessation of prophylactic antibiotics (117, 118). As a result, CF ferrets may not be an ideal system to model the slow progressive lung disease observed in humans, as their disease phenotype develops too quickly. However, a recent study developed homozygous CFTRG551D/G551D ferrets to test effects of in utero treatment with VX-770 (ivacaftor) (119). Prenatal and early postnatal administration ameliorated CF multi-organ disease, posing new research questions around CFTR in early development, the possibility of prenatal modulator therapy, and disease attenuation in CF animals to further study effects of modulator treatment or model mild disease in humans. While neutrophil counts and elastase activity in CF animals trend similarly to human disease, neutrophil reprogramming has yet to be evaluated. Future studies must assess airway neutrophil exocytosis and lung disease severity in CF animals to determine if they are suitable models for characterizing this process in humans.
Primary Airway Epithelial Cells
While animal models allow observation of gross phenotype of disease, in vitro studies permit experimentation in a highly controlled environment and are important for understanding mechanisms of disease at the cellular level. The accepted gold standard for in vitro CF research are patient derived primary airway epithelial cells (pAEC). As a barrier that protects the lung from direct environmental exposure, the airway epithelium has long been recognized for its role in host defense and respiratory disease (120–125). Cells are typically isolated from epithelial brushings of the nose or lower airways, or less frequently from explanted lungs (126, 127). Yields from brushings are variable and ex vivo pAEC have limited proliferative capacity; they become senescent after only a few passages making them difficult to expand in culture (126, 127). The adaptation of conditionally reprogrammed airway epithelial cells (CRAEC) through co-culture with irradiated fibroblast feeder cells has significantly increased passage number capacity of pAEC, while maintaining lineage specific characteristics (128). Additionally, CRAEC can be seeded from co-culture into air-liquid interface culture (ALI) to form a differentiated pseudostratified epithelial layer (128, 129). This has enabled many CF research groups to look to CF primary airway epithelial cell models in order to understand the cellular drivers of progressive lung disease, and more recently to evaluate the efficacy of CFTR modulators in restoring CFTR function in target cells (130, 131). Nasal pAEC are increasingly being used in epithelial CFTR studies, since their growth, differentiation, CFTR activity, and response to modulators are similar to lower airway cells, and have the advantage of being more readily accessible (129). Nasal cells have also been adapted to three dimensional spheroid cultures that are representative of native epithelium and mature more quickly than traditional ALI cultures (132). These spheroids have then been used to quantify CFTR function via spheroid swelling in cultures from CF patients across different mutation classes, to assess individual responses to modulator treatment (133, 134). As such, they have potential as a preclinical screening tool to identify responses to modulator therapies in a personalized medicine approach.
Despite increased airway inflammation in CF patients, there is still debate as to whether the CF airway epithelium is inherently pro-inflammatory (135–137). Baseline expression of neutrophil chemoattractants including IL-8, IL-6, and IL-1β is reported in some studies to be similar in CF vs. non-CF pAEC (138–140), but others report increases in CF cells at baseline (141, 142). Increased airway inflammation could also be a result of dysfunctional CF epithelial innate immunity, a major topic in CF research, as the airway epithelium has an important role in responding to infection and neutrophil recruitment (143–147). Studies have shown IL-8 release and NF-κB activity are increased in CF vs. non-CF pAEC following P. aeruginosa infection (142, 148, 149). CF pAEC have also been shown to display differential gene expression at the transcriptional level compared to non-CF pAEC in response to P. aeruginosa infection, which may be further evidence of CF aberrant immune responses (150, 151). In response to infection with respiratory viruses, studies have also observed increased IL-8 production in CF vs. non-CF pAEC (152, 153), which is analogous to in vivo findings in pediatric CF patients with rhinovirus infection (154). However, other studies have reported no difference in inflammatory cytokine production as a result of in vitro viral infection (155, 156). The filamentous fungi Aspergillus fumigatus is emerging as an important early life CF pathogen increasingly detected in pediatric CF airways (157, 158), with A. fumigatus infection associated with increased air trapping among 5 year old CF patients (159). Two in vitro studies to date have used immortalized cell lines and reported altered cytokine production, though specific data were conflicting (160, 161). Assessing innate immune responses to fungal pathogens in CF will be key to determining treatment priority, but innate immune mechanisms have yet to be corroborated in CF pAEC.
Variability amongst findings from pAEC infection studies could be attributed to the originating cohorts, sampling differences, age and disease severity of subjects, culture methods, and use of differentiated vs. undifferentiated cultures. Additionally, the selection of pathogens can affect outcomes, as pAEC responses can be heterogeneous to individual strains or isolates of the same species (162). Primary cells will continue to be important tools for addressing unanswered questions in CF lung disease, including how epithelial immunity is linked to neutrophil inflammation, how the epithelium responds to fungal and polymicrobial infection, how the airway microbiome affects epithelial homeostasis, and whether CFTR modulators have effects on airway innate immune signaling. Researchers must think carefully about the above factors and how they influence experimental outcomes in pAEC, especially if findings are translated to lung disease pathogenesis in CF patients.
Neutrophil Transmigration to the Airways
The epithelium is not only a barrier to external pathogens, but also presents an obstacle to responding neutrophils. In vitro replication of this mucosal physiology was established by early studies assessing neutrophil transmigration across the intestinal mucosa (163–166). These studies established polarized epithelial cultures onto inverted permeable inserts, which were turned over prior to migration for direct loading of naïve isolated neutrophils and thus model basolateral to apical neutrophil migration. It has since been adapted to characterize transmigration across lung epithelium (167), assess the role of neutrophils in β-catenin mediated airway epithelial repair (168, 169), as well as describe responses to infection with respiratory syncytial virus (RSV) and P. aeruginosa (170–172). The model previously mentioned in this review uses Alvetex™ 3D scaffolds rather than permeable membranes, which better replicate neutrophil swarming (89, 93), to study neutrophil responses to CF sputum. To understand factors driving early neutrophil fate including exocytosis, this same model could be applied with pediatric BALF. However, pediatric ex vivo samples are difficult to obtain, often of limited volume compared to samples from adults. One approach yet to be fully utilized is to apply material from infected CF pAEC as surrogates for human samples. This has multiple benefits. Robust models of pAEC infection responses are well-established and because pAEC can be bio-banked for downstream culture and infection, material can be generated as needed. This approach also facilitates a more focused assessment of factors influencing neutrophil functions, such as epithelial responses to specific infection scenarios.
One caveat of past transmigration studies is the dependence on lung cancer derived cells lines, such as A549, H292, H441, 16HBE, and Calu-3 cells, as a substitute for primary cell derived epithelium. Advantages include easy access to cell lines and robust growth in culture, but at the cost of interpreting epithelial responses during migration through cancer cells. This is critical, since neutrophils directly change airway epithelium via microRNA (173) or exosomes (174) and neoplastic cells may not reproduce CF pAEC responses. As limitations on pAEC culture expansion are overcome, transmigration studies are increasingly incorporating pAEC (175). A remaining challenge is that established methods for differentiating pAEC traditionally employ 0.4 μm pore size inserts, but a 3.0μm pore size or larger is required to permit neutrophil transmigration, which can result in significant loss of primary cells during seeding. One study has managed to address this issue by coating both faces of a transwell insert with extracellular matrix and providing seeded cells with laminins to improve attachment (175). Primary cells differentiated into pseudostratified epithelial layers on a 3.0 μm insert, similar to how they would on a conventional 0.4 μm transwell insert, and permitted neutrophil transmigration upon apical infection with P. aeruginosa (175). Future integration of CF pAEC in models of neutrophil transmigration will be required for studying coordinated immune responses of the CF airway epithelium and recruited neutrophils in a single translational system. If designed with high throughput screening in mind, there is great potential to facilitate much needed pre-clinical testing of anti-inflammatory drugs in CF.
CFTR Modulator Therapy And Airway Inflammation
Depending on their mechanism of action, CFTR modulators are characterized as correctors that improve defective CFTR trafficking to the cell surface, or potentiators that enhance defective CFTR function. Studies of modulators have shown improvements in patients as measured by sweat chloride levels and FEV1, however, efficacy against infection and airway inflammation is poorly investigated. In placebo controlled studies of lumacaftor-ivacaftor and tezacaftor-ivacaftor in CF patients ≥12 years of age, infective pulmonary exacerbations occurred at similar rates in both treatment and placebo groups (176–178). Phase 3 trials of lumacaftor-ivacaftor in CF patients aged 6–11 also found that incidence of infection associated pulmonary exacerbations was similar between patients receiving treatment (18%) and patients receiving a placebo control (19%) (179). The recently FDA approved elexacaftor-tezacaftor-ivacaftor triple therapy roughly halves the incidence of infective pulmonary excacerbations compared to a placebo (180), but the drug is not yet approved for patients under 12 years of age. Multiple studies have shown that administration of modulators reduces bacterial colonization within the first year of treatment and delays acquisition in uncolonized patients; however, bacterial isolates present in the airways prior to treatment persist and may eventually rebound over longer periods (12, 13, 181, 182). Whether CFTR modulators reduce levels of inflammatory cytokines is still not certain, as there is evidence of both reduction and no effect on clinically relevant biomarkers including NE (12, 13). Altogether, current findings suggest that modulator therapy alone may not be sufficient to manage infection and airway inflammation in this population, especially over the long term.
Conclusion
Neutrophils have a major role in CF lung disease but our ability to treat the underlying mechanisms is still limited. Modern approaches are revealing new perspectives on neutrophils as plastic, programmable drivers of airway disease who both respond to and actively shape the local airway environment (Figure 2). These novel neutrophil functions are occurring even in mild and largely asymptomatic pediatric CF lung disease and precede structural lung changes. Even with the advent of combination CFTR modulator therapy, which improves lung function but perhaps not infection and inflammation, continued investigation of initial neutrophil pathological activity is necessary to identify much-needed interventions that can address this problem. Researchers now have available a diverse number of tools for understanding the complex interplay between infection, the airway epithelium, and recruited neutrophils (Figure 1). Moving forward, basic studies will need to consider the advantages of various approaches, caveats, and carefully select appropriate models when exploring the beginnings of CF airway neutrophilic disease.
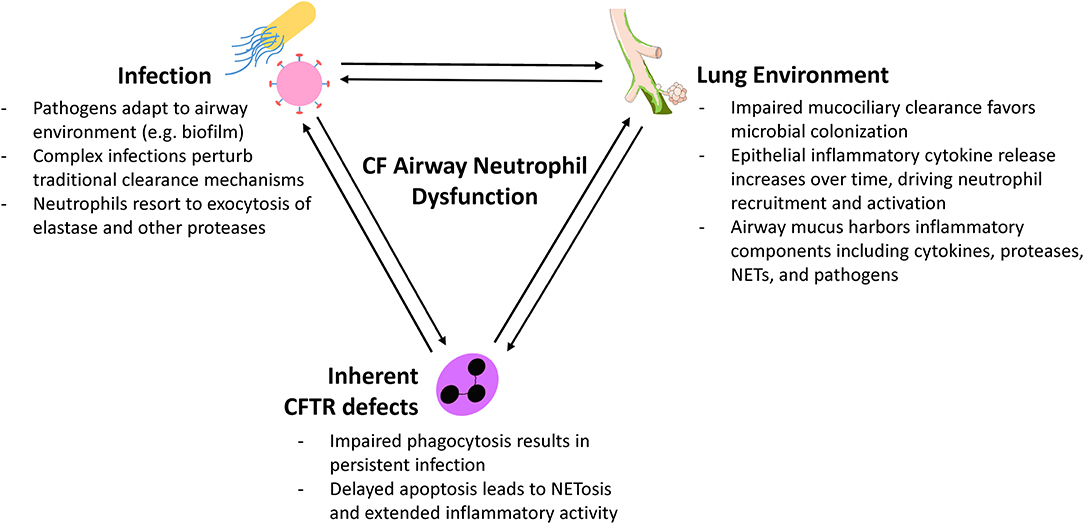
Figure 2. Factors influencing CF airway neutrophil plasticity.
Author Contributions
DL, LG, and AK conceived the review. DL and LG conducted literature review and wrote the manuscript. LG and AK provided critical review.
Funding
This work was funded by the National Health and Medical Research Council (NHMRC). DL is funded by the University of Western Australia, Telethon Kids Institute and NHMRC 1142505. LG is a NHMRC Early Career Fellow 1141479. AK is a Rothwell Family Fellow.
Conflict of Interest
LG and AK are co-investigators with some of the authors cited within the review.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, et al. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros. (2012) 11:363–82. doi: 10.1016/j.jcf.2012.07.003
2. Mayer-Hamblett N, Aitken ML, Accurso FJ, Kronmal RA, Konstan MW, Burns JL, et al. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J Respir Crit Care Med. (2007) 175:822–8. doi: 10.1164/rccm.200609-1354OC
3. Dittrich AS, Kuhbandner I, Gehrig S, Rickert-Zacharias V, Twigg M, Wege S, et al. Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur Respir J. (2018) 51:1701910. doi: 10.1183/13993003.01910-2017
4. Jablonska J, Granot Z. Neutrophil, quo vadis?. J Leukoc Biol. (2017) 102:685–8. doi: 10.1189/jlb.3MR0117-015R
5. Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. (2006) 173:475–82. doi: 10.1164/rccm.200505-840OE
6. Bell SC, Mall MA, Gutierrez H, Macek M, Madge S, Davies JC, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med. (2020) 8:65–124. doi: 10.1016/S2213-2600(19)30337-6
7. Armstrong DS, Hook SM, Jamsen KM, Nixon GM, Carzino R, Carlin JB, et al. Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol. (2005) 40:500–10. doi: 10.1002/ppul.20294
8. Stick SM, Brennan S, Murray C, Douglas T, von Ungern-Sternberg BS, Garratt LW., et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. (2009) 155, 623–8 e1. doi: 10.1016/j.jpeds.2009.05.005
9. Schultz A, Stick S. Early pulmonary inflammation and lung damage in children with cystic fibrosis. Respirology. (2015) 20:569–78. doi: 10.1111/resp.12521
10. Laguna TA, Wagner BD, Williams CB, Stevens MJ, Robertson CE, Welchlin CW, et al. Airway Microbiota in bronchoalveolar lavage fluid from clinically well infants with cystic fibrosis. PLoS ONE. (2016) 11:e0167649. doi: 10.1371/journal.pone.0167649
11. Margaroli C, Garratt LW, Horati H, Dittrich AS, Rosenow T, Montgomery ST, et al. Elastase exocytosis by airway neutrophils is associated with early lung damage in children with cystic fibrosis. Am J Respir Crit Care Med. (2019) 199:873–81. doi: 10.1164/rccm.201803-0442OC
12. Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. (2014) 190:175–84. doi: 10.1164/rccm.201404-0703OC
13. Hisert KB, Heltshe SL, Pope C, Jorth P, Wu X, Edwards RM, et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am J Respir Crit Care Med. (2017) 195:1617–28. doi: 10.1164/rccm.201609-1954OC
14. Kozlowska WJ, Bush A, Wade A, Aurora P, Carr SB, Castle RA, et al. Lung function from infancy to the preschool years after clinical diagnosis of cystic fibrosis. Am J Respir Crit Care Med. (2008) 178:42–9. doi: 10.1164/rccm.200710-1599OC
15. Harrison AN, Regelmann WE, Zirbes JM, Milla CE. Longitudinal assessment of lung function from infancy to childhood in patients with cystic fibrosis. Pediatr Pulmonol. (2009) 44:330–9. doi: 10.1002/ppul.20994
16. Brumback LC, Davis SD, Kerby GS, Kloster M, Johnson R, Castile R, et al. Lung function from infancy to preschool in a cohort of children with cystic fibrosis. Eur Respir J. (2013) 41:60–6. doi: 10.1183/09031936.00021612
17. Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. (2009) 180:146–52. doi: 10.1164/rccm.200901-0069OC
18. Montgomery ST, Dittrich AS, Garratt LW, Turkovic L, Frey DL, Stick SM, et al. Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J Cyst Fibros. (2018) 17:715–22. doi: 10.1016/j.jcf.2018.05.006
19. Esther CR Jr, Muhlebach MS, Ehre C, Hill DB, Wolfgang MC, Kesimer M, et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci Transl Med. (2019) 11:eaav3488. doi: 10.1126/scitranslmed.aav3488
20. Breuer O, Schultz A, Garratt LW, Turkovic L, Rosenow T, Murray CP, et al. Aspergillus infections and progression of structural lung disease in children with cystic fibrosis. Am J Respir Crit Care Med. (2020) 201:688–96. doi: 10.1164/rccm.201908-1585OC
21. Turnbull AR, Pyle CJ, Patel DF, Jackson PL, Hilliard TN, Regamey N, et al. Abnormal pro-gly-pro pathway and airway neutrophilia in pediatric cystic fibrosis. J Cyst Fibros. (2020) 19:40–8. doi: 10.1016/j.jcf.2019.05.017
22. Fu Z, Thorpe M, Akula S, Chahal G, Hellman LT. Extended cleavage specificity of human neutrophil elastase, human proteinase 3, and their distant ortholog clawed frog PR3-three elastases with similar primary but different extended specificities and stability. Front Immunol. (2018) 9:2387. doi: 10.3389/fimmu.2018.02387
23. Jog NR, Rane MJ, Lominadze G, Luerman GC, Ward RA, McLeish KR. The actin cytoskeleton regulates exocytosis of all neutrophil granule subsets. Am J Physiol Cell Physiol. (2007) 292:C1690–700. doi: 10.1152/ajpcell.00384.2006
24. Garratt LW, Sutanto EN, Ling KM, Looi K, Iosifidis T, Martinovich KM, et al. Alpha-1 antitrypsin mitigates the inhibition of airway epithelial cell repair by neutrophil elastase. Am J Respir Cell Mol Biol. (2016) 54:341–9. doi: 10.1165/rcmb.2015-0074OC
25. Pillarisetti N, Williamson E, Linnane B, Skoric B, Robertson CF, Robinson P, et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am J Respir Crit Care Med. (2011) 184:75–81. doi: 10.1164/rccm.201011-1892OC
26. Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med. (2012) 186:857–65. doi: 10.1164/rccm.201203-0507OC
27. Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. (2013) 368:1963–70. doi: 10.1056/NEJMoa1301725
28. Rosenow T, Mok LC, Turkovic L, Berry LJ, Sly PD, Ranganathan S, et al. The cumulative effect of inflammation and infection on structural lung disease in early cystic fibrosis. Eur Respir J. (2019) 54:1801771. doi: 10.1183/13993003.01771-2018
29. Gifford AM, Chalmers JD. The role of neutrophils in cystic fibrosis. Curr Opin Hematol. (2014) 21:16–22. doi: 10.1097/MOH.0000000000000009
30. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence exacerbates pulmonary inflammation in patients with chronic obstructive pulmonary disease. Respiration. (2010) 80:59–70. doi: 10.1159/000268287
31. Fischer BM, Wong JK, Degan S, Kummarapurugu AB, Zheng S, Haridass P, et al. Increased expression of senescence markers in cystic fibrosis airways. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L394–400. doi: 10.1152/ajplung.00091.2012
32. Padrines M, Wolf M, Walz A, Baggiolini M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. (1994) 352:231–5. doi: 10.1016/0014-5793(94)00952-X
33. Pham CT. Neutrophil serine proteases fine-tune the inflammatory response. Int J Biochem Cell Biol. (2008) 40:1317–33. doi: 10.1016/j.biocel.2007.11.008
34. Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Luthi AU, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell. (2011) 44:265–78. doi: 10.1016/j.molcel.2011.07.037
35. Britigan BE, Edeker BL. Pseudomonas and neutrophil products modify transferrin and lactoferrin to create conditions that favor hydroxyl radical formation. J Clin Invest. (1991) 88:1092–102. doi: 10.1172/JCI115408
36. Rubio F, Cooley J, Accurso FJ, Remold-O'Donnell E. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax. (2004) 59:318–23. doi: 10.1136/thx.2003.014902
37. Nordin SL, Jovic S, Kurut A, Andersson C, Gela A, Bjartell A, et al. High expression of midkine in the airways of patients with cystic fibrosis. Am J Respir Cell Mol Biol. (2013) 49:935–42. doi: 10.1165/rcmb.2013-0106OC
38. Devaney JM, Greene CM, Taggart CC, Carroll TP, O'Neill SJ, McElvaney NG. Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Lett. (2003) 544:129–32. doi: 10.1016/S0014-5793(03)00482-4
39. Boxio R, Wartelle J, Nawrocki-Raby B, Lagrange B, Malleret L, Hirche T, et al. Neutrophil elastase cleaves epithelial cadherin in acutely injured lung epithelium. Respir Res. (2016) 17:129. doi: 10.1186/s12931-016-0449-x
40. Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L813–9. doi: 10.1152/ajplung.00435.2004
41. Pruliere-Escabasse V, Clerici C, Vuagniaux G, Coste A, Escudier E, Planes C. Effect of neutrophil elastase and its inhibitor EPI-hNE4 on transepithelial sodium transport across normal and cystic fibrosis human nasal epithelial cells. Respir Res. (2010) 11:141. doi: 10.1186/1465-9921-11-141
42. Le Gars M, Descamps D, Roussel D, Saussereau E, Guillot L, Ruffin M, et al. Neutrophil elastase degrades cystic fibrosis transmembrane conductance regulator via calpains and disables channel function in vitro and in vivo. Am J Respir Crit Care Med. (2013) 187:170–9. doi: 10.1164/rccm.201205-0875OC
43. Martin SL, Moffitt KL, McDowell A, Greenan C, Bright-Thomas RJ, Jones AM, et al. Association of airway cathepsin B and S with inflammation in cystic fibrosis. Pediatr Pulmonol. (2010) 45:860–8. doi: 10.1002/ppul.21274
44. Small DM, Brown RR, Doherty DF, Abladey A, Zhou-Suckow Z, Delaney RJ, et al. Targeting of cathepsin S reduces cystic fibrosis-like lung disease. Eur Respir J. (2019) 53:1801523. doi: 10.1183/13993003.01523-2018
45. Repnik U, Starr AE, Overall CM, Turk B. Cysteine cathepsins activate ELR chemokines and inactivate non-ELR chemokines. J Biol Chem. (2015) 290:13800–11. doi: 10.1074/jbc.M115.638395
46. Taggart CC, Greene CM, Smith SG, Levine RL, McCray PB Jr, O'Neill S, et al. Inactivation of human beta-defensins 2 and 3 by elastolytic cathepsins. J Immunol. (2003) 171:931–7. doi: 10.4049/jimmunol.171.2.931
47. Rogan MP, Taggart CC, Greene CM, Murphy PG, O'Neill SJ, McElvaney NG. Loss of microbicidal activity and increased formation of biofilm due to decreased lactoferrin activity in patients with cystic fibrosis. J Infect Dis. (2004) 190:1245–53. doi: 10.1086/423821
48. Lecaille F, Naudin C, Sage J, Joulin-Giet A, Courty A, Andrault PM, et al. Specific cleavage of the lung surfactant protein A by human cathepsin S may impair its antibacterial properties. Int J Biochem Cell Biol. (2013) 45:1701–9. doi: 10.1016/j.biocel.2013.05.018
49. Andrault PM, Samsonov SA, Weber G, Coquet L, Nazmi K, Bolscher JG, et al. Antimicrobial peptide LL-37 is both a substrate of cathepsins S and K and a selective inhibitor of cathepsin L. Biochemistry. (2015) 54:2785–98. doi: 10.1021/acs.biochem.5b00231
50. Haerteis S, Krappitz M, Bertog M, Krappitz A, Baraznenok V, Henderson I, et al. Proteolytic activation of the epithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflugers Arch. (2012) 464:353–65. doi: 10.1007/s00424-012-1138-3
51. Tan CD, Hobbs C, Sameni M, Sloane BF, Stutts MJ, Tarran R. Cathepsin B contributes to Na+ hyperabsorption in cystic fibrosis airway epithelial cultures. J Physiol. (2014) 592:5251–68. doi: 10.1113/jphysiol.2013.267286
52. Sagel SD, Kapsner RK, Osberg I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr Pulmonol. (2005) 39:224–32. doi: 10.1002/ppul.20165
53. Garratt LW, Sutanto EN, Ling KM, Looi K, Iosifidis T, Martinovich KM, et al. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur Respir J. (2015) 46:384–94. doi: 10.1183/09031936.00212114
54. Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. (2000) 96:2673–81. doi: 10.1182/blood.V96.8.2673
55. Winterbourn CC, Kettle AJ, Hampton MB. Reactive oxygen species and neutrophil function. Annu Rev Biochem. (2016) 85:765–92. doi: 10.1146/annurev-biochem-060815-014442
56. Brown RK, Kelly FJ. Role of free radicals in the pathogenesis of cystic fibrosis. Thorax. (1994) 49:738–42. doi: 10.1136/thx.49.8.738
57. Galli F, Battistoni A, Gambari R, Pompella A, Bragonzi A, Pilolli F, et al. Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim Biophys Acta. (2012) 1822:690–713. doi: 10.1016/j.bbadis.2011.12.012
58. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. (2013) 144:266–73. doi: 10.1378/chest.12-2664
59. Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis. (2011) 6:413–21. doi: 10.2147/COPD.S10770
60. Siddiqui T, Zia MK, Ali SS, Rehman AA, Ahsan H, Khan FH. Reactive oxygen species and anti-proteinases. Arch Physiol Biochem. (2016) 122:1–7. doi: 10.3109/13813455.2015.1115525
61. McElvaney NG. Alpha-1 antitrypsin therapy in cystic fibrosis and the lung disease associated with alpha-1 antitrypsin deficiency. Ann Am Thorac Soc. (2016) 13(Suppl. 2):S191–6. doi: 10.1513/AnnalsATS.201504-245KV
62. Yoshimura K, Nakamura H, Trapnell BC, Chu CS, Dalemans W, Pavirani A, et al. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. (1991) 19:5417–23. doi: 10.1093/nar/19.19.5417
63. Painter RG, Valentine VG, Lanson NA Jr, Leidal K, Zhang Q, Lombard G, et al. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry. (2006) 45:10260–9. doi: 10.1021/bi060490t
64. Bonfield TL, Hodges CA, Cotton CU, Drumm ML. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J Leukoc Biol. (2012) 92:1111–22. doi: 10.1189/jlb.0412188
65. Painter RG, Marrero L, Lombard GA, Valentine VG, Nauseef WM, Wang G. CFTR-mediated halide transport in phagosomes of human neutrophils. J Leukoc Biol. (2010) 87:933–42. doi: 10.1189/jlb.1009655
66. Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, et al. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol. (2008) 83:1345–53. doi: 10.1189/jlb.0907658
67. Zhou Y, Song K, Painter RG, Aiken M, Reiser J, Stanton BA, et al. Cystic fibrosis transmembrane conductance regulator recruitment to phagosomes in neutrophils. J Innate Immun. (2013) 5:219–30. doi: 10.1159/000346568
68. Ng HP, Valentine VG, Wang G. CFTR targeting during activation of human neutrophils. J Leukoc Biol. (2016) 100:1413–24. doi: 10.1189/jlb.4A0316-130RR
69. McKeon DJ, Cadwallader KA, Idris S, Cowburn AS, Pasteur MC, Barker H, et al. Cystic fibrosis neutrophils have normal intrinsic reactive oxygen species generation. Eur Respir J. (2010) 35:1264–72. doi: 10.1183/09031936.00089709
70. Bratcher PE, Rowe SM, Reeves G, Roberts T, Szul T, Harris WT, et al. Alterations in blood leukocytes of G551D-bearing cystic fibrosis patients undergoing treatment with ivacaftor. J Cyst Fibros. (2016) 15:67–73. doi: 10.1016/j.jcf.2015.02.010
71. Pohl K, Hayes E, Keenan J, Henry M, Meleady P, Molloy K, et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood. (2014) 124:999–1009. doi: 10.1182/blood-2014-02-555268
72. McKeon DJ, Condliffe AM, Cowburn AS, Cadwallader KC, Farahi N, Bilton D, et al. Prolonged survival of neutrophils from patients with Delta F508 CFTR mutations. Thorax. (2008) 63:660–1. doi: 10.1136/thx.2008.096834
73. Moriceau S, Lenoir G, Witko-Sarsat V. In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. J Innate Immun. (2010) 2:260–6. doi: 10.1159/000295791
74. Gray RD, Hardisty G, Regan KH, Smith M, Robb CT, Duffin R, et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax. (2018) 73:134–44. doi: 10.1136/thoraxjnl-2017-210134
75. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
76. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
77. Menegazzi R, Decleva E, Dri P. Killing by neutrophil extracellular traps: fact or folklore?. Blood. (2012) 119:1214–6. doi: 10.1182/blood-2011-07-364604
78. Rahman S, Gadjeva M. Does NETosis contribute to the bacterial pathoadaptation in cystic fibrosis?. Front Immunol. (2014) 5:378. doi: 10.3389/fimmu.2014.00378
79. Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
80. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
81. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
82. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386–93. doi: 10.1038/nm.2847
83. Byrd AS, O'Brien XM, Johnson CM, Lavigne LM, Reichner JS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol. (2013) 190:4136–48. doi: 10.4049/jimmunol.1202671
84. Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, et al. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE. (2011) 6:e23637. doi: 10.1371/journal.pone.0023637
85. Manzenreiter R, Kienberger F, Marcos V, Schilcher K, Krautgartner WD, Obermayer A, et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J Cyst Fibros. (2012) 11:84–92. doi: 10.1016/j.jcf.2011.09.008
86. Tirouvanziam R, Gernez Y, Conrad CK, Moss RB, Schrijver I, Dunn CE, et al. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc Natl Acad Sci USA. (2008) 105:4335–9. doi: 10.1073/pnas.0712386105
87. Laval J, Touhami J, Herzenberg LA, Conrad C, Taylor N, Battini JL, et al. Metabolic adaptation of neutrophils in cystic fibrosis airways involves distinct shifts in nutrient transporter expression. J Immunol. (2013) 190:6043–50. doi: 10.4049/jimmunol.1201755
88. Mitchell TC. A GRIM fate for human neutrophils in airway disease. J Leukoc Biol. (2018) 104:657–9. doi: 10.1002/JLB.5CE0418-162R
89. Forrest OA, Ingersoll SA, Preininger MK, Laval J, Limoli DH, Brown MR, et al. Frontline science: pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J Leukoc Biol. (2018) 104:665–75. doi: 10.1002/JLB.5HI1117-454RR
90. McLeish KR, Merchant ML, Creed TM, Tandon S, Barati MT, Uriarte SM, et al. Frontline science: tumor necrosis factor-alpha stimulation and priming of human neutrophil granule exocytosis. J Leukoc Biol. (2017) 102:19–29. doi: 10.1189/jlb.3HI0716-293RR
91. Zhao Y, van Kessel KPM, de Haas CJC, Rogers MRC, van Strijp JAG, Haas PA. Staphylococcal superantigen-like protein 13 activates neutrophils via formyl peptide receptor 2. Cell Microbiol. (2018) 20:e12941. doi: 10.1111/cmi.12941
92. Ibberson CB, Whiteley M. The Staphylococcus aureus transcriptome during cystic fibrosis lung infection. MBio. (2019) 10:e02774–19. doi: 10.1128/mBio.02774-19
93. Grunwell JR, Giacalone VD, Stephenson S, Margaroli C, Dobosh BS, Brown MR, et al. Neutrophil dysfunction in the airways of children with acute respiratory failure due to lower respiratory tract viral and bacterial coinfections. Sci Rep. (2019) 9:2874. doi: 10.1038/s41598-019-39726-w
94. Dorin JR, Dickinson P, Alton EW, Smith SN, Geddes DM, Stevenson BJ, et al. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature. (1992) 359:211–5. doi: 10.1038/359211a0
95. Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science. (1992) 257:1083–8. doi: 10.1126/science.257.5073.1083
96. Colledge WH, Abella BS, Southern KW, Ratcliff R, Jiang C, Cheng SH, et al. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nat Genet. (1995) 10:445–52. doi: 10.1038/ng0895-445
97. McCarron A, Donnelley M, Parsons D. Airway disease phenotypes in animal models of cystic fibrosis. Respir Res. (2018) 19:54. doi: 10.1186/s12931-018-0750-y
98. Hatai H, Lepelley A, Zeng W, Hayden MS, Ghosh S. Toll-like receptor 11 (TLR11) interacts with flagellin and profilin through disparate mechanisms. PLoS ONE. (2016) 11:e0148987. doi: 10.1371/journal.pone.0148987
99. Heeckeren A, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest. (1997) 100:2810–5. doi: 10.1172/JCI119828
100. McMorran BJ, Palmer JS, Lunn DP, Oceandy D, Costelloe EO, Thomas GR, et al. G551D CF mice display an abnormal host response and have impaired clearance of Pseudomonas lung disease. Am J Physiol Lung Cell Mol Physiol. (2001) 281:L740–7. doi: 10.1152/ajplung.2001.281.3.L740
101. Saadane A, Soltys J, Berger M. Acute Pseudomonas challenge in cystic fibrosis mice causes prolonged nuclear factor-kappa B activation, cytokine secretion, and persistent lung inflammation. J Allergy Clin Immunol. (2006) 117:1163–9. doi: 10.1016/j.jaci.2006.01.052
102. van Heeckeren AM, Schluchter MD, Xue W, Davis PB. Response to acute lung infection with mucoid Pseudomonas aeruginosa in cystic fibrosis mice. Am J Respir Crit Care Med. (2006) 173:288–96. doi: 10.1164/rccm.200506-917OC
103. Coleman FT, Mueschenborn S, Meluleni G, Ray C, Carey VJ, Vargas SO, et al. Hypersusceptibility of cystic fibrosis mice to chronic Pseudomonas aeruginosa colonization and lung infection. Proc Natl Acad Sci USA. (2003) 100:1949–54. doi: 10.1073/pnas.0437901100
104. Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, Tuck D, et al. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am J Respir Cell Mol Biol. (2009) 40:295–304. doi: 10.1165/rcmb.2008-0170OC
105. Bruscia EM, Zhang PX, Barone C, Scholte BJ, Homer R, Krause DS, et al. Increased susceptibility of Cftr-/- mice to LPS-induced lung remodeling. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L711–9. doi: 10.1152/ajplung.00284.2015
106. Hasenberg A, Hasenberg M, Mann L, Neumann F, Borkenstein L, Stecher M, et al. Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat Methods. (2015) 12:445–52. doi: 10.1038/nmeth.3322
107. Rosen BH, Chanson M, Gawenis LR, Liu J, Sofoluwe A, Zoso A, et al. Animal and model systems for studying cystic fibrosis. J Cyst Fibros. (2018) 17:S28–34. doi: 10.1016/j.jcf.2017.09.001
108. Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, et al. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. (2008) 118:1571–7. doi: 10.1172/JCI34773
109. Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. (2008) 321:1837–41. doi: 10.1126/science.1163600
110. Sun X, Yan Z, Yi Y, Li Z, Lei D, Rogers CS, et al. Adeno-associated virus-targeted disruption of the CFTR gene in cloned ferrets. J Clin Invest. (2008) 118:1578–83. doi: 10.1172/JCI34599
111. Rogers CS, Abraham WM, Brogden KA, Engelhardt JF, Fisher JT, McCray PB Jr, et al. The porcine lung as a potential model for cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. (2008) 295:L240–63. doi: 10.1152/ajplung.90203.2008
112. Fisher JT, Zhang Y, Engelhardt JF. Comparative biology of cystic fibrosis animal models. Methods Mol Biol. (2011) 742:311–34. doi: 10.1007/978-1-61779-120-8_19
113. Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. (2010) 2:29ra31. doi: 10.1126/scitranslmed.3000928
114. Sathe M, Houwen R. Meconium ileus in Cystic Fibrosis. J Cyst Fibros. (2017) 16(Suppl. 2):S32–9. doi: 10.1016/j.jcf.2017.06.007
115. Fisher JT, Liu X, Yan Z, Luo M, Zhang Y, Zhou W, et al. Comparative processing and function of human and ferret cystic fibrosis transmembrane conductance regulator. J Biol Chem. (2012) 287:21673–85. doi: 10.1074/jbc.M111.336537
116. Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. (2010) 120:3149–60. doi: 10.1172/JCI43052
117. Sun X, Olivier AK, Liang B, Yi Y, Sui H, Evans TI, et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am J Respir Cell Mol Biol. (2014) 50:502–12. doi: 10.1165/rcmb.2013-0261OC
118. Keiser NW, Birket SE, Evans IA, Tyler SR, Crooke AK, Sun X, et al. Defective innate immunity and hyperinflammation in newborn cystic fibrosis transmembrane conductance regulator-knockout ferret lungs. Am J Respir Cell Mol Biol. (2015) 52:683–94. doi: 10.1165/rcmb.2014-0250OC
119. Sun X, Yi Y, Yan Z, Rosen BH, Liang B, Winter MC, et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci Transl Med. (2019) 11:eaau7531. doi: 10.1126/scitranslmed.aau7531
120. Mills PR, Davies RJ, Devalia JL. Airway epithelial cells, cytokines, and pollutants. Am J Respir Crit Care Med. (1999) 160(5 Pt 2):S38–43. doi: 10.1164/ajrccm.160.supplement_1.11
121. Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. (2003) 8:432–46. doi: 10.1046/j.1440-1843.2003.00493.x
122. Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. (2012) 18:684–92. doi: 10.1038/nm.2737
123. Weitnauer M, Mijosek V, Dalpke AH. Control of local immunity by airway epithelial cells. Mucosal Immunol. (2016) 9:287–98. doi: 10.1038/mi.2015.126
124. Mertens TCJ, Karmouty-Quintana H, Taube C, Hiemstra PS. Use of airway epithelial cell culture to unravel the pathogenesis and study treatment in obstructive airway diseases. Pulm Pharmacol Ther. (2017) 45:101–13. doi: 10.1016/j.pupt.2017.05.008
125. De Rose V, Molloy K, Gohy S, Pilette C, Greene CM. Airway epithelium dysfunction in cystic fibrosis and COPD. Mediators Inflamm. (2018) 2018:1309746. doi: 10.1155/2018/1309746
126. Randell SH, Fulcher ML, O'Neal W, Olsen JC. Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol. (2011) 742:285–310. doi: 10.1007/978-1-61779-120-8_18
127. Garratt LW, Sutanto EN, Foo CJ, Ling KM, Looi K, Kicic-Starcevich E, et al. Determinants of culture success in an airway epithelium sampling program of young children with cystic fibrosis. Exp Lung Res. (2014) 40:447–59. doi: 10.3109/01902148.2014.946631
128. Martinovich KM, Iosifidis T, Buckley AG, Looi K, Ling KM, Sutanto EN, et al. Conditionally reprogrammed primary airway epithelial cells maintain morphology, lineage and disease specific functional characteristics. Sci Rep. (2017) 7:17971. doi: 10.1038/s41598-017-17952-4
129. Brewington JJ, Filbrandt ET, LaRosa FJ III, Moncivaiz JD, Ostmann AJ, Strecker LM, et al. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight. (2018) 3:99385. doi: 10.1172/jci.insight.99385
130. Awatade NT, Wong SL, Hewson CK, Fawcett LK, Kicic A, Jaffe A, et al. Human primary epithelial cell models: promising tools in the era of cystic fibrosis personalized medicine. Front Pharmacol. (2018) 9:1429. doi: 10.3389/fphar.2018.01429
131. Castellani S, Di Gioia S, di Toma L, Conese M. Human cellular models for the investigation of lung inflammation and mucus production in cystic fibrosis. Anal Cell Pathol. (2018) 2018:3839803. doi: 10.1155/2018/3839803
132. Brewington JJ, Filbrandt ET, LaRosa FJ III, Moncivaiz JD, Ostmann AJ, Strecker LM, et al. Generation of human nasal epithelial cell spheroids for individualized cystic fibrosis transmembrane conductance regulator study. J Vis Exp. (2018) 134. doi: 10.3791/57492
133. Guimbellot JS, Leach JM, Chaudhry IG, Quinney NL, Boyles SE, Chua M, et al. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight. (2017) 2:95734. doi: 10.1172/jci.insight.95734
134. Brewington JJ, Filbrandt ET, LaRosa FJ III, Ostmann AJ, Strecker LM, Szczesniak RD, et al. Detection of CFTR function and modulation in primary human nasal cell spheroids. J Cyst Fibros. (2018) 17:26–33. doi: 10.1016/j.jcf.2017.06.010
135. Machen TE. Innate immune response in CF airway epithelia: hyperinflammatory?. Am J Physiol Cell Physiol. (2006) 291:C218–30. doi: 10.1152/ajpcell.00605.2005
136. Ratner D, Mueller C. Immune responses in cystic fibrosis: are they intrinsically defective?. Am J Respir Cell Mol Biol. (2012) 46:715–22. doi: 10.1165/rcmb.2011-0399RT
137. Murphy SV, Ribeiro CMP. Cystic fibrosis inflammation: hyperinflammatory, hypoinflammatory, or both?. Am J Respir Cell Mol Biol. (2019) 61:273–4. doi: 10.1165/rcmb.2019-0107ED
138. Becker MN, Sauer MS, Muhlebach MS, Hirsh AJ, Wu Q, Verghese MW, et al. Cytokine secretion by cystic fibrosis airway epithelial cells. Am J Respir Crit Care Med. (2004) 169:645–53. doi: 10.1164/rccm.200207-765OC
139. Wiszniewski L, Jornot L, Dudez T, Pagano A, Rochat T, Lacroix JS, et al. Long-term cultures of polarized airway epithelial cells from patients with cystic fibrosis. Am J Respir Cell Mol Biol. (2006) 34:39–48. doi: 10.1165/rcmb.2005-0161OC
140. Schogler A, Blank F, Brugger M, Beyeler S, Tschanz SA, Regamey N, et al. Characterization of pediatric cystic fibrosis airway epithelial cell cultures at the air-liquid interface obtained by non-invasive nasal cytology brush sampling. Respir Res. (2017) 18:215. doi: 10.1186/s12931-017-0706-7
141. Sajjan U, Keshavjee S, Forstner J. Responses of well-differentiated airway epithelial cell cultures from healthy donors and patients with cystic fibrosis to burkholderia cenocepacia infection. Infect Immun. (2004) 72:4188–99. doi: 10.1128/IAI.72.7.4188-4199.2004
142. Carrabino S, Carpani D, Livraghi A, Di Cicco M, Costantini D, Copreni E, et al. Dysregulated interleukin-8 secretion and NF-kappaB activity in human cystic fibrosis nasal epithelial cells. J Cyst Fibros. (2006) 5:113–9. doi: 10.1016/j.jcf.2005.12.003
143. Conese M, Copreni E, Di Gioia S, De Rinaldis P, Fumarulo R. Neutrophil recruitment and airway epithelial cell involvement in chronic cystic fibrosis lung disease. J Cyst Fibros. (2003) 2:129–35. doi: 10.1016/S1569-1993(03)00063-8
144. Yamamoto K, Ahyi AN, Pepper-Cunningham ZA, Ferrari JD, Wilson AA, Jones MR, et al. Roles of lung epithelium in neutrophil recruitment during pneumococcal pneumonia. Am J Respir Cell Mol Biol. (2014) 50:253–62. doi: 10.1165/rcmb.2013-0114OC
145. Kim ND, Luster AD. The role of tissue resident cells in neutrophil recruitment. Trends Immunol. (2015) 36:547–55. doi: 10.1016/j.it.2015.07.007
146. Parkos CA. Neutrophil-epithelial interactions: a double-edged sword. Am J Pathol. (2016) 186:1404–16. doi: 10.1016/j.ajpath.2016.02.001
147. Brazil JC, Parkos CA. Pathobiology of neutrophil-epithelial interactions. Immunol Rev. (2016) 273:94–111. doi: 10.1111/imr.12446
148. Aldallal N, McNaughton EE, Manzel LJ, Richards AM, Zabner J, Ferkol TW, et al. Inflammatory response in airway epithelial cells isolated from patients with cystic fibrosis. Am J Respir Crit Care Med. (2002) 166:1248–56. doi: 10.1164/rccm.200206-627OC
149. Joseph T, Look D, Ferkol T. NF-kappaB activation and sustained IL-8 gene expression in primary cultures of cystic fibrosis airway epithelial cells stimulated with Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L471–9. doi: 10.1152/ajplung.00066.2004
150. Balloy V, Varet H, Dillies MA, Proux C, Jagla B, Coppee JY, et al. Normal and cystic fibrosis human bronchial epithelial cells infected with Pseudomonas aeruginosa exhibit distinct gene activation patterns. PLoS ONE. (2015) 10:e0140979. doi: 10.1371/journal.pone.0140979
151. Balloy V, Koshy R, Perra L, Corvol H, Chignard M, Guillot L, et al. Bronchial epithelial cells from cystic fibrosis patients express a specific long non-coding RNA signature upon Pseudomonas aeruginosa infection. Front Cell Infect Microbiol. (2017) 7:218. doi: 10.3389/fcimb.2017.00218
152. Zheng S, De BP, Choudhary S, Comhair SA, Goggans T, Slee R, et al. Impaired innate host defense causes susceptibility to respiratory virus infections in cystic fibrosis. Immunity. (2003) 18:619–30. doi: 10.1016/S1074-7613(03)00114-6
153. Sutanto EN, Kicic A, Foo CJ, Stevens PT, Mullane D, Knight DA, et al. Innate inflammatory responses of pediatric cystic fibrosis airway epithelial cells: effects of nonviral and viral stimulation. Am J Respir Cell Mol Biol. (2011) 44:761–7. doi: 10.1165/rcmb.2010-0368OC
154. Kieninger E, Singer F, Tapparel C, Alves MP, Latzin P, Tan HL, et al. High rhinovirus burden in lower airways of children with cystic fibrosis. Chest. (2013) 143:782–90. doi: 10.1378/chest.12-0954
155. Kieninger E, Vareille M, Kopf BS, Blank F, Alves MP, Gisler FM, et al. Lack of an exaggerated inflammatory response on virus infection in cystic fibrosis. Eur Respir J. (2012) 39:297–304. doi: 10.1183/09031936.00054511
156. Dauletbaev N, Das M, Cammisano M, Chen H, Singh S, Kooi C, et al. Rhinovirus load is high despite preserved interferon-beta response in cystic fibrosis bronchial epithelial cells. PLoS ONE. (2015) 10:e0143129. doi: 10.1371/journal.pone.0143129
157. King J, Brunel SF, Warris A. Aspergillus infections in cystic fibrosis. J. Infect. (2016) (72 Suppl):S50–5. doi: 10.1016/j.jinf.2016.04.022
158. Breuer O, Schultz A, Turkovic L, de Klerk N, Keil AD, Brennan S, et al. Changing prevalence of lower airway infections in young children with cystic fibrosis. Am J Respir Crit Care Med. (2019) 200:590–9. doi: 10.1164/rccm.201810-1919OC
159. Harun SN, Wainwright CE, Grimwood K, Hennig S, Australasian Cystic Fibrosis Bronchoalveolar Lavage study G. Aspergillus and progression of lung disease in children with cystic fibrosis. Thorax. (2019) 74:125–131. doi: 10.1136/thoraxjnl-2018-211550
160. Reihill JA, Moore JE, Elborn JS, Ennis M. Effect of Aspergillus fumigatus and Candida albicans on pro-inflammatory response in cystic fibrosis epithelium. J Cyst Fibros. (2011) 10:401–6. doi: 10.1016/j.jcf.2011.06.006
161. Chaudhary N, Datta K, Askin FB, Staab JF, Marr KA. Cystic fibrosis transmembrane conductance regulator regulates epithelial cell response to Aspergillus and resultant pulmonary inflammation. Am J Respir Crit Care Med. (2012) 185:301–10. doi: 10.1164/rccm.201106-1027OC
162. Schogler A, Stokes AB, Casaulta C, Regamey N, Edwards MR, Johnston SL, et al. Interferon response of the cystic fibrosis bronchial epithelium to major and minor group rhinovirus infection. J Cyst Fibros. (2016) 15:332–9. doi: 10.1016/j.jcf.2015.10.013
163. Nash S, Stafford J, Madara JL. Effects of polymorphonuclear leukocyte transmigration on the barrier function of cultured intestinal epithelial monolayers. J Clin Invest. (1987) 80:1104–13. doi: 10.1172/JCI113167
164. Madara JL, Colgan S, Nusrat A, Delp C, Parkos C. A simple approach to measurement of electrical parameters of cultured epithelial monolayers: use in assessing neutrophil-epithelial interactions. J Tissue Culture Methods. (1992) 14:209–15. doi: 10.1007/BF01409013
165. Parkos CA, Colgan SP, Delp C, Arnaout MA, Madara JL. Neutrophil migration across a cultured epithelial monolayer elicits a biphasic resistance response representing sequential effects on transcellular and paracellular pathways. J Cell Biol. (1992) 117:757–64. doi: 10.1083/jcb.117.4.757
166. McCormick BA, Colgan SP, Delp-Archer C, Miller SI, Madara JL. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol. (1993) 123:895–907. doi: 10.1083/jcb.123.4.895
167. Kidney JC, Proud D. Neutrophil transmigration across human airway epithelial monolayers: mechanisms and dependence on electrical resistance. Am J Respir Cell Mol Biol. (2000) 23:389–95. doi: 10.1165/ajrcmb.23.3.4068
168. Zemans RL, Briones N, Suzuki T, Downey GP. Neutrophil migration across cultured lung epithelium induces modulation of the WNT-beta-catenin pathway. Am J Respir Crit Care Med. (2009) 179:A5664. doi: 10.1164/ajrccm-conference.2009.179.1_MeetingAbstracts.A5664
169. Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, et al. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci USA. (2011) 108:15990–5. doi: 10.1073/pnas.1110144108
170. Hurley BP, Siccardi D, Mrsny RJ, McCormick BA. Polymorphonuclear cell transmigration induced by Pseudomonas aeruginosa requires the eicosanoid hepoxilin A3. J Immunol. (2004) 173:5712–20. doi: 10.4049/jimmunol.173.9.5712
171. Kusek ME, Pazos MA, Pirzai W, Hurley BP. In vitro coculture assay to assess pathogen induced neutrophil trans-epithelial migration. J Vis Exp. (2014) 83:e50823. doi: 10.3791/50823
172. Deng Y, Herbert JA, Smith CM, Smyth RL. An in vitro transepithelial migration assay to evaluate the role of neutrophils in respiratory syncytial virus (RSV) induced epithelial damage. Sci Rep. (2018) 8:6777. doi: 10.1038/s41598-018-25167-4
173. Neudecker V, Brodsky KS, Clambey ET, Schmidt EP, Packard TA, Davenport B, et al. Neutrophil transfer of miR-223 to lung epithelial cells dampens acute lung injury in mice. Sci Transl Med. (2017) 9:eaah5360. doi: 10.1126/scitranslmed.aah5360
174. Genschmer KR, Russell DW, Lal C, Szul T, Bratcher PE, Noerager BD, et al. Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell. (2019) 176:113–126 e15. doi: 10.1016/j.cell.2018.12.002
175. Yonker LM, Mou H, Chu KK, Pazos MA, Leung H, Cui D, et al. Development of a primary human co-culture model of inflamed airway mucosa. Sci Rep. (2017) 7:8182. doi: 10.1038/s41598-017-08567-w
176. Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. (2015) 373:1783–4. doi: 10.1056/NEJMoa1409547
177. Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. (2017) 377:2013–23. doi: 10.1056/NEJMoa1709846
178. Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, et al. Tezacaftor/Ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. (2018) 197:214–24. doi: 10.1164/rccm.201704-0717OC
179. Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. (2017) 5:557–67. doi: 10.1016/S2213-2600(17)30215-1
180. Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del Allele. N Engl J Med. (2019) 381:1809–19. doi: 10.1056/NEJMoa1908639
181. Heltshe SL, Mayer-Hamblett N, Burns JL, Khan U, Baines A, Ramsey BW, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis. (2015) 60:703–12. doi: 10.1093/cid/ciu944
Keywords: cystic fibrosis, neutrophil, inflammation, infection, model systems
Citation: Laucirica DR, Garratt LW and Kicic A (2020) Progress in Model Systems of Cystic Fibrosis Mucosal Inflammation to Understand Aberrant Neutrophil Activity. Front. Immunol. 11:595. doi: 10.3389/fimmu.2020.00595
Received: 02 December 2019; Accepted: 13 March 2020;
Published: 07 April 2020.
Edited by:
Christian Herr, Saarland University Hospital, GermanyReviewed by:
Jean-Michel Sallenave, INSERM U1152 Physiopathologie et Epidémiologie des Maladies Respiratoires, FranceMassimo Conese, University of Foggia, Italy
Copyright © 2020 Laucirica, Garratt and Kicic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luke W. Garratt, bHVrZS5nYXJyYXR0QHRlbGV0aG9ua2lkcy5vcmcuYXU=
†These authors share senior authorship