 Rowland S. Osii
Rowland S. Osii Thomas D. Otto1
Thomas D. Otto1 Paul Garside
Paul Garside Francis M. Ndungu
Francis M. Ndungu James M. Brewer
James M. Brewer- 1Institute of Infection, Immunity & Inflammation, University of Glasgow, Glasgow, United Kingdom
- 2KEMRI-CGMRC/Wellcome Trust Research Programme, Kilifi, Kenya
- 3Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
Malaria is caused by apicomplexan parasites of the genus Plasmodium. While infection continues to pose a risk for the majority of the global population, the burden of disease mainly resides in Sub-Saharan Africa. Although immunity develops against disease, this requires years of persistent exposure and is not associated with protection against infection. Repeat infections occur due to the parasite's ability to disrupt or evade the host immune responses. However, despite many years of study, the mechanisms of this disruption remain unclear. Previous studies have demonstrated a parasite-induced failure in dendritic cell (DCs) function affecting the generation of helper T cell responses. These T cells fail to help B cell responses, reducing the production of antibodies that are necessary to control malaria infection. This review focuses on our current understanding of the effect of Plasmodium parasite on DC function, DC-T cell interaction, and T cell activation. A better understanding of how parasites disrupt DC-T cell interactions will lead to new targets and approaches to reinstate adaptive immune responses and enhance parasite immunity.
Introduction
Malaria is caused by the Plasmodium parasite, which affects majority of the world's population. Annually, the disease causes ~228 million cases, resulting in 405,000 deaths. Africa accounts for about 93% of the reported cases and 94% of reported mortality cases occurring in children under the age of 5 (1). Residents in malaria endemic areas are susceptible to repeat malaria infection, with each infection resulting in modification of the hosts immune system. As well as affecting the host response to further infection (2), endemic malaria is also associated with weakened immunity to bystander infections and vaccines (3). Malaria infection has been shown to alter the phenotype and function of dendritic cells (4, 5) B cells (6, 7) and T cells (7–10) causing a disruption in the host immune response.
Plasmodium Life Cycle
Plasmodium has a complex life cycle that occurs in two hosts; the female Anopheles mosquito (sexual reproductive stage) and a vertebrate host (asexual development stage). The latter begins when an infectious female Anopheles mosquito probes the dermis of a mammalian host as it takes a blood meal, releasing a highly motile form of the parasite, sporozoites, from its saliva (Figure 1A) (11, 12). Not all sporozoites manage to reach the blood vessel and those that remain in the dermis are either destroyed or drained into the lymphatics where the host's immune system eliminates them (13, 14). Those that manage to enter the bloodstream circulate and enter the liver through a process known as traversal, to gain access to a suitable hepatocyte (15, 16). Once inside a suitable hepatocyte, the sporozoite forms a parasitophorous vacuole (PV) and undergoes pre-erythrocytic schizogony, forming merozoites that accumulate within the parasitophorous vacuole and bud off the hepatocyte in structures called merosomes, clearing the liver of parasites (Figure 1B). The merosomes enter the bloodstream, releasing the encapsulated merozoites to infect red blood cells (RBCs) (17–19).
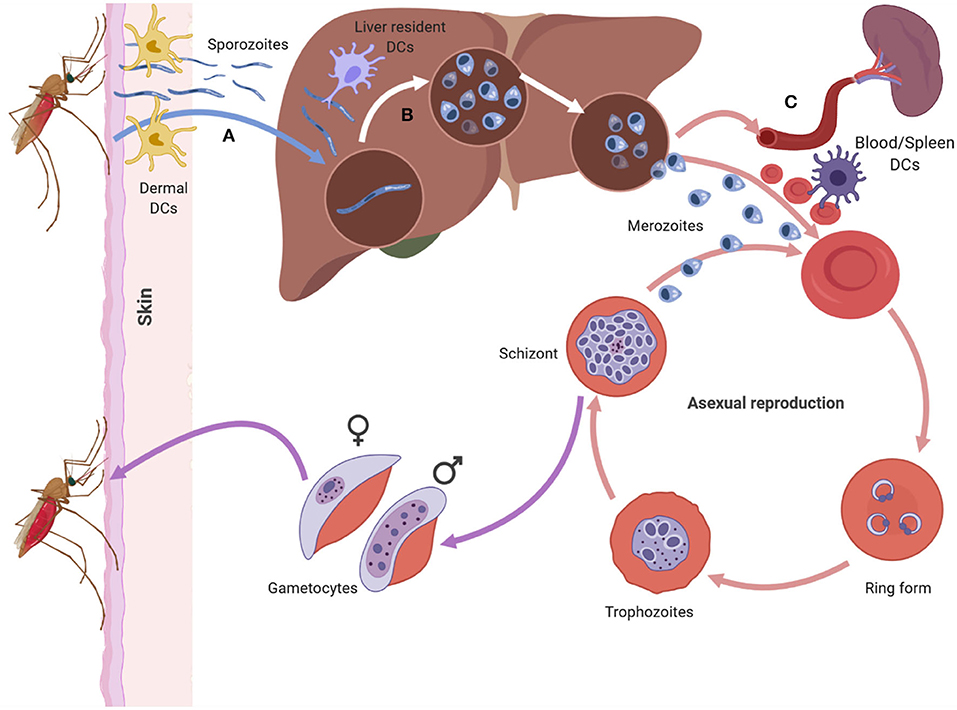
Figure 1. The asexual life cycle of Plasmodium parasite begins when an infected mosquito injects highly motile sporozoites into the skin of the host. The sporozorites enters the bloodstream and migrates to the liver, where it traverses multiple hepatocytes before infecting one. Inside the hepatocyte the sporozoite undergoes pre-erythrocytic schizogony forming merozoites that accumulate and bud off the hepatocyte in structures called merosomes. Merosomes enter the bloodstream and release merozoites which invade RBC, initiating the erythrocytic stage of asexual development. At this stage the parasite develops inside the RBC in distinct forms namely the ring, trophozoite, and schizont form. The schizont, lyses releasing merozoites into the blood stream which reinvade RBCs starting a fresh round of asexual development. After rounds of erythrocytic schizogony some of the asexual parasites develop into gametocytes and are taken up by a mosquito during a blood meal. Dendritic cells can interact with sporozoites in the dermis (A), the liver (B) and the blood and spleen (C). The DCs at each site encounter the parasite in its different forms (Figure was created using BioRender).
In the blood, the free merozoites attach to, and subsequently invade the RBC, initiating the erythrocytic stage of the parasite life cycle. Once inside the RBC, the merozoite matures in three morphologically distinct stages, namely the ring, trophozoite, and schizont stages. During the maturation stages the RBC undergoes a number of structural and functional changes that alter the architecture of the RBC membrane (Figure 1C) (20). Key amongst the structural changes is the expression of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1), a vital parasite protein that is central to P. falciparum pathogenesis (21–23). PfEMP1 is expressed on the surface of parasite infected RBCs (iRBC) and enables iRBCs to sequester and cytoadhere to vascular endothelium, preventing their destruction in the spleen. Apart from the structural changes that occur to the RBC, the parasite also undergoes nuclear division producing merozoites that fill the PV (the schizont stage). The merozoites egress from the iRBC and invade other RBCs initiating another cycle for parasite replication.
After rounds of schizogony, some P. falciparum trophozoites commit to sexual development and form gametocytes. The gametocytes undergo five stages of maturation while being sequestered in the bone marrow. Only stage five gametocytes re-enter circulation and are taken up by a mosquito during a blood meal (24).
Interaction between DCs and Plasmodium parasite occurs at various points during the life cycle of the parasite in a human host (Figure 1). The parasite encounters DCs in the skin (Figure 1A) (13, 25), the liver (Figure 1B) (26, 27), and the blood and spleen (Figure 1C) (4). Tissue resident DCs in each of the sites can phagocytose parasite components and initiate specific immune responses to the parasite.
Dendritic Cells
DCs are mononuclear phagocytic cells that are found in the blood, lymphoid organs and all tissues. They are the most effective professional antigen presenting cells in the body due to their ability to capture, process and present antigen on either major histocompatibility complex (MHC) class I or MCH class II molecules and activate naive CD8 or CD4 T cells (28, 29). DCs are central in initiating and regulating adaptive immune responses and act as a bridge between the innate and adaptive arms of the immune system. DCs differentiate from hematopoietic stem cells (HSC) (30) in the bone marrow to immature DCs, which circulate in blood and home to various peripheral tissues. Immature DCs recognize a range of danger signals such as pathogen-associated molecular patterns (PAMPS) which are found on pathogens and damage associated molecular patterns (DAMPS) which are released by injured host cells (31), through a number of pathogen recognition receptors (PRRs) (32, 33). Ligation of PRRs initiates DC phagocytosis, resulting in ingestion of the invading pathogen and initiation of DC maturation and migration into the lymph node where they present antigens to naive T cells (34). The maturation process results in increased expression of MHC surface molecule coupled with pathogen antigens and costimulatory molecules (CD80, CD86, and CD40), which are key in proliferation and differentiation of naive T cells into effector cells (35). DCs also secrete cytokines and chemokine that attract other immune cells to sights of infection/injury and influence the outcome of T and B cells responses (36).
DCs are lineage negative cells [that is they are defined by the exclusion of T cells (CD3), B Cells (CD19, CD20) natural killer cells (CD56), monocytes (CD14, CD16) and progenitor cells (CD34)] and express MHC class II (HLA-DR) and are broadly classified into either plasmacytoid DCs (pDCs) or conventional DCs (cDCs). In humans, pDCs are characterized by expression of CD123, CD303 (BDCA-2) and CD304 (37) and are known to produce large amounts of type I interferon in response to viruses (38). This is enabled by the high expression levels of toll-like receptor 7 (TLR7) and TLR9, which recognize nucleic acids from viruses, bacteria, and dead cells (39, 40). cDCs specialize in priming and presenting antigen to T cells. They can be further classified into cDC1 and cDC2. cDC1 express BDCA-3/CD141, CLEC9A, and XCR1 and have enhanced ability to cross present antigen (41) to CD8 T cells. cDC2 express BDCA-1/CD1c and have a wide variety of pattern recognition receptors (PRR's) and a good capacity to stimulate naive CD4 T cells but they have a poor ability to cross-present antigens to CD8 T cells compared with cDC1 (37, 40).
DCs are central in any immune response as they sense pathogens and initiate immune responses and are present at various sites during the life cycle of the Plasmodium parasite. As discussed later, the parasite's numerous immune evasion mechanisms interfere with DC function, thus altering downstream immune effector functions and the course of the disease.
T Cells
T cells develop in the thymus from the common lymphoid progenitors which originate from bone marrow derived hematopoietic stem cells (42). After development and maturation, naiveT cells exit the thymus and enter circulation expressing either CD4 or CD8 and an antigen-recognizing T cell receptor (TCR) on their surface. The naive T cells home to secondary lymphoid organs (SLO) where they await a signal from DCs to become activated.
CD8 T cells
Naive CD8 T Cells are activated by recognition of foreign or neoantigens presented by MHC class I molecules on DCs in the secondary lymphoid organs. Additional co-stimulatory signals and cytokines from DCs and/or CD4 T cells help in differentiation and clonal expansion of the T cells (43–46). The activated effector CD8 T cells migrate from the secondary lymphoid organs into circulation and identify their target cells which express cognate antigens on the cell surface bound to MHC class I. MHC class I is expressed on all nucleated cells except red blood cells. The target cells are killed by effector CD8 T cells through cell contact dependent cytolysis by releasing granzyme B and perforin (47–49). Perforin creates pores on the plasma membrane of the target cell; the pores allow granzyme B to enter the target cell and initiate apoptosis resulting in killing of infected cells. After clearing the invading pathogen, antigen specific effector CD8 T cells die off and a small number differentiate into memory CD8 T cells (45, 50).
Antigen specific CD8 T cells have been observed in the peripheral blood of residents from a malaria endemic area (51) and after vaccination of malaria naive individuals with irradiated sporozoites (52). In experimental mouse models of malaria, CD8 T cells specific for sporozoites antigens, liver stage antigens, and blood stage antigens were observed when mice were challenged with radiation attenuated sporozoites (53). It is believed that the priming of CD8 T cells against the pre-erythrocytic stages of Plasmodium occurs in the skin draining lymph nodes when sporozoites are injected into the skin by an infected mosquito (14, 54). These CD8 may offer protection against subsequent Plasmodium infections as incubation time in the liver offers a short window of opportunity for the CD8 T to mount an effective response.
CD4 T cells
CD4 T cells, on the other hand, recognize antigens presented by MHC class II molecules, which are present on antigen presenting cells such as B cells, macrophages, and dendritic cells. CD4 T cells generally provide help to B cells in the germinal center enabling class switching and production of high-affinity antibodies (55). They also aid in CD8 T cell activation by licensing DCs (56–58) or directly signaling CD8 T cells via CD40 (59). They also secrete cytokines such interferon gamma (IFNγ), C-X-C motif ligand 9 (CXCL9), CXCL10 (60) interleukin-2 (IL-2) (61–63), and IL-21 (64) that are key in shaping immune responses. The diverse range of CD4 T cell functions are handled by distinct subsets of cells. The cytokine milieu in the microenvironment during CD4 T cell activation dictates the specific cytokine signaling networks and transcription factor activated for the differentiation of naive CD4 T cells into T cell subsets. The cytokines involved in CD4 T cell differentiation are produced by DCs and other innate immune cells, driving the cells to differentiate into either T-helper 1 (Th1), T-helper 2 (Th2), T-helper 17 (Th17), follicular helper T cell (Tfh), induced T-regulatory (iTreg), or the regulatory type 1 cells (Tr1).
Tfh cells have been a recent focus of interest in malaria immunology. Tfh cells express C-X-C motif receptor 5 (CXCR5) on their surface and are vital in the development of humoral immunity (55). Differentiation of CD4 T cells to Tfh is a multistep step process that first begins with DC interacting with a naive CD4 T cells in the T cells zone (Figure 3). This interaction results in the formation of pre-Tfh cells expressing CXCR5 that migrate to the T-B cell border of the SLO (65). At the T-B cell border and interfollicular zone, pre-Tfh interact with antigen specific B cells to initiate the B cell dependent phase of Tfh differentiation, which is characterized by upregulation of transcription factor B cell lymphoma 6 (Bcl-6) (66) and commits the Tfh lineage. After events at the T-B cell border, the Tfh migrates into the follicle and interacts with B cells forming germinal centers, where B cells undergo affinity maturation and heavy chain class switching, resulting in the production of high-affinity antibodies with enhanced effector functions (67). Tfh differentiation involves a number of cytokines such as IL-6, IL-21 (68), IL-12 (69), IL-27 (70), and TGF-β (71). These cytokines initiate signal transducer and activator of transcription 1 (STAT1), STAT3 (72) and STAT4 (73). The STATs upregulate the transcription factor B cell lymphoma 6 (Bcl-6), the master transcription factor in Tfh differentiation. Apart from cytokines, other signals required during differentiation of Tfh cells include the inducible costimulator (ICOS)- inducible costimulatory ligand (ICOSL) signaling (74, 75) and CD40-CD40L signaling.
CD4 Tfh cells are essential for promoting antibody response that aid in resolving malaria infection (76, 77). In malaria infected humans and mice, Tfh cells adopt a Th1 like phenotype that expresses Tbet+ PD-1+, CXCR5+, CXCR3+, and secretes IFNγ (77, 78). This Tfh phenotype does not provide adequate help to B cells resulting in suboptimal antibody responses. Dysfunctional DCs that are induced by malaria may play a role in initiating this Th1-like phenotype that skews humoral response (Figures 2D,E).
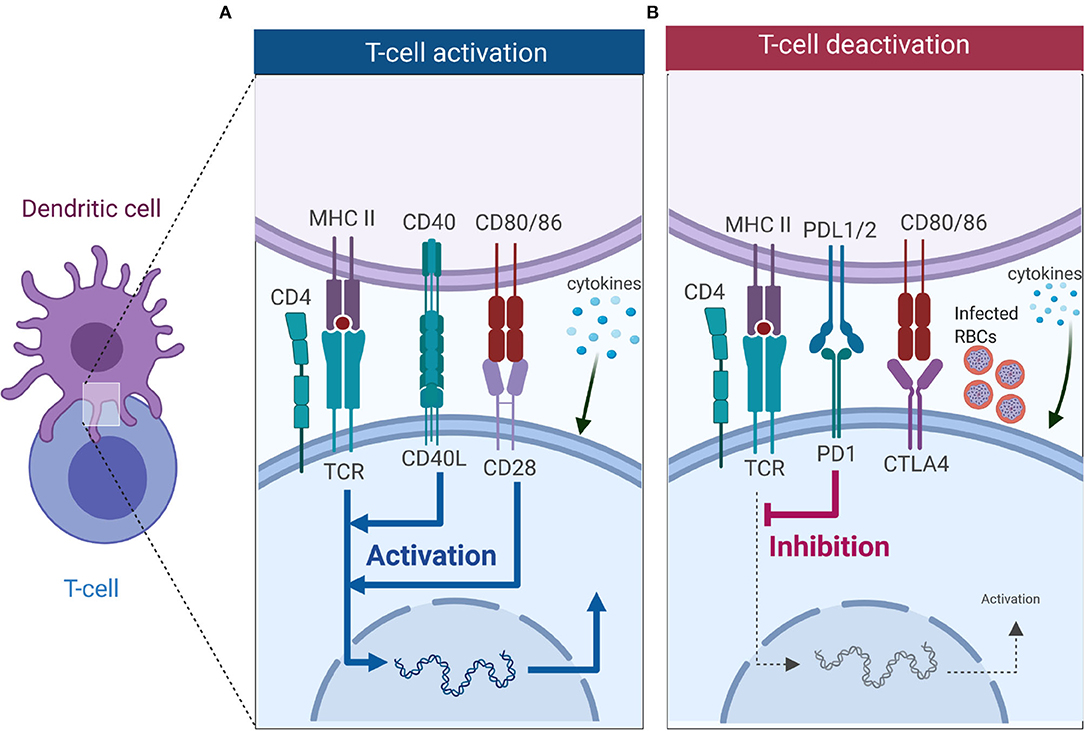
Figure 2. T cell activation or deactivation requires three signals from DCs. (A) T cell activation requires signal 1 in the form of TCR interacting with antigen-MHC complex which is key in innating downward signaling through ITAMs. Interaction of co-stimulatory molecules (interaction of CD40-CD40L, and CD80/86-CD28) form part of signal 2 as they work in tandem with TCR-antigen-MHC complex to enhance TCR signaling and initiate T cell proliferation. Signal 3 comes from DCs in the form of cytokines, and in CD4 T cells it is key in dictating which subset it will differentiate into. (B) Co-inhibitory molecules also form part of the second signal but unlike co-stimulatory molecules they inhibit TCR signaling, thus dampening immune responses. Interaction of PD-1 with PD-L1 inhibits T cell activation, while CTLA4 competes for binding with CD28 to CD80/86 and successful binding of CTLA4 to CD80/86 nullifies CD28-CD80/86 activation signal. Malaria has been shown to induce expression of PD-1, LAG3 and CTLA-4 on CD4 T cells, and this inhibits the activation signal from DCs (Figure was created using BioRender).
The impact of Plasmodium on DC-T cell Interactions
Activation of T cells requires interaction with DCs, which provide three key signals (Figure 2). Signal 1 occurs when T cells recognize cognate peptide antigen presented on either MHC I or MHC II on the surface of DCs via their T cell receptor (TCR). MHC-TCR interactions trigger activation of the T cells and initiates downwards signaling through immunoreceptor tyrosine-based activation motifs (ITAMs) (79). Besides TCR-antigen-MHC complex, a second signal, the costimulatory signal, is required to initiate and sustain T cell activation and proliferation. Co-inhibitory molecules (immune checkpoints) also form part of the second signal, but they downregulate immune responses (Figure 2B) (80, 81). Key costimulatory molecules involved in T cell activation include CD28 (binds to CD80/86 on DCs), ICOS (binds to ICOSL on DC), OX40 (binds to OX40L on DCs), and CD40L (binds to CD40 on DCs), are key in T cell activation, differentiation and survival (Figure 2A). These costimulatory signals work in synergy with the TCR-antigen-MHC complex to enhance the activation of T cells. Co-inhibitory molecules such as cytotoxic T-lymphocyte-associated protein 4 [(CTLA-4), competes for binding to CD80/86 with CD28 on DC], and programmed cell death-1 [(PD-1), binds to PD-1L] work to suppress the activation signal from TCR-antigen-MHC complex (Figure 2B). Once the T cell has received TCR-antigen-MHC complex signaling together with adequate co-stimulation, it receives a third signal in the form of cytokines that are secreted by DCs. As mentioned above, cytokines are important in deciding the fate of CD4 T cell differentiation toward a particular subset. Subsets of CD4 T cells include Th1 type (CD4 T cells exposed to the cytokine IL-12), Th2 (IL-4), Th-17 (IL-6, IL-23), Tfh (IL6, IL21), and iTreg (TGF-β) (Figure 2).
Tfh differentiation is a multistep process that requires signal 1 in the form of antigen presented on MHC II by DCs (Figure 3). This interaction occurs at the T cell zone and involves the costimulatory molecules CD80, CD86, and inducible costimulatory ligand (ICOSL) on DC that interact with CD28 and ICOS to generate signal 2 in T cells. The CD28-CD80/86 interaction results in the upregulation of ICOS on T cells that interacts with ICOSL on DCs. The cytokine (signal 3) produced by DCs that helps in the initial process of Tfh differentiation is IL-12 (82). A combination of CD28-mediated signaling on T cells and IL-12 is adequate to upregulate the expression of Bcl-6, IL-12 also induces IL-21 production in T cells, which acts in an autocrine manner to ensure growth and survival of pre-Tfh. Bcl6 expression upregulates CXCR5 expression allowing the pre-Tfh cells to migrate to the T cell-B cell zone (83). At this zone, the Tfh cell interacts with B via ICOS-ICOSL committing the cell to the Tfh lineage and further upregulating CXCR5 and SAP (67). The CXCR5 and SAP expressing Tfh cells then move into the B cell follicle and form stable, long-lasting interactions with B cells forming germinal center where Tfh cells aid in class switching and generation of long-lived plasma cells that secrete high-affinity antibodies. Germinal center Tfh cells are also involved in the formation of long-lived plasma cells and memory B cells (84, 85).
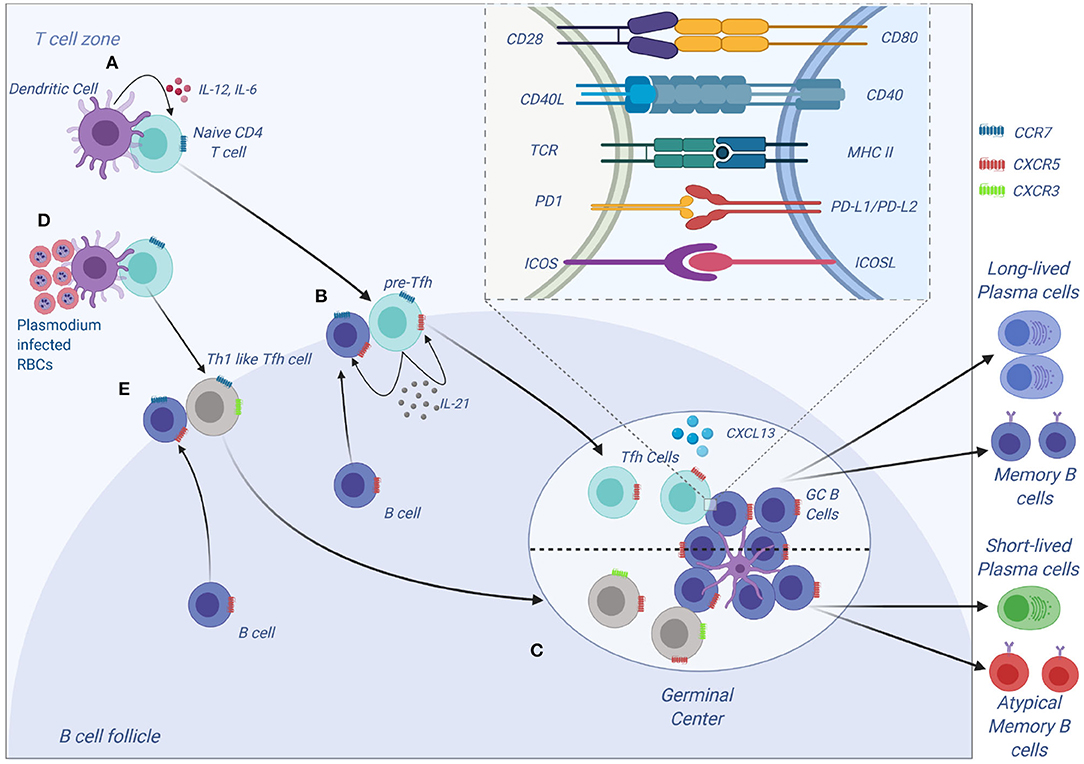
Figure 3. Tfh development and function. (A) Tfh differentiation begins when an activated DC primes a naïve CD4 T cells. Interaction between ICOS on CD4 and ICOS-L on DCs results in upregulation of Bcl-6 and CXCR5 on CD4 T cells. IL-12 and IL-6, help maintain expression of Bcl-6, and IL-12 induces upregulation of IL-21 which is essential for survival of the pre-Tfh cell. Initial CD4 T cell activation also results in upregulation of PD-1. The Bcl-6+CXCR5+ PD-1+ pre–Tfh cell then migrates to the T cell–B cell border. At the same time, antigen-activated B cells upregulate CCR7 and migrates from the B cell follicle to the T cell–B cell border. (B) At the T cell–B cell border, the pre-Tfh cell engages with the B cell. Interaction between antigen-MHC on B cells with TCR on pre-Tfh, and ICOSL (B cell) with ICOS (pre-Tfh cell), fully commits the cell to Tfh lineage. This results in upregulation and maintenance of Bcl-6, SAP, and CXCR5, while downregulating CCR7 on T cells. (C) The Tfh and B cells move deeper into the B cell follicle forming GCs. Tfh cells in the GCs promotes B-cell maturation, class switching and affinity maturation via the cytokines, IL-21 and IL-4, and the molecules, CD40L and PD-1. Both Tfh and GC B-cell are necessary for generation of B-cell memory and long-lived plasma cells. (D,E) Plasmodium-induces polarization of T follicular helper (Tfh) cells to Th1 like phenotytpe that expresses Tbet PD-1, CXCR5, CXCR3 and contributes to the inefficient acquisition of humoral immunity to malaria. Malaria infection in mice and humans induces secretion of Th1-polarizing cytokine that drive the activation of Th1-like Tfh cells that exhibit impaired B cell helper function, thus contributing to germinal center dysfunction and suboptimal antibody responses (Figure was created using BioRender).
P. falciparum Immune Evasion and Suppression of Immunity
P. falciparum is equipped with multiple mechanisms which it uses to evade the host's immune system. These mechanisms include antigenic variation of surface antigens (VSA) expressed on iRBCs such as PfEMP1 which is encoded by the var genes (21), sub-telomeric variable open reading frame (STEVOR) encoded by the stevor genes (86, 87) and repetitive interspersed repeats (RIFIN) encoded by the rif genes (88, 89). Antigenic variation of VSAs normally occurs when the parasite is under intense immune pressure from the host in order to avoid recognition by various immune cells (90, 91). The expression of different VSAs on iRBCs allows the parasite to establish new infections (92). VSAs are key in sequestration and cytoadherence of maturing parasites (trophozoite and schizonts) and rosetting (93, 94). Merozoite surface protein (MSP) polymorphism (95–97) and complement evasion by surface proteins PfMSP3.1 (98), Pf92 (99), and PfGAP50 (100) expressed on merozoites and gametes are other mechanisms used by the parasite to escape elimination by the immune system.
Apart from immune evasion, ongoing Plasmodium infections have been shown to reduce immunogenicity of vaccines in children. Antibody responses to Salmonella typhi and tetanus vaccines were greatly reduced in malaria infected children compared to healthy control and children with other acute illnesses (3). Adults with previous exposure to P. falciparum, showed no response to malaria antigen, regardless of disease severity, and reduced response to non-specific antigens (2). Infection of influenza-immune mice with P. chabaudi resulted in a decrease in influenza specific antibodies and plasma cells resulting in a loss of protective immunity against influenza (101), which recovered several weeks after parasite clearance. This indicates that malaria infections somehow suppress immune function by interfering with the development of adaptive immunity. Ongoing malaria infection reduces immunogenicity to heterologous vaccines and malaria derived antigens. The exact mechanism used to induce this suppression is yet to be uncovered.
The suppression of immune function seen in malaria infection could be attributed to DC/iRBC interaction which alter the maturation state and function of DC in both humans (4, 102, 103) and mice (8, 104). DCs exposed to iRBC in vitro and in vivo have reduced expression of MHC on the surface and are unable to form stable interactions with CD4 helper T cells (104). The DCs also downregulate key costimulatory molecules, such as CD86, CD80, CD40, and secrete IL-10 (105), providing a suppressive environment for CD4 T cell development. This hampers their ability to activate naive CD4 T cells and a failure to generate Tfh cells that are critical in the formation of germinal center and generation of protective antibodies against malaria infection (77, 104). In contrast, other in vitro studies have shown that DCs exposed to iRBC successfully activate T cells, but induce their polarization toward a Th1 phenotype that inhibits commitment to Tfh cell linage, thus affecting humoral responses (103, 106).
What Happens to DCs During a Plasmodium infection?
During the Plasmodium parasite life cycle, different forms of the parasite interact with resident DCs in various organs as it establishes infection. Sporozoites from infectious mosquitoes that are injected into the dermis interact with resident DCs in the skin (107). The sporozoites reach the liver interact with Kupffer cells, hepatocyte, and liver sinusoidal endothelial cells and resident DCs in the liver (108). The blood stage of the parasite interacts with DCs in the blood (4) and spleen (109–111).
DC Interaction with Plasmodium Sporozoites in the Skin and Liver
The skin is the first point sporozoites encounter DC as they are inoculated by infected mosquitoes (Figure 1A). Studies conducted in mice have shown that only a small percentage of inoculated sporozoites leave the site of injection as most end up trapped in the dermis or enter the lymphatic system rather than the blood vessel (13). Other sporozoites infect keratinocytes, hair follicles, and develop into exoerythrocytic forms of the parasite (112). Sporozoites that are trapped in the dermis are phagocytosed by resident DCs which migrate to the skin-draining lymph node and can prime CD4 (113, 114) and CD8 (14) T cell responses. The immune response toward the sporozoite stage of the parasite may protect against subsequent challenges from infected mosquitoes (113).
The sporozoites that mange to enter blood circulation move to the liver and must traverse the sinusoidal barrier to access hepatocytes (Figure 1B) (115). The liver environment is tolerogenic due to the presence of IL-10 and TGF-β which are secreted by Kupffer cells (KC) and liver sinusoidal endothelial cells (LSEC) (116). These cytokines reduce expression levels of MHC class II and costimulatory molecules on the surface of liver resident DCs compared to resident DCs in lymphoid organs and those circulating in the blood (117) thus reducing their capability to activate T cells (118, 119). The tolerogenic environment of the liver could play a role in sporozoite immune evasion as DCs and other immune cells in the liver act to suppress adaptive immune responses which would lead to the elimination of sporozoites (116, 120).
Apart from DCs, the liver has other potential APCs that can present antigens to the adaptive immune system; this includes Kupffer cells (KC), liver sinusoidal endothelial cells (LSEC), and hepatocytes. LSECs are scavenger cells that express MHC class I and II molecules, low levels of CD86, and the adhesion molecules ICAM-1, VCAM-1, and dendritic cell specific intercellular adhesion molecule3-grabbing non-integrin (DC-SIGN). In mice, these cells have the ability to cross present antigens in the liver and activate CD8 T cells, but the T cells are generally tolerized due to the secretion of IL10 and PGE2 by LSECs (121, 122). KCs are resident tissue macrophages found in the liver that express MHC class I and class II molecules, ICAM-1, CD86, CD80, and can activate naive CD4 and CD8 T cells in vitro (123, 124). The role of KC as an APC is controversial as in vitro experiments show that they inhibit T cell activation by secreting IL-10 (125), but activation of KCs via TLR3 increased the expression of MHC class II and their APC function (126). Kuniyasu et al. (127) showed that the liver had the ability to retain adoptively transferred T cells. The T cells proliferated and expanded in the liver, but the expansion was followed by apoptosis, which was initiated by KCs (127). It was later shown that KCs induce T cell apoptosis via the FAS-FAS-L signaling pathway (128).
Hepatocytes express MHC class I and ICAM-1 in their steady state and during inflammation they have been shown to express MHC class II CD40L, CD80 and CD86 and are capable of activating CD8 T cells (129). Their role in generation of malaria liver immunity has been controversial with different studies using mouse models drawing different conclusions of their role in the generation of pre-erythrocytic immunity. Intrasplenic injection of parasite infected hepatocytes in mice resulted in T cell mediated immunity against P. yoelii and P. berghei infections (26), thus showing that hepatocytes are capable of activating T cells. Another study demonstrated that parasite infected hepatocytes undergo apoptosis, thus providing liver DCs with a source of Plasmodium antigens for initiating the adaptive immune response (130). This idea has been challenged and it has been suggested that DCs could obtain Plasmodium antigens directly from viable infected hepatocytes. This is supported by the fact that DCs have the ability to acquire antigens from other live cells and cross present to CD8 T cells (131).
Chakravarty et al. (14) showed that cross presentation of Plasmodium antigens by DCs was key in CD8 T cells activation and this occurred in the skin draining lymph node, not in the liver, and the activated T cells recirculated to the liver (14). Indicating that DCs in the skin that encounter sporozoite play a crucial role in generating T cell mediated liver immunity. Recently Kurup et al. (27) showed that during a malaria infection, a subset of monocyte derived CD11c+ APC infiltrate the liver after hepatocyte infection by Plasmodium parasite and acquire Plasmodium antigens. The monocyte derived CD11c+ APC present the antigens to naive CD8 T cells in the liver draining lymph node, priming them and initiating T cell mediated immunity against Plasmodium infection (27).
While there are still some gaps into how the generation of liver immunity against Plasmodium infection is acquired, it is clear that APCs, especially DCs, play a central role. Hepatocytes may play a part in the generation of liver immunity by providing parasite antigens to DCs but the exact mechanism of this is yet to be uncovered. A better understanding of DC and hepatocyte involvement in the generation of liver immunity is required and also the roles played by KCs and LSECs. The use of humanized mice might provide an opportunity to further investigate skin and liver immunity against P. falciparum (132, 133).
DC Interaction with Plasmodium during the Blood Stage of Malaria
The blood stage of the malaria parasite life cycle provides several opportunities for DC in the blood and spleen to interact with infected RBC (Figure 1C). This stage requires remodeling of the RBC to enable the parasite to survive (134) and results in the expression of parasite antigens on the RBC surface. These antigens, in particular PfEMP, play a key role in immune evasion and vascular sequestration/cytoadherence to avoid splenic clearance (21, 135). It has been suggested that PfEMP1 may be involved in modulation of DC function via interaction with CD36 (4, 136).
Maturation of iRBCs (schizont stage) results in lysis of the iRBCs, releasing merozoites into circulation and the contents of the PV such as the parasites digestive vacuole which contains hemozoin and waste products. The free merozoites have a short window to invade new RBCs (137) and those that fail to invade remain in circulation where they are phagocytosed by immune cells or cleared in the spleen. Parasite waste products and hemozoin do interact with DCs but their overall effect on DC function is contradictory. The effect of hemozoin on DCs has yielded varying results with some studies showing that hemozoin is capable of activating DCs (138) while others showed that DC maturation and function was inhibited by hemozoin (8, 139). The varying results could be due to the different methods that were used to generate hemozoin with contamination by parasite DNA being a potential confounding factor (140).
Overall, the blood stage has an abundance of parasite antigens that DCs can use to mount an immune response. However various immune evasion mechanisms, such as antigenic variation of VSAs (141, 142) and sequestration of mature schizont and trophozoites in blood capillaries (143) thus avoiding splenic clearance (144, 145) the slow acquisition of immunity. DCs at this stage are critical in maintaining an immunological balance between parasite burden and a sufficient immune response. Immune evasion by the parasite could cause an increase in parasite burden resulting in severe pathology, while an excessive and uncontrolled immune response may lead to the development of a severe life threating cerebral malaria (146–148).
Studies of blood stage infections with DC have largely employed DCs prepared from peripheral blood monocytes or isolated from peripheral blood of uninfected individuals (4, 102). Fewer studies have analyzed the phenotype and function of peripheral blood DCs from individuals who are currently undergoing a malaria episode (103, 149, 150). In this context, in vivo mouse models of malaria have been particularly helpful to understand the tissue responses of DC, for example splenic DC and allow temporal analysis of how Plasmodium infection changes DC phenotype (8, 104).
In vitro DC interaction with Plasmodium
In vitro studies have been used to identify the mechanisms used by the parasite to modulate DC function. These studies have either used human monocyte derived dendritic cells (moDCs) or bona fide DCs to assess DC- P. falciparum interactions.
Urban et al. (4) showed that when moDCs were co-cultured with P. falciparum iRBC, at a ratio of 1:100, and later stimulated with lipopolysaccharide (LPS), exhibited a decreased expression of key maturation markers (CD40, CD80, CD86, and CD83) (4). Once moDCs were exposed to iRBC, they lacked the capacity to activate allogeneic T cells (4). This modulation of DC maturation may result from an interaction between CD36 on DCs with PfEMP-1 on iRBC (105). A subsequent study found that a ratio of 1 DC: 100 iRBC inhibited LPS induced moDCs activation, cytokine production, and allogeneic T cell activation regardless of CD36-binding with iRBC (102). The high ratio of DC to iRBC coincided with an increase in apoptotic and necrotic cells, which was observed in both PfEMP1-deficient iRBCs and PfEMP1 expressing iRBCs, this could account for the failure of DCs to respond (102). At low iRBC to moDC ratio (10:1), moDCs made a modest response to LPS induced maturation and retained their ability to secrete cytokines and activate T cells (102). Elliot et al. (102) were unable to point out the mechanism used by P. falciparum to modulate moDC function, although they found that hemozoin, from iRBC lysate, did not inhibit LPS maturation of moDCs (102). The studies show that a dose-dependant relationship exists between iRBC and moDCs inhibition and dose range experiments are an essential part of ensuring experimental reproducibility in the future.
Another study found that at a low ratio of 10 iRBC per moDCs did not trigger the upregulation of HLA-DR, CD83, or CCR7 on moDCS (151), contradicting the study by Elliot et al. (102). At a ratio of 100 iRBCS per moDCs, moDCs were able to secrete IL-1β, IL-6, IL-10, TNF-α, and upregulate the chemokine receptor CXCR4 (151). Exposure of moDCs to schizont lysate resulted in an increase in the expression levels of CD86 while CD80 and HLA-DR levels remained unaffected even at high concentration of schizont extract (106). Exposure to schizont lysate, followed by LPS stimulation, did not affect the maturation of moDCs. The schizont lysate exposed moDCs maintained their ability to differentiate allogeneic T cells into Th1 and regulatory T cells (Treg) that secrete large amounts of IFN-γ. Additionally, the generated Tregs also secreted IL-10 and TGF-β (106).
The different in vitro studies looking at the effect of P. falciparum on moDCs have yielded varying results. This could be attributed to the use of the Plasmodium parasite at different stages of development in the RBC. Another explanation could be that the studies used different experimental methods in the isolation of the Plasmodium infected red blood cells and in the generation of moDCs.
Few studies have examined the effect of P. falciparum on cDCs and pDCs due to their low numbers in peripheral blood. One study examined the effect of P. falciparum on cDC2 and pDCs (103). The co-culture of cDC2 with P. falciparum at a ratio of 1:3 resulted in the upregulation of maturation markers (CD80, CD86, CD40, and HLA-DR) and inflammatory chemokines CCL2, CXCL9 and CXCL10 but did not induce secretion of inflammatory cytokines. Exposure of cDC2 to iRBC did not inhibit cytokine secretion in response to LPS, which was contrary to what was observed with moDCS (103). The low ratio of iRBC to DC may account for this observation as the study did not use a higher ratio of iRBC to cDC2. The cDC2 exposed to iRBC maintained their ability to present antigens and activate naive T cells to polarize them toward a Tfh1 phenotype that secretes IFN-γ (103). The study also found that crosstalk between pDCs and cDC2 was important in shaping immune responses against malaria. The co-culture of pDCs and cDCs resulted in the upregulation of HLA-DR, CD86, and CD40 on pDCs and CD80 and CD86 on cDC2. There was also an increase in the secretion of interferon alpha (IFN-α) by pDCs and chemokines CXCL9 and CXCL10 by cDC2. This cross-talk between these two DCs was contact dependent, suggesting cell to cell interaction is necessary to initiate chemokine secretion (103). The study highlighted the importance of cell to cell interaction which is crucial in trying to understand immune responses in malaria.
In mouse studies using bone marrow derived dendritic cells (BMDCs), P. chabaudi schizonts were shown to be able to activate BMDCs to produce the pro-inflammatory cytokines IL-12 and TNF-α. The P. chabaudi exposed DCs did not inhibit LPS activation, contrary to what was observed with P. falciparum exposed human DCs (152).
Ex vivo DC interaction with Plasmodium
A number of studies have compared peripheral blood DCs in varying malaria transmission settings and different at-risk groups. In Kenya, children hospitalized with either mild or severe malaria were found to have a lower number of DC expressing HLA-DR and a lower number of circulating DCs compared with healthy children (149). A follow up study revealed that the expression levels of HLA-DR was reduced on monocytes and cDC but not on pDC and that DC modulation continued during convalescence. An increase in the frequency of BDCA3+ cDC1 in the peripheral circulation was also observed during the course of the malaria infection (153).
A similar study was conducted in Mali looking at the function of DCs in children with severe malaria from the Dogon and Fulani community. The two communities reside in the same geographical region and are exposed to the same intensity of P. falciparum transmission yet the Fulani are less suspectable to P. falciparum infection (154). DCs from malaria infected children of the Dogon community expressed lower levels of HLA-DR and CD86 on their DCs, while the frequency of BDCA-2+ pDCs and BDCA-3+ cDC1 increased compared to uninfected counterparts. Infected children from the Fulani community exhibited higher levels of HLA-DR and CD86 on their DCs but had a lower number of circulating BDCA-2+ pDCs and BDCA-3+ cDC1 compared to their uninfected counterparts (150). The study also showed that infected children from the Fulani community retained their ability to produce IFN-γ after their PBMC were stimulated with specific TLR ligands at levels that were similar to those of uninfected children. The Dogon children, on the other hand, had low levels of cytokine produced due to TLR impairment which increased parasite burden and development of malaria symptoms (150). This showed that P. falciparum infection resulted in altered DC activation with reduced response to TLR agonists in Dogon children, while in the Fulani children, DC activation and TLR responses were unaffected.
The increase in the number of circulating BDCA-2+ pDCs and BDCA-3+ cDC1 during malaria infection has been attributed to an increase in the amounts of FMS-like tyrosine kinase 3 (Flt3) ligand (Flt3-L) (155). Flt3 is highly expressed on hematopoietic progenitor cells, but the expression is lost as cells commit to lymphoid and myeloid progenitor cells, which gives rise to the various cell lineages but its expression on DCs remains. Flt3 receptor tyrosine kinase and its ligand Flt3-L are known to be key in the development of dendritic cells and maintenance of their numbers (156, 157). Flt3-L production increases during a malaria episode as mast cells become activated and release membrane bound Flt3-L into circulation resulting in an increase in the number of pDCs and CD1c (155).
A few studies have looked at the function of DCs in adults during a malaria episode. A study in Thailand found that adults with both severe and mild malaria had a decreased number of TLR2 expressing cDCs circulating in the periphery and a lower surface expression of TLR9 on pDCs but an increase in the surface expression of TLR2 on cDCs compared with healthy controls (158). There was also a marked reduction in the number of circulating pDCs, this could be attributed to their migration to the secondary lymph nodes, and an increase in serum levels of IFN-α (159). A study conducted in Papua found that adults with acute P. falciparum malaria had a reduced number of circulating pDCs and cDCs, but higher numbers of immature DCs that were HLA-DR+CD11c–CD123– (5). Interestingly both pDCs and cDCs from infected participants were apoptotic as seen by Annexin-V binding. The DCs also expressed low levels of HLA-DR and costimulatory molecules and were unable to adequately capture antigen, resulting in reduced ability to prime naive CD4 T cells (5). These studies are therefore consistent with a role for malaria infection in reducing the number of circulating DCs and their function in antigen presentation and T cell activation.
Controlled human infection model (CHMI) have also been used to assess the function of BDCA-1+ cDC2 and pDCs at varying doses of P. falciparum (160). Healthy volunteers were enrolled into two cohorts; one cohort was inoculated with 150 iRBCs and the other 1,800 iRBCs, participants were treated once parasitaemia reached ≥ 1,000 parasites/ml (160, 161). The expression levels of HLA-DR on BDCA-1+ cDC2 and pDCs in both cohorts were significantly reduced at peak parasitaemia and this effect was still evident on BDCA-1+ cDC2 24 h after anti-malarial treatment. The cohort inoculated with a higher dose of iRBC had a reduced number of circulating BDCA-1+ cDC2 which was attributed to apoptosis of the DCs during the course of the infection, this was evident by the upregulation of caspase-3 (160). The BDCA-1+ cDC2 from this cohort had a defective phagocytic capacity and there was a positive association between HLA-DR expression and phagocytic capacity (160). pDCS on the other hand expressed low levels of CD123 at peak parasitaemia in both cohorts which persisted 24 h after anti-malarial treatment. The number of pDCs in circulation significantly reduced in the s iRBC cohort, this was due to apoptosis of pDCs during the course of infection (161). At peak parasitaemia DCs from the 1,800 iRBC cohort were restimulated ex vivo with TLR ligands and their response measures. On re-stimulation with TLR1/2, TLR4, and TLR7, BDCA-1+ cDC2 failed to upregulate HLA-DR and CD86 but increased TNF secretion (160). While re-stimulation of pDCS with TLR7 and TLR9 resulted in upregulation of HLA-DR, CD123, CD86 on their surface and an increased secretion of IFN-α (161). This shows that malaria infection in naive individuals results in impairment of cDC function but not pDCs function. Indicating that pDCs may play a role during malaria infection and further studies are needed to deduce its role. The altered BDCA-1+ cDC2 also contributed to hampering effector T cells functions, allowing an increase of parasite burden (160).
The various studies above have shown that DC phenotype is altered during a malaria episode resulting in impaired ability to upregulate HLA-DR and the costimulatory molecules CD86 (150, 153, 160). This altered DC phenotype has a reduced phagocytic capacity which impairs its ability to process antigens (160) and adequately stimulate allogeneic T cells (5, 153). The parasite also modulates TLR signaling thereby affecting cytokine secretion (150, 160) resulting in severe pathology. In children, there seems to be a notable increase in the number of circulating BDCA-3+ cDC1s during a malaria episode (150, 153, 155), which was attributed to increases in serum levels of Flt3-L (155), but this effect was not observed in children from Papua (162). In both children and adults, there was a decrease in the number of circulating DCs which was attributed to increased DC apoptosis (5, 159, 161) but also increased DC migration to secondary lymphoid organs may also play a role in reduction on peripheral blood DC numbers. The decrease in peripheral numbers of DCs also corresponded with an increase in IL10 and TNF-α (5, 149, 153), which may play a role in DC loss of function and suppression of T cell function. In these studies, DC function was altered regardless of the severity of malaria infection. The DC phenotype seen in the acute infection in the CHMI study (160), was similar to those seen in naturally exposed individuals, and repeated infection, in naturally exposed individuals, could lead to sustained downregulation of DC function that may impact negatively on the immunity of an individual.
In vivo Mouse Models of Malaria
Mouse models have been extensively used to study DC-Plasmodium interaction. In vitro interaction of P. chabaudi schizonts with mouse bone marrow derived DCs resulted in an increase in the secretion of tumor necrosis factor-α(TNF-α), IL-6, and IL-12p40 and IL-12p70 (152). In mice injected with P. chabaudi, DCs had fully functional cytokine production 6 days after challenge with Plasmodium parasite (163). Further studies demonstrated DCs were able to upregulate co-stimulatory molecules CD40, CD54, CD86 (164) during acute infection, and were able to migrate into T cell areas in the spleen (165). Other studies with P. chabaudi show that during initial stages of murine erythrocyte infection, CD8+ DCs are activated by infected erythrocytes as they expressed high levels of MHC II and costimulatory molecules and initiated a Th1 type of response. This response is short lived as the CD8+ DCs undergo apoptosis and are soon replaced by CD8- DCs with lower expression levels of costimulatory molecules and MHC II (166).
Consistent with the studies above, Millington et al. (8) showed that DCs isolated from the spleen of mice 4 days after P. chabaudi infection were moderately activated as they upregulated surface expression of CD40, CD80, and CD86. However, during convalescence (days 12 and 21 post-infection), DC did not upregulate costimulatory molecules and were refractory to stimulation with LPS or CD40L. When mice infected with P. chabaudi were immunized with ovalbumin (OVA) antigen and LPS, they produced significantly lower levels of OVA-specific IgG compared with uninfected immunized mice, however, this effect was only seen when immunized at days 12 and 21 post infection (not day 4). Thus, initial malaria infection in mice does seem to cause DC activation; however DCs enter a refractory state in following the initial peak of parasitaemia. Similar to convalescent DCs, in vitro bone marrow derived DCs pre-exposed to P. chabaudi were unable to increase expression levels of MHC II and co-stimulatory molecules CD40, CD80, and CD86, and LPS stimulation of these DCs was unable to increase their expression (8).
Further work suggested that hemozoin could also modulate DC function which resulted in impairment of T cell and B cell function. Hemozoin treated DCs retained their capacity to process antigen and present them on MHC class II to naive CD4 T cells. Thus providing the essential signal 1 (peptide-MHC complex) via the T cell receptor (TCR) but these DCs were unable to form stable long lasting clusters with naive T cells, resulting in the generation of dysfunctional T cells (8, 104). These dysfunctional T cells failed to proliferate and produce adequate amount of effector cytokines (IL-2, IL-5, IL-10, IFNγ) (8), and were unable to migrate to B cell areas in the lymph nodes to aid in B cell proliferation and antibody production (8, 104). The short interactions and lack of large clustering observed are known to interfere with the generation of Tfh cells as long sustained DC-T cells interaction is required for commitment of naive CD4 T cells to Tfh cells (167). It is possible the dysfunctional DCs can lead to the generation of exhausted T cells, as a result of the short time of antigen presentation to the T cells in the absence of adequate co-stimulation. The dysfunctional T cells could also lead to the generation of atypical memory B cells which are normally associated with malaria episodes.
Dendritic cells have been shown to play a vital role in the survival of mice during a lethal infection with P. yoelii. Wykes et al. (168) showed that DCs from mice infected with non-lethal P. yoelii infection were fully functional APC and maintained their ability to stimulate T cells, unlike DCs from lethal P. yoelii infection which were not functional. DCs from mice infected with the non-lethal parasite were adoptively transferred into naive mice, which were then infected with lethal infection P. yoelii. These DCs were able to control parasitaemia and aid in survival of the mice by secreting IL-12 (168). This could in part explain the difference in malaria outcomes observed in natural infections.
Downstream Effect of DC Dysregulation
Collectively, the studies above support the hypothesis that during the blood stage of Plasmodium infection, DC function is dysregulated resulting in phenotypically altered DCs that are unable to appropriately activate naive CD4 cells. Furthermore, the failure by naive CD4 T cells to differentiate into CD4 follicular helper T cells results in a failure of B cell help and reduced humoral immunity. The phenotype of the resulting T cell population is unclear; however, P. falciparum infection has been associated with increased expression of the T cell inhibitory receptor programmed cell death-1 (PD-1) on CD4 T cells. This was observed in a cohort of children in Mali (169) and Kenya (7), during an ongoing P. falciparum infection. Apart from an increase in the expression of PD-1 on CD4 T cells, P. falciparum infection was seen to drive an increase in the frequency of atypical memory B cells which was as a result of the exhausted T cell phenotype (7).
Butler et al. (169) using non-lethal P. yoelii infections, also showed that prolonged infection resulted in dysfunctional parasite specific CD4 T cells that expressed exhaustion markers PD-1 and lymphocyte-activation gene-3 (LAG-3) (169). These inhibitory ligands worked in synergy to inhibit T cell function during the Plasmodium infection. The ability of CD4 T cells to produce cytokines deteriorated with prolonged infection, while dual blockade of PD-1 and LAG-3 with monoclonal antibodies restored the number of parasite specific CD4 T cells and their ability to secrete cytokines. It also resulted in an increase in the number of CD4 Tfh cells and plasmablasts, thus improving the anti-parasite humoral response in P. yoelii infected mice (169). These studies show that that malaria induces T cell exhaustion and that PD-1 plays a role in the pathogenesis of malaria.
Apart from the upregulation of the T cell inhibitory receptors programmed cell death-1 (PD-1) and Lymphocyte Activation Gene 3 (LAG3), malaria infection upregulates the production of IFN-γ and IL-10 on CD4 T cells (170, 171). This creates a suppressive environment that polarizes CD4 T cells toward IFN-γ producing Th1 like lineage, suppressing induction of Th2 and Tfh, which are vital in B cell response. This polarization occurs after a single malaria episode and may affect subsequent parasite exposures
T cell exhaustion can be due to persistent antigen exposure, resulting in sustained TCR stimulation by dysfunction DCs, leading to sustained upregulation of PD-1 (172, 173). The inhibitory PD1 signal on T cells works by inhibiting downward signals from TCR and costimulatory molecules and initiates transcription of inhibitory genes (Figure 2B) (174–176). Cytokine signaling also plays a role in T cell exhaustion. Malaria induces secretion of IL-10 from DCs, providing an immunosuppressive environment that skews the development of CD4 T cells and dual blocking IL-10 and PD-1 signaling in mice restores T cell function (177). Transcriptional profile of exhausted T cells greatly varies from effector and memory T cells, indicating that exhaustion is a unique state of T cell differentiation (178–180), that is regulated by the master transcription factor TOX (181). The signaling pathways that lead to the differentiation of exhausted T cells and expression of TOX are yet to be known.
There are still gaps in our current understanding of the intracellular mechanism of PD1 signaling and what its target genes are. The molecular events initiated by downstream IL-10 signaling that shape T cell exhaustion are yet to be known.
Conclusion
The population in malaria endemic areas are known to have a reduced immune response against vaccines (2, 3), and ongoing malaria infections in these individuals reduce pre-existing adaptive immune responses (101). The evidence presented above strongly indicates this is due to dysfunctional DCs that fail to prime effective T cell responses, thus affecting immune responses. There is a potential gap is in our understanding of the effect of antimalarial drugs on the phenotype, function and numbers of DCs and T cells. Whether antimalarial treatment restores DC-T interaction is an area of research is yet to be explored.
This information could address a significant public health challenge in administering malaria vaccines and other vaccines in malaria endemic areas. Malaria infection also induces CD4 T cell exhaustion, through upregulation of negative regulatory molecules such as PD-1 and LAG3 (7, 169), which dampen immune responses. How T cell exhaustion is induced in malaria is still unknown as there is no evidence in literature explaining how and where these cells arise, and if dysfunctional DCs play a role in this, and how the cytokine environment during a malaria episode influence T cell exhaustion. There is a need to better understand the interaction between DC-T cell, the cellular and molecular signals that are involved in the formation of this immune synapse and how malaria affects this interaction. This will aid in developing novel methods that will target the affected molecular pathways and restore DC-T cell interaction and function.
Author Contributions
RO wrote the first draft of the manuscript, which was reviewed and edited by TO, FN, PG, and JB. All authors contributed to the article and approved the submitted version.
Funding
RO is a PhD student supported under FN's MRC/DFID African Research Leadership Award (MR/P020321/1) and the DELTAS Africa Initiative (DEL-15-003). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)'s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa's Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (107769/Z/10/Z) and the UK government. This research received funding from the University of Glasgow, Scottish Funding Council, and the Global Challenges Research Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This manuscript is published with permission from the Director, Kenya Medical Research Institute (KEMRI).
References
2. Ho M, Webster HK, Looareesuwan S, Supanaranond W, Phillips RE, Chanthavanich P, et al. Antigen-specific immunosuppression in human malaria due to Plasmodium falciparum. J Infect Dis. (1986) 153:763–71. doi: 10.1093/infdis/153.4.763
3. Greenwood B, Palit A, Bradley-Moore A, Bryceson A. Immunosuppression in children with malaria. Lancet. (1972) 299:169–72. doi: 10.1016/S0140-6736(72)90569-7
4. Urban BC, Ferguson DJ, Pain A, Willcox N, Plebanski M, Austyn JM, et al. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature. (1999) 400:73–7. doi: 10.1038/21900
5. Pinzon-Charry A, Woodberry T, Kienzle V, McPhun V, Minigo G, Lampah DA, et al. Apoptosis and dysfunction of blood dendritic cells in patients with falciparum and vivax malaria. J Exp Med. (2013) 210:1635. doi: 10.1084/jem.20121972
6. Weiss GE, Crompton PD, Li S, Walsh LA, Moir S, Traore B, et al. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J Immunol. (2009) 183:2176–82. doi: 10.4049/jimmunol.0901297
7. Illingworth J, Butler NS, Roetynck S, Mwacharo J, Pierce SK, Bejon P, et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J Immunol. (2013) 190:1038–47. doi: 10.4049/jimmunol.1202438
8. Millington OR, Di Lorenzo C, Phillips RS, Garside P, Brewer JM. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J Biol. (2006) 5:5. doi: 10.1186/jbiol34
9. Horne-Debets JM, Faleiro R, Karunarathne DS, Liu XQ, Lineburg KE, Poh CM, et al. PD-1 dependent exhaustion of CD8+ T cells drives chronic malaria. Cell Rep. (2013) 5:1204–13. doi: 10.1016/j.celrep.2013.11.002
10. Bediako Y, Ngoi JM, Nyangweso G, Wambua J, Opiyo M, Nduati EW, et al. The effect of declining exposure on T cell-mediated immunity to Plasmodium falciparum–an epidemiological “natural experiment”. BMC Med. (2016) 14:143. doi: 10.1186/s12916-016-0683-6
11. Menard R, Tavares J, Cockburn I, Markus M, Zavala F, Amino R. Looking under the skin: the first steps in malarial infection and immunity. Nat Rev Microbiol. (2013) 11:701–12. doi: 10.1038/nrmicro3111
12. Cowman AF, Healer J, Marapana D, Marsh K. Malaria: biology and disease. Cell. (2016) 167:610–24. doi: 10.1016/j.cell.2016.07.055
13. Amino R, Thiberge S, Martin B, Celli S, Shorte S, Frischknecht F, et al. Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat Med. (2006) 12:220–4. doi: 10.1038/nm1350
14. Chakravarty S, Cockburn IA, Kuk S, Overstreet MG, Sacci JB, Zavala F. CD8+ T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nat Med. (2007) 13:1035–41. doi: 10.1038/nm1628
15. Tavares J, Formaglio P, Thiberge S, Mordelet E, Van Rooijen N, Medvinsky A, et al. Role of host cell traversal by the malaria sporozoite during liver infection. J Exp Med. (2013) 210:905–15. doi: 10.1084/jem.20121130
16. Yang AS, Boddey JA. Molecular mechanisms of host cell traversal by malaria sporozoites. Int J Parasitol. (2017) 47:129–36. doi: 10.1016/j.ijpara.2016.09.002
17. Prudencio M, Rodriguez A, Mota MM. The silent path to thousands of merozoites: the Plasmodium liver stage. Nat Rev Microbiol. (2006) 4:849–56. doi: 10.1038/nrmicro1529
18. Graewe S, Rankin KE, Lehmann C, Deschermeier C, Hecht L, Froehlke U, et al. Hostile takeover by plasmodium: reorganization of parasite and host cell membranes during liver stage egress. PLoS Pathog. (2011) 7:e1002224. doi: 10.1371/journal.ppat.1002224
19. Graewe S, Stanway RR, Rennenberg A, Heussler VT. Chronicle of a death foretold: Plasmodium liver stage parasites decide on the fate of the host cell. FEMS Microbiol Rev. (2012) 36:111–30. doi: 10.1111/j.1574-6976.2011.00297.x
20. Maier AG, Cooke BM, Cowman AF, Tilley L. Malaria parasite proteins that remodel the host erythrocyte. Nat Rev Microbiol. (2009) 7:341–54. doi: 10.1038/nrmicro2110
21. Smith JD, Chitnis CE, Craig AG, Roberts DJ, Hudson-Taylor DE, Peterson DS, et al. Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell. (1995) 82:101–10. doi: 10.1016/0092-8674(95)90056-X
22. Kyes SA, Kraemer SM, Smith JD. Antigenic variation in Plasmodium falciparum: gene organization and regulation of the var multigene family. Eukaryot Cell. (2007) 6:1511–20. doi: 10.1128/EC.00173-07
23. Claessens A, Hamilton WL, Kekre M, Otto TD, Faizullabhoy A, Rayner JC, et al. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of var genes during mitosis. PLoS Genet. (2014) 10:e1004812. doi: 10.1371/journal.pgen.1004812
24. Ngotho P, Soares AB, Hentzschel F, Achcar F, Bertuccini L, Marti M. Revisiting gametocyte biology in malaria parasites. FEMS Microbiol Rev. (2019) 43:401–14. doi: 10.1093/femsre/fuz010
25. Hellmann JK, Münter S, Kudryashev M, Schulz S, Heiss K, Müller A-K, et al. Environmental constraints guide migration of malaria parasites during transmission. PLoS Pathog. (2011) 7:e1002080. doi: 10.1371/journal.ppat.1002080
26. Renia L, Rodrigues MM, Nussenzweig V. Intrasplenic immunization with infected hepatocytes: a mouse model for studying protective immunity against malaria pre-erythrocytic stage. Immunology. (1994) 82:164–8.
27. Kurup SP, Anthony SM, Hancox LS, Vijay R, Pewe LL, Moioffer SJ, et al. Monocyte-derived CD11c(+) cells acquire plasmodium from hepatocytes to prime CD8 T cell immunity to liver-stage malaria. Cell Host Microbe. (2019) 25:565–77 e566. doi: 10.1016/j.chom.2019.02.014
28. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
29. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. (2007) 315:107–11. doi: 10.1126/science.1136080
30. Stockwin LH, McGonagle D, Martin IG, Blair GE. Dendritic cells: immunological sentinels with a central role in health and disease. Immunol Cell Biol. (2000) 78:91–102. doi: 10.1046/j.1440-1711.2000.00888.x
31. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
32. Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. (2005) 17:338–44. doi: 10.1016/j.coi.2005.02.007
33. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
34. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
35. De Smedt T, Pajak B, Muraille E, Lespagnard L, Heinen E, De Baetselier P, et al. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J Exp Med. (1996) 184:1413–24. doi: 10.1084/jem.184.4.1413
36. Blanco P, Palucka A, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytok Growth Factor Rev. (2008) 19:41–52. doi: 10.1016/j.cytogfr.2007.10.004
37. Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. (2017) 356:eaah4573. doi: 10.1126/science.aah4573
38. Palucka K, Banchereau J, Mellman I. Designing vaccines based on biology of human dendritic cell subsets. Immunity. (2010) 33:464–78. doi: 10.1016/j.immuni.2010.10.007
39. Boltjes A, van Wijk F. Human dendritic cell functional specialization in steady-state and inflammation. Front Immunol. (2014) 5:131. doi: 10.3389/fimmu.2014.00131
40. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. (2018) 154:3–20. doi: 10.1111/imm.12888
41. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+(BDCA-3) dendritic cells. (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. (2010) 207:1247–60. doi: 10.1084/jem.20092140
42. Stutman O. Intrathymic and extrathymic T cell maturation. Immunol Rev. (1978) 42:138–84. doi: 10.1111/j.1600-065X.1978.tb00261.x
43. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. (2005) 174:4465–9. doi: 10.4049/jimmunol.174.8.4465
44. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. (2005) 202:637–50. doi: 10.1084/jem.20050821
45. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity. (2007) 27:281–95. doi: 10.1016/j.immuni.2007.07.010
46. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Rα expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. (2010) 32:91–103. doi: 10.1016/j.immuni.2009.11.010
47. Yannelli J, Sullivan J, Mandell G, Engelhard V. Reorientation and fusion of cytotoxic T lymphocyte granules after interaction with target cells as determined by high resolution cinemicrography. J Immunol. (1986) 136:377–82.
48. Peters PJ, Geuze HJ, Van der Donk HA, Slot JW, Griffith JM, Stam NJ, et al. Molecules relevant for T cell-target cell interaction are present in cytolytic granules of human T lymphocytes. Eur J Immunol. (1989) 19:1469–75. doi: 10.1002/eji.1830190819
49. Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. (2001) 15:751–61. doi: 10.1016/S1074-7613(01)00234-5s
50. Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. (2007) 27:393–405. doi: 10.1016/j.immuni.2007.08.007
51. Sedegah M, Sim B, Mason C, Nutman T, Malik A, Roberts C, et al. Naturally acquired CD8+ cytotoxic T lymphocytes against the Plasmodium falciparum circumsporozoite protein. J Immunol. (1992) 149:966–71.
52. Doolan DL, Southwood S, Chesnut R, Appella E, Gomez E, Richards A, et al. HLA-DR-promiscuous T cell epitopes from Plasmodium falciparum pre-erythrocytic-stage antigens restricted by multiple HLA class II alleles. Infect Immun. (2000) 165:1123–37. doi: 10.4049/jimmunol.165.2.1123
53. Epstein JE, Tewari K, Lyke KE, Sim BKL, Billingsley PF, Laurens MB, et al. Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science. (2011) 334:475–80. doi: 10.1126/science.1211548
54. Radtke AJ, Kastenmuller W, Espinosa DA, Gerner MY, Tse SW, Sinnis P, et al. Lymph-node resident CD8alpha+ dendritic cells capture antigens from migratory malaria sporozoites and induce CD8+ T cell responses. PLoS Pathog. (2015) 11:e1004637. doi: 10.1371/journal.ppat.1004637
55. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7loPD-1hi CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. (2013) 39:770–81. doi: 10.1016/j.immuni.2013.09.007
56. Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. (1998) 393:478–80. doi: 10.1038/30996
58. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. (1998) 393:474–8. doi: 10.1038/30989
59. Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. (2002) 297:2060–3. doi: 10.1126/science.1072615
60. Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature. (2009) 462:510–3. doi: 10.1038/nature08511
61. D'Souza WN, Schluns KS, Masopust D, Lefrancois L. Essential role for IL-2 in the regulation of antiviral extralymphoid CD8 T cell responses. J Immunol. (2002) 168:5566–72. doi: 10.4049/jimmunol.168.11.5566
62. D'Souza WN, Lefrancois L. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J Immunol. (2003) 171:5727–35. doi: 10.4049/jimmunol.171.11.5727
63. Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. (2006) 441:890–3. doi: 10.1038/nature04790
64. Kwon H, Thierry-Mieg D, Thierry-Mieg J, Kim HP, Oh J, Tunyaplin C, et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. (2009) 31:941–52. doi: 10.1016/j.immuni.2009.10.008
65. Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Forster R, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. (2002) 416:94–9. doi: 10.1038/416094a
66. Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. (2007) 179:5099–108. doi: 10.4049/jimmunol.179.8.5099
67. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity. (2011) 34:947–60. doi: 10.1016/j.immuni.2011.03.024
68. Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T Cell (Tfh) differentiation. PLoS ONE. (2011) 6:e17739. doi: 10.1371/journal.pone.0017739
69. Powell MD, Read KA, Sreekumar BK, Jones DM, Oestreich KJ. IL-12 signaling drives the differentiation and function of a TH1-derived TFH1-like cell population. Sci Rep. (2019) 9:13991. doi: 10.1038/s41598-019-50614-1
70. Batten M, Ramamoorthi N, Kljavin NM, Ma CS, Cox JH, Dengler HS, et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J Exp Med. 2010) 207:2895–906. doi: 10.1084/jem.20100064
71. Schmitt N, Liu Y, Bentebibel S-E, Munagala I, Bourdery L, Venuprasad K, et al. The cytokine TGF-β co-opts signaling via STAT3-STAT4 to promote the differentiation of human T FH cells. Nat Immunol. (2014) 15:856. doi: 10.1038/ni.2947
72. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol. (2013) 190:3049–53. doi: 10.4049/jimmunol.1203032
73. Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity. (2011) 35:919–31. doi: 10.1016/j.immuni.2011.11.012
74. Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol. (2006) 177:4927–32. doi: 10.4049/jimmunol.177.7.4927
75. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz L-J, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Krüppel-like factor 2. J Exp Med. (2015) 212:217–33. doi: 10.1084/jem.20141432
76. Zander RA, Obeng-Adjei N, Guthmiller JJ, Kulu DI, Li J, Ongoiba A, et al. PD-1 Co-inhibitory and OX40 Co-stimulatory crosstalk regulates helper T cell differentiation and anti-plasmodium humoral immunity. Cell Host Microbe. (2015) 17:628–41. doi: 10.1016/j.chom.2015.03.007
77. Ryg-Cornejo V, Ioannidis LJ, Ly A, Chiu CY, Tellier J, Hill DL, et al. Severe malaria infections impair germinal center responses by inhibiting T follicular helper cell differentiation. Cell Rep. (2016) 14:68–81. doi: 10.1016/j.celrep.2015.12.006
78. Obeng-Adjei N, Portugal S, Tran TM, Yazew TB, Skinner J, Li S, et al. Circulating Th1-cell-type Tfh cells that exhibit impaired B cell help are preferentially activated during acute malaria in children. Cell Rep. (2015) 13:425–39. doi: 10.1016/j.celrep.2015.09.004
79. Letourneur F, Klausner RD. Activation of T cells by a tyrosine kinase activation domain in the cytoplasmic tail of CD3 epsilon. Science. (1992) 255:79–82. doi: 10.1126/science.1532456
80. Thangavelu G, Smolarchuk C, Anderson CC. Co-inhibitory molecules: controlling the effectors or controlling the controllers? Self/nonself . (2010) 1:77–88. doi: 10.4161/self.1.2.11548
81. Okoye IS, Houghton M, Tyrrell L, Barakat K, Elahi S. Coinhibitory receptor expression and immune checkpoint blockade: maintaining a balance in CD8(+) T cell responses to chronic viral infections and cancer. Front Immunol. (2017) 8:1215. doi: 10.3389/fimmu.2017.01215
82. Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through Interleukin-12. Immunity. (2009) 31:158–69. doi: 10.1016/j.immuni.2009.04.016
83. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science. (2009) 325:1001. doi: 10.1126/science.1176676
84. Vinuesa CG, Cyster JG. How T cells earn the follicular rite of passage. Immunity. (2011) 35:671–80. doi: 10.1016/j.immuni.2011.11.001
85. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. (2012) 209:1241–53. doi: 10.1084/jem.20120994
86. Lavazec C, Sanyal S, Templeton TJ. Expression switching in the stevor and Pfmc-2TM superfamilies in Plasmodium falciparum. Mol Microbiol. (2007) 64:1621–34. doi: 10.1111/j.1365-2958.2007.05767.x
87. Niang M, Yan Yam X, Preiser PR. The Plasmodium falciparum STEVOR multigene family mediates antigenic variation of the infected erythrocyte. PLoS Pathog. (2009) 5:e1000307. doi: 10.1371/annotation/c58250db-8cce-40c5-b7ac-42204050069a
88. Fernandez V, Hommel M, Chen Q, Hagblom P, Wahlgren M. Small, clonally variant antigens expressed on the surface of the Plasmodium falciparum–infected erythrocyte are encoded by the rif gene family and are the target of human immune responses. J Exp Med. (1999) 190:1393–404. doi: 10.1084/jem.190.10.1393
89. Lavazec C, Sanyal S, Templeton TJ. Hypervariability within the Rifin, Stevor and Pfmc-2TM superfamilies in Plasmodium falciparum. Nucleic Acids Res. (2006) 34:6696–707. doi: 10.1093/nar/gkl942
90. Gilbert SC, Plebanski M, Gupta S, Morris J, Cox M, Aidoo M, et al. Association of malaria parasite population structure, HLA, and immunological antagonism. Science. (1998) 279:1173–7. doi: 10.1126/science.279.5354.1173
91. Bachmann A, Bruske E, Krumkamp R, Turner L, Wichers JS, Petter M, et al. Controlled human malaria infection with Plasmodium falciparum demonstrates impact of naturally acquired immunity on virulence gene expression. PLOS Pathog. (2019) 15:e1007906. doi: 10.1371/journal.ppat.1007906
92. Marsh K, Howard RJ. Antigens induced on erythrocytes by P. falciparum: expression of diverse and conserved determinants. Science. (1986) 231:150–3. doi: 10.1126/science.2417315
93. Rowe JA, Moulds JM, Newbold CI, Miller LH. P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. (1997) 388:292. doi: 10.1038/40888
94. Smith JD, Rowe JA, Higgins MK, Lavstsen T. Malaria's deadly grip: cytoadhesion of Plasmodium falciparum-infected erythrocytes. Cell Microbiol. (2013) 15:1976–83. doi: 10.1111/cmi.12183
95. Tanabe K, Mackay M, Goman M, Scaife JG. Allelic dimorphism in a surface antigen gene of the malaria parasite Plasmodium falciparum. J Mol Biol. (1987) 195:273–87. doi: 10.1016/0022-2836(87)90649-8
96. Polley SD, Chokejindachai W, Conway DJ. Allele frequency-based analyses robustly map sequence sites under balancing selection in a malaria vaccine candidate antigen. Genetics. (2003) 165:555–61.
97. Pacheco MA, Poe AC, Collins WE, Lal AA, Tanabe K, Kariuki SK, et al. A comparative study of the genetic diversity of the 42kDa fragment of the merozoite surface protein 1 in Plasmodium falciparum and P. vivax. Infect Genet Evol. (2007) 7:180–7. doi: 10.1016/j.meegid.2006.08.002
98. Kennedy AT, Wijeyewickrema LC, Huglo A, Lin C, Pike R, Cowman AF, et al. Recruitment of human C1 esterase inhibitor controls complement activation on blood stage Plasmodium falciparum merozoites. J Immunol. (2017) 198:4728–37. doi: 10.4049/jimmunol.1700067
99. Kennedy AT, Schmidt CQ, Thompson JK, Weiss GE, Taechalertpaisarn T, Gilson PR, et al. Recruitment of factor H as a novel complement evasion strategy for blood-stage Plasmodium falciparum infection. J Immunol. (2016) 196:1239–48. doi: 10.4049/jimmunol.1501581
100. Simon N, Lasonder E, Scheuermayer M, Kuehn A, Tews S, Fischer R, et al. Malaria parasites co-opt human factor H to prevent complement-mediated lysis in the mosquito midgut. Cell Host Microbe. (2013) 13:29–41. doi: 10.1016/j.chom.2012.11.013
101. Ng DHL, Skehel JJ, Kassiotis G, Langhorne J. Recovery of an antiviral antibody response following attrition caused by unrelated infection. PLOS Pathog. (2014) 10:e1003843. doi: 10.1371/journal.ppat.1003843
102. Elliott SR, Spurck TP, Dodin JM, Maier AG, Voss TS, Yosaatmadja F, et al. Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infect Immun. (2007) 75:3621–32. doi: 10.1128/IAI.00095-07
103. Gotz A, Tang MS, Ty MC, Arama C, Ongoiba A, Doumtabe D, et al. Atypical activation of dendritic cells by Plasmodium falciparum. Proc Natl Acad Sci USA. (2017) 114:E10568–77. doi: 10.1073/pnas.1708383114
104. Millington OR, Gibson VB, Rush CM, Zinselmeyer BH, Phillips RS, Garside P. Malaria impairs T cell clustering and immune priming despite normal signal 1 from dendritic cells. PLoS Pathog. (2007) 3:1380–7. doi: 10.1371/journal.ppat.0030143
105. Urban BC, Roberts DJ. Malaria, monocytes, macrophages and myeloid dendritic cells: sticking of infected erythrocytes switches off host cells. Curr Opin Immunol. (2002) 14:458–65. doi: 10.1016/S0952-7915(02)00368-0
106. Clemente AM, Fadigati G, Caporale R, Marchese DG, Castronovo G, Sannella AR, et al. Modulation of the immune and inflammatory responses by Plasmodium falciparum schizont extracts: role of myeloid dendritic cells in effector and regulatory functions of CD4+ lymphocytes. Infect Immun. (2013) 81:1842–51. doi: 10.1128/IAI.01226-12
107. Amino R, Thiberge S, Blazquez S, Baldacci P, Renaud O, Shorte S, et al. Imaging malaria sporozoites in the dermis of the mammalian host. Nat Protoc. (2007) 2:1705–12. doi: 10.1038/nprot.2007.120
108. Frevert U, Engelmann S, Zougbédé S, Stange J, Ng B, Matuschewski K, et al. Intravital Observation of Plasmodium berghei sporozoite infection of the liver. PLOS Biol. (2005) 3:e192. doi: 10.1371/journal.pbio.0030192
109. Helmby H, Jonsson G, Troye-Blomberg M. Cellular changes and apoptosis in the spleens and peripheral blood of mice infected with blood-stage Plasmodium chabaudi chabaudi AS. Infect Immun. (2000) 68:1485–90. doi: 10.1128/IAI.68.3.1485-1490.2000
110. Achtman AH, Khan M, MacLennan IC, Langhorne J. Plasmodium chabaudi chabaudi infection in mice induces strong B cell responses and striking but temporary changes in splenic cell distribution. J Immunol. (2003) 171:317–24. doi: 10.4049/jimmunol.171.1.317
111. Cadman ET, Abdallah AY, Voisine C, Sponaas A-M, Corran P, Lamb T, et al. Alterations of splenic architecture in malaria are induced independently of toll-like receptors 2, 4, and 9 or MyD88 and may affect antibody affinity. Infect Immunity. (2008) 76:3924. doi: 10.1128/IAI.00372-08
112. Gueirard P, Tavares J, Thiberge S, Bernex F, Ishino T, Milon G, et al. Development of the malaria parasite in the skin of the mammalian host. Proc Natl Acad Sci USA. (2010) 107:18640–5. doi: 10.1073/pnas.1009346107
113. Roestenberg M, McCall M, Hopman J, Wiersma J, Luty AJF, Van Gemert GJ, et al. Protection against a malaria challenge by sporozoite inoculation. New Engl J Med. (2009) 361:468–77. doi: 10.1056/NEJMoa0805832
114. Bijker EM, Teirlinck AC, Schats R, Van Gemert G-J, Van De Vegte-Bolmer M, Van Lieshout L, et al. Cytotoxic markers associate with protection against malaria in human volunteers immunized with Plasmodium falciparum sporozoites. J Infect Dis. (2014) 210:1605–15. doi: 10.1093/infdis/jiu293
115. Ishino T, Yano K, Chinzei Y, Yuda M. Cell-passage activity is required for the malarial parasite to cross the liver sinusoidal cell layer. PLoS Biol. (2004) 2:e4. doi: 10.1371/journal.pbio.0020004
116. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. (2006) 43:S54–62. doi: 10.1002/hep.21060
117. Goddard S, Youster J, Morgan E, Adams DH. Interleukin-10 secretion differentiates dendritic cells from human liver and skin. Am J Pathol. (2004) 164:511–9. doi: 10.1016/S0002-9440(10)63141-0
118. Pillarisetty VG, Shah AB, Miller G, Bleier JI, DeMatteo RP. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J Immunol. (2004) 172:1009. doi: 10.4049/jimmunol.172.2.1009
119. Bamboat ZM, Stableford JA, Plitas G, Burt BM, Nguyen HM, Welles AP, et al. Human liver dendritic cells promote T cell hyporesponsiveness. J Immunol. (2009) 182:1901. doi: 10.4049/jimmunol.0803404
120. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
121. Knolle PA, Uhrig A, Hegenbarth S, Loser E, Schmitt E, Gerken G, et al. IL-10 down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules. Clin Exp Immunol. (1998) 114:427–33. doi: 10.1046/j.1365-2249.1998.00713.x
122. Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med. (2000) 6:1348–54. doi: 10.1038/82161
123. Bertolino P, McCaughan GW, Bowen DG. Role of primary intrahepatic T-cell activation in the ‘liver tolerance effect'. Immunol Cell Biol. (2002) 80:84–92. doi: 10.1046/j.0818-9641.2001.01048.x
124. Ebrahimkhani MR, Mohar I, Crispe IN. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology. (2011) 54:1379–87. doi: 10.1002/hep.24508
125. Knoll P, Schlaak J, Uhrig A, Kempf P, zum Büschenfelde KH, Gerken G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide. (LPS) challenge. J Hepatol. (1995) 22:226–9. doi: 10.1016/0168-8278(95)80433-1
126. You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. (2008) 48:978–90. doi: 10.1002/hep.22395
127. Kuniyasu Y, Marfani SM, Inayat IB, Sheikh SZ, Mehal WZ. Kupffer cells required for high affinity peptide-induced deletion, not retention, of activated CD8+ T cells by mouse liver. Hepatology. (2004) 39:1017–27. doi: 10.1002/hep.20153
128. Chen Y, Liu Z, Liang S, Luan X, Long F, Chen J, et al. Role of Kupffer cells in the induction of tolerance of orthotopic liver transplantation in rats. Liver Transpl. (2008) 14:823–36. doi: 10.1002/lt.21450
129. Bertolino P, Trescol-Biemont MC, Rabourdin-Combe C. Hepatocytes induce functional activation of naive CD8+ T lymphocytes but fail to promote survival. Eur J Immunol. (1998) 28:221–36. doi: 10.1002/(SICI)1521-4141(199801)28:01<221::AID-IMMU221>3.0.CO;2-F
130. Leiriao P, Mota MM, Rodriguez A. Apoptotic plasmodium-infected hepatocytes provide antigens to liver dendritic cells. J Infect Dis. (2005) 191:1576–81. doi: 10.1086/429635
131. Harshyne LA, Watkins SC, Gambotto A, Barratt-Boyes SM. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J Immunol. (2001) 166:3717–23. doi: 10.4049/jimmunol.166.6.3717
132. Sack BK, Mikolajczak SA, Fishbaugher M, Vaughan AM, Flannery EL, Nguyen T, et al. Humoral protection against mosquito bite-transmitted Plasmodium falciparum infection in humanized mice. npj Vaccines. (2017) 2:27. doi: 10.1038/s41541-017-0028-2
133. Tyagi RK, Tandel N, Deshpande R, Engelman RW, Patel SD, Tyagi P. Humanized mice are instrumental to the study of Plasmodium falciparum infection. Front Immunol. (2018) 9:2550. doi: 10.3389/fimmu.2018.02550
134. Boddey JA, Cowman AF. Plasmodium nesting: remaking the erythrocyte from the inside out. Annu Rev Microbiol. (2013) 67:243–69. doi: 10.1146/annurev-micro-092412-155730
135. Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, Feldman M, et al. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. (1995) 82:77–87. doi: 10.1016/0092-8674(95)90054-3
136. Urban BC, Willcox N, Roberts DJ. A role for CD36 in the regulation of dendritic cell function. Proc Natl Acad Sci USA. (2001) 98:8750–5. doi: 10.1073/pnas.151028698
137. Gilson PR, Crabb BS. Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int J Parasitol. (2009) 39:91–6. doi: 10.1016/j.ijpara.2008.09.007
138. Coban C, Ishii KJ, Sullivan DJ, Kumar N. Purified malaria pigment (hemozoin) enhances dendritic cell maturation and modulates the isotype of antibodies induced by a DNA vaccine. Infect Immunity. (2002) 70:3939. doi: 10.1128/IAI.70.7.3939-3943.2002
139. Skorokhod OA, Alessio M, Mordmüller B, Arese P, Schwarzer E. Hemozoin (malarial pigment) inhibits differentiation and maturation of human monocyte-derived dendritic cells: a peroxisome proliferator-activated receptor-γ-mediated effect. J Immunol. (2004) 173:4066. doi: 10.4049/jimmunol.173.6.4066
140. Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. Innate sensing of malaria parasites. Nat Rev Immunol. (2014) 14:744–57. doi: 10.1038/nri3742
141. Escalante AA, Lal AA, Ayala FJ. Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics. (1998) 149:189–202.
142. Wilson KL, Xiang SD, Plebanski M. A model to study the impact of polymorphism driven liver-stage immune evasion by malaria parasites, to help design effective cross-reactive vaccines. Front Microbiol. (2016) 7:303. doi: 10.3389/fmicb.2016.00303
143. Kraemer SM, Smith JD. A family affair: var genes, PfEMP1 binding, and malaria disease. Curr Opin Microbiol. (2006) 9:374–80. doi: 10.1016/j.mib.2006.06.006
144. Demar M, Legrand E, Hommel D, Esterre P, Carme B. Plasmodium falciparum malaria in splenectomized patients: two case reports in French Guiana and a literature review. Am J Trop Med Hyg. (2004) 71:290–3. doi: 10.4269/ajtmh.2004.71.290
145. Bachmann A, Esser C, Petter M, Predehl S, Von Kalckreuth V, Schmiedel S, et al. Absence of erythrocyte sequestration and lack of multicopy gene family expression in Plasmodium falciparum from a splenectomized malaria patient. PLoS ONE. (2009) 4:e7459. doi: 10.1371/journal.pone.0007459
146. Dewalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, et al. Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J Immunol. (2007) 178:6033–7. doi: 10.4049/jimmunol.178.10.6033
147. Piva L, Tetlak P, Claser C, Karjalainen K, Renia L, Ruedl C. Cutting edge: Clec9A+ dendritic cells mediate the development of experimental cerebral malaria. J Immunol. (2012) 189:1128–32. doi: 10.4049/jimmunol.1201171
148. Gowda DC, Wu X. Parasite recognition and signaling mechanisms in innate immune responses to malaria. Front Immunol. (2018) 9:3006. doi: 10.3389/fimmu.2018.03006
149. Urban BC. Peripheral blood dendritic cells in children with acute Plasmodium falciparum malaria. Blood. (2001) 98:2859–61. doi: 10.1182/blood.V98.9.2859
150. Arama C, Giusti P, Boström S, Dara V, Traore B, Dolo A, et al. Interethnic differences in antigen-presenting cell activation and TLR responses in malian children during Plasmodium falciparum malaria. PLoS ONE. (2011) 6:e18319. doi: 10.1371/journal.pone.0018319
151. Giusti P, Urban BC, Frascaroli G, Albrecht L, Tinti A, Troye-Blomberg M, et al. Plasmodium falciparum-infected erythrocytes and beta-hematin induce partial maturation of human dendritic cells and increase their migratory ability in response to lymphoid chemokines. Infect Immun. (2011) 79:2727–36. doi: 10.1128/IAI.00649-10
152. Seixas E, Cross C, Quin S, Langhorne J. Direct activation of dendritic cells by the malaria parasite, Plasmodium chabaudi chabaudi. Eur J Immunol. (2001) 31:2970–8. doi: 10.1002/1521-4141(2001010)31:10<2970::AID-IMMU2970>3.0.CO;2-S
153. Urban BC, Cordery D, Shafi MJ, Bull PC, Newbold CI, Williams TN, et al. The frequency of BDCA3-positive dendritic cells is increased in the peripheral circulation of Kenyan children with severe malaria. Infect Immunity. (2006) 74:6700–6. doi: 10.1128/IAI.00861-06
154. Dolo A, Modiano D, Maiga B, Daou M, Dolo G, Guindo H, et al. Difference in susceptibility to malaria between two sympatric ethnic groups in Mali. Am J Trop Med Hyg. (2005) 72:243–8. doi: 10.4269/ajtmh.2005.72.243
155. Guermonprez P, Helft J, Claser C, Deroubaix S, Karanje H, Gazumyan A, et al. Inflammatory Flt3l is essential to mobilize dendritic cells and for T cell responses during Plasmodium infection. Nat Med. (2013) 19:730–8. doi: 10.1038/nm.3197
156. Karsunky H, Merad M, Cozzio A, Weissman IL, Manz MG. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med. (2003) 198:305–13. doi: 10.1084/jem.20030323
157. Waskow C, Liu K, Darrasse-Jèze G, Guermonprez P, Ginhoux F, Merad M, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. (2008) 9:676–83. doi: 10.1038/ni.1615
158. Loharungsikul S, Troye-Blomberg M, Amoudruz P, Pichyangkul S, Yongvanitchit K, Looareesuwan S, et al. Expression of toll-like receptors on antigen-presenting cells in patients with falciparum malaria. Acta tropica. (2008) 105:10–5. doi: 10.1016/j.actatropica.2007.08.002
159. Pichyangkul S, Yongvanitchit K, Kum-Arb U, Hemmi H, Akira S, Krieg AM, et al. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a toll-like receptor 9-dependent pathway. J Immunol. (2004) 172:4926–33. doi: 10.4049/jimmunol.172.8.4926
160. Loughland JR, Minigo G, Burel J, Tipping PE, Piera KA, Amante FH, et al. Profoundly reduced CD1c+ myeloid dendritic cell HLA-DR and CD86 expression and increased tumor necrosis factor production in experimental human blood-stage malaria infection. Infect Immunity. (2016) 84:1403–12. doi: 10.1128/IAI.01522-15
161. Loughland JR, Minigo G, Sarovich DS, Field M, Tipping PE, Montes De Oca M, et al. Plasmacytoid dendritic cells appear inactive during sub-microscopic Plasmodium falciparum blood-stage infection, yet retain their ability to respond to TLR stimulation. Sci Rep. (2017) 7:2596. doi: 10.1038/s41598-017-02096-2
162. Kho S, Marfurt J, Noviyanti R, Kusuma A, Piera KA, Burdam FH, et al. Preserved dendritic cell HLA-DR expression and reduced regulatory T cell activation in asymptomatic Plasmodium falciparum and P. vivax infection. Infect Immun. (2015) 83:3224–32. doi: 10.1128/IAI.00226-15
163. Ing R, Segura M, Thawani N, Tam M, Stevenson MM. Interaction of mouse dendritic cells and malaria-infected erythrocytes: uptake, maturation, and antigen presentation. J Immunol. (2006) 176:441. doi: 10.4049/jimmunol.176.1.441
164. Perry JA, Rush A, Wilson RJ, Olver CS, Avery AC. Dendritic cells from malaria-infected mice are fully functional APC. J Immunol. (2004) 172:475–82. doi: 10.4049/jimmunol.172.1.475
165. Leisewitz AL, Rockett KA, Gumede B, Jones M, Urban B, Kwiatkowski DP. Response of the splenic dendritic cell population to malaria infection. Infect Immun. (2004) 72:4233–9. doi: 10.1128/IAI.72.7.4233-4239.2004
166. Sponaas A-M, Cadman ET, Voisine C, Harrison V, Boonstra A, O'Garra A, et al. Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J Exp Med. (2006) 203:1427–33. doi: 10.1084/jem.20052450
167. Benson RA, MacLeod MK, Hale BG, Patakas A, Garside P, Brewer JM. Antigen presentation kinetics control T cell/dendritic cell interactions and follicular helper T cell generation in vivo. Elife. (2015) 4:e06994. doi: 10.7554/eLife.06994
168. Wykes MN, Liu XQ, Beattie L, Stanisic DI, Stacey KJ, Smyth MJ, et al. Plasmodium strain determines dendritic cell function essential for survival from malaria. PLoS Pathog. (2007) 3:e96. doi: 10.1371/journal.ppat.0030096
169. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. (2012) 13:188–95. doi: 10.1038/ni.2180
170. Jagannathan P, Eccles-James I, Bowen K, Nankya F, Auma A, Wamala S, et al. IFNγ/IL-10 co-producing cells dominate the CD4 response to malaria in highly exposed children. PLOS Pathogens. (2014) 10:e1003864. doi: 10.1371/journal.ppat.1003864
171. Villegas-Mendez A, Inkson CA, Shaw TN, Strangward P, Couper KN. Long-lived CD4+IFN-γ+T cells rather than short-lived CD4+IFN-γ+IL-10+T cells initiate rapid IL-10 production to suppress anamnestic T cell responses during secondary malaria infection. J Immunol. (2016) 197:3152–64. doi: 10.4049/jimmunol.1600968
172. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. (2013) 14:1212–8. doi: 10.1038/ni.2762
173. Honda T, Egen JG, Lämmermann T, Kastenmüller W, Torabi-Parizi P, Germain RN. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity. (2014) 40:235–47. doi: 10.1016/j.immuni.2013.11.017
174. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
175. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. (2009) 229:114–25. doi: 10.1111/j.1600-065X.2009.00767.x
176. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
177. Brooks DG, Ha S-J, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci USA. (2008) 105:20428–33. doi: 10.1073/pnas.0811139106
178. Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
179. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity. (2014) 40:289–302. doi: 10.1016/j.immuni.2014.01.005
180. Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. (2012) 37:1130–44. doi: 10.1016/j.immuni.2012.08.021
Keywords: dendritic cells, T cells, malaria, DC-T cell interaction, T helper cells, Tfh
Citation: Osii RS, Otto TD, Garside P, Ndungu FM and Brewer JM (2020) The Impact of Malaria Parasites on Dendritic Cell–T Cell Interaction. Front. Immunol. 11:1597. doi: 10.3389/fimmu.2020.01597
Received: 27 January 2020; Accepted: 16 June 2020;
Published: 24 July 2020.
Edited by:
Yasuyuki Goto, The University of Tokyo, JapanReviewed by:
Elizabeth Helen Aitken, The University of Melbourne, AustraliaGaurav Gupta, NIIT University, India
Copyright © 2020 Osii, Otto, Garside, Ndungu and Brewer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James M. Brewer, amFtZXMuYnJld2VyQGdsYXNnb3cuYWMudWs=