 Marta Dratwa1*
Marta Dratwa1* Barbara Wysoczańska
Barbara Wysoczańska Piotr Łacina
Piotr Łacina Katarzyna Bogunia-Kubik
Katarzyna Bogunia-Kubik- 1Laboratory of Clinical Immunogenetics and Pharmacogenetics, Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wroclaw, Poland
- 2Department of Computer Engineering, Faculty of Electronics, Wrocław University of Science and Technology, Wroclaw, Poland
Telomerase reverse transcriptase (TERT) is a catalytic subunit of telomerase. Telomerase complex plays a key role in cancer formation by telomere dependent or independent mechanisms. Telomere maintenance mechanisms include complex TERT changes such as gene amplifications, TERT structural variants, TERT promoter germline and somatic mutations, TERT epigenetic changes, and alternative lengthening of telomere. All of them are cancer specific at tissue histotype and at single cell level. TERT expression is regulated in tumors via multiple genetic and epigenetic alterations which affect telomerase activity. Telomerase activity via TERT expression has an impact on telomere length and can be a useful marker in diagnosis and prognosis of various cancers and a new therapy approach. In this review we want to highlight the main roles of TERT in different mechanisms of cancer development and regulation.
Introduction
In most human cancers, telomerase is reactivated during carcinogenesis by expression of the catalytic subunit telomerase reverse transcriptase (TERT). TERT plays a key role in cancer formation, ensuring chromosomal stability by maintaining telomere length, and allowing cells to avert senescence. It constitutes a limiting factor for formation of the telomerase complex in cancer cells (1). TERT is one of two major components of the larger telomerase complex, which extends telomeres by adding specific short repetitive DNA sequences. These tandem repeats are bound by the shelterin complex, which is composed of six proteins: telomere repeat factor 1 and 2 (TRF1, TRF2), protection of telomeres 1 (POT1), TRF1-interacting nuclear protein 2 (TIN2), tripeptidyl peptidase I (TPP1), and repressor/activator protein 1 (RAP1) (Figure 1) (2). The Shelterin complex plays a fundamental role in protecting chromosome ends and in telomere length regulation (3, 4).
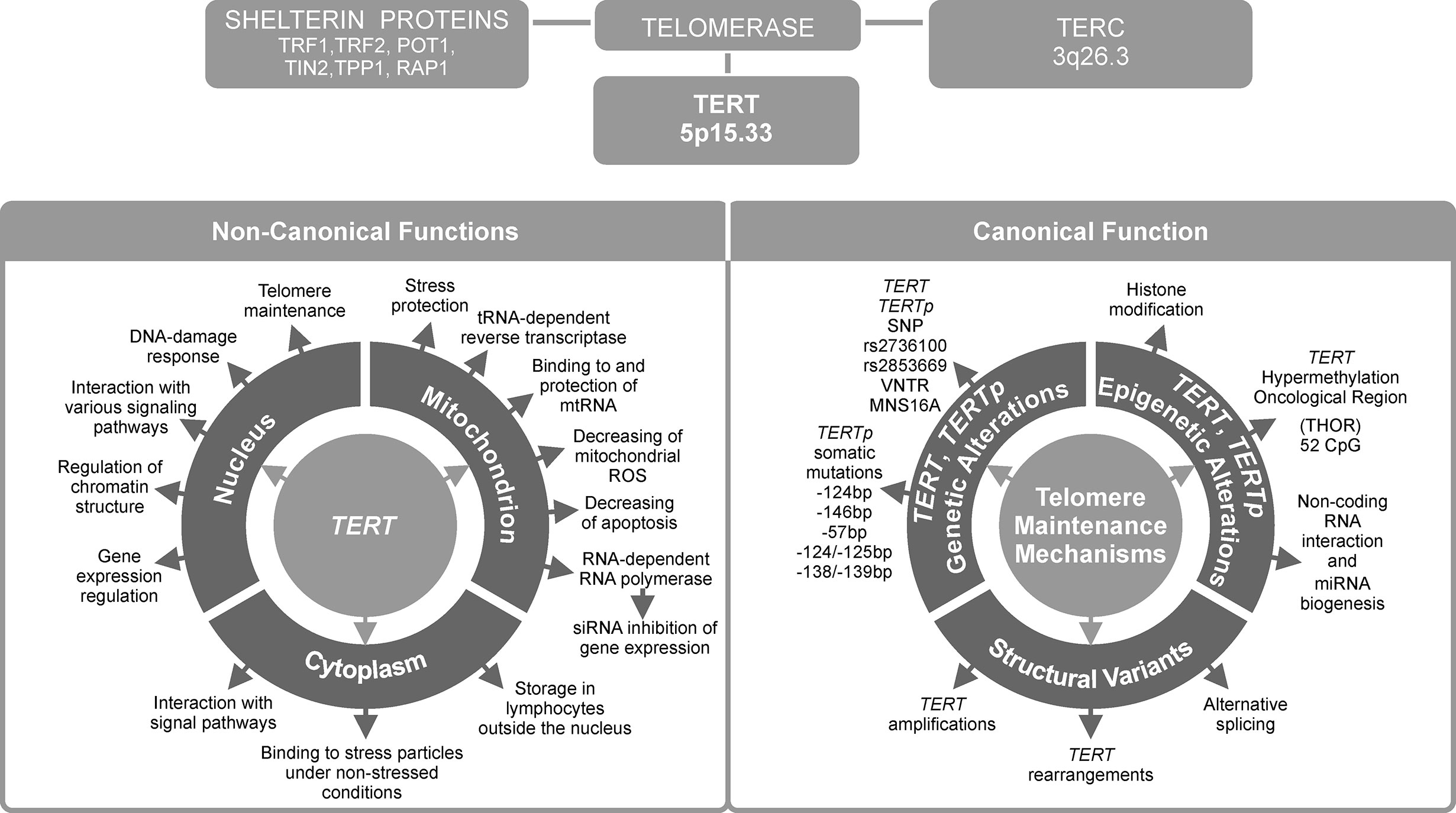
Figure 1 Telomerase reverse transcriptase (TERT) is the most important telomerase subunit and plays a major role in telomerase activity and in other telomere-unrelated processes in cancer development. Telomerase is a complex reverse transcriptase that comprises, besides TERT, an RNA template for telomere repeats (TERC), and a group of proteins called shelterin complex (upper panel). While the primary function of TERT is telomere lengthening (canonical function, lower panel, on the right), there are also other, telomere-unrelated functions (non-canonical functions, lower panel, on the left).
The TERT gene is situated at chromosome 5p15.33 in humans, and is an integral and essential part of the telomerase holoenzyme. TERT gene is 42 kb long and consists of 15 introns and 16 exons with a 260 bp promoter core (5). The reverse transcriptase domain is encoded by 5–9 exons. The TERT transcript can be spliced into 22 isoforms (6). TERT promoter (TERTp) region contains GC boxes that bind the zinc finger transcription factor Sp1, which increases TERT transcription, and E-boxes that bind both transcriptional enhancers and repressors. TERTp lacks a TATA box but it contains binding sites for many different transcription factors (7).
Another major component of the telomerase complex is telomerase RNA component (TERC). It is an RNA sequence, which functions as a template for synthesis of telomeres by TERT. These two main components of telomerase are accompanied by a host of auxiliary proteins, including dyskerin (DKC1), telomerase Cajal body protein 1 (TCAB1), non-histone chromosome protein 2 (NHP2), nucleolar protein 10 (NOP10), glycine arginine rich 1 (GAR1), heat shock protein 90 (HSP90) and serine and arginine rich splicing factor 11 (SRSF11) (8). This complex is essential for maintaining telomere homeostasis, which is crucial in regulation of aging and cancer development (9).
Over 80% of tumors adopt various regulatory strategies, known as telomere maintenance mechanisms (TMMs). They maintain telomere length by reactivating telomerase, and therefore are known as TERT canonical functions (10). Individual TMMs are specific for cancer type, tissue histotype, and cell lines. The most important TMMs are (1) TERT gene rearrangements and TERT and TERC gene amplification, (2) TERTp somatic mutations, (3) epigenetic alterations, (4) transcription factor binding, (5) polymorphic variants within TERT gene body and TERTp, and (6) alternative splicing (Figure 1). Each of these mechanisms will be described in detail in subsequent sections of this manuscript.
Approximately 10–15% of tumor cells acquire immortality through a telomerase-independent mechanism, namely alternative lengthening of telomeres (ALT) (11). On the other hand, the so called non-defined telomere maintenance mechanism (NDTMM) are activated when both telomerase (or TERT) expression and ALT are absent (10, 12). While telomere lengthening is considered a major function of telomerase, it can also modulate expression of various genes, such as nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) and Wnt/β-catenin signaling pathway genes (13, 14). Such alternative, non-telomere-related roles are known as non-canonical functions of TERT. They will be presented in the last two chapters of this review (Figure 1), together with potential consequences of TERT telomere-unrelated functions for the development of anti-cancer strategies and applications of TERT as a potential therapeutic target.
Chromosomal Rearrangements
Chromosomal rearrangements are a type of mutation that results in a change in chromosome structure. They may involve duplications, amplifications, insertions, interchromosomal changes, inverted orientations, or deletions (15). A concept associated with chromosomal rearrangements is copy number variation (CNV). CNV describes the fact that some sections of the genome may be repeated and the number of these repeats may be different between individuals. CNVs involve 50 bp to over 1,000,000 bp fragments of gene regulatory regions (16). They are associated with gene expression and phenotype by affecting gene copy number (17). Chromosomal rearrangements may affect TERT gene copy number and are a known TMM. They may involve insertion of active enhancers close to the TERT gene and increasing TERT expression. A common process is TERT amplification, which can arise from telomere dysfunction (18). It results from a dysfunctional telomere, promoting fusion of chromosome ends, and subsequently forming a dicentric chromosome (19). Several studies showed that chromosomal rearrangements at the TERT locus may be associated with cancer development and as was observed, e.g., in the case of neuroblastoma (20–22). Furthermore, a major study specifically focusing on TERT gene amplification found it to occur in many cancers, such as esophageal, ovarian cancer, and squamous cell carcinoma (12). In addition, other authors found telomerase activity to be the highest in tumors with TERT amplification (22, 23). Gay-Bellile et al. observed increased number of TERT gene copies in breast cancer cells, and upregulation of TERT gene was associated with worse prognosis in breast cancer, thyroid carcinoma, and lung adenocarcinoma (24). This suggests that TERT rearrangement could be a critical step in cancer development.
TERT Promoter Hot-Spot Mutations
TERT somatic mutations are the most common non-coding mutations in human cancer cells. While they are documented to occur in the coding region, they are far more common in the promoter region. Some TERTp mutations were shown to affect TERT expression, telomere length and telomerase activity by abrogating telomerase silencing (25). TERTp mutations occur in specific clinical and phenotypic subtypes of various cancers and cell lines, and recurrent mutations have been identified in 19% of cancers (26). In cancer cells, TERTp mutations are generally associated with higher TERT expression level.
The two most common TERTp mutations are C>T transitions, located at -124 bp, and -146 bp from the transcription start site (TSS). They are also referred to as C228T and C250T, respectively (27, 28). These mutations result in an 11 bp nucleotide fragment providing a new consensus binding site for E-twenty-six (ETS) transcription factors (29). Many other somatic mutations were detected that occur in the TERTp in cancer, although less frequently than C228T and C250T and they also may contribute to increased TERT transcription. A group of CC>TT substitutions, located at −124/−125 and −138/−139 bp relative to the TSS, result in an ETS binding site in skin cancers (30). In melanoma patients, the −138/−139 mutation correlated with more adverse survival (31). In basal cell carcinoma, Maturo et al. observed additional TERTp alterations other than the recurrent TERTp hotspot mutations (32).
TERTp mutations were found in several tumor types with different frequencies. Generally, two types of tumors can be distinguished: those with low and high proliferative potential (33). Tumors with high levels of TERTp mutation, such as, melanoma, glioblastoma, bladder cancer or hepatocellular carcinoma (somatic mutation levels of 64–80%, ~84%, ~65%, and 32–45%, respectively) are characterized by low proliferative potential (28, 33–36). Tumors with low or undetectable level of TERTp mutation have high proliferative potential, e.g., breast cancer 0.9% (37), testicular cancer 3% (38), intestinal cancer (34) and acute myeloid leukemia and non-Hodgkin’s lymphoma (39, 40). It is important to note that TERTp mutation was not detected in hematological cell lines cultured in vitro (41), as well as in a group of patients with hematological malignances, with the exception of mantle cell lymphoma patients (42). In the case of cancers with low proliferative potential, TERTp mutation is considered a late tumorigenic event (33). In some other cancers, e.g., basal cell carcinoma, TERTp mutations may appear as a result of environmental factors, such as contact with carcinogens, in which case it is considered as an early tumorigenic event (10, 26). TERTp mutations are thought to contribute to tumorigenesis in two distinct phases. In the first phase, TERTp mutations heal the shortest telomeres, thus extending life span of cells containing them, but they fail to avert general telomere shortening. This leads to the second phase, where the critically short telomeres result in genomic instability, causing further increase in telomerase expression needed for continued cell proliferation (43).
Another interesting aspect of TERTp mutation is the possible cooperation with mutations, such as those in genes coding for BRAF, FGFR3, and IDH (44–48). BRAF is a serine/threonine kinase and its mutation results in activation of the mitogen-activated protein kinase (MAPK) and/or phosphatidylinositol 3-kinase–serine threonine protein kinase (PI3K-AKT) pathways. This leads to upregulation of the ETS system and induction of TERT expression. Out of a variety of BRAF mutations, V600E (a glutamic acid to valine substitution) is the most frequent. This mutation leads to increased GABPA-GABPB complex formation and activation of TERT expression (29, 49). Co-existence of TERTp mutation and V600E is associated with poor prognosis in patients with thyroid cancer, particularly papillary thyroid cancer (8, 50). Fibroblast growth factor receptor 3 (FGFR3) is another example of genetic alterations interacting with TERTp. Its mutation is well described in urothelial carcinoma (51). FGFR3 belongs to the tyrosine kinase receptor family and stimulates the RAS-mitogen-activated protein kinase and PI3K-AKT pathways. TERTp and FGFR3 mutations are more often present together than alone (47). Co-occurrence of these mutations may support creation of tumors with poor prognosis (10). Additionally, tumors with TERTp and/or FGFR3 mutations had shorter telomeres when compared to tumors without these mutations (47). Malignant gliomas, acute myeloid leukemia and cholangiocarcinoma, are often associated with mutations in isocitrate dehydrogenase 1 and/or 2 (IDH1 and/or IDH2) (52). These somatic mutations occur at arginine residues of the IDH active site (namely, IDH1R132H, IDH2R140Q, and IDH2R172K) (53). According to Diplas et al., TERTp and IDH mutation status can be used together to classify over 80% of all diffuse gliomas (54). A previous study suggested that presence of TERTp mutation and additional 1p/19q co-deletion and also mutation within the IDH gene led to a better response to chemotherapy and better outcome in glioma patients (55).
In conclusion, TERTp mutation status, alone or in combination with mutations in other genes, can be used to characterize distinguish various types of tumors, as well as predict prognosis and outcome. While TERTp mutation status appears to significantly impact cancer development, some cancers, such as prostate, lung, breast, colorectal, and hematological malignancies display telomerase activity, even though they contain few TERTp mutations (24, 39, 40, 56, 57). Consequently, other undefined or epigenetic mechanisms of TERT-upregulating are expected to exist.
Epigenetic Modifications
DNA Methylation
Epigenetics describes stable, and possibly heritable changes in activity and expression, which are not associated with any underlying changes in DNA sequence (58). DNA methylation is a common epigenetic mechanism that is essential for regulation of gene expression. It occurs primarily at non-coding regions of DNA characterized by high frequency of CG repeats. Such regions, called CpG islands, are most commonly found in gene promoters. 60–70% of genes contain promoters with these CpG islands (59).
Tissue-specific DNA hypo- or hypermethylation is considered to be important in regulation of gene expression during development. Such tissue-specific DNA hypermethylation is present at promoters rich in CpG islands (60, 61). Promoter DNA methylation is ubiquitous in human cells and is one of the most commonly encountered mechanisms of gene expression regulation. Promoter methylation generally causes gene silencing by interfering with transcription factor bindings sites. Therefore, promoters of actively transcribed genes are normally unmethylated (62). However, DNA hypermethylation may occur at introns/exons (rather than promoters) of actively transcribed genes, as well as at intra- and intergenic enhancers (63). Having an important role in tissue-specific regulation of transcription, DNA hypermethylation may be considered as a marker for a broad variety of diseases and cancers (64, 65).
Promoter methylation is also a major regulatory element of TERT expression, correlating both with TERT mRNA levels and telomerase activity (66). An approximately 300 bp part of TERTp situated on either site of the TSS is unmethylated in actively transcribed TERT. However, Castelo-Branco et al. and, more recently, Lee et al. documented that hypermethylation of the TERT gene correlates with telomerase activity in different types of cancers (67–69). A study on patients with pediatric brain tumors brought to light a new group of 5 CpG islands located upstream of the TSS, which were hypermethylated and correlated with TERT expression. On the other hand, healthy tissues without TERT expression did not have this hypermethylation (59). This pattern is counter to the generally established functions of DNA methylation (63). Lee et al. discovered that it is due to presence of a new, larger region known as the TERT Hypermethylated Oncological Region (THOR). It is located distal to the TSS and is composed of 52 CpG islands (69, 70). This means that there are two regions of TERTp regarding methylation status in telomerase-positive cells: the unmethylated proximal TERT core promoter, which is where transcription factors are usually bound, and the hypermethylated THOR, located further away from the core promoter (67, 69, 71) (Figure 2). The unusual nature of THOR methylation is due to it acting as a transcription repressor in its unmethylated state. Recently, several authors documented an association between THOR hypermethylation and cancer progression coupled with TERT upregulation in pancreatic and gastric cancers (72, 73). Interestingly, both THOR and the TERTp region proximal to TSS were mostly unmethylated in normal thyroid tissue (49).
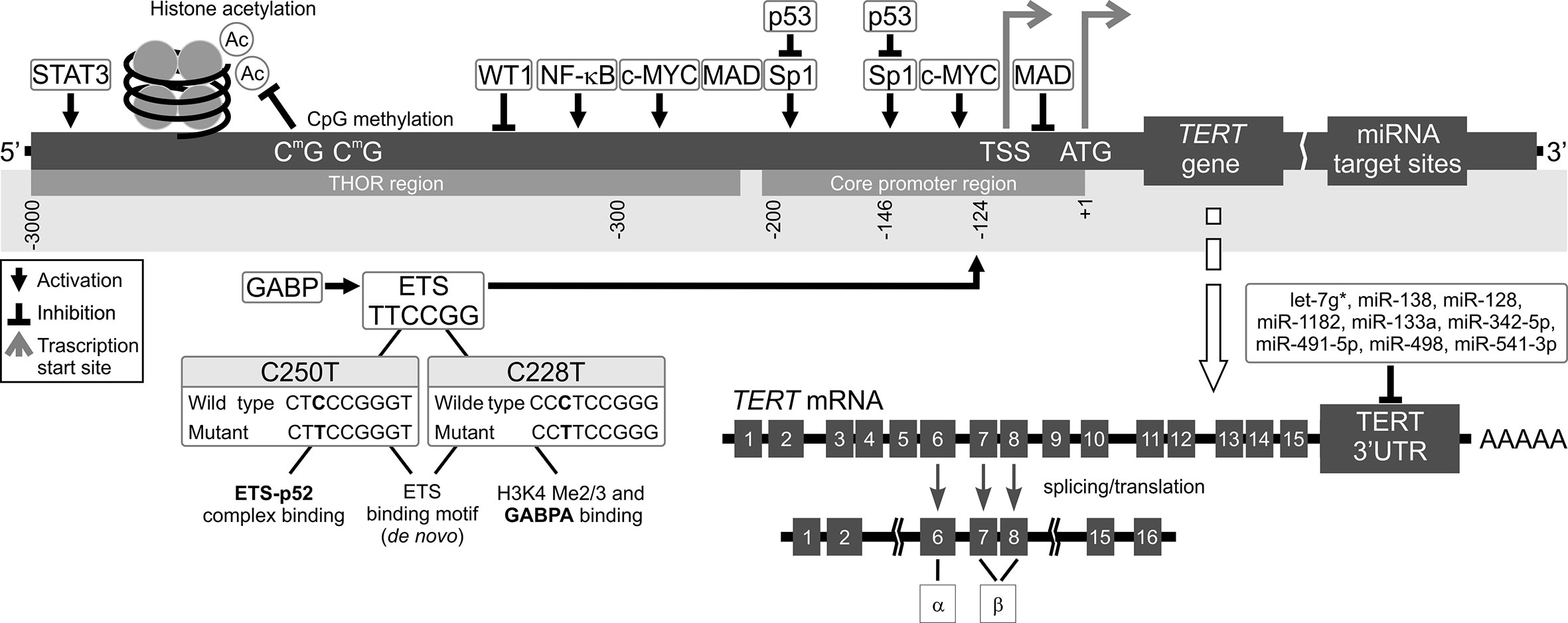
Figure 2 Mechanisms of TERT transcription regulation. The figure shows various mechanisms regulating TERT expression at the transcriptional level. Transcription factors: activators (e.g., c-MYC, SP1, STAT3, NF-κB, and ETS), repressors (e.g., MAD, p53, and WT1), and their respective binding sites are shown. Binding of these transcriptional agents to TERT could be controlled by DNA methylation (CpG sites) in the TERT Hypermethylation Oncological Region (THOR). Two main hotspot mutations within TERTp, -146C > T (C250T) and -124C > T (C228T) upstream of the transcription start site (TSS) generate new E-twenty-six (ETS) binding sites, leading to GABP recruitment and, eventually, TERT transcription. Alternatively spliced variants of TERT, which do not have telomerase activity, could be also generated. Most tissues and organs express no or very low levels of TERT mRNA, dependent on histone markers that are correlated with passive or active transcription in many cells. The figure also shows different miRNAs at the 3’UTR that inhibit translation of TERT.
Regarding TERTp mutation status, it appears that it does interfere with effects of THOR hypermethylation in cancers where TERTp mutation is common. Furthermore, presence of both of these factors may have a synergistic effect on TERT expression. In a study on urothelial bladder cancer patients, co-occurrence of THOR hypermethylation and TERTp mutation was a marker of higher risk of disease recurrence and progression (74). Likewise, a study on melanoma patients showed a similar effect on reduced recurrence-free survival (75). These and other examples show that TERTp mutation coupled with THOR hypermethylation is a better marker of disease progression than TERTp mutation alone. Nevertheless, it should be noted that THOR hypermethylation does not associate with progression in a small group of cancers such as esophageal cancer, meningioma or pituitary adenoma (76).
Another interesting issue is the possible interplay between TERTp mutation, methylation, and histone modifications, which constitute yet another epigenetic mechanism affecting chromatin accessibility. A study by Stern et al. on monoallelic cancers showed that cancers without a specific TERTp mutation at −124 from the TSS had promoter hypermethylation, which was accompanied by repressive histone H3K27me3 methylation, leading to gene inactivation. They hypothesized that presence of this mutation coupled with low TERTp methylation discourages H3K27me3 histone methylation in transcriptionally active TERT (70). Interestingly, one study showed that TERTp hypermethylation was present in both melanoma and normal skin cells. However, only in melanoma cells with TERTp mutation did this hypermethylation correspond to increased TERT expression and chromatin accessibility (77). A further study by McKelvey et al. on thyroid cancer cell lines heterozygous for TERTp mutation demonstrated conclusively that TERTp methylation was allele-specific, whereby TERTp with mutation was significantly less methylated than wildtype promoter. Moreover, MYC, a transcription activator, bound only to the hypomethylated mutated TERTp, resulting in monoallelic expression (MAE) in heterozygous cells (29). MAE is one of two TERT expression categories as described by Huang et al., the other being biallelic expression (BAE, both alleles transcriptionally active). These two expression patterns appeared to be specific for many cancers, although some cancers exhibited variation between MAE and BAE in differed cell lines (34). However, a later study by Rowland et al. showed that this simple classification into MAE and BAE-specific cancer cell lines does not sufficiently describe the complex nature of TERT expression. In a study conducted on a single cell-level, they found great heterogeneity in TERT expression between various cells, within both the cell lines described as MAE, and those described as BAE by Huang et al. (78).
micro-RNA
Most recent studies focus on TERT regulation at the transcriptional level. Meanwhile, post-transcriptional regulation by microRNAs (miRNAs), has not been expensively studied. miRNAs are a class of small non-coding RNAs (~22–24 nucleotides) (79). miRNA recognition sites are typically located in 3′ untranslated regions (3′UTRs) of mRNA (Figures 1 and 2). miRNAs binding to 3’UTR generally silences the transcript, thus reducing gene expression. miRNAs are ubiquitous elements of gene regulation, and control many different biological processes. In cancer, miRNAs function as gene regulatory molecules, acting as tumor suppressors or oncogenic drivers (18, 80).
Various miRNAs are known as regulators of TERT. In particular let-7g-3p, miR-128, miR-133a, miR-138-5p, miR-498, miR-541-3p, and miR-1182, downregulate expression of TERT and telomerase activation (18, 81). Functional analyses indicated that overexpression of miR-138-5p and miR-422a significantly inhibit TERT expression through interaction with TERT 3’UTR in colorectal cancer cells (79, 82). Moreover, miR-138-5p represses TERT protein expression in human anaplastic thyroid carcinoma and cervical cancer cells (79, 83). Likewise, miR-1182, miR-1266, miR-532, miR-1207-5p, and miR-3064 suppress gastric, bladder, ovarian cancer growth and invasion by binding to the TERT 3’UTR (10, 79, 84, 85). Furthermore, miR-128 was found to control TERT expression in HeLa and teratoma cell lines (81, 86).
miRNAs can also regulate TERT indirectly by controlling expression of various transcription factors. Accordingly, c-MYC, a major regulator of TERT, was regulated by miR-494 and miR-1294 in esophageal squamous cell carcinoma and pancreatic cancer. Additionally, c-MYC and FoxM1 were targeted by a known tumor suppressor, miR-34a, causing senescence in cells (18). Interestingly, the study of Lassmann et al. suggested that TERT is able to regulate miRNA levels at the early phase of miRNA processing. They demonstrated that deletion of TERT resulted in a decrease of most mature miRNAs (87).
Transcription Factors
Transcriptional Activators
TERTp contains binding sites for a huge number of transcriptional activators and repressors that directly or indirectly regulate gene expression. Multiple pathways, such as RAS/RAF/MEK/MAPK, PI3K/Akt/mTOR, IKK/NF-κB, transforming growth factor β/Smads, PKC, and the JAK-STAT pathway regulate TERT expression and telomerase enzymatic activity (88). In fact, most transcription factors have been identified as possible TERT gene regulators, such as protein kinases, growth factors, and oncogenic proteins. Canonical positive regulators of TERT transcription include the oncogene c-MYC, Sp1, NF-κB, STAT family of proteins, AP-2, and GSC. These activatory transcription factors will be described in detail in the following section.
MYC encodes a basic helix-loop-helix leucine zipper (bHLH-LZ) transcriptional factor called c-MYC (89, 90). The MYC gene family regulates expression of genes implicated in many processes, such as proliferation, cell growth, differentiation, self-renewal, apoptosis (91, 92). It is essential for embryonic development and it is expressed in normal somatic cells. There are several ways for healthy cells to control MYC levels, such as targeted degradation by the ubiquitin-proteasome system (92). Chromosome translocations, gene amplification, retroviral insertion or mutations of MYC gene are tumorigenic in mice and correlate with development of most human cancers (93, 94). c-MYC functions is dependent on heterodimerization with MAX (90, 95). While MYC gene contains a transcription activation domain, no such regulatory domain has been reported for MAX (96). The c-MYC/MAX heterodimers can bind to specific DNA sequences located within the core promoter region, known as E-box motifs (5′-CACGTG-3′), thus activating various genes (90, 92). c-MYC activates telomerase by inducing expression of TERT (90, 94). In addition, TERT is responsible for maintenance of c-MYC levels and regulates c-MYC proteasomal degradation (97).
The core promoter of TERT also contains specificity protein 1 (Sp1) binding sites that are necessary for TERT expression. Sp1 belongs to the family of nuclear proteins called Sp/KLF (specificity protein/Krüppel-like factor) that binds GC-(GGGGCGGGG) and GT-(GGTGTGGGG) rich elements (98, 99). It is one of the best characterized transcriptional activators of housekeeping genes and other TATA-less genes (89, 99). Sp1 regulates processes such as inflammation, carcinogenesis, senescence, hormonal activation, apoptosis and angiogenesis (98). Transcriptional activity of Sp1 is regulated by a few post-translational modifications (glycosylation, acetylation, phosphorylation) and by direct interaction with other proteins, including other transcription factors, nuclear factors, oncogenes, and tumor suppressors. Sp1-silencing completely inhibits telomerase activity by suppressing TERT expression, leading to apoptosis. Furthermore, mutations in Sp1 binding sites (GC‐boxes) significantly decrease transcriptional activity of TERTp, suggesting that Sp1 protein is involved in TERT transcription (100). Some reports indicated that cooperation between Sp1 and c-MYC drives cell type-specific TERT expression. This is further substantiated by the fact that normal cells have lower levels of Sp1 and c-MYC than cancer cells. However, Sp1 would be a weak candidate for a biomarker of cancer‐specific TERT expression because of its ubiquitous expression in normal cells (89, 100).
NF-κB is well known for playing a major role in inflammation, tumorigenesis, cytokine and chemokine expression, stress regulation, cell division and transformation (101, 102). NF-κB regulates expression of apoptosis inhibitors. The NF-κB signaling pathway is a master regulator of TERT activation in cancer cells. It initiates expression of TERT by binding to either of two potential motifs in TERTp (101). Additionally, TERT can directly regulate expression of NF-κB-dependent genes through binding to the p65 subunit. Studies have demonstrated that telomerase can directly regulate recruitment to promoters of NF-κB target genes, such as those encoding interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF‐α) that are critical for inflammation and cancer progression (103).
The signal transducer and activator of transcription (STAT) family of cytoplasmic proteins are direct mediators of signaling from the extracellular environment to the nucleus (104). Seven STAT proteins have been identified as STAT 1–4, 5A, 5B, and 6 (105–107). They are normally inactive, but can be activated by phosphorylation. Of the seven human STAT encoding genes, STAT3 has drawn the most interest for its association with a wide variety of human cancers (104, 108).
In addition, these proteins are able to regulate TERT expression in tumor and normal cells (104). TERTp contains binding sites STAT3 and is overexpressed in prostate, breast, head, neck, and hematologic cancers, which implicates STAT3 as an important anticancer target (105).
The adipocyte protein 2 (AP-2) family of transcription factors contains five isoforms: AP-2α, AP-2β, AP-2γ, AP-2δ, and AP-2ϵ (109, 110). They are encoded by the FABP4 gene. These isoforms have a major role in gene regulation and have different biological functions. They are required for morphogenesis during embryonic development (109). AP-2β specifically binds to the TERTp and activates telomerase in human cancer cells, but not normal cells. Two E-box sites in a 320-bp region of TERTp (320 bp upstream of the translational ATG site) have been observed to regulate promoter activity in human rhabdomyosarcoma cells (110).
A recent showed that goosecoid homebox protein (GSC) may be a new potential activator of TERT expression (49). It is normally involved in embryonic development and interacts with TGF-β and Wnt/β-catenin signaling pathways, which are implicated in tumor invasion (111). It was found to be overexpressed and to correlate with metastasis in patients with breast carcinoma (112), and was also associated with poor prognosis and chemoresistance in ovarian carcinoma (111). An analysis of TERTp areas with locally decreased methylation in thyroid cancer cells revealed a GSC biding site. GSC is a TERT activator and was variously expressed in both thyroid cancer and normal thyroid cells. Additionally, GSC was overexpressed in thyroid cancer (49).
Transcriptional Repressors
Transcriptional repressors are proteins that attach to DNA at specific silencer sites and block transcription of nearby genes. In the following section, we are going to briefly discuss repressors that have been shown to downregulate TERT transcription, such as MAD1/2, p53, WT1, CTCF, and MZF-2.
The mitotic checkpoint is a crucial mechanism in maintaining chromosomal stability. It guarantees precise chromosome segregation by delaying separation of replicated sister chromatids (113, 114). Mitotic arrest deficient 1 (MAD1) is a major element of the mitotic checkpoint, and it recruits its binding partner MAD2 to nuclear pores (113, 115). During mitosis, MAD1 localizes to unattached kinetochores, where it serves as a docking site for MAD2. Kinetochore-bound MAD1–MAD2 act as a catalyst for conformational change of free MAD2 (114, 116). MAD1 upregulation serves as a marker of poor prognosis, as it tends to be overexpressed in cancers (116). Upregulation of MAD1 leads to chromosomal instability and resistance to microtubule poisons that are currently used as chemotherapeutic agents (116). MAD1 is recognized as an important cellular antagonist of c-MYC (117, 118). In addition, c-MYC and MAD1 are involved in regulation of TERT expression because they bind to the same promoter sites (E-box) to activate TERT expression (119). There two E-boxes in TERTp, and both of them constitute binding sites for c-MYC/MAX or MAD1/MAX heterodimers (117, 120). A switch from c-MYC/MAX to MAD1/MAX, triggers decrease in H3 and H4 histone acetylation at TERTp (119, 120).
p53 is the best known human tumor suppressor which is a member of a larger p53 family of tumor suppressors (121, 122). Other than p53, this family also includes p63 and p73 (123, 124). p53 acts primarily as an inducer of cell cycle arrest, cell differentiation, senescence, and apoptosis in response to numerous intrinsic and extrinsic stress signals (122, 125). It has a major role in the control of genomic stability, DNA replication, and DNA repair. The p53 encoding TP53 gene is mutated in approximately 50% of human cancers. TERTp contains two p53 binding motifs (123). Several findings showed that p53 suppresses telomerase activity by inhibiting TERT expression (125). This inhibition may be caused indirectly, by an interaction between Sp1 and overexpressed p53 (125, 126). Furthermore, this inhibition of TERT could be possibly independent of other p53 functions, such as those associated with apoptosis (125).
Another protein implicated in inhibition of TERT is the Wilms’ tumor 1 (WT1) tumor suppressor (125). It contains four zinc fingers and an RNA-binding protein that directs the development of several organs (heart, diaphragm) and genitourinary tissues (127, 128). It is normally expressed in kidney, testes, ovaries, and spleen (129). Most neoplasms, including lung carcinomas, renal cell carcinoma, pediatric sarcomas, and breast, ovarian, colon, melanoma, and pancreas cancers, and exhibit a possible oncogenic activity of WT1 (130, 131). In addition, it is overexpressed in most acute myeloid leukemia patients, and is considered to be an independent marker of minimal residual disease (132). A WT1 binding site is located in TERTp (−352 upstream of the TSS), and its mutation significantly reduced telomerase activity and TERT mRNA expression in 293 embryonic kidney cells but not in HeLa cells (1, 89, 125). Additionally, WT1 inhibited TERT transcription during differentiation. This inactivation may influence activation of telomerase in the tumorigenesis phase. Furthermore, WT1 binding to TERTp suppresses c-MYC level at both protein, and mRNA level (1, 2).
CCCTC-binding factor (CTCF) is a zinc finger transcription factor which is ubiquitously expressed in human (133). Its binding sites are located in the first two exons of the TERT gene, and are located in a CpG island. Earlier studies showed that CTCF does not bind to TERT in telomerase-positive cells, which is correlated with methylation of exon 1 in these cells (134). Hypermethylation in this exonic region is common in most cancers, and CTCF is considered a major TERT repressor in normal cells. Methylation at specific CpG dinucleotides in exon 1 results in a change in secondary structure of DNA and creation of a four-strand structure known as G-quadruplex, which disrupts CTCF binding (135). Interestingly, CTCF was observed not to bind to TERT in normal thyroid tissue despite the presence of methylation, while thyroid cancer cell lines exhibited both partial methylation and CTCF binding (49).
The myeloid zinc finger protein (MZF)-2 is a Krüppel-like C2H2 zinc finger protein expressed predominantly in myeloid progenitor cells and involved in growth, differentiation, and tumorigenesis (136). The mechanisms involved in MZF-2-induced suppression of TERTp activity are still unclear (137). There are multiple binding sites for MZF-2 within the TERTp region, and upregulation of MZF-2 inhibits TERTp activity. This suggests a role for MZF-2 in transcriptional downregulation of TERT (125, 137).
TERT Gene Polymorphisms
Single Nucleotide Polymorphism
Single nucleotide polymorphisms (SNPs) have been described as being associated with increased risk for developing various cancers. They may be located both in intronic and exonic sequences of TERT, as well as in TERTp. Some common TERT SNPs found may modify survival and prognosis of certain cancers. A number of studies have recently been conducted to identify new SNP loci related to telomere length, which have shown a relationship between the risk of disease, its severity and the survival time in various cancers (138–140). In this section, we will discuss four common TERT polymorphisms that may be associated with gene expression.
Located at intron 2 of the TERT gene, rs2736100 A>C is a important non-coding SNP (141, 142). It has been associated with multiple cancers, especially with lung adenocarcinoma, which is characterized by significantly increased TERT gene expression, telomerase activity and gene copy number (143). Other solid cancers that are associated with this SNP include gliomas, bladder cancer, melanoma. rs2736100 has been identified as a major predisposing factor to sporadic and familial myeloproliferative neoplasms (MPNs), independently of the major diagnostic and molecular MPN subtypes. The C allele of rs2736100 and JAK2 46/1(GGCC) haplotype are major factors predisposing to MPN (141–143). Interestingly, the two alleles of rs2736100 seem to be associated with different types of diseases. While the C allele is primarily associated with cancers, the A allele, which is linked to shorter telomeres, is generally associated with predisposition to degenerative diseases (144). Furthermore, rs2736100 C is linked to increased blood cell count in the Japanese population (145).
Another TERT SNP, rs2853669 A>G is located in the TERTp region. It obstructs an ETS2 binding site, located close to an E-box. Previous studies showed that TERTp mutations creating a putative binding site for ETS, resulted in TERT upregulation and increased telomerase activity, while mutations at the ETS2 binding site suppressed c-MYC binding to the E-box (146, 147). Studies on rs2853669 showed that it is significantly associated with poor survival and increased cancer risk rate in hepatocellular carcinoma patients (146). In contrast, it was also observed to correlate with improved survival in patients with clear cell renal cell carcinoma, melanoma and glioblastoma (148). The C variant of this functional polymorphism results in decreased telomerase activity. Several studies suggest that rs2853669, in the presence of certain TERTp mutations, may also affect development of cancers (149). It was reported that it could influence telomere length and telomerase activity (150, 151). Furthermore, a study by Rachakonda et al. demonstrated that, in patients with urothelial bladder carcinoma, TERT rs2853669 may correlate with survival, prognosis, and tumor recurrence (152).
The two SNPs described above were located in non-coding regions of TERT, there are however also SNPs situated in exonic regions, of which rs2736098 G>A is a notable example. It is a synonymous A305A substitution located in exon 2, and was found to correlate with telomere length (153). Genotype GG was found to be associated with longer telomeres and decreased cancer susceptibility in patients with renal cell carcinoma (154). In another study, Xiao et al. showed that Chinese males harboring allele rs2736098 A had a greater risk of developing lung cancer than those with allele G (155). Allele A was also found to be significantly associated with risk of bladder cancer in the North Indian population (156). Further studies showed that it may impact risk for many other cancers, such as breast, esophageal, prostate, and basal cell carcinoma (153, 157).
Variable Number of Tandem Repeats Polymorphism
It was demonstrated that TERT may be regulated via a variable number of tandem repeats (VNTR) polymorphism named MNS16A (Figure 1). It is located upstream of promoter region of an antisense TERT transcript. Depending on the number tandem repeats, promoter activity is affected differently. There are two MNS16A variant alleles: short (S) and long (L). The L allele correlates with higher promoter activity in the antisense strand and increased expression of the antisense TERT transcript. This increased expression of antisense TERT leads to silencing of functional TERT (158). As a result, the S allele is associated with higher telomerase activity, while LL homozygotes have lower telomerase activity (158). Our previous work showed that the S variant was more frequent in non-Hodgkin’s B-cell lymphoma patients how did not respond to treatment, as well as those with intermediate/high International Prognostic Index (159). In contrast, the S variant was less frequent in chronic lymphocytic leukemia patients with high disease stage (160).
Alternative Splicing
TERT regulation is a multifarious process, which involves not only the transcriptional mechanisms described in the previous sections, but also posttranscriptional ones. This includes pre-mRNA alternative splicing of the TERT gene (161–163). There as many as 22 potential alternative splicing sites in the TERT gene, but the function of many of them is unclear (164–168). One of the most commonly studied splicing sites are deletions at two sites, α and β (Figure 2). The β splice site results in a major deletion (182 bp) and creates a non-functional, truncated protein. The α splice site generates a smaller (36 bp) deletion, which produces an impaired protein. Both of these splice sites result in TERT proteins that are incapable of telomere elongation (169–172). In many cancers, the full length TERT transcript (α+β+) correlated with tumor development and shorter survival in patients (173). However, the α variant alone is known to cause decreased telomerase activity and shorter telomeres, while the β splice variant was reported to not only inhibit telomerase activity but also the ability of cancer cells to induce apoptosis (174, 175). Another splice TERT variant may be generated by a deletion of exons 4–13, resulting in an inactive protein lacking its catalytic domain. This deletion was observed in both telomerase-negative and -positive cells, and was associated with increased cell proliferation (6).
Involvement of TERT in Non-Telomere-Related Mechanisms
In the previous sections, we described TERT regulation and telomerase reactivation mechanisms that are involved in telomere maintenance. Telomere-related functions of TERT, also known as canonical, may likewise entail prevention of chromosome fusions (176, 177). However, telomerase also has non-canonical (telomere-independent) roles (Figure 1). These roles can be grouped into two broad categories: a) involving telomerase activity but not telomere elongation and b) involving neither telomere elongation nor telomerase activity (177). The telomere-independent roles contribute to the regulation of metabolic mechanisms, epigenetic regulation of chromatin, stress response, RNA silencing, signal transduction pathways (Wnt and c-MYC signaling pathways), enhanced mitochondrial function, cell adhesion, and migration (176, 178, 179).
TERT is found in cytoplasm and mitochondria, alongside its usual nuclear localization (176, 180) (Figure 1). In humans, mice and rats, TERT contains two specific targeting sequences that regulate its transport in and out of organelles: a nuclear targeting signal sequence, and a mitochondrial targeting sequence (181). In inactive CD4+ lymphocytes, TERT is mainly cytoplasmic but after activation it is transported to the nucleus in a process controlled by the kinase Akt (182). Additionally, shuttling TERT out of the nucleus may be promoted by oxidative stress, and this mechanism is dependent on phosphorylation of tyrosine 707 by Src kinase. Translocation of TERT into mitochondria improves mitochondrial potential which eventually leading to cancer cell survival (183). The extra-nuclear TERT functionalities are generally thought of as non-telomere related, i.e. non-canonical, and will be described below (179).
Cytoplasmic TERT exhibits many functions, including interacting with signaling pathways such as Wnt/β-catenin signaling. In addition, TERT binds to stress particles under non-stress conditions, and in lymphocytes, it is stored outside the nucleus without stimulation. TERT may also form a part of a TERT–NF-κB subunit p65 complex, which can move from the cytoplasm to the nucleus in multiple myeloma cells, upon TNF‐α induction (184). NF-κB, in turn, controls expression of a variety of genes involved in inflammation, immune responses, and cell differentiation (179). Zhou et al. demonstrated that the endoplasmic reticulum transiently activates the expression of TERT in cancer cell lines (185).
As much as 10–20% of total TERT is localized in mitochondria (176, 179). Therein, TERT binds to mitochondrial DNA (mtDNA) and improves respiratory chain activity, protecting mitochondrion from environmental damage and decreasing reactive oxygen species (mtROS) production (180, 186). mtROS production leads to mitochondrial damage and telomere shortening. Neutralization of mtROS does not recover the mitochondrial function but reduces telomere shortening (187). Additionally, telomere and mitochondrial disfunction is mediated by p53, which induces growth arrest, senescence and apoptosis in cells (188). TERT import depends on membrane potential and it is located close to the inner membrane (181). TERT binds to mtDNA in the region coding for NADH ubiquinone oxidoreductase subunits 1 (ND1) and 2 (ND2) and protects mtDNA from environmental damage (181). Mitochondrial TERT plays a role in decreasing apoptosis and improving mitochondrial membrane potential. Furthermore, it has unusual DNA- and RNA-dependent RNA polymerase activities, upon interaction with tRNAs (189). TERT can also interact with mitochondrial RNA processing endoribonuclease (RMRP) and use the RNA-dependent RNA polymerase to synthesize dsRNA. Mutations in RMRP can interfere with RMRP-TERT binding, contributing to pleiotropic syndrome cartilage–hair hypoplasia (190).
TERT as a Potential Therapeutic Target
The unique feature of telomerase is its low or nonexistent expression in somatic cells, but overexpression in most cancer cells (191). Thus, telomerase and other telomere components offer a highly attractive diagnostic and prognostic biomarker of cancer and a target for development of therapeutics. Several strategies have been devised to target telomerase functions: telomerase inhibition, telomerase peptide vaccines, and suicide gene therapy. Epigenetic processes were suggested as another promising target for therapeutic purposes (192). Some of these are already used in treatment of patients as part of clinical trials (193).
TERT inhibition has been regarded as a promising therapeutic strategy, as earlier in vitro studies showed that TERT silencing cell proliferation (194, 195). An early approach was to design compounds that would interact with DNA at the 3’ overhang, stabilizing telomeric G-quadruplex secondary structures, and thus blocking telomerase access to DNA. Telomestatin, BRACO-19, RHPS4, TMPyP4 are some of the most commonly studied G-quadruplex binding proteins (191, 196, 197). Telomestatin (OBP-301) is a natural product isolated from Streptomyces anulatus (198). The primary mechanism of telomestatin action involves a highly specific interaction with the G-quadruplex to stabilize its structure (199). These DNA-binding compounds are now less popular due to discovery of better molecular strategies, such as targeting the TERT active site directly. Studies on such inhibitors led to discovery of 2-[[(E)-3-naphthalen-2-ylbut-2-enoyl]amino]benzoic acid (BIBR1532), which inhibits telomerase by binding non-competitively to the TERT active site (197, 200). This binding leads to increased oxidative stress and decreased nitrogen monoxide bioavailability in favor of H2O2. However, BIBR1532 has not yet progressed to clinical tests (201). Aside from synthetic compound, various naturally occurring compounds, such as allicin (from garlic), curcumin (from turmeric), silibinin (from thistle), and epigallocathechin gallate (EGCG, from tea) were found to have telomerase inhibitory properties (202). A synthetic, more stable derivative of EGCG, MST-312, was shown to inhibit telomerase in various cancer, although its mechanism of action remains unknown (203–205).
Some peptide vaccines can possibly target the telomerase active site (199). GV1001 (KAEL-GemVax Co. Ltd., Gangnam-gu Seoul, Republic of Korea) is the only such vaccine to enter clinical trials (206). Its structure is based on a peptide sequence from TERT active site and it capable of binding multiple HLA class II molecules. It functions by stimulating tumor-reactive CD8+ and CD4+ T-cell immunity specific for TERT (199, 207, 208). GV1001 is used in treatment of patients with advanced stage melanoma, lung, hepatocellular carcinoma and pancreatic cancer (196). Two other TERT-based peptide vaccines, p540 and p675 were also observed to elicit TERT-specific cytotoxic T cell HLA-A*02:01- restricted immunity (208, 209). Other TERT-based vaccines are composed of more than one separate peptide sequence. An example of such a vaccine is GX301, composed of four peptides. This multi-peptide character means that it recognizes more HLA haplotypes, binding to both class I and II HLA molecules (210). GX301 is currently (October 2020) in phase II of a clinical trial on patients with prostate cancer (211). GRNVAC1 is a dendritic cell vaccine, which was created by transfecting dendritic cells with mRNA encoding TERT-chimeric protein, and then returning the transfected cells to the patient (196). These cells would then target telomerase-expressing tumor cells. The clinical trial is in phase I/II, and the vaccine is currently used in treatment of patients with metastatic prostate cancer (196, 207).
Another strategy are the suicide gene therapies. They include oncolytic virotherapy, the predominantly used strategy to treat cancer, which has potential to specifically lyse the tumor, and not healthy cells. This approach involves adenoviruses replicating selectively in cancer cells, and subsequently killing them (212). This viral system relies on the highly active TERC/TERT promoter controlling expression of a bacterial protein nitroreductase. Neither this nor any other suicide gene therapy has entered into clinical trials (193).
Recent studies increasingly suggest that epigenetic mechanisms may be targeted in new therapeutic strategies. Chidamide, an inhibitor of the enzyme histone deacetylase, was shown to decrease telomerase expression through miR-129-3p up-regulation in non-small cell lung cancer cells. This leads to subsequent ROS accumulation and subsequent cell cycle arrest (213). Epigenetic mechanisms may also be exploited in potential therapies using personalized approach. A study on effects of all-trans retinoic acid (ATRA) in treatment of ovarian carcinoma patients showed that the efficacy of therapy correlated inversely with methylation level of TERTp. This was of particular interest in a large subgroup of serous ovarian carcinoma patients, who had hypomethylated TERTp, and could therefore be treated effectively with ATRA (214).
As shown by the examples described above, telomerase is an attractive target for cancer immunotherapy. The main advantage of TERT is its high cancer-specific expression. Results from clinical trials have been encouraging, because of the safety and good tolerability of telomerase inhibitors (215). As a final point, it should be noted that using just one type immunotherapy may not suffice to eliminate cancer cells. Therefore, new studies should focus on strategies integrating various types of therapies (216).
Summary
TERT is normally actively transcribed only in early embryonic development and in cells with high proliferative potential, while it is inactive in most somatic cells in adults. However, in most cancers, TERT undergoes reactivation, and by extending telomeres (the canonical function of TERT) it contributes to cancer formation and progression. There are many regulatory mechanisms involved in telomerase reactivation and adjustment of TERT expression, among which TERTp mutation is perhaps the most important. Other major TERT regulation mechanisms (also known as telomere maintenance mechanisms) are: chromosome rearrangements, methylation, miRNA interference, binding of transcription factors, genetic polymorphism, and alternative splicing. Some of these mechanisms may interact with each other, having a synergistic effect on TERT expression. Aside from the better-known telomere lengthening function, TERT also has many secondary, telomere-independent roles (non-canonical functions of TERT). Taking in to account its major importance in cancer, TERT has become a target of various therapeutic strategies in cancer treatment and continues to be an interesting object of research.
The following features of TERT described in this manuscript can be highlighted:
● TERT is a functional catalytic protein subunit of telomerase, which lengthens telomeres by adding short DNA repeats, consequently averting chromosomal instability;
● Its regulation is a multifarious process where both transcriptional and posttranscriptional mechanisms are involved;
● TERT is also a major component of various oncogenic signaling pathways, and its overexpression often contributes to tumorigenesis;
● TERT gene is often overexpressed in cancers, and this overexpression can be induced by a variety of mechanisms, such as: TERT gene amplification, TERT gene polymorphism, TERTp mutation and methylation, and miRNA interference, alternative splicing of the TERT;
● Aside from its primary nuclear localization, TERT can also be transported to cytoplasm and mitochondria;
● It has many non-canonical, i.e. telomere-unrelated, functions these include: interaction with signaling pathways, stress protection, regulation of chromatin structure, binding to and protection of mitochondrial DNA;
● TERT and its gene may also act as an attractive target for therapeutic interventions with a diagnostic and prognostic impact.
Author Contributions
MD and KB-K contributed to the conception and design of the review, drafted, and finalized the manuscript. BW contributed to the conception and design of the review and its draft version. PŁ contributed to the writing, reviewing, and editing of the final version of the manuscript. TK contributed to the final version of the manuscript and drew all the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the TARGETTELO project No. STRATEGMED3/306853 from the National Centre for Research and Development, Warsaw, Poland.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Daniel M, Peek GW, Tollefsbol TO. Regulation of the Human Catalytic Subunit of Telomerase (hTERT). Gene (2012) 498:135–46. doi: 10.1016/j.gene.2012.01.095
2. Ramlee MK, Wang JW, WX T, Li S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes (Basel) (2016) 7:50. doi: 10.3390/genes7080050
3. Martínez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer (2011) 11:161–76. doi: 10.1038/nrc3025
4. Aramburu T, Plucinsky S, Skordalakes E. POT1-TPP1 telomere length regulation and disease. Comput Struct Biotechnol J (2020) 18:1939–46. doi: 10.1016/j.csbj.2020.06.040
5. Akincilar SC, Unal B, Tergaonkar V. Reactivation of telomerase in cancer. Cell Mol Life Sci (2016) 73:1659–70. doi: 10.1007/s00018-016-2146-9
6. Hrdličková R, Nehyba J, Bose HR Jr. Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation. Mol Cell Biol (2012) 32:4283–96. doi: 10.1128/MCB.00550-12
7. Takakura M, Kyo S, Kanaya T, Hirano H, Takeda J, Yutsudo M, et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res (1999) 59:551–7.
8. Trybek T, Kowalik A, Góźdź S, Kowalska A. Telomeres and telomerase in oncogenesis. Oncol Lett (2020) 20:1015–27. doi: 10.3892/ol.2020.11659
9. Li Y, Tergaonkar V. Noncanonical functions of telomerase: implications in telomerase-targeted cancer therapies. Cancer Res (2014) 74:1639–44. doi: 10.1158/0008-5472.CAN-13-3568
10. Gaspar TB, Sá A, Lopes JM, Sobrinho-Simões M, Soares P, Vinagre J. Telomere Maintenance Mechanisms in Cancer. Genes (Basel) (2018) 9:241. doi: 10.3390/genes9050241
11. Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet (2010) 11, 319–330. doi: 10.1038/nrg2763
12. Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet (2017) 49:349–57. doi: 10.1038/ng.3781
13. Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature (2009) 460:66–72. doi: 10.1038/nature08137
14. Ghosh A, Saginc G, Leow SC, Khattar E, Shin EM, Yan TD, et al. Telomerase directly regulates NF-κB dependent transcription. Nat Cell Biol (2012) 14:1270–81. doi: 10.1038/ncb2621
15. Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature (2009) 462:1005–10. doi: 10.1038/nature08645
16. Hovhannisyan G, Harutyunyan T, Aroutiounian R, Liehr T. DNA Copy Number Variations as Markers of Mutagenic Impact. Int J Mol Sci (2019) 20:4723. doi: 10.3390/ijms20194723
17. Lew AR, Kellermayer TR, Sule BP, Szigeti K. Copy Number Variations in Adult-onset Neuropsychiatric Diseases. Curr Genomics (2018) 19:420–30. doi: 10.2174/1389202919666180330153842
18. Leão R, Apolónio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: clinical impacts in cancer. J BioMed Sci (2018) 25:22. doi: 10.1186/s12929-018-0422-8
19. Albertson DG. Gene amplification in cancer. Trends Genet (2006) 22:447–55. doi: 10.1016/j.tig.2006.06.007
20. Zhang F, Cheng D, Wang S, Zhu J. Human Specific Regulation of the Telomerase Reverse Transcriptase Gene. Genes (Basel) (2016) 7:30. doi: 10.3390/genes7070030
21. George SL, Parmar V, Lorenzi F, Marshall LV, Jamin Y, Poon E, et al. Novel therapeutic strategies targeting telomere maintenance mechanisms in high-risk neuroblastoma. J Exp Clin Cancer Res (2020) 39:78. doi: 10.1186/s13046-020-01582-2
22. Srinivas N, Rachakonda S, Kumar R. Telomeres and Telomere Length: A General Overview. Cancers (Basel) (2020) 12:558. doi: 10.3390/cancers12030558
23. Yuan X, Larsson C, Xu D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene (2019) 38:6172–83. doi: 10.1038/s41388-019-0872-9
24. Gay-Bellile M, Véronèse L, Combes P, Eymard-Pierre E, Kwiatkowski F, Dauplat MM, et al. TERT promoter status and gene copy number gains: effect on TERT expression and association with prognosis in breast cancer. Oncotarget (2017) 8:77540–51. doi: 10.18632/oncotarget.20560
25. Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, et al. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science (2017) 357:1416–20. doi: 10.1126/science.aao0535
26. Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun (2013) 4:2185. doi: 10.1038/ncomms3185
27. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science (2013) 339:959–61. doi: 10.1126/science.1230062
28. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Levi A Garraway LA, et al. Highly recurrent TERT promoter mutations in human melanoma. Science (2013) 339:957–9. doi: 10.1126/science.1229259
29. McKelvey BA, Gilpatrick T, Wang Y, Timp W, Umbricht CB, Zeiger MA. Characterization of Allele-Specific Regulation of Telomerase Reverse Transcriptase in Promoter Mutant Thyroid Cancer Cell Lines. Thyroid (2020) 30:1470–81. doi: 10.1089/thy.2020.0055
30. Nagore E, Heidenreich B, Rachakonda S, Garcia-Casado Z, Requena C, Soriano V, et al. TERT promoter mutations in melanoma survival. Int J Cancer (2016) 139:75–84. doi: 10.1002/ijc.30042
31. Andrés-Lencina JJ, Rachakonda S, García-Casado Z, Srinivas N, Skorokhod A, Requena C, et al. TERT promoter mutation subtypes and survival in stage I and II melanoma patients. Int J Cancer (2019) 144:1027–36. doi: 10.1002/ijc.31780
32. Maturo MG, Rachakonda S, Heidenreich B, Pellegrini C, Srinivas N, Requena C, et al. Coding and noncoding somatic mutations in candidate genes in basal cell carcinoma. Sci Rep (2020) 10:8005. doi: 10.1038/s41598-020-65057-2
33. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA (2013) 110:6021–6. doi: 10.1073/pnas.1303607110
34. Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F, et al. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis (2015) 4:e176. doi: 10.1038/oncsis.2015.39
35. Kinde I, Munari E, Faraj SF, Hruban RH, Schoenberg M, Bivalacqua T, et al. TERT promoter mutations occur early in urothelial neoplasia and are biomarkers of early disease and disease recurrence in urine. Cancer Res (2013) 73:7162–7. doi: 10.1158/0008-5472.CAN-13-2498
36. Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun (2013) 4:2218. doi: 10.1038/ncomms3218
37. Shimoi T, Yoshida M, Kitamura Y, Yoshino T, Kawachi A, Shimomura A, et al. TERT promoter hotspot mutations in breast cancer. Breast Cancer (2018) 25:292–6. doi: 10.1007/s12282-017-0825-5
38. Carcano FM, Vidal DO, van Helvoort Lengert A, Neto CS, Queiroz L, Marques H, et al. Hotspot TERT promoter mutations are rare events in testicular germ cell tumors. Tumour Biol J (2016) 37:4901–7. doi: 10.1007/s13277-015-4317-y
39. Mosrati MA, Willander K, Falk IJ, Hermanson M, Höglund M, Stockelberg D, et al. Association between TERT promoter polymorphisms and acute myeloid leukemia risk and prognosis. Oncotarget (2015) 6:25109–20. doi: 10.18632/oncotarget.4668
40. Lam G, Xian RR, Li Y, Burns KH, Beemon KL. Lack of TERT Promoter Mutations in Human B-Cell Non-Hodgkin Lymphoma. Genes (Basel) (2016) 7:E93. doi: 10.3390/genes7110093
41. Dratwa M, Wysoczanska B, Turlej E, Anisiewicz A, Maciejewska M, Wietrzyk J, et al. Heterogeneity of telomerase reverse transcriptase mutation and expression, telomerase activity and telomere length across human cancer cell lines cultured in vitro. Exp Cell Res (2020) 396:112298. doi: 10.1016/j.yexcr.2020.112298
42. Panero J, Alves-Paiva RM, Roisman A, Santana-Lemos BA, Falcão RP, Oliveira G, et al. Acquired TERT promoter mutations stimulate TERT transcription in mantle cell lymphoma. Am J Hematol (2016) 91:481–5. doi: 10.1002/ajh.24324
43. Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife (2015) 4:e07918. doi: 10.7554/eLife.07918
44. Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, et al. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol (2014) 32:2718–26. doi: 10.1200/JCO.2014.55.5094
45. Xing M, Alzahrani AS, Carson KA, Shong YK, Kim TY, Viola D, et al. Association between BRAF V600E mutation and recurrence of papillary thyroid cancer. J Clin Oncol (2015) 33:42–50. doi: 10.1200/JCO.2014.56.8253
46. Liu R, Bishop J, Zhu G, Zhang T, Ladenson PW, Xing M. Mortality Risk Stratification by Combining BRAF V600E and TERT Promoter Mutations in Papillary Thyroid Cancer: Genetic Duet of BRAF and TERT Promoter Mutations in Thyroid Cancer Mortality. JAMA Oncol (2017) 3:202–8. doi: 10.1001/jamaoncol.2016.3288
47. Hosen I, Rachakonda PS, Heidenreich B, de Verdier PJ, Ryk C, Steineck G, et al. Mutations in TERT promoter and FGFR3 and telomere length in bladder cancer. Int J Cancer (2015) 137:1621–9. doi: 10.1002/ijc.29526
48. Vuong HG, Altibi AMA, Duong UNP, Ngo HTT, Pham TQ, Chan AK, et al. TERT promoter mutation and its interaction with IDH mutations in glioma: Combined TERT promoter and IDH mutations stratifies lower-grade glioma into distinct survival subgroups-A meta-analysis of aggregate data. Crit Rev Oncol Hematol (2017) 120:1–9. doi: 10.1016/j.critrevonc.2017.09.013
49. Avin BA, Wang Y, Gilpatrick T, Workman RE, Lee I, Timp W, et al. Characterization of human telomerase reverse transcriptase promoter methylation and transcription factor binding in differentiated thyroid cancer cell lines. Genes Chromosomes Cancer (2019) 58:530–40. doi: 10.1002/gcc.22735
50. Pestana A, Vinagre J, Sobrinho-Simões M, Soares P. TERT biology and function in cancer: beyond immortalization. J Mol Endocrinol (2017) 58:R129–46. doi: 10.1530/JME-16-0195
51. Saing Kim YS, Kim K, Kwon GY, Lee SJ, Park SH. ibroblast growth factor receptor 3 (FGFR3) aberrations in muscle-invasive urothelial carcinoma. BMC Urol (2018) 18:68. doi: 10.1186/s12894-018-0380-1
52. Killela PJ, Pirozzi CJ, Healy P, Reitman ZJ, Lipp E, Rasheed BA, et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget (2014) 5:1515–25. doi: 10.18632/oncotarget.1765
53. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discovery Med (2016) 21:373–80.
54. Diplas BH, Liu H, Yang R, Hansen LJ, Zachem AL, Zhao F, et al. Sensitive and rapid detection of TERT promoter and IDH mutations in diffuse gliomas. Neuro Oncol (2019) 21:440–50. doi: 10.1093/neuonc/noy167
55. Ozturk-Isik E, Cengiz S, Ozcan A, Yakicier C, Danyeli AE, Pamir MN, et al. Identification of IDH and TERTp mutation status using 1 H-MRS in 112 hemispheric diffuse gliomas. J Magn Reson Imaging (2020) 51:1799–809. doi: 10.1002/jmri.26964
56. Ma X, Gong R, Wang R, Pan Y, Cai D, Pan B, et al. Recurrent TERT promoter mutations in non-small cell lung cancers. Lung Cancer (2014) 86:369–73. doi: 10.1016/j.lungcan.2014.10.009
57. Stoehr R, Taubert H, Zinnall U, Giedl J, Gaisa NT, Burger M, et al. Frequency of TERT Promoter Mutations in Prostate Cancer. Pathobiology (2015) 82:53–7. doi: 10.1159/000381903
59. Lewis KA, Tollefsbol TO. Regulation of the Telomerase Reverse Transcriptase Subunit through Epigenetic Mechanisms. Front Genet (2016) 7:83. doi: 10.3389/fgene.2016.00083
60. Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res (2013) 23:555–67. doi: 10.1101/gr.147942.112
61. Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet (2007) 39:457–66. doi: 10.1038/ng1990
62. Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer (2011) 11:726–34. doi: 10.1038/nrc3130
63. Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nature (2017) 543:72–7. doi: 10.1038/nature21373
64. Ehrlich M, Ehrlich KC. DNA cytosine methylation and hydroxymethylation at the borders. Epigenomics (2014) 6:563–6. doi: 10.2217/epi.14.48
65. Li L, Gao Y, Wu Q, Wu Q, Cheng ASL, Yip KY, et al. New guidelines for DNA methylome studies regarding 5-hydroxymethylcytosine for understanding transcriptional regulation. Genome Res (2019) 29:543–53. doi: 10.1101/gr.240036.118
66. Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer (2002) 101:335–41. doi: 10.1002/ijc.10593
67. Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipmanet T, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol (2013) 14:534–42. doi: 10.1016/S1470-2045(13)70110-4
68. Castelo-Branco P, Leão R, Lipman T, Campbell B, Lee D, Price A, et al. A cancer specific hypermethylation signature of the TERT promoter predicts biochemical relapse in prostate cancer: a retrospective cohort study. Oncotarget (2016) 7:57726–36. doi: 10.18632/oncotarget.10639
69. Lee DD, Leão R, Komosa M, Gallo M, Zhang CH, Lipman T, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest (2019) 129:223–9. doi: 10.1172/JCI121303
70. Stern JL, Paucek RD, Huang FW, Ghandi M, Nwumeh R, Costello JC, et al. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT. Cell Rep (2017) 21:3700–7. doi: 10.1016/j.celrep.2017.12.001
71. Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res (2007) 67:194–201. doi: 10.1158/0008-5472.CAN-06-3396
72. Wang N, Kjellin H, Sofiadis A, Fotouhi O, Juhlin CC, Bäckdahl M, et al. Genetic and epigenetic background and protein expression profiles in relation to telomerase activation in medullary thyroid carcinoma. Oncotarget (2016) 7:21332–46. doi: 10.18632/oncotarget.7237
73. Kumari A, Srinivasan R, Vasishta RK, Wig JD. Positive regulation of human telomerase reverse transcriptase gene expression and telomerase activity by DNA methylation in pancreatic cancer. Ann Surg Oncol (2009) 16:1051–9. doi: 10.1245/s10434-009-0333-8
74. Leão R, Lee D, Figueiredo A, Hermanns T, Wild P, Komosa M, et al. Combined genetic and epigenetic alterations of the TERT promoter affect clinical and biological behavior of bladder cancer. Int J Cancer (2019) 144:1676–84. doi: 10.1002/ijc.31935
75. Seynnaeve B, Lee S, Borah S, Park Y, Pappo A, Kirkwood JM, et al. Genetic and epigenetic alterations of TERT are associated with inferior outcome in adolescent and young adult patients with melanoma. Sci Rep (2017) 7:45704. doi: 10.1038/srep45704
76. Lee DD, Komosa M, Nunes NM, Tabori U. DNA methylation of the TERT promoter and its impact on human cancer. Curr Opin Genet Dev (2020) 60:17–24. doi: 10.1016/j.gde.2020.02.003
77. Salgado C, Roelse C, Nell R, Gruis N, van Doorn R, van der Velden P. Interplay between TERT promoter mutations and methylation culminates in chromatin accessibility and TERT expression. PloS One (2020) 15:e0231418. doi: 10.1371/journal.pone.0231418
78. Rowland TJ, Dumbović G, Hass EP, Rinn JL, Cech TR. Single-cell imaging reveals unexpected heterogeneity of telomerase reverse transcriptase expression across human cancer cell lines. Proc Natl Acad Sci U S A (2019) 116:18488–97. doi: 10.1073/pnas.1908275116
79. Chai L, Kang XJ, Sun ZZ, Zeng MF, Yu SR, Ding Y, et al. MiR-497-5p, miR-195-5p and miR-455-3p function as tumor suppressors by targeting hTERT in melanoma A375 cells. Cancer Manag Res (2018) 10:989–1003. doi: 10.2147/CMAR.S163335
80. Ohira T, Naohiro S, Nakayama Y, Osaki M, Okada F, Oshimura M, et al. miR-19b regulates hTERT mRNA expression through targeting PITX1 mRNA in melanoma cells. Sci Rep (2015) 5:8201. doi: 10.1038/srep08201
81. Schrank Z, Khan N, Osude C, Singh S, Miller RJ, Merrick C, et al. Oligonucleotides Targeting Telomeres and Telomerase in Cancer. Molecules (2018) 23:2267. doi: 10.3390/molecules23092267
82. Wang X, Zhao Y, Cao W, Wang C, Sun B, Chen J, et al. miR-138-5p acts as a tumor suppressor by targeting hTERT in human colorectal cancer. Int J Clin Exp Pathol (2017) 10:11516–25.
83. Koziel JE, Fox MJ, Steding CE, Sprouse AA, Herbert BS. Medical genetics and epigenetics of telomerase. J Cell Mol Med (2011) 15:457–67. doi: 10.1111/j.1582-4934.2011.01276.x
84. Chen L, Lü MH, Zhang D, Hao NB, Fan YH, Wu YY, et al. miR-1207-5p and miR-1266 suppress gastric cancer growth and invasion by targeting telomerase reverse transcriptase. Cell Death Dis (2014) 5:e1034. doi: 10.1038/cddis.2013.553
85. Zhang XL, Xu LL, Wang F. Hsa_circ_0020397 regulates colorectal cancer cell viability, apoptosis and invasion by promoting the expression of the miR-138 targets TERT and PD-L1. Cell Biol Int (2017) 41:1056–64. doi: 10.1002/cbin.10826
86. Guzman H, Sanders K, Idica A, Bochnakian A, Jury D, Daugaard I. miR-128 inhibits telomerase activity by targeting TERT mRNA. Oncotarget (2018) 9:13244–53. doi: 10.18632/oncotarget.24284
87. Lassmann T, Maida Y, Tomaru Y, Yasukawa M, Ando Y, Kojima M, et al. Telomerase reverse transcriptase regulates microRNAs. Int J Mol Sci (2015) 16:1192–208. doi: 10.3390/ijms16011192
88. Yamada O, Kawauchi K. The role of the JAK-STAT pathway and related signal cascades in telomerase activation during the development of hematologic malignancies. JAKSTAT (2013) 2:e25256. doi: 10.4161/jkst.25256
89. Cukusić A, Skrobot Vidacek N, Sopta M, Rubelj I. Telomerase Regulation at the Crossroads of Cell Fate. Cytogenet Genome Res (2008) 122:263–72. doi: 10.1159/000167812
90. Zheng L, Suzuki H, Nakajo Y, Nakano A, Kato M. Regulation of c-MYC Transcriptional Activity by Transforming Growth Factor-Beta 1-stimulated Clone 22. Cancer Sci (2018) 109:395–402. doi: 10.1111/cas.13466
91. Zhang Y, Zhang A, Shen C, Zhang B, Rao Z, Wang R, et al. 1E21 Acts as a Negative Feedback Regulator of c-Myc induced hTERT Transcription During Tumorigenesis. Oncol Rep (2014) 32:1273–80. doi: 10.3892/or.2014.3287
92. Farrell AS, Sears RC. MYC Degradation. Cold Spring Harb Perspect Med (2014) 4:a014365. doi: 10.1101/cshperspect.a014365
93. Dilshara MG, Gedara R, Jayasooriya PT, Choi YH, Kim GY. Camptothecin Induces c-Myc- And Sp1-mediated hTERT Expression in LNCaP Cells: Involvement of Reactive Oxygen Species and PI3K/Akt. Food Chem Toxicol (2019) 127:53–60. doi: 10.1016/j.fct.2019.03.001
94. Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, et al. Direct activation of TERT transcription by c-MYC. Nat Genet (1999) 21:220–4. doi: 10.1038/6010
95. Koh CM, Khattar E, Leow SC, Liu CY, Muller J, Ang WX, et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest (2015) 125:2109–22. doi: 10.1172/JCI79134
96. Farina A, Faiola F, Martinez E. Reconstitution of an E box-binding Myc:Max complex with recombinant full-length proteins expressed in Escherichia coli. Protein Expr Purif (2004) 34:215–22. doi: 10.1016/j.pep.2003.11.021
97. Pestana A, Vinagre J, Sobrinho-Simões M, Soares P. TERT Biology and Function in Cancer: Beyond Immortalisation. J Mol Endocrinol (2017) 58:R129–46. doi: 10.1530/JME-16-0195
98. Chang WC, Hung JJ. Functional role of post-translational modifications of Sp1 in tumorigenesis. J BioMed Sci (2012) 19:94. doi: 10.1186/1423-0127-19-94
99. Vellingiri B, Iyer M, Subramaniam MD, Jayaramayya K, Siama Z, Giridharan B. Understanding the Role of the Transcription Factor Sp1 in Ovarian Cancer: from Theory to Practice. Int J Mol Sci (2020) 21:1153. doi: 10.3390/ijms21031153
100. Jie MM, Chang X, Zeng S, Liu C, Liao GB, Wu YR, et al. Diverse Regulatory Manners of Human Telomerase Reverse Transcriptase. Cell Commun Signal (2019) 17:63. doi: 10.1186/s12964-019-0372-0
101. Li Y, Zhou QL, Sun W, Chandrasekharan P, Cheng HS, Ying Z, et al. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol (2015) 17:1327–38. doi: 10.1038/ncb3240
102. Tong L, Tergaonkar V. Rho Protein GTPases and Their Interactions With NFκB: Crossroads of Inflammation and Matrix Biology. Biosci Rep (2014) 34:e00115. doi: 10.1042/BSR20140021
103. Ding D, Zhou J, Wang M, Cong YS. Implications of telomere-independent activities of telomerase reverse transcriptase in human cancer. FEBS J (2013) 280:3205–11. doi: 10.1111/febs.12258
104. Konnikova L, Simeone MC, Kruger MM, Kotecki M, Cochran BH. Signal Transducer and Activator of Transcription 3 (STAT3) Regulates Human Telomerase Reverse Transcriptase (hTERT) Expression in Human Cancer and Primary Cells. Cancer Res (2005) 65:6516–20. doi: 10.1158/0008-5472.CAN-05-0924
105. Chau MN, El Touny LH, Jagadeesh S, Banerjee PP. Physiologically Achievable Concentrations of Genistein Enhance Telomerase Activity in Prostate Cancer Cells via the Activation of STAT3. Carcinogenesis (2007) 28:2282–90. doi: 10.1093/carcin/bgm148
106. Hao Y, Yang X, Chen C, Wang Y, Wang X, Li M, et al. STAT3 signalling pathway is involved in the activation of microglia induced by 2.45 GHz electromagnetic fields. Int J Radiat Biol (2010) 86:27–36. doi: 10.3109/09553000903264507
107. Bromberg J, Darnell JE Jr. The Role of STATs in Transcriptional Control and Their Impact on Cellular Function. Oncogene (2000) 19:2468–73. doi: 10.1038/sj.onc.1203476
108. de Haas N, de Koning C, di Blasio S, Flórez-Grau G, de Vries IJM, Hato SV. STAT Family Protein Expression and Phosphorylation State during moDC Development Is Altered by Platinum-Based Chemotherapeutics. J Immunol Res (2019) 4:1–12, 7458238. doi: 10.1155/2019/7458238
109. Wei CW, C-ch L, YI Y, Lin C-Y, Lin P-CH, Wu M-T, et al. n-Butylidenephthalide induced apoptosis in the A549 human lung adenocarcinoma cell line by coupled down-regulation of AP-2α and telomerase activity. Acta Pharmacol Sin (2009) 30:1297–306. doi: 10.1038/aps.2009.124
110. Deng WG, Jayachandran G, Wu G, Xu K, Roth JA, Ji L. Tumor-specific Activation of Human Telomerase Reverses Transcriptase Promoter Activity by Activating Enhancer-binding Protein-2 in Human Lung Cancer Cells. J Biol Chem (2007) 282:26460–70. doi: 10.1074/jbc.M610579200
111. Kang KW, Lee MJ, Song JA, Jeong JY, Kim YK, Lee C, et al. Overexpression of goosecoid homeobox is associated with chemoresistance and poor prognosis in ovarian carcinoma. Oncol Rep (2014) 32:189–98. doi: 10.3892/or.2014.3203
112. Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC, Weinberg RA. The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci U S A (2006) 103:18969–74. doi: 10.1073/pnas.0608636103
113. Wan J, Block S, Scribano CM, Thiry R, Esbona K, Audhya A, et al. Mad1 destabilizes p53 by preventing PML from sequestering MDM2. Nat Commun (2019) 10:1540. doi: 10.1038/s41467-019-09471-9
114. Luo Y, Ahmad E, Liu ST. MAD1: Kinetochore Receptors and Catalytic Mechanisms. Front Cell Dev Biol (2018) 6:51. doi: 10.3389/fcell.2018.00051
115. Raich N, Mahmoudi S, Emre D, Karess RE. Mad1 influences interphase nucleoplasm organization and chromatin regulation in Drosophila. Open Biol (2018) 8:180166. doi: 10.1098/rsob.180166
116. Ryan SD, Britigan EMC, Zasadil LM, Audhya WA, Roopra A, Weavera BA. Up-regulation of the mitotic checkpoint component Mad1 causes chromosomal instability and resistance to microtubule poisons. Proc Natl Acad Sci U S A (2012) 109:E2205–14. doi: 10.1073/pnas.1201911109
117. Lüscher B. MAD1 and its life as a MYC antagonist: An update. Eur J Cell Biol (2012) 91:506–14. doi: 10.1016/j.ejcb.2011.07
118. Fujiki T, Udono M, Kadooka K, Yamashita S, Miura T, Shirahata S, et al. Regulatory Mechanisms of Human and Mouse Telomerase Reverse Transcriptase Gene Transcription: Distinct Dependency on c-Myc. Cytotechnology (2010) 62:333–9. doi: 10.1007/s10616-010-9276-y
119. Meeran SM, Patel SN, Chan T-H, Tollefsbol TO. A Novel Prodrug of epigallocatechin-3-gallate: Differential Epigenetic hTERT Repression in Human Breast Cancer Cells. Cancer Prev Res (Phila) (2011) 4:1243–54. doi: 10.1158/1940-6207.CAPR-11-0009
120. Xu D, Popov N, Hou M, Wang Q, Björkholm M, Gruber A, et al. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc Natl Acad Sci U S A (2001) 98:3826–31. doi: 10.1073/pnas.071043198
121. Horikawa I, Barrett JC. Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis (2003) 24:1167–76. doi: 10.1093/carcin/bgg085
122. Berkers CR, Maddocks ODK, Cheung EC, Mor I, Vousden KH. Metabolic Regulation by p53 Family Members. Cell Metab (2013) 18:617–33. doi: 10.1016/j.cmet.2013.06.019
123. Aylon Y, Oren M. New plays in the p53 theater. Curr Opin Genet Dev (2011) 21:86–92. doi: 10.1016/j.gde.2010.10.002
124. Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 Family: Guardians of Maternal Reproduction. Nat Rev Mol Cell Biol (2011) 12:259–65. doi: 10.1038/nrm3086
125. Cong Y-S, Wright WE, Shay JW. Human Telomerase and Its Regulation. Microbiol Mol Biol Rev (2002) 66:407–25. doi: 10.1128/MMBR.66.3.407-425.2002
126. Horikawa I, Michishita E, Barrett JC. Regulation of hTERT transcription: a target of cellular and viral mechanisms for immortalization and carcinogenesis. Cytotechnology (2004) 45:23–32. doi: 10.1007/s10616-004-5122-4
127. Toska E, Roberts SGE. Mechanisms of transcriptional regulation by WT1 (Wilms’ tumour 1). Biochem J (2014) 461:15–32. doi: 10.1042/BJ20131587
128. Eozenou C, Gonen N, Sol Touzon M, Jorgensen A, Yatsenko SA, Fusee L, et al. Testis formation in XX individuals resulting from novel pathogenic variants in Wilms’ tumor 1 (WT1) gene. PNAS (2020) 117:13680–8. doi: 10.1073/pnas.1921676117
129. Liu L, Lai S, Andrews LG, Tollefsbol TO. Genetic and Epigenetic Modulation of Telomerase Activity in Development and Disease. Gene (2004) 340:1–10. doi: 10.1016/j.gene.2004.06.011
130. Ariyaratana S, Loeb DM. The Role of the Wilms Tumour Gene (WT1) in Normal and Malignant Haematopoiesis. Expert Rev Mol Med (2007) 9:1–17. doi: 10.1017/S1462399407000336
131. Han Y, San-Marina S, Yang L, Khoury H, Minden MD. The zinc finger domain of Wilms’ tumor 1 suppressor gene (WT1) behaves as a dominant negative, leading to abrogation of WT1 oncogenic potential in breast cancer cells. Breast Cancer Res (2007) 9:R43. doi: 10.1186/bcr1743
132. Ayatollahi H, Sadeghian MH, Naderi M, Jafarian AH, Shams SF, Motamedirad N. Quantitative assessment of Wilms tumor 1 expression by real-time quantitative polymerase chain reaction in patients with acute myeloblastic leukemia. J Res Med Sci (2017) 22:54. doi: 10.4103/jrms.JRMS_448_16
133. Renaud S, Loukinov D, Bosman FT, Lobanenkov V, Benhattar J. CTCF binds the proximal exonic region of hTERT and inhibits its transcription. Nucleic Acids Res (2005) 33:6850–60. doi: 10.1093/nar/gki989
134. Renaud S, Loukinov D, Abdullaev Z, Guilleret I, Bosman FT, Lobanenkov V, et al. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res (2007) 35:1245–56. doi: 10.1093/nar/gkl1125
135. Li PT, Wang ZF, Chu IT, Kuan YM, Li MH, Huang MC, et al. Expression of the human telomerase reverse transcriptase gene is modulated by quadruplex formation in its first exon due to DNA methylation. J Biol Chem (2017) 292:20859–70. doi: 10.1074/jbc.M117.808022
136. Ogawa H, Murayama A, Nagata S, Fukunaga R. Regulation of Myeloid Zinc Finger Protein 2A Transactivation Activity Through Phosphorylation by Mitogen-Activated Protein Kinases. J Biol Chem (2003) 278:2921–7. doi: 10.1074/jbc.M207615200
137. Fujimoto K, Kyo S, Takakura M, Kanaya T, Kitagawa Y, Itoh H, et al. Identification and characterization of negative regulatory elements of the human telomerase catalytic subunit (hTERT) gene promoter: possible role of MZF-2 in transcriptional repression of hTERT. Nucleic Acids Res (2000) 28:2557–62. doi: 10.1093/nar/28.13.2557
138. Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, et al. Identification of seven loci affecting mean telomere length and their asso-ciation with disease. Nat Genet (2013) 45:422–27, 427e1-2. doi: 10.1038/ng.2528
139. Ding H, Yan F, Zhou L-L, Ji X-H, Gu X-N, Tang Z-W, et al. Association between previously identified loci affecting telomere length and coronary heart disease (CHD) in Han Chinese population. Clin Interv Aging (2014) 9:857–61. doi: 10.18632/oncotarget.10754
140. Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet (2013) 45:371–384e2. doi: 10.1038/ng.2566
141. Hong T, Luo M, Liu Q. The TERT rs2736100 Polymorphism and Susceptibility to Myeloproliferative Neoplasms: A Systematic Review and Meta-Analysis. Genet Test Mol Biomarkers (2020) 24:181–7. doi: 10.1089/gtmb.2019.0277
142. Jäger R, Harutyunyan AS, Rumi E, Pietra D, Berg T, Olcaydu D, et al. Common germline variation at the TERT locus contributes to familial clustering of myeloproliferative neoplasms. Am J Hematol (2014) 89:1107–10. doi: 10.1002/ajh.23842
143. Trifa AP, Bănescu C, Tevet M, Bojan A, Dima D, Urian L, et al. TERT rs2736100 A>C SNP and JAK2 46/1 haplotype significantly contribute to the occurrence of JAK2 V617F and CALR mutated myeloproliferative neoplasms - a multicentric study on 529 patients. Br J Haematol (2016) 174:218–26. doi: 10.1111/bjh.14041
144. Snetselaar R, van Oosterhout MFM, Grutters JC, van Moorsel CHM. Telomerase Reverse Transcriptase Polymorphism rs2736100: A Balancing Act between Cancer and Non-Cancer Disease, a Meta-Analysis. Front Med (Lausanne) (2018) 5:41. doi: 10.3389/fmed.2018.00041
145. Matsuguma M, Yujiri T, Yamamoto K, Kajimura Y, Tokunaga Y, Tanaka M, et al. TERT and JAK2 polymorphisms define genetic predisposition to myeloproliferative neoplasms in Japanese patients. Int J Hematol (2019) 110:690–8. doi: 10.1007/s12185-019-02742-7
146. Vinothkumar V, Arun K, Arunkumar G, Revathidevi S, Ramani R, Bhaskar LVKS, et al. Association between functional TERT promoter polymorphism rs2853669 and cervical cancer risk in South Indian women. Mol Clin Oncol (2020) 12:485–94. doi: 10.3892/mco.2020.2003
147. Hosen I, Rachakonda PS, Heidenreich B, Sitaram RT, Ljungberg B, Roos G, et al. TERT promoter mutations in clear cell renal cell carcinoma. Int J Cancer (2015) 136:2448–52. doi: 10.1002/ijc.29279
148. Ozturk MB, Li Y, Tergaonkar V. Current Insights to Regulation and Role of Telomerase in Human Diseases. Antioxid (Basel) (2017) 6:17. doi: 10.3390/antiox6010017
149. Shen N, Lu Y, Wang X, Peng J, Zhu Y, Cheng L. Association between rs2853669 in TERT gene and the risk and prognosis of human cancer: a systematic review and meta-analysis. Oncotarget (2017) 8:50864–72. doi: 10.18632/oncotarget.15140
150. Baird DM. Variation at the TERT locus and predisposition for cancer. Exp Rev Mol Med (2010) 12:e16. doi: 10.1017/S146239941000147X
151. Yoo SS, Do SK, Choi JE, Lee SY, Lee J, Cha, et al. TERT Polymorphism rs2853669 Influences on Lung Cancer Risk in the Korean Population. J Korean Med Sci (2015) 30:1423–8. doi: 10.3346/jkms.2015.30.10.1423
152. Rachakonda PS, Hosen I, de Verdier PJ, Fallah M, Heidenreich B, Ryk C, et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci U S A (2013) 110:17426–31. doi: 10.1073/pnas.1310522110
153. Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, et al. Sequence variants at the TERT-CLPTM1L locus associated with many cancer type. Nat Genet (2009) 41:221–7. doi: 10.1038/ng.296
154. de Martino M, Taus C, Lucca I, Hofbauer SL, Haitel A, Shariat SF, et al. Association of human telomerase reverse transcriptase gene polymorphisms, serum levels, and telomere length with renal cell carcinoma risk and pathology. Mol Carcinog (2016) 55:1458–66. doi: 10.1002/mc.22388
155. Xiao X, He W. Genetic polymorphisms in the TERT-CLPTM1L region and lung cancer susceptibility in Chinese males. Oncol Lett (2017) 14:1588–94. doi: 10.3892/ol.2017.6289
156. Singh V, Jaiswal PK, Mittal RD. Replicative study of GWAS TP63C/T, TERTC/T, and SLC14A1C/T with susceptibility to bladder cancer in North Indians. Urol Oncol (2014) 32:1209–14. doi: 10.1016/j.urolonc.2014.05.013
157. Li T, Xian Y, Tian T, Zhuang X, Chu M. New evidence of TERT rs2736098 polymorphism and cancer risk: an updated meta-analysis. J BUON (2016) 21:491–7.
158. Wang L, Soria JC, Chang YS, Lee HY, Wei Q, Mao L. Association of a functional tandem repeats in the downstream of human telomerase gene and lung cancer. Oncogene (2003) 22:7123–9. doi: 10.1038/sj.onc.1206852
159. Wysoczanska B, Wrobel T, Dobrzynska O, Mazur G, Bogunia-Kubik K. Role of the functional MNS16A VNTR-243 variant of the human telomerase reverse transcriptase gene in progression and response to therapy of patients with non-Hodgkin’s B-cell lymphomas. Int J Immunogenet (2015) 42:100–5. doi: 10.1111/iji.12182
160. Wysoczanska B, Dratwa M, Gebura K, Mizgala J, Mazur G, Wrobel T, et al. Variability within the human TERT gene, telomere length and predisposition to chronic lymphocytic leukemia. Onco Targets Ther (2019) 12:4309–20. doi: 10.2147/OTT.S198313
161. Palma M, Parker A, Hojjat-Farsangi M, Forster J, Kokhaei P, Hansson L, et al. Telomere length and expression of human telomerase reverse transcriptase splice variants in chronic lymphocytic leukemia. Exp Hematol (2013) 41:615–26. doi: 10.1016/j.exphem.2013.03.008
162. Dong W, Qian Y, Yang L. Telomerase, hTERT and splice variants in patients with myelodysplastic syndromes. Leuk Res (2014) 38:830–5. doi: 10.1016/j.leukres.2014.04.008
163. Dong W, Wu L, Sun H, Ren X, Epling-Burnette PK, Yang L. MDS shows a higher expression of hTERT and alternative splice variants in unactivated T-cells. Oncotarget (2016) 7:71904–14. doi: 10.18632/oncotarget.12115
164. Kilian A, Bowtell DD, Abud HE, Hime GR, Venter DJ, Keese PK, et al. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet (1997) 1997) 6:2011–9. doi: 10.1093/hmg/6.12.2011
165. Wick M, Zubov D, Hagen G. Genomic organization and promoter characterization of the gene encoding the human telomerase reverse transcriptase (hTERT). Gene (1999) 232:97–106. doi: 10.1016/s0378-1119(99)00108-0
166. Hisatomi H, Ohyashiki K, Ohyashiki JH, Nagao K, Kanamaru T, Hirata H, et al. Expression profile of a gamma-deletion variant of the human telomerase reverse transcriptase gene. Neoplasia (2003) 5:193–7. doi: 10.1016/S1476-5586(03)80051-9
167. Saeboe-Larssen S, Fossberg E, Gaudernack G. Characterization of novel alternative splicing sites in human telomerase reverse transcriptase (hTERT): analysis of expression and mutual correlation in mRNA isoforms from normal and tumour tissues. BMC Mol Biol (2006) 7:26. doi: 10.1186/1471-2199-7-26
168. Amor S, Remy S, Dambrine G, Le Vern Y, Rasschaert D, Laurent S. Alternative splicing and nonsense-mediated decay regulate telomerase reverse transcriptase (TERT) expression during virus-induced lymphomagenesis in vivo. BMC Cancer (2010) 10:571. doi: 10.1186/1471-2407-10-571
169. Fan Y, Liu Z, Fang X, Ge Z, Ge N, Jia Y, et al. Differential expression of full-length telomerase reverse transcriptase mRNA and telomerase activity between normal and malignant renal tissues. Clin Cancer Res (2005) 11:4331–7. doi: 10.1158/1078-0432.CCR-05-0099
170. Ulaner GA, Hu JF, Vu TH, Giudice LC, Hoffman AR. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res (1998) 58:4168–72.
171. Wang N, Xu D, Sofiadis A, Hoog A, Vukojevic V, Backdahl M, et al. Telomerase-dependent and independent telomere maintenance and its clinical implications in medullary thyroid carcinoma. J Clin Endocrinol Metab (2014) 99(8):E1571–9. doi: 10.1210/jc.2014-1158
172. Krams M, Hero B, Berthold F, Parwaresch R, Harms D, Rudolph P. Full-length telomerase reverse transcriptase messenger RNA is an independent prognostic factor in neuroblastoma. Am J Pathol (2003) 162:1019–26. doi: 10.1016/S0002-9440(10)63896-5
173. Brinkman BMN. Splice variants as cancer biomarkers. Clin Biochem (2004) 37:584–94. doi: 10.1016/j.clinbiochem.2004.05.015
174. Colgin LM, Wilkinson C, Englezou A, Kilian A, Robinson MO, Reddel RR. The hTERTalpha splice variant is a dominant negative inhibitor of telomerase activity. Neoplasia (2000) 2:426–32. doi: 10.1038/sj.neo.7900112
175. Listerman I, Sun J, Gazzaniga FS, Lukas JL, Blackburn EH. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res (2013) 73:2817–28. doi: 10.1158/0008-5472.CAN-12-3082
176. Romaniuk A, Paszel Jaworska A, Totoń E, Lisiak N, Hołysz H, Królak A, et al. The non-canonical functions of telomerase: to turn off or not to turn off. Mol Biol Rep (2019) 46:1401–11. doi: 10.1007/s11033-018-4496-x
177. Parkinson EK, Fitchett C, Cereser B. Dissecting the non-canonical functions of telomerase. Cytogenet Genome Res (2008) 122:273–80. doi: 10.1159/000167813
178. Nassir N. A telomerase with novel non-canonical roles: TERT controls cellular aggregation and tissue size in Dictyostelium. PloS Genet (2019) 15:e1008188. doi: 10.1371/journal.pgen.1008188
179. Saretzki G. Extra-telomeric Functions of Human Telomerase: Cancer, Mitochondria and Oxidative Stress. Curr Pharm Des (2014) 20:6386–403. doi: 10.2174/1381612820666140630095606
180. Ségal-Bendirdjian E, Geli V. Non-canonical Roles of Telomerase: Unraveling the Imbroglio. Front Cell Dev Biol (2019) 7:332. doi: 10.3389/fcell.2019.00332
181. Lionaki E, Gkikas I, Tavernarakis N. Differential Protein Distribution between the Nucleus and Mitochondria: Implications in Aging. Front Genet (2016) 7:162. doi: 10.3389/fgene.2016.00162
182. Kovalenko OA, Caron MJ, Ulema P, Medrano C, Thomas AP, Kimura M, et al. A mutant telomerase defective in nuclear-cytoplasmic shuttling fails to immortalize cells and is associated with mitochondrial dysfunction. Aging Cell (2010) 9:203–19. doi: 10.1111/j.1474-9726.2010.00551.x
183. Jaiswal RK, Kumar P, Yadava PK. Telomerase and its extracurricular activities. Cell Mol Biol Lett (2013) 18:538–54. doi: 10.2478/s11658-013-0105-0
184. Akiyama M, Hideshima T, Hayashi T, Tai Y-T, Mitsiades CS, Mitsiades N, et al. Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res (2003) 63:18–21.
185. Zhou J, Mao B, Zhou Q, Ding D, Wang M, Guo P, et al. Endoplasmic reticulum stress activates telomerase. Aging Cell (2014) 13:197–200. doi: 10.1111/acel.12161
186. Moro L. Mitochondrial Dysfunction in Aging and Cancer. J Clin Med (2019) 8(11):1983. doi: 10.3390/jcm8111983
187. Billard P, Poncet DA. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. Int J Mol Sci (2019) 20:4959. doi: 10.3390/ijms20194959
188. Bernardes de Jesus B, Blasco MA. Telomerase at the intersection of cancer and aging. Trends Genet (2013) 29:513–20. doi: 10.1016/j.tig.2013.06.007
189. Chiodi I, Mondello C. Telomere-independent functions of telomerase in nuclei, cytoplasm, and mitochondria. Front Oncol (2012) 2:133. doi: 10.3389/fonc.2012.00133
190. Wu L, Fidan K, Um JY, Ahn KS. Telomerase: Key regulator of inflammation and cancer. Pharmacol Res (2020) 155:104726. doi: 10.1016/j.phrs.2020.104726
191. Ivancich M, Schrank Z, Wojdyla L, Leviskas B, Kuckovic A, Sanjali A. Treating Cancer by Targeting Telomeres and Telomerase. Antioxid (Basel) (2017) 6:15. doi: 10.3390/antiox6010015
192. Rahman MM, Brane AC, Tollefsbol TO. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells (2019) 8:1214. doi: 10.3390/cells8101214
193. Xu Y, Goldkorn A. Telomere and Telomerase Therapeutics in Cancer. Genes (Basel) (2016) 7:22. doi: 10.3390/genes7060022
194. Baginski M, Serbakowska K. In silico design of telomerase inhibitors. Drug Discovery Today (2020) 25:1213–22. doi: 10.1016/j.drudis.2020.04.024
195. Li S, Rosenberg JE, Donjacour AA, Botchkina IL, Hom YK, Cunha GR, et al. Rapid Inhibition of Cancer Cell Growth Induced by Lentiviral Delivery and Expression of Mutant-Template Telomerase RNA and Anti-telomerase Short-Interfering RNA. Cancer Res (2004) 64:4833–40. doi: 10.1158/0008-5472.CAN-04-0953
196. Ruden M, Puri N. Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev (2013) 39:444–56. doi: 10.1016/j.ctrv.2012.06.007
197. Konieczna N, Romaniuk-Drapała A, Lisiak N, Totoń E, Paszel-Jaworska A, Kaczmarek M, et al. Telomerase Inhibitor TMPyP4 Alters Adhesion and Migration of Breast-Cancer Cells MCF7 and MDA-MB-231. Int J Mol Sci (2019) 20:2670. doi: 10.3390/ijms20112670
198. Mulholland K, Wu C. Binding of Telomestatin to a Telomeric G-Quadruplex DNA Probed by All-Atom Molecular Dynamics Simulations with Explicit Solvent. J Chem Inf Model (2016) 56:2093–102. doi: 10.1021/acs.jcim.6b00473
199. Man RJ, Chen LW, Zhu HL. Telomerase inhibitors: a patent review (2010-2015). Expert Opin Ther Pat (2016) 26:679–88. doi: 10.1080/13543776.2016.1181172
200. National Center for Biotechnology Information. PubChem Compound Summary for CID 9927531 (2020). Available at: https://pubchem.ncbi.nlm.nih.gov/compound/bibr-1532 (Accessed September 28, 2020).
201. Kailashiya C, Sharma HB, Kailashiya J. Telomerase based anticancer immunotherapy and vaccines approaches. Vaccine (2017) 35:5768–75. doi: 10.1016/j.vaccine.2017.09.011
202. Mutlu AG, Şentürk M. Telomerase Inhibitors and Activators: Pharmaceutical Importance. In: Enzyme Inhibitors and Activators. London, UK: IntechOpen (2017). p. 125–38. doi: 10.5772/65933
203. Ameri Z, Ghiasi S, Farsinejad A, Hassanshahi G, Ehsan M, Fatemi A. Telomerase inhibitor MST-312 induces apoptosis of multiple myeloma cells and down-regulation of anti-apoptotic, proliferative and inflammatory genes. Life Sci (2019) 228:66–71. doi: 10.1016/j.lfs.2019.04.060
204. Gurung RL, Lim HK, Venkatesan S, Lee PSW, Hande MP. Targeting DNA-PKcs and telomerase in brain tumour cells. Mol Cancer (2014) 13:232. doi: 10.1186/1476-4598-13-232
205. Serrano D, Bleau AM, Fernandez-Garcia I, Fernandez-Marcelo T, Iniesta P, Ortiz-de-Solorzano C, et al. Inhibition of telomerase activity preferentially targets aldehyde dehydrogenase-positive cancer stem-like cells in lung cancer. Mol Cancer (2011) 10:96. doi: 10.1186/1476-4598-10-96
206. Jäger K, Walter M. Therapeutic Targeting of Telomerase. Genes (Basel) (2016) 7:39. doi: 10.3390/genes7070039
207. Shay JW, Keith WN. Targeting telomerase for cancer therapeutics. Br J Cancer (2008) 98:677–83. doi: 10.1038/sj.bjc.6604209
208. Hunger RE, Lang KK, Markowski CJ, Trachsel S, Møller M, Eriksen JA. Vaccination of patients with cutaneous melanoma with telomerase-specific peptides. Cancer Immunol Immunother (2011) 60:1553–64. doi: 10.1007/s00262-011-1061-z
209. Huang G, Geng J, Wang R, Chen L. Identification of a new cytotoxic T-cell epitope p675 of human telomerase reverse transcriptase. Cancer Biother Radiopharm (2012) 27:600–5. doi: 10.1089/cbr.2012.1193
210. Fenoglio D, Parodi A, Lavieri R, Kalli F, Ferrera F, Tagliamacco A, et al. Immunogenicity of GX301 cancer vaccine: Four (telomerase peptides) are better than one. Hum Vaccin Immunother (2015) 11:838–50. doi: 10.1080/21645515.2015.1012032
211. ClinicalTrials.Gov Database. (2020). Available at: https://clinicaltrials.gov/ct2/show/NCT02293707 (Accessed October 3, 2020).
212. Shanmugaraj B, Priya LB, Mahalakshmi B, Subbiah S, Hu R-M, Velmurugan BK, et al. Bacterial and viral vectors as vaccine delivery vehicles for breast Cancer therapy. Life Sci (2020) 250:117550. doi: 10.1016/j.lfs.2020.117550
213. Wu YF, Ou CC, Chien PJ, Chang HY, Ko JL, Wang BY. Chidamide-induced ROS accumulation and miR-129-3p-dependent cell cycle arrest in non-small lung cancer cells. Phytomedicine (2019) 56:94–102. doi: 10.1016/j.phymed.2018.09.218
214. Losi L, Lauriola A, Tazzioli E, Gozzi G, Scurani L, D’Arca D, et al. Involvement of epigenetic modification of TERT promoter in response to all-trans retinoic acid in ovarian cancer cell lines. J Ovarian Res (2019) 12:62. doi: 10.1186/s13048-019-0536-y
215. Saraswati AP, Relitti N, Brindisi M, Gemma S, Zisterer D, Butini S, et al. Raising the bar in anticancer therapy: recent advances in, and perspectives on, telomerase inhibitors. Drug Discovery Today (2019) 24:1370–88. doi: 10.1016/j.drudis.2019.05.015
Keywords: telomerase reverse transcriptase, cancer progression, TERTp mutations, telomere maintenance mechanisms, TERT structural variants, TERT epigenetic alterations, TERT transcriptional activators and repressors
Citation: Dratwa M, Wysoczańska B, Łacina P, Kubik T and Bogunia-Kubik K (2020) TERT—Regulation and Roles in Cancer Formation. Front. Immunol. 11:589929. doi: 10.3389/fimmu.2020.589929
Received: 31 July 2020; Accepted: 16 October 2020;
Published: 19 November 2020.
Edited by:
Katherine Chiappinelli, George Washington University, United StatesReviewed by:
Yu-Sheng Cong, Hangzhou Normal University, ChinaMartha Allen Zeiger, National Institutes of Health (NIH), United States
Copyright © 2020 Dratwa, Wysoczańska, Łacina, Kubik and Bogunia-Kubik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Dratwa, bWFydGEuZHJhdHdhQGhpcnN6ZmVsZC5wbA==; Katarzyna Bogunia-Kubik, a2F0YXJ6eW5hLmJvZ3VuaWEta3ViaWtAaGlyc3pmZWxkLnBs