 Michela Terri1,2†
Michela Terri1,2† Flavia Trionfetti1,2†
Flavia Trionfetti1,2† Claudia Montaldo2
Claudia Montaldo2 Marco Cordani3
Marco Cordani3 Marco Tripodi1,2,4
Marco Tripodi1,2,4 Manuel Lopez-Cabrera5
Manuel Lopez-Cabrera5 Raffaele Strippoli1,2*
Raffaele Strippoli1,2*- 1Department of Molecular Medicine, Sapienza University of Rome, Rome, Italy
- 2National Institute for Infectious Diseases L. Spallanzani, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
- 3instituto Madrileño de Estudios Avanzados en Nanociencia (IMDEA) Nanociencia, Madrid, Spain
- 4Istituto Pasteur Italia-Fondazione Cenci Bolognetti, Sapienza University of Rome, Rome, Italy
- 5Programa de Homeostasis de Tejidos y Organos, Centro de Biología Molecular “Severo Ochoa”-Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain
Peritoneal fibrosis is characterized by abnormal production of extracellular matrix proteins leading to progressive thickening of the submesothelial compact zone of the peritoneal membrane. This process may be caused by a number of insults including pathological conditions linked to clinical practice, such as peritoneal dialysis, abdominal surgery, hemoperitoneum, and infectious peritonitis. All these events may cause acute/chronic inflammation and injury to the peritoneal membrane, which undergoes progressive fibrosis, angiogenesis, and vasculopathy. Among the cellular processes implicated in these peritoneal alterations is the generation of myofibroblasts from mesothelial cells and other cellular sources that are central in the induction of fibrosis and in the subsequent functional deterioration of the peritoneal membrane. Myofibroblast generation and activity is actually integrated in a complex network of extracellular signals generated by the various cellular types, including leukocytes, stably residing or recirculating along the peritoneal membrane. Here, the main extracellular factors and the cellular players are described with emphasis on the cross-talk between immune system and cells of the peritoneal stroma. The understanding of cellular and molecular mechanisms underlying fibrosis of the peritoneal membrane has both a basic and a translational relevance, since it may be useful for setup of therapies aimed at counteracting the deterioration as well as restoring the homeostasis of the peritoneal membrane.
Introduction
Peritoneum is a serosal membrane forming the lining of the abdominal cavity. Peritoneum is a first line of defense against microorganisms and tumor cells. Moreover, peritoneum constitutes a slippery non-adhesive surface allowing frictionless movements of the viscera in the abdominal cavity. Peritoneum is composed of a continuous monolayer of cells of mesodermal origin, the mesothelial cells (MCs). MCs cover a submesothelial region made of a thin layer of connective tissue composed mainly of bundles of collagen fibers with few fibroblasts, macrophages (MØs), mast cells, and hematic and lymphatic vessels (1, 2).
Peritoneal fibrosis is the end point of a progressive alteration of the peritoneal membrane due to a wide array of inflammatory and infectious events, many of which are directly related to clinical practices (3). A main cause of peritoneal fibrosis is, in fact, peritoneal dialysis (PD). PD is a form of renal replacement alternative to the hemodialysis, where peritoneal membrane is used as a dialysis membrane in therapeutic procedures for the treatment of end-stage renal disease. Currently, peritoneal dialysis (PD) accounts for around 10% of all forms of renal replacement therapy worldwide (4). During PD practice, signs of fibrosis are found in 50 to 80% of patients within one or two years of PD (3, 5).
Peritoneal fibrosis represents an important cause of PD discontinuation, together with peritonitis and death due to cardiovascular complications. PD is also a risk factor for the onset of encapsulating peritoneal sclerosis (EPS), the most serious complication of PD, with potentially fatal manifestation (6). EPS is a syndrome characterized by loss of ultrafiltration function, anorexia, weight loss, diarrhea, intestinal obstruction, inflammation, peritoneal thickening, fibrin deposition, sclerosis, calcification and encapsulation (7). However, peritoneum during PD practice often presents only limited complications and many patients develop a simple peritoneal sclerosis (SPS), characterized by thickening of the peritoneum, calcification, presence of inflammatory elements, angiogenesis and dilatation of blood and lymphatic vessels in the absence of systemic disease, and whose alterations are at least in part reversible after discontinuation of PD.
Besides fibrosis during PD practice, peritoneum is directly implicated in the genesis of post-surgical intra-abdominal adhesions (peritoneal adhesions, PAs), which are fibrous bands tethering organs to one another or to the parietal peritoneal wall, leading to a significant cause of post-surgical morbidity and posing a major public health challenge (8). Their primary sequelae include bowel obstruction, female infertility, ectopic gestation, chronic abdominal and pelvic pain, poor quality of life, and death. It is estimated that ~93% of patients undergoing abdominal surgery develop adhesions and about 20% require re-hospitalization for adhesion-related complications (9, 10).
Finally, the insurgence of peritoneal fibrosis has a clinical relevance also for peritoneal metastases. In this context, metastatic tumors (generally ovary or colon cancers) instruct a fibrotic response in the peritoneal membrane, generating areas where tumor spreading and dissemination are facilitated (11–13). Although fibrosis related to peritoneal tumors is the object of increasing interest, due to its intrinsic specificities, this review article will not deal with this topic.
The induction of peritoneal fibrosis is a complex pathological event where peritoneal cells sense the pro-fibrotic stimuli and secrete extracellular mediators leading to the recruitment of circulating leukocytes playing a role in induction and amplification of the inflammatory response. The generation of myofibroblasts, cells of heterogenous origin with the ability of producing and remodeling the extracellular matrix proteins (ECM) is central for fibrosis onset. At the same time, the nature of the stimuli imparts signals promoting the resolution of the inflammatory state, with phagocytosis of dead cells and removal of debris. In this context, an implication of adaptive immunity has been proven relevant in its cross talk with peritoneal stroma or innate immunity components.
Therefore, the onset of peritoneal fibrosis is the final result of a tight network of signals between stromal resident and immune recirculating leukocytes, whose understanding may lead to a better medical containment of this deleterious pathologic event.
There is now plenty of information on the role of the non-immune components of peritoneal membrane (MCs, fibroblasts, endothelium) and activities of innate and adaptive immunity have been described by relevant studies (14); the main underlying intracellular mechanisms have been reviewed elsewhere (3, 15). The aim of this review article is to create a comprehensive synthetic description of how different signals from both stromal cells and immune system components are integrated and how cellular components are mutually influenced during the induction of peritoneal fibrosis.
Peritoneal Fibrosis: Multiple Ingredients for One Cake
Peritoneal fibrosis onset is the final result of complex interactions between external stimuli, intrinsic properties of the peritoneal membrane, and subsequent activities of the local innate-adaptive immune system. A flowchart describing the stimuli discussed in this chapter and the main peritoneal stromal responses is shown in Figure 1.
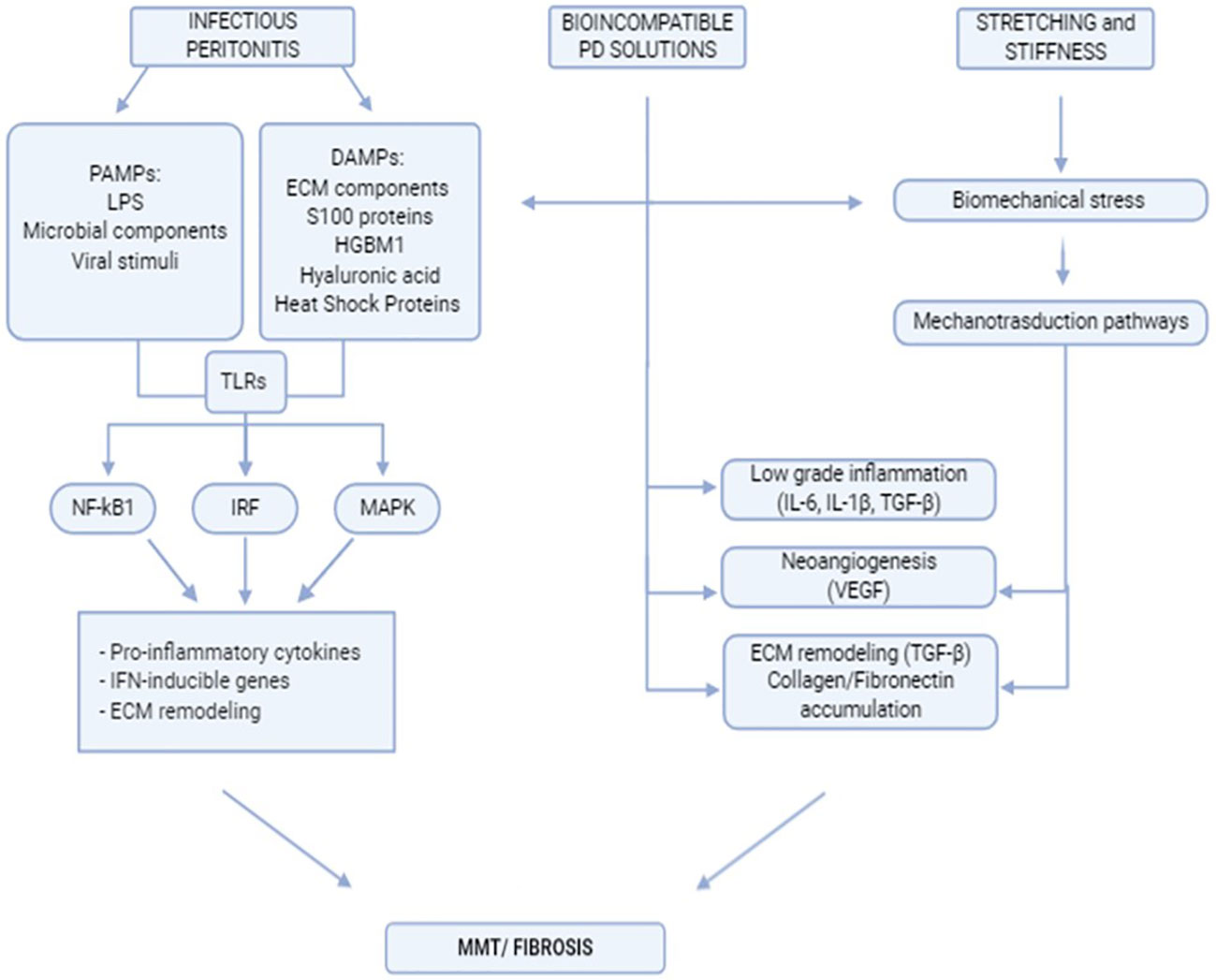
Figure 1 Flowchart indicating the main extracellular stimuli promoting peritoneal fibrosis and subsequent mesothelial cell response.
Infectious Peritonitis
Peritonitis is a main cause of fibrosis induction in peritoneum. Peritonitis onset is one of the most serious complication of PD: it induces angiogenesis and fibrosis and is a major cause of morbidity and mortality in PD patients (16). Repeated episodes of peritonitis are often a cause of discontinuation of PD and may precede induction of EPS (17).
Many microorganisms may infect peritoneum; the peritoneal membrane is contiguous to the intestine which harbor bacteria than can leak towards the peritoneal cavity. Moreover, medical actions such as catheter positioning and maintenance, practice of peritoneal dialysis and abdominal surgery may favor the entry of microorganisms in the peritoneum space.
The majority of peritonitis episodes in PD can be ascribed to Gram-positive bacteria of the skin and, to a minor degree, to Gram-negative bacteria presumably originating from the enteric flora (18).
Compared to bacteria, there are limited reports on the role of viruses. The suspect of virus infection occurs when cultures from peritonitis appear negative, an event occurring around 20% of the cases: however, virus infection is not diagnosed by standard tests (19).
Similarly to pericardium, coxackievirus B1 infection has been reported in peritoneum and it is characterized by the presence of monocytosis in PD effluent (20). Also, less studied are peritonitides caused by fungal infections. They constitute a serious complication of PD and account for between 1 and 15% of all PD-associated peritonitis episodes. The majority of these FP episodes are caused by Candida species such as Candida albicans (21, 22).
Mechanism of Inflammatory Response: Exogenous TLR Ligands
It is believed that the damage to the peritoneal membrane by infectious agents is mediated mainly by innate pattern recognition receptors (PRRs) on peritoneum, which include Toll-like receptors (TLRs), RIG-I-like receptors, NOD-like receptors, and C-type lectin receptors. The intracellular signaling cascades triggered by these PRRs lead to transcriptional expression of inflammatory mediators that coordinate the elimination of pathogens and infected cells (23).
Pathogens are recognized by PRRs through the interaction with molecules conserved among microbial species, which are called pathogen-associated molecular patterns (PAMPs). Besides PAMPs, PRRs also recognize endogenous molecules released from damaged cells, termed damage-associated molecular patterns (DAMPs) (24).
Among PRRs, TLRs play a critical role in innate immune responses by specifically recognizing molecular patterns from a wide range of microorganisms, including bacteria, fungi and viruses. TLRs are responsible for sensing invading pathogens outside of the cell and in intracellular endosomes and lysosomes (23). 10 different TLRs in humans and 12 in mice have been so far identified. Each of them recognizes different molecular patterns of microorganisms and self-components.
Human MCs respond to bacterial ligands through a specific subset of TLRs (i.e. TLR1, TLR2, TLR3, TLR5 and TLR6).
Gram positive bacteria are recognized by TLR2 and TLR5 (25), both singularly and cross-talking to better counteract microbial infections (26).
TLR2 recognizes an array of microbial molecules in part by hetero-dimerization with other TLRs (e.g. TLR1 and TLR6) or unrelated receptors (e.g. Dectin-1, CD36 and CD14). TLR activation triggers nuclear factor-kappa B (NF-κB), interferon regulatory factor (IRF) and mitogen-activated protein kinase (MAPK) signaling leading to altered gene expression, including pro-inflammatory cytokine and IFN-inducible genes (27).
TLR5 recognizes flagellin, a flagellum component in many motile bacteria (28). TLR5 expression on MCs may therefore be a critical signal of flagellated bacteria’s invasion into the peritoneal cavity. Translocation of intestinal bacteria is a potential cause of infection in PD patients, along with access through the intraperitoneal catheter, and many flagellated bacteria are Gram-negative species, with a poor outcome in PD associated peritonitis (25).
Gram-negative bacteria induce responses through TLR4, initially identified as responsible for the recognition of lipopolysaccharide (LPS). Differently from murine MCs, human MCs do not directly respond to TLR4. However, TLR4 is present in MØ stably residing in the peritoneal membrane and their response may contribute to inflammation leading to fibrosis.
Recent studies have shown that the modulation of TLR2 and TLR4 activity through specific antibodies or soluble Toll-like receptor 2 (sTLR2), a TLR2 inhibitor, is able to cause a substantial reduction of inflammatory parameters to inhibit fibrosis development in an experimental model of S. epidermidis infection (29).
A set of TLRs, comprising TLR3, TLR7, TLR8, and TLR9, act in the intracellular space in order to recognize nucleic acids derived from viruses and bacteria, as well as endogenous nucleic acids in pathogenic contexts (23). These TLRs respond by activating the production of type I IFNs and pro-inflammatory cytokines. Viral stimuli are recognized by the intracellular TLR3, which is functionally expressed in MCs (30). While for several exogenous TLRs the signaling pathway depends on MyD88, known as the inductor of the early phase response in MØs, TLR3, specifically, acts thought TRIF that plays an essential role in inducing a NF-κB mediated fibrosis and a late phase immune response activation (31, 32).
In human MCs, TLR3 is also involved in the regulation of the final common pathway of inflammation and fibrosis acting on matrix-remodeling proteins. In particular, TLR3 is correlated in time- and dose-dependent upregulation of MMP9 and TIMP1 (33).
Mechanisms of Inflammatory Response: Endogenous TLR Ligands
In addition to PAMPs, TLR mediated response can be stimulated by endogenous TLR molecules, inducing sterile inflammatory processes (34, 35). Many endogenous TLRs derive from ECM components, such as fibronectin or fibrinogen or ECM interacting proteins such as tenascin-C (36, 37).
Proteins with various functions may serve as endogenous TLRs such as cardiac myosin, S100 proteins, HGBM1 (38) (39–41). While the last protein may interact with several TLRs, the majority of these ligands are direct agonists of TLR2 and TLR4 (42, 43). Interestingly, exposure to PD fluids promotes the expression of Hsp60, Hsp70 and hyaluronic acid (HA), all TLR2 and TLR4 ligands, by leukocytes and MCs, thus driving an inflammatory response in the absence of infectious stimuli (see below) (44). Accordingly, treatment with soluble TLR2 (sTLR2) reduces pro-inflammatory and fibrotic response in mice exposed to PD fluids. These discoveries open to future clinical trials testing the clinical efficacy of these compound in patients undergoing long term PD (44).
Bioincompatibility of PD Solutions
The partial bioincompatibility of fluids used for the practice of PD may act as pro-fibrotic stimuli causing progressive morphological changes and leading to functional alterations that may cause ultrafiltration failure, discontinuation of PD and increased risk of developing EPS.
Traditional PD solutions, in fact, are hyperosmotic, hyperglycemic and acid. These solutions contain sodium, chloride, calcium, magnesium, lactate and a high concentration of glucose. Low pH in these solutions counteracts glucose oxidation that may release in the solution toxic glucose degradation products (GDPs) during the sterilization process. Moreover, glucose and reactive carbonyl compounds can form Advanced Glycation End-products (AGEs), binding to free amino groups on proteins or lipids (45, 46). The high osmolarity and the high glucose concentration favor ultrafiltration and toxin elimination by keeping the electrolyte balance (47).
All these factors may promote a low-grade inflammatory status in the peritoneal membrane, characterized by increase of inflammatory and profibrotic cytokine production such as IL-6, IL-1β, TGF-β1, VEGF, acceleration in TIMP release, causing a loss of balance in ECM remodeling and an accumulation of collagen and fibronectin. The same factors have a cytotoxic effect on MCs inducing mesothelial denudation of the peritoneal membrane and a decrease in the intercellular junctional proteins levels, causing hyperpermeability (48). In vitro evidence has demonstrated that the so called ‘bioincompatible’ PD fluid may induce apoptosis of MCs (49).
More recently, in vitro and in vivo studies have demonstrated that high glucose peritoneal dialysis solutions (HGPDS) may cause apoptosis and autophagy of MCs. However, further efforts will be necessary for the full understanding of the role of these mechanisms in the genesis of fibrosis (50).
This variety of stimuli also promotes a process known as mesothelial to mesenchymal transition (MMT) (see below) contributing to matrix deposition, increased stiffness and fibrosis (51). These cellular and molecular alterations parallel the induction of numerous morphological changes in the peritoneal membrane (PM) including increased thickness of the submesothelial space, vascular changes with subendothelial hyalinization, luminal narrowing or obliteration, increased density of blood vessels (52).
Clinically, these changes reflect an increase in small solute transport due to neoangiogenesis that extends the peritoneal surface area (a blood vessel density related parameter) and ultrafiltration reduction due to fibrosis and thickness of the submesothelial zone (53, 54). In certain cases, the simple peritoneal sclerosis common in peritoneum of PD patients can lead to EPS (7).
In order to mitigate the side effects of traditional PD solutions, a second generation of so called ‘biocompatible’ PD fluids has been designed that can be divided in two main groups: PD solutions with neutral pH, low GDPs and PD solutions where glucose is replaced with glucose polymers (icodextrin) or amino acids (55–57). The functionality of PD solutions is debated. It has been reported that these solutions better preserve the residual renal function and diuresis with a decrease in peritonitis frequency (3, 58). In vitro and in vivo effects of traditional versus biocompatible PD fluids are summarized in Table 1.
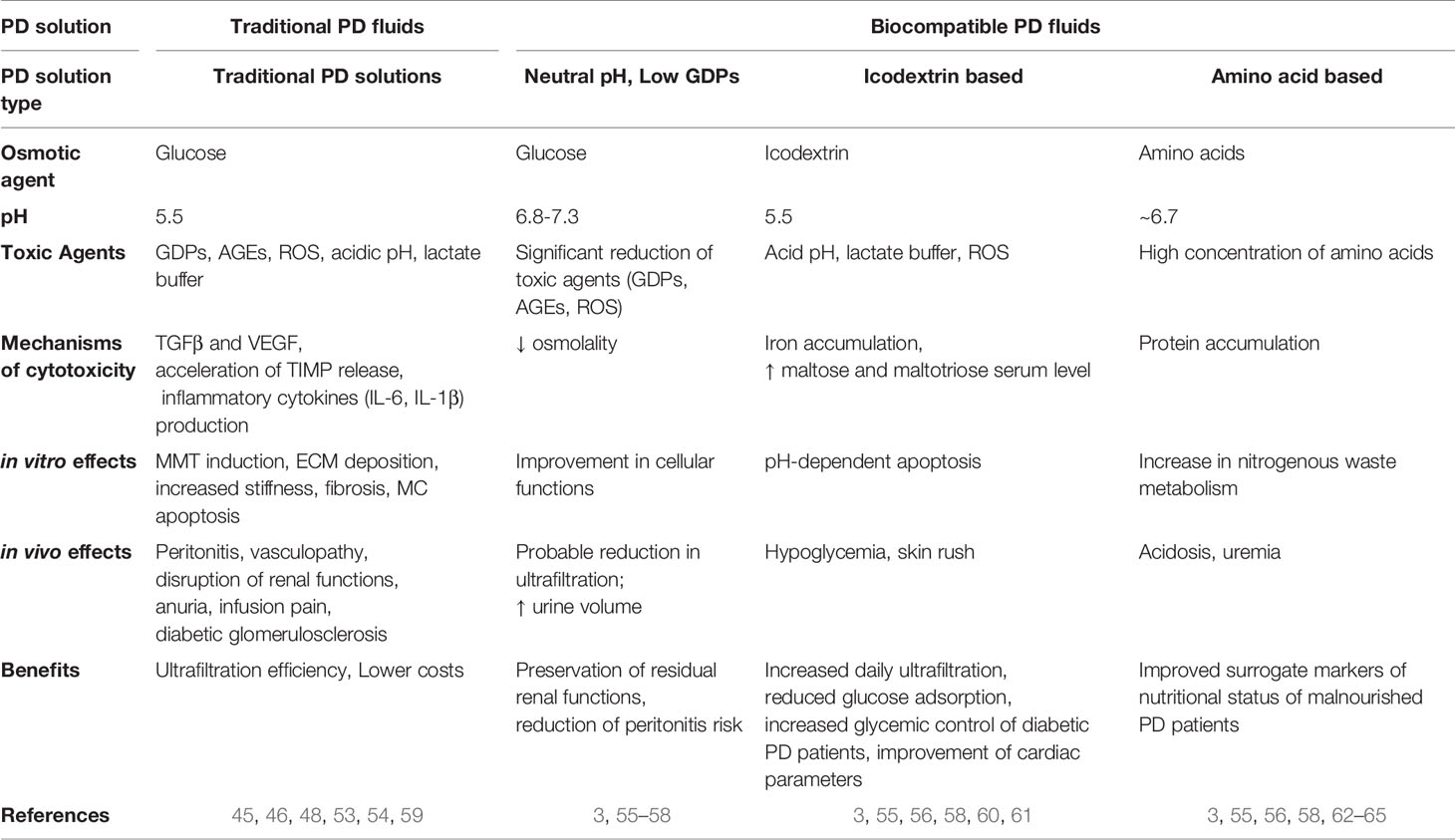
Table 1 Table comparing the main characteristics of traditional versus biocompatible PD with emphasis on in vitro/in vivo mechanisms of toxicity.
However, the effectiveness and the long-term benefits are currently being analyzed and there is not a definitive consensus on the benefits of this treatment, especially in the long term (66–69).
Icodextrin is a glucose polymer with a high molecular weight. PD solutions based on the use of icodextrin seem to increase peritoneal ultrafiltration, to reduce glucose absorption and to improve cardiac parameter (60). The use of a glucose polymer as an osmotic agent is particularly interesting as a glucose substitute in diabetic subjects. The reduced carbohydrate load also seems to provide a long-term metabolic advantage in terms of lipid control (61). However, icodextrin can interfere with blood glucose measurement by providing falsely elevated results. It can also cause hypersensitivity reactions, and it is more expensive than other PD solutions.
Solutions containing amino acids have been produced to improve the nutritional status of subjects on PD. PD causes a significant loss of protein in the dialysate, estimated to be 2–4 g of amino acids per day. Amino acid 1.1% solutions were found to be effective osmotic agents (62). In some studies, they have improved the nutritional status of malnourished PD patients (63). Common side effects include worsening of acidosis and an increase in blood urea linked to the increase in nitrogenous waste metabolism.
Glucose has been partially replaced by two osmometabolic agents, xylitol and L-carnitine. Treatment with this new formulation resulted a higher cell viability, better preservation of the integrity of the mesothelial layer, and reduced release of pro-inflammatory cytokines, as reported in a recent in vitro study (70).
Another field of investigation is the search of immunomodulators that may be added to mitigate the cellular effect of prolonged PD treatment. A recent discovery is the immunomodulatory effect of alanyl-glutamine (AlaGln) supplementation in PD solutions. This treatment seems to ameliorate peritoneal inflammation status and to improve healthy peritoneum biomarkers as well as tight junction organization and functionality (64, 65).
PM Damage by Biomechanical Cues: Stiffness and Stretching
Besides extracellular biochemical mediators, a vast body of evidence has demonstrated a role for biomechanical forces in mediating cell physiopathological responses.
Changes in biomechanical features of the extracellular matrix (ECM), such as ECM stiffness, can modify cell state and are major promoters of a fibrotic response (71). Beyond ECM stiffness, the sensing of mechanical stretching is characteristic of organs and tissues exposed to continuous variations of dynamic cues, such as respiratory and abdominal movements or the cyclic blood circulation pulse wave. In cells with epithelial features, the effects of cellular stretching have been analyzed especially on tissues composed of monocellular layers, such as lung epithelial cells and endothelium (72, 73). Biomechanical forces affect signal transduction (mechanotransduction) with a consequent impact on cellular behavior (74).
During exposure to PD fluid, the PM experiences continuous biomechanical cues. PD practice requires the injection of large PD solution volume (2 l). This causes mechanical stress by swelling the abdominal cavity, involving mechanical stretching of MCs. Other mechanical perturbations may arise from the trauma of the peritoneal membrane after abdominal laparotomies (75).
MCs upon exposure to cellular stretch in vitro increase the expression of VEGF and of TGF-β1 (76). It has been recently demonstrated that exposure of MCs to linear cyclic stretch in vitro leads to several cellular modifications corresponding to bona fide MMT induction. The experimental data are summarized in a model where a cross-talk between biomechanical and biochemical signals result in the induction of MMT (77).
Biomechanical forces are also involved in the formation of PAs, with a key contribution by MCs. It is believed that in in vivo conditions besides the mechanical tension, also hypoxia and activation of coagulation contribute to the formation of the fibrotic response leading to PAs formation (78).
Cellular Players of Peritoneal Fibrosis
Stromal Components of the PM: MCs, Endothelial Cells and Stromal Fibroblasts as a Source of Myofibroblasts
Peritoneal MCs constitute a monolayer of cells with an epithelial-like cobblestone shape covering in a continuum the peritoneal cavity. MCs originate from mesoderm during the gastrulation, and their differentiation is controlled by the transcription factor WT1, which is commonly used for lineage tracing experiments (79, 80). Despite their mesodermal origin, MCs show a cobblestone morphology and actually coexpress in basal conditions epithelial and mesenchymal markers (51, 59, 81).
MCs express tight and adherent junction related molecules such as ZO-1, occludin, claudins and E-cadherin, which is expressed both in plasma membrane and in cytoplasm (82). Moreover, these cells express epithelial intermediate filament proteins such as cytokeratins (8–18) that play an important role in maintaining cellular structural integrity. At the same time, MCs constitutively express also mesenchymal intermediate filaments such as vimentin and desmin (51).
The coexistence of both epithelial and mesenchymal markers may be linked to the characteristic plasticity and to the ability of these cells to acquire mesenchymal-like features in response to a variety of pro-inflammatory/profibrotic stimuli. Almost all the pro-inflammatory factors described in the previous sections may promote, although with different intensity, induction of MMT in MCs. This dedifferentiation process culminates with the acquisition of morphological and functional features making these cells indistinguishable from myofibroblasts of other origin (see below) (Table 2).
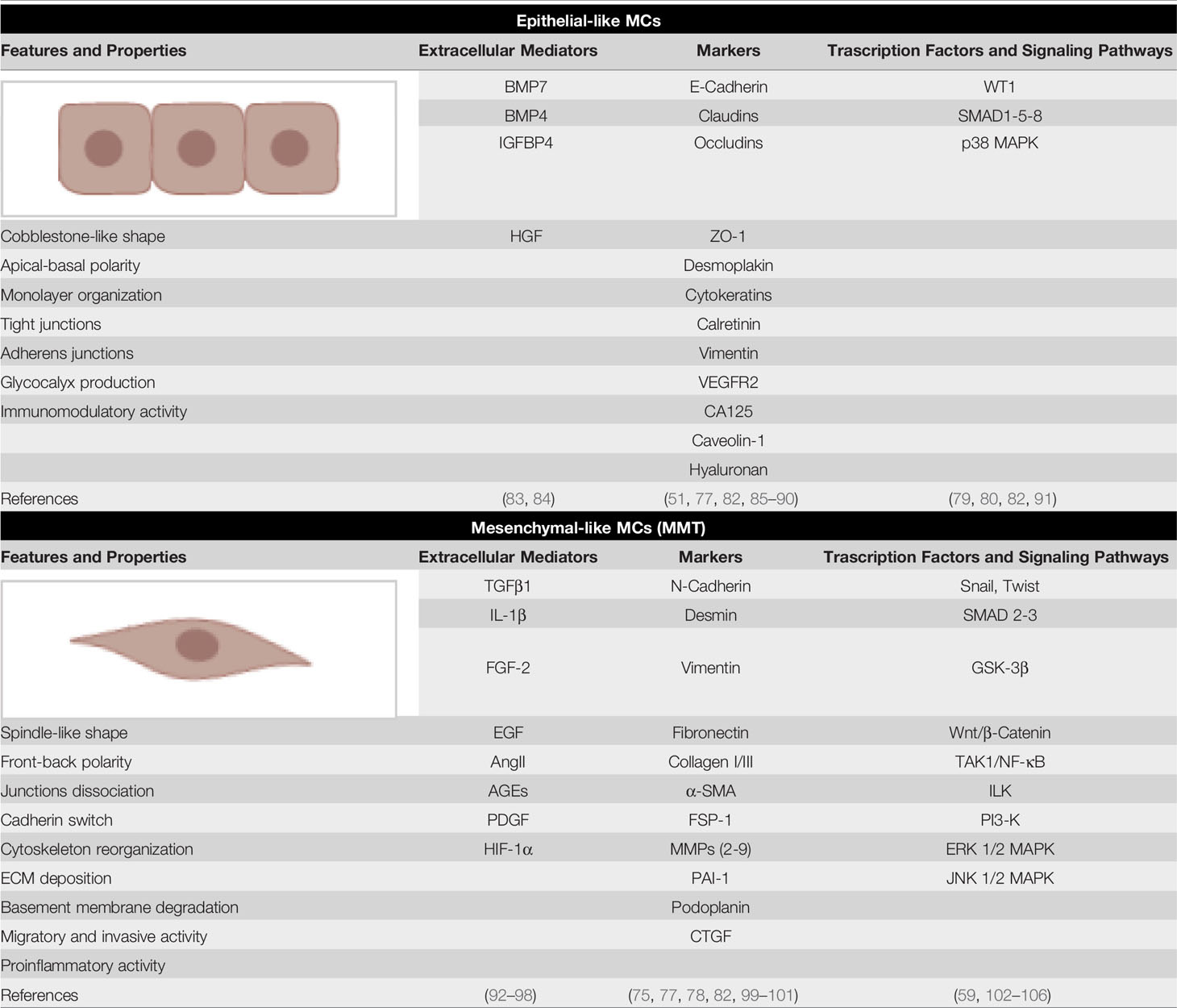
Table 2 Epithelial-like and mesenchymal markers of MCs. The main extracellular regulators of MC plasticity, molecular markers and signaling pathways implicated are shown.
The secretion of TGF-β by MCs or by other cells such as MØs is central for a full induction of the MMT program. Once transdifferentiated, MCs may invade the submesothelial stroma where they proliferate and produce cytokines and ECM proteins directly promoting peritoneal fibrosis.
The profibrotic activity of TGF-β1 is counteracted by members of the BMP family, such as IGFBP4, BMP4 and BMP7, secreted by the same MCs (83, 84).
Interestingly, transdifferentiated MCs tend to acquire a new stability. This behaviour is different from that of cells with a stronger epithelial identity that rapidly recover epithelial features once the transdifferentiating stimulus has been removed (107). The maintenance of mesenchymal features in MCs has been linked to epigenetic changes, and epigenetic modulation may both influence mesothelial differentiation and promote the recovery of a “epithelial-like” phenotype from in vivo transdifferentiated cells (99, 108).
High throughput experiments have demonstrated that induction of MMT from different stimuli induces the acquisition of common dedifferentiation features characterized by the expression of signatures of profibrotic and pro-inflammatory cytokines such as TGFβ, VEGF, and IL-6 (29, 77, 83, 109). In fact, activated MCs are major producers of TGF-β1, VEGF and IL-6, whose concentrations are elevated especially during peritonitis and have been associated with ultrafiltration decline and protein loss (18, 110). The secretion of these cytokines impacts fibrosis, angiogenesis and the inflammatory response.
The ability of MCs to generate myofibroblasts has been a highly debated topic in the previous years. Lineage tracing experiments performed to demonstrate the mesothelial origin of peritoneal myofibroblasts have given contrasting results, with the more recent studies suggesting the existence of a population of MCs origin invading the submesothelial space (83, 111, 112).
Moreover, the coexpression of bona fide MMT markers such as αSMA and fsp1 absent in epithelial-like MCs with mesothelial/epithelial markers has been demonstrated in vivo both in peritoneum after PD and in peritoneal adhesion (75, 77, 78, 82).
In the peritoneum of mice exposed to PD fluid, the relative contribution of the myofibroblasts-generating cellular populations, including resident dermal fibroblasts, endothelial cells, bone marrow derived cells and MCs, has been quantified (113). As in other organs, endothelial cells contribute to peritoneal fibrosis through a process of endothelial to mesenchymal transition (EndMT) (82, 114). Also, bone marrow derived progenitors, such as mesenchymal stem cells and fibrocytes may generate peritoneal myofibroblasts (115).
MCs may in different ways influence the fibrotic process. Besides being a main source of TGF-β1, activated MCs produce abundant amounts of fibronectin and collagens, may rearrange the ECM through the expression of contractile proteins (αSMA) and produce various metalloproteinases (MMPs), such as MMP2, MMP9 and MMP14 as well as MMP inhibitors such as TIMP1 and PAI1 (77, 99, 100). Besides directly impacting the fibrotic process, the production of inflammatory cytokines and chemokines stimulates other stromal cells and components of innate and adaptive immunity (see below). Thus, MCs are candidates for cellular interventions aimed at restoring the continuity of the monolayer and to warrant the peritoneal function.
Leukocyte Subpopulations Implicated in the Fibrotic Response
Peritoneum as a Lymphatic Organ: The Role of FALCs
Due to its unique localization in the abdominal cavity and its huge extension, peritoneum is a favorite site for encountering with antigens and for the generation of the subsequent immune response. Recirculating leukocytes patrol the peritoneal cavity in uninflamed peritoneum in addition to stably resident leukocyte populations constituted mainly by macrophages and mast cells.
Besides conventional lymph nodes, peritoneum hosts unique anatomic structures called milky spots or fat-associated lymphoid clusters (FALCs), which are clusters of leukocytes localized especially in omentum and endowed with the ability to collect fluids, particulates, and cells from the peritoneal cavity. Their frequency and size increase in the omentum of patients undergoing PD (116, 117). FALCs are mainly composed of MØs, MCs and B1 cells. B1 cells consist in a subset of B cells that can be distinguished from conventional B (B2) cells by expression of distinct cell-surface markers and antigen receptors that can bind common bacterial epitopes. B1 cells have the potential to produce natural antibodies that provide a first protection to bacterial infections (118). Intestinal leakage or the intraperitoneal delivery of microorganisms lead to rapid activation of B1 cells and promote T cell-independent antibody responses (119).
The chemokine CXCL13, of mesothelial origin, controls the localization of B1 cells into FALCs (120). Another chemokine, CCL19, is produced by other structural components of FALCs called FALCs fibroblast reticular cells (FALCs FRCs) and it is relevant for monocyte recruitment during inflammation. The cross-talk between CCL19 producing FALCs FRCs and inflammatory monocytes promote T cell dependent-B cell immune responses (121) (Figure 2A). Thus, FALCs play a main role both in the regulation of PMN and mononuclear cell recruitment in the first phase of inflammation, as well as in the subsequent induction of the adaptive immunity.
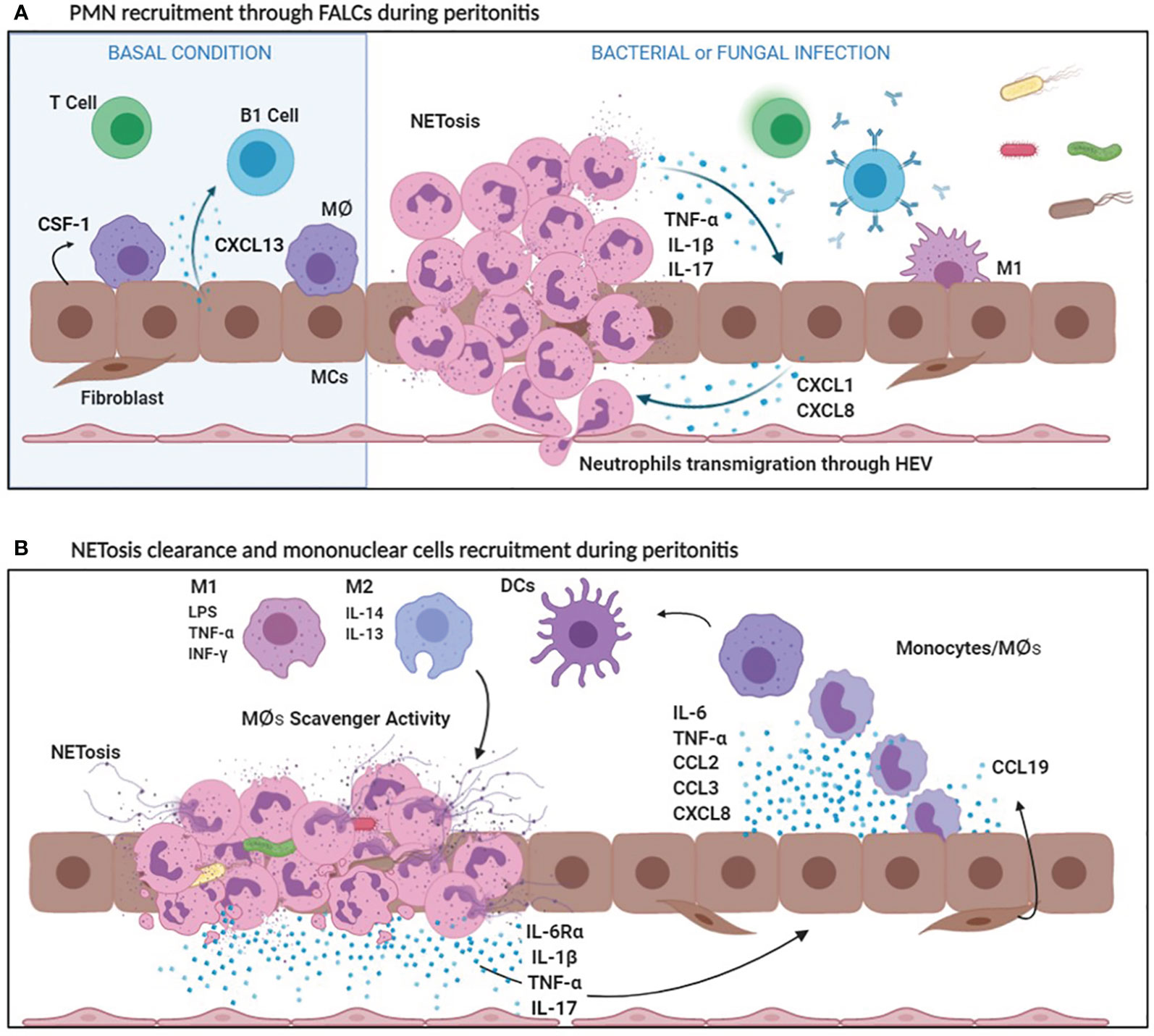
Figure 2 (A) PMN recruitment through FALCs during peritonitis. In basal conditions, MCs secrete CXCL13, which attracts B1 cells in FALCs and CSF1, a specific MØ growth factor. Bacterial and fungal infections stimulate the production of CXCL1 and CXCL8, by MCs. Bacterial products, CXCL1 and CXCL8 promote the recruitment of a first wave of PMNs entering the peritoneal cavity through FALCs. PMNs cause an initial inflammatory response secreting inflammatory cytokines (IL-1ß, TNF-α). Afterwards, NETosis helps in sequestering microorganisms in FALCs. (B) NETosis clearance and mononuclear cell recruitment during peritonitis. Bacterial products, as well as IL-1ß, stimulate the production of IL-6, TNF-α, CCL2, CCL3, and CXCL8 by MCs. IL-6Ra shedding by PMNs promotes a peripheral IL-6 response (transignaling). Cytokines and chemokines released during the inflammatory process favor mononuclear recruitment and differentiation. Mononuclear phagocytes differentiate in Macrophages (MØs) and dendritic cells (DCs). Among MØs, M1 subtype is endowed with pro-inflammatory and cytotoxic properties, whereas M2 MØs have an anti-inflammatory activity. Moreover, M2 MØs play a key role in the clearance of neutrophils debris due to scavenger activity.
Leukocyte Recruitment During Peritonitis: From Neutrophils to Mononuclear Cells
Infectious peritonitis offers a favorite experimental model to study the interactions between immune system and the peritoneal stroma (122). Infection with Gram positive bacteria such as Staphylococcal spp. or with cell-free components such as LPS or zymosan, mimicking Gram negative or fungal infection, respectively, promotes a first wave of polymorphonuclear neutrophils recruited by chemoattractants of bacterial origin and by chemokines such as CXCL1 and CXCL8 produced mainly by MCs and omental fibroblasts. Neutrophils can use high endothelial venules present in FALCs to enter the peritoneal cavity under the guidance of CXCL1 (120).
Neutrophil influx causes an initial inflammatory response due to the accumulation of neutrophil-secreted proteases and reactive oxygen species. Once they entered the peritoneum, neutrophils undergo NETosis, which consists in the release of necrotic cell DNA forming a net of aggregated neutrophils able to trap and sequester microorganisms in FALCs, thus limiting their spreading (123). Interestingly, HMGB1 produced during the inflammatory response promotes NETosis and trap formation through interaction with TLR4 (124) (Figure 2B).
The production of CXCL1 and CXCL8 by the peritoneal stroma is enhanced by inflammatory cytokines such as IL-1β and to a lesser extent, TNFα (125). MCs stimulated by LPS or IL-1β also produce a number of cytokines and chemokines including IL-6, TNFα, CCL2, CCL3, that favor mononuclear cell recruitment and activation (126). The first wave of neutrophils is then replaced by a mononuclear infiltrate.
Neutrophils take part in this process secreting a shed form of IL-6 receptor, IL-6Rα. Through a mechanism called transignaling, the local increase of IL-6Rα promotes an IL-6-mediated neutrophil clearance subsequent to mononuclear cell recruitment (127, 128). Apoptotic neutrophils are phagocytosed by MØs and to a lesser extent by the same MCs (129). Necrotic neutrophils and NETs promote the infiltration of mature MØs recruited also by locally produced chemokines such as CXCL8 and CCL2 (130).
Neutrophil influx and clearance are also regulated by two other cytokines, IL-17 and IFN-γ. IL-17 is secreted mainly by various leukocyte subpopulations, including neutrophils, Th17 and γδ T lymphocytes and its expression correlates with the duration of the PD treatment and with the extent of peritoneal inflammation and fibrosis (131, 132). IFN-γ production by Th1 lymphocytes and NK cells (see below) contributes with IL-6 in favoring an initial neutrophil recruitment and subsequent clearance (133).
IL-17 promotes CXCL1 production by MCs through expression and activation of the transcription factor Sp1, whereas IFN-γ through STAT1 activation limits Sp1 induced CXCL1 production. Breaking of this homeostatic cross regulation may lead to excessive or ineffective recruitment of neutrophils during peritonitis with subsequent damages in the PM (131).
Inflammation, Scavenging and Antigen Presentation: Monocytes/MØs and Dendritic Cells
Tissue mononuclear phagocytes, comprised mainly of MØs and dendritic cells (DCs), are key tissue-resident components of the peritoneal immune system. Their roles include induction of the inflammatory response, pathogen clearance, tissue repair, and antigen presentation.
MØs are the major resident immune population in the PM. At the same time, monocytes/MØs are the predominant cell types found in dialysis effluent (134, 135).
Resident MØs form the first line of defense against peritoneal infection in peritonitis. Once the inflammatory response is initiated, monocytes follow a first wave of leukocytes composed mainly by PMN neutrophils.
MØs are generally classified into two functional subtypes. Classical active MØs, also called M1 MØs (representative markers: iNOS and CD80) are defined by their pro-inflammatory and cytotoxic properties, while M2 MØs (representative markers: CD163, CD206 and Arg1) are characterized by anti-inflammatory and scavenging properties (136). However, MØ M1 and M2 subtypes should be considered as the extreme points of a continuum of different cellular populations acting in in different physio-pathological contexts (137, 138).
M1 polarization typically involves IFN-γ with a TLR agonist, such as LPS. M1 MØs, through production of IL-1β and TNF-α, are capable of amplifying the first phase of the inflammatory process and of recruiting other leukocytes into the peritoneum via the creation of a gradient of chemotactic cytokines, such as CXCL8, CCL2 and CCL5. This process is also facilitated by a cytokine driven up-regulation of adhesion molecule expression (ICAM-1 and VCAM-1) on the surface of MCs. At the same time, the generation of M2 MØs, which is sustained by IL-4 and IL-13, plays a role in the resolution of inflammation through the production of soluble anti-inflammatory mediators, and the clearance of debris such as apoptotic or necrotic products, due to their scavenger activity (137).
In the model of peritonitis induced by zymosan, mimicking fungal infection, an infiltration of both M1 and M2 MØs occurs. In this context, MØs are involved in the clearance of debris resulting from neutrophil apoptosis. Both M1 and M2 MØs recognize and endocytose dead cellular debris through apoptosis inhibitor of macrophage (AIM, also called CD5L), a member of the scavenger receptor cysteine-rich superfamily (22) (Figure 3A).
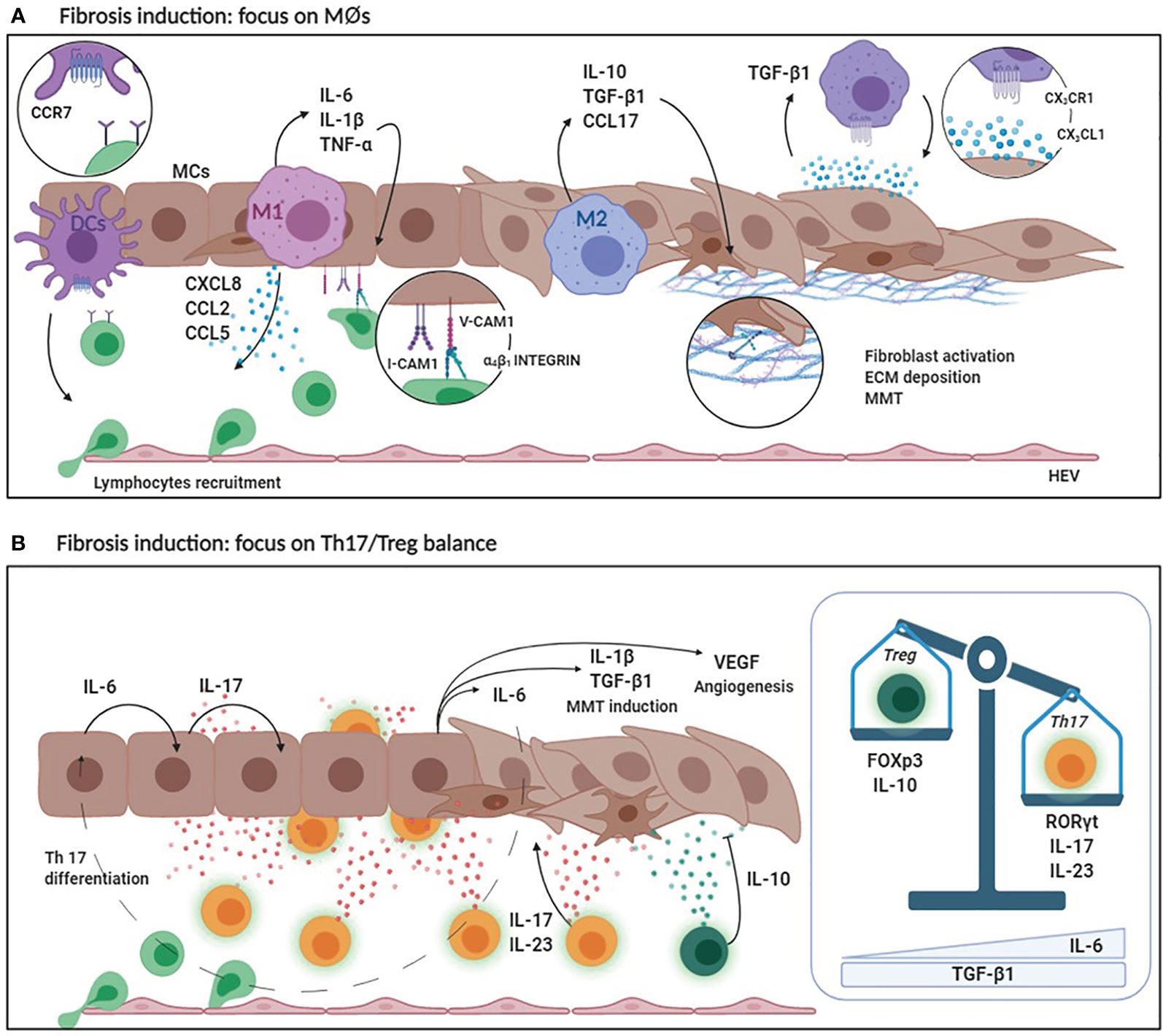
Figure 3 (A) Fibrosis induction: focus on MØs. DCs secrete high levels of CCR7 favoring lymphocyte recruitment. Pro-inflammatory M1 MØs secrete CCL2, CCL3 and CXCL8 that are chemoattractant for lymphocytes and monocytes. At the same time, M1 MØs produce inflammatory cytokines such as IL-6, IL-1ß and TNF-α that enhance the expression of adhesion molecules (V-CAM1/I-CAM1) by MCs promoting leukocyte adhesion. M2 MØs produce anti-inflammatory cytokines (IL-10) and lymphocyte chemoattracting chemokines (CCL17). Predominance of M2 MØ response leads to an increased TGF-ß1 secretion that induces MMT of MCs with up-regulation of ECM protein production. Moreover, MCs secreting CX3CL1 recruit MØs expressing CX3CR1. Receptor/ligand interaction determines a positive loop that promote, in turn, CX3CL1 and TGF-ß1 expression. (B) Fibrosis induction: focus on Th17/Treg balance. The production of IL-6 by MØs and MCs during the inflammatory process promotes IL-17 production by the peritoneal stroma which, in combination with IL-23, promotes the differentiation of Th17 lymphocytes. IL-17 promotes IL-1ß, TGF-ß1, VEGF and IL-6 production causing MMT induction and neoangiogenesis. Th17 abundance affects the activity of regulatory T lymphocytes. Treg lineage has an anti-inflammatory activity (due to IL-10 production) and protects the peritoneal membrane by mediating tolerance mechanisms. High levels of IL-6 and TGF-ß1 determine the predominance of Th17 over Treg with consequent peritoneal damage and fibrosis.
Experimental evidence analysing samples from PD patients demonstrates that the majority of peritoneal MØs phenotypically and functionally resemble in vitro polarized M2 MØs (139, 140).
In this context, a dysregulated M2 MØs response may promote the development of fibrosis and the decrease of functionality of the PM through the production of a number of extracellular factors.
The production of TGF-β1 by M2 MØs mediates the induction of MMT in MCs and the proliferation and activation of submesothelial fibroblasts, a process leading to ECM production, rearrangement, angiogenesis, and fibrosis. Moreover, MMP9 and CCL18 secretion is increased in both PD effluents and PM biopsies of PD patients (139). MMP9 plays a role in the activation of latent TGF-β1, whereas CCL18 levels have been associated with poor ultrafiltration capability and with development of EPS (139). M2 MØs may favor a fibrotic response also producing CCL17, which promotes migration, proliferation and collagen production by submesothelial fibroblasts (141).
However, besides M2, some evidence points towards an active role of the M1 subtype in the genesis of peritoneal fibrosis. The inhibition of the protein kinase C beta pathway promotes peritoneal damage and fibrosis via M1 MØs polarization in a murine model of PD (142). Also, in vivo approaches of MØ chemical deletion followed by reperfusion pointed towards an active role of the M1 subpopulation in the genesis of fibrosis, as suggested also by an in vitro study (143, 144).
The presence of MØs in the PM is tightly dependent on a cross talk with MCs. In basal conditions, MØs require and interact with MCs present in FALCs and secreting colony stimulating factor 1 (CSF1), a specific MØ growth factor (145). In inflammatory conditions and during PD, fractalkine (CX3CL1), a chemokine secreted by MCs, recruits and activates MØs expressing CX3CR1 in the peritoneal wall, promoting the fibrotic process. A positive feedback loop is induced where direct interaction with CX3CR1-expressing MØs promotes expression of CX3CL1 and TGF-β1 by MCs. In turn, TGF-β1 upregulates CX3CR1 expression in MØs (146).
Besides MØs endowed with pro-inflammatory or scavenger abilities, migrating mononuclear cells may differentiate in DC subsets, characterized by CD1c positivity and different profiles with respect to CD14 positive cells. In particular, CD1c positive cells have upregulated costimulatory molecules, CD80 and CD86, important for antigen presentation and T-cell activation, and CCR7, favoring migration to secondary lymphoid organs such as local lymph nodes where antigen presentation may occur (134). Interestingly, severe and recurrent episodes of peritonitis were associated with significantly higher numbers of peritoneal neutrophils, MØs as well as higher ratio of MØs to DCs than the successfully treated ones (134). The functional role of MØs has been analyzed with pharmacological but not with genetic or immunological approaches. Chemical depletion of MØs using clodronate attenuated peritoneal thickening and suppressed TGF-β1, VEGF expression and MMT induction in a model of peritoneal fibrosis induced by chlorhexidine gluconate in rats (147). Accordingly, depletion of MØs limited fibrosis in a mouse model of PD fluid exposure (143).
Mastocytes
Mastocytes or mast cells are predominantly localized at sites that have direct contact with the external environment, such as the skin, airways, and intestine, where they function as sentinel cells in host defense and as main inducer of type I hypersensitivity and of the allergic response (148).
While mast cells have been implicated in fibrogenesis, angiogenesis, and the immune response against bacteria in various organs such as kidney and in lung, only a few studies have dealt on the role of mastocytes in peritoneal fibrosis (149, 150).
Mechanistically, mast cells secrete various mediators of inflammation such as histamine, platelet-activating factor, prostaglandins, thromboxane, leukotriene, chymase. Moreover, the secretion of cytokines such as TGF-β1 and IL-17 directly contributes to peritoneal fibrosis (149, 151, 152)
The number of mast cells was significantly higher in the fibrotic peritoneum of rats with chronic renal failure (CRF rats). Tranilast, an anti-allergic drug with an activity of mast cell stabilizer, was demonstrated to block the progression of peritoneal fibrosis in CRF rats (153).
An interesting study performed on mast cell-deficient rats demonstrated that mast cells promoted the increase of the omental thickness and omental adhesion formation favoring leukocyte recruitment (154).
Results in human peritoneal disease are controversial. A first study showed reduced numbers of mast cells in samples from PD patients (155), whereas increased mastocytes numbers have been found in samples from different inflammatory and fibrotic peritoneal diseases, including PD and EPS (156). Thus, although evidence suggests that these cells may amplify the inflammatory response during peritoneal damage, their functional role has not so far been demonstrated.
Natural Killer Cells
Natural killer (NK) cells are a specialized lymphocyte subpopulation that play a significant role during viral infections and in tumor immune surveillance through direct killing of virus infected or tumor cells or by production of cytokines and chemokines. NK cells recirculate throughout the peritoneal cavity and are present in the peritoneal fluid. Moreover, in uninflamed peritoneum, a resident NK cell population isolated in mice was able to secrete IFN-γ, GM-CSF, and TNF-α and endowed with killing ability (157).
During an acute inflammatory process such as peritonitis, NK cells produce inflammatory cytokines such as TNFα and IL-6. Moreover, through production of IFN-γ and TGF-β1 these cells may directly orchestrate the fibrotic process.
In other organs, NK cells actively contribute to the genesis of the fibrotic damage. Tubulointerstitial human CD56bright NK cells correlate with loss of kidney function and with induction of fibrosis and chronic kidney disease progression, mechanistically linked to increased NK cell-mediated IFN-γ production (158).
Interestingly, besides its potential in the amplification of the inflammatory response, NK cells appear to have a role in the resolution of inflammation in antigen-dependent peritonitis promoting neutrophil apoptosis (159). Previous results confirm that NK cells are capable of inducing apoptosis of neutrophils (160).
Studies performed in humans are limited to adoptive transfer of activated NK cells in an autologous NK cells setting used in a frame of tumor therapy. It was demonstrated that administration of NK cells in combination with IL-2 in patients with malignancies caused peritoneal fibrosis (161). Thus, although their role is potentially relevant, no definitive information is reported about NK cells in the genesis of peritoneal fibrosis.
T Lymphocytes: A Balance of Mutually Influencing Subpopulations
Besides the components of the innate immunity, the activity of different T lymphocyte subsets is fundamental for the regulation of the inflammatory response in the genesis of peritoneal fibrosis and it could provide molecular targets to control peritoneal damages. The relevance of the adaptive immunity in peritoneal fibrosis is demonstrated by the use of lymphocyte-deficient mice. In RAG-deficient mice, lacking mature T and B lymphocytes, treatment with zymosan induced an exaggerated inflammatory response with increased PMN infiltration (162). Accordingly, the use of another experimental system demonstrated a role of adaptive immunity in limiting PMN and MØ recruitment (163).
More generally, these approaches suggest that a network of mutual interactions occurs between peritoneal stroma, innate and adaptive immunity effectors during the genesis of peritoneal fibrosis.
The composition of peritoneal fluid lymphocytes varies with respect to blood lymphocytes. In particular, B lineage comprises only around 2% of the total fluid, and T leukocyte subpopulations are differently represented (164). Moreover, changes in the consistency of T lymphocyte subpopulations occur during peritoneal inflammation and during the practice of PD.
With respect to the balance between CD4+ T-helper 1(Th1) and T-helper 2 (Th2) subpopulations, it has been demonstrated that during episodes of acute peritonitis, the immune response is predominantly directed to the induction of Th1 cells (165).
On the other hand, the Th2 subset rapidly expands with the practice of PD (166). The Th1/Th2 ratio could be evaluated by measuring IFN-γ (Th1 subset) and IL-4 (Th2 subset) levels both in circulating and peritoneum-derived Th lymphocytes. In PD patients, the IFN-γ/IL-4 ratio is significantly reduced, indicating a negative effect of bioincompatible fluids towards the Th1 cell subset. Interestingly, this effect could be avoided using more biocompatible fluids containing bicarbonate-buffered and icodextrin, that may reestablish a more physiological Th1/Th2 balance and a reduced peritonitis rate (167).
A recently characterized leukocyte subpopulation, Th17 lymphocytes have been demonstrated as the main driver of peritoneal fibrosis (132). The expression and the activity of this lymphocytic subset is linked to the production of IL-17. Besides Th17, other leukocytes, including CD4+ and γδ T lymphocytes, neutrophils, and mast cells may secrete this cytokine during exposure to PD fluids or during peritonitis (168).
The strong stimulation of Th17 response during these pathological conditions is due to both exogenous and endogenous factors. Exogenous factors are represented by bacteria and their derivatives entering the peritoneal cavity through PD catheter or via intestinal translocation. These bacteria stimulate TLR’s response by the peritoneal stroma, which leads to an upregulation of IL-6 levels, promoting IL-17 production and subsequent differentiation of Th17 lymphocytes (169). Similarly to bacterial derivatives, also factors related to PD fluid such as AGEs expressed in conventional lactate-based PD solution with low pH and high GDP contents are able to stimulate the Th17 response (170) (Figure 3B).
IL-17 contributes to the host defense against bacteria and fungi (171). It promotes neutrophil recruitment favoring the release by MCs of chemotactic factors specifically attracting neutrophils. Moreover, the IL-17 response favors the secretion of a network of cytokines and chemokines including IL-1β, IL-6, CCL2 and TGF-β1. In peritoneum, IL-17 favors through different mechanisms the secretion of VEGF by MCs, promoting angiogenesis (168). Most importantly, repeated intraperitoneal administration of exogenous IL-17 led to increased expression of several fibrosis-related genes, whereas its neutralization with anti-IL-17 alleviated the extent of peritoneal fibrosis (132).
The consistence of Th17 population in the peritoneum directly affects the activity of another T cell subset, regulatory T lymphocytes (Treg) (14). Tregs are suppressors of activated T cell expansion, their activity is anti-inflammatory and favors the induction of tolerance (172). IL-6, in combination with TGF-β1, is the main cytokine involved in the helper 17/regulatory T (Th17/Treg) balance. The predominance of IL-6 favors the generation of Th17 lymphocytes, which produce inflammatory cytokines. On the other hand, TGF-β1 in the absence of IL-6 promotes the Treg lineage, able to maintain peripheral tolerance and produce anti-inflammatory mediators such as IL-10, which has been linked to protection of the peritoneal membrane from inflammatory damage (173).
Interestingly, the plasma membrane receptor CD69 appears to control Th17/Treg balance. The exacerbated peritoneal fibrosis observed in CD69−/− mice could be alleviated by the blockade of IL-17 (174). Mechanistically, it was demonstrated that CD69 directly interacting with Jak3/STAT5 blocks Th17 differentiation (175).
Besides shaping the immune response, these changes may impact MC plasticity: it was demonstrated that IL-17 itself is able to induce EMT in bronchial cells while inducing peritoneal fibrosis in vivo (132, 176). In contrast, low levels of IL-6 may promote Treg differentiation, which is associated to high IL-10 expression, leading to the establishment of an anti-inflammatory state and possibly MMT reversal (14, 177–179).
In case of peritoneal dialyzed patients, the predominassnce of Th17 over Treg favors fibrosis development and PM failure instead of Treg-mediated tolerance. Currently, the modulation of the expression of the cytokines involved in Th17/Treg balance through recombinant antibodies or cytokines is an attracting field for the design of new therapies aimed at counteracting peritoneal MMT and fibrosis.
A bridge between the adaptive and the innate arm of the immune system is constituted by Mucosal-associated invariant T (MAIT) cells. These cells are different from conventional T cells, since they do not react through major histocompatibility complex (MHC) (180). Peritoneal MAITs (pMAITs) provide a marker for systemic inflammation during spontaneous bacterial peritonitis (SBP), since they are configured to respond pro-inflammatory chemotactic signals sensed by CCR5, CXCR3 and CCR6 (181). pMAITs are a source of IL-17 (102). Clinical data indicated a specific immune activation of pMAIT, driven by CD69 expression and correlated to disease severity (181).
Conclusions
The cellular and molecular mechanisms described above witness the complexity of the physiopathologic response occurring in the inflamed peritoneum.
A study published almost 25 years ago by Topley et al. identified the relationship between MØs and MCs as a key factor in the response of peritoneum to infections, whose dysregulation was causal to ultrafiltration failure and fibrosis in PD patients (182). At that time, it was already clear that MCs under MØ-driven stimuli may produce a number of extracellular mediators including arachidonic acid derivatives, cytokines and chemokines promoting the amplification of the inflammatory response. Since then, the understanding of cellular and molecular mechanisms underlying has evolved considerably. A relevant discovery has been the characterization of different MØ subpopulations implicated in the secretion of pro-inflammatory mediators, phagocytosis, apoptotic debris removal and scavenging activity. Another major breakthrough has been the identification of the ability of MCs and other cells to undergo deep dedifferentiation processes culminating in the generation of myofibroblasts. Moreover, a relevant concept that has emerged in these years is that the relationship between MØs and MCs is not unidirectional: MCs play an active role in influencing MØ recruitment, survival and differentiation due to the synthesis of extracellular mediators acting specifically on MØs. Last, this dialogue is not limited to MØs and MCs: stromal driven signals such as IL-17, IL-6 and TGF-β1 shape the Th17/Treg balance, thus impacting the fibrotic response.
Although the fibrotic process has common marks in all the organs, peritoneum fibrosis has specific features due to the anatomic localization and the cellular components forming this organ. The anatomic localization favors the encounter with microorganisms through unique structures (i.e. the FALCs) With regard to peritoneal stroma, the characteristic plasticity of MCs, their ability to transdifferentiate and to become indistinguishable from myofibroblasts, makes the difference with respect to other organs, such as the liver, where parenchyma cells (hepatocytes) may give little direct contribution in the genesis of fibrosis.
These new discoveries related to cellular communication and cellular plasticity may have an impact in future therapeutic strategies. Future directions aimed at improving peritoneal viability and thus duration of PD therapy may be focused at further improving PD fluid biocompatibility, as well at using inhibitory monoclonal antibodies, in line with recent advances with therapy of inflammatory/profibrotic diseases. Also specific therapies aimed at supporting MC viability and regulating peritoneal immune system and immune cell/MC interactions may give a contribution. Deepening the analysis of cellular and molecular mechanisms underlying peritoneal fibrosis may shed light on our understanding of how we can preserve the long-term function of the PM as a dialysing organ, but also to treat other forms of peritoneal fibrosis such as post-surgical adhesions or tumor related-peritoneal fibrosis.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
MTe wrote the manuscript and composed the figures. FT wrote the manuscript and organized the tables. CM wrote the manuscript. MC wrote the manuscript and composed the figures. MTr critically reviewed the manuscript. ML critically reviewed the manuscript and provided a general scheme of interpretation. RS conceived and wrote the manuscript, and critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Sapienza University of Rome RG11916B6A9C42C7 to MT. IMPROVE-PD project that has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement number 812699 to ML-C. Spanish Ministry of Science and Innovation/Fondo Europeo de Desarrollo Regional (PID2019-110132RB-I00/AEI/10.13039/501100011033) to ML-C.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr Loredana Cifaldi and Dr Nicoletta Mancianti for critically reviewing this manuscript. Figures were created with BioRender.com.
References
1. Mutsaers SE, Birnie K, Lansley S, Herrick SE, Lim CB, Prele CM. Mesothelial cells in tissue repair and fibrosis. Front Pharmacol (2015) 6:113. doi: 10.3389/fphar.2015.00113
2. Di Paolo N, Sacchi G. Atlas of peritoneal histology. Perit Dial Int (2000) 20 Suppl 3:S5–96. doi: 10.1177/089686080002003S02
3. Zhou Q, Bajo MA, Del Peso G, Yu X, Selgas R. Preventing peritoneal membrane fibrosis in peritoneal dialysis patients. Kidney Int (2016) 90(3):515–24. doi: 10.1016/j.kint.2016.03.040
4. Cho Y, Johnson DW. Peritoneal dialysis-related peritonitis: towards improving evidence, practices, and outcomes. Am J Kidney Dis (2014) 64:278–89. doi: 10.1053/j.ajkd.2014.02.025
5. Jagirdar RM, Bozikas A, Zarogiannis SG, Bartosova M, Schmitt CP, Liakopoulos V. Encapsulating Peritoneal Sclerosis: Pathophysiology and Current Treatment Options. Int J Mol Sci (2019) 20(22):5765. doi: 10.3390/ijms20225765
6. Tseng CC, Chen JB, Wang IK, Liao SC, Cheng BC, Wu AB, et al. Incidence and outcomes of encapsulating peritoneal sclerosis (EPS) and factors associated with severe EPS. PloS One (2018) 13(1):e0190079. doi: 10.1371/journal.pone.0190079
7. Danford CJ, Lin SC, Smith MP, Wolf JL. Encapsulating peritoneal sclerosis. World J Gastroenterol (2018) 24:3101–11. doi: 10.3748/wjg.v24.i28.3101
8. Liakakos T, Thomakos N, Fine PM, Dervenis C, Young RL. Peritoneal adhesions: etiology, pathophysiology, and clinical significance. Recent advances in prevention and management. Dig Surg (2001) 18(4):260–73. doi: 10.1159/000050149
9. Ten Broek RP, Issa Y, Van Santbrink EJ, Bouvy ND, Kruitwagen RF, Jeekel J, et al. Burden of adhesions in abdominal and pelvic surgery: systematic review and met-analysis. BMJ (2013) 347:f5588. doi: 10.1136/bmj.f5588
10. Mutsaers SE, Prele CM, Pengelly S, Herrick SE. Mesothelial cells and peritoneal homeostasis. Fertil Steril (2016) 106(5):1018–24. doi: 10.1016/j.fertnstert.2016.09.005
11. Sandoval P, Jimenez-Heffernan JA, Rynne-Vidal A, Perez-Lozano ML, Gilsanz A, Ruiz-Carpio V, et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J Pathol (2013) 231(4):517–31. doi: 10.1002/path.4281
12. Rynne-Vidal A, Au-Yeung CL, Jimenez-Heffernan JA, Perez-Lozano ML, Cremades-Jimeno L, Barcena C, et al. Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J Pathol (2017) 242(2):140–51. doi: 10.1002/path.4889
13. Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL, et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J Clin Invest (2014) 124:4614–28. doi: 10.1172/JCI74778
14. Liappas G, Gonzalez-Mateo GT, Majano P, Sanchez-Tomero JA, Ruiz-Ortega M, Rodrigues Diez R, et al. T Helper 17/Regulatory T Cell Balance and Experimental Models of Peritoneal Dialysis-Induced Damage. BioMed Res Int (2015) 2015:416480. doi: 10.1155/2015/416480
15. Strippoli R, Moreno-Vicente R, Battistelli C, Cicchini C, Noce V, Amicone L, et al. Molecular Mechanisms Underlying Peritoneal EMT and Fibrosis. Stem Cells Int (2016) 2016:3543678. doi: 10.1155/2016/3543678
16. Mehrotra R, Devuyst O, Davies SJ, Johnson DW. The Current State of Peritoneal Dialysis. J Am Soc Nephrol (2016) 27:3238–52. doi: 10.1681/ASN.2016010112
17. Nochaiwong S, Ruengorn C, Koyratkoson K, Thavorn K, Awiphan R, Chaisai C, et al. A Clinical Risk Prediction Tool for Peritonitis-Associated Treatment Failure in Peritoneal Dialysis Patients. Sci Rep (2018) 8(1):14797. doi: 10.1038/s41598-018-33196-2
18. Goodlad C, George S, Sandoval S, Mepham S, Parekh G, Eberl M, et al. Measurement of innate immune response biomarkers in peritoneal dialysis effluent using a rapid diagnostic point-of-care device as a diagnostic indicator of peritonitis. Kidney Int (2020) 97:1253–9. doi: 10.1016/j.kint.2020.01.044
19. Aufricht C, Beelen R, Eberl M, Fischbach M, Fraser D, Jorres A, et al. Biomarker research to improve clinical outcomes of peritoneal dialysis: consensus of the European Training and Research in Peritoneal Dialysis (EuTRiPD) network. Kidney Int (2017) 92:824–35. doi: 10.1016/j.kint.2017.02.037
20. Pauwels S, De Moor B, Stas K, Magerman K, Gyssens IC, Van Ranst M, et al. Coxsackievirus B1 peritonitis in a patient treated with continuous ambulatory peritoneal dialysis: a case report and brief review of the literature. Clin Microbiol Infect (2012) 18(10):E431–434. doi: 10.1111/j.1469-0691.2012.03985.x
21. Miles R, Hawley CM, Mcdonald SP, Brown FG, Rosman JB, Wiggins KJ, et al. Predictors and outcomes of fungal peritonitis in peritoneal dialysis patients. Kidney Int (2009) 76(6):622–8. doi: 10.1038/ki.2009.202
22. Tomita T, Arai S, Kitada K, Mizuno M, Suzuki Y, Sakata F, et al. Apoptosis inhibitor of macrophage ameliorates fungus-induced peritoneal injury model in mice. Sci Rep (2017) 7(1):6450. doi: 10.1038/s41598-017-06824-6
23. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
24. Roh JS, Sohn DH. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw (2018) 18(4):e27. doi: 10.4110/in.2018.18.e27
25. Colmont CS, Raby AC, Dioszeghy V, Lebouder E, Foster TL, Jones SA, et al. Human peritoneal mesothelial cells respond to bacterial ligands through a specific subset of Toll-like receptors. Nephrol Dialysis Transplant (2011) 26:4079–U4614. doi: 10.1093/ndt/gfr217
26. Van Aubel RA, Keestra AM, Krooshoop DJ, Van Eden W, Van Putten JP. Ligand-induced differential cross-regulation of Toll-like receptors 2, 4 and 5 in intestinal epithelial cells. Mol Immunol (2007) 44(15):3702–14. doi: 10.1016/j.molimm.2007.04.001
27. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
28. Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol (2001) 167:1882–5. doi: 10.4049/jimmunol.167.4.1882
29. Raby AC, Gonzalez-Mateo GT, Williams A, Topley N, Fraser D, Lopez-Cabrera M, et al. Targeting Toll-like receptors with soluble Toll-like receptor 2 prevents peritoneal dialysis solution-induced fibrosis. Kidney Int (2018) 94(2):346–62. doi: 10.1016/j.kint.2018.03.014
30. Wornle M, Sauter M, Kastenmuller K, Ribeiro A, Roeder M, Schmid H, et al. Novel role of toll-like receptor 3, RIG-I and MDA5 in poly (I:C) RNA-induced mesothelial inflammation. Mol Cell Biochem (2009) 322(1-2):193–206. doi: 10.1007/s11010-008-9957-4
31. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (2003) 301(5633):640–3. doi: 10.1126/science.1087262
32. Doyle S, Vaidya S, O’connell R, Dadgostar H, Dempsey P, Wu T, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity (2002) 17:251–63. doi: 10.1016/S1074-7613(02)00390-4
33. Merkle M, Ribeiro A, Sauter M, Ladurner R, Mussack T, Sitter T, et al. Effect of activation of viral receptors on the gelatinases MMP-2 and MMP-9 in human mesothelial cells. Matrix Biol (2010) 29(3):202–8. doi: 10.1016/j.matbio.2009.12.002
34. Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol (2004) 76(3):514–9. doi: 10.1189/jlb.0304127
35. Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med (2010) 14(11):2592–603. doi: 10.1111/j.1582-4934.2010.01127.x
36. Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem (2001) 276:10229–33. doi: 10.1074/jbc.M100099200
37. Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol (2001) 167(5):2887–94. doi: 10.4049/jimmunol.167.5.2887
38. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, Van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med (2007) 13(9):1042–9. doi: 10.1038/nm1638
39. Croce K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation (2009) 120:427–36. doi: 10.1161/CIRCULATIONAHA.108.814582
40. Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med (2009) 15(7):774–80. doi: 10.1038/nm.1987
41. Zhang P, Cox CJ, Alvarez KM, Cunningham MW. Cutting edge: cardiac myosin activates innate immune responses through TLRs. J Immunol (2009) 183(1):27–31. doi: 10.4049/jimmunol.0800861
42. Tang AH, Brunn GJ, Cascalho M, Platt JL. Pivotal advance: endogenous pathway to SIRS, sepsis, and related conditions. J Leukoc Biol (2007) 82(2):282–5. doi: 10.1189/jlb.1206752
43. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
44. Raby AC, Labeta MO. Preventing Peritoneal Dialysis-Associated Fibrosis by Therapeutic Blunting of Peritoneal Toll-Like Receptor Activity. Front Physiol (2018) 9:1692. doi: 10.3389/fphys.2018.01692
45. Linden T, Cohen A, Deppisch R, Kjellstrand P, Wieslander A. 3,4-Dideoxyglucosone-3-ene (3,4-DGE): a cytotoxic glucose degradation product in fluids for peritoneal dialysis. Kidney Int (2002) 62(2):697–703. doi: 10.1046/j.1523-1755.2002.00490.x
46. Mortier S, Faict D, Schalkwijk CG, Lameire NH, De Vriese AS. Long-term exposure to new peritoneal dialysis solutions: Effects on the peritoneal membrane. Kidney Int (2004) 66(3):1257–65. doi: 10.1111/j.1523-1755.2004.00879.x
47. Mcintyre CW. Update on peritoneal dialysis solutions. Kidney Int (2007) 71(6):486–90. doi: 10.1038/sj.ki.5002109
48. Yung S, Chan TM. Pathophysiological changes to the peritoneal membrane during PD-related peritonitis: the role of mesothelial cells. Mediators Inflammation (2012) 2012:484167. doi: 10.1155/2012/484167
49. Yang AH, Chen JY, Lin YP, Huang TP, Wu CW. Peritoneal dialysis solution induces apoptosis of mesothelial cells. Kidney Int (1997) 51(4):1280–8. doi: 10.1038/ki.1997.175
50. Wu J, Xing C, Zhang L, Mao H, Chen X, Liang M, et al. Autophagy promotes fibrosis and apoptosis in the peritoneum during long-term peritoneal dialysis. J Cell Mol Med (2018) 22(2):1190–201. doi: 10.1111/jcmm.13393
51. Lopez-Cabrera M. Mesenchymal conversion of mesothelial cells is a key event in the pathophysiology of the peritoneum during peritoneal dialysis. Adv Med (2014) 2014:17. doi: 10.1155/2014/473134
52. Williams JD, Craig KJ, Topley N, Von Ruhland C, Fallon M, Newman GR, et al. Morphologic changes in the peritoneal membrane of patients with renal disease. J Am Soc Nephrol (2002) 13(2):470–9.
53. Davies SJ. Longitudinal relationship between solute transport and ultrafiltration capacity in peritoneal dialysis patients. Kidney Int (2004) 66:2437–45. doi: 10.1111/j.1523-1755.2004.66021.x
54. Davies SJ, Mushahar L, Yu Z, Lambie M. Determinants of peritoneal membrane function over time. Semin Nephrol (2011) 31:172–82. doi: 10.1016/j.semnephrol.2011.01.006
55. Schmitt CP, Aufricht C. Is there such a thing as biocompatible peritoneal dialysis fluid? Pediatr Nephrol (2017) 32(10):1835–43. doi: 10.1007/s00467-016-3461-y
56. Dousdampanis P, Musso CG, Trigka K. Icodextrin and peritoneal dialysis: advantages and new applications. Int Urol Nephrol (2018) 50:495–500. doi: 10.1007/s11255-017-1647-2
57. Cho Y, Johnson DW, Badve SV, Craig JC, Strippoli GF, Wiggins KJ. The impact of neutral-pH peritoneal dialysates with reduced glucose degradation products on clinical outcomes in peritoneal dialysis patients. Kidney Int (2013b) 84:969–79. doi: 10.1038/ki.2013.190
58. Yohanna S, Alkatheeri AM, Brimble SK, Mccormick B, Iansavitchous A, Blake PG, et al. Effect of Neutral-pH, Low-Glucose Degradation Product Peritoneal Dialysis Solutions on Residual Renal Function, Urine Volume, and Ultrafiltration: A Systematic Review and Meta-Analysis. Clin J Am Soc Nephrol (2015) 10(8):1380–8. doi: 10.2215/CJN.05410514
59. Yanez-Mo M, Lara-Pezzi E, Selgas R, Ramirez-Huesca M, Dominguez-Jimenez C, Jimenez-Heffernan JA, et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med (2003) 348(5):403–13. doi: 10.1056/NEJMoa020809
60. Cho Y, Johnson DW, Badve S, Craig JC, Strippoli GF, Wiggins KJ. Impact of icodextrin on clinical outcomes in peritoneal dialysis: a systematic review of randomized controlled trials. Nephrol Dial Transplant (2013a) 28:1899–907. doi: 10.1093/ndt/gft050
61. Li PK, Culleton BF, Ariza A, Do JY, Johnson DW, Sanabria M, et al. Randomized, controlled trial of glucose-sparing peritoneal dialysis in diabetic patients. J Am Soc Nephrol (2013) 24(11):1889–900. doi: 10.1681/ASN.2012100987
62. Shockley TR, Martis L, Tranaeus AP. New solutions for peritoneal dialysis in adult and pediatric patients. Perit Dial Int (1999) 19 Suppl 2:S429–434. doi: 10.1177/089686089901902S70
63. Kopple JD, Bernard D, Messana J, Swartz R, Bergstrom J, Lindholm B, et al. Treatment of malnourished CAPD patients with an amino acid based dialysate. Kidney Int (1995) 47(4):1148–57. doi: 10.1038/ki.1995.164
64. Vychytil A, Herzog R, Probst P, Ribitsch W, Lhotta K, Machold-Fabrizii V, et al. A randomized controlled trial of alanyl-glutamine supplementation in peritoneal dialysis fluid to assess impact on biomarkers of peritoneal health. Kidney Int (2018) 94(6):1227–37. doi: 10.1016/j.kint.2018.08.031
65. Bartosova M, Herzog R, Ridinger D, Levai E, Jenei H, Zhang C, et al. Alanyl-Glutamine Restores Tight Junction Organization after Disruption by a Conventional Peritoneal Dialysis Fluid. Biomolecules (2020) 10:1–17. doi: 10.3390/biom10081178
66. Htay H, Johnson DW, Wiggins KJ, Badve SV, Craig JC, Strippoli GF, et al. Biocompatible dialysis fluids for peritoneal dialysis. Cochrane Database Syst Rev (2018) 10:CD007554. doi: 10.1002/14651858.CD007554.pub3
67. Johnson DW, Brown FG, Clarke M, Boudville N, Elias TJ, Foo MW, et al. The effects of biocompatible compared with standard peritoneal dialysis solutions on peritonitis microbiology, treatment, and outcomes: the balANZ trial. Perit Dial Int (2012) 32:497–506. doi: 10.3747/pdi.2012.00052
68. Schaefer B, Bartosova M, Macher-Goeppinger S, Sallay P, Voros P, Ranchin B, et al. Neutral pH and low-glucose degradation product dialysis fluids induce major early alterations of the peritoneal membrane in children on peritoneal dialysis. Kidney Int (2018) 94(2):419–29. doi: 10.1016/j.kint.2018.02.022
69. Bartosova M, Schmitt CP. Biocompatible Peritoneal Dialysis: The Target Is Still Way Off. Front Physiol (2018) 9:1853. doi: 10.3389/fphys.2018.01853
70. Piccapane F, Bonomini M, Castellano G, Gerbino A, Carmosino M, Svelto M, et al. A Novel Formulation of Glucose-Sparing Peritoneal Dialysis Solutions with l-Carnitine Improves Biocompatibility on Human Mesothelial Cells. Int J Mol Sci (2020) 22(1):1–16. doi: 10.3390/ijms22010123
71. Santos A, Lagares D. Matrix Stiffness: the Conductor of Organ Fibrosis. Curr Rheumatol Rep (2018) 20(1):2. doi: 10.1007/s11926-018-0710-z
72. Jufri NF, Mohamedali A, Avolio A, Baker MS. Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vasc Cell (2015) 7:8. doi: 10.1186/s13221-015-0033-z
73. Heise RL, Stober V, Cheluvaraju C, Hollingsworth JW, Garantziotis S. Mechanical stretch induces epithelial-mesenchymal transition in alveolar epithelia via hyaluronan activation of innate immunity. J Biol Chem (2011) 286:17435–44. doi: 10.1074/jbc.M110.137273
74. Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature (2011) 475:316–23. doi: 10.1038/nature10316
75. Sandoval P, Jimenez-Heffernan JA, Guerra-Azcona G, Perez-Lozano ML, Rynne-Vidal A, Albar-Vizcaino P, et al. Mesothelial-to-mesenchymal transition in the pathogenesis of post-surgical peritoneal adhesions. J Pathol (2016) 239:48–59. doi: 10.1002/path.4695
76. He Z, Potter R, Li X, Flessner M. Stretch of human mesothelial cells increases cytokine expression. Adv Perit Dial (2012) 28:2–9.
77. Strippoli R, Sandoval P, Moreno-Vicente R, Rossi L, Battistelli C, Terri M, et al. Caveolin1 and YAP drive mechanically induced mesothelial to mesenchymal transition and fibrosis. Cell Death Dis (2020) 11(8):647. doi: 10.1038/s41419-020-02822-1
78. Tsai JM, Sinha R, Seita J, Fernhoff N, Christ S, Koopmans T, et al. Surgical adhesions in mice are derived from mesothelial cells and can be targeted by antibodies against mesothelial markers. Sci Transl Med (2018) 10(469):1-16. doi: 10.1126/scitranslmed.aan6735
79. Gulyas M, Hjerpe A. Proteoglycans and WT1 as markers for distinguishing adenocarcinoma, epithelioid mesothelioma, and benign mesothelium. J Pathol (2003) 199:479–87. doi: 10.1002/path.1312
80. Wilm B, Munoz-Chapuli R. The Role of WT1 in Embryonic Development and Normal Organ Homeostasis. Methods Mol Biol (2016) 1467:23–39. doi: 10.1007/978-1-4939-4023-3_3
81. Yang AH, Chen JY, Lin JK. Myofibroblastic conversion of mesothelial cells. Kidney Int (2003) 63:1530–9. doi: 10.1046/j.1523-1755.2003.00861.x
82. Strippoli R, Loureiro J, Moreno V, Benedicto I, Lozano MLP, Barreiro O, et al. Caveolin-1 deficiency induces a MEK-ERK1/2-Snail-1-dependent epithelial-mesenchymal transition and fibrosis during peritoneal dialysis. EMBO Mol Med (2015) 7(1):102–23. doi: 10.15252/emmm.201404127
83. Namvar S, Woolf AS, Zeef L, Wilm T, Wilm B, Herrick SE. Functional molecules in mesothelial-to-mesenchymal transition revealed by transcriptome analyses. J Pathol (2018) 245:491–501. doi: 10.1002/path.5101
84. Loureiro J, Schilte M, Aguilera A, Albar-Vizcaino P, Ramirez-Huesca M, Perez-Lozano ML, et al. BMP-7 blocks mesenchymal conversion of mesothelial cells and prevents peritoneal damage induced by dialysis fluid exposure. Nephrol Dial Transplant (2010) 25(4):1098–108. doi: 10.1093/ndt/gfp618
85. Wu YJ, Parker LM, Binder NE, Beckett MA, Sinard JH, Griffiths CT, et al. The mesothelial keratins: a new family of cytoskeletal proteins identified in cultured mesothelial cells and nonkeratinizing epithelia. Cell (1982) 31(3 Pt 2):693–703. doi: 10.1016/0092-8674(82)90324-5
86. Chung-Welch N, Patton WF, Yen-Patton GP, Hechtman HB, Shepro D. Phenotypic comparison between mesothelial and microvascular endothelial cell lineages using conventional endothelial cell markers, cytoskeletal protein markers and in vitro assays of angiogenic potential. Differentiation (1989) 42:44–53. doi: 10.1111/j.1432-0436.1989.tb00606.x
87. Ito T, Yorioka N, Yamamoto M, Kataoka K, Yamakido M. Effect of glucose on intercellular junctions of cultured human peritoneal mesothelial cells. J Am Soc Nephrol (2000) 11:1969–79.
88. Ando T, Jordan P, Wang Y, Jennings MH, Harper MH, Houghton J, et al. Homogeneity of mesothelial cells with lymphatic endothelium: expression of lymphatic endothelial markers by mesothelial cells. Lymphat Res Biol (2005) 3:117–25. doi: 10.1089/lrb.2005.3.117
89. Yung S, Chan TM. Pathophysiology of the peritoneal membrane during peritoneal dialysis: the role of hyaluronan. J BioMed Biotechnol (2011) 2011:180594. doi: 10.1155/2011/180594
90. Aroeira LS, Aguilera A, Selgas R, Ramirez-Huesca M, Perez-Lozano ML, Cirugeda A, et al. Mesenchymal conversion of mesothelial cells as a mechanism responsible for high solute transport rate in peritoneal dialysis: role of vascular endothelial growth factor. Am J Kidney Dis (2005) 46:938–48. doi: 10.1053/j.ajkd.2005.08.011
91. Strippoli R, Benedicto I, Foronda M, Perez-Lozano ML, Sanchez-Perales S, Lopez-Cabrera M, et al. p38 maintains E-cadherin expression by modulating TAK1-NF-kappa B during epithelial-to-mesenchymal transition. J Cell Sci (2010) 123(pt 24):4321–31. doi: 10.1242/jcs.071647
92. Leung JC, Chan LY, Tam KY, Tang SC, Lam MF, Cheng AS, et al. Regulation of CCN2/CTGF and related cytokines in cultured peritoneal cells under conditions simulating peritoneal dialysis. Nephrol Dial Transplant (2009) 24:458–69. doi: 10.1093/ndt/gfn524
93. Hong FY, Bao JF, Hao J, Yu Q, Liu J. Methylglyoxal and advanced glycation end-products promote cytokines expression in peritoneal mesothelial cells via MAPK signaling. Am J Med Sci (2015) 349:105–9. doi: 10.1097/MAJ.0000000000000394
94. Morishita Y, Ookawara S, Hirahara I, Muto S, Nagata D. HIF-1alpha mediates Hypoxia-induced epithelial-mesenchymal transition in peritoneal mesothelial cells. Ren Fail (2016) 38:282–9. doi: 10.3109/0886022X.2015.1127741
95. Patel P, West-Mays J, Kolb M, Rodrigues JC, Hoff CM, Margetts PJ. Platelet derived growth factor B and epithelial mesenchymal transition of peritoneal mesothelial cells. Matrix Biol (2010) 29:97–106. doi: 10.1016/j.matbio.2009.10.004
96. Noh H, Ha H, Yu MR, Kim YO, Kim JH, Lee HB. Angiotensin II mediates high glucose-induced TGF-beta1 and fibronectin upregulation in HPMC through reactive oxygen species. Perit Dial Int (2005) 25:38–47. doi: 10.1177/089686080502500110
97. Yang WS, Kim BS, Lee SK, Park JS, Kim SB. Interleukin-1beta stimulates the production of extracellular matrix in cultured human peritoneal mesothelial cells. Perit Dial Int (1999) 19:211–20. doi: 10.1177/089686089901900306
98. Offner FA, Feichtinger H, Stadlmann S, Obrist P, Marth C, Klingler P, et al. Transforming growth factor-beta synthesis by human peritoneal mesothelial cells. Induction by interleukin-1. Am J Pathol (1996) 148:1679–88.
99. Rossi L, Battistelli C, De Turris V, Noce V, Zwergel C, Valente S, et al. HDAC1 inhibition by MS-275 in mesothelial cells limits cellular invasion and promotes MMT reversal. Sci Rep (2018) 8(1):8492. doi: 10.1038/s41598-018-26319-2
100. Ma C, Tarnuzzer RW, Chegini N. Expression of matrix metalloproteinases and tissue inhibitor of matrix metalloproteinases in mesothelial cells and their regulation by transforming growth factor-beta1. Wound Repair Regener (1999) 7(6):477–85. doi: 10.1046/j.1524-475X.1999.00477.x
101. Saed GM, Zhang W, Chegini N, Holmdahl L, Diamond MP. Alteration of type I and III collagen expression in human peritoneal mesothelial cells in response to hypoxia and transforming growth factor-beta1. Wound Repair Regener (1999) 7(6):504–10. doi: 10.1046/j.1524-475X.1999.00504.x
102. Xiao X, Cai J. Mucosal-Associated Invariant T Cells: New Insights into Antigen Recognition and Activation. Front Immunol (2017) 8:1540. doi: 10.3389/fimmu.2017.01540
103. Boren J, Shryock G, Fergis A, Jeffers A, Owens S, Qin W, et al. Inhibition of Glycogen Synthase Kinase 3beta Blocks Mesomesenchymal Transition and Attenuates Streptococcus pneumonia-Mediated Pleural Injury in Mice. Am J Pathol (2017) 187:2461–72. doi: 10.1016/j.ajpath.2017.07.007
104. Davidson B, Nielsen S, Christensen J, Asschenfeldt P, Berner A, Risberg B, et al. The role of desmin and N-cadherin in effusion cytology: a comparative study using established markers of mesothelial and epithelial cells. Am J Surg Pathol (2001) 25:1405–12. doi: 10.1097/00000478-200111000-00008
105. Strippoli R, Benedicto I, Perez Lozano ML, Pellinen T, Sandoval P, Lopez-Cabrera M, et al. Inhibition of transforming growth factor-activated kinase 1 (TAK1) blocks and reverses epithelial to mesenchymal transition of mesothelial cells. PloS One (2012) 7:e31492. doi: 10.1371/journal.pone.0031492
106. Li CX, Ren YQ, Jia XT, Liang PY, Lou WJ, He LJ, et al. Twist overexpression promoted epithelial-to-mesenchymal transition of human peritoneal mesothelial cells under high glucose. Nephrol Dialysis Transplant (2012) 27:4119–24. doi: 10.1093/ndt/gfs049
107. De Santis Puzzonia M, Cozzolino AM, Grassi G, Bisceglia F, Strippoli R, Guarguaglini G, et al. TGFbeta Induces Binucleation/Polyploidization in Hepatocytes through a Src-Dependent Cytokinesis Failure. PloS One (2016) 11:e0167158. doi: 10.1371/journal.pone.0167158
108. Snitow M, Lu M, Cheng L, Zhou S, Morrisey EE. Ezh2 restricts the smooth muscle lineage during mouse lung mesothelial development. Development (2016) 143(20):3733–41. doi: 10.1242/dev.134932
109. Ruiz-Carpio V, Sandoval P, Aguilera A, Albar-Vizcaino P, Perez-Lozano ML, Gonzalez-Mateo GT, et al. Genomic reprograming analysis of the Mesothelial to Mesenchymal Transition identifies biomarkers in peritoneal dialysis patients. Sci Rep (2017) 7:44941. doi: 10.1038/srep44941
110. Lambie M, Chess J, Donovan KL, Kim YL, Do JY, Lee HB, et al. Independent effects of systemic and peritoneal inflammation on peritoneal dialysis survival. J Am Soc Nephrol (2013) 24(12):2071–80. doi: 10.1681/ASN.2013030314
111. Lua I, Li Y, Pappoe LS, Asahina K. Myofibroblastic Conversion and Regeneration of Mesothelial Cells in Peritoneal and Liver Fibrosis. Am J Pathol (2015) 185(12):3258–73. doi: 10.1016/j.ajpath.2015.08.009
112. Chen YT, Chang YT, Pan SY, Chou YH, Chang FC, Yeh PY, et al. Lineage tracing reveals distinctive fates for mesothelial cells and submesothelial fibroblasts during peritoneal injury. J Am Soc Nephrol (2014) 25:2847–58. doi: 10.1681/ASN.2013101079
113. Loureiro J, Aguilera A, Selgas R, Sandoval P, Albar-Vizcaino P, Perez-Lozano ML, et al. Blocking TGF-beta1 protects the peritoneal membrane from dialysate-induced damage. J Am Soc Nephrol (2011) 22:1682–95. doi: 10.1681/ASN.2010111197
114. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, Mcmullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med (2007) 13(8):952–61. doi: 10.1038/nm1613
115. Witowski J, Kawka E, Rudolf A, Jorres A. New developments in peritoneal fibroblast biology: implications for inflammation and fibrosis in peritoneal dialysis. BioMed Res Int (2015) 2015:134708. doi: 10.1155/2015/134708
116. Beelen RH, Oosterling SJ, Van Egmond M, Van Den Born J, Zareie M. Omental milky spots in peritoneal pathophysiology (spots before your eyes). Perit Dial Int (2005) 25:30–2. doi: 10.1177/089686080502500107
117. Rangel-Moreno J, Moyron-Quiroz JE, Carragher DM, Kusser K, Hartson L, Moquin A, et al. Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens. Immunity (2009) 30(5):731–43. doi: 10.1016/j.immuni.2009.03.014
118. Mebius RE. Lymphoid organs for peritoneal cavity immune response: milky spots. Immunity (2009) 30(5):670–2. doi: 10.1016/j.immuni.2009.04.005
119. Ansel KM, Harris RB, Cyster JG. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity (2002) 16:67–76. doi: 10.1016/S1074-7613(01)00257-6
120. Jackson-Jones LH, Smith P, Portman JR, Magalhaes MS, Mylonas KJ, Vermeren MM, et al. Stromal Cells Covering Omental Fat-Associated Lymphoid Clusters Trigger Formation of Neutrophil Aggregates to Capture Peritoneal Contaminants. Immunity (2020) 52:700–715 e706. doi: 10.1016/j.immuni.2020.03.011
121. Perez-Shibayama C, Gil-Cruz C, Cheng HW, Onder L, Printz A, Morbe U, et al. Fibroblastic reticular cells initiate immune responses in visceral adipose tissues and secure peritoneal immunity. Sci Immunol (2018) 3(26):1–12. doi: 10.1126/sciimmunol.aar4539
122. Laurin LP, Brissette MJ, Lepage S, Cailhier JF. Regulation of experimental peritonitis: a complex orchestration. Nephron Exp Nephrol (2012) 120(1):e41–46. doi: 10.1159/000334169
123. Buechler MB, Turley SJ. Neutrophils Follow Stromal Omens to Limit Peritoneal Inflammation. Immunity (2020) 52:578–80. doi: 10.1016/j.immuni.2020.03.015
124. Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol (2013) 304(5):L342–349. doi: 10.1152/ajplung.00151.2012
125. Witowski J, Tayama H, Ksiazek K, Wanic-Kossowska M, Bender TO, Jorres A. Human peritoneal fibroblasts are a potent source of neutrophil-targeting cytokines: a key role of IL-1beta stimulation. Lab Invest (2009) 89(4):414–24. doi: 10.1038/labinvest.2009.1
126. Riese J, Denzel C, Zowe M, Mehler C, Hohenberger W, Haupt W. Secretion of IL-6, monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha, and TNFalpha by cultured intact human peritoneum. Eur Surg Res (1999) 31(3):281–8. doi: 10.1159/000008704
127. Fielding CA, Mcloughlin RM, Mcleod L, Colmont CS, Najdovska M, Grail D, et al. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol (2008) 181:2189–95. doi: 10.4049/jimmunol.181.3.2189
128. Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol (2005) 175:3463–8. doi: 10.4049/jimmunol.175.6.3463
129. Tirado M, Koss W. Differentiation of mesothelial cells into macrophage phagocytic cells in a patient with clinical sepsis. Blood (2018) 132(4):1460. doi: 10.1182/blood-2018-07-859991
130. Wang J, Kubes P. A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell (2016) 165(3):668–78. doi: 10.1016/j.cell.2016.03.009
131. Catar RA, Chen L, Cuff SM, Kift-Morgan A, Eberl M, Kettritz R, et al. Control of neutrophil influx during peritonitis by transcriptional cross-regulation of chemokine CXCL1 by IL-17 and IFN-gamma. J Pathol (2020) 251:175–86. doi: 10.1002/path.5438
132. Rodrigues-Diez R, Aroeira LS, Orejudo M, Bajo MA, Heffernan JJ, Rodrigues-Diez RR, et al. IL-17A is a novel player in dialysis-induced peritoneal damage. Kidney Int (2014) 86(2):303–15. doi: 10.1038/ki.2014.33
133. Mcloughlin RM, Witowski J, Robson RL, Wilkinson TS, Hurst SM, Williams AS, et al. Interplay between IFN-gamma and IL-6 signaling governs neutrophil trafficking and apoptosis during acute inflammation. J Clin Invest (2003) 112(4):598–607. doi: 10.1172/JCI17129
134. Liao CT, Andrews R, Wallace LE, Khan MW, Kift-Morgan A, Topley N, et al. Peritoneal macrophage heterogeneity is associated with different peritoneal dialysis outcomes. Kidney Int (2017) 91(5):1088–103. doi: 10.1016/j.kint.2016.10.030
135. Kubicka U, Olszewski WL, Tarnowski W, Bielecki K, Ziolkowska A, Wierzbicki Z. Normal human immune peritoneal cells: subpopulations and functional characteristics. Scand J Immunol (1996) 44(2):157–63. doi: 10.1046/j.1365-3083.1996.d01-297.x
136. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
137. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
138. Ruytinx P, Proost P, Van Damme J, Struyf S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front Immunol (2018) 9:1930. doi: 10.3389/fimmu.2018.01930
139. Bellon T, Martinez V, Lucendo B, Del Peso G, Castro MJ, Aroeira LS, et al. Alternative activation of macrophages in human peritoneum: implications for peritoneal fibrosis. Nephrol Dial Transplant (2011) 26:2995–3005. doi: 10.1093/ndt/gfq771
140. Lin J, Kong Q, Hao W, Hu W. High glucose contributes to the polarization of peritoneal macrophages to the M2 phenotype in vivo and in vitro. Mol Med Rep (2020) 22(1):127–34. doi: 10.3892/mmr.2020.11130
141. Chen YT, Hsu H, Lin CC, Pan SY, Liu SY, Wu CF, et al. Inflammatory macrophages switch to CCL17-expressing phenotype and promote peritoneal fibrosis. J Pathol (2020) 250:55–66. doi: 10.1002/path.5350
142. Balzer MS, Helmke A, Ackermann M, Casper J, Dong L, Hiss M, et al. Protein kinase C beta deficiency increases glucose-mediated peritoneal damage via M1 macrophage polarization and up-regulation of mesothelial protein kinase C alpha. Nephrol Dial Transplant (2019) 34:947–60. doi: 10.1093/ndt/gfy282
143. Li Q, Zheng M, Liu Y, Sun W, Shi J, Ni J, et al. A pathogenetic role for M1 macrophages in peritoneal dialysis-associated fibrosis. Mol Immunol (2018) 94:131–9. doi: 10.1016/j.molimm.2017.12.023
144. Shi J, Li Q, Sheng M, Zheng M, Yu M, Zhang L. The Role of TLR4 in M1 Macrophage-Induced Epithelial-Mesenchymal Transition of Peritoneal Mesothelial Cells. Cell Physiol Biochem (2016) 40(6):1538–48. doi: 10.1159/000453204
145. Ivanov S, Gallerand A, Gros M, Stunault MI, Merlin J, Vaillant N, et al. Mesothelial cell CSF1 sustains peritoneal macrophage proliferation. Eur J Immunol (2019) 49:2012–8. doi: 10.1002/eji.201948164
146. Helmke A, Nordlohne J, Balzer MS, Dong L, Rong S, Hiss M, et al. CX3CL1-CX3CR1 interaction mediates macrophage-mesothelial cross talk and promotes peritoneal fibrosis. Kidney Int (2019) 95:1405–17. doi: 10.1016/j.kint.2018.12.030
147. Kushiyama T, Oda T, Yamada M, Higashi K, Yamamoto K, Oshima N, et al. Effects of liposome-encapsulated clodronate on chlorhexidine gluconate-induced peritoneal fibrosis in rats. Nephrol Dial Transplant (2011) 26(10):3143–54. doi: 10.1093/ndt/gfr068
148. Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med (2012) 18:693–704. doi: 10.1038/nm.2755
149. Gunjigake K, Kinoshita J, Yamaguchi T, Saito H, Fujimori D, Horiike T, et al. Interleukin-17A derived from mast cells contributes to fibrosis in gastric cancer with peritoneal dissemination. Gastric Cancer (2020) 24:31–44 (2021). doi: 10.1007/s10120-020-01092-2
150. Wygrecka M, Dahal BK, Kosanovic D, Petersen F, Taborski B, Von Gerlach S, et al. Mast cells and fibroblasts work in concert to aggravate pulmonary fibrosis: role of transmembrane SCF and the PAR-2/PKC-alpha/Raf-1/p44/42 signaling pathway. Am J Pathol (2013) 182(6):2094–108. doi: 10.1055/s-0033-1334523
151. Overed-Sayer C, Rapley L, Mustelin T, Clarke DL. Are mast cells instrumental for fibrotic diseases? Front Pharmacol (2013) 4:174. doi: 10.3389/fphar.2013.00174
152. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol (2011) 187(1):490–500. doi: 10.4049/jimmunol.1100123
153. Kazama I, Baba A, Endo Y, Toyama H, Ejima Y, Matsubara M, et al. Mast cell involvement in the progression of peritoneal fibrosis in rats with chronic renal failure. Nephrol (Carlton) (2015) 20(9):609–16. doi: 10.1111/nep.12489
154. Zareie M, Fabbrini P, Hekking LH, Keuning ED, Ter Wee PM, Beelen RH, et al. Novel role for mast cells in omental tissue remodeling and cell recruitment in experimental peritoneal dialysis. J Am Soc Nephrol (2006) 17(12):3447–57. doi: 10.1681/ASN.2005111173
155. Jimenez-Heffernan JA, Bajo MA, Perna C, Del Peso G, Larrubia JR, Gamallo C, et al. Mast cell quantification in normal peritoneum and during peritoneal dialysis treatment. Arch Pathol Lab Med (2006) 130:1188–92. doi: 10.5858/2006-130-1188-MCQINP
156. Alscher DM, Braun N, Biegger D, Fritz P. Peritoneal mast cells in peritoneal dialysis patients, particularly in encapsulating peritoneal sclerosis patients. Am J Kidney Dis (2007) 49:452–61. doi: 10.1053/j.ajkd.2006.11.040
157. Gonzaga R, Matzinger P, Perez-Diez A. Resident peritoneal NK cells. J Immunol (2011) 187:6235–42. doi: 10.4049/jimmunol.1101540
158. Law BMP, Wilkinson R, Wang X, Kildey K, Lindner M, Rist MJ, et al. Interferon-gamma production by tubulointerstitial human CD56(bright) natural killer cells contributes to renal fibrosis and chronic kidney disease progression. Kidney Int (2017) 92(1):79–88. doi: 10.1016/j.kint.2017.02.006
159. Anuforo OUU, Bjarnarson SP, Jonasdottir HS, Giera M, Hardardottir I, Freysdottir J. Natural killer cells play an essential role in resolution of antigen-induced inflammation in mice. Mol Immunol (2018) 93:1–8. doi: 10.1016/j.molimm.2017.10.019
160. Thoren FB, Riise RE, Ousback J, Della Chiesa M, Alsterholm M, Marcenaro E, et al. Human NK Cells induce neutrophil apoptosis via an NKp46- and Fas-dependent mechanism. J Immunol (2012) 188:1668–74. doi: 10.4049/jimmunol.1102002
161. Steis RG, Urba WJ, Vandermolen LA, Bookman MA, Smith JW,2, Clark JW, et al. Intraperitoneal lymphokine-activated killer-cell and interleukin-2 therapy for malignancies limited to the peritoneal cavity. J Clin Oncol (1990) 8(10):1618–29. doi: 10.1200/JCO.1990.8.10.1618
162. Rajakariar R, Lawrence T, Bystrom J, Hilliard M, Colville-Nash P, Bellingan G, et al. Novel biphasic role for lymphocytes revealed during resolving inflammation. Blood (2008) 111(8):4184–92. doi: 10.1182/blood-2007-08-108936
163. Kipari T, Watson S, Houlberg K, Lepage S, Hughes J, Cailhier JF. Lymphocytes modulate peritoneal leukocyte recruitment in peritonitis. Inflammation Res (2009) 58(9):553–60. doi: 10.1007/s00011-009-0019-5
164. Hartman J, Maassen V, Rieber P, Fricke H. T lymphocytes from normal human peritoneum are phenotypically different from their counterparts in peripheral blood and CD3- lymphocyte subsets contain mRNA for the recombination activating gene RAG-1. Eur J Immunol (1995) 25:2626–31. doi: 10.1002/eji.1830250933
165. Brauner A, Hylander B, Wretlind B. Tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-1 receptor antagonist in dialysate and serum from patients on continuous ambulatory peritoneal dialysis. Am J Kidney Dis (1996) 27:402–8. doi: 10.1016/S0272-6386(96)90364-9
166. Yokoyama T, Nitta K, Futatsuyama K, Hayashi T, Honda K, Uchida K, et al. Identification of T helper cell subsets in continuous ambulatory peritoneal dialysis patients. Nephron (2001) 89(2):215–8. doi: 10.1159/000046070
167. Libetta C, Esposito P, Sepe V, Guastoni C, Zucchi M, Meloni F, et al. Effects of different peritoneal dialysis fluids on the TH1/TH2 balance. Eur Cytokine Netw (2011) 22(1):24–31. doi: 10.1684/ecn.2011.0280
168. Witowski J, Kamhieh-Milz J, Kawka E, Catar R, Jorres A. IL-17 in Peritoneal Dialysis-Associated Inflammation and Angiogenesis: Conclusions and Perspectives. Front Physiol (2018) 9:1694. doi: 10.3389/fphys.2018.01694
169. Wang HH, Lee TY, Lin CY. Kinetics and involvement of interleukin-17 in the outcome of peritonitis in nondiabetic patients undergoing peritoneal dialysis. J Chin Med Assoc (2011) 74(1):11–5. doi: 10.1016/j.jcma.2011.01.002
170. Vila Cuenca M, Keuning ED, Talhout W, Paauw NJ, Van Ittersum FJ, Ter Wee PM, et al. Differences in peritoneal response after exposure to low-GDP bicarbonate/lactate-buffered dialysis solution compared to conventional dialysis solution in a uremic mouse model. Int Urol Nephrol (2018) 50(6):1151–61. doi: 10.1007/s11255-018-1872-3
171. Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine (2013) 64:477–85. doi: 10.1016/j.cyto.2013.07.022
172. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122
173. Sandoval P, Loureiro J, Gonzalez-Mateo G, Perez-Lozano ML, Maldonado-Rodriguez A, Sanchez-Tomero JA, et al. PPAR-gamma agonist rosiglitazone protects peritoneal membrane from dialysis fluid-induced damage. Lab Invest (2010) 90(10):1517–32. doi: 10.1038/labinvest.2010.111
174. Liappas G, Gonzalez-Mateo GT, Sanchez-Diaz R, Lazcano JJ, Lasarte S, Matesanz-Marin A, et al. Immune-Regulatory Molecule CD69 Controls Peritoneal Fibrosis. J Am Soc Nephrol (2016) 27(12):3561–76. doi: 10.1681/ASN.2015080909
175. Martin P, Gomez M, Lamana A, Cruz-Adalia A, Ramirez-Huesca M, Ursa MA, et al. CD69 association with Jak3/Stat5 proteins regulates Th17 cell differentiation. Mol Cell Biol (2010) 30(20):4877–89. doi: 10.1128/MCB.00456-10
176. Ji X, Li J, Xu L, Wang W, Luo M, Luo S, et al. IL4 and IL-17A provide a Th2/Th17-polarized inflammatory milieu in favor of TGF-beta1 to induce bronchial epithelial-mesenchymal transition (EMT). Int J Clin Exp Pathol (2013) 6:1481–92.
177. Fielding CA, Jones GW, Mcloughlin RM, Mcleod L, Hammond VJ, Uceda J, et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity (2014) 40:40–50. doi: 10.1016/j.immuni.2013.10.022
178. Jones GW, Mcloughlin RM, Hammond VJ, Parker CR, Williams JD, Malhotra R, et al. Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J Immunol (2010) 184(4):2130–9. doi: 10.4049/jimmunol.0901528
179. Katz S, Zsiros V, Kiss AL. Under inflammatory stimuli mesenteric mesothelial cells transdifferentiate into macrophages and produce pro-inflammatory cytokine IL-6. Inflammation Res (2019) 68:525–8. doi: 10.1007/s00011-019-01247-7
180. Godfrey DI, Uldrich AP, Mccluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol (2015) 16:1114–23. doi: 10.1038/ni.3298
181. Ibidapo-Obe O, Stengel S, Kose-Vogel N, Quickert S, Reuken PA, Busch M, et al. Mucosal-Associated Invariant T Cells Redistribute to the Peritoneal Cavity During Spontaneous Bacterial Peritonitis and Contribute to Peritoneal Inflammation. Cell Mol Gastroenterol Hepatol (2020) 9:661–77. doi: 10.1016/j.jcmgh.2020.01.003
Keywords: peritoneal fibrosis, mesothelial cells, peritonitis, innate immunity, T cell subpopulations, pro-inflammatory cytokines
Citation: Terri M, Trionfetti F, Montaldo C, Cordani M, Tripodi M, Lopez-Cabrera M and Strippoli R (2021) Mechanisms of Peritoneal Fibrosis: Focus on Immune Cells–Peritoneal Stroma Interactions. Front. Immunol. 12:607204. doi: 10.3389/fimmu.2021.607204
Received: 16 September 2020; Accepted: 19 January 2021;
Published: 29 March 2021.
Edited by:
Marianna Nicoletta Rossi, Bambino Gesù Children Hospital (IRCCS), ItalyReviewed by:
Steven Eugene Mutsaers, University of Western Australia, AustraliaAntonio Facchiano, Institute of Dermatology Immaculate (IRCCS), Italy
Sibylle Von Vietinghoff, Hannover Medical School, Germany
Copyright © 2021 Terri, Trionfetti, Montaldo, Cordani, Tripodi, Lopez-Cabrera and Strippoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raffaele Strippoli, cmFmZmFlbGUuc3RyaXBwb2xpQHVuaXJvbWExLml0
†These authors have contributed equally to this work