 Siti Muhamad Nur Husna1
Siti Muhamad Nur Husna1 Hern-Tze Tina Tan1
Hern-Tze Tina Tan1 Norasnieda Md Shukri2,3
Norasnieda Md Shukri2,3 Noor Suryani Mohd Ashari1,2
Noor Suryani Mohd Ashari1,2 Kah Keng Wong1,2*
Kah Keng Wong1,2*- 1Department of Immunology, School of Medical Sciences Malaysia, Universiti Sains Malaysia, Kubang Kerian, Malaysia
- 2Hospital Universiti Sains Malaysia, Kubang Kerian, Malaysia
- 3Department of Otorhinolaryngology, Head and Neck Surgery, School of Medical Sciences, Universiti Sains Malaysia, Kubang Kerian, Malaysia
Allergic rhinitis (AR) is a common disorder affecting up to 40% of the population worldwide and it usually persists throughout life. Nasal epithelial barrier constitutes the first line of defense against invasion of harmful pathogens or aeroallergens. Cell junctions comprising of tight junctions (TJs), adherens junctions, desmosomes and hemidesmosomes form the nasal epithelial barrier. Impairment of TJ molecules plays causative roles in the pathogenesis of AR. In this review, we describe and discuss the components of TJs and their disruption leading to development of AR, as well as regulation of TJs expression by epigenetic changes, neuro-immune interaction, epithelial-derived cytokines (thymic stromal lymphopoietin, IL-25 and IL-33), T helper 2 (Th2) cytokines (IL-4, IL-5, IL-6 and IL-13) and innate lymphoid cells. These growing evidence support the development of novel therapeutic approaches to restore nasal epithelial TJs expression in AR patients.
Introduction
Allergic rhinitis (AR) is a global health problem affecting 10–40% of the population worldwide and it usually persists throughout life (1, 2). AR is a symptomatic disorder of the nose induced by an immunoglobulin E (IgE)-mediated inflammation after allergen exposure of the membranes lining the nose, and it is usually accompanied by classical symptoms such as nasal itching, sneezing, rhinorrhea, and nasal congestion (1, 3, 4). Epithelial cells play important roles as physical barrier to prevent the entry of allergens, pathogens and other foreign particles (5). Tight junctions (TJs) comprise of cell-cell adhesion complexes between epithelial cells required for epithelial barrier function. Previous studies have reported that impairment of nasal epithelial is one of the underlying causes of AR (6, 7). Differential expression of TJ molecules has also been found in distinct inflammatory phenotypes of allergic airway inflammation in mice compared to controls (8). These findings suggest that TJs disruption plays causative roles in the development of allergy through increased exposure of nasal tissues to environmental allergens (9). In this review, we present and discuss the association of TJs disruption in AR, as well as regulation of TJs expression by epigenetic changes, neuro-immune interaction, epithelium-derived cytokines, T helper 2 (Th2) cytokines and innate lymphoid cells (ILCs).
Nasal Epithelial Barrier and Tight Junctions
Nasal Epithelial Barrier
Epithelial barrier is the first line of defense where an intact mucosal barrier is crucial in protecting the host immune system from the exposure of harmful pathogens (10). This epithelium also plays vital roles in regulating both innate and adaptive mucosal immunity through activation of functional molecules (e.g. pro-inflammatory cytokines, growth factors and chemokines) (11). Epithelial cells also secrete antimicrobial substances known as antimicrobial peptides (AMPs) such as lysozyme, defensins (α and β), lactoferrin and S-100 proteins, and they are essential in the first line defense to hinder the entry of pathogens (12, 13). Mucociliary clearance conducted by ciliated epithelial cells involves trapping of microbes and microparticles in mucus layer secreted by glands, goblet cells or ciliated cells from the nasal cavity into the esophagus (14).
Apical junctional complexes (AJCs) connect epithelial cells to one another and they consist of TJs, adherens junction, desmosomes and hemidesmosomes (15). TJs are the most apically located epithelial junctions composed of over 40 proteins either as transmembrane proteins or cytoplasmic actin-binding proteins (16). TJs function in regulating homeostasis of ions, water and certain macromolecules (17, 18). Thus, TJs are crucial in producing rate-limiting barrier to inhaled pathogens. Adherens junctions are essential for cell adhesion (19), cell proliferation and differentiation (16). Desmosomes are in close connectivity with adherens junctions (10), and they play key roles in maintaining intercellular cohesion and cellular integrity (20, 21). Lastly, hemidesmosomes are responsible to facilitate the stable adhesion of the basal epithelial cells to the basement membrane (22–24), and to link the extracellular matrix to the intermediate filaments of the actin cytoskeleton.
Tight Junctions (TJs)
TJs in the epithelial cells consist of three primary constituents of transmembrane proteins namely occludin (OCLN), claudin (CLDN) and junctional adhesion molecules (JAMs) (25). OCLN (~65 kDa) is the first identified integral membrane protein present in both epithelial and endothelial cells (26, 27). OCLN has two extracellular loops, N- and C-terminal cytoplasmic domains. The C-terminal is important for the barrier formation of TJs which it directly interacts with zonula occludens-1 (ZO-1), and the N-terminus is involved in the regulation of paracellular permeability (5).
The CLDN family contains more than 25 protein members (28). CLDNs are four-transmembrane spanning proteins consisting of a short cytoplasmic N-terminal, two extracellular loops and a C-terminal cytoplasmic domain. CLDNs serve as cellular gate to restrict the elements to pass through the epithelial (5, 10). The C-terminal of CLDNs is vital for stability and interactions with ZO-1 (28). CLDNs can be divided into two groups based on their effects on airway epithelial barrier namely the pore- and barrier-forming CLDNs (10). Pore-forming CLDNs comprise of CLDN2, CLDN10, CLDN10b and CLDN17, while barrier-forming CLDNs are CLDN1, CLDN3, CLDN4 and CLDN7 (29, 30). The unique functional groups and co-expression of CLDN family members are thought to determine the selectivity and build the strength of TJs (30).
The JAM family of proteins comprise of classical JAMs (JAM-A, JAM-B and JAM-C) and four related proteins (JAM-4, JAML, CAR and ESAM). JAMs are type I transmembrane glycoproteins composed of two immunoglobulin-like domains i.e. one transmembrane domain and one cytoplasmic tail of variable length containing a PDZ domain that can interact with ZO (31). They have a significant role in the regulation of cell polarity and endothelium permeability (31).
Disruption of Tight Junctions in AR
Occludin
Breakdown of nasal epithelial integrity is attributable to reduced expression of TJ molecules, and both OCLN mRNA and protein expressions are decreased in the nasal epithelium of AR patients compared to controls (32). Nasal epithelial barrier function has been known to be impaired in HDM-induced AR patients (33). Lower OCLN mRNA expression also occurred in HDM-induced AR patients compared to non-allergic controls (33, 34). Moreover, reduced ordered arrangement of OCLN was found in AR patients and the defect might expedite the passage of allergens and environmental pro-inflammatory agents through nasal epithelial barrier (33). This was demonstrated by reduced trans-tissue resistance and increased passage to fluorescein isothiocyanate (FITC)-dextran 4 kDa (FD4) in the tissues.
A lower mRNA expression of OCLN was present in the nasal biopsies of AR patients compared to healthy subjects and idiopathic rhinitis (IR) patients (7). This was supported by immunofluorescent imaging of OCLN from paraffin-embedded mucosal biopsy specimens of AR patients which displayed a severely disrupted layer and irregular pattern of OCLN expression compared to controls (7). OCLN expression also showed a decreased pattern in the mouse model of HDM-induced allergic airway inflammation with increased FD4 levels and bronchoalveolar lavage (BAL) albumin levels (7). Furthermore, HLA-DR- and CD11c-positive dendritic cells (DCs) penetrated beyond OCLN in the epithelium of the nasal mucosa of AR patients (35). OCLN is found at the uppermost layer of pseudostratified columnar epithelium of the nasal mucosa. Hence, these results indicated that DCs may gain access to antigens beyond epithelial TJs in the human nasal mucosa of AR.
Claudins
Previous studies have shown the defect in epithelial barrier due to decreased expression of CLDNs that contribute to AR in both patients and animal models. Recently, we demonstrated a decreased expression of CLDN3 and CLDN7 in nasal epithelial cells (NECs) of HDM-induced AR patients compared to non-allergic controls (34). CLDN1, CLDN3, CLDN7 and CLDN12 mRNA and protein expressions were significantly decreased in nasal epithelium of AR patients compared to controls subject (32). In pollen-induced AR mouse model, mice were sensitized and nasally challenged with Japanese cheddar (JC) or ragweed pollens with or without recombinant human (rh) Cystatin SN (an endogenous cysteine protease inhibitor) to investigate the effects of rhCystatin SN on the epithelial barrier in vivo (36). CLDN1 expression in the nasal mucosa was reduced in JC and ragweed-challenged mice, and only CLDN1 expression in rhCystatin SN-treated-JC-challenged mice was maintained (36). This indicates that Cystatin SN specifically inhibits JC-induced but not ragweed-induced nasal TJ disruption through inhibition of protease activities.
In AR patients, the mRNA expression of CLDN1 and CLDN4 in mucosal biopsies of AR patients was decreased compared to the healthy controls (7). Lower mRNA expression of CLDN1, CLDN4, CLDN7, CLDN8, CLDN12, CLDN13 and CLDN14 were also detected in the nasal mucosa of AR patients (35). HLA-DR+ and CD11c+ DCs expressed CLDN1 and invaded beyond OCLN in the epithelium of the nasal mucosa with AR but not in subjects without AR (35). These findings demonstrate that antigens beyond epithelial TJ in the human nasal mucosa of AR can be accessed by DCs (35). Downregulation of CLDN4 protein expression in AR nasal epithelium has also been observed by immunohistochemical staining (37).
Environmental Factors and Disruption of TJS in AR
Air Pollution and Disruption of TJs in AR
Environmental factors such as urban locations, air pollutants and presence of airborne allergens play contributive roles to the disruption of TJs in AR. We recently demonstrated that OCLN and CLDN7 mRNA expressions were significantly reduced in AR patients sensitized to HDMs compared with non-allergic controls, and lower OCLN or CLDN7 expression was associated with urban locations or exposure to second-hand smoke, respectively (34). This is comparable with recent findings that air pollution represents a risk factor for AR onset whereby epithelial barrier can be negatively affected by air pollutant such as diesel exhaust particles (DEPs) and fine particulate matter ≤2.5 μm (PM2.5) causing sinonasal diseases (38–41).
AR rat exposed to PM2.5 had significantly increased AR symptoms (i.e. number of sneezes and nasal rubs) and exhibited disorderedly arrangement of nasal mucosa epithelium (42), and a substantial increase in goblet cell hyperplasia and collagen deposition were also observed compared with control rats (43). The exposure of DEPs to primary nasal epithelial cells (PNECs) in air liquid interface (ALI) culture and exposure of PM2.5 to human nasal epithelial cell (HNEC) line (RPMI 2650 cells) significantly reduced the expression of TJ molecules including OCLN, ZO-1 and CLDN1 (38–40). It also reduced the transepithelial resistance (TER) of the cells and increased their permeability as monitored by fluorescently-labeled dextran permeability in both studies (38, 39). DEP disrupted TJs through a reactive oxygen species (ROS)‐mediated pathway, leading to increased permeability of NECs (40).
ROS suppresses TJ proteins expression through p38 MAPK, p65 NF-κB, and Akt signaling pathways independent of IL-8 and through inhibition of tyrosine phosphatase (44, 45). Cultured human epithelial cells exposed to urban PM (UPM) showed a decrease in cell viability i.e. detached and shrunken epithelial cells with condensed nuclei (44, 46). The exposed cell demonstrated a significantly amplified ROS pathway leading to decreased ZO-1, OCLN, CLDN1 and E-cadherin expression compared to unexposed cells (44, 46).
Treatment of cultured human epithelial cells with ROS scavenger N-acetylcysteine (NAC) or Akt inhibitor (MK-2206) reversed the effects of UPM. Through Akt inhibition, it decreased UPM-induced ROS formation and p38 and p65 protein phosphorylation, and restored the expression of ZO-1 and E-cadherin (44). Meanwhile, the mucosal epithelium was observed to be arranged in a more orderly manner when treated with ursolic acid (UA) (a pentacyclic triterpene extract from natural plants) in AR model exposed to PM2.5 (42), and decrease in number and size of goblet cells in epithelial layer, expression of MUC5AC and proportion of collagen deposition areas (43). UA possesses anti-inflammatory, antioxidant and anti-fibrotic properties where it is capable of modulating the MAPK and NF-κB signaling pathways (47).
House Dust Mites and Disruption of TJs in AR
HDM is the most common allergens causing allergic sensitization among AR patients and mostly affecting the 25–35 age group without significant differences in both genders (48–52). The major species of HDM causing allergy include Dermatophagoides pteronyssinus (Der p) and Dermatophagoides farinae (Der f) (53–55). HDM allergen is highly associated with the disruption of epithelial barrier where they have proteolytic activity that can cleave the epithelial TJs protein. Der p 1 (i.e. a HDM cysteine proteinase allergen) has been reported to cleave extracellular domain sites of OCLN and in CLDN1 proteins, resulted in amplified epithelial permeability that allowed the passage of Der p 1 through the epithelial barrier (33, 56, 57). Inhibition of the protease activity of Der p 1 as a therapeutic approach to reduce HDM-induced barrier dysfunction has been proposed (58). Treatment of cultured primary HNECs in vitro with Der p 1 showed markedly decrease in CLDN1, resulted in significantly increased FD4 epithelial permeability (32).
Breakdown of epithelial barrier can also be caused by proteases via fungi exposure that facilitates the access of pathogens and directly activates immune cells (59, 60). The serine proteases in fungi such as Alternaria has been reported to decrease the mRNA and protein expression of TJs including ZO-1, OCLN and CLDN1 in PNECs (61). Reduction in transepithelial resistance and increase of ROS was demonstrated in the study (61). Exposure of Der p1 to HNECs line (RPMI 2650 cells) and in vivo AR model (rats) downregulated the expression of both mRNA and protein levels of OCLN, CLDN1, ZO-1 and JAM-A as well as increased TER and FD4 permeability compared to control group (62).
Epithelial Cell-derived Cytokines in AR
The onset of AR is also triggered by disrupted sinonasal epithelium through production of the inflammatory epithelium-derived cytokines TSLP, IL-25 and IL-33. These cytokines are key regulatory factors that connect epithelial–mesenchymal communications and elicit pathological modifications in the airway (63). Receptors expressed on the surface of epithelial cells such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) have the ability to identify structurally conservative pathogen-associated molecular patterns (PAMPs) in pathogens and induce innate and adaptive immune response. TSLP, IL-25 and IL-33 are important in the PAMP-TLR/NLR interaction (64), and each cytokine is released into the sinonasal environment upon exposure to allergens.
TSLP and TJs Disruption in AR
TSLP is an IL-7-like cytokine that potently induces deregulation of Th2 responses, a hallmark feature in allergic inflammatory diseases such as asthma, AD and AR (65–67). TSLP mRNA expression was significantly increased in the nasal mucosa of AR patients compared with controls (68). Treatment with TSLP also rapidly enhanced the barrier function of cultured HNECs together with an increase of TJ proteins CLDN1, CLDN4, CLDN7 and OCLN (68). The nasal epithelial-derived TSLP not only activates DCs but also preserves the epithelial barrier via upregulation of TJ proteins during the early stage of AR (68). High expression of TSLP was found in epithelium of AR patients with recruitment and infiltration of CD11c+ DCs (68).
TSLP was significantly upregulated in sensitized and nasally-challenged mouse model of AR (69, 70), and the expression of TSLP was suppressed by HDACi sodium butyrate (SoB) with improvements in AR clinical symptoms (69). However, TSLP expression was abolished in mast cell-deficient WBB6F1-W/Wv mice and Fc receptor γ chain (FcγR)-deficient mice (where the IgE receptor FcϵRI was not present on mast cells in these mice) compared to controls (70). This suggests that direct stimulation of epithelial cells by antigens alone may not be sufficient to induce TSLP expression in the nasal epithelium, and epithelial TSLP expression is regulated by mast cells via FcϵRI.
IL-25, IL-33, Innate Lymphoid Cells (ILCs) and TJs Dysfunction in AR
Both of the epithelial-derived cytokines IL-25 and IL-33 are pro-inflammatory. IL-25, also known as IL-17E, was first identified as a Th2 cell-derived cytokine (71). The interaction between IL-25 and IL-17RA/B leads to the activation and upregulation of transcription factors (e.g. NFκB, STAT6, GATA3, and NFATC1). This results in the activation and polarization of memory Th2 cells, leading to the secretion of Th2 cytokines such as IL-4, IL-5, and IL-13 (63). Increased production of IL-25 was induced by dsRNA in HNECs of AR patients (72). IL-33 is most likely released through cell necrosis or injury in mucosal epithelial cells (73), and IL-33 plays central roles in the initiation of Th2 cytokines and chemokines responses in AR. IL-33 expression was increased in the serum of AR patients and IL-33 single nucleotide polymorphism (SNP) occurs in AR patients (74). IL-33 knockout murine model of ragweed pollen-specific AR showed a decrease in eosinophil accumulation, reduced ability to mount an IgE response, as well as declined expression of Th2 cytokines compared to controls. Importantly, ragweed-immunized IL-33 knockout mice showed a significant reduction in the frequency of sneezing (75).
ILCs are immune cells of lymphoid lineage where one of their main functions involves the protection of mucosal barrier (76). ILCs play important roles in the development and progression of allergic diseases including AR (77). Type 2 ILC (ILC2)-mediated immune microenvironment are characterized by the production of both IL-25 and IL-33 as well as other cytokines (e.g. IL-13), and presence of mast cells and histiocytes. ILCs especially ILC2s are enriched in barrier tissues such as the skin, lung, and intestine and where they regulate immune responses surrounding the epithelium and sensitization to allergens (78).
Increased IL-13-producing ILC2s were found in the blood of patients with HDM allergy, cat-sensitized adults and in grass allergic patients and in the nasal fluid upon grass pollen challenge (79–81). IL-33 is thought to be a major cytokine that mainly activates ILC2s and signifies the rationale to utilize IL-33-induced lung inflammation models (82). Recently, it has been demonstrated that ILC2s facilitate bronchial epithelial barrier disruption via downregulation of the TJ barrier proteins (i.e. OCLN) through IL-13 in asthma (82). However, the influence of ILC2s on the nasal epithelial barrier has not been examined in AR and it warrants future investigations. For further information on the roles of TSLP, IL-25 and IL-33 in innate and adaptive immune responses, reviews from Hong and colleagues (63), and Hammad and Lambrect (83) are recommended.
Th2 Cytokines in AR
Th2 cytokines not only enhance inflammatory cell activation but also regulate epithelial cell barrier in allergic disease (i.e. AR, AD [atopic dermatitis], eosinophilic esophagitis, asthma and chronic rhinosinusitis) by reducing expression of TJs in epithelial cells (7, 10, 65, 84–86). The cytokines may also be released within the sinonasal microenvironment including sinonasal epithelial cells, altering TJs composition resulting in the “tight” barrier properties of TJ proteins switched to “leaky” properties (87, 88).
IL-4 is the paradigmatic cytokine involved in type-2 immune responses and plays a critical role in the development of Th2 cells and subsequent allergic reactions. Treatment with anti- interleukin-4 receptor α (IL-4Rα) monoclonal antibodies to nasal secretion of AR patients successfully restored the Th2-induced epithelial barrier dysfunction (7). It was supported by in vivo findings where anti-IL-4 treatment in HDM-challenged mice prevented the loss of OCLN and ZO-1 mRNA expression (7). IL-4 was found to disrupt epithelial integrity in vitro in PNECs with reduced ZO-1 and OCLN expression (33).
Therapeutic effects of the HDACi SoB on mice with AR and Trichostatin A (TSA) on nasal lavage fluid of AR patients resulted in significantly decreased serum levels of IL-4 and IL-10, and IL-4 and IL-5, respectively (69, 89). Both inhibitors further improved clinical symptoms and SoB enhanced the nasal mucosa epithelial morphology in AR mice model (69, 89). IL-4, IL-5 and IL-13 were significantly higher in the BAL fluid of HDM-induced mice, and treatment with HDACi (JNJ-26481585) significantly reduced the interleukins to levels in non-allergic saline control mice (90). In terms of IL-6, IL6 SNP (rs1800795) was linked with an increased risk of AR (91) and positively associated with the AR severity (92). The production of IL-6 by cultured NECs of AR was stimulated by alarmin protein high-mobility group box 1 (HMGB1), and anti-toll-like receptor 4 (anti-TLR4) blocking antibody significantly inhibited HMGB1-induced secretion of IL-6 (93).
Addition of IL-4 and IL-13 to reconstructed human epidermis cells resulted in downregulation of CLDN1 expression (85). Gene expression of IL13 was detected in the epithelial compartment of the nasal mucosa of perennial AR patients but not observed in normal subjects (94). IL-13 was regulated by microRNA (miR)-143 where overexpression of miR-143 significantly decreased the expression of pro-inflammatory factors (e.g. eotaxin and mucin) responsible for producing nasal symptoms (95). Taken together, these suggest that local nasal provocation is attributable to systemic overproduction of Th2 cytokines.
Neuroimmune and Epithelial Interaction in AR
Pathophysiology of allergic diseases involves a reciprocal regulation between neural and immune systems where both systems work synergistically to detect and respond to harmful stimuli. Neuronal cell types are usually found at skin and mucosal barrier surfaces, forming neuronal‐immune cell network (96, 97), and have been shown to regulate mucosal immunity and mucosal barrier integrity (98, 99). Neurogenic inflammation activates local release of neuropeptides and neurotransmitters such as substance P (SP), neurokinin A, neuromedin U (NMU), calcitonin gene-related peptide (CGRP), acetylcholine and norepinephrine upon activation of sensory nerve endings. This includes regulation of itch, cough, sneezing, bronchoconstriction vasodilation, plasma extravasation, recruitment of leukocytes and degranulation of mast cells (100–102).
CGRP and SP are increased in the airways of AR patients (103) and nasal secretions of idiopathic rhinitis (IR) patients (104), respectively. Neuronal ILC2s that selectively express NMU receptor 1 (NMUR1) are pro-inflammatory when exposed to NMU, and NMU is increased in the presence of IL‐25, IL‐33, and TSLP (105). In particular, NMU could activate ILC2s, and co-administration of NMU with IL-25 amplifies allergic inflammation (105).
Solitary chemosensory cells (SCCs) are found in healthy sinus cavity and turbinate tissues of the human upper airway where they function as epithelial sentinels by detecting pathogenic metabolites and initiate protective immune defense (106). Increased number of SCCs was found in the sinonasal epithelia of chronic rhinosinusitis with nasal polyps patients (107). SCCs include tuft cells (i.e. bottle-shaped with apical microvilli), and they are found in the nose, trachea and proximal airways. Tuft cells have close proximity with nerve fibre and can promote protective respiratory reflexes such as sneezing, release neurotransmitters (such as acetylcholine), eicosanoids and cytokines (e.g. IL-25 and TSLP) (108, 109). Tuft cell-derived IL-25 can initiate type-2 immune responses by activating IL-13 production in ILC2s. SCCs also utilize bitter taste receptors and canonical taste transduction pathways that play crucial roles as sentinels of respiration (110).
Neuronal transient receptor potential (TRPs) expressed in epithelial cells and endothelial cells are involved in inflammatory process and have been shown to regulate the permeability of cellular barriers in several tissues (111–113). TRP vanilloid (TRPV) channels are a group of non-selective cation channels associated with the transmission of sensory information (114). Overexpression of TRPV1 in nasal secretions were observed in IR patients (104). In the nose, nerve growth factor secreted by eosinophils can sensitize TRPV1 and TRP ankyrin 1 (TRPA1) in sensory nerve endings resulting in increased SP content and induced dendrite sprouting (115). Seasonal AR stimulated with TRPV1 intranasally with capsaicin experienced elevated symptoms and pain (116, 117). TRPV1 also mediated acidity-induced barrier dysfunction by disrupting CLDN3 and CLDN4 in human bronchial epithelial cells (16HBE) in vitro (118). Hence, barrier function in AR might be disrupted by increased expression or activity of epithelial TRPV1. Moreover, increased TRPV4 expression was shown in epithelial cells of AR patients compared with normal controls and in cultured epithelial cells stimulated by Th2 cytokines (IL-4 and IL-13) (119). Decreased E-cadherin and ZO-1 expression was demonstrated in epithelial cells of AR patients exposed to HDM allergen and TRPV4 agonist (GSK1016790A), suggesting possible roles of TRPV4 in the pathogenesis of allergen-induced epithelial barrier disruption in AR (119).
Epigenetics Changes in Regulating Epithelial Barrier in AR
In recent years, more attention has focused on multi-omics approaches such as epigenomics in developing precision medicine (101). Epigenetic alterations include DNA methylation, histone modifications and regulation of non-coding RNAs that affect gene transcription by changing the structural conformation and accessibility of genes without altering the gene sequence (120, 121). In AR, epigenetics disruption has been implicated in Th1/Th2 subsets, dendritic cells function, mast cells and macrophages activation that have been well discussed (122, 123). Recently, the role of microRNAs (miRNAs), a group of short non-coding regulatory RNAs that target mRNAs for cleavage causing translational repression, is implicated in regulating epithelial barrier of AR (124). A number of miRNAs are differentially expressed in AR and asthma patients including miR-125b, miR-16, miR-299-5p, miR-126, miR-206 and miR-133b (125).
Exposure of Der p1 to HNECs (RPMI 2650) increased miR-125b expression by increasing the expression and activation of CXCR4 which downregulated FoxP3 expression (62). The expression levels of OCLN, CLDN1, ZO-1, and JAM-A were significantly lower in the miR-125b-overexpressed group and vice versa in the miR-125b inhibitor group compared with the control group. The autophagy inhibitor 3-MA restored TJ proteins expression in the miR-125b-overexpressed group. These findings was demonstrated in both in vitro (RPMI 2650) and in vivo (rat AR model) (62). This study demonstrates that the CXCR4/miR-125b/FoxP3 axis may play pathogenic roles in AR development by promoting autophagy in epithelial cells leading to breakdown of the epithelial barrier.
Lastly, epigenetic regulation can occur via histone posttranslational modifications (PTMs) such as acetylation (123). Histone deacetylases (HDACs), an enzymes responsible for removing acetyl group from lysine residues of target proteins and block genes transcription by allowing DNA to be wrapped by histones and promote chromatin condensation (9, 126, 127). HDACs are associated with defects in epithelial barrier of AR (128). Higher expression of HDAC1 protein in nasal epithelium of AR patients was associated with disruption of CLDN4 (129). Treatment with HDAC inhibitor (HDACi) JNJ-26481585 successful restored ZO molecules structure in NECs of AR patients (90).
Epigenetic Memory
Memory of previous exposure to inflammatory stimuli is not exclusive to the hematopoietic lineage. Upon exposure to inflammatory stimuli, epithelial cells sense the signals such as from microbe-associated molecular pattern (MAMP) and transmit the information to immune cells which then facilitate chromatin remodeling of epithelial cells, resulting in increased chromatin accessibility while embedding a memory of the experience within their chromatin (130). This novel function of epithelial cells is known as epigenetic memory where epigenetic priming of enhancer regions induce stronger responses to secondary stimuli and strengthen their sensitivity towards future encounter (131).
Naik et al. demonstrated a prolonged memory to acute inflammation in mouse enable skin epithelial stem cells (EpSCs) to hasten barrier restoration after subsequent tissue damage (132). Accelerated wound healing process was observed in mice exposed to various inflammatory stimuli (e.g. imiquimod, abrasion wounding or Candida infection) compared to naïve mice, even 180 days later. This was possible because at 30 days after the exposure, most chromatin changes were still maintained in EpSCs and increased chromatin accessibility in genes encoding molecules involved in inflammation, interleukin signaling, oxidative stress response and proliferation (132).
The treatment with an anti-IL4Rα monoclonal antibody (mAb) targeting IL-4 and IL-13 receptors in patients with polyp showed preservation of several disease-associated genes including CTNNB1, a key WNT mediator that influences basal cell proliferation and differentiation (133). This explained how anti-IL4Rα mAb was capable in reducing nasal polyp burden overtime. Meanwhile, tissue-resident memory (TRM) cells could be maintained in barrier tissues for prolonged periods, suggesting their roles in preserving epithelial barrier (134). It was found that 80% of the transcriptomes in lung epithelial cells were dependent on CD4+ TRM T cells including eosinophil infiltration during host defense in pneumonia and maintained even when T cells were depleted (135). CD8+ TRM cells were also found to be maintained in the lung airways independently of tissue-circulating effector memory T (TEM) cells via a homeostatic proliferation mechanism (136).
The structure of nasal epithelial barrier, their associations with ILCs and regulation of TJs expression by cytokines, nerves activation and epigenetic changes, are presented in Figure 1. In addition, Table 1 summarizes the TJs expression in AR patients versus normal subjects, or in murine AR models.
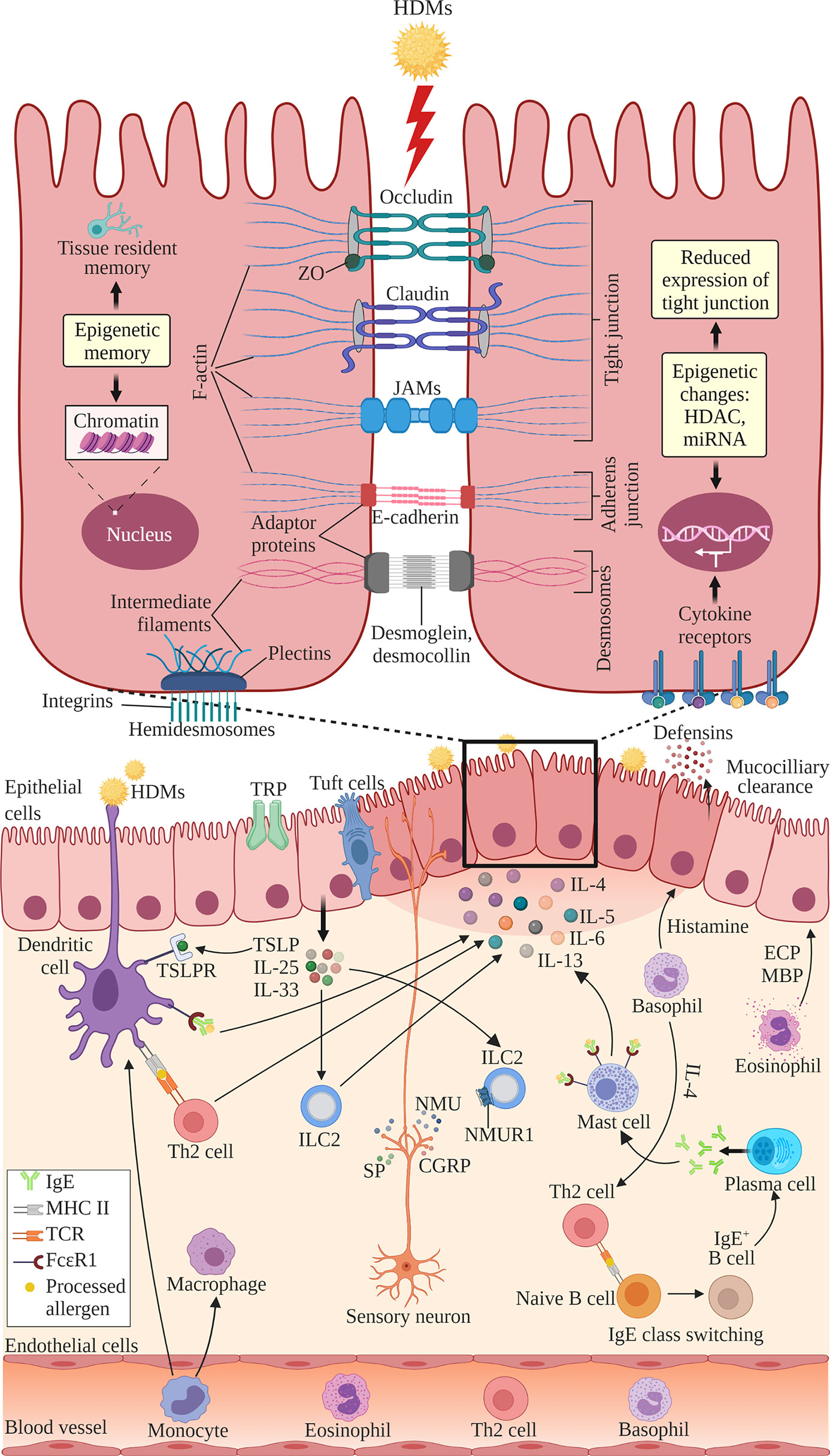
Figure 1 The structure of nasal epithelial barrier comprises of tight junction (TJ), adherens junction, desmosomes and hemidesmosomes. TJs are composed of occludin, claudin and JAMs that span the intercellular space and intracellular adaptor proteins ZO. Adherens junctions are composed of E-cadherins and adaptor proteins. Desmosomes consist of desmoglein and desmocollin proteins that bind internal adaptor proteins. F-actin and intermediate filaments act as cytoskeleton for these cell junctions. Hemidesmosomes comprise of plectins that link to the intermediate filaments and integrins, the transmembrane linkers of extracellular matrix and actin cytoskeleton. Epithelial cells secrete antimicrobial substances such as defensins and conduct mucociliary clearance. Epithelial activation by airborne allergens during allergic response in AR leads to the activation of epithelial cells and release of epithelial-derived cytokines TSLP, IL-25 and IL-33. This triggers subsequent activation of ILC2s and production of Th2 cytokines such as IL-4, IL-5, IL-6 and IL-13. Released cytokines promote DCs where they present antigens and activate naïve B cells to induce IgE class switching and maturation into plasma cells, which produce IgE. Secreted IgE binds the FcϵRI receptor on submucosal mast cells, leading to the release of preformed mediators such as histamine and inflammatory cytokines. In the late allergic phase, recruited eosinophils and basophils release mediators that further contribute to AR symptoms via epithelial damage and microvascular leaking. Blood-derived monocytes differentiate into DCs and macrophages that promote allergic responses. The overproduction of Th2 cytokines by a variety of cells suppresses the transcription of TJ molecules causing the breakdown in the nasal epithelial barrier of AR patients. Sensory neuron release NMU, SP and CGRP upon activation at sensory nerve endings. NMUR1 expressed on ILC2 intensifies the inflammatory response in the presence of IL‐25, IL‐33, and TSLP. TRP and tuft cells also have roles in regulating epithelial barrier. Epithelial barrier is also regulated by epigenetic changes through histone modification by HDACs and miRNAs. Epithelial cells exhibit epigenetic memory by embedding the memory of previous encounters within their chromatin, and tissue-resident memory cells preserve the epithelial barrier. DC, dendritic cell; ECP, eosinophil cationic protein; MBP, major basic protein; MHC, major histocompatibility complex; IgE, immunoglobulin E; ILC2, type 2 innate lymphoid cell; JAMs, junctional adhesion molecules; TCR, T cell receptor; Th2, T helper 2; TSLP, thymic stromal lymphopoietin; TSLPR, thymic stromal lymphopoietin receptor; ZO, zonula occludens; HDAC, histone deacetylases; miR, microRNA; NMU, neuromedin U; NMUR1, neuromedin U receptor 1; SP, substance P; CGRP, calcitonin gene-related peptide; TRP, transient receptor potential. Created with BioRender.com.
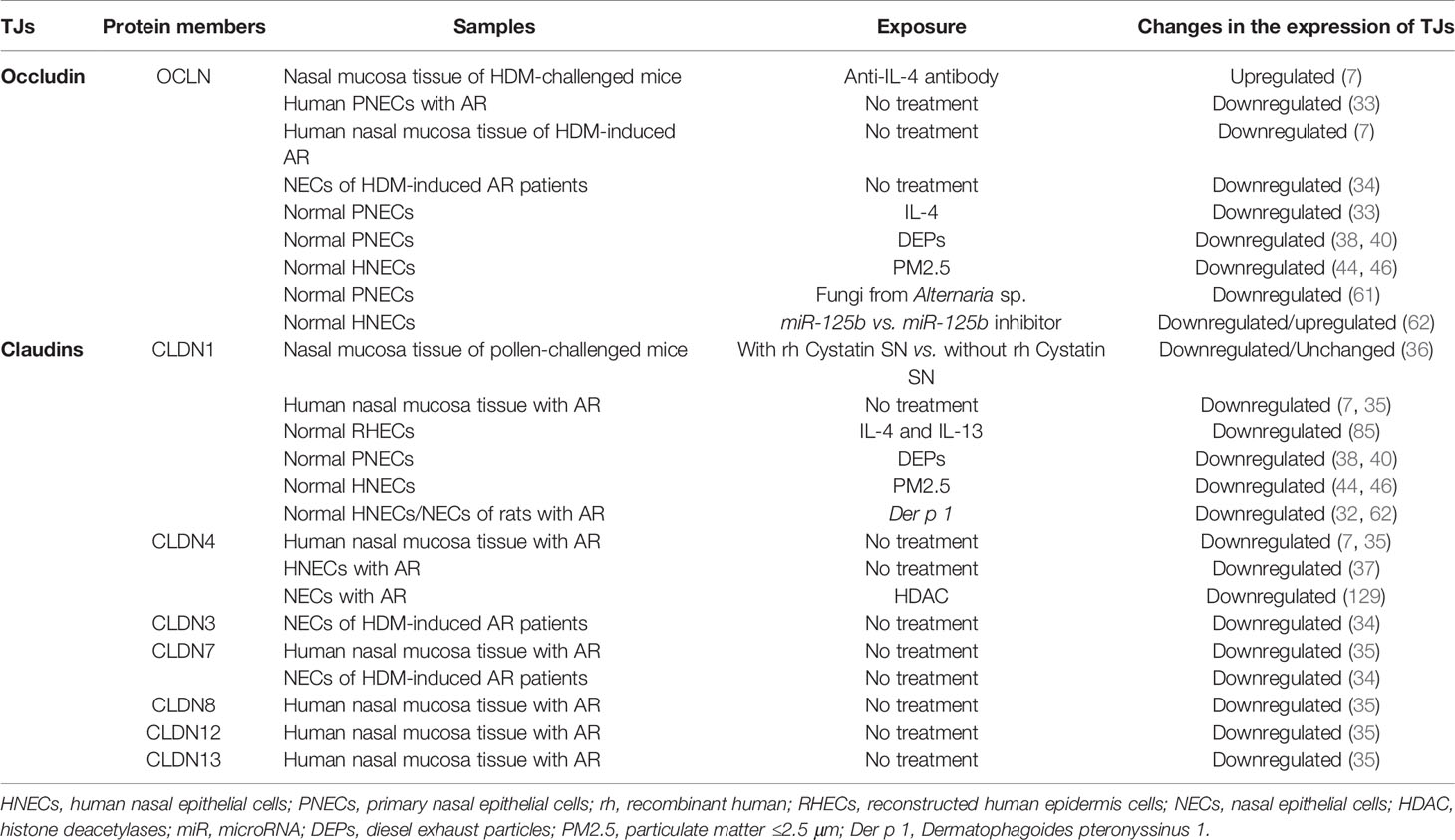
Table 1 TJ molecules expression in AR patients versus normal subjects, or in murine AR models.
Conclusions and Perspectives
Impairment of nasal epithelial barrier through the loss of TJs expression contributes to the development of AR. The pro-inflammatory, epithelial-derived cytokines TSLP, IL-25 and IL-33 play critical roles in AR inflammation. These cytokines activate and expand ILC2s upon encounter with allergens which subsequently induce the release of other Th2 cytokines leading to TJs breakdown in nasal epithelial of AR patients. Thus, targeted therapy against these cytokines is a therapeutic opportunity for the disease. For instance, tezepelumab, a human monoclonal therapeutic antibody that targets TSLP, is currently being evaluated for the treatment of airway allergic diseases including asthma and AD (63). Although therapeutic inhibition of TSLP or IL-33 has not been investigated in clinical trials of AR patients, murine model studies have shown that knockout of these epithelium-derived cytokines could reduce AR manifestations (75, 137). In addition, future studies should also examine the potential disruption of other components of the cell junctions such as desmosomes in AR. In conclusion, barrier defects are the results of multiple exogenous (e.g. air pollution, HDMs) and endogenous (e.g. cytokines, neuroimmune–epithelial interaction, epigenetics) triggers trapping the epithelium in a diseased state contributing to AR development, and protection of the nasal epithelial barrier integrity through restoring TJs expression is a promising therapeutic approach for AR patients.
Author Contributions
SMNH, HTT, NSMA and KKW conceived the manuscript. SMNH and KKW designed the manuscript, wrote and revised the manuscript. SMNH prepared the figure and table. HTT, NSMA and NMS edited and revised the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by Universiti Sains Malaysia (USM) grants comprising of the Research University Grant (1001/PPSP/8012349) awarded to KKW, Research University Grant (1001.PPSP.8012285) awarded to NSMA and USM Fellowship Scheme awarded to SMNH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bousquet J, Khaltaev N, Cruz AA, Denburg J, Fokkens WJ, Togias A, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 Update (in Collaboration With the World Health Organization, GA(2)LEN and Allergen). Allergy (2008) 63(Suppl 86):S8–160. doi: 10.1111/j.1398-9995.2007.01620.x
2. Brozek JL, Bousquet J, Agache I, Agarwal A, Bachert C, Bosnic-Anticevich S, et al. Allergic Rhinitis and its Impact on Asthma (Aria) guidelines-2016 Revision. J Allergy Clin Immunol (2017) 140:950–8. doi: 10.1016/j.jaci.2017.03.050
3. Ashari NSM, Amin SNSM, Hamid WZWA, Rahman AA, Muhammad I. Determination of Interleukin 31 (Il-31) Serum Levels According to Severity of Allergic Rhinitis. Bangladesh J Med Sci (2015) 14:359–62. doi: 10.3329/bjms.v14i4.20393
4. Azid NA, Sani MM, Zamry AA, Ashari NSM, Tan TH-T, Wong KK, et al. Total IgE Levels and Their Relevance in the Diagnosis of Allergy Among Malaysian Population in the North-East Region of Peninsular Malaysia. J BioMed Clin Sci (2019) 4:1–7.
5. Fukuoka A, Yoshimoto T. Barrier Dysfunction in the Nasal Allergy. Allergol Int (2018) 67:18–23. doi: 10.1016/j.alit.2017.10.006
6. Vareille M, Kieninger E, Edwards MR, Regamey N. The Airway Epithelium: Soldier in the Fight Against Respiratory Viruses. Clin Microbiol Rev (2011) 24:210–29. doi: 10.1128/CMR.00014-10
7. Steelant B, Seys SF, Van Gerven L, Van Woensel M, Farre R, Wawrzyniak P, et al. Histamine and T Helper Cytokine-Driven Epithelial Barrier Dysfunction in Allergic Rhinitis. J Allergy Clin Immunol (2018) 141:951–63.e8. doi: 10.1016/j.jaci.2017.08.039
8. Tan HT, Hagner S, Ruchti F, Radzikowska U, Tan G, Altunbulakli C, et al. Tight Junction, Mucin, and Inflammasome-Related Molecules are Differentially Expressed in Eosinophilic, Mixed, and Neutrophilic Experimental Asthma in Mice. Allergy (2019) 74:294–307. doi: 10.1111/all.13619
9. Sarah COS, Shukri NM, Ashari NSM, Wong KK. Zonula Occludens and Nasal Epithelial Barrier Integrity in Allergic Rhinitis. PeerJ (2020) 8:e9834. doi: 10.7717/peerj.9834
10. Steelant B, Seys SF, Boeckxstaens G, Akdis CA, Ceuppens JL, Hellings PW. Restoring Airway Epithelial Barrier Dysfunction: A New Therapeutic Challenge in Allergic Airway Disease. Rhinology (2016) 54:195–205. doi: 10.4193/Rhin15.376
11. Toppila-Salmi S, van Drunen CM, Fokkens WJ, Golebski K, Mattila P, Joenvaara S, et al. Molecular Mechanisms of Nasal Epithelium in Rhinitis and Rhinosinusitis. Curr Allergy Asthma Rep (2015) 15:495. doi: 10.1007/s11882-014-0495-8
12. Yang D, Oppenheim JJ. Antimicrobial Proteins Act as “Alarmins” in Joint Immune Defense. Arthritis Rheum (2004) 50:3401–3. doi: 10.1002/art.20604
13. Radek K, Gallo R. Antimicrobial Peptides: Natural Effectors of the Innate Immune System. Semin Immunopathol (2007) 29(1):27–43. doi: 10.1007/s00281-007-0064-5
14. Kumpitsch C, Koskinen K, Schopf V, Moissl-Eichinger C. The Microbiome of the Upper Respiratory Tract in Health and Disease. BMC Biol (2019) 17:87. doi: 10.1186/s12915-019-0703-z
15. Guttman JA, Finlay BB. Tight Junctions as Targets of Infectious Agents. Biochim Biophys Acta (2009) 1788:832–41. doi: 10.1016/j.bbamem.2008.10.028
16. Campbell HK, Maiers JL, DeMali KA. Interplay Between Tight Junctions & Adherens Junctions. Exp Cell Res (2017) 358:39–44. doi: 10.1016/j.yexcr.2017.03.061
17. Georas SN, Rezaee F. Epithelial Barrier Function: At the Front Line of Asthma Immunology and Allergic Airway Inflammation. J Allergy Clin Immunol (2014) 134:509–20. doi: 10.1016/j.jaci.2014.05.049
18. Kirschner N, Rosenthal R, Furuse M, Moll I, Fromm M, Brandner JM. Contribution of Tight Junction Proteins to Ion, Macromolecule, and Water Barrier in Keratinocytes. J Invest Dermatol (2013) 133:1161–9. doi: 10.1038/jid.2012.507
19. Bruser L, Bogdan S. Adherens Junctions on the Move-Membrane Trafficking of E-Cadherin. Cold Spring Harb Perspect Biol (2017) 9. doi: 10.1101/cshperspect.a029140
20. Rubsam M, Broussard JA, Wickstrom SA, Nekrasova O, Green KJ, Niessen CM. Adherens Junctions and Desmosomes Coordinate Mechanics and Signaling to Orchestrate Tissue Morphogenesis and Function: An Evolutionary Perspective. Cold Spring Harb Perspect Biol (2018) 10. doi: 10.1101/cshperspect.a029207
21. Hatzfeld M, Keil R, Magin TM. Desmosomes and Intermediate Filaments: Their Consequences for Tissue Mechanics. Cold Spring Harb Perspect Biol (2017) 9. doi: 10.1101/cshperspect.a029157
22. Jindal S, Chockalingam S, Ghosh SS, Packirisamy G. Connexin and Gap Junctions: Perspectives From Biology to Nanotechnology Based Therapeutics. Transl Res (2021). doi: 10.1016/j.trsl.2021.02.008
23. Walko G, Castanon MJ, Wiche G. Molecular Architecture and Function of the Hemidesmosome. Cell Tissue Res (2015) 360:363–78. doi: 10.1007/s00441-014-2061-z
24. Wang W, Zuidema A, Te Molder L, Nahidiazar L, Hoekman L, Schmidt T, et al. Hemidesmosomes Modulate Force Generation Via Focal Adhesions. J Cell Biol (2020) 219. doi: 10.1083/jcb.201904137
25. Siliciano JD, Goodenough DA. Localization of the Tight Junction Protein, ZO-1, is Modulated by Extracellular Calcium and Cell-Cell Contact in Madin-Darby Canine Kidney Epithelial Cells. J Cell Biol (1988) 107:2389–99. doi: 10.1083/jcb.107.6.2389
26. Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: A Novel Integral Membrane Protein Localizing at Tight Junctions. J Cell Biol (1993) 123:1777–88. doi: 10.1083/jcb.123.6.1777
27. Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, et al. Epithelial Transport and Barrier Function in Occludin-Deficient Mice. Biochim Biophys Acta (2005) 1669:34–42. doi: 10.1016/j.bbamem.2005.01.008
28. Gunzel D, Yu AS. Claudins and the Modulation of Tight Junction Permeability. Physiol Rev (2013) 93:525–69. doi: 10.1152/physrev.00019.2012
29. Li J, Zhuo M, Pei L, Rajagopal M, Yu AS. Comprehensive Cysteine-Scanning Mutagenesis Reveals Claudin-2 Pore-Lining Residues With Different Intrapore Locations. J Biol Chem (2014) 289:6475–84. doi: 10.1074/jbc.M113.536888
30. Krause G, Protze J, Piontek J. Assembly and Function of Claudins: Structure-function Relationships Based on Homology Models and Crystal Structures. Semin Cell Dev Biol (2015) 42:3–12. doi: 10.1016/j.semcdb.2015.04.010
31. Garrido-Urbani S, Bradfield PF, Imhof BA. Tight Junction Dynamics: The Role of Junctional Adhesion Molecules (Jams). Cell Tissue Res (2014) 355:701–15. doi: 10.1007/s00441-014-1820-1
32. Wang M, Jun KM, Xue HM, Zhi-Qun SM, Li LM, Qing L, et al. Protease-Activated Receptor-2 Decreased Zonula Occlidens-1 and Claudin-1 Expression and Induced Epithelial Barrier Dysfunction in Allergic Rhinitis. Am J Rhinol Allergy (2021) 35:26–35. doi: 10.1177/1945892420932486
33. Steelant B, Farre R, Wawrzyniak P, Belmans J, Dekimpe E, Vanheel H, et al. Impaired Barrier Function in Patients With House Dust Mite-Induced Allergic Rhinitis is Accompanied by Decreased Occludin and Zonula Occludens-1 Expression. J Allergy Clin Immunol (2016) 137:1043–53.e5. doi: 10.1016/j.jaci.2015.10.050
34. Husna SMN, Sarah COS, Tan H-TT, Shukri NM, Ashari NSM, Wong KK. Reduced Occludin and Claudin-7 Expression is Associated With Urban Locations and Exposure to Second-Hand Smoke in Allergic Rhinitis Patients. Sci Rep (2021) 11:1–12. doi: 10.1038/s41598-020-79208-y
35. Takano K, Kojima T, Go M, Murata M, Ichimiya S, Himi T, et al. Hla-DR- and CD11c-positive Dendritic Cells Penetrate Beyond Well-Developed Epithelial Tight Junctions in Human Nasal Mucosa of Allergic Rhinitis. J Histochem Cytochem (2005) 53:611–9. doi: 10.1369/jhc.4A6539.2005
36. Fukuoka A, Matsushita K, Morikawa T, Adachi T, Yasuda K, Kiyonari H, et al. Human Cystatin SN is an Endogenous Protease Inhibitor That Prevents Allergic Rhinitis. J Allergy Clin Immunol (2019) 143:1153–62.e12. doi: 10.1016/j.jaci.2018.06.035
37. Kaneko Y, Konno T, Kohno T, Kakuki T, Miyata R, Ohkuni T. Et Al, Induction of Airway Progenitor Cells Via p63 and KLF11 by Rho-kinase Inhibitor Y27632 in hTERT-human Nasal Epithelial Cells. Am J Transl Res (2019) 11:599–611.
38. Kim N, Han DH, Suh MW, Lee JH, Oh SH, Park MK. Effect of Lipopolysaccharide on Diesel Exhaust Particle-Induced Junctional Dysfunction in Primary Human Nasal Epithelial Cells. Environ Pollut (2019) 248:736–42. doi: 10.1016/j.envpol.2019.02.082
39. Zhao R, Guo Z, Zhang R, Deng C, Xu J, Dong W, et al. Nasal Epithelial Barrier Disruption by Particulate Matter </=2.5 Mum Via Tight Junction Protein Degradation. J Appl Toxicol (2018) 38:678–87. doi: 10.1002/jat.3573
40. Fukuoka A, Matsushita K, Morikawa T, Takano H, Yoshimoto T. Diesel Exhaust Particles Exacerbate Allergic Rhinitis in Mice by Disrupting the Nasal Epithelial Barrier. Clin Exp Allergy (2016) 46:142–52. doi: 10.1111/cea.12597
41. Park M, Lee JS, Park MK. The Effects of Air Pollutants on the Prevalence of Common Ear, Nose, and Throat Diseases in South Korea: A National Population-Based Study. Clin Exp Otorhinolaryngol (2019) 12:294–300. doi: 10.21053/ceo.2018.00612
42. Sun N, Han Z, Wang H, Guo Z, Deng C, Dong W, et al. Effects of Ursolic Acid on the Expression of Th1-Th2-related Cytokines in a Rat Model of Allergic Rhinitis After Pm2.5 Exposure. Am J Rhinol Allergy (2020) 34:587–96. doi: 10.1177/1945892420913430
43. Sun N, Deng C, Zhao Q, Han Z, Guo Z, Wang H, et al. Ursolic Acid Alleviates Mucus Secretion and Tissue Remodeling in Rat Model of Allergic Rhinitis After Pm2.5 Exposure. Am J Rhinol Allergy (2021) 35:272–9. doi: 10.1177/1945892420953351
44. Lee DC, Choi H, Oh JM, Lee J, Lee J, Lee HY, et al. Urban Particulate Matter Regulates Tight Junction Proteins by Inducing Oxidative Stress Via the Akt Signal Pathway in Human Nasal Epithelial Cells. Toxicol Lett (2020) 333:33–41. doi: 10.1016/j.toxlet.2020.07.017
45. Kim KA, Jung JH, Kang IG, Choi YS, Kim ST. Ros Is Involved in Disruption of Tight Junctions of Human Nasal Epithelial Cells Induced by HRV16. Laryngoscope (2018) 128:E393–401. doi: 10.1002/lary.27510
46. Xian M, Ma S, Wang K, Lou H, Wang Y, Zhang L, et al. Particulate Matter 2.5 Causes Deficiency in Barrier Integrity in Human Nasal Epithelial Cells. Allergy Asthma Immunol Res (2020) 12:56–71. doi: 10.4168/aair.2020.12.1.56
47. Ma J-Q, Ding J, Zhang L, Liu C-M. Ursolic Acid Protects Mouse Liver Against CCl4-induced Oxidative Stress and Inflammation by the MAPK/NF-κb Pathway. Environ Toxicol Pharmacol (2014) 37:975–83. doi: 10.1016/j.etap.2014.03.011
48. Ho TM, Murad S, Kesavapillai R, Singaram SP. Prevalence of Allergy to Some Inhalants Among Rhinitis Patients in Malaysia. Asian Pac J Allergy Immunol (1995) 13:11–6.
49. Liam CK, Loo KL, Wong CM, Lim KH, Lee TC. Skin Prick Test Reactivity to Common Aeroallergens in Asthmatic Patients With and Without Rhinitis. Respirology (2002) 7:345–50. doi: 10.1046/j.1440-1843.2002.00409.x
50. Lee JH, Kim SC, Choi H, Jung CG, Ban GY, Shin YS, et al. A Retrospective Study of Clinical Response Predictors in Subcutaneous Allergen Immunotherapy With House Dust Mites for Allergic Rhinitis. Allergy Asthma Immunol Res (2018) 10:18–24. doi: 10.4168/aair.2018.10.1.18
51. Sani MM, Ashari NSM, Abdullah B, Wong KK, Musa KI, Mohamud R, et al. Reduced Cd4+ Terminally Differentiated Effector Memory T Cells In Moderate-Severe House Dust Mites Sensitized Allergic Rhinitis Patients. Asian Pac J Allergy Immunol (2018) 37:138–46. doi: 10.12932/AP-191217-0220
52. Wang IJ, Tung TH, Tang CS, Zhao ZH. Allergens, Air Pollutants, and Childhood Allergic Diseases. Int J Hyg Environ Health (2016) 219:66–71. doi: 10.1016/j.ijheh.2015.09.001
53. Lim FL, Hashim Z, Than LT, Md Said S, Hisham Hashim J, Norback D. Asthma, Airway Symptoms and Rhinitis in Office Workers in Malaysia: Associations With House Dust Mite (Hdm) Allergy, Cat Allergy and Levels of House Dust Mite Allergens in Office Dust. PloS One (2015) 10:e0124905. doi: 10.1371/journal.pone.0124905
54. Majdiah W, Khaiza NY, Suzina SAH, Maraina CC, Suryani N. Association Between Specific IgE Levels and Severity of Symptoms Among Patients With Rhinitis in North East Malaysia. Int Med J (2011) 18:348–9.
55. Ashari NSM. Allergen Specific Immunoglobulin E Assay in Identification of Allergens in Allergic Diseases Patients. Int Med J (2009) 16:25–9.
56. Platts-Mills TA, Woodfolk JA. Allergens and Their Role in the Allergic Immune Response. Immunol Rev (2011) 242:51–68. doi: 10.1111/j.1600-065X.2011.01021.x
57. London NR Jr, Ramanathan M Jr. The Role of the Sinonasal Epithelium in Allergic Rhinitis. Otolaryngol Clin North Am (2017) 50:1043–50. doi: 10.1016/j.otc.2017.08.002
58. John RJ, Rusznak C, Ramjee M, Lamont AG, Abrahamson M, Hewitt EL. Functional Effects of the Inhibition of the Cysteine Protease Activity of the Major House Dust Mite Allergen Der P 1 by a Novel Peptide-Based Inhibitor. Clin Exp Allergy (2000) 30:784–93. doi: 10.1046/j.1365-2222.2000.00840.x
59. Shin SH, Lee YH, Jeon CH. Protease-Dependent Activation of Nasal Polyp Epithelial Cells by Airborne Fungi Leads to Migration of Eosinophils and Neutrophils. Acta Otolaryngol (2006) 126:1286–94. doi: 10.1080/00016480500395179
60. Schleimer RP, Berdnikovs S. Etiology of Epithelial Barrier Dysfunction in Patients With Type 2 Inflammatory Diseases. J Allergy Clin Immunol (2017) 139:1752–61. doi: 10.1016/j.jaci.2017.04.010
61. Shin SH, Ye MK, Lee DW, Che MH. Alternaria-Induced Barrier Dysfunction of Nasal Epithelial Cells: Role of Serine Protease and Reactive Oxygen Species. Int Forum Allergy Rhinol (2018) 9(5):514–21. doi: 10.1002/alr.22266
62. Zheng J, Zeng M, Nian J-B, Zeng L-Y, Fu Z, Huang Q-J, et al. The CXCR4/miR-125b/FoxP3 Axis Regulates the Function of the Epithelial Barrier Via Autophagy in Allergic Rhinitis. Am J Trans Res (2020) 12:2570.
63. Hong H, Liao S, Chen F, Yang Q, Wang DY. Role of IL-25, Il-33, and TSLP in Triggering United Airway Diseases Toward Type 2 Inflammation. Allergy (2020) 75:2794–804. doi: 10.1111/all.14526
64. Patel NN, Kohanski MA, Maina IW, Workman AD, Herbert DBR, Cohen NA. Sentinels at the Wall: Epithelial-Derived Cytokines Serve as Triggers of Upper Airway Type 2 Inflammation. Int Forum Allergy Rhinol (2019) 9(1):93–9. doi: 10.1002/alr.22206
65. Holgate ST. Epithelium Dysfunction in Asthma. J Allergy Clin Immunol (2007) 120:1233–44. doi: 10.1016/j.jaci.2007.10.025
66. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human Epithelial Cells Trigger Dendritic Cell Mediated Allergic Inflammation by Producing TSLP. Nat Immunol (2002) 3:673–80. doi: 10.1038/ni805
67. Ying S, O’Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic Stromal Lymphopoietin Expression is Increased in Asthmatic Airways and Correlates With Expression of Th2-attracting Chemokines and Disease Severity. J Immunol (2005) 174:8183–90. doi: 10.4049/jimmunol.174.12.8183
68. Kamekura R, Kojima T, Koizumi J, Ogasawara N, Kurose M, Go M, et al. Thymic Stromal Lymphopoietin Enhances Tight-Junction Barrier Function of Human Nasal Epithelial Cells. Cell Tissue Res (2009) 338:283–93. doi: 10.1007/s00441-009-0855-1
69. Wang J, Wen L, Wang Y, Chen F. Therapeutic Effect of Histone Deacetylase Inhibitor, Sodium Butyrate, on Allergic Rhinitis In Vivo. DNA Cell Biol (2016) 35:203–8. doi: 10.1089/dna.2015.3037
70. Miyata M, Hatsushika K, Ando T, Shimokawa N, Ohnuma Y, Katoh R, et al. Mast Cell Regulation of Epithelial TSLP Expression Plays an Important Role in the Development of Allergic Rhinitis. Eur J Immunol (2008) 38:1487–92. doi: 10.1002/eji.200737809
71. Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. Il-25 Induces IL-4, Il-5, and IL-13 and Th2-associated Pathologies In Vivo. Immunity (2001) 15:985–95. doi: 10.1016/s1074-7613(01)00243-6
72. Xu G, Zhang L, Wang D, Xu R, Liu Z, Han D, et al. Opposing Roles of IL-17A and IL-25 in the Regulation of TSLP Production in Human Nasal Epithelial Cells. Allergy (2010) 65:581–9. doi: 10.1111/j.1398-9995.2009.02252.x
73. Moussion C, Ortega N, Girard J-P. The IL-1-like Cytokine IL-33 is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PloS One (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
74. Sakashita M, Yoshimoto T, Hirota T, Harada M, Okubo K, Osawa Y, et al. Association of Serum interleukin-33 Level and the interleukin-33 Genetic Variant With Japanese Cedar Pollinosis. Clin Exp Allergy (2008) 38:1875–81. doi: 10.1111/j.1365-2222.2008.03114.x
75. Haenuki Y, Matsushita K, Futatsugi-Yumikura S, Ishii KJ, Kawagoe T, Imoto Y, et al. A Critical Role of IL-33 in Experimental Allergic Rhinitis. J Allergy Clin Immunol (2012) 130:184–94.e11. doi: 10.1016/j.jaci.2012.02.013
76. Fan H, Wang A, Wang Y, Sun Y, Han J, Chen W, et al. Innate Lymphoid Cells: Regulators of Gut Barrier Function and Immune Homeostasis. J Immunol Res (2019) 2019:2525984. doi: 10.1155/2019/2525984
77. Yu Q, Guo Y, Li X, Li C, Tan W, Fan X, et al. ILC 2 Frequency and Activity are Inhibited by Glucocorticoid Treatment Via STAT Pathway in Patients With Asthma. Allergy (2018) 73:1860–70. doi: 10.1111/all.13438
78. Kortekaas Krohn I, Shikhagaie MM, Golebski K, Bernink J, Breynaert C, Creyns B, et al. Emerging Roles of Innate Lymphoid Cells in Inflammatory Diseases: Clinical Implications. Allergy (2018) 73:837–50. doi: 10.1111/all.13340
79. Fan D, Wang X, Wang M, Wang Y, Zhang L, Li Y, et al. Allergen-Dependent Differences in ILC2s Frequencies in Patients With Allergic Rhinitis. Allergy Asthma Immunol Res (2016) 8:216–22. doi: 10.4168/aair.2016.8.3.216
80. Doherty TA, Scott D, Walford HH, Khorram N, Lund S, Baum R, et al. Allergen Challenge in Allergic Rhinitis Rapidly Induces Increased Peripheral Blood Type 2 Innate Lymphoid Cells That Express CD84. J Allergy Clin Immunol (2014) 133:1203–5.e7. doi: 10.1016/j.jaci.2013.12.1086
81. Dhariwal J, Cameron A, Trujillo-Torralbo M-B, Del Rosario A, Bakhsoliani E, Paulsen M, et al. Mucosal Type 2 Innate Lymphoid Cells are a Key Component of the Allergic Response to Aeroallergens. Am J Respir Crit Care Med (2017) 195:1586–96. doi: 10.1164/rccm.201609-1846OC
82. Sugita K, Steer CA, Martinez-Gonzalez I, Altunbulakli C, Morita H, Castro-Giner F, et al. Type 2 Innate Lymphoid Cells Disrupt Bronchial Epithelial Barrier Integrity by Targeting Tight Junctions Through IL-13 in Asthmatic Patients. J Allergy Clin Immunol (2018) 141:300–10.e11. doi: 10.1016/j.jaci.2017.02.038
83. Hammad H, Lambrecht B. Dendritic Cells and Airway Epithelial Cells at the Interface Between Innate and Adaptive Immune Responses. Allergy (2011) 66:579–87. doi: 10.1111/j.1398-9995.2010.02528.x
84. Heijink IH, Nawijn MC, Hackett TL. Airway Epithelial Barrier Function Regulates the Pathogenesis of Allergic Asthma. Clin Exp Allergy (2014) 44:620–30. doi: 10.1111/cea.12296
85. Gruber R, Bornchen C, Rose K, Daubmann A, Volksdorf T, Wladykowski E, et al. Diverse Regulation of Claudin-1 and Claudin-4 in Atopic Dermatitis. Am J Pathol (2015) 185:2777–89. doi: 10.1016/j.ajpath.2015.06.021
86. Travers J, Rochman M, Miracle CE, Cohen JP, Rothenberg ME. Linking Impaired Skin Barrier Function to Esophageal Allergic Inflammation Via IL-33. J Allergy Clin Immunol (2016) 138:1381–3. doi: 10.1016/j.jaci.2016.09.001
87. London NR Jr, Tharakan A, Ramanathan M Jr. The Role of Innate Immunity and Aeroallergens in Chronic Rhinosinusitis. Adv Otorhinolaryngol (2016) 79:69–77. doi: 10.1159/000445132
88. Capaldo CT, Nusrat A. Cytokine Regulation of Tight Junctions. Biochim Biophys Acta (2009) 1788:864–71. doi: 10.1016/j.bbamem.2008.08.027
89. Cho JS, Kang JH, Han IH, Um JY, Park IH, Lee SH, et al. Antiallergic Effects of Trichostatin A in a Murine Model of Allergic Rhinitis. Clin Exp Otorhinolaryngol (2015) 8:243–9. doi: 10.3342/ceo.2015.8.3.243
90. Steelant B, Wawrzyniak P, Martens K, Jonckheere AC, Pugin B, Schrijvers R, et al. Blocking Histone Deacetylase Activity as a Novel Target for Epithelial Barrier Defects in Patients With Allergic Rhinitis. J Allergy Clin Immunol (2019) 144:1242–53.e7. doi: 10.1016/j.jaci.2019.04.027
91. Zhao N, Liu HJ, Sun YY, Li YZ. Role of Interleukin-6 Polymorphisms in the Development of Allergic Rhinitis. Genet Mol Res (2016) 15:14–7. doi: 10.4238/gmr.15016987
92. Zhao H, Li H, Zhou C, Shi W, Zhang J, Hao Y, et al. Aqueous Nasal Spray of Chitosan Oligosaccharide Ameliorates Perennial Allergic Rhinitis by Affecting Serum Levels of Interleukin-6 and Interleukin-10. Int J Clin Exp Med (2018) 11:539–48.
93. Shimizu S, Kouzaki H, Kato T, Tojima I, Shimizu T. HMGB1-TLR4 Signaling Contributes to the Secretion of Interleukin 6 and Interleukin 8 by Nasal Epithelial Cells. Am J Rhinol Allergy (2016) 30:167–72. doi: 10.2500/ajra.2016.30.4300
94. Pawankar RU, Okuda M, Hasegawa S, Suzuki K, Yssel H, Okubo K, et al. Interleukin-13 Expression in the Nasal Mucosa of Perennial Allergic Rhinitis. Am J Respir Crit Care Med (1995) 152:2059–67. doi: 10.1164/ajrccm.152.6.8520776
95. Teng Y, Zhang R, Liu C, Zhou L, Wang H, Zhuang W, et al. miR-143 Inhibits interleukin-13-induced Inflammatory Cytokine and Mucus Production in Nasal Epithelial Cells From Allergic Rhinitis Patients by Targeting IL13Ralpha1. Biochem Biophys Res Commun (2015) 457:58–64. doi: 10.1016/j.bbrc.2014.12.058
96. Chavan SS, Tracey KJ. Essential Neuroscience in Immunology. J Immunol (2017) 198:3389–97. doi: 10.4049/jimmunol.1601613
97. Chesné J, Cardoso V, Veiga-Fernandes H. Neuro-Immune Regulation of Mucosal Physiology. Mucosal Immunol (2019) 12:10–20. doi: 10.1038/s41385-018-0063-y
98. Sticlaru L, Stăniceanu F, Cioplea M, Nichita L, Bastian A, Micu G, et al. Neuroimmune Cross-Talk in Helicobacter Pylori Infection: Analysis of Substance P and Vasoactive Intestinal Peptide Expression in Gastric Enteric Nervous System. J Immunoassay Immunochem (2018) 39:660–71. doi: 10.1080/15321819.2018.1529683
99. Veiga-Fernandes H, Mucida D. Neuro-Immune Interactions at Barrier Surfaces. Cell (2016) 165:801–11. doi: 10.1016/j.cell.2016.04.041
100. López-Requena A, Boonen B, Van Gerven L, Hellings PW, Alpizar YA, Talavera K. 15 Roles of Neuronal Trp. Neurobiol TRP Channels (2017) 277.
101. Breiteneder H, Diamant Z, Eiwegger T, Fokkens WJ, Traidl-Hoffmann C, Nadeau K, et al. Future Research Trends in Understanding the Mechanisms Underlying Allergic Diseases for Improved Patient Care. Allergy (2019) 74:2293–311. doi: 10.1111/all.13851
102. Feng CH, Miller MD, Simon RA. The United Allergic Airway: Connections Between Allergic Rhinitis, Asthma, and Chronic Sinusitis. Am J Rhinol Allergy (2012) 26:187–90. doi: 10.2500/ajra.2012.26.3762
103. Mosimann BL, White MV, Hohman RJ, Goldrich MS, Kaulbach HC, Kaliner MA, et al. Calcitonin Gene-Related Peptide, and Vasoactive Intestinal Peptide Increase in Nasal Secretions After Allergen Challenge in Atopic Patients. J Allergy Clin Immunol (1993) 92:95–104. doi: 10.1016/0091-6749(93)90043-f
104. Van Gerven L, Steelant B, Hellings P. Nasal Hyperreactivity in Rhinitis: A Diagnostic and Therapeutic Challenge. Allergy (2018) 73:1784–91. doi: 10.1111/all.13453
105. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour R-EE, Nyman J, Dionne D, et al. The Neuropeptide NMU Amplifies ILC2-driven Allergic Lung Inflammation. Nature (2017) 549:351–6. doi: 10.1038/nature24029
106. Chen J, Larson ED, Anderson CB, Agarwal P, Frank DN, Kinnamon SC, et al. Expression of Bitter Taste Receptors and Solitary Chemosensory Cell Markers in the Human Sinonasal Cavity. Chem Senses (2019) 44:483–95. doi: 10.1093/chemse/bjz042
107. Kohanski MA, Workman AD, Patel NN, Hung LY, Shtraks JP, Chen B, et al. Solitary Chemosensory Cells are a Primary Epithelial Source of IL-25 in Patients With Chronic Rhinosinusitis With Nasal Polyps. J Allergy Clin Immunol (2018) 142:460–9 e7. doi: 10.1016/j.jaci.2018.03.019
108. Schneider C, O’Leary CE, Locksley RM. Regulation of Immune Responses by Tuft Cells. Nat Rev Immunol (2019) 19:584–93. doi: 10.1038/s41577-019-0176-x
109. Von Moltke J, Ji M, Liang H-E, Locksley RM. Tuft-Cell-Derived IL-25 Regulates an Intestinal ILC2–epithelial Response Circuit. Nature (2016) 529:221–5. doi: 10.1038/nature16161
110. Finger TE, Böttger B, Hansen A, Anderson KT, Alimohammadi H, Silver WL. Solitary Chemoreceptor Cells in the Nasal Cavity Serve as Sentinels of Respiration. Proc Natl Acad Sci USA (2003) 100:8981–6. doi: 10.1073/pnas.1531172100
111. Kojima T, Go M, Takano K, Kurose M, Ohkuni T, Koizumi J, et al. Regulation of Tight Junctions in Upper Airway Epithelium. BioMed Res Int (2013) 2013:947072. doi: 10.1155/2013/947072
112. Akazawa Y, Yuki T, Yoshida H, Sugiyama Y, Inoue S. Activation of TRPV4 Strengthens the Tight-Junction Barrier in Human Epidermal Keratinocytes. Skin Pharmacol Physiol (2013) 26:15–21. doi: 10.1159/000343173
113. Janssen DA, Jansen CJ, Hafmans TG, Verhaegh GW, Hoenderop JG, Heesakkers JP, et al. TRPV4 Channels in the Human Urogenital Tract Play a Role in Cell Junction Formation and Epithelial Barrier. Acta Physiol (Oxf) (2016) 218:38–48. doi: 10.1111/apha.12701
114. Millqvist E. TRP Channels and Temperature in Airway Disease—Clinical Significance. Temperature (Austin) (2015) 2:172–7. doi: 10.1080/23328940.2015.1012979
115. Backaert W, Steelant B, Hellings PW, Talavera K, Van Gerven L. A TRiP Through the Roles of Transient Receptor Potential Cation Channels in Type 2 Upper Airway Inflammation. Curr Allergy Asthma Rep (2021) 21:20. doi: 10.1007/s11882-020-00981-x
116. Alenmyr L, Högestätt E, Zygmunt P, Greiff L. TRPV1-Mediated Itch in Seasonal Allergic Rhinitis. Allergy (2009) 64:807–10. doi: 10.1111/j.1398-9995.2009.01937.x
117. Kowalski M, Dietrich-miłobędzki A, Majkowska-Wojciechowska B, Jarzębska M. Nasal Reactivity to Capsaicin in Patients With Seasonal Allergic Rhinitis During and After the Pollen Season. Allergy (1999) 54:804–10. doi: 10.1034/j.1398-9995.1999.00897.x
118. Xu R, Li Q, Zhou J, Zhou X-D, Perelman JM, Kolosov VP. The Degradation of Airway Tight Junction Protein Under Acidic Conditions is Probably Mediated by Transient Receptor Potential Vanilloid 1 Receptor. Biosci Rep (2013) 33. doi: 10.1042/BSR20130087
119. Lee K, Byun J, Kim B, Yeon J, Tai J, Lee SH, et al. Trpv4-Mediated Epithelial Junction Disruption in Allergic Rhinitis Triggered by House Dust Mites. Am J Rhinol Allergy (2020) 1945892420964169. doi: 10.1177/1945892420964169
120. Moheimani F, Hsu AC, Reid AT, Williams T, Kicic A, Stick SM, et al. The Genetic and Epigenetic Landscapes of the Epithelium in Asthma. Respir Res (2016) 17:119. doi: 10.1186/s12931-016-0434-4
121. Loxham M, Davies DE, Blume C. Epithelial Function and Dysfunction in Asthma. Clin Exp Allergy (2014) 44:1299–313. doi: 10.1111/cea.12309
122. Yang J, Zhong W, Xue K, Wang Z. Epigenetic Changes: An Emerging Potential Pharmacological Target in Allergic Rhinitis. Int Immunopharmacol (2019) 71:76–83. doi: 10.1016/j.intimp.2019.03.004
123. Akhtari M, Mahmoudi M. Prognostic Epigenetics: Chapter 6 - Epigenetic Biomarkers of Asthma and Allergic Disorders. Amsterdam: Elsevier (2019) p. 139–69.
124. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. MicroRNAs in Apoptosis, Autophagy and Necroptosis. Oncotarget (2015) 6:8474. doi: 10.18632/oncotarget.3523
125. Panganiban RP, Wang Y, Howrylak J, Chinchilli VM, Craig TJ, August A, et al. Circulating microRNAs as Biomarkers in Patients With Allergic Rhinitis and Asthma. J Allergy Clin Immunol (2016) 137:1423–32. doi: 10.1016/j.jaci.2016.01.029
126. Jiang J, Liu JQ, Li J, Li M, Chen HB, Yan H, et al. Trek1 Contributes to Maintaining Nasal Epithelial Barrier Integrity. Sci Rep (2015) 5:9191. doi: 10.1038/srep09191
127. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone Deacetylases as Regulators of Inflammation and Immunity. Trends Immunol (2011) 32:335–43. doi: 10.1016/j.it.2011.04.001
128. Wawrzyniak P, Wawrzyniak M, Wanke K, Sokolowska M, Bendelja K, Ruckert B, et al. Regulation of Bronchial Epithelial Barrier Integrity by Type 2 Cytokines and Histone Deacetylases in Asthmatic Patients. J Allergy Clin Immunol (2017) 139:93–103. doi: 10.1016/j.jaci.2016.03.050
129. Kaneko Y, Kohno T, Kakuki T, Takano KI, Ogasawara N, Miyata R, et al. The Role of Transcriptional Factor p63 in Regulation of Epithelial Barrier and Ciliogenesis of Human Nasal Epithelial Cells. Sci Rep (2017) 7:10935. doi: 10.1038/s41598-017-11481-w
130. Larsen SB, Cowley CJ, Fuchs E. Epithelial Cells: Liaisons of Immunity. J Allergy Clin Immunol (2020) 62:45–53. doi: 10.1016/j.coi.2019.11.004
131. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent Enhancers Activated by Stimulation in Differentiated Cells. Cell (2013) 152:157–71. doi: 10.1016/j.cell.2012.12.018
132. Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, et al. Inflammatory Memory Sensitizes Skin Epithelial Stem Cells to Tissue Damage. Nature (2017) 550:475–80. doi: 10.1038/nature24271
133. Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic Inflammatory Memory in Human Respiratory Epithelial Progenitor Cells. Nature (2018) 560:649–54. doi: 10.1038/s41586-018-0449-8
134. Takamura S. Niches for the Long-Term Maintenance of Tissue-Resident Memory T Cells. Front Immunol (2018) 9:1214. doi: 10.3389/fimmu.2018.01214
135. Shenoy AT, Wasserman GA, Arafa EI, Wooten AK, Smith NM, Martin IM, et al. Lung CD4+ Resident Memory T Cells Remodel Epithelial Responses to Accelerate Neutrophil Recruitment During Pneumonia. Mucosal Immunol (2020) 13:334–43. doi: 10.1038/s41385-019-0229-2
136. Takamura S, Kato S, Motozono C, Shimaoka T, Ueha S, Matsuo K, et al. Interstitial-Resident Memory CD8(+) T Cells Sustain Frontline Epithelial Memory in the Lung. J Exp Med (2019) 216:2736–47. doi: 10.1084/jem.20190557
Keywords: allergic rhinitis, tight junction, TSLP, IL-25, IL-33, innate lymphoid cells, Th2 cytokines, epigenetic
Citation: Nur Husna SM, Tan H-TT, Md Shukri N, Mohd Ashari NS and Wong KK (2021) Nasal Epithelial Barrier Integrity and Tight Junctions Disruption in Allergic Rhinitis: Overview and Pathogenic Insights. Front. Immunol. 12:663626. doi: 10.3389/fimmu.2021.663626
Received: 03 February 2021; Accepted: 22 April 2021;
Published: 21 May 2021.
Edited by:
Tomohiro Yoshimoto, Hyogo College of Medicine, JapanReviewed by:
Carmen Rondon, Regional University Hospital of Malaga, SpainBrecht Steelant, KU Leuven, Belgium
Copyright © 2021 Nur Husna, Tan, Md Shukri, Mohd Ashari and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kah Keng Wong, a2Foa2VuZ0B1c20ubXk=