 Tamiris Lameira Bittencourt1
Tamiris Lameira Bittencourt1 Rhana Berto da Silva Prata1
Rhana Berto da Silva Prata1 Bruno Jorge de Andrade Silva2
Bruno Jorge de Andrade Silva2 Mayara Garcia de Mattos Barbosa3
Mayara Garcia de Mattos Barbosa3 Margareth Pretti Dalcolmo4
Margareth Pretti Dalcolmo4 Roberta Olmo Pinheiro1*
Roberta Olmo Pinheiro1*- 1Leprosy Laboratory, Oswaldo Cruz Institute, Oswaldo Cruz Foundation (Fiocruz), Rio de Janeiro, Brazil
- 2Division of Dermatology, David Geffen School of Medicine, Los Angeles, CA, United States
- 3Department of Surgery, University of Michigan, Ann Arbor, MI, United States
- 4Helio Fraga Reference Center, Sergio Arouca National School of Public Health, Fiocruz, Rio de Janeiro, Brazil
Pathogenic mycobacteria species may subvert the innate immune mechanisms and can modulate the activation of cells that cause disease in the skin. Cutaneous mycobacterial infection may present different clinical presentations and it is associated with stigma, deformity, and disability. The understanding of the immunopathogenic mechanisms related to mycobacterial infection in human skin is of pivotal importance to identify targets for new therapeutic strategies. The occurrence of reactional episodes and relapse in leprosy patients, the emergence of resistant mycobacteria strains, and the absence of effective drugs to treat mycobacterial cutaneous infection increased the interest in the development of therapies based on repurposed drugs against mycobacteria. The mechanism of action of many of these therapies evaluated is linked to the activation of autophagy. Autophagy is an evolutionary conserved lysosomal degradation pathway that has been associated with the control of the mycobacterial bacillary load. Here, we review the role of autophagy in the pathogenesis of cutaneous mycobacterial infection and discuss the perspectives of autophagy as a target for drug development and repurposing against cutaneous mycobacterial infection.
Introduction
Pathogenic mycobacteria species subvert the innate immune system barriers and modulate the activation of phagocytes to cause disease not only in the respiratory tract but also in soft tissues and skin, sometimes resulting in disseminated infection (1). Cutaneous mycobacterial infections may cause different clinical manifestations, such as cutaneous manifestations of Mycobacterium tuberculosis (M. tuberculosis) infection, Buruli ulcer caused by M. ulcerans and other related slowly growing mycobacteria, leprosy caused by M. leprae and M. lepromatosis, and cutaneous infections caused by rapidly growing mycobacteria such as M. abscessus subsp. abscessus, M. abscessus subsp. bolletti, M. abscessus subsp. massiliense, M. chelonae and M. fortuitum (1–9). Among patients with advanced immunosuppression, M. avium-intracellulare complex, the M. haemophilum, and M. kansasii may cause cutaneous or disseminated disease. Mycobacterial infections of the skin and subcutaneous tissue are associated with important stigma, deformity, and disability. The treatment for cutaneous mycobacterial infections depends on the specific pathogen, whereas for rapidly growing mycobacteria, the official treatment guidelines recommend carrying out susceptibility tests for antibacterial drugs of different classes (10, 11). Management often includes use of multiple antibiotics for several months (12). Treatment options for cutaneous tuberculosis follow the same recommendations for the treatment of other forms of TB, being limited to conventional oral therapy and surgical intervention for severe forms, such as lupus vulgaris (13, 14). The therapeutic regimen is based on the combination of isoniazid, rifampicin, pyrazinamide, ethambutol and streptomycin according to the needs of each individual. In most cases, skin manifestations result from hematogenous dissemination or are a direct extension from the focus of the infection (14, 15). In addition, treatment of leprosy is performed with multidrug therapy (MDT) and comprises 6 or 12 doses, depending on the clinical form. There is not a consensus for the treatment of cutaneous infections caused by non-tuberculous mycobacteria. Recently, much effort has been made to develop more effective therapies by modulating host responses to mycobacteria (i.e., host-directed therapy).
After recognition by skin cells, mycobacteria may use a wide range of strategies to escape the microbicidal activity of skin host cells. Some of these immune escape mechanisms are the inhibition of the maturation of phagolysosomes, inhibition of the acidification of phagolysosomes, bacterial escape to reside in the cytosol, modulation of host cell metabolism, inhibition of oxidative stress, and inhibition of apoptosis and autophagy associated with increased type 1 interferon (IFN) expression and inflammasome activation (16–23).
Autophagy is an intracellular catabolic process that may contribute to the removal of invading pathogens via a lysosomal degradation pathway. The activation of autophagy by diverse drugs or agents may represent a promising treatment strategy against mycobacterial diseases. In this review, we discuss the current knowledge of, advances and perspectives on new therapeutic strategies targeting autophagy against mycobacterial infections in the skin.
Overview of Autophagy Machinery on Skin Cells
Autophagy is a homeostatic mechanism highly conserved evolutionarily and dependent on the lysosome action (24). It is responsible for the cellular catabolism of dysfunctional organelles, components of the cytoplasm and, more recently, invading pathogens, thus determining the maintenance of homeostasis and adaptation of the cell to stress (25, 26). Autophagy has been described as having a primary role in physiological cellular processes such as development and growth, in the senescence process, and immune defense (25, 27–29). Based on the way the autophagy target is taken to the lysosome, its final destination of degradation, autophagy was didactically classified into three forms: macroautophagy, microautophagy, and chaperone-mediated autophagy. In this review, we will exclusively address the action and manipulation of the macroautophagy pathway.
Only a small amount of research has considered the impact of autophagy on the pathogenesis of skin diseases, including diseases caused by mycobacteria. Skin is the largest organ of the body and it is not only the first line of defense against numerous insults but it is also the site whereas some infectious, including mycobacterial diseases, may manifest.
Autophagy is considered an effector tool of the immune system since it is a relevant pathway of elimination and recognition of pathogens by the immune system (30). As well to cellular homeostasis, autophagy works to eliminate intracellular pathogens, including some pathogens associated with skin diseases, such as Streptococcus pyogenes from group A (31, 32), Staphylococcus aureus (33, 34), M. leprae (35, 36), M. marinum (37, 38), and M. tuberculosis (39–42). Through a process called xenophagy, which plays a principal role in innate immune defense, intracellular pathogens are directed to the autophagosome and then to the lysosomal degradation pathway (43, 44). Xenophagy is the process of eliminating intracellular pathogens through autophagic machinery, being a unique type of macroautophagy/selective autophagy that targets invasive pathogens, being an important defense mechanism against infectious diseases (45, 46).
Few studies have focused on deciphering autophagy machinery in skin cells, such as: keratinocytes, skin fibroblasts, melanocytes, Langerhans cells, dendritic cells, mast cells, neutrophils, NK and B cells. The current knowledge regarding skin cell autophagy during mycobacterial diseases is based mainly in studies with cell lineage and dermal macrophages.
Briefly, after pathogen recognition by host cells, the first step is the formation of the isolation membrane, which starts to grow and expand in size until sequestration and the surrounding of the target and finally closure to form the autophagosome. Subsequently, autophagosomes fuse with lysosomes to generate autolysosomes through elimination and recycling the sequestered charges via the lysosomal proteases (Figure 1) (28). A large number of proteins have been identified as highly relevant in different stages of control and action in autophagic flow. Several cell types have autophagy as an effector mechanism for homeostatic/immune functions as skin cells like keratinocytes and macrophages (Figure 1) (47).
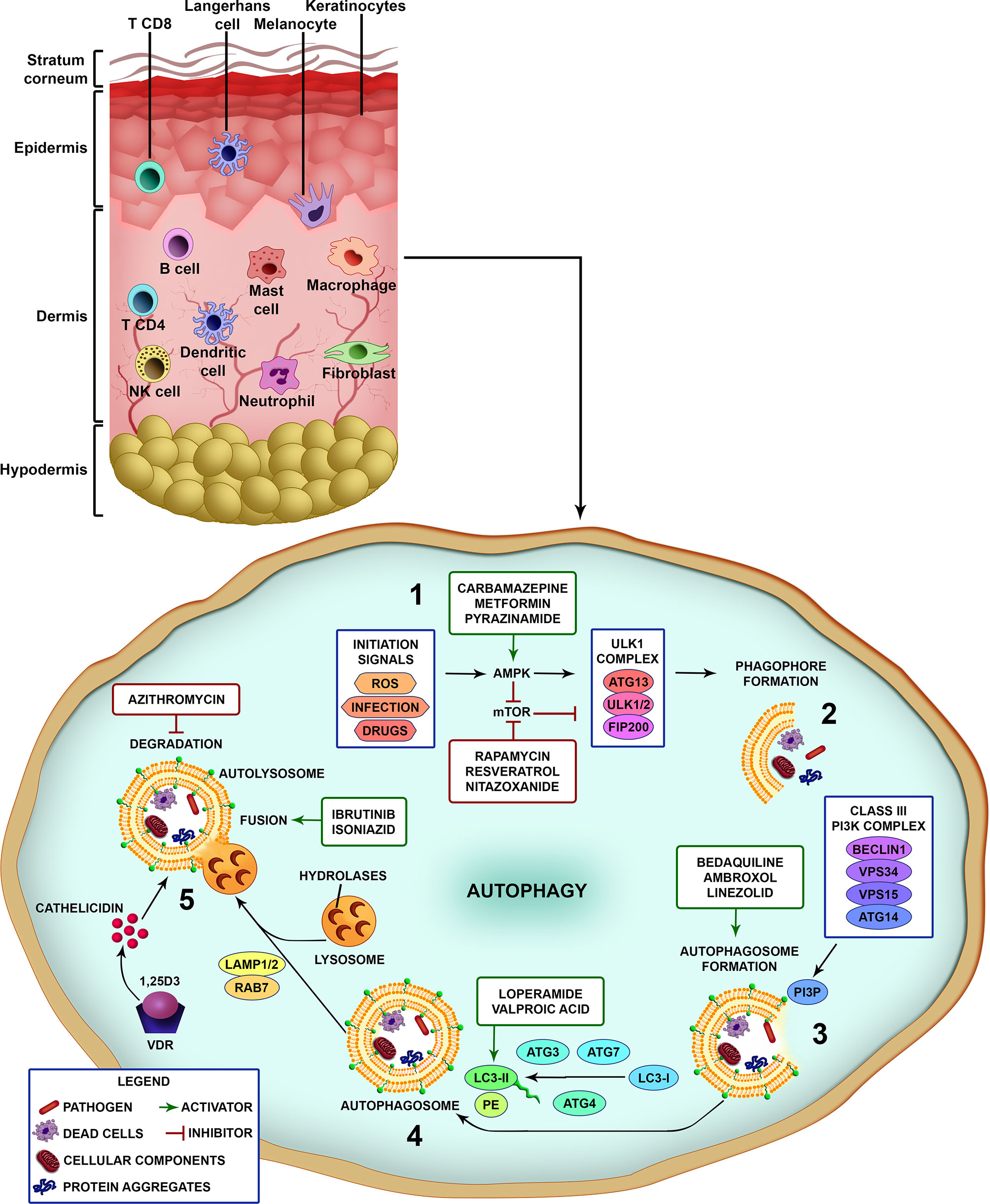
Figure 1 Different steps of the autophagic pathway targeted by autophagy-modulating drugs. A schematic view of the different cell types populating the skin. Vertebrate skin is comprised of two major compartments: the epidermis and the dermis. The superficial part of the epidermis, known as the stratum corneum, is composed of dead keratinocytes and acts as a barrier. The epidermis is composed mainly of keratinocytes with few melanocytes. The major immune cells in this compartment include Langerhans cells (LCs) and CD8 T-cells. The dermis is composed of fibroblasts, NK cells, T-cells (CD4 αβ, and γδ), B cells, dermal dendritic cells, macrophages, mast cells, and neutrophils (non-exhaustive list). The knowledge of skin cell autophagy is mainly based in studies with dermal macrophages. Briefly, (1) autophagy is inhibited by mTOR and activated by AMPK. mTOR is inhibited by the autophagy-initiation signals as metabolic stress, ROS, infection and drugs, and leads to the activation of AMPK. After AMPK activation, the ULK1 complex (ATG13, ULK1/2, FIP200) initiates the phagophore formation (2), involving the targets (pathogens, dead cells, cellular components and organelles, protein aggregates), which in turn activates the Class III PI3K complex (Beclin 1, VPS34, VPS15, ATG14) (3). This complex completes the autophagosome maturation and elongation by forming PI3P in the omegasome membrane and recruiting downstream ubiquitin-like conjugation systems that convert LC3-I to LC3-II (4). Fully formed autophagosomes then fuse with lysosomes (autolysosomes), degrade the sequestered cargo via lysosomal hydrolases and recycle macromolecule components (5). Several drugs can interfere with the autophagic pathway by inhibiting or activating different parts of the process (see also Table 1). Drugs as rapamycin, resveratrol and nitazoxanide, that inhibit mTOR, or carbamazepine, metformin and pyrazinamide, that activate AMPK, induce autophagy. Bedaquiline, ambroxol and linezolid increase the formation of autophagosomes. Loperamide and valproic acid increase the colocalization of LC3-decorated autophagosomes with M. tuberculosis. Ibrutinib and isoniazid facilitate the fusion of phagosome and lysosome. Vitamin D3 (1,25D3) induces the expression of antimicrobial peptides as cathelicidin and upregulates the expression of Beclin 1 and ATG5, that are pivotal for the autophagosome formation. On the other hand, azithromycin was demonstrated to inhibit the acidification of the autolysosome impairing M. abscessus degradation.
A wide variety of signals regulates the activation of autophagy. The induction of autophagy can occur through the recognition of microbial factors that are ubiquitinated and recognized by autophagy cargo adaptor proteins (these include p62 (sequestosome 1), NBR1 (neighbor of BRCA1 gene 1 protein), NDP52 (calcium binding and coiled-coil domain 2), optineurin and galectin) or can occur by the production of reactive oxygen radicals and IFN-γ-mediated proteolysis, and autophagosome formation (43, 48–52). The autophagy pathway may be negatively regulated by PI3K (phosphoinositide 3-kinase)/Akt (protein kinase B)/mTOR (target of rapamycin in mammals) signalling (53). In contrast, the mitogen-activated protein kinase pathway (MAPK) can induce autophagy (54, 55).
Autophagy as an Innate Immune Mechanism Against Mycobacterial Diseases
There is a strong relationship between autophagy signals and pattern recognition receptors, such as TLR (Toll-Like Receptors) that include TLR3, TLR4, TLR5, TLR6, TLR9, and the heterodimers TLR1/2, TLR7/8 that are capable of activating autophagy in macrophages, dendritic cells, and neutrophils (56–58). This activation occurs via signaling of the adaptor proteins MyD88 (myeloid differentiation factor 88) and TRIF (TIR-domain-containing adapter-inducing interferon-β). Xu and colleagues (59) demonstrated that after the stimulation of TLR4, positive LC3 (microtubule-associated protein 1A/1B-light chain 3) aggregates form in the macrophage cytoplasm and increase mycobacterial elimination through autophagy. Interestingly, for the LC3-aggregates induction, via TLR4 induction, it is necessary to activate the protein TRIF, as well as other proteins like RIP1 (receptor-interacting protein 1) and p38 for autophagic induction (56, 59). TLR4 acts as a pro-autophagic receptor in TRIF-dependent pathways. TLR4 induces the production of TNF (tumor necrosis factor) by a mechanism that is mediated both by reactive oxygen species (ROS) and nitrogen intermediates (i.e. nitric oxide), and by p38 and MAPK and the inhibition of these components may lead to total autophagy inactivity (60–62). Studies have shown that in LPS (lipopolysaccharide)-TLR4-mediated autophagy, activation of the transcription factor Nrf2 (nuclear factor erythroid 2–related factor 2) occurs, which leads to increased p62 transcription and formation of aggresome-like induced structures (ALIS) with subsequent autophagic degradation (63, 64), showing the ability of this receptor to link innate immunity with cellular oxidative response or adaptive immunity.
It is known that TLR receptors are of great importance for the activation of dendritic cells (DCs) and their subsequent maturation, some of these receptors such as TLR4 and TLR2 are already described as inducing an innate response against M. tuberculosis (65–67). Khan and colleagues (68) observed that the co-stimulus of CD40 and TLR4 leads to the production of pro-inflammatory cytokines such as IL-6, IL-12 and TNF, autophagy and death of mycobacteria. Interestingly, when they evaluated this co-stimulus as an adjunct to anti-TB therapy, they observed an increase in vivo and in vitro of the deadly potential of anti-TB drugs. Shin and colleagues (69) showed that stimulation of TLR2/1/CD14 by mycobacterial lipoprotein LpqH can activate antibacterial autophagy by activating vitamin D receptor signaling and inducing cathelicidin. They suggested that the TLR2/1/CD14-Ca2+-AMPK (Adenosine monophosphate-activated protein kinase)-p38 MAPK pathways contribute to cathelicidin-dependent expression, which played an important role in LpqH-induced autophagy. A study comparing the induction of autophagy by different species of mycobacteria found that non-pathogenic mycobacteria, such as M. smegmatis, induce a more robust autophagy response than M. tuberculosis (strain H37Rv) (70). The group observed a decrease in LC3-II protein expression when the TLR2 receptor was blocked, as well as a reduction in the colocalization of LC3 with M. smegmatis ΔpmmB (lipoglycan deficient mutant), suggesting the participation of TLR2 in the activation of autophagy during infection with M. smegmatis (70). M. smegmatis can also be recognized by NOD2 (nucleotide-binding oligomerization domain-containing protein 2) and dectin-2 receptors (71).
In addition to the TLR receptors, another group of innate receptors was the nucleotide-binding oligomerization domain (NLRs). It has already been described that the presence of the NOD2 receptor is capable of synergistically amplify the production of pro-inflammatory cytokines and their bactericidal activity (72). In previous studies, Khan and colleagues (73) have demonstrated that after the induction of both receptors, an increase in the bactericidal capacity of DCs in vitro was observed and they required a much lower dose of the drug to kill M. tuberculosis, in addition, activated DCs induced a more effective T cell response in vivo with an increase in autophagy (73, 74). Since pathogenic mycobacteria can modulate the autophagy machinery in skin cells, we hypothesize that autophagy may be a target for new therapeutic strategies against mycobacterial infections in the skin.
Autophagy-Targeting Therapeutics Upon Mycobacterial Infection
Despite the efficacy of anti-TB treatment based on classic isoniazid and rifampicin, limitations in terms of drug resistance, duration of treatment, associated with the use of a complex treatment regimen (75), made the researchers use another strategy in the treatment of different bacterial disease. Besides, unlike infections caused by M. tuberculosis and M. leprae for which there is a well-established therapeutic regimen, there are no standardized and effective regimens for the treatment of non-tuberculosis mycobacteria (NTMs) (10). A promising strategy in the treatment of infectious diseases is the use of host-directed therapy. It works as an adjuvant therapy, which aims to enhance the main components of the host’s antimycobacterial effector mechanisms (76–79). Several studies on immunity, host-pathogen interactions, and host-directed interventions have shown that the antimycobacterial action of anti-TB drugs (standardized scheme) is associated with the induction of autophagy (40). Thus, several drugs used in the clinical area to treat infectious diseases may have their action through the autophagic process.
We previously showed that xenophagy is a crucial mechanism in the leprosy outcome. A functional autophagy pathway driven by IFN-γ and Beclin 1 in skin lesion macrophages was associated with the self-healing paucibacillary tuberculoid form of the disease, whereas a BCL2 (apoptosis regulator Bcl-2)-mediated block of Beclin 1 autophagy axis was linked to the progressive multibacillary lepromatous pole (35). While macrophages patrol the dermis, the human epidermis is enriched for Langerhans cells (LC). Langerhans cells restricted human immunodeficiency virus (HIV)-1 infection through the capture of viral particles by langerin and subsequent internalization into Birbeck granules and targeting of HIV-1 for destruction in the TRIM5 (tripartite motif-containing protein 5) auto lysosomal pathway (80), which in turn is induced by IFN-γ (81). In M. leprae-infected LC, the antimicrobial activity induced by IFN-γ treatment is achieved through autophagy, which improves the degradation of M. leprae-containing phagolysosomes and fine-tunes LC’s power to present antigens for T cells in a CD1a-restricted manner (82). Thus, IFN-γ therapy or a drug targeting autophagy on skin cells could be favorable to the clinical management of leprosy and other skin-related mycobacteriosis such as fish-tank granuloma (83) and Buruli ulcer (1), as well as outbreak associated postsurgical and tattoo ink infections caused by rapidly growing mycobacteria (2, 4). Indeed, the acid-fast bacilli clearance in the skin of multibacillary leprosy patients is accelerated when multidrug therapy is used along with an intradermal treatment with recombinant human IFN-γ (84).
Cell-based studies in leprosy have predominantly focused on dermal cells such as macrophages, neutrophils and T cells. In the dermis, macrophages are an important cell type that promote Th1-type responses, but there is evidence about the involvement of the epidermis in the development of reactional episodes (85) which are acute inflammatory episodes that can occur before, during or after the release of multidrug therapy, being responsible for the cases of disability caused by the disease (86). The relevance of autophagy as a drug target is not only restricted to the control of M. leprae infection but also to its potential to regulate the exacerbated inflammation associated with leprosy reactional episodes, as autophagy tempers inflammation by hijacking active inflammasomes for destruction (87). The downregulation of autophagy observed in skin lesion macrophages of multibacillary leprosy patients also predicts the reversal reaction onset. This impairment of the autophagic pathway correlates with the activation of NLRP3 (NALP3; NACHT, LRR and PYD domains-containing protein 3) inflammasome and IL-1β production, which drive the inflammatory status found in multibacillary patients when undergoing reversal reaction (36). On the other hand, due to Th2→Th1 shift and increased IFN-γ production, autophagy levels are restored in lepromatous patients when the reversal reaction episode is established, which in turn help to reduce the bacillary load in skin cells (35). Therefore, leprosy lesion skin cells can earn a dual benefit from the use of autophagy as a platform for drug development; both inflammasome and antimicrobial optimal activities can be reached by modulating autophagy to a certain level. However, some bacterial pathogens inhibit autophagosome maturation and promote bacterial replication, such as M. tuberculosis (88, 89). Given the background, Silva and colleagues (35) demonstrated that live but not dead M. leprae can inhibit the autophagic flux in macrophages, which indicates a requirement for an active mycobacterial ESX-1 secretion system.
The ESX-1 secretion system is also involved in the targeting of M. marinum by LC3; however, ubiquitination does not seem to be necessary for this process (83). Legionella pneumophila and Coxiella burnetii also developed strategies to explore or subvert autophagy (88). Kim and colleagues (42) demonstrated that M. abscessus (UC22 – rough variant) induces autophagy and inhibits autophagic flow in murine macrophages. Also as observed, the lipid components of the clinical isolate UC22, which is highly virulent, play a critical role in the formation of the autophagosome. These data suggest that virulent M. abscessus can survive and grow within autophagosomes, preventing autophagosome-lysosome fusion and clearance from cells (42). A study demonstrates the role of lactoferrin, an antimicrobial peptide, in the autophagy of macrophages infected with M. avium. D-lactoferrin inhibits intracellular growth of M. avium and, at the same time, leads to structural changes in infected macrophages leading to increased lysosomal content and increased numbers of autophagic vesicles (90).
P-aminosalicylate, one of oldest drugs used against tuberculosis, inhibits the assimilation of iron (91). Depletion of iron is strongly associated with increased expression and accumulation of regulated in DNA damage and development 1 (REDD1), which inhibits mTOR activation, decrease phosphorylation of Akt and TSC2 (tuberous sclerosis complex 2) (92, 93). Iron depletion was also shown to increase the activation of HIF-1α (hypoxia-inducible factor) and AMPK and induce autophagy (92, 94).
Zinc has been shown to be a positive regulator of autophagy in several different cell types and conditions, increasing the production of ROS, the formation and turnover of autophagosomes and cellular clearance (95–101). Nevertheless, zinc depletion was found to induce non-selective autophagy in yeast to release zinc recycled from zinc-rich proteins (91, 102, 103), demonstrating the key role of autophagy on zinc homeostasis. Zinc chelation was found to arrest autophagy and impair lysosomal acidification (95, 104). Phosphorylation of ERK1/2 is necessary for the regulation of zinc-induced autophagy by either activating the Beclin 1-PI3K complex or by promoting disassembly of mTOR complex but the mechanisms in which zinc modulates autophagy are still not completely understood (95, 99, 105). Uncoupling of autophagy and zinc homeostasis in the airway epithelial cells was demonstrated to be a fundamental mechanism in the pathogenesis of chronic obstructive pulmonary disease (106). In TB, previous studies have shown that zinc levels in the peripheral blood decrease with age and during active disease but are improved after the beginning of treatment with anti-TB drugs (107–111). Oral zinc supplementation in Brazilian children exposed to adults with pulmonary TB was demonstrated to increase the positivity of tuberculin test (PPD) and induration size, decreasing false negative results (112). It is postulated that zinc supplementation could correct asymptomatic zinc deficiencies, improve the effect of autophagy-mediated therapy in TB, as well as giving a booster to immunity (109, 111, 112). There are currently several studies associating autophagy and infection by bacteria, including studies showing the different strategies developed by bacteria to inhibit the host’s autophagic responses (113–117), as well as studies that show that the activation of autophagy by starvation or by treatment with rapamycin restricts bacterial growth and is capable of improving cell resistance to infection (39, 40, 118–120). The therapeutic benefit of pharmacological agents that can modulate autophagy must be considered since a diverse variety of pathogens using autophagic machinery has been described in their favor. It is primary to understand whether the pathogen exploits this pathway as a whole (systemically) or just part of components to increase its intracellular replication and/or survival. Besides, it is necessary to consider whether the drug will act on all autophagic pathways or only on a specific component, which may, or may not, be used to replicate for the pathogen. For example, intracellular Brucella abortus (B. abortus) survives by promoting the formation of vacuoles containing B. abortus, which requires the activity of the autophagy initiation proteins PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3), ULK1 (serine/threonine-protein kinase ULK1), ATG (autophagy-related protein) 14L (Barkor; Beclin 1-associated autophagy-related key regulator), and Beclin 1, but not the autophagy activity stretching proteins ATG16L1, ATG4B, ATG5, ATG7 and LC3-II (121). In this condition, the use of inhibitors of the autophagy protein conjugation systems or inhibitors of autophagosome maturation would not have a protective effect against the survival of this bacterium. Still in this context, it is important to consider those patients who are affected by infections (for example, TB) that can be eliminated if autophagy is regulated positively, but who are co-infected with pathogens that use the autophagic pathway in their favor, such as concomitant infections with the Hepatitis B virus and HIV (122). Under other conditions, the co-infected patient is favored by autophagic activation, as is the case of patients with cystic fibrosis (CF) who are treated with cysteamine. The autophagic stimulus mediated by cysteamine in macrophages of cystic fibrosis (with the CFTRdel506 mutation) patients favors the clearance of Pseudomonas aeruginosa, a bacterium that frequently infects the lungs of CF patients (123). Therefore, it is primary to understand the differences between each stimulus, pathogen, and the type of cell under study so that the use of this route as a target for the development of antimycobacterial drugs can be advanced.
Treatments Inducing Autophagy During Tuberculous Mycobacterial Infection
When autophagy studies were started, the only drug that was able to chronically induce this pathway was rapamycin. There is evidence of its antimycobacterial activity, where it has been observed that it significantly inhibits infection by M. kansasii, M. avium, Bacillus Calmette–Guérin (BCG), and virulent strains of M. tuberculosis (124, 125). However, the adverse effects of rapamycin (which were not associated with autophagy induction) made this drug unattractive for use. Several drugs are capable of inducing autophagy and treating mycobacterial diseases, some examples are summarized in Table 1 and their activities are illustrated in Figure 1.
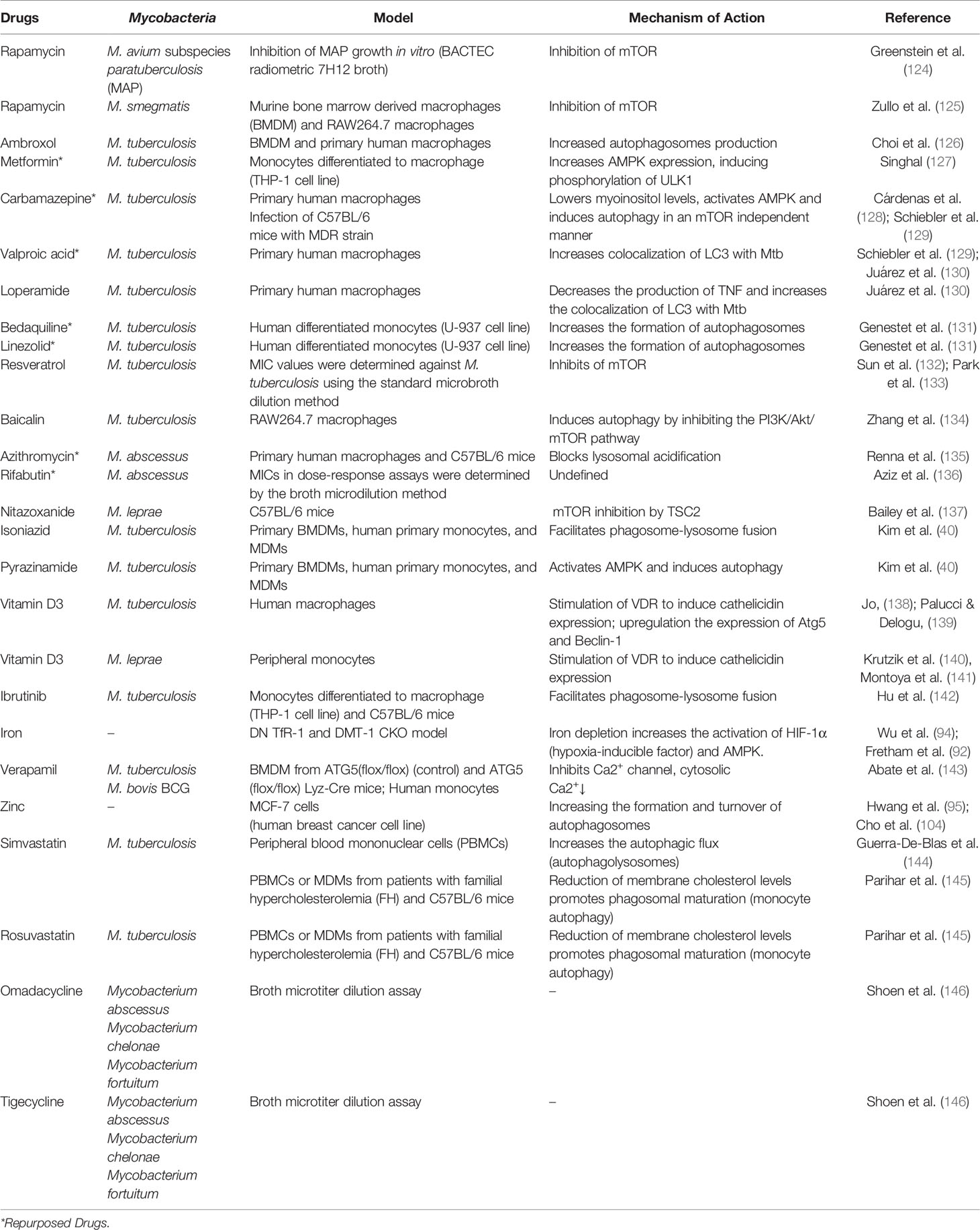
Table 1 Therapeutic strategies of drug repositioning targeting autophagy of host cells against mycobacterial diseases.
Among the various drugs described in the literature with pro-autophagic properties, ambroxol (126), metformin (127), verapamil (143), carbamazepine (128, 129), valproic acid (129, 130), and loperamide (130) are already approved for clinical use in different pathologies. The strategy of using drugs with a known safety profile for new indications related to autophagy is attractive because they do not need to undergo a complete toxicological assessment (18, 147, 148).
Regarding the pro-autophagic property of ambroxol, it has been shown to potentiate the antimicrobial activity of rifampicin in the murine model in trials for TB (126). The antidiabetic drug metformin reduced the intracellular growth of M. tuberculosis in a manner dependent on AMPK. Also, metformin was able to induce reactive mitochondrial oxygen species and facilitate phagosome-lysosome fusion (127). However, a more recent study failed to show the improvement in the bacterial activity of antituberculosis drugs by metformin in the murine model (149). This data makes us reflect on the importance of considering whether the anti-TB drug may or may not alter the pharmacokinetics of the repositioning drug. The use of rifampicin in this more recent study (149) may have altered the pharmacokinetics of metformin. Besides, it is also prudent to pay attention to the differences in the experimental design carried out to assess the effectiveness of the therapy, which can be combined (149) or used alone (monotherapy) (127).
Initial studies that evaluated the effect of verapamil and its analogs on macrophages infected with M. tuberculosis showed that the structural analog KSV21 had an additive effect on the inhibitory antimicrobial activity of Isoniazid and Rifampicin (143). In addition, the antibiotics isoniazid and pyrazinamide, two first-line cocktail drugs used to treat TB, exert their antimycobacterial activity through autophagy (40).
Recently, the impact of linezolid and bedaquiline on the intra-macrophagic behavior of M. tuberculosis has been reported. It was observed that the anti-Mtb effect of these new drugs occurred via activation of autophagy and increased formation of autolysosomes in infected macrophages (131). Bedaquiline induces metabolic stress in M. tuberculosis, which results in the accumulation of NADH (nicotinamide adenine dinucleotide), followed by the generation of ROS (subsequently generating ROS by the bacteria) (150). Although not directly proven, ROS can trigger autophagy activation and be responsible for antibiotic-induced death of M. tuberculosis (151).
Resveratrol has also been studied for its antioxidant effect and its role in inducing autophagy. Regarding the antioxidant effect, resveratrol can increase the activity of antioxidant enzymes and works by eliminating free radicals (152, 153). Resveratrol has inhibitory activity on the mTOR molecule (133, 154). Other studies have shown antibacterial properties, mainly activity against gram-positive bacteria, flavonoid, and resveratrol (132). Still, on drugs capable of stimulating the autophagic death of M. tuberculosis, the anticonvulsant drug carbamazepine was able to induce autophagy in mice infected with the multidrug-resistant M. tuberculosis strain, resulting in a decrease in their bacterial load and improvement in pulmonary pathology (129). It was observed that carbamazepine induces antimicrobial autophagy due to decreased levels of Myoinositol (by blocking myoinositol uptake) into a pathway independent of mTOR. Furthermore, it was seen that this drug also activates AMPK (128). In that same study, the group described the induction of autophagy by the drug valproic acid, another anticonvulsant drug (129), which favored the increase in the co-localization of LC3 with M. tuberculosis, an effect similar to that observed after treatment with anti-diarrhea medication loperamide (130). Unlike carbamazepine, which activates AMPK, the induction of autophagy by baicalin in macrophages infected by M. tuberculosis occurred through inhibition of the PI3K/Akt/mTOR pathway. Additionally, baicalin showed a suppressive effect on the activation of the NLRP3 inflammasome via PI3K/Akt/NF-κB (nuclear factor-κB), as well as reduced the levels of the pro-inflammatory cytokine IL-1β (134). Both the induction of autophagy and the inhibition of NF-κB contribute to limit the activation of the NLRP3 inflammasome. Autophagy can limit the activation of the inflammasome indirectly or directly. Indirectly, it can reduce endogenous stimuli that favor the activation of the inflammasome (155, 156) and can directly inhibit the autophagic degradation of inflammasome components (87, 157).
Fluvastatin is a statin class drug currently used to treat hypercholesteromia and prevent cardiovascular disease, by blocking the enzyme hydroxy-methyl-glutaryl-CoA (HMG-CoA) reductase, which catalyzes a key step in cholesterol synthesis. Fluvastatin was demonstrated to be effective in targeting not only the mycobacteria but also increasing the ability of the host cells to eliminate M. tuberculosis infection (158). Other statins, including simvastatin and rosuvastatin were also demonstrated to control M. tuberculosis infection by promoting phagosomal maturation and autophagy (145).
Some studies demonstrated the protective role of autophagy in excessive inflammation during M. tuberculosis infection (159). Based on these studies, we conclude that autophagy plays an important role in the fight against TB, by direct killing of the pathogen, while also avoiding excessive inflammatory damage. This makes an antimycobacterial agent that has autophagy as a pharmacological target, a promising candidate to assist in therapy directed at the host.
Role of Autophagy in Therapeutic Approaches for NTMs and Skin Diseases
The treatment of nontuberculous mycobacteriosis is not very rewarding. Currently, the proposed therapeutic regimen for infection with NTMs is based on the use of macrolides (clarithromycin or azithromycin), ethambutol, and rifamycins (160). Azithromycin is a potent antibiotic and is often prescribed for prophylaxis and treatment regimens of mycobacterial infections (10). However, one study reported that long-term use of azithromycin by adults with CF increased the risk of infection with M. abscessus. That was observed because the therapeutic dosage of azithromycin impaired autophagic degradation (135). That is, these data emphasize the importance of autophagy in the host’s response to infection by NTMs.
The challenge of treating lung diseases caused by M. abscessus is related to antibiotic resistance, including all first-line drugs for anti-TB treatment (161, 162). Even rifampicin, which has bactericidal activity against M. tuberculosis and M. leprae, has low potency against M. abscessus. Although rifampicin is part of the treatment regimens established for M. kansasii and Mycobacterium avium complex infections, it is not recommended against M. abscessus (163, 164). Recently, rifabutin (of the rifamycin group) was shown, through its bactericidal activity, to be effective against strains of clinical isolates from the three subspecies of the M. abscessus complex (subsp. abscessus, subsp. massiliense, and subsp. bolletii) (136). Recently, the in vitro activity of omadacycline and tigecycline against clinical isolates of M. abscessus, M. chelonae and M. fortuitum was evaluated (146). Omadacycline, a new tetracycline analog, approved for the treatment of acute bacterial skin and skin structure infections (ABSSSI) (165) showed activity against the three clinical isolates (146). There are reports that these microorganisms have been identified in postoperative infections caused by mycobacteria, including the three opportunistic pathogens: M. fortuitum (166), M. abscessus (167) and M. chelonae (168). Postoperative infections have been reported after orthopedic, laparoscopic, ophthalmic procedures and cosmetic operations (mainly liposuction, abdominoplasty, rhinoplasty) (169, 170). M. chelonae can cause localized skin infection after being accidentally inoculated from the environment (pedicure beds, water heaters, and tattoo parlors) (171, 172). In immunocompromised patients, the infection caused by this mycobacterium can manifest itself as a disseminated skin disease. A case report demonstrated M. chelonae skin and soft tissue infection in a patient with chronic lymphocytic leukemia (LLC) who was using ibrutinib, an oral drug, which acts by inhibiting Bruton tyrosine kinase (BTK) for the treatment of various malignant B-cell diseases (173, 174). After 6 months of therapy with ibrutinib, the 85-year-old man developed skin lesions on his arms and legs (175). Fiocari and colleagues (176) showed that ibrutinib promotes an M2 phenotype by modifying the function of macrophages/monocytes in the LLC. Taken together, these results showed that ibrutinib can have detrimental consequences on the microbicidal response in patients treated with ibrutinib. On the other hand, a more current study reported the impact of the drug ibrutinib on the intra-macrophagic behavior of M. tuberculosis. It was observed that the anti-TB effect of this medication occurred via activation of autophagy and facilitates phagosome-lysosome fusion in infected macrophages (142).
Nitazoxanide has also been studied for its role in inducing autophagy. The use of nitazoxanide in C57BL/6 mice infected with M. leprae showed a bactericidal action similar to that of rifampicin, an antibiotic used in the therapeutic regimen against leprosy (137). Based on this study, nitazoxanide (NTZ) may be an effective option for the treatment of leprosy (137).
The epidermis is composed mainly by keratinocytes, which contributes to the defense responses against various stimuli in the environment (177). Numerous findings indicate that autophagy plays an important role in the biology and pathology of keratinocytes (177). It has already been seen that calcipotriol, a vitamin D analog, has the ability to induce autophagy in keratinocytes (178). Analogous vitamin D molecules have been used to treat different skin diseases, such as psoriasis, lamellar ichthyosis and epidermolytic hyperkeratosis (179). The autophagic pathway converges with the vitamin D3-cathelicidin pathway, which is preferably seen in the paucibacillary form of leprosy (140, 141). Vitamin D3 induces autophagy via cathelicidin in macrophages infected with M. tuberculosis, with cathelicidin being required for IFNγ-mediated antimicrobial activity (180, 181). Also, 1,25(OH)2D3-induced LL-37 (C-terminal antimicrobial peptide) enhances the colocalization of mycobacterial phagosomes and autophagosomes (182). Vitamin D3 has been used successfully in the treatment of patients with TB (183). Vitamin D3 could be one of the components for the treatment of leprosy and other chronic infectious diseases in which the cellular immune response is unregulated (184, 185). Vitamin D prevents tissue damage through the negative regulation of perforin, granzyme B and granulisine in cytotoxic T lymphocytes (186).
Many species of mycobacteria that cause skin infections are considered to have a natural ability to acquire resistance to antibiotics and to have a significant reduction in sensitivity to antibiotics, which makes treatment efficacy more difficult by increasing failure rates (187, 188). Thus, using therapies directed at the host, such as those that induce autophagy, to inhibit bacterial cell release and form biofilms or bacterial media can increase the effectiveness of currently available antibiotics, i.e. azithromycin (135) and verapamil (143, 189) already mentioned in the text, as well as, Carvacrol (190–193), Tetracycline (146, 194, 195), Thioridazine (196–199) and, Mefloquine (200, 201).
Conclusion
This review describes the potential of host cell autophagy as a target for the development of new strategies against mycobacterial diseases. There are few studies focusing on skin cell autophagy during mycobacterial infections but in this review we summarized autophagy mechanisms in some cells most relevant to skin mycobacterial diseases. In addition, drug repurposing presents itself as a promising perspective in the control of infections caused by mycobacteria, being used in isolation or complementary to existing treatments. Some challenges still need to be faced, such as the understanding of the mechanisms used by different species of mycobacteria to induce autophagy, the evaluation of host cell autophagy by different clinical isolates, including resistant strains, the impact of a therapy directed at the host cell in cases where there is co-infection and, finally, if the use of a drug in combination with current therapeutic regimens will have a beneficial effect on bacillary load.
Author Contributions
TB, RP, BS, MG, and RP wrote the manuscript. TB, RP, and MG made the table and the figure. RP and MD provided intellectual output in the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We thank CAPES, FAPERJ, and CNPq funding institutions for all their financial support. This study was partially supported by the Coordination for the Improvement of Higher Education Personnel (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES) - Finance Code 001. National Council for Scientific and Technological Development (CNPq) - Finance Code 303834/2017-0. Rio de Janeiro Carlos Chagas Filho Research Foundation (FAPERJ) - Finance Code E-26/010.002231/2019.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Franco-Paredes C, Marcos LA, Henao-Martínez AF, Rodríguez-Morales AJ, Villamil-Gómez WE, Gotuzzo E, et al. Cutaneous Mycobacterial Infections. Clin Microbiol Rev (2018) 32(1):e00069–18. doi: 10.1128/CMR.00069-18
2. Duarte RS, Lourenço MCS, Fonseca LDS, Leão SC, Amorim EDLT, Rocha ILL, et al. Epidemic of Postsurgical Infections Caused by Mycobacterium Massiliense. J Clin Microbiol (2009) 47(7):2149–55. doi: 10.1128/JCM.00027-09
3. Khan FA, Khakoo R. Nontuberculous Mycobacterial Cutaneous Infections: An Updated Review. Cutis (2011) 88(4):194–200.
4. Kennedy BS, Bedard B, Younge M, Tuttle D, Ammerman E, Ricci J, et al. Outbreak of Mycobacterium Chelonae Infection Associated With Tattoo Ink. New Engl J Med (2012) 367(11):1020–4. doi: 10.1056/nejmoa1205114
5. Gervais P, Manuel O, Jaton K, Giulieri S. Skin Infections Due to Rapid-Growing Mycobacteria. Rev Med Suisse (2014) 10(427):931–4.
6. Pinheiro RO, Schmitz V, Silva BJ, de A, Dias AA, de Souza BJ, et al. Innate Immune Responses in Leprosy. Front Immunol (2018) 9:518. doi: 10.3389/fimmu.2018.00518
7. de Macedo CS, Lara FA, Pinheiro RO, Schmitz V, de Berrêdo-Pinho M, Pereira GM, et al. New Insights Into the Pathogenesis of Leprosy: Contribution of Subversion of Host Cell Metabolism to Bacterial Persistence, Disease Progression, and Transmission. F1000Research (2020) 9:F1000 Faculty Rev–70. doi: 10.12688/f1000research.21383.1
8. Johansen MD, Herrmann JL, Kremer L. Non-Tuberculous Mycobacteria and the Rise of Mycobacterium Abscessus. Nat Rev Microbiol (2020) 18(7):392–407. doi: 10.1038/s41579-020-0331-1
9. Costa LL, Veasey JV. Diagnosis of Cutaneous Tuberculosis (Lymph Node Scrofuloderma) Using the Xpert MTB/RIF® Method. Anais Bras Dermatol (2021) 96(1):82–4. doi: 10.1016/j.abd.2020.01.009
10. Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, et al. An Official ATS/IDSA Statement: Diagnosis, Treatment, and Prevention of Nontuberculous Mycobacterial Diseases. Am J Respir Crit Care Med (2007) 175(4):367–416. doi: 10.1164/rccm.200604-571ST
11. Forbes BY, Hall GS, Miller MB, Novak S, Rowlinson MC, Salfinger M, et al. Practice Guidelines for Clinical Microbiology Laboratories: Mycobacteria. Clin Microbiol Rev (2018) 31(2):e00038–17. doi: 10.1128/CMR.00038-17
12. Wang SH, Pancholi P. Mycobacterial Skin and Soft Tissue Infection. Curr Infect Dis Rep (2014) 16(11):438. doi: 10.1007/s11908-014-0438-5
13. Barbagallo J, Tager P, Ingleton R, Hirsch RJ, Weinberg JM. Cutaneous Tuberculosis: Diagnosis and Treatment. Am J Clin Dermatol (2002) 3(5):319–28. doi: 10.2165/00128071-200203050-00004
14. Zyl LV, du Plessis J, Vilijoen J. Cutaneous Tuberculosis Overview and Current Treatment Regimens. Tuberculosis (2015) 95(6):629–38. doi: 10.1016/j.tube.2014.12.006
15. Bravo FG, Gotuzzo E. Cutaneous Tuberculosis. Clinics Dermatol (2007) 25(2):173–80. doi: 10.1016/j.clindermatol.2006.05.005
16. Rahman SA, Singh Y, Kohli S, Ahmad J, Ehtesham NZ, Tyagi AK, et al. Comparative Analyses of Nonpathogenic, Opportunistic, and Totally Pathogenic Mycobacteria Reveal Genomic and Biochemical Variabilities and Highlight the Survival Attributes of Mycobacterium Tuberculosis. mBio (2014) 5(6):e02020. doi: 10.1128/mbio.02020-14
17. Carranza C, Chavez-Galan L. Several Routes to the Same Destination: Inhibition of Phagosome-Lysosome Fusion by Mycobacterium Tuberculosis. Am J Med Sci (2019) 357(3):184–94. doi: 10.1016/j.amjms.2018.12.003
18. Kim YS, Silwal P, Kim SY, Yoshimori T, Jo EK. Autophagy-Activating Strategies to Promote Innate Defense Against Mycobacteria. Exp Mol Med (2019) 51(12):1–10. doi: 10.1038/s12276-019-0290-7
19. Kim JK, Silwal P, Jo EK. Host-Pathogen Dialogues in Autophagy, Apoptosis, and Necrosis During Mycobacterial Infection. Immune Netw (2020) 20(5):e37. doi: 10.4110/in.2020.20.e37
20. Chai Q, Wang L, Liu CH, Ge B. New Insights Into the Evasion of Host Innate Immunity by Mycobacterium Tuberculosis. Cell Mol Immunol (2020) 17(9):901–13. doi: 10.1038/s41423-020-0502-z
21. Ankley L, Thomas S, Olive AJ. Fighting Persistence: How Chronic Infections With Mycobacterium Tuberculosis Evade T Cell-Mediated Clearance and New Strategies to Defeat Them. Infect Immun (2020) 88(7):e00916–19. doi: 10.1128/IAI.00916-19
22. Bernard EM, Fearns A, Bussi C, Santucci P, Peddie CJ, Lai RJ, et al. M. Tuberculosis Infection of Human iPSC-derived Macrophages Reveals Complex Membrane Dynamics During Xenophagy Evasion. J Cell Sci (2020) 134(5):jcs252973. doi: 10.1242/jcs.252973
23. Naeem MA, Ahmad W, Tyagi R, Akram Q, Younus M, Liu X. Stealth Strategies of Mycobacterium Tuberculosis for Immune Evasion. Curr Issues Mol Biol (2021) 41:597–616. doi: 10.21775/cimb.041.597
24. Deter RL, Baudhuin P, De Duve C. Participation of Lysosomes in Cellular Autophagy Induced in Rat Liver by Glucagon. J Cell Biol (1967) 35(2):C11–6. doi: 10.1083/jcb.35.2.c11
25. Klionsky DJ, Emr SD. Autophagy as a Regulated Pathway of Cellular Degradation. Science (2000) 290(5497):1717–21. doi: 10.1126/science.290.5497.1717
26. Wileman T. Autophagy as a Defence Against Intracellular Pathogens. Essays Biochem (2013) 55(1):153–63. doi: 10.1042/BSE0550153
27. Tsukamoto S, Kuma A, Mizushima N. The Role of Autophagy During the Oocyte-to-Embryo Transition. Autophagy (2008) 4(8):1076–8. doi: 10.4161/auto.7065
28. Gottlieb RA, Carreira RS. Mitophagy as a Way of Life. Am J Physiol Cell Physiol (2010) 299(2):C203–210. doi: 10.1152/ajpcell.00097.2010
29. Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, et al. Autophagy in Stem Cells. Autophagy (2013) 9(6):830–49. doi: 10.4161/auto.24132
30. Deretic V, Levine B. Autophagy Balances Inflammation in Innate Immunity. Autophagy (2018) 14(2):243–51. doi: 10.1080/15548627.2017.1402992
31. Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy Defends Cells Against Invading Group A Streptococcus. Science (2004) 306(5698):1037–40. doi: 10.1126/science.1103966
32. Nakajima S, Aikawa C, Nozawa T, Minowa-Nozawa A, Toh H, Nakagawa I. Bcl-Xl Affects Group a Streptococcus-Induced Autophagy Directly, by Inhibiting Fusion Between Autophagosomes and Lysosomes, and Indirectly, by Inhibiting Bacterial Internalization Via Interaction With Beclin 1-UVRAG. PloS One (2017) 12(1):e0170138. doi: 10.1371/journal.pone.0170138
33. Muñoz-Sánchez S, van der Vaart M, Meijer AH. Autophagy and Lc3-Associated Phagocytosis in Zebrafish Models of Bacterial Infections. Cells (2020) 9(11):2372. doi: 10.3390/cells9112372
34. Prajsnar TK, Serba JJ, Dekker BM, Gibson JF, Masud S, Fleming A, et al. The Autophagic Response to Staphylococcus Aureus Provides an Intracellular Niche in Neutrophils. Autophagy (2020) 17(4):888–902. doi: 10.1080/15548627.2020.1739443
35. Silva BJ, de A, Barbosa MG deM, Andrade PR, Ferreira H, Nery JA daC, et al. Autophagy Is an Innate Mechanism Associated With Leprosy Polarization. PloS Pathog (2017a) 13(1):1–29. doi: 10.1371/journal.ppat.1006103
36. de Mattos Barbosa MG, de Andrade Silva BJ, Assis TQ, Prata RB, da S, Ferreira H, et al. Autophagy Impairment is Associated With Increased Inflammasome Activation and Reversal Reaction Development in Multibacillary Leprosy. Front Immunol (2018) 9:1223. doi: 10.3389/fimmu.2018.01223
37. Chen Z, Shao X, Wang C, Hua Mh, Wang Cn, Wang X, et al. Mycobacterium Marinum Infection in Zebrafish and Microglia Imitates the Early Stage of Tuberculous Meningitis. J Mol Neurosci (2018) 64(2):321–30. doi: 10.1007/s12031-018-1026-1
38. Zhang R, Varela M, Vallentgoed W, Forn-Cuni G, van der Vaart M, Meijer AH. The Selective Autophagy Receptors Optineurin and p62 are Both Required for Zebrafish Host Resistance to Mycobacterial Infection. PloS Pathog (2019) 15(2):e1007329. doi: 10.1371/journal.ppat.1007329
39. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a Defense Mechanism Inhibiting BCG and Mycobacterium Tuberculosis Survival in Infected Macrophages. Cell (2004) 119(6):753–66. doi: 10.1016/j.cell.2004.11.038
40. Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, et al. Host Cell Autophagy Activated by Antibiotics Is Required for Their Effective Antimycobacterial Drug Action. Cell Host Microbe (2012) 11(5):457–68. doi: 10.1016/j.chom.2012.03.008
41. Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The Cytosolic Sensor Cgas Detects Mycobacterium Tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe (2015) 17(6):811–9. doi: 10.1016/j.chom.2015.05.004
42. Kim JK, Lee HM, Park KS, Shin DM, Kim TS, Kim YS, et al. MIR144* Inhibits Antimicrobial Responses Against Mycobacterium Tuberculosis in Human Monocytes and Macrophages by Targeting the Autophagy Protein DRAM2. Autophagy (2017b) 13(2):423–41. doi: 10.1080/15548627.2016.1241922
43. Virgin HW, Levine B. Autophagy Genes in Immunity. Nat Immunol (2009) 10(5):461–70. doi: 10.1038/ni.1726
44. Levine B, Mizushima N, Virgin HW. Autophagy in Immunity and Inflammation. Nature (2011) 469(7330):323–35. doi: 10.1038/nature09782
45. Ravenhill BJ, Boyle KB, von Muhlinen N, Ellison CJ, Masson GR, Otten EG, et al. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol Cell (2019) 74(2):320–329.e6. doi: 10.1016/j.molcel.2019.01.041
46. Wen X, Klionsky DJ. How Bacteria can Block Xenophagy: An Insight From Salmonella. Autophagy (2020) 16(2):193–4. doi: 10.1080/15548627.2019.1666580
47. Puleston DJ, Simon AK. Autophagy in the Immune System. Immunology (2013) 141(1):1–8. doi: 10.1111/imm.12165
48. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science (2011) 333(6039):228–33. doi: 10.1126/science.1205405
49. Oh JE, Lee HK. Modulation of Pathogen Recognition by Autophagy. Front Immunol (2012) 3:44. doi: 10.3389/fimmu.2012.00044
50. Paik S, Jo EK. An Interplay Between Autophagy and Immunometabolism for Host Defense Against Mycobacterial Infection. Front Immunol (2020) 11:603951. doi: 10.3389/fimmu.2020.603951
51. Randow F, Youle RJ. Self and Nonself: How Autophagy Targets Mitochondria and Bacteria. in. Cell Host Microbe (2014) 15(4):403–11. doi: 10.1016/j.chom.2014.03.012
52. Da Silva Prata RB, de Mattos Barbosa MG, de Andrade Silva BJ, Oliveira JAP, Bittencourt TL, et al. Macrophages in the Pathogenesis of Leprosy. In: Macrophage activation - Biology and Disease. United Kingdom: Intech Open (2019), p. 1–19. doi: 10.5772/INTECHOPEN.88754 IntechOpen.
53. Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J. The Role of PI3K/AKT/mTOR Pathway in the Modulation of Autophagy and the Clearance of Protein Aggregates in Neurodegeneration. Cell Signal (2014) 26(12):2694–701. doi: 10.1016/j.cellsig.2014.08.019
54. Krishna M, Narang H. The Complexity of Mitogen-Activated Protein Kinases (Mapks) Made Simple. Cell Mol Life Sci (2008) 65(22):3525–44. doi: 10.1007/s00018-008-8170-7
55. Zhou YY, Li Y, Jiang WQ, Zhou LF. MAPK/JNK Signalling: A Potential Autophagy Regulation Pathway. Biosci Rep (2015) 35(3):1–10. doi: 10.1042/BSR20140141
56. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-Like Receptors Control Autophagy. EMBO J (2008) 27(7):1110–21. doi: 10.1038/emboj.2008.31
57. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
58. Pellegrini JM, Sabbione F, Morelli MP, Tateosian NL, Castello FA, Amiano NO, et al. Neutrophil Autophagy During Human Active Tuberculosis is Modulated by SLAMF1. Autophagy (2020) 16:1–10. doi: 10.1080/15548627.2020.1825273
59. Xu Y, Jagannath C, Liu X, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-Like Receptor 4 Is a Sensor for Autophagy Associated With Innate Immunity. Immunity (2007) 27(1):135–44. doi: 10.1016/j.immuni.2007.05.022
60. Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of Reactive Oxygen Species in Toll-Like Receptor 4-Dependent Activation of NF-κb. J Immunol (2004) 172(4):2522–9. doi: 10.4049/jimmunol.172.4.2522
61. Zhou M, Xu W, Wang J, Yan J, Shi Y, Zhang C, et al. Boosting mTOR-dependent Autophagy Via Upstream TLR4-MyD88-MAPK Signalling and Downstream NF-κb Pathway Quenches Intestinal Inflammation and Oxidative Stress Injury. EBioMedicine (2018) 35:345–60. doi: 10.1016/j.ebiom.2018.08.035
62. Hasan A, Akhter N, Al-Roub A, Thomas R, Kochumon S, Wilson A, et al. Tnf-α in Combination With Palmitate Enhances IL-8 Production Via the MyD88- Independent TLR4 Signaling Pathway: Potential Relevance to Metabolic Inflammation. Int J Mol Sci (2019) 20(17):4112. doi: 10.3390/ijms20174112
63. Fujita KI, Srinivasula SM. TLR4-Mediated Autophagy in Macrophages is a p62-dependent Type of Selective Autophagy of Aggresome-Like Induced Structures (ALIS). Autophagy (2011a) 7(5):552–4. doi: 10.4161/auto.7.5.15101
64. Fujita KI, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated Induction of p62 Controls Toll-like receptor-4-driven Aggresome-Like Induced Structure Formation and Autophagic Degradation. Proc Natl Acad Sci USA (2011b) 108(4):1427–32. doi: 10.1073/pnas.1014156108
65. Shinya E, Owaki A, Norose Y, Sato S, Takahashi H. Quick Method of Multimeric Protein Production for Biologically Active Substances Such as Human GM-CSF (Hgm-CSF). Biochem Biophys Res Commun (2009) 386(1):40–4. doi: 10.1016/j.bbrc.2009.05.125
66. Pompei L, Jang S, Zamlynny B, Ravikumar S, McBride A, Hickman SP, et al. Disparity in IL-12 Release in Dendritic Cells and Macrophages in Response to Mycobacterium Tuberculosis is Due to Use of Distinct TLRs. J Immunol (2007) 178(8):5192–9. doi: 10.4049/jimmunol.178.8.5192
67. Drage MG, Pecora ND, Hise AG, Febbraio M, Silverstein RL, Golenbock DT, et al. TLR2 and its Co-Receptors Determine Responses of Macrophages and Dendritic Cells to Lipoproteins of Mycobacterium Tuberculosis. Cell Immunol (2009) 258(1):29–37. doi: 10.1016/j.cellimm.2009.03.008
68. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. Stimulation Through CD40 and TLR-4 is an Effective Host Directed Therapy Against Mycobacterium Tuberculosis. Front Immunol (2016a) 7:386. doi: 10.3389/fimmu.2016.00386
69. Shin DM, Yuk JM, Lee HM, Lee SH, Son JW, Harding CV, et al. Mycobacterial Lipoprotein Activates Autophagy Via TLR2/1/CD14 and a Functional Vitamin D Receptor Signalling. Cell Microbiol (2010) 12(11):1648–65. doi: 10.1111/j.1462-5822.2010.01497
70. Bah A, Lacarrière C, Vergne I. Autophagy-Related Proteins Target Ubiquitin-Free Mycobacterial Compartment to Promote Killing in Macrophages. Front Cell Infect Microbiol (2016) 6:53. doi: 10.3389/fcimb.2016.00053
71. Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim YG, Magalhes JG, et al. Nod1 and Nod2 Direct Autophagy by Recruiting ATG16L1 to the Plasma Membrane At the Site of Bacterial Entry. Nat Immunol (2010) 11(1):55–62. doi: 10.1038/ni.1823
72. Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, De Jong DJ, Langenberg DML, et al. NOD2 and Toll-Like Receptors are Nonredundant Recognition Systems of Mycobacterium Tuberculosis. PloS Pathog (2005) 1(3):279–85. doi: 10.1371/journal.ppat.0010034
73. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. NOD-2 and TLR-4 Signaling Reinforces the Efficacy of Dendritic Cells and Reduces the Dose of TB Drugs Against Mycobacterium Tuberculosis. J Innate Immun (2016b) 8(3):228–42. doi: 10.1159/000439591
74. Khan N, Vidyarthi A, Pahari S, Negi S, Aqdas M, Nadeem S, et al. Signaling Through NOD-2 and TLR-4 Bolsters the T Cell Priming Capability of Dendritic Cells by Inducing Autophagy. Sci Rep (2016c) 6:19084. doi: 10.1038/srep19084
75. Zumla A, Rao M, Parida SK, Keshavjee S, Cassell G, Wallis R, et al. Inflammation and Tuberculosis: Host-directed Therapies. in. J Internal Med (2015) 277(4):373–87. doi: 10.1111/joim.12256
76. Hawn TR, Matheson AI, Maley SN, Vandal O. Host-Directed Therapeutics for Tuberculosis: can We Harness the Host? Microbiol Mol Biol Rev (2013) 77(4):608–27. doi: 10.1128/mmbr.00032-13
77. Zumla A, Rao M, Wallis RS, Kaufmann SHE, Rustomjee R, Mwaba P, et al. Host-directed Therapies for Infectious Diseases: Current Status, Recent Progress, and Future Prospects. Lancet Infect Dis (2016) 16(4):e47–63. doi: 10.1016/S1473-3099(16)00078-5
78. Machelart A, Song OR, Hoffmann E, Brodin P. Host-Directed Therapies Offer Novel Opportunities for the Fight Against Tuberculosis. Drug Discovery Today (2017) 22(8):1250–7. doi: 10.1016/j.drudis.2017.05.005
79. Yang CS. Advancing Host-Directed Therapy for Tuberculosis. Microb Cell (2017) 4(3):105–7. doi: 10.15698/mic2017.03.565
80. Ribeiro CMS, Sarrami-Forooshani R, Setiawan LC, Zijlstra-Willems EM, Van Hamme JL, Tigchelaar W, et al. Receptor Usage Dictates HIV-1 Restriction by Human TRIM5α in Dendritic Cell Subsets. Nature (2016) 540(7633):448–52. doi: 10.1038/nature20567
81. Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, Johansen T, et al. TRIM-Mediated Precision Autophagy Targets Cytoplasmic Regulators of Innate Immunity. J Cell Biol (2015) 210(6):973–89. doi: 10.1083/jcb.201503023
82. Dang AT, Teles RMB, Liu PT, Choi A, Legaspi A, Sarno EN, et al. Autophagy Links Antimicrobial Activity With Antigen Presentation in Langerhans Cells. JCI Insight (2019) 4(8):e126955. doi: 10.1172/jci.insight.126955
83. Lerena MC, Colombo MI. Mycobacterium Marinum Induces a Marked LC3 Recruitment to its Containing Phagosome That Depends on a Functional ESX-1 Secretion System. Cell Microbiol (2011) 13(6):814–35. doi: 10.1111/j.1462-5822.2011.01581.x
84. Sampaio E, Malta A, Sarno E, Kaplan G. Effect of rhuIFN-gamma Treatment in Multibacillary Leprosy Patients. Int J Lepr Other Mycobact Dis (1996) 64(3):268–73.
85. Cogen AL, Walker SL, Roberts CH, Hagge DA, Neupane KD, Khadge S, et al. Human Beta-Defensin 3 Is Up-Regulated in Cutaneous Leprosy Type 1 Reactions. PloS Neglect Trop Dis (2012) 6(11):e1869. doi: 10.1371/journal.pntd.0001869
86. Scollard DM, Adams LB, Gillis TP, Krahenbuhl JL, Truman RW, Williams DL. The Continuing Challenges of Leprosy. Clin Microbiol Rev (2006) 19(2):338–81. doi: 10.1128/CMR.19.2.338-381.2006
87. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of Autophagy by Inflammatory Signals Limits IL-1β Production by Targeting Ubiquitinated Inflammasomes for Destruction. Nat Immunol (2012) 13(3):255–63. doi: 10.1038/ni.2215
88. Campoy E, Colombo MI. Autophagy Subversion by Bacteria. Curr Topics Microbiol Immunol (2009) 335:227–50. doi: 10.1007/978-3-642-00302-8_11
89. Cemma M, Brumell JHH. Interactions of Pathogenic Bacteria With Autophagy Systems. Curr Biol (2012) 22(13):R540–5. doi: 10.1016/j.cub.2012.06.001
90. Silva T, Moreira AC, Nazmi K, Moniz T, Vale N, Rangel M, et al. Lactoferricin Peptides Increase Macrophages’ Capacity to Kill Mycobacterium Avium. mSphere (2017b) 2(4):301–17. doi: 10.1128/mSphere
91. Periyasamy KM, Ranganathan UD, Tripathy SP, Bethunaickan R. Vitamin D – A Host Directed Autophagy Mediated Therapy for Tuberculosis. Mol Immunol (2020) 127:238–44. doi: 10.1016/j.molimm.2020.08.007
92. Fretham SJB, Carlson ES, Georgieff MK. Neuronal-Specific Iron Deficiency Dysregulates Mammalian Target of Rapamycin Signaling During Hippocampal Development in Nonanemic Genetic Mouse Models. J Nutr (2013) 143(3):260–6. doi: 10.3945/jn.112.168617
93. Watson A, Lipina C, McArdle HJ, Taylor PM, Hundal HS. Iron Depletion Suppresses mTORC1-directed Signalling in Intestinal Caco-2 Cells Via Induction of REDD1. Cell Signal (2016) 28(5):412–24. doi: 10.1016/j.cellsig.2016.01.014
94. Wu Y, Li X, Xie W, Jankovic J, Le W, Pan T. Neuroprotection of Deferoxamine on Rotenone-Induced Injury Via Accumulation of HIF-1 Alpha and Induction of Autophagy in SH-SY5Y Cells. Neurochem Int (2010) 57(3):198–205. doi: 10.1016/j.neuint.2010.05.008
95. Hwang JJ, Ha NK, Kim J, Cho D, Mi JK, Kim Y, et al. Zinc (II) Ion Mediates Tamoxifen-Induced Autophagy and Cell Death in MCF-7 Breast Cancer Cell Line. Biometals (2010) 23(6):997–1013. doi: 10.1007/s10534-010-9346-9
96. Lee S, Koh J. Roles of Zinc and Metallothionein-3 in Oxidative Stress-Induced Lysosomal Dysfunction, Cell Death, and Autophagy in Neurons and Astrocytes. Mol Brain (2010) 3(1):30. doi: 10.1186/1756-6606-3-30
97. Kim KW, Speirs CK, Jung DK, Lu B. The Zinc Ionophore PCI-5002 Radiosensitizes Non-Small Cell Lung Cancer Cells by Enhancing Autophagic Cell Death. J Thorac Oncol (2011) 6(9):1542–52. doi: 10.1097/JTO.0b013e3182208fac
98. Hung H, Huang W, Pan C. Dopamine-and Zinc-Induced Autophagosome Formation Facilitates PC12 Cell Survival. Cell Biol Toxicol (2013) 29(6):415–29. doi: 10.1007/s10565-013-9261-2
99. Liuzzi JP, Yoo C. Role of Zinc in the Regulation of Autophagy During Ethanol Exposure in Human Hepatoma Cells. Biol Trace Element Res (2013) 156(1-3):350–6. doi: 10.1007/s12011-013-9816-3
100. Pan R, Timmins GS, Liu W, Liu KJ. Autophagy Mediates Astrocyte Death During Zinc-Potentiated Ischemia–Reperfusion Injury. Biol Trace Element Res (2015) 166(1):89–95. doi: 10.1007/s12011-015-0287-6
101. Popp L, Segatori L. Zinc Oxide Particles Induce Activation of the Lysosome–Autophagy System. ACS Omega (2019) 4(1):573–81. doi: 10.1021/acsomega.8b01497
102. Kawamata T, Horie T, Matsunami M, Sasaki M, Ohsumi Y. Zinc Starvation Induces Autophagy in Yeast. J Biol Chem (2017) 292(20):8520–30. doi: 10.1074/jbc.M116.762948
103. Ding B, Zhong Q. Zinc Deficiency: An Unexpected Trigger for Autophagy. J Biol Chem (2017) 292(20):8531–2. doi: 10.1074/jbc.H116.762948
104. Cho YH, Lee SH, Lee SJ, Kim HN, Koh JY. A Role of Metallothionein-3 in Radiation-Induced Autophagy in Glioma Cells. Sci Rep (2020) 10(1):2015. doi: 10.1038/s41598-020-58237-7
105. Liuzzi JP, Guo L, Yoo C, Stewart TS. Zinc and Autophagy. Biometals (2014) 27(6):1087–96. doi: 10.1007/s10534-014-9773-0
106. Roscioli E, Tran HB, Jersmann H, Nguyen PT, Hopkins E, Lester S, et al. The Uncoupling of Autophagy and Zinc Homeostasis in Airway Epithelial Cells as a Fundamental Contributor to COPD. Am J Physiol Lung Cell Mol Physiol (2017) 313(3):L453–65. doi: 10.1152/ajplung.00083.2017
107. Taneja DP. Observations on Serum Zinc in Patients of Pulmonary Tuberculosis. J Indian Med Assoc (1990) 88(10):280–1.
108. Ray M, Kumar L, Prasad R. Plasma Zinc Status in Indian Childhood Tuberculosis: Impact of Antituberculosis Therapy. Int J Tuberculosis Lung Dis (1998) 2(9):719–25.
109. Karyadi E, Schultink W, Nelwan RH, Gross R, Amin Z, Dolmans WM, et al. Poor Micronutrient Status of Active Pulmonary Tuberculosis Patients in Indonesia. J Nutr (2000) 130(12):2953–8. doi: 10.1093/jn/130.12.2953
110. Koyanagi A, Kuffó D, Gresely L, Shenkin A, Cuevas LE. Relationships Between Serum Concentrations of C-reactive Protein and Micronutrients, in Patients With Tuberculosis. Ann Trop Med Parasitol (2004) 98(4):391–9. doi: 10.1179/000349804225003424
111. Ghulam H, Kadri SM, Manzoor A, Waseem Q, Aatif MS, Khan GQ, et al. Status of Zinc in Pulmonary Tuberculosis. J Infect Dev Ctries (2009) 3(5):365–8. doi: 10.3855/jidc.244
112. Cuevas LE, Almeida LM, Mazunder P, Paixão AC, Silva AM, Maciel L, et al. Effect of Zinc on the Tuberculin Response of Children Exposed to Adults With Smear-Positive Tuberculosis. Ann Trop Paediatr (2002) 22(4):313–9. doi: 10.1179/027249302125001967
113. Yoshikawa Y, Ogawa M, Hain T, Chakraborty T, Sasakawa C. Listeria Monocytogenes ActA is a Key Player in Evading Autophagic Recognition. Autophagy (2009) 5(8):1220–1. doi: 10.4161/auto.5.8.10177
114. Shahnazari S, Namolovan A, Mogridge J, Kim PK, Brumell JH. Bacterial Toxins can Inhibit Host Cell Autophagy Through cAMP Generation. Autophagy (2011) 7(9):957–65. doi: 10.4161/auto.7.9.16435
115. Tattoli I, Sorbara MT, Philpott DJ, Girardin SE. Bacterial Autophagy: The Trigger, the Target and the Timing. Autophagy (2012) 8(12):1848–50. doi: 10.4161/auto.21863
116. Dong N, et al. Structurally Distinct Bacterial TBC-like Gaps Link Arf Gtpase to Rab1 Inactivation to Counteract Host Defenses. Cell (2012) 150(5):1029–41. doi: 10.1016/j.cell.2012.06.050
117. O’Keeffe KM, Wilk MM, Leech JM, Murphy AG, Laabei M, Monk IR, et al. Manipulation of Autophagy in Phagocytes Facilitates Staphylococcus Aureus Bloodstream Infection. Infect Immun (2015) 83(9):3445–57. doi: 10.1128/IAI.00358-15
118. Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in Autophagy Favour Adherent-Invasive Escherichia Coli Persistence Within Macrophages Leading to Increased Pro-Inflammatory Response. Cell Microbiol (2012) 14(6):791–807. doi: 10.1111/j.1462-5822.2012.01768.x
119. Kuo SY, Castoreno AB, Aldrich LN, Lassen KG, Goel G, Dančík V, et al. Small-molecule Enhancers of Autophagy Modulate Cellular Disease Phenotypes Suggested by Human Genetics. Proc Natl Acad Sci USA (2015) 112(31):E4281–7. doi: 10.1073/pnas.1512289112
120. Miao Y, Li G, Zhang X, Xu H, Abraham SN. A TRP Channel Senses Lysosome Neutralization by Pathogens to Trigger Their Expulsion. Cell (2015) 161(6):1306–19. doi: 10.1016/j.cell.2015.05.009
121. Starr T, Child R, Wehrly TD, Hansen B, Hwang S, López-Otin C, et al. Selective Subversion of Autophagy Complexes Facilitates Completion of the Brucella Intracellular Cycle. Cell Host Microbe (2012) 11(1):33–45. doi: 10.1016/j.chom.2011.12.002
122. Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, et al. Subversion of Cellular Autophagy Machinery by Hepatitis B Virus for Viral Envelopment. J Virol (2011) 85(13):6319–33. doi: 10.1128/JVI.02627-10
123. Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, et al. Cysteamine Re-Establishes the Clearance of Pseudomonas Aeruginosa by Macrophages Bearing the Cystic Fibrosis-Relevant F508del-CFTR Mutation. Cell Death Dis (2017) 8(1):e2544. doi: 10.1038/cddis.2016.476
124. Greenstein RJ, Su L, Juste RA, Brown ST. On the Action of Cyclosporine A, Rapamycin and Tacrolimus on M. Avium Including Subspecies Paratuberculosis. PloS One (2008) 3(6):e2496. doi: 10.1371/journal.pone.0002496
125. Zullo AJ, Jurcic Smith KL, Lee S. Mammalian Target of Rapamycin Inhibition and Mycobacterial Survival are Uncoupled in Murine Macrophages. BMC Biochem (2014) 15(1):4. doi: 10.1186/1471-2091-15-4
126. Choi SW, Gu Y, Peters RS, Salgame P, Ellner JJ, Timmins GS, et al. Ambroxol Induces Autophagy and Potentiates Rifampin Antimycobacterial Activity. Antimicrobial Agents Chemother (2018) 62(9):e01019–18. doi: 10.1128/aac.01019-18
127. Singhal A, Jie L, Kumar P, Hong GS, Leow MKS, Paleja B, et al. Metformin as Adjunct Antituberculosis Therapy. Sci Trans Med (2014) 6(263):263ra159. doi: 10.1126/scitranslmed.3009885
128. Cárdenas-Rodríguez N, Coballase-Urrutia E, Rivera-Espinosa L, Romero-Toledo A, Sampieri AI, Ortega-Cuellar D, et al. Modulation of Antioxidant Enzymatic Activities by Certain Antiepileptic Drugs (Valproic Acid, Oxcarbazepine, and Topiramate): Evidence in Humans and Experimental Models. Oxid Med Cell Longevity (2013) 2013:598493. doi: 10.1155/2013/598493
129. Schiebler M, Brown K, Hegyi K, Newton SM, Renna M, Hepburn L, et al. Functional Drug Screening Reveals Anticonvulsants as Enhancers of mTOR-Independent Autophagic Killing of Mycobacterium Tuberculosis Through Inositol Depletion. EMBO Mol Med (2015) 7(2):127–39. doi: 10.15252/emmm.201404137
130. Juárez E, Carranza C, Sánchez G, González M, Chávez J, Sarabia C, et al. Loperamide Restricts Intracellular Growth of Mycobacterium Tuberculosis in Lung Macrophages. Am J Respir Cell Mol Biol (2016) 55(6):837–47. doi: 10.1165/rcmb.2015-0383OC
131. Genestet C, Bernard-Barret F, Hodille E, Ginevra C, Ader F, Goutelle S, et al. Antituberculous Drugs Modulate Bacterial Phagolysosome Avoidance and Autophagy in Mycobacterium Tuberculosis-Infected Macrophages. Tuberculosis (2018) 111:67–70. doi: 10.1016/j.tube.2018.05.014
132. Sun D, Hurdle JG, Lee R, Lee R, Cushman M, Pezzuto JM. Evaluation of Flavonoid and Resveratrol Chemical Libraries Reveals Abyssinone II as a Promising Antibacterial Lead. ChemMedChem (2012) 7(9):1541–5. doi: 10.1002/cmdc.201200253
133. Park D, Jeong H, Lee MN, Koh A, Kwon O, Yang YR, et al. Resveratrol Induces Autophagy by Directly Inhibiting mTOR Through ATP Competition Dohyun Park1. Sci Rep (2016) 23:6–21772. doi: 10.1038/srep21772
134. Zhang Q, Sun J, Wang Y, He W, Wang L, Zheng Y, et al. Antimycobacterial and Anti-Inflammatory Mechanisms of Baicalin Via Induced Autophagy in Macrophages Infected With Mycobacterium Tuberculosis. Front Microbiol (2017) 8:2142. doi: 10.3389/fmicb.2017.02142
135. Renna M, Schaffner C, Brown K, Shang S, Tamayo MH, Hegyi K, et al. Azithromycin Blocks Autophagy and may Predispose Cystic Fibrosis Patients to Mycobacterial Infection. J Clin Invest (2011) 121(9):3554–63. doi: 10.1172/JCI46095
136. Aziz DB, Low JL, Wu ML, Gengenbacher M, Teo JWP, Dartois V, et al. Rifabutin Is Active Against Mycobacterium Abscessus Complex. Antimicrobial Agents Chemother (2017) 61(6):e00155–17. doi: 10.1128/AAC.00155-17
137. Bailey MA, Na H, Duthie MS, Gillis TP, Lahiri R, Parish T. Nitazoxanide is Active Against Mycobacterium Leprae. PloS One (2017) 12(8):e0184107. doi: 10.1371/journal.pone.0184107
138. Jo EK. Innate Immunity to Mycobacteria: Vitamin D and Autophagy. Cell Microbiol (2010) 12(8):1026–35. doi: 10.1111/j.1462-5822.2010.01491.x
139. Palucci I, Delogu G. Host Directed Therapies for Tuberculosis: Futures Strategies for an Ancient Disease. Chemotherapy (2018) 63(3):172–80. doi: 10.1159/000490478
140. Krutzik SR, Hewison M, Liu PT, Robles JA, Stenger S, Adams JS, et al. Il-15 Links Tlr2/1-Induced Macrophage Differentiation to the Vitamin D-Dependent Antimicrobial Pathway. J Immunol (2008) 181(10):7115–20. doi: 10.4049/jimmunol.181.10.7115
141. Montoya D, Cruz D, Teles RMB, Lee DJ, Ochoa MT, Krutzik SR, et al. Divergence of Macrophage Phagocytic and Antimicrobial Programs in Leprosy. Cell Host Microbe (2009) 6(4):343–53. doi: 10.1016/j.chom.2009.09.002
142. Hu Y, Wen Z, Liu S, Cai Y, Guo J, Xu Y, et al. Ibrutinib Suppresses Intracellular Mycobacterium Tuberculosis Growth by Inducing Macrophage Autophagy. J Infect (2020) 80(6):e19–26. doi: 10.1016/j.jinf.2020.03.003
143. Abate G, Ruminiski PG, Kumar M, Singh K, Hamzabegovic F, Hoft DF, et al. New Verapamil Analogs Inhibit Intracellular Mycobacteria Without Affecting the Functions of Mycobacterium-Specific T Cells. Antimicrobial Agents Chemother (2016) 60(3):1216–25. doi: 10.1128/AAC.01567-15
144. Guerra-De-Blas PDC, Bobadilla-Del-Valle M, Sada-Ovalle I, Estrada-Garcia I, Torres-Gonzalez P, Lopez-Saavedra A, et al. Simvastatin Enhances the Immune Response Against Mycobacterium Tuberculosis. Front Microbiol (2019) 10:2097. doi: 10.3389/fmicb.2019.02097 doi: 10:2097.
145. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin Therapy Reduces the Mycobacterium Tuberculosis Burden in Human Macrophages and in Mice by Enhancing Autophagy and Phagosome Maturation. J Infect Dis (2014) 209(5):754–63. doi: 10.1093/infdis/jit550
146. Shoen C, Benaroch D, Sklaney M, Cynamon M. In Vitro Activities of Omadacycline Against Rapidly Growing Mycobacteria. Antimicrobial Agents Chemother (2019) 63(5):e02522–18. doi: 10.1128/AAC.02522-18
147. Sundaramurthy V, Barsacchi R, Samusik N, Marsico G, Gilleron J, Kalaidzidis I, et al. Integration of Chemical and RNAi Multiparametric Profiles Identifies Triggers of Intracellular Mycobacterial Killing. Cell Host Microbe (2013) 13(2):129–42. doi: 10.1016/j.chom.2013.01.008
148. Stanley RE, Ragusa MJ, Hurley JH. The Beginning of the End: How Scaffolds Nucleate Autophagosome Biogenesis. Trends Cell Biol (2014) 24(1):73–81. doi: 10.1016/j.tcb.2013.07.008
149. Dutta NK, Pinn ML, Karakousis PC. Metformin Adjunctive Therapy Does Not Improve the Sterilizing Activity of the First-Line Antitubercular Regimen in Mice. Antimicrobial Agents Chemother (2017) 61(8):e00652–17. doi: 10.1128/AAC.00652-17
150. Bhat SA, Iqbal IK, Kumar A. Imaging the NADH: NAD+ Homeostasis for Understanding the Metabolic Response of Mycobacterium to Physiologically Relevant Stresses. Front Cell Infect Microbiol (2016) 6:145. doi: 10.3389/fcimb.2016.00145
151. Piccaro G, Pietraforte D, Giannoni F, Mustazzolu A, Fattorini L. Rifampin Induces Hydroxyl Radical Formation in Mycobacterium Tuberculosis. Antimicrobial Agents Chemother (2014) 58(12):7527–33. doi: 10.1128/AAC.03169-14
152. Alarcón De La Lastra C, Villegas I. Resveratrol as an Antioxidant and Pro-Oxidant Agent: Mechanisms and Clinical Implications. Biochem Soc Trans (2007) 35(5):1156–60. doi: 10.1042/BST0351156
153. Kuršvietienė L, Stanevičienė I, Mongirdienė A, Bernatonienė J. Multiplicity of Effects and Health Benefits of Resveratrol. Med (Kaunas) (2016) 52(3):148–55. doi: 10.1016/j.medici.2016.03.003
154. Liu M, Wilk SA, Wang A, Zhou L, Wang RH, Ogawa W, et al. Resveratrol Inhibits mTOR Signaling by Promoting the Interaction Between mTOR and DEPTOR. J Biol Chem (2010) 285(47):36387–94. doi: 10.1074/jbc.M110.169284
155. Nakahira K, Haspel JA, Rathinam VAK, Lee SJ, Dolinay T, Lam HC, et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
156. Zhou R, Yazdi AS, Menu P, Tschopp J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature (2011) 469(7329):221–6. doi: 10.1038/nature09663
157. Harris J, Lang T, Thomas JPW, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and Inflammasomes. Mol Immunol (2017) 86:10–5. doi: 10.1016/j.molimm.2017.02.013
158. Battah B, Chemi G, Butini S, Campiani G, Brogi S, Delogu G, et al. A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs With Potential Multi-Targeting Profiles. Molecules (2019) 24(23):4373. doi: 10.3390/molecules24234373
159. Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy Protects Against Active Tuberculosis by Suppressing Bacterial Burden and Inflammation. Proc Natl Acad Sci USA (2012) 109(46):3168–76. doi: 10.1073/pnas.1210500109
160. Griffith DE. Treatment of Mycobacterium Avium Complex (Mac). Semin Respir Crit Care Med (2018) 39(3):351–61. doi: 10.1055/s-0038-1660472
161. Luthra S, Rominski A, Sander P. The Role of Antibiotic-Target-Modifying and Antibiotic-Modifying Enzymes in Mycobacterium Abscessus Drug Resistance. Front Microbiol (2018) 9:2179. doi: 10.3389/fmicb.2018.02179
162. Wu ML, Aziz DB, Dartois V, Dick T. NTM Drug Discovery: Status, Gaps and the Way Forward. Drug Discovery Today (2018) 23(8):1502–19. doi: 10.1016/j.drudis.2018.04.001
163. Chopra S, Matsuyama K, Hutson C, Madrid P. Identification of Antimicrobial Activity Among FDA-approved Drugs for Combating Mycobacterium Abscessus and Mycobacterium Chelonae. J Antimicrob Chemother (2011) 66(7):1533–6. doi: 10.1093/jac/dkr154
164. Pang H, Li G, Zhao X, Liu H, Wan K, Yu P. Drug Susceptibility Testing of 31 Antimicrobial Agents on Rapidly Growing Mycobacteria Isolates From China. BioMed Res Int (2015) 2015:419392. doi: 10.1155/2015/419392
165. O’Riordan W, Green S, Overcash JS, Puljiz I, Metallidis S, Gardovskis J, et al. Omadacycline for Acute Bacterial Skin and Skin-Structure Infections. New Engl J Med (2019) 380(6):528–38. doi: 10.1056/nejmoa1800170
166. Celdrán A, Esteban J, Mañas J, Granizo JJ. Wound Infections Due to Mycobacterium Fortuitum After Polypropylene Mesh Inguinal Hernia Repair. J Hosp Infect (2007) 66(4):374–7. doi: 10.1016/j.jhin.2007.05.006
167. Murillo J, Torres J, Bofill L, Rios-Fabra A, Irausquin E, Istúriz R. Skin and Wound Infection by Rapidly Growing Mycobacteria: An Unexpected Complication of Liposuction and Liposculpture. The Venezuelan Collaborative Infectious and Tropical Diseases Study Group - Pubmed. Arch Dermatol (2000) 136(11):1347–52. doi: 10.1001/archderm.136.11.1347
168. Brickman M, Parsa AA, Parsa FD. Mycobacterium Cheloneae Infection After Breast Augmentation. Aesthet Plast Surg (2005) 29(2):116–8. doi: 10.1007/s00266-004-0023-7
169. Mauriello JA. Atypical Mycobacterial Infection of the Periocular Region After Periocular and Facial Surgery. Ophthalmic Plast Reconstructive Surg (2003) 19(3):182–8. doi: 10.1097/01.IOP.0000064994.09803.CB
170. Gravante G, Caruso R, Araco A, Cervelli V. Infections After Plastic Procedures: Incidences, Etiologies, Risk Factors, and Antibiotic Prophylaxis. Aesthet Plast Surg (2008) 32(2):243–51. doi: 10.1007/s00266-007-9068-8
171. Goldman J, Caron F, De Quatrebarbes J, Pestel-Caron M, Courville P, Doré MX, et al. Infections From Tattooing: Outbreak of Mycobacterium Chelonae in France. BMJ (2010) 341:c5483. doi: 10.1136/bmj.c5483
172. Falkinham JO. Nontuberculous Mycobacteria From Household Plumbing of Patients With Nontuberculous Mycobacteria Disease. Emerg Infect Dis (2011) 17(3):419–24. doi: 10.3201/eid1703.101510
173. Khan M, Gibbons JL, Ferrajoli A. Spotlight on Ibrutinib and its Potential in Frontline Treatment of Chronic Lymphocytic Leukemia. OncoTargets Ther (2017) 10:1909–14. doi: 10.2147/OTT.S98689
174. Tran PN, O’Brien S. The Safety of Bruton’s Tyrosine Kinase Inhibitors for the Treatment of Chronic Lymphocytic Leukemia. Expert Opin Drug Saf (2017) 16(9):1079–88. doi: 10.1080/14740338.2017.1344213
175. Dousa KM, Babiker A, Van Aartsen D, Shah N, Bonomo RA, Johnson JL, et al. Ibrutinib Therapy and Mycobacterium Chelonae Skin and Soft Tissue Infection. Open Forum Infect Dis (2018) 5(7):ofy168. doi: 10.1093/ofid/ofy168
176. Fiorcari S, Maffei R, Audrito V, Martinelli S, Hacken E, Zucchini P, et al. Ibrutinib Modifies the Function of Monocyte/Macrophage Population in Chronic Lymphocytic Leukemia. Oncotarget (2016) 7(40):65968–81. doi: 10.18632/oncotarget.11782
177. Li L, Chen X, Gu H. The Signaling Involving in Autophagy Machinery in Keratinocytes and Therapeutic Approaches for Skin Diseases. Oncotarget (2016) 7(31):50682–97. doi: 10.18632/oncotarget.9330
178. Wang RC, Levine B. Calcipotriol Induces Autophagy in Hela Cells and Keratinocytes. J Invest Dermatol (2011) 131(4):990–3. doi: 10.1038/jid.2010.423
179. Ito K, Koga M, Shibayama Y, Tatematsu S, Nakayama J, Imafuku S. Proactive Treatment With Calcipotriol Reduces Recurrence of Plaque Psoriasis. J Dermatol (2016) 43(4):402–5. doi: 10.1111/1346-8138.13158
180. Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, et al. Vitamin D3 Induces Autophagy in Human Monocytes/Macrophages Via Cathelicidin. Cell Host Microbe (2009) 6(3):231–43. doi: 10.1016/j.chom.2009.08.004
181. Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, et al. Vitamin D is Required for IFN-γ-Mediated Antimicrobial Activity of Human Macrophages. Sci Trans Med (2011) 3(104):104ra102. doi: 10.1126/scitranslmed.3003045
182. Vickers NJ. Animal Communication: When I’m Calling You, Will You Answer Too? Curr Biol (2017) 27(14):R713–5. doi: 10.1016/j.cub.2017.05.064
183. Martineau AR, Honecker FU, Wilkinson RJ, Griffiths CJ. Vitamin D in the Treatment of Pulmonary Tuberculosis. J Steroid Biochem Mol Biol (2007) 103(3–5):793–8. doi: 10.1016/j.jsbmb.2006.12.052
184. Selvaraj P. Vitamin D, Vitamin D Receptor, and Cathelicidin in the Treatment of Tuberculosis. Vitam Horm (2011) 86:307–25. doi: 10.1016/B978-0-12-386960-9.00013-7
185. Liu PT, Wheelwright M, Teles R, Komisopoulou E, Edfeldt K, Ferguson B, et al. MicroRNA-21 Targets the Vitamin D-dependent Antimicrobial Pathway in Leprosy. Nat Med (2012) 18(2):267–73. doi: 10.1038/nm.2584
186. Afsal K, Selvaraj P, Harishankar M. 1, 25-Dihydroxyvitamin D3 Downregulates Cytotoxic Effector Response in Pulmonary Tuberculosis. Int Immunopharmacol (2018) 62:251–60. doi: 10.1016/j.intimp.2018.07.018
187. Sanguinetti M, Ardito F, Fiscarelli E, La Sorda M, D’Argenio P, Ricciotti G, et al. Fatal Pulmonary Infection Due to Multidrug-Resistant Mycobacterium Abscessus in a Patient With Cystic Fibrosis. J Clin Microb (2001) 39(2):816–9. doi: 10.1128/JCM.39.2.816-819.2001
188. Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. Mycobacterium Abscessus: A New Antibiotic Nightmare. J Antimicrob Chemother (2012) 67(4):810–8. doi: 10.1093/jac/dkr578
189. Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel Targets for Huntington’s Disease in an mTOR-independent Autophagy Pathway. Nat Chem Biol (2008) 4(5):295–305. doi: 10.1038/nchembio.79
190. Nowotarska SW, Nowotarski K, Grant IR, Elliott CT, Friedman M, Situ C. Mechanisms of Antimicrobial Action of Cinnamon and Oregano Oils, Cinnamaldehyde, Carvacrol, 2,5-Dihydroxybenzaldehyde, and 2-Hydroxy-5-Methoxybenzaldehyde Against Mycobacterium Avium Subsp. Paratuberculosis (Map). Foods (2017) 6(9):72. doi: 10.3390/foods6090072
191. Potoènjak I, Gobin I, Domitroviæ R. Carvacrol Induces Cytotoxicity in Human Cervical Cancer Cells But Causes Cisplatin Resistance: Involvement of MEK–ERK Activation. Phytother Res (2018) 32(6):1090–7. doi: 10.1002/ptr.6048
192. Spalletta S, Flati V, Toniato E, Di Gregorio J, Marino A, Pierdomenico L, et al. Carvacrol Reduces Adipogenic Differentiation by Modulating Autophagy and ChREBP Expression. PloS One (2018) 13(11):e0206894. doi: 10.1371/journal.pone.0206894
193. Marini E, Di Giulio M, Ginestra G, Magi G, Di Lodovico S, Marino A, et al. Efficacy of Carvacrol Against Resistant Rapidly Growing Mycobacteria in the Planktonic and Biofilm Growth Mode. PloS One (2019) 14(7):e0219038. doi: 10.1371/journal.pone.0219038
194. Brüning A, Brem GJ, Vogel M, Mylonas I. Tetracyclines Cause Cell Stress-Dependent ATF4 Activation and mTOR Inhibition. Exp Cell Res (2014) 320(2):281–9. doi: 10.1016/j.yexcr.2013.11.012
195. Kaushik A, Ammerman NC, Martins O, Parrish NM, Nuermberger EL. In Vitro Activity of New Tetracycline Analogs Omadacycline and Eravacycline Against Drug-Resistant Clinical Isolates of Mycobacterium Abscessus. Antimicrob Agents Chemother (2019) 63(6):e470–19. doi: 10.1128/aac.00470-19
196. Rodrigues L, Wagner D, Viveiros M, Sampaio D, Couto I, Vavra M, et al. Thioridazine and Chlorpromazine Inhibition of Ethidium Bromide Efflux in Mycobacterium Avium and Mycobacterium Smegmatis. J Antimicrob Chemother (2008) 61(5):1076–82. doi: 10.1093/jac/dkn070
197. Deshpande D, Srivastava S, Musuka S, Gumbo T. Thioridazine as Chemotherapy for Mycobacterium Avium Complex Diseases. Antimicrob Agents Chemother (2016) 60(8):4652–8. doi: 10.1128/aac.02985-15
198. Seervi M, Rani A, Sharma AK, Santhosh, Kumar TR. ROS Mediated ER Stress Induces Bax-Bak Dependent and Independent Apoptosis in Response to Thioridazine. Biomed Pharmacother (2018) 106:200–9. doi: 10.1016/j.biopha.2018.06.123
199. Chu CW, Ko HJ, Chou CH, Cheng TS, Cheng HW, Liang YH, et al. Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/β-Catenin Signaling Pathway in Glioma Cells. Int J Mole Sci (2019) 20(3):473. doi: 10.3390/ijms20030473
200. Bermudez LE, Kolonoski P, Wu M, Aralar PA, Inderlied CB, Young LS. Mefloquine Is Active In Vitro and In Vivo Against Mycobacterium Avium Complex. Antimicrob Agents Chemother (1999) 43(8):1870–4. doi: 10.1128/aac.43.8.1870
Keywords: autophagy, skin, mycobacteria, drug development, skin cells
Citation: Bittencourt TL, da Silva Prata RB, de Andrade Silva BJ, de Mattos Barbosa MG, Dalcolmo MP and Pinheiro RO (2021) Autophagy as a Target for Drug Development Of Skin Infection Caused by Mycobacteria. Front. Immunol. 12:674241. doi: 10.3389/fimmu.2021.674241
Received: 01 March 2021; Accepted: 28 April 2021;
Published: 25 May 2021.
Edited by:
Esther Christina De Jong, Academic Medical Center, NetherlandsReviewed by:
Eun-Kyeong Jo, Chungnam National University, South KoreaMirian Nacagami Sotto, University of São Paulo, Brazil
Copyright © 2021 Bittencourt, da Silva Prata, de Andrade Silva, de Mattos Barbosa, Dalcolmo and Pinheiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberta Olmo Pinheiro, cm9iZXJ0YW9sbW9AZ21haWwuY29t