 Tihong Shao1,2†
Tihong Shao1,2† Xiaodong Shi3†Shanpeng Yang4Wei Zhang5Xiaohu Li6Jingwei Shu6Shehabaldin Alqalyoobi7Amir A. Zeki8
Xiaodong Shi3†Shanpeng Yang4Wei Zhang5Xiaohu Li6Jingwei Shu6Shehabaldin Alqalyoobi7Amir A. Zeki8 Patrick S. Leung2*
Patrick S. Leung2* Zongwen Shuai1*
Zongwen Shuai1*- 1Department of Rheumatology and Immunology, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 2Division of Rheumatology/Allergy and Clinical Immunology, University of California, Davis, Davis, CA, United States
- 3Rheumatology, First Hospital of Jilin University, Changchun, China
- 4Department of Pharmacy, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 5Department of Pathology, The First Affiliated Hospital (Yijishan Hospital) of Wannan Medical College, Wuhu, China
- 6Department of Radiology, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 7Internal Medicine - Pulmonary, Critical Care, and Sleep Medicine, Brody School of Medicine, Greenville, NC, United States
- 8University of California (U.C.), Davis, Lung Center, Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Internal Medicine, U.C. Davis School of Medicine, University of California, Davis, Davis, CA, United States
Connective tissue disease (CTD) related interstitial lung disease (CTD-ILD) is one of the leading causes of morbidity and mortality of CTD. Clinically, CTD-ILD is highly heterogenous and involves rheumatic immunity and multiple manifestations of respiratory complications affecting the airways, vessels, lung parenchyma, pleura, and respiratory muscles. The major pathological features of CTD are chronic inflammation of blood vessels and connective tissues, which can affect any organ leading to multi-system damage. The human lung is particularly vulnerable to such damage because anatomically it is abundant with collagen and blood vessels. The complex etiology of CTD-ILD includes genetic risks, epigenetic changes, and dysregulated immunity, which interact leading to disease under various ill-defined environmental triggers. CTD-ILD exhibits a broad spectra of clinical manifestations: from asymptomatic to severe dyspnea; from single-organ respiratory system involvement to multi-organ involvement. The disease course is also featured by remissions and relapses. It can range from stability or slow progression over several years to rapid deterioration. It can also present clinically as highly progressive from the initial onset of disease. Currently, the diagnosis of CTD-ILD is primarily based on distinct pathology subtype(s), imaging, as well as related CTD and autoantibodies profiles. Meticulous comprehensive clinical and laboratory assessment to improve the diagnostic process and management strategies are much needed. In this review, we focus on examining the pathogenesis of CTD-ILD with respect to genetics, environmental factors, and immunological factors. We also discuss the current state of knowledge and elaborate on the clinical characteristics of CTD-ILD, distinct pathohistological subtypes, imaging features, and related autoantibodies. Furthermore, we comment on the identification of high-risk patients and address how to stratify patients for precision medicine management approaches.
Introduction
Connective tissue disease (CTD) is a heterogeneous group of inflammatory disorders that can affect bone, cartilage, tendons, ligaments, muscle, joints, blood vessels, and even specific organs. Many CTDs such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Sjogren’s syndrome (SS), polymyositis (PM)/dermatomyositis (DM), systemic sclerosis (SSc), and mixed connective tissue disease (MCTD) are autoimmune mediated. The major pathological features of autoimmune mediated CTD are chronic inflammation of blood vessels and connective tissues, which can affect any organ leading to multi-system damage.
The human lung is particularly vulnerable to such damage because anatomically it is abundant with collagen and blood vessels that are essential for metabolic, endocrine, and immune functions. Various components of the respiratory system including the airways, vessels, parenchyma, pleura, and respiratory muscles may also be involved (1). In such cases, this manifests clinically as pulmonary interstitial diseases, pulmonary vascular diseases, diffuse alveolar hemorrhage, bronchiolitis, pulmonary parenchymal nodules, pleural lesions or effusions, respiratory muscle weakness, and aspiration pneumonia. Understanding the diverse clinical manifestations and high mortality of interstitial lung disease (ILD) in patients with CTD is important and highly relevant to the practice of rheumatology.
The heterogeneity in disease severity, underlying mechanisms, and clinical manifestations of CTD-ILD can be perplexing. Clinical and research-based rheumatologists are faced with several challenges in the diagnosis and management of CTD-ILD: (a) despite extensive effort, the precise mechanisms that drive CTD-ILD remain unclear; (b) it is easy to miss or misdiagnose patients when they present with pulmonary involvement but without clear immunological manifestations; (c) there is no standard protocol for evaluating a given patient’s condition and assessing disease progression, e.g. when we confirm that the patient is deteriorating, there are no effective methods or biomarkers to determine whether the patient’s deterioration is due to the progression of ILD or other reasons, such as infection or drug-induced causes; (d) CTD-ILD is difficult to treat. CTD-ILD has a more favorable prognosis than idiopathic interstitial pneumonia (IIP) because it can be treated with glucocorticoid (GC) and immunosuppressive agents (2, 3). However, the side effects of these medications, treatment dose(s), and clinical course can vary greatly between patients. Further, once the patient progresses to pulmonary fibrosis, the prognosis becomes less optimistic. Owing to the complexity of treatment, tailoring treatment protocols for CTD-ILD requires vigorous effort and a multidisciplinary team approach often including close collaboration with the patient’s pulmonologist (4, 5).
Pathogenesis of CTD-ILD
ILD refers to a group of heterogeneous non-neoplastic diseases belonging to the category of diffuse parenchymal lung diseases (DPLDs) that affect alveolar epithelial cells, pulmonary capillary endothelial cells, basement membrane, perivascular, and lymphoid tissues. CTD-related ILD (CTD-ILD) can be similar to the IIPs [e.g. idiopathic pulmonary fibrosis (IPF)], especially when the lung is the only organ involved, or the lung injury happened in connective tissues prior to the extrapulmonary manifestations. Genetics (6), environment (7, 8), and immunological factors (9, 10) could be involved in the pathogenesis of CTD-ILD (Figure 1). Here, we discuss our current understanding of genetic predisposition, the environment, and immune regulation of CTD-ILD.
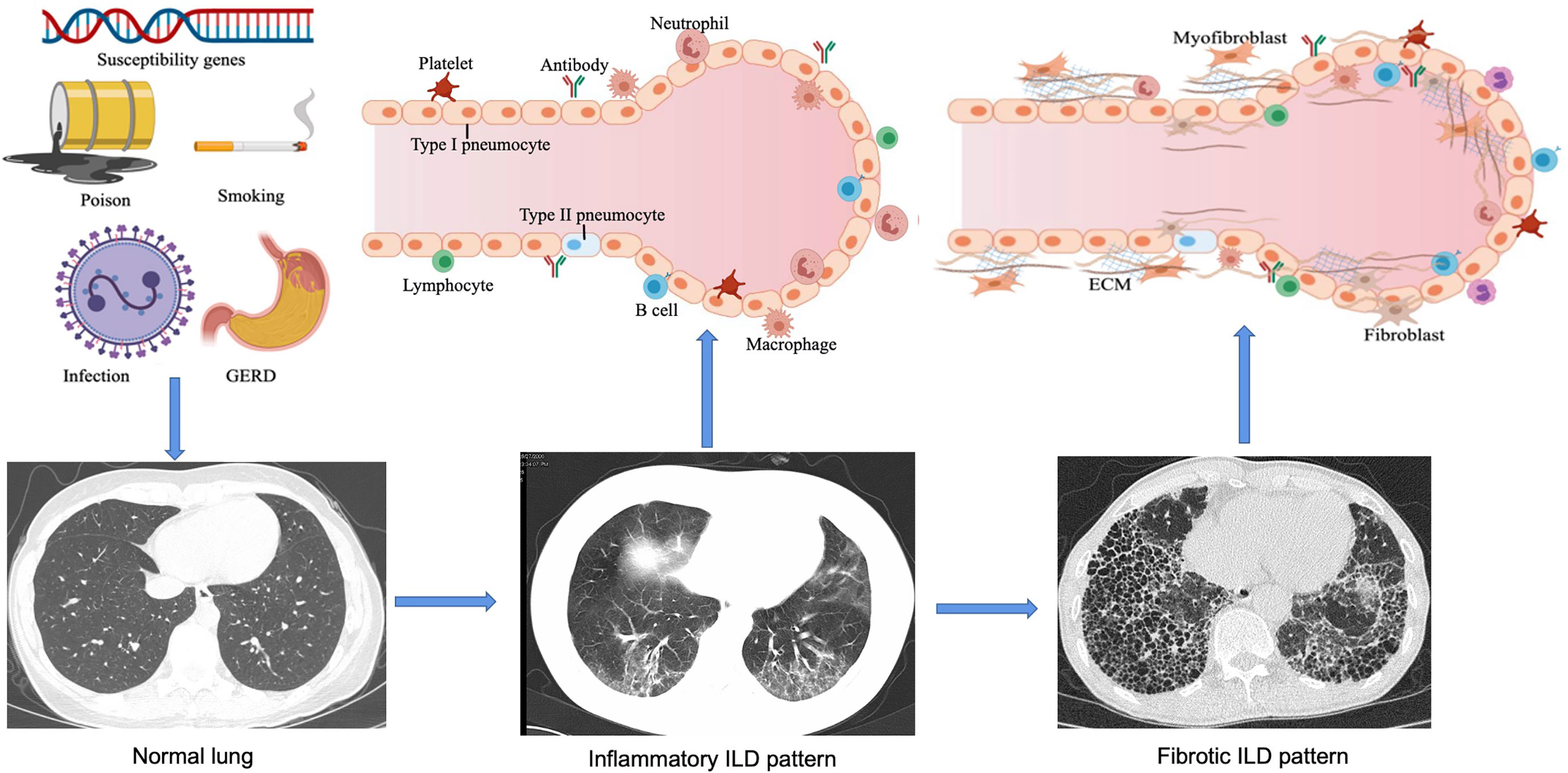
Figure 1 Pathogenesis and development of ILD. In genetically susceptible individuals, external factors such as smoking, environmental chemicals, infections and gastroesophageal reflux disease (GERD) can lead to epithelial cell injury and aberrant repair, alveolar macrophage activation, neutrophil recruitment, and oxidative stress. Over time, increased ECM turnover will result in the development of fibrosis. With these exposures the host's immune tolerance is broken leading to chronic inflammation from cellular and humoral autoimmunity, endothelial cell dysfunction, granuloma formation, and alveolar macrophage activation thus further aggravating inflammation. As the disease progresses, interstitial pneumonia changes from an early alveolitis (increased alveolar space content of inflammatory products, reflecting inflammatory ILD pattern) to a transition period (thickened alveolar septum and the deposition of collagen fibers, significantly reduced capillary beds), and eventually ending in alveolar structural destruction and fibrosis. This figure is created with MedPeer.
Genetics
The application of high throughput genetic analysis has led to the identification of a number of genetic loci that are associated with the risk of developing CTD-ILD and worse prognosis (11–13). We will discuss major findings regarding the effects of genetic susceptibility and its association with the poor prognosis seen in CTD- ILD. Rare pathogenic mutations in telomere maintenance genes and chromosome-protected terminal telomere shortening are related to pulmonary fibrosis. Newton et al. have examined telomere-related variants in patients with ILD (14). Patients with rare telomere-related variants TERT, TERC, PARN, or RTEL1 exhibit various forms of pulmonary fibrosis, ranging from IPF, interstitial pneumonia with autoimmune features (IPAF), to CTD-ILD. Interestingly, there exists statistical correlations in the mean diagnostic age of patients with different gene mutations. TERC mutation carriers were diagnosed at the earliest age (51 years old) and had a higher incidence of hematological comorbidities. The age of diagnosis was highest in PARN, followed by RTEL1, TERT and TERC, and is consistent with the order of average telomere length (14). Studies have reported that shorter leukocyte telomere length is associated with faster decline in lung function and shorter duration of transplant-free survival in patients with IPAF and CTD-ILD (11–13).
The strongest risk factor for the development of IPF has been identified as the gain-of-function of the MUC5B promoter variant rs35705950, which is observed in more than two-thirds of patients with IPF and accounts for 30% to 35% of the risk of developing disease (15–23). Several studies have demonstrated that MUC5B minor alleles correlate with the deterioration of lung function and survival rate of IPAF and CTD-ILD (11–13). Compared with non-CTD-ILD controls, the frequency of MUC5B minor allele frequency is higher in CTD-ILD, especially the RA-ILD subgroup (24). Notably, Juge et al. examined the effects of MUC5B promoter variant RS35705950 on RA-ILD and showed that the MUC5B promoter variant was associated with RA-ILD, with a characteristic interstitial pneumonia imaging pattern (6). Genetically driven MUC5B overexpression of MUC5B protein can hinder cilia clearance or disrupt normal lung repair mechanisms (25). Collectively, these studies support that MUC5B is involved in the pathogenesis of CTD-ILD and may be a therapeutic target.
Similar to familial IPF patients, data from exome-sequencing revealed the presence of TTR, RTL1, PARN, or SFTPC mutations in RA-ILD patients, indicating the contribution of IPF-linked genes in RA-ILD susceptibility (26). In addition to common genetic features, RA-ILD and IPF patients have overlapping clinical features, such as older age, and higher occurrence in males and in cigarette smokers (27, 28). Cumulative evidence has also indicated that a number of genetic loci are associated with susceptibility to SSc-ILD, including CTGF (also known as CCN2, encoding connective tissue growth factor), CD247 and IRF5 (29–32). Fingerlin et al. reported that two HLA alleles in the high linkage disequilibrium are associated with pulmonary fibrosis (DRB1 * 15:01 and DQB1 * 06:02) (33, 34). These susceptibility genes are similar to the previous ILD-related loci associated with PM/DM (34–36). A list of CTD-ILD susceptibility genes currently identified in the literature is shown in the Table 1.
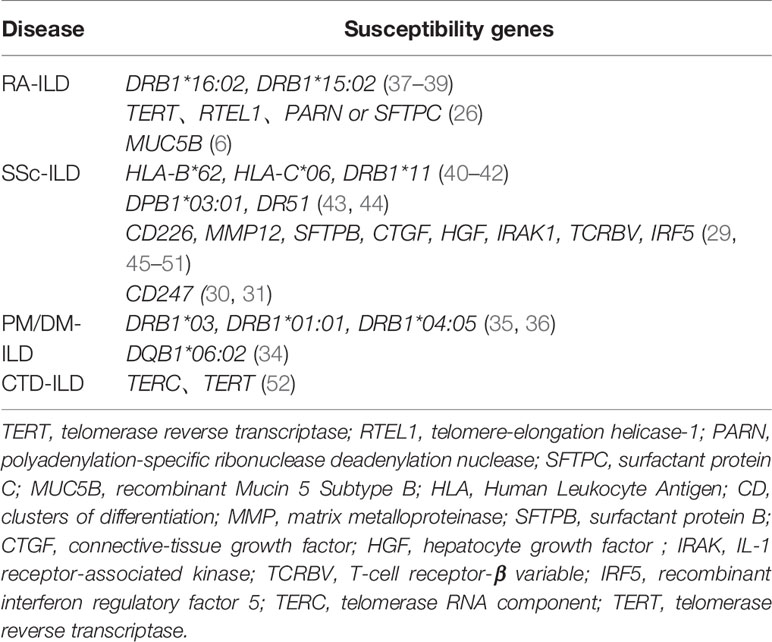
Table 1 Susceptibility genes in CTD-ILD.
There are epigenetic mechanisms including DNA methylation, post-translational histone modification, and non-coding RNA in IPF. The differences in their DNA methylation pattern may influence the expressions of many target genes and microRNAs (miRNAs), as well as the regulatory sites of genes involved in IPF (53, 54). Based on comparative analysis of genome-wide DNA methylation together with gene expression patterns in lung tissues from IPF patients and normal controls, Sanders et al. demonstrated that ZNF467 and CLDN5 with hypermethylation are down-regulated, whereas TP53INP1 and DDAH1 with hypomethylation are up-regulated in IPF (55). Studies on histone modifications mainly revealed the involvement of epithelial-mesenchymal transition (EMT), apoptosis, and the prostaglandin E2 pathway (56). Histone deacetylase inhibitors can eliminate the differentiation of fibroblast-myofibroblasts induced by transforming growth factor-β1 (TGF-β1), restore the expression of surfactant protein-C in alveolar epithelial type II cells, and mitigate bleomycin-induced pulmonary fibrosis (57, 58). Histone deacetylase inhibition can also increase Fas expression, which exhibited low level expression in fibroblasts from both IPF patients and mice with experimental pulmonary fibrosis, and restore sensitivity to Fas-mediated apoptosis, indicating the key role of histone modification in the development of anti-apoptotic fibroblasts (59). Changes in miRNA profiles have been observed in IPF patients and mouse models of fibrosis, including the down-regulation of some microRNAs, such as let-7, mir-29 and mir-30, members of the miR-200 family, and upregulation of miRNAs, such as mir-155 and mir-21. Regulating the expression of miRNAs can attenuate or aggravate IPF, which exploits a new era for a miRNA-mediated therapeutic approach to the treatment of IPF (60–64). There is increasing evidence to support the involvement of epigenetics in the pathogenesis of IPF, however, there are limited studies on the correlation between epigenetics and CTD-ILD. Therefore, relevant studies are needed to address this relationship.
Environmental Factors
Multiple environmental factors including gastroesophageal reflux disease (GERD), infections (7, 65), environmental chemicals, toxic substances, drugs (66–68), and tobacco smoke are associated with inflammatory lung injury (8, 69, 70). The prevalence of GERD in ILD can be as high as 94% (71–73). Animal studies have shown that chronic aspiration leads to pulmonary fibrosis (74). It is postulated that GERD-associated chronic micro-aspiration induces repetitive lung injury, resulting in pneumonitis, increased epithelial permeability, fibrotic hyperplasia, and ultimately pulmonary fibrosis (75). A murine model of aspiration-induced lung injury model exhibited extensive collagen deposition by the second week (76), and revealed reflux containing bile acids, elevated TGF-β levels, and prominent fibroblast proliferation (77). On the contrary, meta-regression analysis adjusted for smoking suggested chronic micro-aspiration in GERD is not associated with IPF (78). In addition to the contribution from acidic stomach contents, Helicobacter pylori in gastric juice can also cause lung injury, and thereby, promote progressive pulmonary fibrosis (79, 80).
Although Epstein-Barr virus is a prime suspect, other viruses and bacteria (e.g., retroviruses, parvoviruses, mycobacteria, Mycoplasma species, and Borrelia species) have also been implicated in inflammatory lung injury (7).
Particulate matter and toxic chemicals in tobacco smoke can activate immune cells, recruit inflammatory cells, and lead to the influx of various immune cells into the lungs, and this in concert can eventually lead to ILD (81, 82). However, the effect of smoking on CTD-ILD is unclear. Among various autoimmune diseases, RA is most definitely associated with smoking. Epidemiological studies have demonstrated that people exposed to tobacco smoke are at a higher risk of developing seropositive RA, and in predisposed individuals, smoking can promote the production of anti-cyclic citrullinated peptide (anti-CCP) antibodies (83–85). Notably, increased prevalence of emphysema and decreased survival have been noted in patients with SSc who smoke heavily, indicating the adverse effects of smoking in SSc (86–88).
More than 600 drugs have been reported to cause severe pulmonary injury (See pneumotox.com for a list of drugs that have been reported to cause lung toxicity). Multiple drugs used in treating cardiovascular diseases, anti-inflammatory, antimicrobial, and cancer immunotherapies as well nonbiologic and biologic disease-modifying anti-rheumatic drugs (DMARDs) have also been associated with severe lung injury (89). Anti-rheumatic drug-induced ILD (DILD) is not uncommon and can be driven via dose-dependent toxicity and immune-mediated allergic reaction (90). Risk factors of DILD include genetic susceptibility (91), age, sex, smoking, underlying lung disease such as pre-existing ILD, bronchiectasis, chronic obstructive pulmonary disease, dosage of drugs, and interactions with concomitant drugs and previous treatment, such as chest radiotherapy (68, 92, 93). Clinically, it is difficult to distinguish DLID from other interstitial pneumonias. Multiple imaging patterns can result from the same drug, and vice versa (68). Similarly, it is challenging to diagnose DILD due to presenting signs and symptoms that are often very similar to other ILDs.
The diagnosis of DILD is based on the following: (a) an exposure to the causative agent and presenting concomitant respiratory signs and symptoms which are consistent with previous reports, (b) ruling out other causes of lung damage including infection, cancerous lymphangitis, radiotherapy-induced pneumonitis, congestive heart failure, and exacerbation of pre-existing ILD, (c) alleviation of symptoms after discontinuation of the offending drug and relapse after reapplication. Rheumatologists may face several challenging clinical scenarios including the development of initial symptoms after drug withdrawal, or continued aggravation of clinical symptoms despite drug discontinuation. In addition, when a patient develops ILD during the treatment of a rheumatologic disorder, it is difficult to determine whether it is drug-induced or whether it is complicated by CTD. Similarly, when CTD-ILD patients progress or worsen during treatment, it is difficult to determine whether this is due to a drug side effect or the natural progression of disease.
Importantly, clinicians need to be aware of what medications are associated with DILD. The main nonbiological DMARDs include gold (94, 95), penicillamine (96), sulfasalazine (97), tacrolimus (98, 99), methotrexate (MTX) (93, 100–102), and leflunomide (103–105), and the biological agents mainly include anti-TNF, anti-CD20, and cytokine monoclonal antibodies (106, 107). Although MTX-induced ILD is well-established by many studies, there is some emerging conflicting evidence suggesting no association between MTX and RA-ILD (108). For example, a study of MTX use and the risk of ILD in RA patients demonstrated that there was no further increase in risk associated with MTX treatment (109). Other studies have also reported similar findings suggesting that MTX may delay the onset of ILD (110, 111). However, the overall picture and recommendation indicates that exposure to any of the aforementioned drugs could potentially lead to DILD. Therefore, the alert clinician must be aware of this possibility because early recognition could lead to the earlier initiation of therapy.
Immunological Factors
Both innate and adaptive immune system are potential culprits for the pathogenesis of CTD-ILD. For example, B cells contribute to autoimmune ILD (112) with studies showing the presence of extensive B cell infiltrations in lung tissue samples of SSc-ILD patients (9). Compared with IIP, RA-ILD is distinguished by its prominent increase in CD4+ cells and follicular B cell hyperplasia in the lung (113, 114). In patients with RA-ILD and SSc-ILD, T cells release fibrogenic mediators which subsequently stimulate fibroblasts and prime the fibrotic response (115). In SSc-ILD, alveolar macrophages become M2 polarized upon induction by the Th2 cytokines IL-4 and IL-10, suggesting that the M2/Th2 pathway is involved in the pathogenesis and development of SSc-ILD (116). Autoantibodies are also associated with CTD-ILD, with some antibodies occasionally related to the course and severity of the disease, reinforcing the notion that humoral immunity is involved in the pathogenesis of CTD-ILD (117, 118). This is discussed in greater detail in the CTD-ILD related autoantibody section below.
Toll-like receptors (TLRs), key components of innate immunity, have multi-faceted effects on ILD in patients with CTD. TLRs have been proposed as markers of ILD progression (10). Correlation studies showed that TLR2 (119) and TLR9 (120) are profibrotic while TLR3 (121) is anti-fibrotic in pulmonary fibrosis. On the other hand, TLR4 can be either profibrotic (122) or anti-fibrotic (123) depending on the micro-environment. TLR2, TLR3 mRNA in bronchoalveolar lavage fluid (BALF) T-lymphocytes and peripheral blood monocytes, are over-expressed in CTD-ILD compared with healthy controls, suggesting that TLRs may be involved in the pathogenesis of CTD-ILD (124–126). The contribution of other innate players on CTD-ILD remains to be explored. Understanding the mechanistic roles of immune cell activities in CTD-ILD will help in the development of innovative and novel therapeutic approaches.
In genetically predisposed individuals, the pathogenesis of CTD-ILD involves recurrent alveolar injury and dysfunctional healing which are key causative mechanisms in the development of pulmonary fibrosis. Pulmonary fibroblasts are activated to produce extracellular matrix as inflammatory cells enter and infiltrate the lung interstitial and alveolar spaces. This leads to an imbalance of collagen formation and degradation, resulting in collagen over-accumulation in the lung (127). Epithelial and mesenchymal cells, as well as components of the innate and adaptive immune system, lead to a favorable microenvironment that promotes disease pathogenesis (128). These factors together, contribute to the chronic inflammation, gradual destruction of functional lung parenchyma, replacement by collagen, thus, ultimately leading to pulmonary fibrosis, respiratory failure, and early mortality.
Clinical Characteristics of CTD-ILD
Clinical manifestations of CTD-ILD include constitutional and respiratory symptoms, but these are rather non-specific. The common constitutional symptoms include fatigue, fever, and weight loss. The most common respiratory symptoms include exertional dyspnea, exercise intolerance, and dry (or non-productive) cough with slow progression over the time. Other concomitant symptoms may include chest pain, palpitations, tachypnea, and hemoptysis. In addition to pulmonary involvement, CT-ILD can also involve the mucocutaneous, musculoskeletal, neurological, gastrointestinal, cardiac, and hematologic systems.
Generally speaking, compared with IIP, CTD-ILD patients are more likely to be younger, female, and non-smokers. However, the exact frequency of CTD-ILD is not known. Although the incidence and prevalence vary between studies, it is estimated that 10 to 90% of patients with CTD will have evidence of pulmonary involvement during their lifetime. The types of pulmonary manifestations may vary by underlying CTD diagnosis (Table 2). The prevalence and mortality of ILD for each CTD are different, and the prevalence of ILD secondary to various CTDs varies as follows: 1 to 15% in SLE (106), 6.5 to 33% in RA (107, 108), 19.9 to 86% in PM/DM (109, 110), 86% in anti-Jo-1 positive patients (111), 40 to 91% in SSc (112, 113), 47 to 90% in MCTD based on radiologic feature (108, 114), and 9 to 20% in SS (115). Related reports indicate the mortality of ILD is 20% in RA-ILD (116, 117), 12 to 44% in PM/DM (118), and a 10-year mortality of up to 40% in SSc (119). In patients with RA and SSc, the 5-year mortality is 3-fold higher than that without ILD (120, 121).
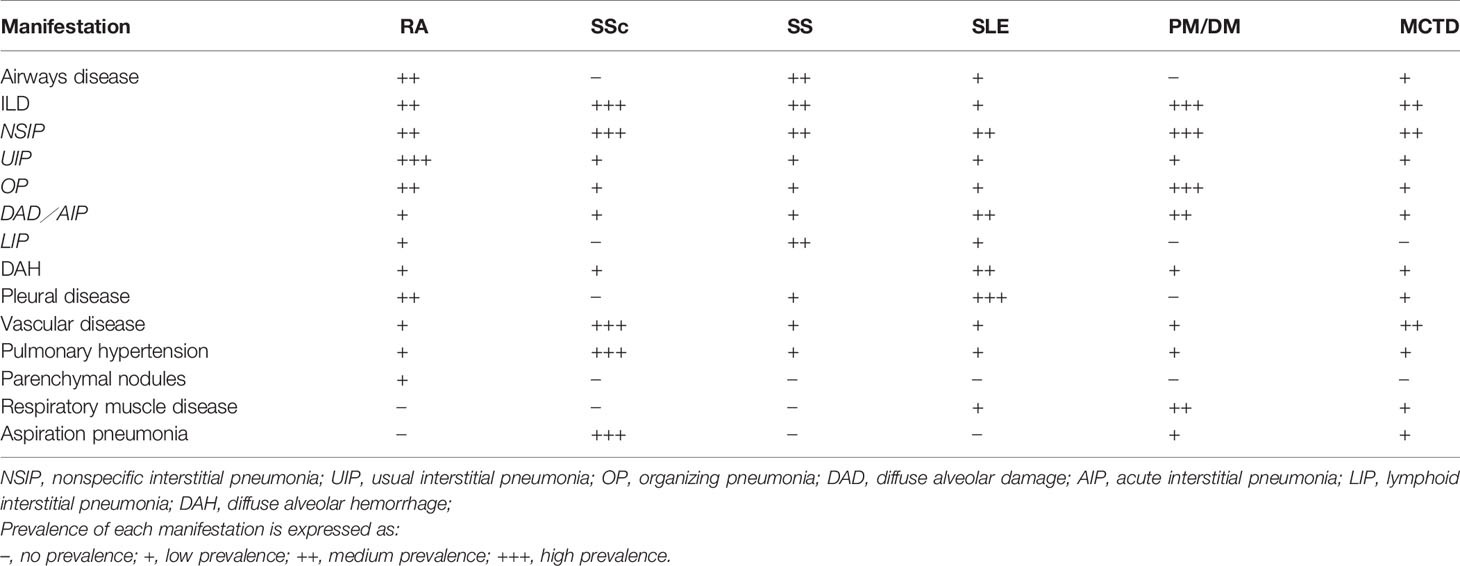
Table 2 Characteristics of lung involvement in different CTD-ILD.
Histological Classification of CTD-ILD
Histologically, CTD-ILD can be divided into 7 types including: usual interstitial pneumonia (UIP), nonspecific interstitial pneumonia (NSIP), desquamative interstitial pneumonia (DIP), respiratory bronchiolitis (RB), organizing pneumonia (OP), diffuse alveolar damage (DAD), and lymphoid interstitial pneumonia (LIP) (129, 130). These histological classifications provide a more comprehensive diagnosis of CTD presenting with ILD. We note that NSIP is the most common histopathologic type in CTD-ILD (with the exception of RA), however, UIP is the most common in IIP (131). In addition, the frequency and severity of fibroblastic lesions in CTD-ILD is lower than IPF-UIP (132). We also note that the coexistence of UIP and NSIP patterns is one of the most significant features that distinguishes CTD-UIP from IPF-UIP (133). The frequency of ILD pathological subtype from different underlying CTDs is shown in Table 2 (134–137). Although the pathological classification of CTD-ILD is identical to that of IIP, some histopathologic features such as extensive plasma cell infiltration, increased lymphoid aggregates, and more germinal centers are considered to be characteristic histologic features of CTD-ILD as compared with IIP (138–140). Indeed, the clinical characteristics, therapeutic response to GC and immunosuppressive agents, and prognosis of CTD-ILD vary according to the pathological subtypes (Table 3).
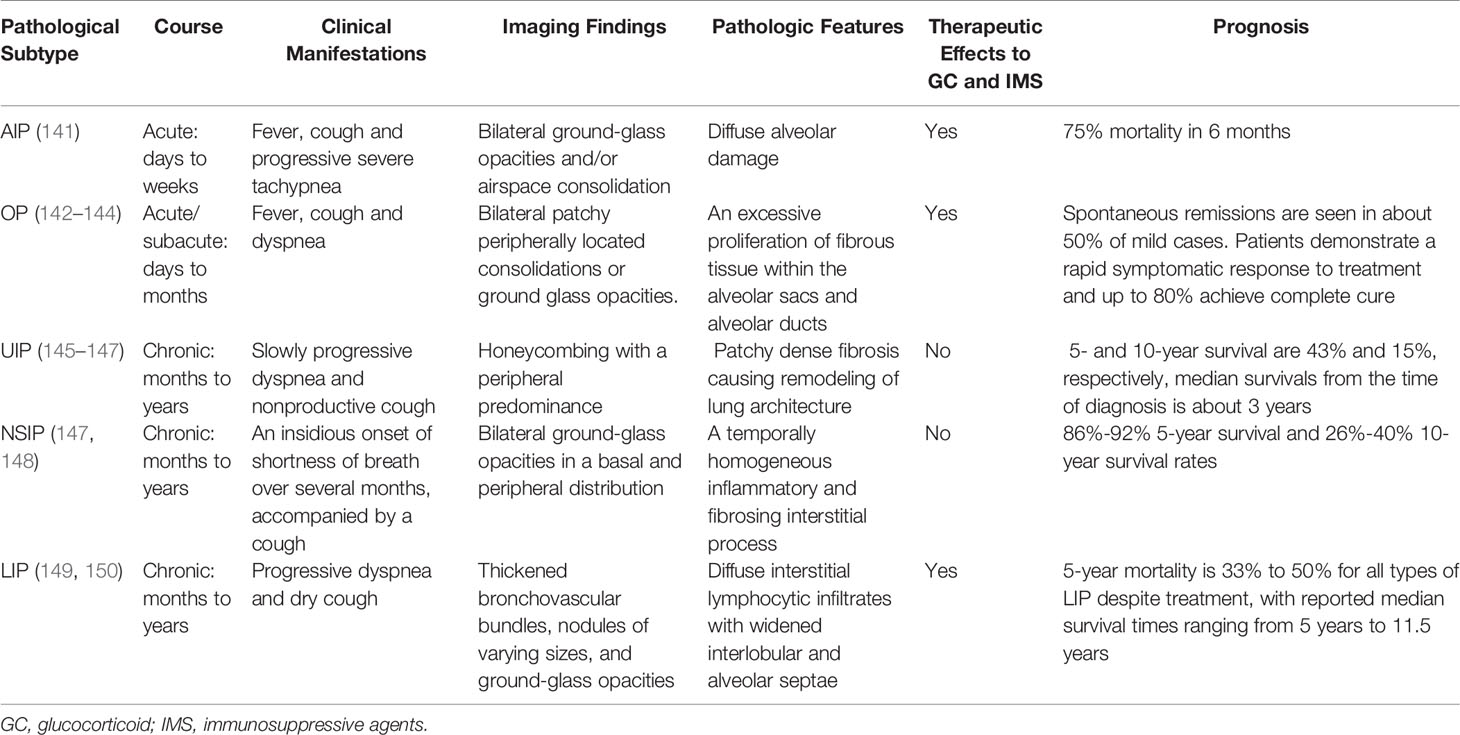
Table 3 Clinical Characteristics, Response to Therapy and Prognosis of CTD-ILD Subtypes.
Imaging Features of CTD-ILD
Radiologically, high resolution computed topography (HRCT) scans can be effectively used to diagnose and identify disease and assess disease improvement or progression. CTD-ILD may manifest as a focal or a diffuse pulmonary abnormality, especially at the periphery of the lung, such as reticulation, ground-glass opacities (GGOs) (which refers to focal or diffuse veil-like opacification of the lung), and nodules. Imaging findings of the different kinds of pulmonary lesions vary with specific diseases and histopathologic patterns observed (151, 152). In addition to their respective characteristic imaging manifestations, certain radiological clues support the diagnosis of CTD-ILD (Table 3 and Figure 2). For example, (a) In the combined NSIP-OP pattern (130, 153), CTDs such as idiopathic inflammatory myopathies (IIMs) or anti-synthetase syndrome (ASS) should be suspected when the fibrosis at the lung bases overlaps with an OP pattern. (b) In the combined DAD-IIP pattern, when the DAD is superimposed on another IIP pattern it may indicate the presence of CTD. Sometimes ASS manifests as acute respiratory failure with DAD superimposed on underlying IIP, however, this is not specific to ASS (151, 154). Atypical interstitial pneumonia may be due to unclassifiable or mixed imaging findings (155).
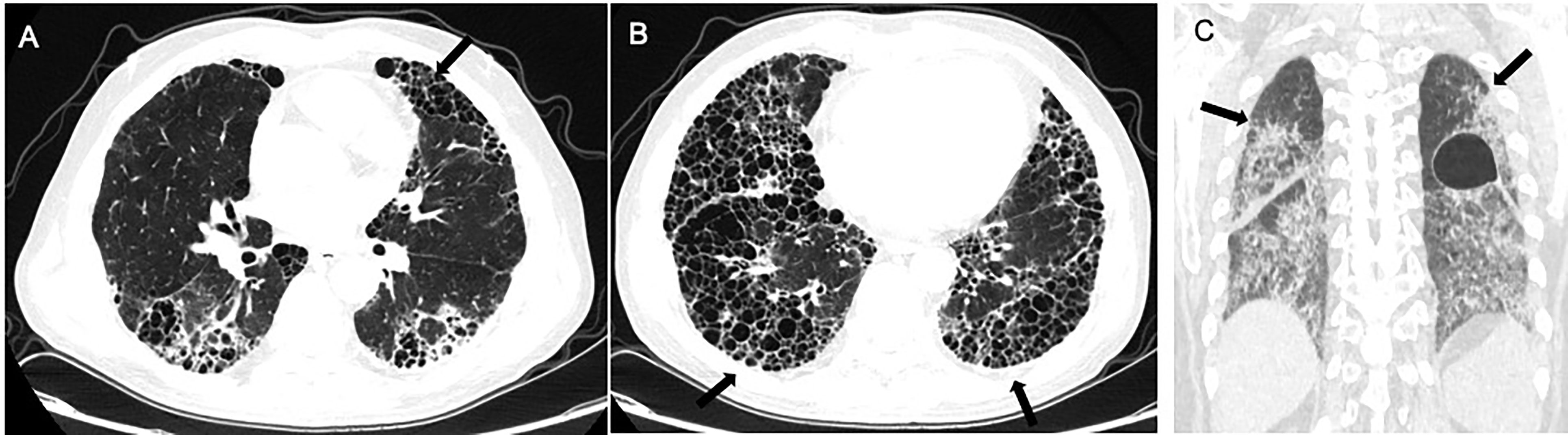
Figure 2 Radiological imaging pattern of CTD-ILD. (A) “anterior upper lobe” sign. (B) “exuberant honeycombing” sign. (C) “straight-edge” sign. All these signs are indicated by arrows.
Chung et al. compared the CT manifestations of CTD-UIP and IPF-UIP, and found that there are three imaging manifestations with high specificity but low sensitivity for CTD-ILD, including (i) the “anterior upper lobe” sign and concomitant lower lobe involvement, (ii) “exuberant honeycombing” sign constituting greater than 70% of fibrotic portions of the lung, and (iii) “straight-edge” sign indicating lung basal fibrosis with sharp demarcation in the craniocaudal plane (155).
Finally, some extrapulmonary signs on HRCT support the diagnosis of CTD-ILD, such as esophageal or pericardial abnormalities, features of pulmonary arterial hypertension, evidence of airway disease, findings suggestive of bone and joint involvement, and soft-tissue calcifications (156).
Clinical Significane of CTD-ILD Related Autoantibodies
There are multiple autoantibodies in the sera of patients with CTD, many of which are associated with interstitial lung injury (Table 4). Among the SSc-ILD-related antibodies, anti-topoisomerase antibodies are more likely to be associated with pulmonary fibrosis, while anti-RNA polymerase III antibodies are less likely to be associated with pulmonary fibrosis (157). Other antibodies have also been linked to increased lung fibrosis risk in SSc, including anti-U11/U12 ribonucleoprotein (RNP) antibodies, or anti-Th/To-RNP antibodies (158). Anti-U11/U12 RNP may be related to the severity of ILD (159–162). The correlation between anti-Scl-70 and the severity of ILD is unclear, however, anti-Ro52 antibody is associated with ILD and poor prognosis in SSc. The ANA of nucleolar pattern is also associated with pulmonary fibrosis in patients with SSc whereas anticentromere antibodies (ACA) are not. ANA patterns can be used to predict the risk of pulmonary fibrosis in patients with SSc (163).
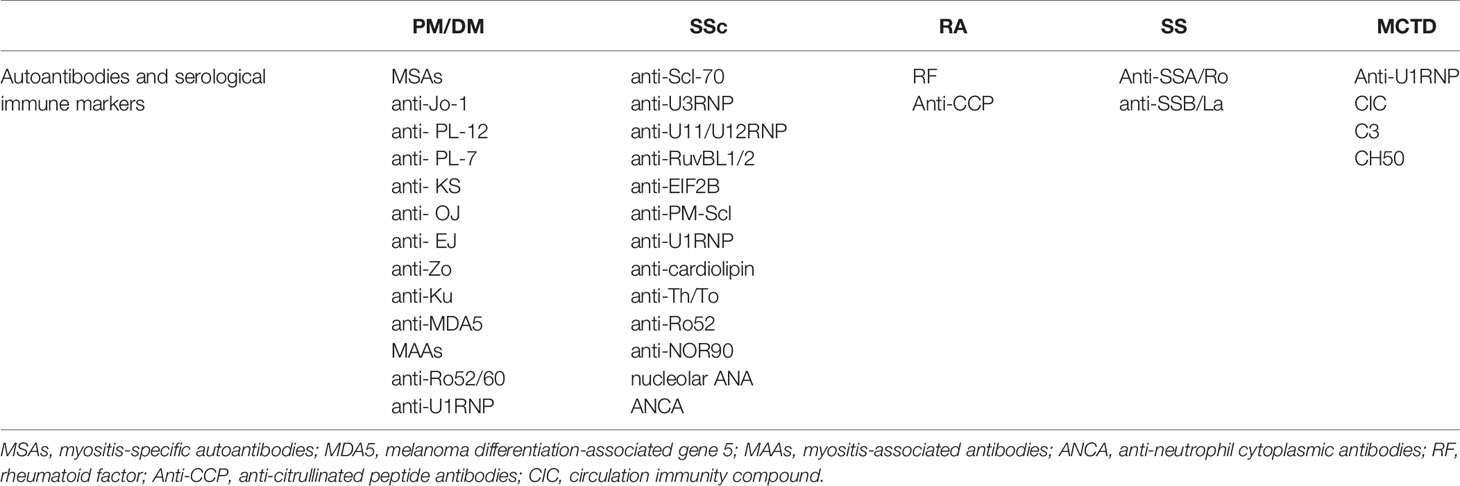
Table 4 Autoantibodies and serological immune markers associated with CTD-ILD.
In patients with autoinflammatory myopathy, myositis-specific autoantibodies (MSAs) and myositis-associated antibodies (MAAs) are associated with IIM-ILD, and those with anti-Ku antibodies have a higher risk of lung involvement (161). Single factor Cox hazards analysis showed that the presence of anti-aminoacyl-transfer RNA synthetase (ARS) antibodies indicates a better prognosis, and the presence of anti-synthetase antibodies might be used as a prognostic marker for PM/DM ILD patients. Sabbagh et al. discovered that patients with adolescent myositis with anti-Ro52 were more likely to develop ILD, have more severe disease, and have a worse prognosis (164). A related study demonstrated that anti-Ro52 antibodies and anti-Jo1 antibodies are usually present together (165), and anti-Ro52 antibody titers correlate with ILD severity (166). Compared to anti-Jo1 autoantibodies alone, adult patients with both autoantibodies were more prone to severe ILD, poorer response to various immunosuppressive drugs, and lower survival rates (167–169). Specifically targeted to scaffold attachment factor B (SAFB), anti-SAFB antibodies were detected in a small number of patients with SSc and/or PM/DM, and ILD. Anti-SAFB antibodies may be a novel CTD-related autoantibody associated with ILD (170).
Elevation of RF and anti-CCP antibodies in the serum and BALF are considered risk factors for RA-ILD (171–174). While anti-SSA/Ro and anti-SSB/La are associated with SS-ILD, it is interesting that the specificity of anti-La antibodies in lung involvement is higher than that of anti-Ro antibodies. Anti-U1-RNP, immune complex, complement C3 factor, and CH50 are highly expressed in MCTD-ILD. Anti-Ro52 antibodies are associated with MCTD-ILD (175). Anti-endothelial cell antibodies (AECAs) are associated with a high incidence of pulmonary fibrosis and severe diffusion abnormalities (176). In addition to these autoantibodies, other ILD-associated CTD autoantibodies and serological immune markers are listed in Table 4 (177).
Identifying High-Risk Patients
Rheumatologists should be vigilant about CTD-ILD, including the timely identification of high-risk patients. ILD is characterized by non-productive cough, fever, a gradual onset of exertional dyspnea, and fine bibasilar inspiratory crackles (“velcro” crackles). These signs and symptoms are non-specific and can be seen in a variety of pulmonary and/or heart diseases, and approximately 5% of patients have no symptoms when ILD is serendipitously diagnosed. One important consideration involves patients with negative autoantibodies and no extrapulmonary immune features who are eventually diagnosed with CTD-ILD after long-term follow-up. Since diagnosis of these patients can be challenging and delayed, regular assessments are required during clinic follow-ups to make the correct diagnosis. In a study on 1,044 Chinese CTD-ILD patients, 43.8% of them had a negative autoantibody serological test at the time of initial admission, however 25.1% seroconversions and 18.7% persistent negatives were found on subsequent follow-ups. In the latter group, most of these patients were finally diagnosed with CTD-ILD because of their emerging extrapulmonary features and/or need for lung biopsy (178).
Pulmonary dysfunction mainly manifests as a restrictive pattern with a decrease in total lung capacity (TLC), forced vital capacity (FVC), residual volume (RV), functional residual capacity (FRC), and diffusion capacity of carbon monoxide (DLCO). These pulmonary function test (PFT) findings reflect a restrictive ventilator defect due to pulmonary interstitial fibrous tissue hyperplasia leading to increased diffusion distance, and decreased diffusion capacity.
An emerging phenotype called “progressive fibrosing-ILD” (PF-ILD) that is characterized by significant decline in FVC (relative decline of ≥5-10%) and DLCO (relative decline of ≥5-15%) over a period of time ranging between 6 to 24 months, is associated with increased mortality (179, 180). Other criteria included worsening symptoms and increased fibrotic changes on HRCT (181). CTD-ILD can present with a PF-ILD phenotype if they meet the above criteria (179, 181). A recent decline in FVC and DLCO are independent predictors of decreased survival rate in SSc-ILD (157, 182–184).
On thoracic imaging, loss of lung volume, parenchymal reticulations, and GGOs are common. Typical pulmonary HRCT findings in patients with CTD-ILD include GGOs, fiber strips, sub-pleural interlobular septal thickening, small nodules, traction bronchiectasis, subpleural arc shadow, honeycomb lung changes (mainly concentrated in the middle and lower lungs), and cystic formations. Some specific imaging features are associated with an increased likelihood of progression and risk of death in interstitial lung abnormalities. For example, an increased extent of lung fibrosis on HRCT and definitive signs of fibrosis (e.g. pulmonary parenchymal architectural distortion) predict the highest risk of progression. Subpleural reticular marks suggest increased likelihood of progression as well as honeycombing and traction bronchiectasis, while centrilobular nodules may suggest a lower likelihood of progression. Both ‘probable UIP’ and ‘UIP patterns’ are related to increased risk of death (185). In addition, disease progression on imaging is associated with increasing age and MUC5B genotype copies (186, 187). Interestingly, computer-based computed tomography analysis (CALIPER) indicates that pulmonary vessel volume is an independent predictor of mortality in CTD-ILD patients (188).
Serum markers have also been investigated in the diagnosis and prognosis of ILD (189, 190). The presence of the same biomarkers suggests that CTD-ILD and IPF share a common pathophysiological process or mechanism (191, 192). In CTD-ILD, different biomarkers have been associated with worse outcome such as Krebs von den Lungen-6 (KL-6), cancer antigen 19-9 (CA 19-9), cancer antigen 125 (CA 125), vascular cell adhesion molecule-1 (VCAM-1), and C-X-C motif chemokine ligand 13 (CXCL13) (193). SSc-ILD prognostic biomarkers included, in addition to the above biomarkers, surfactant protein-D (SP-D), surfactant protein-A (SP-A), chitinase-3-like protein 1 (YKL-40), matrix metalloproteinases 12 (MMP12), tissue inhibitor of metalloproteinase-1 (TIMP-1), 16-kDa Clara cell secretory protein (CC16), (Tenascin C), C-C motif chemokine ligand 2 and 8 (CCL2 and CCL18), interleukins 6 and 2 (IL-6 and IL-2), C reactive protein (CRP), C-X-C motif chemokine ligand 4 and 10 (CXCL4 and CXCL10), and fractalkine (CX3CL1) (193).
Most importantly, rheumatologists must consider the clinical characteristics and radiographic findings of their patients and use serum biomarkers to aid their diagnostic work-up, and where appropriate, help in prognostication. KL-6 has the strongest value in diagnosing IPF and CTD-ILD, followed by SP-D, and MMPs as the most meaningful tools for IPF diagnosis. KL-6, SP-D, and chemokine ligand 18 (CCL18) have a high sensitivity but are not specific in diagnosing SSc-ILD, and CCL18 can predict the deterioration of IPF and SSc-ILD where CCL18 has a higher predictive value (194–208).
Assessment and Treatment of CTD-ILD
The correct and timely diagnosis of CTD-ILD is necessary in order to delivery appropriate therapy. Once the diagnosis of CTD-ILD is established and extent of disease progression is assessed, then prognosis can be determined. An individualized treatment regime can then be initiated with regular clinic follow-ups (3, 209, 210). Indications to use GC and immunosuppressive agents depends on the primary disease, systemic activity, reversibility, and ILD clinical course. ILD can be divided into main IIP, rare IIP, and unclassified IIP (211). Main IIP can be further sub-divided into acute IIP (days to weeks), mainly AIP and OP; subacute IIP (weeks to months), mainly OP; and chronic IIP (months to years), mainly UIP and NSIP. The rare IIP is typically LIP. Patients with acute and subacute phase IIP need timely initiation of GC treatment combined with immunosuppressive therapy. For patients with chronic phase IIP such as honeycombed lung, high-dose GC and immunosuppressive therapy may not be beneficial. In this case, anti-pulmonary fibrosis treatment such as pirfenidone and nintedanib (212–214) may be considered.
The prognosis of CTD-ILD depends on ILD classification. The more urgent the course is, the better the effect of GC and immunosuppressive agents. On the other hand, the slower the course of the disease, as in NSIP and UIP, the poorer the efficacy of GC and immunosuppressive agents and the worse the prognosis. Considering the selection of immunosuppressive agents, there is currently no uniform management guideline for CTD-ILD. Rheumatologists should carefully consider the actual situation of each patient according to their underlying CTD, disease severity, the rate of disease progression (215–218) when selecting immunosuppressive agents [i.e. cyclophosphamide (CYC) (219–222), mycophenolic mofetil (223–225), azathioprine (226, 227), cyclosporine (228), tacrolimus (229, 230), and CD20 monoclonal antibody (231, 232)].
Several new therapeutic agents have been reported for the treatment of CTD-ILD, including Tripterygium wilfordii Hook F (233), tocilizumab, and abatacept. A clinical study reported the therapeutic efficacy of Tripterygium wilfordii Hook F as being comparable to CYC in the treatment of SSc-ILD when used only for maintenance therapy, but not for induction therapy (233). Biologics are also increasingly becoming available to treat ILD. Based on the rationale that elevated circulating IL-6 is predictive of progression in SSc-ILD (234), and the promising results from clinical trials (235–237), the FDA has approved tocilizumab in adult patients with SSc-ILD (218). Current evidence also indicates the promising efficacy and safety of abatacept in treating RA-ILD patients (238, 239).
In terms of anti-fibrosis therapy, the United States FDA has approved nintedanib, an inhibitor of multiple tyrosine kinases, for use in CTD-ILD with PF-ILD phenotype and SSc-ILD (218, 240). Two large randomized clinical trials (SENSCIS and INBUILD) showed that nintedanib reduced the annual rate of loss of FVC (212, 240). CTD-ILD with the PF-ILD phenotype and SSc-ILD patients who still exhibit disease progression after being treated with MMF or CYC may benefit from the addition of nintedanib to standard treatment (212, 240, 241). Although a pilot study has shown that administration of pirfenidone was associated with a reduction in dyspnea and an increase in vital capacity in SSc-ILD (242), other studies have not demonstrated a significant effect of pirfenidone (243), and therefore, the evidence for pirfenidone in these groups is less convincing (244). Other treatments include IVIG, plasmapheresis (245), and anti-reflux drugs. For end-stage or refractory cases, two promising novel therapeutic strategies such as autologous hematopoietic stem-cell transplantation and lung transplantation may be considered (246).
Clinical deterioration during routine follow-up should prompt the treating rheumatologists to consider the possible underlying causes: Acute exacerbation of CTD-ILD? Drug-induced ILD? Infection? Of note, when considering whether a patient’s presentation is due to acute exacerbation of CTD-ILD or infection, it is critically important to rule out infection before initiating immunosuppressive treatment (247–252). The exact cause of a patient’s exacerbation should be determined as expeditiously as possible. However, it is also possible that multiple concomitant factors are causing the exacerbation. Notably, it is important to recognize that infections and acute exacerbation of CTD-ILD can mimic one another, can coexist, and can promote each other (65, 248, 253–255).
A comprehensive clinical evaluation is required including: (a) evaluation of the patient’s occupation or living environment, (b) analysis of current or prior medication use, (c) systemic analysis of the patient’s symptoms, signs, imaging characteristics, and infection screening, and (d) evaluation of the patient’s immune function status (including neutrophils, humoral immunity, cellular immunity levels and functional status). The first step is to remove any suspicious drugs, then followed by initiation of specific therapy such as GC (as long as infection has been ruled out). If the diagnosis is still uncertain after this evaluation, for patients with mild disease, the diagnosis should be confirmed with more extensive or invasive examination. If the diagnosis remains elusive, then antimicrobial therapy may be considered first. If this approach is ineffective, then empiric treatment with a GC may be considered. For critically ill patients, treatment with combination antibiotics and a GC is generally recommended albeit clinical evidence is limited.
Supportive care including cessation of cigarette smoking, use of supplemental oxygen, annual influenza vaccination, and pneumococcal vaccination, should all be considered. In addition to treating the underlying disease, it is also necessary to treat comorbidities such as GERD, pulmonary hypertension, and sleep apnea. Consultation with a gastroenterologist, cardiologist, and pulmonologist is often necessary to formulate an appropriate treatment plan for the patient. Prognosis is related to the patient’s underlying disease, type of ILD, response to treatment, related comorbidities, as well as the patient’s education level and compliance with medical therapy (Table 3). The treating rheumatologist needs to actively educate patients, effectively communicate with patients and their families (including end-of-life considerations), and work with patients to develop individualized treatment plans.
Future Directions
The clinical incidence of CTD-ILD is high. Each type of CTD-ILD has its distinct clinical characteristics, therapeutic response, and prognosis. In general, the lung is one of the important organs involved in CTD where symptoms can first arise and where lung function is an independent prognostic indicator in CTD. In light of the heterogeneity and complexity of CTD-ILD and its clinical manifestations and presentations, a multi-disciplinary collaborative effort with other clinical specialists is often necessary to further our understanding of CTD-ILD and to develop individualized treatment plans.
Our collective goal is to improve the early diagnosis and treatment of CTD-ILD in order to improve the prognosis and survival of patients. Looking into the near future, advanced research technologies using high-throughput genomics, proteomics, and metabolomics together with artificial intelligence will further pave the way and provide insight in identifying relevant mechanistic pathways and molecular targets for drug development and disease interventions.
Author Contributions
TS, XS, PL, and ZS wrote the main manuscript text and prepared all figures. XL and JS provided the image data. SY, WZ, SA, AZ, PL, and ZS jointly supervised this work. All authors contributed to the article and approved the submitted version.
Funding
The Key Research and Development Projects of Anhui Province (1804h08020228).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary
AECAs: Anti-endothelial cell antibodies
anti-CCP: anti-cyclic citrullinated peptide
ARS: anti-aminoacyl-transfer RNA synthetase
ASS: anti-synthetase syndrome
BALF: bronchoalveolar lavage fluid
BECs: bronchial epithelial cells
CCL18: chemokine ligand 18
COP: cryptogenic organizing pneumonia
CTD: Connective tissue disease
CYC: cyclophosphamide
DAD: diffuse alveolar damage
DILD: drugs induced ILD
DIP: desquamative interstitial pneumonia
DLCO: diffusion capacity of the lung for carbon monoxide
DM: dermatomyositis
DMARDs: disease-modifying anti-rheumatic drugs
DPLDs: diffuse parenchymal lung diseases
EMT: epithelial-mesenchymal transition
FVC: forced vital capacity
GC: glucocorticoid
GERD: gastroesophageal reflux disease
GGOs: ground-glass opacities
HDAC: histone deacetylase
HRCT: high resolution computed topography
IIMs: idiopathic inflammatory myopathies
IIP: idiopathic interstitial pneumonia
ILD: interstitial lung disease
IPAF: interstitial pneumonia with autoimmune features
IPF: idiopathic pulmonary fibrosis
KL-6: Krebs von den Lungen-6
LIP: lymphoid interstitial pneumonia
LTL: leukocyte telomere length
MAAs: myositis-associated antibodies
MCTD: mixed connective tissue disease
MMPs: matrix metalloproteinases
MSAs: myositis-specific autoantibodies
MTX: methotrexate
NSIP: nonspecific interstitial pneumonia
OP: organizing pneumonia
PF-ILD: progressive fibrosing-ILD
PM: polymyositis
RA: rheumatoid arthritis
RB: respiratory bronchiolitis
RNP: ribonucleoprotein
RV: residual volume
SAFB: scaffold attachment factor B
SLE: systemic lupus erythematosus
SP-D: surfactant protein-D
SS: Sjogren’s syndrome
SSc: systemic sclerosis
TGF-β: transforming growth factor-β
TLC: total lung capacity
TLRs: Toll-like receptors
TREM-1: Triggering receptor expressed on myeloid cells type 1
UIP: usual interstitial pneumonia
ZEB1: zinc finger E-box-homeobox 1
References
1. Crestani B. The Respiratory System in Connective Tissue Disorders. Allergy (2005) 60:715–34. doi: 10.1111/j.1398-9995.2005.00761.x
2. Mira-Avendano I, Abril A, Burger CD, Dellaripa PF, Fischer A, Gotway MB, et al. Interstitial Lung Disease and Other Pulmonary Manifestations in Connective Tissue Diseases. Mayo Clin Proc (2019) 94:309–25. doi: 10.1016/j.mayocp.2018.09.002
3. Mathai SC, Danoff SK. Management of Interstitial Lung Disease Associated With Connective Tissue Disease. Bmj (2016) 352:h6819. doi: 10.1136/bmj.h6819
4. Korsten P, Konig MF, Tampe B, Mirsaeidi M. Interstitial Lung Disease in the Context of Systemic Disease: Pathophysiology, Treatment and Outcomes. Front Med (2020) 7:1138. doi: 10.3389/fmed.2020.644075
5. Sebastiani M, Faverio P, Manfredi A, Cassone G, Vacchi C, Stainer A, et al. Interstitial Pneumonia With Autoimmune Features: Why Rheumatologist-Pulmonologist Collaboration is Essential. Biomedicines (2020) 9:17. doi: 10.3390/biomedicines9010017
6. Juge PA, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B Promoter Variant and Rheumatoid Arthritis With Interstitial Lung Disease. N Engl J Med (2018) 379:2209–19. doi: 10.1056/NEJMoa1801562
7. Azadeh N, Limper AH, Carmona EM, Ryu JH. The Role of Infection in Interstitial Lung Diseases: A Review. Chest (2017) 152:842–52. doi: 10.1016/j.chest.2017.03.033
8. Kumar A, Cherian SV, Vassallo R, Yi ES, Ryu JH. Current Concepts in Pathogenesis, Diagnosis, and Management of Smoking-Related Interstitial Lung Diseases. Chest (2018) 154:394–408. doi: 10.1016/j.chest.2017.11.023
9. Lafyatis R, O’Hara C, Feghali-Bostwick CA, Matteson E. B Cell Infiltration in Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheum (2007) 56:3167–8. doi: 10.1002/art.22847
10. Karampitsakos T, Woolard T, Bouros D, Tzouvelekis A. Toll-Like Receptors in the Pathogenesis of Pulmonary Fibrosis. Eur J Pharmacol (2017) 808:35–43. doi: 10.1016/j.ejphar.2016.06.045
11. Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, et al. Effect of Telomere Length on Survival in Patients With Idiopathic Pulmonary Fibrosis: An Observational Cohort Study With Independent Validation. Lancet Respir Med (2014) 2:557–65. doi: 10.1016/s2213-2600(14)70124-9
12. Dai J, Cai H, Li H, Zhuang Y, Min H, Wen Y, et al. Association Between Telomere Length and Survival in Patients With Idiopathic Pulmonary Fibrosis. Respirology (2015) 20:947–52. doi: 10.1111/resp.12566
13. Snetselaar R, van Batenburg AA, van Oosterhout MFM, Kazemier KM, Roothaan SM, Peeters T, et al. Short Telomere Length in IPF Lung Associates With Fibrotic Lesions and Predicts Survival. PloS One (2017) 12:e0189467. doi: 10.1371/journal.pone.0189467
14. Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, et al. Telomere-Related Lung Fibrosis is Diagnostically Heterogeneous But Uniformly Progressive. Eur Respir J (2016) 48:1710–20. doi: 10.1183/13993003.00308-2016
15. Seibold MA, Wise AL, Speer MC, Steele MP, Schwartz DA. A Common Muc5b Promoter Polymorphism and Pulmonary Fibrosis. New Engl J Med (2011) 364:1503–12. doi: 10.1056/NEJMoa1013660
16. Zhang Y, Noth I, Garcia JGN, Kaminski N. A Variant in the Promoter of MUC5B and Idiopathic Pulmonary Fibrosis. N Engl J Med (2011) 364:1576–7. doi: 10.1056/NEJMc1013504
17. Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-Wide Association Study Identifies Multiple Susceptibility Loci for Pulmonary Fibrosis. Nat Genet (2013) 45:613–20. doi: 10.1038/ng.2609
18. Noth I, Zhang Y, Ma S-F, Flores C, Barber M, Huang Y, et al. Genetic Variants Associated With Idiopathic Pulmonary Fibrosis Susceptibility and Mortality: A Genome-Wide Association Study. Lancet Respir Med (2013)1:309–17. doi: 10.1016/S2213-2600(13)70045-6
19. Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C, et al. The MUC5B Variant is Associated With Idiopathic Pulmonary Fibrosis But Not With Systemic Sclerosis Interstitial Lung Disease in the European Caucasian Population. PloS One (2013) 8:e70621. doi: 10.1371/journal.pone.0070621
20. Horimasu Y, Ohshimo S, Bonella F, Tanaka S, Ishikawa N, Hattori N, et al. MUC5B Promoter Polymorphism in Japanese Patients With Idiopathic Pulmonary Fibrosis. Respirology (2015) 20:439–44. doi: 10.1111/resp.12466
21. Stock CJ, Sato H, Fonseca C, Banya WAS, Molyneaux PL, Adamali H, et al. Mucin 5B Promoter Polymorphism is Associated With Idiopathic Pulmonary Fibrosis But Not With Development of Lung Fibrosis in Systemic Sclerosis or Sarcoidosis. Thorax (2013) 68:436–41. doi: 10.1136/thoraxjnl-2012-201786
22. Lee MG, Lee YH. A Meta-Analysis Examining the Association Between the MUC5B Rs35705950 T/G Polymorphism and Susceptibility to Idiopathic Pulmonary Fibrosis. Inflammation Res (2015) 64:463–70. doi: 10.1007/s00011-015-0829-6
23. Van DV, Joanne J, Snetselaar R, Kazemier KM, ten Klooster L, Grutters JC, et al. Effect of Muc5b Promoter Polymorphism on Disease Predisposition and Survival in Idiopathic Interstitial Pneumonias. Respirology (2016) 21:712–7. doi: 10.1111/resp.12728
24. Newton CA, Oldham JM, Ley B, Anand V, Adegunsoye A, Liu G, et al. Telomere Length and Genetic Variant Associations With Interstitial Lung Disease Progression and Survival. Eur Respir J (2019) 53:1801641. doi: 10.1183/13993003.01641-2018
25. Evans CM, Fingerlin TE, Schwarz MI, Lynch D, Kurche J, Warg L, et al. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol Rev (2016) 96:1567–91. doi: 10.1152/physrev.00004.2016
26. Juge PA, Borie R, Kannengiesser C, Gazal S, Dieudé P. Shared Genetic Predisposition in Rheumatoid Arthritis-Interstitial Lung Disease and Familial Pulmonary Fibrosis. Eur Respir J (2017) 49:1602314. doi: 10.1136/annrheumdis-2017-eular.5237
27. Kelly CA, Saravanan V, Nisar M, Arthanari S, Woodhead FA, Price-Forbes AN, et al. Rheumatoid Arthritis-Related Interstitial Lung Disease: Associations, Prognostic Factors and Physiological and Radiological Characteristics—A Large Multicentre UK Study. Rheumatology (2014) 53:1676–82. doi: 10.1093/rheumatology/keu165
28. Doyle TJ, Dellaripa PF, Batra K, Frits ML, Iannaccone CK, Hatabu H, et al. Functional Impact of a Spectrum of Interstitial Lung Abnormalities in Rheumatoid Arthritis. Chest (2014) 146:41–50. doi: 10.1378/chest.13-1394
29. Fonseca C, Lindahl GE, Ponticos M, Sestini P, Abraham DJ. A Polymorphism in the CTGF Promoter Region Associated With Systemic Sclerosis. N Engl J Med (2007) 357:1210–20. doi: 10.1056/NEJMoa067655
30. Bossini-Castillo L, Simeon CP, Beretta L, Broen JC, Vonk MC, Ríos-Fernández R, et al. A Multicenter Study Confirms CD226 Gene Association With Systemic Sclerosis-Related Pulmonary Fibrosis. Arthritis Res Ther (2012) 14:1–7. doi: 10.1186/ar3809
31. Gorlova O, Martin JE, Rueda B, Koeleman BP, Ying J, Teruel M, et al. Identification of Novel Genetic Markers Associated With Clinical Phenotypes of Systemic Sclerosis Through a Genome-Wide Association Strategy. PloS Genet (2011) 7:e1002178. doi: 10.1371/journal.pgen.1002178
32. Stock CJW, Renzoni EA. Genetic Predictors of Systemic Sclerosis-Associated Interstitial Lung Disease: A Review of Recent Literature. Eur J Hum Genet (2018) 26:765–77. doi: 10.1038/s41431-018-0104-8
33. Furukawa H, Oka S, Shimada K, Tsuchiya N, Tohma S. Genetics of Interstitial Lung Disease: Vol De Nuit (Night Flight). Clin Med Insights Circ Respir Pulm Med (2015) 9:1–7. doi: 10.4137/ccrpm.s23283
34. Fingerlin TE, Zhang W, Yang IV, Ainsworth HC, Russell PH, Blumhagen RZ, et al. Genome-Wide Imputation Study Identifies Novel HLA Locus for Pulmonary Fibrosis and Potential Role for Auto-Immunity in Fibrotic Idiopathic Interstitial Pneumonia. BMC Genet (2016) 17:74. doi: 10.1186/s12863-016-0377-2
35. Chinoy H, Salway F, Fertig N, Shephard N, Tait BD, Thomson W, et al. In Adult Onset Myositis, the Presence of Interstitial Lung Disease and Myositis Specific/Associated Antibodies are Governed by HLA Class II Haplotype, Rather Than by Myositis Subtype. Arthritis Res Ther (2005) 8:R13. doi: 10.1186/ar1862
36. Gono T, Kawaguchi Y, Kuwana M, Sugiura T, Furuya T, Takagi K, et al. Brief Report: Association of HLA–DRB1* 0101/* 0405 With Susceptibility to Anti–Melanoma Differentiation–Associated Gene 5 Antibody–Positive Dermatomyositis in the Japanese Population. Arthritis Rheum (2012) 64:3736–40. doi: 10.1002/art.34657
37. Migita K, Nakamura T, Koga T, Eguchi K. Hla-DRB1 Alleles and Rheumatoid Arthritis-Related Pulmonary Fibrosis. J Rheumatol (2010) 37:205–7. doi: 10.3899/jrheum.090303
38. Furukawa H, Oka S, Shimada K, Sugii S, Ohashi J, Matsui T, et al. Association of Human Leukocyte Antigen With Interstitial Lung Disease in Rheumatoid Arthritis: A Protective Role for Shared Epitope. PloS One (2012) 7:e33133. doi: 10.1371/journal.pone.0033133
39. Mori S, Koga Y, Sugimoto M. Different Risk Factors Between Interstitial Lung Disease and Airway Disease in Rheumatoid Arthritis. Respir Med (2012) 106:1591–9. doi: 10.1016/j.rmed.2012.07.006
40. Gladman DD, Kung TN, Siannis F, Pellett F, Farewell VT, Lee P. HLA Markers for Susceptibility and Expression in Scleroderma. J Rheumatol (2005) 32:1481–7. doi: 10.1097/01.rhu.0000173620.95740.e2
41. Simeón CP, Fonollosa V, Tolosa C, Palou E, Selva A, Solans R, et al. Association of HLA Class II Genes With Systemic Sclerosis in Spanish Patients. J Rheumatol (2009) 36:2733–6. doi: 10.3899/jrheum.090377
42. Tikly M, Rands A, McHugh N, Wordsworth P, Welsh K. Human Leukocyte Antigen Class II Associations With Systemic Sclerosis in South Africans. Tissue Antigens (2004) 63:487–90. doi: 10.1111/j.0001-2815.2004.00199.x
43. Wang J, Guo X, Yi L, Guo G, Tu W, Wu W, et al. Association of HLA-DPB1 With Scleroderma and its Clinical Features in Chinese Population. PloS One (2014) 9:e87363. doi: 10.1371/journal.pone.0087363
44. Odani T, Yasuda S, Ota Y, Fujieda Y, Kon Y, Horita T, et al. Up-Regulated Expression of HLA-DRB5 Transcripts and High Frequency of the HLA-DRB5* 01: 05 Allele in Scleroderma Patients With Interstitial Lung Disease. Rheumatology (2012) 51:1765–74. doi: 10.1093/rheumatology/kes149
45. Bossini-Castillo L, Simeon CP, Beretta L, Broen JC, Vonk MC, Ríos-Fernández R, et al. A Multicenter Study Confirms CD226 Gene Association With Systemic Sclerosis-Related Pulmonary Fibrosis. Arthritis Res Ther (2012) 14:R85. doi: 10.1186/ar3809
46. Manetti M, Ibba-Manneschi L, Fatini C, Guiducci S, Cuomo G, Bonino C, et al. Association of a Functional Polymorphism in the Matrix metalloproteinase-12 Promoter Region With Systemic Sclerosis in an Italian Population. J Rheumatol (2010) 37:1852–7. doi: 10.3899/jrheum.100237
47. Sumita Y, Sugiura T, Kawaguchi Y, Baba S, Soejima M, Murakawa Y, et al. Genetic Polymorphisms in the Surfactant Proteins in Systemic Sclerosis in Japanese: T/T Genotype at 1580 C/T (Thr131Ile) in the SP-B Gene Reduces the Risk of Interstitial Lung Disease. Rheumatology (2008) 47:289–91. doi: 10.1093/rheumatology/kem355
48. Hoshino K, Satoh T, Kawaguchi Y, Kuwana M. Association of Hepatocyte Growth Factor Promoter Polymorphism With Severity of Interstitial Lung Disease in Japanese Patients With Systemic Sclerosis. Arthritis Rheum (2011) 63:2465–72. doi: 10.1002/art.30415
49. Dieude P, Bouaziz M, Guedj M, Riemekasten G, Airo P, Müller M, et al. Evidence of the Contribution of the X Chromosome to Systemic Sclerosis Susceptibility: Association With the Functional IRAK1 196Phe/532Ser Haplotype. Arthritis Rheum (2011) 63:3979–87. doi: 10.1002/art.30640
50. Bredemeier M, Chies JAB, Wieck A, Capobianco KG, Pitrez EH, Rohde LEP, et al. TCRBV20S1 and TCRBV3S1 Gene Segment Polymorphisms in Systemic Sclerosis. J Rheumatol (2008) 35:1058–63. doi: 10.1097/RHU.0b013e3181778cb5
51. Sharif R, Mayes MD, Tan FK, Gorlova OY, Hummers LK, Shah AA, et al. IRF5 Polymorphism Predicts Prognosis in Patients With Systemic Sclerosis. Ann Rheum Dis (2012) 71:1197–202. doi: 10.1136/annrheumdis-2011-200901
52. Adegunsoye A, Vij R, Noth I. Integrating Genomics Into Management of Fibrotic Interstitial Lung Disease. Chest (2019) 155:1026–40. doi: 10.1016/j.chest.2018.12.011
53. Evans IC, Barnes JL, Garner IM, Pearce DR, Maher TM, Shiwen X, et al. Epigenetic Regulation of Cyclooxygenase-2 by Methylation of c8orf4 in Pulmonary Fibrosis. Clin Sci (Lond) (2016) 130:575–86. doi: 10.1042/cs20150697
54. Luo QK, Zhang H, Li L. Research Advances on DNA Methylation in Idiopathic Pulmonary Fibrosis. Adv Exp Med Biol (2020) 1255):73–81. doi: 10.1007/978-981-15-4494-1_6
55. Sanders YY, Ambalavanan N, Halloran B, Zhang X, Liu H, Crossman DK, et al. Altered DNA Methylation Profile in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2012) 186:525–35. doi: 10.1164/rccm.201201-0077OC
56. Zhuang W, Li Z, Dong X, Zhao N, Liu Y, Wang C, et al. Schisandrin B Inhibits TGF-β1-Induced Epithelial-Mesenchymal Transition in Human A549 Cells Through Epigenetic Silencing of ZEB1. Exp Lung Res (2019) 45:157–66. doi: 10.1080/01902148.2019.1631906
57. Ota C, Yamada M, Fujino N, Motohashi H, Tando Y, Takei Y, et al. Histone Deacetylase Inhibitor Restores Surfactant Protein-C Expression in Alveolar-Epithelial Type II Cells and Attenuates Bleomycin-Induced Pulmonary Fibrosis In Vivo. Exp Lung Res (2015) 41:422–34. doi: 10.3109/01902148.2015.1060275
58. Guo W, Shan B, Klingsberg RC, Qin X, Lasky JA. Abrogation of TGF-beta1-induced Fibroblast-Myofibroblast Differentiation by Histone Deacetylase Inhibition. Am J Physiol Lung Cell Mol Physiol (2009) 297:L864–70. doi: 10.1152/ajplung.00128.2009
59. Huang SK, Scruggs AM, Donaghy J, Horowitz JC, Zaslona Z, Przybranowski S, et al. Histone Modifications are Responsible for Decreased Fas Expression and Apoptosis Resistance in Fibrotic Lung Fibroblasts. Cell Death Dis (2013) 4:e621. doi: 10.1038/cddis.2013.146
60. Pandit KV, Milosevic J, Kaminski N. MicroRNAs in Idiopathic Pulmonary Fibrosis. Transl Res (2011) 157:191–9. doi: 10.1016/j.trsl.2011.01.012
61. Yang S, Banerjee S, de Freitas A, Sanders YY, Ding Q, Matalon S, et al. Participation of miR-200 in Pulmonary Fibrosis. Am J Pathol (2012) 180:484–93. doi: 10.1016/j.ajpath.2011.10.005
62. Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, et al. Inhibition and Role of let-7d in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2010) 182:220–9. doi: 10.1164/rccm.200911-1698OC
63. Pottier N, Maurin T, Chevalier B, Puisségur M-P, Lebrigand K, Robbe-Sermesant K, et al. Identification of Keratinocyte Growth Factor as a Target of microRNA-155 in Lung Fibroblasts: Implication in Epithelial-Mesenchymal Interactions. PloS One (2009) 4:e6718. doi: 10.1371/journal.pone.0006718
64. Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, et al. miR-21 Mediates Fibrogenic Activation of Pulmonary Fibroblasts and Lung Fibrosis. J Exp Med (2010) 207:1589–97. doi: 10.1084/jem.20100035
65. Moore BB, Moore TA. Viruses in Idiopathic Pulmonary Fibrosis. Etiology and Exacerbation. Ann Am Thoracic Soc (2015) 12:S186–92. doi: 10.1513/AnnalsATS.201502-088AW
66. Roubille C, Haraoui B. Interstitial Lung Diseases Induced or Exacerbated by DMARDS and Biologic Agents in Rheumatoid Arthritis: A Systematic Literature Review. Semin Arthritis Rheum (2014) 43:613–26. doi: 10.1016/j.semarthrit.2013.09.005
67. Mahmood T, Cuevas J, Huizar I, Nugent K. The Effect of Disease Modifying Drugs on the Lung in Patients With Rheumatoid Arthritis. Southwest Respir Crit Care Chronicles (2016) 4:3–11. doi: 10.12746/swrccc2016.0413.166
68. Skeoch S, Weatherley N, Swift AJ, Oldroyd A, Johns C, Hayton C, et al. Drug-Induced Interstitial Lung Disease: A Systematic Review. J Clin Med (2018) 7:356. doi: 10.3390/jcm7100356
69. Margaritopoulos GA, Harari S, Caminati A, Antoniou KM. Smoking-Related Idiopathic Interstitial Pneumonia: A Review. Respirology (2016) 21:57–64. doi: 10.1111/resp.12576
70. Caminati A, Graziano P, Sverzellati N, Harari S. Smoking-Related Interstitial Lung Diseases. Pathologica (2010) 102:525–36.
71. Tobin RW, Pope CE, Pellegrini CA, Emond MJ, J. Sillery and G. Raghu: Increased Prevalence of Gastroesophageal Reflux in Patients With Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (1998) 158:1804–8. doi: 10.1164/ajrccm.158.6.9804105
72. Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in Idiopathic Pulmonary Fibrosis Patients: A Systematic Literature Review. Eur Respir J (2015) 46:1113–30. doi: 10.1183/13993003.02316-2014
73. Raghu G, Freudenberger T, Yang S, Curtis J, Spada C, Hayes J, et al. High Prevalence of Abnormal Acid Gastro-Oesophageal Reflux in Idiopathic Pulmonary Fibrosis. Eur Respir J (2006) 27:136–42. doi: 10.1183/09031936.06.00037005
74. Appel JZ, Lee SM, Hartwig MG, Li B, Hsieh C-C, Cantu E, et al. Characterization of the Innate Immune Response to Chronic Aspiration in a Novel Rodent Model. Respir Res (2007) 8:87. doi: 10.1186/1465-9921-8-87
75. Johannson KA, Strâmbu I, Ravaglia C, Grutters JC, Valenzuela C, Mogulkoc N, et al. Antacid Therapy in Idiopathic Pulmonary Fibrosis: More Questions Than Answers? Lancet Respir Med (2017) 5:591–8. doi: 10.1016/S2213-2600(17)30219-9
76. Amigoni M, Bellani G, Scanziani M, Masson S, Bertoli E, Radaelli E, et al. Lung Injury and Recovery in a Murine Model of Unilateral Acid Aspiration: Functional, Biochemical, and Morphologic Characterization. J Am Soc Anesthesiologists (2008) 108:1037–46. doi: 10.1097/ALN.0b013e318173f64f
77. Perng D-W, Chang K-T, Su K-C, Wu Y-C, Wu M-T, Hsu W-H, et al. Exposure of Airway Epithelium to Bile Acids Associated With Gastroesophageal Reflux Symptoms: A Relation to Transforming Growth Factor-β1 Production and Fibroblast Proliferation. Chest (2007) 132:1548–56. doi: 10.1378/chest.07-1373
78. Méthot DB, Leblanc É, Lacasse Y. Meta-Analysis of Gastroesophageal Reflux Disease and Idiopathic Pulmonary Fibrosis. Chest (2019) 155:33–43. doi: 10.1016/j.chest.2018.07.038
79. Adriani A, Repici A, Hickman I, Pellicano R. Helicobacter Pylori Infection and Respiratory Diseases: Actual Data and Directions for Future Studies. Minerva Med (2014) 105:1–8. doi: 10.3325/cmj.2014.55.83
80. Ibrahim W. Helicobacter Pylori Eradication in the Management of Idiopathic Pulmonary Fibrosis. Eur Respir J (2007) 30:395–6. doi: 10.1183/09031936.00033907
81. Zanetti F, Giacomello M, Donati Y, Carnesecchi S, Frieden M, Barazzone-Argiroffo C. Nicotine Mediates Oxidative Stress and Apoptosis Through Cross Talk Between NOX1 and Bcl-2 in Lung Epithelial Cells. Free Radic Biol Med (2014) 76:173–84. doi: 10.1016/j.freeradbiomed.2014.08.002
82. Stevenson CS, Docx C, Webster R, Battram C, Hynx D, Giddings J, et al. Comprehensive Gene Expression Profiling of Rat Lung Reveals Distinct Acute and Chronic Responses to Cigarette Smoke Inhalation. Am J Physiol Lung Cell Mol Physiol (2007) 293:L1183–93. doi: 10.1152/ajplung.00105.2007
83. Criswell LA, Merlino LA, Cerhan JR, Mikuls TR, Mudano AS, Burma M, et al. Cigarette Smoking and the Risk of Rheumatoid Arthritis Among Postmenopausal Women: Results From the Iowa Women’s Health Study. Am J Med (2002) 112:465–71. doi: 10.1016/s0002-9343(02)01051-3
84. Stolt P, Bengtsson C, Nordmark B, Lindblad S, Lundberg I, Klareskog L, et al. Quantification of the Influence of Cigarette Smoking on Rheumatoid Arthritis: Results From a Population Based Case-Control Study, Using Incident Cases. Ann Rheum Dis (2003) 62:835–41. doi: 10.1136/ard.62.9.835
85. Liu X, Tedeschi SK, Barbhaiya M, Leatherwood CL, Speyer CB, Lu B, et al. Impact and Timing of Smoking Cessation on Reducing Risk of Rheumatoid Arthritis Among Women in the Nurses’ Health Studies. Arthritis Care Res (Hoboken) (2019) 71:914–24. doi: 10.1002/acr.23837
86. Margaritopoulos GA, Antoniou KM, Hansell D, Rubens MB, Desai S, Siafakas NM, et al. The Role of Smoking in the Pathogenesis of Systemic Sclerosis Associated Lung Fibrosis. In: American Thoracic Society International Conference. (2012). p. A6607–7.
87. Hudson M, Lo E, Lu Y, Hercz D, Baron M, Steele R. Cigarette Smoking in Patients With Systemic Sclerosis. Arthritis Rheum (2011) 63:230–8. doi: 10.1002/art.30071
88. Hissaria P, Roberts-Thomson PJ, Lester S, Ahern MJ, Smith MD, Walker JG. Cigarette Smoking in Patients With Systemic Sclerosis Reduces Overall Survival: Comment on the Article by Hudson et al. Arthritis Rheum (2011) 63:1758–9. doi: 10.1002/art.30352
89. Schwaiblmair M, Behr W, Haeckel T, Märkl B, Foerg W, Berghaus T. Drug Induced Interstitial Lung Disease. Open Respir Med J (2012) 6:63. doi: 10.2174/1874306401206010063
90. Matsuno O. Drug-Induced Interstitial Lung Disease: Mechanisms and Best Diagnostic Approaches. Respir Res (2012) 13:39. doi: 10.1186/1465-9921-13-39
91. Furukawa H, Oka S, Shimada K, Tsuchiya N, Tohma S, Consortium R. A.-I. L. D. S. HLA-A* 31: 01 and Methotrexate-Induced Interstitial Lung Disease in Japanese Rheumatoid Arthritis Patients: A Multidrug Hypersensitivity Marker? Ann Rheum Dis (2013) 72:153–5. doi: 10.1136/annrheumdis-2012-201944
92. Nakamura H, Kanazawa M. Definition and Pathogenesis of Drug-Induced Lung Injury: What Is DLI? In: Drug Induced Lung Injury Springer. Springer Nature Singapore Pte Ltd (2018). pp. 3–12. doi: 10.1007/978-981-10-4466-3_1
93. Alarcón GS, Kremer JM, Macaluso M, Weinblatt ME, Cannon GW, Palmer WR, et al. Risk Factors for Methotrexate-Induced Lung Injury in Patients With Rheumatoid Arthritis. A Multicenter, Case-Control Study. Methotrexate-Lung Study Group. Ann Intern Med (1997) 127:356–64. doi: 10.7326/0003-4819-127-5-199709010-00003
94. Sinha A, Silverstone EJ, O’Sullivan MM. Gold-Induced Pneumonitis: Computed Tomography Findings in a Patient With Rheumatoid Arthritis. Rheumatol (Oxford) (2001) 40:712–4. doi: 10.1093/rheumatology/40.6.712
95. Lansdown AB. GOLD: Human Exposure and Update on Toxic Risks. Crit Rev Toxicol (2018) 48:596–614. doi: 10.1080/10408444.2018.1513991
96. Scott DL, Bradby GV, Aitman TJ, Zaphiropoulos GC, Hawkins CF. Relationship of Gold and Penicillamine Therapy to Diffuse Interstitial Lung Disease. Ann Rheum Dis (1981) 40:136–41. doi: 10.1136/ard.40.2.136
97. Hamadeh MA, Atkinson J, Smith LJ. Sulfasalazine-Induced Pulmonary Disease. Chest (1992) 101:1033–7. doi: 10.1378/chest.101.4.1033
98. Koike R, Tanaka M, Komano Y, Sakai F, Sugiyama H, Nanki T, et al. Tacrolimus-Induced Pulmonary Injury in Rheumatoid Arthritis Patients. Pulm Pharmacol Ther (2011) 24:401–6. doi: 10.1016/j.pupt.2011.01.016
99. Sasaki T, Nakamura W, Inokuma S, Matsubara E. Characteristic Features of Tacrolimus-Induced Lung Disease in Rheumatoid Arthritis Patients. Clin Rheumatol (2016) 35:541–5. doi: 10.1007/s10067-015-2865-6
100. Conway R, Low C, Coughlan RJ, O’Donnell MJ, Carey JJ. Methotrexate Use and Risk of Lung Disease in Psoriasis, Psoriatic Arthritis, and Inflammatory Bowel Disease: Systematic Literature Review and Meta-Analysis of Randomised Controlled Trials. Bmj (2015) 350:h1269. doi: 10.1136/bmj.h1269
101. Conway R, Low C, Coughlan RJ, O’Donnell MJ, Carey JJ. Methotrexate and Lung Disease in Rheumatoid Arthritis: A Meta-Analysis of Randomized Controlled Trials. Arthritis Rheumatol (2014) 66:803–12. doi: 10.1002/art.38322
102. Inokuma S, Kono H, Kohno Y, Hiramatsu K, Ito K, Shiratori K, et al. Methotrexate-Induced Lung Injury in Patients With Rheumatoid Arthritis Occurs With Peripheral Blood Lymphocyte Count Decrease. Ann Rheum Dis (2006) 65:1113–4. doi: 10.1136/ard.2005.045211
103. Chikura B, Lane S, Dawson JK. Clinical Expression of Leflunomide-Induced Pneumonitis. Rheumatol (Oxford) (2009) 48:1065–8. doi: 10.1093/rheumatology/kep050
104. Sato T, Inokuma S, Sagawa A, Matsuda T, Takemura T, Otsuka T, et al. Factors Associated With Fatal Outcome of Leflunomide-Induced Lung Injury in Japanese Patients With Rheumatoid Arthritis. Rheumatol (Oxford) (2009) 48:1265–8. doi: 10.1093/rheumatology/kep227
105. Sawada T, Inokuma S, Sato T, Otsuka T, Saeki Y, Takeuchi T, et al. Leflunomide-Induced Interstitial Lung Disease: Prevalence and Risk Factors in Japanese Patients With Rheumatoid Arthritis. Rheumatol (Oxford) (2009) 48:1069–72. doi: 10.1093/rheumatology/kep052
106. Lioté H, Lioté F, Séroussi B, Mayaud C, Cadranel J. Rituximab-Induced Lung Disease: A Systematic Literature Review. Eur Respir J (2010) 35:681–7. doi: 10.1183/09031936.00080209
107. Curtis JR, Sarsour K, Napalkov P, Costa LA, Schulman KL. Incidence and Complications of Interstitial Lung Disease in Users of Tocilizumab, Rituximab, Abatacept and Anti-Tumor Necrosis Factor α Agents, a Retrospective Cohort Study. Arthritis Res Ther (2015) 17:319. doi: 10.1186/s13075-015-0835-7
108. Fragoulis GE, Conway R, Nikiphorou E. Methotrexate and Interstitial Lung Disease: Controversies and Questions. A Narrative Review of the Literature. Rheumatol (Oxford) (2019) 58:1900–6. doi: 10.1093/rheumatology/kez337
109. Ibfelt EH, Jacobsen RK, Kopp TI, Cordtz RL, Jakobsen AS, Seersholm N, et al. Methotrexate and Risk of Interstitial Lung Disease and Respiratory Failure in Rheumatoid Arthritis: A Nationwide Population-Based Study. Rheumatol (Oxford) (2021) 60:346–52. doi: 10.1093/rheumatology/keaa327
110. Dawson J, Graham D, Desmond J, Fewins H, Lynch* M. Investigation of the Chronic Pulmonary Effects of Low-Dose Oral Methotrexate in Patients With Rheumatoid Arthritis: A Prospective Study Incorporating HRCT Scanning and Pulmonary Function Tests. Rheumatology (2002) 41:262–7. doi: 10.1093/rheumatology/41.3.262
111. Kiely P, Busby AD, Nikiphorou E, Sullivan K, Walsh DA, Creamer P, et al. Is Incident Rheumatoid Arthritis Interstitial Lung Disease Associated With Methotrexate Treatment? Results From a Multivariate Analysis in the ERAS and ERAN Inception Cohorts. BMJ Open (2019) 9:e028466. doi: 10.1136/bmjopen-2018-028466
112. Deane KD, Nicolls MR. Developing Better Biomarkers for Connective Tissue Disease-Associated Interstitial Lung Disease: Citrullinated hsp90 Autoantibodies in Rheumatoid Arthritis. Arthritis Rheum (2013) 65:864–8. doi: 10.1002/art.37878
113. Turesson C, Matteson EL, Colby TV, Vuk-Pavlovic Z, Vassallo R, Weyand CM, et al. Increased Cd4+ T Cell Infiltrates in Rheumatoid Arthritis-Associated Interstitial Pneumonitis Compared With Idiopathic Interstitial Pneumonitis. Arthritis Rheum (2005) 52:73–9. doi: 10.1002/art.20765
114. Atkins SR, Turesson C, Myers JL, Tazelaar HD, Ryu JH, Matteson EL, et al. Morphologic and Quantitative Assessment of CD20+ B Cell Infiltrates in Rheumatoid Arthritis-Associated Nonspecific Interstitial Pneumonia and Usual Interstitial Pneumonia. Arthritis Rheum (2006) 54:635–41. doi: 10.1002/art.21758
115. Wollin L, Ostermann A, Williams C. Nintedanib Inhibits Pro-Fibrotic Mediators From T Cells With Relevance to Connective Tissue Disease-Associated Interstitial Lung Disease. In: Eur Respir Soc (2017) 50:PA903. doi: 10.1183/1393003.congress-2017.PA903
116. Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, et al. Alternatively Activated Alveolar Macrophages in Pulmonary Fibrosis—Mediator Production and Intracellular Signal Transduction. Clin Immunol (2010) 137:89–101. doi: 10.1016/j.clim.2010.06.017
117. Bonella F, Costabel U. Biomarkers in Connective Tissue Disease-Associated Interstitial Lung Disease. Semin Respir Crit Care Med (2014) 35:181–200. doi: 10.1055/s-0034-1371527
118. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-Based Guidelines for Diagnosis and Management. Am J Respir Crit Care Med (2011) 183:788–824. doi: 10.1164/rccm.2009-040GL
119. Yang HZ, Cui B, Liu HZ, Chen ZR, Yan HM, Hua F, et al. Targeting TLR2 Attenuates Pulmonary Inflammation and Fibrosis by Reversion of Suppressive Immune Microenvironment. J Immunol (2009) 182:692–702. doi: 10.4049/jimmunol.182.1.692
120. Meneghin A, Choi ES, Evanoff HL, Kunkel SL, Martinez FJ, Flaherty KR, et al. TLR9 is Expressed in Idiopathic Interstitial Pneumonia and its Activation Promotes In Vitro Myofibroblast Differentiation. Histochem Cell Biol (2008) 130:979–92. doi: 10.1007/s00418-008-0466-z
121. O’Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, et al. The Toll-like Receptor 3 L412F Polymorphism and Disease Progression in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2013) 188:1442–50. doi: 10.1164/rccm.201304-0760OC
122. He Z, Gao Y, Deng Y, Li W, Chen Y, Xing S, et al. Lipopolysaccharide Induces Lung Fibroblast Proliferation Through Toll-like Receptor 4 Signaling and the Phosphoinositide3-kinase-Akt Pathway. PloS One (2012) 7:e35926. doi: 10.1371/journal.pone.0035926
123. Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, et al. Hyaluronan and TLR4 Promote surfactant-protein-C-positive Alveolar Progenitor Cell Renewal and Prevent Severe Pulmonary Fibrosis in Mice. Nat Med (2016) 22:1285–93. doi: 10.1038/nm.4192
124. Margaritopoulos GA, Antoniou KM, Karagiannis K, Samara KD, Lasithiotaki I, Vassalou E, et al. Investigation of Toll-like Receptors in the Pathogenesis of Fibrotic and Granulomatous Disorders: A Bronchoalveolar Lavage Study. Fibrogenesis Tissue Repair (2010) 3:20. doi: 10.1186/1755-1536-3-20
125. Samara KD, Antoniou KM, Karagiannis K, Margaritopoulos G, Lasithiotaki I, Koutala E, et al. Expression Profiles of Toll-like Receptors in non-Small Cell Lung Cancer and Idiopathic Pulmonary Fibrosis. Int J Oncol (2012) 40:1397–404. doi: 10.3892/ijo.2012.1374
126. Papanikolaou IC, Boki KA, Giamarellos-Bourboulis EJ, Kotsaki A, Kagouridis K, Karagiannidis N, et al. Innate Immunity Alterations in Idiopathic Interstitial Pneumonias and Rheumatoid Arthritis-Associated Interstitial Lung Diseases. Immunol Lett (2015) 163:179–86. doi: 10.1016/j.imlet.2014.12.004
127. Jindal SK, Agarwal R. Autoimmunity and Interstitial Lung Disease. Curr Opin Pulm Med (2005) 11:438–46. doi: 10.1097/01.mcp.0000170522.71497.61
128. Wells AU, Denton CP. Interstitial Lung Disease in Connective Tissue Disease—Mechanisms and Management. Nat Rev Rheumatol (2014) 10:728. doi: 10.1038/nrrheum.2014.149
129. Doyle TJ, Dellaripa PF. Lung Manifestations in the Rheumatic Diseases. Chest (2017) 152:1283–95. doi: 10.1016/j.chest.2017.05.015
130. Travis WD, Costabel U, Hansell DM, King TE Jr., Lynch DA, Nicholson AG, et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med (2013) 188:733–48. doi: 10.1164/rccm.201308-1483ST
131. Nagai S, Handa T, Tabuena R, Kitaichi M, Izumi T. Nonspecific Interstitial Pneumonia: A Real Clinical Entity? Clin Chest Med (2004) 25:705–15, vi. doi: 10.1016/j.ccm.2004.04.009
132. Cipriani NA, Strek M, Noth I, Gordon IO, Charbeneau J, Krishnan JA, et al. Pathologic Quantification of Connective Tissue Disease-Associated Versus Idiopathic Usual Interstitial Pneumonia. Arch Pathol Lab Med (2012) 136:1253–8. doi: 10.5858/arpa.2012-0102-OA
133. Cipriani NA, Strek M, Noth I, Gordon IO, Charbeneau J, Krishnan JA, et al. Pathologic Quantification of Connective Tissue Disease–Associated Versus Idiopathic Usual Interstitial Pneumonia. Arch Pathol Lab Med (2012) 136:1253–8. doi: 10.5858/arpa.2012-0102-OA
134. Tzelepis GE, Toya SP, Moutsopoulos HM. Occult Connective Tissue Diseases Mimicking Idiopathic Interstitial Pneumonias. Eur Respir J (2008) 31:11–20. doi: 10.1183/09031936.00060107
135. Parambil JG, Myers JL, Lindell RM, Matteson EL, Ryu JH. Interstitial Lung Disease in Primary Sjogren Syndrome. Chest (2006) 130:1489–95. doi: 10.1378/chest.130.5.1489
136. Kim EJ, Collard HR, King TE Jr. : Rheumatoid Arthritis-Associated Interstitial Lung Disease: The Relevance of Histopathologic and Radiographic Pattern. Chest (2009) 136:1397–405. doi: 10.1378/chest.09-0444
137. Shi JH, Liu HR, Xu WB, Feng RE, Zhang ZH, Tian XL, et al. Pulmonary Manifestations of Sjogren’s Syndrome. Respiration (2009) 78:377–86. doi: 10.1159/000214841
138. Song JW, Do KH, Kim MY, Jang SJ, Colby TV, Kim DS. Pathologic and Radiologic Differences Between Idiopathic and Collagen Vascular Disease-Related Usual Interstitial Pneumonia. Chest (2009) 136:23–30. doi: 10.1378/chest.08-2572
139. Fischer A, West SG, Swigris JJ, Brown KK, du Bois RM. Connective Tissue Disease-Associated Interstitial Lung Disease: A Call for Clarification. Chest (2010) 138:251–6. doi: 10.1378/chest.10-0194
140. Kim HC, Song JS, Park S, Yoon HY, Lim SY, Chae EJ, et al. Histologic Features Suggesting Connective Tissue Disease in Idiopathic Pulmonary Fibrosis. Sci Rep (2020) 10:21137. doi: 10.1038/s41598-020-78140-5
141. Bouros D, Nicholson A, Polychronopoulos V, Du Bois R. Acute Interstitial Pneumonia. Eur Respir J (2000) 15:412–8. doi: 10.1034/j.1399-3003.2000.15b31.x
142. Chandra D, Maini R, Hershberger DM. Cryptogenic Organizing Pneumonia. In: Statpearls. Treasure Island (FL: StatPearls Publishing Copyright © (2021) StatPearls Publishing LLC. (2021).
143. Zare Mehrjardi M, Kahkouee S, Pourabdollah M. Radio-Pathological Correlation of Organizing Pneumonia (OP): A Pictorial Review. Br J Radiol (2017) 90:20160723. doi: 10.1259/bjr.20160723
144. Onishi Y, Kawamura T, Higashino T, Mimura R, Tsukamoto H, Sasaki S. Clinical Features of Acute Fibrinous and Organizing Pneumonia: An Early Histologic Pattern of Various Acute Inflammatory Lung Diseases. PloS One (2021) 16:e0249300. doi: 10.1371/journal.pone.0249300
145. Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr., et al. Acute Exacerbations of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2007) 176:636–43. doi: 10.1164/rccm.200703-463pp
146. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic Pulmonary Fibrosis. Nat Rev Dis Primers (2017) 3:17074. doi: 10.1038/nrdp.2017.74
147. Travis WD, Matsui K, Moss J, Ferrans VJ. Idiopathic Nonspecific Interstitial Pneumonia: Prognostic Significance of Cellular and Fibrosing Patterns: Survival Comparison With Usual Interstitial Pneumonia and Desquamative Interstitial Pneumonia. Am J Surg Pathol (2000) 24:19. doi: 10.1097/00000478-200001000-00003
148. Travis WD, Hunninghake G, King TE Jr., Lynch DA, Colby TV, Galvin JR, et al. Idiopathic Nonspecific Interstitial Pneumonia: Report of an American Thoracic Society Project. Am J Respir Crit Care Med (2008) 177:1338–47. doi: 10.1164/rccm.200611-1685oc
149. Swigris JJ, Berry GJ, Raffin TA, Kuschner WG. Lymphoid Interstitial Pneumonia: A Narrative Review. Chest (2002) 122:2150–64. doi: 10.1378/chest.122.6.2150
150. Panchabhai TS, Farver C, Highland KB. Lymphocytic Interstitial Pneumonia. Clin Chest Med (2016) 37:463–74. doi: 10.1016/j.ccm.2016.04.009
151. Henry TS, Little BP, Veeraraghavan S, Bhalla S, Elicker BM. The Spectrum of Interstitial Lung Disease in Connective Tissue Disease. J Thoracic Imaging (2016) 31:65–77. doi: 10.1097/RTI.0000000000000191
152. Bryson T, Sundaram B, Khanna D, Kazerooni EA. Connective Tissue Disease-Associated Interstitial Pneumonia and Idiopathic Interstitial Pneumonia: Similarity and Difference. Semin Ultrasound CT MR (2014) 35:29–38. doi: 10.1053/j.sult.2013.10.010
153. Debray M-P, Borie R, Revel M-P, Naccache J-M, Khalil A, Toper C, et al. Interstitial Lung Disease in Anti-Synthetase Syndrome: Initial and Follow-Up CT Findings. Eur J Radiol (2015) 84:516–23. doi: 10.1016/j.ejrad.2014.11.026
154. Tillie-Leblond I, Wislez M, Valeyre D, Crestani B, Rabbat A, Israel-Biet D, et al. Interstitial Lung Disease and anti-Jo-1 Antibodies: Difference Between Acute and Gradual Onset. Thorax (2008) 63:53–9. doi: 10.1136/thx.2006.069237
155. Chung JH, Cox CW, Montner SM, Adegunsoye A, Oldham JM, Husain AN, et al. CT Features of the Usual Interstitial Pneumonia Pattern: Differentiating Connective Tissue Disease–Associated Interstitial Lung Disease From Idiopathic Pulmonary Fibrosis. Am J Roentgenol (2018) 210:307–13. doi: 10.2214/AJR.17.18384
156. Chawla A, Lim TC, Krishnan V, Tsen CG. Imaging of Interstitial Lung Diseases. In: Thoracic Imaging. Singapore: Springer (2019). p. 361–423.
157. Nihtyanova SI, Schreiber BE, Ong VH, Rosenberg D, Moinzadeh P, Coghlan JG, et al. Prediction of Pulmonary Complications and Long-Term Survival in Systemic Sclerosis. Arthritis Rheumatol (2014) 66:1625–35. doi: 10.1002/art.38390
158. Nihtyanova SI, Denton CP. Autoantibodies as Predictive Tools in Systemic Sclerosis. Nat Rev Rheumatol (2010) 6:112. doi: 10.1038/nrrheum.2009.238
159. Yamakawa H, Hagiwara E, Kitamura H, Yamanaka Y, Ikeda S, Sekine A, et al. Clinical Features of Idiopathic Interstitial Pneumonia With Systemic Sclerosis-Related Autoantibody in Comparison With Interstitial Pneumonia With Systemic Sclerosis. PloS One (2016) 11:e0161908. doi: 10.1371/journal.pone.0161908
160. Wodkowski M, Hudson M, Proudman S, Walker J, Stevens W, Nikpour M, et al. Monospecific Anti-Ro52/TRIM21 Antibodies in a Tri-Nation Cohort of 1574 Systemic Sclerosis Subjects: Evidence of an Association With Interstitial Lung Disease and Worse Survival. Clin Exp Rheumatol (2015) 33:S131–5. doi: 10.1016/j.canrad.2006.09.088
161. Cotton CV, Spencer LG, New RP, Cooper RG. The Utility of Comprehensive Autoantibody Testing to Differentiate Connective Tissue Disease Associated and Idiopathic Interstitial Lung Disease Subgroup Cases. Rheumatology (2017) 56:1264–71. doi: 10.1093/rheumatology/kew320
162. Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. The Prevalence and Clinical Significance of Anti-U1 Rna Antibodies in Patients With Systemic Sclerosis. J Invest Dermatol (2003) 120:204–10. doi: 10.1046/j.1523-1747.2003.12028.x
163. Hesselstrand and R. The Association of Antinuclear Antibodies With Organ Involvement and Survival in Systemic Sclerosis. Rheumatology (2003) 42:534–40. doi: 10.1093/rheumatology/keg170
164. Sabbagh S, Pinal-Fernandez I, Kishi T, Targoff IN, Miller FW, Rider LG, et al. Anti-Ro52 Autoantibodies are Associated With Interstitial Lung Disease and More Severe Disease in Patients With Juvenile Myositis. Ann Rheum Dis (2019) 78:988–95. doi: 10.1136/annrheumdis-2018-215004
165. Pinal-Fernandez I, Casal-Dominguez M, Huapaya JA, Albayda J, Paik JJ, Johnson C, et al. A Longitudinal Cohort Study of the Anti-Synthetase Syndrome: Increased Severity of Interstitial Lung Disease in Black Patients and Patients With anti-PL7 and anti-PL12 Autoantibodies. Rheumatology (2017) 56:999–1007. doi: 10.1093/rheumatology/kex021
166. Bauhammer J, Blank N, Max R, Lorenz H-M, Wagner U, Krause D, et al. Rituximab in the Treatment of Jo1 Antibody–Associated Antisynthetase Syndrome: Anti-Ro52 Positivity as a Marker for Severity and Treatment Response. J Rheumatol (2016) 43:1566–74. doi: 10.3899/jrheum.150844
167. La Corte R, Lo Mo naco A, Locaputo A, Dolzani F, Trotta F. In Patients With Antisynthetase Syndrome the Occurrence of anti-Ro/SSA Antibodies Causes a More Severe Interstitial Lung Disease. Autoimmunity (2006) 39:249–53. doi: 10.1080/08916930600623791
168. Váncsa A, Csípő I, Németh J, Dévényi K, Gergely L, Dankó K. Characteristics of Interstitial Lung Disease in SS-A Positive/Jo-1 Positive Inflammatory Myopathy Patients. Rheumatol Int (2009) 29:989–94. doi: 10.1007/s00296-009-0884-9
169. Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF, et al. Short-Term and Long-Term Outcome of Anti-Jo1-positive Patients With anti-Ro52 Antibody. Semin Arthritis Rheum (2012) 41:890–9. doi: 10.1016/j.semarthrit.2011.09.008
170. Takeuchi A, Matsushita T, Kaji K, Okamoto Y, Yasui M, Hirata M, et al. Autoantibody to Scaffold Attachment Factor B (SAFB): A Novel Connective Tissue Disease-Related Autoantibody Associated With Interstitial Lung Disease. J Autoimmun (2017) 76:101–7. doi: 10.1016/j.jaut.2016.09.006
171. Doyle TJ, Patel AS, Hatabu H, Nishino M, Wu G, Osorio JC, et al. Detection of Rheumatoid Arthritis–Interstitial Lung Disease is Enhanced by Serum Biomarkers. Am J Respir Crit Care Med (2015) 191:1403–12. doi: 10.1164/rccm.201411-1950OC
172. Kelly CA, Saravanan V, Nisar M, Arthanari S, Woodhead FA, Price-Forbes AN, et al. Rheumatoid Arthritis-Related Interstitial Lung Disease: Associations, Prognostic Factors and Physiological and Radiological Characteristics–a Large Multicentre UK Study. Rheumatol (Oxford) (2014) 53:1676–82. doi: 10.1093/rheumatology/keu165
173. Doyle TJ, Patel AS, Hatabu H, Nishino M, Wu G, Osorio JC, et al. Detection of Rheumatoid Arthritis-Interstitial Lung Disease Is Enhanced by Serum Biomarkers. Am J Respir Crit Care Med (2015) 191:1403–12. doi: 10.1164/rccm.201411-1950OC
174. Gochuico BR, Avila NA, Chow CK, Novero LJ, Wu H-P, Ren P, et al. Progressive Preclinical Interstitial Lung Disease in Rheumatoid Arthritis. Arch Internal Med (2008) 168:159–66. doi: 10.1001/archinternmed.2007.59
175. Gunnarsson R, El-Hage F, Aalokken TM, Reiseter S, Lund MB, Garen T, et al. Associations Between anti-Ro52 Antibodies and Lung Fibrosis in Mixed Connective Tissue Disease. Rheumatol (Oxford) (2016) 55:103–8. doi: 10.1093/rheumatology/kev300
176. Magro CM, Waldman WJ, Knight DA, Allen JN, Nadasdy T, Frambach GE, et al. Idiopathic Pulmonary Fibrosis Related to Endothelial Injury and Antiendothelial Cell Antibodies. Hum Immunol (2006) 67:284–97. doi: 10.1016/j.humimm.2006.02.026
177. Perelas A, Silver RM, Arrossi AV, Highland KB. Systemic Sclerosis-Associated Interstitial Lung Disease. Lancet Respir Med (2020) 8:304–20. doi: 10.1016/s2213-2600(19)30480-1
178. Hu Y, Wang LS, Wei YR, Du SS, Du YK, He X, et al. Clinical Characteristics of Connective Tissue Disease-Associated Interstitial Lung Disease in 1,044 Chinese Patients. Chest (2016) 149:201–8. doi: 10.1378/chest.15-1145
179. Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing Interstitial Lung Diseases: Knowns and Unknowns. Eur Respir Rev (2019) 28:151. doi: 10.1183/16000617.0100-2018
180. Wong AW, Ryerson CJ, Guler SA. Progression of Fibrosing Interstitial Lung Disease. Respir Res (2020) 21:32. doi: 10.1186/s12931-020-1296-3
181. Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, et al. Presentation, Diagnosis and Clinical Course of the Spectrum of Progressive-Fibrosing Interstitial Lung Diseases. Eur Respir Rev (2018) 27:180076. doi: 10.1183/16000617.0076-2018
182. Volkmann ER, Tashkin DP, Sim M, Li N, Goldmuntz E, Keyes-Elstein L, et al. Short-Term Progression of Interstitial Lung Disease in Systemic Sclerosis Predicts Long-Term Survival in Two Independent Clinical Trial Cohorts. Ann Rheum Dis (2019) 78:122–30. doi: 10.1136/annrheumdis-2018-213708
183. Panopoulos S, Bournia V-K, Konstantonis G, Fragiadaki K, Sfikakis PP, Tektonidou MG. Predictors of Morbidity and Mortality in Early Systemic Sclerosis: Long-Term Follow-Up Data From a Single-Centre Inception Cohort. Autoimmun Rev (2018) 17:816–20. doi: 10.1016/j.autrev.2018.02.008
184. Goh NS, Hoyles RK, Denton CP, Hansell DM, Renzoni EA, Maher TM, et al. Short-Term Pulmonary Function Trends are Predictive of Mortality in Interstitial Lung Disease Associated With Systemic Sclerosis. Arthritis Rheumatol (2017) 69:1670–8. doi: 10.1002/art.40130
185. Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, et al. Diagnostic Criteria for Idiopathic Pulmonary Fibrosis: A Fleischner Society White Paper. Lancet Respir Med (2018) 6:138–53. doi: 10.1016/S2213-2600(17)30433-2
186. Putman RK, Gudmundsson G, Axelsson GT, Hida T, Honda O, Araki T, et al. Imaging Patterns are Associated With Interstitial Lung Abnormality Progression and Mortality. Am J Respir Crit Care Med (2019) 200:175–83. doi: 10.1164/rccm.201809-1652OC
187. Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, et al. MUC5B Promoter Polymorphism and Interstitial Lung Abnormalities. New Engl J Med (2013) 368:2192–200. doi: 10.1056/NEJMoa1216076
188. Jacob J, Bartholmai BJ, Rajagopalan S, Brun AL, Egashira R, Karwoski R, et al. Evaluation of Computer-Based Computer Tomography Stratification Against Outcome Models in Connective Tissue Disease-Related Interstitial Lung Disease: A Patient Outcome Study. BMC Med (2016) 14:190. doi: 10.1186/s12916-016-0739-7
189. Elhai M, Avouac J, Allanore Y. Circulating Lung Biomarkers in Idiopathic Lung Fibrosis and Interstitial Lung Diseases Associated With Connective Tissue Diseases: Where do We Stand? Semin Arthritis Rheum (2020) 50:480–91. doi: 10.1016/j.semarthrit.2020.01.006
190. Jee AS, Sahhar J, Youssef P, Bleasel J, Adelstein S, Nguyen M, et al. Review: Serum Biomarkers in Idiopathic Pulmonary Fibrosis and Systemic Sclerosis Associated Interstitial Lung Disease - Frontiers and Horizons. Pharmacol Ther (2019) 202:40–52. doi: 10.1016/j.pharmthera.2019.05.014
191. Podolanczuk AJ, Wong AW, Saito S, Lasky JA, Ryerson CJ, Eickelberg O. Update in Interstitial Lung Disease 2020. Am J Respir Crit Care Med (2021). doi: 10.1164/rccm.202103-0559UP
192. Alqalyoobi S, Adegunsoye A, Linderholm A, Hrusch C, Cutting C, Ma S-F, et al. Circulating Plasma Biomarkers of Progressive Interstitial Lung Disease. Am J Respir Crit Care Med (2020) 201:250–3. doi: 10.1164/rccm.201907-1343LE
193. Inoue Y, Kaner RJ, Guiot J, Maher TM, Tomassetti S, Moiseev S, et al. Diagnostic and Prognostic Biomarkers for Chronic Fibrosing Interstitial Lung Diseases With a Progressive Phenotype. Chest (2020) 158:646–59. doi: 10.1016/j.chest.2020.03.037
194. Hamai K, Iwamoto H, Ishikawa N, Horimasu Y, Masuda T, Miyamoto S, et al. Comparative Study of Circulating Mmp-7, CCL18, Kl-6, SP-A, and SP-D as Disease Markers of Idiopathic Pulmonary Fibrosis. Dis Markers (2016) 2016):4759040. doi: 10.1155/2016/4759040
195. Ohnishi H, Yokoyama A, Kondo K, Hamada H, Abe M, Nishimura K, et al. Comparative Study of KL-6, Surfactant protein-A, Surfactant protein-D, and Monocyte Chemoattractant Protein-1 as Serum Markers for Interstitial Lung Diseases. Am J Respir Crit Care Med (2002) 165:378–81. doi: 10.1164/ajrccm.165.3.2107134
196. Satoh H, Kurishima K, Ishikawa H, Ohtsuka M. Increased Levels of KL-6 and Subsequent Mortality in Patients With Interstitial Lung Diseases. J Intern Med (2006) 260:429–34. doi: 10.1111/j.1365-2796.2006.01704.x
197. Ohshimo S, Ishikawa N, Horimasu Y, Hattori N, Hirohashi N, Tanigawa K, et al. Baseline KL-6 Predicts Increased Risk for Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Respir Med (2014) 108:1031–9. doi: 10.1016/j.rmed.2014.04.009
198. Ishii H, Kushima H, Kinoshita Y, Fujita M, Watanabe K. The Serum KL-6 Levels in Untreated Idiopathic Pulmonary Fibrosis can Naturally Decline in Association With Disease Progression. Clin Respir J (2018) 12:2411–8. doi: 10.1111/crj.12946
199. Elhai M, Hoffmann-Vold AM, Avouac J, Pezet S, Cauvet A, Leblond A, et al. Performance of Candidate Serum Biomarkers for Systemic Sclerosis–Associated Interstitial Lung Disease. Arthritis Rheumatol (2019) 71:972–82. doi: 10.1002/art.40815
200. Kumánovics G, Görbe E, Minier T, Simon D, Berki T, Czirják L. Follow-Up of Serum KL-6 Lung Fibrosis Biomarker Levels in 173 Patients With Systemic Sclerosis. Clin Exp Rheumatol (2014) 32:S–138-44.
201. Benyamine A, Heim X, Resseguier N, Bertin D, Gomez C, Ebbo M, et al. Elevated Serum Krebs Von Den Lungen-6 in Systemic Sclerosis: A Marker of Lung Fibrosis and Severity of the Disease. Rheumatol Int (2018) 38:813–9. doi: 10.1007/s00296-018-3987-3
202. Yamaguchi K, Iwamoto H, Sakamoto S, Horimasu Y, Masuda T, Miyamoto S, et al. Serial Measurements of KL-6 for Monitoring Activity and Recurrence of Interstitial Pneumonia With anti-aminoacyl-tRNA Synthetase Antibody: A Retrospective Cohort Study. Med (Baltimore) (2018) 97:e13542. doi: 10.1097/md.0000000000013542
203. Hoffmann-Vold AM, Tennøe AH, Garen T, Midtvedt Ø, Abraityte A, Aaløkken TM, et al. High Level of Chemokine CCL18 Is Associated With Pulmonary Function Deterioration, Lung Fibrosis Progression, and Reduced Survival in Systemic Sclerosis. Chest (2016) 150:299–306. doi: 10.1016/j.chest.2016.03.004
204. Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, et al. MMP1 and MMP7 as Potential Peripheral Blood Biomarkers in Idiopathic Pulmonary Fibrosis. PloS Med (2008) 5:e93. doi: 10.1371/journal.pmed.0050093
205. Tzouvelekis A, Herazo-Maya JD, Slade M, Chu JH, Deiuliis G, Ryu C, et al. Validation of the Prognostic Value of MMP-7 in Idiopathic Pulmonary Fibrosis. Respirology (2017) 22:486–93. doi: 10.1111/resp.12920
206. Greene KE, King TE Jr., Kuroki Y, Bucher-Bartelson B, Hunninghake GW, Newman LS, et al. Serum Surfactant Proteins-a and -D as Biomarkers in Idiopathic Pulmonary Fibrosis. Eur Respir J (2002) 19:439–46. doi: 10.1183/09031936.02.00081102
207. Bonhomme O, André B, Gester F, de Seny D, Moermans C, Struman I, et al. Biomarkers in Systemic Sclerosis-Associated Interstitial Lung Disease: Review of the Literature. Rheumatol (Oxford) (2019) 58:1534–46. doi: 10.1093/rheumatology/kez230
208. Kolb M, Bondue B, Pesci A, Miyazaki Y, Song JW, Bhatt NY, et al. Acute Exacerbations of Progressive-Fibrosing Interstitial Lung Diseases. Eur Respir Rev (2018) 27:180071. doi: 10.1183/16000617.0071-2018
209. Chartrand S, Fischer A. Management of Connective Tissue Disease–associated Interstitial Lung Disease. Rheumatic Dis Clinics (2015) 41:279–94. doi: 10.1016/j.rdc.2015.01.002
210. Jee AS, Corte TJ. Current and Emerging Drug Therapies for Connective Tissue Disease-Interstitial Lung Disease (CTD-ILD). Drugs (2019) 79:1511–28. doi: 10.1007/s40265-019-01178-x
211. Zaizen Y, Fukuoka J. Pathology of Idiopathic Interstitial Pneumonias. Surg Pathol Clinics (2020) 13:91–118. doi: 10.1016/j.path.2019.11.006
212. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med (2019) 380:2518–28. doi: 10.1056/NEJMoa1903076
213. Li T, Guo L, Chen Z, Gu L, Sun F, Tan X, et al. Pirfenidone in Patients With Rapidly Progressive Interstitial Lung Disease Associated With Clinically Amyopathic Dermatomyositis. Sci Rep (2016) 6:33226. doi: 10.1038/srep33226
214. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in Patients With Unclassifiable Progressive Fibrosing Interstitial Lung Disease: A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet Respir Med (2020) 8:147–57. doi: 10.1016/s2213-2600(19)30341-8
215. Dellaripa PF, Miller M. Interstitial Lung Disease in Dermatomyositis and Polymyositis: Treatment. UpToDate Waltham, MA: Topic (2015), 4357.
216. de Carvalho CRR, Deheinzelin D, Kairalla RA, King TE Jr, Hollingsworth H. Interstitial Lung Disease Associated With Sjögren’s Syndrome: Management and Prognosis. UpToDate (2020).
218. Varga J, Montesi S. Treatment and Prognosis of Interstitial Lung Disease in Systemic Sclerosis (Scleroderma). UpToDate (2021).
219. Barnes H, Holland AE, Westall GP, Goh NS, Glaspole IN. Cyclophosphamide for Connective Tissue Disease-Associated Interstitial Lung Disease. Cochrane Database Syst Rev (2018) 1:Cd010908. doi: 10.1002/14651858.CD010908.pub2
220. Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide Versus Placebo in Scleroderma Lung Disease. New Engl J Med (2006) 354:2655–66. doi: 10.1056/NEJMoa055120
221. Khanna D, Tashkin DP, Denton CP, Lubell MW, Vazquez-Mateo C, Wax S. Ongoing Clinical Trials and Treatment Options for Patients With Systemic Sclerosis-Associated Interstitial Lung Disease. Rheumatol (Oxford) (2019) 58:567–79. doi: 10.1093/rheumatology/key151
222. Sullivan KM, McSweeney PA, Nash RA. Cyclophosphamide in Scleroderma Lung Disease. N Engl J Med (2006) 355:1173–4; author reply 1174. doi: 10.1056/NEJMc061920
223. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate Mofetil Versus Oral Cyclophosphamide in Scleroderma-Related Interstitial Lung Disease (SLS II): A Randomised Controlled, Double-Blind, Parallel Group Trial. Lancet Respir Med (2016) 4:708–19. doi: 10.1016/s2213-2600(16)30152-7
224. Zaheen A, Stanbrook MB, Anand A. Mycophenolate Mofetil for Scleroderma-Related Interstitial Lung Disease. Lancet Respir Med (2016) 4:e53. doi: 10.1016/s2213-2600(16)30318-6
225. Volkmann ER, Tashkin DP, Li N, Roth MD, Khanna D, Hoffmann-Vold AM, et al. Mycophenolate Mofetil Versus Placebo for Systemic Sclerosis-Related Interstitial Lung Disease: An Analysis of Scleroderma Lung Studies I and II. Arthritis Rheumatol (Hoboken NJ) (2017) 69:1451–60. doi: 10.1002/art.40114
226. Oldham JM, Lee C, Valenzi E, Witt LJ, Adegunsoye A, Hsu S, et al. Azathioprine Response in Patients With Fibrotic Connective Tissue Disease-Associated Interstitial Lung Disease. Respir Med (2016) 121:117–22. doi: 10.1016/j.rmed.2016.11.007
227. Paone C, Chiarolanza I, Cuomo G, Ruocco L, Vettori S, Menegozzo M, et al. Twelve-Month Azathioprine as Maintenance Therapy in Early Diffuse Systemic Sclerosis Patients Treated for 1-Year With Low Dose Cyclophosphamide Pulse Therapy. Clin Exp Rheumatol (2007) 25:613–6. doi: 10.1002/art.22744
228. Labirua-Iturburu A, Selva-O’Callaghan A, Martínez-Gómez X, Trallero-Araguás E, Labrador-Horrillo M, Vilardell-Tarrés M. Calcineurin Inhibitors in a Cohort of Patients With Antisynthetase-Associated Interstitial Lung Disease. Clin Exp Rheumatol (2013) 31:436–9. doi: 10.1016/j.jbspin.2012.09.027
229. Takada K, Katada Y, Ito S, Hayashi T, Kishi J, Itoh K, et al. Impact of Adding Tacrolimus to Initial Treatment of Interstitial Pneumonitis in Polymyositis/Dermatomyositis: A Single-Arm Clinical Trial. Rheumatol (Oxford) (2020) 59:1084–93. doi: 10.1093/rheumatology/kez394
230. Tsuji H, Nakashima R, Hosono Y, Imura Y, Yagita M, Yoshifuji H, et al. Multicenter Prospective Study of the Efficacy and Safety of Combined Immunosuppressive Therapy With High-Dose Glucocorticoid, Tacrolimus, and Cyclophosphamide in Interstitial Lung Diseases Accompanied by Anti-Melanoma Differentiation-Associated Gene 5-Positive Dermatomyositis. Arthritis Rheumatol (2020) 72:488–98. doi: 10.1002/art.41105
231. Keir GJ, Maher TM, Ming D, Abdullah R, de Lauretis A, Wickremasinghe M, et al. Rituximab in Severe, Treatment-Refractory Interstitial Lung Disease. Respirology (2014) 19:353–9. doi: 10.1111/resp.12214
232. Sharp C, McCabe M, Dodds N, Edey A, Mayers L, Adamali H, et al. Rituximab in Autoimmune Connective Tissue Disease-Associated Interstitial Lung Disease. Rheumatol (Oxford) (2016) 55:1318–24. doi: 10.1093/rheumatology/kew195
233. Yang L, Wang Q, Hou Y, Zhao J, Li M, Xu D, et al. The Chinese Herb Tripterygium Wilfordii Hook F for the Treatment of Systemic Sclerosis-Associated Interstitial Lung Disease: Data From a Chinese Eustar Center. Clin Rheumatol (2020) 39:813–21. doi: 10.1007/s10067-019-04784-y
234. De Lauretis A, Sestini P, Pantelidis P, Hoyles R, Hansell DM, Goh NS, et al. Serum Interleukin 6 is Predictive of Early Functional Decline and Mortality in Interstitial Lung Disease Associated With Systemic Sclerosis. J Rheumatol (2013) 40:435–46. doi: 10.3899/jrheum.120725
235. Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, et al. Tocilizumab in Systemic Sclerosis: A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Respir Med (2020) 8:963–74. doi: 10.1016/s2213-2600(20)30318-0
236. Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, et al. Safety and Efficacy of Subcutaneous Tocilizumab in Adults With Systemic Sclerosis (faSScinate): A Phase 2, Randomised, Controlled Trial. Lancet (2016) 387:2630–40. doi: 10.1016/S0140-6736(16)00232-4
237. Khanna D, Denton CP, Lin CJ, van Laar JM, Frech TM, Anderson ME, et al. Safety and Efficacy of Subcutaneous Tocilizumab in Systemic Sclerosis: Results From the Open-Label Period of a Phase II Randomised Controlled Trial (Fasscinate). Ann Rheum Dis (2018) 77:212–20. doi: 10.1136/annrheumdis-2017-211682
238. Vicente-Rabaneda EF, Atienza-Mateo B, Blanco R, Cavagna L, Ancochea J, Castañeda S, et al. Efficacy and Safety of Abatacept in Interstitial Lung Disease of Rheumatoid Arthritis: A Systematic Literature Review. Autoimmun Rev (2021) 20:102830. doi: 10.1016/j.autrev.2021.102830
239. Fernández-Díaz C, Castañeda S, Melero-González RB, Ortiz-Sanjuán F, Juan-Mas A, Carrasco-Cubero C, et al. Abatacept in Interstitial Lung Disease Associated With Rheumatoid Arthritis: National Multicenter Study of 263 Patients. Rheumatol (Oxford) (2020) 59:3906–16. doi: 10.1093/rheumatology/keaa621
240. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SL, Inoue Y, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. New Engl J Med (2019) 381:1718–27. doi: 10.1056/NEJMoa1908681
241. Varga J, K. T Jr. Prognosis and Treatment of Interstitial Lung Disease in Systemic Sclerosis (Scleroderma). UpToDate Waltham, MA: Topic (2011) 4375.
242. Miura Y, Saito T, Fujita K, Tsunoda Y, Tanaka T, Takoi H, et al. Clinical Experience With Pirfenidone in Five Patients With Scleroderma-Related Interstitial Lung Disease. Sarcoidosis Vasculitis Diffuse Lung Diseases: Off J WASOG (2014) 31:235–8.
243. Acharya N, Sharma SK, Mishra D, Dhooria S, Dhir V, Jain S. Efficacy and Safety of Pirfenidone in Systemic Sclerosis-Related Interstitial Lung Disease-a Randomised Controlled Trial. Rheumatol Int (2020) 40:703–10. doi: 10.1007/s00296-020-04565-w
244. Erre GL, Sebastiani M, Manfredi A, Gerratana E, Atzeni F, Passiu G, et al. Antifibrotic Drugs in Connective Tissue Disease-Related Interstitial Lung Disease (CTD-ILD): From Mechanistic Insights to Therapeutic Applications. Drugs Context (2021) 10:2020-8-6. doi: 10.7573/dic.2020-8-6
245. Shirakashi M, Nakashima R, Tsuji H, Tanizawa K, Handa T, Hosono Y, et al. Efficacy of Plasma Exchange in anti-MDA5-positive Dermatomyositis With Interstitial Lung Disease Under Combined Immunosuppressive Treatment. Rheumatol (Oxford) (2020) 59:3284–92. doi: 10.1093/rheumatology/keaa123
246. van Bijnen S, de Vries-Bouwstra J, van den Ende CH, Boonstra M, Kroft L, Geurts B, et al. Predictive Factors for Treatment-Related Mortality and Major Adverse Events After Autologous Haematopoietic Stem Cell Transplantation for Systemic Sclerosis: Results of a Long-Term Follow-Up Multicentre Study. Ann Rheum Dis (2020) 79:1084–9. doi: 10.1136/annrheumdis-2020-217058
247. Kreuter M, Polke M, Walsh SLF, Krisam J, Collard HR, Chaudhuri N, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis: International Survey and Call for Harmonisation. Eur Respir J (2020) 55:1901760. doi: 10.1183/13993003.01760-2019
248. Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am J Respir Crit Care Med (2016) 194:265–75. doi: 10.1164/rccm.201604-0801CI
249. Wootton SC, Kim DS, Kondoh Y, Chen E, Lee JS, Song JW, et al. Viral Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2011) 183:1698–702. doi: 10.1164/rccm.201010-1752OC
250. Manfredi A, Sebastiani M, Cerri S, Vacchi C, Tonelli R, Della Casa G, et al. Acute Exacerbation of Interstitial Lung Diseases Secondary to Systemic Rheumatic Diseases: A Prospective Study and Review of the Literature. J Thoracic Dis (2019) 11:1621. doi: 10.21037/jtd.2019.03.28
251. Tachikawa R, Tomii K, Ueda H, Nagata K, Nanjo S, Sakurai A, et al. Clinical Features and Outcome of Acute Exacerbation of Interstitial Pneumonia: Collagen Vascular Diseases-Related Versus Idiopathic. Respiration (2012) 83:20–7. doi: 10.1159/000329893
252. Suda T, Kaida Y, Nakamura Y, Enomoto N, Fujisawa T, Imokawa S, et al. Acute Exacerbation of Interstitial Pneumonia Associated With Collagen Vascular Diseases. Respir Med (2009) 103:846–53. doi: 10.1016/j.rmed.2008.12.019
253. Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, et al. Lung Microbiome and Disease Progression in Idiopathic Pulmonary Fibrosis: An Analysis of the COMET Study. Lancet Respir Med (2014) 2:548–56. doi: 10.1016/s2213-2600(14)70069-4
254. Molyneaux PL, Cox MJ, Wells AU, Kim HC, Ji W, Cookson WO, et al. Changes in the Respiratory Microbiome During Acute Exacerbations of Idiopathic Pulmonary Fibrosis. Respir Res (2017) 18:29. doi: 10.1186/s12931-017-0511-3
Keywords: connective tissue disease, interstitial lung disease, genetics, environmental exposure, autoantibodies, signs and symptoms, risk assessment, therapeutics
Citation: Shao T, Shi X, Yang S, Zhang W, Li X, Shu J, Alqalyoobi S, Zeki AA, Leung PS and Shuai Z (2021) Interstitial Lung Disease in Connective Tissue Disease: A Common Lesion With Heterogeneous Mechanisms and Treatment Considerations. Front. Immunol. 12:684699. doi: 10.3389/fimmu.2021.684699
Received: 23 March 2021; Accepted: 17 May 2021;
Published: 07 June 2021.
Edited by:
A. Richard Kitching, Monash University, AustraliaReviewed by:
Lynn Fussner, The Ohio State University, United StatesBeatriz Tejera Segura, Insular University Hospital of Gran Canaria, Spain
Copyright © 2021 Shao, Shi, Yang, Zhang, Li, Shu, Alqalyoobi, Zeki, Leung and Shuai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zongwen Shuai, YW11c2h1YWl6d0AxNjMuY29t; Patrick S. Leung, cHNsZXVuZ0B1Y2RhdmlzLmVkdQ==
†These authors have contributed equally to this work and share first authorship