 Konstantinos Ntatsoulis1†
Konstantinos Ntatsoulis1† Theodoros Karampitsakos2†
Theodoros Karampitsakos2† Eliza Tsitoura3†
Eliza Tsitoura3† Elli-Anna Stylianaki1Alexios N. Matralis1Argyrios Tzouvelekis2*
Elli-Anna Stylianaki1Alexios N. Matralis1Argyrios Tzouvelekis2* Katerina Antoniou3*
Katerina Antoniou3* Vassilis Aidinis1*
Vassilis Aidinis1*- 1Institute of Bio-Innovation, Biomedical Sciences Research Center Alexander Fleming, Athens, Greece
- 2Department of Respiratory Medicine, School of Medicine, University of Patras, Patras, Greece
- 3Laboratory of Molecular & Cellular Pneumonology, Department of Respiratory Medicine, School of Medicine, University of Crete, Heraklion, Greece
Severe COVID-19 is characterized by acute respiratory distress syndrome (ARDS)-like hyperinflammation and endothelial dysfunction, that can lead to respiratory and multi organ failure and death. Interstitial lung diseases (ILD) and pulmonary fibrosis confer an increased risk for severe disease, while a subset of COVID-19-related ARDS surviving patients will develop a fibroproliferative response that can persist post hospitalization. Autotaxin (ATX) is a secreted lysophospholipase D, largely responsible for the extracellular production of lysophosphatidic acid (LPA), a pleiotropic signaling lysophospholipid with multiple effects in pulmonary and immune cells. In this review, we discuss the similarities of COVID-19, ARDS and ILDs, and suggest ATX as a possible pathologic link and a potential common therapeutic target.
COVID-19
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection leads to the development of Coronavirus Disease 2019 (COVID-19), proclaimed pandemic on March 11, 2020 (1, 2). Upon airborne, mainly, CoV-2 transmission, the viral spike (S) glycoprotein mediates viral entry via binding to angiotensin-converting enzyme 2 (ACE2), supported by the transmembrane protease serine protease 2 (TMPRSS2) that proteolytically processes the S protein (3, 4). Infected cells in the lung, as detected with immunocytochemistry, include the upper airway bronchiolar epithelium and submucosal gland epithelium, as well as alveolar epithelial cells and macrophages (1). ACE2 is distributed mainly in the intestine, heart, kidney, as well as the lung, where alveolar epithelial type II cells are the major expressing cells. TMPRSS2 is highly expressed in several tissues; in the lung is co- expressed with ACE2 in nasal epithelial cells and alveolar epithelial type II cells, which might partially explain the tissue tropism of CoV-2 infection (3, 4).
CoV-2 infection is either asymptomatic or causes only mild respiratory diseases (non-pneumonia or mild pneumonia) in most individuals. However, a significant number of elderly individuals, frequently with comorbidities (such as cardiovascular diseases, diabetes, obesity), will develop a more severe form of the disease and will require hospital care (1, 2). COVID-19 most common clinical manifestations include fever, fatigue and dry cough, and dyspnea in severe cases (5). Severe COVID-19 associated histopathological changes are found mainly in the lungs, characterized by diffused alveolar damage (DAD), hyaline membranes and fibrin deposits, as well as severe endothelial injury, capillary microthrombi and exudative inflammation (6–12). A systematic review of published case reports and studies identified three main COVID-19 histological patterns: epithelial (85%), vascular (59%) and fibrotic (22%), with a frequent overlap (60%), whereas the epithelial and vascular patterns were present in all stages of severe COVID-19 (13).
COVID-19, ARDS and Pulmonary Fibrosis
The rapid replication of SARS- CoV-2 and the associated epithelial cell death may, depending on the underlying genetic, inflammatory or metabolic context, trigger alveolar macrophages to produce excessive amounts of cytokines (such as TNF, IL-1b, IL-6, MIP1, IFN-γ and VEGF), a “cytokine storm”, associated with systemic infections such as sepsis or immunotherapies aftermath (14). The highly divert cytokine profile of COVID-19 hyperinflammation resembles, in some cases, other cytokine release syndromes, such as macrophage activation syndrome (15), although it is more heterogeneous and less robust, both quantitively (levels) and qualitatively (number of elevated cytokines). Noteworthy, IL-6 was found to be the most consistently upregulated cytokine and among the most overall predictive biomarkers (16, 17). In turn, the excessive production of cytokines further induces lung injury and Acute Respiratory Distress syndrome (ARDS), leading frequently to respiratory and multi organ failure and death (18).
ARDS develops most commonly in the setting of bacterial and viral pneumonias, or non-pulmonary sepsis, and is characterized by focal epithelial damage and excessive alveolocapillary permeability, leading to interstitial and alveolar edema and hypoxemia in the acute phase (18). Many severe COVID-19 patients will develop ARDS with impaired gas exchange and characteristic CT findings; however, the combination of multiple pathogenetic stimuli in COVID-19-induced ARDS results in a highly heterogeneous, “atypical” clinical appearance that has stimulated considerable controversy (2, 19–24). Nevertheless, excessive inflammation and endothelial dysfunction are among the top candidate pathologic events linking ARDS and COVID-19 (21, 25, 26) and markers of endothelial dysfunction have been recently correlated with COVID-19 mortality (27). Moreover, endothelial dysfunction is also a major characteristic of the most common comorbidities of COVID-19 that are associated with worse prognosis, hypertension, diabetes and obesity (21, 25).
The initial acute exudative inflammatory phase of ARDS is followed by a proliferative phase characterized by alveolar epithelial cell hyperplasia (18). A subset of acute ARDS survivors will further develop a fibroproliferative response, including fibroblast accumulation, deposition of collagen and other lung extracellular matrix (ECM) components (28), the magnitude of which was associated with ARDS duration (29). Moreover, and although invasive mechanical ventilation has revolutionized the management of ARDS, ventilator associated lung injury is considered as an additional contributor to pulmonary fibrosis in ventilated ARDS patients (30). Accordingly, a literature review of published histopathological analyses of COVID-19 lungs postmortem indicated, beyond DAD and hyaline membranes, the frequent presence of pulmonary fibrosis (31), while abnormal pulmonary architecture and functions have been reported in many recovering COVID-19 patients (32–34), suggesting persisting fibrotic abnormalities, pending large-scale and long-term follow up studies. Finally, CoV-2 infection per se has been reported to induce the expression of different pro-fibrotic factors including TGFβ (35). On the other hand, patients with interstitial lung diseases (ILD) had increased odds for ARDS development and severe COVID-19 (12, 36–39), while COVID-19-related acute exacerbation of ILDs had worse prognosis than non-COVID ILD acute exacerbations (40), thus suggesting pulmonary fibrosis both as a disease risk and a possible complication of COVID-19.
Autotaxin (ATX; ENPP2) and COVID-19
ATX is a secreted glycoprotein that can be detected in most biological fluids, including blood and bronchoalveolar lavage fluid (BALF) (41). A large percentage (~40%) of serum ATX is thought to originate from the adipose tissue (42), while the damaged liver has been suggested as an additional possible source of serum ATX (43). High ATX expression has been reported from endothelial cells in high endothelial venules (HEVs) (44, 45), however their expected relative contribution to circulating levels should be low. Inflammatory macrophages have been also reported to express ATX upon inflammation (46–48), thus contributing to BALF ATX levels (46).
ATX is a constitutively active lysophospholipase D, that catalyzes the extracellular hydrolysis of lysophosphatidylcholine (LPC) to lysophosphatidic acid (LPA) (49). LPC is synthesized from fatty acids or membrane phosphatidylcholine (PC) by phospholipase A2 (PLA2) enzymes and is highly abundant in the plasma, associated with oxidized low-density lipoprotein (oxLDL) and albumin (50, 51). LPA is a growth factor-like signaling phospholipid with numerous effects in most cell types through its G-protein coupled receptors (LPAR1-6) (51–53). ATX has been suggested to bind to cell surface integrins (54–56), thus avoiding clearance, as well as localizing LPA production to its adjacent receptors, that exhibit widespread distribution and overlapping functions (51, 57).
Viral infections have been shown to increase systemic ATX levels, including HCV, HIV and HBV (43, 58, 59), while LPA has been also shown to directly affect HCV viral infection and replication (43, 60). Increased ENPP2 mRNA expression was detected in nasopharyngeal swabs from COVID-19 patients, likely from immune cells (61), suggesting that ATX/LPA might stimulate viral infections, that could include SARS-CoV-2.
Increased serum/plasma ATX levels have been also reported in different diseases, including different forms of cancer, liver diseases, as well as respiratory diseases such as asthma and idiopathic pulmonary fibrosis (IPF) (Table 1) (41, 51). A variety of methods has been utilized, while reported levels exhibit remarkable heterogeneity, with no apparent consensus on healthy levels (Table 1). Increased ATX serum levels were recently reported in non-surviving ARDS patients, where ATX serum levels were shown to be an independent prognostic factor for 28 day mortality, outperforming the established SOFA/APACHE scores (62). Plasma ATX levels correlated with mortality also in a cohort of patients with severe sepsis (63), suggesting a role for ATX/LPA in systemic hyperinflammation. ATX serum levels in ARDS correlated with the increased IL-6/IL-8 serum levels (62), further supporting an interplay of ATX/LPA with inflammation, as previously suggested in breast cancer (64). ATX serum levels also correlated with the severity of lung injury (62), while increased ATX BALF levels upon endotoxin-induced acute lung injury (ALI) (65, 66), and ventilator-induced lung injury (VILI) in mice (67) have been reported. Moreover, ATX BALF levels in ARDS patients were positively associated with inflammatory and fibrotic mediators (IL-6, IL-8, TNF-α, MMP-7, fibronectin, OSM, and SPARC), suggesting that ATX may also have a role in the fibrotic component of ARDS (62). In line with the above, increased ATX levels have been detected in IPF patients and fibrotic animal models (46, 68), where results from genetic and pharmacologic studies have established a pro-fibrotic role for ATX (46, 69–72). Increased serum ATX levels were very recently detected also in COVID-19 patients hospitalized in the intensive care unit (ICU) as compared with less severe patients hospitalized in the clinic (61), thus adding ATX expression to the commonalities of COVID-19, ARDS and pulmonary fibrosis, and suggesting ATX as a possible pathologic link.
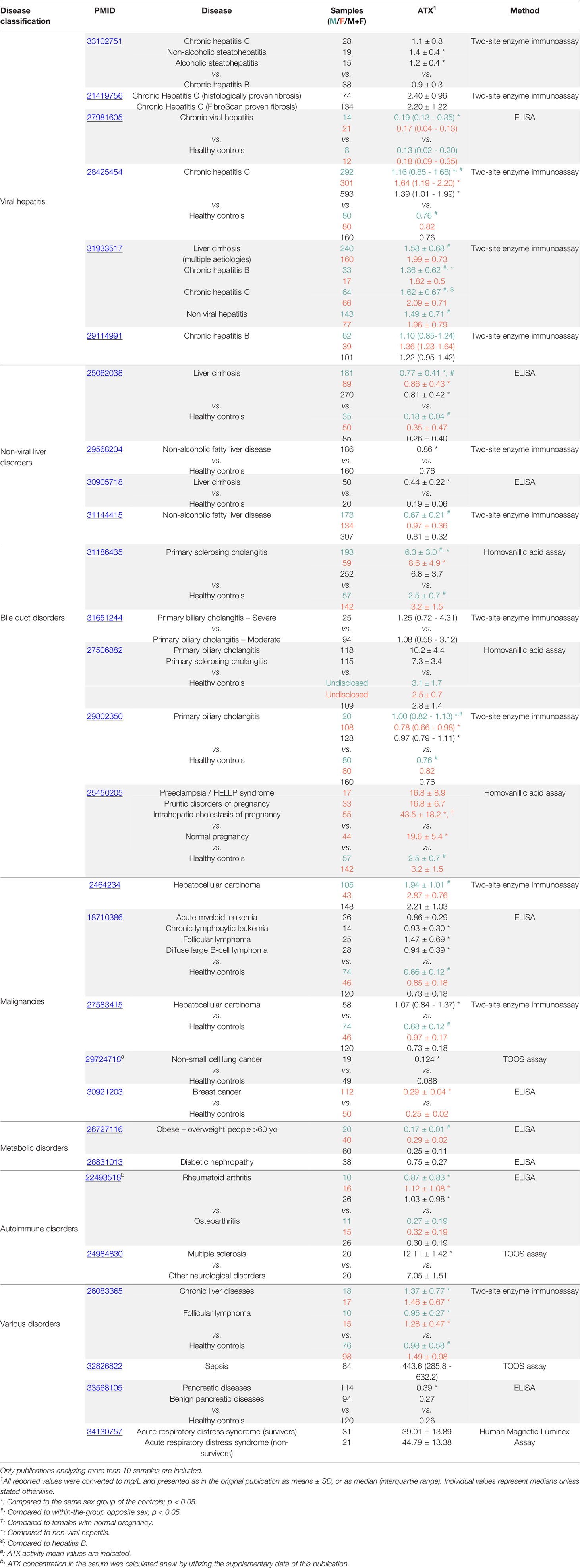
Table 1 Autotaxin serum levels in patients of different inflammatory diseases and cancer.
ATX levels in severe COVID-19 patients correlated with the increased IL-6 serum levels (61), as recently also shown in ARDS (62), as well in acute-on-chronic liver failure (ACLF) patients (73), suggesting interdependent regulation of expression. Accordingly, IL-6 has been reported to stimulate ATX expression from adipocytes (73) and human dermal fibroblasts (74). Vice versa, LPA has been reported to stimulate the expression of IL-6 from synovial fibroblasts (75, 76) and dermal fibroblasts (74), suggesting an ATX/LPA/IL-6 expression loop. Among the different components of the cytokine storm, IL-6 is the most predictive biomarker in COVID-19 (16, 17), correlating with respiratory failure and the need for mechanical ventilation (77), as well as with mortality risk (78).
Beyond hyperinflammation, and/or as its consequence, endothelial dysfunction is a major characteristic of COVID-19/ARDS (21, 25, 26). The increased ATX levels that were detected in severe COVID-19 patients correlated with markers of endothelial dysfunction (sP-sel, sICAM) (61) that have been independently correlated in the same samples with COVID-19 mortality (27). Similarly, ATX correlated with angiopoietin-2 levels and mortality in severe septic patients (63). In support for a major role of ATX/LPA on vascular homeostasis, ATX expression and LPA signaling have been shown necessary for the embryonic development of the vascular (and neural) system in mice (79–81). In adult mice, in studies unraveling the molecular mechanisms of SARS-CoV and MERS-CoV pathogenesis in the Collaborative Cross mice, Enpp2, the gene encoding ATX, has been reported to be a high priority candidate gene for pulmonary hemorrhage (82, 83). More importantly, LPAR1 null mice were reported to be protected from bleomycin (BLM)-induced pulmonary fibrosis, attributed to fibroblast accumulation and reduced vascular leak (68), as well as from Candida albicans water-soluble fraction (CAWS)-induced vasculitis, attributed to reduced CXCL1/IL-8-mediated neutrophil infiltration (84). Noteworthy, the stability of LPAR1 in the context of acute lung injury in mice has been proposed to be regulated by ubiquitination (84).
LPA Signaling in Pulmonary and Immune Cells
Overall, any ATX effect will rely on its local levels (locally produced and/or extravasated) and its possible cell surface attachment, the local availability of LPC, the cell-specific expression profile of LPA receptors, as well as of the expression of the transmembrane lipid phosphate phosphatases (PLPP1-3; PPA2 A-C) that catabolize LPA (41, 85, 86). In this context, the possible effects of increased ATX levels can be deduced from the corresponding effects of LPA in the relative cells in the tissue microenvironment in question.
A plethora of LPA effects on pulmonary non-immune cells in vitro have been reported, as previously reviewed (87–89) and as summarized at Table 2. These include the promotion of apoptosis and the secretion of chemotactic signals (IL-8, MCP-1, CXCL1) from epithelial cells, the integrin-mediated activation of TGFβ on epithelial and smooth muscle cells, the modulation of permeability, leukocyte adhesion and cytokine secretion from endothelial cells, and the chemoattraction and accumulation of myofibroblasts (Table 2). LPAR1 has been reported as the main receptor mediating these effects, involving different well-known G-protein mediated pathways (Table 2). Moreover, LPA has been reported to transactivate different growth factors including TGFβ, PDGF and EGF that activate similar signal transduction pathways, while LPA was reported to signal also via RAGE (Table 2), further increasing the pleiotropic complexity of LPA signaling in the lung.
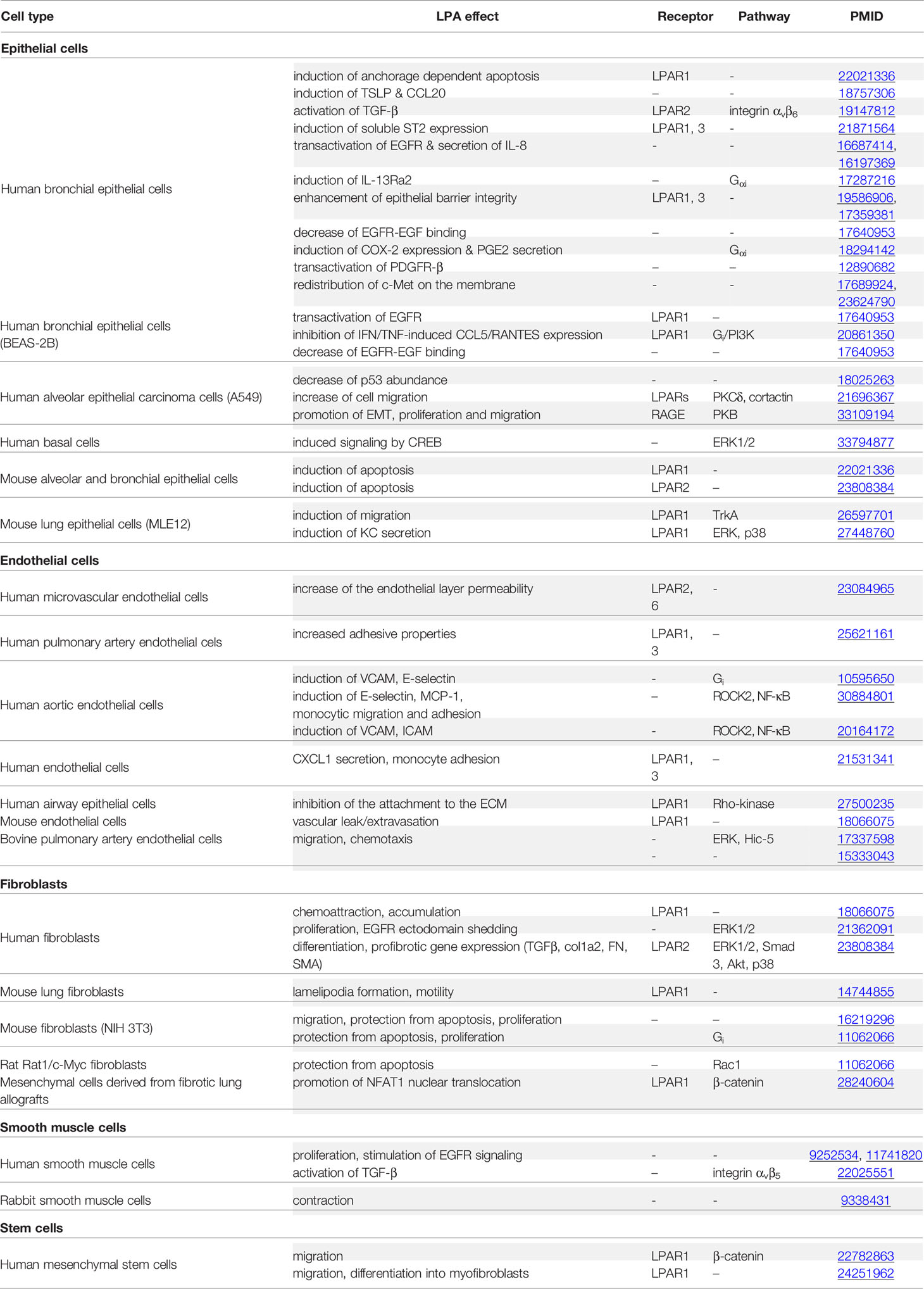
Table 2 Reported Lysophosphatidic acid (LPA) effects on pulmonary, non-immune, cells.
The effects of ATX and LPA signaling on the regulation of immune cells have been previously reviewed in detail (87–92). Briefly, high ATX expression from ECs in HEVs in lymph nodes has been reported (44, 45), where ATX has been suggested to facilitate lymphocyte homing via the promotion of the adhesion (44), transmigration and motility of lymphocytes (45, 93, 94). Intriguingly, LPA signaling has been proposed to intersect with sphingosine phosphate (S1P) signaling (95), a closely related phospholipid (96), that has been shown to affect lymphocyte egress from the lymph nodes (97).
Non-withstanding the effects of ATX/LPA on lymphocyte homeostasis, highly pertinent for both ILD/IPF and COVID-19, a role for ATX/LPA on the homeostasis of the monocyte phagocyte system is emerging. Macrophages are central players in the pathogenesis of both IPF (98, 99) and COVID-19 (15, 100, 101), exhibiting remarkable heterogeneity and spatiotemporal plasticity. LPA has been suggested to stimulate the expression of macrophage chemotactic factors from ECs, such as monocyte chemoattractant protein-1 (MCP-1) (102) and CXCL1 (103), thus promoting both monocyte migration as well as adhesion to ECs (102–104). Beyond LPA-induced macrophage chemoattraction and adhesion to ECs, inflammatory macrophages per se have been reported to express ATX upon BLM-induced pulmonary inflammation and fibrosis, while IPF macrophages have been shown to stain for ATX (46). scRNAseq analysis of BALF cells from COVID-19 patients indicated a predominance of macrophages (100, 101), where ENPP2 mRNA expression was detected in monocyte-derived alveolar macrophages (Mo-AMs) (61), that have been shown to drive the development of BLM-induced pulmonary fibrosis in mice (105). In turn, accumulating evidence indicates that LPA co-stimulate macrophage maturation and/or activation (47, 106–109), suggesting an autocrine role of ATX/LPA in macrophage pathologic responses. Moreover, LPA has been suggested to stimulate oxLDL uptake and foam cell formation (110, 111), linking macrophages and ATX/LPA with hyperlipidaemia and cardiovascular diseases (112), major comorbidities of COVID-19.
While LPA promotes bone marrow derived monocyte (CD11b+) activation (F4/80 expression) in vitro as potently as M-CSF (106), LPA has been also reported to co-stimulate the GM-CSF/IL-4-induced conversion of monocytes to DCs (113, 114). Moreover, LPA has been also reported to modulate the activity of TCF4 (115), a decisive transcription factor in plasmacytoid dendritic cells (pDCs) development and homeostasis (116). Increased ENPP2 expression was detected in COVID-19 DCs and pDCs, correlating with markers of immature DCs (61), while an anti-inflammatory role of LPA has been previously proposed for DCs via LPAR2 (117), suggesting that ATX/LPA could be also involved in suppression of DC responses in COVID-19.
Therefore, increases of ATX levels and LPA local production in ARDS, ILD/IPF and COVID-19 can exacerbate numerous pathogenic responses in the lung, likely in co-ordination with other pathologic inflammatory and fibrotic factors.
Pharmacologic Targeting of ATX as an Additional Therapeutic Option in COVID-19
Dexamethasone (Dex), the first line of defense against systemic inflammation, has been proven effective in COVID-19 patients requiring oxygen or ventilation (118, 119), the only approved single therapy against severe COVID-19. Remarkably, Dex treatment of ventilated COVID-19 patients attenuated serum ATX levels, suggesting that the therapeutic effects of Dex include the suppression of ATX expression (61) and that ATX can be druggable.
The exacerbated production of IL-6 and other storm cytokines, where present, is considered among the leading causes of COVID-19/ARDS-related mortality, and therefore many clinical trials have been conducted targeting storm cytokines or their receptors, with inconsistent results, spurring controversial opinions on the use of systemic anti-inflammatory drugs (120). ATX and IL-6 levels were shown to correlate in ACLF (73), ARDS (62) and COVID-19 (61) patients, suggesting that simultaneous inhibition of both IL-6 and ATX may be an effective therapeutic strategy for COVID-19, as previously suggested in systemic sclerosis (74).
The antifibrotic compounds pirfenidone and nintedanib, approved for IPF, have shown efficacy in fibrotic lung diseases other than IPF (121–125). Therefore, since COVID-19 and IPF share disease severity risk factors, such as sex/age and comorbidities, existing and developing anti-fibrotic compounds have been suggested as additional therapeutic options in COVID-19 (126–128). Among them, GLPG1690 (Table 3 and Figure 1) targets ATX and, together with the standard of care treatment (pirfenidone or nintedanib), has entered phase III international clinical trials (ISABELA 1 and 2, NCT03711162 and NCT03733444) (129). Given the above, the same or a similar regime might also prove effective in COVID-19.
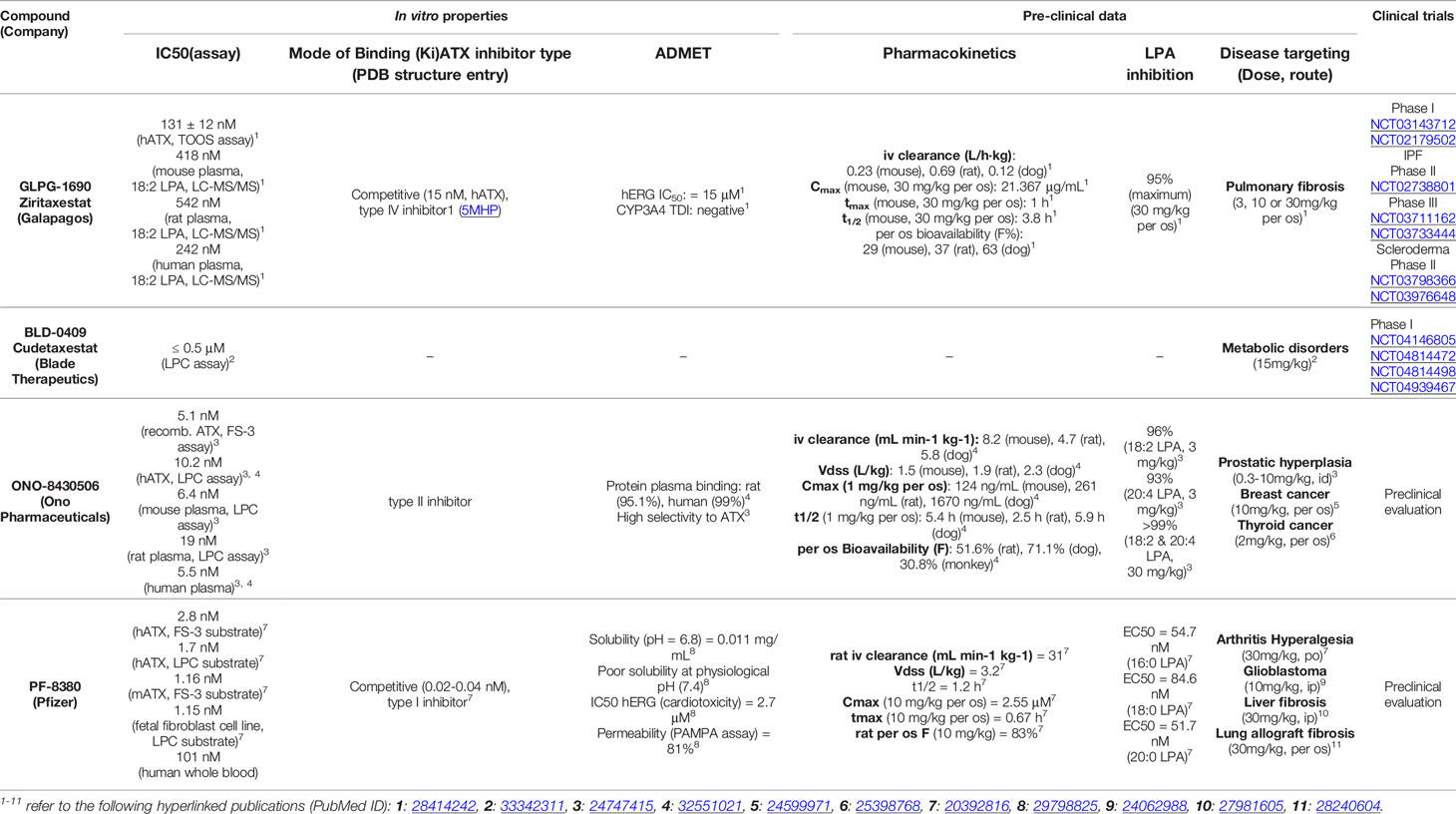
Table 3 Representative small molecules targeting ATX in late-stage preclinical and clinical development.
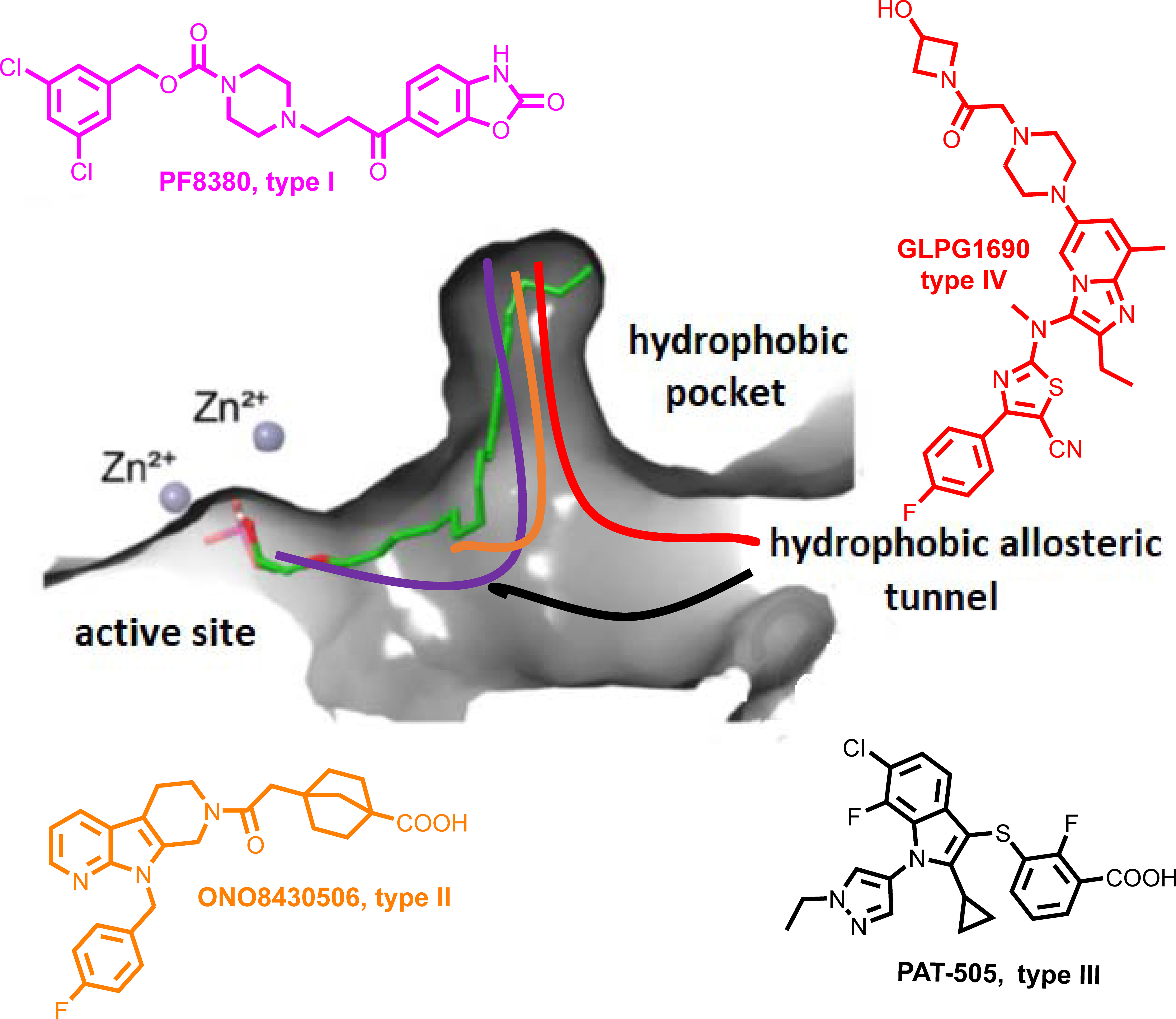
Figure 1 Schematic representation of the mode of binding (color coded) to ATX by the 4 different types of ATX inhibitors (I-IV). The mode of binding of LPA is also displayed (in green).
The crystal structure of ATX has been solved (55, 130, 131), allowing a deep understanding of its structure and function relationship (132) (Figure 1), and thus promoting rational drug design. Given the establishment of ATX as a therapeutic target in IPF, as well as the promising results from the initial clinical trials, a plethora of ATX inhibitors have been developed (133, 134); the ones at late-stage development as shown in Table 3 and their mode in binding at Figure 1.
Inducible genetic deletion of ATX in adult life, resulting in 70-80% decreases in serum ATX levels and mRNA expression levels in different tissues, did not have any appreciable effects in gross pathophysiology of major organs (135), suggesting that the bulk of ATX activity in mice is dispensable for adult life. Moreover, potent (IC50 2 nM), long term (3 weeks) pharmacological inhibition of ATX with PF-8380 (120 mg/Kg - 4 times the effective concentration; PO; bid) had no effects in survival or gross pathology of major organs (135), suggesting that ATX pharmaceutical targeting is safe and well tolerated in mice. In humans, GLPG1690 was reported to be well tolerated in a phase 1 randomized clinical trial (NCT02179502), safe and efficacious in a phase 2a randomized placebo-controlled clinical trial (NCT02738801), supporting ATX inhibition as a novel treatment for IPF (136, 137). In addition, administration of BBT-877, another orally available small molecule inhibitor targeting ATX (IC50 ~6.7 nM), to healthy volunteers in a phase I clinical trial (NCT03830125), did not reveal severe adverse events (138, 139). However, the GLPG1690 phase III clinical trial was recently discontinued on account of “low benefit to risk ratio“. Likewise, BBT-877’s scheduled phase II clinical trial was also postponed due to “toxicity concerns“. Since the relative results are not announced yet, it is not known if the toxicity was imposed from the compounds themselves or their target. Nevertheless, several new candidates are emerging, while possible compound toxicity can be eliminated with targeted modifications or bypassed via inhaled administration.
Conclusions
The increased levels of ATX in ILDs/IPF, ARDS and COVID-19 add yet another commonality between them and suggest that LPA signaling is involved in their pathogenesis, including the amplification of vascular damage, the regulation of the immune system and the promotion of fibrosis. Therefore, the therapeutic targeting of ATX in IPF and fibrotic diseases could be also applied in COVID-19, alone or together with approved anti-fibrotic, anti-rheumatic and anti-viral drugs, especially given its predicted short-term administration, as well as the emergency nature and unmet medical need for the treatment of COVID-19 severe cases.
Author Contributions
KN, TK, ET, ES and AM drafted the paper. AT and KA critically commented on the draft version. VA finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work has been co-financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call Research – Create – Innovate (project code: T1EDK-0049. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hu B, Guo H, Zhou P, Shi Z-L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol (2020) 19(3):141–54. doi: 10.1038/s41579-020-00459-7
2. Osuchowski MF, Winkler MS, Skirecki T, Cajander S, Shankar-Hari M, Lachmann G, et al. The COVID-19 Puzzle: Deciphering Pathophysiology and Phenotypes of a New Disease Entity. Lancet Respir Med (2021) 9(6):622–42. doi: 10.1016/S2213-2600(21)00218-6
3. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80.e8. doi: 10.1016/j.cell.2020.02.052
4. Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 Entry Factors Are Highly Expressed in Nasal Epithelial Cells Together With Innate Immune Genes. Nat Med (2020) 26(5):681–7. doi: 10.1038/s41591-020-0868-6
5. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. Jama (2020) 323(11):1061–9. doi: 10.1001/jama.2020.1585
6. Guan W-J, Ni Z-Y, Hu Y, Liang W-H, Ou C-Q, He J-X, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382(18):1708–20. doi: 10.1101/2020.02.06.20020974
7. Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical Course and Outcomes of Critically Ill Patients With SARS-CoV-2 Pneumonia in Wuhan, China: A Single-Centered, Retrospective, Observational Study. Lancet Respir Med (2020) 8(5):475–81. doi: 10.1016/S2213-2600(20)30079-5
8. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet (2020) 395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7
9. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. Jama (2020) 323(13):1239–42. doi: 10.1001/jama.2020.2648
10. Torres Acosta MA, Singer BD. Pathogenesis of COVID-19-Induced ARDS: Implications for an Ageing Population. Eur Respir J (2020) 56(3)10:2002049. doi: 10.1183/13993003.02049-2020
11. Shanmugam C, Mohammed AR, Ravuri S, Luthra V, Rajagopal N, Karre S. COVID-2019 - A Comprehensive Pathology Insight. Pathol Res Pract (2020) 216(10):153222. 11. doi: 10.1016/j.prp.2020.153222
12. Bussani R, Schneider E, Zentilin L, Collesi C, Ali H, Braga L, et al. Persistence of Viral RNA, Pneumocyte Syncytia and Thrombosis Are Hallmarks of Advanced COVID-19 Pathology. EBioMedicine (2020) 61:103104. doi: 10.1016/j.ebiom.2020.103104
13. Polak SB, Van Gool IC, Cohen D, von der Thüsen JH, van Paassen J. A Systematic Review of Pathological Findings in COVID-19: A Pathophysiological Timeline and Possible Mechanisms of Disease Progression. Mod Pathol (2020) 33(11):2128–38. doi: 10.1038/s41379-020-0603-3
14. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
15. Merad M, Martin JC. Pathological Inflammation in Patients With COVID-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol (2020) 20(6):355–62. doi: 10.1038/s41577-020-0331-4
16. Zhang Z-L, Hou Y-L, Li D-T, Li F-Z. Laboratory Findings of COVID-19: A Systematic Review and Meta-Analysis. Scand J Clin Lab Invest (2020) 80(6):441–7. doi: 10.1080/00365513.2020.1768587
17. Ulhaq ZS, Soraya GV. Interleukin-6 as a Potential Biomarker of COVID-19 Progression. Med Mal Infect (2020) 50(4):382–3. doi: 10.1016/j.medmal.2020.04.002
18. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute Respiratory Distress Syndrome. Nat Rev Dis Primers (2019) 5(1):18. doi: 10.1038/s41572-019-0069-0
19. Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D. COVID-19 Does Not Lead to a "Typical" Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med (2020) 201(10):1299–300. doi: 10.1164/rccm.202003-0817LE
20. Ziehr DR, Alladina J, Petri CR, Maley JH, Moskowitz A, Medoff BD, et al. Respiratory Pathophysiology of Mechanically Ventilated Patients With COVID-19: A Cohort Study. Am J Respir Crit Care Med (2020) 201(12):1560–4. doi: 10.1164/rccm.202004-1163LE
21. Mangalmurti NS, Reilly JP, Cines DB, Meyer NJ, Hunter CA, Vaughan AE. COVID-19-Associated Acute Respiratory Distress Syndrome Clarified: A Vascular Endotype? Am J Respir Crit Care Med (2020) 202(5):750–3. doi: 10.1164/rccm.202006-2598LE
22. Tobin MJ. Pondering the Atypicality of ARDS in COVID-19 Is a Distraction for the Bedside Doctor. Intensive Care Med (2021) 47(3):361–2. doi: 10.1007/s00134-020-06340-8
23. Goligher EC, Ranieri VM, Slutsky AS. Is Severe COVID-19 Pneumonia a Typical or Atypical Form of ARDS? And Does it Matter? Intensive Care Med (2021) 47(1):83–5. doi: 10.1007/s00134-020-06320-y
24. Bos LDJ. COVID-19-Related Acute Respiratory Distress Syndrome: Not So Atypical. Am J Respir Crit Care Med (2020) 202(4):622–4. doi: 10.1164/rccm.202004-1423LE
25. Huertas A, Montani D, Savale L, Pichon J, Tu L, Parent F, et al. Endothelial Cell Dysfunction: A Major Player in SARS-CoV-2 Infection (COVID-19)? Eur Respir J (2020) 56(1):2001634. doi: 10.1183/13993003.01634-2020
26. Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial Damage in Acute Respiratory Distress Syndrome. Int J Mol Sci (2020) 21(22):8793. doi: 10.3390/ijms21228793
27. Vassiliou AG, Keskinidou C, Jahaj E, Gallos P, Dimopoulou I, Kotanidou A, et al. ICU Admission Levels of Endothelial Biomarkers as Predictors of Mortality in Critically Ill COVID-19 Patients. Cells (2021) 10(1):186. doi: 10.3390/cells10010186
28. Burnham EL, Janssen WJ, Riches DW, Moss M, Downey GP. The Fibroproliferative Response in Acute Respiratory Distress Syndrome: Mechanisms and Clinical Significance. Eur Respir J (2014) 43(1):276–85. doi: 10.1183/09031936.00196412
29. Thille AW, Esteban A, Fernández-Segoviano P, Rodriguez JM, Aramburu JA, Vargas-Errázuriz P, et al. Chronology of Histological Lesions in Acute Respiratory Distress Syndrome With Diffuse Alveolar Damage: A Prospective Cohort Study of Clinical Autopsies. Lancet Respir Med (2013) 1(5):395–401. doi: 10.1016/S2213-2600(13)70053-5
30. Cabrera-Benitez NE, Laffey JG, Parotto M, Spieth PM, Villar J, Zhang H, et al. Mechanical Ventilation-Associated Lung Fibrosis in Acute Respiratory Distress Syndrome: A Significant Contributor to Poor Outcome. Anesthesiology (2014) 121(1):189–98. doi: 10.1097/ALN.0000000000000264
31. Maiese A, Manetti AC, La Russa R, Di Paolo M, Turillazzi E, Frati P, et al. Autopsy Findings in COVID-19-Related Deaths: A Literature Review. Forensic Sci Med Pathol (2020) 17(2):279–96. doi: 10.1007/s12024-020-00310-8
32. Mo X, Jian W, Su Z, Chen M, Peng H, Peng P, et al. Abnormal Pulmonary Function in COVID-19 Patients at Time of Hospital Discharge. Eur Respir J (2020) 55(6):2001217. doi: 10.1183/13993003.01217-2020
33. Gassel RJJV, Bels JLM, Raafs A, Bussel BCTV, Poll MCGVD, Simons SO, et al. High Prevalence of Pulmonary Sequelae at 3 Months After Hospital Discharge in Mechanically Ventilated Survivors of COVID-19. Am J Respir Crit Care Med (2021) 203(3):371–4. doi: 10.1164/rccm.202010-3823LE
34. Myall KJ, Mukherjee B, Castanheira AM, Lam JL, Benedetti G, Mak SM, et al. Persistent Post-COVID-19 Interstitial Lung Disease. An Observational Study of Corticosteroid Treatment. Ann Am Thorac Soc (2021) 18(5):799–806. doi: 10.1513/AnnalsATS.202008-1002OC
35. McDonald LT. Healing After COVID-19: Are Survivors at Risk for Pulmonary Fibrosis? Am J Physiol Lung Cell Mol Physiol (2021) 320(2):L257–l65. doi: 10.1152/ajplung.00238.2020
36. Esposito AJ, Menon AA, Ghosh AJ, Putman RK, Fredenburgh LE, El-Chemaly SY, et al. Increased Odds of Death for Patients With Interstitial Lung Disease and COVID-19: A Case-Control Study. Am J Respir Crit Care Med (2020) 202(12):1710–3. doi: 10.1164/rccm.202006-2441LE
37. Drake TM, Docherty AB, Harrison EM, Quint JK, Adamali H, Agnew S, et al. Outcome of Hospitalization for COVID-19 in Patients With Interstitial Lung Disease. An International Multicenter Study. Am J Respir Crit Care Med (2020) 202(12):1656–65. doi: 10.1164/rccm.202007-2794OC
38. Beltramo G, Cottenet J, Mariet AS, Georges M, Piroth L, Tubert-Bitter P, et al. Chronic Respiratory Diseases Are Predictors of Severe Outcome in COVID-19 Hospitalised Patients: A Nationwide Study. Eur Respir J (2021) 2004474. doi: 10.1183/13993003.04474-2020
39. Drake TM, Docherty AB, Harrison EM, Quint JK, Adamali H, Agnew S, et al. Outcome of Hospitalization for COVID-19 in Patients with Interstitial Lung Disease. An International Multicenter Study. Am J Respir Crit Care Med (2020) 202(12):1656–65. doi: 10.1164/rccm.202007-2794OC.
40. Kondoh Y, Kataoka K, Ando M, Awaya Y, Ichikado K, Kataoka M, et al. COVID-19 and Acute Exacerbation of Interstitial Lung Disease. Respir Investig (2021) 59(5):675–8. doi: 10.1016/j.resinv.2021.06.007
41. Magkrioti C, Galaris A, Kanellopoulou P, Stylianaki EA, Kaffe E, Aidinis V. Autotaxin and Chronic Inflammatory Diseases. J Autoimmun (2019) 104:102327. doi: 10.1016/j.jaut.2019.102327
42. Dusaulcy R, Rancoule C, Gres S, Wanecq E, Colom A, Guigne C, et al. Adipose-Specific Disruption of Autotaxin Enhances Nutritional Fattening and Reduces Plasma Lysophosphatidic Acid. J Lipid Res (2011) 52(6):1247–55. doi: 10.1194/jlr.M014985
43. Kaffe E, Katsifa A, Xylourgidis N, Ninou I, Zannikou M, Harokopos V, et al. Hepatocyte Autotaxin Expression Promotes Liver Fibrosis and Cancer. Hepatology (2017) 65(4):1369–83. doi: 10.1002/hep.28973
44. Kanda H, Newton R, Klein R, Morita Y, Gunn MD, Rosen SD. Autotaxin, an Ectoenzyme That Produces Lysophosphatidic Acid, Promotes the Entry of Lymphocytes Into Secondary Lymphoid Organs. Nat Immunol (2008) 9(4):415–23. doi: 10.1038/ni1573
45. Bai Z, Cai L, Umemoto E, Takeda A, Tohya K, Komai Y, et al. Constitutive Lymphocyte Transmigration Across the Basal Lamina of High Endothelial Venules Is Regulated by the Autotaxin/Lysophosphatidic Acid Axis. J Immunol (2013) 190(5):2036–48. doi: 10.4049/jimmunol.1202025
46. Oikonomou N, Mouratis MA, Tzouvelekis A, Kaffe E, Valavanis C, Vilaras G, et al. Pulmonary Autotaxin Expression Contributes to the Pathogenesis of Pulmonary Fibrosis. Am J Respir Cell Mol Biol (2012) 47(5):566–74. doi: 10.1165/rcmb.2012-0004OC
47. Ninou I, Sevastou I, Magkrioti C, Kaffe E, Stamatakis G, Thivaios S, et al. Genetic Deletion of Autotaxin From CD11b+ Cells Decreases the Severity of Experimental Autoimmune Encephalomyelitis. PloS One (2020) 15(4):e0226050. doi: 10.1371/journal.pone.0226050
48. Kim SJ, Howe C, Mitchell J, Choo J, Powers A, Oikonomopoulos A, et al. Autotaxin Loss Accelerates Intestinal Inflammation by Suppressing TLR4-Mediated Immune Responses. EMBO Rep (2020) 21(10):e49332. doi: 10.15252/embr.201949332
49. Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, et al. Autotaxin has Lysophospholipase D Activity Leading to Tumor Cell Growth and Motility by Lysophosphatidic Acid Production. J Cell Biol (2002) 158(2):227–33. doi: 10.1083/jcb.200204026
50. Perrakis A, Moolenaar WH. Autotaxin: Structure-Function and Signaling. J Lipid Res (2014) 55(6):1010–18. doi: 10.1194/jlr.R046391
51. Barbayianni E, Kaffe E, Aidinis V, Kokotos G. Autotaxin, A Secreted Lysophospholipase D, as a Promising Therapeutic Target in Chronic Inflammation and Cancer. Prog Lipid Res (2015) 58:76–96. doi: 10.1016/j.plipres.2015.02.001
52. Aikawa S, Hashimoto T, Kano K, Aoki J. Lysophosphatidic Acid as a Lipid Mediator With Multiple Biological Actions. J Biochem (2015) 157(2):81–9. doi: 10.1093/jb/mvu077
53. Benesch MG, Tang X, Venkatraman G, Bekele RT, Brindley DN. Recent Advances in Targeting the Autotaxin-Lysophosphatidate-Lipid Phosphate Phosphatase Axis In Vivo. J BioMed Res (2016) 30(4):272–84. doi: 10.7555/JBR.30.20150058
54. Fulkerson Z, Wu T, Sunkara M, Kooi CV, Morris AJ, Smyth SS. Binding of Autotaxin to Integrins Localizes Lysophosphatidic Acid Production to Platelets and Mammalian Cells. J Biol Chem (2011) 286(40):34654–63. doi: 10.1074/jbc.M111.276725
55. Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, et al. Structural Basis of Substrate Discrimination and Integrin Binding by Autotaxin. Nat Struct Mol Biol (2011) 18(2):198–204. doi: 10.1038/nsmb.1980
56. Leblanc R, Lee SC, David M, Bordet JC, Norman DD, Patil R, et al. Interaction of Platelet-Derived Autotaxin With Tumor Integrin Alphavbeta3 Controls Metastasis of Breast Cancer Cells to Bone. Blood (2014) 124(20):3141–50. doi: 10.1182/blood-2014-04-568683
57. Yung YC, Stoddard NC, Chun J. LPA Receptor Signaling: Pharmacology, Physiology, and Pathophysiology. J Lipid Res (2014) 55(7):1192–214. doi: 10.1194/jlr.R046458
58. Kostadinova L, Shive CL, Anthony DD. Elevated Autotaxin and LPA Levels During Chronic Viral Hepatitis and Hepatocellular Carcinoma Associate With Systemic Immune Activation. Cancers (Basel) (2019) 11(12):1867. doi: 10.3390/cancers11121867
59. Joshita S, Ichikawa Y, Umemura T, Usami Y, Sugiura A, Shibata S, et al. Serum Autotaxin Is a Useful Liver Fibrosis Marker in Patients With Chronic Hepatitis B Virus Infection. Hepatol Res (2018) 48(4):275–85. doi: 10.1111/hepr.12997
60. Farquhar MJ, Humphreys IS, Rudge SA, Wilson GK, Bhattacharya B, Ciaccia M, et al. Autotaxin-Lysophosphatidic Acid Receptor Signalling Regulates Hepatitis C Virus Replication. J Hepatol (2017) 66(5):919–29. doi: 10.1016/j.jhep.2017.01.009
61. Nikitopoulou I, Fanidis D, Ntatsoulis K, Moulos P, Mpekoulis G, Evangelidou M, et al. Increased Autotaxin Levels in Severe COVID-19, Correlating With IL-6 Levels, Endothelial Dysfunction Biomarkers, and Impaired Functions of Dendritic Cells. medRxiv (2021) 2021.07.30.21261361. doi: 10.1101/2021.07.30.21261361
62. Gao L, Li X, Wang H, Liao Y, Zhou Y, Wang K, et al. Autotaxin Levels in Serum and Bronchoalveolar Lavage Fluid Are Associated With Inflammatory and Fibrotic Biomarkers and the Clinical Outcome in Patients With Acute Respiratory Distress Syndrome. J Intensive Care (2021) 9(1):44. doi: 10.1186/s40560-021-00559-3
63. Sexton T, Chalhoub G, Ye S, Morris W, Annabathula R, Dugan A, et al. Autotaxin Activity Predicts 30-Day Mortality in Sepsis Patients and Correlates With Platelet Count and Vascular Dysfunction. Shock (2020) 54(6):738–43. doi: 10.1097/SHK.0000000000001569
64. Benesch MG, Tang X, Dewald J, Dong WF, Mackey JR, Hemmings DG, et al. Tumor-Induced Inflammation in Mammary Adipose Tissue Stimulates a Vicious Cycle of Autotaxin Expression and Breast Cancer Progression. FASEB J (2015) 29(9):3990–4000. doi: 10.1096/fj.15-274480
65. Mouratis MA, Magkrioti C, Oikonomou N, Katsifa A, Prestwich GD, Kaffe E, et al. Autotaxin and Endotoxin-Induced Acute Lung Injury. PloS One (2015) 10(7):e0133619. doi: 10.1371/journal.pone.0133619
66. Zhao J, He D, Su Y, Berdyshev E, Chun J, Natarajan V, et al. Lysophosphatidic Acid Receptor 1 Modulates Lipopolysaccharide-Induced Inflammation in Alveolar Epithelial Cells and Murine Lungs. Am J Physiol Lung Cell Mol Physiol (2011) 301(4):L547–56. doi: 10.1152/ajplung.00058.2011
67. Nikitopoulou I, Ninou I, Manitsopoulos N, Dimopoulou I, Orfanos SE, Aidinis V, et al. A Role for Bronchial Epithelial Autotaxin in Ventilator-Induced Lung Injury. Intensive Care Med Exp (2021) 9(1):12. doi: 10.1186/s40635-021-00379-7
68. Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, et al. The Lysophosphatidic Acid Receptor LPA1 Links Pulmonary Fibrosis to Lung Injury by Mediating Fibroblast Recruitment and Vascular Leak. Nat Med (2008) 14(1):45–54. doi: 10.1038/nm1685
69. Ninou I, Kaffe E, Muller S, Budd DC, Stevenson CS, Ullmer C, et al. Pharmacologic Targeting of the ATX/LPA Axis Attenuates Bleomycin-Induced Pulmonary Fibrosis. Pulm Pharmacol Ther (2018) 52:32–40. doi: 10.1016/j.pupt.2018.08.003
70. Ninou I, Magkrioti C, Aidinis V. Autotaxin in Pathophysiology and Pulmonary Fibrosis. Front Med (Lausanne) (2018) 5:180. doi: 10.3389/fmed.2018.00180
71. Gierse JK, Thorarensen A, Beltey K, Bradshaw-Pierce E, Cortes-Burgos L, Hall T, et al. A Novel Autotaxin Inhibitor Reduces Lysophosphatidic Acid Levels in Plasma and the Site of Inflammation. J Pharmacol Exp Ther (2010) 334(1):310–7. doi: 10.1124/jpet.110.165845
72. Tager AM. Autotaxin Emerges as a Therapeutic Target for Idiopathic Pulmonary Fibrosis: Limiting Fibrosis by Limiting Lysophosphatidic Acid Synthesis. Am J Respir Cell Mol Biol (2012) 47(5):563–5. doi: 10.1165/rcmb.2012-0235ED
73. Sun S, Wang R, Song J, Guan M, Li N, Zhang X, et al. Blocking Gp130 Signaling Suppresses Autotaxin Expression in Adipocytes and Improves Insulin Sensitivity in Diet-Induced Obesity. J Lipid Res (2017) 58: (11):2102–13. doi: 10.1194/jlr.M075655
74. Castelino FV, Bain G, Pace VA, Black KE, George L, Probst CK, et al. An Autotaxin/Lysophosphatidic Acid/Interleukin-6 Amplification Loop Drives Scleroderma Fibrosis. Arthritis Rheumatol (Hoboken NJ) (2016) 68(12):2964–74. doi: 10.1002/art.39797
75. Nikitopoulou I, Oikonomou N, Karouzakis E, Sevastou I, Nikolaidou-Katsaridou N, Zhao Z, et al. Autotaxin Expression From Synovial Fibroblasts Is Essential for the Pathogenesis of Modeled Arthritis. J Exp Med (2012) 209(5):925–33. doi: 10.1084/jem.20112012
76. Lei L, Su J, Chen J, Chen W, Chen X, Peng C. The Role of Lysophosphatidic Acid in the Physiology and Pathology of the Skin. Life Sci (2019) 220:194–200. doi: 10.1016/j.lfs.2018.12.040
77. Herold T, Jurinovic V, Arnreich C, Lipworth BJ, Hellmuth JC, von Bergwelt-Baildon M, et al. Elevated Levels of IL-6 and CRP Predict the Need for Mechanical Ventilation in COVID-19. J Allergy Clin Immunol (2020) 146(1):128–36.e4. doi: 10.1016/j.jaci.2020.05.008
78. Guirao JJ, Cabrera CM, Jiménez N, Rincón L, Urra JM. High Serum IL-6 Values Increase the Risk of Mortality and the Severity of Pneumonia in Patients Diagnosed With COVID-19. Mol Immunol (2020) 128:64–8. doi: 10.1016/j.molimm.2020.10.006
79. Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M, et al. Autotaxin Stabilizes Blood Vessels and Is Required for Embryonic Vasculature by Producing Lysophosphatidic Acid. J Biol Chem (2006) 281(35):25822–30. doi: 10.1074/jbc.M605142200
80. van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradere JP, et al. Autotaxin, a Secreted Lysophospholipase D, Is Essential for Blood Vessel Formation During Development. Mol Cell Biol (2006) 26(13):5015–22. doi: 10.1128/MCB.02419-05
81. Fotopoulou S, Oikonomou N, Grigorieva E, Nikitopoulou I, Paparountas T, Thanassopoulou A, et al. ATX Expression and LPA Signalling Are Vital for the Development of the Nervous System. Dev Biol (2010) 339(2):451–64. doi: 10.1016/j.ydbio.2010.01.007
82. Gralinski LE, Menachery VD, Morgan AP, Totura AL, Beall A, Kocher J, et al. Allelic Variation in the Toll-Like Receptor Adaptor Protein Ticam2 Contributes to SARS-Coronavirus Pathogenesis in Mice. G3 (Bethesda) (2017) 7(6):1653–63. doi: 10.1534/g3.117.041434
83. Gralinski LE, Ferris MT, Aylor DL, Whitmore AC, Green R, Frieman MB, et al. Genome Wide Identification of SARS-CoV Susceptibility Loci Using the Collaborative Cross. PloS Genet (2015) 11(10):e1005504. doi: 10.1371/journal.pgen.1005504
84. Zhao J, Wei J, Dong S, Bowser RK, Zhang L, Jacko AM, et al. Destabilization of Lysophosphatidic Acid Receptor 1 Reduces Cytokine Release and Protects Against Lung Injury. EBioMedicine (2016) 10:195–203. doi: 10.1016/j.ebiom.2016.07.020
85. Ren H, Panchatcharam M, Mueller P, Escalante-Alcalde D, Morris AJ, Smyth SS. Lipid Phosphate Phosphatase (LPP3) and Vascular Development. Biochim Biophys Acta (2013) 1831(1):126–32. doi: 10.1016/j.bbalip.2012.07.012
86. Tang X, Brindley DN. Lipid Phosphate Phosphatases and Cancer. Biomolecules (2020) 10(9):1263. doi: 10.3390/biom10091263
87. Magkrioti C, Aidinis V. ATX and LPA Signalling in Lung Pathophysiology. World J Respirol (2013) 3(3):77–103. doi: 10.5320/wjr.v3.i3.77
88. Shea BS, Tager AM. Role of the Lysophospholipid Mediators Lysophosphatidic Acid and Sphingosine 1-Phosphate in Lung Fibrosis. Proc Am Thorac Soc (2012) 9(3):102–10. doi: 10.1513/pats.201201-005AW
89. Zhao Y, Natarajan V. Lysophosphatidic Acid (LPA) and Its Receptors: Role in Airway Inflammation and Remodeling. Biochim Biophys Acta (2013) 1831(1):86–92. doi: 10.1016/j.bbalip.2012.06.014
90. Kim SJ, Moon HG, Park GY. The Roles of Autotaxin/Lysophosphatidic Acid in Immune Regulation and Asthma. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865(5):158641. doi: 10.1016/j.bbalip.2020.158641
91. Knowlden S, Georas SN. The Autotaxin-LPA Axis Emerges as a Novel Regulator of Lymphocyte Homing and Inflammation. J Immunol (2014) 192(3):851–7. doi: 10.4049/jimmunol.1302831
92. Lee SC, Dacheux MA, Norman DD, Balazs L, Torres RM, Augelli-Szafran CE, et al. Regulation of Tumor Immunity by Lysophosphatidic Acid. Cancers (Basel) (2020) 12(5):1202. doi: 10.3390/cancers12051202
93. Knowlden SA, Capece T, Popovic M, Chapman TJ, Rezaee F, Kim M, et al. Regulation of T Cell Motility In Vitro and In Vivo by LPA and LPA2. PloS One (2014) 9(7):e101655. doi: 10.1371/journal.pone.0101655
94. Katakai T, Kondo N, Ueda Y, Kinashi T. Autotaxin Produced by Stromal Cells Promotes LFA-1-Independent and Rho-Dependent Interstitial T Cell Motility in the Lymph Node Paracortex. J Immunol (2014) 193(2):617–26. doi: 10.4049/jimmunol.1400565
95. Hisano Y, Kono M, Cartier A, Engelbrecht E, Kano K, Kawakami K, et al. Lysolipid Receptor Cross-Talk Regulates Lymphatic Endothelial Junctions in Lymph Nodes. J Exp Med (2019) 216(7):1582–98. doi: 10.1084/jem.20181895
96. Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the Sphingosine-1-Phosphate Axis in Cancer, Inflammation and Beyond. Nat Rev Drug Discov (2013) 12(9):688–702. doi: 10.1038/nrd4099
97. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte Egress From Thymus and Peripheral Lymphoid Organs Is Dependent on S1P Receptor 1. Nature (2004) 427(6972):355–60. doi: 10.1038/nature02284
98. Wynn Thomas A, Vannella Kevin M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
99. Desai O, Winkler J, Minasyan M, Herzog EL. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front Med (2018) 5:43. doi: 10.3389/fmed.2018.00043
100. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
101. Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M, Guzman ER, et al. Circuits Between Infected Macrophages and T Cells in SARS-CoV-2 Pneumonia. Nature (2021) 590(7847):635–41. doi: 10.1038/s41586-020-03148-w
102. Shimada H, Rajagopalan LE. Rho-Kinase Mediates Lysophosphatidic Acid-Induced IL-8 and MCP-1 Production via P38 and JNK Pathways in Human Endothelial Cells. FEBS Lett (2010) 584(13):2827–32. doi: 10.1016/j.febslet.2010.04.064
103. Zhou Z, Subramanian P, Sevilmis G, Globke B, Soehnlein O, Karshovska E, et al. Lipoprotein-Derived Lysophosphatidic Acid Promotes Atherosclerosis by Releasing CXCL1 From the Endothelium. Cell Metab (2011) 13(5):592–600. doi: 10.1016/j.cmet.2011.02.016
104. Takeda Y, Matoba K, Kawanami D, Nagai Y, Akamine T, Ishizawa S, et al. ROCK2 Regulates Monocyte Migration and Cell to Cell Adhesion in Vascular Endothelial Cells. Int J Mol Sci (2019) 20(6):1331. doi: 10.3390/ijms20061331
105. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-Derived Alveolar Macrophages Drive Lung Fibrosis and Persist in the Lung Over the Life Span. J Exp Med (2017) 214(8):2387–404. doi: 10.1084/jem.20162152
106. Ray R, Rai V. Lysophosphatidic Acid Converts Monocytes Into Macrophages in Both Mice and Humans. Blood (2017) 129(9):1177–83. doi: 10.1182/blood-2016-10-743757
107. Santos-Nogueira E, Lopez-Serrano C, Hernandez J, Lago N, Astudillo AM, Balsinde J, et al. Activation of Lysophosphatidic Acid Receptor Type 1 Contributes to Pathophysiology of Spinal Cord Injury. J Neurosci (2015) 35(28):10224–35. doi: 10.1523/JNEUROSCI.4703-14.2015
108. Kaya B, Doñas C, Wuggenig P, Diaz OE, Morales RA, Melhem H, et al. Lysophosphatidic Acid-Mediated GPR35 Signaling in CX3CR1(+) Macrophages Regulates Intestinal Homeostasis. Cell Rep (2020) 32(5):107979. doi: 10.1016/j.celrep.2020.107979
109. Fransson J, Gómez-Conde AI, Romero-Imbroda J, Fernández O, Leyva L, de Fonseca FR, et al. Activation of Macrophages by Lysophosphatidic Acid Through the Lysophosphatidic Acid Receptor 1 as a Novel Mechanism in Multiple Sclerosis Pathogenesis. Mol Neurobiol (2021) 58(2):470–82. doi: 10.1007/s12035-020-02130-x
110. Chang CL, Hsu HY, Lin HY, Chiang W, Lee H. Lysophosphatidic Acid-Induced Oxidized Low-Density Lipoprotein Uptake Is Class A Scavenger Receptor-Dependent in Macrophages. Prostaglandins Other Lipid Mediat (2008) 87(1-4):20–5. doi: 10.1016/j.prostaglandins.2008.05.002
111. An D, Hao F, Zhang F, Kong W, Chun J, Xu X, et al. CD14 Is a Key Mediator of Both Lysophosphatidic Acid and Lipopolysaccharide Induction of Foam Cell Formation. J Biol Chem (2017) 292(35):14391–400. doi: 10.1074/jbc.M117.781807
112. Schober A, Siess W. Lysophosphatidic Acid in Atherosclerotic Diseases. Br J Pharmacol (2012) 167(3):465–82. doi: 10.1111/j.1476-5381.2012.02021.x
113. Martino A, Volpe E, Baldini PM. The Influence of Lysophosphatidic Acid on the Immunophenotypic Differentiation of Human Monocytes Into Dendritic Cells. Haematologica (2006) 91(9):1273–4. doi: 10.3324/%x
114. Chen R, Roman J, Guo J, West E, McDyer J, Williams MA, et al. Lysophosphatidic Acid Modulates the Activation of Human Monocyte-Derived Dendritic Cells. Stem Cells Dev (2006) 15(6):797–804. doi: 10.1089/scd.2006.15.797
115. Guo L, He P, No YR, Yun CC. Krüppel-Like Factor 5 Incorporates Into the β-Catenin/TCF Complex in Response to LPA in Colon Cancer Cells. Cell Signal (2015) 27(5):961–8. doi: 10.1016/j.cellsig.2015.02.005
116. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription Factor E2-2 Is an Essential and Specific Regulator of Plasmacytoid Dendritic Cell Development. Cell (2008) 135(1):37–48. doi: 10.1016/j.cell.2008.09.016
117. Emo J, Meednu N, Chapman TJ, Rezaee F, Balys M, Randall T, et al. Lpa2 Is a Negative Regulator of Both Dendritic Cell Activation and Murine Models of Allergic Lung Inflammation. J Immunol (2012) 188(8):3784–90. doi: 10.4049/jimmunol.1102956
118. Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, et al. Dexamethasone in Hospitalized Patients With Covid-19. N Engl J Med (2021) 384(8):693–704. doi: 10.1056/NEJMoa2021436
119. Sterne JAC, Murthy S, Diaz JV, Slutsky AS, Villar J, Angus DC, et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-Analysis. Jama (2020) 324(13):1330–41. doi: 10.1001/jama.2020.17023
120. Abdin SM, Elgendy SM, Alyammahi SK, Alhamad DW, Omar HA. Tackling the Cytokine Storm in COVID-19, Challenges and Hopes. Life Sci (2020) 257:118054. doi: 10.1016/j.lfs.2020.118054
121. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. N Engl J Med (2019) 380(26):2518–28. doi: 10.1056/NEJMoa1903076
122. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med (2019) 381(18):1718–27. doi: 10.1056/NEJMoa1908681
123. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in Patients With Unclassifiable Progressive Fibrosing Interstitial Lung Disease: A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet Respir Med (2020) 8(2):147–57. doi: 10.1016/S2213-2600(19)30341-8
124. King TE Jr., Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A Phase 3 Trial of Pirfenidone in Patients With Idiopathic Pulmonary Fibrosis. N Engl J Med (2014) 370(22):2083–92. doi: 10.1056/NEJMoa1402582
125. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N Engl J Med (2014) 370(22):2071–82. doi: 10.1056/NEJMoa1402584
126. Spagnolo P, Balestro E, Aliberti S, Cocconcelli E, Biondini D, Casa GD, et al. Pulmonary Fibrosis Secondary to COVID-19: A Call to Arms? Lancet Respir Med (2020) 8(8):750–2. doi: 10.1016/S2213-2600(20)30222-8
127. George PM, Wells AU, Jenkins RG. Pulmonary Fibrosis and COVID-19: The Potential Role for Antifibrotic Therapy. Lancet Respir Med (2020) 8(8):807–15. doi: 10.1016/S2213-2600(20)30225-3
128. Vasarmidi E, Tsitoura E, Spandidos DA, Tzanakis N, Antoniou KM. Pulmonary Fibrosis in the Aftermath of the COVID-19 Era (Review). Exp Ther Med (2020) 20(3):2557–60. doi: 10.3892/etm.2020.8980
129. Maher TM, Kreuter M, Lederer DJ, Brown KK, Wuyts W, Verbruggen N, et al. Rationale, Design and Objectives of Two Phase III, Randomised, Placebo-Controlled Studies of GLPG1690, a Novel Autotaxin Inhibitor, in Idiopathic Pulmonary Fibrosis (ISABELA 1 and 2). BMJ Open Respir Res (2019) 6(1):e000422. doi: 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A4597
130. Nishimasu H, Okudaira S, Hama K, Mihara E, Dohmae N, Inoue A, et al. Crystal Structure of Autotaxin and Insight Into GPCR Activation by Lipid Mediators. Nat Struct Mol Biol (2011) 18(2):205–12. doi: 10.1038/nsmb.1998
131. Moolenaar WH, Perrakis A. Insights Into Autotaxin: How to Produce and Present a Lipid Mediator. Nat Rev Mol Cell Biol (2011) 12(10):674–9. doi: 10.1038/nrm3188
132. Salgado-Polo F, Perrakis A. The Structural Binding Mode of the Four Autotaxin Inhibitor Types That Differentially Affect Catalytic and Non-Catalytic Functions. Cancers (2019) 11(10):1577. doi: 10.3390/cancers11101577
133. Matralis AN, Afantitis A, Aidinis V. Development and Therapeutic Potential of Autotaxin Small Molecule Inhibitors: From Bench to Advanced Clinical Trials. Med Res Rev (2018) 39(3):976–1013. doi: 10.1002/med.21551
134. Castagna D, Budd DC, Macdonald SJ, Jamieson C, Watson AJ. Development of Autotaxin Inhibitors: An Overview of the Patent and Primary Literature. J Med Chem (2016) 59(12):5604–21. doi: 10.1021/acs.jmedchem.5b01599
135. Katsifa A, Kaffe E, Nikolaidou-Katsaridou N, Economides AN, Newbigging S, McKerlie C, et al. The Bulk of Autotaxin Activity Is Dispensable for Adult Mouse Life. PloS One (2015) 10(11):e0143083. doi: 10.1371/journal.pone.0143083
136. van der Aar E, Desrivot J, Dupont S, Heckmann B, Fieuw A, Stutvoet S, et al. Safety, Pharmacokinetics, and Pharmacodynamics of the Autotaxin Inhibitor GLPG1690 in Healthy Subjects: Phase 1 Randomized Trials. J Clin Pharmacol (2019) 59(10):1366–78. doi: 10.1002/jcph.1424
137. Maher TM, van der Aar EM, Van de Steen O, Allamassey L, Desrivot J, Dupont S, et al. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of GLPG1690, a Novel Autotaxin Inhibitor, to Treat Idiopathic Pulmonary Fibrosis (FLORA): A Phase 2a Randomised Placebo-Controlled Trial. Lancet Respir Med (2018) 6(8):627–35. doi: 10.1016/S2213-2600(18)30181-4
138. Lee G, Kang SU, Ryou J-H, Lim J-J, Lee Y-H. Late Breaking Abstract - BBT-877, A Potent Autotaxin Inhibitor in Clinical Development to Treat Idiopathic Pulmonary Fibrosis. Eur Respir J (2019) 54(suppl 63):PA1293. doi: 10.1183/13993003.congress-2019.PA1293
139. Lee G, Kang SU, Ryou JH, Lim JJ, Lee DY, Kwon HJ, et al. (2019). BBT-877, a Potent Autotaxin Inhibitor in Clinical Development to Treat Idiopathic Pulmonary Fibrosis. A108 PATHOPHYSIOLOGY IN DIFFUSE PARENCHYMAL LUNG DISEASES, in: American Thoracic Society International Conference Abstracts, pp. A2577–A. American Thoracic Society.
Keywords: COVID-19, ARDS, pulmonary fibrosis, Autotaxin, lysophosphatidic acid
Citation: Ntatsoulis K, Karampitsakos T, Tsitoura E, Stylianaki E-A, Matralis AN, Tzouvelekis A, Antoniou K and Aidinis V (2021) Commonalities Between ARDS, Pulmonary Fibrosis and COVID-19: The Potential of Autotaxin as a Therapeutic Target. Front. Immunol. 12:687397. doi: 10.3389/fimmu.2021.687397
Received: 29 March 2021; Accepted: 13 August 2021;
Published: 04 October 2021.
Edited by:
Tanima Bose, Ludwig Maximilian University of Munich, GermanyReviewed by:
Mohd Wajid Ali Khan, University of Hail, Saudi ArabiaRosalinda Sorrentino, University of Salerno, Italy
Copyright © 2021 Ntatsoulis, Karampitsakos, Tsitoura, Stylianaki, Matralis, Tzouvelekis, Antoniou and Aidinis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Argyrios Tzouvelekis, YXR6b3V2ZWxla2lzQHVwYXRyYXMuZ3I=; Katerina Antoniou, a2FudG9uaW91QG1lZC51b2MuZ3I=; Vassilis Aidinis, Vi5BaWRpbmlzQEZsZW1pbmcuZ3I=
†These authors have contributed equally to this work