 Satish kumar R. Noonepalle
Satish kumar R. Noonepalle Lidia Karabon
Lidia Karabon Katherine B. Chiappinelli
Katherine B. Chiappinelli Alejandro Villagra
Alejandro Villagra- 1Department of Biochemistry and Molecular Medicine, GW Cancer Center, School of Medicine and Health Sciences, George Washington University, Washington DC, United States
- 2Department of Experimental Therapy, Ludwik Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wroclaw, Poland
- 3Department of Microbiology, Immunology, and Tropical Medicine, GW Cancer Center, School of Medicine and Health Sciences, George Washington University, Washington DC, United States
Editorial on the Research Topic
Genetic and Epigenetic Control of Immune Responses
Introduction
Cancer, traditionally viewed as a disease driven by genomic alterations, is now perceived as an accumulation of genetic mutations as well as global epigenetic changes to the chromatin that regulate gene expression (1, 2). Genetic alterations to either tumor suppressors or oncogenes can result in dramatic gene expression changes leading to cancer; however, changes in the epigenome are rather subtle. Despite similar genomic sequences in all the cell types, the epigenome can vary considerably, resulting in distinct gene expression patterns and, therefore, distinct cellular functions. Epigenetic modifications to chromatin include DNA methylation, histone modifications, nucleosome positioning, and non-coding RNAs that can regulate access of DNA to transcription factors and other cis-regulatory elements, thereby affecting gene expression (3). It is now recognized that genetic and epigenetic components complement each other to drive tumor initiation and progression (4). Recent technical advances in high throughput sequencing have improved understanding of the epigenomic landscape at a higher resolution. Massive datasets and databases, including the encyclopedia of DNA elements (ENCODE) (5), The Cancer Genome Atlas (TCGA) ((https://www.cancer.gov/tcga), the NIH Roadmap Epigenomics Mapping Consortium (6), the Epigenome browser (7), have enhanced our ability to understand the interplay between cancer cells, tumor microenvironment (TME) and immune cells. Therefore, a new classification of molecular epigenetic modifications is needed to differentiate “cancer intrinsic” and “cancer extrinsic” mechanisms influencing anti-tumor immune responses. The schematic shows a graphical representation of various intrinsic and extrinsic factors affecting the cancer cells and thereby regulate the TME (Figure 1). The International Human Epigenome Consortium (IHEC) provides high-resolution reference epigenomes of major primary human cell types (8). Based on the data from these projects, genetic and epigenetic crosstalk in cells is evident, and it has led to the identification of novel biomarkers and the development of novel therapeutic strategies. The articles in this Research Topic on Genetic and Epigenetic Control of Immune Responses address both cell-intrinsic and cell-extrinsic mechanisms controlling the immune response to tumors.
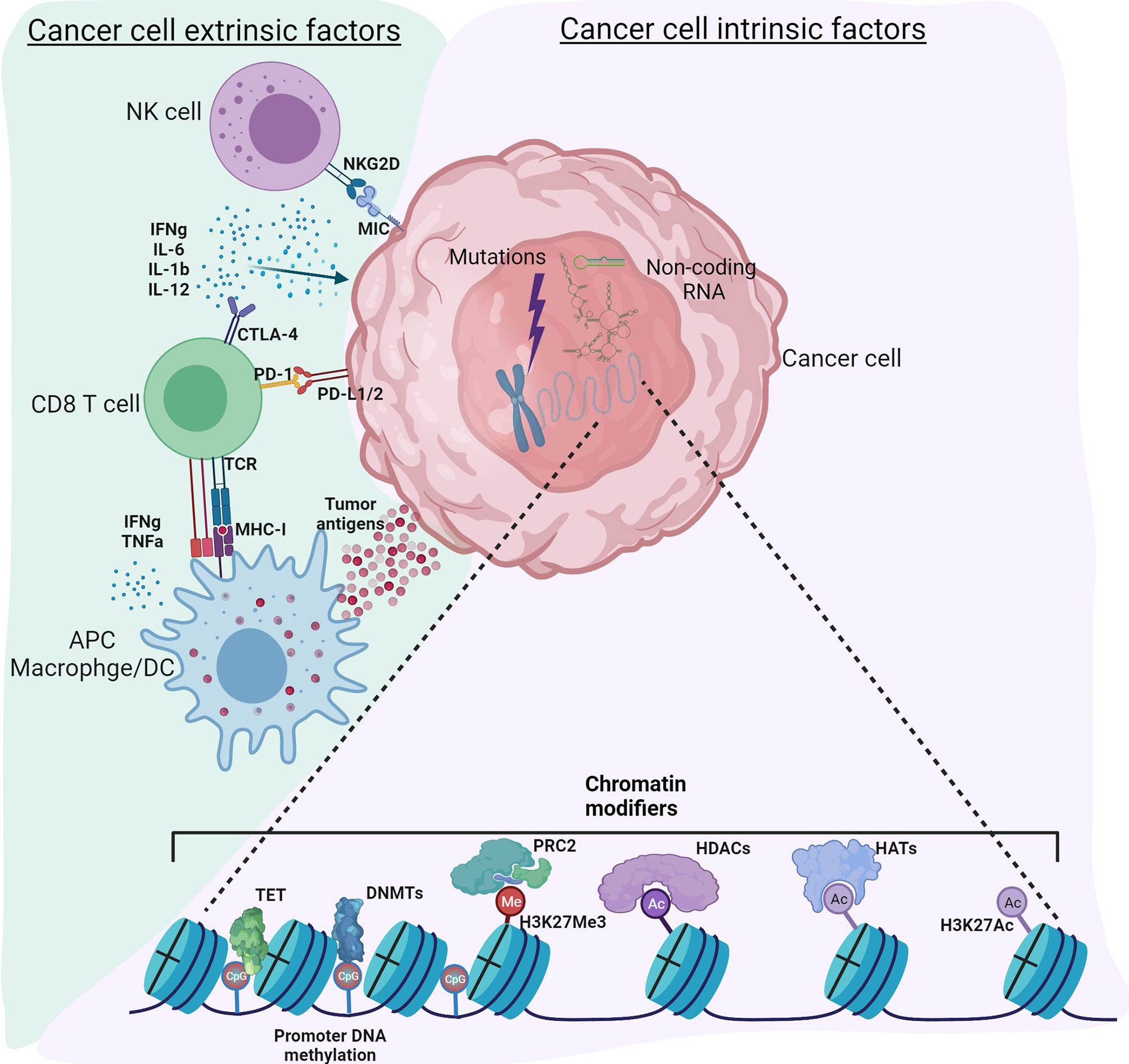
Figure 1 Tumor intrinsic and extrinsic factors regulating immune responses in the tumor microenvironment. Cancer cell intrinsic factors such as genomic mutations, chromatin modifiers and non-coding RNA regulate tumor initiation, propagation as well as immunogenicity. Epigenetic modifications such as DNA methylation, histone acetylation and methylation regulate gene expression. Non-coding RNA including long non-coding RNA, microRNA, circular non-coding RNA regulate gene transcription as well as mRNA stability. Other mechanisms intrinsic to cancer cells include expression of immunosuppressive cytokines to facilitate escape from anti-tumor immunity, expression of immunosuppressive molecules such as PD-L1 and PD-L2, suppression of antigen processing and presentation machinery and tumor associated antigens. Cancer cell extrinsic factors include tumor infiltrated immune cells, fibroblasts, stromal cells and endothelial cells. Extrinsic factors also include secretory factors such as cytokines, chemokines, metabolites, growth factors and immune checkpoint molecules. Tumor associated antigens presented by antigen presenting cells such as macrophages, dendritic cells activate CD8 T-cells for effective anti-tumor immunity. However, immune checkpoint molecules expressed by cancer cells regulate the inflammatory status of the tumor tamping down the inflammation. Use of epigenetic modifiers such as DNMT inhibitors, HDAC inhibitors, BET inhibitors etc. can alter these reversible modifications to enhance anti-tumor immunity. Figure created with BioRender.com.
Several articles utilized publicly available databases to investigate the relationship between tumor and immune cells in the microenvironment and, consequently, response to immune therapies. In this Research Topic, Zhuang et al. investigated the relationship between immune-related genes and the outcome of lung squamous cell carcinoma (LUSC) using datasets from TCGA and defined an immune gene risk index. The immune gene risk index served as an independent prognostic factor indicating that infiltration of neutrophils and dendritic cells was strongly associated with the high-risk group. Another prognostic model in this Research Topic designed by Xu J et al., with machine learning, indicated that LYN, C3, COPG2IT1, LA.DQA1 and NFRSF17 may serve as novel biomarkers to assess the prognosis of patients with non-small cell lung cancer and ICB therapy. Systematic analysis of skin cutaneous melanoma data from TCGA by Qu et al. indicated that ETS family member ETV7 transcription factor expression was downregulated in melanoma tumors and associated with poor prognosis. Further gene enrichment analysis and immune profile analysis indicated that ETV7 regulates differentiation and activation of T cells (Qu et al.).
Regarding immune-related features, the triggering receptor expressed on myeloid cells 2 (TREM2), a transmembrane receptor of the immunoglobulin superfamily, played an important role in tumor progression by modulating immune responses of TAMs in the TME (9). Meta-analysis of TREM2 by Cheng et al., across 33 different cancer types from various databases, including TCGA, indicated that TREM2 gene expression negatively correlated with the level of infiltration of most immune cells but positively correlated with infiltration levels of M1 and M2 macrophages in 6 different cancer types. Integrative pan-can analysis of cancer datasets by Xu W-X et al. concluded that Lipocalin 2 (LCN2), a novel innate immune protein, is a potential biomarker for immune infiltration and poor prognosis in cancers. Analysis of TCGA datasets for Hepatitis B virus-related hepatocellular carcinoma also indicated a significant difference in prognosis based on immune phenotypes associated with higher expression of metabolic and stem cell-related genes (Zhang et al.). Based on the aforementioned research studies in this Research Topic, it can be concluded that cancer genomic and transcriptomic databases have revolutionized the computational approach to understanding the tumor microenvironment.
Due to the reversible nature of epigenetic changes, epigenomic modulators as therapeutic agents are gaining more attention to influence the TME towards antitumor immunity. In this editorial, we will highlight the latest studies on genetic and epigenetic factors that influence the fate of cancer cells and immune cells as well as the factors that shape the TME. Finally, we will discuss unique epigenetic profiles of cancer cells and immune cells with exciting possibilities that link the TME and changes in the gene expression profiles of the immune cells. Finally, we will explore the possibility of using epigenetic modifiers as drug targets either alone or in combination with immunotherapies.
Tumor Immunology and Cancer Immunotherapy
The immune system plays a major role in both the eradication and the establishment of tumors. Active immune surveillance by the innate immune system can identify and eliminate nascent tumor cells, eradicating cancer (10). However, the establishment of cancer indicates that tumor cells successfully evade host immune defenses through a process called immunoediting, which is divided into three phases: elimination, equilibrium, and escape (11). An early study indicated that IFNγ producing lymphocytes prevented tumorigenesis in mice with an intact immune system. However, tumors that developed by escaping immune detection were less immunogenic than those developed in immunodeficient mice, supporting the concept of cancer immunoediting (12). Recent advances in immunotherapy have revolutionized cancer therapies. The basic premise of cancer immunotherapies is to potentiate the ability of T-cells to recognize and eliminate cancer cells. Based on the infiltration of immune cells, tumors can be classified as hot or cold tumors. Tumors with poorly infiltrated immune cells are often called “cold tumors” whereas tumors with inflammation resulting from heavy infiltration of immune cells are called “hot tumors” (13). Patients with so-called cold tumors are non-responsive to immunotherapy resulting in primary resistance.
On the other hand, patients exhibiting an initial response to immunotherapy can eventually acquire resistance due to immunoediting (1). The differentiation of naïve T-cells into anti-tumor effector T-cells, as shown in the schematic, begins with the formation of an immune synapse between the T-cell receptor (TCR) and the antigenic peptide presented by major histocompatibility complex (MHC) of antigen-presenting cells (APCs), which serves as signal 1 (14). Signal 2 involves binding co-stimulatory molecules like the interaction between CD28 and B7 molecules for complete T-cell activation. Lack of signal 2 despite effective antigen presentation leads to T-cell anergy (15). This step is tightly regulated by countering immune checkpoint molecules to prevent prolonged activation and autoimmunity (16). Finally, signal 3 involves cytokine stimulation that facilitates proliferation and clonal expansion of effector T-cells (17).
The impetus for cancer immunotherapy was derived from ipilimumab’s phase 3 clinical study, which blocks cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), an immune checkpoint molecule, to potentiate T-cell mediated anti-tumor response. Anti-CTLA-4 treatment improved overall survival in a large cohort of metastatic melanoma patients (18). CTLA-4, due to its high affinity for B7 ligands, competes with CD28 and inhibits T-cell proliferation and IL-2 secretion (19). Another immune checkpoint molecule expressed by reactive T-cells is programmed death 1 (PD-1), and its ligands PD-L1 and PD-L2 expressed by tumors cells and other immune cells when engaged result in T cell dysfunction (20). The subsequent discovery of other immune checkpoint molecules such as lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), T cell immunoglobulin and ITIM domain (TIGIT), and V-domain Ig suppressor of T cell activation (VISTA) has led to the exploration of ICB therapy for numerous cancer types (21).
Epigenetic modifications play a critical role in regulating the expression of immune checkpoint molecules. DNA methylation (5-methylcytosine) is an epigenetic silencing mark associated with hemimethylated CpG palindrome sequences (approximately 1 kb CpG-rich regions) known as CpG islands (22). Analysis of breast tumor tissue and normal breast tissue for the expression of immune checkpoint molecules indicated that CpG islands in the promoter regions of PD-1, CTLA-4, and TIM-3 were hypomethylated (loss of DNA methylation) in tumor tissues compared with normal tissues. CpG islands of PD-L1 and LAG-3 were hypomethylated, whereas TIGIT was hypermethylated in both normal and breast tumor tissues. This methylation data was inversely correlated with gene expression. H3K9me3 and H3K27me3 marks were reduced in the promoter loci of PD-1, CTLA-4, TIM-3, and LAG-3 in breast tumor tissues suggesting that epigenetic mechanisms affecting the cancer cell side of the equation can affect immune cells and vice versa (23). In the presented Research Topic Wagner et al. further provided an extensive analysis of immune checkpoint molecules and inherited variations as a marker for cancer risk and indicate that the variants in genes encoding these molecules might be considered as low-risk variants (OR<2) for cancer development, which has been well documented by numerous reports for CTLA-4, PDCD1, PD-L1 genes, while still more studies are needed for BTLA, TIM3, LAG3 and TIGIT.
Cancer Intrinsic Epigenetic Mechanisms
As described by Hanahan and Weinberg, Hallmarks of cancer comprise six cellular processes that include sustaining proliferative signaling, resisting cell death, enabling replicative immortality, evading growth suppressors, inducing angiogenesis, and activation of invasion and metastasis (1). Underlying genomic instability further expedites the ability of cancer cells to attain these hallmarks. Epigenomic deregulation adds another layer of complexity to tumorigenesis (24); for example, genome-wide DNA hypomethylation can induce genomic instability (25). Stage IV metastatic colon cancer patients often have tumors with defective DNA mismatch repair and high microsatellite instability (MSI-High) (26). In this Research Topic Lin et al. report that analysis of gene expression and mutational data of colon and rectum adenocarcinomas from TCGA reveals that MSI-High tumors had higher infiltration of immune cell, expression of immune-related genes, and better immunogenicity than MSI-Low or microsatellite stable tumors. Therefore, patients with MSI-High colorectal cancer having MSI-High respond better to ICB therapy (27). Chromosomal instability is mediated by the loss of telomeres which protect the ends of chromosomes and prevent chromosome fusions (28). The expression of telomerase reverse transcriptase (TERT), a catalytic subunit of telomerase, regulated by multiple genetic and epigenetic alterations that affect tumors’ telomerase activity is presented as well in this Research Topic (Dratwa et al.). Therefore, tumor cell intrinsic genomic features mentioned above can significantly influence the initiation and propagation of cancer cells.
In this issue, a comprehensive analysis of the mutational status of tumor suppressor genes that include TP53, CDKN2A, PTEN, RB1, BRCA1, BRCA2, and immune-related gene expression in lung squamous cell carcinoma and lung adenocarcinoma samples from the TCGA database indicated that infiltration of immune cells was suppressed by tumors harboring mutations to the tumor suppressor genes (Kim et al.). This underscores the impact of the mutational status of tumor cells in shaping the TME and potentially dictating the usage of immunotherapeutic strategies in patients with mutations of tumor suppressor genes. Another study in this Research Topic by Wu et al. identified a prognostic TP53 associated immune signature in muscle-invasive bladder cancer based on differentially expressed immune-related genes between patients with or without TP53 mutations (29). The TP53 associated immune signature identified a high-risk group of patients characterized by increased infiltration of immunosuppressive regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages. This high-risk group of patients also had higher expression of CTLA4, LAG3, PDCD1, TIGIT, and HAVCR2, suggesting that they were more likely to respond to anti-PD-1 and neoadjuvant chemotherapy (Wu et al.).
A global survey of tyrosine kinase signaling identified c-ros oncogene 1 (ROS1) chromosomal rearrangements non-small-cell lung cancer (NSCLC) (30). ROS1 fusion NSCLCs are sensitive to crizotinib, a receptor tyrosine kinase MET inhibitor (31) but eventually develop resistance (32), and the effect of ROS1 fusion on ICB therapy is not known. In this Research Topic, Cai et al. reported that ROS1 fusion upregulated PD-L1 through activation of ROS1-SHP2 pathway using ROS1 fusion and crizotinib-resistant cell lines, suggesting that oncogenic driver mutations play a direct role in the expression of checkpoint PD-L1 molecule and facilitate the immune escape of NSCLC tumors.
Another mechanism of immune escape is downregulation of MHC-I molecules of antigen presentation machinery (33) through promoter hypermethylation (34), binding of polycomb repressive complex 2 (PRC2) at H3K27me3 repressive marks (35). Somatic mutations or loss of expression of the beta 2-microglobulin (B2M) gene in lung cancer cells can result in defective MHC class I expression, allowing the cancer cells to escape recognition by cytotoxic T cells (36). Due to genomic instability, mutated proteins expressed by tumor cells function as tumor-associated antigens (TAAs). In colorectal premalignant polyps, an estimated 11,000 genomic events per cell were detected (37). Most importantly, epigenetic driver mutations often dictate the success of immunotherapeutic approaches. For example, ARID1A driver mutations resulted in condensed chromatin of IFN responsive genes, reduced T-cell infiltration, and thereby anemic anti-tumor immunity (38). Epigenetic mechanisms in tumor cells also control the immune status of the TME as polycomb repressive complex 2 (PRC2) repressed the expression of Th-1 chemokines CXCL9 and CXCL10, resulting in poor T-cell infiltration in colon cancer (39), highlighting the complex interplay between tumor cells and immune cells in the TME. In the triple-negative breast cancer (TNBC) subtype with hypomethylated IDO1 gene promoter compared to the estrogen receptor-positive breast cancer subtype with hypermethylated IDO1 gene promoter resulted in the expression of IDO1 enzyme in the presence of activated CD8 T-cells, suggesting a counteractive mechanism employed by tumor cells to escape anti-tumor immunity (40, 41). Metabolic deregulation is often associated with epigenomic deregulation in cancer cells (42). Myeloproliferative neoplasms are not driven by BCR-ABL mutations in hematological malignancies, rather due to somatic mutations in JAK2 and exhibit metabolic vulnerabilities due to a high dependence on glucose metabolism. Here, Sharma et al. discuss the role of histone methylation and acetylation in metabolic deregulation of myeloproliferative neoplasms.
Moreover, in this Research Topic, increasing evidence indicates the role of non-coding RNA, including long non-coding RNAs (lncRNAs), in the immunomodulation of the TME (43). Hu et al. analyzed the gene expression data of patients with adenocarcinoma of esophagogastric junction from TCGA, identified 1470 differentially expressed lncRNAs, and narrowed them to an immune-related risk signature that can effectively predict the response to immunotherapy and chemotherapy. Li J-P et al. reported a seven lncRNA signature out of 331 immune-related genes (AC022784-1, NKILA, AC026355-1, AC068338-3, LINC01843, SYNPR-AS1, and AC123595-1) as a predictive model to forecast the progression of lung adenocarcinoma. Fang et al. discuss the regulatory roles of circular RNAs in the context of tumor immunology and immunotherapy.
Cancer Extrinsic Epigenetic Mechanisms
Cancer extrinsic mechanisms constitute factors contributing to tumor initiation and progression not by the tumor cells but due to factors such as tumor-associated immune cells, stromal cells, and fibroblasts. These cells can directly influence epigenetic outcomes in tumor cells by secreting factors such as cytokines, chemokines, metabolites, growth factors, and other soluble factors. As shown in the schematic, we will discuss the role of predominant immune cells in the TME, including tumor-associated macrophages and infiltrated lymphocytes.
Macrophages
Macrophages are a heterogeneous population of cells that play a critical role in enforcing both innate and adaptive arms of the immune system. Macrophages are terminally differentiated from circulating monocytes with their origin in the bone marrow, whereas evidence indicates that tissue macrophages progenitors are derived from the yolk sac and fetal liver during early embryonic hematopoiesis (44) with self-renewal capabilities (45). An extremely simplistic classification of macrophages based on the phenotypes is pro-inflammatory or classically activated (M1) macrophages and anti-inflammatory or alternatively activated (M2) macrophages (46). The M1/M2 nomenclature is originally linked to Th1/Th2 lymphocytes producing IFNγ or IL4 for activation of M1 or M2 macrophages, respectively. Macrophages are highly adaptable cells capable of responding to cues from the microenvironment and exhibit properties that make it difficult to strictly assign M1 or M2 phenotypes. Such plasticity of phenotypes demands remarkable changes in the epigenome resulting in distinct gene expression patterns (47). In the context of the TME, pro-inflammatory M1 macrophages are attributed to the anti-tumor activity, whereas anti-inflammatory M2 macrophages are deemed to be tumor-promoting. The complexity is even higher with tumor-associated macrophages (TAMs).
Macrophages differentiate from monocytes in the presence of colony-stimulating factors, resulting in significant gene expression changes (48). Epigenetic profiling of monocyte to macrophage differentiation uncovered approximately 8000 dynamic regions associated with at least 11000 DNase I hypersensitive sites suggesting a profound remodeling of chromatin (49). Differentiation was associated with demethylation catalyzed by ten-eleven translocation (TET) methylcytosine dioxygenases enzymes (50) of at least 114 Differentially methylated regions (DMRs) belonging to genes of the ERBB2, PDGFRβ, CXCR4, and PIK3 signaling pathways. Demethylated DMRs were also nucleosome depleted and enriched with activating histone marks H3K27ac and H3K4me1 in macrophages compared to monocytes (51). Exposure of macrophages to TLR ligands such as LPS and/or Th-1 cytokines such as IFNγ leads to M1 polarization by activating several epigenetic modifiers leading to transcription of pro-inflammatory cytokine genes mediated through the NF-κB (52), STAT1, HIF1α, IRF4 (53), and MAPK (54) pathways. Enhancer regions of loci that encode inflammatory genes are poised for gene expression with an open chromatin state marked by H3K4me1 and binding of macrophage lineage determining PU.1 and C/EBP transcription factors (55, 56). Upon activation signal, these cells readily express pro-inflammatory cytokines. Another epigenetic modifier, histone lysine methyltransferase EZH2, a member of the repressive PRC2 complex, increases H3K27me3 marks on the SOCS3 gene leading to suppression of SOCS3 gene expression in activated M1 macrophages (57). Similarly, DNMT1-mediated hypermethylation of SOCS1 resulted in decreased expression of SOCS1 and therefore increased expression of LPS-induced pro-inflammatory TNF and IL-6 cytokines in macrophages (58). Both SOCS1 and SOCS3 are negative regulators of the JAK/STAT pathway (59). On the contrary, repressive mechanisms mediated by negative histone marks H3K9me3, H3K27me3, H4K20me3, and repressors that bind inflammatory loci exist to prevent uncontrolled and chronic inflammation (60, 61). Jmjd3, an H3K27 demethylase deficiency, affected trimethylation of Irf4, a key transcription factor that regulated M2 macrophage polarization (62).
Lymphocytes
IL-17 producing Th-17 cells, a subset of CD4 T-cells, are usually associated with proinflammatory function. Binding of Cxxc finger protein 1 (Cxxc1), a transcription factor with high affinity to unmethylated CpG sites at the IL6R gene promoter, retained H3K4me3 marks and regulated IL-6Rα expression, which mediates IL-6/STAT3 pathway and thereby controls the fate of CD4 differentiation towards T-regs or Th-17 CD4 T-cells (63). Lymphocytes isolated from HDAC11 knockout mice exhibited increased expression of Eomes and Tbet transcription factors and displayed enhanced proliferation, increased production of pro-inflammatory cytokines, and effector molecule expression suggesting that HDAC11 acts as a negative epigenetic regulator of T-cell effector function and phenotype (64). HDAC11 is shown to regulate the expression of OX40L and IL-10 producing T-regs in Hodgkin lymphoma (65). Entinostat, a synthetic benzamide-derived Class I HDAC inhibitor (66), enhanced NK cells’ ability to kill cancer cells by increasing the expression of MIC in tumor cells and NKG2D in primary human NK cells (67).
Inflammasomes are multiprotein complexes that sense danger signals emanated from cancer cells and mediate a pro-inflammatory response resulting in cell death through activation of cysteine proteases called caspases (68). Deregulation of inflammasome and chronic inflammation often damages healthy tissue along with tumor cells (69). Epigenetic mechanisms such as DNA methylation (70) and epigenetic readers such as BRD4 regulate NLRP3 gene expression (71). In this Research Topic, Zhong et al. reported that activation of NLRP3 inflammasome in an IL-1β dependent manner in AML cells promoted proliferation of leukemia cells by inhibiting apoptosis, resulting in resistance to chemotherapy. Another epigenetic mechanism to regulate inflammation described in this issue is mediated by microRNAs. MiR-124, which targets STAT3 and subsequently binding of STAT3 to the IL17 gene promoter to activate it, in a Citrobacter rodentium infection and AOM/DSS induced colon cancer murine model, was shown to inhibit TH17 cell polarization and blocked colitis-related cancer (Lin et al.). Additionally, Zhang et al. reported that targeting miR-148b-5p in gastric cancer tumors reprogrammed the metabolic properties and altered the TME by shifting the lymphocyte and myeloid populations and rendered them sensitive to chemotherapeutic agents. These studies indicate modulation of tumor infiltrated immune cell properties and function through genetic and epigenetic mechanisms thereby affecting the TME.
New Immunotherapeutic Opportunities With Pharmacological Inhibitors of Epigenetic Modifiers
The reversible nature of epigenetic marks has immense implications in the prevention and treatment of cancers. When combined with immunotherapeutic approaches, they provide an opportunity to design targeted therapies to affect a positive clinical outcome. Epigenetic modifiers have been tested in various preclinical and clinical studies with varying degrees of success. Saleh et al. provide a comprehensive review of epigenetic modifications on the regulation of immune checkpoint molecules, therapeutic approaches to epigenetic modifiers as therapy in clinical trials.
DNMT inhibitors, 5-azacytidine, and decitabine degrade and inhibit DNA methyltransferases, resulting in hypomethylation and re-expression of tumor suppressor genes such as CDKN2A in cancer cells (72, 73). DNMT inhibitors also increase the expression of PD-L1 and PD-L2 in melanoma. A similar effect was observed with class I HDAC inhibitors and, when combined with anti-PD1 therapy, suppressed the tumor progression and improved survival (74). As mentioned earlier, DNA methylation and repressive histone marks regulate the expression of immune checkpoint molecules in breast cancer and colorectal cancer (23, 75). In ovarian cancer cells, treatment with DNMT inhibitors through upregulation of previously hypermethylated endogenous retroviruses activated cytosolic sensing of double-stranded RNA, causing a type I interferon response and apoptosis (76). Combining DNMT inhibitor with HDAC6 inhibitor resulted in increased type I interferon response, leading to profound cytokine and chemokine expression and higher expression of the MHC I antigen presentation complex in human and mouse ovarian cancer cell lines (77). Approval of DNMT inhibitors for hematological malignancies has renewed interest in epigenetic therapy despite limited success in solid tumors (78, 79). A combination of low dose azacytidine and HDAC inhibitor entinostat in patients with non-small cell lung cancer resulted in durable responses and improved long-term survival (80).
Genetic abrogation or pharmacological inhibition of HDAC6 in melanoma cells decreased cell proliferation by inducing G1-cell cycle arrest without triggering apoptosis. This was also associated with increased expression of TAAs and MHC-1 molecules, indicating a greater role of HDAC6 in modulating anti-tumor immunity (81). HDAC6 is also reported to interact with STAT3 to regulate the expression of immunosuppressive cytokine, IL-10, by binding to the IL10 gene promoter in antigen-presenting cells (82). Furthermore, inhibition of HDAC6 decreased STAT3 mediated expression of PD-L1 in primary melanoma samples and a panel of melanoma cell lines (83). Finally, using a murine melanoma model, pre-treatment with HDAC6 inhibitor prior to anti-PD1 immunotherapy resulted in decreased pro-tumor macrophages associated with increased infiltration of effector T-cells in the TME, providing evidence to the potential use of epigenetic modifiers as therapeutic agents for immunotherapy (84). In this Research Topic, similar results are presented in CLL whereby inhibition of HDAC6 augmented anti-PD1 and anti-PDL1 immunotherapy by increasing cytotoxic CD8 T-cell phenotype (Maharaj et al.). HDAC inhibitors also enhanced the expression of T-cell chemokines CXCL9 and CXCL10 and augmented anti-PD-1 immunotherapy response in lung adenocarcinoma (85).
Future of Epigenetic Therapies
One of the downsides to using pharmacological approaches to inhibit or activate epigenetic modifiers is the lack of targeted effects, resulting in undesirable global changes that can discourage their usage as long-term cancer therapies. For example, treatment with HDAC inhibitors, despite having better toxicity profiles than traditional chemotherapeutic agents, caused patients’ suffering from gastrointestinal, hematological, and cardiac effects (86). Despite the limitations, epigenetic modifiers have been tested in the clinic in combination immunotherapeutic strategies. Several examples are as follows: BET inhibitor and atezolizumab (anti-PD-L1) combination is in a phase Ib open label trial in patients with advanced ovarian cancer or triple negative breast cancer (ClinicalTrials.gov Identifier: NCT0329217). Vorinostat which is an FDA-approved drug for cutaneous T-cell lymphoma is currently is under consideration to assess the early signals anti-tumor activity in combination with pembrolizumab (anti-PD1) in patients with advanced prostate, renal or urothelial cell carcinoma (ClinicalTrials.gov Identifier: NCT02619253). Phase I clinical trail to study the side effects and identify the best dose of class I HDAC inhibitor entinostat and nivolumab (anti-PD1) when given together with ipilimumab (anti-CTLA4) in treating patients with metastatic or unrescetable solid tumors that have spread to lymph nodes or other organ sites in human epidermal growth factor receptor 2 (HER2)-negative breast cancer patients (ClinicalTrials.gov Identifier: NCT02453620). In this Research Topic, Saleh et al. has provided an extensive list of latest clinical trials highlighting the increasing prominence of epigenetic drugs as immunomodulators of TME.
However, this issue can be addressed by developing highly selective isoform-specific HDAC inhibitors. Chronic, high doses of DNMT induce chromosomal instability and induce tumors in mice (87, 88). Therefore, a better approach would be to target a specific locus of chromatin to influence a desirable outcome. Targeted epigenetic modifications can be achieved by combining sequence-specific DNA binding domains with an epigenetic modifier. An early study showed that using synthetic zinc finger proteins fused to a library of about 223 yeast chromatin regulators can target specific locus (89).
Another example is an engineered transcriptional repressor fused to the catalytic domain of DNA methyltransferase (90). However, this in itself is a limiting factor as targeting different DNA sequences will require corresponding site-specific DNA binding domains. Other technologies include transcription activator-like effectors (TALEs) and RNA-guided clustered regularly interspaced short palindromic repeat (CRISPR) associated protein (Cas9) for precise epigenome editing (91, 92). With the advent of CRISPR technologies, we are entering a new frontier of targeted epigenomic therapies for cancer treatment.
Author Contributions
SN wrote the manuscript and prepared the illustration. AV supervised and guided in preparing the manuscript. KC and LK reviewed and provided input and suggestions in writing the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
4. You JS, Jones PA. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell (2012) 22:9–20.
5. Consortium EP. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature (2012) 489:57–74.
6. Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol (2010) 28:1045–8.
7. Li D, Hsu S, Purushotham D, Sears RL, Wang T. WashU Epigenome Browser Update 2019. Nucleic Acids Res (2019) 47:W158–W65.
8. Stunnenberg HG, International Human Epigenome C, Hirst M. The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell (2016) 167:1145–9.
9. Nakamura K, Smyth MJ. TREM2 Marks Tumor-Associated Macrophages. Signal Transduct Target Ther (2020) 5:233.
10. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat Immunol (2002) 3:991–8.
11. Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annu Rev Immunol (2004) 22:329–60.
12. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and Lymphocytes Prevent Primary Tumour Development and Shape Tumour Immunogenicity. Nature (2001) 410:1107–11.
13. Kelderman S, Schumacher TN, Haanen JB. Acquired and Intrinsic Resistance in Cancer Immunotherapy. Mol Oncol (2014) 8:1132–9.
14. Dustin ML, Choudhuri K. Signaling and Polarized Communication Across the T Cell Immunological Synapse. Annu Rev Cell Dev Biol (2016) 32:303–25.
16. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12:252–64.
17. Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, et al. Inflammatory Cytokines Provide a Third Signal for Activation of Naive CD4+ and CD8+ T Cells. J Immunol (1999) 162:3256–62.
18. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363:711–23.
19. Krummel MF, Allison JP. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J Exp Med (1995) 182:459–65.
20. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers, and Combinations. Sci Trans Med (2016) 8:328rv4.
21. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel Immune Checkpoint Targets: Moving Beyond PD-1 and CTLA-4. Mol Cancer (2019) 18:155.
22. Jones PA. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat Rev Genet (2012) 13:484–92.
23. Sasidharan Nair V, El Salhat H, Taha RZ, John A, Ali BR, Elkord E. DNA Methylation and Repressive H3K9 and H3K27 Trimethylation in the Promoter Regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 Genes in Human Primary Breast Cancer. Clin Epigenet (2018) 10:78.
24. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic Plasticity and the Hallmarks of Cancer. Science (2017) 357:eaal2380.
25. Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA Hypomethylation Leads to Elevated Mutation Rates. Nature (1998) 395:89–93.
26. Kawakami H, Zaanan A, Sinicrope FA. Microsatellite Instability Testing and its Role in the Management of Colorectal Cancer. Curr Treat Options Oncol (2015) 16:30.
27. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol (2020) 38:1–10.
29. Wu X, Lv D, Cai C, Zhao Z, Wang M, Chen W, et al. A TP53-Associated Immune Prognostic Signature for the Prediction of Overall Survival and Therapeutic Responses in Muscle-Invasive Bladder Cancer. Front Immunol (2020) 11:590618.
30. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global Survey of Phosphotyrosine Signaling Identifies Oncogenic Kinases in Lung Cancer. Cell (2007) 131:1190–203.
31. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-Rearranged Non-Small-Cell Lung Cancer. N Engl J Med (2014) 371:1963–71.
32. Facchinetti F, Loriot Y, Kuo MS, Mahjoubi L, Lacroix L, Planchard D, et al. Crizotinib-Resistant ROS1 Mutations Reveal a Predictive Kinase Inhibitor Sensitivity Model for ROS1- and ALK-Rearranged Lung Cancers. Clin Cancer Res (2016) 22:5983–91.
33. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol (2021) 12:636568.
34. Ye Q, Shen Y, Wang X, Yang J, Miao F, Shen C, et al. Hypermethylation of HLA Class I Gene is Associated With HLA Class I Down-Regulation in Human Gastric Cancer. Tissue Antigens (2010) 75:30–9.
35. Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell (2019) 36:385–401.e8.
36. Chen HL, Gabrilovich D, Virmani A, Ratnani I, Girgis KR, Nadaf-Rahrov S, et al. Structural and Functional Analysis of Beta2 Microglobulin Abnormalities in Human Lung and Breast Cancer. Int J Cancer (1996) 67:756–63.
37. Stoler DL, Chen N, Basik M, Kahlenberg MS, Rodriguez-Bigas MA, Petrelli NJ, et al. The Onset and Extent of Genomic Instability in Sporadic Colorectal Tumor Progression. Proc Natl Acad Sci USA (1999) 96:15121–6.
38. Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, et al. Epigenetic Driver Mutations in ARID1A Shape Cancer Immune Phenotype and Immunotherapy. J Clin Invest (2020) 130:2712–26.
39. Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res (2016) 76:275–82.
40. Dewi DL, Mohapatra SR, Blanco Cabanes S, Adam I, Somarribas Patterson LF, Berdel B, et al. Suppression of Indoleamine-2,3-Dioxygenase 1 Expression by Promoter Hypermethylation in ER-Positive Breast Cancer. Oncoimmunology (2017) 6:e1274477.
41. Noonepalle SK, Gu F, Lee EJ, Choi JH, Han Q, Kim J, et al. Promoter Methylation Modulates Indoleamine 2,3-Dioxygenase 1 Induction by Activated T Cells in Human Breast Cancers. Cancer Immunol Res (2017) 5:330–44.
42. Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic Control of Epigenetics in Cancer. Nat Rev Cancer (2016) 16:694–707.
43. Pi YN, Qi WC, Xia BR, Lou G, Jin WL. Long Non-Coding RNAs in the Tumor Immune Microenvironment: Biological Properties and Therapeutic Potential. Front Immunol (2021) 12:697083.
44. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, et al. Adult Langerhans Cells Derive Predominantly From Embryonic Fetal Liver Monocytes With a Minor Contribution of Yolk Sac-Derived Macrophages. J Exp Med (2012) 209:1167–81.
45. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-Resident Macrophages Self-Maintain Locally Throughout Adult Life With Minimal Contribution From Circulating Monocytes. Immunity (2013) 38:792–804.
46. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41:14–20.
47. Chen S, Yang J, Wei Y, Wei X. Epigenetic Regulation of Macrophages: From Homeostasis Maintenance to Host Defense. Cell Mol Immunol (2020) 17:36–49.
48. Hume DA, Freeman TC. Transcriptomic Analysis of Mononuclear Phagocyte Differentiation and Activation. Immunol Rev (2014) 262:74–84.
49. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic Programming of Monocyte-to-Macrophage Differentiation and Trained Innate Immunity. Science (2014) 345:1251086.
50. Kong L, Tan L, Lv R, Shi Z, Xiong L, Wu F, et al. A Primary Role of TET Proteins in Establishment and Maintenance of De Novo Bivalency at CpG Islands. Nucleic Acids Res (2016) 44:8682–92.
51. Wallner S, Schroder C, Leitao E, Berulava T, Haak C, Beisser D, et al. Epigenetic Dynamics of Monocyte-to-Macrophage Differentiation. Epigenet Chromatin (2016) 9:33.
52. von Knethen A, Callsen D, Brune B. NF-kappaB and AP-1 Activation by Nitric Oxide Attenuated Apoptotic Cell Death in RAW 264.7 Macrophages. Mol Biol Cell (1999) 10:361–72.
53. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic Reprogramming Mediated by the Mtorc2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity (2016) 45:817–30.
54. Bode JG, Ehlting C, Haussinger D. The Macrophage Response Towards LPS and its Control Through the P38(MAPK)-STAT3 Axis. Cell signalling (2012) 24:1185–94.
55. De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, et al. Jmjd3 Contributes to the Control of Gene Expression in LPS-Activated Macrophages. EMBO J (2009) 28:3341–52.
56. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell (2010) 38:576–89.
57. Qiao Y, Kang K, Giannopoulou E, Fang C, Ivashkiv LB. IFN-Gamma Induces Histone 3 Lysine 27 Trimethylation in a Small Subset of Promoters to Stably Silence Gene Expression in Human Macrophages. Cell Rep (2016) 16:3121–9.
58. Cheng C, Huang C, Ma TT, Bian EB, He Y, Zhang L, et al. SOCS1 Hypermethylation Mediated by DNMT1 is Associated With Lipopolysaccharide-Induced Inflammatory Cytokines in Macrophages. Toxicol Lett (2014) 225:488–97.
59. Croker BA, Kiu H, Nicholson SE. SOCS Regulation of the JAK/STAT Signalling Pathway. Semin Cell Dev Biol (2008) 19:414–22.
60. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. A Selective Jumonji H3K27 Demethylase Inhibitor Modulates the Proinflammatory Macrophage Response. Nature (2012) 488:404–8.
61. Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, et al. Control of Proinflammatory Gene Programs by Regulated Trimethylation and Demethylation of Histone H4K20. Mol Cell (2012) 48:28–38.
62. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 Axis Regulates M2 Macrophage Polarization and Host Responses Against Helminth Infection. Nat Immunol (2010) 11:936–44.
63. Lin F, Meng X, Guo Y, Cao W, Liu W, Xia Q, et al. Epigenetic Initiation of the TH17 Differentiation Program is Promoted by Cxxc Finger Protein 1. Sci Adv (2019) 5:eaax1608.
64. Woods DM, Woan KV, Cheng F, Sodre AL, Wang D, Wu Y, et al. T-Cells Lacking HDAC11 Have Increased Effector Functions and Mediate Enhanced Alloreactivity in a Murine Model. Blood (2017) 130(2):146–55.
65. Buglio D, Khaskhely NM, Voo KS, Martinez-Valdez H, Liu YJ, Younes A. HDAC11 Plays an Essential Role in Regulating OX40 Ligand Expression in Hodgkin Lymphoma. Blood (2011) 117:2910–7.
66. Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, et al. Determination of the Class and Isoform Selectivity of Small-Molecule Histone Deacetylase Inhibitors. Biochem J (2008) 409:581–9.
67. Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, et al. The Narrow-Spectrum HDAC Inhibitor Entinostat Enhances NKG2D Expression Without NK Cell Toxicity, Leading to Enhanced Recognition of Cancer Cells. Pharm Res (2015) 32:779–92.
68. Guo H, Callaway JB, Ting JP. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat Med (2015) 21:677–87.
69. Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in Cancer: A Double-Edged Sword. Protein Cell (2014) 5:12–20.
70. Wei M, Wang L, Wu T, Xi J, Han Y, Yang X, et al. NLRP3 Activation Was Regulated by DNA Methylation Modification During Mycobacterium Tuberculosis Infection. BioMed Res Int (2016) 2016:4323281.
71. Tan YF, Wang M, Chen ZY, Wang L, Liu XH. Inhibition of BRD4 Prevents Proliferation and Epithelial-Mesenchymal Transition in Renal Cell Carcinoma via NLRP3 Inflammasome-Induced Pyroptosis. Cell Death Dis (2020) 11:239.
72. Zhu WG, Dai Z, Ding H, Srinivasan K, Hall J, Duan W, et al. Increased Expression of Unmethylated CDKN2D by 5-Aza-2’-Deoxycytidine in Human Lung Cancer Cells. Oncogene (2001) 20:7787–96.
73. Schneider-Stock R, Diab-Assef M, Rohrbeck A, Foltzer-Jourdainne C, Boltze C, Hartig R, et al. 5-Aza-Cytidine is a Potent Inhibitor of DNA Methyltransferase 3a and Induces Apoptosis in HCT-116 Colon Cancer Cells via Gadd45- and P53-Dependent Mechanisms. J Pharmacol Exp Ther (2005) 312:525–36.
74. Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy With PD-1 Blockade. Cancer Immunol Res (2015) 3:1375–85.
75. Sasidharan Nair V, Toor SM, Taha RZ, Shaath H, Elkord E. DNA Methylation and Repressive Histones in the Promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and Galectin-9 Genes in Human Colorectal Cancer. Clin Epigenet (2018) 10:104.
76. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell (2015) 162:974–86.
77. Moufarrij S, Srivastava A, Gomez S, Hadley M, Palmer E, Austin PT, et al. Combining DNMT and HDAC6 Inhibitors Increases Anti-Tumor Immune Signaling and Decreases Tumor Burden in Ovarian Cancer. Sci Rep (2020) 10:3470.
78. Cowan LA, Talwar S, Yang AS. Will DNA Methylation Inhibitors Work in Solid Tumors? A Review of the Clinical Experience With Azacitidine and Decitabine in Solid Tumors. Epigenomics (2010) 2:71–86.
79. Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized Controlled Trial of Azacitidine in Patients With the Myelodysplastic Syndrome: A Study of the Cancer and Leukemia Group B. J Clin oncol: Off J Am Soc Clin Oncol (2002) 20:2429–40.
80. Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination Epigenetic Therapy has Efficacy in Patients With Refractory Advanced Non-Small Cell Lung Cancer. Cancer Discovery (2011) 1:598–607.
81. Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, Knox T, et al. Targeting Histone Deacetylase 6 Mediates a Dual Anti-Melanoma Effect: Enhanced Antitumor Immunity and Impaired Cell Proliferation. Mol Oncol (2015) 9:1447–57.
82. Cheng F, Lienlaf M, Wang HW, Perez-Villarroel P, Lee C, Woan K, et al. A Novel Role for Histone Deacetylase 6 in the Regulation of the Tolerogenic STAT3/IL-10 Pathway in APCs. J Immunol (2014) 193:2850–62.
83. Lienlaf M, Perez-Villarroel P, Knox T, Pabon M, Sahakian E, Powers J, et al. Essential Role of HDAC6 in the Regulation of PD-L1 in Melanoma. Mol Oncol (2016) 10:735–50.
84. Knox T, Sahakian E, Banik D, Hadley M, Palmer E, Noonepalle S, et al. Selective HDAC6 Inhibitors Improve Anti-PD-1 Immune Checkpoint Blockade Therapy by Decreasing the Anti-Inflammatory Phenotype of Macrophages and Down-Regulation of Immunosuppressive Proteins in Tumor Cells. Sci Rep (2019) 9:6136.
85. Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res (2016) 22:4119–32.
86. Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals (Basel) (2010) 3:2751–67.
87. Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal Instability and Tumors Promoted by DNA Hypomethylation. Science (2003) 300:455.
88. Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of Tumors in Mice by Genomic Hypomethylation. Science (2003) 300:489–92.
89. Keung AJ, Bashor CJ, Kiriakov S, Collins JJ, Khalil AS. Using Targeted Chromatin Regulators to Engineer Combinatorial and Spatial Transcriptional Regulation. Cell (2014) 158:110–20.
90. Amabile A, Migliara A, Capasso P, Biffi M, Cittaro D, Naldini L, et al. Inheritable Silencing of Endogenous Genes by Hit-And-Run Targeted Epigenetic Editing. Cell (2016) 167:219–32.e14.
91. Gao X, Tsang JC, Gaba F, Wu D, Lu L, Liu P. Comparison of TALE Designer Transcription Factors and the CRISPR/dCas9 in Regulation of Gene Expression by Targeting Enhancers. Nucleic Acids Res (2014) 42:e155.
Keywords: tumor microenvironment, tumor immunology, tumor immune escape mechanisms, checkpoint inhibition therapy, genetic and epigenetic alterations, DNA methylation, HDAC inhibitor
Citation: Noonepalle SkR, Karabon L, Chiappinelli KB and Villagra A (2021) Editorial: Genetic and Epigenetic Control of Immune Responses. Front. Immunol. 12:775101. doi: 10.3389/fimmu.2021.775101
Received: 13 September 2021; Accepted: 16 September 2021;
Published: 05 October 2021.
Copyright © 2021 Noonepalle, Karabon, Chiappinelli and Villagra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alejandro Villagra, YXZpbGxhZ3JhQGd3dS5lZHU=