 Binshan Zhang
Binshan Zhang Guolan Xing*
Guolan Xing*- Department of nephrology, First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Objective: The pathogenesis of thrombotic microangiopathy (TMA) in lupus nephritis (LN) remains complicated. This study aimed to detect the deposition of complement lectin pathway (LP) and alternative pathway (AP) components in renal tissues, then evaluate the clinicopathological characteristics and risk factors for renal survival between patients with or without TMA in LN cohorts.
Methods: We included 79 patients with biopsy-proven LN-associated TMA and matched the same number of LN patients without TMA as the control group. The deposition of mannose binding lectin (MBL), MBL-associated serine proteases 1/3 (MASP1/3), complement factor B (CFB), complement factor D (CFD), C4d, and von Willebrand factor (VWF) in renal tissue was assessed by immunohistochemistry and immunofluorescence. Besides, co-localization of C5b-9 and CD34 was detected by confocal microscopy.
Results: In our retrospective cohort, the incidence of acute kidney injury (30% vs. 14%, p = 0.013), acute hemodialysis (35% vs. 5%, p < 0.001), and interstitial fibrosis (43% vs. 13%, p < 0.001) is higher in the TMA, compared with the control group. Despite aggressive steroids pulse, plasma exchange, and immunosuppressive therapy among TMA group, they still had significantly inferior 3-year renal survival rates (68% vs. 89%, p = 0.002) than those in the non-TMA group. COX regression analysis identified that TMA (HR 4.807, 95% CI [2.052, 11.263], p < 0.001) is a risk factor in LN. MBL, MASP1/3, CFB, CFD, C4d, and VWF deposited along the glomerulus among LN, while TMA had stronger staining intensity and deposition. The co-localized expression of CD34 and C5b-9 in the endothelial cells was also observed in the renal tissues.
Conclusions: TMA is an independent risk factor for renal survival in LN patients. Moreover, LP and AP activation are involved in the pathogenesis of LN-associated TMA.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the overproduction of autoantibodies, which has various clinical manifestations and affects multiple tissues and organs, of which the renal involvement is the most important predictor of morbidity and mortality (1, 2). The histological damage in lupus nephritis (LN) is often associated with different treatment responses and outcomes (3). In addition to common glomerulonephritis, renal vasculopathies closely related to prognosis, including vascular immune complex deposits, arterial sclerosis, noninflammatory necrotizing vasculopathy, thrombotic microangiopathy (TMA), and true renal vasculitis, have attracted great attention (4–6). TMA-associated with LN has a high probability of developing end-stage renal disease (ESRD) and death (7, 8).
The etiology of LN is complicated and include extrarenal and intrarenal factors, such as inheritance, sex hormone, virus, immunity, and environment (9). Hypocomplementemia is often thought to be related to the activity of renal disease. However, the effects of deficiency and over activation of the complement system in SLE and TMA have not been fully elucidated. Complement activation can be initiated through 3 different pathways. Immune complexes deposited in the glomerulus and peritubular capillaries activate the complement classical pathway (CP), which is thought to be the dominant pathway in LN (9). Interestingly, C1q deficiency is strongly relevant to development of SLE (10). The lectin pathway (LP) is activated when mannose binding lectin (MBL), as one of the pattern recognition molecules, binds to bacterial carbohydrate motifs and engage the MBL-associated serine proteases (MASPs). MASPs are indispensable for LP and alternative pathway (AP) activation. MASP-1 autoactivates firstly, then it activates MASP-2 (11). MASP-2 subsequently cleave C2 and C4 to form the C3 convertase (C4b2a) (12). Furthermore, MASP-3 can exclusively activate pro-factor D in normal resting human blood to regulate AP activity (13, 14). Complement factor D (CFD) cleaves factor B to generate the AP convertase (C3bBb) (12). Three complement pathways eventually form the C5b-9 membrane attack complex to promote cells lysis (15).
Growing evidence suggests that LP and AP are involved in the pathogenesis of LN and TMA (16). It has been reported that most LN patients presented MBL deposition and had higher urinary protein than those negative (17). MBL2 gene polymorphism also influences vulnerability to SLE (18). Recently, J Laurence et al. discovered that elevated MASP-2 levels in TMA patients are associated with microvascular endothelial cell (MVEC) damage and can be restrained by narsoplimab (anti-MASP-2 antibody) in vitro (19). CFD deficiency in MRL/lpr mice has a protective effect on renal disease due to the lack of AP activation (20). Additionally, AP overactivation leading to MVEC damage and microvascular thrombosis is a common mechanism that induces TMA (21).
The complement component deposition of LP and AP in TMA-associated with LN patients has not been studied before. Therefore, this research detected the expression of relevant indicators in renal tissues to explore the pathogenesis of LN with TMA. In addition, we analyzed the clinicopathological characteristics and risk factors affecting prognosis between patients with or without TMA in LN cohorts.
Materials and methods
Patients selection
A total of 2130 Chinese underwent renal biopsy at The First Affiliated Hospital of Zhengzhou University between January 2012 and July 2021 were enrolled. Of the 2130 individuals, patients who fulfilled the following criteria were defined as LN-TMA: (1) meet the 2019 European League Against Rheumatism/American College of Rheumatologic Classification Criteria (EULAR/ACR) for the diagnosis of SLE (22). (2) LN was diagnosed according to The 2018 International Society of Nephrology/Renal Pathology Society (ISN/PRS) pathological classification (23). (3) evaluation of TMA pathological features under light microscopy (LM) and electron microscopy (EM). The exclusion criteria were as follows: (1) The total number of glomeruli was less than 10 under LM. (2) inadequate clinical information data (no information on proteinuria, serum creatinine, etc.). (3) insufficient time or loss to follow-up. Ultimately, 79 patients met the criteria of this research. According to gender, age, and LN classes, we matched the same number of LN patients without TMA as the control group. The retrospective study protocol was approved by the ethics committees of The First Affiliated Hospital of Zhengzhou University.
Clinical and laboratory data
The collection of complete clinical information of enrolled patients was performed at the time of renal biopsy and listed below: (1) General information: age, gender, duration of SLE/LN, systolic and diastolic blood pressure (SBP and DBP), leukocytes, hemoglobin (Hb), platelets (PLT), 24-h urinary protein, hematuria, serum albumin (Alb), serum creatinine (SCr), total cholesterol and triglyceride; (2) LN-related specific indicators: serum C3 and C4 levels, C-reactive protein, erythrocyte sedimentation rate (ESR), antinuclear antibody(ANA), anti-dsDNA antibody (A-dsDNA), fever, rash, mucosal ulcers, arthritis, serositis, neurological symptoms, the SLE Disease Activity Index (SLEDAI) (24), acute kidney injury(AKI), acute hemodialysis and acute heart failure (AHF). The above serological examinations which were measured in a fasting state and performed in the central laboratory in our hospital are routinely tested in clinical practice. During the follow-up period, the interval between patients visit is 3-6 months. The above parameters were collected during each visit.
Renal histopathology
The paraffin-embedded kidney tissues were stained with hematoxylin-eosin (HE), periodic acid-schiff (PAS), masson, and periodic acid-silver metheramine (PASM), respectively. Three-micrometer cryostat sections were stained with anti-human IgG, IgM, IgA, C3, C4, and C1q for immunofluorescence (IF). A routine read of every renal biopsy specimen was performed by two professional pathologists through LM, IF, and EM, and reported according to the 2018 ISN/PRS classification (23).
TMA is divided into acute and chronic lesions, which involve the glomeruli, renal tubules, and interstitial blood vessels. Under the LM, glomerular manifests as capillary thrombi, swelling of endothelial cells, and mesangiolysis, which can lead to lumen stenosis or even occlusion. Ischemic shrinkage of the glomerulus occurs when the vascular lesions are severely involved. Arterial lesions are the most characteristic pathological manifestation of TMA. Arterioles may show arteriolar thrombi, fibrinoid necrosis, and subintimal myxoid edema in the acute phase. The typical “onion skin” pattern lesion is formed in the advanced stage (Figure 1). By EM, the swollen endothelial cells, widening subendothelial space, thickened glomerular basement membrane, and microthrombosis can be seen (5, 23, 25) (Figure 2).
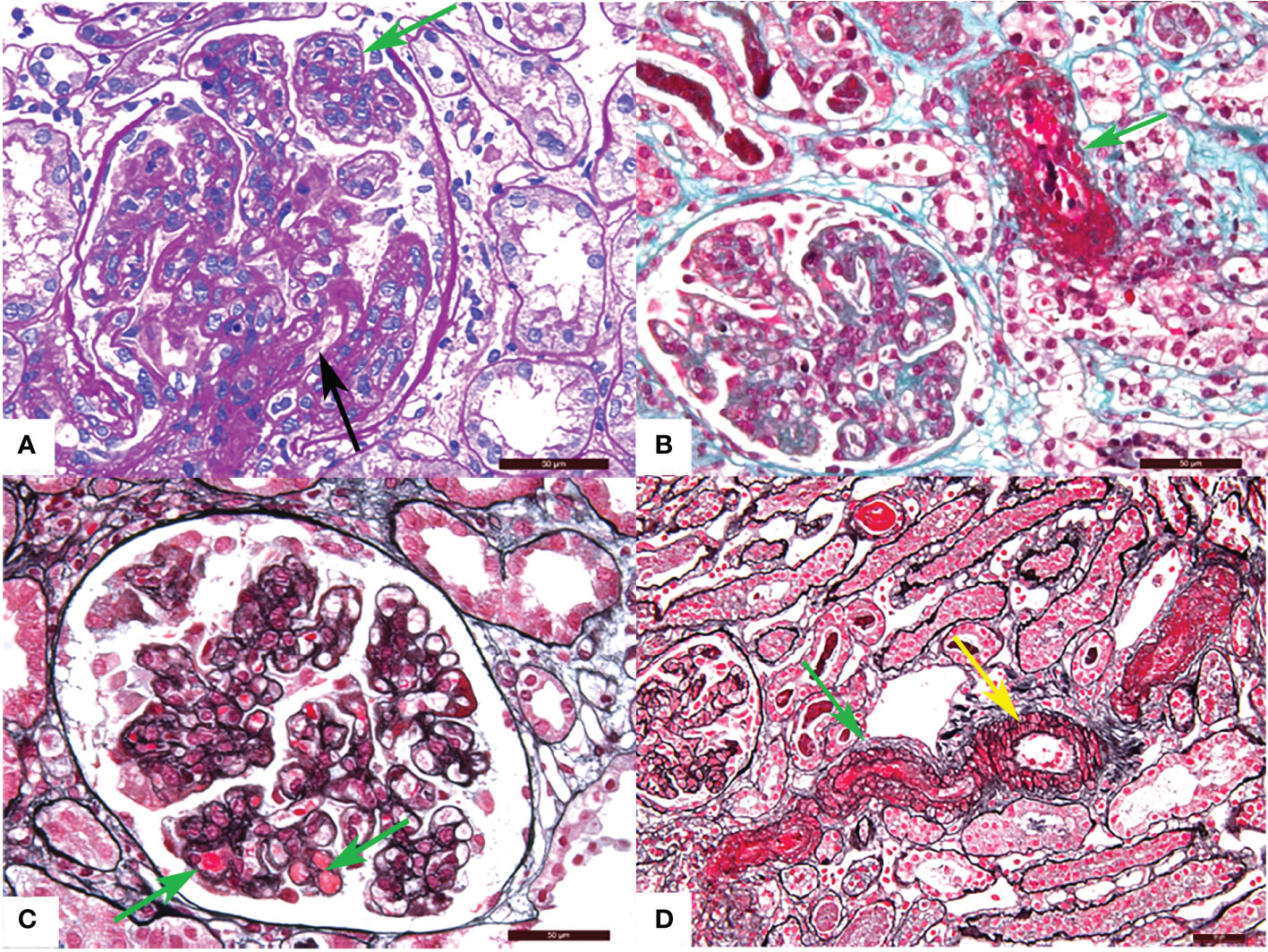
Figure 1 Representative light microscopy images of renal thrombotic microangiopathy (TMA). (A) Proliferation of glomerular mesangial cell and endothelial cell (green arrow), thickening of glomerular basement membrane and formation of the double track sign (black arrow) (PAS). (B) Fibrinous necrosis of the arteriole (green arrow) (Masson). (C) Microthrombosis in the glomerular capillary (green arrow) (PASM-Masson). (D) Arteriolar thrombosis (green arrow) and intimal myxoid oedema (yellow arrow) (PASM-Masson). PAS, periodic acid-schiff. PASM, periodic acid-silver methenamine. Scale bars represent 50μm.

Figure 2 Representative electron microscopy changes of renal thrombotic microangiopathy. (A) The dense deposition (blue arrow) and mild, moderate widening of the loose layer (red arrow) in the basement membrane. (B) The formation of the double track sign (yellow arrow) and dense deposition in the basement membrane (blue arrow). Scale bars represent 2μm.
Immunosuppressive protocol and adjunctive therapies
Hydroxychloroquine (HCQ) is recommended for all LN patients without specific contraindications. Renin-angiotensin system inhibitors (RASi) can be used to control blood pressure and proteinuria. Under the standard induction period, glucocorticoids combined with cyclophosphamide (CYC) or mycophenolate mofetil (MMF), or calcineurin inhibitors (CNI) were used to treat patients with active Class III/IV ± V LN. Promising biological agents belimumab also can be chosen. Plasma exchange (PEX) was used in patients who showed clinical or pathological evidence of systemic TMA.
Definitions
The starting point of follow-up was the time of kidney biopsy and was concluded when the last follow-up or end-point event occurred, which was defined as ESRD, regular dialysis, and death. Complete remission (CR) was defined as proteinuria < 0.5g/day and stable renal function, the latter indicated by a serum creatinine level not higher than 115% of baseline value. Partial remission (PR) was defined as decreasing proteinuria > 50% of the baseline value and < 3.5g/day and stable renal function. Those who did not meet CR or PR criteria were defined as treatment failure (26).
Detection of renal MBL, MASP1/3, C4d, CFB, and CFD by immunofluorescence
Six patients classified as IV ± V LN with or without TMA were chosen for immunofluorescence and immunohistochemistry. Frozen kidney tissue sections were incubated with rabbit anti-human MBL polyclonal antibodies (1:20 dilution, Abcam, Cambridge, UK), rabbit anti-human MASP1/3 polyclonal antibodies (1:20 dilution; Proteintech, Wuhan, China), rabbit anti-human C4d polyclonal antibodies (1:300 dilution; Quidel, America), rabbit anti-human CFB polyclonal antibodies (1:20 dilution, Abcam, Cambridge, UK), and rabbit anti-human CFD polyclonal antibodies (1:20 dilution; Proteintech, Wuhan, China) for 2 hours at 37°C in a moist chamber. After washing these sections, FITC-conjugated affinipure secondary antibodies (1:30 dilution; Proteintech, Wuhan, China) were incubated for 50 minutes at 37°C. Ultimately, these slides were examined by the fluorescence microscope (Nikon, Tokyo, Japan). Semi-quantitative scoring of the results was performed by the professional pathologist under the same fluorescence photography conditions. 0 refers to no positive staining. 1, 2, and 3 refers to weak, moderate, and intense glomerular staining, respectively.
Co-express of renal C5b-9 and CD34 detected by double-staining immunofluorescence
Add rabbit anti-human C5b-9 polyclonal antibodies (1:500 dilution; Abcam, Cambridge, UK) and mouse anti-human CD34 monoclonal antibodies (1:50 dilution; Proteintech, Wuhan, China) to frozen kidney tissue sections and incubated at 37°C for 2 hours. Then FITC/Cy3-conjugated affinipure secondary antibodies (1:30 dilution; Proteintech, Wuhan, China) were incubated at 37°C for 50 minutes. These sections were viewed eventually by a confocal laser scanning microscope and scored semi-quantitatively.
Detection of VWF by immunohistochemistry
The minimal change disease (MCD) patients confirmed by renal biopsy were used as negative controls. Paraffin-embedded kidney biopsies were cut in 3 micrometer thick sections placed on slides, heat, deparaffinized by xylene, hydrated by gradient descent alcohol, recovered by microwave heating in citrate buffer (pH 6.0), and incubated in 0.3% hydrogen peroxide phosphate-buffered saline to inactivate endogenous peroxidase. Next, sections were stained with rabbit polyclonal antibodies for von Willebrand factor (VWF) (1:1200 dilution, Abcam, Cambridge, UK) overnight at 4°C in a moist chamber. Biotinylated secondary antibodies (1:100 dilution; Zhongshan Golden Bridge, Beijing, China) were incubated for 20 min at 37°C and an avidin-biotin-peroxidase complex (1:20 dilution; Zhongshan Golden Bridge, Beijing, China) was applied. Finally, diaminobenzidine was used as chromogen and hematoxylin as counterstaining. The staining for VWF was observed and assessed semi-quantitatively.
Statistical analysis
Continuous variables were expressed as mean (standard deviation, SD) or median (range). Categorical variables were expressed as frequency (percentage). Normally distributed data were analyzed by Student’s t-test, while non-normal variables were compared by the Mann-Whitney U test appropriate. Categorical variables were compared using the x2 test or Fisher’s exact test when appropriate. Renal survival rates were estimated by the Kaplan-Meier method. Risk factors for renal outcomes were analyzed by multivariate Cox-regression analysis. All statistical analyses were performed by SPSS 25.0 (IBM Corporation, NY, USA) and Prism 8.0. P-values < 0.05 (two-tailed) were considered statistically significant.
Results
Patients’ general clinical characteristics and laboratory markers
104 patients with kidney biopsy showing LN and TMA were re-examined by two pathologists. 8 cases were excluded because the absence of clinical data. 17 cases were excluded because no evidence of TMA was observed under EM. Eventually, 79 LN patients showing renal TMA and 79 matched non-TMA controls were included in the analysis. The clinical characteristics of LN with or without TMA at the time of biopsy are listed in Table 1.
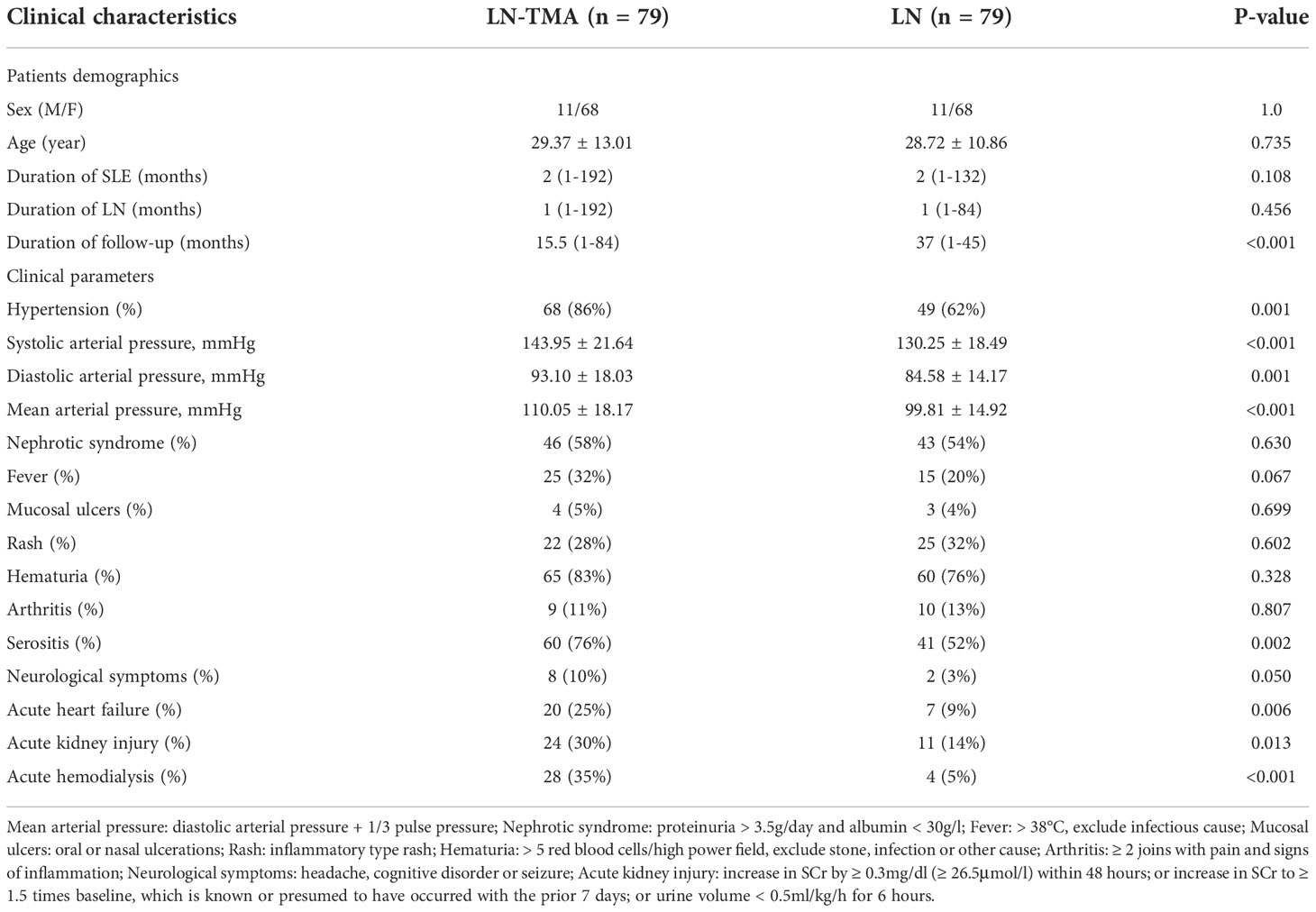
Table 1 Clinical characteristics of lupus nephritis patients with or without renal thrombotic microangiopathy.
As is shown in Table 1, of our 79 patients with renal TMA, females were accounted for 68 while males for 11, and aged 29.37 ± 13.01 years at the time of biopsy. Their SLE duration was 2 (range, 1~192) months and LN duration was 2 (range, 1~132) months. TMA patients presented a higher prevalence rate for hypertension, compared with non-TMA groups (86% vs. 62%, p = 0.001). In addition, patients with renal TMA showed higher systolic arterial pressure and diastolic arterial pressure (143.95 ± 21.64 mmHg and 93.10 ± 18.03 mmHg respectively) compared with non-TMA controls (130.25 ± 18.49 mmHg and 84.58 ± 14.17 mmHg respectively) (p < 0.001 and = 0.001 respectively). The incidence of serositis (76%), acute kidney injury (30%), and acute heart failure (25%) are higher in the cases with TMA (p = 0.002, 0.013, and 0.006 respectively). There are 28 (35%) lupus nephritis with TMA cases requiring acute hemodialysis compared to 4 (5%) non-TMA patients (p < 0.001). Neuropsychiatric SLE in TMA and non-TMA patients were 8 cases and 2 cases respectively (10% vs. 3%, p = 0.050). However, there was no difference in the prevalence of nephrotic syndrome, fever, mucosal ulcers, rash, hematuria, and arthritis between the two groups (p > 0.05, for all).
The comparison of baseline serological markers was also summarized in Table 2. SLE often affects the hematological system when it is active. Therefore, TMA cases that had higher SLEDAI scores (17.51 ± 4.75, compared with 15.08 ± 4.44 in non-TMA controls) had more serious anemia (p < 0.001) and lower platelets (p = 0.006). And microangiopathic hemolytic anemia (MAHA) was observed in 12 (15%) LN-TMA patients. In addition, more than half of TMA cases (59%) had a renal failure at the time of biopsy, while only 19% of LN patients had elevated serum creatinine. No statistical significance was observed in these serological indexes between LN patients with or without TMA, including proteinuria, albumin, C3 or C4 level, ESR, CRP, ANA antibody profiles, and antiphospholipid antibody (APLA). (p > 0.05, for all).
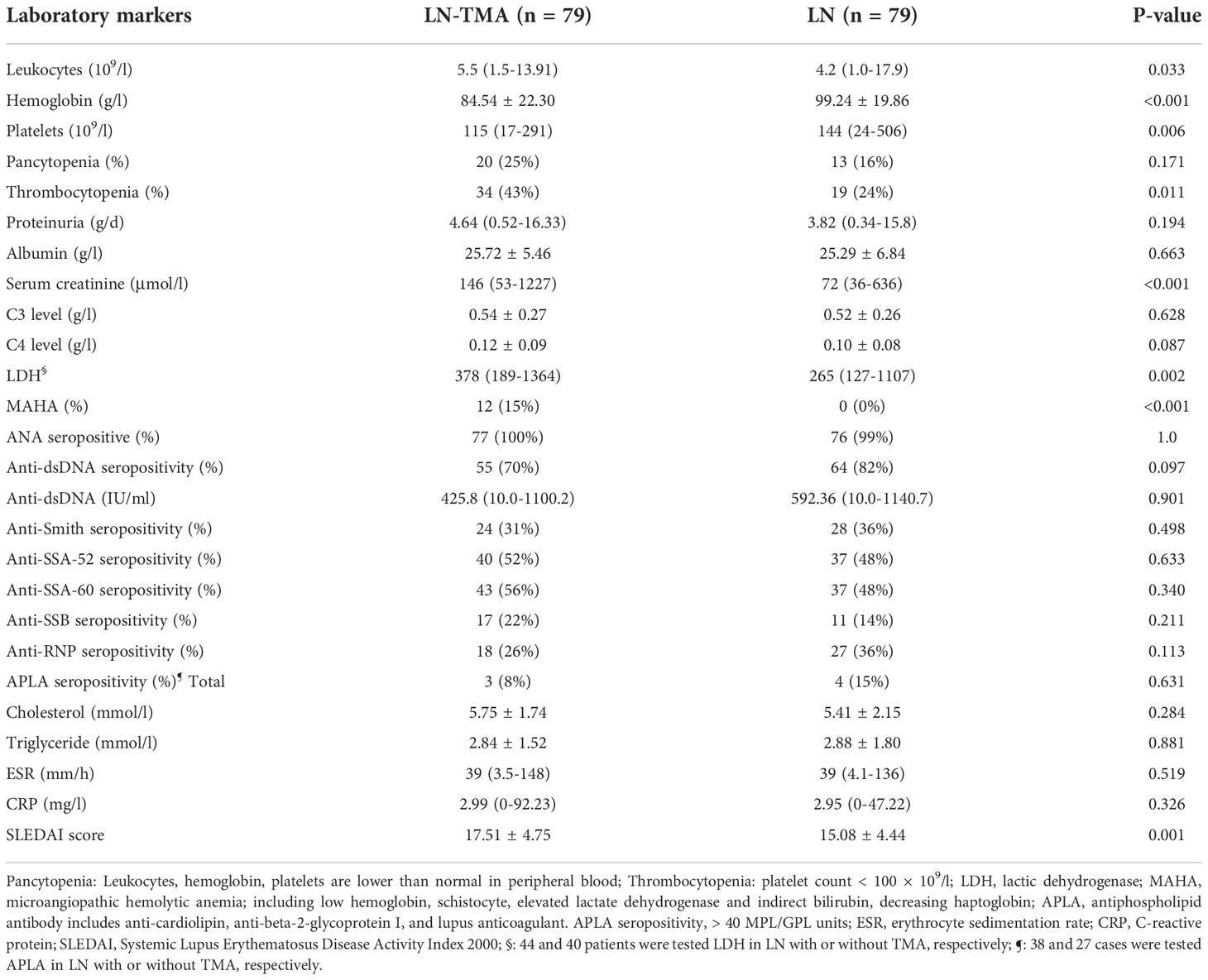
Table 2 Laboratory parameters of lupus nephritis patients with or without renal thrombotic microangiopathy.
Renal histological features
The renal histological features are detailed in Table 3. In our LN-TMA group, classes III, IV, III+V, and IV+V accounted for 8%, 52%, 10%, and 30% of cases; respectively (Figure 3). The activity index between the two groups was not statistically significant. However, the median scores of chronicity index were higher in the renal TMA group compared with the non-TMA group (p = 0.002). Among the CI-related indicators, the LN-TMA group had a higher proportion of fibrous crescents, tubular atrophy, and interstitial fibrosis (p = 0.001, 0.007, and < 0.001 respectively). The positive staining of the IgG, IgM, IgA, C3, C1q using immunofluorescence is denominated as “full house” pattern, which is common in LN and TMA.
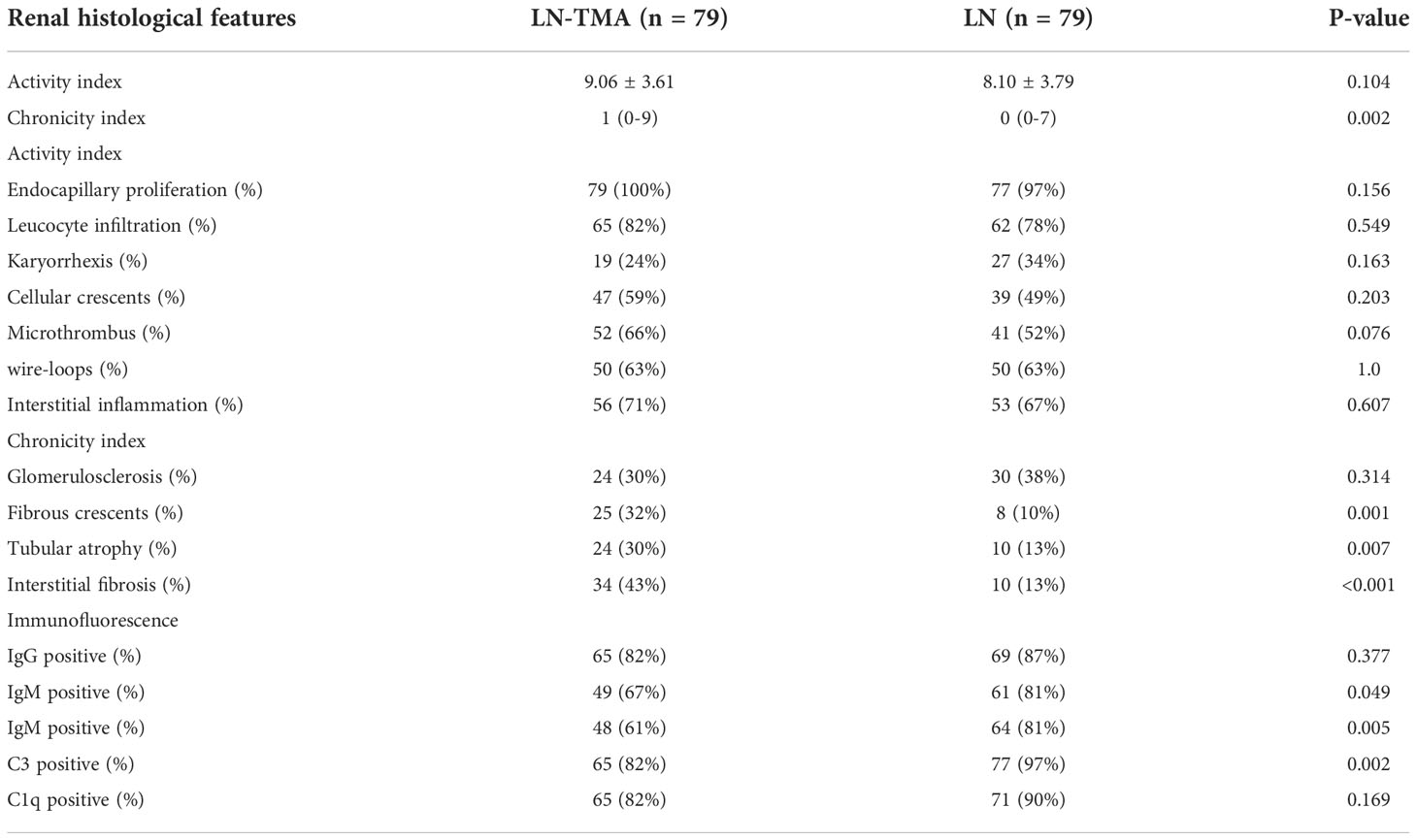
Table 3 Renal histological features in lupus nephritis patients with or without thrombotic microangiopathy.
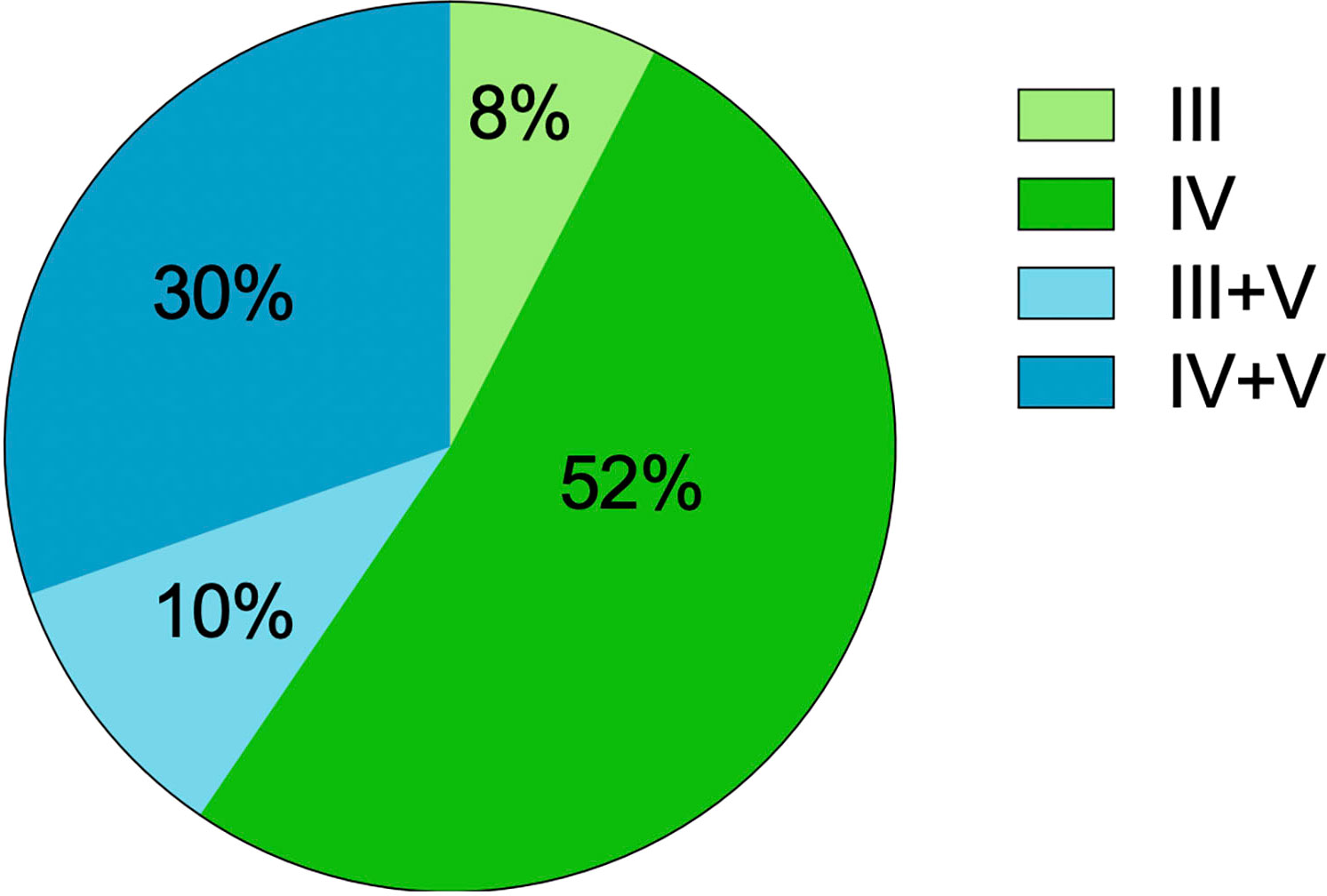
Figure 3 The distribution of lupus nephritis classes with TMA.
Treatment protocols
All patients received oral prednisone. High-dose intravenous methylprednisolone pulse therapy was administered to 41 patients with TMA, compared with 26 cases without TMA (52% vs. 33%, p = 0.016, Table 4). Among 79 LN with TMA patients, 14 received PEX, much higher than the LN group (18% vs. 1%, p < 0.001). More TMA individuals chose CYC as induction therapy (27% vs. 13%, p = 0.028). However, there was no significant difference in the application of MMF and CNI between the two groups in the initial induction program.

Table 4 Treatment of lupus nephritis patients with or without renal thrombotic microangiopathy.
Clinical outcomes
After the median follow-up of 15.5 (range, 1~84) months and 37 (range, 1~45) months in LN with or without TMA respectively, LN-TMA patients achieved inferior complete remission (CR) rate compared with non-TMA controls (22% vs. 63%, and 38% vs. 68%, at 6 and 12 months respectively, p < 0.001 for both, Table 5). Partial remission (PR) at 6 months of the TMA group was significantly lower than that of non-TMA controls (33% vs. 15% respectively, p = 0.008). However, the difference at 12 months did not reach statistical significance (20% vs. 15% respectively, p = 0.41). The patients in the LN-TMA group had significantly lower 2-year renal survival (73% vs. 90%, p = 0.008) and 3-year renal survival (68% vs. 89%, p = 0.002) rates than those in the non-TMA group. Nine individuals in the TMA groups developed the end-stage renal disease during the first month of treatment and seven died. While in the control group, one person needed regular dialysis and one died within the first month.
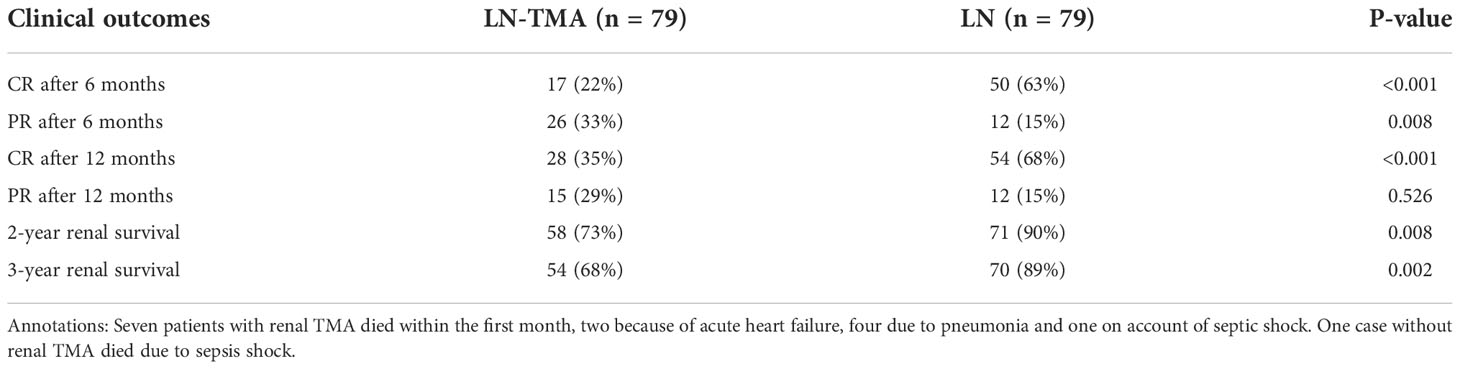
Table 5 Clinical outcomes of lupus nephritis patients with or without renal thrombotic microangiopathy.
The risk factors
We used a COX proportional hazards model to analyze the risk factors which affect the prognosis of LN. Univariate COX regression analysis identified that TMA is a risk factor for renal survival in LN patients (HR 4.807, 95% CI [2.052, 11.263], p < 0.001). Hypertension (HR 12.044, 95% CI [1.643, 88.312], p = 0.014) is usually accompanied by a dismal prognosis, as did serositis (HR 2.634, 95% CI [1.083, 6.409], p = 0.033), acute kidney injury (HR 2.371, 95% CI [1.16, 4.849], p = 0.018), acute hemodialysis (HR 22.100, 95% CI [9.292, 52.560], p < 0.001), acute heart failure (HR 5.052, 95% CI [2.513, 10.154], p < 0.001), thrombocytopenia (HR 2.590, 95% CI [1.303, 5.148], p = 0.007) and interstitial fibrosis (HR 4.099, 95% CI [2.040, 8.238], p < 0.001) in LN. However, using RASi (HR 0.229, 95% CI [0.101, 0.519], p < 0.001) could improve renal survival rate. After adjusting for the influencing factors in the Table 6, multivariate Cox analysis found that acute hemodialysis (HR 7.089, 95% CI [1.130, 44.454], p = 0.037), acute heart failure (HR 3.605, 95% CI [1.186, 10.956], p = 0.024), and serum creatinine (HR 1.003, 95% CI [1.001, 1.006], p = 0.015) remained independent risk factors for renal outcome.
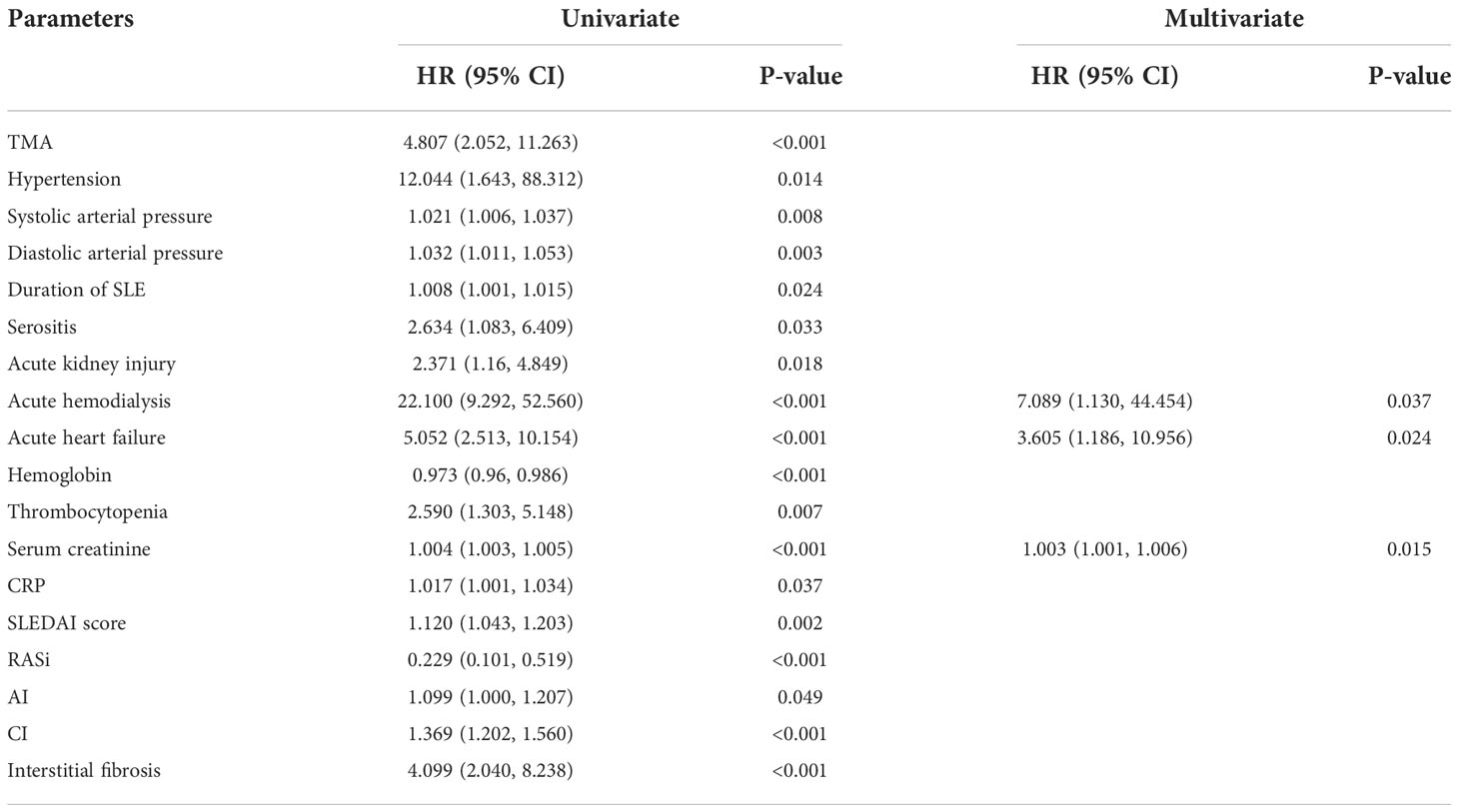
Table 6 Risk factors for renal survival determined by univariate/multivariate COX proportional hazard analysis in lupus nephritis.
Moreover, we used a COX proportional hazards model to evaluate the risk factors which affect the prognosis of LN-TMA (Table 7). Univariate COX regression analysis showed that acute hemodialysis (HR 19.473, 95% CI [5.572, 65.921], p < 0.001), acute heart failure (HR 3.525, 95% CI [1.605, 7.745], p = 0.002), thrombocytopenia (HR 2.627, 95% CI [1.170, 5.902], p = 0.019), glomerulosclerosis (HR 2.305, 95% CI [1.049, 5.064], p = 0.038) are risk factors for renal survival in LN-TMA patients. After adjusting the influencing factors in Table 7, acute hemodialysis (HR 8.719, 95% CI [1.319, 57.611], p = 0.025) was also an independent factor for renal survival.
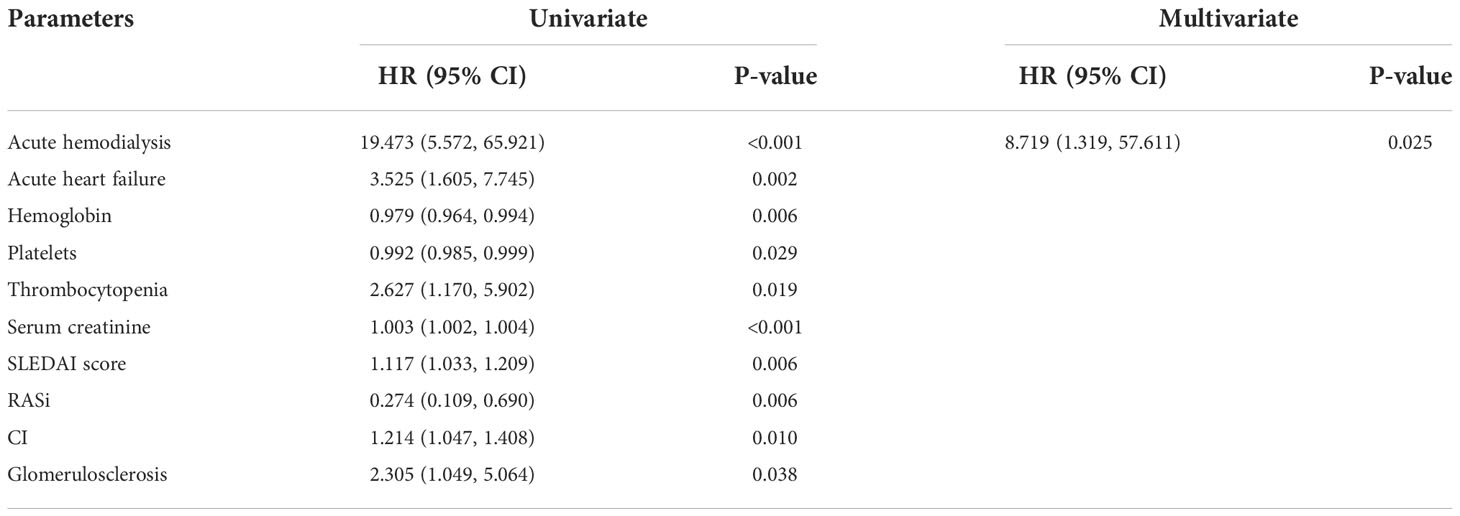
Table 7 Risk factors for renal survival determined by univariate/multivariate COX proportional hazard analysis in LN-TMA.
Survival analysis
As is shown in Figure 4, LN patients with renal TMA presented an inferior renal survival rate compared with non-TMA controls (p < 0.001, Figure 4A). TMA often involves the hematological system, manifesting as thrombocytopenia and anemia. The result showed that the prognosis of patients who had thrombocytopenia was worse (p = 0.009, Figure 4B). Similarly, TMA patients who showed Hb ≤ 90g/l had a poor prognosis (p = 0.002, Figure 4C). And using RASi is a protective factor for LN (p < 0.001, Figure 4D).
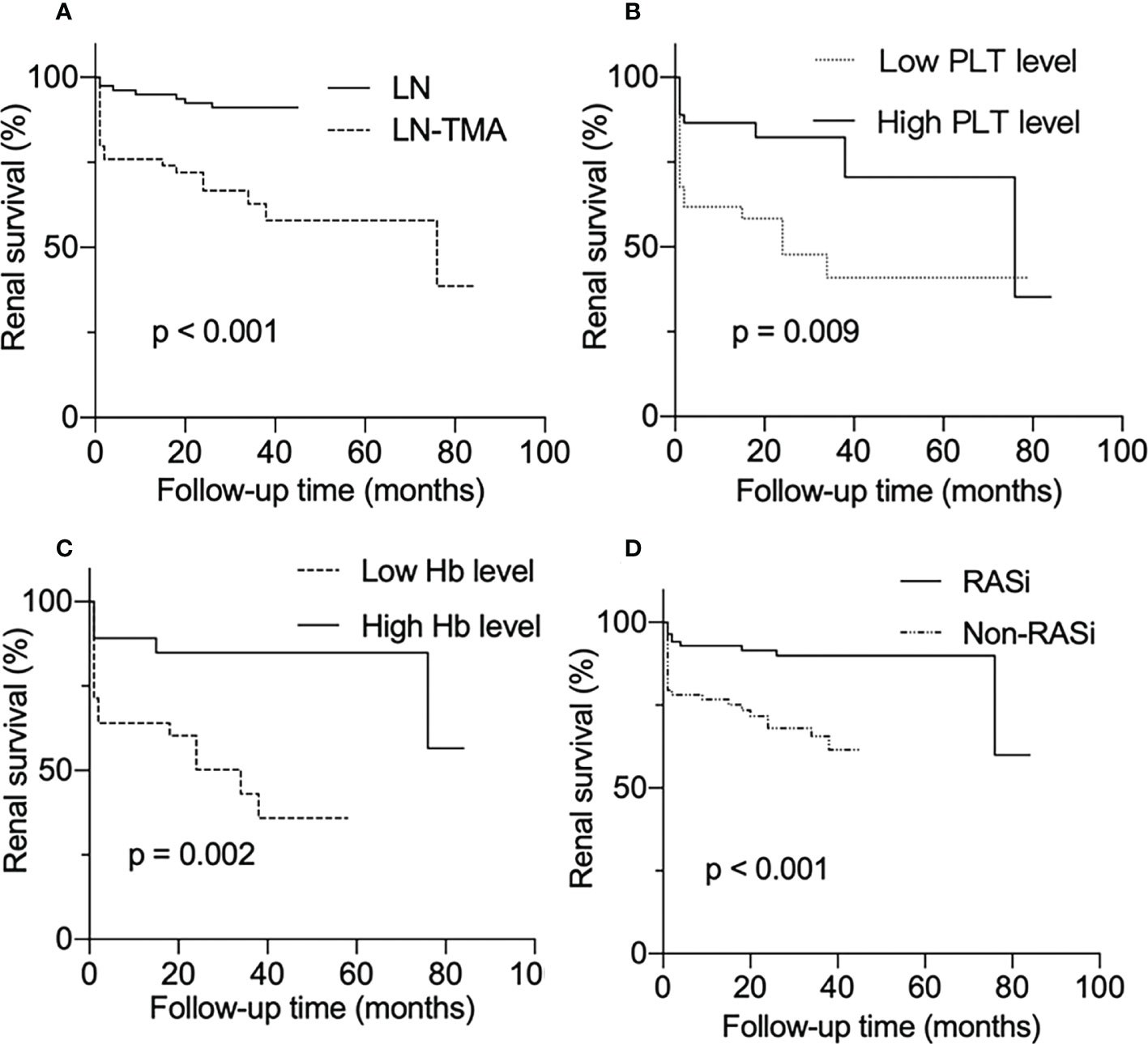
Figure 4 Renal survival rates of lupus nephritis patients. (A) With or without renal thrombotic microangiopathy among lupus nephritis patients. (B) Divided into low and high PLT level groups according to whether the platelet count is less than 100 × 109/l among LN-TMA group. (C) TMA group was separated into low Hb and high Hb level according to whether Hb was less than 90g/l. (D) With or without RASi among LN patients.
The expression of MBL, MASP1/3, C4d, CFB, CFD, C5b-9, CD34, and VWF in kidney tissue
The LP activation elements MBL, MASP1/3, and the AP activation modules CFB, CFD deposited along the glomerulus and blood vessels (Figure 5). Compared with alone LN, the immunofluorescence staining intensity of patients with TMA was higher (Supplementary Figure 1), while the above complement components were not detected in the renal tissue of MCD patients. It is well known that activating the CP is the principal way to involve SLE progression (9). C4d is a fragment of C4 produced during complement activation and is considered as a symbol of classical and lectin pathway activation (27). Expression of C4d was strong in the LN group, especially in the renal arterials of TMA (Figure 6).
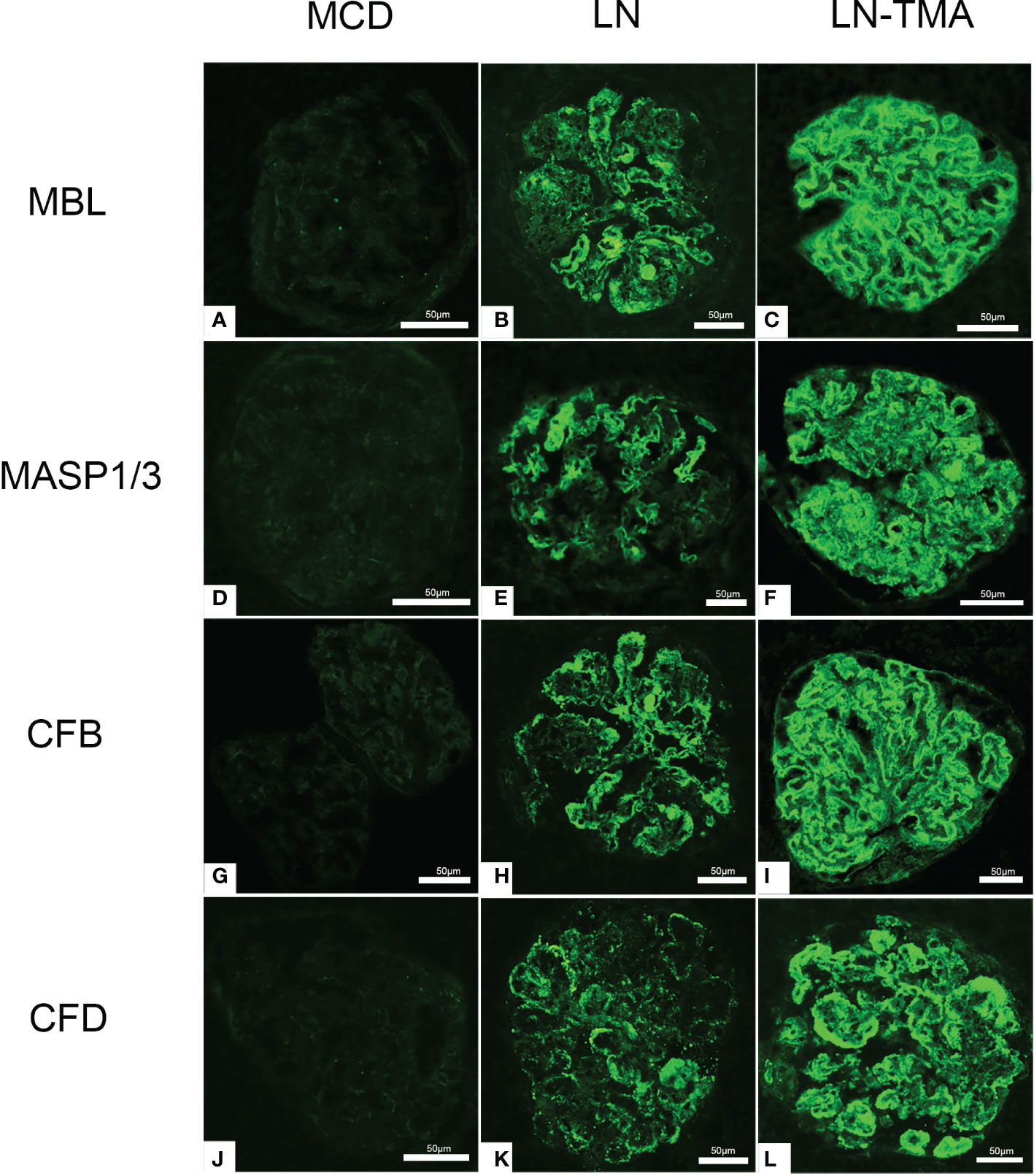
Figure 5 Immunofluorescence staining for MBL (A-C), MASP1/3 (D-F), CFB (G-I), and CFD (J-L) among MCD, LN, and TMA groups. (A, D, G, J) Staining for MBL, MASP1/3, CFB, and CFD was negative in MCD renal tissues. (B, E, H, K) Moderate granular positive staining for MBL, MASP1/3, CFB, and CFD along the glomerular capillary and mesangial region in LN renal tissues. (C, F, I, L) Strong granular positive staining for MBL, MASP1/3, CFB, and CFD along the glomerular capillary and mesangial region in TMA renal tissues. Scale bars represent 50μm.
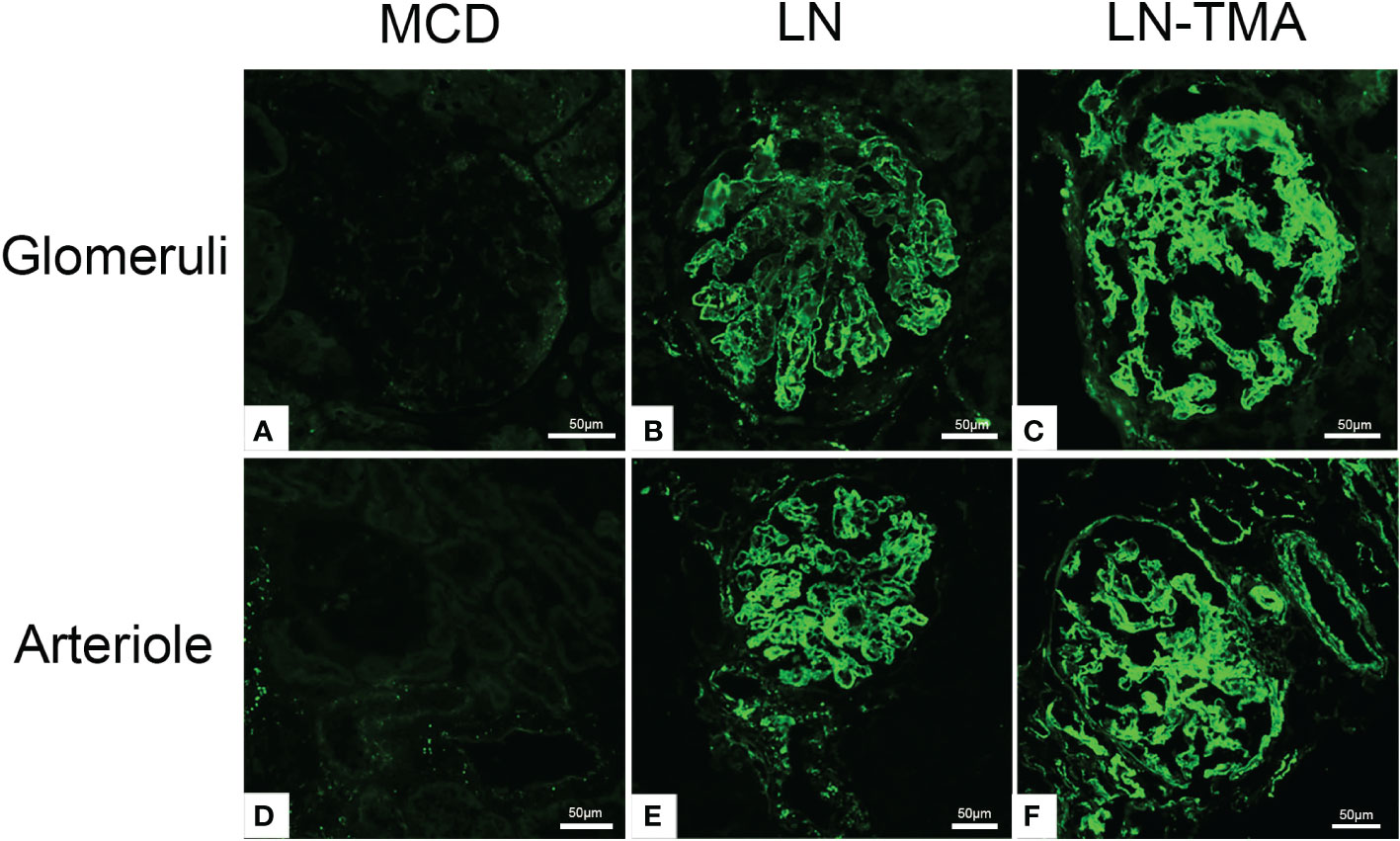
Figure 6 Frozen sections for C4d using immunofluorescence among MCD (A, D), LN (B, E), and TMA (C, F) patients. (A, D) Staining for C4d was negative in MCD renal tissues. (B, E) Moderate granular positive staining for C4d along the glomerular capillary and arteriole in LN renal tissues. (C, F) Strong granular positive staining for C4d along the glomerular capillary and arteriole in TMA renal tissues. Scale bars represent 50μm.
Using double-staining immunofluorescence, the co-localized expression of terminal complex of complement C5b-9 and endothelial marker CD34 in the glomerular and arterioles was observed in the renal tissue of LN patients with or without TMA (Figure 7). No positivity was observed for any of the studied indicators in MCD. The strong expression of VWF was detected in LN and TMA by immunohistochemistry, whereas weak staining was observed in MCD kidneys (Figure 8).
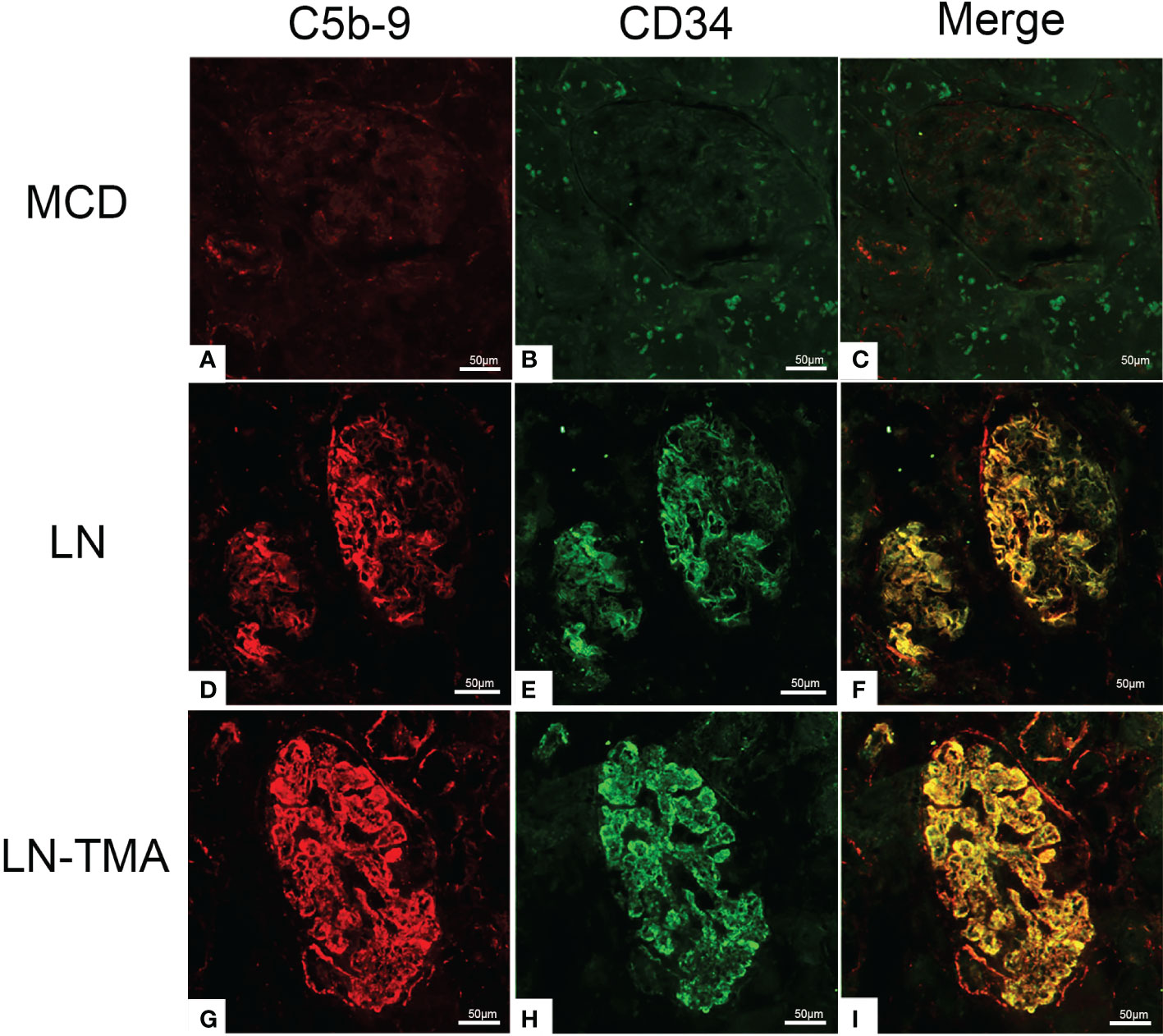
Figure 7 Frozen sections for C5b-9, CD34, and co-localization using immunofluorescence among MCD (A-C), LN (D-F), and TMA (G-I) patients. (A-C) Deposition of C5b-9 (red), CD34 (green), and co-localization (yellow) in MCD was negative. (D-F) Deposition of C5b-9 (red), CD34 (green), and co-localization (yellow) along the glomerular capillary and arteriole in LN was moderate staining. (G-I) Deposition of C5b-9 (red), CD34 (green), and co-localization (yellow) along the glomerular capillary and arteriole in TMA was strong staining. Scale bars represent 50μm.
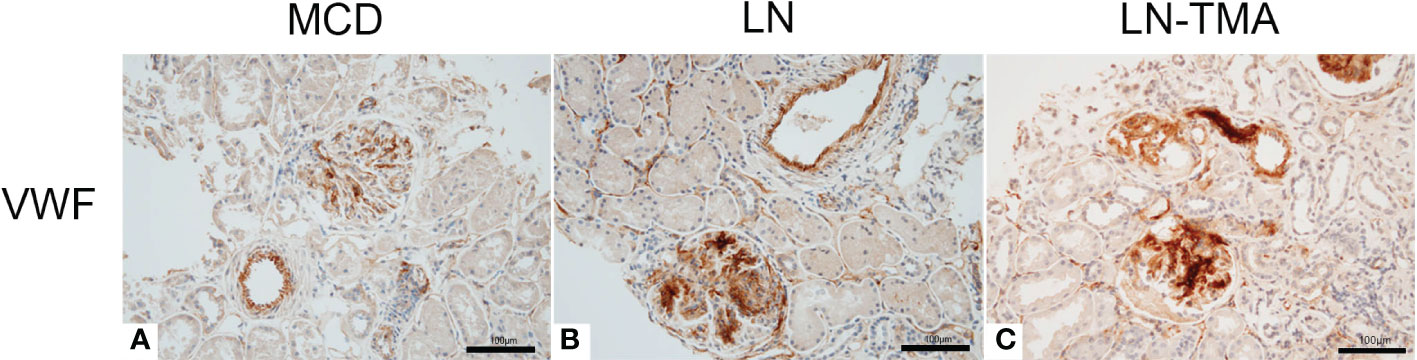
Figure 8 Paraffin sections for VWF (A-C) by immunohistochemistry among MCD, LN, and TMA patients. (A-C) The expression of VWF along the glomerular capillary and arteriole was strong, moderate, and weak in MCD (A), LN (B), and TMA (C) groups, respectively. Scale bars represent 100μm.
Discussion
The incidence of TMA in LN kidney biopsies series range between 0.6% and 24.3% (7, 28, 29). In our retrospective cohort, we identified 79 renal TMA among 2130 biopsy-proven LN patients, occurring at a prevalence of 3.7%. In addition to ethnicity, genetics and environment, the significant variation may be also related to clinicians and pathologists deepening the understanding of TMA.
Our analysis results showed that patients with both renal TMA and LN had severer kidney injury features and poorer renal survival rates. Clinical characteristics including hypertension, serositis, acute kidney injury, acute hemodialysis, acute heart failure, anemia, thrombocytopenia, serum creatinine, and SLEDAI score; as well as renal histological indicators including chronicity index and interstitial fibrosis, are not only worse in TMA than those without TMA, but worthy variables to predict renal outcome in LN. Consistent with other previous studies, TMA was an independent risk factor for renal survival in LN patients (6, 7, 30–32).
TMA is characterized by thrombocytopenia, MAHA, and organ injury (33). The incidence of MAHA in TMA patients fluctuates from 8% to 60%, which is a valuable serum marker to assist in diagnosis and correlate with renal prognosis (34, 35). There were more TMA patients requiring acute hemodialysis at presentation due to elevated serum creatinine levels and various serious complications. Endocapillary lesions could be reversed by appropriate immunosuppression, which is related to renal function recovery (23). While interstitial fibrosis is the chronic damage that usually caused progressive renal insufficiency.
Renal vasculopathy in SLE are common, mainly due to the production of autoantibodies to form immune complex-induced vascular inflammation. There are many etiologies of renal TMA in lupus nephritis, including antiphospholipid syndrome, thrombotic thrombocytopenic purpura, malignant hypertension, pregnancy, systemic sclerosis overlap, drugs, and infections (26, 33). In addition, Christine et al. have revealed the importance of inherited and acquired disorders of complement in SLE and TMA (36). These gene mutations, including complement factor H, complement factor I, complement factor B, thrombomodulin, and MCP/CD46, contribute to occurring TMA (37).The clinical manifestations and treatment regimens caused by various causes are distinct (38, 39). KDIGO guidelines recommend PEX, glucocorticoids, rituximab, or caplacizumab in patients with ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type I motif, member 13) activity < 10% (26). However, ADAMTS13 activity was not detected in this cohort. When antiphospholipid antibodies were positive and ADAMTS13 activity was normal, antiphospholipid syndrome nephropathy was considered and started anticoagulant therapy (26). We found that 6 cases in the TMA group were positive for antiphospholipid antibodies. Two additional patients presented with overlap syndrome, both overlapping with ANCA-associated vasculitis. In addition, no transplantation, malignancy, or drug-related TMAs were observed.
It is well known that the dysregulation of complement system is inextricably linked with SLE and TMA. Compared with LN alone, patients with TMA had superior serum Ba and C5b-9 levels (40). Besides, the plasma concentrations of MBL, MASP-1 and MASP-3 in SLE patients were higher than those in healthy controls (41). A study from Japan found that LN patients with MBL, L-ficolin, and properdin deposition had more urinary protein excretion. Meanwhile, patients with CFB and factor H deposition had the worse interstitial fibrosis (42). The MASP1 gene encodes MASP-1 and MASP-3, which are obtained by alternative splicing and translation of mRNA. Exons 1-8 and 10-11 encode the five N-terminal domains shared by MASP1 and MASP3. Exons 12 and 13-18 encode the linker and protease domains of MASP3 and MASP1, respectively (14). The role of MASP-1 in LP activation has been well understood, while Takahashi et al. found that sera from MASP1 gene knockout lupus-prone MRL/lpr mice (Masp1/3-/- MRL/lpr mice) had little-to-no activation of both the LP and AP with zymogen forms of CFD (43). Furthermore, adding specific inhibitors for MASP-1 and MASP-3 to the plasma, CFD activation of the former was not affected, while the latter factor D existed as a precursor. Hence, MASP-3 was seen as a direct pro-CFD activator in resting blood (14). The influence of AP dysregulation on LN has been validated in murine models. Genetic deficiency of factor H in MRL/lpr mice presented marked albuminuria and azotemia. On the contrary, CFB deficient MRL/lpr mice had ameliorative clinical manifestations (20, 44, 45). The spectrum of C4d staining in glomerulus and arterioles was usual in both our and former studies (7, 32, 46, 47). Previous research found that C4d was a common denominator in TMA patients and arteriolar C4d deposition had prognostic value for the renal outcomes (32, 48). Vascular endothelial cell injury is a key link in the development of SLE and TMA (49). Co-localized expression of C5b-9 and CD34 confirmed the damage of complement activation to endothelial cells (12). The ultralarge VWF multimers secreted from damaged endothelial cells are released into the circulation and adhere to platelets to form microthrombi, leading to tissue ischemia, platelet consumption, and schistocytes (50, 51). Consequently, VWF deposition is strongly positive in TMA kidney tissues.
Despite treatment with glucocorticoids, immunosuppressive agents, and plasma exchange, the 3-year renal survival rate of TMA patients in this study was only 68%. Of the fourteen people underwent plasma exchange, six developed ESRD or died within the first month, suggesting that it is urgent to find novel biologic agents (52). 93% of TMA associated with LN patients had a favorable renal outcome under the premise of anti-C5 monoclonal antibody, eculizumab (53, 54). Moreover, Avacopan as an orally administered small-molecule C5a receptor inhibitor may be useful in LN patients (55). Narsoplimab, a monoclonal antibody against MASP-2, can inhibit LP activation and alleviate endothelial damage. Results from a clinical trial showed that it significantly improved remission and survival rate in patients with hematopoietic stem cell transplant-associated thrombotic microangiopathy (NCT047906), but its efficacy and safety in LN are still being tested (NCT02682407). LNP023, an invertible binding inhibitor of CFB, could alleviate pathological injury in the kidneys of MRL/lpr mice (56). In addition, CFB cleavage is abrogated in aHUS (atypical hemolytic uremic syndrome) patients’ serum when using danicopan, which is an oral CFD inhibitor (57).
There are some limitations in this study. Firstly, these enrolled individuals were mainly from Northern China, which is a single-center, retrospective research. Moreover, detecting complement-related autoantibody activity or gene mutations was needed. Furthermore, it is regrettable not to detect LP and AP-related complement components in serum and urine.
In conclusion, LN-associated TMA patients usually have more serious clinicopathological manifestations and inferior renal survival rates. LP and AP activation may play a crucial role in the pathogenesis of TMA in LN patients. This suggests that complement inhibition drugs can be widely used as novel therapeutic approaches in the future.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by The first affiliated hospital of zhengzhou university. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
GX designed the study, BZ searched eligible literature, processing data and participated in the statistical analysis. BZ and GX drafted and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grant from National Natural Science Fund of China (81870480).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1081942/full#supplementary-material
References
1. Gregersen JW, Jayne DR. B-cell depletion in the treatment of lupus nephritis. Nat Rev Nephrol (2012) 8(9):505–14. doi: 10.1038/nrneph.2012.141
2. Tian J, Zhang D, Yao X, Huang Y, Lu Q. Global epidemiology of systemic lupus erythematosus: A comprehensive systematic analysis and modelling study. Ann Rheum Dis (2022). doi: 10.1136/ard-2022-223035
3. Maria NI, Davidson A. Protecting the kidney in systemic lupus erythematosus: From diagnosis to therapy. Nat Rev Rheum (2020) 16(5):255–67. doi: 10.1038/s41584-020-0401-9
4. Tsokos GC. Systemic lupus erythematosus. N Engl J Med (2011) 365(22):2110–21. doi: 10.1056/NEJMra1100359
5. Appel GB, Pirani CL, D’Agati V. Renal vascular complications of systemic lupus erythematosus. J Am Soc Nephrol (1994) 4(8):1499–515. doi: 10.1681/asn.V481499
6. Wu LH, Yu F, Tan Y, Qu Z, Chen MH, Wang SX, et al. Inclusion of renal vascular lesions in the 2003 Isn/Rps system for classifying lupus nephritis improves renal outcome predictions. Kidney Int (2013) 83(4):715–23. doi: 10.1038/ki.2012.409
7. Song D, Wu LH, Wang FM, Yang XW, Zhu D, Chen M, et al. The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res Ther (2013) 15(1):R12. doi: 10.1186/ar4142
8. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med (2014) 371(7):654–66. doi: 10.1056/NEJMra1312353
9. Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol (2013) 24(9):1357–66. doi: 10.1681/asn.2013010026
10. Macedo AC, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol (2016) 7:55. doi: 10.3389/fimmu.2016.00055
11. Héja D, Kocsis A, Dobó J, Szilágyi K, Szász R, Závodszky P, et al. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease masp-1 as the exclusive activator of masp-2. Proc Natl Acad Sci USA (2012) 109(26):10498–503. doi: 10.1073/pnas.1202588109
12. Thurman JM. Complement and the kidney: An overview. Adv Chronic Kidney Dis (2020) 27(2):86–94. doi: 10.1053/j.ackd.2019.10.003
13. Dobó J, Szakács D, Oroszlán G, Kortvely E, Kiss B, Boros E, et al. Masp-3 is the exclusive pro-factor d activator in resting blood: The lectin and the alternative complement pathways are fundamentally linked. Sci Rep (2016) 6:31877. doi: 10.1038/srep31877
14. Oroszlán G, Kortvely E, Szakács D, Kocsis A, Dammeier S, Zeck A, et al. Masp-1 and masp-2 do not activate pro-factor d in resting human blood, whereas masp-3 is a potential activator: Kinetic analysis involving specific masp-1 and masp-2 inhibitors. J Immunol (2016) 196(2):857–65. doi: 10.4049/jimmunol.1501717
15. Mathern DR, Heeger PS. Molecules great and small: The complement system. Clin J Am Soc Nephrol (2015) 10(9):1636–50. doi: 10.2215/cjn.06230614
16. Satyam A, Hisada R, Bhargava R, Tsokos MG, Tsokos GC. Intertwined pathways of complement activation command the pathogenesis of lupus nephritis. Transl Res (2022) 245:18–29. doi: 10.1016/j.trsl.2022.03.005
17. Nisihara RM, Magrini F, Mocelin V, Messias-Reason IJ. Deposition of the lectin pathway of complement in renal biopsies of lupus nephritis patients. Hum Immunol (2013) 74(8):907–10. doi: 10.1016/j.humimm.2013.04.030
18. Lee YH, Lee HS, Choi SJ, Ji JD, Song GG. The association between the mannose-binding lectin codon 54 polymorphism and systemic lupus erythematosus: A meta-analysis update. Mol Biol Rep (2012) 39(5):5569–74. doi: 10.1007/s11033-011-1361-6
19. Elhadad S, Chapin J, Copertino D, Van Besien K, Ahamed J, Laurence J. Masp2 levels are elevated in thrombotic microangiopathies: Association with microvascular endothelial cell injury and suppression by anti-Masp2 antibody narsoplimab. Clin Exp Immunol (2021) 203(1):96–104. doi: 10.1111/cei.13497
20. Elliott MK, Jarmi T, Ruiz P, Xu Y, Holers VM, Gilkeson GS. Effects of complement factor d deficiency on the renal disease of Mrl/Lpr mice. Kidney Int (2004) 65(1):129–38. doi: 10.1111/j.1523-1755.2004.00371.x
21. Noris M, Mescia F, Remuzzi G. Stec-hus, atypical hus and ttp are all diseases of complement activation. Nat Rev Nephrol (2012) 8(11):622–33. doi: 10.1038/nrneph.2012.195
22. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. European League against Rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheum (2019) 71(9):1400–12. doi: 10.1002/art.40930
23. Bajema IM, Wilhelmus S, Alpers CE, Bruijn JA, Colvin RB, Cook HT, et al. Revision of the international society of Nephrology/Renal pathology society classification for lupus nephritis: Clarification of definitions, and modified national institutes of health activity and chronicity indices. Kidney Int (2018) 93(4):789–96. doi: 10.1016/j.kint.2017.11.023
24. Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the sledai. a disease activity index for lupus patients. the committee on prognosis studies in sle. Arthritis Rheum (1992) 35(6):630–40. doi: 10.1002/art.1780350606
25. Ding Y, Tan Y, Qu Z, Yu F. Renal microvascular lesions in lupus nephritis. Ren Fail (2020) 42(1):19–29. doi: 10.1080/0886022x.2019.1702057
26. KDIGO. Clinical practice guideline for the management of glomerular diseases. Kidney Int (2021) 100(4s):S1–s276. doi: 10.1016/j.kint.2021.05.021
27. Walport MJ. Complement. first of two parts. N Engl J Med (2001) 344(14):1058–66. doi: 10.1056/nejm200104053441406
28. Li C, Yap DYH, Chan G, Wen YB, Li H, Tang C, et al. Clinical outcomes and clinico-pathological correlations in lupus nephritis with kidney biopsy showing thrombotic microangiopathy. J Rheum (2019) 46(11):1478–84. doi: 10.3899/jrheum.180773
29. Descombes E, Droz D, Drouet L, Grünfeld JP, Lesavre P. Renal vascular lesions in lupus nephritis. Med (Baltimore) (1997) 76(5):355–68. doi: 10.1097/00005792-199709000-00003
30. Austin HA 3rd, Muenz LR, Joyce KM, Antonovych TA, Kullick ME, Klippel JH, et al. Prognostic factors in lupus nephritis. contribution of renal histologic data. Am J Med (1983) 75(3):382–91. doi: 10.1016/0002-9343(83)90338-8
31. Contreras G, Pardo V, Cely C, Borja E, Hurtado A, de la Cuesta C, et al. Factors associated with poor outcomes in patients with lupus nephritis. Lupus (2005) 14(11):890–5. doi: 10.1191/0961203305lu2238oa
32. Ding Y, Yu X, Wu L, Tan Y, Qu Z, Yu F. The spectrum of C4d deposition in renal biopsies of lupus nephritis patients. Front Immunol (2021) 12:654652. doi: 10.3389/fimmu.2021.654652
33. Brocklebank V, Wood KM, Kavanagh D. Thrombotic microangiopathy and the kidney. Clin J Am Soc Nephrol (2018) 13(2):300–17. doi: 10.2215/cjn.00620117
34. Hu WX, Liu ZZ, Chen HP, Zhang HT, Li LS, Liu ZH. Clinical characteristics and prognosis of diffuse proliferative lupus nephritis with thrombotic microangiopathy. Lupus (2010) 19(14):1591–8. doi: 10.1177/0961203310376523
35. Banfi G, Bertani T, Boeri V, Faraggiana T, Mazzucco G, Monga G, et al. Renal vascular lesions as a marker of poor prognosis in patients with lupus nephritis. gruppo italiano per lo studio Della nefrite lupica (Gisnel). Am J Kidney Dis (1991) 18(2):240–8. doi: 10.1016/s0272-6386(12)80885-7
36. Józsi M, Heinen S, Hartmann A, Ostrowicz CW, Hälbich S, Richter H, et al. Factor h and atypical hemolytic uremic syndrome: Mutations in the c-terminus cause structural changes and defective recognition functions. J Am Soc Nephrol (2006) 17(1):170–7. doi: 10.1681/asn.2005080868
37. Grumach AS, Kirschfink M. Are complement deficiencies really rare? overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol (2014) 61(2):110–7. doi: 10.1016/j.molimm.2014.06.030
38. Yu F, Haas M, Glassock R, Zhao MH. Redefining lupus nephritis: Clinical implications of pathophysiologic subtypes. Nat Rev Nephrol (2017) 13(8):483–95. doi: 10.1038/nrneph.2017.85
39. Scheen M, Adedjouma A, Esteve E, Buob D, Abisror N, Planche V, et al. Kidney disease in antiphospholipid antibody syndrome: Risk factors, pathophysiology and management. Autoimmun Rev (2022) 21(5):103072. doi: 10.1016/j.autrev.2022.103072
40. Mejia-Vilet JM, Gómez-Ruiz IA, Cruz C, Méndez-Pérez RA, Comunidad-Bonilla RA, Uribe-Uribe NO, et al. Alternative complement pathway activation in thrombotic microangiopathy associated with lupus nephritis. Clin Rheum (2021) 40(6):2233–42. doi: 10.1007/s10067-020-05499-1
41. Troldborg A, Thiel S, Laska MJ, Deleuran B, Jensenius JC, Stengaard-Pedersen K. Levels in plasma of the serine proteases and associated proteins of the lectin pathway are altered in patients with systemic lupus erythematosus. J Rheum (2015) 42(6):948–51. doi: 10.3899/jrheum.141163
42. Sato N, Ohsawa I, Nagamachi S, Ishii M, Kusaba G, Inoshita H, et al. Significance of glomerular activation of the alternative pathway and lectin pathway in lupus nephritis. Lupus (2011) 20(13):1378–86. doi: 10.1177/0961203311415561
43. Takahashi M, Ishida Y, Iwaki D, Kanno K, Suzuki T, Endo Y, et al. Essential role of mannose-binding lectin-associated serine protease-1 in activation of the complement factor d. J Exp Med (2010) 207(1):29–37. doi: 10.1084/jem.20090633
44. Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, et al. Modulation of renal disease in Mrl/Lpr mice genetically deficient in the alternative complement pathway factor b. J Immunol (2000) 164(2):786–94. doi: 10.4049/jimmunol.164.2.786
45. Bao L, Haas M, Quigg RJ. Complement factor h deficiency accelerates development of lupus nephritis. J Am Soc Nephrol (2011) 22(2):285–95. doi: 10.1681/asn.2010060647
46. Batal I, Liang K, Bastacky S, Kiss LP, McHale T, Wilson NL, et al. Prospective assessment of C4d deposits on circulating cells and renal tissues in lupus nephritis: A pilot study. Lupus (2012) 21(1):13–26. doi: 10.1177/0961203311422093
47. Shen Y, Chen XW, Sun CY, Dai M, Yan YC, Yang CD. Association between anti-Beta2 glycoprotein I antibodies and renal glomerular C4d deposition in lupus nephritis patients with glomerular microthrombosis: A prospective study of 155 cases. Lupus (2010) 19(10):1195–203. doi: 10.1177/0961203310368409
48. Chua JS, Baelde HJ, Zandbergen M, Wilhelmus S, van Es LA, de Fijter JW, et al. Complement factor C4d is a common denominator in thrombotic microangiopathy. J Am Soc Nephrol (2015) 26(9):2239–47. doi: 10.1681/asn.2014050429
49. Park MH, Caselman N, Ulmer S, Weitz IC. Complement-mediated thrombotic microangiopathy associated with lupus nephritis. Blood Adv (2018) 2(16):2090–4. doi: 10.1182/bloodadvances.2018019596
50. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood (2017) 129(21):2836–46. doi: 10.1182/blood-2016-10-709857
51. Zheng XL. Adamts13 and Von willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med (2015) 66:211–25. doi: 10.1146/annurev-med-061813-013241
52. Parra Sánchez AR, Voskuyl AE, van Vollenhoven RF. Treat-to-Target in systemic lupus erythematosus: Advancing towards its implementation. Nat Rev Rheum (2022) 18(3):146–57. doi: 10.1038/s41584-021-00739-3
53. Wright RD, Bannerman F, Beresford MW, Oni L. A systematic review of the role of eculizumab in systemic lupus erythematosus-associated thrombotic microangiopathy. BMC Nephrol (2020) 21(1):245. doi: 10.1186/s12882-020-01888-5
55. Jayne DRW, Merkel PA, Schall TJ, Bekker P. Avacopan for the treatment of anca-associated vasculitis. N Engl J Med (2021) 384(7):599–609. doi: 10.1056/NEJMoa2023386
56. Chen K, Deng Y, Shang S, Tang L, Li Q, Bai X, et al. Complement factor b inhibitor Lnp023 improves lupus nephritis in Mrl/Lpr mice. BioMed Pharmacother (2022) 153:113433. doi: 10.1016/j.biopha.2022.113433
57. Aradottir SS, Kristoffersson AC, Roumenina LT, Bjerre A, Kashioulis P, Palsson R, et al. Factor d inhibition blocks complement activation induced by mutant factor b associated with atypical hemolytic uremic syndrome and membranoproliferative glomerulonephritis. Front Immunol (2021) 12:690821. doi: 10.3389/fimmu.2021.690821
Keywords: lupus nephritis, thrombotic microangiopathy, lectin pathway, alternative pathway, complement
Citation: Zhang B and Xing G (2022) Thrombotic microangiopathy mediates poor prognosis among lupus nephritis via complement lectin and alternative pathway activation. Front. Immunol. 13:1081942. doi: 10.3389/fimmu.2022.1081942
Received: 27 October 2022; Accepted: 28 November 2022;
Published: 13 December 2022.
Edited by:
Dong-Qing Ye, Anhui Medical University, ChinaReviewed by:
Masaru Kato, Hokkaido University, JapanKoshy Nithin Thomas, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI), India
Copyright © 2022 Zhang and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guolan Xing, eGdsQHp6dS5lZHUuY24=