 Yizhang Mo
Yizhang Mo Kebing Chen
Kebing Chen- Department of Spine Surgery, The Sixth Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
High mobility group box 1 (HMGB1) has dual functions as a nonhistone nucleoprotein and an extracellular inflammatory cytokine. In the resting state, HMGB1 is mainly located in the nucleus and regulates key nuclear activities. After spinal cord injury, HMGB1 is rapidly expressed by neurons, microglia and ependymal cells, and it is either actively or passively released into the extracellular matrix and blood circulation; furthermore, it also participates in the pathophysiological process of spinal cord injury. HMGB1 can regulate the activation of M1 microglia, exacerbate the inflammatory response, and regulate the expression of inflammatory factors through Rage and TLR2/4, resulting in neuronal death. However, some studies have shown that HMGB1 is beneficial for the survival, regeneration and differentiation of neurons and that it promotes the recovery of motor function. This article reviews the specific timing of secretion and translocation, the release mechanism and the role of HMGB1 in spinal cord injury. Furthermore, the role and mechanism of HMGB1 in spinal cord injury and, the challenges that still need to be addressed are identified, and this work will provide a basis for future studies.
1 Introduction
High mobility group box 1 (HMGB1), also known as amphotericin or HMG1, is a nonhistone chromatin binding protein first discovered in the 1960s. HMGB1 shows high electrophoretic mobility when run on polyacrylamide gels, hence its name (1, 2). HMGB1 is highly conserved in evolution, and the HMGB1 in rodents shares 99% homology with that in humans (3–5). HMGB1 is also expressed partially in the cytoplasm because it shuttles back and forth from the nucleus (6). HMGB1 has the dual functions of a nonhistone nucleoprotein and an extracellular inflammatory cytokine. HMGB1 binds extensively to DNA in the nucleus and participates in transcriptional regulation, DNA replication and repair, telomere maintenance and nucleosome assembly (7). Extracellular HMGB1 is passively released or actively secreted by necrotic tissue or stress cells. As a chemokine or cytokine, it binds to pattern recognition receptors (PRRs) to create a damage-associated molecular pattern (DAMP) (6–12). HMGB1 plays an important role in many diseases, including traumatic shock, fatty liver disease, septicaemia, autoimmune diseases, and cancer (8, 11, 13–15).
In recent years, the role of HMGB1 in spinal cord injury (SCI) has attracted a great deal of attention. SCI is characterized by sensory, motor and autonomic nerve dysfunction (16) mediated by complex and diverse pathophysiological processes, including neuroinflammation, neuronal death, glial scar formation and axonal regeneration (17–20). The concentration of HMGB1 in the injured area increases rapidly and lasts for a long time (21–31), and it not only aggravates injury by playing the role of an inflammatory cytokine, but it also promotes the recovery of the injured spinal cord.
This article reviews the research progress of studies investigating the expression and release of HMGB1 after SCI, its effect on the injured spinal cord and its potential therapeutic mechanism.
2 Main text
2.1 The structure of HMGB1 and the lower function of the resting state
The HMGB1 protein is a highly conserved nuclear protein that consists of 215 amino acids and has a molecular weight of approximately 30 kDa. Structurally, HMGB1 is divided into the following three functional regions: the A-box, the B-box and the acidic C-terminus. The A-box and B-box are composed of 80-90 amino acid residues with similar amino acid repeat sequences and nonspecific DNA-binding sites, and the B-box is the functional structural region that causes inflammation (32, 33); however, the A-box has a certain antagonistic effect on the B-box (34). The acidic C-terminus containing aspartic acid and glutamate is mainly involved in regulating the binding of HMGB1 to DNA and mediating gene transcription and chromosome unwinding (35). The B-box domain has two key binding sites, Toll-like receptor 4 (TLR4) and a receptor for advanced glycation end (RAGE) products, which regulate the release of proinflammatory cytokines (36, 37). HMGB1 has two nuclear localization sequences (NLSs); one is in the A-box, the other is between the B-box and the C-terminal tail, and the nuclear export signal (NES) is contained in the DNA-binding domain (38).
During the resting state of HMGB1, it is located mainly in the nucleus and it regulates key nuclear activities, including transcription, replication, DNA repair and nucleosome formation, all of which are important for maintaining steady-state cellular function (7, 38).
Collectively, these results show that HMGB1 is a highly conserved protein with proinflammatory and anti-inflammatory potential. In the resting state, HMGB1 is located mainly in the nucleus and regulates physiological processes there; however, there are no related studies on the structural and functional changes to HMGB1 in the nucleus after SCI.
2.2 Expression of HMGB1 after SCI
After SCI, HMGB1 is expressed and released by neurons, microglia/macrophages, and ependymal cells (21–23). After injury, the expression of HMGB1 is rapidly upregulated and released into the extracellular matrix and circulating blood, which lasts for a long period of time. In a rat experiment, the expression of HMGB1 mRNA and HMGB1 protein was found to be upregulated 2-6 hours after SCI, the peak level was reached 1-3 days after injury, and this enhanced level lasted for 28 days after SCI (21, 23, 39, 40); however, the number of HMGB1 positive cells in the spinal cord of injured rats was the highest 48 hours after injury (41). In addition, Fan et al. found that the concentration of HMGB1 in serum increased significantly 3 days after SCI (42). In a model of SCI using neurons in vitro, we found that after injury, the concentration of HMGB1 in the culture medium immediately increased to 5 ng and 17 ng/ml at 6 and 12 hours, respectively, and reached 19 ng/ml HMGB1 at 24 hours (27). Interestingly, in addition to the increased expression of HMGB1 in the acute and subacute phases after SCI, the level of HMGB1 also increased significantly in the chronic phase. Papatheodorou et al. found that HMBG1 levels also increased significantly in patients 5 or more years after SCI (43). However, the findings of Fang et al. in zebrafish experiments suggested different expression patterns of HMGB1, and these results contradict the continuous increase in HMGB1 found by most studies. The level of HMGB1 mRNA increased twofold at 4 hours after SCI, decreased 12 hours and 11 days after injury, and increased again 21 days after injury (22).
Collectively, these results show that the expression of HMGB1 increases rapidly after SCI and lasts for a long time, even throughout the acute, subacute, and chronic stages, implying that HMGB1 may affect the severity of SCI and the process of recovery. In addition, the expression of HMGB1 was still upregulated in the chronic phase after injury, and its specific mechanism and effect still need to be explored. Moreover, the specific mechanism of the new model of HMGB1 expression remains to be further validated. Reguarding the role of HMGB1, we speculate that the first increase in HMGB1 expression is beneficial to protect injured neurons, then the decrease in HMGB1 expression is beneficial to reduce inflammation and further reduce neuronal death, and then the continuous increase in expression helps promote nerve regeneration and recovery of the injured spinal cord.
2.3 Translocation time of HMGB1 after SCI
After SCI, the expression of HMGB1 increases rapidly, translocates from the nucleus to the cytoplasm; furthermore, it may be released into the extracellular matrix through cytoplasmic vacuoles. Two hours after SCI, the nuclear level of HMGB1 increased significantly, and HMGB1 in the nucleus was gradually released into the cytoplasm. Six hours after injury, the cytoplasmic level of HMGB1 increased significantly. HMGB1 is located mainly in the nucleus of neurons in the early stages after SCI, and then it translocates to the cytoplasm a few hours later (21, 27, 44). Interestingly, in a zebrafish SCI model, Fang et al. found that HMGB1 was in the cytoplasm of motoneurons 4 hours after injury; however, 12 hours after injury, the cytoplasmic HMGB1 level in motoneurons decreased. On the 21st day of SCI, the level of HMGB1 in the cytoplasm decreased, but HMGB1 was again detected in the nucleus. HMGB1 exists in the motoneuron nucleus and it translocates to the cytoplasm after injury; furthermore, its expression is downregulated in motoneurons (22). The most significant change in HMGB1 translocation after SCI in mice was in the macrophages at the lesion centre and near the lesion boundary. HMGB1 could not even be detected in the nucleus, while it was still found in small cytoplasmic vacuoles, implying that HMGB1 may be packaged in the cell to be secreted into the extracellular space (23).
Taken together, these results indicate that HMGB1 increased in the nucleus and was released from the nucleus to the cytoplasm and extracellular matrix; however, the specific time and node of translocation have not been clarified.
2.4 Possible mechanism of HMGB1 release after SCI
The mechanism of secretion and release of HMGB1 in SCI has not been specifically explained, but it is generally thought that HMGB1 mainly comes from the secretion of inflammatory cells and the release of dead neurons (21–23, 45, 46).
In SCI, HMGB1 may carry out nuclear and cytoplasmic transport through acetylation, phosphorylation and methylation of the nuclear localization sites (NLS). In a study of T. thermophila CU 427 and CU 428, two NLSs were found to control the nuclear localization of HMGB1 in the steady state (47). Posttranslational modifications of the NLS site, including acetylation, phosphorylation and methylation, regulate the ability of HMGB1 to be transported to the cytoplasm during cellular stress (48–51). In a study of mouse fibroblasts, HeLa cells and Saos-2 cells, it was found that excessive acetylation of lysine at the NLS site was essential for the translocation of HMGB1 from the nucleus to the cytoplasm in monocytes stimulated by LPS, TNF or IL-1. Inhibiting the peracetylation of the NLS, can inhibit the translocation of HMGB1 from the nucleus to the cytoplasm and block the translocation of HMGB1 from the cytoplasm to the nucleus (2, 48, 52). In addition, the NLS site of HMGB1 was found to be phosphorylated and translocated to the cytoplasm of mouse macrophages stimulated by TNF (53). In neutrophils, the methylation of the lysine site of HMGB1 NLS was found to weaken the DNA-binding activity of HMGB1, resulting in the passive diffusion of HMGB1 out of the nucleus (49).
In SCI, the mechanism of extracellular secretion of HMGB1 is related to inflammation. Inflammation can induce many kinds of cells to secrete HMGB1 (54, 55); however, HMGB1 cannot be actively secreted through the conventional endoplasmic reticulum-Golgi secretion pathway utilized by most soluble secretory proteins (56, 57). At present, scholars have proposed two forms of active release of HMGB1 (58). One is stimulation and activation of the target cells, causing HMGB1 to be secreted into the outer space of the cells (48, 59). The second is packaging HMGB1 into intracellular vesicles, and then releasing HMGB1 outside the cell after the vesicles fuse with the cell membrane (60, 61). The latter is consistent with HMGB1 being in cytoplasmic vacuoles in macrophages after SCI (23).
HMGB1 translocation and release during aseptic inflammation can be regulated by calcium-mediated signal transduction (50, 62, 63). Phosphorylation and release of HMGB1 are regulated by activation of calcium-mediated protein kinases, especially calmodulin-dependent protein kinases (CaMKKs) (64–67). Calcium signal inhibitors inhibit HMGB1 secretion and protect animals in various disease models (50, 68). In addition, heat shock protein family A (Hsp70) member 1A (HSPA 1A, also known as HSP72) can block HMGB1 secretion in macrophages by inhibiting the interaction between HMGB1 and XPO1 (69). In different inflammation and injury models, peroxisome proliferator activated receptor (PPAR) binds to specific ligands and activates the transcription of PPAR target genes. The secretion of HMGB1 in activated macrophages is negatively regulated by PPAR (70). In contrast, JAK-regulated STAT1 and STAT3 activation plays an active role in the expression, modification and/or release of HMGB1 (71–75), while extracellular HMGB1 can trigger the activation of the STAT1 and STAT3 pathways (74–78). In addition, MAPK family members and inflammatory bodies promote the release of HMGB1 in different inflammatory and injury models (79–82). Deficiency of complement 5a receptor 2 (C5aR2) limits the activation of NLRP3 inflammatory bodies and the release of HMGB1 in vitro (83).
Expression of the inflammatory cytokines TNF and NF-κB were upregulated after SCI. TNF knockout or a TNF neutralization antibody directly inhibited TNF and partially inhibited HMGB1 release induced by IFN-γ and LPS in macrophages, indicating that the secretion of HMGB1 is partially mediated by a TNF-dependent mechanism (84). In addition, the NF-κB pathway is involved in the release of HMGB1, and the inhibition of the classical NF-κB pathway limits the secretion of HMGB1 in activated immune cells; however, the target gene of NF-κB that causes the secretion of HMGB1 is still unknown (85–87). Furthermore, the involvement of TNF (the classical NF-κB target gene) in the NF-κB dependent release of HMGB1 cannot be ruled out.
After SCI, HMGB1 can be passively released after various types of cell death in response to various stimuli or injuries. In addition, the release of many intracellular substances (cathepsin, antioxidant enzymes, DNase, caspases) after cell death can also promote the secretion of HMGB1 by inflammatory cells (81, 88–100).
In summary, although the secretion and release mechanism of HMGB1 in SCI has not been specifically described, the release mechanism of HMGB1 described in other fields if of great value. In SCI, HMGB1 may be actively released by inflammatory cells or passively released by necrotic neurons, and intracellular substances released by necrotic cells may further induce the active secretion of HMGB1 (Figure 1).
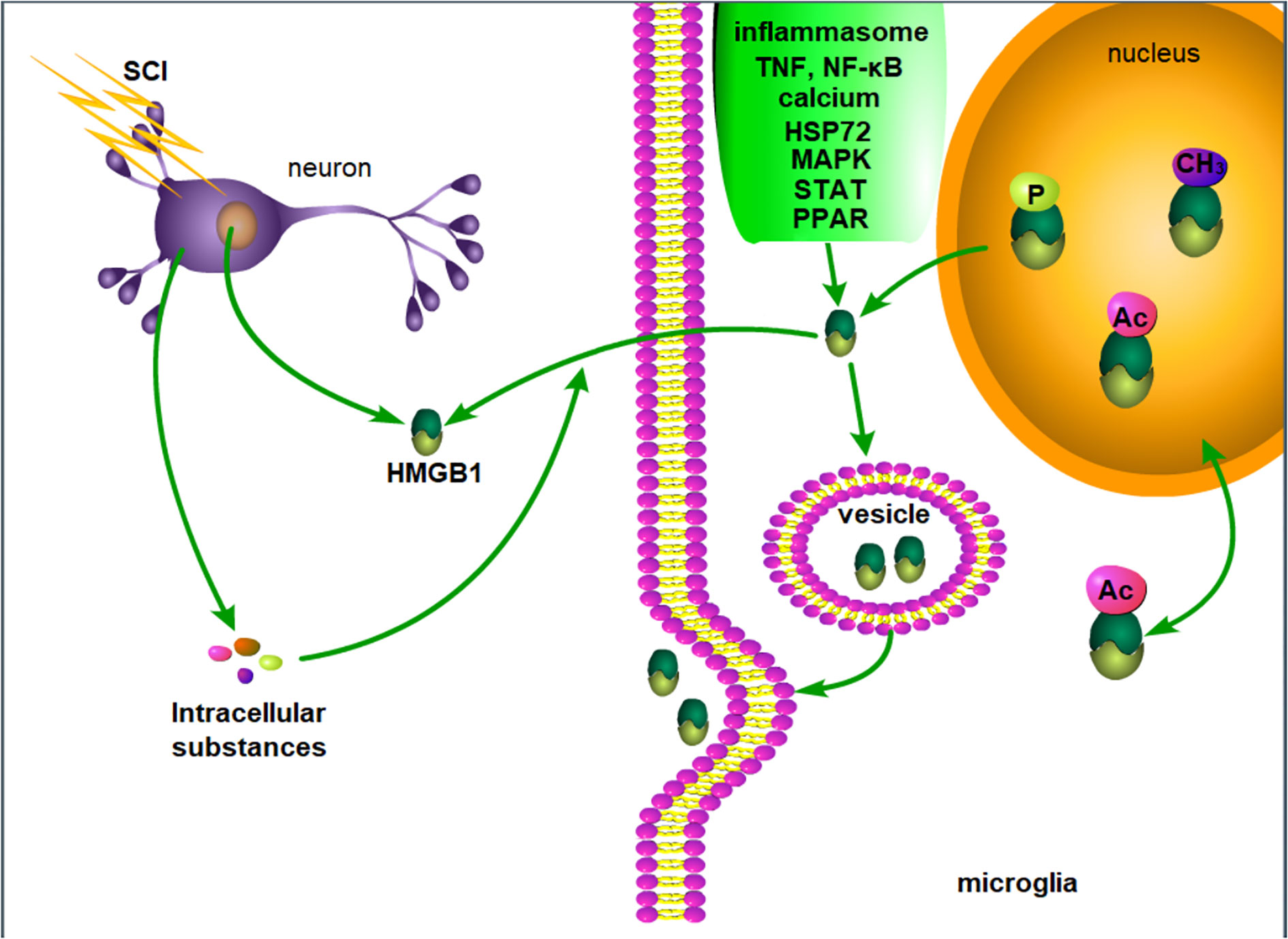
Figure 1 Possible mechanism of HMGB1 release after SCI. After SCI, injured neurons passively release HMGB1, and the intracellular substances released by necrotic neurons and inflammation lead to the active release of HMGB1 by microglia. P, phosphorylation. Ac, acetylation. CH3, methylation.
2.5 HMGB1 may regulate cell migration after SCI
Although the role of that HMGB1 plays in inducing cell migration in SCI has not been reported, in other diseases, HMGB1 has been found to induce cell migration by activating RAGE or activating the CXCR4 receptor through a heterologous complex with CXCL12. Many studies have shown that HMGB1, as a potential host cell-derived chemokine, promotes the migration of nerve processes and many cell types. These cells include smooth muscle cells, myoblasts, tumour cells, hepatic stellate cells, stem cells, endothelial cells, keratinocytes, monocytes, dendritic cells and neutrophils (101–117).
RAGE is necessary for HMGB1-mediated cell migration. HMGB1 triggers RAGE to induce the transcription of chemokine genes such as CCL3, CCL4 and CXCL12, while HMGB1-induced migration can quickly be blocked by anti-RAGE or anti-HMGB1 neutralizing antibodies, confirming the importance of RAGE in the process of migration (102, 118–121). Recent studies have shown that the triggering of RAGE by HMGB1 induces the transcription of chemokine genes such as CCL3, CCL4 and CXCL12, and subsequently it participates in cell migration (118, 119, 121). In addition, HMGB1 can induce cell migration by forming heterocomplexes with CXCL12 is mediated by CXCR4 receptors, and the HMGB1-CXCL12 complex is more effective in inducing monocyte migration than CXCL12 alone (109, 122–124). By activating CXCR4 receptors, HMGB1-CXCL12 complexes recruit leukocytes from the circulation, and then induce leukocytes to activate and secrete cytokines and chemokines, thus promoting inflammation (125, 126). Interestingly, HMGB1 not only stimulates but also inhibits migration in some cells. For example, exogenous HMGB1 selectively inhibits VEGF-induced cell migration in pulmonary artery endothelial cells (HPAECs), but does not inhibit human umbilical vein endothelial cells (HUVECs). In addition, the IRF3-dependent TLR4 pathway is necessary for HMGB1-mediated inhibition of migration in HPAECs (115).
Collectively, these data show that HMGB1 can promote the migration of a variety of cells, including inflammatory cells and stem cells. Although the chemotaxis of HMGB1 has not been studied in SCI, it remains possible that HMGB1 could induce inflammatory cell migration to aggravate the inflammatory response or promote injury recovery by inducing stem cell migration following SCI.
2.6 HMGB1 induces an inflammatory response after SCI
HMGB1 aggravates the inflammatory response after SCI. We found that after SCI, HMGB1 and its receptors were significantly colocalized in the white matter of the injured rat spinal cord (21). After HMGB1 injection, the focus of activated microglia was obviously in the ventral horn of the spinal cord (23), but the lesion area of anti-HMGB1 mAb-treated mice was significantly smaller than the lesion area of untreated mice (127–129). HMGB1 is thought to aggravate SCI. To further investigate its specific mechanism, Nakajo et al. found that anti-HMGB1 mAb treatment could protect the BSCB from damage caused by SCI, reduce the level of AQP4 protein in the spinal cord, and inhibit the swelling of the injured spinal cord. Anti-HMGB1 mAb inhibits inflammation after SCI in the early acute phase and it can prevent BSCB destruction directly or indirectly by inhibiting the expression of inflammatory cytokines and MMP in SCI model mice. Anti-HMGB1 mAb can relieve SCI, oedema and demyelination, thus promoting the recovery of the spinal cord (27, 128).
After SCI, HMGB1 induces inflammation mainly by activating microglia rather than astrocytes. Colocalization of microglial activation and obvious neuronal loss occurs in the spinal cord of rats with HMGB1 injection (23). HMGB1 has been reported to induce inflammation by activating microglia/macrophages (21, 44). In addition, further studies found that HMGB1 contributes to the development of the neurotoxic inflammatory macrophages (M1) phenotype after SCI (23). The mRNA levels of TNF-α, iNOS and CD86 increased significantly in microglia treated with HMGB1, which provided direct evidence for the activation of microglia to the M1 phenotype by HMGB1. HMGB1 or RAGE inhibition can inhibit the activation of macrophages/microglia to the M1 phenotype and promote the activation of macrophages/microglia to the M2 phenotype after SCI (27, 42, 130). Moreover, recombinant HMGB1 promoted the migration of BV2 microglia, while the anti-HMGB1 polyclonal antibody weakened the migration of BV2 microglia. The secretion of HMGB1 after SCI has been reported to recruit microglia to participate in the inflammatory response (27); however, HMGB1 failed to activate the classical inflammatory signalling pathway of primary astrocytes, indicating that astrocytes may not be induced by HMGB1.
After SCI, HMGB1 regulates the expression of inflammatory factors and the inflammatory response through RAGE and TLR2/4. HMGB1,TNF-α and RAGE are expressed in the same apoptotic neurons after SCI (41). There was a significant correlation between the levels of HMGB1 and NF-κB and the expression of TLR4 and NF-κB protein after SCI (131). HMGB1, TNF-α and RAGE are expressed in the same apoptotic neurons, and the temporal expression patterns of RAGE and HMGB-1 are similar (21, 41). The expression of RAGE, TNF-α, NF-κB IFN-γ, IL-1α, IL-6 and IL-17 increased in microglia treated with HMGB1, while inhibition of HMGB1 decreased the levels of RAGE, TNF-α, NF-κB IFN-γ, IL-1α, IL-6 and IL-17 (27, 42). HMGB1 induced inflammation by activating TLR2/4 or RAGE, activating its downstream pathway and upregulating the levels of TNF-α and NF-κB (28, 132–136). Interestingly, Wang et al. found that the administration of recombinant HMGB1 did not increase the levels of inflammatory cytokines TNF-α and IL-1β, while blocking RAGE reduced the induction of cytokines induced by LPS. It has been proposed that HMGB1/RAGE does not directly increase proinflammatory cytokines, and the effect of RAGE on cytokines may be related to pathogen-related interactions (137).
Collectively, these results shown that the expression of HMGB1 is upregulated after SCI, the inflammatory response is aggravated by regulating the activation of microglia to the M1 phenotype, and the expression of inflammatory factors is regulated by RAGE and TLR2/4. However, the studies of Wang et al. indicate different possibilities, and the specific mechanism remains to be further validated. In addition, these studies investigated the role of HMGB1 in only acute and subacute SCI. However, as the expression of HMGB1 is still upregulated in the chronic phase of SCI, its specific role needs to be further explored. Finally, extracellular HMGB1 can aggravate the inflammatory response after SCI, but the specific changes in nuclear HMGB1 after SCI are not clear, and its specific role needs to be understood.
2.7 Effect of HMGB1 on injured neurons after SCI
After SCI, HMGB1 promotes neuronal death by promoting nerve inflammation. After HMGB1 injection, focus of activated microglia and the area of neuronal loss were determined. Coculture of macrophages stimulated by HMGB1 with neurons showed a decrease in axonal growth (23). Inhibition of the HMGB1/TLR4/NF-κB signalling pathway can inhibit neuroinflammation and apoptosis in SCI, reduce the damage, oedema and demyelination, improve the survival rate of host neurons, and promote the recovery of the spinal cord (24, 27, 28, 127, 128, 138–140) Interestingly, Song et al. found that transplantation of HMGB1-preconditioned neural stem cells can promote neuronal survival after SCI in rats, promote the connection between relay neurons and motor neurons; furthermore, the newly formed neural circuits greatly improved motor recovery (25, 30). These findings are consistent with the findings of Wang et al. in cerebral ischemia-reperfusion injury. HMGB1 preconditioning can significantly reduce neurological deficits, infarct size, brain swelling, apoptosis and blood-brain barrier permeability in rats with cerebral ischemia-reperfusion injury (141). In addition, RAGE blockade was shown to not be conducive to neuronal survival after SCI (137), and consistent with the findings of Huttunen et al. HMGB1 can promote neuronal survival and axonal growth by activating RAGE and increasing the expression of the anti-apoptotic protein Bcl2 (142–145). HMGB1 is thought to be beneficial for the survival of injured spinal cord neurons.
In summary, numerous studies have shown that HMGB1 can exacerbate neuronal injury by aggravating neuroinflammation. Some studies have also suggested that HMGB1 is conducive to the survival and recovery of injured neurons. However, the specific mechanism by which HMGB1 protects injured neurons and promotes injury recovery is not clear, and this does not rule out the possibility that HMGB1 indirectly injures neurons by promoting the inflammatory response and protects neurons through direct action.
2.8 HMGB1 induces neuronal regeneration after SCI
After SCI, HMGB1 promotes the growth of neuronal axons and induces the differentiation of neural stem cells. Fang et al. found that HMGB1 increased significantly and the number of perivascular motoneurons increased significantly 6th days after SCI. After inhibition of HMGB1, the number of detected axons decreased significantly. HMGB1 is thought to participate in axonal regeneration and promote motor recovery (22). Further in vitro experiments showed that HMGB1, especially when close to the neuronal cell body, significantly increased neuronal axonal growth (23). In addition, HMGB1 can also promote neuronal differentiation. HMGB1 can induce the differentiation of neural stem cells through the ERK signalling pathway and the RAGE signalling pathway, while antagonism of these pathways can reduce the expression of marker molecules in mature neurons (25, 137). These results indicate that HMGB1 can induce the differentiation and maturation of neural stem cells through the ERK signalling pathway and the RAGE signalling pathway. Interestingly, Palumbo et al. found that HMGB1 can induce stem cells to migrate across the endothelial barrier in vitro and in vivo (104). Thus, the role of recruiting stem cells in the recovery of SCI remains to be explored.
Collectively, these results show that HMGB1 can promote the growth of neuronal axons and induce the differentiation of neural stem cells after SCI; although, the specific mechanism needs to be further explored. In addition, in the chronic stage of SCI, the upregulation of HMGB1 may be involved in regulating neuronal regeneration and promoting motor recovery.
2.9 HMGB1: A potential target for clinical treatment after SCI
After SCI, HMGB1 aggravates the damage by inducing inflammation, and after treatment with hyperbaric oxygen, shikorin, glycyrrhizin, Higenamine, ethyl pyruvate, glycyrrhizin, Catalpol, Dihydrotanshinone I, mir-34a, anti-HMGB1 mAb, etc., HMGB1 can reduce inflammation and reduce spinal cord oedema, protect spinal cord neurons and promote functional recovery after SCI by downregulating the expression of HMGB1 and NF-κB (24, 26, 30, 42, 127–129, 131, 146–153). Among these studies, Higenamine induced an increase in M2 macrophages and saw an enhanced the anti-inflammatory effect (147). miR-34a, miR-129-5p, Catalpol and shikonin-induced HMGB1 downregulate the NF-κB signalling pathway involved in the decreased expression of TLR4 (24, 131, 148, 149, 151). Dihydrotanshinone I was shown to protects against SCI in vivo through the HMGB1/TLR4/NOX4 pathway. Ethyl pyruvate or glycyrrhizin we shown to reduce spinal cord oedema by reducing the expression of AQP4 in the spinal cord of rats with SCI (129). Furthermore, glycyrrhizin reduced the area of the cystic cavity and glial scar formation (153). However, as mentioned earlier, HMGB1 does not only induce inflammation to aggravate SCI, but it also promotes neuronal regeneration. Transplantation of neural stem cells pre-treated with HMGB1 promotes neuronal survival after SCI in rats. The motor recovery of rats in the HMGB1 combination with neural stem cell group was better than the motor recovery of rats in the single transplantation group and in HMGB1 group (25) (Table 1).
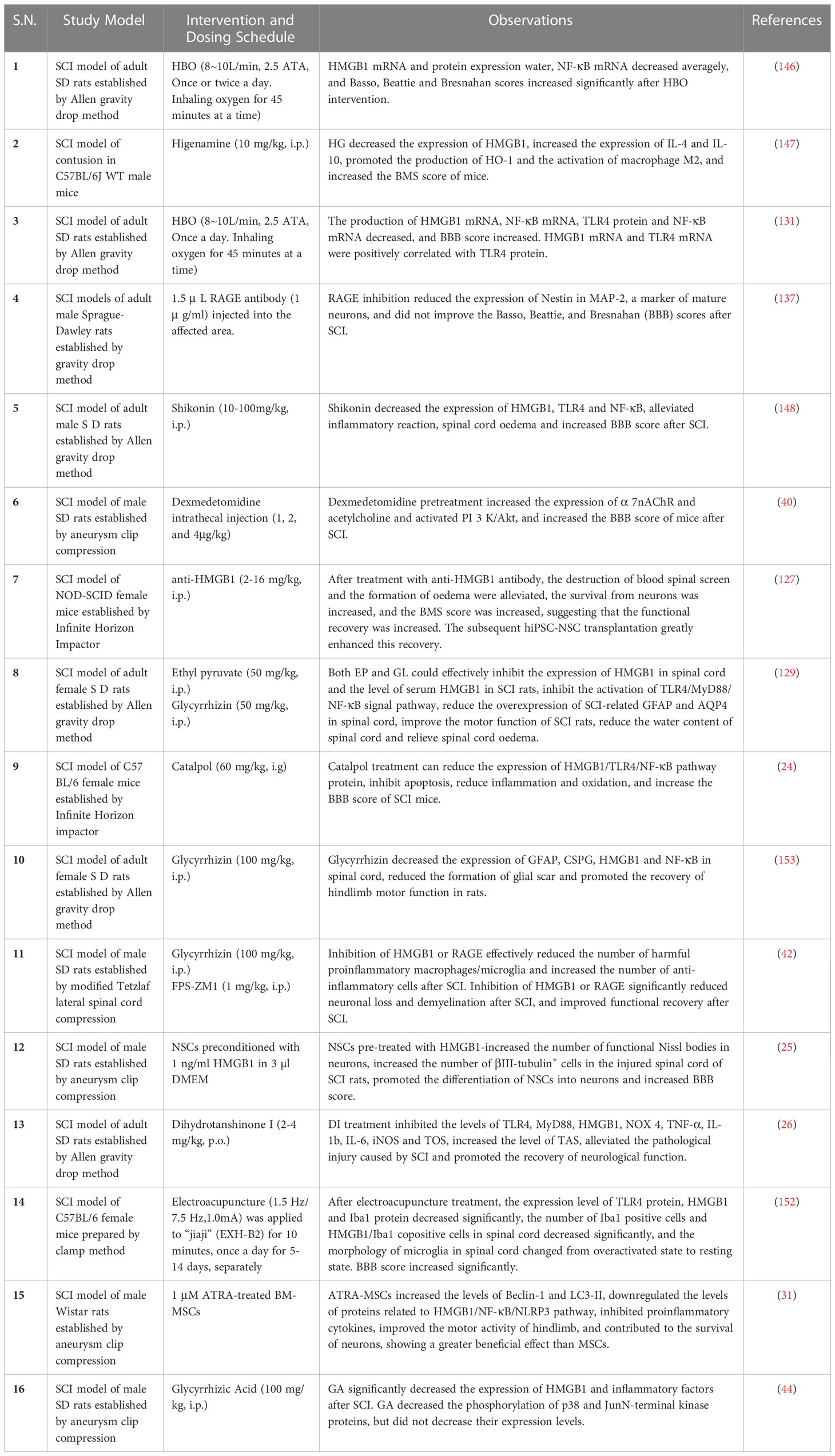
Table 1 Summary of targeted HMGB1 treatment of SCI.
In summary, HMGB1 plays a multifunctional role in SCI. Inhibition of HMGB1 inhibits nerve inflammation and protects the spinal cord; furthermore, HMGB1 can be used to protect neurons, promote neuronal regeneration and induce neuronal differentiation to promote the recovery of SCI. This information will provide new theoretical support and therapeutic targets for SCI; however, at present, research on treating SCI by targeting HMGB1 is not sufficient, and its specific application scheme still needs to be further explored.
3 Discussion
The HMGB1 protein is a highly conserved nonhistone binding nuclear protein. Structurally, HMGB1 is divided into the following three functional regions: the A-box, the B-box and the acidic C-terminus, with similar amino acid repeat sequences and nonspecific DNA-binding sites; the B-box is the functional structural region that causes inflammation, the A-box has a certain antagonistic effect on the B-box, and the C-terminal is involved mainly in regulating the binding affinity of HMGB1 and DNA, mediating gene transcription and chromosome unwinding. In the resting state, HMGB1 is located primarily in the nucleus and it regulates key nuclear activities, including transcription, replication, DNA repair and nucleosome formation, all of which are important for maintaining steady-state cellular function; however, there are no related studies on the structural and functional changes in HMGB1 after SCI.
After SCI, HMGB1 is expressed and released by neurons, microglia/macrophages and ependymal cells. After injury, the expression of HMGB1 is rapidly upregulated and released into the extracellular matrix and circulating blood, where it remains for a long time. Interestingly, in addition to the increased expression of HMGB1 in the acute and subacute phases after SCI, the level of HMGB1 also increased significantly in the chronic phases. The expression of HMGB1 increases after SCI, and it runs through the acute, subacute and chronic stages of SCI, indicating that HMGB1 may affect the severity and recovery process of SCI. In addition, the expression of HMGB1 is still upregulated in the chronic phase after injury, and its specific mechanism and effect still need to be explored. HMGB1 increased in the nucleus and it was then released from the nucleus to the cytoplasm and extracellular matrix; however, the specific time and node of translocation have not been clarified.
In SCI, the secretion and release mechanism of HMGB1 has not been specifically described, but it is generally thought that most HMGB1 comes from the secretion of inflammatory cells and the release of dead neurons. Inflammation can induce many kinds of cells to secrete HMGB1; however, HMGB1 cannot be actively secreted through the conventional endoplasmic reticulum-Golgi secretion pathway utilized by most soluble secretory proteins. Scholars have proposed two mechanisms for secretion. Among these, one mechanism hypothesizes that HMGB1 is packaged into intracellular vesicles, and then HMGB1 is released out of cells after the fusion of vesicles and the cytoplasmic membrane, consistent with the cytoplasmic vacuoles of HMGB1 observed in macrophages after SCI.
After SCI, HMGB1 may promote the active release of inflammatory cells through calcium-mediated signal transduction in inflammation, HSPA1A inhibition of the interaction between HMGB1 and XPO1, MAPK family members, inflammatory bodies, activation of STAT1 and STAT3 regulated by JAK, and upregulated expression of TNF and NF-κB. HMGB1 may also be passively released by necrotic neurons, and the intracellular substances released by necrotic cells can also induce further active secretion of HMGB1.
HMGB1 that is secreted and released into the extracellular space can induce the migration of inflammatory cells, aggravate the inflammatory response by regulating the activation of microglia to the M1 phenotype, and regulate the expression of inflammatory factors through RAGE and TLR2/4. However, the studies of Wang et al. show different results, and the specific mechanism needs to be further explored. In addition, the above studies address the role of HMGB1 only in acute and subacute SCI; however, the expression of HMGB1 is still upregulated in the chronic stage of SCI. Its specific role needs to be further explored, and the possibility that HMGB1 promotes the recovery of SCI cannot be ruled out. Moreover, extracellular HMGB1 can aggravate the inflammatory response after SCI, but the specific changes in nuclear HMGB1 after SCI are not clear, and its specific role remains to be understood.
HMGB1 can aggravate neuronal inflammation and lead to neuronal death, and it is also beneficial to neuronal survival and recovery. However, the specific mechanism by which HMGB1 protects injured neurons and promotes injury recovery is not clear, and possibility that HMGB1 injures neurons indirectly by promoting the inflammatory response, while protecting neurons through direct action has not been excluded. In addition, further studies have found that HMGB1 can promote the growth of neuronal axons and induce the migration and differentiation of neural stem cells. The specific mechanism needs to be further explored. In the chronic stage of SCI, the upregulation of HMGB1 may be involved in regulating neuronal regeneration and promoting motor recovery.
HMGB1 plays a multifunctional role in SCI. Inhibiting HMGB1 inhibits nerve inflammation and protects the spinal cord; furthermore, it can be used to protect neurons, promote neuronal regeneration and induce neuronal differentiation to promote the recovery of SCI. These findings provide a new theoretical basis for studying SCI and new therapeutic targets for SCI treatment. However, at present, research into the treatment of SCI based on targeting HMGB1 is not sufficient, and its specific application scheme still needs to be further defined.
For future research, we suggest focusing on the different roles of HMGB1 in the nucleus and in the extracellular space after SCI, clarifying the specific secretion and release mechanism, and determining the spatiotemporal relationship of HMGB1 expression, the protective effect on neurons, and the mechanism of promoting differentiation.
In summary, HMGB1 plays a complex role in SCI, and its mechanism remains to be understood. Additional research will provide a new theoretical basis and therapeutic target for the treatment of SCI.
Author contributions
Conceptualization, data curation, writing-original draft preparation, writing-review and editing: all authors; supervision: KC. Funding acquisition: KC. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (82072513 to KC), Science and Technology Programme of Guangzhou (202102080182 to KC).
Acknowledgments
We wish to thank the National Natural Science Foundation of China, the Science and Technology Programme of Guangzhou, for their generous support. We also wish to thank Guojun Qi for his guidance on this article and the relevant authors in the citations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Einck L, Bustin M. The intracellular distribution and function of the high mobility group chromosomal proteins. Exp Cell Res (1985) 156(2):295–310. doi: 10.1016/0014-4827(85)90539-7
2. Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science (2001) 293(5535):1653–7. doi: 10.1126/science.1062374
3. Ferrari S, Ronfani L, Calogero S, Bianchi ME. The mouse gene coding for high mobility group 1 protein (HMG1). J Biol Chem (1994) 269(46):28803–8. doi: 10.1016/S0021-9258(19)61977-0
4. Gariboldi M, De Gregorio L, Ferrari S, Manenti G, Pierotti MA, Bianchi ME, et al. Mapping of the Hmg1 gene and of seven related sequences in the mouse. Mamm Genome (1995) 6(9):581–5. doi: 10.1007/BF00352361
5. Wen L, Huang JK, Johnson BH, Reeck GR. A human placental cDNA clone that encodes nonhistone chromosomal protein HMG-1. Nucleic Acids Res (1989) 17(3):1197–214. doi: 10.1093/nar/17.3.1197
6. Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, et al. HMGB1 in health and disease. Mol Aspects Med (2014) 40:1–116. doi: 10.1016/j.mam.2014.05.001
7. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol (2005) 5(4):331–42. doi: 10.1038/nri1594
8. Andersson U, Yang H, Harris H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol (2018) 38:40–8. doi: 10.1016/j.smim.2018.02.011
9. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol (2010) 28:367–88. doi: 10.1146/annurev.immunol.021908.132603
10. Aucott H, Lundberg J, Salo H, Klevenvall L, Damberg P, Ottosson L, et al. Neuroinflammation in response to intracerebral injections of different HMGB1 redox isoforms. J Innate Immun (2018) 10(3):215–27. doi: 10.1159/000487056
11. Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal (2011) 14(7):1315–35. doi: 10.1089/ars.2010.3356
12. Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: endogenous danger signaling. Mol Med (2008) 14(7-8):476–84. doi: 10.2119/2008-00034.Klune
13. Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol (2015) 6:93. doi: 10.3389/fphys.2015.00093
14. Okamoto K, Tamura T, Sawatsubashi Y. Sepsis and disseminated intravascular coagulation. J Intensive Care (2016) 4:23. doi: 10.1186/s40560-016-0149-0
15. Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res (2016) 111:534–44. doi: 10.1016/j.phrs.2016.06.031
16. Quadri SA, Farooqui M, Ikram A, Zafar A, Khan MA, Suriya SS, et al. Recent update on basic mechanisms of spinal cord injury. Neurosurgical Rev (2018) 43(2):425–41. doi: 10.1007/s10143-018-1008-3
17. Ge MH, Tian H, Mao L, Li DY, Lin JQ, Hu HS, et al. Zinc attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury by activating Nrf2/GPX4 defense pathway. CNS Neurosci Ther (2021) 27(9):1023–40. doi: 10.1111/cns.13657
18. Han B, Liang W, Hai Y, Liu Y, Chen Y, Ding H, et al. Elucidating the potential mechanisms underlying distraction spinal cord injury-associated neuroinflammation and apoptosis. Front Cell Dev Biol (2022) 10:839313. doi: 10.3389/fcell.2022.839313
19. Parker D. The functional properties of synapses made by regenerated axons across spinal cord lesion sites in lamprey. Neural Regener Res (2022) 17(10):2272–7. doi: 10.4103/1673-5374.335828
20. Zhou G, Wang Z, Han S, Chen X, Li Z, Hu X, et al. Multifaceted roles of cAMP signaling in the repair process of spinal cord injury and related combination treatments. Front Mol Neurosci (2022) 15:808510. doi: 10.3389/fnmol.2022.808510
21. Chen KB, Uchida K, Nakajima H, Yayama T, Hirai T, Rodriguez Guerrero A, et al. High-mobility group box-1 and its receptors contribute to proinflammatory response in the acute phase of spinal cord injury in rats. Spine (Phila Pa 1976) (2011) 36(25):2122–9. doi: 10.1097/BRS.0b013e318203941c
22. Fang P, Pan HC, Lin SL, Zhang WQ, Rauvala H, Schachner M, et al. HMGB1 contributes to regeneration after spinal cord injury in adult zebrafish. Mol Neurobiol (2014) 49(1):472–83. doi: 10.1007/s12035-013-8533-4
23. Kigerl KA, Kigerl KA, Lai W, Wallace LM, Yang H, Popovich PG.. High mobility group box-1 (HMGB1) is increased in injured mouse spinal cord and can elicit neurotoxic inflammation. Brain Behav Immun (2018) 72:22–33. doi: 10.1016/j.bbi.2017.11.018
24. Xia H, Wang D, Guo X, Wu K, Huang F, Feng Y. Catalpol protects against spinal cord injury in mice through regulating MicroRNA-142-Mediated HMGB1/TLR4/NF-kappaB signaling pathway. Front Pharmacol (2020) 11:630222. doi: 10.3389/fphar.2020.630222
25. Xue X, Zhang L, Yin X, Chen XX, Chen ZF, Wang CX, et al. Transplantation of neural stem cells preconditioned with highmobility group box 1 facilitates functional recovery after spinal cord injury in rats. Mol Med Rep (2020) 22(6):4725–33. doi: 10.3892/mmr.2020.11565
26. Yu L, Qian J. Dihydrotanshinone I alleviates spinal cord injury via suppressing inflammatory response, oxidative stress and apoptosis in rats. Med Sci Monit (2020) 26:e920738. doi: 10.12659/MSM.920738
27. Chen KB, Chang MM, Wang SL, Li YX, Wang YX, Xu ZG, et al. High mobility group box-1 serves a pathogenic role in spinal cord injury via the promotion of pro-inflammatory cytokines. J Leukoc Biol (2021) 110(6):1131–42. doi: 10.1002/JLB.3MA0721-007R
28. Niu F, Pan S. MicroRNA-488 inhibits neural inflammation and apoptosis in spinal cord injury through restraint on the HMGB1/TLR4/NF-kappaB signaling pathway. Neuroreport (2021) 32(12):1017–26. doi: 10.1097/WNR.0000000000001680
29. Song HH, Song TC, Yang T, Sun CS, He BQ, Li H, et al. High mobility group box 1 mediates inflammatory response of astrocytes via cyclooxygenase 2/prostaglandin E2 signaling following spinal cord injury. Neural Regener Res (2021) 16(9):1848–55. doi: 10.4103/1673-5374.303039
30. Zhu Y, Uezono N, Yasui T, Nakajo M, Nagai T, Wang D, et al. Combinatrial treatment of anti-high mobility group box-1 monoclonal antibody and epothilone b improves functional recovery after spinal cord contusion injury. Neurosci Res (2021) 172:13–25. doi: 10.1016/j.neures.2021.04.002
31. Gholaminejhad M, Jameie SB, Abdi M, Abolhassani F, Mohammed I, Hassanzadeh G. All-trans retinoic acid-preconditioned mesenchymal stem cells improve motor function and alleviate tissue damage after spinal cord injury by inhibition of HMGB1/NF-kappaB/NLRP3 pathway through autophagy activation. J Mol Neurosci (2022) 72(5):947–62. doi: 10.1007/s12031-022-01977-0
32. Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta (2010) 1799(1-2):101–13. doi: 10.1016/j.bbagrm.2009.09.008
33. Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med (2003) 9(1-2):37–45. doi: 10.1007/BF03402105
34. Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, et al. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res (2007) 13(10):2836–48. doi: 10.1158/1078-0432.CCR-06-1953
35. Ueda T, Chou H, Kawase T, Shirakawa H, Yoshida M. Acidic c-tail of HMGB1 is required for its target binding to nucleosome linker DNA and transcription stimulation. Biochemistry (2004) 43(30):9901–8. doi: 10.1021/bi035975l
36. Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, Rauvala H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res (2002) 62(16):4805–11.
37. Diener KR, Al-Dasooqi N, Lousberg EL, Hayball JD. The multifunctional alarmin HMGB1 with roles in the pathophysiology of sepsis and cancer. Immunol Cell Biol (2013) 91(7):443–50. doi: 10.1038/icb.2013.25
38. Xue J, Suarez JS, Minaai M, Li S, Gaudino G, Pass HI, et al. HMGB1 as a therapeutic target in disease. J Cell Physiol (2021) 236(5):3406–19. doi: 10.1002/jcp.30125
39. Didangelos A, Puglia M, Iberl M, Sanchez-Bellot C, Roschitzki B, Bradbury EJ. High-throughput proteomics reveal alarmins as amplifiers of tissue pathology and inflammation after spinal cord injury. Sci Rep (2016) 6:21607. doi: 10.1038/srep21607
40. Rong H, Zhao Z, Feng J, Lei Y, Wu H, Sun R, et al. The effects of dexmedetomidine pretreatment on the pro- and anti-inflammation systems after spinal cord injury in rats. Brain Behav Immun (2017) 64:195–207. doi: 10.1016/j.bbi.2017.03.006
41. Kawabata H, Setoguchi T, Yone K, Souda M, Yoshida H, et al. High mobility group box 1 is upregulated after spinal cord injury and is associated with neuronal cell apoptosis. Spine (Phila Pa 1976) (2010) 35(11):1109–15. doi: 10.1097/BRS.0b013e3181bd14b6
42. Fan H, Tang HB, Chen Z, Wang HQ, Zhang L, Jiang Y, et al. Inhibiting HMGB1-RAGE axis prevents pro-inflammatory macrophages/microglia polarization and affords neuroprotection after spinal cord injury. J Neuroinflamm (2020) 17(1):295. doi: 10.1186/s12974-020-01973-4
43. Papatheodorou A, Stein A, Bank M, Sison CP, Gibbs K, Davies P, et al. High-mobility group box 1 (HMGB1) is elevated systemically in persons with acute or chronic traumatic spinal cord injury. J Neurotrauma (2017) 34(3):746–54. doi: 10.1089/neu.2016.4596
44. Wu Z, Zhang Z, Wang Z, Zhu H, Li M. MiR-181a-5p alleviates the inflammatory response of PC12 cells by inhibiting high-mobility group box-1 protein expression. World Neurosurg (2022) 162:e427–35. doi: 10.1016/j.wneu.2022.03.025
45. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418(6894):191–5. doi: 10.1038/nature00858
46. Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep (2004) 5(8):825–30. doi: 10.1038/sj.embor.7400205
47. Schulman IG, et al. Macronuclei and micronuclei in tetrahymena thermophila contain high-mobility-group-like chromosomal proteins containing a highly conserved eleven-amino-acid putative DNA-binding sequence. Mol Cell Biol (1991) 11(1):166–74. doi: 10.1128/mcb.11.1.166-174.1991
48. Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J (2003) 22(20):5551–60. doi: 10.1093/emboj/cdg516
49. Ito I, Fukazawa J, Yoshida M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem (2007) 282(22):16336–44. doi: 10.1074/jbc.M608467200
50. Zhang X, Wheeler D, Tang Y, Guo L, Shapiro RA, Ribar TJ, et al. Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J Immunol (2008) 181(7):5015–23. doi: 10.4049/jimmunol.181.7.5015
51. Tang Y, Zhao X, Antoine D, Xiao X, Wang H, Andersson U, et al. Regulation of posttranslational modifications of HMGB1 during immune responses. Antioxid Redox Signal (2016) 24(12):620–34. doi: 10.1089/ars.2015.6409
52. Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 c-terminal domain. Cell (1997) 90(4):595–606. doi: 10.1016/S0092-8674(00)80521-8
53. Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol (2006) 177(11):7889–97. doi: 10.4049/jimmunol.177.11.7889
54. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285(5425):248–51. doi: 10.1126/science.285.5425.248
55. Cai X, Biswas I, Panicker SR, Giri H, Rezaie AR. Activated protein c inhibits lipopolysaccharide-mediated acetylation and secretion of high-mobility group box 1 in endothelial cells. J Thromb Haemost (2019) 17(5):803–17. doi: 10.1111/jth.14425
56. Kwak MS, Kim HS, Lee B, Kim YH, Son M, Shin JS. Immunological significance of HMGB1 post-translational modification and redox biology. Front Immunol (2020) 11:1189. doi: 10.3389/fimmu.2020.01189
57. Palade G. Intracellular aspects of the process of protein synthesis. Science (1975) 189(4200):347–58. doi: 10.1126/science.1096303
58. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin d. Nat Commun (2020) 11(1):4561. doi: 10.1038/s41467-020-18443-3
59. Lee WJ, Song SY, Roh H, Ahn HM, Na Y, Kim J, et al. Profibrogenic effect of high-mobility group box protein-1 in human dermal fibroblasts and its excess in keloid tissues. Sci Rep (2018) 8(1):8434. doi: 10.1038/s41598-018-26501-6
60. Lu B, Wang C, Wang M, Li W, Chen F, Tracey KJ, et al. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: an updated review. Expert Rev Clin Immunol (2014) 10(6):713–27. doi: 10.1586/1744666X.2014.909730
61. Pisetsky DS. The expression of HMGB1 on microparticles released during cell activation and cell death in vitro and in vivo. Mol Med (2014) 20:158–63. doi: 10.2119/molmed.2014.00014
62. Peng HH, Liu YJ, Ojcius DM, Lee CM, Chen RH, Huang PR, et al. Mineral particles stimulate innate immunity through neutrophil extracellular traps containing HMGB1. Sci Rep (2017) 7(1):16628. doi: 10.1038/s41598-017-16778-4
63. Tian T, Yao D, Zheng L, Zhou Z, Duan Y, Liu B, et al. Sphingosine kinase 1 regulates HMGB1 translocation by directly interacting with calcium/calmodulin protein kinase II-delta in sepsis-associated liver injury. Cell Death Dis (2020) 11(12):1037. doi: 10.1038/s41419-020-03255-6
64. Zhao P, Ye T, Yan X, Hu X, Liu P, Wang X. HMGB1 release by H2O2-induced hepatocytes is regulated through calcium overload and 58-f interference. Cell Death Discovery (2017) 3:17008. doi: 10.1038/cddiscovery.2017.8
65. Chen S, Dong Z, Yang P, Wang X, Jin G, Yu H, et al. Hepatitis b virus X protein stimulates high mobility group box 1 secretion and enhances hepatocellular carcinoma metastasis. Cancer Lett (2017) 394:22–32. doi: 10.1016/j.canlet.2017.02.011
66. Quan H, Bae HB, Hur YH, Lee KH, Lee CH, Jang EA, et al. Stearoyl lysophosphatidylcholine inhibits LPS-induced extracellular release of HMGB1 through the G2A/calcium/CaMKKbeta/AMPK pathway. Eur J Pharmacol (2019) 852:125–33. doi: 10.1016/j.ejphar.2019.02.038
67. Li W, Deng M, Loughran PA, Yang M, Lin M, Yang C, et al. LPS induces active HMGB1 release from hepatocytes into exosomes through the coordinated activities of TLR4 and caspase-11/GSDMD signaling. Front Immunol (2020) 11:229. doi: 10.3389/fimmu.2020.00229
68. Shin JH, Kim ID, Kim SW, Lee HK, Jin Y, Park JH, et al. Ethyl pyruvate inhibits HMGB1 phosphorylation and release by chelating calcium. Mol Med (2015) 20:649–57. doi: 10.2119/molmed.2014.00039
69. Tang D, Kang R, Xiao W, Wang H, Calderwood SK, Xiao X.. The anti-inflammatory effects of heat shock protein 72 involve inhibition of high-mobility-group box 1 release and proinflammatory function in macrophages. J Immunol (2007) 179(2):1236–44. doi: 10.4049/jimmunol.179.2.1236
70. Hernandez-Quiles M, Broekema MF, Kalkhoven E. PPARgamma in metabolism, immunity, and cancer: Unified and diverse mechanisms of action. Front Endocrinol (Lausanne) (2021) 12:624112. doi: 10.3389/fendo.2021.624112
71. Zhou S, Lu H, Chen R, Tian Y, Jiang Y, Zhang S, et al. Angiotensin II enhances the acetylation and release of HMGB1 in RAW264.7 macrophage. Cell Biol Int (2018) 42(9):1160–9. doi: 10.1002/cbin.10984
72. Hao J, Zhang YJ, Lv X, Xu N, Liu QJ, Zhao S, et al. IFN-gamma induces lipogenesis in mouse mesangial cells via the JAK2/STAT1 pathway. Am J Physiol Cell Physiol (2013) 304(8):C760–7. doi: 10.1152/ajpcell.00352.2012
73. Park EJ, Kim YM, Kim HJ, Chang KC. Degradation of histone deacetylase 4 via the TLR4/JAK/STAT1 signaling pathway promotes the acetylation of high mobility group box 1 (HMGB1) in lipopolysaccharide-activated macrophages. FEBS Open Bio (2018) 8(7):1119–26. doi: 10.1002/2211-5463.12456
74. Imbaby S, Matsuda N, Tomita K, Hattori K, Palikhe S, Yokoo H, et al. Beneficial effect of STAT3 decoy oligodeoxynucleotide transfection on organ injury and mortality in mice with cecal ligation and puncture-induced sepsis. Sci Rep (2020) 10(1):15316. doi: 10.1038/s41598-020-72136-x
75. Wu Y, Xu J, Xu J, Zheng W, Chen Q, Jiao D. Study on the mechanism of JAK2/STAT3 signaling pathway-mediated inflammatory reaction after cerebral ischemia. Mol Med Rep (2018) 17(4):5007–12. doi: 10.3892/mmr.2018.8477
76. Wang G, Zhang J, Dui D, Ren H, Liu J. High mobility group box 1 induces the activation of the janus kinase 2 and signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway in pancreatic acinar cells in rats, while AG490 and rapamycin inhibit their activation. Bosn J Basic Med Sci (2016) 16(4):307–12. doi: 10.17305/bjbms.2016.1442
77. Guo HF, Liu SX, Zhang YJ, Liu QJ, Hao J, Gao LX. High mobility group box 1 induces synoviocyte proliferation in rheumatoid arthritis by activating the signal transducer and activator transcription signal pathway. Clin Exp Med (2011) 11(2):65–74. doi: 10.1007/s10238-010-0116-3
78. Conti L, Lanzardo S, Arigoni M, Antonazzo R, Radaelli E, Cantarella D. The noninflammatory role of high mobility group box 1/Toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J (2013) 27(12):4731–44. doi: 10.1096/fj.13-230201
79. Ma Y, Zhang Z, Chen R, Shi R, Zeng P, Chen R. NRP1 regulates HMGB1 in vascular endothelial cells under high homocysteine condition. Am J Physiol Heart Circ Physiol (2019) 316(5):H1039–h1046. doi: 10.1152/ajpheart.00746.2018
80. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PloS One (2009) 4(10):e7446. doi: 10.1371/journal.pone.0007446
81. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol (2010) 185(7):4385–92. doi: 10.4049/jimmunol.1000803
82. Barlan AU, Griffin TM, McGuire KA, Wiethoff CM. Adenovirus membrane penetration activates the NLRP3 inflammasome. J Virol (2011) 85(1):146–55. doi: 10.1128/JVI.01265-10
83. Yu S, Wang D, Huang L, Zhang Y, Luo R, Adah D, et al. The complement receptor C5aR2 promotes protein kinase r expression and contributes to NLRP3 inflammasome activation and HMGB1 release from macrophages. J Biol Chem (2019) 294(21):8384–94. doi: 10.1074/jbc.RA118.006508
84. Chen G, Li J, Ochani M, Rendon-Mitchell B, Qiang X, Susarla S, et al. Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14- and TNF-dependent mechanisms. J Leukoc Biol (2004) 76(5):994–1001. doi: 10.1189/jlb.0404242
85. Lin F, Pei L, Zhang Q, Han W, Jiang S, Lin Y, et al. Ox-LDL induces endothelial cell apoptosis and macrophage migration by regulating caveolin-1 phosphorylation. J Cell Physiol (2018) 233(10):6683–92. doi: 10.1002/jcp.26468
86. Hiramoto S, Tsubota M, Yamaguchi K, Okazaki K, Sakaegi A, Toriyama Y, et al. Cystitis-related bladder pain involves ATP-dependent HMGB1 release from macrophages and its downstream H2S/Cav3.2 signaling in mice. Cells (2020) 9(8):1748. doi: 10.3390/cells9081748
87. Wang CM, Jiang M, Wang HJ. Effect of NFkappaB inhibitor on highmobility group protein B1 expression in a COPD rat model. Mol Med Rep (2013) 7(2):499–502. doi: 10.3892/mmr.2012.1181
88. Castro RC, Gonçales RA, Zambuzi FA, Frantz FG. Notch signaling pathway in infectious diseases: role in the regulation of immune response. Inflammation Res (2021) 70(3):261–74. doi: 10.1007/s00011-021-01442-5
89. Khambu B, Huda N, Chen X, Antoine DJ, Li Y, et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J Clin Invest (2018) 128(6):2419–35. doi: 10.1172/JCI91814
90. Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med (2006) 203(7):1637–42. doi: 10.1084/jem.20052203
91. Morinaga Y, Yanagihara K, Nakamura S, Hasegawa H, Seki M, Izumikawa K, et al. Legionella pneumophila induces cathepsin b-dependent necrotic cell death with releasing high mobility group box1 in macrophages. Respir Res (2010) 11:158. doi: 10.1186/1465-9921-11-158
92. Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis (2018) 9(6):597. doi: 10.1038/s41419-017-0090-8
93. Brambilla L, Martorana F, Guidotti G, Rossi D. Dysregulation of astrocytic HMGB1 signaling in amyotrophic lateral sclerosis. Front Neurosci (2018) 12:622. doi: 10.3389/fnins.2018.00622
94. Yon JM, Kim YB, Park D. The ethanol fraction of white rose petal extract abrogates excitotoxicity-induced neuronal damage in vivo and in vitro through inhibition of oxidative stress and proinflammation. Nutrients (2018) 10(10):1375. doi: 10.3390/nu10101375
95. Lo Coco D, Veglianese P, Allievi E, Bendotti C. Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of ALS. Neurosci Lett (2007) 412(1):73–7. doi: 10.1016/j.neulet.2006.10.063
96. Kang R, Livesey KM, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 as an autophagy sensor in oxidative stress. Autophagy (2011) 7(8):904–6. doi: 10.4161/auto.7.8.15704
97. Tang D, Kang R, Livesey KM, Zeh HJ 3rd, Lotze MT. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid Redox Signal (2011) 15(8):2185–95. doi: 10.1089/ars.2010.3666
98. Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med (2012) 18(6):911–7. doi: 10.1038/nm.2749
99. Yamada Y, Fujii T, Ishijima R, Tachibana H, Yokoue N, Takasawa R, et al. The release of high mobility group box 1 in apoptosis is triggered by nucleosomal DNA fragmentation. Arch Biochem Biophys (2011) 506(2):188–93. doi: 10.1016/j.abb.2010.11.011
100. Yamada Y, Fujii T, Ishijima R, Tachibana H, Yokoue N, Takasawa R, et al. DR396, an apoptotic DNase gamma inhibitor, attenuates high mobility group box 1 release from apoptotic cells. Bioorg Med Chem (2011) 19(1):168–71. doi: 10.1016/j.bmc.2010.11.037
101. Fages C, Nolo R, Huttunen HJ, Eskelinen E, Rauvala H. Regulation of cell migration by amphoterin. J Cell Sci (2000) 113(Pt 4):611–20. doi: 10.1242/jcs.113.4.611
102. Degryse B, Bonaldi T, Scaffidi P, Müller S, Resnati M, Sanvito F, et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol (2001) 152(6):1197–206. doi: 10.1083/jcb.152.6.1197
103. Degryse B, de Virgilio M. The nuclear protein HMGB1, a new kind of chemokine? FEBS Lett (2003) 553(1-2):11–7. doi: 10.1016/S0014-5793(03)01027-5
104. Palumbo R, Bianchi ME. High mobility group box 1 protein, a cue for stem cell recruitment. Biochem Pharmacol (2004) 68(6):1165–70. doi: 10.1016/j.bcp.2004.03.048
105. Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A, et al. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol (2004) 164(3):441–9. doi: 10.1083/jcb.200304135
106. Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, et al. Regulation of monocyte migration by amphoterin (HMGB1). Blood (2004) 104(4):1174–82. doi: 10.1182/blood-2003-10-3536
107. Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol (2007) 81(1):84–91. doi: 10.1189/jlb.0306171
108. Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires mac-1-integrin. EMBO J (2007) 26(4):1129–39. doi: 10.1038/sj.emboj.7601552
109. Palumbo R, Galvez BG, Pusterla T, De Marchis F, Cossu G, Marcu KB, et al. Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-kappaB activation. J Cell Biol (2007) 179(1):33–40. doi: 10.1083/jcb.200704015
110. Pedrazzi M, Patrone M, Passalacqua M, Ranzato E, Colamassaro D, Sparatore B, et al. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J Immunol (2007) 179(12):8525–32. doi: 10.4049/jimmunol.179.12.8525
111. Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol (2007) 81(1):59–66. doi: 10.1189/jlb.0306180
112. Palumbo R, De Marchis F, Pusterla T, Conti A, Alessio M, Bianchi ME. Src family kinases are necessary for cell migration induced by extracellular HMGB1. J Leukoc Biol (2009) 86(3):617–23. doi: 10.1189/jlb.0908581
113. Ranzato E, Patrone M, Pedrazzi M, Burlando B. HMGb1 promotes scratch wound closure of HaCaT keratinocytes via ERK1/2 activation. Mol Cell Biochem (2009) 332(1-2):199–205. doi: 10.1007/s11010-009-0192-4
114. van Zoelen MA, Yang H, Florquin S, Meijers JC, Akira S, Arnold B, et al. Role of toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock (2009) 31(3):280–4. doi: 10.1097/SHK.0b013e318186262d
115. Bauer EM, Shapiro R, Billiar TR, Bauer PM. High mobility group box 1 inhibits human pulmonary artery endothelial cell migration via a toll-like receptor 4- and interferon response factor 3-dependent mechanism(s). J Biol Chem (2013) 288(2):1365–73. doi: 10.1074/jbc.M112.434142
116. Chen HG, Xie KL, Han HZ, Wang WN, Liu DQ, Wang GL, et al. Heme oxygenase-1 mediates the anti-inflammatory effect of molecular hydrogen in LPS-stimulated RAW 264.7 macrophages. Int J Surg (2013) 11(10):1060–6. doi: 10.1016/j.ijsu.2013.10.007
117. Rauvala H, Rouhiainen A. Physiological and pathophysiological outcomes of the interactions of HMGB1 with cell surface receptors. Biochim Biophys Acta (2010) 1799(1-2):164–70. doi: 10.1016/j.bbagrm.2009.11.012
118. Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med (2000) 192(4):565–70. doi: 10.1084/jem.192.4.565
119. Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature (2000) 405(6784):354–60. doi: 10.1038/35012626
120. Andersson U, Harris HE. The role of HMGB1 in the pathogenesis of rheumatic disease. Biochim Biophys Acta (2010) 1799(1-2):141–8. doi: 10.1016/j.bbagrm.2009.11.003
121. Penzo M, Molteni R, Suda T, Samaniego S, Raucci A, Habiel DM, et al. Inhibitor of NF-kappa b kinases alpha and beta are both essential for high mobility group box 1-mediated chemotaxis [corrected]. J Immunol (2010) 184(8):4497–509. doi: 10.4049/jimmunol.0903131
122. Kew RR, Penzo M, Habiel DM, Marcu KB. The IKKalpha-dependent NF-kappaB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J Immunol (2012) 188(5):2380–6. doi: 10.4049/jimmunol.1102454
123. Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med (2012) 209(3):551–63. doi: 10.1084/jem.20111739
124. Salanga CL, Handel TM. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp Cell Res (2011) 317(5):590–601. doi: 10.1016/j.yexcr.2011.01.004
125. Shakir M, Tang D, Zeh HJ, Tang SW, Anderson CJ, Bahary N, et al. The chemokine receptors CXCR4/CXCR7 and their primary heterodimeric ligands CXCL12 and CXCL12/high mobility group box 1 in pancreatic cancer growth and development: finding flow. Pancreas (2015) 44(4):528–34. doi: 10.1097/MPA.0000000000000298
126. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood (2003) 101(7):2652–60. doi: 10.1182/blood-2002-05-1300
127. Uezono N, Zhu Y, Fujimoto Y, Yasui T, Matsuda T, Nakajo M, et al. Prior treatment with anti-high mobility group box-1 antibody boosts human neural stem cell transplantation-mediated functional recovery after spinal cord injury. Stem Cells (2018) 36(5):737–50. doi: 10.1002/stem.2802
128. Nakajo M, Uezono N, Nakashima H, Wake H, Komiya S, Nishibori M, et al. Therapeutic time window of anti-high mobility group box-1 antibody administration in mouse model of spinal cord injury. Neurosci Res (2019) 141:63–70. doi: 10.1016/j.neures.2018.03.004
129. Sun L, Li M, Ma X, Zhang L, Song J, Lv C, et al. Inhibiting high mobility group box-1 reduces early spinal cord edema and attenuates astrocyte activation and aquaporin-4 expression after spinal cord injury in rats. J Neurotrauma (2019) 36(3):421–35. doi: 10.1089/neu.2018.5642
130. Wu Z, Wang Z, Xie Z, Zhu H, Li C, Xie S, et al. Glycyrrhizic acid attenuates the inflammatory response after spinal cord injury by inhibiting high mobility group box-1 protein through the p38/Jun n-terminal kinase signaling pathway. World Neurosurg (2022) 158:e856–64. doi: 10.1016/j.wneu.2021.11.085
131. Kang N, Hai Y, Yang J, Liang F, Gao CJ. Hyperbaric oxygen intervention reduces secondary spinal cord injury in rats via regulation of HMGB1/TLR4/NF-κB signaling pathway. Int J Clin Exp Pathol (2015) 8(2):1141–53.
132. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem (2004) 279(9):7370–7. doi: 10.1074/jbc.M306793200
133. Paudel YN, Angelopoulou E, Piperi C, Balasubramaniam VRMT, Othman I, Shaikh MF. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur J Pharmacol (2019) 858:172487. doi: 10.1016/j.ejphar.2019.172487
134. van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis (2008) 11(1):91–9. doi: 10.1007/s10456-008-9093-5
135. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol (2011) 29:139–62. doi: 10.1146/annurev-immunol-030409-101323
136. Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol (2012) 8(4):195–202. doi: 10.1038/nrrheum.2011.222
137. Wang H, Mei X, Cao Y, Liu C, Zhao Z, Guo Z, et al. HMGB1/Advanced glycation end products (RAGE) does not aggravate inflammation but promote endogenous neural stem cells differentiation in spinal cord injury. Sci Rep (2017) 7(1):10332. doi: 10.1038/s41598-017-10611-8
138. Kim D, Murray M, Simansky KJ. The serotonergic 5-HT(2C) agonist m-chlorophenylpiperazine increases weight-supported locomotion without development of tolerance in rats with spinal transections. Exp Neurol (2001) 169(2):496–500. doi: 10.1006/exnr.2001.7660
139. Nothias JM, Mitsui T, Shumsky JS, Fischer I, Antonacci MD, Murray M. Combined effects of neurotrophin secreting transplants, exercise, and serotonergic drug challenge improve function in spinal rats. Neurorehabil Neural Repair (2005) 19(4):296–312. doi: 10.1177/1545968305281209
140. Ghosh M, Pearse DD. The role of the serotonergic system in locomotor recovery after spinal cord injury. Front Neural Circuits (2014) 8:151. doi: 10.3389/fncir.2014.00151
141. Wang C, Liu XX, Huang KB, Yin SB, Wei JJ, Hu YF, et al. Preconditioning with recombinant high-mobility group box 1 induces ischemic tolerance in a rat model of focal cerebral ischemia-reperfusion. J Neurochem (2016) 137(4):576–88. doi: 10.1111/jnc.13611
142. Passalacqua M, Patrone M, Picotti GB, Del Rio M, Sparatore B, Melloni E, et al. Stimulated astrocytes release high-mobility group 1 protein, an inducer of LAN-5 neuroblastoma cell differentiation. Neuroscience (1998) 82(4):1021–8. doi: 10.1016/s0306-4522(97)00352-7
143. Huttunen HJ, Kuja-Panula J, Rauvala H. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J Biol Chem (2002) 277(41):38635–46. doi: 10.1074/jbc.M202515200
144. Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem (2000) 275(51):40096–105. doi: 10.1074/jbc.M006993200
145. Hayakawa K, Qiu J, Lo EH. Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Ann N Y Acad Sci (2010) 1207:50–7. doi: 10.1111/j.1749-6632.2010.05728.x
146. Yang J, Liu X, Zhou Y, Wang G, Gao C, Su Q. Hyperbaric oxygen alleviates experimental (spinal cord) injury by downregulating HMGB1/NF-kappaB expression. Spine (Phila Pa 1976) (2013) 38(26):E1641–8. doi: 10.1097/BRS.0000000000000005
147. Zhang Z, Li M, Wang Y, Wu J, Li J. Higenamine promotes M2 macrophage activation and reduces Hmgb1 production through HO-1 induction in a murine model of spinal cord injury. Int Immunopharmacol (2014) 23(2):681–7. doi: 10.1016/j.intimp.2014.10.022
148. Bi Y, Zhu Y, Zhang M, Zhang K, Hua X, Fang Z, et al. Effect of shikonin on spinal cord injury in rats via regulation of HMGB1/TLR4/NF-kB signaling pathway. Cell Physiol Biochem (2017) 43(2):481–91. doi: 10.1159/000480474
149. Yang Y, Wang J, Yang Q, Wu S, Yang Z, Zhu H, et al. Shikonin inhibits the lipopolysaccharide-induced release of HMGB1 in RAW264.7 cells via IFN and NF-kappaB signaling pathways. Int Immunopharmacol (2014) 19(1):81–7. doi: 10.1016/j.intimp.2014.01.003
150. Zhou J, Shuang O, Li J, Cai Z, Wu C, Wang W. miR-34a alleviates spinal cord injury via TLR4 signaling by inhibiting HMGB-1. Exp Ther Med (2019) 17(3):1912–8. doi: 10.3892/etm.2018.7102
151. Wan G, An Y, Tao J, Wang Y, Zhou Q, Yang R, et al. MicroRNA-129-5p alleviates spinal cord injury in mice via suppressing the apoptosis and inflammatory response through HMGB1/TLR4/NF-kappaB pathway. Biosci Rep (2020) 40(3):BSR20193315. doi: 10.1042/BSR20193315
152. Zhao HD, Huang SQ, Tang CL, Dai N, Dai P, Tan YL. [Influence of electroacupuncture on locomotor function and expression of spinal HMGB1 and TLR4 in mice with spinal cord injury]. Zhen Ci Yan Jiu (2021) 46(4):259–65. doi: 10.13702/j.1000-0607.200490
Keywords: HMGB1, spinal cord injury, inflammation, neuron, mechanism
Citation: Mo Y and Chen K (2023) Review: The role of HMGB1 in spinal cord injury. Front. Immunol. 13:1094925. doi: 10.3389/fimmu.2022.1094925
Received: 10 November 2022; Accepted: 19 December 2022;
Published: 12 January 2023.
Edited by:
Bo Li, Department of Orthopedics, Sun Yat-sen University, ChinaCopyright © 2023 Mo and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kebing Chen, Y2hrYmluZ0BtYWlsLnN5c3UuZWR1LmNu