 Jun Dai1,2,3†
Jun Dai1,2,3† Huan Wang2†
Huan Wang2† Ying Liao2Lei Tan2
Ying Liao2Lei Tan2 Yingjie Sun2Cuiping Song2
Yingjie Sun2Cuiping Song2 Weiwei Liu2
Weiwei Liu2 Xusheng Qiu2*
Xusheng Qiu2* Chan Ding2,4*
Chan Ding2,4*- 1College of Animal Science and Technology, Guangxi University, Nanning, China
- 2Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Shanghai, China
- 3Experimental Animal Center, Zunyi Medical University, Zunyi City, China
- 4Jiangsu Co-Innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou University, Yangzhou, China
Host cholesterol metabolism remodeling is significantly associated with the spread of human pathogenic coronaviruses, suggesting virus-host relationships could be affected by cholesterol-modifying drugs. Cholesterol has an important role in coronavirus entry, membrane fusion, and pathological syncytia formation, therefore cholesterol metabolic mechanisms may be promising drug targets for coronavirus infections. Moreover, cholesterol and its metabolizing enzymes or corresponding natural products exert antiviral effects which are closely associated with individual viral steps during coronavirus replication. Furthermore, the coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 infections are associated with clinically significant low cholesterol levels, suggesting cholesterol could function as a potential marker for monitoring viral infection status. Therefore, weaponizing cholesterol dysregulation against viral infection could be an effective antiviral strategy. In this review, we comprehensively review the literature to clarify how coronaviruses exploit host cholesterol metabolism to accommodate viral replication requirements and interfere with host immune responses. We also focus on targeting cholesterol homeostasis to interfere with critical steps during coronavirus infection.
Introduction
Coronaviruses are enveloped viruses with non-segmented, single-stranded, positive sense RNA genomes (1). They belong to the Nidovirales order in the Coronaviridae, and are divided into four types: α, β, γ, and δ (1, 2). Coronavirus subfamily members are widespread in infected birds and mammals and some respiratory and intestinal diseases (3–5). Currently, at least seven coronaviruses are known to infect humans, including respiratory syndrome coronavirus 2, human coronavirus OC43, human coronavirus NL63, human coronavirus 229E, severe acute respiratory syndrome coronavirus, human coronavirus HKU1, and Middle East respiratory syndrome coronavirus (6). Coronavirus diversity is due to the low fidelity of RNA-dependent RNA polymerase during viral coding which produces approximately 10−3–10−5 substitutions/site/year (7). Previous evidence indicated that coronaviruses undergo rapid recombination which creates new strains with altered virulence (8). Recent studies reported molecular and serological evidence of the active transmission of SARS-CoV-2-associated coronavirus (SC2R-CoV) in bats in Southeast Asia (9). Closely related coronaviruses are found in distantly related animals; the consequences of this species barrier jump may be devastating and lead to serious disease and death, e.g., SARS-CoV and MERS-CoV are zoonotic viruses that have crossed the species barrier via bats/palm civets and dromedary camels, respectively (10). Beta-coronavirus spill over from Hipposideridae to Rhinolophidae, and then from Rhinolophidae to civets and humans (11). Swine Acute Diarrhea Syndrome CoV is derived from the species Rhinolophus bat coronavirus HKU2 which potentially infects rodents (12, 13). Mechanistically, the species barrier jump is believed to be due to a failure in specific interactions between the viral spike protein receptor binding domain (RBD) and the host receptor, angiotensin converting enzyme 2 (ACE2) (14). This mechanism demonstrated a major tendency to jump from animals to humans.
The COVID-19 pandemic caused by SARS-CoV-2 is the latest example of a major threat to human health (15). This marks the third time a highly pathogenic coronavirus was transmitted to humans from animals (16, 17). SARS-CoV-2 is believed to have originated in bats, however the intermediate host species and the transmission mode remains unclear (18, 19). Studies confirmed that SARS-CoV-2 replicates more easily in ferrets and cats when compared with dogs, pigs, chickens, and ducks (20). The main pathophysiological feature of SARS-CoV-2 is the excessive production of inflammatory factors, leading to systemic inflammation and multiple organ dysfunction syndrome, with an acute impact on the cardiovascular system and lung fibrosis (21–24). Since SARS-CoV-2 belongs to β-coronavirus family and is not a common human pathogen, humans lack a natural immunity to SARS-CoV-2 (25). Unfortunately, the development of novel coronavirus vaccines commenced too late to effectively control the first infection wave (26). Thus far, no specific, highly effective antiviral therapies are available. The disease has rapidly spread to more than 200 countries and territories (27–30). According to World Health Organization statistics, as of February 11th 2022, the total number of COVID-19 cases worldwide had reached 402,044,502, of which 5,770,023 had died (https://www.who.int/emergencies/diseases/novel-coronavirus-2019). SARS-CoV-2 is estimated to have 2–4 times more affinity for ACE2 than the SARS virus (31). Multiple mutations have been identified in the viral S1 subunit, of which three are in the RBD. This not only increases RBD affinity for ACE2 but improves viral escape from the immune system (32). Currently (September 2021), several SARS-CoV-2 delta variants have become more infectious due to mutations in the S protein RBD, and they are also ORF8-deficient. These variants have rapidly spread globally, including outbreaks in the UK (33–36), Taiwan (37), Southeast Asia (38), Germany (39), France (40), USA (41), Poland (42), and Italy (43). Strikingly, ORF8-deficient variants had spread among domestic mink and pangolin in Denmark, and were detected in humans (44, 45). Thus, ORF8-deficient variants in unknown animal reservoirs pose great challenges to human public health and safety (46), thus monitoring such SARS-CoV-2 variants is critical for the prevention and control of the COVID-19 pandemic (36).
Previous studies reported that COVID-19 severity was related to several risk factors, for example, obesity, old age, and underlying disease (47–49). A recent study suggested that when compared to the normal population, cholesterol levels were significantly lower in COVID-19 patients (50). Furthermore, high-density lipoprotein cholesterol was lower in patients with severe and critical disease than in patients with moderate or mild disease, in a study on cholesterol metabolism in mild, moderate, severe, and critical COVID-19 patients (51). Age-specific COVID-19-associated death data from 45 countries showed that the infection fatality ratio was lowest among 5–9-year-old children, with a log-linear increase by age in individuals over 30 years old (52). In Spanish subjects over 75 years old, the lethality rate approached 36% in hospitalized patients, far higher than for younger groups under the same conditions in hospitals, despite having s similar course to younger individuals (53). In addition, Richter and Sohrabi studied obese factors in COVID-19 patients when compared with the normal population. In general, they observed that obese patients were twice as likely to develop COVID-19 as those with a normal weight range (54, 55). A study of clinical characteristics on 393 patients with COVID-19 in New York City, they found that respiratory failure, a severe clinical symptom of COVID-19, was more common among the obese patient subgroup, comprising 35.8% of the patients studied in New York. In addition, they also found that after advanced age, obesity was the most common risk factor leading to severe disease and death from COVID-19 (56). In addition to these factors, male sex, diabetes, smoking, hypertension, and cardiovascular disease also affect COVID-19 risk severity.
Importantly, transcriptomics data indicated that host cholesterol metabolism affects virus replication (57). Cholesterol content in the plasma membrane is extremely high and is important for biochemical and biophysical functions (58). As a unique feature of mammalian membranes, host cell cholesterol is targeted by pathogens (cytosolic bacteria and viruses) for entry and egress (59–63), however, a small number of coronavirus strains are distinct in terms of their dependence on cholesterol (64). Notwithstanding, a strong relationship between cholesterol and coronavirus replication is widely documented in the literature; in some instances, cholesterol is vital for coronavirus entry, membrane fusion, translation, pathological syncytia formation and vascular pathology (65–68) (Figure 1). Cholesterol metabolism may be hijacked by enveloped viruses to provide raw materials for virus particle replication, assembly, and maturation (69), e.g., Hepatitis C virus (HCV), human cytomegalovirus, and Epstein-Barr virus (70–72). For coronaviruses, cholesterol and other specific lipid requirements are required for viral replication scaffolds (73–75). Also, previous studies reported significant associations between cholesterol homeostasis and type I interferon (IFN) responses (76). Viral infections may induce host cells to alter the expression of cholesterol metabolizing enzymes and metabolites, and similarly, cholesterol metabolism can also regulate host antiviral responses (77, 78). Therefore, weaponizing host cholesterol metabolism dysregulation against coronavirus infectivity could be an effective antiviral strategy (79, 80).
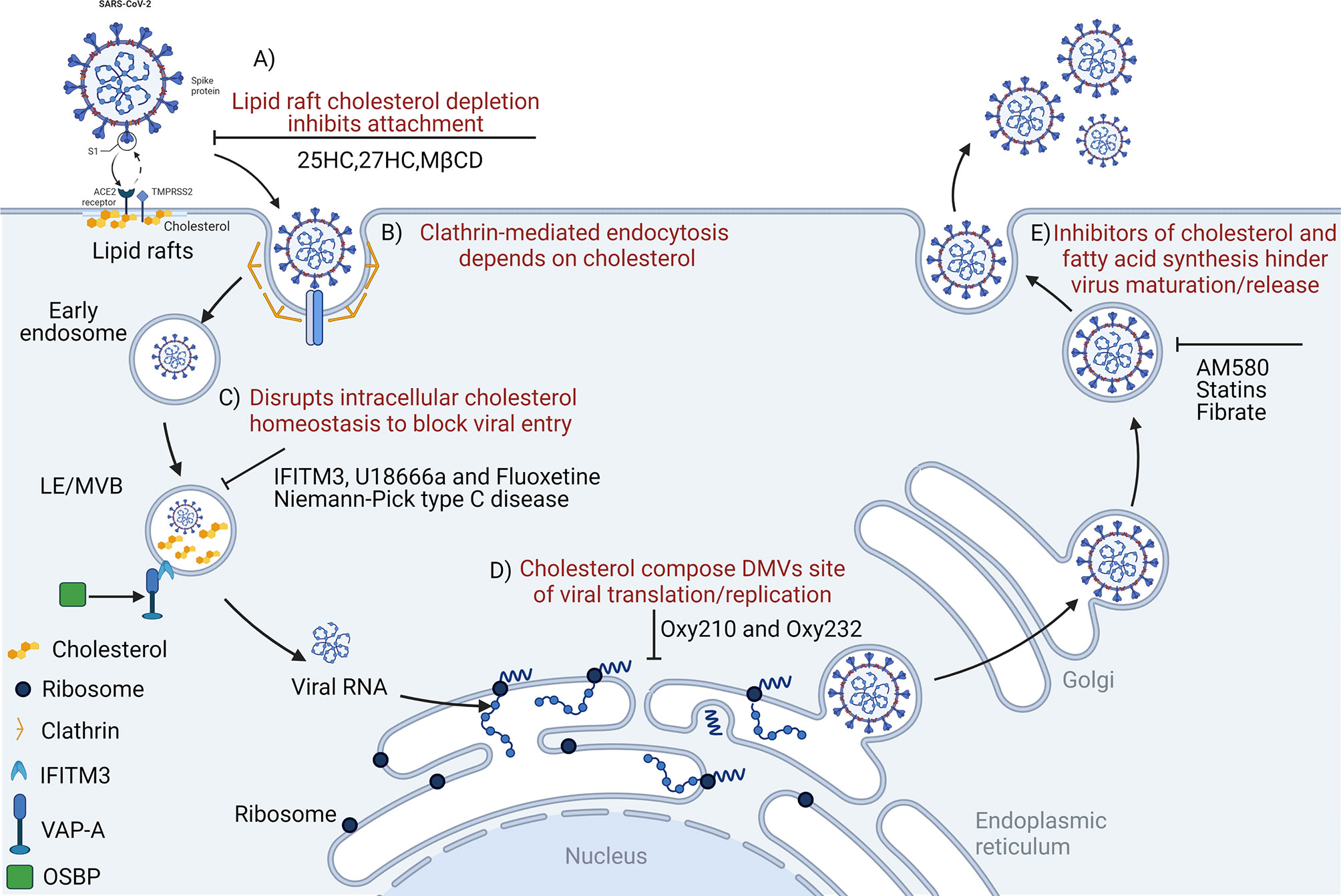
Figure 1 Disrupting cholesterol homeostasis interferes with critical steps during coronavirus infection. Cholesterol is important for coronavirus attachment (A), endocytosis (B), membrane fusion (C), translation/replication (D), and maturation/release (E). 25HC, 25-hydroxycholesterol; MβCD, methyl-beta-cyclodextrin; LE/MVB, late Endosomes/Multivesicular Bodies; IFITM3, interferon-induced transmembrane protein-3; U18666a, an intra-cellular cholesterol transport inhibitor; DMVs, double-membrane vesicles; OSBP, oxysterol-binding protein; VAP-A, vesicle-membrane-protein-associated protein A; Oxy210, semi-synthetic oxysterols; Oxy232, semi-synthetic oxysterols; AM580, a selective retinoic acid receptor-α agonist.
Thus, cholesterol could be an important tool for the in-depth exploration of COVID-19 pathophysiology (81). The disruption of cholesterol homeostasis may interfere with critical steps of coronavirus infection, therefore understanding cholesterol functions during COVID-19 pathogenesis may generate improved prognostics and therapeutics (27). Recently, Daniloski et al. conducted a large-scale screen of > 20,000 drugs potentially used to treat COVID-19, and identified cholesterol biosynthesis pathway induction as a possible mechanism of viral inhibition (82, 83). The pharmacological inhibition of phosphatidylinositol kinases and cholesterol homeostasis reduced replication of all three coronaviruses. These findings provide important insights for an increased understanding of the coronavirus life cycle and the development of host-directed therapies (84). This review focuses on the latest scientific evidence and clarifies how coronaviruses manipulate host cholesterol metabolism to meet their own replication needs and impair host IFN responses. In addition, targeting and altering cholesterol levels in host cell membranes, and interfering with intracellular cholesterol metabolism pathways may be effective strategies in preventing early coronavirus cell entry and subsequent translation and replication. These approaches could provide foundations for the design of anti-coronavirus drug and treatment strategies (79, 85).
The Role of Cholesterol Metabolism During the Coronavirus Life Cycle
Molecular Mechanisms of Coronavirus Entry
Coronavirus entry into host cells is key for virus replication cycles and evading host antiviral responses (86, 87). However, coronaviruses enter cells by two ways: 1) when sufficient proteases are present on plasma membranes, viruses exploit this by fusing with the cell through the “early pathway” via the plasma membrane, and 2) in the absence of extracellular proteases, endosomal proteases activate the viral S protein to gain cell entry via the endosomal pathway (88) which is sensitive to the pH of endosome/lysosome pathways (89). Moreover, the infection efficiency of SARS-CoV in the cell in the “early pathway” is 100-1000 times higher than the endosomal pathway (90). Previous research reported that many coronaviruses, including infectious bronchitis virus (IBV), porcine hemagglutinating encephalomyelitis virus (PHEV), porcine epidemic diarrhea virus (PEDV), and feline coronavirus (FCoV) enter host cells via the endocytic pathway and then pass-through endosomal compartments via multivesicular bodies (MVBs) to enter the cytoplasm (91–94). The coronavirus S protein plays a key role in early viral infection stages and is necessary for viral entry and chemotaxis in hosts (95). This protein is a type I homotrimeric transmembrane fusion glycoprotein, composed of S1 and S2 subunits with different functions. The RBD of the S1 subunit recognizes the cell receptor, ACE2, which determines cell homogeneity and pathogenicity of coronaviruses. The S2 subunit mediates virus and host cell membrane fusion via a wide range of conformational rearrangements (96–98). Moreover, the S1 subunit of SARS-2-S also binds to cholesterol and possibly high-density lipoprotein (HDL) components to enhance in vitro viral uptake; this mechanism is mediated by the HDL scavenger receptor B type 1 (99). In addition, the SARS-CoV-2 spike protein S1/S2 boundary sequence has key roles in regulating viral entry and spread within the cell (96). More importantly, S1/S2 border sequence deletion switches SARS-CoV-2 from the plasma membrane to the endosomal fusion pathway, significantly reducing viral transmission efficiency in hamsters (100). In particular, the S1/S2 boundary sequence contains a furin protease cleavage site which pre-activates the S protein for membrane fusion, reducing SARS-CoV-2 dependence on plasma membrane proteases (e.g., TMPRSS2), thereby efficiently improving cell entry (101). When compared with other coronaviruses, murine coronavirus (MHV) is unique; its receptor binding has dual roles when gaining cell entry: the S protein N-terminal domain binds to the host receptor protein, CEACAM1a (102), then MHV uses a zinc metalloprotease for invasion and cell-cell fusion (103). This not only promotes MHV attachment to host cells, but also promotes MHV fusion with the host membrane (104). More specifically, the CEACAM1 receptor or a pH 8 induces conformational changes in the MHV S glycoprotein at 37°C. This conformational change is more conducive to triggering membrane fusion without the need to activate cleavage between S1 and S2 in advance (105).
Niemann-Pick disease type C (NPC) is a lysosomal storage disorder (106) caused by deficient lipid efflux from the late endosome/lysosome (LE/L) and induces intracellular cholesterol synthesis and transport disorders to impair viral SARS-CoV-2 infectivity via several lipid-dependent mechanisms (79). By intervening in the NPC1 pathway, SARS-CoV-2 is blocked from entering the host cell from the plasma membrane or endosomes/lysosomes, thus viral infectivity is weakened (107). The Ebola virus requires a functional NPC1 protein to complete its replication cycle, however, it is unclear if this is true for coronaviruses (108). Studies reported that SARS-CoV particle transport through endosomes to NPC1 positive compartments of the lysosomal system was necessary for successful infection (109). Mingo et al. showed that Ebola virus reaching NPC1-positive LE/Ls was the rate-limiting step in determining viral infection (109). Furthermore, although SARS-CoV does not require NPC1 for entry, its entry into the cytoplasm begins after colocalization with NPC1 (109). Therefore, pharmacological interventions targeting lysosomal functions could induce transient NPC1-like cells and biochemical phenotypes, which could constitute a possible rationale for COVID-19 treatment (110). Drugs such as fluoxetine not only damage LE/L acidification but also accumulate cholesterol in these compartments (111).
Host Cholesterol in Coronavirus Entry
Coronavirus enters host cells mainly via plasma membrane fusion or endocytosis (112, 113). Lipid rafts participate in endocytosis-mediated processes, and function as platform and docking sites for coronavirus entry and genome release (114, 115). Cholesterol is an important component of lipid rafts; increased lipid raft formation is benefitted by increased cholesterol levels (116). Early coronavirus infection depends on lipid rafts (117) which may harbor ACE2 receptors for the SARS-CoV-2 S protein (118–121), permitting membrane rearrangements to facilitate transmissible gastroenteritis virus (TGEV) entry (122). In addition, lipid rafts act as attachment factors to promote IBV absorption before it enters the cell (119). MHV entry and membrane fusion also require lipid rafts (123). Membrane cholesterol consumption inhibits SARS-CoV-2 and other coronaviruses from fusing with cells, preventing viral entry (124, 125). By reducing plasma membrane cholesterol levels and changing lipid raft-dependent ACE2 and TMPRSS2 activities, these processes interfere with viral internalization by host cells (87, 110, 126). Therefore, cholesterol depletion from cellular membranes using e.g., methyl-β-cyclodextrin (MβCD) eliminates cholesterol in lipid rafts and significantly reduces clathrin-dependent endocytosis to significantly eliminate IBV, TGEV, and SARS-CoV infectivity (Figure 1A) (117, 119, 127). In addition, SARS-CoV-2 pathogenicity was significantly dependent on TMPRSS2 (128). Contributions of human ACE2 and TMPRSS2 in determining host-pathogen interaction of COVID-19 (129). The SARS-CoV-2 Omicron variant showed less efficient replication and fusion activity when compared with the Delta variant in TMPRSS2-expressed cells (130). Omicron infection was not enhanced by TMPRSS2 but was largely mediated by the endocytic pathway. The differences in pathway entry between variants may have impacted on clinical manifestation or disease severity (130). In addition, anti-androgens target TMPRSS2 and reduce SARS-CoV-2 virus entry in lung cells (131), which may at least in part explain why men with COVID-19 have a worse prognosis when compared to women (132). SARS-CoV-2 cell entry inhibition via TMPRSS2 was facilitated by camostat, nafamostat mesylate and alpha-1 antitrypsin (133, 134). It is therefore possible that inhibiting androgen signaling by anti-androgens could reduce TMPRSS2 expression in the lung, and concomitantly reduce viral entry. For this reason, anti-androgens are proposed as treatment options for COVID-19 (135, 136).
Infectivity is also reduced by depleting cholesterol from the viral envelope as in TGEV (127). Similarly, plasma membrane cholesterol depletion is also triggered by ACE2 displacement from lipid rafts to non-raft membrane domains, thereby reducing efficient SARS-CoV cell entry (120). Previous studies reported that 27-hydroxycholesterol (27HC) accumulation in lipid rafts caused the rapid consumption of lipid raft cholesterol, interrupted cell signal transduction in lipid raft membrane microdomains, and specifically inhibited IL-6-JAK-STAT3 signaling (137, 138). It is worth emphasizing that lipid raft destruction due to cholesterol consumption may be the main reason for inhibiting extracellular signal-regulated kinase (ERK) signaling and activation inhibition (139). Since the Raf/MEK/ERK pathway is involved in the modulation of various important cellular functions, numerous DNA and RNA viruses coopt this pathway for efficient viral propagation (140). The ERK pathway is known to be modulated during PEDV infection (141). In our previous research, we reported that IBV infection activated ERK1/2 signaling and that up-regulation of the phosphatase, DUSP6 formed a negative regulation loop (142). ERK activation is necessary for PEDV and porcine deltacoronavirus (PDCoV) replication, the suppression of viral protein expression, and viral RNA transcription via ERK activation inhibition (140, 143). Also, the negative regulation of the Raf/MEK/ERK signaling pathway by the MEK inhibitor, U0126 or DUSP6 upregulation significantly impairs MHV and IBV progeny production (142, 144), respectively. However, the exact mechanism whereby ERK activity regulates the replication cycle of PEDV during infection remains unclear. Therefore, the targeted regulation of lipid raft cholesterol levels may be a host defense strategy against coronavirus infection (145).
Host Cholesterol in Viral Fusion
Along with binding to host cell receptors, viral envelope fusion with host cell membranes is critical in establishing successful coronavirus infection, especially for viral gene delivery into the cytoplasm. Coronaviruses enter cells by fusing directly with the cell surface or internalization via endosomal membranes (146). In general, the viral envelope contains specific cholesterol quantities; cholesterol is an important component of lipid rafts, and the fusion of viruses and host plasma membranes is affected by the ratio of membrane cholesterol to fatty acids (147). Genome-wide clustered regularly interspaced short palindromic repeats (CRISPR) screening revealed that cholesterol metabolism was a key host pathway promoting coronavirus (SARS-CoV-2, HCoV-229E, and HCoV-OC43) infections (84), whereas cholesterol dysregulation reduced viral invasion (82, 84, 100, 148). In addition, coronavirus enters cells either via fusion or endocytosis (149) via clathrin-mediated mechanisms in a cholesterol dependent manner (Figure 1B) (92). Cellular cholesterol homeostasis regulation, especially in endosomal compartments, exerts a significant impact on the entry stage of viral infection (108). It is because that coronavirus or coronavirus-containing MVBs via the endosomal cathepsin activate viral S protein to mediate the cytoplasmic release of viral nucleic acid, and artificially destroying the homeostasis of cholesterol in the endosomal membrane will inhibit this invasion step (91, 150). Therefore, the virus reprograms cholesterol metabolism to promote virus replication, or specific infection-induced host defense responses. Targeting cholesterol metabolism pathways in cells could be a potential target for interfering with “viral cargo”, and may be used as an intervention to inhibit coronavirus membrane fusion in the endosome (79). Many coronaviruses, including IBV, PHEV, PEDV, and FCoV pass through endosomal compartment via MVBs to enter the cytoplasm (91–94). In a previous study, IBV membrane fusion was induced in the LE/L after 1 hour post infection (91). The accumulation of cholesterol and oxidized sterols in late endosomes and MVBs also impaired virus functions, hindered viral membrane fusion, and subsequent replication (79, 151). Therefore, the destruction of cholesterol homeostasis to block viral entry exemplifies the importance of cholesterol during viral infections (150). Cholesterol function during viral invasion was extensively studied in several coronaviruses, including SARS-CoV (120), PEDV (152), MHV (123, 153), PDCoV (154), and IBV (117, 119).
Coronavirus infections may be significantly restricted by IFN-induced transmembrane proteins (IFITMs) (155). These proteins significantly inhibit endosome membrane fusion and are driven by the viral S protein (156). IFITMs inhibit viral membrane fusion before hemifusion occurs, by reducing membrane fluidity and imparting positive spontaneous curvature to outer cell membrane leaflets (157). IFITM phosphorylation status and carboxy-terminal amino acid residues are key factors determining human coronavirus entry, including, HCoV-NL63, SARS-CoV, MERS-CoV, HCoV-OC43, and MERS-CoV (158). These functional units may pass it interacts with the virus and/or host cell components of the virus entry site to regulate the fusion of the virus envelope and cell membrane (158)The vesicle membrane-associated protein A (VAPA) and oxysterol-binding protein (OSBP) jointly regulate intracellular cholesterol balance (150). IFITM3, as a member of the IFITM protein family, hinders binding of OSBP and VAPA, which not only causes abnormal cholesterol accumulation in late endosomes, but also increases membrane hardness and inhibits viral nucleic acid release (Figure 1C) (150).
Cholesterol Metabolism Is Involved in Coronavirus Translation/Replication
As a positive-strand RNA virus, after internalization and un-coating, coronavirus first uses its own genomic RNA as a template to replicate and produce the polyproteins, pp1a and pp1ab via cap-dependent translation, and then via autoproteolytic cleavage, 15–16 nonstructural proteins (NSPs) (159). NSPs induce the rearrangement of cholesterol-rich lipid rafts on cell membranes, forming double-membrane vesicles (DMVs) in the cytoplasm, thereby anchoring viral replication transcription complexes (160). DMVs act as efficient replication sites for coronavirus genomic RNA and provide a safe site for viral RNA replication and translation (161). Cholesterol is also enriched in DMVs and constitutes the viral replication site of DMVs with fatty acids (80, 162). DMVs destroyed by Oxy210 (semi-synthetic oxysterol) significantly inhibit SARS-COV-2 replication in vitro (163) (Figure 1D). Thus, disruption of lipid rafts may affect viral replication and transcriptional synthesis. Recently, it was reported that intracellular cholesterol biosynthesis and transport systems were related to virus replication (164–166). Cellular cholesterol is derived from the biosynthesis and cellular uptake of low-density lipoprotein (167–169). U18666A is a cationic amphiphilic drug affecting cholesterol biosynthesis and intracellular transport (170). Previous studies reported that cholesterol was involved in the viral life cycle of type I FCoV infection (64), and that U18666A induced cholesterol accumulation via NPC1 dysfunction and type I FCoV replication inhibition (144, 171, 172).
Host Cholesterol May Not Be Involved in Coronavirus Assembly and Release
After un-coating, translation, and genome replication, virus particles assemble in the endoplasmic reticulum (ER)-Golgi intermediate compartment and are coordinated by the M protein (159, 173). For most coronaviruses, virus assembly sites contain highly active enzymes involved in the cholesterol biosynthesis pathway; these include, cholesterol-synthesizing enzyme, 3-hydroxy-3-methyl glutaryl coenzyme A reductase, and mevalonate diphospho decarboxylase (165, 174, 175). Several studies indicated that many enveloped viruses, such as human immunodeficiency virus, Dengue, Zika, and alphavirus contain cholesterol in the virion, and that viral proteins involved in virus particle assembly and budding are related to cholesterol (78, 176). Furthermore, different cholesterol levels in hosts generate different envelope cholesterol levels in alphaviruses (177). When compared with mayaro virus particles from mosquito cells, virus particle envelopes from vertebrate cells have higher cholesterol levels (178). In terms of coronaviruses, Simons et al. found that although MHV- S protein was localized to the Golgi, that contained cholesterol and lipid rafts, the assembled and released of MHV is not associated with cholesterol (167). But, cholesterol involvement in virus assembly and budding has mainly focused on viruses budding from cell membranes, however, studies on viruses budding from intracellular membranes are rare (167). Moreover, the different functional roles of cholesterol in enveloped RNA virus stability, infectivity, and assembly are not entirely clear (177, 179). Therefore, coronavirus assembly and budding may not necessarily use cholesterol on Golgi membranes, thus specific mechanisms require further study.
Cholesterol Metabolizing Enzymes and Metabolites Combat Coronavirus Infectivity
Coronavirus infection induces host cells to alter the expression of certain cholesterol metabolizing enzymes and metabolites which may exert antiviral effects (Figure 2A) (77). Indeed, both 25-hydroxycholesterol (25HC) and 27-hydroxycholesterol (27HC) are physiologically produced by the enzymatic oxidation of cholesterol and may be used to inhibit enveloped and non-enveloped human viruses (183, 184) and highly pathogenic viruses, including Zika (185), mammalian reovirus (186), Lassa virus (187), encephalomyocarditis virus (188), porcine reproductive and respiratory syndrome virus (189). A recent study reported that the 25HC treatment of mice infected with SARS-CoV-2 significantly reduced virus numbers in the lungs and trachea (148). On the one hand, cholesterol is transformed into 25HC by Cholesterol 25-Hydroxylase (CH25H). By obstructing membrane fusion, 25HC exhibits extensive anti-coronavirus activity (125, 184). Similarly, the internalization of 25HC aggregates in late endosomes may inhibit spike protein-catalyzed membrane fusion of SARS‐CoV‐2 by blocking cholesterol export (124); however, CH25H consumes available cholesterol on the plasma membrane to suppress virus-cell fusion (125). These data indicate that membrane-modifying oxysterols are possible antiviral therapeutics, thereby inhibiting SARS-CoV-2 and other coronaviruses (Figure 1A) (153). However, it is possible to obstruct PDCoV proliferation using CH25H which acts as a host restriction factor, but this inhibition is not entirely dependent on its enzymatic activity (190). The junction adhesion molecule-A and the cation independent isoform of the mannose-6-phosphate receptor are two key replication molecules common to all viruses that use adhesion molecules and the endosomal pathway to enter and diffuse target cells. Both molecules are downregulated by 25HC and 27HC (191). Previous studies suggested that SARS-CoV-2 propagation in cultured cells was inhibited by various cholesterol molecules, including natural oxysterols, 7-ketocholesterol, 22(R)-hydroxycholesterol, 24(S)-hydroxycholesterol, and 27HC (Figures 2A-e, c) (163). At effective concentrations, 25HC, 7-dehydrocholesterol (7DHC), and 27HC were non-toxic natural products, with potentially curative applications for emerging virus infections, such as SARS-CoV-2 (192), human immunodeficiency virus, Ebola virus, Nipah virus, Rift Valley fever virus, and Zika (153).
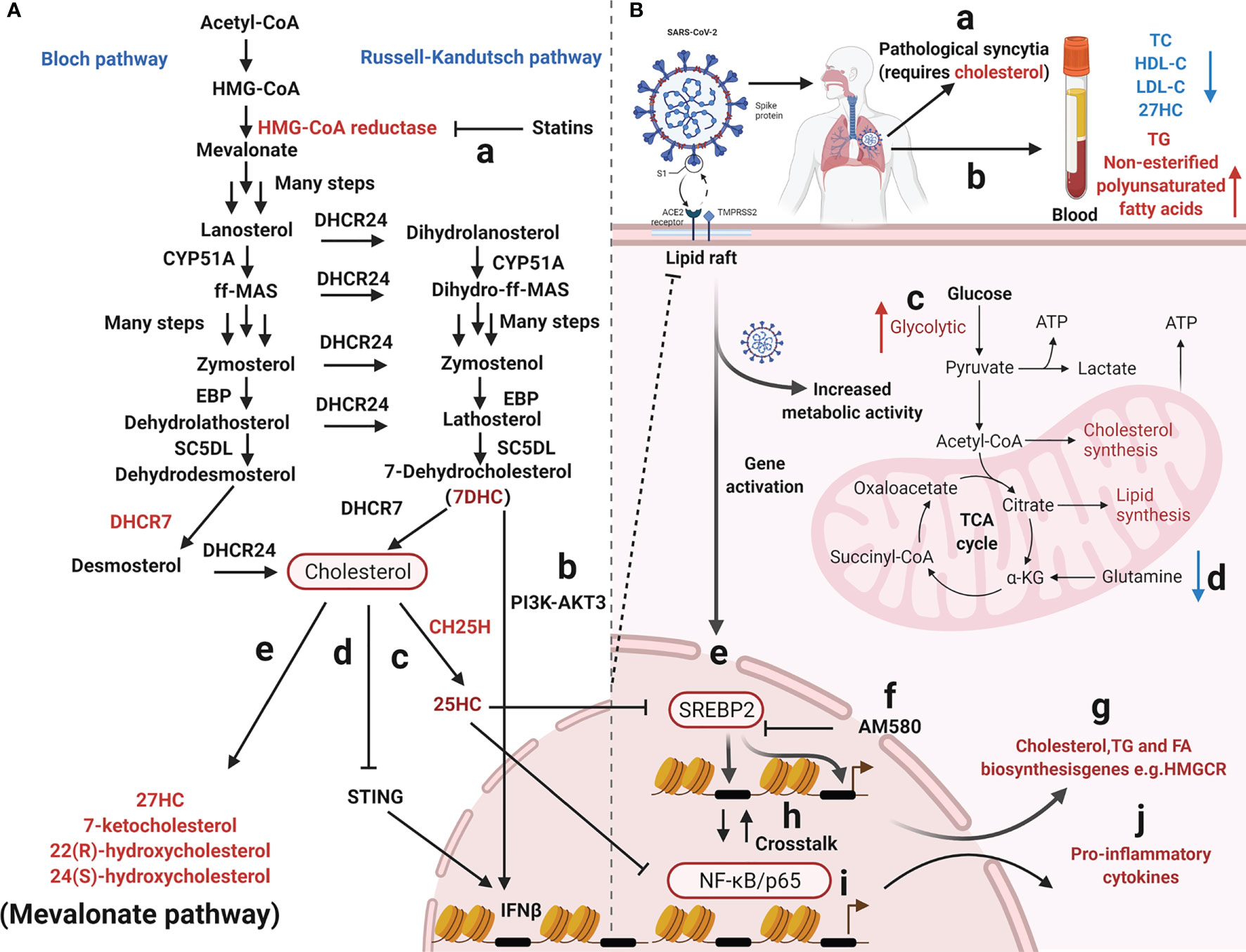
Figure 2 Overview of mevalonate pathway and coronavirus infection. (A) Cholesterol metabolizing enzymes and metabolites act against viral infectivity. Red type represents cholesterol metabolizing enzymes or corresponding natural products which may be used as drug targets or directly to exert antiviral effects. (a) HMG-CoA reductase regulates cholesterol biosynthesis and is targeted by statins. (b, c, d) The sterol metabolic network participates in interferon (IFN) antiviral responses. (e, f) SARS-CoV-2 propagation in cultured cells is inhibited by various cholesterol molecules and semi-synthetic oxysterols. The detailed steps of the cholesterol synthesis pathway can be found in (180–182). (B) Cholesterol metabolism reprogramming and antiviral responses after viral infection. (a) Cholesterol promotes pathological syncytial formation during SARS-COV-2 infection. (b) Serum TC, TG, and non-esterified polyunsaturated fatty acid levels are remodeled in COVID-19 patients. (c, d) SARS-CoV-2 infection increases glucose entry into the TCA cycle via increased pyruvate carboxylase expression and reduced oxidative glutamine metabolism, while maintaining reductive carboxylation. (f) SREBP-dependent lipidomic reprogramming is a broad-spectrum antiviral target, AM580 strongly inhibits coronavirus replication by interacting with SREBP-2. (e, h, g, i, j) COVID-19-activated SREBP-2 disturbs cholesterol biosynthesis, leading to a cytokine storm. Importantly, SREBP-2 activity is regulated by crosstalk between cholesterol consumption and NF-κB expression via several inflammatory response processes induced by SARS-CoV-2 infection. Red arrows represent upregulation and blue arrows represent downregulation. Acetyl-CoA, Acetyl-Coenzyme A; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; EBP, Δ(7)-isomerase; DHCR24, 3-β-hydroxysteroid-Δ-24-reductase; SC5DL, Sterol C5-desaturase; DHCR7, 7-dehydrocholesterol reductase; CH25H, cholesterol-25-hydroxylase; 25HC, 25-hydroxycholesterol; 27HC, 27-hydroxycholesterol; IFNβ, Interferon-β; TC, Total cholesterol; HDL-C, High-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TG, Triglyceride; ATP, Adenosine triphosphate; SREBP-2, Sterol regulatory element-binding protein 2; AM580, a selective RARα agonist; HMGCR, 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase; NF-κB, Nuclear transcription factor-κB.
The Sterol Metabolic Network Participates in Host-Immune Responses
All coronaviruses have a similar infection mechanism, which successfully manipulates host cell functions. One strategy to suppress the host innate immune response to evade antiviral responses, is shielding RNA intermediates in replication organelles (193, 194). In fact, coronaviruses with +RNA genomes, which duplicate solely in the cytoplasmic matrix, and modify the inner membranes of cells to form virus duplication bases, also known as “replication factories” or “replication organelles”. While varying in morphology and membrane composition, these structures appear to centralize viral replication machinery, intermediates, and products in membrane-bound vesicles or invaginations, and are beyond the reach of innate immune sensors in the cytosol. Thus, viral infection outcomes are determined by metabolic interactions between hosts and viruses (195). Cholesterol is a crucial component of cell membranes and lipid rafts. Cholesterol metabolism contributes to the formation of immune synapses and downstream signal transmission (196).The host’s defenses against virus infection requires IFN-mediated cholesterol biosynthesis and the formation of immune synapses, and also host innate immune metabolic regulators as potential antiviral strategies (197).
Two molecules, sterol regulatory element-binding protein-2 (SREBP-2) and 3-hydroxy-3-methyglutaryl CoA reductase (HMGCR), have significant roles in the cholesterol biosynthetic pathway; SREBP-2 is the master transcriptional regulator of cholesterol biosynthesis (Figures 2B-e, g) and HMGCR is a rate-limiting enzyme for cholesterol synthesis (198). SREBP-2 cleavage and HMGCR degradation are two major feedback regulatory mechanisms governing cholesterol biosynthesis (Figures 2A-a) (58). Recently, the sterol metabolic network was shown to participate in interferon (IFN) antiviral responses (76, 199). Studies have reported that the IFN regulatory loop mechanism downregulates sterol biosynthesis, linking innate immune responses to viral infection, via sterol metabolism regulation (171). After viral infection, the infected cells produce high IFN levels, thereby reducing enzyme expression in the cholesterol pathway (171). On the one hand, the sterol metabolic network is involved in IFN antiviral responses (76) with reduced flux through the mevalonate pathway leading to upregulation of type I IFN responses (172) (Figures 2A-b, d). IFN-γ induces proteasomal degradation of HMG-CoA reductase and the rapid proteasomal elimination of HMG-CoA reductase by IFN-γ in primary macrophages which requires endogenous 25HC synthesis (200). On the other hand, cholesterol metabolism and mevalonate pathways are crucial for regulator T-cells which efficiently drive regulatory T cell proliferation and enhance and stabilize their suppressive capacity (201, 202). In particular, LKB1 triggered activation of the mevalonate pathway by upregulating IFN-γ and IL-17A levels, which were essential for the stabilization of T regulatory cells (201). Cholesterol is required for SARS-CoV-2 to form pathological syncytia which is believed to help replicate and evade host immune responses (67). For example, cholesterol biosynthesis pathways are affected by SARS-CoV which regulate levels of SREBP2, S1 protein, peroxisome proliferators-activated receptors γ (PPARγ), diacylglycerol acyltransferase-1or cholesterol efflux regulatory protein (203–205). COVID-19-activated SREBP2 disturbs cholesterol biosynthesis (Figures 2B-e), leading to a cytokine storm (50) (Figures 2 B-j). Importantly, SREBP-2 activity is regulated by crosstalk between cholesterol consumption and nuclear factor κ-B (NF-κB) expression from various inflammatory response processes induced by SARS-CoV-2 infection (Figures 2B-h) (50). In addition, a metabolic configuration is induced by SREBPs where glucose is metabolized via the citrate malate shuttle, thus enabling natural killer cell growth, proliferation, and function (206). Therefore, SREBP-dependent lipidomic reprogramming may be viewed as a broad-spectrum antiviral target (203). 25HC exerts inflammatory properties and significantly attenuates proteolytic processing of SREBP2 (Figures 2A-c), thereby inhibiting the isoprenoid branch of the mevalonate pathway (207). 25HC also amplifies inflammatory signals (207), with growing evidence suggesting it has a broad impact on innate and adaptive immunity (195, 208–213), including antiviral immunity, inflammasome activation, and antibody class switching (214), In addition, lung-selective 25HC nano-therapeutics may function as inhibitors of COVID-19-mediated cytokine storms (215). IFN-β production may be regulated by targeting the 7-dehydrocholesterol reductase (DHCR7) and adding 7-dehydrocholesterol (7-DHC), an intermediate natural product in the cholesterol metabolism pathway (77) (Figures 2A-b). Moreover, it is possible to enhance anti-viral immunity by promoting serine/threonine kinase 3 (AKT3) activation (77); a positive feedback loop is formed via type I IFN signaling and 7DHC accumulation to amplify innate immune responses and control viral infection by activating AKT3. Also, the emergence of highly pathogenic viruses may be inhibited by DHCR7 inhibitors and 7-DHC (216). York et al. suggested that a reduction in cholesterol biosynthesis was a key event in inducing antiviral responses in virus-infected cells (172) and that decreased cholesterol biosynthesis facilitated anti-viral signaling by the stimulator of interferon gene (STING) in the ER (Figures 2A-d) (216). Although cholesterol is a crucial component of immune cell membranes, cholesterol accumulation in lymphoid organs promoted T cell priming and stimulated the production of the B cell growth factors, Baff and April (217). Ito et al. (2016) reported that defects in cholesterol metabolism in CD11c+ immune cells resulted in impaired antigen presentation and ultimately autoimmune disease (218). Excessive cholesterol may exert immune dysfunction and promote excessive pulmonary and systemic inflammatory responses (219).
Cholesterol-Modifying Drugs Inhibit Coronavirus Replication
Recently, several commonly prescribed medications were shown to interfere with sterol biosynthesis, including haloperidol, aripiprazole, cariprazine, fluoxetine, trazodone, and amiodarone (220). Cholesterol-modifying drugs exert anti-viral roles by reducing the absorption or synthesis of systemic cholesterol or directly changing cholesterol levels in target cell membranes (219) (Figure 1A). It is possible to alter the SARS-CoV-2 cycle in vitro and in vivo using various cholesterol-modifying drugs (e.g., AM580 is a selective retinoic acid receptor alpha (RAR-α) agonist, fibrates, and statins) which hinder fatty acid and cholesterol synthesis (Figure 1E) (221). In fact, cholesterol-binding agents, including statins or MβCD, affect cholesterol and destroy lipid rafts, thereby damaging coronavirus adhesion and binding properties (119, 222). Moreover, these compounds also block key downstream virus infectivity molecules, reduce proinflammatory tumor necrosis factor-α (TNF-α) and IL-6 levels, and/or affect autophagic processes in viral replication and clearance (222). It is worth noting that cholesterol, fatty acids, cytosolic phospholipase A2α (cPLA2α), and fatty acid synthase contribute to SARS-CoV-2 DMV formation (204, 223). For example, fenofibrate (reduces triglyceride and low-density lipoprotein cholesterol levels inhibit SARS-CoV-2 replication and pathogenesis by affecting lipid metabolism pathways in the lung cells of patients with COVID-19 (224). AM580 is a retinoid derivative which interacts with N-terminal SREBP to block lipogenic transactivation (203). Statins reduce intra- and extra-cellular cholesterol by targeting HMGCR (Figures 2A-a) (225, 226), thereby affecting viral infection, immunity, and inflammation (219). Statins may also limit inflammation by altering HMGCR mediators in the cholesterol biosynthesis pathway (227, 228). These anti-inflammatory properties are considered statin’s core protective effects in cardiovascular disease, in addition to lowering cholesterol levels (225). Wang et al. suggested that high cholesterol levels increased entry of pseudotyped SARS-CoV-2 and the infection of virus particles, and more of the receptor ACE2 can be recruited to the internalization site (229). Statins ability to decrease lipids, enhance protective immune responses, and exert anti-inflammatory properties are beneficial during SARS-CoV-2 infections (219). Statin therapy was previously reported to increase blood clearance rates in chronic HCV infections and reduce mortality and intubation requirements during influenza infection (118). Statins, especially pitavastatin, may significantly inhibit activity of SARS-CoV-2’s main protease, Mpro, which has a greater binding energy than proteases or polymerase inhibitors (230). Decreasing cellular cholesterol may also trigger the intake of more cholesterol from the blood, reducing serum HDL-cholesterol (HDL-C) and LDL-C levels. As cholesterol-lowering drugs, statins are widely used in cardiovascular and metabolic diseases (231, 232). They inhibit inflammation by reducing cholesterol and phospholipid deposition in blood vessels. Thus, anti-inflammatory molecules provide protective effects in cardiovascular diseases, and do not just lower cholesterol (225).
Cholesterol as a Potential Marker for Monitoring COVID-19
SARS-CoV-2 infection reshapes cholesterol metabolism via gene activation and increased host metabolism activity (23) (Figure 2B). Briefly, SARS-CoV-2 infection disturbs cholesterol biosynthesis by activating SREBP-2 and affecting glucose or glutamine metabolism (50, 233) (Figures 2B-e, c, d). Clinical data has also indicated that lipid disorders may facilitate increased COVID-19 mediated pathogenicity, therefore lowering cholesterol levels may inhibit SARS-CoV-2 replication and viral loads in patients (51, 65). When compared with healthy individuals, patients with dyslipidemia-related diseases are more likely to be infected by SARS-CoV-2 (234, 235). Also, SARS-CoV-2 infection caused some COVID-19 patients to have lower serum cholesterol levels (e.g., 27HC, total cholesterol, high density lipoprotein cholesterol, and low-density lipoprotein cholesterol), while triglyceride and non-esterified polyunsaturated fatty acid levels were up-regulated (Figures 2B-b) (27, 236–238). In particular, decreased serum HDL-C levels are positively correlated with COVID-19 infection severity (239). As infection worsens, serum TC and HDL are lowered, but upon recovery, cholesterol levels return to normal (65, 239–243). This may be due to SARS-CoV-2 S proteins affecting HDL functions by removing lipids from HDL and remodeling its composition/structure (243), potentially affecting virus clearance in infected patients (244). Thus, serum cholesterol and lipoprotein marker monitoring may have an important clinical value for COVID-19 risk prediction (115). Increased triglyceride/HDL-C ratios may be useful for the early identification of patients with high risk and poor outcomes (245, 246). Moreover, in patients with severe disease, significantly elevated serum HDL levels are associated with favorable outcomes (112). HDL-C levels decrease significantly in critically ill COVID-19 patients and are negatively correlated with C-reactive protein and IL-6 levels, however lymphocyte levels are increased with increased HDL-C levels, which positively correlate with the COVID-19 severity (247). Therefore, LDL-C levels may be used as predictors of COVID-19 progression and risk assessment (248).
SREBP is a membrane junction protein attached to the ER and nuclear envelope (249); it regulates the effective synthesis of fat and cholesterol and plays important roles in maintaining energy homeostasis (Figures 2B-g) (250, 251). The SREBP protein family regulate lipid cholesterol and fatty acid gene expression via mitogen-activated protein kinase (MAPK) signaling (252). A recent study reported that SREBP-2 C-terminal fragment was detected for the first time in the blood of patients with COVID-19. Based on data from clinical samples, SREBP-2 C-term was suggested as a reference indicator to assess disease severity after SARS-CoV-2 infection (50). SREBP-2-dependent lipidomic reprogramming is a broad-spectrum antiviral target, with SREBP-2 activation correlating with COVID-19-induced cytokine storm activation (235). AM580 strongly inhibits coronavirus replication by interacting with SREBP-2 (203, 204).
Conclusions
Viruses are intracellular parasitic pathogens. They exploit host nutrients and metabolites to accommodate their survival and are highly adaptable molecules in escaping host antiviral responses. Therefore, interventions in host specific metabolic pathways could become potential antiviral targets (80, 203, 205, 253). Potential cholesterol-modifying drugs exert broad-spectrum antiviral effects by inhibiting activities of key rate-limiting enzymes in the mevalonate pathway, and also SREBP proteins which regulate host cholesterol homeostasis, thereby affecting coronavirus entry, membrane fusion, and pathological syncytia formation (204). Thus, cholesterol metabolism disorder is a double-edged sword; it affects the normal physiological functions of cells, however, weaponizing cholesterol dysregulation in local cell environments such as lipid rafts or endosomes could inhibit coronavirus replication. Therefore, the development of selective cholesterol-modifying drugs targeting key cellular components such as lipid rafts and endosomes in infected cells could be a promising antiviral strategy for the early stages of coronavirus infection.
Author Contributions
CD and XQ conceived the review concept and drafted the article. JD and HW wrote the original draft and prepared figures. YL, LT, YS, CS, and WL edited and reviewed the manuscript. All authors read and agreed to the final published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 32030108), the Foundation of Key Laboratory of Veterinary Biotechnology (No. shklab202001), and Natural Science Foundation of Shanghai (Grant No. 21ZR1476800 & 20ZR1469400). We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript. Figures were drafted using BioRender.com. Adapted from “ZIKV Infection Cycle” and “Mechanisms behind the induction of trained Immunity” by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
References
1. Drosten C, Günther S, Preiser W, van der Werf S, Brodt H-R, Becker S, et al. Identification of a Novel Coronavirus in Patients With Severe Acute Respiratory Syndrome. N Engl J Med (2003) 348(20):1967–76. doi: 10.1056/NEJMoa030747
2. Isaacs D, Flowers D, Clarke JR, Valman HB, Macnaughton MR. Epidemiology of Coronavirus Infections. Arch Dis Childhood (1983) 58(7):500–3. doi: 10.1136/adc.58.7.500
3. Yang D, Leibowitz JL. The Structure and Functions of Coronavirus Genomic 3' and 5' Ends. Virus Res (2015) 206:120–33. doi: 10.1016/j.virusres.2015.02.025
4. Cui J, Li F, Shi Z-L. Origin and Evolution of Pathogenic Coronaviruses. Nat Rev Microbiol (2019) 17(3):181–92. doi: 10.1038/s41579-018-0118-9
5. Song Z, Xu Y, Bao L, Zhang L, Yu P, Qu Y, et al. From Sars to Mers, Thrusting Coronaviruses Into the Spotlight. Viruses (2019) 11(1):59. doi: 10.3390/v11010059
6. Chan JF-W, Lau SK-P, Woo PC-Y. The Emerging Novel Middle East Respiratory Syndrome Coronavirus: The "Knowns" and "Unknowns". J Formos Med Assoc (2013) 112(7):372–81. doi: 10.1016/j.jfma.2013.05.010
7. Belouzard S, Millet JK, Licitra BN, Whittaker GR. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses (2012) 4(6):1011–33. doi: 10.3390/v4061011
8. Kirtipal N, Bharadwaj S, Kang SG. From Sars to Sars-Cov-2, Insights on Structure, Pathogenicity and Immunity Aspects of Pandemic Human Coronaviruses. Infect Genet Evol (2020) 85:104502. doi: 10.1016/j.meegid.2020.104502
9. Wacharapluesadee S, Tan CW, Maneeorn P, Duengkae P, Zhu F, Joyjinda Y, et al. Evidence for Sars-Cov-2 Related Coronaviruses Circulating in Bats and Pangolins in Southeast Asia. Nat Commun (2021) 12(1):972. doi: 10.1038/s41467-021-21240-1
10. Tortorici MA, Veesler D. Structural Insights Into Coronavirus Entry:. Adv Virus Res (2019) 105:93–116. doi: 10.1016/bs.aivir.2019.08.002
11. Gouilh MA, Puechmaille SJ, Gonzalez J-P, Teeling E, Kittayapong P, Manuguerra J-C. Sars-Coronavirus Ancestor's Foot-Prints in South-East Asian Bat Colonies and the Refuge Theory. Infect Genet Evol (2011) 11(7):1690–702. doi: 10.1016/j.meegid.2011.06.021
12. Yang Y-L, Qin P, Wang B, Liu Y, Xu G-H, Peng L, et al. Broad Cross-Species Infection of Cultured Cells by Bat Hku2-Related Swine Acute Diarrhea Syndrome Coronavirus and Identification of Its Replication in Murine Dendritic Cells Highlight Its Potential for Diverse Interspecies Transmission. J Virol (2019) 93(24):e01448-19. doi: 10.1128/JVI.01448-19
13. Zhou P, Fan H, Lan T, Yang X-L, Shi W-F, Zhang W, et al. Fatal Swine Acute Diarrhoea Syndrome Caused by an Hku2-Related Coronavirus of Bat Origin. Nature (2018) 556(7700):255–8. doi: 10.1038/s41586-018-0010-9
14. Li F. Structural Analysis of Major Species Barriers Between Humans and Palm Civets for Severe Acute Respiratory Syndrome Coronavirus Infections. J Virol (2008) 82(14):6984–91. doi: 10.1128/JVI.00442-08
15. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A Novel Coronavirus From Patients With Pneumonia in China, 2019. N Engl J Med (2020) 382(8):727–33. doi: 10.1056/NEJMoa2001017
16. V'Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus Biology and Replication: Implications for Sars-Cov-2. Nat Rev Microbiol (2021) 19(3):155–70. doi: 10.1038/s41579-020-00468-6
17. Asselah T, Durantel D, Pasmant E, Lau G, Schinazi RF. Covid-19: Discovery, Diagnostics and Drug Development. J Hepatol (2021) 74(1):168–84. doi: 10.1016/j.jhep.2020.09.031
18. Yang Y, Peng F, Wang R, Yange M, Guan K, Jiang T, et al. The Deadly Coronaviruses: The 2003 Sars Pandemic and the 2020 Novel Coronavirus Epidemic in China. J Autoimmun (2020) 109:102434. doi: 10.1016/j.jaut.2020.102434
19. Yan Y, Shin WI, Pang YX, Meng Y, Lai J, You C, et al. The First 75 Days of Novel Coronavirus (Sars-CoV-2) Outbreak: Recent Advances, Prevention, and Treatment. Int J Environ Res Public Health (2020) 17(7):2323. doi: 10.3390/ijerph17072323
20. Shi J, Wen Z, Zhong G, Yang H, Wang C, Huang B, et al. Susceptibility of Ferrets, Cats, Dogs, and Other Domesticated Animals to Sars-Coronavirus 2. Science (2020) 368(6494):1016–20. doi: 10.1126/science.abb7015
21. Azevedo RB, Botelho BG, JVGd H, Ferreira LVL, Junqueira de Andrade LZ, Oei SSML, et al. Covid-19 and the Cardiovascular System: A Comprehensive Review. J Hum Hypertens (2021) 35(1):4–11. doi: 10.1038/s41371-020-0387-4
22. Stasi C, Fallani S, Voller F, Silvestri C. Treatment for Covid-19: An Overview. Eur J Pharmacol (2020) 889:173644. doi: 10.1016/j.ejphar.2020.173644
23. Xu J, Xu X, Jiang L, Dua K, Hansbro PM, Liu G. Sars-CoV-2 Induces Transcriptional Signatures in Human Lung Epithelial Cells That Promote Lung Fibrosis. Respir Res (2020) 21(1):182. doi: 10.1186/s12931-020-01445-6
24. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. Sars-CoV-2 Binds Platelet Ace2 to Enhance Thrombosis in Covid-19. J Hematol Oncol (2020) 13(1):120. doi: 10.1186/s13045-020-00954-7
25. Xiang Z, Liu J, Shi D, Chen W, Li J, Yan R, et al. Glucocorticoids Improve Severe or Critical Covid-19 by Activating Ace2 and Reducing Il-6 Levels. Int J Biol Sci (2020) 16(13):2382–91. doi: 10.7150/ijbs.47652
26. Awadasseid A, Wu Y, Tanaka Y, Zhang W. Current Advances in the Development of Sars-CoV-2 Vaccines. Int J Biol Sci (2021) 17(1):8–19. doi: 10.7150/ijbs.52569
27. Rezaei A, Neshat S, Heshmat-Ghahdarijani K. Alterations of Lipid Profile in Covid-19: A Narrative Review. Curr Probl Cardiol (2021) 47(3):100907. doi: 10.1016/j.cpcardiol.2021.100907
28. Dong E, Du H, Gardner LM. An Interactive Web-Based Dashboard to Track Covid-19 in Real Time. Lancet Infect Dis (2020) 20(5):533–4. doi: 10.1016/S1473-3099(20)30120-1
29. Kumar S, Singh B, Kumari P, Kumar PV, Agnihotri G, Khan S, et al. Identification of Multipotent Drugs for Covid-19 Therapeutics With the Evaluation of Their Sars-CoV2 Inhibitory Activity. Comput Struct Biotechnol J (2021) 19:1998–2017. doi: 10.1016/j.csbj.2021.04.014
30. Majumder J, Minko T. Recent Developments on Therapeutic and Diagnostic Approaches for Covid-19. AAPS J (2021) 23(1):14. doi: 10.1208/s12248-020-00532-2
31. Nguyen HL, Lan PD, Thai NQ, Nissley DA, O'Brien EP, Li MS. Does Sars-Cov-2 Bind to Human Ace2 More Strongly Than Does Sars-CoV? J Phys Chem B (2020) 124(34):7336–47. doi: 10.1021/acs.jpcb.0c04511
32. Khateeb J, Li Y, Zhang H. Emerging Sars-CoV-2 Variants of Concern and Potential Intervention Approaches. Crit Care (2021) 25(1):244. doi: 10.1186/s13054-021-03662-x
33. Harrington D, Kele B, Pereira S, Couto-Parada X, Riddell A, Forbes S, et al. Confirmed Reinfection With Sars-CoV-2 Variant Voc-202012/01. Clin Infect Dis (2021) 73(10):1946–7. doi: 10.1093/cid/ciab014
34. Sheikh A, McMenamin J, Taylor B, Robertson C. Sars-CoV-2 Delta Voc in Scotland: Demographics, Risk of Hospital Admission, and Vaccine Effectiveness. Lancet (2021) 397(10293):2461–2. doi: 10.1016/S0140-6736(21)01358-1
35. Leung K, Shum MH, Leung GM, Lam TT, Wu JT. Early Transmissibility Assessment of the N501y Mutant Strains of Sars-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill (2021) 26(1):2002106. doi: 10.2807/1560-7917.ES.2020.26.1.2002106
36. Pereira F. Sars-CoV-2 Variants Combining Spike Mutations and the Absence of Orf8 May Be More Transmissible and Require Close Monitoring. Biochem Biophys Res Commun (2021) 550:8–14. doi: 10.1016/j.bbrc.2021.02.080
37. Gong Y-N, Tsao K-C, Hsiao M-J, Huang C-G, Huang P-N, Huang P-W, et al. Sars-CoV-2 Genomic Surveillance in Taiwan Revealed Novel Orf8-Deletion Mutant and Clade Possibly Associated With Infections in Middle East. Emerg Microbes Infect (2020) 9(1):1457–66. doi: 10.1080/22221751.2020.1782271
38. Islam OK, Al-Emran HM, Hasan MS, Anwar A, Jahid MIK, Hossain MA. Emergence of European and North American Mutant Variants of Sars-CoV-2 in South-East Asia. Transbound Emerg Dis (2021) 68(2):824–32. doi: 10.1111/tbed.13748
39. Jungnick S, Hobmaier B, Mautner L, Hoyos M, Haase M, Baiker A, et al. Detection of the New Sars-CoV-2 Variants of Concern B.1.1.7 and B.1.351 in Five Sars-CoV-2 Rapid Antigen Tests (Rats), Germany, March 2021. Euro Surveill (2021) 26(16):2100413. doi: 10.2807/1560-7917.ES.2021.26.16.2100413
40. Bal A, Destras G, Gaymard A, Stefic K, Marlet J, Eymieux S, et al. Two-Step Strategy for the Identification of Sars-CoV-2 Variant of Concern 202012/01 and Other Variants With Spike Deletion H69-V70, France, August to December 2020. Euro Surveill (2021) 26(3):2100008. doi: 10.2807/1560-7917.ES.2021.26.3.2100008
41. Galloway SE, Paul P, MacCannell DR, Johansson MA, Brooks JT, MacNeil A, et al. Emergence of Sars-CoV-2 B.1.1.7 Lineage - United States, December 29, 2020-January 12, 2021. MMWR Morb Mortal Wkly Rep (2021) 70(3):95–9. doi: 10.15585/mmwr.mm7003e2
42. Charkiewicz R, Nikliński J, Biecek P, Kiśluk J, Pancewicz S, Moniuszko-Malinowska AM, et al. The First Sars-CoV-2 Genetic Variants of Concern (Voc) in Poland: The Concept of a Comprehensive Approach to Monitoring and Surveillance of Emerging Variants. Adv Med Sci (2021) 66(2):237–45. doi: 10.1016/j.advms.2021.03.005
43. Loconsole D, Sallustio A, Accogli M, Leaci A, Sanguedolce A, Parisi A, et al. Investigation of an Outbreak of Symptomatic Sars-CoV-2 Voc 202012/01-Lineage B.1.1.7 Infection in Healthcare Workers, Italy. Clin Microbiol Infect (2021) 27(8):1174.e1–.4. doi: 10.1016/j.cmi.2021.05.007
44. Pereira F. Sars-CoV-2 Variants Lacking Orf8 Occurred in Farmed Mink and Pangolin. Gene (2021) 784:145596. doi: 10.1016/j.gene.2021.145596
45. Sharun K, Tiwari R, Natesan S, Dhama K. Sars-CoV-2 Infection in Farmed Minks, Associated Zoonotic Concerns, and Importance of the One Health Approach During the Ongoing Covid-19 Pandemic. Vet Q (2021) 41(1):50–60. doi: 10.1080/01652176.2020.1867776
46. Sharun K, Dhama K, Pawde AM, Gortázar C, Tiwari R, Bonilla-Aldana DK, et al. Sars-CoV-2 in Animals: Potential for Unknown Reservoir Hosts and Public Health Implications. Vet Q (2021) 41(1):181–201. doi: 10.1080/01652176.2021.1921311
47. Apicella M, Campopiano MC, Mantuano M, Mazoni L, Coppelli A, Del Prato S. Covid-19 in People With Diabetes: Understanding the Reasons for Worse Outcomes. Lancet Diabetes Endocrinol (2020) 8(9):782–92. doi: 10.1016/S2213-8587(20)30238-2
48. Mostaza JM, Salinero-Fort MA, Cardenas-Valladolid J, Rodriguez-Artalejo F, Díaz-Almiron M, Vich-Pérez P, et al. Pre-Infection Hdl-Cholesterol Levels and Mortality Among Elderly Patients Infected With Sars-CoV-2. Atherosclerosis (2022) 341:13–9. doi: 10.1016/j.atherosclerosis.2021.12.009
49. Drucker DJ. Diabetes, Obesity, Metabolism, and Sars-CoV-2 Infection: The End of the Beginning. Cell Metab (2021) 33(3):479–98. doi: 10.1016/j.cmet.2021.01.016
50. Lee W, Ahn JH, Park HH, Kim HN, Kim H, Yoo Y, et al. Covid-19-Activated Srebp2 Disturbs Cholesterol Biosynthesis and Leads to Cytokine Storm. Signal Transduct Target Ther (2020) 5(1):186. doi: 10.1038/s41392-020-00292-7
51. Hilser JR, Han Y, Biswas S, Gukasyan J, Cai Z, Zhu R, et al. Association of Serum Hdl-Cholesterol and Apolipoprotein A1 Levels With Risk of Severe Sars-CoV-2 Infection. J Lipid Res (2021) 62:100061. doi: 10.1016/j.jlr.2021.100061
52. O'Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, Paireau J, et al. Age-Specific Mortality and Immunity Patterns of Sars-CoV-2. Nature (2021) 590(7844):140–5. doi: 10.1038/s41586-020-2918-0
53. Mostaza JM, García-Iglesias F, González-Alegre T, Blanco F, Varas M, Hernández-Blanco C, et al. Clinical Course and Prognostic Factors of Covid-19 Infection in an Elderly Hospitalized Population. Arch Gerontol Geriatr (2020) 91:104204. doi: 10.1016/j.archger.2020.104204
54. Richter FC, Alrubayyi A, Teijeira Crespo A, Hulin-Curtis S. Impact of Obesity and Sars-CoV-2 Infection: Implications for Host Defence - A Living Review. Oxford Open Immunol (2021) 2(1):iqab001. doi: 10.1093/oxfimm/iqab001
55. Sohrabi Y, Dos Santos JC, Dorenkamp M, Findeisen H, Godfrey R, Netea MG, et al. Trained Immunity as a Novel Approach Against Covid-19 With a Focus on Bacillus Calmette-Guérin Vaccine: Mechanisms, Challenges and Perspectives. Clin Transl Immunol (2020) 9(12):e1228. doi: 10.1002/cti2.1228
56. Goyal P, Choi JJ, Pinheiro LC, Schenck EJ, Chen R, Jabri A, et al. Clinical Characteristics of Covid-19 in New York City. N Engl J Med (2020) 382(24):2372–4. doi: 10.1056/NEJMc2010419
57. Wang X, Zhao Y, Yan F, Wang T, Sun W, Feng N, et al. Viral and Host Transcriptomes in Sars-CoV-2 Infected Human Lung Cells. J Virol (2021) 95(18):e0060021. doi: 10.1128/JVI.00600-21
58. Luo J, Yang H, Song B-L. Mechanisms and Regulation Of Cholesterol Homeostasis. Nat Rev Mol Cell Biol (2020) 21(4):225–45. doi: 10.1038/s41580-019-0190-7
59. Andrews NW. Cholesterol Access Aids Pathogen Spread. Nat Microbiol (2020) 5(7):881–2. doi: 10.1038/s41564-020-0744-7
60. Hu B, Höfer CT, Thiele C, Veit M. Cholesterol Binding to the Transmembrane Region of a Group 2 Hemagglutinin (Ha) of Influenza Virus Is Essential for Virus Replication, Affecting Both Virus Assembly and Ha Fusion Activity. J Virol (2019) 93(15):e00555-19. doi: 10.1128/JVI.00555-19
61. Liu KN, Boxer SG. Target Membrane Cholesterol Modulates Single Influenza Virus Membrane Fusion Efficiency But Not Rate. Biophys J (2020) 118(10):2426–33. doi: 10.1016/j.bpj.2020.03.021
62. Lee J, Kreutzberger AJB, Odongo L, Nelson EA, Nyenhuis DA, Kiessling V, et al. Ebola Virus Glycoprotein Interacts With Cholesterol to Enhance Membrane Fusion and Cell Entry. Nat Struct Mol Biol (2021) 28(2):181–9. doi: 10.1038/s41594-020-00548-4
63. Dang EV, Madhani HD, Vance RE. Cholesterol in Quarantine. Nat Immunol (2020) 21(7):716–7. doi: 10.1038/s41590-020-0712-7
64. Takano T, Satomi Y, Oyama Y, Doki T, Hohdatsu T. Differential Effect of Cholesterol on Type I and Ii Feline Coronavirus Infection. Arch Virol (2016) 161(1):125–33. doi: 10.1007/s00705-015-2655-0
65. Cao X, Yin R, Albrecht H, Fan D, Tan W. Cholesterol: A New Game Player Accelerating Vasculopathy Caused by Sars-CoV-2? Am J Physiol Endocrinol Metab (2020) 319(1):E197–202. doi: 10.1152/ajpendo.00255.2020
66. Meher G, Bhattacharjya S, Chakraborty H. Membrane Cholesterol Modulates Oligomeric Status and Peptide-Membrane Interaction of Severe Acute Respiratory Syndrome Coronavirus Fusion Peptide. J Phys Chem B (2019) 123(50):10654–62. doi: 10.1021/acs.jpcb.9b08455
67. Sanders DW, Jumper CC, Ackerman PJ, Bracha D, Donlic A, Kim H, et al. Sars-CoV-2 Requires Cholesterol for Viral Entry and Pathological Syncytia Formation. Elife (2021) 10:e65962. doi: 10.7554/eLife.65962
68. Thorp EB, Gallagher TM. Requirements for Ceacams and Cholesterol During Murine Coronavirus Cell Entry. J Virol (2004) 78(6):2682–92. doi: 10.1128/JVI.78.6.2682-2692.2004
69. Heaton NS, Randall G. Multifaceted Roles for Lipids in Viral Infection. Trends Microbiol (2011) 19(7):368–75. doi: 10.1016/j.tim.2011.03.007
70. Syed GH, Amako Y, Siddiqui A. Hepatitis C Virus Hijacks Host Lipid Metabolism. Trends Endocrinol Metab (2010) 21(1):33–40. doi: 10.1016/j.tem.2009.07.005
71. Del Campo JA, Romero-Gómez M. Modulation of Host Lipid Metabolism by Hepatitis C Virus: Role of New Therapies. World J Gastroenterol (2015) 21(38):10776–82. doi: 10.3748/wjg.v21.i38.10776
72. Lange PT, Lagunoff M, Tarakanova VL. Chewing the Fat: The Conserved Ability of DNA Viruses to Hijack Cellular Lipid Metabolism. Viruses (2019) 11(2):119. doi: 10.3390/v11020119
73. Cheng K, Riva L, Sinha S, Pal LR, Nair NU, Martin-Sancho L, et al. Genome-Scale Metabolic Modeling Reveals Sars-CoV-2-Induced Host Metabolic Reprogramming and Identifies Metabolic Antiviral Targets. bioRxiv (2021). doi: 10.1101/2021.01.27.428543
74. Balgoma D, Gil-de-Gómez L, Montero O. Lipidomics Issues on Human Positive Ssrna Virus Infection: An Update. Metabolites (2020) 10(9):356. doi: 10.3390/metabo10090356
75. Luquain-Costaz C, Rabia M, Hullin-Matsuda F, Delton I. Bis(Monoacylglycero)Phosphate, an Important Actor in the Host Endocytic Machinery Hijacked by Sars-CoV-2 and Related Viruses. Biochimie (2020) 179:247–56. doi: 10.1016/j.biochi.2020.10.018
76. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, et al. The Transcription Factor Stat-1 Couples Macrophage Synthesis of 25-Hydroxycholesterol to the Interferon Antiviral Response. Immunity (2013) 38(1):106–18. doi: 10.1016/j.immuni.2012.11.004
77. Xiao J, Li W, Zheng X, Qi L, Wang H, Zhang C, et al. Targeting 7-Dehydrocholesterol Reductase Integrates Cholesterol Metabolism and Irf3 Activation to Eliminate Infection. Immunity (2020) 52(1):109–22.e6. doi: 10.1016/j.immuni.2019.11.015
78. Osuna-Ramos JF, Reyes-Ruiz JM, Del Ángel RM. The Role of Host Cholesterol During Flavivirus Infection. Front Cell Infect Microbiol (2018) 8:388. doi: 10.3389/fcimb.2018.00388
79. Sturley SL, Rajakumar T, Hammond N, Higaki K, Márka Z, Márka S, et al. Potential Covid-19 Therapeutics From a Rare Disease: Weaponizing Lipid Dysregulation to Combat Viral Infectivity. J Lipid Res (2020) 61(7):972–82. doi: 10.1194/jlr.R120000851
80. Proto MC, Fiore D, Piscopo C, Pagano C, Galgani M, Bruzzaniti S, et al. Lipid Homeostasis and Mevalonate Pathway in Covid-19: Basic Concepts and Potential Therapeutic Targets. Prog Lipid Res (2021) 82:101099. doi: 10.1016/j.plipres.2021.101099
81. Yan B, Chu H, Yang D, Sze K, Lai P, Yuan S, et al. Characterization of the Lipidomic Profile of Human Coronavirus-Infected Cells: Implications for Lipid Metabolism Remodeling Upon Coronavirus Replication. Viruses (2019) 11(1):73. doi: 10.3390/v11010073
82. Daniloski Z, Jordan TX, Wessels H-H, Hoagland DA, Kasela S, Legut M, et al. Identification of Required Host Factors for Sars-CoV-2 Infection in Human Cells. Cell (2021) 184(1):92–105.e16. doi: 10.1016/j.cell.2020.10.030
83. Hoagland DA, Clarke D, Moeller R, Han Y, Tenoever BR. Modulating the Transcriptional Landscape of Sars-CoV-2 as an Effective Method for Developing Antiviral Compounds. (2020). doi: 10.1101/2020.07.12.199687
84. Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM, et al. Genetic Screens Identify Host Factors for Sars-CoV-2 and Common Cold Coronaviruses. Cell (2021) 184(1):106–19.e14. doi: 10.1016/j.cell.2020.12.004
85. Lorizate M, Kräusslich H-G. Role of Lipids in Virus Replication. Cold Spring Harb Perspect Biol (2011) 3(10):a004820. doi: 10.1101/cshperspect.a004820
86. Taylor MP, Koyuncu OO, Enquist LW. Subversion of the Actin Cytoskeleton During Viral Infection. Nat Rev Microbiol (2011) 9(6):427. doi: 10.1038/nrmicro2574
87. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. Sars-CoV-2 Cell Entry Depends on Ace2 and Tmprss2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80.e8. doi: 10.1016/j.cell.2020.02.052
88. Matsuyama S, Taguchi F. Two-Step Conformational Changes in a Coronavirus Envelope Glycoprotein Mediated by Receptor Binding and Proteolysis. J Virol (2009) 83(21):11133–41. doi: 10.1128/JVI.00959-09
89. Li X, Zhu W, Fan M, Zhang J, Peng Y, Huang F, et al. Dependence of Sars-CoV-2 Infection on Cholesterol-Rich Lipid Raft and Endosomal Acidification. Comput Struct Biotechnol J (2021) 19:1933–43. doi: 10.1016/j.csbj.2021.04.001
90. Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. Protease-Mediated Enhancement of Severe Acute Respiratory Syndrome Coronavirus Infection. Proc Natl Acad Sci USA (2005) 102(35):12543–7. doi: 10.1073/pnas.0503203102
91. Wang H, Yuan X, Sun Y, Mao X, Meng C, Tan L, et al. Infectious Bronchitis Virus Entry Mainly Depends on Clathrin Mediated Endocytosis and Requires Classical Endosomal/Lysosomal System. Virology (2019) 528:118–36. doi: 10.1016/j.virol.2018.12.012
92. Li Z, Zhao K, Lan Y, Lv X, Hu S, Guan J, et al. Porcine Hemagglutinating Encephalomyelitis Virus Enters Neuro-2a Cells Via Clathrin-Mediated Endocytosis in a Rab5-, Cholesterol-, and Ph-Dependent Manner. J Virol (2017) 91(23):e01083-17. doi: 10.1128/JVI.01083-17
93. Wei X, She G, Wu T, Xue C, Cao Y. Pedv Enters Cells Through Clathrin-, Caveolae-, and Lipid Raft-Mediated Endocytosis and Traffics Via the Endo-/Lysosome Pathway. Vet Res (2020) 51(1):10. doi: 10.1186/s13567-020-0739-7
94. Takano T, Wakayama Y, Doki T. Endocytic Pathway of Feline Coronavirus for Cell Entry: Differences in Serotype-Dependent Viral Entry Pathway. Pathogens (2019) 8(4):300. doi: 10.3390/pathogens8040300
95. Kirchdoerfer RN, Wang N, Pallesen J, Wrapp D, Turner HL, Cottrell CA, et al. Stabilized Coronavirus Spikes Are Resistant to Conformational Changes Induced by Receptor Recognition or Proteolysis. Sci Rep (2018) 8(1):15701. doi: 10.1038/s41598-018-34171-7
96. Belouzard S, Chu VC, Whittaker GR. Activation of the Sars Coronavirus Spike Protein Via Sequential Proteolytic Cleavage at Two Distinct Sites. Proc Natl Acad Sci USA (2009) 106(14):5871–6. doi: 10.1073/pnas.0809524106
97. Perlman S, Netland J. Coronaviruses Post-Sars: Update on Replication and Pathogenesis. Nat Rev Microbiol (2009) 7(6):439–50. doi: 10.1038/nrmicro2147
98. Li F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu Rev Virol (2016) 3(1):237–61. doi: 10.1146/annurev-virology-110615-042301
99. Wei C, Wan L, Yan Q, Wang X, Zhang J, Yang X, et al. Hdl-Scavenger Receptor B Type 1 Facilitates Sars-CoV-2 Entry. Nat Metab (2020) 2(12):1391–400. doi: 10.1038/s42255-020-00324-0
100. Zhu Y, Feng F, Hu G, Wang Y, Yu Y, Zhu Y, et al. A Genome-Wide Crispr Screen Identifies Host Factors That Regulate Sars-CoV-2 Entry. Nat Commun (2021) 12(1):961. doi: 10.1038/s41467-021-21213-4
101. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell Entry Mechanisms of Sars-CoV-2. Proc Natl Acad Sci USA (2020) 117(21):11727–34. doi: 10.1073/pnas.2003138117
102. Tsai JC, Zelus BD, Holmes KV, Weiss SR. The N-Terminal Domain of the Murine Coronavirus Spike Glycoprotein Determines the Ceacam1 Receptor Specificity of the Virus Strain. J Virol (2003) 77(2):841–50. doi: 10.1128/JVI.77.2.841-850.2003
103. Phillips JM, Gallagher T, Weiss SR. Neurovirulent Murine Coronavirus Jhm.Sd Uses Cellular Zinc Metalloproteases for Virus Entry and Cell-Cell Fusion. J Virol (2017) 91(8):e01564-16. doi: 10.1128/JVI.01564-16
104. Shang J, Wan Y, Liu C, Yount B, Gully K, Yang Y, et al. Structure of Mouse Coronavirus Spike Protein Complexed With Receptor Reveals Mechanism for Viral Entry. PloS Pathog (2020) 16(3):e1008392. doi: 10.1371/journal.ppat.1008392
105. Zelus BD, Schickli JH, Blau DM, Weiss SR, Holmes KV. Conformational Changes in the Spike Glycoprotein of Murine Coronavirus Are Induced at 37 Degrees C Either by Soluble Murine Ceacam1 Receptors or by Ph 8. J Virol (2003) 77(2):830–40. doi: 10.1128/JVI.77.2.830-840.2003
106. Devlin C, Pipalia NH, Liao X, Schuchman EH, Maxfield FR, Tabas I. Improvement in Lipid and Protein Trafficking in Niemann-Pick C1 Cells by Correction of a Secondary Enzyme Defect. Traffic (2010) 11(5):601–15. doi: 10.1111/j.1600-0854.2010.01046.x
107. Li G, Su B, Fu P, Bai Y, Ding G, Li D, et al. Npc1-Regulated Dynamic of Clathrin-Coated Pits Is Essential for Viral Entry. Sci China Life Sci (2021) 65(2):41–61. doi: 10.1007/s11427-021-1929-y
108. Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, et al. Ebola Virus Entry Requires the Cholesterol Transporter Niemann-Pick C1. Nature (2011) 477(7364):340–3. doi: 10.1038/nature10348
109. Mingo RM, Simmons JA, Shoemaker CJ, Nelson EA, Schornberg KL, D'Souza RS, et al. Ebola Virus and Severe Acute Respiratory Syndrome Coronavirus Display Late Cell Entry Kinetics: Evidence That Transport to Npc1+ Endolysosomes Is a Rate-Defining Step. J Virol (2015) 89(5):2931–43. doi: 10.1128/JVI.03398-14
110. Ballout RA, Sviridov D, Bukrinsky MI, Remaley AT. The Lysosome: A Potential Juncture Between Sars-CoV-2 Infectivity and Niemann-Pick Disease Type C, With Therapeutic Implications. FASEB J (2020) 34(6):7253–64. doi: 10.1096/fj.202000654R
111. Schloer S, Brunotte L, Goretzko J, Mecate-Zambrano A, Korthals N, Gerke V, et al. Targeting the Endolysosomal Host-Sars-CoV-2 Interface by Clinically Licensed Functional Inhibitors of Acid Sphingomyelinase (Fiasma) Including the Antidepressant Fluoxetine. Emerg Microbes Infect (2020) 9(1):2245–55. doi: 10.1080/22221751.2020.1829082
112. Wei C, Wan L, Zhang Y, Fan C, Zhong H. Cholesterol Metabolism–Impact for Sars-CoV-2 Infection Prognosis, Entry, and Antiviral Therapies. (2020)doi: 10.1101/2020.04.16.20068528
113. Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, et al. Sars Coronavirus Entry Into Host Cells Through a Novel Clathrin- and Caveolae-Independent Endocytic Pathway. Cell Res (2008) 18(2):290–301. doi: 10.1038/cr.2008.15
114. Baglivo M, Baronio M, Natalini G, Beccari T, Chiurazzi P, Fulcheri E, et al. Natural Small Molecules as Inhibitors of Coronavirus Lipid-Dependent Attachment to Host Cells: A Possible Strategy for Reducing Sars-CoV-2 Infectivity? Acta BioMed (2020) 91(1):161–4. doi: 10.23750/abm.v91i1.9402
115. Kočar E, Režen T, Rozman D. Cholesterol, Lipoproteins, and Covid-19: Basic Concepts and Clinical Applications. Biochim Biophys Acta Mol Cell Biol Lipids (2021) 1866(2):158849. doi: 10.1016/j.bbalip.2020.158849
116. Morgan PK, Fang L, Lancaster GI, Murphy AJ. Hematopoiesis Is Regulated by Cholesterol Efflux Pathways and Lipid Rafts: Connections With Cardiovascular Diseases. J Lipid Res (2020) 61(5):667–75. doi: 10.1194/jlr.TR119000267
117. Li G-M, Li Y-G, Yamate M, Li S-M, Ikuta K. Lipid Rafts Play an Important Role in the Early Stage of Severe Acute Respiratory Syndrome-Coronavirus Life Cycle. Microbes Infect (2007) 9(1):96–102. doi: 10.1016/j.micinf.2006.10.015
118. Radenkovic D, Chawla S, Pirro M, Sahebkar A, Banach M. Cholesterol in Relation to Covid-19: Should We Care About It? J Clin Med (2020) 9(6):1909. doi: 10.3390/jcm9061909
119. Guo H, Huang M, Yuan Q, Wei Y, Gao Y, Mao L, et al. The Important Role of Lipid Raft-Mediated Attachment in the Infection of Cultured Cells by Coronavirus Infectious Bronchitis Virus Beaudette Strain. PloS One (2017) 12(1):e0170123. doi: 10.1371/journal.pone.0170123
120. Lu Y, Liu DX, Tam JP. Lipid Rafts Are Involved in Sars-CoV Entry Into Vero E6 Cells. Biochem Biophys Res Commun (2008) 369(2):344–9. doi: 10.1016/j.bbrc.2008.02.023
121. Glende J, Schwegmann-Wessels C, Al-Falah M, Pfefferle S, Qu X, Deng H, et al. Importance of Cholesterol-Rich Membrane Microdomains in the Interaction of the S Protein of Sars-Coronavirus With the Cellular Receptor Angiotensin-Converting Enzyme 2. Virology (2008) 381(2):215–21. doi: 10.1016/j.virol.2008.08.026
122. Yin J, Glende J, Schwegmann-Wessels C, Enjuanes L, Herrler G, Ren X. Cholesterol Is Important for a Post-Adsorption Step in the Entry Process of Transmissible Gastroenteritis Virus. Antiviral Res (2010) 88(3):311–6. doi: 10.1016/j.antiviral.2010.10.002
123. Choi KS, Aizaki H, Lai MMC. Murine Coronavirus Requires Lipid Rafts for Virus Entry and Cell-Cell Fusion But Not for Virus Release. J Virol (2005) 79(15):9862–71. doi: 10.1128/JVI.79.15.9862-9871.2005
124. Zang R, Case JB, Yutuc E, Ma X, Shen S, Gomez Castro MF, et al. Cholesterol 25-Hydroxylase Suppresses Sars-CoV-2 Replication by Blocking Membrane Fusion. Proc Natl Acad Sci USA (2020) 117(50):32105–13. doi: 10.1073/pnas.2012197117
125. Wang S, Li W, Hui H, Tiwari SK, Zhang Q, Croker BA, et al. Cholesterol 25-Hydroxylase Inhibits Sars-CoV-2 and Other Coronaviruses by Depleting Membrane Cholesterol. EMBO J (2020) 39(21):e106057. doi: 10.15252/embj.2020106057
126. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural Basis for the Recognition of Sars-CoV-2 by Full-Length Human Ace2. Science (2020) 367(6485):1444–8. doi: 10.1126/science.abb2762
127. Ren X, Glende J, Yin J, Schwegmann-Wessels C, Herrler G. Importance of Cholesterol for Infection of Cells by Transmissible Gastroenteritis Virus. Virus Res (2008) 137(2):220–4. doi: 10.1016/j.virusres.2008.07.023
128. Abbasi AZ, Kiyani DA, Hamid SM, Saalim M, Fahim A, Jalal N. Spiking Dependence of Sars-CoV-2 Pathogenicity on Tmprss2. J Med Virol (2021) 93(7):4205–18. doi: 10.1002/jmv.26911
129. Senapati S, Banerjee P, Bhagavatula S, Kushwaha PP, Kumar S. Contributions of Human Ace2 and Tmprss2 in Determining Host–Pathogen Interaction of Covid-19. J Genet (2021) 100(1):12. doi: 10.1007/s12041-021-01262-w
130. Zhao H, Lu L, Peng Z, Chen L-L, Meng X, Zhang C, et al. Sars-CoV-2 Omicron Variant Shows Less Efficient Replication and Fusion Activity When Compared With Delta Variant in Tmprss2-Expressed Cells. Emerg Microbes Infect (2022) 11(1):277–83. doi: 10.1080/22221751.2021.2023329
131. Leach DA, Andrea M, Zwacka R, Giottis S, Yates L, Lloyd C, et al. Antiandrogens Target Tmprss2 and Reduce Sars-CoV-2 Virus Entry in Lung Cells. J Endocrine Soc (2021) 5(1):A60–1. doi: 10.1210/jendso/bvab048.123
132. Leach DA, Mohr A, Giotis ES, Cil E, Brooke GN. The Antiandrogen Enzalutamide Downregulates Tmprss2 and Reduces Cellular Entry of Sars-CoV-2 in Human Lung Cells. Nat Commun (2021) 12(1):4068. doi: 10.1038/s41467-021-24342-y
133. Hempel T, Raich L, Olsson S, Azouz NP, Noe F. Molecular Mechanism of Sars-CoV-2 Cell Entry Inhibition Via Tmprss2 by Camostat and Nafamostat Mesylate. (2020). doi: 10.1101/2020.07.21.214098
134. Wettstein L, Weil T, Conzelmann C, Müller J. Alpha-1 Antitrypsin Inhibits Tmprss2 Protease Activity and Sars-CoV-2 Infection. Nat Commun (2021) 12(1):1726. doi: 10.1038/s41467-021-21972-0
135. McCoy J, Wambier C.G, Vano-Galvan S, Shapiro J, Sinclair R, Ramos P.M, et al. Racial Variations in Covid-19 Deaths May Be Due to Androgen Receptor Genetic Variants Associated With Prostate Cancer and Androgenetic Alopecia. Are Anti-Androgens a Potential Treatment for Covid-19? J Cosmetic Dermatol (2020) 19(7):1542–3. doi: 10.1111/jocd.13455
136. Montopoli M, Zumerle S, Vettor R, Rugge M, Alimonti A. Androgen-Deprivation Therapies for Prostate Cancer and Risk of Infection by Sars-CoV-2: A Population-Based Study (N = 4532). Ann Oncol (2020) 31(8): 1040–5. doi: 10.1016/j.annonc.2020.04.479
137. Ragot K, Mackrill JJ, Zarrouk A, Nury T, Aires V, Jacquin A, et al. Absence of Correlation Between Oxysterol Accumulation in Lipid Raft Microdomains, Calcium Increase, and Apoptosis Induction on 158n Murine Oligodendrocytes. Biochem Pharmacol (2013) 86(1):67–79. doi: 10.1016/j.bcp.2013.02.028
138. Dambal S, Alfaqih M, Sanders S, Maravilla E, Ramirez-Torres A, Galvan GC, et al. 27-Hydroxycholesterol Impairs Plasma Membrane Lipid Raft Signaling as Evidenced by Inhibition of Il6-Jak-Stat3 Signaling in Prostate Cancer Cells. Mol Cancer Res (2020) 18(5):671–84. doi: 10.1158/1541-7786.MCR-19-0974
139. Yan J, Li Q-F, Wang L-S, Wang H, Xiao F-J, Yang Y-F, et al. Methyl-β-Cyclodextrin Induces Programmed Cell Death in Chronic Myeloid Leukemia Cells and, Combined With Imatinib, Produces a Synergistic Downregulation of Erk/Spk1 Signaling. Anticancer Drugs (2012) 23(1):22–31. doi: 10.1097/CAD.0b013e32834a099c
140. Jeon JH, Lee YJ, Lee C. Porcine Deltacoronavirus Activates the Raf/Mek/Erk Pathway to Promote Its Replication. Virus Res (2020) 283:197961. doi: 10.1016/j.virusres.2020.197961
141. Lee C, Kim Y, Jeon JH. Jnk and P38 Mitogen-Activated Protein Kinase Pathways Contribute to Porcine Epidemic Diarrhea Virus Infection. Virus Res (2016) 222:1–12. doi: 10.1016/j.virusres.2016.05.018
142. Wang H, Liu D, Sun Y, Meng C, Tan L, Song C, et al. Upregulation of Dusp6 Impairs Infectious Bronchitis Virus Replication by Negatively Regulating Erk Pathway and Promoting Apoptosis. Vet Res (2021) 52(1):7. doi: 10.1186/s13567-020-00866-x
143. Kim Y, Lee C. Extracellular Signal-Regulated Kinase (Erk) Activation Is Required for Porcine Epidemic Diarrhea Virus Replication. Virology (2015) 484:181–93. doi: 10.1016/j.virol.2015.06.007
144. Cai Y, Liu Y, Zhang X. Suppression of Coronavirus Replication by Inhibition of the Mek Signaling Pathway. J Virol (2007) 81(2):446–56. doi: 10.1128/JVI.01705-06
145. Zhou QD, Chi X, Lee MS, Hsieh WY, Mkrtchyan JJ, Feng A-C, et al. Interferon-Mediated Reprogramming of Membrane Cholesterol to Evade Bacterial Toxins. Nat Immunol (2020) 21(7):746–55. doi: 10.1038/s41590-020-0695-4
146. Bosch BJ, van der Zee R, de Haan CAM, Rottier PJM. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J Virol (2003) 77(16):8801–11. doi: 10.1128/JVI.77.16.8801-8811.2003
147. Daya M, Cervin M, Anderson R. Cholesterol Enhances Mouse Hepatitis Virus-Mediated Cell Fusion. Virology (1988) 163(2):276–83. doi: 10.1016/0042-6822(88)90267-X
148. Zu S, Deng Y-Q, Zhou C, Li J, Li L, Chen Q, et al. 25-Hydroxycholesterol Is a Potent Sars-CoV-2 Inhibitor. Cell Res (2020) 30(11):1043–5. doi: 10.1038/s41422-020-00398-1
149. Orlowski S, Mourad J-J, Gallo A, Bruckert E. Coronaviruses, Cholesterol and Statins: Involvement and Application for Covid-19. Biochimie (2021) 189:51–64. doi: 10.1016/j.biochi.2021.06.005
150. Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, et al. The Antiviral Effector Ifitm3 Disrupts Intracellular Cholesterol Homeostasis to Block Viral Entry. Cell Host Microbe (2013) 13(4):452–64. doi: 10.1016/j.chom.2013.03.006
151. Chevallier J, Chamoun Z, Jiang G, Prestwich G, Sakai N, Matile S, et al. Lysobisphosphatidic Acid Controls Endosomal Cholesterol Levels. J Biol Chem (2008) 283(41):27871–80. doi: 10.1074/jbc.M801463200
152. Zhang Y, Song Z, Wang M, Lan M, Zhang K, Jiang P, et al. Cholesterol 25-Hydroxylase Negatively Regulates Porcine Intestinal Coronavirus Replication by the Production of 25-Hydroxycholesterol. Vet Microbiol (2019) 231:129–38. doi: 10.1016/j.vetmic.2019.03.004
153. Liu S-Y, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, et al. Interferon-Inducible Cholesterol-25-Hydroxylase Broadly Inhibits Viral Entry by Production of 25-Hydroxycholesterol. Immunity (2013) 38(1):92–105. doi: 10.1016/j.immuni.2012.11.005
154. Jeon JH, Lee C. Cholesterol Is Important for the Entry Process of Porcine Deltacoronavirus. Arch Virol (2018) 163(11):3119–24. doi: 10.1007/s00705-018-3967-7
155. Shi G, Kenney AD, Kudryashova E, Zani A, Zhang L, Lai KK, et al. Opposing Activities of Ifitm Proteins in Sars-CoV-2 Infection. EMBO J (2021) 40(3):e106501. doi: 10.15252/embj.2020106501
156. Wrensch F, Winkler M, Pöhlmann S. Ifitm Proteins Inhibit Entry Driven by the Mers-Coronavirus Spike Protein: Evidence for Cholesterol-Independent Mechanisms. Viruses (2014) 6(9):3683–98. doi: 10.3390/v6093683
157. Li K, Markosyan RM, Zheng Y-M, Golfetto O, Bungart B, Li M, et al. Ifitm Proteins Restrict Viral Membrane Hemifusion. PloS Pathog (2013) 9(1):e1003124. doi: 10.1371/journal.ppat.1003124
158. Zhao X, Sehgal M, Hou Z, Cheng J, Shu S, Wu S, et al. Identification of Residues Controlling Restriction Versus Enhancing Activities of Ifitm Proteins on Entry of Human Coronaviruses. J Virol (2018) 92(6):e01535-17. doi: 10.1128/JVI.01535-17
159. Masters PS. The Molecular Biology of Coronaviruses. Adv Virus Res (2006) 66:193–292. doi: 10.1016/S0065-3527(06)66005-3
160. Fung TS, Liu DX. Human Coronavirus: Host-Pathogen Interaction. Annu Rev Microbiol (2019) 73:529–57. doi: 10.1146/annurev-micro-020518-115759
161. Paul D, Bartenschlager R. Architecture and Biogenesis of Plus-Strand Rna Virus Replication Factories. World J Virol (2013) 2(2):32–48. doi: 10.5501/wjv.v2.i2.32
162. Stoeck IK, Lee J-Y, Tabata K, Romero-Brey I, Paul D, Schult P, et al. Hepatitis C Virus Replication Depends on Endosomal Cholesterol Homeostasis. J Virol (2018) 92(1):e01196-17. doi: 10.1128/JVI.01196-17
163. Ohashi H, Wang F, Stappenbeck F, Tsuchimoto K, Kobayashi C, Saso W, et al. Identification of Anti-Severe Acute Respiratory Syndrome-Related Coronavirus 2 (Sars-CoV-2) Oxysterol Derivatives In Vitro. Int J Mol Sci (2021) 22(6):3163. doi: 10.3390/ijms22063163
164. Aizaki H, Morikawa K, Fukasawa M, Hara H, Inoue Y, Tani H, et al. Critical Role of Virion-Associated Cholesterol and Sphingolipid in Hepatitis C Virus Infection. J Virol (2008) 82(12):5715–24. doi: 10.1128/jvi.02530-07
165. Mackenzie JM, Khromykh AA, Parton RG. Cholesterol Manipulation by West Nile Virus Perturbs the Cellular Immune Response. Cell Host Microbe (2007) 2(4):229–39. doi: 10.1016/j.chom.2007.09.003
166. Zheng Y-H, Plemenitas A, Fielding CJ, Peterlin BM. Nef Increases the Synthesis of and Transports Cholesterol to Lipid Rafts and Hiv-1 Progeny Virions. Proc Natl Acad Sci USA (2003) 100(14):8460–5. doi: 10.1073/pnas.1437453100
167. Simons K, Ikonen E. How Cells Handle Cholesterol. Science (New York NY) (2000) 290(5497):1721–6. doi: 10.1126/science.290.5497.1721
168. Bashore AC, Liu M, Key C-CC, Boudyguina E, Wang X, Carroll CM, et al. Targeted Deletion of Hepatocyte Abca1 Increases Plasma Hdl (High-Density Lipoprotein) Reverse Cholesterol Transport Via the Ldl (Low-Density Lipoprotein) Receptor. Arterioscler Thromb Vasc Biol (2019) 39(9):1747–61. doi: 10.1161/ATVBAHA.119.312382
169. Pirmoradi L, Seyfizadeh N, Ghavami S, Zeki AA, Shojaei S. Targeting Cholesterol Metabolism in Glioblastoma: A New Therapeutic Approach in Cancer Therapy. J Investig Med (2019) 67(4):715–9. doi: 10.1136/jim-2018-000962
170. Cenedella RJ. Cholesterol Synthesis Inhibitor U18666a and the Role of Sterol Metabolism and Trafficking in Numerous Pathophysiological Processes. Lipids (2009) 44(6):477–87. doi: 10.1007/s11745-009-3305-7
171. Fessler MB. The Intracellular Cholesterol Landscape: Dynamic Integrator of the Immune Response. Trends Immunol (2016) 37(12):819–30. doi: 10.1016/j.it.2016.09.001
172. York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I Ifn Signaling. Cell (2015) 163(7):1716–29. doi: 10.1016/j.cell.2015.11.045
173. Klumperman J, Locker JK, Meijer A, Horzinek MC, Geuze HJ, Rottier PJ. Coronavirus M Proteins Accumulate in the Golgi Complex Beyond the Site of Virion Budding. J Virol (1994) 68(10):6523–34. doi: 10.1128/jvi.68.10.6523-6534.1994
174. Rothwell C, Lebreton A, Young Ng C, Lim JYH, Liu W, Vasudevan S, et al. Cholesterol Biosynthesis Modulation Regulates Dengue Viral Replication. Virology (2009) 389(1-2):8–19. doi: 10.1016/j.virol.2009.03.025
175. Soto-Acosta R, Mosso C, Cervantes-Salazar M, Puerta-Guardo H, Medina F, Favari L, et al. The Increase in Cholesterol Levels at Early Stages After Dengue Virus Infection Correlates With an Augment in Ldl Particle Uptake and Hmg-Coa Reductase Activity. Virology (2013) 442(2):132–47. doi: 10.1016/j.virol.2013.04.003
176. Graham DRM, Chertova E, Hilburn JM, Arthur LO, Hildreth JEK. Cholesterol Depletion of Human Immunodeficiency Virus Type 1 and Simian Immunodeficiency Virus With Beta-Cyclodextrin Inactivates and Permeabilizes the Virions: Evidence for Virion-Associated Lipid Rafts. J Virol (2003) 77(15):8237–48. doi: 10.1128/JVI.77.15.8237-8248.2003
177. Sousa IP, Carvalho CAM, Ferreira DF, Weissmüller G, Rocha GM, Silva JL, et al. Envelope Lipid-Packing as a Critical Factor for the Biological Activity and Stability of Alphavirus Particles Isolated From Mammalian and Mosquito Cells. J Biol Chem (2011) 286(3):1730–6. doi: 10.1074/jbc.M110.198002
178. Sousa IP, Carvalho CAM, Gomes AMO. Current Understanding of the Role of Cholesterol in the Life Cycle of Alphaviruses. Viruses (2020) 13(1):35. doi: 10.3390/v13010035
179. Martín-Acebes MA, Jiménez de Oya N, Saiz J-C. Lipid Metabolism as a Source of Druggable Targets for Antiviral Discovery Against Zika and Other Flaviviruses. Pharmaceuticals (Basel) (2019) 12(2):97. doi: 10.3390/ph12020097
180. Mitsche MA, Mcdonald JG, Hobbs HH, Cohen JC. Flux Analysis of Cholesterol Biosynthesis in Vivo Reveals Multiple Tissue and Cell-Type Specific Pathways. eLife (2015) 4:e07999. doi: 10.7554/eLife.07999
181. Skubic C, Vovk I, Rozman D, Kriman M. Simplified Lc-Ms Method for Analysis of Sterols in Biological Samples. Molecules (2020) 25(18):4116. doi: 10.3390/molecules25184116
182. Ačimovič J, Rozman D. Steroidal Triterpenes of Cholesterol Synthesis. Molecules (2013) 18(4):4002–17. doi: 10.3390/molecules18044002
183. Civra A, Francese R, Gamba P, Testa G, Cagno V, Poli G, et al. 25-Hydroxycholesterol and 27-Hydroxycholesterol Inhibit Human Rotavirus Infection by Sequestering Viral Particles Into Late Endosomes. Redox Biol (2018) 19:318–30. doi: 10.1016/j.redox.2018.09.003
184. Zhao J, Chen J, Li M, Chen M, Sun C. Multifaceted Functions of Ch25h and 25hc to Modulate the Lipid Metabolism, Immune Responses, and Broadly Antiviral Activities. Viruses (2020) 12(7):727. doi: 10.3390/v12070727
185. Li C, Deng Y-Q, Wang S, Ma F, Aliyari R, Huang X-Y, et al. 25-Hydroxycholesterol Protects Host Against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity (2017) 46(3):446–56. doi: 10.1016/j.immuni.2017.02.012
186. Doms A, Sanabria T, Hansen JN, Altan-Bonnet N, Holm GH. 25-Hydroxycholesterol Production by the Cholesterol-25-Hydroxylase Interferon-Stimulated Gene Restricts Mammalian Reovirus Infection. J Virol (2018) 92(18):e01047-18. doi: 10.1128/JVI.01047-18
187. Shrivastava-Ranjan P, Bergeron É, Chakrabarti AK, Albariño CG, Flint M, Nichol ST, et al. 25-Hydroxycholesterol Inhibition of Lassa Virus Infection Through Aberrant Gp1 Glycosylation. mBio (2016) 7(6):e01808. doi: 10.1128/mBio.01808-16
188. Li S, Li L, Zhu H, Shi M, Fan H, Gao Y, et al. Cholesterol 25-Hydroxylase Inhibits Encephalomyocarditis Virus Replication Through Enzyme Activity-Dependent and Independent Mechanisms. Vet Microbiol (2020) 245:108658. doi: 10.1016/j.vetmic.2020.108658
189. Ke W, Fang L, Tao R, Li Y, Jing H, Wang D, et al. Porcine Reproductive and Respiratory Syndrome Virus E Protein Degrades Porcine Cholesterol 25-Hydroxylase Via the Ubiquitin-Proteasome Pathway. J Virol (2019) 93(20):e00767-19. doi: 10.1128/JVI.00767-19
190. Ke W, Wu X, Fang P, Zhou Y, Fang L, Xiao S. Cholesterol 25-Hydroxylase Suppresses Porcine Deltacoronavirus Infection by Inhibiting Viral Entry. Virus Res (2021) 295:198306. doi: 10.1016/j.virusres.2021.198306
191. Civra A, Colzani M, Cagno V, Francese R, Leoni V, Aldini G, et al. Modulation of Cell Proteome by 25-Hydroxycholesterol and 27-Hydroxycholesterol: A Link Between Cholesterol Metabolism and Antiviral Defense. Free Radic Biol Med (2020) 149:30–6. doi: 10.1016/j.freeradbiomed.2019.08.031
192. Qayyum S, Mohammad T, Slominski RM, Hassan MI, Tuckey RC, Raman C, et al. Vitamin D and Lumisterol Novel Metabolites Can Inhibit Sars-CoV-2 Replication Machinery Enzymes. Am J Physiol Endocrinol Metab (2021) 321(2):E246–E51. doi: 10.1152/ajpendo.00174.2021
193. Nagy PD, Strating JRPM, van Kuppeveld FJM. Building Viral Replication Organelles: Close Encounters of the Membrane Types. PloS Pathog (2016) 12(10):e1005912. doi: 10.1371/journal.ppat.1005912
194. Nelemans T, Kikkert M. Viral Innate Immune Evasion and the Pathogenesis of Emerging Rna Virus Infections. Viruses (2019) 11(10):961. doi: 10.3390/v11100961
195. Singaravelu R, Srinivasan P, Pezacki JP. Armand-Frappier Outstanding Student Award — the Emerging Role of 25-Hydroxycholesterol in Innate Immunity 1. Can J Microbiol (2015) 61(8):521. doi: 10.1139/cjm-2015-0292
196. Bietz A, Zhu H, Xue M, Xu C. Cholesterol Metabolism in T Cells. Front Immunol (2017) 8:1664. doi: 10.3389/fimmu.2017.01664
197. Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, et al. Host Defense Against Viral Infection Involves Interferon Mediated Down-Regulation of Sterol Biosynthesis. PloS Biol (2011) 9(3):e1000598. doi: 10.1371/journal.pbio.1000598
198. Chen L, Ma M-Y, Sun M, Jiang L-Y, Zhao X-T, Fang X-X, et al. Endogenous Sterol Intermediates of the Mevalonate Pathway Regulate Hmgcr Degradation and Srebp-2 Processing. J Lipid Res (2019) 60(10):1765–75. doi: 10.1194/jlr.RA119000201
199. O'Neill LAJ. How Low Cholesterol Is Good for Anti-Viral Immunity. Cell (2015) 163(7):1572–4. doi: 10.1016/j.cell.2015.12.004
200. Lu H, Talbot S, Robertson KA, Watterson S, Forster T, Roy D, et al. Rapid Proteasomal Elimination of 3-Hydroxy-3-Methylglutaryl-Coa Reductase by Interferon-Γ in Primary Macrophages Requires Endogenous 25-Hydroxycholesterol Synthesis. Steroids (2015) 99(Pt B):219–29. doi: 10.1016/j.steroids.2015.02.022
201. Timilshina M, You Z, Lacher SM, Acharya S, Jiang L, Kang Y, et al. Activation of Mevalonate Pathway Via Lkb1 Is Essential for Stability of T Cells. Cell Rep (2019) 27(10):2948–61.e7. doi: 10.1016/j.celrep.2019.05.020
202. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. Mtorc1 Couples Immune Signals and Metabolic Programming to Establish T(Reg)-Cell Function. Nature (2013) 499(7459):485–90. doi: 10.1038/nature12297
203. Yuan S, Chu H, Chan JF-W, Ye Z-W, Wen L, Yan B, et al. Srebp-Dependent Lipidomic Reprogramming as a Broad-Spectrum Antiviral Target. Nat Commun (2019) 10(1):120. doi: 10.1038/s41467-018-08015-x
204. Abu-Farha M, Thanaraj TA, Qaddoumi MG, Hashem A, Abubaker J, Al-Mulla F. The Role of Lipid Metabolism in Covid-19 Virus Infection and as a Drug Target. Int J Mol Sci (2020) 21(10):3544. doi: 10.3390/ijms21103544
205. Dias SSG, Soares VC, Ferreira AC, Sacramento CQ, Fintelman-Rodrigues N, Temerozo JR, et al. Lipid Droplets Fuel Sars-CoV-2 Replication and Production of Inflammatory Mediators. PloS Pathog (2020) 16(12):e1009127. doi: 10.1371/journal.ppat.1009127
206. Assmann N, O'Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-Controlled Glucose Metabolism Is Essential for Nk Cell Functional Responses. Nat Immunol (2017) 18(11):1197–206. doi: 10.1038/ni.3838
207. Gold ES, Diercks AH, Podolsky I, Podyminogin RL, Askovich PS, Treuting PM, et al. 25-Hydroxycholesterol Acts as an Amplifier of Inflammatory Signaling. Proc Natl Acad Sci USA (2014) 111(29):10666–71. doi: 10.1073/pnas.1404271111
208. Cyster JG, Dang EV, Reboldi A, Yi T. 25-Hydroxycholesterols in Innate and Adaptive Immunity. Nat Rev Immunol (2014) 14(11):731–43. doi: 10.1038/nri3755
209. Wong MY, Lewis M, Doherty JJ, Shi Y, Cashikar AG, Amelianchik A, et al. 25-Hydroxycholesterol Amplifies Microglial Il-1β Production in an Apoe Isoform-Dependent Manner. J Neuroinflamm (2020) 17(1):192. doi: 10.1186/s12974-020-01869-3
210. Madenspacher JH, Morrell ED, Gowdy KM, McDonald JG, Thompson BM, Muse G, et al. Cholesterol 25-Hydroxylase Promotes Efferocytosis and Resolution of Lung Inflammation. JCI Insight (2020) 5(11):e137189. doi: 10.1172/jci.insight.137189
211. Emgård J, Kammoun H, García-Cassani B, Chesné J, Parigi SM, Jacob J-M, et al. Oxysterol Sensing Through the Receptor Gpr183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity (2018) 48(1):120–32.e8. doi: 10.1016/j.immuni.2017.11.020
212. Koarai A, Yanagisawa S, Sugiura H, Ichikawa T, Kikuchi T, Furukawa K, et al. 25-Hydroxycholesterol Enhances Cytokine Release and Toll-Like Receptor 3 Response in Airway Epithelial Cells. Respir Res (2012) 13:63. doi: 10.1186/1465-9921-13-63
213. Pokharel SM, Shil NK, Gc JB, Colburn ZT, Tsai S-Y, Segovia JA, et al. Integrin Activation by the Lipid Molecule 25-Hydroxycholesterol Induces a Proinflammatory Response. Nat Commun (2019) 10(1):1482. doi: 10.1038/s41467-019-09453-x
214. Abrams ME, Johnson KA, Perelman SS, Zhang L-S, Endapally S, Mar KB, et al. Oxysterols Provide Innate Immunity to Bacterial Infection by Mobilizing Cell Surface Accessible Cholesterol. Nat Microbiol (2020) 5(7):929–42. doi: 10.1038/s41564-020-0701-5
215. Kim H, Lee HS, Ahn JH, Hong KS, Jang JG, An J, et al. Lung-Selective 25-Hydroxycholesterol Nanotherapeutics as a Suppressor of Covid-19-Associated Cytokine Storm. Nano Today (2021) 38:101149. doi: 10.1016/j.nantod.2021.101149
216. Chi X, Bensinger SJ. (Sterol)Ized Immunity: Could Pi3k/Akt3 Be the Answer? Immunity (2020) 52(1):4–6. doi: 10.1016/j.immuni.2019.12.017
217. Ito A, Hong C, Oka K, Salazar JV, Diehl C, Witztum JL, et al. Cholesterol Accumulation in Cd11c Immune Cells Is a Causal and Targetable Factor in Autoimmune Disease. Immunity (2016) 45(6):1311–26. doi: 10.1016/j.immuni.2016.11.008
218. Widenmaier SB, Hotamışlıgil GS. Immune Cell Intolerance for Excess Cholesterol. Immunity (2016) 45(6):1186–8. doi: 10.1016/j.immuni.2016.12.006
219. Schmidt NM, Wing P, Mckeating JA, Maini MK. Cholesterol-Modifying Drugs in Covid-19. Oxford Open Immunol (2020) 1):1. doi: 10.1093/oxfimm/iqaa001
220. Korade Z, Heffer M, Mirnics K. Medication Effects on Developmental Sterol Biosynthesis. Mol Psychiatry (2021) 27(1):490–501. doi: 10.1038/s41380-021-01074-5
221. Yuan S, Chan CC-Y, Chik KK-H, Tsang JO-L, Liang R, Cao J, et al. Broad-Spectrum Host-Based Antivirals Targeting the Interferon and Lipogenesis Pathways as Potential Treatment Options for the Pandemic Coronavirus Disease 2019 (Covid-19). Viruses (2020) 12(6):628. doi: 10.3390/v12060628
222. Sorice M, Misasi R, Riitano G, Manganelli V, Martellucci S, Longo A, et al. Targeting Lipid Rafts as a Strategy Against Coronavirus. Front Cell Dev Biol (2020) 8:618296. doi: 10.3389/fcell.2020.618296
223. Strating JR, van Kuppeveld FJ. Viral Rewiring of Cellular Lipid Metabolism to Create Membranous Replication Compartments. Curr Opin Cell Biol (2017) 47:24–33. doi: 10.1016/j.ceb.2017.02.005
224. Pawar A, Pal A, Goswami K, Squitti R, Rongiolettie M. Molecular Basis of Quercetin as a Plausible Common Denominator of Macrophage-Cholesterol-Fenofibrate Dependent Potential Covid-19 Treatment Axis. Results Chem (2021) 3:100148. doi: 10.1016/j.rechem.2021.100148
225. Jain MK, Ridker PM. Anti-Inflammatory Effects of Statins: Clinical Evidence and Basic Mechanisms. Nat Rev Drug Discovery (2005) 4(12):977–87. doi: 10.1038/nrd1901
226. Spann NJ, Glass CK. Sterols and Oxysterols in Immune Cell Function. Nat Immunol (2013) 14(9):893–900. doi: 10.1038/ni.2681
227. Sirtori CR. The Pharmacology of Statins. Pharmacol Res (2014) 88:3–11. doi: 10.1016/j.phrs.2014.03.002
228. Carmena R, Betteridge DJ. Diabetogenic Action of Statins: Mechanisms. Curr Atheroscler Rep (2019) 21(6):23. doi: 10.1007/s11883-019-0780-z
229. Wang H, Yuan Z, Pavel MA, Jablonski SM, Jablonski J, Hobson R, et al. The Role of High Cholesterol in Age-Related Covid19 Lethality. bioRxiv (2021). doi: 10.1101/2020.05.09.086249
230. Reiner Ž, Hatamipour M, Banach M, Pirro M, Al-Rasadi K, Jamialahmadi T, et al. Statins and the Covid-19 Main Protease: Evidence on Direct Interaction. Arch Med Sci (2020) 16(3):490–6. doi: 10.5114/aoms.2020.94655
231. Oesterle A, Laufs U, Liao JK. Pleiotropic Effects of Statins on the Cardiovascular System. Circ Res (2017) 120(1):229–43. doi: 10.1161/CIRCRESAHA.116.308537
232. Mitchell JD, Fergestrom N, Gage BF, Paisley R, Moon P, Novak E, et al. Impact of Statins on Cardiovascular Outcomes Following Coronary Artery Calcium Scoring. J Am Coll Cardiol (2018) 72(25):3233–42. doi: 10.1016/j.jacc.2018.09.051
233. Mullen PJ, Garcia G, Purkayastha A, Matulionis N, Schmid EW, Momcilovic M, et al. Sars-CoV-2 Infection Rewires Host Cell Metabolism and Is Potentially Susceptible to Mtorc1 Inhibition. Nat Commun (2021) 12(1):1876. doi: 10.1038/s41467-021-22166-4
234. Schwarz B, Sharma L, Roberts L, Peng X, Bermejo S, Leighton I, et al. Cutting Edge: Severe Sars-CoV-2 Infection in Humans Is Defined by a Shift in the Serum Lipidome, Resulting in Dysregulation of Eicosanoid Immune Mediators. J Immunol (2021) 206(2):329–34. doi: 10.4049/jimmunol.2001025
235. Sohrabi Y, Reinecke H, Godfrey R. Altered Cholesterol and Lipid Synthesis Mediates Hyperinflammation in Covid-19. Trends Endocrinol Metab (2021) 32(3):132–4. doi: 10.1016/j.tem.2021.01.001
236. Nguyen M, Bourredjem A, Piroth L, Bouhemad B, Jalil A, Pallot G, et al. High Plasma Concentration of Non-Esterified Polyunsaturated Fatty Acids Is a Specific Feature of Severe Covid-19 Pneumonia. Sci Rep (2021) 11(1):10824. doi: 10.1038/s41598-021-90362-9
237. Tanaka S, De Tymowski C, Assadi M, Zappella N, Jean-Baptiste S, Robert T, et al. Lipoprotein Concentrations Over Time in the Intensive Care Unit Covid-19 Patients: Results From the Apocovid Study. PloS One (2020) 15(9):e0239573. doi: 10.1371/journal.pone.0239573
238. Marcello A, Civra A, Milan Bonotto R, Nascimento Alves L, Rajasekharan S, Giacobone C, et al. The Cholesterol Metabolite 27-Hydroxycholesterol Inhibits Sars-CoV-2 and Is Markedly Decreased in Covid-19 Patients. Redox Biol (2020) 36:101682. doi: 10.1016/j.redox.2020.101682
239. Hu X, Chen D, Wu L, He G, Ye W. Declined Serum High Density Lipoprotein Cholesterol Is Associated With the Severity of Covid-19 Infection. Clin Chim Acta; Int J Clin Chem (2020) 510:105–10. doi: 10.1016/j.cca.2020.07.015
240. Hu X, Chen D, Wu L, He G, Ye W. Low Serum Cholesterol Level Among Patients With Covid-19 Infection in Wenzhou, China. Soc Sci Electronic Publishing (2020) 21. doi: 10.2139/ssrn.3544826
241. Wang G, Zhang Q, Zhao X, Dong H, Wu C, Wu F, et al. Low High-Density Lipoprotein Level Is Correlated With the Severity of Covid-19 Patients: An Observational Study. Lipids Health Dis (2020) 19(1):204. doi: 10.1186/s12944-020-01382-9
242. Ressaire Q, Dudoignon E, Moreno N, Coutrot M, Dépret F. Low Total Cholesterol Blood Level Is Correlated With Pulmonary Severity in Covid-19 Critical Ill Patients. Anaesth Crit Care Pain Med (2020) 39(6):733–5. doi: 10.1016/j.accpm.2020.07.015
243. Correa Y, Waldie S, Thépaut M, Micciula S, Moulin M, Fieschi F, et al. Sars-CoV-2 Spike Protein Removes Lipids From Model Membranes and Interferes With the Capacity of High Density Lipoprotein to Exchange Lipids. J Colloid Interface Sci (2021) 602:732–9. doi: 10.1016/j.jcis.2021.06.056
244. Ding X, Zhang J, Liu L, Yuan X, Zang X, Lu F, et al. High-Density Lipoprotein Cholesterol as a Factor Affecting Virus Clearance in Covid-19 Patients. Respir Med (2020) 175:106218. doi: 10.1016/j.rmed.2020.106218
245. Zhang B, Dong C, Li S, Song X, Wei W, Liu L. Triglyceride to High-Density Lipoprotein Cholesterol Ratio Is an Important Determinant of Cardiovascular Risk and Poor Prognosis in Coronavirus Disease-19: A Retrospective Case Series Study. Diabetes Metab Syndr Obes (2020) 13:3925–36. doi: 10.2147/DMSO.S268992
246. Scalsky RJ, Chen Y-J, Desai K, O'Connell JR, Perry JA, Hong CC. Baseline Cardiometabolic Profiles and Sars-CoV-2 Risk in the Uk Biobank. medRxiv (2020). doi: 10.1101/2020.07.25.20161091
247. Wei X, Zeng W, Su J, Wan H, Yu X, Cao X, et al. Hypolipidemia Is Associated With the Severity of Covid-19. J Clin Lipidol (2020) 14(3):297–304. doi: 10.1016/j.jacl.2020.04.008
248. Fan J, Wang H, Ye G, Cao X, Xu X, Tan W, et al. Letter to the Editor: Low-Density Lipoprotein Is a Potential Predictor of Poor Prognosis in Patients With Coronavirus Disease 2019. Metabolism (2020) 107:154243. doi: 10.1016/j.metabol.2020.154243
249. Sheng Z, Otani H, Brown MS, Goldstein JL. Independent Regulation of Sterol Regulatory Element-Binding Proteins 1 and 2 in Hamster Liver. Proc Natl Acad Sci USA (1995) 92(4):935–8. doi: 10.1073/pnas.92.4.935
250. Brown MS, Goldstein JL. The Srebp Pathway: Regulation of Cholesterol Metabolism by Proteolysis of a Membrane-Bound Transcription Factor. Cell (1997) 89(3):331–40. doi: 10.1016/S0092-8674(00)80213-5
251. Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, et al. Srebp-1, a Basic-Helix-Loop-Helix-Leucine Zipper Protein That Controls Transcription of the Low Density Lipoprotein Receptor Gene. Cell (1993) 75(1):187–97. doi: 10.1016/S0092-8674(05)80095-9
252. Horton JD, Goldstein JL, Brown MS. Srebps: Activators of the Complete Program of Cholesterol and Fatty Acid Synthesis in the Liver. J Clin Invest (2002) 109(9):1125–31. doi: 10.1172/JCI0215593
Keywords: coronavirus, cholesterol, metabolism dysregulation, immune response, therapy
Citation: Dai J, Wang H, Liao Y, Tan L, Sun Y, Song C, Liu W, Qiu X and Ding C (2022) Coronavirus Infection and Cholesterol Metabolism. Front. Immunol. 13:791267. doi: 10.3389/fimmu.2022.791267
Received: 08 October 2021; Accepted: 21 March 2022;
Published: 21 April 2022.
Edited by:
Vishwanath Venketaraman, Western University of Health Sciences, United StatesReviewed by:
Jiansheng Huang, Vanderbilt University Medical Center, United StatesPeter Van Der Voort, University Medical Center Groningen, Netherlands
Damjana Rozman, University of Ljubljana, Slovenia
Copyright © 2022 Dai, Wang, Liao, Tan, Sun, Song, Liu, Qiu and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xusheng Qiu, eHNxaXUxOTgxQHNodnJpLmFjLmNu; Chan Ding, c2hvdmVsZGVlbkBzaHZyaS5hYy5jbg==
†These authors share first authorship