 Elisabeth Taucher
Elisabeth Taucher Iurii Mykoliuk
Iurii Mykoliuk Joerg Lindenmann
Joerg Lindenmann Freyja-Maria Smolle-Juettner
Freyja-Maria Smolle-Juettner- 1Division of Pulmonology, Department of Internal Medicine, Medical University Graz, Graz, Austria
- 2Division of Thoracic Surgery, Department of Surgery, Medical University Graz, Graz, Austria
Cigarette smoking is reported in about one third of adults worldwide. A strong relationship between cigarette smoke exposure and chronic obstructive pulmonary disease (COPD) as well as lung cancer has been proven. However, about 15% of lung cancer cases, and between one fourth and one third of COPD cases, occur in never-smokers. The effects of cigarette smoke on the innate as well as the adaptive immune system have been widely investigated. It is assumed that certain immunologic features contribute to lung cancer and COPD development in the absence of smoking as the major risk factor. In this article, we review different immunological aspects of lung cancer and COPD with a special focus on non-smoking related risk factors.
1 Introduction
Tobacco smoking is reported in about one third of the adult population worldwide (1, 2). Smoke from tobacco is made up of various toxic and carcinogenic compounds, as for instance nicotine, nitrogen oxides and cadmium or carbon monoxide (3, 4). A relation of cigarette smoke exposure to cancers, respiratory-, cardiovascular-, infectious- and neurologic diseases has been proven (5–8). Immune function is considerably altered by smoking, which promotes the secretion of inflammatory mediators, comprising pro-, but also antiinflammatory cytokines (2, 9–12). Chronic inflammation and autoimmunity are two among many systemic effects of smoking (2, 3, 11, 13).
Lung cancer, as the number one cause of cancer-related deaths worldwide, is mainly a consequence of tobacco smoking (14–16). Still, there is a subgroup of lung cancer patients who have never smoked, and they differ from lung cancer patients who have smoked with respect to molecular markers and prognosis (15, 17, 18). About 10-15% of lung cancer cases occur in the never-smoking population (19–21). Known risk factors for lung cancer in never-smokers include second-hand smoking, indoor air pollution, occupational exposure to certain chemicals and a genetic predisposition (22, 23). However, certain immunological aspects may contribute to the susceptibility of never-smokers who develop lung cancer as well.
Chronic obstructive pulmonary disease (COPD) counts among the leading causes of morbidity and mortality worldwide, with the worldwide incidence still increasing (24, 25). Apart from tobacco exposure, which constitutes a major risk factor, other risk factors have also been outlined, including exposure to smoke from biomass fuel, occupational exposure to gases and dusts, a history of respiratory-tract infections in the early childhood or history of tuberculosis as well as air pollution (25, 26). Cigarette smoking is acknowledged as the single most important risk factor for COPD, yet, between one fourth and one third of COPD cases are attributed to lifetime never-smokers (27–30). Like in lung cancer, it remains unclear why certain non-smoking individuals develop COPD.
In this article, we review possible immunological mechanisms leading to lung cancer or COPD in the never-smoking population.
1.1 The Immune System in Elderly Patients
Lung cancer and COPD affect manly the elderly population (31, 32). In the elderly, the propensity for aberrant immune function increases significantly (33). The aging process goes along with changes of the innate, as well as the adaptive immune function (34, 35). Cancer and autoimmune disorders are encountered in elderly patients more frequently, which is partly due to age-related immunological changes, termed immunosenescence (36). Changes of the innate immunity include dendritic cell function, natural killer cells, neutrophil function and macrophages (37). Dendritic cells generally decrease in quantity during the aging process, like for example Langerhans cells in the skin or plasmocytes (38). Aged dendritic cells are characterized by dysfunction of their mitochondria, leading to a reduced ATP turnover and coupling efficiency as well as greater reactive oxygen species (ROS) production (39). Immunosenescene also leads to a dysfunctional maturation and function of natural killer cells (40). Changes in natural killer cell properties that are aging-related comprise a slower response to inflammatory conditions and a general increase of bacterial and fungal infections in the elderly population, which is due to the decline in natural killer cell quantity (41). During aging, spontaneous neutrophil apoptosis, mediated by the increased secretion of proinflammatory cytokines such as INK-1, occurs less frequently (42). Immunosenescence also impairs neutrophil migration to the lung tissue, as demonstrated in a mouse model (43). Insufficient neutrophil migration in elderly patients is most probably due to an increased constitutive PI3K activation, since lower rates of PI3K activation are normally observed in young individuals (44). Macrophages, like other cell types in aging persons, feature telomere shortening, which leads to a decreased GM-CSF-dependent proliferation of these cells, resulting from a decreased phosphorylation of STAT5. In aged mice, it was demonstrated that macrophages became increasingly susceptible to oxidative stress, explaining why DNA damage is caused by ROS to a much greater extent in old people, paving the way for malignant transformation (45). In elderly people, macrophages produce considerably lower amounts of cytokines such as TNF-α and IL-6, and show a reduced expression of the B7 receptor, resulting in a reduced activation of T-cells (46).
The adaptive immune system is also affected by immunosenescence, altering the function of B- and T-lymphocytes. Humoral immunity is impaired in aging patients, because of a relative increase of the memory B cells (IgG(+)IgD(-)CD27(-), double negative, DN) population, leading to a classical low-level chronic inflammatory microenvironment. Naïve/memory B-cell populations in older adults feature a different receptor expression, which has been discussed using the term “inflamm-aging” (47). Interestingly, naïve B-lymphocytes from young persons need very strong signals in order to be activated in vitro, whereas B-cells from elderly patients can be activated when only physiologically stimulated (48). The plasma cell count in the bloodstream of elderly patients was also observed to decline, as compared to young individuals (49). T-lymphocytes feature an altered functionality and distribution in old patients, which is why the susceptibility for infections and other diseases increases with age (50). A decline in naïve T-lymphocyte output is due to an age-related regression of the thymus (51). Another effect of thymus regression in the elderly is a reduced T-lymphocyte diversity (52).
Taken together, the above listed data shows distinct immunological differences in elderly, as compared to young patients. The increased prevalence of lung cancer and COPD in old subjects, smokers and non-smokers alike, is probably a synergistic event of cumulative noxa and immunosenescence.
2 The Effect of Smoking on the Immune Landscape
Smoking alters the immune response in different ways, paving the way for chronic bronchial inflammation and consecutively, lung cancer and COPD. In this chapter, we give an overview on the impacts of chronic cigarette smoke exposure on the immune system.
T-lymphocytes form one major subset of immune cells who mediate the adaptive immune response (2). Naïve T-lymphocytes may be activated and differentiated into effector T-cells, memory- and regulatory T-cells (53). The profound effect of tobacco smoking on T-cell function, including their secretion of proinflammatory mediators, has been shown by various studies (54, 55). One study was performed to analyze the occurrence of T-cells in bronchoalveolar lavage (BAL) fluid and in peripheral blood from non-smokers (n=40) and smokers (n=40) who had a normal lung function, compared with the result in COPD patients (n=38). CD8+ BAL cells were more abundant in smokers than in non-smokers (56). Meanwhile, CD4+ T-cells were found to be lower in the blood as well as the BAL fluid in the smoking group (56). Another study demonstrated a disruption of T-helper cell homeostasis in smokers who developed COPD when compared with healthy non-smokers (57). Second-hand smoking also affects the prevalence of T-cells, leading to an increase of CD3+ T-cells while active smoking increases CD8+ T-cells and lowers CD4+ T-cells (58). Moreover, in circulating T-cell subpopulations, the percentage of Th17 cells is increased in COPD patients, and the percentage of Th1 cells is also increased in COPD patients, and in current smokers without COPD as well (59). In a mouse model where mice were exposed to tobacco smoke for a minimum of six months, a significant elevation of Th1 and Th17 cells was observed (54). Taken together, murine as well as human studies indicate an increase of Th1 and Th17 cell subsets to result from chronic tobacco smoke exposure (2).
Cytotoxic T-lymphocytes (CD8+ cells) exert a main function in host immune defense, killing infected or otherwise impaired cells. In CD8 knockout mice, exposure to chronic cigarette smoke did not lead to an immune response, inflammation or emphysema (60). Furthermore, it was shown that IP-10 from CD8+ cells promoted elastase production from macrophages, consecutively resulting in elastin fragmentation and lung injury (60). Based on this result, it is assumed that CD8+ cells play a major role in the development of COPD. Another study demonstrated human CD8+ cells from COPD patients or from smokers without COPD to express more toll like receptor (TLR)4 and TLR9 proteins than controls. An enhancement in cytokine expression and general activation of circulating CD8+ T cells was also a direct effect of cigarette smoke exposure (61). A study on mice with pulmonary emphysema showed cigarette smoke to increase the percentage of IL-21+ Th17 and IL-21R+ CD8+ T-cells in peripheral blood, and to upregulate their expression of IL-17 and IL-21. Consecutively, perforin and granzyme B were upregulated in the CD8+ cells as well, showing a regulation of CD8+ T-cell function by Th17 cells in lung emphysema (62).
Also, regulatory T-cell function is disrupted by cigarette smoke exposure, as it was demonstrated in a study on BAL fluid from COPD patients or healthy smokers (63). An analysis of women smoking actively or passively during pregnancy showed a reduced regulatory T-cell count in the umbilical cord blood, leading to a higher amount of atopic dermatitis or food allergies in infancy (64).
According to epidemiologic studies, a higher prevalence of memory B-cells in the peripheral blood and memory IgG+ B-cells in the lung results from chronic smoking, when compared to non-smokers (65, 66). Cigarette smoke exposure downregulated B220+CD34− pre-B-cells and/or B220+CD34+ pro-B-cells in the bone marrow of mice (67, 68).
Memory T-cells (CD3+CD45RO+, CD4+CD45RO+) and class-switched memory B-cells are elevated as a consequence of chronic cigarette smoke exposure (65, 69–71). Interestingly, other data suggests the opposite effect of cigarette smoking on memory T-cells, namely a significant reduction of CD3+CD45RO+ and CD4+CD45RO+ memory T-cells in the blood of children exposed to second-hand smoke, accompanying with augmented percentages of CD3+ and CD4+CD45RA+ naive T-cells (58).
Not surprisingly, cigarette smoking has been linked to aberrations in the innate immune system as well (72–74). One study showed that exposure to cigarette smoke led to an elevation of IL-33 secretion from epithelial cells and changed the expression profile of IL-33 cognate receptor ST2 in various immune cells (75). In the same study, an enhanced proinflammatory response of macrophages and natural killer (NK) cells in case of inflammation, due to an augmented expression of ST2 was shown (75). Cigarette smoking caused inflammatory responses mediated by neutrophils and monocytes in a murine model, despite activated CD4+ T-cells being present in the murine lungs (76). This finding indicates that the innate immunity alone may cause acute inflammation as a response to smoke stimulation (76). It has been shown that TLRs are significantly involved in inflammatory response mechanisms caused by smoking (77). For instance, an analysis of patients suffering from periodontitis showed smoking to upregulate mRNA expression of TLR2 and TLR4 in the gingival tissue (78, 79). The results of studies investigating the effect of cigarette smoking on dendritic cells, however, are contradictory, because it has been found that cigarette smoke can suppress as well as promote dendritic cell development both in humans and mice (2, 80, 81). The differences in the immune response of smokers and non-smokers were illustrated by a study on the response of primary bronchial epithelial cells to pseudomonas aeruginosa lipopolysaccharide (82). Primary bronchial epithelial cells from smokers and non-smokers were compared in vitro. Bronchial epithelial cells from 16 patients with COPD, 10 healthy smokers and 9 non-smokers were cultured and exposed to cigarette smoke, prior to stimulation with pseudomonas aeruginosa lipopolysaccharide. It was found that more IL-8 and IL-6 was released from the COPD cultures than from cells of healthy smokers or non-smokers. Interestingly, pre-treatment with cigarette smoke reduced the release of IL-8 from the COPD patients’ bronchial epithelial cells, however, led to an increase of IL-8 release in the cells of smokers without airway obstruction and of non-smokers. After cigarette smoke treatment, TLR4 expression, mitogen activated protein kinase (MAPK) and NF-kB activation went down in COPD cultures, but not in the other two groups (82). It is therefore concluded that exposure to tobacco smoke decreases the inflammatory response to pseudomonas aeruginosa lipopolysaccharide in patients with COPD, but not in non-smokers or smokers without airflow limitation (82).
Mounting evidence suggests that the MAPK signaling pathway is strongly involved in COPD pathogenesis, contributing to a chronic inflammatory state by cell chemotaxis, remodeling of the bronchi and alveoli, insensitivity to corticosteroid treatment and airflow limitation (83, 84). The knowledge about dysfunctional MAPK signaling in vivo in COPD led to the investigation of MAPK inhibitors as possible COPD therapeutics, aiming to resolve in particular the aberrant neutrophil apoptosis observed in COPD (85, 86). Also for NF-kB, an involvement in COPD/chronic airway inflammation has been demonstrated in vivo (87). Therefore, NF-kB has increasingly become the focus of interest, outlining it as a potential therapeutic target in COPD treatment, and moreover, in the context of COVID-19 pneumonia (88).
The above listed data shows the profound impact cigarette smoking has on the immune function, affecting the innate as well as the adaptive immune response. Certain immunological properties, resulting from risk factors of pulmonary inflammation other than cigarette smoke, may specifically contribute to COPD and lung cancer in lifetime non-smokers.
3 Non-Smoking Related Risk Factors and Their Impact on the Immune Response
3.1 Air Pollution and Oxidative Stress
3.1.1 The Impact of Air Pollution and Oxidative Stress on COPD Development
Air pollution is one of the main causes of COPD in lifetime non-smokers. Acute airway obstruction, exacerbations of asthma and COPD and a general increase in emergency department visits have been reported as a consequence of exposure to particulate air pollution (89, 90). Rudell et al. published a study where the adverse effect of diesel exhaust on the airways was investigated (90). 10 healthy and never-smoking individuals were exposed to diluted diesel exhaust without a ceramic particle trap for one hour, three times, with several weeks between exposures. 24 hours after the exposures, bronchoalveolar lavage was performed. The particle trap reduced the number of particles found in the BAL fluid. However, diesel exhaust caused a significant increase in neutrophil count in the lavage fluid. In vitro, diesel exhaust led to a reduction of phagocytosis by alveolar macrophages after diesel exhaust exposure with or without particle trap. In the absence of the particle trap, phagocytosis was reduced to a greater extent. Diesel exhaust also led to a migration of alveolar macrophages into the airspace, and a reduction in CD3+ CD25+ cells was observed (90). These immunologic events may contribute considerably to chronic airway inflammation in non-smokers upon chronic exposure to outdoor air pollution.
Oxidative stress is a main predisposing factor for the development of COPD (91). In smokers, antioxidant capacity is obviously reduced due to cigarette smoking, but also in non-smokers ROS from outdoor air pollution can lead to immunological changes that endorse COPD development. In COPD patients, alveolar macrophages are highly activated when compared to healthy subjects. These macrophages release increased amounts of ROS in the form of superoxide radical and hydrogen peroxide (92). Activated neutrophils from the peripheral blood from patients with COPD exert similar effects, releasing increased quantities of ROS as well, especially during COPD exacerbations. Markers of oxidative stress, like hydrogen peroxide, carbon monoxide and myeloperoxidase, as well as markers of oxidative tissue damage such as 8-isoprostane and carbonyl stress were shown to be upregulated in the exhaled breath from COPD patients (93–96). More than 50 cytokines and chemokines were demonstrated to be associated with COPD. Intracellular signaling pathways which are triggered by – or themselves drive – the release of inflammatory mediators, are susceptible to oxidative stress (91). Continuous inflammation in the lung tissue contributes considerably to COPD progression, and therefore a resolution of the inflammatory response is equally as important in COPD treatment as its induction. In this context, the clearance of apoptotic cells by phagocytosis plays a key role. However, in COPD patients, phagocytosis is impaired, which contributes to ongoing chronic lung inflammation (97, 98). Oxidative stress by ambient air pollution or other sources impacts the phagocytosis capacity, either intracellularly through changes in cytoskeletal organization (99), or extracellularly by the carbonylation of tissue proteins, leading to a competition for the same pattern recognition receptors (PRRs) expressed on alveolar macrophage surfaces that normally recognize and clear carbonyl-modified proteins and apoptotic cell detritus (100). Carbonylation caused by ROS was shown to modify and impair the function of PRRs as well, further impeding phagocytosis (101). Moreover, oxidative stress is responsible for the suppression of proinflammatory genes by corticosteroids in COPD patients (102). Carbonylation and nitration caused by air pollution leads to a reduced activity and expression of histone deacetylase 2 (HDAC2), a prime transcriptional corepressor of activated inflammatory genes and the antiinflammatory properties of corticosteroids (103, 104). According to these data, oxidative stress from air pollution can cause an increase in the expression of inflammatory genes, a failure to resolve the inflammatory response, insensitivity to corticosteroids and a diminished ability to induce endogenous antioxidant defense mechanisms. All these factors contribute to a more rapid aging of the lung (105), and may ultimately cause COPD in never-smoking subjects exposed to constant air pollution.
A study by Wooding et al. was conducted to show the immunologic mechanisms of COPD caused by outdoor air pollution in non-smokers (106). It has been shown previously that neutrophils are recruited to the lung as a consequence of diesel exhaust exposure. 18 individuals, amongst them never-smokers (n=7), ex-smokers (n=4) and patients with mild to moderate COPD (n=7), were included into this analysis. Following two hours exposure to diesel exhaust and two hours exposure of filtered air on separate occasions, neutrophil function in the blood was measured. The amount of circulating neutrophils was reduced after diesel exhaust exposure as compared to filtered air (106). The proportion of neutrophil extracellular traps in the lung was increased in all participants of this study. The authors confirmed these findings with in vitro experiments, showing that diesel exhaust particles led to an increase of neutrophil extracellular traps in isolated neutrophils. These data indicate the formation of neutrophil extracellular traps as a distinct mechanism contributing to airway obstruction and COPD formation in non-smokers.
3.1.2 Air Pollution and Lung Cancer
Apart from COPD and chronic respiratory inflammation, a high risk of lung carcinogenesis has been attributed to the exposure to airborne particulate matter (PM) and ozone. PM are a group of different compounds, where the particle core is variable and there is a vast array of surface-related features, like heavy metals (107, 108). Ambient air pollution with PM under the size of 2.5 μm in diameter has been found to be a causative factor for lung tumors (108–110). Particles from air pollution can penetrate into the airways and persist there as particle deposits. PM from combustion sources cause the generation of ROS, leading to DNA damage. Most notably in this context are transition metals with redox capacity, persistent free radicals and redox-cycling of quinones, which may be activated to ROS, causing bulky adducts or strand breaks of the cellular DNA (111, 112). The effects of diesel exhaust particles have been investigated in numerous studies, showing that chronic exposure results in oxidative stress and radical-induced oxidative lesions. 8-Oxo-7,8-dihydroguanine (8-oxoGua) can be used as a biomarker for oxidative stress, measured by high-performance liquid chromatography (HPLC) and gas chromatography–mass spectrometry (GC-MS) in biomonitoring studies (108, 113–115).
Ozone has been reported as a pulmonary irritant, causing redox effects and leading to chronic oxidative stress in the lung tissue (116). Ozone exposure leads to an increased 8-hydroxy-2′-deoxyguanosine (8-OHdG) and heme oxygenase-1 (HO-1) expression in alveolar macrophages (117). According to epidemiological studies, individuals who are exposed to combined air pollutants, e.g. ozone in addition to second-hand smoke or PM, are at a considerably increased risk of pulmonary diseases due to chronic oxidative stress and inflammation (118). Oxidative stress is the key factor in PM-related airway disease, since ROS caused by inhalation of PM are involved in lipid peroxidation, DNA damage and oxidative damage of enzymes (119, 120). Antioxidants are crucial to counteract damage which is – to some degree – continuously done by ROS. Antioxidants allow for a maintenance of the redox balance, mediating biotic and antibiotic stress response and impact gene expression (108, 121). ROS created from airborne PM were demonstrated to activate MAPK family members as well as transcription factors, such as NF-κB and AP-1 (activator protein 1). By activation of these molecules, apoptosis, proliferation, differentiation and ultimately, malignant transformation are affected (122). Notably, ROS are not only generated as a direct effect of airborne PM, but also by target pulmonary epithelial cells or macrophages upon contact with PM (123). Resident macrophages from the lungs and alveolar spaces can release ROS after phagocytosis of inhaled PM, which leads to signal cascades contributing to inflammation and other pathobiological damage (124). Phagocyte-generated ROS comprise, amongst others, superoxide anion (O2−·), nitric oxide, free radical (NO·) and hydrogen peroxide (H2O2) (125). Macrophages are the prime cellular source of NO· in the lungs, while neutrophils form the second largest group. The enzyme inducible nitric oxide synthase (iNOS) serves as an immunological defense mechanism, generating NO· radicals from L-Arginine, NADPH and oxygen (126). ROS and RNS are essential for host defense, yet, they can cause considerable damage to nucleic acids, cellular lipids and proteins. A vast range of diseases is associated with an increase in ROS, like cardiovascular disease, neurological disorders, chronic inflammatory conditions, pulmonary fibrosis and cancer (127). Carcinogenesis is promoted by ROS because of DNA base modifications, rearrangement of DNA sequences, miscoding of DNA lesions, gene duplications and the activation of oncogenes (128). ROS also cause indirect oncogenic effects that lead to cancer, like the upregulation of hypoxia response genes, e.g. elevated levels of hypoxia-inducible transcription factor (HIF-1α) (129).
Oxidative stress results from constant exposure to ambient air pollution, and can be a causative factor for lung carcinogenesis in never-smokers. A study by Ito et al. was specifically designed to evaluate, why the prevalence of primary lung cancer has been increasing in never-smokers despite the decrease in smokers in the developed world (130). They investigated the impact of oxidative stress, possibly caused by outdoor air pollution, in never-smoking individuals with primary lung cancer. Never-smokers, smokers with more than 20 pack years and patients with benign lung diseases were included into the analysis. Serum oxidative stress and antioxidant capacity (AOC) were examined, and the oxidative damage on DNA in the lung tissue was examined. AOC was found to be significantly lower in never-smokers than in smokers. DNA damage was most often found in smokers, and also never-smokers with lung cancer were found to have significantly more DNA damage than subjects with benign lung tumors (130). The authors conclude that a low AOC in never-smokers with lung cancer might explain why some individuals are especially susceptible for lung cancer due to air pollution, even in the absence of smoking as a prime risk factor. The relation of long-term exposure to air pollution with lung cancer has been widely acknowledged. Yet, there is also a short-term association between air pollution and lung cancer mortality, which was examined in a Chinese study (131). Data on the daily prevalence of PM with a diameter <2.5 μm (PM2.5), and PM with a diameter <10 μm (PM10), sulfur dioxide (SO2), and ozone (O3), was assessed. These data were correlated to lung cancer mortality by time-series generalized linear models, correcting for meteorological factors as a possible confounder. According to this study, there is a positive association of the concentrations of PM2.5 and PM10, as well as SO2 with lung cancer mortality on a given day (131). Moreover, daily ozone exposure correlated with short-term lung cancer mortality as well. It was found that each 10 μg/m3 increase of any pollutant was linked to an excess risk of mortality, with a 7.16% increased risk of mortality at the highest concentrations. This study also highlights the important role of oxidative stress caused by air pollution in lung cancer, not only promoting carcinogenesis but also mortality from preexisting lung cancer. Another analysis from China was conducted to investigate the association between spatial air pollution and lung cancer incidence (132). SO2 had the greatest effect on lung cancer incidence in the north of China. In the south, all investigated pollutants (PM2.5, PM10, SO2, NO2, CO and O3) were significantly linked to an increased lung cancer incidence (132). The authors of this study also point out that smoking has a significant synergistic negative effect together with air pollution (132). Air pollution and the consecutive oxidative stress and associated immunological changes should therefore be seen as important contributing factors to lung cancer particularly in never-smokers, while increasing the lung cancer risk even further in the smoking population.
Oxidative stress from ambient air pollution or smoking seems to be the most widely investigated form in the context of lung cancer and COPD. Still, other sources of free radicals, like for instance, nutrition- or exercise-related ones, may alter the immune landscape and contribute to lung disease (133). Moderate exercise seems to be beneficial in the treatment of lung cancer and COPD (134), however, it must be kept in mind that strenuous activity as such leads to increments in ROS (135) and may actually be harmful. Inadequate or excessive nutrient intake also promotes or causes oxidative stress, disrupts oxidative homeostasis and activates molecular pathways that alter the immune landscape in a pro-carcinogenic fashion (136). Nutritional oxidative imbalance has been outlined as a risk factor for cancer, however, the exact immunological aspects in this context remain to be elucidated in further detail.
3.2 Occupational Exposure to Chemicals
3.2.1 COPD as a Result of Exposure to Occupational Lung Irritants
Chronic pulmonary inflammation and COPD may also occur as a response to inhaled toxins, like mycotoxins and airborne particles of asbestos, silica and heavy metals (137). These toxins cause cell injury, which, as mentioned previously, poses a risk for lung carcinogenesis. In addition, cellular response to toxic particles leads to the initiation of immune-mediated repair processes. Consecutively, sustained inflammation and lung tissue remodeling leads to COPD. Especially in developing nations, burn biomass fuels, domestic fires and toxic particles from occupational exposure are a major risk factor for COPD (138). Lung inflammation as a response to toxins is a complex process, involving epithelial cells lining the airways and alveoli and immune cells from the bloodstream (137). Immune cells also secrete cytokines and chemokines, as well as growth factors, that endorse tissue restructuring and fibrosis (139, 140). During the acute inflammatory phase, neutrophils migrate to the lungs as first-line response, producing ROS, secreting serine proteases, matrix metalloproteinases and other proinflammatory enzymes. Theses secondary molecules lead to alveolar destruction which may result in COPD (141).
3.2.2 Occupational Lung Carcinogens
In subjects with a history of silica and asbestos exposure, follow-up studies have suggested an increased risk of lung cancer later in life (142, 143). As early as the 1960s it was shown that workers from the textile industry exposed to high amounts of asbestos dust carried a 10-fold increased risk of lung cancer compared to the general population (144). In a European multi-center study, it was confirmed that exposure to silica dust leads to a two-fold increased lung cancer risk (145).
Generally, chronic exposure to silica and asbestos causes pneumoconiosis, also known as silicosis and asbestosis, ultimately leading to progressive lung fibrosis (146, 147). Silica and asbestos particles, after inhalation, are taken up by alveolar macrophages, resulting in an increase in the amount of collagenic fibroblasts and fibrous tissue remodeling surrounding the inhaled particles (148–150). The impact of silica dust leads to aberrations in antitumor immunity, contributing to the increased lung cancer risk in subjects with chronic silica dust exposure. The most critical change happens on the level of the macrophage, as shown for example by Rakhmilevich et al. (151, 152). In this study it was demonstrated that macrophages can be activated by CD40 ligation, consecutively exerting direct cytotoxicity against cancer cells. Anti-CD40 monoclonal antibody treatment could block tumor growth effectively, however, this effect was annihilated by silica exposure (151, 152). The reduction in macrophage count by silica could therefore promote lung carcinogenesis in non-smokers. Another effect leading to reduced antitumor immunity in non-smokers exposed to silica dust is the induction of macrophage and neutrophil infiltration into lung tissue, resulting in an enhanced secretion of cytokines, chemokines and ROS (150). In a murine model, injected silica led to a variety of immunologic changes, including the recruitment of splenic macrophages and neutrophils, reductions in B-lymphocyte levels as well as alterations in T-lymphocyte abundance (153). Silica also caused a five-fold increase in IL-12 release, and antigen presentation to T-cells in vitro, as well as the priming of antigen-specific T- and B-lymphocytes, was largely inhibited. Therefore, despite an abundance of IL-12, macrophage functions which are mandatory for antitumor immune response are blocked due to silica (154, 155). It has been acknowledged that silica dust exposure causes FasL overexpression in lung tissue (156, 157). Therefore, it is assumed that FasL leads to an increase of macrophage apoptosis in subjects who are exposed to silica dust, allowing for an unimpeded proliferation of cells transformed by silica (150). Fas/FasL affects macrophage function, rendering them unable to activate neither themselves, nor other cells that are critical to recognize and remove malignant cells. The interference with normal macrophage function is one key mechanism by which lung carcinogenesis is endorsed in non-smokers exposed to silica dust. According to the current literature, FasL overexpression due to silica leads to an increased release of TNF-α and other proinflammatory cytokines. However, despite newly-recruited immune cells due to cytokine release, no effective defense against already transformed cells is possible. This can be explained by an increased presence of transforming growth factor-β (TGF-β), resulting in a shift of FasL from an inflammatory function to a suppressive one, as shown by Chen et al. (158). In this study it was demonstrated that an increment in TGF-β blocked neutrophil activation by FasL and led to a reduced recognition of tumor cells by immune cells (158). Silica dust leads to an increased TGF-β expression, as shown by several studies (159, 160). It is thus assumed that the tumor-promoting effect of silica dust is mediated by TGF-β, mitigating the function of FasL towards a decreased release of cytokines or chemokines that are crucial for antitumor immunity, and causing a shift in FasL-associated activities from an inflammatory to a suppressive function, so that the removal of silica-induced transformed cells is largely impeded (150).
Chronic occupational exposure to asbestos and log-term inhalation of asbestos fibers has been linked to lung-, and specifically pleural carcinogenesis (161). The function of NK-cells and CD8+ cytotoxic T-lymphocytes is altered by asbestos fibers (162, 163), and since these cell types constitute the first-line anticarcinogenic defense mechanism, directly killing malignant cells (164, 165), antitumor immune response of asbestos-exposed individuals is impaired, rendering them especially at risk for lung and pleural carcinogenesis after a latency period of 30-40 years (162, 166). A study on the human NK-cell line YT-A1 showed a significantly reduced expression of two NK-cell specific activating receptors and of the serine protease granzyme A usually secreted by NK-cells to directly kill cancer cells, after continuous exposure to asbestos (150, 167). Asbestos also reduced the expression of the activating NK-cell receptor NKp46 in NK-cells stemming from mesothelioma patients (150, 167). A study on the effect of asbestos fibers on T-lymphocyte function, where a T-lymphocyte cell line was exposed to high asbestos dosages, showed a progressive time- and dose-dependent apoptosis of the T-cells in culture (150). Moreover, an activation of the mitochondrial apoptosis pathway was demonstrated, in conjunction with ROS production and proapoptotic signaling via p38 and c-Jun N-terminal kinase, as well as the activation of caspases-9 and -3 (150). In line with this observations, similar effects on alveolar epithelial cells as well as pleural mesothelial cells were seen upon exposure to asbestos in vitro (168–171). Maeda et al. also examined the long-term effect of asbestos exposure on T-cells in vitro. They found that after one year of chronic exposure to the asbestos agent chrysotile, the cells acquired resistance to asbestos-induced apoptosis. In addition, an increased expression and secretion of IL-10 and of the anti-apoptotic molecule Bcl-2 was observed (150). The T-lymphocyte cell line with acquired asbestos resistance also featured an enhanced TGF-β production along with a reduced production of IFN-γ, TNF-α, and IL-6.
Aside from silica and asbestos, several other occupational agents have been classified as group 1 carcinogenic agents for lung cancer by the International Agency for Research on Cancer (172, 173). Table 1 summarizes these carcinogens as well as the respective pathomechanisms in relation to lung cancer (adapted from Algranti et al. and Siemiatyck et al.) (173, 174).
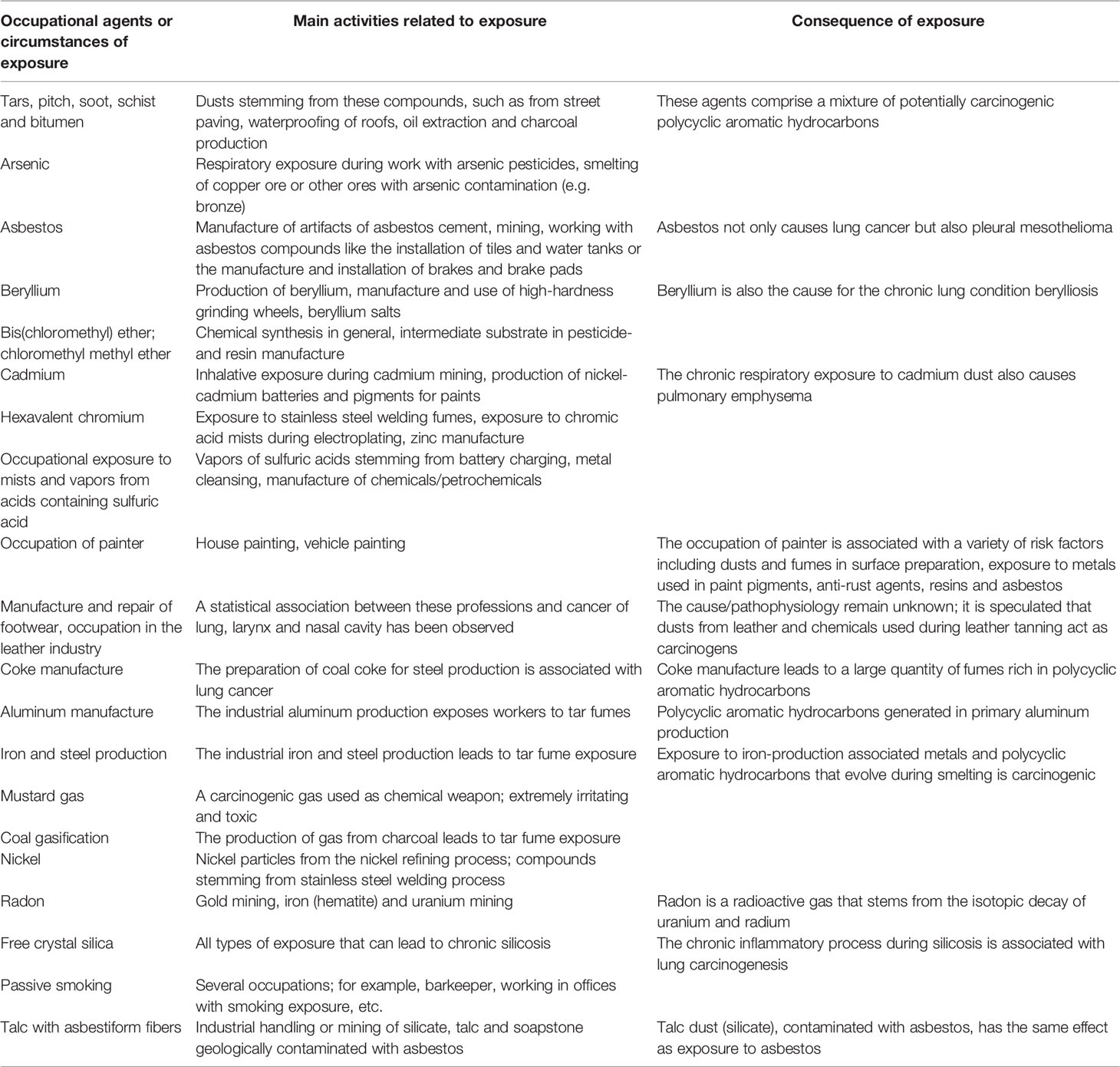
Table 1 Group 1 occupational carcinogens for lung cancer.
3.3 Pulmonary Fibrosis as a Predisposing Factor for Lung Cancer
Mounting evidence suggests that fibrotic lung conditions also predispose individuals to lung carcinogenesis. This was first assumed based on autopsy studies, where coexisting interstitial lung disease and lung cancer have been found (175). Idiopathic pulmonary fibrosis is the most common form of fibrotic lung disease (176). Many patients with pulmonary fibrosis also have a history of smoking, rendering them especially susceptible for lung cancer. Still, non-smokers with fibrotic lung disease seem to have a higher lung cancer risk as well, as idiopathic pulmonary fibrosis increases lung cancer risk by 7-20%, regardless of other risk factors (177). Fibrosis of the lung originates from perpetuated and excessive connective tissue remodeling in response to recurring alveolar microinjuries. Excessive deposition of components of the extracellular matrix leads to an irreversible remodeling of lung tissue (178, 179). Fibroblasts, during excessive and aberrant wound healing, respond by hyperproliferation and change to a pro-fibrotic phenotype resistant to apoptosis (178, 179). Activated fibroblasts are highly responsive to growth factors and cytokines, e.g. TGF-β, connective tissue growth factor, platelet-derived growth factor and IL-6 (179, 180). This chronic low-level inflammatory process leads to an increased risk of lung carcinogenesis in patients with pulmonary fibrosis. T helper cells type 1 and 2 (Th1/Th2) play an important role in the inflammatory phase of pulmonary fibrosis (181). It was demonstrated in a mouse model that depletion of T-lymphocytes by means of an anti-CD3 monoclonal antibody impeded the fibrotic lung remodeling (182). The role of regulatory T-cells in fibrotic lung disease has not been completely clarified. A reduction of regulatory T-cells in the peripheral blood and BAL of pulmonary fibrosis patients was shown in on study, while other data showed an increase of regulatory T-cells (183). A pro-fibrotic impact of regulatory T-cells was shown in one study, increasing TGF-β1 release and collagen deposit (184). However, in late stages of lung fibrosis, regulatory T-cells acted anti-fibrotic (184). It may be that regulatory T-cell dysfunction contributes not only to pulmonary fibrosis but also to lung carcinogenesis. Particularly in the early stages of tumor development regulatory T-cells play a crucial role. A mouse model of mutant Kras-driven lung adenocarcinoma showed that Kras transgenic mice who lacked FoxP3+ regulatory T-cells were 75% less likely to develop lung tumors (185). Tobacco smoke led to an increase in FoxP3+ lymphocytes, according to the mouse study, which poses one among many factors contributing to smoking-related lung cancer. The depletion of regulatory T-cells led to tumor cell death at early stages of lung carcinogenesis according to another mouse study (186), resulting in elevated levels of granzyme A and B, perforin and IFN-γ in infiltrating CD8+ T-cells. Correspondingly, Xiong et al. demonstrated that regulatory T-cell depletion was a protective factor in radiation-induced lung fibrosis (187). Viewing the data on regulatory T-cells and lung fibrosis, it is evident that they can exert pro- as well as anti-fibrotic roles, dependent on the stage of pulmonary fibrosis and the mutual interaction with other T-cells. Th9 and Th22 T-cells, producing IL-9 and IL-22 were also involved in fibrotic remodeling of the lung, exerting pro- and anti-fibrotic properties similarly to regulatory T-cells (188–190). Macrophages, as a source of proinflammatory and pro-fibrotic cytokines, have been linked to lung fibrogenesis as well (191). The role of macrophages in lung carcinogenesis has been widely investigated, showing the important role macrophages play in this context. Macrophages make up the majority of tumor-infiltrating immune cells and are a key link from inflammation to lung cancer (192). Two hypotheses on the effect of macrophages and lung carcinogenesis have emerged: An extrinsic pathway driven by inflammatory conditions increasing the risk for lung cancer, as well as an intrinsic one driven by genetic changes (192). Tumors triggered by chronic inflammation at distinct tissue sites are characterized by infiltrating leukocytes, predominantly macrophages, cytokines, chemokines and growth factors. Tumor-infiltrating macrophages secrete ROS and nitrogen intermediates, inducing DNA damage. In response to tissue damage at sites of chronic inflammation, inflammatory cytokines are released to recruit cells that initiate tissues repair. However, the cytokines endorse tumorigenesis themselves by inhibiting key enzymes (193). Two types of tumor-associated macrophages are known today: canonically activated M1 and alternatively activated M2 macrophages. Both in lung cancer and in lung fibrosis, these two subtypes contribute considerably to disease progression, exerting beneficial as well as harmful functions (194, 195). M2 macrophages have been known to accumulate in fibrotic lungs and have been linked to pro-fibrotic activities (194). Conversely, anti-fibrotic roles have been reported for macrophages as well, mediated by diverse mechanisms such as scavenging of proinflammatory cellular debris, digestion of extracellular matrix components and by secretion of mediators inducing myofibroblast apoptosis (196). The effects of macrophages both in fibrotic lung disease and in lung cancer may explain why lung fibrosis is linked to lung cancer, and why changes in the lung microenvironment associated with lung fibrosis render the patients more susceptible for lung cancer as well.
Neutrophils play a role in chronic inflammation in general, and their role in lung carcinogenesis as well as in lung fibrosis has been commonly acknowledged (197, 198). Levels of IL-8/CXCL8, which is a key chemotactic factor for neutrophil function, are increased in patients with pulmonary fibrosis (199), and the number of neutrophils in the BAL fluid correlated well with G-CSF levels, an important neutrophil growth factor (200). Airway neutrophils are generally activated at a higher level in idiopathic pulmonary fibrosis, compared with healthy individuals, which is demonstrated by increased levels of their main proteolytic product, neutrophil elastase (201). Neutrophil elastase is a protease of neutrophil extracellular traps which are formed as a response to chronic lung inflammation. One mouse study showed that neutrophil elastase proteolytically remodeled laminin in lung tissue, leading to the proliferation of dormant lung cancer cells via the activation of integrin α3β1 signaling (202). Tissue remodeling processes in lung fibrosis, mediated by neutrophils, could therefore facilitate concomitant lung carcinogenesis. Summing up all the above-mentioned data, a great variety of immunological changes can be linked to fibrotic lung disease. Many of these changes are generally associated with chronic inflammation or tissue remodeling, paving the way for malignant transformation of cells. The correlation of lung fibrosis with an increased risk of lung cancer – particularly in never-smokers – can partly be explained by these distinct immunological changes.
Mycotoxins and fungal spores are especially prevalent in damp buildings or at farms, in the malt and wood industry, i.e. all occupations that involve handling moldy materials (203). Different fungi are sources of mycotoxins as secondary metabolites, including trichothecenes synthesized by the genera Fusarium, Myrothecium, Trichoderma, Trichothecium, Cephalosporium, Verticimonosporium, and Stachybotrys (204). Trichothecenes lead to a suppression of lymphocyte-related immune response and stimulate the production of IL-1β by macrophages (205). Additionally, these toxins inhibit protein synthesis by targeting the ribosome, impairment of the mitochondria and activation of MAPKs (206). Trichothecenes also upregulate gene expression related to the inflammatory response, which leads to further ribosome damage through inflammatory cytokines (207, 208). Deoxynivalenol is a trichothecene which is found in contaminated cereal grains. It may exert cytotoxic effects and inflammation synergistically with particulate matter, inducing the inflammatory response (209). Animal studies have demonstrated that upon exposure, mycotoxins not only damage the lung but are distributed to the liver, kidney and spleen (210). Consecutively, alveolar macrophages and neutrophils are recruited, promoting pulmonary hemorrhage, cytokine secretion and ultimately, damage to various organs (211). Notable, even after ingestion of mycotoxins, the lung tissue is injured in response (212).
Ricin is a ribosome-inactivating protein found in beans of the castor plant Ricinus communis. Ricin aerosol does not occur naturally but may be used as a biological weapon because of its high toxicity. Ricin is used as a compound of immunotoxins to target cancer cells (213). Ricin intoxication leads to a strong inflammatory cascade, i.e. the migration of neutrophils to the airways, causing apoptosis and necrosis of lung epithelial cells (214). Conversely to cigarette smoke, ricin also causes apoptosis of alveolar macrophages (214). While severe ricin poisoning leads to interstitial pneumonia, alveolar edema, respiratory failure and death within days, a sublethal ricin dosage leads to lung fibrosis and pulmonary hemorrhage (215). Like cigarette smoke and mycotoxins, ricin increases the expression of proinflammatory genes in airway epithelial cells and pulmonary macrophages (137). Both ricin and mycotoxins are factors predisposing exposed individuals to fibrotic lung disease. The above-listed inflammatory mechanisms contribute not only to fibrosis, but to DNA damage of lung epithelial cells as well, which may lead to lung cancer in the long term. Lastly, also non-biological chemicals can lead to lung inflammation when inhaled. These include volatile organic compounds from household items, office supplies and craft materials, e.g. formaldehyde, benzene and perchloroethylene, and chronic exposure results in damage of the lung epithelium, fibrotic remodeling and malignant transformation (216).
3.4 Second-Hand Smoke, COPD and Lung Cancer
Second-hand smoke counts among the risk factors for COPD in non- and never-smokers (217). Similarly, second-hand smoke, also termed “environmental tobacco smoke”, has been classified as a main pulmonary carcinogen (218). Second-hand smoke is a composition of mainstream- and sidestream smoke, wich are mostly similar in quality and composition (219), yet, sidestream smoke arises at lower burning temperature and the quantities of its chemical compounds in vapor and particular phase (i.e. the quantity of semi-volatile or non-volatile agents) differ from the quantities in mainstream smoke (220). Certain carcinogens, such as aromatic amines, are more abundant in sidestream smoke (220, 221). Second-hand smoke, once released into the air, has the potential to aggregate and mix with other types of air pollutants (222). Smokers are generally exposed to a much larger quantity of smoke-related carcinogens (221). Interestingly, sidestream smoke was associated with a higher potency to induce mouse skin tumors, which gave rise to the assumption that second-hand smoke in non-smoking subjects might even be more carcinogenic than mainstream smoke inhaled by smokers (223).
Since data on second-hand smoke, COPD and lung cancer is gathered epidemiologically, the evidence linking second-hand smoke to these conditions remains weak (224), and a direct biological link is difficult to outline. In a mouse study, it was shown that alveolar macrophage recruitment was increased after exposure to second-hand smoke in a dose-dependent fashion (217). Moreover, an increased expression of alveolar macrophage markers and an increase of the COPD-associated pro-inflammatory markers CCL2 and TNFalpha was also observed in this study (217), showing a direct causative effect of second-hand smoke in COPD.
In lung carcinogenesis, second-hand smoke is an established risk factor (22, 225), however, attempts to epidemiologically confirm the associated risk have only brought forth disputable evidence (226). In COPD and lung cancer, the uncertainties in risk assessment of second-hand smoke exposure have arisen, because exposure assessment is difficult to standardize, given temporal variabilities in source, formulation and concentration of effluent second-hand smoke (227). However, for lung cancer, the risk does significantly increase due to second-hand smoke with many years of latency (228). Novel studies have focused on outlining second-hand smoke biomarkers with the aim to measure exposure over a relevant time period which augments the risk of lung carcinogenesis (221, 229). Human epidemiologic studies on second-hand smoke and lung cancer have been observational (226), correlating the incidence of lung cancer in long-term second-hand smoke exposed individuals, such as family members of smokers, to the level of second-hand smoke exposure (230). Rodent studies on the carcinogenic potential have been performed, allowing for a better standardization, however, animals are usually treated with considerably higher doses of smoke than humans would be exposed to in real life (231). Moreover, animals with smoke exposure often respond with appetite suppression and the consecutive inadequate caloric intake may further contribute to tumor initiation (231). Thus, it is difficult to exactly quantify the carcinogenic potential of passive smoking.
Very few data exists on the immunologic changes induced by second-hand smoke. Presumably this is due to the assumption that the changes are very similar to those caused by active smoking. One mouse study investigated in utero and early-life passive smoke exposure, and the resulting immunologic changes (232). Changes in plasma cytokine concentrations and in immune cell populations, i.e. decreased percentages of B-cells and increased percentages of myeloid cells were observed in the bone marrow of smoke-exposed versus control mice. In the spleen of passive smoke exposed mice, the abundance of B-cells decreased and that of T-cells increased, whereas myeloid cells were significantly lower in the peripheral blood in smoke exposed mice (232). The authors of this study conclude that the immune changes resulting from second-hand smoke in utero and in early life are particularly threatening, contributing to carcinogenesis by lowering host defense mechanisms. In a review article about the immunologic changes of second-hand smoke in humans, it becomes evident that the literature is yet too limited to draw conclusions on how exactly the immunologic changes induced by second-hand smoke differ from that of active smoking (233). Environmental smoke exposure may induce hypersensitivity, leading to asthma and may furthermore suppress immunoregulatory mechanisms. Some studies even failed to detect any immunological changes in passive smoke-exposed individuals (233). Thus, more research is clearly warranted on this topic.
3.5 Pulmonary Infections and the Lung Microbiome Are Related to Lung Carcinogenesis
There is evidence for an increased risk of lung cancer due to a history of pulmonary infections caused by Mycobacterium tuberculosis (234). This evidence is supported by large cohort studies, like for instance a Chinese analysis amongst individuals with a history of tuberculosis infection. A positive correlation with lung adenocarcinoma as well as squamous cell carcinoma has been shown (235). According to previous data, the risk of lung cancer is increased by about 50% after a prior diagnosis of tuberculosis (236). The more recently the infection was diagnosed, the greater the risk of lung cancer became (236). Moreover, it is discussed whether a chronic pulmonary colonization with Chlamydia pneumoniae led to an increment in the risk of lung cancer as well (237). As shown by serologic testing, a chronic Chlamydia pneumoniae infection resulted in impaired apoptosis of the infected cells, because IL-10, a prime immunosuppressive cytokine, was activated, resulting in reduced antitumor immune response (238). Moreover, an upregulation of IL-8, mediated by Chlamydia pneumoniae, has been reported to increase the risk of malignancy, endorsing angiogenesis and cell proliferation, as shown in a mouse model (239).
The lung microbiome in general seems to impact the likelihood of lung cancer, in smokers and non-smokers alike (240). Apart from chronic inflammation and bacterial and viral lung infections, even periodontal disease and not only pathogenic but also opportunistic microorganisms can drive lung carcinogenesis in the long term, as shown for Haemophilus influenzae, Enterobacter spp., E. coli, Pneumococcus (241), Legionella (242) and Moraxella genera (137, 243). These pathogens can increase the likelihood of lung cancer, with an association even with certain histological subtypes (240). Klebsiella, Rhodoferax, Acidovorax, Comamonas and Polarmonas genera, for example, are frequently found in the lung microbiome of small-cell lung cancer (SCLC) patients, but not in patients with adenocarcinoma (244). It is proposed, that in some cases not the infection itself increases the risk for subsequent lung cancer, but the significant disruption of the lung microbial landscape. Alpha diversity of the lung microbiome, namely the number of different microbes in one habitat, is usually low in lung cancer patients. Interestingly, the beta diversity, meaning the diversity of microbes between habitats is not different when comparing healthy lungs and those from lung cancer patients (245). In recent years, the importance of the microbiome as an indicator of malignant transformation was recognized increasingly. A study on the lung microbiome, comparing samples from patients with benign and malignant tumors via next generation sequencing, showed the genera Veillonella and Megasphaera to be potential lung cancer biomarkers (243). Moreover, a correlation between Acidovorax genus and SCLC has been shown (244), and Pseudomonas is associated with lung adenocarcinoma (240, 246). Within the last decades, the molecular mechanisms by which microbes endorse malignant transformation, have been investigated in greater detail. The microbial carcinogenic effects comprise bacterial toxins, inflammatory stimuli from immune cells and the direct effects of microbes on epithelial cells. A study on mice with a dysfunctional lung microbiome and impaired function of γδT17 T-cells due to long-term antibiotic treatment, showed an increased risk for Lewis lung carcinoma. The total bacterial count of the lung microbiome in these mice decreased dramatically, showing that the commensal microbiome is indeed crucial for anti-tumor immune cell functions (247). Also bacterial toxins change the immune landscape in a way that may contribute to tumorigenesis. For instance, cytolethal distending toxin (CDT), cytotoxic necrotizing factor 1, and Bacteroides fragilis toxin can damage the DNA repair system, allowing for malignant transformation (248–250). In a murine model of A427 non-small cell lung cancer (NSCLC) it was demonstrated that the toxin of Cyanobacteria decreases CD36 protein levels and increases the concentration of PARP1, an enzyme profoundly linked to tumor development. In the mouse model, these results were proven with bacteria-positive lung cancer (251). Moreover, heat-inactivated E. coli bacteria were found to enhance migration and metastatic dissemination of NSCLC cells in vivo (252). Apart from these direct effects of microorganisms on lung cancer development, it was also shown that a shift in the lung microbiome causes ROS rates to increase. Higher amount of ROS lead to an increase in DNA damage in the long term and predispose to tumor development. Interestingly, TP53 mutated lung tumors are known to be associated with the colonization with Acidovorax genus in the tissue microenvironment (244).
A disruption of the lung microbiome, triggered by lung infections or possibly chronic exposure to carcinogenic dusts or second-hand smoke, may therefore be an important factor predisposing non- and never-smokers for lung cancer.
4 Conclusion
The prime risk factor for lung cancer and COPD is tobacco smoking. However, in non- and never-smokers, these diseases also occur because of other noxa not related to tobacco smoke. When comparing the effects of smoking on the immune landscape with the damage on lung tissue caused by ambient air pollutants, oxidative stress in general, occupational toxins and DNA damage as a side effect of lung fibrosis or pulmonary infections, the complex changes of the immune landscape are partly overlapping. Recently it was discovered that the lung microbiome also has a profound effect on the immune response, carcinogenesis and tissue remodeling, which remains to be investigated in greater detail. It can be said that smoking has a synergistic adverse effect when co-occurring with the above-mentioned other risk factors.
Table 2 contains the most important bullet points of this manuscript, summarizing non-smoking related risk factors for lung cancer and COPD (Table 2).
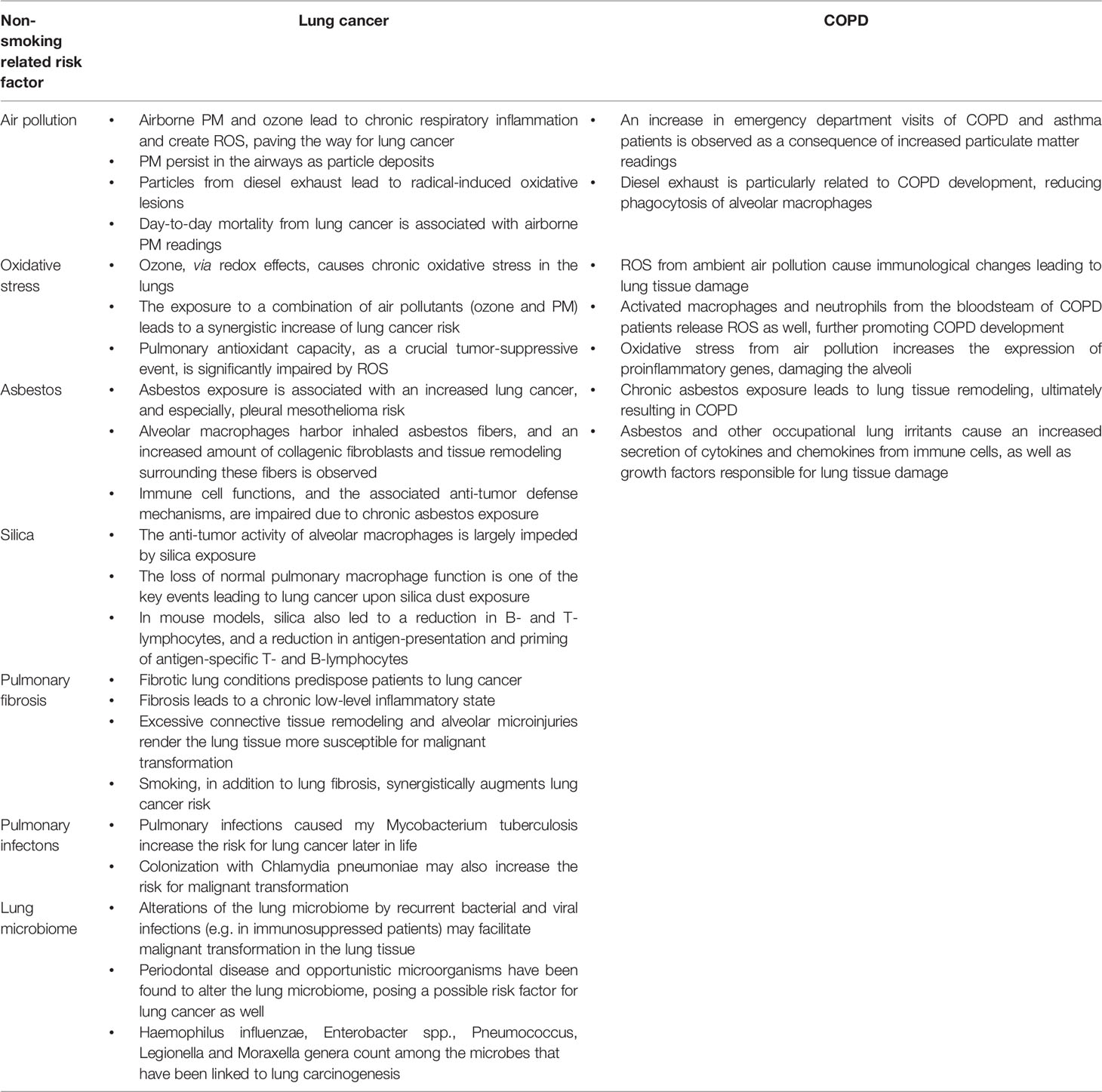
Table 2 Take-home messages of this review article.
Author Contributions
ET, concept and structure of this article and main part of manuscript writing. IM, assistance in writing and proofreading. JL, in-depth proofreading, assistance in writing and continuous input during the writing process. F-MS-J, in-depth proofreading, final remarks and revision. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Qiu F, Liang CL, Liu H, Zeng YQ, Hou S, Huang S, et al. Impacts of Cigarette Smoking on Immune Responsiveness: Up and Down or Upside Down? Oncotarget (2017) 8:268–84. doi: 10.18632/ONCOTARGET.13613
3. Rennard SI. Cigarette Smoke in Research. Am J Respir Cell Mol Biol (2004) 31:479–80. doi: 10.1165/RCMB.F284
4. Talhout R, Schulz T, Florek E, van Benthem J, Wester P, Opperhuizen A. Hazardous Compounds in Tobacco Smoke. Int J Environ Res Public Health (2011) 8:613–28. doi: 10.3390/IJERPH8020613
5. Cataldo JK, Prochaska JJ, Glantz SA. Cigarette Smoking is a Risk Factor for Alzheimer’s Disease: An Analysis Controlling for Tobacco Industry Affiliation. J Alzheimer’s Disease : JAD (2010) 19:465–80. doi: 10.3233/JAD-2010-1240
6. Warren GW, Cummings KM. Tobacco and Lung Cancer: Risks, Trends, and Outcomes in Patients With Cancer. American Society of Clinical Oncology Educational Book. American Society of Clinical Oncology (2013) p. 359–64. doi: 10.14694/EDBOOK_AM.2013.33.359
7. Warren GW, Sobus S, Gritz ER. The Biological and Clinical Effects of Smoking by Patients With Cancer and Strategies to Implement Evidence-Based Tobacco Cessation Support. Lancet Oncol (2014) 15:e568–80. doi: 10.1016/S1470-2045(14)70266-9
8. Mainali P, Pant S, Rodriguez AP, Deshmukh A, Mehta JL. Tobacco and Cardiovascular Health. Cardiovasc Toxicol (2015) 15:107–16. doi: 10.1007/S12012-014-9280-0
9. Hagiwara E, Takahashi KI, Okubo T, Ohno S, Ueda A, Aoki A, et al. Cigarette Smoking Depletes Cells Spontaneously Secreting Th(1) Cytokines in the Human Airway. Cytokine (2001) 14:121–6. doi: 10.1006/CYTO.2001.0860
10. Meuronen A, Majuri ML, Alenius H, Mäntylä T, Wolff H, Piirilä P, et al. Decreased Cytokine and Chemokine mRNA Expression in Bronchoalveolar Lavage in Asymptomatic Smoking Subjects. Respiration; Int Rev Thorac Dis (2008) 75:450–8. doi: 10.1159/000114855
11. Gonçalves RB, Coletta RD, Silvério KG, Benevides L, Casati MZ, da Silva JS, et al. Impact of Smoking on Inflammation: Overview of Molecular Mechanisms. Inflammation research : Off J Eur Histamine Res Soc . [et al.] (2011) 60:409–24. doi: 10.1007/S00011-011-0308-7
12. Friedrichs B, Neumann U, Schüller J, Peck MJ. Cigarette-Smoke-Induced Priming of Neutrophils From Smokers and non-Smokers for Increased Oxidative Burst Response is Mediated by TNF-α. Toxicol Vitro : an Int J Published Assoc BIBRA (2014) 28:1249–58. doi: 10.1016/J.TIV.2014.06.007
13. Lee J, Taneja V, Vassallo R. Cigarette Smoking and Inflammation: Cellular and Molecular Mechanisms. J Dental Res (2012) 91:142–9. doi: 10.1177/0022034511421200
14. Jemal A, Thun MJ, Ries LAG, Howe HL, Weir HK, Center MM, et al. Annual Report to the Nation on the Status of Cance-2005, Featuring Trends in Lung Cancer, Tobacco Use, and Tobacco Control. J Natl Cancer Institute (2008) 100:1672–94. doi: 10.1093/JNCI/DJN389
15. Rivera GA, Wakelee H. Lung Cancer in Never Smokers. Adv Exp Med Biol (2016) 893:43–57. doi: 10.1007/978-3-319-24223-1_3
16. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: Cancer J Clin (2018) 68:394–424. doi: 10.3322/CAAC.21492
17. Dutu T, Michiels S, Fouret P, Penault-Llorca F, Validire P, Benhamou S, et al. Differential Expression of Biomarkers in Lung Adenocarcinoma: A Comparative Study Between Smokers and Never-Smokers. Ann Oncology : Off J Eur Soc Med Oncol (2005) 16:1906–14. doi: 10.1093/ANNONC/MDI408
18. Bryant A, Cerfolio RJ. Differences in Epidemiology, Histology, and Survival Between Cigarette Smokers and Never-Smokers Who Develop non-Small Cell Lung Cancer. Chest (2007) 132:185–92. doi: 10.1378/CHEST.07-0442
19. Thun MJ, Henley SJ, Burns D, Jemal A, Shanks TG, Calle EE. Lung Cancer Death Rates in Lifelong Nonsmokers. J Natl Cancer Institute (2006) 98:691–9. doi: 10.1093/JNCI/DJJ187
20. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer Statistic. CA: Cancer J Clin (2008) 58:71–96. doi: 10.3322/CA.2007.0010
21. Samet JM, Avila-Tang E, Boffetta P, Hannan LM, Olivo-Marston S, Thun MJ, et al. Lung Cancer in Never Smokers: Clinical Epidemiology and Environmental Risk Factors. Clin Cancer research : an Off J Am Assoc Cancer Res (2009) 15:5626–45. doi: 10.1158/1078-0432.CCR-09-0376
22. Vineis P, Airoldi L, Veglia F, Olgiati L, Pastorelli R, Autrup H, et al. Environmental Tobacco Smoke and Risk of Respiratory Cancer and Chronic Obstructive Pulmonary Disease in Former Smokers and Never Smokers in the EPIC Prospective Study. BMJ (Clinical Res ed.) (2005) 330:277–80. doi: 10.1136/BMJ.38327.648472.82
23. Clément-Duchêne C, Vignaud JM, Stoufflet A, Bertrand O, Gislard A, Thiberville L, et al. Characteristics of Never Smoker Lung Cancer Including Environmental and Occupational Risk Factors. Lung Cancer (Amsterdam Netherlands) (2010) 67:144–50. doi: 10.1016/J.LUNGCAN.2009.04.005
24. Tashkin DP, Murray RP. Smoking Cessation in Chronic Obstructive Pulmonary Disease. Respir Med (2009) 103:963–74. doi: 10.1016/J.RMED.2009.02.013
25. Rycroft CE, Heyes A, Lanza L, Becker K. Epidemiology of Chronic Obstructive Pulmonary Disease: A Literature Review. Int J Chronic Obstructive Pulmonary Dis (2012) 7:457–94. doi: 10.2147/COPD.S32330
26. Whittemore AS, Perlin SA, DiCiccio Y. Chronic Obstructive Pulmonary Disease in Lifelong Nonsmokers: Results From NHANES. Am J Public Health (1995) 85:702–6. doi: 10.2105/AJPH.85.5.702
27. Behrendt CE. Mild and Moderate-to-Severe COPD in Nonsmokers: Distinct Demographic Profiles. Chest (2005) 128:1239–44. doi: 10.1378/CHEST.128.3.1239
28. Celli BR, Halbert RJ, Nordyke RJ, Schau B. Airway Obstruction in Never Smokers: Results From the Third National Health and Nutrition Examination Survey. Am J Med (2005) 118:1364–72. doi: 10.1016/J.AMJMED.2005.06.041
29. Lamprecht B, Schirnhofer L, Kaiser B, Buist S, Studnicka M. Non-Reversible Airway Obstruction in Never Smokers: Results From the Austrian BOLD Study. Respir Med (2008) 102:1833–8. doi: 10.1016/J.RMED.2008.07.007
30. Lamprecht B, McBurnie MA, Vollmer WM, Gudmundsson G, Welte T, Nizankowska-Mogilnicka E, et al. COPD in Never Smokers: Results From the Population-Based Burden of Obstructive Lung Disease Study. Chest (2011) 139:752–63. doi: 10.1378/CHEST.10-1253
31. Schwartz AG, Cote ML. Epidemiology of Lung Cancer. Adv Exp Med Biol (2016) 893:21–41. doi: 10.1007/978-3-319-24223-1_2
32. Ntritsos G, Franek J, Belbasis L, Christou MA, Markozannes G, Altman P, et al. Gender-Specific Estimates of COPD Prevalence: A Systematic Review and Meta-Analysis. Int J Chronic Obstructive Pulmonary Dis (2018) 13:1507–14. doi: 10.2147/COPD.S146390
33. Weyand CM, Yang Z, Goronzy JJ. T-Cell Aging in Rheumatoid Arthritis. Curr Opin Rheumatol (2014) 26:93–100. doi: 10.1097/BOR.0000000000000011
34. Lutz CT, Quinn LBS. Sarcopenia, Obesity, and Natural Killer Cell Immune Senescence in Aging: Altered Cytokine Levels as a Common Mechanism. Aging (2012) 4:535–46. doi: 10.18632/AGING.100482
35. Golomb L, Sagiv A, Pateras IS, Maly A, Krizhanovsky V, Gorgoulis VG, et al. Age-Associated Inflammation Connects RAS-Induced Senescence to Stem Cell Dysfunction and Epidermal Malignancy. Cell Death Differentiation (2015) 22:1764–74. doi: 10.1038/CDD.2015.21
36. Bueno V, Sant’Anna OA, Lord JM. Ageing and Myeloid-Derived Suppressor Cells: Possible Involvement in Immunosenescence and Age-Related Disease. Age (Dordrecht Netherlands) (2014) 36. doi: 10.1007/S11357-014-9729-X
37. Fuentes E, Fuentes M, Alarcón M, Palomo I. Immune System Dysfunction in the Elderly. Anais da Academia Bras Cienc (2017) 89:285–99. doi: 10.1590/0001-3765201720160487
38. Agrawal A, Gupta S. Impact of Aging on Dendritic Cell Functions in Humans. Ageing Res Rev (2011) 10:336–45. doi: 10.1016/J.ARR.2010.06.004
39. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of Phagocytic and Antigen Cross-Presenting Capacity in Aging Dendritic Cells Is Associated With Mitochondrial Dysfunction. J Immunol (Baltimore Md. : 1950) (2015) 195:2624–32. doi: 10.4049/JIMMUNOL.1501006
40. Shehata HM, Hoebe K, Chougnet CA. The Aged Nonhematopoietic Environment Impairs Natural Killer Cell Maturation and Function. Aging Cell (2015) 14:191–9. doi: 10.1111/ACEL.12303
41. Hazeldine J, Lord JM. The Impact of Ageing on Natural Killer Cell Function and Potential Consequences for Health in Older Adults. Ageing Res Rev (2013) 12:1069–78. doi: 10.1016/J.ARR.2013.04.003
42. Fortin CF, Larbi A, Dupuis G, Lesur O, Fülöp T. GM-CSF Activates the Jak/STAT Pathway to Rescue Polymorphonuclear Neutrophils From Spontaneous Apoptosis in Young But Not Elderly Individuals. Biogerontology (2007) 8:173–87. doi: 10.1007/S10522-006-9067-1
43. Chen MM, Palmer JL, Plackett TP, Deburghgraeve CR, Kovacs EJ. Age-Related Differences in the Neutrophil Response to Pulmonary Pseudomonas Infection. Exp Gerontol (2014) 54:42–6. doi: 10.1016/J.EXGER.2013.12.010
44. Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N, et al. Phosphoinositide 3-Kinase Inhibition Restores Neutrophil Accuracy in the Elderly: Toward Targeted Treatments for Immunosenescence. Blood (2014) 123:239–48. doi: 10.1182/BLOOD-2013-08-519520
45. Sebastián C, Herrero C, Serra M, Lloberas J, Blasco MA, Celada A. Telomere Shortening and Oxidative Stress in Aged Macrophages Results in Impaired STAT5a Phosphorylation. J Immunol (Baltimore Md. : 1950) (2009) 183:2356–64. doi: 10.4049/JIMMUNOL.0901131
46. van Duin D, Allore HG, Mohanty S, Ginter S, Newman FK, Belshe RB, et al. Prevaccine Determination of the Expression of Costimulatory B7 Molecules in Activated Monocytes Predicts Influenza Vaccine Responses in Young and Older Adults. J Infect Dis (2007) 195:1590–7. doi: 10.1086/516788
47. Bulati M, Buffa S, Martorana A, Candore G, Lio D, Caruso C, et al. Trafficking Phenotype and Production of Granzyme B by Double Negative B Cells (IgG(+)IgD(-)CD27(-)) in the Elderly. Exp Gerontol (2014) 54:123–9. doi: 10.1016/J.EXGER.2013.12.011
48. Buffa S, Pellicanò M, Bulati M, Martorana A, Goldeck D, Caruso C, et al. A Novel B Cell Population Revealed by a CD38/CD24 Gating Strategy: CD38(-)CD24 (-) B Cells in Centenarian Offspring and Elderly People. Age (Dordrecht Netherlands) (2013) 35:2009–24. doi: 10.1007/S11357-012-9488-5
49. Pritz T, Lair J, Ban M, Keller M, Weinberger B, Krismer M, et al. Plasma Cell Numbers Decrease in Bone Marrow of Old Patients. Eur J Immunol (2015) 45:738–46. doi: 10.1002/EJI.201444878
50. Vasudev A, Tan Tze Ying C, Ayyadhury S, Joo Puan K, Kumar Andiappan A, Shwe Zin Nyunt M, et al. γ/δ T Cell Subsets in Human Aging Using the Classical α/β T Cell Model. J Leukocyte Biol (2014) 96:647–55. doi: 10.1189/JLB.5A1213-650RR
51. Garcia GG, Miller RA. Age-Dependent Defects in TCR-Triggered Cytoskeletal Rearrangement in CD4+ T Cells. J Immunol (Baltimore Md. : 1950) (2002) 169:5021–7. doi: 10.4049/JIMMUNOL.169.9.5021
52. Palmer DB. The Effect of Age on Thymic Function. Front Immunol (2013) 4:316. doi: 10.3389/FIMMU.2013.00316
53. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T Cell Subsets and Their Signature Cytokines in Autoimmune and Inflammatory Diseases. Cytokine (2015) 74:5–17. doi: 10.1016/J.CYTO.2014.09.011
54. Harrison OJ, Foley J, Bolognese BJ, Long E, Podolin PL, Walsh PT. Airway Infiltration of CD4+ CCR6+ Th17 Type Cells Associated With Chronic Cigarette Smoke Induced Airspace Enlargement. Immunol Lett (2008) 121:13–21. doi: 10.1016/J.IMLET.2008.07.011
55. Baskara I, Kerbrat S, Dagouassat M, Nguyen HQ, Guillot-Delost M, Surenaud M, et al. Cigarette Smoking Induces Human CCR6 + Th17 Lymphocytes Senescence and VEGF-A Secretion. Sci Rep (2020) 10. doi: 10.1038/S41598-020-63613-4
56. Forsslund H, Mikko M, Karimi R, Grunewald J, Wheelock Å.M, Wahlström J, et al. Distribution of T-Cell Subsets in BAL Fluid of Patients With Mild to Moderate COPD Depends on Current Smoking Status and Not Airway Obstruction. Chest (2014) 145:711–22. doi: 10.1378/CHEST.13-0873
57. Zhang MQ, Wan Y, Jin Y, Xin JB, Zhang JC, Xiong XZ, et al. Cigarette Smoking Promotes Inflammation in Patients With COPD by Affecting the Polarization and Survival of Th/Tregs Through Up-Regulation of Muscarinic Receptor 3 and 5 Expression. PloS One (2014) 9. doi: 10.1371/JOURNAL.PONE.0112350
58. Vardavas CI, Plada M, Tzatzarakis M, Marcos A, Warnberg J, Gomez-Martinez S, et al. Passive Smoking Alters Circulating Naïve/Memory Lymphocyte T-Cell Subpopulations in Children. Pediatr Allergy Immunology : Off Publ Eur Soc Pediatr Allergy Immunol (2010) 21:1171–8. doi: 10.1111/J.1399-3038.2010.01039.X
59. Vargas-Rojas MI, Ramírez-Venegas A, Limón-Camacho L, Ochoa L, Hernández-Zenteno R, Sansores RH. Increase of Th17 Cells in Peripheral Blood of Patients With Chronic Obstructive Pulmonary Disease. Respir Med (2011) 105:1648–54. doi: 10.1016/J.RMED.2011.05.017
60. Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T Cells are Required for Inflammation and Destruction in Cigarette Smoke-Induced Emphysema in Mice. J Immunol (Baltimore Md. : 1950) (2007) 178:8090–6. doi: 10.4049/JIMMUNOL.178.12.8090
61. Nadigel J, Préfontaine D, Baglole CJ, Maltais F, Bourbeau J, Eidelman DH, et al. Cigarette Smoke Increases TLR4 and TLR9 Expression and Induces Cytokine Production From CD8(+) T Cells in Chronic Obstructive Pulmonary Disease. Respir Res (2011) 12. doi: 10.1186/1465-9921-12-149
62. Duan MC, Huang Y, Zhong XN, Tang HJ. Th17 Cell Enhances CD8 T-Cell Cytotoxicity via IL-21 Production in Emphysema Mice. Mediators Inflamm (2012) 2012. doi: 10.1155/2012/898053
63. Barceló B, Pons J, Ferrer JM, Sauleda J, Fuster A, Agustí AGN. Phenotypic Characterisation of T-Lymphocytes in COPD: Abnormal CD4+CD25+ Regulatory T-Lymphocyte Response to Tobacco Smoking. Eur Respir J (2008) 31:555–62. doi: 10.1183/09031936.00010407
64. Hinz D, Bauer M, Röder S, Olek S, Huehn J, Sack U, et al. Cord Blood Tregs With Stable FOXP3 Expression are Influenced by Prenatal Environment and Associated With Atopic Dermatitis at the Age of One Year. Allergy (2012) 67:380–9. doi: 10.1111/J.1398-9995.2011.02767.X
65. Brandsma CA, Hylkema MN, Geerlings M, van Geffen WH, Postma DS, Timens W, et al. Increased Levels of (Class Switched) Memory B Cells in Peripheral Blood of Current Smokers. Respir Res (2009) 10. doi: 10.1186/1465-9921-10-108
66. Brandsma CA, Kerstjens HAM, van Geffen WH, Geerlings M, Postma DS, Hylkema MN, et al. Differential Switching to IgG and IgA in Active Smoking COPD Patients and Healthy Controls. Eur Respir J (2012) 40:313–21. doi: 10.1183/09031936.00011211
67. Fusby JS, Kassmeier MD, Palmer VL, Perry GA, Anderson DK, Hackfort BT, et al. Cigarette Smoke-Induced Effects on Bone Marrow B-Cell Subsets and CD4+:CD8+ T-Cell Ratios are Reversed by Smoking Cessation: Influence of Bone Mass on Immune Cell Response to and Recovery From Smoke Exposure. Inhalation Toxicol (2010) 22:785–96. doi: 10.3109/08958378.2010.483258
68. Palmer VL, Kassmeier MD, Willcockson J, Akhter MP, Cullen DM, Swanson PC. N-Acetylcysteine Increases the Frequency of Bone Marrow Pro-B/pre-B Cells, But Does Not Reverse Cigarette Smoking-Induced Loss of This Subset. PloS One (2011) 6. doi: 10.1371/JOURNAL.PONE.0024804
69. Chavance M, Perrot JY, Annesi I. Smoking, CD45R0+ (Memory), and CD45RA+ (Naive) CD4+ T Cells. Am Rev Respir Dis (1993) 148:237–40. doi: 10.1164/AJRCCM/148.1.237
70. Tanigawa T, Araki S, Nakata A, Kitamura F, Yasumoto M, Sakurai S, et al. Increase in Memory (CD4+CD29+ and CD4+CD45RO+) T and Naive (CD4+CD45RA+) T-Cell Subpopulations in Smokers. Arch Environ Health (1998) 53:378–83. doi: 10.1080/00039899809605724
71. Nakata A, Takahashi M, Irie M, Fujioka Y, Haratani T, Araki S. Relationship Between Cumulative Effects of Smoking and Memory CD4+ T Lymphocyte Subpopulations. Addictive Behav (2007) 32:1526–31. doi: 10.1016/J.ADDBEH.2006.11.007
72. Soler P, Moreau A, Basset F, Hance AJ. Cigarette Smoking-Induced Changes in the Number and Differentiated State of Pulmonary Dendritic Cells/Langerhans Cells. Am Rev Respir Dis (1989) 139:1112–7. doi: 10.1164/AJRCCM/139.5.1112
73. Prescott SL, Noakes PS. Maternal Smoking in Pregnancy: Do the Effects on Innate (Toll-Like Receptor) Function Have Implications for Subsequent Allergic Disease? Allergy Asthma Clin Immunology : Off J Can Soc Allergy Clin Immunol (2007) 3. doi: 10.1186/1710-1492-3-1-10
74. Doz E, Noulin N, Boichot E, Guénon I, Fick L, le Bert M, et al. Cigarette Smoke-Induced Pulmonary Inflammation is TLR4/MyD88 and IL-1r1/MyD88 Signaling Dependent. J Immunol (Baltimore Md. : 1950) (2008) 180:1169–78. doi: 10.4049/JIMMUNOL.180.2.1169
75. Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N, et al. Cigarette Smoke Silences Innate Lymphoid Cell Function and Facilitates an Exacerbated Type I Interleukin-33-Dependent Response to Infection. Immunity (2015) 42:566–79. doi: 10.1016/J.IMMUNI.2015.02.011
76. Botelho FM, Gaschler GJ, Kianpour S, Zavitz CCJ, Trimble NJ, Nikota JK, et al. Innate Immune Processes are Sufficient for Driving Cigarette Smoke-Induced Inflammation in Mice. Am J Respir Cell Mol Biol (2010) 42:394–403. doi: 10.1165/RCMB.2008-0301OC
77. Kawai T, Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat Immunol (2010) 11:373–84. doi: 10.1038/NI.1863
78. Pace E, Giarratano A, Ferraro M, Bruno A, Siena L, Mangione S, et al. TLR4 Upregulation Underpins Airway Neutrophilia in Smokers With Chronic Obstructive Pulmonary Disease and Acute Respiratory Failure. Hum Immunol (2011) 72:54–62. doi: 10.1016/J.HUMIMM.2010.09.009
79. Fatemi K, Radvar M, Rezaee A, Rafatpanah H, Azangoo Khiavi H, Dadpour Y, et al. Comparison of Relative TLR-2 and TLR-4 Expression Level of Disease and Healthy Gingival Tissue of Smoking and non-Smoking Patients and Periodontally Healthy Control Patients. Aust Dental J (2013) 58:315–20. doi: 10.1111/ADJ.12089
80. Robays LJ, Lanckacker EA, Moerloose KB, Maes T, Bracke KR, Brusselle GG, et al. Concomitant Inhalation of Cigarette Smoke and Aerosolized Protein Activates Airway Dendritic Cells and Induces Allergic Airway Inflammation in a TLR-Independent Way. J Immunol (Baltimore Md. (2009) 1950) 183:2758–66. doi: 10.4049/JIMMUNOL.0802204
81. Botelho FM, Nikota JK, Bauer CMT, Morissette MC, Iwakura Y, Kolbeck R, et al. Cigarette Smoke-Induced Accumulation of Lung Dendritic Cells is Interleukin-1α-Dependent in Mice. Respir Res (2012) 13. doi: 10.1186/1465-9921-13-81
82. Comer DM, Kidney JC, Ennis M, Elborn JS. Airway Epithelial Cell Apoptosis and Inflammation in COPD, Smokers and Nonsmokers. Eur Respir J (2013) 41:1058–67. doi: 10.1183/09031936.00063112
83. Chung KF. P38 Mitogen-Activated Protein Kinase Pathways in Asthma and COPD. Chest (2011) 139:1470–9. doi: 10.1378/CHEST.10-1914
84. Ratcliffe MJ, Dougall IG. Comparison of the Anti-Inflammatory Effects of Cilomilast, Budesonide and a P38 Mitogen Activated Protein Kinase Inhibitor in COPD Lung Tissue Macrophages. BMC Pharmacol Toxicol (2012) 13. doi: 10.1186/2050-6511-13-15
85. Yang H, Long F, Zhang Y, Yu R, Zhang P, Li W, et al. 1α,25-Dihydroxyvitamin D3 Induces Neutrophil Apoptosis Through the P38 MAPK Signaling Pathway in Chronic Obstructive Pulmonary Disease Patients. PloS One (2015) 10. doi: 10.1371/JOURNAL.PONE.0120515
86. Bewley MA, Belchamber KBR, Chana KK, Budd RC, Donaldson G, Wedzicha JA, et al. Differential Effects of P38, MAPK, PI3K or Rho Kinase Inhibitors on Bacterial Phagocytosis and Efferocytosis by Macrophages in COPD. PloS One (2016) 11. doi: 10.1371/JOURNAL.PONE.0163139
87. Alharbi KS, Fuloria NK, Fuloria S, Rahman SB, Al-Malki WH, Javed Shaikh MA, et al. Nuclear Factor-Kappa B and its Role in Inflammatory Lung Disease. Chemico-biological Interact (2021) 345. doi: 10.1016/J.CBI.2021.109568
88. Hariharan A, Hakeem AR, Radhakrishnan S, Reddy MS, Rela M. The Role and Therapeutic Potential of NF-Kappa-B Pathway in Severe COVID-19 Patients. Inflammopharmacology (2021) 29:91–100. doi: 10.1007/S10787-020-00773-9
89. Dockery DW, Pope CA, Xu X, Spengler JD, Ware JH, Fay ME, et al. An Association Between Air Pollution and Mortality in Six U.S. Cities. N Engl J Med (1993) 329:1753–9. doi: 10.1056/NEJM199312093292401
90. Rudell B, Blomberg A, Helleday R, Ledin MC, Lundbäck B, Stjernberg N, et al. Bronchoalveolar Inflammation After Exposure to Diesel Exhaust: Comparison Between Unfiltered and Particle Trap Filtered Exhaust. Occup Environ Med (1999) 56:527–34. doi: 10.1136/OEM.56.8.527
91. Kirkham PA, Barnes PJ. Oxidative Stress in COPD. Chest (2013) 144:266–73. doi: 10.1378/CHEST.12-2664
92. Schaberg T, Klein U, Rau M, Eller J, Lode H. Subpopulations of Alveolar Macrophages in Smokers and Nonsmokers: Relation to the Expression of CD11/CD18 Molecules and Superoxide Anion Production. Am J Respir Crit Care Med (1995) 151:1551–8. doi: 10.1164/AJRCCM.151.5.7735614
93. Paredi P, Kharitonov SA, Leak D, Ward S, Cramer D, Barnes PJ. Exhaled Ethane, a Marker of Lipid Peroxidation, is Elevated in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2000) 162:369–73. doi: 10.1164/AJRCCM.162.2.9909025
94. Paredi P, Kharitonov SA, Barnes PJ. Analysis of Expired Air for Oxidation Products. Am J Respir Crit Care Med (2002) 166. doi: 10.1164/RCCM.2206012
95. Montuschi P. Exhaled Breath Condensate Analysis in Patients With COPD. Clinica Chimica acta; Int J Clin Chem (2005) 356:22–34. doi: 10.1016/J.CCCN.2005.01.012
96. Paggiaro PL, Bartoli ML, Novelli F, Costa F, Malagrin L, Melosini L, et al. Malondialdehyde in Exhaled Breath Condensate as a Marker of Oxidative Stress in Different Pulmonary Diseases. Mediators Inflamm (2011) 2011. doi: 10.1155/2011/891752
97. Vandivier RW, Henson PM, Douglas IS. Burying the Dead: The Impact of Failed Apoptotic Cell Removal (Efferocytosis) on Chronic Inflammatory Lung Disease. Chest (2006) 129:1673–82. doi: 10.1378/CHEST.129.6.1673
98. Donnelly LE, Barnes PJ. Defective Phagocytosis in Airways Disease. Chest (2012) 141:1055–62. doi: 10.1378/CHEST.11-2348
99. Richens TR, Linderman DJ, Horstmann SA, Lambert C, Xiao YQ, Keith RL, et al. Cigarette Smoke Impairs Clearance of Apoptotic Cells Through Oxidant-Dependent Activation of RhoA. Am J Respir Crit Care Med (2009) 179:1011–21. doi: 10.1164/RCCM.200807-1148OC
100. Kirkham PA, Spooner G, Rahman I, Rossi AG. Macrophage Phagocytosis of Apoptotic Neutrophils is Compromised by Matrix Proteins Modified by Cigarette Smoke and Lipid Peroxidation Products. Biochem Biophys Res Commun (2004) 318:32–7. doi: 10.1016/J.BBRC.2004.04.003
101. Bozinovski S, Vlahos R, Zhang Y, Lah LC, Seow HJ, Mansell A, et al. Carbonylation Caused by Cigarette Smoke Extract is Associated With Defective Macrophage Immunity. Am J Respir Cell Mol Biol (2011) 45:229–36. doi: 10.1165/RCMB.2010-0272OC
102. Barnes PJ, Adcock IM, Ito K. Histone Acetylation and Deacetylation: Importance in Inflammatory Lung Diseases. Eur Respir J (2005) 25:552–63. doi: 10.1183/09031936.05.00117504
103. Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative Stress Reduces Histone Deacetylase 2 Activity and Enhances IL-8 Gene Expression: Role of Tyrosine Nitration. Biochem Biophys Res Commun (2004) 315:240–5. doi: 10.1016/J.BBRC.2004.01.046
104. Meja KK, Rajendrasozhan S, Adenuga D, Biswas SK, Sundar IK, Spooner G, et al. Curcumin Restores Corticosteroid Function in Monocytes Exposed to Oxidants by Maintaining HDAC2. Am J Respir Cell Mol Biol (2008) 39:312–23. doi: 10.1165/RCMB.2008-0012OC
105. Ito K, Barnes PJ. COPD as a Disease of Accelerated Lung Aging. Chest (2009) 135:173–80. doi: 10.1378/CHEST.08-1419
106. Wooding DJ, Ryu MH, Li H, Alexis NE, Pena O, Carlsten C. Acute Air Pollution Exposure Alters Neutrophils in Never-Smokers and at-Risk Humans. Eur Respir J (2020) 55. doi: 10.1183/13993003.01495-2019
107. Shi T, Knaapen AM, Begerow J, Birmili W, Borm PJA, Schins RPF. Temporal Variation of Hydroxyl Radical Generation and 8-Hydroxy-2’-Deoxyguanosine Formation by Coarse and Fine Particulate Matter. Occup Environ Med (2003) 60:315–21. doi: 10.1136/OEM.60.5.315
108. Valavanidis A, Fiotakis K, Vlachogianni T. Airborne Particulate Matter and Human Health: Toxicological Assessment and Importance of Size and Composition of Particles for Oxidative Damage and Carcinogenic Mechanisms. J Environ Sci Health Part C Environ Carcinog Ecotoxicol Rev (2008) 26:339–62. doi: 10.1080/10590500802494538
109. Nagai H, Toyokuni S. Biopersistent Fiber-Induced Inflammation and Carcinogenesis: Lessons Learned From Asbestos Toward Safety of Fibrous Nanomaterials. Arch Biochem Biophysics (2010) 502:1–7. doi: 10.1016/J.ABB.2010.06.015
110. Chuang HC, Fan CW, Chen KY, Chang-Chien GP, Chan CC. Vasoactive Alteration and Inflammation Induced by Polycyclic Aromatic Hydrocarbons and Trace Metals of Vehicle Exhaust Particles. Toxicol Lett (2012) 214:131–6. doi: 10.1016/J.TOXLET.2012.08.012
111. Squadrito GL, Cueto R, Dellinger B, Pryor WA. Quinoid Redox Cycling as a Mechanism for Sustained Free Radical Generation by Inhaled Airborne Particulate Matter. Free Radical Biol Med (2001) 31:1132–8. doi: 10.1016/S0891-5849(01)00703-1
112. Risom L, Møller P, Loft S. Oxidative Stress-Induced DNA Damage by Particulate Air Pollution. Mutat Res (2005) 592:119–37. doi: 10.1016/J.MRFMMM.2005.06.012
113. Wu LL, Chiou CC, Chang PY, Wu JT. Urinary 8-OHdG: A Marker of Oxidative Stress to DNA and a Risk Factor for Cancer, Atherosclerosis and Diabetics. Clinica Chimica acta; Int J Clin Chem (2004) 339:1–9. doi: 10.1016/J.CCCN.2003.09.010
114. Pilger A, Rüdiger HW. 8-Hydroxy-2’-Deoxyguanosine as a Marker of Oxidative DNA Damage Related to Occupational and Environmental Exposures. Int Arch Occup Environ Health (2006) 80:1–15. doi: 10.1007/S00420-006-0106-7
115. Ohyama M, Otake T, Adachi S, Kobayashi T, Morinaga K. A Comparison of the Production of Reactive Oxygen Species by Suspended Particulate Matter and Diesel Exhaust Particles With Macrophages. Inhalation Toxicol (2007) 19 Suppl1:157–60. doi: 10.1080/08958370701496103
116. Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA Damage and Disease: Induction, Repair and Significance. Mutat Res (2004) 567:1–61. doi: 10.1016/J.MRREV.2003.11.001
117. Sunil VR, Vayas KN, Massa CB, Gow AJ, Laskin JD, Laskin DL. Ozone-Induced Injury and Oxidative Stress in Bronchiolar Epithelium are Associated With Altered Pulmonary Mechanics. Toxicological Sciences : an Off J Soc Toxicol (2013) 133:309–19. doi: 10.1093/TOXSCI/KFT071
118. Sunil VR, Patel-Vayas K, Shen J, Laskin JD, Laskin DL. Classical and Alternative Macrophage Activation in the Lung Following Ozone-Induced Oxidative Stress. Toxicol Appl Pharmacol (2012) 263:195–202. doi: 10.1016/J.TAAP.2012.06.009
119. Gurgueira SA, Lawrence J, Coull B, Krishna Murthy GG, González-Flecha B. Rapid Increases in the Steady-State Concentration of Reactive Oxygen Species in the Lungs and Heart After Particulate Air Pollution Inhalation. Environ Health Perspect (2002) 110:749–55. doi: 10.1289/EHP.02110749
120. Li N, Xia T, Nel AE. The Role of Oxidative Stress in Ambient Particulate Matter-Induced Lung Diseases and its Implications in the Toxicity of Engineered Nanoparticles. Free Radical Biol Med (2008) 44:1689–99. doi: 10.1016/J.FREERADBIOMED.2008.01.028
121. Foyer CH, Noctor G. Redox Homeostasis and Antioxidant Signaling: A Metabolic Interface Between Stress Perception and Physiological Responses. Plant Cell (2005) 17:1866–75. doi: 10.1105/TPC.105.033589
122. Terzano C, Di Stefano F, Conti V, Graziani E, Petroianni A. Air Pollution Ultrafine Particles: Toxicity Beyond the Lung. Eur Rev Med Pharmacol Sci (2010) 14:809–21.
123. DosReis GA, Borges VM. Role of Fas-Ligand Induced Apoptosis in Pulmonary Inflammation and Injury. Curr Drug Targets Inflamm Allergy (2003) 2:161–7. doi: 10.2174/1568010033484287
124. Martin LD, Krunkosky TM, Dye JA, Fischer BM, Jiang NF, Rochelle LG, et al. The Role of Reactive Oxygen and Nitrogen Species in the Response of Airway Epithelium to Particulates. Environ Health Perspect (1997) 105(Suppl 5):1301–7. doi: 10.1289/EHP.97105S51301
125. Paola Rosanna D, Salvatore C. Reactive Oxygen Species, Inflammation, and Lung Diseases. Curr Pharm Design (2012) 18:3889–900. doi: 10.2174/138161212802083716
126. Knaapen AM, Güngör N, Schins RPF, Borm PJA, van Schooten FJ. Neutrophils and Respiratory Tract DNA Damage and Mutagenesis: A Review. Mutagenesis (2006) 21:225–36. doi: 10.1093/MUTAGE/GEL032
127. Brieger K, Schiavone S, Miller FJ, Krause KH. Reactive Oxygen Species: From Health to Disease. Swiss Med Weekly (2012) 142. doi: 10.4414/SMW.2012.13659
128. Waris G, Ahsan H. Reactive Oxygen Species: Role in the Development of Cancer and Various Chronic Conditions. J Carcinog (2006) 5. doi: 10.1186/1477-3163-5-14
129. Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-Inducible Factor-1alpha is a Key Regulator of Metastasis in a Transgenic Model of Cancer Initiation and Progression. Cancer Res (2007) 67:563–72. doi: 10.1158/0008-5472.CAN-06-2701
130. Ito K, Yano T, Morodomi Y, Yoshida T, Kohno M, Haro A, et al. Serum Antioxidant Capacity and Oxidative Injury to Pulmonary DNA in Never-Smokers With Primary Lung Cancer. Anticancer Res (2012) 32:1063–7.
131. Wang N, Mengersen K, Tong S, Kimlin M, Zhou M, Wang L, et al. Short-Term Association Between Ambient Air Pollution and Lung Cancer Mortality. Environ Res (2019) 179. doi: 10.1016/J.ENVRES.2019.108748
132. Xing DF, Xu CD, Liao XY, Xing TY, Cheng SP, Hu MG, et al. Spatial Association Between Outdoor Air Pollution and Lung Cancer Incidence in China. BMC Public Health (2019) 19. doi: 10.1186/S12889-019-7740-Y
133. Filaire E, Dupuis C, Galvaing G, Aubreton S, Laurent H, Richard R, et al. Lung Cancer: What are the Links With Oxidative Stress, Physical Activity and Nutrition. Lung Cancer (Amsterdam Netherlands) (2013) 82:383–9. doi: 10.1016/J.LUNGCAN.2013.09.009
134. Li X, Li S, Yan S, Wang Y, Wang X, Sihoe ADL, et al. Impact of Preoperative Exercise Therapy on Surgical Outcomes in Lung Cancer Patients With or Without COPD: A Systematic Review and Meta-Analysis. Cancer Manage Res (2019) 11:1765–77. doi: 10.2147/CMAR.S186432
135. Ost M, Coleman V, Kasch J, Klaus S. Regulation of Myokine Expression: Role of Exercise and Cellular Stress. Free Radical Biol Med (2016) 98:78–89. doi: 10.1016/J.FREERADBIOMED.2016.02.018
136. Saha SK, Lee S, Won J, Choi HY, Kim K, Yang GM, et al. Correlation Between Oxidative Stress, Nutrition, and Cancer Initiation. Int J Mol Sci (2017) 18. doi: 10.3390/IJMS18071544
137. Wong JL, Evans SE. Bacterial Pneumonia in Patients With Cancer: Novel Risk Factors and Management. Clinics Chest Med (2017) 38:263–77. doi: 10.1016/J.CCM.2016.12.005
138. Laumbach RJ, Kipen HM. Respiratory Health Effects of Air Pollution: Update on Biomass Smoke and Traffic Pollution. J Allergy Clin Immunol (2012) 129:3–11. doi: 10.1016/J.JACI.2011.11.021
139. Hellermann GR, Nagy SB, Kong X, Lockey RF, Mohapatra SS. Mechanism of Cigarette Smoke Condensate-Induced Acute Inflammatory Response in Human Bronchial Epithelial Cells. Respir Res (2002) 3. doi: 10.1186/RR172
140. Herfs M, Hubert P, Poirrier AL, Vandevenne P, Renoux V, Habraken Y, et al. Proinflammatory Cytokines Induce Bronchial Hyperplasia and Squamous Metaplasia in Smokers: Implications for Chronic Obstructive Pulmonary Disease Therapy. Am J Respir Cell Mol Biol (2012) 47:67–79. doi: 10.1165/RCMB.2011-0353OC
141. Meijer M, Rijkers GT, van Overveld FJ. Neutrophils and Emerging Targets for Treatment in Chronic Obstructive Pulmonary Disease. Expert Rev Clin Immunol (2013) 9:1055–68. doi: 10.1586/1744666X.2013.851347
142. Smith AH, Lopipero PA, Barroga VR. Meta-Analysis of Studies of Lung Cancer Among Silicotics. Epidemiol (Cambridge Mass.) (1995) 6:617–24. doi: 10.1097/00001648-199511000-00010
143. Brims FJH, Kong K, Harris EJA, Sodhi-Berry N, Reid A, Murray CP, et al. Pleural Plaques and the Risk of Lung Cancer in Asbestos-Exposed Subjects. Am J Respir Crit Care Med (2020) 201:57–62. doi: 10.1164/RCCM.201901-0096OC
144. Knox JF, Holmes S, Doll R, Hill ID. Mortality From Lung Cancer and Other Causes Among Workers in an Asbestos Textile Factory. Br J Ind Med (1968) 25:293–303. doi: 10.1136/OEM.25.4.293
145. Cassidy A, Mannetje A, van Tongeren M, Field JK, Zaridze D, Szeszenia-Dabrowska N, et al. Occupational Exposure to Crystalline Silica and Risk of Lung Cancer: A Multicenter Case-Control Study in Europe. Epidemiol (Cambridge Mass.) (2007) 18:36–43. doi: 10.1097/01.EDE.0000248515.28903.3C
146. Singh N, Davis GS. Review: Occupational and Environmental Lung Disease. Curr Opin pulmonary Med (2002) 8:117–25. doi: 10.1097/00063198-200203000-00007
147. Cohen RAC, Patel A, Green FHY. Lung Disease Caused by Exposure to Coal Mine and Silica Dust. Semin Respir Crit Care Med (2008) 29:651–61. doi: 10.1055/S-0028-1101275
148. Nishimura Y, Nishiike-Wada T, Wada Y, Miura Y, Otsuki T, Iguchi H. Long-Lasting Production of TGF-Beta1 by Alveolar Macrophages Exposed to Low Doses of Asbestos Without Apoptosis. Int J Immunopathol Pharmacol (2007) 20:661–71. doi: 10.1177/039463200702000402
149. Hamilton RF, Thakur SA, Holian A. Silica Binding and Toxicity in Alveolar Macrophages. Free Radical Biol Med (2008) 44:1246–58. doi: 10.1016/J.FREERADBIOMED.2007.12.027
150. Maeda M, Nishimura Y, Kumagai N, Hayashi H, Hatayama T, Katoh M, et al. Dysregulation of the Immune System Caused by Silica and Asbestos. J Immunotoxicol (2010) 7:268–78. doi: 10.3109/1547691X.2010.512579
151. Buhtoiarov IN, Lum HD, Berke G, Sondel PM, Rakhmilevich AL. Synergistic Activation of Macrophages via CD40 and TLR9 Results in T Cell Independent Antitumor Effects. J Immunol (Baltimore Md. : 1950) (2006) 176:309–18. doi: 10.4049/JIMMUNOL.176.1.309
152. Lum HD, Buhtoiarov IN, Schmidt BE, Berke G, Paulnock DM, Sondel PM, et al. In Vivo CD40 Ligation can Induce T-Cell-Independent Antitumor Effects That Involve Macrophages. J Leukocyte Biol (2006) 79:1181–92. doi: 10.1189/JLB.0405191
153. Rao TD, Frey AB. Administration of Silica Sensitizes Lipopolysaccharide Responsiveness of Murine Macrophages But Inhibits T and B Cell Priming by Inhibition of Antigen Presenting Function. Immunol Investigations (1998) 27:181–99. doi: 10.3109/08820139809089455
154. Su W, Ito T, Oyama T, Kitagawa T, Yamori T, Fujiwara H, et al. The Direct Effect of IL-12 on Tumor Cells: IL-12 Acts Directly on Tumor Cells to Activate NF-kappaB and Enhance IFN-Gamma-Mediated STAT1 Phosphorylation. Biochem Biophys Res Commun (2001) 280:503–12. doi: 10.1006/BBRC.2000.4150
155. Weiss JM, Subleski JJ, Wigginton JM, Wiltrout RH. Immunotherapy of Cancer by IL-12-Based Cytokine Combinations. Expert Opin Biol Ther (2007) 7:1705–21. doi: 10.1517/14712598.7.11.1705
156. Borges VM, Falcão H, Leite-Júnior JH, Alvim L, Teixeira GP, Russo M, et al. Fas Ligand Triggers Pulmonary Silicosis. J Exp Med (2001) 194:155–63. doi: 10.1084/JEM.194.2.155
157. Corsini E, Giani A, Peano S, Marinovich M, Galli CL. Resistance to Silica-Induced Lung Fibrosis in Senescent Rats: Role of Alveolar Macrophages and Tumor Necrosis Factor-α (TNF). Mech Ageing Dev (2004) 125:145–6. doi: 10.1016/j.mad.2003.11.002
158. Chen JJ, Sun Y, Nabel GJ. Regulation of the Proinflammatory Effects of Fas Ligand (CD95L). Sci (New York N.Y.) (1998) 282:1714–7. doi: 10.1126/SCIENCE.282.5394.1714
159. Jagirdar J, Begín R, Dufresne A, Goswami S, Lee TC, Rom WN. Transforming Growth Factor-Beta (TGF-Beta) in Silicosis. Am J Respir Crit Care Med (1996) 154:1076–81. doi: 10.1164/AJRCCM.154.4.8887610
160. Barbarin V, Arras M, Misson P, Delos M, McGarry B, Phan SH, et al. Characterization of the Effect of Interleukin-10 on Silica-Induced Lung Fibrosis in Mice. Am J Respir Cell Mol Biol (2004) 31:78–85. doi: 10.1165/RCMB.2003-0299OC
161. Markowitz S. Asbestos-Related Lung Cancer and Malignant Mesothelioma of the Pleura: Selected Current Issues. Semin Respir Crit Care Med (2015) 36:334–46. doi: 10.1055/S-0035-1549449
162. Nicholson WJ. The Carcinogenicity of Chrysotile Asbestos–a Review. Ind Health (2001) 39:57–64. doi: 10.2486/INDHEALTH.39.57
163. Niklinski J, Niklinska W, Chyczewska E, Laudanski J, Naumnik W, Chyczewski L, et al. The Epidemiology of Asbestos-Related Diseases. Lung Cancer (Amsterdam Netherlands) (2004) 45(Suppl 1). doi: 10.1016/J.LUNGCAN.2004.04.008
164. Snyder J, Alexander-Miller M, Berzofsky J, Belyakov I. Molecular Mechanisms and Biological Significance of CTL Avidity. Curr HIV Res (2003) 1:287–94. doi: 10.2174/1570162033485230
165. Raulet DH, Guerra N. Oncogenic Stress Sensed by the Immune System: Role of Natural Killer Cell Receptors. Nat Rev Immunol (2009) 9:568–80. doi: 10.1038/NRI2604
166. Gruber UF. Asbestos-Related Benign Disease and Cancer: Symptoms and Treatment. Anti-cancer Drugs (1990) 1:187–97. doi: 10.1097/00001813-199012000-00012
167. Nishimura Y, Miura Y, Maeda M, Kumagai N, Murakami S, Hayashi H, et al. Impairment in Cytotoxicity and Expression of NK Cell- Activating Receptors on Human NK Cells Following Exposure to Asbestos Fibers. Int J Immunopathol Pharmacol (2009) 22:579–90. doi: 10.1177/039463200902200304
168. BéruBé KA, Quinlan TR, Fung H, Magae J, Vacek P, Taatjes DJ, et al. Apoptosis is Observed in Mesothelial Cells After Exposure to Crocidolite Asbestos. Am J Respir Cell Mol Biol (1996) 15:141–7. doi: 10.1165/AJRCMB.15.1.8679218
169. Fung H, Kow YW, van Houten B, Mossman BT. Patterns of 8-Hydroxydeoxyguanosine Formation in DNA and Indications of Oxidative Stress in Rat and Human Pleural Mesothelial Cells After Exposure to Crocidolite Asbestos. Carcinogenesis (1997) 18:825–32. doi: 10.1093/CARCIN/18.4.825
170. Ollikainen T, Puhakka A, Kahlos K, Linnainmaa K, Kinnula VL. Modulation of Cell and DNA Damage by Poly(ADP)ribose Polymerase in Lung Cells Exposed to H(2)O(2) or Asbestos Fibres. Mutat Res (2000) 470:77–84. doi: 10.1016/S1383-5718(00)00093-0
171. Aljandali A, Pollack H, Yeldandi A, Li Y, Weitzman SA, Kamp DW. Asbestos Causes Apoptosis in Alveolar Epithelial Cells: Role of Iron-Induced Free Radicals. J Lab Clin Med (2001) 137:330–9. doi: 10.1067/MLC.2001.114826
172. International Agency for Research on Cancer Agents Classified by The IARC Monographs Vol. 1-100. Lyon: IARC. Available at: http://monographs.iarc.fr/ENG/Classification/index.php (Accessed February 2022).
173. Siemiatycki J, Richardson L, Straif K, Latreille B, Lakhani R, Campbell S, et al. Listing Occupational Carcinogens. Environ Health Perspect (2004) 112:1447–59. doi: 10.1289/EHP.7047
174. Algranti E, Buschinelli JTP, de Capitani EM. Occupational Lung Cancer. Jornal Brasileiro Pneumologia : Publicacao Oficial da Sociedade Bras Pneumologia e Tisilogia (2010) 36:784–94. doi: 10.1590/S1806-37132010000600017
175. Haddad R, Massaro D. Idiopathic Diffuse Interstitial Pulmonary Fibrosis (Fibrosing Alveolitis), Atypical Epithelial Proliferation and Lung Cancer. Am J Med (1968) 45:211–9. doi: 10.1016/0002-9343(68)90039-9
176. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-Based Guidelines for Diagnosis and Management. Am J Respir Crit Care Med (2011) 183:788–824. doi: 10.1164/RCCM.2009-040GL
177. Ballester B, Milara J, Cortijo J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int J Mol Sci (2019) 20. doi: 10.3390/IJMS20030593
178. Wynn TA. Integrating Mechanisms of Pulmonary Fibrosis. J Exp Med (2011) 208:1339–50. doi: 10.1084/JEM.20110551
179. Fernandez IE, Eickelberg O. The Impact of TGF-β on Lung Fibrosis: From Targeting to Biomarkers. Proc Am Thorac Soc (2012) 9:111–6. doi: 10.1513/PATS.201203-023AW
180. Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune Mechanisms in Pulmonary Fibrosis. Am J Respir Cell Mol Biol (2016) 55:309–22. doi: 10.1165/RCMB.2016-0121TR
181. Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T Lymphocytes in Pulmonary Fibrosis. J Leukocyte Biol (2008) 83:237–44. doi: 10.1189/JLB.0707504
182. Sharma SK, MacLean JA, Pinto C, Kradin RL. The Effect of an Anti-CD3 Monoclonal Antibody on Bleomycin-Induced Lymphokine Production and Lung Injury. Am J Respir Crit Care Med (1996) 154:193–200. doi: 10.1164/AJRCCM.154.1.8680680
183. Galati D, de Martino M, Trotta A, Rea G, Bruzzese D, Cicchitto G, et al. Peripheral Depletion of NK Cells and Imbalance of the Treg/Th17 Axis in Idiopathic Pulmonary Fibrosis Patients. Cytokine (2014) 66:119–26. doi: 10.1016/J.CYTO.2013.12.003
184. Boveda-Ruiz D, D’Alessandro-Gabazza CN, Toda M, Takagi T, Naito M, Matsushima Y, et al. Differential Role of Regulatory T Cells in Early and Late Stages of Pulmonary Fibrosis. Immunobiology (2013) 218:245–54. doi: 10.1016/J.IMBIO.2012.05.020
185. Granville CA, Memmott RM, Balogh A, Mariotti J, Kawabata S, Han W, et al. A Central Role for Foxp3+ Regulatory T Cells in K-Ras-Driven Lung Tumorigenesis. PloS One (2009) 4. doi: 10.1371/JOURNAL.PONE.0005061
186. Ganesan A-P, Johansson M, Ruffell B, Beltran A, Lau J, Jablons DM, et al. Tumor-Infiltrating Regulatory T Cells Inhibit Endogenous Cytotoxic T Cell Responses to Lung Adenocarcinoma. J Immunol (Baltimore Md. : 1950) (2013) 191:2009–17. doi: 10.4049/JIMMUNOL.1301317
187. Xiong S, Guo R, Yang Z, Xu L, Du L, Li R, et al. Treg Depletion Attenuates Irradiation-Induced Pulmonary Fibrosis by Reducing Fibrocyte Accumulation, Inducing Th17 Response, and Shifting IFN-γ, IL-12/IL-4, IL-5 Balance. Immunobiology (2015) 220:1284–91. doi: 10.1016/J.IMBIO.2015.07.001
188. Arras M, Huaux F, Vink A, Delos M, Coutelier JP, Many MC, et al. Interleukin-9 Reduces Lung Fibrosis and Type 2 Immune Polarization Induced by Silica Particles in a Murine Model. Am J Respir Cell Mol Biol (2001) 24:368–75. doi: 10.1165/AJRCMB.24.4.4249
189. van den Brûle S, Heymans J, Havaux X, Renauld JC, Lison D, Huaux F, et al. Profibrotic Effect of IL-9 Overexpression in a Model of Airway Remodeling. Am J Respir Cell Mol Biol (2007) 37:202–9. doi: 10.1165/RCMB.2006-0397OC
190. Simonian PL, Wehrmann F, Roark CL, Born WK, O’Brien RL, Fontenot AP. γδ T Cells Protect Against Lung Fibrosis via IL-22. J Exp Med (2010) 207:2239–53. doi: 10.1084/JEM.20100061
191. Barron L, Wynn TA. Fibrosis is Regulated by Th2 and Th17 Responses and by Dynamic Interactions Between Fibroblasts and Macrophages. American Journal of Physiology. Gastrointestinal Liver Physiol (2011) 300. doi: 10.1152/AJPGI.00414.2010
192. Conway EM, Pikor LA, Kung SHY, Hamilton MJ, Lam S, Lam WL, et al. Macrophages, Inflammation, and Lung Cancer. Am J Respir Crit Care Med (2016) 193:116–30. doi: 10.1164/RCCM.201508-1545CI
193. Maeda H, Akaike T. Nitric Oxide and Oxygen Radicals in Infection, Inflammation, and Cancer. Biochem Biokhimiia (1998) 63:1007–19.
194. Pechkovsky D, Prasse A, Kollert F, Engel KMY, Dentler J, Luttmann W, et al. Alternatively Activated Alveolar Macrophages in Pulmonary Fibrosis-Mediator Production and Intracellular Signal Transduction. Clin Immunol (Orlando Fla.) (2010) 137:89–101. doi: 10.1016/J.CLIM.2010.06.017
195. Sun L, Louie MC, Vannella KM, Wilke CA, Levine AM, Moore BB, et al. New Concepts of IL-10-Induced Lung Fibrosis: Fibrocyte Recruitment and M2 Activation in a CCL2/CCR2 Axis. Am J Physiol Lung Cell Mol Physiol (2011) 300. doi: 10.1152/AJPLUNG.00122.2010
196. Murray PJ, Wynn TA. Protective and Pathogenic Functions of Macrophage Subsets. Nat Rev Immunol (2011) 11:723–37. doi: 10.1038/NRI3073
197. Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE. Baseline BAL Neutrophilia Predicts Early Mortality in Idiopathic Pulmonary Fibrosis. Chest (2008) 133:226–32. doi: 10.1378/CHEST.07-1948
198. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-Associated Neutrophils Stimulate T Cell Responses in Early-Stage Human Lung Cancer. J Clin Invest (2014) 124:5466–80. doi: 10.1172/JCI77053
199. Xaubet A, Agustí C, Luburich P, Barberá JA, Carrión M, Ayuso MC, et al. Interleukin-8 Expression in Bronchoalveolar Lavage Cells in the Evaluation of Alveolitis in Idiopathic Pulmonary Fibrosis. Respir Med (1998) 92:338–44. doi: 10.1016/S0954-6111(98)90118-4
200. Ashitani Ji, Mukae H, Taniguchi H, Ihi T, Kadota Ji, Kohno S, et al. Granulocyte-Colony Stimulating Factor Levels in Bronchoalveolar Lavage Fluid From Patients With Idiopathic Pulmonary Fibrosis. Thorax (1999) 54:1015–20. doi: 10.1136/THX.54.11.1015
201. Obayashi Y, Yamadori I, Fujita J, Yoshinouchi T, Ueda N, Takahara J. The Role of Neutrophils in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Chest (1997) 112:1338–43. doi: 10.1378/CHEST.112.5.1338
202. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil Extracellular Traps Produced During Inflammation Awaken Dormant Cancer Cells in Mice. Sci (New York N.Y.) (2018) 361. doi: 10.1126/SCIENCE.AAO4227
203. Eduard W. Fungal Spores: A Critical Review of the Toxicological and Epidemiological Evidence as a Basis for Occupational Exposure Limit Setting. Crit Rev Toxicol (2009) 39:799–864. doi: 10.3109/10408440903307333
204. Bennett JW, Klich M. Mycotoxins. Clin Microbiol Rev (2003) 16:497–516. doi: 10.1128/CMR.16.3.497-516.2003
205. Kankkunen P, Rintahaka J, Aalto A, Leino M, Majuri M-L, Alenius H, et al. Trichothecene Mycotoxins Activate Inflammatory Response in Human Macrophages. J Immunol (Baltimore Md. (2009) 1950) 182:6418–25. doi: 10.4049/JIMMUNOL.0803309
206. Rocha O, Ansari K, Doohan FM. Effects of Trichothecene Mycotoxins on Eukaryotic Cells: A Review. Food Additives Contaminants (2005) 22:369–78. doi: 10.1080/02652030500058403
207. Ouyang YL, Azcona-olivera JI, Pestka JJ. Effects of Trichothecene Structure on Cytokine Secretion and Gene Expression in Murine CD4+ T-Cells. Toxicology (1995) 104:187–202. doi: 10.1016/0300-483X(95)03147-8
208. Meky FA, Hardie LJ, Evans SW, Wild CP. Deoxynivalenol-Induced Immunomodulation of Human Lymphocyte Proliferation and Cytokine Production. Food Chem Toxicology : an Int J Published Br Ind Biol Res Assoc (2001) 39:827–36. doi: 10.1016/S0278-6915(01)00029-1
209. Capasso L, Longhin E, Caloni F, Camatini M, Gualtieri M. Synergistic Inflammatory Effect of PM10 With Mycotoxin Deoxynivalenol on Human Lung Epithelial Cells. Toxicon : Off J Int Soc Toxinol (2015) 104:65–72. doi: 10.1016/J.TOXICON.2015.08.008
210. Amuzie CJ, Harkema JR, Pestka JJ. Tissue Distribution and Proinflammatory Cytokine Induction by the Trichothecene Deoxynivalenol in the Mouse: Comparison of Nasal vs. Oral Exposure. Toxicology (2008) 248:39–44. doi: 10.1016/J.TOX.2008.03.005
211. Lichtenstein Rosenblum JH, Molina RM, Donaghey TC, Amuzie CJ, Pestka JJ, Coull BA, et al. Pulmonary Responses to Stachybotrys Chartarum and its Toxins: Mouse Strain Affects Clearance and Macrophage Cytotoxicity. Toxicological Sciences : an Off J Soc Toxicol (2010) 116:113–21. doi: 10.1093/TOXSCI/KFQ104
212. Liu C, Shen H, Yi L, Shao P, Soulika AM, Meng X, et al. Oral Administration of Aflatoxin G₁ Induces Chronic Alveolar Inflammation Associated With Lung Tumorigenesis. Toxicol Lett (2015) 232:547–56. doi: 10.1016/J.TOXLET.2014.11.002
213. Barta SK, Zou Y, Schindler J, Shenoy N, Bhagat TD, Steidl U, et al. Synergy of Sequential Administration of a Deglycosylated Ricin A Chain-Containing Combined Anti-CD19 and Anti-CD22 Immunotoxin (Combotox) and Cytarabine in a Murine Model of Advanced Acute Lymphoblastic Leukemia. Leukemia Lymphoma (2012) 53:1999–2003. doi: 10.3109/10428194.2012.679267
214. Brown RFR, White DE. Ultrastructure of Rat Lung Following Inhalation of Ricin Aerosol. Int J Exp Pathol (1997) 78:267–76. doi: 10.1046/J.1365-2613.1997.300363.X
215. Roy CJ, Hale M, Hartings JM, Pitt L, Duniho S. Impact of Inhalation Exposure Modality and Particle Size on the Respiratory Deposition of Ricin in BALB/c Mice. Inhalation Toxicol (2003) 15:619–38. doi: 10.1080/08958370390205092
216. Liang J, Zhao H, Yao L, Tang H, Dong H, Wu Y, et al. Phosphatidylinositol 3-Kinases Pathway Mediates Lung Caspase-1 Activation and High Mobility Group Box 1 Production in a Toluene-Diisocyanate Induced Murine Asthma Model. Toxicol Lett (2015) 236:25–33. doi: 10.1016/J.TOXLET.2015.04.011
217. Woodruff PG, Ellwanger A, Solon M, Cambier CJ, Pinkerton KE, Koth LL. Alveolar Macrophage Recruitment and Activation by Chronic Second Hand Smoke Exposure in Mice. COPD (2009) 6:86–94. doi: 10.1080/15412550902751738
218. Jinot J, Bayard S. Respiratory Health Effects of Passive Smoking: Lung Cancer and Other Disorders (1992). Available at: https://books.google.com/books?hl=de&lr=&id=uUnCzSqEn7EC&oi=fnd&pg=PR9&ots=dQMwUPyest&sig=HK37S7L6SABYUM6OIgP9JLhTeR8 (Accessed January 29, 2022).
219. Hecht SS. Cigarette Smoking: Cancer Risks, Carcinogens, and Mechanisms. Langenbeck’s Arch Surg (2006) 391:603–13. doi: 10.1007/S00423-006-0111-Z
220. Adams JD, O’mara-adams KJ, Hoffmann D. Toxic and Carcinogenic Agents in Undiluted Mainstream Smoke and Sidestream Smoke of Different Types of Cigarettes. Carcinogenesis (1987) 8:729–31. doi: 10.1093/CARCIN/8.5.729
221. Besaratinia A, Pfeifer GP. Second-Hand Smoke and Human Lung Cancer. Lancet Oncol (2008) 9:657–66. doi: 10.1016/S1470-2045(08)70172-4
222. National Research Council: Committee on Passive Smoking - Google Scholar. Available at: https://scholar.google.com/scholar_lookup?title=Committee+on+passive+smoking+board+on+environmental+studies+and+toxicology&publication_year=1986& (Accessed January 31, 2022).
223. Mohtashamipur E, Mohtashamipur A, Germann PG, Ernst H, Norpoth K, Mohr U. Comparative Carcinogenicity of Cigarette Mainstream and Sidestream Smoke Condensates on the Mouse Skin. J Cancer Res Clin Oncol (1990) 116:604–8. doi: 10.1007/BF01637081
224. Robbins AS, Abbey DE, Lebowitz MD. Passive Smoking and Chronic Respiratory Disease Symptoms in non-Smoking Adults. Int J Epidemiol (1993) 22:809–17. doi: 10.1093/IJE/22.5.809
225. Kirkland LR. Environmental Tobacco Smoke and Lung Cancer in Nonsmoking Women. JAMA: J Am Med Assoc (1995) 273:519–20. doi: 10.1001/jama.1995.03520310011006
226. Brownson RC, Figgs LW, Caisley LE. Epidemiology of Environmental Tobacco Smoke Exposure. Oncogene (2002) 21:7341–8. doi: 10.1038/SJ.ONC.1205809
227. Hecht SS. Carcinogen Derived Biomarkers: Applications in Studies of Human Exposure to Secondhand Tobacco Smoke. Tobacco Control (2004) 13 Suppl 1. doi: 10.1136/TC.2002.002816
228. Sato M, Shames DS, Gazdar AF, Minna JD. A Translational View of the Molecular Pathogenesis of Lung Cancer. J Thorac Oncology : Off Publ Int Assoc Study Lung Cancer (2007) 2:327–43. doi: 10.1097/01.JTO.0000263718.69320.4C
229. Hecht SS. A Biomarker of Exposure to Environmental Tobacco Smoke (ETS) and Ernst Wynder’s Opinion About ETS and Lung Cancer. Prev Med (2006) 43:256–60. doi: 10.1016/J.YPMED.2006.07.020
230. Metabolites of a tobacco-specific lung carcinogen in the urine of elementary school-aged children - Google Suche. Available at: https://www.google.com/search?q=Metabolites+of+a+tobacco-specific+lung+carcinogen+in+the+urine+of+elementary+school-aged+children&rlz=1C1PRFI_enAT888AT888&oq=Metabolites+of+a+tobacco-specific+lung+carcinogen+in+the+urine+of+elementary+school-aged+children&aqs=chrome..69i57j69i61l2.426j0j4&sourceid=chrome&ie=UTF-8 (Accessed January 31, 2022).
231. Coggins CRE. An Updated Review of Inhalation Studies With Cigarette Smoke in Laboratory Animals. Int J Toxicol (2007) 26:331–8. doi: 10.1080/10915810701490190
232. Snijders AM, Zhou M, Whitehead TP, Fitch B, Pandey P, Hechmer A, et al. In Utero and Early-Life Exposure to Thirdhand Smoke Causes Profound Changes to the Immune System. Clin Sci (London England : 1979) (2021) 135:1053–63. doi: 10.1042/CS20201498
233. Johnson JD, Houchens DP, Kluwe WM, Craig DK, Fisher GL. Effects of Mainstream and Environmental Tobacco Smoke on the Immune System in Animals and Humans: A Review. Crit Rev Toxicol (1990) 20:369–95. doi: 10.3109/10408449009089870
234. O’Callaghan DS, O’Donnell D, O’Connell F, O’Byrne KJ. The Role of Inflammation in the Pathogenesis of non-Small Cell Lung Cancer. J Thorac Oncology: Off Publ Int Assoc Study Lung Cancer (2010) 5:2024–36. doi: 10.1097/JTO.0B013E3181F387E4
235. Brenner A, Wang Z, Kleinerman RA, Wang L, Zhang S, Metayer C, et al. Previous Pulmonary Diseases and Risk of Lung Cancer in Gansu Province, China. Int J Epidemiol (2001) 30:118–24. doi: 10.1093/IJE/30.1.118
236. Zheng W, Blot WJ, Liao ML, Wang ZX, Levin LI, Zhao JJ, et al. Lung Cancer and Prior Tuberculosis Infection in Shanghai. Br J Cancer (1987) 56:501–4. doi: 10.1038/BJC.1987.233
237. Littman AJ, Jackson LA, Vaughan TL. Chlamydia Pneumoniae and Lung Cancer: Epidemiologic Evidence. Cancer Epidemiol Biomarkers Prevention : Publ Am Assoc Cancer Res Cosponsored by Am Soc Prev Oncol (2005) 14:773–8. doi: 10.1158/1055-9965.EPI-04-0599
238. Geng Y, Shane RB, Berencsi K, Gonczol E, Zaki MH, Margolis DJ, et al. Chlamydia Pneumoniae Inhibits Apoptosis in Human Peripheral Blood Mononuclear Cells Through Induction of IL-10. J Immunol (Baltimore Md. : 1950) (2000) 164:5522–9. doi: 10.4049/JIMMUNOL.164.10.5522
239. Arenberg DA, Kunkel SL, Polverini PJ, Glass M, Burdick MD, Strieter RM. Inhibition of Interleukin-8 Reduces Tumorigenesis of Human non-Small Cell Lung Cancer in SCID Mice. J Clin Invest (1996) 97:2792–802. doi: 10.1172/JCI118734
240. Kovaleva O. v., Romashin D, Zborovskaya IB, Davydov MM, Shogenov MS, Gratchev A. Human Lung Microbiome on the Way to Cancer. J Immunol Res (2019) 2019. doi: 10.1155/2019/1394191
241. Laroumagne S, Salinas-Pineda A, Hermant C, Murris M, Gourraud PA, Do C, et al. Incidence and Characteristics of Bronchial Colonisation in Patient With Lung Cancer: A Retrospective Study of 388 Cases. Rev Des Maladies Respiratoires (2011) 28:328–35. doi: 10.1016/J.RMR.2010.05.020
242. Yu G, Gail MH, Consonni D, Carugno M, Humphrys M, Pesatori AC, et al. Characterizing Human Lung Tissue Microbiota and its Relationship to Epidemiological and Clinical Features. Genome Biol (2016) 17. doi: 10.1186/S13059-016-1021-1
243. Jiang LX, Ren HY, Zhou HJ, Zhao SH, Hou BY, Yan JP, et al. Simultaneous Detection of 13 Key Bacterial Respiratory Pathogens by Combination of Multiplex PCR and Capillary Electrophoresis. Biomed Environ Sciences : BES (2017) 30:549–61. doi: 10.3967/BES2017.074
244. Greathouse KL, White JR, Vargas AJ, Bliskovsky V, Beck JA, von Muhlinen N, et al. Interaction Between the Microbiome and TP53 in Human Lung Cancer. Genome Biol (2018) 19. doi: 10.1186/S13059-018-1501-6
245. Morgan XC, Huttenhower C. Chapter 12: Human Microbiome Analysis. PloS Comput Biol (2012) 8. doi: 10.1371/JOURNAL.PCBI.1002808
246. Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the Lung Microbiome in the “Healthy” Smoker and in COPD. PloS One (2011) 6. doi: 10.1371/JOURNAL.PONE.0016384
247. Cheng M, Qian L, Shen G, Bian G, Xu T, Xu W, et al. Microbiota Modulate Tumoral Immune Surveillance in Lung Through a γδt17 Immune Cell-Dependent Mechanism. Cancer Res (2014) 74:4030–41. doi: 10.1158/0008-5472.CAN-13-2462
248. Nešić D, Hsu Y, Stebbins CE. Assembly and Function of a Bacterial Genotoxin. Nature (2004) 429:429–33. doi: 10.1038/NATURE02532
249. Travaglione S, Fabbri A, Fiorentini C. The Rho-Activating CNF1 Toxin From Pathogenic E. Coli: A Risk Factor for Human Cancer Development? Infect Agents Cancer (2008) 3. doi: 10.1186/1750-9378-3-4
250. Yaghoobi H, Bandehpour M, Kazemi B. Apoptotic Effects of the B Subunit of Bacterial Cytolethal Distending Toxin on the A549 Lung Cancer Cell Line. Asian Pacific J Cancer Prevention : APJCP (2016) 17:299–304. doi: 10.7314/APJCP.2016.17.S3.299
251. Apopa PL, Alley L, Penney RB, Arnaoutakis K, Steliga MA, Jeffus S, et al. PARP1 Is Up-Regulated in Non-Small Cell Lung Cancer Tissues in the Presence of the Cyanobacterial Toxin Microcystin. Front Microbiol (2018) 9:1757. doi: 10.3389/FMICB.2018.01757
Keywords: immune landscape, COPD, lung cancer, smoker, never-smoker
Citation: Taucher E, Mykoliuk I, Lindenmann J and Smolle-Juettner F-M (2022) Implications of the Immune Landscape in COPD and Lung Cancer: Smoking Versus Other Causes. Front. Immunol. 13:846605. doi: 10.3389/fimmu.2022.846605
Received: 31 December 2021; Accepted: 28 February 2022;
Published: 21 March 2022.
Edited by:
Shyamasree Ghosh, National Institute of Science Education and Research (NISER), IndiaReviewed by:
Krishna Prahlad Maremanda, Texas A&M University, United StatesVenkataramana Sidhaye, Johns Hopkins University, United States
Copyright © 2022 Taucher, Mykoliuk, Lindenmann and Smolle-Juettner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisabeth Taucher, ZWxpc2FiZXRoLnRhdWNoZXJAbWVkdW5pZ3Jhei5hdA==