 Mark Mensink1
Mark Mensink1 Thi Ngoc Minh Tran2,3†
Thi Ngoc Minh Tran2,3† Esther A. Zaal2†
Esther A. Zaal2† Ellen Schrama1
Ellen Schrama1 Celia R. Berkers2,3
Celia R. Berkers2,3 Jannie Borst1*
Jannie Borst1* Sander de Kivit1
Sander de Kivit1- 1Department of Immunology and Oncode Institute, Leiden University Medical Center, Leiden, Netherlands
- 2Division of Cell Biology, Metabolism & Cancer, Department Biomolecular Health Sciences, Faculty of Veterinary Medicine, Utrecht University, Utrecht, Netherlands
- 3Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research, Utrecht University, Utrecht, Netherlands
CD4+ conventional T cells (Tconvs) mediate adaptive immune responses, whereas regulatory T cells (Tregs) suppress those responses to safeguard the body from autoimmunity and inflammatory diseases. The opposing activities of Tconvs and Tregs depend on the stage of the immune response and their environment, with an orchestrating role for cytokine- and costimulatory receptors. Nutrient availability also impacts T-cell functionality via metabolic and biosynthetic processes that are largely unexplored. Many data argue that costimulation by Tumor Necrosis Factor Receptor 2 (TNFR2) favors support of Treg over Tconv responses and therefore TNFR2 is a key clinical target. Here, we review the pertinent literature on this topic and highlight the newly identified role of TNFR2 as a metabolic regulator for thymus-derived (t)Tregs. We present novel transcriptomic and metabolomic data that show the differential impact of TNFR2 on Tconv and tTreg gene expression and reveal distinct metabolic impact on both cell types.
Introduction
Adaptive immunity is controlled by the opposing activities of Tconvs and Tregs. The Treg lineage is hallmarked by expression of the master transcriptional regulator FOXP3 that, in conjunction with other transcription factors, dictates Treg function (1, 2). CD4+ T cells may gain FOXP3 expression during thymic development and thus become tTregs that have a T-cell receptor (TCR) repertoire focused on self-antigens. Alternatively, CD4+ Tconvs may gain FOXP3 expression and convert into peripherally induced (p)Tregs during responses to foreign antigens (1, 2). In most tissues, specialized tTreg subsets reside that respond and adapt to Tconv responses (3, 4). Tregs prevent autoimmunity, maintain tissue homeostasis and limit inflammation. Aberrant Treg function may therefore contribute to immune disorders or cancer (5–7). To improve immunotherapy of such diseases, we must understand the signals that control Tconv and Treg responses. These responses are initiated in lymphoid organs, but continue and often persist in many non-lymphoid tissues in health and disease (3, 4). Both T-cell types are activated by engagement of their TCR, which together with costimulatory- and cytokine receptor stimulation leads to proliferation and effector differentiation (8). Besides these signals that are primarily delivered by dendritic cells (DCs), other environmental signals such as nutrient availability impact T-cell responses (9). Especially in rapidly proliferating cells, nutrients provide the metabolic building blocks for biosynthetic pathways (10). Therefore, it can be envisioned that the interplay of immune receptors and nutrient metabolism is important for the activity of Tconvs and Tregs.
Tconvs and Tregs share numerous immunoregulatory receptors. These may differentially impact the response of each cell type, due to differential wiring of signal transduction pathways (11) and other physiological differences between the cell types. Altered signaling in Tregs is for example due to constitutive low expression of STAT4, which disallows pro-inflammatory cytokine expression (11), or due to binding of FOXP3 to NFAT, which disallows IL-2 production (12). Accumulating data emphasize the selective importance of the T-cell costimulatory receptor TNFR2 for promoting Treg as opposed to Tconv responses. In mouse models, TNFR2-stimulated Treg responses were shown to protect from autoimmunity and graft-versus-host disease (13–17). These in vivo studies independently demonstrated that TNFR2 expression on Tregs is essential to maintain the expression of Treg signature molecules such as FOXP3, CD25 and CTLA-4, that are important for suppressive function. We recently reported that TNFR2 costimulation causes a glycolytic switch in activated human tTregs, providing the first evidence that TNFR2 can regulate cell metabolism (18). Given the great interest in antibody-based TNFR2 targeting in immune-related diseases, it is important to understand the consequences of TNFR2 costimulation for Tconv and Treg responses. At present, data suggest that TNFR2 preferentially promotes Treg over Tconv responses. Here, we review the validity of this assumption and present recent data from our own laboratory that highlight the response of tTregs and Tconvs to TNFR2 costimulation at the metabolic level. TNFR2 differentially regulates metabolism of tTregs and Tconvs, which may have implications for physiology, cell therapy and drug targeting.
Rationale for TNFR2 Targeting in Immune-Related Diseases
TNF(R) Targeting in Disease
Tumor Necrosis Factor (TNF) is a cytokine involved in a plethora of biological processes and is well known for its pro-inflammatory properties (19). Its involvement in the pathogenesis of autoimmune and chronic inflammatory diseases such as rheumatoid arthritis has led to the development and broad clinical application of TNF-blocking agents, including the antibodies infliximab, adalimumab and golimumab and the soluble IgG1-TNFR2 fusion protein etanercept (20, 21). Despite the successes of TNF-blocking therapies, these treatments are ineffective for part of the patients, can have side effects and can even exacerbate the disease (22–25). Around 30% of rheumatoid arthritis patients withdraw from treatment with a TNF-blocking agent for these reasons in the first year of therapy (26). This calls for a better understanding of the mechanistic consequences of TNF inhibition.
TNF is initially expressed on the cell surface as a transmembrane molecule and cleaved to be released in its soluble form (27–29). Either soluble or transmembrane TNF can activate TNFR1 (TNFRSF1A), while transmembrane TNF primarily activates TNFR2 (TNFRSF1B) (30, 31). TNFR1 is a ubiquitously expressed receptor, while TNFR2 has a more restricted tissue distribution (https://www.proteinatlas.org/). Both receptors link to TRAF signaling adaptors that can mediate cell survival, chemokine and cytokine signaling, but only TNFR1 has a death domain that can bind TRADD and mediate cell death by apoptosis or necroptosis (19, 32, 33).
It is already known for decades that TNF can have both pro-inflammatory and anti-inflammatory effects (32, 34–37). Neutralization of TNF in immune-related diseases aims to block the pro-inflammatory effects of TNF–TNFR1 signaling (38). However, such intervention will also block the anti-inflammatory properties of TNF, which seem to largely result from TNFR2 signaling into Tregs (32, 37, 39). Thus, drugs interfering with TNF function may restrain Treg responses and therefore be pro- instead of anti-inflammatory. Drugs that specifically modulate either TNFR1 or TNFR2 activity will therefore be more suited to combat inflammatory and autoimmune diseases and transplant rejection, as has been reviewed comprehensively (38, 40–45).
The Role of TNFR2 on Tregs and Tconvs
In human peripheral blood, TNFR2 is expressed by more than 80% of Tregs that are identified as FOXP3+CD25hi cells (46). TNFR2 is upregulated on human and mouse Tregs upon TCR stimulation, indicating that TNFR2 is particularly expressed on effector Tregs as opposed to resting or naïve Tregs (18, 47). On Tregs, TNFR2 supports FOXP3 expression and proliferation and maintains their suppressive activity (13, 18, 47–51). Specific polymorphisms in the human TNFRSF1B gene of which one was shown to impair TNFR2 signaling (52) are associated with inflammatory bowel disease, ankylosing spondylitis, lupus and/or rheumatoid arthritis, supporting the idea that TNFR2 protects against these diseases (40, 53–58). In mouse models, further evidence has been gathered that support this notion. Wild-type but not TNFR2-deficient Tregs could inhibit experimental colitis in mice (13). In experimental autoimmune encephalomyelitis (EAE), a mouse model for multiple sclerosis, Treg-restricted TNFR2 deficiency led to a reduction in expression of Treg signature molecules and suppressive function, which was associated with disease exacerbation (14). Another study supports the notion that Tregs require TNFR2 expression to suppress EAE (15). Tregs also selectively expanded upon TNFR2 agonism and reduced graft-versus-host disease in a TNFR2-dependent manner in allogeneic hematopoietic stem cell transplantation in mice (16, 17). Whereas promoting TNFR2 activity on Tregs could be promising in autoimmune diseases and transplantation, TNFR2 inhibition is considered in cancer therapy (59). High TNFR2 expression is observed on certain cancers and on tumor-associated Tregs (59–63). TNFR2 agonism and antagonism are currently being explored in both autoimmune diseases and cancer, potentially opening new treatment avenues (62, 64–66).
TNFR2 functions as a costimulatory receptor not only on Tregs, but also on Tconvs by lowering the threshold of T-cell activation and increasing survival and proliferation (67–70). TNFR2 expression is upregulated on Tconvs upon TCR activation and is associated with resistance to Treg-mediated suppression in mice, as determined by increased proliferation and effector cytokine production (47, 71). TNFR2-deficient Tconvs failed to induce colitis in mice, which was linked to reduced proliferation and less IFN-γ production (72). Together, these findings indicate that TNFR2 has costimulatory effects on both Tconvs and Tregs, but that under specific conditions, Treg responsiveness prevails. Here we offer our recent experimental findings resulting from unbiased “omics” approaches that give new insights into the effects of TNFR2 agonism on both tTregs and Tconvs. We believe that this information can stimulate new research directions with clinical relevance, in particular regarding the impact of environmental metabolic cues on the Treg/Tconv balance and functional outcomes.
We present an unbiased, side-by-side analysis of the consequences of TNFR2 costimulation on human Tconvs and tTregs at the level of gene expression and cell metabolism. The Treg lineage consists of tTregs and pTregs (73, 74), but pTregs are largely restricted to mucosal surfaces and the maternal-fetal interface (75, 76). The majority of Tregs in vivo are tTregs, which control systemic and tissue-specific autoimmunity (3, 77). Therefore, we focused on tTregs that were isolated to high purity from blood as described before (18, 78).
Effects of TNFR2 on Tregs and Tconvs According to Our Recent Work
Transcriptomics Identifies Responses of Tconvs and tTregs to TNFR2 Costimulation
Contrary to Tconvs, Tregs rely on costimulation to initiate proliferation after in vitro triggering of the TCR/CD3 complex by anti-CD3 antibody (79). In addition, Tregs require exogenous IL-2 in such assays, since they cannot produce this cytokine after activation, unlike Tconvs (80, 81). Accordingly, in our experimental setting, Tconvs already started to proliferate upon stimulation of the TCR/CD3 complex alone, whereas tTregs required additional costimulation by either CD28 or TNFR2 (Figure 1A) (18). We used a comparative setting of Tconvs and tTregs that had been pre-expanded, rested and restimulated via CD3 alone, CD3/CD28 or CD3/TNFR2. After 24 h, cells were harvested for transcriptome analysis (18) (Supplementary Methods).
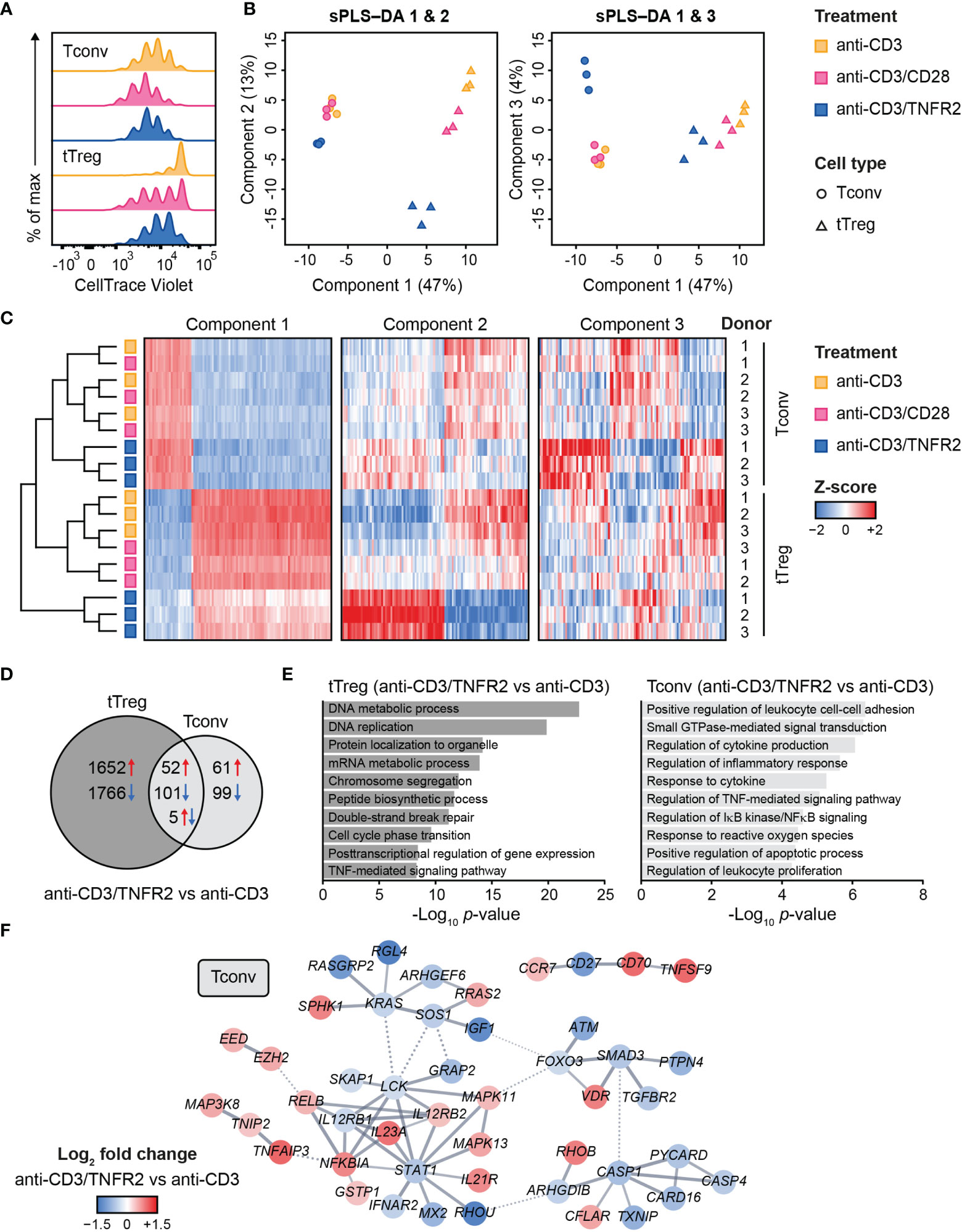
Figure 1 Transcriptome analysis of human Tconvs and tTregs highlights distinct responses upon CD3, CD3/CD28 or CD3/TNFR2 stimulation. (A) Tconvs and tTregs were sorted and pre-expanded for 2 weeks, then labeled with CellTrace Violet and restimulated for 96 h to assess cell proliferation by flow cytometry. For color legend of stimuli, see panel (B) Tconvs and tTregs were sorted and pre-expanded for 2 weeks, restimulated as indicated for 24 h and subjected to transcriptomics. sPLS–DA shows clustering of indicated sample groups by components 1–3. (C) Representative heat map of the sPLS–DA results, showing hierarchical clustering of indicated sample groups and relative expression levels (color-coded z-scores) of the top 100 transcripts per component 1–3. (D) Venn diagram depicting the number of up- (red) and downregulated (blue) transcripts in CD3/TNFR2- versus CD3-stimulated cells, including unique and shared changes in tTregs and Tconvs (p < 0.05, log2 fold change > 0.32 or < -0.32). (E) GO Biological Process enrichment analysis of the differentially expressed genes shown in (D). (F) STRING network of genes involved in the enriched processes for Tconvs in (E), only including genes with high-confidence associations. Markov Cluster Algorithm (MCL) clustering (82) was performed and inter-cluster associations are displayed as dotted lines. Genes were colored based on log2 fold change. Further details are described in the Supplementary Methods.
We aimed to globally identify the most discriminative transcripts that separated the sample groups, with focus on the effects of TNFR2 or CD28 costimulation. Therefore, we performed sparse partial least squares discriminant analysis (sPLS–DA), which involves supervised clustering due to awareness of the sample classes (Figure 1B) (83). Component 1 (47%) distinguished tTregs from Tconvs regardless of costimulation (Figure 1B). Component 2 (13%) discriminated TNFR2-costimulated tTregs from CD3(/CD28)-stimulated tTregs and did the same, albeit more modestly, for Tconvs (Figure 1B, left panel). Component 3 (4%) highlighted a unique reaction of Tconvs to TNFR2 costimulation (Figure 1B, right panel). A representative heat map including transcripts with the highest contribution to components 1–3 showed that TNFR2-costimulated Tconvs and tTregs both expressed genes that set them apart from their CD3- or CD3/CD28-stimulated counterparts (Figure 1C, Supplementary Table 1). However, the transcripts that contributed most to component 2 indicated a greater impact of TNFR2 costimulation on the transcriptome of tTregs versus that of Tconvs. The transcriptomes of CD3- or CD3/CD28-stimulated tTregs and Tconvs clustered more closely together, especially in Tconvs, likely due to the convergence of CD3- and CD28 signaling pathways (84).
In terms of numbers of differentially expressed genes following TNFR2 costimulation, a Venn diagram revealed in an unbiased manner that tTregs respond more strongly to TNFR2 costimulation than Tconvs (Figure 1D). Gene Ontology (GO) enrichment analysis showed that processes related to mitosis were highly overrepresented in TNFR2-costimulated tTregs (Figure 1E, left panel). In TNFR2-costimulated Tconvs, GO and STRING network analyses indicated activation of TNFR family costimulatory pathways (CD70/TNFSF7, TNFSF9, RELB, MAPKs) and cytokine pathways (IL-12RB2, IL-21R, IL-23A) and inhibition of apoptosis (CFLAR/cFLIP, TNFAIP3) and inflammation (CASP-1 and -4, PYCARD, CARD16) (Figure 1E, right panel; Figure 1F). These data indicate that human Tconvs and tTregs both respond to TNFR2 costimulation, in a different manner, although the breadth of the transcriptomic changes is much larger in tTregs (18).
TNFR2 Costimulation Regulates Metabolism in tTregs and Tconvs
Activated T cells rely on metabolic programs that support energy supply and biosynthesis needed for their rapid proliferation and effector functions (9, 10). Tconvs that are activated via the TCR/CD3 complex exhibit an mTOR-driven upregulation of glycolysis, which is promoted by CD28 costimulation (85, 86). Divergent results have been reported for Tregs (87–94), which may be explained by the complexity of the cell populations studied. For example, pTregs and tTregs are often not discriminated and cell populations often contain Tconv contamination. The impact of TNFR family members on cell metabolism has not been studied in any depth thus far. We recently reported that TNFR2 impacts T-cell metabolism. Specifically, we showed that upon TCR/CD3-mediated activation, human tTregs undergo an mTOR-driven glycolytic switch after TNFR2 costimulation, but not after CD28 costimulation, even though they enter cell division in both settings (18). As compared to TCR/CD3-activated glycolytic Tconvs, TNFR2-costimulated glycolytic tTregs show a net lactate retention and an increased flux of glucose-derived carbon into the tricarboxylic acid (TCA) cycle. Glycolysis proved essential to maintain FOXP3 expression and suppressive function in TNFR2-costimulated tTregs (18). Besides glucose, the amino acid glutamine is essential for T-cell proliferation and function (10). In Tconvs, glutamine uptake and catabolism is enhanced upon CD3/CD28 stimulation and is crucial for proliferation, differentiation and cytokine production (95–99). Glutamine is initially metabolized to glutamate and α-ketoglutarate, which can be further processed in the TCA cycle to support mitochondrial oxidative phosphorylation and the biosynthetic processes needed for cell proliferation.
Cell metabolism is often regulated at the posttranslational level and, therefore, transcriptome analyses do not predict metabolic activity well (100). For this reason, we performed metabolomics to examine the metabolic processing of glucose and glutamine by TCR/CD3-activated tTregs and Tconvs in response to CD28 versus TNFR2 costimulation. We fed the cells [13C6]-glucose or [13C5]-glutamine and compared by mass spectrometry the processing of these nutrients into intermediates of the TCA cycle and nucleotide synthesis pathways (Supplementary Methods). Tracing of [13C6]-glucose confirmed our previous finding (18) that TNFR2 costimulation promotes the influx of glucose-derived 13C into the TCA cycle in tTregs in particular (Figure 2A). Incorporation of 13C into citrate, α-ketoglutarate (both TCA cycle) or aspartate was increased in TNFR2-costimulated tTregs, whereas the increase was less pronounced in Tconvs (Figure 2A). Aspartate is a precursor for pyrimidine nucleotide synthesis, which can be analyzed by measuring UTP. Tracing of [13C5]-glutamine revealed that TNFR2 costimulation promoted both in Tconvs and tTregs incorporation of glutamine-derived 13C into α-ketoglutarate, aspartate and the nucleotide UTP (Figure 2B).
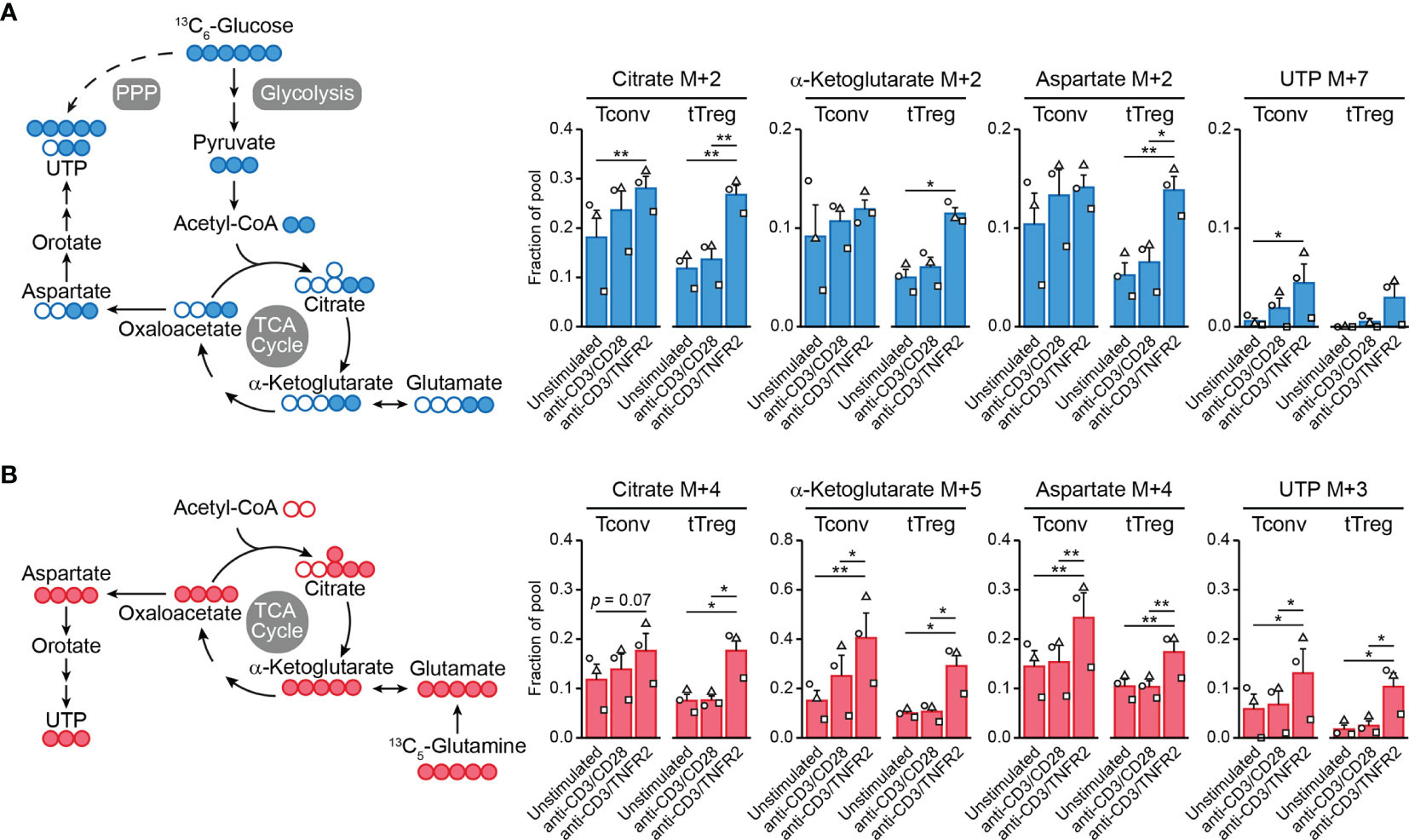
Figure 2 TNFR2 costimulation regulates Tconv and tTreg metabolism. Tconvs and tTregs were sorted as in Figure 1 and pre-expanded for 1 week. Subsequently, cells were restimulated as indicated for 24 h in the presence of IL-2 and either [13C6]-glucose (A) or [13C5]-glutamine (B) and analyzed by liquid chromatography–mass spectrometry (LC–MS) to trace 13C-labeled metabolites. (A, B) Left panels: schematic diagrams of the fate of 13C in metabolic pathways following [13C6]-glucose uptake (blue) or [13C5]-glutamine uptake (red), including glycolysis, the TCA cycle, the pentose phosphate pathway (PPP) and nucleotide synthesis. (A, B) Right panels: quantifications of 13C-labeled (M+) citrate, α-ketoglutarate (both TCA cycle), aspartate and UTP (both nucleotide synthesis) as fractions of the pool. M+7 was shown for UTP in the [13C6]-glucose tracer experiment, as incorporated 13C originated from aspartate as well as PPP-derived ribose. Two-way ANOVA with Tukey’s post hoc test was used for statistical analysis (*p < 0.05, **p < 0.01). Data are presented as mean ± SEM and data points are depicted as unique symbols per donor. Additional information is described in the Supplementary Methods.
This is the first evidence that TNFR2 affects metabolism both in tTregs and Tconvs, with remodeling of glutamine metabolism as a commonality between both cell types and the enhancement of glycolysis and coupled TCA cycle events being unique for tTregs. Glutamine metabolism appears to promote nucleotide biosynthesis in both tTregs and Tconvs after TNFR2 costimulation, which can sustain cell proliferation. However, glutamine can also regulate effector differentiation: in CD4+ T cells it supports Th17 differentiation via glutamate and the antioxidant glutathione and impairs Th1 differentiation via α-ketoglutarate that acts as a cofactor for histone- and DNA demethylases (98). In this way, glutamine metabolism may support epigenetic reprogramming to maintain or direct tTreg and/or Tconv functionality under conditions of TNFR2 costimulation. This hypothesis remains to be proven, but we have already shown that glycolysis is essential to maintain FOXP3 expression and suppressive functions of tTregs after TNFR2 costimulation (18).
Discussion
Although TNF biology is very complex, TNF blockade is widely applied in disease and often successful. It has been well-established that TNFR1 and TNFR2 have distinct tissue distributions and signaling functions and that TNFR1 can be classified as pro-inflammatory, while TNFR2 is more anti-inflammatory. This realization fosters the approach of selective TNFR1- or TNFR2 agonism or antagonism in therapy of human diseases. Particularly, selective TNFR2 agonism to promote Treg responses and thus attenuate autoimmunity, inflammation and transplant rejection is highlighted in recent literature. Many data, including in vivo agonism of TNFR2 in mouse models argue that Tregs rather than Tconvs are the main responders to TNFR2 agonism. Even so, TNFR2 can also costimulate the proliferative response of Tconvs. We add here a side-by-side unbiased analysis of the response of human Tconvs and tTregs to TNFR2 costimulation at the transcriptomic level. This analysis highlights that both TCR/CD3-activated cell types have an inherent gene expression pattern that is discrepant between the cell types and does not change much upon CD28 or TNFR2 costimulation. We also observe a gene set that responds similarly to TNFR2 costimulation in tTregs and Tconvs, and a gene set that differentially responds between the cell types. These reactions are unique to TNFR2 costimulation and not shared with CD28 costimulation. These data suggest that the responder cells adopt a unique functionality upon TNFR2 costimulation that warrants further investigation. Furthermore, we highlight TNFR2 as a metabolic regulator—not only for tTregs, but also for Tconvs—as shown by a common upregulation of glutamine metabolism. It will also be of interest to investigate how this affects Tconv and tTreg responses.
Since tTregs are the major controllers of autoimmunity (77), we studied the response of tTregs to TNFR2 costimulation. The exact role of TNFR2 on pTreg responses remains to be elucidated. TNFR2 expression was not required for in vitro-generated, TGF-β-induced pTregs to suppress colitis in mice (101). Moreover, TNF blockade ameliorated EAE due to increased pTreg numbers, presumably by releasing an inhibitory effect of TNFR2 on TGF-β-induced FOXP3 expression (102). However, a contrasting study reports that TNF–TNFR2 signaling increases TGF-β-induced pTreg differentiation and suppressive function in vitro and in vivo (103). It remains to be unraveled whether the (metabolic) responses of human pTregs to TNFR2 costimulation are unique or more skewed towards the responses of Tconvs or tTregs.
Why Tregs as opposed to Tconvs predominate the response to TNFR2 agonists in vivo is not clear. One reason may be that TNFR2 expression is higher on Tregs than on Tconvs (18, 46, 47). By binding newly produced cell surface TNF, TNFR2hi Tregs may outcompete TNFR2lo Tconvs from responding to this ligand. By binding transmembrane TNF, Tregs may also prevent TNF from being shed and from activating TNFR1 on multiple cell types. It can be envisioned that Tregs in this way provide negative feedback to dampen inflammation (104). Certainly during priming in secondary lymphoid organs, proliferating Tregs and Tconvs are in close proximity. The precise contexts of TNFR2 costimulation remain to be resolved, but both CD4+ and CD8+ Tconvs (105, 106) as well as Tregs (107) can express transmembrane TNF. In a graft-versus-host disease model, TNF produced by Tconvs proved to be crucial for Treg responses (17). Activated Tconvs also produce IL-2 that Tregs need for their proliferation (81). Therefore, it is likely that Tconvs can invite their own suppression by Tregs via transmembrane TNF. Subsequent TNFR2-induced Treg proliferation may shift the balance—a numbers game—towards immunosuppression. In addition to Tconvs, monocytes (108) and tolerogenic monocyte-derived dendritic cells (109) can express transmembrane TNF, but how this impacts Treg or Tconv responses is not yet known.
After priming, Tregs relocate to non-lymphoid tissues, where they encounter Tconvs and other cell types. It has been shown that Tregs adapt their differentiation state in peripheral tissues to that of locally resident effector T cells, by responding to specific cytokines and gaining expression of lineage-determining transcription factors such as T-bet or GATA-3 in addition to FOXP3 (1–3). Since metabolic activity depends on nutrient availability in the environment, it is tempting to speculate that TNFR2 costimulation endows Tconvs and Tregs with a degree of metabolic flexibility that allows them to facilitate survival, replication and functionality in metabolically changing environments, such as sites of inflammation or cancer. TNFR2 is also involved in the function of myeloid-derived suppressor cells (110), mesenchymal stem cells (111) and multiple cell types of the central nervous system, including oligodendrocytes (112), their precursor cells (113) and microglia (114). The interesting commonality between these cell types and Tregs is that they possess immunomodulatory or tissue-regenerative properties. Similar to Tregs, these TNFR2-expressing cell types may exhibit a negative feedback mechanism to suppress the pathological effects of excessive TNF–TNFR1 signaling, e.g. in neurological disorders (37). Future studies are required to establish common TNFR2-induced (metabolic) responses of these cell types and how such responses may support their protective functions.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GEO under accession code GSE138604.
Ethics Statement
The studies involving human participants were reviewed and approved by the internal ethical board of Sanquin, Amsterdam, The Netherlands. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MM designed and performed cell stimulation and transcriptomics experiments and data analysis, prepared figures and wrote the manuscript. EZ performed metabolomics and data analysis and prepared figures. TT performed transcriptomics data analysis and prepared figures. ES assisted with cell isolation. CB and JB contributed to study design and writing the manuscript. SK designed and performed cell stimulation and metabolomics experiments and contributed to study design and writing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was financially supported by Oncode, Strategic Funds from LUMC and grant ICI-00025 from the Institute for Chemical Immunology, funded by ZonMW Gravitation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the staff of the flow cytometry facility and genomics facility at the Netherlands Cancer Institute and the flow cytometry facility at Leiden University Medical Center for their technical assistance. We also thank Lonneke Nouwen for support in transcriptomics analysis, Jeroen Jansen at the Utrecht Metabolism Expertise Centre for technical assistance, and Dr Sarantos Kostidis and Dr Martin Giera from the Center for Proteomics and Metabolomics at Leiden University Medical Center for insightful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.881166/full#supplementary-material
References
1. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
2. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
3. Liston A, Gray DH. Homeostatic Control of Regulatory T Cell Diversity. Nat Rev Immunol (2014) 14(3):154–65. doi: 10.1038/nri3605
4. Munoz-Rojas AR, Mathis D. Tissue Regulatory T Cells: Regulatory Chameleons. Rev Immunol (2021) 21(9):597–611. doi: 10.1038/s41577-021-00519-w
5. Dominguez-Villar M, Hafler DA. Regulatory T Cells in Autoimmune Disease. Nat Immunol (2018) 19(7):665–73. doi: 10.1038/s41590-018-0120-4
6. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX Syndrome to FOXP3 Mutation: A Lesson on Immune Dysregulation. Ann N Y Acad Sci (2018) 1417(1):5–22. doi: 10.1111/nyas.13011
7. Plitas G, Rudensky AY. Regulatory T Cells in Cancer. Annu Rev Canc Biol (2020) 4:459–77. doi: 10.1146/annurev-cancerbio-030419-033428
8. Kumar P, Bhattacharya P, Prabhakar BS. A Comprehensive Review on the Role of Co-Signaling Receptors and Treg Homeostasis in Autoimmunity and Tumor Immunity. J Autoimmun (2018) 95:77–99. doi: 10.1016/j.jaut.2018.08.007
9. Chapman NM, Boothby MR, Chi H. Metabolic Coordination of T Cell Quiescence and Activation. Nat Rev Immunol (2020) 20(1):55–70. doi: 10.1038/s41577-019-0203-y
10. Buck MD, O'Sullivan D, Pearce EL. T Cell Metabolism Drives Immunity. J Exp Med (2015) 212(9):1345–60. doi: 10.1084/jem.20151159
11. Cuadrado E, van den Biggelaar M, de Kivit S, Chen YY, Slot M, Doubal I, et al. Proteomic Analyses of Human Regulatory T Cells Reveal Adaptations in Signaling Pathways That Protect Cellular Identity. Immunity (2018) 48(5):1046–59.e6. doi: 10.1016/j.immuni.2018.04.008
12. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, et al. FOXP3 Controls Regulatory T Cell Function Through Cooperation With NFAT. Cell (2006) 126(2):375–87. doi: 10.1016/j.cell.2006.05.042
13. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 Is Critical for the Stabilization of the CD4+Foxp3+ Regulatory T. Cell Phenotype in the Inflammatory Environment. J Immunol (2013) 190(3):1076–84. doi: 10.4049/jimmunol.1202659
14. Atretkhany KN, Mufazalov IA, Dunst J, Kuchmiy A, Gogoleva VS, Andruszewski D, et al. Intrinsic TNFR2 Signaling in T Regulatory Cells Provides Protection in CNS Autoimmunity. Proc Natl Acad Sci USA (2018) 115(51):13051–6. doi: 10.1073/pnas.1807499115
15. Ronin E, Pouchy C, Khosravi M, Hilaire M, Gregoire S, Casrouge A, et al. Tissue-Restricted Control of Established Central Nervous System Autoimmunity by TNF Receptor 2-Expressing Treg Cells. Proc Natl Acad Sci USA (2021) 118(13):e2014043118. doi: 10.1073/pnas.2014043118
16. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous TNFR2 Activation Protects From Acute GvHD via Host T Reg Cell Expansion. J Exp Med (2016) 213(9):1881–900. doi: 10.1084/jem.20151563
17. Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of GVHD by Regulatory T Cells Depends on TNF Produced by T Cells and TNFR2 Expressed by Regulatory T Cells. Blood (2016) 128(12):1651–9. doi: 10.1182/blood-2016-02-700849
18. de Kivit S, Mensink M, Hoekstra AT, Berlin I, Derks RJE, Both D, et al. Stable Human Regulatory T Cells Switch to Glycolysis Following TNF Receptor 2 Costimulation. Nat Metab (2020) 2(10):1046–61. doi: 10.1038/s42255-020-00271-w
19. Wajant H, Pfizenmaier K, Scheurich P. Tumor Necrosis Factor Signaling. Cell Death Differ (2003) 10(1):45–65. doi: 10.1038/sj.cdd.4401189
20. Choy EH, Panayi GS. Cytokine Pathways and Joint Inflammation in Rheumatoid Arthritis. N Engl J Med (2001) 344(12):907–16. doi: 10.1056/NEJM200103223441207
21. Monaco C, Nanchahal J, Taylor P, Feldmann M. Anti-TNF Therapy: Past, Present and Future. Int Immunol (2015) 27(1):55–62. doi: 10.1093/intimm/dxu102
22. Kary S, Worm M, Audring H, Huscher D, Renelt M, Sorensen H, et al. New Onset or Exacerbation of Psoriatic Skin Lesions in Patients With Definite Rheumatoid Arthritis Receiving Tumour Necrosis Factor Alpha Antagonists. Ann Rheum Dis (2006) 65(3):405–7. doi: 10.1136/ard.2005.037424
23. Ramos-Casals M, Brito-Zeron P, Munoz S, Soria N, Galiana D, Bertolaccini L, et al. Autoimmune Diseases Induced by TNF-Targeted Therapies: Analysis of 233 Cases. Med (Baltimore) (2007) 86(4):242–51. doi: 10.1097/MD.0b013e3181441a68
24. Ramos-Casals M, Brito-Zeron P, Cuadrado MJ, Khamashta MA. Vasculitis Induced by Tumor Necrosis Factor-Targeted Therapies. Curr Rheumatol Rep (2008) 10(6):442–8. doi: 10.1007/s11926-008-0072-z
25. Ko JM, Gottlieb AB, Kerbleski JF. Induction and Exacerbation of Psoriasis With TNF-Blockade Therapy: A Review and Analysis of 127 Cases. J Dermatol Treat (2009) 20(2):100–8. doi: 10.1080/09546630802441234
26. Soliman MM, Ashcroft DM, Watson KD, Lunt M, Symmons DP, Hyrich KL, et al. Impact of Concomitant Use of DMARDs on the Persistence With Anti-TNF Therapies in Patients With Rheumatoid Arthritis: Results From the British Society for Rheumatology Biologics Register. Ann Rheum Dis (2011) 70(4):583–9. doi: 10.1136/ard.2010.139774
27. Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A Novel Form of TNF/cachectin Is a Cell Surface Cytotoxic Transmembrane Protein: Ramifications for the Complex Physiology of TNF. Cell (1988) 53(1):45–53. doi: 10.1016/0092-8674(88)90486-2
28. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A Metalloproteinase Disintegrin That Releases Tumour-Necrosis Factor-Alpha From Cells. Nature (1997) 385(6618):729–33. doi: 10.1038/385729a0
29. Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a Disintegrin Metalloproteinase That Processes Precursor Tumour-Necrosis Factor-Alpha. Nature (1997) 385(6618):733–6. doi: 10.1038/385733a0
30. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The Transmembrane Form of Tumor Necrosis Factor Is the Prime Activating Ligand of the 80 kDa Tumor Necrosis Factor Receptor. Cell (1995) 83(5):793–802. doi: 10.1016/0092-8674(95)90192-2
31. Grell M, Wajant H, Zimmermann G, Scheurich P. The Type 1 Receptor (CD120a) Is the High-Affinity Receptor for Soluble Tumor Necrosis Factor. Proc Natl Acad Sci USA (1998) 95(2):570–5. doi: 10.1073/pnas.95.2.570
32. Tseng WY, Huang YS, Lin HH, Luo SF, McCann F, McNamee K, et al. TNFR Signalling and Its Clinical Implications. Cytokine (2018) 101:19–25. doi: 10.1016/j.cyto.2016.08.027
33. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated Necrosis: The Expanding Network of Non-Apoptotic Cell Death Pathways. Nat Rev Mol Cell Biol (2014) 15(2):135–47. doi: 10.1038/nrm3737
34. Jacob CO, McDevitt HO. Tumour Necrosis Factor-Alpha in Murine Autoimmune 'Lupus' Nephritis. Nature (1988) 331(6154):356–8. doi: 10.1038/331356a0
35. Gordon C, Ranges GE, Greenspan JS, Wofsy D. Chronic Therapy With Recombinant Tumor Necrosis Factor-Alpha in Autoimmune NZB/NZW F1 Mice. Clin Immunol Immunopathol (1989) 52(3):421–34. doi: 10.1016/0090-1229(89)90157-8
36. Jacob CO, Aiso S, Michie SA, McDevitt HO, Acha-Orbea H. Prevention of Diabetes in Nonobese Diabetic Mice by Tumor Necrosis Factor (TNF): Similarities Between TNF-Alpha and Interleukin 1. Proc Natl Acad Sci USA (1990) 87(3):968–72. doi: 10.1073/pnas.87.3.968
37. Probert L. TNF and its Receptors in the CNS: The Essential, the Desirable and the Deleterious Effects. Neuroscience (2015) 302:2–22. doi: 10.1016/j.neuroscience.2015.06.038
38. an Hauwermeiren F, Vandenbroucke RE, Libert C. Treatment of TNF Mediated Diseases by Selective Inhibition of Soluble TNF or TNFR1. Cytokine Growth Factor Rev (2011) 22(5-6):311–9. doi: 10.1016/j.cytogfr.2011.09.004
39. Chen X, Oppenheim JJ. TNF-Alpha: An Activator of CD4+FoxP3+TNFR2+ Regulatory T Cells. Curr Dir Autoimmun (2010) 11:119–34. doi: 10.1159/000289201
40. Faustman D, Davis M. TNF Receptor 2 Pathway: Drug Target for Autoimmune Diseases. Nat Rev Drug Discov (2010) 9(6):482–93. doi: 10.1038/nrd3030
41. Zou H, Li R, Hu H, Hu Y, Chen X. Modulation of Regulatory T Cell Activity by TNF Receptor Type II-Targeting Pharmacological Agents. Front Immunol (2018) 9:594. doi: 10.3389/fimmu.2018.00594
42. Medler J, Wajant H. Tumor Necrosis Factor Receptor-2 (TNFR2): An Overview of an Emerging Drug Target. Expert Opin Ther Targets (2019) 23(4):295–307. doi: 10.1080/14728222.2019.1586886
43. Wajant H, Beilhack A. Targeting Regulatory T Cells by Addressing Tumor Necrosis Factor and Its Receptors in Allogeneic Hematopoietic Cell Transplantation and Cancer. Front Immunol (2019) 10:2040. doi: 10.3389/fimmu.2019.02040
44. Fischer R, Kontermann RE, Pfizenmaier K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front Cell Dev Biol (2020) 8:401. doi: 10.3389/fcell.2020.00401
45. Salomon BL. Insights Into the Biology and Therapeutic Implications of TNF and Regulatory T Cells. Nat Rev Rheumatol (2021) 17(8):487–504. doi: 10.1038/s41584-021-00639-6
46. Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. Co-Expression of TNFR2 and CD25 Identifies More of the Functional CD4+FOXP3+ Regulatory T Cells in Human Peripheral Blood. Eur J Immunol (2010) 40(4):1099–106. doi: 10.1002/eji.200940022
47. Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ. Interaction of TNF With TNF Receptor Type 2 Promotes Expansion and Function of Mouse CD4+CD25+ T Regulatory Cells. J Immunol (2007) 179(1):154–61. doi: 10.4049/jimmunol.179.1.154
48. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting Edge: Expression of TNFR2 Defines a Maximally Suppressive Subset of Mouse CD4+CD25+FoxP3+ T Regulatory Cells: Applicability to Tumor-Infiltrating T Regulatory Cells. J Immunol (2008) 180(10):6467–71. doi: 10.4049/jimmunol.180.10.6467
49. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous Expansion of Human T-Regulatory Cells via Tumor Necrosis Factor Receptor 2. Sci Rep (2013) 3:3153. doi: 10.1038/srep03153
50. He X, Landman S, Bauland SC, van den Dolder J, Koenen HJ, Joosten I. A TNFR2-Agonist Facilitates High Purity Expansion of Human Low Purity Treg Cells. PloS One (2016) 11(5):e0156311. doi: 10.1371/journal.pone.0156311
51. Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive Activity of Human Regulatory T Cells Is Maintained in the Presence of TNF. Nat Med (2016) 22(1):16–7. doi: 10.1038/nm.4019
52. Till A, Rosenstiel P, Krippner-Heidenreich A, Mascheretti-Croucher S, Croucher PJ, Schafer H, et al. The Met-196 -> Arg Variation of Human Tumor Necrosis Factor Receptor 2 (TNFR2) Affects TNF-Alpha-Induced Apoptosis by Impaired NF-kappaB Signaling and Target Gene Expression. J Biol Chem (2005) 280(7):5994–6004. doi: 10.1074/jbc.M411541200
53. Li D, Silverberg MS, Haritunians T, Dubinsky MC, Landers C, Stempak JM, et al. TNFRSF1B Is Associated With ANCA in IBD. Inflammation Bowel Dis (2016) 22(6):1346–52. doi: 10.1097/MIB.0000000000000771
54. Sashio H, Tamura K, Ito R, Yamamoto Y, Bamba H, Kosaka T, et al. Polymorphisms of the TNF Gene and the TNF Receptor Superfamily Member 1B Gene are Associated With Susceptibility to Ulcerative Colitis and Crohn’s Disease, Respectively. Immunogenetics (2002) 53(12):1020–7. doi: 10.1007/s00251-001-0423-7
55. Chatzikyriakidou A, Georgiou I, Voulgari PV, Drosos AA. The Role of Tumor Necrosis Factor (TNF)-Alpha and TNF Receptor Polymorphisms in Susceptibility to Ankylosing Spondylitis. Clin Exp Rheumatol (2009) 27(4):645–8.
56. Komata T, Tsuchiya N, Matsushita M, Hagiwara K, Tokunaga K. Association of Tumor Necrosis Factor Receptor 2 (TNFR2) Polymorphism With Susceptibility to Systemic Lupus Erythematosus. Tissue Antigens (1999) 53(6):527–33. doi: 10.1034/j.1399-0039.1999.530602.x
57. Barton A, John S, Ollier WE, Silman A, Worthington J. Association Between Rheumatoid Arthritis and Polymorphism of Tumor Necrosis Factor Receptor II, But Not Tumor Necrosis Factor Receptor I, in Caucasians. Arthritis Rheum (2001) 44(1):61–5. doi: 10.1002/1529-0131(200101)44:1<61::AID-ANR9>3.0.CO;2-Q
58. Canet LM, Filipescu I, Caliz R, Lupianez CB, Canhao H, Escudero A, et al. Genetic Variants Within the TNFRSF1B Gene and Susceptibility to Rheumatoid Arthritis and Response to Anti-TNF Drugs: A Multicenter Study. Pharmacogenet Genomics (2015) 25(7):323–33. doi: 10.1097/FPC.0000000000000140
59. Vanamee ES, Faustman DL. TNFR2: A Novel Target for Cancer Immunotherapy. Trends Mol Med (2017) 23(11):1037–46. doi: 10.1016/j.molmed.2017.09.007
60. Govindaraj C, Scalzo-Inguanti K, Madondo M, Hallo J, Flanagan K, Quinn M, et al. Impaired Th1 Immunity in Ovarian Cancer Patients Is Mediated by TNFR2+ Tregs Within the Tumor Microenvironment. Clin Immunol (2013) 149(1):97–110. doi: 10.1016/j.clim.2013.07.003
61. Govindaraj C, Tan P, Walker P, Wei A, Spencer A, Plebanski M. Reducing TNF Receptor 2+ Regulatory T Cells via the Combined Action of Azacitidine and the HDAC Inhibitor, Panobinostat for Clinical Benefit in Acute Myeloid Leukemia Patients. Clin Cancer Res (2014) 20(3):724–35. doi: 10.1158/1078-0432.CCR-13-1576
62. Yang M, Tran L, Torrey H, Song Y, Perkins H, Case K, et al. Optimizing TNFR2 Antagonism for Immunotherapy With Tumor Microenvironment Specificity. J Leukoc Biol (2020) 107(6):971–80. doi: 10.1002/JLB.5AB0320-415RRRRR
63. Moatti A, Cohen JL. The TNF-Alpha/TNFR2 Pathway: Targeting a Brake to Release the Anti-Tumor Immune Response. Front Cell Dev Biol (2021) 9:725473. doi: 10.3389/fcell.2021.725473
64. Case K, Tran L, Yang M, Zheng H, Kuhtreiber WM, Faustman DL. TNFR2 Blockade Alone or in Combination With PD-1 Blockade Shows Therapeutic Efficacy in Murine Cancer Models. J Leukoc Biol (2020) 107(6):981–91. doi: 10.1002/JLB.5MA0420-375RRRRR
65. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, et al. Targeting TNFR2 With Antagonistic Antibodies Inhibits Proliferation of Ovarian Cancer Cells and Tumor-Associated Tregs. Sci Signal (2017) 10(462):eaaf8608. doi: 10.1126/scisignal.aaf8608
66. Torrey H, Kuhtreiber WM, Okubo Y, Tran L, Case K, Zheng H, et al. A Novel TNFR2 Agonist Antibody Expands Highly Potent Regulatory T Cells. Sci Signal (2020) 13(661):eaba9600. doi: 10.1126/scisignal.aba9600
67. Kim EY, Teh HS. TNF Type 2 Receptor (P75) Lowers the Threshold of T Cell Activation. J Immunol (2001) 167(12):6812–20. doi: 10.4049/jimmunol.167.12.6812
68. Kim EY, Priatel JJ, Teh SJ, Teh HS. TNF Receptor Type 2 (P75) Functions as a Costimulator for Antigen-Driven T Cell Responses In Vivo. J Immunol (2006) 176(2):1026–35. doi: 10.4049/jimmunol.176.2.1026
69. Aspalter RM, Eibl MM, Wolf HM. Regulation of TCR-Mediated T Cell Activation by TNF-RII. J Leukoc Biol (2003) 74(4):572–82. doi: 10.1189/jlb.0303112
70. Soloviova K, Puliaiev M, Haas M, Via CS. In Vivo Maturation of Allo-Specific CD8 CTL and Prevention of Lupus-Like Graft-Versus-Host Disease is Critically Dependent on T Cell Signaling Through the TNF P75 Receptor But Not the TNF P55 Receptor. J Immunol (2013) 190(9):4562–72. doi: 10.4049/jimmunol.1300091
71. Chen X, Hamano R, Subleski JJ, Hurwitz AA, Howard OM, Oppenheim JJ. Expression of Costimulatory TNFR2 Induces Resistance of CD4+FoxP3- Conventional T Cells to Suppression by CD4+FoxP3+ Regulatory T Cells. J Immunol (2010) 185(1):174–82. doi: 10.4049/jimmunol.0903548
72. Chen X, Nie Y, Xiao H, Bian Z, Scarzello AJ, Song NY, et al. TNFR2 Expression by CD4 Effector T Cells is Required to Induce Full-Fledged Experimental Colitis. Sci Rep (2016) 6:32834. doi: 10.1038/srep32834
73. Ohkura N, Kitagawa Y, Sakaguchi S. Development and Maintenance of Regulatory T Cells. Immunity (2013) 38(3):414–23. doi: 10.1016/j.immuni.2013.03.002
74. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ Regulatory T Cells in Immune Tolerance. Annu Rev Immunol (2012) 30:733–58. doi: 10.1146/annurev-immunol-020711-075043
75. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic Generation of Regulatory T Cells in Placental Mammals Mitigates Maternal-Fetal Conflict. Cell (2012) 150(1):29–38. doi: 10.1016/j.cell.2012.05.031
76. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically Generated Regulatory T Cells Control Mucosal TH2 Inflammation. Nature (2012) 482(7385):395–9. doi: 10.1038/nature10772
77. Burzyn D, Benoist C, Mathis D. Regulatory T Cells in Nonlymphoid Tissues. Nat Immunol (2013) 14(10):1007–13. doi: 10.1038/ni.2683
78. Opstelten R, de Kivit S, Slot MC, van den Biggelaar M, Iwaszkiewicz-Grzes D, Gliwinski M, et al. GPA33: A Marker to Identify Stable Human Regulatory T Cells. J Immunol (2020) 204(12):3139–48. doi: 10.4049/jimmunol.1901250
79. Golovina TN, Mikheeva T, Suhoski MM, Aqui NA, Tai VC, Shan X, et al. CD28 Costimulation Is Essential for Human T Regulatory Expansion and Function. J Immunol (2008) 181(4):2855–68. doi: 10.4049/jimmunol.181.4.2855
80. Li L, Godfrey WR, Porter SB, Ge Y, June CH, Blazar BR, et al. CD4+CD25+ Regulatory T-Cell Lines From Human Cord Blood Have Functional and Molecular Properties of T-Cell Anergy. Blood (2005) 106(9):3068–73. doi: 10.1182/blood-2005-04-1531
81. Cheng G, Yu A, Malek TR. T-Cell Tolerance and the Multi-Functional Role of IL-2R Signaling in T-Regulatory Cells. Immunol Rev (2011) 241(1):63–76. doi: 10.1111/j.1600-065X.2011.01004.x
82. Van Dongen S. Graph Clustering Via a Discrete Uncoupling Process. SIAM J Matrix Anal Appl (2008) 30(1):121–41. doi: 10.1137/040608635
83. Le Cao KA, Boitard S, Besse P. Sparse PLS Discriminant Analysis: Biologically Relevant Feature Selection and Graphical Displays for Multiclass Problems. BMC Bioinf (2011) 12:253. doi: 10.1186/1471-2105-12-253
84. Acuto O, Michel F. CD28-Mediated Co-Stimulation: A Quantitative Support for TCR Signalling. Nat Rev Immunol (2003) 3(12):939–51. doi: 10.1038/nri1248
85. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity (2002) 16(6):769–77. doi: 10.1016/s1074-7613(02)00323-0
86. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep (2018) 22(6):1509–21. doi: 10.1016/j.celrep.2018.01.040
87. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
88. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
89. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25(6):1282–93 e7. doi: 10.1016/j.cmet.2016.12.018
90. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-Like Receptor Signaling Balance Treg Cell Anabolic Metabolism for Suppression. Nat Immunol (2016) 17(12):1459–66. doi: 10.1038/ni.3577
91. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 Drives Oxidative Phosphorylation and Protection From Lipotoxicity. JCI Insight (2017) 2(3):e89160. doi: 10.1172/jci.insight.89160
92. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis Controls the Induction of Human Regulatory T Cells by Modulating the Expression of FOXP3 Exon 2 Splicing Variants. Nat Immunol (2015) 16(11):1174–84. doi: 10.1038/ni.3269
93. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The Proteomic Landscape of Human Ex Vivo Regulatory and Conventional T Cells Reveals Specific Metabolic Requirements. Immunity (2016) 44(3):712. doi: 10.1016/j.immuni.2016.02.022
94. Priyadharshini B, Loschi M, Newton RH, Zhang JW, Finn KK, Gerriets VA, et al. Cutting Edge: TGF-Beta and Phosphatidylinositol 3-Kinase Signals Modulate Distinct Metabolism of Regulatory T Cell Subsets. J Immunol (2018) 201(8):2215–9. doi: 10.4049/jimmunol.1800311
95. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine Uptake and Metabolism are Coordinately Regulated by ERK/MAPK During T Lymphocyte Activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
96. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of Amino-Acid Transport by Antigen Receptors Coordinates the Metabolic Reprogramming Essential for T Cell Differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
97. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and Mtorc1 Kinase Activation. Immunity (2014) 40(5):692–705. doi: 10.1016/j.immuni.2014.04.007
98. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell (2018) 175(7):1780–95.e19. doi: 10.1016/j.cell.2018.10.001
99. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The Transcription Factor Myc Controls Metabolic Reprogramming Upon T Lymphocyte Activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
100. Donati S, Sander T, Link H. Crosstalk Between Transcription and Metabolism: How Much Enzyme Is Enough for a Cell? Wiley Interdiscip Rev Syst Biol Med (2018) 10(1):e1396. doi: 10.1002/wsbm.1396
101. Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, et al. Natural But Not Inducible Regulatory T Cells Require TNF-Alpha Signaling for In Vivo Function. J Immunol (2011) 186(12):6779–87. doi: 10.4049/jimmunol.1003868
102. Zhang Q, Cui F, Fang L, Hong J, Zheng B, Zhang JZ. TNF-Alpha Impairs Differentiation and Function of TGF-Beta-Induced Treg Cells in Autoimmune Diseases Through Akt and Smad3 Signaling Pathway. J Mol Cell Biol (2013) 5(2):85–98. doi: 10.1093/jmcb/mjs063
103. Yang S, Xie C, Chen Y, Wang J, Chen X, Lu Z, et al. Differential Roles of TNFalpha-TNFR1 and TNFalpha-TNFR2 in the Differentiation and Function of CD4(+)Foxp3(+) Induced Treg Cells In Vitro and In Vivo Periphery in Autoimmune Diseases. Cell Death Dis (2019) 10(1):27. doi: 10.1038/s41419-018-1266-6
104. Chen X, Oppenheim JJ. Contrasting Effects of TNF and Anti-TNF on the Activation of Effector T Cells and Regulatory T Cells in Autoimmunity. FEBS Lett (2011) 585(23):3611–8. doi: 10.1016/j.febslet.2011.04.025
105. Grinberg-Bleyer Y, Saadoun D, Baeyens A, Billiard F, Goldstein JD, Gregoire S, et al. Pathogenic T Cells Have a Paradoxical Protective Effect in Murine Autoimmune Diabetes by Boosting Tregs. J Clin Invest (2010) 120(12):4558–68. doi: 10.1172/JCI42945
106. Joedicke JJ, Myers L, Carmody AB, Messer RJ, Wajant H, Lang KS, et al. Activated CD8+ T Cells Induce Expansion of Vbeta5+ Regulatory T Cells via TNFR2 Signaling. J Immunol (2014) 193(6):2952–60. doi: 10.4049/jimmunol.1400649
107. Urbano PCM, Koenen H, Joosten I, He X. An Autocrine TNFalpha-Tumor Necrosis Factor Receptor 2 Loop Promotes Epigenetic Effects Inducing Human Treg Stability In Vitro. Front Immunol (2018) 9:573. doi: 10.3389/fimmu.2018.00573
108. Nguyen DX, Ehrenstein MR. Anti-TNF Drives Regulatory T Cell Expansion by Paradoxically Promoting Membrane TNF-TNF-RII Binding in Rheumatoid Arthritis. J Exp Med (2016) 213(7):1241–53. doi: 10.1084/jem.20151255
109. Kleijwegt FS, Laban S, Duinkerken G, Joosten AM, Zaldumbide A, Nikolic T, et al. Critical Role for TNF in the Induction of Human Antigen-Specific Regulatory T Cells by Tolerogenic Dendritic Cells. J Immunol (2010) 185(3):1412–8. doi: 10.4049/jimmunol.1000560
110. Polz J, Remke A, Weber S, Schmidt D, Weber-Steffens D, Pietryga-Krieger A, et al. Myeloid Suppressor Cells Require Membrane TNFR2 Expression for Suppressive Activity. Immun Inflammation Dis (2014) 2(2):121–30. doi: 10.1002/iid3.19
111. Yan L, Zheng D, Xu RH. Critical Role of Tumor Necrosis Factor Signaling in Mesenchymal Stem Cell-Based Therapy for Autoimmune and Inflammatory Diseases. Front Immunol (2018) 9:1658. doi: 10.3389/fimmu.2018.01658
112. Madsen PM, Desu HL, de Rivero Vaccari JP, Florimon Y, Ellman DG, Keane RW, et al. Oligodendrocytes Modulate the Immune-Inflammatory Response in EAE via TNFR2 Signaling. Brain Behav Immun (2020) 84:132–46. doi: 10.1016/j.bbi.2019.11.017
113. Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF Alpha Promotes Proliferation of Oligodendrocyte Progenitors and Remyelination. Nat Neurosci (2001) 4(11):1116–22. doi: 10.1038/nn738
Keywords: TNFR2, regulatory T cell, conventional T cell, metabolism, transcriptomics, therapy
Citation: Mensink M, Tran TNM, Zaal EA, Schrama E, Berkers CR, Borst J and de Kivit S (2022) TNFR2 Costimulation Differentially Impacts Regulatory and Conventional CD4+ T-Cell Metabolism. Front. Immunol. 13:881166. doi: 10.3389/fimmu.2022.881166
Received: 22 February 2022; Accepted: 04 May 2022;
Published: 30 June 2022.
Edited by:
Xin Chen, University of Macau, ChinaReviewed by:
Ken Oestreich, The Ohio State University, United StatesSarah Dimeloe, University of Birmingham, United Kingdom
Benoit L. Salomon, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2022 Mensink, Tran, Zaal, Schrama, Berkers, Borst and de Kivit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jannie Borst, ai5nLmJvcnN0QGx1bWMubmw=
†These authors have contributed equally to this work