 Giuseppe Ancona1
Giuseppe Ancona1 Laura Alagna1
Laura Alagna1 Claudia Alteri2,3
Claudia Alteri2,3 Emanuele Palomba1,4*Anna Tonizzo1,4Andrea Pastena1,4Antonio Muscatello1
Emanuele Palomba1,4*Anna Tonizzo1,4Andrea Pastena1,4Antonio Muscatello1 Andrea Gori1,4*
Andrea Gori1,4* Alessandra Bandera1,4
Alessandra Bandera1,4- 1Infectious Diseases Unit, Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 2Department of Oncology and Hemato-Oncology, University of Milan, Milan, Italy
- 3Multimodal Research Area, Bambino Gesù Children Hospital (IRCCS), Rome, Italy
- 4Department of Pathophysiology and Transplantation, Centre for Multidisciplinary Research in Health Science (MACH), University of Milan, Milan, Italy
The gut microbiota plays a crucial role in human health and disease. Gut dysbiosis is known to be associated with increased susceptibility to respiratory diseases and modifications in the immune response and homeostasis of the lungs (the so-called gut-lung axis). Furthermore, recent studies have highlighted the possible role of dysbiosis in neurological disturbances, introducing the notion of the “gut-brain axis.” During the last 2 years, several studies have described the presence of gut dysbiosis during coronavirus disease 2019 (COVID-19) and its relationship with disease severity, SARS-CoV-2 gastrointestinal replication, and immune inflammation. Moreover, the possible persistence of gut dysbiosis after disease resolution may be linked to long-COVID syndrome and particularly to its neurological manifestations. We reviewed recent evidence on the association between dysbiosis and COVID-19, investigating the possible epidemiologic confounding factors like age, location, sex, sample size, the severity of disease, comorbidities, therapy, and vaccination status on gut and airway microbial dysbiosis in selected studies on both COVID-19 and long-COVID. Moreover, we analyzed the confounding factors strictly related to microbiota, specifically diet investigation and previous use of antibiotics/probiotics, and the methodology used to study the microbiota (α- and β-diversity parameters and relative abundance tools). Of note, only a few studies focused on longitudinal analyses, especially for long-term observation in long-COVID. Lastly, there is a lack of knowledge regarding the role of microbiota transplantation and other therapeutic approaches and their possible impact on disease progression and severity. Preliminary data seem to suggest that gut and airway dysbiosis might play a role in COVID-19 and in long-COVID neurological symptoms. Indeed, the development and interpretation of these data could have important implications for future preventive and therapeutic strategies.
1 Introduction
1.1 COVID-19, long-COVID, and gastrointestinal disease during SARS-CoV-2 infection
Coronavirus disease 2019 (COVID-19) is a highly contagious infectious disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus, a novel RNA beta-coronavirus, with more than 663 million cases and 6.71 million deaths worldwide documented until 20 January 2023 (1). COVID-19 is mainly a respiratory illness, ranging from asymptomatic, mild-moderate, severe, and critical illness (2), especially affecting elderly subjects with underlying medical conditions (3).
After COVID-19, some patients may experience persistent symptoms or other conditions that are colloquially referred to as long-COVID. The Centers for Disease Control and Prevention have defined post-COVID conditions as new, returning, or ongoing symptoms that people experience ≥4 weeks after being infected with SARS-CoV-2 (4). The prevalence of these conditions varies widely from 5% to 80%, and the most frequently reported symptoms are fatigue, cough, shortness of breath, and chest pain (2, 5). Furthermore, half of the patients report persistent neurological symptoms at 6 months, the most frequent being “brain fog” and cognitive changes, described in up to one-third of subjects (6).
With regard to the gastrointestinal (GI) tract involvement, early reports from Wuhan showed that 2% to 10% of patients with acute COVID-19 had GI symptoms including nausea and diarrhea (7), but more recent metaanalyses reported a higher prevalence, up to 20% of patients (8). SARS-CoV-2 virus has been detected in anal swabs and stool samples in almost 50% of patients with COVID-19, suggesting that the digestive tract might be an extrapulmonary site for virus replication and activity (9), through ACE2 receptors binding with spike protein-S.
1.2 Gut microbiota and its role in health and disease
The human gut microbiota harbors up to 1014 resident microorganisms, including bacteria, archaea, viruses, fungi, and other eucaryotes, with bacteria being the most abundant microorganisms at the gut level. The most represented phyla at the gut level are Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, Verrucomicrobia, and Fusobacteria (10). An increase in bacteria has been documented from duodenum to colon, with a decrease in facultative anaerobic Bacilli (Firmicutes) and Enterobacterales (Proteobacteria) taxa and an increase in obligate anaerobic bacteria, especially Bacteroidia (Bacteroidetes) and Clostridia (Firmicutes) classes (11, 12).
Gut microbiota is crucial for several functions, such as energy extraction from the diet, vitamin and short-chain fat acids (SCFAs) production, and immunomodulation, with the regulation of TH17 and T reg balance (13–15). A complex equilibrium exists among prebiotics, like microbiota accessible carbohydrates (MAC), probiotics, and postbiotics, like their products, SCFAs (16, 17), with involvement of several networks between gut microbiota and other body sites through axes (i.e., gut-lung, gut-liver, gut-brain axis), influencing processes in health and disease.
An unbalance of the crucial homeostasis between Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria phyla (Figure 1) is often associated with a change in the numbers of microbes and/or diversity of the microbiota; such a condition is defined as dysbiosis (18). Recently, a new definition of dysbiosis has been suggested, based on a model represented in several diseases, defined by the increase in facultative anaerobic bacteria, like Bacilli class and Enterobacterales order, and a parallel decrease in obligate anaerobic bacteria, such as propionate and butyrate-producing bacteria (BPBs) (11).
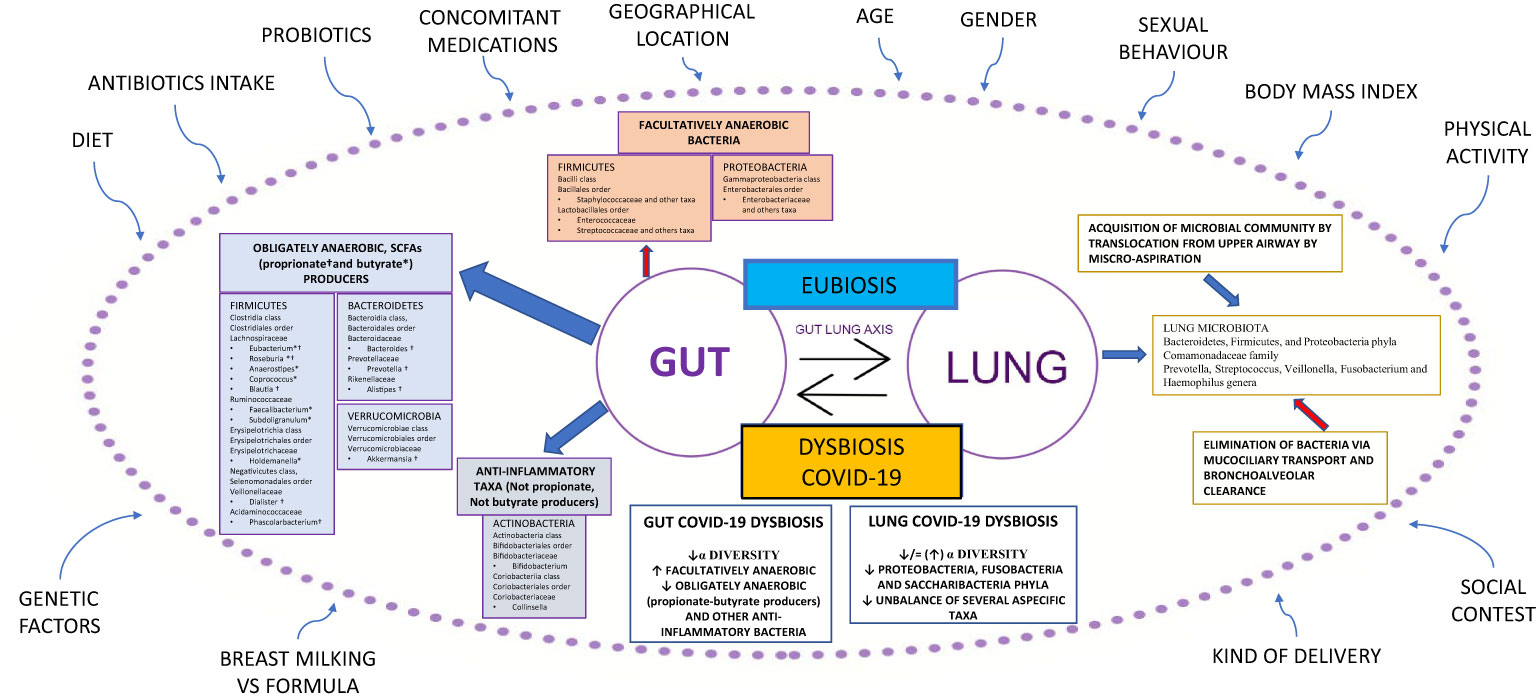
Figure 1 Gut-lung axis microbiota in COVID-19. This figure shows a summary of the gut-lung axis and its alterations during COVID-19. Left: the gut microbiota taxa obligately anaerobic short-chain fatty acids (propionate and butyrate) producers and anti-inflammatory taxa, not propionate and butyrate producers. Upper: facultatively anaerobic bacteria. Right: the homeostasis of the lung microbiota, resulting from acquisition (blue arrow) and elimination (red arrow) clearance. Bottom: the most significant alterations detected in gut and lung microbiota during COVID-19. All around, the confounding factors are strictly related to Microbiota. BMI: Body Mass Index; ° indicates propionate-producing bacteria, * indicates butyrate-producing bacteria, Upward arrows “↑”: increase; Downward arrows “↓”: decrease.
Gut microbiota dysbiosis can have a role in several disease models affecting the lung, brain, liver, and heart (19).
In the last decade, research on lung microbiota and its pathogenetic link to pulmonary conditions has significantly improved. Previously, the lung has been considered a sterile organ; however, numerous studies have demonstrated the presence of bacterial DNA in the lower respiratory tract in healthy individuals. The lung microbiota of healthy subjects is characterized by the presence of differentiated ecological niches belonging to Bacteroidetes, Firmicutes, and Proteobacteria phyla and Prevotella, Streptococcus, Veillonella, Fusobacterium, and Haemophilus genera (20). Its balance is the result of acquisition and clearance (Figure 1). Many other factors contribute to this complex mechanism, such as the immune system (innate and adaptive immune recognition, secretory IgA), in addition to various exogenous components such as diet, environmental biodiversity, and drug treatments, in particular antibiotics (21).
Chronic respiratory diseases are often characterized by an imbalance between microbial immigration and elimination in the lung. Moreover, the presence of chronic inflammation results in the alteration of physicochemical proprieties that facilitate the growth of select species in the microbial community, such as microorganisms from the Proteobacteria phylum, that are linked to a proinflammatory state (22). It is important to emphasize that lung and gut microbiota are in close communication with each other through the circulation of soluble metabolites (i.e., peptidoglycan or LPS) transported by the blood (21). These peptides are recognized by host cells that express pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs) and Nod-like receptors (NLRs). The interplay between lung and gut microbiota, defined as the gut-lung axis, has been demonstrated in different animal models (23–26).
Further studies are needed to better understand the complex gut-lung interplay and characterize the gut microbial metabolites (i.e., indole derivative, niacin, polyamines, urolithin, and pyruvic acid) that act as immunomodulants and might have a possible impact on respiratory health (27, 28).
Another captivating field of microbiota studies is related to its connection with the brain through the so-called gut-brain axis, which is thought to be a bidirectional system. On one side, there is the involvement of microbiota-derived metabolites on the blood–brain barrier like SCFAs, tryptophan, and linoleic acid metabolites as well as cytokines produced at the gut level; on the other side, the brain controls gut activity through the neuroendocrine and parasympathetic systems (i.e., regulation of intestinal permeability through the vagus nerve) (29). Such connections have been studied in animal models: physiological aging affects gut microbiota in mouse models through cognitive frailty (30).
Gut microbiota dysbiosis seems to play a role in several neurodegenerative and psychiatric disorders (31), as well as in other neurological conditions (32). For example, damage to the GI barrier is a possible pathological pattern for depression disorders; moreover, increased LPS and microbiota-cytokine production seems to be related to Alzheimer’s disease (29).
The relationship between gut microbiota and the brain could be deeper and more complex: alteration of the hypothalamic “master clock” could impact the diurnal environmental fluctuations and lead to dysbiosis-related metabolic disorders like obesity and/or diabetes (33). Furthermore, gut dysbiosis could determine sleep disturbances (sleep loss, alteration of circadian rhythm), eventually leading to fatigue (34). Following this hypothesis, the gut microbiota, which is mostly influenced by diet, could represent a link between the immune and endocrine systems through brain function and the host metabolism (35).
High-fat food intake can indeed damage the GI barrier, affecting both the “intestinal epithelial barrier” (characterized by the mucus layer and the epithelial cells) (36) and the “gut vascular barrier,” regulated by the expression of plasmalemma vesicle-associated protein-1 (PV1). This condition, known as “leaky gut,” can favor microbial translocation to the liver (37), leading to hepatic and systemic disease.
Finally, another example of the role of dysbiosis in disease has been studied in the cardiological setting, where the increased production of trimethylamine (and its metabolite-liver trimethylamine-N-oxide) by gut microbiota has been linked to the development of cardiovascular disease (29).
2 Gut microbiota dysbiosis in acute COVID-19
2.1 Study characteristics and confounding factors
We identified 22 studies on gut microbiota in COVID-19 patients published in a 2-year window period between 03 January 2020 and 03 January 2022 (Table 1A).
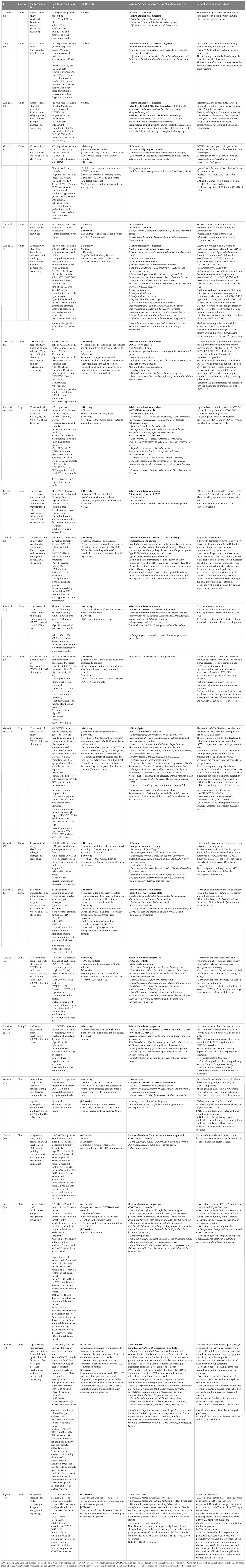
Table 1A Selected studies on gut microbiota and COVID-19.
To critically revise the studies, we first considered all the variables potentially influencing the final observations: study design, location, material source, microbial technology used, sample size, and patient characteristics—age, body mass index (BMI), gender, sexual behaviors, COVID-19 severity index, comorbidities, recent previous use of antibiotics/probiotics, diet, and lifestyle.
The cross-sectional study design was the most common. Less than half of studies (45%) had a longitudinal/prospective design, 20% of which focused on long-COVID-19.
The study location was a critical factor: most studies (19/22, 86%) were set in Asia (18 in China, one in South Korea), and three of 22 (14%) in Europe; no other geographic regions were represented.
Lifestyle and diet were not analyzed, even though both factors are crucial elements in shaping microbial core composition (32, 59, 60).
The material source was a fecal sample in 19/22 (86%) studies, while three of 22 (14%) were based on anal swab analysis. Most studies (12/22) used next-generation sequencing (NGS) technology through ribosomal-S16-DNA hypervariable region sequencing (V4 or V3–V4 regions preferred) to analyze microbiota; shotgun metagenomic sequencing was used in seven of 22 studies, whereas one study used multi-omics methodologies (55), one study nanopore technology (38), and another used quantitative PCR (39).
Regarding patients’ characteristics, all studies included both men and women, but no studies considered sexual behavior, although its impact on microbiota core is known in several disease models (61, 62). Only one-third of studies (seven of 22) included BMI data, and control groups, when included, were often matched for BMI. Fifty percent of the subjects in the studies, 50% were aged 50 or younger.
The small sample size was a limit reported by several authors, with a total number of enrolled subjects below 40 in almost two-thirds of studies 13/21 (62%). The COVID-19 severity index was reported by most studies, with high heterogeneity in the works analyzed.
Scarce data were available on comorbidities and concomitant medications; hypertension was the most commonly reported, followed by diabetes.
No data were generally reported on COVID-19 vaccine status for subjects enrolled after the introduction of the vaccine; only one study investigated the microbiota changes in two groups of patients vaccinated with two different vaccines (58). During hospitalization, both antibiotics and/or antiretroviral treatments and probiotics were administered in several studies; however, these data were not critically investigated in most published studies.
2.2 Microbiota analysis
After assessing the possible confounding factors, we compared the gut microbiota features according to two ecological measures, α-diversity and β-diversity, in association with relative abundance results.
In humans, α-diversity measures the level of diversity within individual samples; it includes several indexes gathered in two groups: richness indexes (Faith index, Observed and Chao-1 index) and evenness indexes (Shannon index, Peliou’s evenness, Simpson, and inverse Simpson indexes) (63, 64).
In parallel to other disease models, α-diversity at the gut level, more frequently described with richness indexes (like Chao-1), resulted in a global reduction in all COVID-19 patients compared to controls (see details in Table 1A). An interesting study observed this reduction already in the acute phase of the disease (48). On the contrary, Yeoh et al. (43) did not report alterations in α-diversity indexes, even though they enrolled most COVID-19 patients with a mild or moderate severity index (90% of patients).
In a Korean longitudinal analysis performed on patients who were asymptomatic or affected by the mild disease, an increase in α-diversity (Peliou’s evenness) was observed in the recovered subgroups compared to infected patients (51). Interestingly, Xu et al. (46) observed a trend toward increased bacterial diversity from the early to late stages of COVID-19 in a 35-day longitudinal analysis of inpatients with mild disease. Furthermore, the same study described an interesting synchronous restoration of microbiota in both gut and upper airways, suggesting a possible role of the gut-lung axis.
Moreira-Rosario et al. (53) described a reduced α-diversity gradient trend (Shannon index) from mild to severe COVID-19 patients, and Chen et al. (48) showed how richness was not restored to a normal level even after 6 months in 30 COVID-19 patients (one-third with severe disease), although a trend toward healthy controls was noticed.
β-Diversity measures the level of diversity (or dissimilarity) between samples, mostly by using a Permanova analysis (65, 66). All the studies showed a difference between COVID-19 patients and controls, in general, and according to different severity index categories.
Mazzarelli et al. (44) have shown a difference in β-diversity among patients hospitalized in regular wards compared to ICU patients and hospitalized no-COVID-19 controls, although no data on prior antibiotic intake was gathered. Regarding this aspect, two studies (9, 43) compared microbiota composition in COVID-19 patient subgroups (with and without antibiotics) with healthy controls, confirming a separation among groups, with high heterogeneity revealed in the antibiotic subgroup.
Regarding relative abundance analysis, several studies described a significant reduction in Firmicutes members, especially for BPBs (both Lachnospiraceae and Ruminococcaeae families, mostly Faecalibacterium prausnitzii) in COVID-19 patients compared to no-COVID controls, while discordant data have been reported about Erysipelotrichaceae and Veillonellaceae taxa.
Conversely, several facultative anaerobic bacteria like members of the Bacilli class, resulted in increased growth, mostly in the Enterococcaceae family as well as Streptococcaceae and Lactobacillaceae (Table 1). Contrasting data have been described regarding the Bacteroidetes phylum during COVID-19, with some works reporting an increase in Bacteroidetes phylum with a consequent reduction of the Firmicutes/Bacteroidetes ratio (53) as opposed to other studies reporting a reduction in taxa belonging to this phylum. Other factors, like diet and/or antibiotics, could play a role in these findings, highlighting the importance of assess for confounding factors when considering the study results.
Reduction in the Actinobacteria phylum, including the Bifidobacterium genus and Collinsella genus (recently associated with SARS-CoV-2-ACE2 binding inhibition), represents another significant finding in COVID-19 studies (67). The Bifidobacterium genus was found to be increased only in three studies (notably, in one study, a probiotic including this taxon was administered (56)), while the Collinsella genus resulted was increased in a few other studies (40, 45, 49); the reason for this last difference is not clear. Proteobacteria resulted increased in almost all studies performed on COVID-19 patients, although some authors have described an increase in Enterococcaceae/Enterobacteriaceae ratio (39), probably linked to the use of antibiotics. Finally, the Akkermansia genus (Verrucomicrobia), a propionate-producing bacterium genus with anti-inflammatory features, resulted in reduced COVID-19 (but not in all studies). To note, the severity of COVID-19 disease seems to emphasize differences in the relative abundance of gut microbiota, although most studies included asymptomatic/mild/moderate categories.
3 Airway microbiota dysbiosis in acute COVID-19
We analyzed 13 studies on airway microbiota changes during SARS-CoV-2 infection, mostly comparing COVID-19 patients with healthy subjects and/or patients with different respiratory diseases (Table 1B).
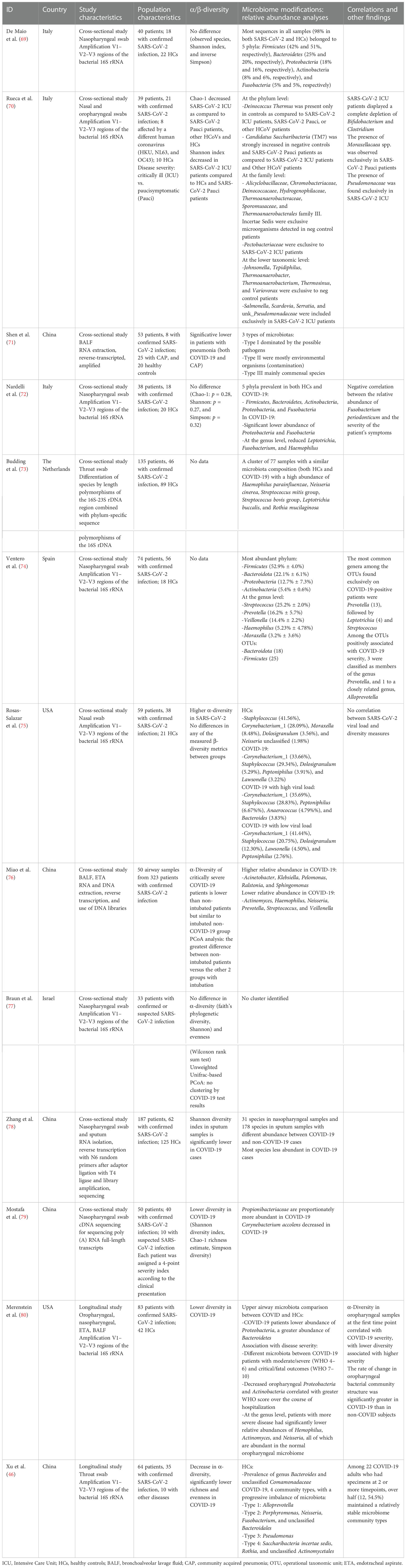
Table 1B Selected studies on airway microbiota and COVID-19.
Nasopharyngeal swabs were the most studied material, with the exception of three studies analyzing samples from the lower respiratory tract, such as bronchoalveolar lavage fluid and endotracheal aspirate. Bacterial communities were prevalently mapped by amplification of 16S gene hypervariable regions, with only a few studies employing genome sequencing. Eighty percent of the studies were set in China or Europe (five studies each). No data on possible confounding factors such as diet, BMI, relevant comorbidity, and antibiotic/antiviral consumption were investigated.
Overall, patients with SARS-CoV-2 infection showed diminished diversity in airway microbiota composition, by means of Shannon, Simpson, and Chao-1 indexes, when compared to both healthy subjects (46, 69, 70, 75, 77–79) and patients with community-acquired pneumonia (70).
A similar reduction in diversity measures is reported in critically ill COVID-19 patients, as opposed to subject with milder symptoms, other coronavirus infections, and healthy subjects (69). Interestingly, a reduction in diversity and greater difference at principal coordinate analysis (PCoA) is observed in patients needing mechanical ventilation compared to non-intubated patients regardless of SARS-CoV-2 infection (75). Such data suggest that COVID-19 impacts airway microbiota diversity mostly in severe infections, and this imbalance is strongly biased by other confounding factors such as intubation.
Of note, a number of the report showed no significant differences between COVID-19 patients and the control group in both bacterial richness and diversity/evenness indexes (observed species, Shannon index, and inverse Simpson index) (68, 71, 76). These findings can be partially explained by the heterogeneous population included in the studies and by the different methods used to sequence bacterial communities and assess diversity.
Curiously, Rosas-Salazar et al. (74) observed higher overall α-diversity in SARS-CoV-2-infected subjects compared to healthy controls, with no significant differences in any of the measured β-diversity.
COVID-19 severity correlates to α-diversity in oropharyngeal samples at the first time point, with lower diversity associated with higher disease severity (79). However, no significant association between high versus low SARS-CoV-2 viral load and any of the α-diversity or β-diversity metrics was observed (74).
In the studies analyzed, the airway microbiota of healthy individuals is characterized by the predominance of Bacteroidetes and Comamonadaceae taxa (46, 68), and no specific microbiota pattern has been found in COVID-19 patients. However, some peculiar alterations in relative composition have been observed.
Reduced abundance in Proteobacteria and Fusobacteria phyla is reported in subjects with SARS-CoV-2 infection as compared to controls, and decreased oropharyngeal Proteobacteria and Actinobacteria phyla correlate with greater disease severity (71, 79). At the genus level, patients with more severe diseases have significantly lower relative abundances of Haemophilus, Actinomyces, and Neisseria, all of which are abundant in the normal oropharyngeal microbiome (74, 79). Interestingly, Fusobacterium periodonticum is less represented in COVID-19 patients, negatively correlating with the severity of symptoms (71). A possible explanation is that these bacteria could modulate sialic acid metabolism and regulate ACE expression, impacting SARS-CoV-2 binding to the epithelium of the respiratory tract, as shown for other intestinal microorganisms (71, 80).
Conversely, COVID-19 patients show a high abundance of Saccharibacteria (formerly known as TM7), Streptococcus mitis group, Streptococcus bovis group, and Rothia mucilaginosa taxa (46, 72, 73), the latter often associated with cancer and bacteremia (81).
Significant changes among operational taxonomic unit (OTU) abundances are also reported, with decreased complexity of coabundance networks in severe COVID-19. OTUs associated with higher disease severity are members of the genus Prevotella and Veillonella. Particularly, it has been postulated that Prevotella spp. can worsen disease progression by activating immune signaling pathways that modulate inflammation (73).
Critically ill COVID-19 patients display a complete depletion of Bifidobacterium and Clostridium genera, with the presence of Salmonella, Scardovia, Serratia, and Pectobacteriaceae taxa. In these subjects, there is also a relative abundance of the Pseudomonaceae family, known to be associated with pathogenic conditions such as severe acute respiratory syndromes (69). Another characteristic of the airway microbiota in severe COVID-19 patients is low diversity and more richness in non-fermenting bacteria like Acinetobacter, Pelomonas, Ralstonia, and Sphingomonas genera. As mentioned before, these changes might be attributed to intubation and mechanical ventilation rather than COVID-19 pneumonia per se (75).
Interestingly, similar characteristics of an imbalanced microbiota with an enrichment of proinflammatory Enterobacteriaceae are found in patients with other respiratory diseases (46).
To date, there is scarce data coming from longitudinal studies on airway microbiota in SARS-CoV-2 infection. Analyzing throat swabs from 64 patients, 35 of which with confirmed infection, Xu et al. (46) postulated that a peculiar microbial community might represent the progressive imbalance of the respiratory microbiota. Interestingly, even though over half COVID-19 patients analyzed maintained relatively stable microbiome community types, 70% of the subjects experienced a gradual decrease of microbial diversity, with the enrichment of opportunistic pathogenic bacteria such as Saccharibacteria and Rothia and a reduction of Alloprevotella. This shift toward dysbiosis shows how impaired homeostasis of inflammation pathways, a hallmark of the advanced stage of SARS-CoV-2 infection, affects microbial communities and can represent a biomarker of disease progression.
4 Microbiota dysbiosis in long-COVID
4.1 Microbiota changes in long-COVID
Few studies tried to investigate α-diversity alterations during long-COVID: in this setting, Zhuo et al. (52) reported a reduced Shannon index in a 15-patient cohort, followed up for 3 months with at least one persistent COVID-19 symptom. Coherently with these findings, in a 6-month follow-up, Liu et al. (57) have confirmed in long-COVID patients both a persistently reduced α-diversity (Shannon and Chao-1 indexes) and different gut microbiota clusters compared to controls. Notably, the subgroup who had COVID-19 at baseline without developing long-COVID did not show the same dysbiosis pattern. Reduced BPBs were reported in both COVID-19 subgroups compared to controls, but only in the long-COVID subgroup the microbial composition was different compared to controls at 6-month follow-up (Table 1A). Interestingly, the authors found no correlation between viral load in the gut and respiratory levels and long-COVID development at 6 months, nor did they find any effect of previous antibiotic intake. On the contrary, in the long-COVID subgroup, increased fecal relative abundance of opportunistic pathogens was positively associated with fatigue, respiratory and neuropsychiatric symptoms, while decreased other anti-inflammatory/BPB taxa was negatively correlated with long-COVID at 6 months. Coherently, Zhuo et al. (52) described both a negative correlation between some taxa (Faecalibacterium prausnitzii, Intestinimonas butyriproducens) and chronic respiratory symptoms as well as a positive correlation between Proteobacteria members and long-COVID symptoms.
4.2 Microbiota role in neurological and pulmonary symptoms
Persistent dysbiosis in long-COVID and its pathogenic role still need to be studied in humans, while rodent and non-human primate animal models of COVID-19 already showed long-term changes in both lung and gut microbiome (82, 83). The influence of gut microbiota on neurological symptoms, via the gut-brain axis, has been investigated in the animal model since the early decades of the new millennium. In murine models, Bercik at al. suggested that gut microbiota could influence the behavior of mice (84). Recently, Carloni et al. identified a closing in the choroid plexus vascular barrier during gut inflammation, suggesting a link between intestinal inflammation and neurologic/psychiatric symptoms, like a deficit in short-term memory and anxiety-like behavior (85). Moreover, a recent review summarized three different arms of inflammation for the gut-brain axis in a non-COVID-19 setting, where the systemic humoral pathway, cellular immune pathway, and neuronal pathway are involved (86). By translating these inflammatory patterns to the long-COVID setting, where gut dysbiosis persists at least after 6 months of follow-up, we can conclude that this microbial imbalance plays a role in maintaining both a chronic inflammatory status at the gut level and favoring the development of neurological/neuropsychiatric symptoms, as seen in the animal models mentioned above. However, it is not clear which immunologic pathway is dominant during long-COVID. It is plausible that several factors could coexist in the same disease model: (a) reduction in BPBs leading the butyrate loss linked to neuropsychiatric disorders (87); (b) development of the cytokine release syndrome during COVID-19, in particular with increased kynurenine:tryptophan ratio, already linked to depression syndrome (88); and (c) changes in l-DOPA production, regulated by ACE2 activation at the gut level (89).
There is still a lack of evidence on the role of microbiota dysbiosis in respiratory symptoms during long-COVID. Shortness of breath, frequently experienced by subjects after recovery from primary SARS-CoV-2 infection, could represent a clinical manifestation of the fibrosis secondary to chronic inflammation of lung parenchyma, leading to reduced total lung capacity. Such a condition is already linked to gut dysbiosis in non-COVID patients, as described in a recent review (90).
5 Relationship between gut dysbiosis, fecal SARS-CoV-2 replication, and immune-inflammation in COVID-19
It is well known that some microbial species can modulate ACE2 receptor expression and/or prevent SARS-CoV-2-ACE2 binding (67). Moreover, some studies found that the gut microbiota composition of COVID-19 patients, especially during hospitalization, is correlated with plasma concentrations of several cytokines, chemokines, and inflammation markers, suggesting that the gut microbiota could play a role in modulating host immune response and potentially influence disease severity and outcomes (43).
Interestingly, Zhuo et al. (50) studied α-diversity in a COVID-19 cohort stratified according to the presence of fever, discovering that COVID-19 patients with fever have shown a trend in reduced Chao-1 index compared to patients without fever, and similarly a β-diversity separation measured with Bray–Curtis. A negative correlation between PBPs and both inflammatory markers (9, 39, 43) and viral gut SARS-CoV-2 replication (40) was reported, despite the presence of GI disease and/or virological clearance. Interestingly, Zuo et al. (9) have discovered a negative correlation between Bacteroides taxa and fecal SARS-CoV-2 load and a positive correlation between Erysipelotrichaceae taxa and fecal SARS-CoV-2 replication. In contrast, Moreira-Rosario et al. (53) failed to see an association between fecal RNA viral replication and COVID-19 severity.
Wu et al. (46) reported a positive correlation between fecal SARS-CoV-2 replication and P. copri, E. dolichum taxa and a negative correlation between SARS-CoV-2 replication and other taxa like Streptococcus, Dialister, Alistipes, Ruminococcus, Clostridium, Bifidobacterium, and Haemophylus genera.
Finally, a longitudinal interventional study implementing fecal microbiota transplantation (FMT) in COVID-19 (45) described modulation of both gut microbiota core and peripheral lymphocyte subsets, with an increase in healthy taxa associated with a reduction in peripheral naïve B cells and an increase in memory B cells.
Data coming from clinical trials enrolling COVID-19 patients analyzing other possible drugs modulating gut microbiota, such as probiotics, are still scarce and not conclusive (91).
6 Conclusion
Microbiota homeostasis plays a role in human health and disease, and that applies to SARS-CoV-2 infection as well. During the last 2 years, several studies reported dysbiosis in COVID-19 patients for both gut and lung microbial composition. The main microbiota alterations that have been observed during COVID-19 were (a) significant reduction in α-diversity, already during the early phase of the disease and especially at the gut level, with a gradient from mild to severe clinical categories; (b) different β-diversity composition of microbiota core, characterized by a profile with higher facultative anaerobic bacteria and lower obligate anaerobic bacteria; and (c) possible connections between gut dysbiosis and peripheral inflammation markers, such as cytokines.
Data from longitudinal analyses currently available do not clearly show whether gut dysbiosis in COVID-19 ends with a complete functional restoration or if it does persist, posing the physiopathological premises for long-COVID. Indeed, a prolonged alteration of gut microbiota following the primary infection could contribute to causing some of the neurological and respiratory symptoms reported via the gut-brain and gut-lung axis. Further longitudinal studies are needed to characterize these conditions and assess the impact of prior comorbidity on the natural history of dysbiosis in SARS-CoV-2 infection.
Moreover, a knowledge gap regarding the role of FMT and other therapeutic approaches emerged, reinforcing the necessity for new evidence on the interaction of microbiota with host immunity. Such information is paramount to developing microbiota interventions aimed at improving COVID-19 and long-COVID outcomes.
Author contributions
Conceptualization: GA, LA, EP, and AB. Data analysis: GA, LA, EP, AT, and AP. Editing and supervision: AM, CA, AG, and AB. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partially funded by the Italian Ministry of Health—Current Research IRCCS, the Fondazione Cariplo 2021-4236 LLC Network project, the Fondazione Bolton Hope Onlus “PREP-COVID” project, and the Associazione Nazionale per la Lotta contro l’AIDS (ANLAIDS). The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication. All authors declare no other competing interests.
Acknowledgments
We would like to acknowledge all the nurses, doctors, and clinical trial staff of the Infectious Diseases Unit of Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. National Institutes of Health. Treatment guidelines panel. Coronavirus disease 2019 (COVID-19). (2021) 2019:1–243.
3. Rosenthal N, Cao Z, Gundrum J, Sianis J, Safo S. Risk factors associated with in-hospital mortality in a US national sample of patients with COVID-19. JAMA Netw Open (2020) 3(12):1–14. doi: 10.1001/jamanetworkopen.2020.29058
5. Sykes DL, Holdsworth L, Jawad N, Gunasekera P, Morice AH, Crooks MG. Post-COVID-19 symptom burden: What is long-COVID and how should we manage it? Lung (2021) 199(2):113–9. doi: 10.1007/s00408-021-00423-z
6. Stefanou M-I, Palaiodimou L, Bakola E, Smyrnis N, Papadopoulou M, Paraskevas GP, et al. Neurological manifestations of long-COVID syndrome: A narrative review. Ther Adv Chronic Dis. Tsivgoulis MG and G. (2022) 13:20406223221076890. doi: 10.1177/20406223221076890
7. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
8. Cheung KS, Hung IFN, Chan PPY, Lung KC, Tso E, Liu R, et al. Gastrointestinal manifestations of SARS-CoV-2 infection and virus load in fecal samples from a Hong Kong cohort: Systematic review and meta-analysis. Gastroenterology (2020) 159(1):81–95. doi: 10.1053/j.gastro.2020.03.065
9. Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H, et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology (2020) 159(3):944–955.e8. doi: 10.1053/j.gastro.2020.05.048
10. Rajilić-Stojanović M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev (2014) 38(5):996–1047. doi: 10.1111/1574-6976.12075
11. Lee JY, Tsolis RM, Bäumler AJ. The microbiome and gut homeostasis. Sci (80- ). (2022) 377(6601). doi: 10.1126/science.abp9960
12. Macpherson AJ, McCoy KD. Stratification and compartmentalisation of immunoglobulin responses to commensal intestinal microbes. Semin Immunol (2013) 25(5):358–63. doi: 10.1016/j.smim.2013.09.004
13. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol (2017) 19(1):29–41. doi: 10.1111/1462-2920.13589
14. Durack J, Lynch SV. The gut microbiome: Relationships with disease and opportunities for therapy. J Exp Med (2019) 216(1):20–40. doi: 10.1084/jem.20180448
15. Adak A, Khan MR. An insight into gut microbiota and its functionalities. Cell Mol Life Sci (2019) 76(3):473–93. doi: 10.1007/s00018-018-2943-4
16. Suez J, Cohen Y, Valdés-Mas R, Mor U, Dori-Bachash M, Federici S, et al. Personalized microbiome-driven effects of non-nutritive sweeteners on human glucose tolerance. Cell (2022) 185(18):3307–3328.e19. doi: 10.1016/j.cell.2022.07.016
17. Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-induced extinctions in the gut microbiota compound over generations. In: Nature. (2016) p:212–5. doi: 10.1038/nature16504
18. Berg G, Rybakova D, Fischer D, Cernava T, Vergès MCC, Charles T, et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome (2020) 8(1):1–22. doi: 10.1186/s40168-020-00875-0
19. Hou K, Wu ZX, Chen XY, Wang JQ, Zhang D, Xiao C, et al. Microbiota in health and diseases. Signal Transduct Target Ther (2022) 7(1):135. doi: 10.1038/s41392-022-00974-4
20. Mathieu E, Escribano-Vazquez U, Descamps D, Cherbuy C, Langella P, Riffault S, et al. Paradigms of lung microbiota functions in health and disease, particularly, in asthma. Front Physiol (2018) 9:1168. doi: 10.3389/fphys.2018.01168
21. Wypych TP, Wickramasinghe LC, Marsland BJ. The influence of the microbiome on respiratory health. Nat Immunol (2019) 20(10):1279–90. doi: 10.1038/s41590-019-0451-9
22. Mouraux S, Bernasconi E, Pattaroni C, Koutsokera A, Aubert JD, Claustre J, et al. Airway microbiota signals anabolic and catabolic remodeling in the transplanted lung. J Allergy Clin Immunol (2018) 141(2):718–729.e7. doi: 10.1016/j.jaci.2017.06.022
23. Sze MA, Tsuruta M, Yang SWJ, Oh Y, Man SFP, Hogg JC, et al. Changes in the bacterial microbiota in gut, blood, and lungs following acute LPS instillation into mice lungs. PloS One (2014) 9(10):e111228. doi: 10.1371/journal.pone.0111228
24. Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, et al. Microbiota regulates immune defense against respiratory tract influenza a virus infection. Proc Natl Acad Sci USA 2011108(13):5354–9. doi: 10.1073/pnas.1019378108
25. Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science (2018) 359(6371):114–9. doi: 10.1126/science.aam5809
26. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350(6263):981–5. doi: 10.1126/science.aac9593
27. Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun (2019) 10(1):1–18. doi: 10.1038/s41467-018-07859-7
28. Zelante T, Iannitti RG, Cunha C, DeLuca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39(2):372–85. doi: 10.1016/j.immuni.2013.08.003
29. Gomaa EZ. Human gut microbiota/microbiome in health and diseases: A review. Antonie van Leeuwenhoek Int J Gen Mol Microbiol (2020) 113(12):2019–40. doi: 10.1007/s10482-020-01474-7
30. Ratto D, Roda E, Romeo M, Venuti MT, Desiderio A, Lupo G, et al. The many ages of microbiome–Gut–Brain axis. Nutrients (2022) 14(14):1–22. doi: 10.3390/nu14142937
31. Dinan TG, Cryan JF. Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol (2017) 595(2):489–503. doi: 10.1113/JP273106
32. Schroeder BO, Bäckhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med (2016) 22(10):1079–89. doi: 10.1038/nm.4185
33. Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell (2014) 159(3):514–29. doi: 10.1016/j.cell.2014.09.048
34. Matenchuk BA, Mandhane PJ, Kozyrskyj AL. Sleep, circadian rhythm, and gut microbiota. Sleep Med Rev (2020) 53:101340. doi: 10.1016/j.smrv.2020.101340
35. Kaczmarek JL, Musaad SMA, Holscher HD. Time of day and eating behaviors are associated with the composition and function of the human gastrointestinal microbiota. Am J Clin Nutr (2017) 106(5):1220–31. doi: 10.3945/ajcn.117.156380
36. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol (2019) 71(6):1216–28. doi: 10.1016/j.jhep.2019.08.005
37. Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Sci (80- ). (2015) 350(6262):830–4. doi: 10.1126/science.aad0135
38. Yu L, Tong Y, Shen G, Fu A, Lai Y, Zhou X, et al. Immunodepletion with hypoxemia: A potential high risk subtype of coronavirus disease 2019. medRxiv (2020). doi: 10.1101/2020.03.03.20030650v1.abstract
39. Tang L, Gu S, Gong Y, Li B, Lu H, Li Q, et al. Clinical significance of the correlation between changes in the major intestinal bacteria species and COVID-19 severity. Engineering (2020) 6(10):1178–84. doi: 10.1016/j.eng.2020.05.013
40. Zuo T, Liu Q, Zhang F, Lui GCY, Tso EYK, Yeoh YK, et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut (2021) 70(2):276–84. doi: 10.1136/gutjnl-2020-322294
41. Gu S, Chen Y, Wu Z, Chen Y, Gao H, Lv L, et al. Alterations of the gut microbiota in patients with COVID-19 or H1N1 influenza silan. Journals Gerontol Ser A Biol Sci Med Sci (2020) 0813:1–11. doi: 10.1093/cid/ciaa709
42. Tao W, Zhang G, Wang X, Guo M, Zeng W, Xu Z, et al. Analysis of the intestinal microbiota in COVID-19 patients and its correlation with the inflammatory factor IL-18. Med Microecol (2020) 5(September):100023. doi: 10.1016/j.medmic.2020.100023
43. Yeoh YK, Zuo T, Lui GCY, Zhang F, Liu Q, Li AYL, et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut (2021) 70(4):698–706. doi: 10.1136/gutjnl-2020-323020
44. Mazzarelli A, Giancola ML, Farina A, Marchioni L, Rueca M, Gruber CEM, et al. 16S rRNA gene sequencing of rectal swab in patients affected by COVID-19. PloS One (2021) 16:1–15. doi: 10.1371/journal.pone.0247041
45. Liu F, Ye S, Zhu X, He X, Wang S, Li Y, et al. Gastrointestinal disturbance and effect of fecal microbiota transplantation in discharged COVID-19 patients. J Med Case Rep (2021) 15(1):1–9. doi: 10.1186/s13256-020-02583-7
46. Xu R, Lu R, Zhang T, Wu Q, Cai W, Han X, et al. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun Biol (2021) 4(1):1–11. doi: 10.1038/s42003-021-01796-w
47. Ren Z, Wang H, Cui G, Lu H, Wang L, Luo H, et al. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut (2021) 70(7):1253–65. doi: 10.1136/gutjnl-2020-323826
48. Chen Y, Gu S, Chen Y, Lu H, Shi D, Guo J, et al. Six-month follow-up of gut microbiota richness in patients with COVID-19. Gut (2022) 71(1):222–5. doi: 10.1136/gutjnl-2021-324090
49. Gaibani P, D’Amico F, Bartoletti M, Lombardo D, Rampelli S, Fornaro G, et al. The gut microbiota of critically ill patients with COVID-19. Front Cell Infect Microbiol (2021) 11(June):1–11. doi: 10.3389/fcimb.2021.670424
50. Zhou Y, Shi X, Fu W, Xiang F, He X, Yang B, et al. Gut microbiota dysbiosis correlates with abnormal immune response in moderate covid-19 patients with fever. J Inflammation Res (2021) 14:2619–31. doi: 10.2147/JIR.S311518
51. Kim HN, Joo EJ, Lee CW, Ahn KS, Kim HL, Park D, et al. Reversion of gut microbiota during the recovery phase in patients with asymptomatic or mild covid-19: Longitudinal study. Microorganisms (2021) 9(6):1–16. doi: 10.3390/microorganisms9061237
52. Zhou Y, Zhang J, Zhang D, Ma WL, Wang X. Linking the gut microbiota to persistent symptoms in survivors of COVID-19 after discharge. J Microbiol (2021) 59(10):941–8. doi: 10.1007/s12275-021-1206-5
53. Moreira-Rosário A, Marques C, Pinheiro H, Araújo JR, Ribeiro P, Rocha R, et al. Gut microbiota diversity and c-reactive protein are predictors of disease severity in COVID-19 patients. Front Microbiol (2021) 12:1–13. doi: 10.3389/fmicb.2021.705020
54. Wu Y, Cheng X, Jiang G, Tang H, Ming S, Tang L, et al. Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. NPJ Biofilms Microbiomes (2021) 7(1):90. doi: 10.1038/s41522-021-00262-z
55. He F, Zhang T, Xue K, Fang Z, Jiang G, Huang S, et al. Fecal multi-omics analysis reveals diverse molecular alterations of gut ecosystem in COVID-19 patients. Anal Chim Acta (2021) 1180:338881. doi: 10.1016/j.aca.2021.338881
56. Li S, Yang S, Zhou Y, Disoma C, Dong Z, Du A, et al. Microbiome profiling using shotgun metagenomic sequencing identified unique microorganisms in COVID-19 patients with altered gut microbiota. Front Microbiol (2021) 12(October). doi: 10.3389/fmicb.2021.712081
57. Liu Q, Mak JWY, Su Q, Yeoh YK, Lui GCY, Ng SSS, et al. Gut microbiota dynamics in a prospective cohort of patients with post-acute COVID-19 syndrome. Gut (2022) 71(3):544–52. doi: 10.1136/gutjnl-2021-325989
58. Ng SC, Peng Y, Zhang L, Mok CK, Zhao S, Li A, et al. Gut microbiota composition is associated with SARS-CoV-2 vaccine immunogenicity and adverse events. Gut (2022) 71(6):1106–16. doi: 10.1136/gutjnl-2021-326563
59. Zmora N, Suez J, Elinav E. You are what you eat: Diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol (2019) 16(1):35–56. doi: 10.1038/s41575-018-0061-2
60. Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med (2017) 15(1):1–17. doi: 10.1186/s12967-017-1175-y
61. Noguera-Julian M, Rocafort M, Guillén Y, Rivera J, Casadellà M, Nowak P, et al. Gut microbiota linked to sexual preference and HIV infection. EBioMedicine (2016) 5:135–46. doi: 10.1016/j.ebiom.2016.01.032
62. Neff CP, Krueger O, Xiong K, Arif S, Nusbacher N, Schneider JM, et al. Fecal microbiota composition drives immune activation in HIV-infected individuals. EBioMedicine (2018) 30:192–202. doi: 10.1016/j.ebiom.2018.03.024
63. Hill TCJ, Walsh KA, Harris JA, Moffett BF. Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol (2003) 43(1):1–11. doi: 10.1111/j.1574-6941.2003.tb01040.x
64. He Y, Zhou BJ, Deng GH, Jiang XT, Zhang H, Zhou HW. Comparison of microbial diversity determined with the same variable tag sequence extracted from two different PCR amplicons. BMC Microbiol (2013) 13(1):1. doi: 10.1186/1471-2180-13-208
65. Gregorius HR. Effective numbers in the partitioning of biological diversity. J Theor Biol (2016) 409:133–47. doi: 10.1016/j.jtbi.2016.08.037
66. Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol (2007) 73(5):1576–85. doi: 10.1128/AEM.01996-06
67. Hirayama M, Nishiwaki H, Hamaguchi T, Ito M, Ueyama J, Maeda T, et al. Intestinal collinsella may mitigate infection and exacerbation of COVID-19 by producing ursodeoxycholate. PloS One (2021) 16(11 November):1–11. doi: 10.1371/journal.pone.0260451
68. De Maio F, Posteraro B, Ponziani FR, Cattani P, Gasbarrini A, Sanguinetti M. Nasopharyngeal microbiota profiling of SARS-CoV-2 infected patients. Biol Proced Online. (2020) 22(1):20–3. doi: 10.1186/s12575-020-00131-7
69. Rueca M, Fontana A, Bartolini B, Piselli P, Mazzarelli A, Copetti M, et al. Investigation of nasal/oropharyngeal microbial community of covid-19 patients by 16s rdna sequencing. Int J Environ Res Public Health (2021) 18(4):1–12. doi: 10.3390/ijerph18042174
70. Shen Z, Xiao Y, Kang L, Ma W, Shi L, Zhang L, et al. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin Infect Dis (2020) 71(15):713–20. doi: 10.1093/cid/ciaa203
71. Nardelli C, Gentile I, Setaro M, Di Domenico C, Pinchera B, Buonomo AR, et al. Nasopharyngeal microbiome signature in COVID-19 positive patients: Can we definitively get a role to fusobacterium periodonticum? Front Cell Infect Microbiol (2021) 11:1–7. doi: 10.3389/fcimb.2021.625581
72. Budding A, Sieswerda E, Wintermans B, Bos M. An Age Dependent Pharyngeal Microbiota Signature Associated with SARS-CoV-2 Infection. (2020). doi: 10.2139/ssrn.3582780
73. Ventero MP, Cuadrat RRC, Vidal I, Andrade BGN, Molina-Pardines C, Haro-Moreno JM, et al. Nasopharyngeal microbial communities of patients infected with SARS-CoV-2 that developed COVID-19. Front Microbiol (2021) 12:1–10. doi: 10.3389/fmicb.2021.637430
74. Rosas-Salazar C, Kimura KS, Shilts MH, Strickland BA, Freeman MH, Wessinger BC, et al. SARS-CoV-2 infection and viral load are associated with the upper respiratory tract microbiome. J Allergy Clin Immunol (2021) 147(4):1226–1233.e2. doi: 10.1016/j.jaci.2021.02.001
75. Miao Q, Ma Y, Ling Y, Jin W, Su Y, Wang Q, et al. Evaluation of superinfection, antimicrobial usage, and airway microbiome with metagenomic sequencing in COVID-19 patients: A cohort study in shanghai. J Microbiol Immunol Infect (2021) 54(5):808–15. doi: 10.1016/j.jmii.2021.03.015
76. Braun T, Halevi S, Hadar R, Efroni G, Glick Saar E, Keller N, et al. SARS-CoV-2 does not have a strong effect on the nasopharyngeal microbial composition. Sci Rep [Internet]. (2021) 11(1):8922. doi: 10.1038/s41598-021-88536-6
77. Zhang H, Ai JW, Yang W, Zhou X, He F, Xie S, et al. Metatranscriptomic characterization of coronavirus disease 2019 identified a host transcriptional classifier associated with immune signaling. Clin Infect Dis (2021) 73(3):376–85. doi: 10.1093/cid/ciaa663
78. Mostafa HH, Fissel JA, Fanelli B, Bergman Y, Gniazdowski V, Dadlani M, et al. Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect covid-19 patients. MBio (2020) 11(6):1–13. doi: 10.1128/mBio.01969-20
79. Merenstein C, Liang G, Whiteside SA, Cobián-Güemes AG, Merlino MS, Taylor LJ, et al. Signatures of COVID-19 severity and immune response in the respiratory tract microbiome. ASM J MBio (2021) 12(4). doi: 10.1128/mBio.01777-21
80. Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, et al. Mining the human gut microbiota for immunomodulatory organisms. Cell (2017) 168(5):928–943.e11. doi: 10.1016/j.cell.2017.01.022
81. Ramanan P, Barreto JN, Osmon DR, Tosh PK. Rothia bacteremia: A 10-year experience at Mayo clinic, Rochester, Minnesota. J Clin Microbiol (2014) 52(9):3184–9. doi: 10.1128/JCM.01270-14
82. Bernard-Raichon L, Venzon M, Klein J, Axelrad JE, Zhang C, Sullivan AP, et al. Gut microbiome dysbiosis in antibiotic-treated COVID-19 patients is associated with microbial translocation and bacteremia. Nat Commun (2022) 13(1):1–13. doi: 10.1038/s41467-022-33395-6
83. Seibert B, Cáceres CJ, Cardenas-Garcia S, Carnaccini S, Geiger G, Rajao DS, et al. Mild and severe SARS-CoV-2 infection induces respiratory and intestinal microbiome changes in the K18-hACE2 transgenic mouse model. Microbiol Spectr (2021) 9(1):e00536-21. doi: 10.1128/Spectrum.00536-21
84. Bercik P, Denou E, Collins J, Jackson W, Lu J, Jury J, et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology (2011) 141(2):599–609. doi: 10.1053/j.gastro.2011.04.052
85. Carloni S, Bertocchi A, Mancinelli S, Bellini M, Erreni M, Borreca A, et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science (2021) 374(6566):439–48. doi: 10.1126/science.abc6108
86. Agirman G, Yu KB, Hsiao EYT. Signaling inflammation across the gut-brain axis. (2021) 1092:1087–92.
87. Sajdel-Sulkowska EM. Neuropsychiatric ramifications of COVID-19: Short-chain fatty acid deficiency and disturbance of microbiota-Gut-Brain axis signaling. BioMed Res Int (2021) 2021:7880448. doi: 10.1155/2021/7880448
88. Xiao N, Nie M, Pang H, Wang B, Hu J, Meng X, et al. Integrated cytokine and metabolite analysis reveals immunometabolic reprogramming in COVID-19 patients with therapeutic implications. Nat Commun (2021) 12(1):1–13. doi: 10.1038/s41467-021-21907-9
89. Nataf S, Pays L. Molecular insights into sars-cov2-induced alterations of the gut/brain axis. Int J Mol Sci (2021) 22(19):10440. doi: 10.3390/ijms221910440
90. Drakopanagiotakis F, Stavropoulou E, Tsigalou C, Nena E, Steiropoulos P. The role of the microbiome in connective-Tissue-Associated interstitial lung disease and pulmonary vasculitis. Biomed (2022) 10(12):3195. doi: 10.3390/biomedicines10123195
Keywords: microbiota, microbiome, gut-brain-axis, gut-lung-axis, dysbiosis, COVID-19, long Covid, SARS-CoV-2
Citation: Ancona G, Alagna L, Alteri C, Palomba E, Tonizzo A, Pastena A, Muscatello A, Gori A and Bandera A (2023) Gut and airway microbiota dysbiosis and their role in COVID-19 and long-COVID. Front. Immunol. 14:1080043. doi: 10.3389/fimmu.2023.1080043
Received: 25 October 2022; Accepted: 13 February 2023;
Published: 08 March 2023.
Edited by:
Sergio Serrano-Villar, Ramón y Cajal University Hospital, SpainReviewed by:
Yean Kong Yong, Xiamen University, MalaysiaMahesh Mohan, Texas Biomedical Research Institute, United States
Copyright © 2023 Ancona, Alagna, Alteri, Palomba, Tonizzo, Pastena, Muscatello, Gori and Bandera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emanuele Palomba, ZW1hbnVlbGUucGFsb21iYUB1bmltaS5pdA==; Andrea Gori, YW5kcmVhLmdvcmlAdW5pbWkuaXQ=