 Yi Ye
Yi Ye Qiying Xu
Qiying Xu Tana Wuren
Tana Wuren- 1Research Center for High Altitude Medicine, Qinghai University, Xining, China
- 2High-Altitude Medicine Key Laboratory of the Ministry of Education, Xining, China
- 3Qinghai Provincial Key Laboratory for Application of High-Altitude Medicine, Xining, China
- 4Qinghai-Utah Key Laboratory of High-Altitude Medicine, Xining, China
Hypoxic pulmonary hypertension (HPH) is a complicated vascular disorder characterized by diverse mechanisms that lead to elevated blood pressure in pulmonary circulation. Recent evidence indicates that HPH is not simply a pathological syndrome but is instead a complex lesion of cellular metabolism, inflammation, and proliferation driven by the reprogramming of gene expression patterns. One of the key mechanisms underlying HPH is hypoxia, which drives immune/inflammation to mediate complex vascular homeostasis that collaboratively controls vascular remodeling in the lungs. This is caused by the prolonged infiltration of immune cells and an increase in several pro-inflammatory factors, which ultimately leads to immune dysregulation. Hypoxia has been associated with metabolic reprogramming, immunological dysregulation, and adverse pulmonary vascular remodeling in preclinical studies. Many animal models have been developed to mimic HPH; however, many of them do not accurately represent the human disease state and may not be suitable for testing new therapeutic strategies. The scientific understanding of HPH is rapidly evolving, and recent efforts have focused on understanding the complex interplay among hypoxia, inflammation, and cellular metabolism in the development of this disease. Through continued research and the development of more sophisticated animal models, it is hoped that we will be able to gain a deeper understanding of the underlying mechanisms of HPH and implement more effective therapies for this debilitating disease.
1 Introduction
Hypoxic pulmonary hypertension (HPH) (1), also known as WHO group III pulmonary hypertension (PH), can be caused by exposure to high altitudes or chronic lung diseases that lead to systemic hypoxia, which is a heterogeneous group of diseases that leads to a continuous increase in pulmonary artery pressure (PAP) and significant progression of pulmonary vascular remodeling in the presence of alveolar hypoxia (2).
With a combination of targeted drugs (3) such as phosphodiesterase-5 inhibitors, endothelin receptor antagonists, prostacyclin analogs, and prostacyclin receptor agonists, patients with pulmonary arterial hypertension (PAH, i.e., WHO group I PH) have shown a significant improvement in activity tolerance and disease prognosis. However, clinical trials of these drugs have not shown any beneficial effects in patients with HPH (4, 5). Therefore, clarifying the underlying pathogenic mechanisms in this group of patients will help to improve targeted therapies.
To date, researchers have begun to study the role of perivascular inflammation in the onset of HPH, and there is mounting evidence that hypoxia, immune-cell inflammation, and metabolic imbalance in vascular cells play a role in its progression and development. High altitude or hypoxic exposure can also lead to PH as they affect immune homeostasis and regulatory activity.
This review presents evidence of immune-inflammatory dysregulation occurring in pulmonary arteries during chronic hypoxia exposure, including hypoxia-induced hyperproliferation/hypertrophy of resident vascular cells, infiltration of immune cells, and expression of pro-inflammatory factors. It also emphasizes that metabolic reprogramming under the hypoxic microenvironment triggers functional changes in the inflammatory response and immune cell activity, suggesting the importance of hypoxia in the initiation and progression of pulmonary vascular lesions and inflammation.
In this review, we provide evidence of immune-inflammatory disruption in pulmonary arteries upon chronic exposure to hypoxia, characterized by hyperproliferation/hypertrophy of resident vascular cells, in addition to the recruitment of immune cells and upregulation of pro-inflammatory agents. These results indicate the significance of hypoxia in the development and progression of pulmonary vascular lesions and inflammation (Figure 1).
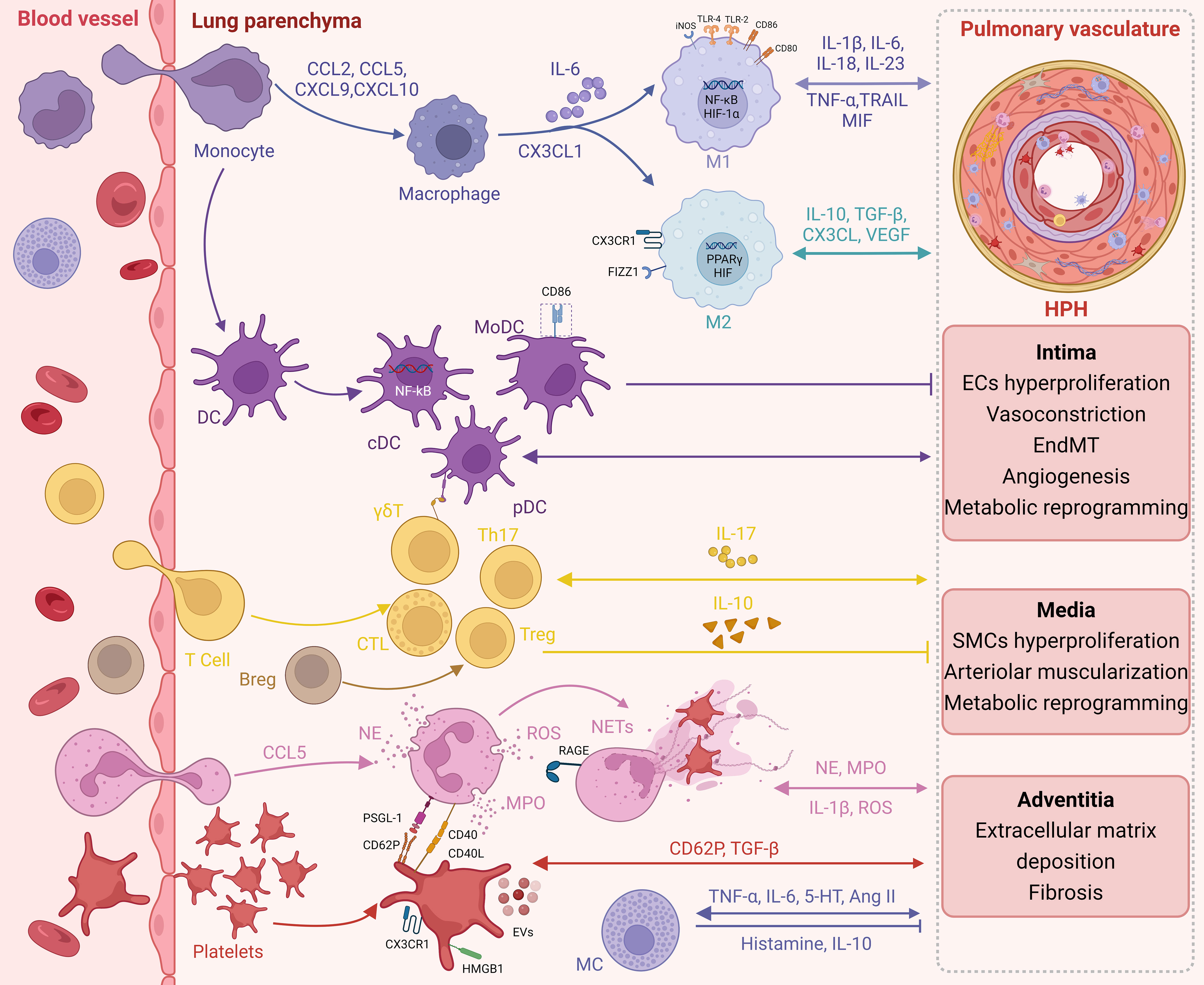
Figure 1 Immunoinflammation in HPH. Infiltration of circulating immune cells and expression of pro-inflammatory factors during chronic hypoxic exposure promote immune inflammatory dysregulation in the pulmonary arteries. In turn, long-term resident vascular cell dysfunction further promotes an immune inflammatory response.
2 Evidence for inflammatory infiltration in HPH
The presence of inflammation in HPH has been shown in histological results (6); inflammatory infiltrates consisting of macrophages, mast cells (MCs), neutrophils, platelets, T- and B-lymphocytes, and dendritic cells have been found around pulmonary vascular lesions. These studies indicate that perivascular inflammation could significantly correlate with intimal and medial fractional thickness, leading to increased PAP (7) and contributing to the structural remodeling of pulmonary circulation.
Oxygen deprivation in the lung tissue is the basis of HPH vasculopathy. Hypoxia inducible factor (HIF) is a major transcription factor involved in oxygen-sensing homeostasis, which contributes to the pathogenesis of HPH (8). HIF is responsible for the activation of signaling pathways in pulmonary arterial endothelial cells (PAECs) (9) and pulmonary artery smooth muscle cells (PASMCs), which have a direct influence on cell proliferation (10); it also orchestrates inflammatory dysregulation in response to hypoxia levels, which in turn induces profound changes in endothelial and smooth muscle cell phenotypes.
HPH is caused by chronic hypoxia, which can be primarily or secondarily caused by a lack of oxygen in the cellular microenvironment owing to an inflammatory response. HIF promotes aggregation of inflammatory cells and release of inflammatory mediators. Immune cells exposed to a hypoxic environment leak from the oxygen-enriched blood to the site of inflammation, which activates and stabilizes HIF. For example, when macrophages and neutrophils infiltrate HPH, HIF-1α is known to increase macrophage proliferation, chemotaxis, and infiltration, and drive the release of cytokines (11). Under hypoxia, the inhibition of mitochondrial oxidative phosphorylation and altered electron transport chain causes an increase in macrophage reactive oxygen species (ROS) production, which activates and stabilizes HIF-1α (12). HIF-1α also increases neutrophil extracellular traps (NETs) formation (13) and neutrophil survival by inhibiting apoptosis and triggering the nuclear factor NF-κB signaling pathway (14). Activated neutrophils stimulate the expression of HIF-1α and HIF-2α (15).
In contrast, activation of HIF-centered hypoxic signaling pathways can initiate immune inflammatory responses. The key regulators of transcriptional responses in hypoxia and inflammation are HIF and NF-κB (16), respectively, both of which promote positive feedback loops in pulmonary vascular remodeling. The p65 and p50 subunits of NF-κB efficiently bind to the HIF-1α promoter thereby enhancing HIF-1α (17) expression. Jin et al. (18) found that, in response to hypoxia, endothelial cells can efficiently secrete TNF-α, which in turn leads to the activation of HIF through the NF-κB pathway, ultimately boosting vascular endothelial growth factor (VEGF) production. After a reduction in NF-κB pathway participant expression, the TNF-α/NF-κB/HIF signaling cascade was inhibited, causing HIF-dependent inactivation of downstream events and ultimately hypoxia-induced inhibition of angiogenesis. HIF-1 increases its expression by binding to the promoter region of CD146. Therefore, an increase in CD146 levels stimulates the HIF-1 transcriptional pathway through NF-B activation (19), leading to hypoxia-induced cell proliferation and anti-apoptosis. Thus, elevated CD146 in PASMC correlates with the severity of PH. CD146-mediated cross-regulation of HIF-1α with NF-κB promotes the synthetic phenotype of PASMCs. Chronic hypoxia triggers pulmonary vasculopathy and NF-κB expression, and NF-κB inhibition reverses hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy (20).
There is a strong link between hypoxia and the HIF immune inflammatory response; therefore, HIF-mediated immune cell dysregulation and HIF/NF-κB crosstalk are jointly involved in the vascular pathology of HPH. The formation of HPH begins with hypoxia-triggered pulmonary vascular inflammatory dysfunction, which is a cause rather than a consequence of the development of PH. Along with increased perivascular aggregation and intravascular infiltration of immune cells, hypoxia-induced vasoconstriction and hyperproliferation are exacerbated by elevated circulating cytokines and chemokines (21), highlighting the importance of their involvement in vasculopathy in the pathogenesis of HPH.
3 Immune cells and inflammatory factors in HPH
Recent research indicates that inflammatory and vascular cells interact in vascular lesions associated with HPH. Inflammatory cells not only participate in the pathological process of HPH through direct contact with pulmonary vascular cells but also release cytokines and chemokines to contribute to the exaggerated contractility and proliferation of vascular cells.
3.1 Macrophages
The most prominent type of inflammatory cells in the alveolar and pulmonary vessel walls is macrophages (22), which play a role in the vascular remodeling process associated with chronic hypoxia. Given the tendency of macrophages to be recruited to areas of poor vascularity with low oxygen tension (23), the primary function of macrophages in the regulation of HPH (24) deserves further investigation.
Many studies have revealed early and persistent accumulation of macrophages in perivascular lesions (24, 25). The significance of macrophages in PAP (26) and pulmonary vascular remodeling has been validated using targeted interventions. Macrophage depletion normalized mean pulmonary artery pressure (mPAP) and restored right ventricular systolic pressure (RVSP) in bone morphogenetic protein type-II receptor (BMPR-II) knockout mouse models subjected to chronic hypoxia (27).
Macrophages can adapt to their immune microenvironment by undergoing polarized activation, which may be roughly classified into two types: conventionally activated macrophages (M1) and alternatively activated macrophages (M2) (28). By encouraging the release of pro-inflammatory molecules, M1 enhances inflammation, whereas M2 stimulates proliferation and angiogenesis (29). HPH patients and animal models activated both M1 and M2, but M2 predominated (25). Inducing inflammation and pulmonary vascular remodeling, M2 accelerates PAH development (30). Macrophage polarization was affected by sex disparity, and macrophage-depleted HPH mice confirmed that HPH progression was associated with M2 predominance in the lungs of male mice (31), and that this M1/M2 ratio imbalance was mediated by interleukin 6 (IL-6) (32).
It is essential for the development of HPH that macrophages are recruited early and activated in hypoxic lungs, leading to the destruction of blood vessels and their remodeling (21). In hypoxia, chemokines such as fractalkine/CX3CL1, which cause the adhesion of CX3CR1 expressing immune cells (particularly macrophages) to PAECs, lead to dysfunction of PAECs and initiation of a perivascular inflammatory response (33). By influencing monocyte recruitment, macrophage polarization, and PASMCs proliferation in the lungs, CX3CR1 plays a significant role in the development of HPH (24). This was demonstrated by the progression of PH being slowed in mice lacking the CX3CR1 gene (25).
Under hypoxic exposure, activated PAECs trigger the production of IL-6 (34), Chemokine (C-C motif) ligand 2 (CCL2) (35), matrix metalloproteinase (MMP)-9 (36), platelet-derived growth factor-B (37), and leukotriene B4 (LTB4) (38), and the accumulation of these known inflammatory mediators may produce a pro-inflammatory microenvironment that is detrimental to the integrity of the pulmonary vascular bed, thereby further worsening vascular remodeling.
The aggregation of macrophages surrounding pulmonary artery vessels is a hallmark of HPH. Macrophage-mediated lung inflammation is a prime cause of lung remodeling and, by extension, of right ventricular remodeling. New therapeutic options for HPH require a better understanding of the role of macrophages in immunological responses to the disease.
3.2 Mast cells
Accumulation of mast cells (MCs) around blood vessels was found to be positively associated with right ventricular hypertrophy in chronically hypoxic pigs, rats, and sheep (39). Further research has found that hypoxia induces an increase in MCs in the lungs of rats and humans, leading to pulmonary vascular remodeling and HPH (40, 41).
Hypoxia causes recruitment, activation, and degranulation of MCs, releasing compounds that promote PH and pulmonary vascular remodeling (42), such as tumor necrosis factor alpha (TNF-α), IL-6 (43), trypsin, chymotrypsin, 5-HT, and Ang II. Targeting MCs in PH may reduce PAP and pulmonary vascular remodeling (44). However, MCs can also reduce inflammation and suppress the immune system (45). Recovery from chronic hypoxia is associated with the reversal of pulmonary vascular remodeling, which is linked to an increase in collagenase in pulmonary MCs and collagenolytic and elastolytic activities in lung tissue (46). More research is needed to understand the dual involvement of MCs in increasing inflammation and dampening immune responses in HPH.
3.3 Neutrophils
Neutrophils have long been thought to be terminally differentiated cells, but little attention has been paid to their function in the pathophysiology of HPH. As first-line effectors of host defense, they need the capacity to detect low oxygen tension in the environment and adapt by altering a number of crucial processes (47). Cells may adapt by increasing their degranulation in response to low oxygen levels. A variety of regulatory inputs may alter neutrophils to alter HIF activity at hypoxic inflammatory sites (48). This suggests that neutrophils play a larger role in the pathophysiology of HPH than previously thought.
It has been shown that PH patients and animal models, have significantly elevated levels of neutrophils (49), associated with clinical indices of disease progression such as increased pulmonary vascular resistance (7) and decreased 6-minute walk distance (6 MWD) (50). Recent research has pointed to the neutrophil-to-lymphocyte ratio (NLR) as a possible inflammatory indicator that may be used as a marker of systemic inflammation in a variety of conditions (51), including PH. NLR and pulmonary vascular resistance are strongly correlated in individuals with PAH (52) or sarcoidosis-associated PH (53), also known as WHO Group V. Similarly, NLR predicts preoperative pulmonary vascular resistance and surgical mortality in patients with chronic thromboembolic PH (54), also known as WHO Group IV.
In both pre- and clinical studies of PH, more neutrophils were found in blood and lung tissues. However, the increase in neutrophils was different in each PH group, with the most noticeable change occurring in HPH (55). The activation of recruited neutrophils leads to further recruitment of cellular mediators, secretion of cytokines, and release of neutrophil elastase (NE) (56) and myeloperoxidase (MPO) (57).
Neutrophils infiltrating the pulmonary vasculature of mice with chronic hypoxia-induced PH were able to secrete IL-1β (58), and knockdown of IL-1β receptors in mice or use of IL-1β receptor inhibitors attenuated the persistent elevation of PAP in an animal model of PH, resulting in a significant reduction in RVSP (59). In PASMCs, IL-1β can inhibit the conversion of ATP to cAMP by downregulating the activity of adenylate cyclase (60). IL-1β modulates the growth of PASMCs through the IL-1R1/MyD88 pathway (61). IL-1β released by neutrophils may directly control hypoxia-stimulated vasoconstriction and remodeling in pulmonary arteries. In the pathological condition of HPH, neutralization of IL-1β, inhibition of IL-1β signaling, or suppression of upstream pathways regulating IL-1β secretion may be effective in attenuating disease progression.
Nickel et al (56) observed increased NE activity in the pulmonary vasculature of a rat model of SU5416/hypoxia (Su/Hx)-induced PH and that inhibition of NE activity by application of elafin (an endogenous NE inhibitor) improved endothelial function, reduced small pulmonary artery remodeling, and decreased PAP, emphasizing that inhibition of NE activity offers new insights for the therapy of HPH. By reducing MPO in rats with Su/Hx-induced PH, MPO exacerbates pulmonary vasoconstriction and pulmonary vascular remodeling by activating the RhoA/Rho kinase pathway (57), suggesting that MPO may be a precipitating factor in HPH.
Recent studies have shown a functional relationship between NETs and inflammatory angiogenesis in vitro and in vivo and a correlation between their formation and pulmonary vascular remodeling (62). NETs may promote pulmonary vascular remodeling via multiple mechanisms, and their mediated imbalance in pulmonary vascular homeostasis may have implications for a variety of different types of PH diseases. In hypoxia, dysfunction of pulmonary vascular wall cells leads to increased production of ROS (63), which NETs are dependent on (62).
Elevated cytokine levels in HPH enhance neutrophil population and function. Neutrophils, after being recruited to lung tissue during HPH, are not only involved in perivascular inflammation (64) but also mediate pulmonary vascular remodeling through the degranulation and release of cytokines and proteases, as well as the formation of NETs (65).
3.4 Platelets
As the second most abundant cell population in peripheral blood, platelets are involved not only in the hemostatic process but also in the repair of endothelial cells, promotion of cell growth, and the development of inflammatory diseases (66). The immune function of platelets depends on the precise balance between pathogenic and hemostatic effects and their thrombogenic actions. Several studies have shown that platelets are involved in the onset and development of HPH and are associated with its prognosis of HPH (67–70). Platelet distribution width (PDW), a measure of platelet activation, has been proposed as a risk predictor of PAH (71). In patients with severe HPH, in-hospital PDW was independently related to all-cause death, suggesting that PDW may be a predictive indicator for platelet activation and severity (72). The platelet-to-lymphocyte ratio (PLR) is a novel inflammatory marker that can be used to predict inflammation and mortality in various diseases. As a combination of platelets and lymphocytes, the PLR has been shown to correlate with the severity and prognosis of HPH (73).
As markers of platelet surface activation, P-selectin (also known as CD62p) and glycoprotein (GP)IIb/IIIa are highly expressed in the Su/Hx mouse model (74), clearly demonstrates the activation of platelets in HPH. P-selectin can bind to NEDD9 to facilitate hypoxia-sensitive pulmonary vascular remodeling through platelet-endothelial cell adhesion (75), a new mediator of HIF-1 dependent PAECs (76). Additionally, activated platelet supernatant promotes PASMCs metabolic reprogramming via the release of transforming growth factor-β (TGF-β) (74).
Platelets and neutrophils interact through the binding of multiple ligands/receptors, involvement of chemokines, and extracellular vesicle-mediated effects to promote mutual activation (77) at the tissue and systemic levels. HMGB1/HMGB1 receptors (69, 70), TLR4, and RAGE (78) levels were significantly higher in HPH, implicating an underlying platelet-neutrophil interaction in the pathogenesis of HPH. The binding of the chemokine fractalkine to the platelet-specific receptor CX3CR1 stimulates neutrophil activation and causes perivascular inflammation, leading to abnormal constriction of small pulmonary arteries, vascular remodeling, and increased PAP. The neutrophil chemokines CXCL4 and CCL5 were significantly reduced in PH mice with reduced platelet specificity (79). Among neutrophil–platelet interactions, the modulation of NETs is of great interest (80). Although few studies have been conducted on platelet-neutrophil interactions in PH disease progression, the current evidence highlights the dual-mediated inflammatory response and prothrombotic state as an underlying part of HPH disease progression.
As a result, platelets contribute to the formation and progression of HPH through aggregation, activation, and promotion of thrombosis, leading to increased pulmonary vascular resistance through secretion of various pro-vasoconstriction factors, growth factors, inflammatory mediators, and production of EVs, causing pulmonary artery constriction and proliferation of vascular component cells through recruitment of various inflammatory cells (especially neutrophils) to promote local injury. Our previous study confirmed that in high-altitude polycythemia, a type of altitude sickness, hypoxia leads to an increase in platelet activation and procoagulant factors (81), again supporting platelet involvement in HPH.
3.5 T Cells, B Cells, and DCs
Based on their function and surface markers, T lymphocytes can be classified as helper T cells (Th cells), regulatory T cells (Tregs), and cytotoxic T cells (CTLs). Th17 cells lead to an increase in perivascular T cells and are involved in the development of HPH due to their anti-apoptotic effects (82) and their ability to enhance the proliferation and migration of PASMCs (83). Th17 cell development was also observed. The Th1 subset produces inflammatory mediators and colocalizes with Th17 in the pulmonary vascular region (84), likely contributing to HPH progression through mutual recruitment. Tregs can protect mice from HPH by inhibiting the inflammatory response, promoting the release of anti-inflammatory factors such as IL-10 (85) and reversing the effect of hypoxia on the cell cycle (86). Hypoxia is known to mediate pulmonary vascular remodeling via activation of the Akt/Erk signaling pathway (87). Tregs also downregulate the phosphorylation of Akt and ERK (86), thereby improving hemodynamic parameters and vascular remodeling in HPH. Tregs and macrophages potentially interact in HPH; activated macrophages can increase LTB4 secretion in the absence of Tregs, leading to changes in vascular cell phenotype (88). Tregs suppress adaptive immune responses; there is an imbalance in the Th17/Treg ratio in patients with PH associated with chronic obstructive pulmonary disease (COPD-PH), and the Th17/Treg balance axis trends towards the Th17 cell lineage (89). The pathophysiology of PH is associated with the dysregulation of circulating CTLs and their granulysin, but its function is not well understood (90, 91). Therefore, T lymphocytes play a significant role in the progression of HPH through their interactions with different signaling pathways and their ability to promote or inhibit inflammation; however, further research is needed to fully understand the role of T cells as therapeutic targets.
The role of B cells in HPH was previously understood to merely regulate antibody production (92). However, recent studies have suggested that a subset called regulatory B cells (Bregs) has additional functions including immunosuppression (93). Bregs have been shown to prevent HPH by regulating the immune balance of Tfh/Tfr and inhibiting the proliferation of PASMCs under hypoxic conditions (94).
Dendritic cells (DCs) play a role in regulating the inflammatory response and have been found in lung samples from IPAH patients and their corresponding animal models (95). Subgroups of DCs have varying effects on the pulmonary vascular immune microenvironment. For example, activated conventional dendritic cells (cDCs) promote inflammation via the NF-κB pathway and development of PH (96). PAH patients show reduced expression of the co-stimulatory factor CD86 on monocyte-derived dendritic cells (MoDCs), leading to impaired function in the PAH-initiated immune or immune tolerance state (97). Plasmacytoid dendritic cells (pDCs) are increased in the lungs of PAH patients (98) and likely act in the tissue immune microenvironment of PH through crosstalk with γδT cells (99).
4 Cytokines, chemokines, and HPH
In hypoxia, dysfunction of pulmonary vascular wall cells and activation of inflammatory cells leads to an increased release of pro-inflammatory mediators. Hypoxia may be a link between cytokines and chemokines and the vascular pathology of HPH. Table 1 provides a summary of the evidence for the involvement of various cytokines/chemokines in the pathogenesis of HPH, including both preclinical (animal models of disease) and clinical findings.
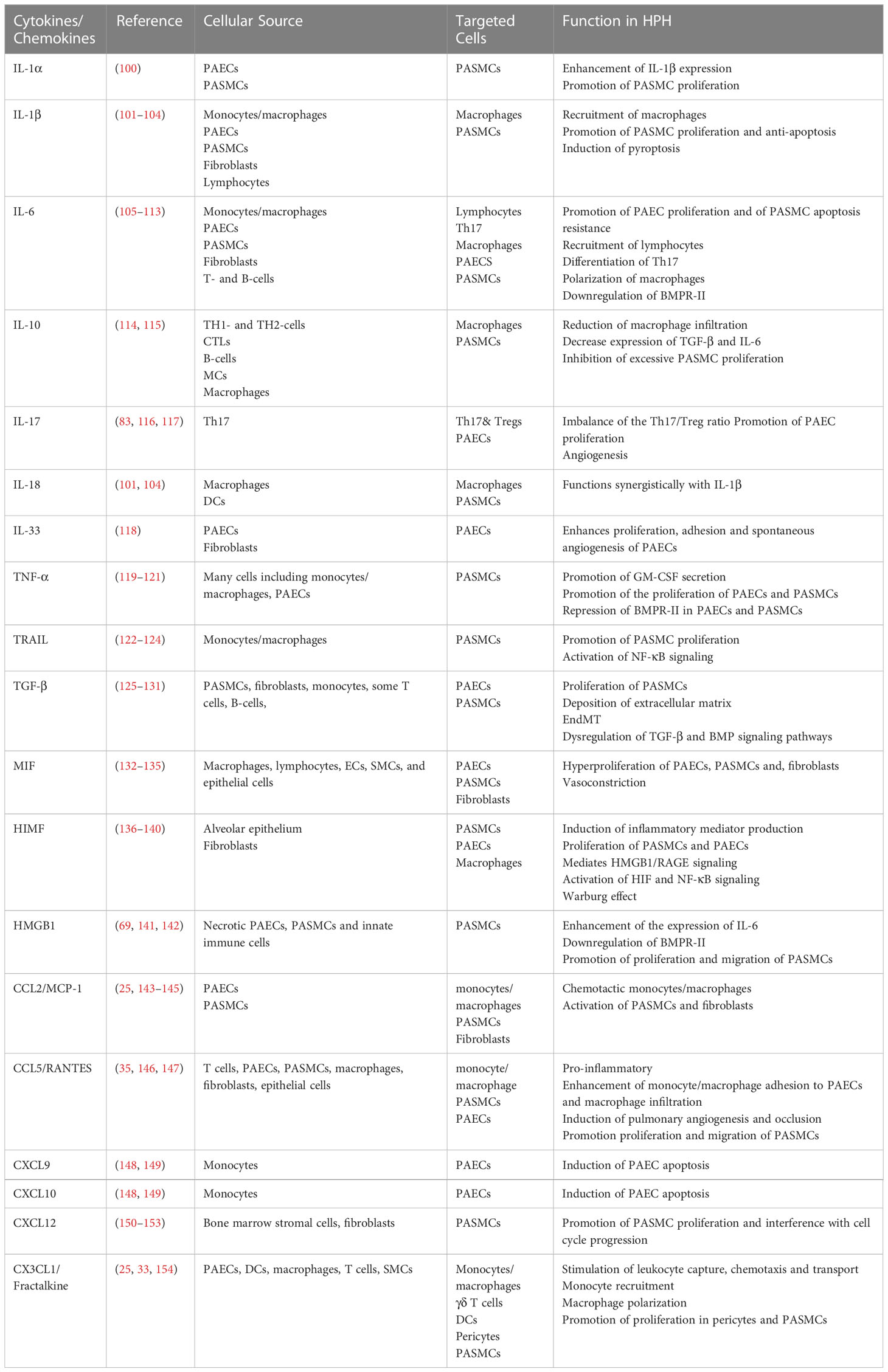
Table 1 Cytokines and chemokines associated with pulmonary vascular inflammation.
4.1 Cytokines
The IL-1 family of cytokines plays a crucial role in the inflammatory response, particularly in promoting excessive proliferation of PASMCs (155). In animal models of HPH, higher levels of IL-1α and IL-1R1 (specific receptors of IL-1a, IL-1b, and IL-1Ra) have been detected in lung tissues, lesions in pulmonary vessels, and serum (100). Treatment with IL-1R1 knockout or anakinra, an IL-1R1 antagonist, protected mice from HPH.
IL-1β and IL-18 (101) have been shown to regulate the recruitment of pulmonary perivascular macrophages, resist hypoxia-induced pyroptosis (102), and synergistically activate the caspase-1/STAT3 pathway, promoting macrophage-based inflammatory cell infiltration (103). Furthermore, serum levels of IL-1β and IL-18 (104) are reliable predictors of pulmonary vascular remodeling and HPH risk. IL-33 has also been found to enhance HPAEC proliferation, adhesion, and spontaneous angiogenesis in an HIF-1α-dependent manner, resulting in vascular remodeling and HPH (118).
IL-6 is one of the most significant players in the pathophysiology of HPH and is highly expressed in plasma (105), lung tissue, and the vascular media of PASMC from HPH patients and animal models (106). Lung-specific IL-6 overexpressing mice developed PH spontaneously under chronic hypoxic conditions and show distal small arterial muscularization. Animals with knocked out IL-6 receptors (107), as well as mice lacking IL-21 receptors, are resistant to HPH (34). After exposure to hypoxia, there was a boost in IL-6 mRNA levels in the lungs, and delivery of recombinant IL-6 protein induced vascular remodeling and enhanced the response of HPH (108).
IL-6 signals via glycoprotein 130 (gp130) and membrane-bound IL-6 receptor (mIL-6R) or soluble IL-6 receptor (sIL-6R), referred to as classic or trans-signaling, respectively. This induces anti-inflammatory and pro-inflammatory responses, thereby initiating its pleiotropic effects (109). Recent studies suggest that IL-6 trans-signaling contributes to HPH by (1) IL-6 induced excessive proliferation of PAECs and proliferative apoptotic resistant phenotype of PASMCs in the distal pulmonary vascular wall in HPH through upregulation of vascular endothelial growth factor receptor 2 (VEGFR2), MMP-9 expression, and lymphocyte recruitment (110); (2) BMPR-II was found to be significantly downregulated in rodents exposed to hypoxia (111); IL-6 is responsible for BMPR-II downregulation through the transcriptional activator STAT3-microRNA cluster 17/92 pathway (112); and (3) IL-6 potentiates hypoxia-induced PASMC migration induced by TH17 cells and promotes the infiltration of M2 macrophages around lesional pulmonary vessels (113).
This suggests a role for IL-6 in the regulation of pulmonary vascular inflammation and myelination in HPH through immune and inflammatory responses mediated by pro-proliferative anti-apoptotic mechanisms.
IL-10 is linked to the pathogenesis and progression of COPD-PH, and a decrease in IL-10 levels is a risk factor for the development of COPD-PH (114). Previous studies have shown that exogenous IL-10 prevents proliferative vasculopathy, PASMCs proliferation, and chemokine expression in vivo by inhibiting inflammatory cell infiltration to prevent Monocrotaline-induced PH (115). The pathogenic mechanism of IL-10 in HPH requires further investigation. Th17 induces an altered vascular cell phenotype in HPH by a mechanism of inflammatory cell infiltration and vascular cell proliferation (82), and the Th17-produced pro-inflammatory cytokine IL-17 expression is upregulated in both bronchodilator/COPD-PH patients and HPH model mice (116). Hypoxia upregulates IL-17 expression (117), which mediates pulmonary vascular remodeling by affecting cell proliferation, angiogenesis, and adhesion function of PAECs in HPH (83).
TNF-α, a pro-inflammatory cytokine, was significantly elevated in the medial layer of the pulmonary artery in patients with COPD-PH (156) and in the Su/Hx rat model (119), confirming the involvement of TNF-α as an inflammatory indicator of HPH pathology. TNF-α drives PAECs and PASMCs proliferation in HPH by inhibiting pyruvate dehydrogenase (PDH) activity (120), suppressing BMPR-II, and altering NOTCH signaling. Rats treated with the TNF-α antagonist, recombinant TNF-α receptor II, and IgG Fc fusion protein (rhTNFRFc) (121) showed some improvement in pulmonary hemodynamics and lung inflammation, which may provide alternative therapeutic targets for HPH treatment.
TNF-related apoptosis-inducing ligand, also known as TRAIL, is a potent stimulator of pulmonary vascular remodeling in human cells and rodent models. In rodent models of HPH, TRAIL depletion or blockade is associated with reduced pulmonary artery remodeling and reduced PASMC proliferation (122). TRAIL knockdown has a similar protective effect in HPH mouse model (123). In addition, TRAIL/TRAIL receptors transduce pro-inflammatory signals through the activation of NF-κB signaling (124), thus participating in the inflammatory response of HPH.
TGF-β is a highly polytropic cytokine that regulates inflammatory processes, particularly in the cardiovascular system. Activation of TGF-β is both necessary and sufficient for the development of PH in chronically hypoxia-exposed mice (125). According to research, increased TGF-β signaling (126) and decreased BMP signaling (127) are involved in the occurrence and development of HPH by promoting PASMCs proliferation (128), extracellular matrix deposition (129) and EndMT (130). These processes were dominated by PASMCs and PAECs. TGF-β is a crucial regulator of pulmonary vascular remodeling and inflammation in addition to cardiac hypertrophy and fibrosis. Selective TGF-β ligand blockade (131) in multiple experimental HPH models improves hemodynamics and remodeling as well as RVSP, right ventricular function, and survival.
Macrophage migration inhibitory factor (MIF) is a multifunctional cytokine that regulates inflammation and immune responses. MIF expression was increased in PASMCs and PAECs of HPH rats (132). MIF enhances hypoxia-induced pulmonary vasoconstriction (133) in addition to participating in hypoxia-induced pulmonary vascular remodeling by promoting the proliferation of ECs (134), SMCs, and fibroblasts (135).
Hypoxia-induced mitogenic factor (HIMF/RELM/FIZZ1) expression was positively correlated with elevated PAP in the remodeled pulmonary vascular system (136). HIMF stimulates pulmonary vascular cells to produce high levels of pro-inflammatory mediators (137), such as HIF-1, IL-6, MCP-1, and ROS, which enhance perivascular immune cell recruitment. Among them, HIF-1 is a major transcription factor involved in HIMF-induced pulmonary vascular remodeling and the development of PH. HIMF has been shown to (1) directly stimulate proliferation of SMCs in HPH through activation of the MAPK signaling pathway (138); (2) stimulate intrapulmonary EC activation and proliferation via the HIF-1/VEGF signaling pathway; (3) promote the Warburg effect through activation of the NF-κB/HIF-1α pathway (139); and (4) drive the high-mobility group protein 1 (HMGB1)/RAGE axis to promote EC-SMC vascular interactions in HPH (70). In addition, HIMF has angiogenic and vasoconstrictive properties that augment PAP and vascular resistance (140). This indicates that HIMF play a role in lung inflammation and vascular remodeling.
HMGB1 is frequently considered to be an atypical inflammatory cytokine. In experimental HPH, HMGB1 levels in serum (141) and pulmonary arteries were markedly elevated, which was associated with mPAP (68). Administration of exogenous recombinant HMGB1 worsened HPH, while treatment with HMGB1 inhibitors significantly attenuated the progression of HPH by recovering hemodynamic parameters, pulmonary vascular remodeling, and BMPR-II signaling pathways (69). Studies have demonstrated that HMGB1 promotes the proliferation and migration of PASMCs by inhibiting BMPR-II signaling and enhancing the expression of the pro-inflammatory factor IL-6 (142), thereby exacerbating the progression of HPH. Therefore, HMGB1 may be a potential therapeutic target for the prevention of vascular remodeling and pathology in HPH.
4.2 Chemokines
Dysregulated signaling of chemokines and their ligands has recently been linked to HPH development. In an animal model of chronic hypoxic PH, increased CCL2 production by pulmonary vascular cells contributes to increased chemotactic activity of monocytes/macrophages, which in turn activates outer membrane fibroblasts (143), stimulates PASMCs migration and proliferation, and induces pathological vascular remodeling. CCR2, a homologous receptor for CCL2, is also elevated in the lung tissue of HPH (25). CCR2 deficiency mediates pulmonary vascular wall lesions, and in the presence of hypoxia, results in more severe pathological changes owing to the activation of Notch signaling (144). However, Florentin et al (145) demonstrated that there was decreased pulmonary inflammation and reduced pulmonary vascular remodeling in CCR2-deficient mice, despite having no effect on hemodynamics. Despite these seemingly contradictory data, it is clear that the CCL2/CCR2 axis is a key signaling mechanism implicated in the pathological phase of PAH.
Increased CCL5 expression has been detected in several animal models of PH, including CH- (146), Su/Hx-, and HIMF-treated PH mice (147). CCL5 deletion significantly attenuated the development of HPH, as CCL5 deletion restored the function of BMP-mediated PAECs and reversed pulmonary artery angiogenesis and vascular occlusion. CCL5 has an upregulated receptor called CCR5, which is abundant in HPH in both vascular cells and lung tissues. Interestingly, the crosstalk between CCR5 and CCR2 mediates the collaboration between macrophages and PASMC, promoting inflammatory cell infiltration and PASMC migration and proliferation during PAH development (35).
The Y chromosome is protective against the development of hypoxia-induced PH in mice undergoing gonadectomy, as expression of the Y chromosome gene Uty suppresses CXCL9 and CXCL10, thereby attenuating the triggering of endothelial apoptosis and PH (148), whereas suppression of CXCL9 and CXCL10 reverses disease progression in multiple experimental models of HPH (149).
Total lung tissue expression of CXCL12 and its receptor, CXC chemokine receptor 4 (CXCR4), is elevated in animals subjected to chronic hypoxia (150). Progenitor cells, which modulate HPH and vascular remodeling, are mobilized and recruited to the pulmonary vascular system in both of these (151). CXCR7 is another receptor for CXCL12, and the CXCR7 antagonist CCX771 attenuates PH in newborn mice exposed to prolonged hypoxia by decreasing pulmonary vasculature remodeling and increasing oxygen delivery (152). However, treatment with CCX771 did not protect mice from developing chronic hypoxia-induced PH (150), suggesting that the pulmonary vascular microenvironment has a significant impact on CXCR7 function in HPH. In addition, the HIF-2α/CXCL12 axis is important for regulating pulmonary vascular homeostasis and pulmonary fibrosis. MK-6482 therapy, a Hif-2α inhibitor, attenuates HPH by reducing CXCL12 (153) production.
The inflammatory chemokine CX3CL1/fractalkine converts and activates CX3CR1+ leukocytes, such as monocytes/macrophages, γδT lymphocytes, and DCs, and the infiltration and accumulation of these immune cells in the lungs contributes to structural disruption and remodeling of the pulmonary vasculature. CX3CL1/Fractalkine expression was significantly upregulated in pulmonary microvascular endothelial cells from high-altitude individuals, patients with obstructive lung disease, and mice (33). CX3CL1 promotes the proliferation of pericytes and smooth muscle cells, which in turn aggravates pulmonary small-vessel myelination, causing pulmonary hemodynamic changes and RV hypertrophy (154). In addition, CX3CL1 can worsen HPH by regulating monocyte recruitment and macrophage polarization (25).
Briefly, cytokines, chemokines, and their receptors are involved in the progression of HPH and right ventricular remodeling. In addition, some cytokines are associated with disease severity and prognosis. Targeting these mediators may be a promising therapeutic strategy for HPH.
5 HIF linking inflammation and metabolism
Metabolic reprogramming (157) refers to the process by which tumor cells regulate their anabolic and catabolic metabolism to obtain the energy and substances required to survive in an extreme microenvironment. This process primarily involves the regulation of metabolic pathways involving sugars, lipids, and amino acids.
The Warburg effect, which represents metabolic reprogramming, is present in rapidly proliferating cells, such as many types of immune cells, and determines the role of immune cells in inflammation (158). Metabolic dysregulation of pulmonary vascular cells and immune cells is closely related to vascular lesions and structural changes in the right ventricle (159) Notably, HIF is a key reprogramming factor for inflammatory cell metabolism, and stabilization of HIF signaling contributes to the metabolic phenotypic shift in immune cells observed in PH.
Depending on their environment, immune cells with a strong plurality can polarize into a variety of functionally distinct phenotypes, with different polarization directions corresponding to different energy metabolism patterns. To react appropriately to various immunological responses, particularly in hypoxic inflammatory tissues, these cells adjust their specific energy demands under the regulation of HIF. Changes in energy supply patterns and cellular signaling pathways resulting from metabolic reprogramming promote pulmonary vasoconstriction and structural remodeling from multiple perspectives. Metabolism is an important node in HPH pathogenesis.
Immunoinflammation and metabolic reprogramming are causally and mutually regulated, and HIF-1α may be a linking target of metabolic and inflammatory responses, with all three regulating the development of HPH (Figure 2).
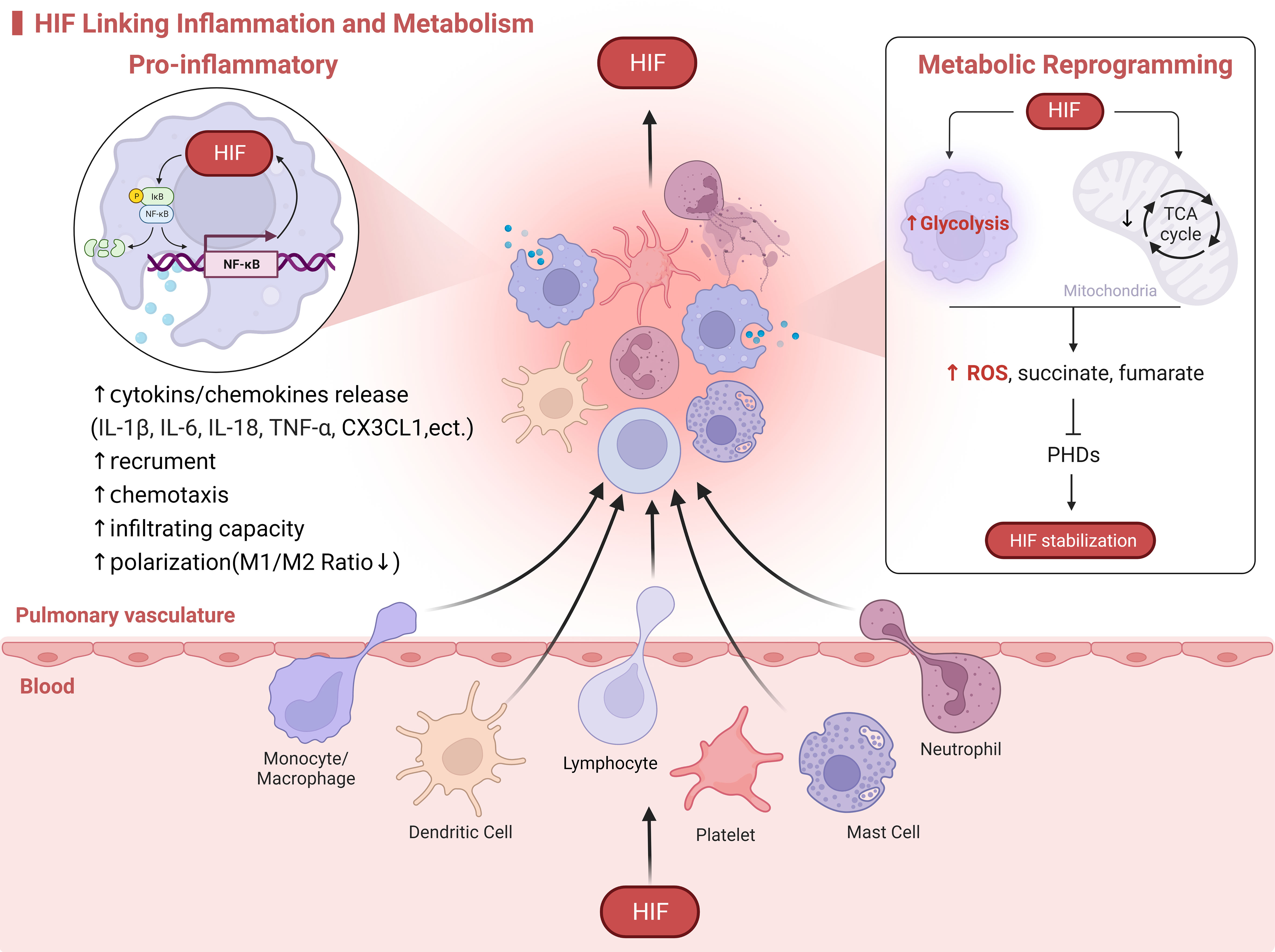
Figure 2 HIF Linking Inflammation and Metabolism. HIF serves as a molecular bridge in the immunoinflammatory and metabolism. On one hand, HIF promotes the activation of immune cells, metabolic reprogramming, and the release of inflammatory mediators. On the other hand, the exposure of immune cells to hypoxia from oxygen-deprived environments at sites of inflammation activates HIF. For example, in the case of macrophages infiltrating HPH, HIF drives the release of cytokines through crosstalk with the NF-κB signaling pathway. This enhances macrophage recruitment, chemotaxis, and polarization. Additionally, in response to hypoxia, mitochondrial oxidative phosphorylation is inhibited while glycolysis is enhanced. This leads to an increased production of ROS and metabolites such as succinate and fumarate by macrophages, which in turn activate and stabilize HIF.
6 Limitations of animal models
Hypoxia induces persistent vasoconstriction, pulmonary vascular remodeling, and development of PH during prolonged exposure. Hypoxic stress is a key driver of the pathogenesis of HPH. As a result, hypoxia is frequently utilized to induce PH in animal models and produce pathological features in cells (160). The small size, low cost, ease of maintenance, reproducibility, and consistency of rodent models mean that they are currently the primary source for understanding the molecular mechanisms of PH. The available HPH model construction methods include the chronic hypoxia model and the Su/Hx model.
Chronic hypoxia consistently induces PH in rodents (161), with both chronic sustained hypoxia (CSH) and chronic intermittent hypoxia (CIH) related to changes in PH and pulmonary vascular remodelling (162). Chronic hypoxia rarely causes muscular medial pulmonary artery hypertrophy and occlusive neointimal lesions and induces only mild-to-moderate PH (163). The PH and histological lesions are reversed when rodents return to normoxia. Therefore, the CSH model is well adapted to study less severe forms of PH, such as those associated with chronic lung disease and high altitude. Hence, it is a classical model for identifying cellular modifications that contribute to the pathogenesis of HPH. The degree of pulmonary vascular remodeling in response to hypoxia increases with the phylogenic order of the species (rat > mouse) (6). Compared to rats, pulmonary circulation in mice is relatively blunted in response to chronic persistent hypoxic stimuli, whereas PAP and pulmonary vascular remodeling induced by hypoxia are significantly lower (164). The microarray results suggest that this may be due to the fact that gene expression patterns are not identical between mouse and rat lung tissues (165).
CIH, which mimics the hypoxia-reoxygenation cycle characteristic of obstructive sleep apnea (166) has been found to cause PH in rodents (167, 168); however, the mechanisms underlying the differences between the effects of CIH in animal models and humans remain to be investigated.
The degree of hypoxia used in rodents is much more severe than that in humans, but a single hypoxic insult only causes minor vascular remodeling, which does not accurately mimic the severity of the disease in humans. Sugen 5416, an inhibitor of VEGFR2, combined with hypoxic stimulation, induces more severe PH pathological changes with an increased second hit, which contains inflammatory and vascular occlusive factors (169). Pathologically, SU5416 blunts VEGF receptors, leading to PAECs apoptosis (170). Disturbed by hypoxia, endothelial cell subpopulations and PASMCs proliferate further, and the combination of these actions leads to pathological vascular remodeling in the pulmonary circulation, elevated PA pressure, and right ventricular remodeling. Similar to PAH patients, Su/Hx rodent models have persistent and progressive PH (171), which differs from animals exposed to chronic hypoxia alone. Therefore, it may be more appropriate for the Su/Hx model to simulate a preclinical model of PAH rather than that of HPH.
In chronically hypoxic animals, several preclinical therapies were able to successfully prevent and reverse PH, but the majority of these treatments did not alleviate HPH. Therefore, the search for more robust animal models of HPH continues, and genetically engineered animals are promising tools for future pulmonary vascular studies.
7 Conclusion
In conclusion, the evidence points to the hypothesis that the pathophysiology of HPH is significantly influenced by immune response and inflammation. Histological analysis of HPH reveals how resident pulmonary vascular cells are affected by the extensive accumulation of immune cells and secretion of pro-inflammatory mediators. There is a causal connection between hypoxia and PH, and hypoxia interacts with the metabolic remodeling of immune cells and control of inflammation through the HIF signaling pathway. The establishment of a precise mechanism of action in pulmonary vascular pathology will open up a variety of novel targets for the intervention of HPH.
Author contributions
YY conceived and drafted the manuscript. QX drew a graphic abstract. TW revised and supervised the manuscript and provided the financial support. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Science and Technology Department of Qinghai Province (Grant No. 2021-ZJ-961Q) and the Qinghai Provincial Key Laboratory for Application of High-Altitude Medicine (Grant No. 2022-ZJ-Y15).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing and BioRender for the predrawn icons.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J (2019) 53(1):1801913. doi: 10.1183/13993003.01913-2018
2. McGettrick M, Peacock A. Group 3 pulmonary hypertension: challenges and opportunities. Glob Cardiol Sci Pract (2020) 2020:e202006. doi: 10.21542/gcsp.2020.6
3. Gupta T, Shariati F, Krim SR. A simplified diagnostic and therapeutic approach to pulmonary hypertension. Curr Probl Cardiol (2022) 47:100857. doi: 10.1016/j.cpcardiol.2021.100857
4. Prins KW, Duval S, Markowitz J, Pritzker M, Thenappan T. Chronic use of PAH-specific therapy in world health organization group III pulmonary hypertension: a systematic review and meta-analysis. Pulm Circ (2017) 7:145–55. doi: 10.1086/690017
5. Chen X, Tang S, Liu K, Li Q, Kong H, Zeng Xb, et al. Therapy in stable chronic obstructive pulmonary disease patients with pulmonary hypertension: a systematic review and meta-analysis. J Thorac Dis (2015) 7:309–19. doi: 10.3978/j.issn.2072-1439.2015.02.08
6. Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol (2013) 75:23–47. doi: 10.1146/annurev-physiol-030212-183802
7. Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med (2012) 186:261–72. doi: 10.1164/rccm.201201-0164OC
8. Pullamsetti SS, Mamazhakypov A, Weissmann N, Seeger W, Savai R. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest (2020) 130:5638–51. doi: 10.1172/JCI137558
9. Nicolls MR, Mizuno S, Taraseviciene-Stewart L, Farkas L, Drake JI, Al Husseini A, et al. New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm Circ (2012) 2:434–42. doi: 10.4103/2045-8932.105031
10. Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med (2014) 189:314–24. doi: 10.1164/rccm.201302-0302OC
11. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity (2014) 41:518–28. doi: 10.1016/j.immuni.2014.09.008
12. Canton M, Sánchez-Rodríguez R, Spera I, Venegas FC, Favia M, Viola A, et al. Reactive oxygen species in macrophages: sources and targets. Front Immunol (2021) 12:734229. doi: 10.3389/fimmu.2021.734229
13. Khawaja AA, Chong DLW, Sahota J, Mikolasch TA, Pericleous C, Ripoll VM, et al. Identification of a novel HIF-1α-α(M)β(2) integrin-NET axis in fibrotic interstitial lung disease. Front Immunol (2020) 11:2190. doi: 10.3389/fimmu.2020.02190
14. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med (2005) 201:105–15. doi: 10.1084/jem.20040624
15. Thompson AA, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood (2014) 123:366–76. doi: 10.1182/blood-2013-05-500207
16. D'Ignazio L, Batie M, Rocha S. Hypoxia and inflammation in cancer, focus on HIF and NF-κB. Biomedicines (2017) 5(2):21. doi: 10.3390/biomedicines5020021
17. Jiang Y, Zhu Y, Wang X, Gong J, Hu C, Guo B, et al. Temporal regulation of HIF-1 and NF-κB in hypoxic hepatocarcinoma cells. Oncotarget (2015) 6:9409–19. doi: 10.18632/oncotarget.3352
18. Jin F, Zheng X, Yang Y, Yao G, Ye L, Doeppner TR, et al. Impairment of hypoxia-induced angiogenesis by LDL involves a HIF-centered signaling network linking inflammatory TNFα and angiogenic VEGF. Aging (Albany NY) (2019) 11:328–49. doi: 10.18632/aging.101726
19. Luo Y, Teng X, Zhang L, Chen J, Liu Z, Chen X, et al. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat Commun (2019) 10:3551. doi: 10.1038/s41467-019-11500-6
20. Li L, Wei C, Kim IK, Janssen-Heininger Y, Gupta S. Inhibition of nuclear factor-κB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension (2014) 63:1260–9. doi: 10.1161/HYPERTENSIONAHA.114.03220
21. Voelkel NF, Mizuno S, Bogaard HJ. The role of hypoxia in pulmonary vascular diseases: a perspective. Am J Physiol Lung Cell Mol Physiol (2013) 304:L457–65. doi: 10.1152/ajplung.00335.2012
22. Florentin J, Dutta P. Origin and production of inflammatory perivascular macrophages in pulmonary hypertension. Cytokine (2017) 100:11–5. doi: 10.1016/j.cyto.2017.08.015
23. Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, et al. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation (2011) 123:1986–95. doi: 10.1161/CIRCULATIONAHA.110.978627
24. Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol (2006) 168:659–69. doi: 10.2353/ajpath.2006.050599
25. Amsellem V, Abid S, Poupel L, Parpaleix A, Rodero M, Gary-Bobo G, et al. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 chemokine systems in hypoxic pulmonary hypertension. Am J Respir Cell Mol Biol (2017) 56:597–608. doi: 10.1165/rcmb.2016-0201OC
26. Žaloudíková M, Vytášek R, Vajnerová O, Hniličková O, Vízek M, Hampl V, et al. Depletion of alveolar macrophages attenuates hypoxic pulmonary hypertension but not hypoxia-induced increase in serum concentration of MCP-1. Physiol Res (2016) 65:763–8. doi: 10.33549/physiolres.933187
27. West JD, Chen X, Ping L, Gladson S, Hamid R, Lloyd JE, et al. Adverse effects of BMPR2 suppression in macrophages in animal models of pulmonary hypertension. Pulm Circ (2019) 10:2045894019856483. doi: 10.1177/2045894019856483
28. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
29. Yang Y, Guo Z, Chen W, Wang X, Cao M, Han X, et al. M2 macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2F2. Mol Ther (2021) 29:1226–38. doi: 10.1016/j.ymthe.2020.11.024
30. Fan Y, Hao Y, Gao D, Li G, Zhang Z. Phenotype and function of macrophage polarization in monocrotaline-induced pulmonary arterial hypertension rat model. Physiol Res (2021) 70:213–26. doi: 10.33549/physiolres.934456
31. Zawia A, Arnold ND, West L, Pickworth JA, Turton H, Iremonger J, et al. Altered macrophage polarization induces experimental pulmonary hypertension and is observed in patients with pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol (2021) 41:430–45. doi: 10.1161/ATVBAHA.120.314639
32. Yang L, Guo P, Wang P, Wang W, Liu J. IL-6/ERK signaling pathway participates in type I IFN-programmed, unconventional M2-like macrophage polarization. Sci Rep (2023) 13:1827. doi: 10.1038/s41598-022-23721-9
33. Zhang J, Hu H, Palma NL, Harrison JK, Mubarak KK, Carrie RD, et al. Hypoxia-induced endothelial CX3CL1 triggers lung smooth muscle cell phenotypic switching and proliferative expansion. Am J Physiol Lung Cell Mol Physiol (2012) 303:L912–22. doi: 10.1152/ajplung.00014.2012
34. Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci USA (2015) 112:E2677–86. doi: 10.1073/pnas.1424774112
35. Abid S, Marcos E, Parpaleix A, Amsellem V, Breau M, Houssaini A, et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur Respir J (2019) 54(4):1802308. doi: 10.1183/13993003.02308-2018
36. Liu Y, Zhang H, Yan L, Du W, Zhang M, Chen H, et al. MMP-2 and MMP-9 contribute to the angiogenic effect produced by hypoxia/15-HETE in pulmonary endothelial cells. J Mol Cell Cardiol (2018) 121:36–50. doi: 10.1016/j.yjmcc.2018.06.006
37. Ntokou A, Dave JM, Kauffman AC, Sauler M, Ryu C, Hwa J, et al. Macrophage-derived PDGF-b induces muscularization in murine and human pulmonary hypertension. JCI Insight (2021) 6(6):e139067. doi: 10.1172/jci.insight.139067
38. Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, et al. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med (2013) 5:200ra117. doi: 10.1126/scitranslmed.3006674
39. Tucker A, McMurtry IF, Alexander AF, Reeves JT, Grover RF. Lung mast cell density and distribution in chronically hypoxic animals. J Appl Physiol Respir Environ Exerc Physiol (1977) 42:174–8. doi: 10.1152/jappl.1977.42.2.174
40. Mungall IP. Hypoxia and lung mast cells: influence of disodium cromoglycate. Thorax (1976) 31:94–100. doi: 10.1136/thx.31.1.94
41. Chao J, Wood JG, Gonzalez NC. Alveolar hypoxia, alveolar macrophages, and systemic inflammation. Respir Res (2009) 10:54. doi: 10.1186/1465-9921-10-54
42. Gulliksson M, Carvalho RF, Ullerås E, Nilsson G. Mast cell survival and mediator secretion in response to hypoxia. PLoS One (2010) 5:e12360. doi: 10.1371/journal.pone.0012360
43. Wang X, Lin L, Chai X, Wu Y, Li Y and Liu X. Hypoxic mast cells accelerate the proliferation, collagen accumulation and phenotypic alteration of human lung fibroblasts. Int J Mol Med (2020) 45:175–85. doi: 10.3892/ijmm.2019.4400
44. Banasová A, Maxová H, Hampl V, Vízek M, Povýsilová V, Novotná J, et al. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration (2008) 76:102–7. doi: 10.1159/000121410
45. Galli SJ, Grimbaldeston M, Tsai M. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat Rev Immunol (2008) 8:478–86. doi: 10.1038/nri2327
46. Maxová H, Vasilková M, Novotná J, Vajnerová O, Bansová A, Vízek M, et al. Prevention of mast cell degranulation by disodium cromoglycate delayed the regression of hypoxic pulmonary hypertension in rats. Respiration (2010) 80:335–9. doi: 10.1159/000312403
47. Lodge KM, Cowburn AS, Li W, Condliffe AM. The impact of hypoxia on neutrophil degranulation and consequences for the host. Int J Mol Sci (2020) 21(4):1183. doi: 10.3390/ijms21041183
48. Dölling M, Eckstein M, Singh J, Schauer C, Schoen J, Shan X, et al. Hypoxia promotes neutrophil survival after acute myocardial infarction. Front Immunol (2022) 13:726153. doi: 10.3389/fimmu.2022.726153
49. Liang S, Desai AA, Black SM, Tang H. Cytokines, chemokines, and inflammation in pulmonary arterial hypertension. Adv Exp Med Biol (2021) 1303:275–303. doi: 10.1007/978-3-030-63046-1_15
50. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation (2010) 122:920–7. doi: 10.1161/CIRCULATIONAHA.109.933762
51. Buonacera A, Stancanelli B, Colaci M, Malatino L. Neutrophil to lymphocyte ratio: an emerging marker of the relationships between the immune system and diseases. Int J Mol Sci (2022) 23(7):3636. doi: 10.3390/ijms23073636
52. Mirsaeidi M, Mortaz E, Omar HR, Camporesi EM, Sweiss N. Association of neutrophil to lymphocyte ratio and pulmonary hypertension in sarcoidosis patients. Tanaffos (2016) 15:44–7.
53. Yanartas M, Kalkan ME, Arslan A, Tas SG, Koksal C, Bekiroglu N, et al. Neutrophil/Lymphocyte ratio can predict postoperative mortality in patients with chronic thromboembolic pulmonary hypertension. Ann Thorac Cardiovasc Surg (2015) 21:229–35. doi: 10.5761/atcs.oa.14-00190
54. Yogeswaran A, Tello K, Lund J, Klose H, Harbaum L, Sommer N, et al. Risk assessment in pulmonary hypertension based on routinely measured laboratory parameters. J Heart Lung Transpl (2022) 41:400–10. doi: 10.1016/j.healun.2021.10.018
55. Kimishima Y, Misaka T, Yokokawa T, Wada K, Ueda K, Sugimoto K, et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nat Commun (2021) 12:6177. doi: 10.1038/s41467-021-26435-0
56. Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, et al. Elafin reverses pulmonary hypertension via caveolin-1-Dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med (2015) 191:1273–86. doi: 10.1164/rccm.201412-2291OC
57. Klinke A, Berghausen E, Friedrichs K, Molz S, Lau D, Remane L, et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular rho-kinase. JCI Insight (2018) 3(11):e97530. doi: 10.1172/jci.insight.97530
58. Cero FT, Hillestad V, Sjaastad I, Yndestad A, Aukrust P, Ranheim T, et al. Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol (2015) 309:L378–87. doi: 10.1152/ajplung.00342.2014
59. Voelkel NF, Tuder RM, Bridges J, Arend WP. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol (1994) 11:664–75. doi: 10.1165/ajrcmb.11.6.7946395
60. Itoh A, Nishihira J, Makita H, Miyamoto K, Yamaguchi E, Nishimura M. Effects of IL-1beta, TNF-alpha, and macrophage migration inhibitory factor on prostacyclin synthesis in rat pulmonary artery smooth muscle cells. Respirology (2003) 8:467–72. doi: 10.1046/j.1440-1843.2003.00491.x
61. Yang PS, Kim DH, Lee YJ, Lee SE, Kang WJ, Chang HJ, et al. Glycyrrhizin, inhibitor of high mobility group box-1, attenuates monocrotaline-induced pulmonary hypertension and vascular remodeling in rats. Respir Res (2014) 15:148. doi: 10.1186/s12931-014-0148-4
62. Sharma S, Hofbauer TM, Ondracek AS, Chausheva S, Alimohammadi A, Artner T, et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood (2021) 137:1104–16. doi: 10.1182/blood.2020005861
63. Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol (2013) 3:1011–34. doi: 10.1002/cphy.c120024
64. Rose F, Hattar K, Gakisch S, Grimminger F, Olschewski H, Seeger W, et al. Increased neutrophil mediator release in patients with pulmonary hypertension–suppression by inhaled iloprost. Thromb Haemost (2003) 90:1141–9. doi: 10.1160/TH03-03-0173
65. Florentin J, Zhao J, Tai YY, Vasamsetti SB, O'Neil SP, Kumar R, et al. Interleukin-6 mediates neutrophil mobilization from bone marrow in pulmonary hypertension. Cell Mol Immunol (2021) 18:374–84. doi: 10.1038/s41423-020-00608-1
66. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res (2018) 122:337–51. doi: 10.1161/CIRCRESAHA.117.310795
67. Bauer EM, Chanthaphavong RS, Sodhi CP, Hackam DJ, Billiar TR, Bauer PM. Genetic deletion of toll-like receptor 4 on platelets attenuates experimental pulmonary hypertension. Circ Res (2014) 114:1596–600. doi: 10.1161/CIRCRESAHA.114.303662
68. Bauer EM, Shapiro R, Zheng H, Ahmad F, Ishizawar D, Comhair SA, et al. High mobility group box 1 contributes to the pathogenesis of experimental pulmonary hypertension via activation of toll-like receptor 4. Mol Med (2013) 18:1509–18. doi: 10.2119/molmed.2012.00283
69. Wang J, Tian XT, Peng Z, Li WQ, Cao YY, Li Y, et al. HMGB1/TLR4 promotes hypoxic pulmonary hypertension via suppressing BMPR2 signaling. Vascul Pharmacol (2019) 117:35–44. doi: 10.1016/j.vph.2018.12.006
70. Lin Q, Fan C, Gomez-Arroyo J, Van Raemdonck K, Meuchel LW, Skinner JT, et al. HIMF (Hypoxia-induced mitogenic factor) signaling mediates the HMGB1 (High mobility group box 1)-dependent endothelial and smooth muscle cell crosstalk in pulmonary hypertension. Arterioscler Thromb Vasc Biol (2019) 39:2505–19. doi: 10.1161/ATVBAHA.119.312907
71. Zheng YG, Yang T, Xiong CM, He JG, Liu ZH, Gu Q, et al. Platelet distribution width and mean platelet volume in idiopathic pulmonary arterial hypertension. Heart Lung Circ (2015) 24:566–72. doi: 10.1016/j.hlc.2014.11.025
72. Wang L, Shen L, Zhao YL, Pudasaini B, Zhao QH, Gong SG, et al. Survival in severe pulmonary hypertension due to chronic lung disease: influence of in-hospital platelet distribution width. Pulm Circ (2021) 11:20458940211026484. doi: 10.1177/20458940211026484
73. Song YJ, Kwon JH, Kim JY, Kim BY, Cho KI. The platelet-to-lymphocyte ratio reflects the severity of obstructive sleep apnea syndrome and concurrent hypertension. Clin Hypertens (2015) 22:1. doi: 10.1186/s40885-015-0036-3
74. Zhu Y, Shu D, Gong X, Lu M, Feng Q, Zeng XB, et al. Platelet-derived TGF (Transforming growth factor)-β1 enhances the aerobic glycolysis of pulmonary arterial smooth muscle cells by PKM2 (Pyruvate kinase muscle isoform 2) upregulation. Hypertension (2022) 79:932–45. doi: 10.1161/HYPERTENSIONAHA.121.18684
75. Wang J, Yang K, Yuan JX. NEDD9, a hypoxia-upregulated mediator for pathogenic platelet-endothelial cell interaction in pulmonary hypertension. Am J Respir Crit Care Med (2021) 203:1455–8. doi: 10.1164/rccm.202101-0007ED
76. Alba GA, Samokhin AO, Wang RS, Zhang YY, Wertheim BM, Arons E, et al. NEDD9 is a novel and modifiable mediator of platelet-endothelial adhesion in the pulmonary circulation. Am J Respir Crit Care Med (2021) 203:1533–45. doi: 10.1164/rccm.202003-0719OC
77. Rossaint J, Kühne K, Skupski J, Van Aken H, Looney MR, Hidalgo A, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun (2016) 7:13464. doi: 10.1038/ncomms13464
78. Jia D, He Y, Zhu Q, Liu H, Zuo C, Chen G, et al. RAGE-mediated extracellular matrix proteins accumulation exacerbates HySu-induced pulmonary hypertension. Cardiovasc Res (2017) 113:586–97. doi: 10.1093/cvr/cvx051
79. Delaney C, Davizon-Castillo P, Allawzi A, Posey J, Gandjeva A, Neeves K, et al. Platelet activation contributes to hypoxia-induced inflammation. Am J Physiol Lung Cell Mol Physiol (2021) 320:L413–l421. doi: 10.1152/ajplung.00519.2020
80. Baptista de Barros Ribeiro Dourado LP, Santos M, Moreira-Gonçalves D. Nets, pulmonary arterial hypertension, and thrombo-inflammation. J Mol Med (Berl) (2022) 100:713–22. doi: 10.1007/s00109-022-02197-0
81. Wang Z, Tenzing N, Xu Q, Liu H, Ye Y, Wen Y, et al. Apoptosis is one cause of thrombocytopenia in patients with high-altitude polycythemia. Platelets (2023) 34:2157381. doi: 10.1080/09537104.2022.2157381
82. Maston LD, Jones DT, Giermakowska W, Howard TA, Cannon JL, Wang W, et al. Resta TC and Gonzalez bosc LV. central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol (2017) 312:L609–l624. doi: 10.1152/ajplung.00531.2016
83. Wang L, Liu J, Wang W, Qi X, Wang Y, Tian B, et al. Targeting IL-17 attenuates hypoxia-induced pulmonary hypertension through downregulation of β-catenin. Thorax (2019) 74:564–78. doi: 10.1136/thoraxjnl-2018-211846
84. Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med (2008) 205:799–810. doi: 10.1084/jem.20071258
85. Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res (2011) 109:867–79. doi: 10.1161/CIRCRESAHA.110.236927
86. Chu Y, Xiangli X, Xiao W. Regulatory T cells protect against hypoxia-induced pulmonary arterial hypertension in mice. Mol Med Rep (2015) 11:3181–7. doi: 10.3892/mmr.2014.3106
87. Nakahara M, Ito H, Skinner JT, Lin Q, Tamosiuniene R, Nicolls MR, et al. Johns RA and yamaji-kegan k. the inflammatory role of dysregulated IRS2 in pulmonary vascular remodeling under hypoxic conditions. Am J Physiol Lung Cell Mol Physiol (2021) 321:L416–l428. doi: 10.1152/ajplung.00068.2020
88. Qian J, Tian W, Jiang X, Tamosiuniene R, Sung YK, Shuffle EM, et al. Leukotriene B4 activates pulmonary artery adventitial fibroblasts in pulmonary hypertension. Hypertension (2015) 66:1227–39. doi: 10.1161/HYPERTENSIONAHA.115.06370
89. Zhu R, Xie X, Wang N, Chen L, Hong Y. The T helper type 17/regulatory T cell imbalance was associated with ras-GTPase overexpression in patients with pulmonary hypertension associated with chronic obstructive pulmonary disease. Immunology (2019) 157:304–11. doi: 10.1111/imm.13084
90. Edwards AL, Gunningham SP, Clare GC, Hayman MW, Smith M, Frampton CM, et al. Professional killer cell deficiencies and decreased survival in pulmonary arterial hypertension. Respirology (2013) 18:1271–7. doi: 10.1111/resp.12152
91. Perros F, Cohen-Kaminsky S, Gambaryan N, Girerd B, Raymond N, Klingelschmitt I, et al. Cytotoxic cells and granulysin in pulmonary arterial hypertension and pulmonary veno-occlusive disease. Am J Respir Crit Care Med (2013) 187:189–96. doi: 10.1164/rccm.201208-1364OC
92. Colvin KL, Cripe PJ, Ivy DD, Stenmark KR, Yeager ME. Bronchus-associated lymphoid tissue in pulmonary hypertension produces pathologic autoantibodies. Am J Respir Crit Care Med (2013) 188:1126–36. doi: 10.1164/rccm.201302-0403OC
93. Rosser EC, Mauri C. Regulatory b cells: origin, phenotype, and function. Immunity (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
94. Li C, Liu P, Yao H, Zhu H, Zhang S, Meng F, et al. Regulatory b cells protect against chronic hypoxia-induced pulmonary hypertension by modulating the Tfh/Tfr immune balance. Immunology (2023) 168(4):580–96. doi: 10.1111/imm.13589
95. Perros F, Dorfmüller P, Souza R, Durand-Gasselin I, Mussot S, Mazmanian M, et al. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur Respir J (2007) 29:462–8. doi: 10.1183/09031936.00094706
96. Koudstaal T, van Hulst JAC, Das T, Neys SFH, Merkus D, Bergen IM, et al. DNGR1-cre-mediated deletion of Tnfaip3/A20 in conventional dendritic cells induces pulmonary hypertension in mice. Am J Respir Cell Mol Biol (2020) 63:665–80. doi: 10.1165/rcmb.2019-0443OC
97. Wang W, Yan H, Zhu W, Cui Y, Chen J, Wang X, et al. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J Clin Immunol (2009) 29:705–13. doi: 10.1007/s10875-009-9322-8
98. Marsh LM, Jandl K, Grünig G, Foris V, Bashir M, Ghanim B, et al. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur Respir J (2018) 51(1):1701214. doi: 10.1183/13993003.01214-2017
99. Girard P, Ponsard B, Charles J, Chaperot L, Aspord C. Potent bidirectional cross-talk between plasmacytoid dendritic cells and γδT cells through BTN3A, type I/II IFNs and immune checkpoints. Front Immunol (2020) 11:861. doi: 10.3389/fimmu.2020.00861
100. Parpaleix A, Amsellem V, Houssaini A, Abid S, Breau M, Marcos E, et al. Role of interleukin-1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur Respir J (2016) 48:470–83. doi: 10.1183/13993003.01448-2015
101. Udjus C, Cero FT, Halvorsen B, Behmen D, Carlson CR, Bendiksen BA, et al. Caspase-1 induces smooth muscle cell growth in hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol (2019) 316:L999–l1012. doi: 10.1152/ajplung.00322.2018
102. Hu S, Wang L, Xu Y, Li F, Wang T. Disulfiram attenuates hypoxia-induced pulmonary hypertension by inhibiting GSDMD cleavage and pyroptosis in HPASMCs. Respir Res (2022) 23:353. doi: 10.1186/s12931-022-02279-0
103. Rong W, Liu C, Li X, Wan N, Wei L, Zhu W, et al. Caspase-8 promotes pulmonary hypertension by activating macrophage-associated inflammation and IL-1β (Interleukin 1β) production. Arterioscler Thromb Vasc Biol (2022) 42:613–31. doi: 10.1161/ATVBAHA.121.317168
104. Morisawa D, Hirotani S, Oboshi M, Nishimura K, Sawada H, Eguchi A, et al. Interleukin-18 disruption suppresses hypoxia-induced pulmonary artery hypertension in mice. Int J Cardiol (2016) 202:522–4. doi: 10.1016/j.ijcard.2015.09.118
105. Eddahibi S, Chaouat A, Tu L, Chouaid C, Weitzenblum E, Housset B, et al. Interleukin-6 gene polymorphism confers susceptibility to pulmonary hypertension in chronic obstructive pulmonary disease. Proc Am Thorac Soc (2006) 3:475–6. doi: 10.1513/pats.200603-038MS
106. Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, et al. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res (2009) 10:6. doi: 10.1186/1465-9921-10-6
107. Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest (2018) 128:1956–70. doi: 10.1172/JCI96462
108. Golembeski SM, West J, Tada Y, Fagan KA. Interleukin-6 causes mild pulmonary hypertension and augments hypoxia-induced pulmonary hypertension in mice. Chest (2005) 128:572s–3s. doi: 10.1378/chest.128.6_suppl.572S-a
109. Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci (2012) 8:1237–47. doi: 10.7150/ijbs.4989
110. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res (2009) 104:236–44. doi: 10.1161/CIRCRESAHA.108.182014
111. Takahashi H, Goto N, Kojima Y, Tsuda Y, Morio Y, Muramatsu M, et al. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol (2006) 290:L450–8. doi: 10.1152/ajplung.00206.2005
112. Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, et al. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res (2009) 104:1184–91. doi: 10.1161/CIRCRESAHA.109.197491
113. Maston LD, Jones DT, Giermakowska W, Resta TC, Ramiro-Diaz J, Howard TA, et al. Interleukin-6 trans-signaling contributes to chronic hypoxia-induced pulmonary hypertension. Pulm Circ (2018) 8:2045894018780734. doi: 10.1177/2045894018780734
114. Yang D, Wang L, Jiang P, Kang R, Xie Y. Correlation between hs-CRP, IL-6, IL-10, ET-1, and chronic obstructive pulmonary disease combined with pulmonary hypertension. J Healthc Eng (2022) 2022:3247807. doi: 10.1155/2022/3247807
115. Ito T, Okada T, Miyashita H, Nomoto T, Nonaka-Sarukawa M, Uchibori R, et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res (2007) 101:734–41. doi: 10.1161/CIRCRESAHA.107.153023
116. Li C, Liu PP, Tang DD, Song R, Zhang YQ, Lei S and Wu SJ. Targeting the RhoA-ROCK pathway to regulate T-cell homeostasis in hypoxia-induced pulmonary arterial hypertension. Pulm Pharmacol Ther (2018) 50:111–22. doi: 10.1016/j.pupt.2018.04.004
117. Liu J, Deng Y, Fan Z, Xu S, Wei L, Huang X, et al. Construction and analysis of the abnormal lncRNA-miRNA-mRNA network in hypoxic pulmonary hypertension. Biosci Rep (2021) 41(8):BSR20210021. doi: 10.1042/BSR20210021
118. Liu J, Wang W, Wang L, Chen S, Tian B, Huang K, et al. IL-33 initiates vascular remodelling in hypoxic pulmonary hypertension by up-regulating HIF-1α and VEGF expression in vascular endothelial cells. EBioMedicine (2018) 33:196–210. doi: 10.1016/j.ebiom.2018.06.003
119. Hurst LA, Dunmore BJ, Long L, Crosby A, Al-Lamki R, Deighton J, et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat Commun (2017) 8:14079. doi: 10.1038/ncomms14079
120. Sutendra G, Dromparis P, Bonnet S, Haromy A, McMurtry MS, Bleackley RC, et al. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med (Berl) (2011) 89:771–83. doi: 10.1007/s00109-011-0762-2
121. Wang Q, Zuo XR, Wang YY, Xie WP, Wang H, Zhang M. Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-α antagonists via the suppression of TNF-α expression and NF-κB pathway in rats. Vascul Pharmacol (2013) 58:71–7. doi: 10.1016/j.vph.2012.07.006
122. Dawson SH, Arnold ND, Pickworth JA, Francis SE, Lawrie A. TRAIL deficient mice are protected from Sugen/Hypoxia induced pulmonary arterial hypertension. Diseases (2014) 2:260–73. doi: 10.3390/diseases2030260
123. Hameed AG, Arnold ND, Chamberlain J, Pickworth JA, Paiva C, Dawson S, et al. Inhibition of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) reverses experimental pulmonary hypertension. J Exp Med (2012) 209:1919–35. doi: 10.1084/jem.20112716
124. Song S, Choi K, Ryu SW, Kang SW, Choi C. TRAIL promotes caspase-dependent pro-inflammatory responses via PKCδ activation by vascular smooth muscle cells. Cell Death Dis (2011) 2:e223. doi: 10.1038/cddis.2011.103
125. Kumar R, Mickael C, Kassa B, Gebreab L, Robinson JC, Koyanagi DE, et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes schistosoma- and hypoxia-induced pulmonary hypertension. Nat Commun (2017) 8:15494. doi: 10.1038/ncomms15494
126. Gong K, Xing D, Li P, Aksut B, Ambalavanan N, Yang Q, et al. Hypoxia induces downregulation of PPAR-γ in isolated pulmonary arterial smooth muscle cells and in rat lung via transforming growth factor-β signaling. Am J Physiol Lung Cell Mol Physiol (2011) 301:L899–907. doi: 10.1152/ajplung.00062.2011
127. Tielemans B, Delcroix M, Belge C, Quarck R. TGFβ and BMPRII signalling pathways in the pathogenesis of pulmonary arterial hypertension. Drug Discov Today (2019) 24:703–16. doi: 10.1016/j.drudis.2018.12.001
128. Zhang N, Dong M, Luo Y, Zhao F, Li Y. Danshensu prevents hypoxic pulmonary hypertension in rats by inhibiting the proliferation of pulmonary artery smooth muscle cells via TGF-β-smad3-associated pathway. Eur J Pharmacol (2018) 820:1–7. doi: 10.1016/j.ejphar.2017.12.010
129. Ambalavanan N, Nicola T, Hagood J, Bulger A, Serra R, Murphy-Ullrich J, et al. Transforming growth factor-beta signaling mediates hypoxia-induced pulmonary arterial remodeling and inhibition of alveolar development in newborn mouse lung. Am J Physiol Lung Cell Mol Physiol (2008) 295:L86–95. doi: 10.1152/ajplung.00534.2007
130. Mammoto T, Muyleart M, Konduri GG, Mammoto A. Twist1 in hypoxia-induced pulmonary hypertension through transforming growth factor-β-Smad signaling. Am J Respir Cell Mol Biol (2018) 58:194–207. doi: 10.1165/rcmb.2016-0323OC
131. Yung LM, Nikolic I, Paskin-Flerlage SD, Pearsall RS, Kumar R, Yu PB. A selective transforming growth factor-β ligand trap attenuates pulmonary hypertension. Am J Respir Crit Care Med (2016) 194:1140–51. doi: 10.1164/rccm.201510-1955OC
132. Zhang B, Shen M, Xu M, Liu LL, Luo Y, Xu DQ, et al. Role of macrophage migration inhibitory factor in the proliferation of smooth muscle cell in pulmonary hypertension. Mediators Inflamm (2012) 2012:840737. doi: 10.1155/2012/840737
133. Zhang B, Luo Y, Liu ML, Wang J, Xu DQ, Dong MQ, et al. Macrophage migration inhibitory factor contributes to hypoxic pulmonary vasoconstriction in rats. Microvasc Res (2012) 83:205–12. doi: 10.1016/j.mvr.2011.09.014
134. Le Hiress M, Tu L, Ricard N, Phan C, Thuillet R, Fadel E, et al. Proinflammatory signature of the dysfunctional endothelium in pulmonary hypertension. role of the macrophage migration inhibitory Factor/CD74 complex. Am J Respir Crit Care Med (2015) 192:983–97. doi: 10.1164/rccm.201402-0322OC
135. Zhang Y, Talwar A, Tsang D, Bruchfeld A, Sadoughi A, Hu M, et al. Macrophage migration inhibitory factor mediates hypoxia-induced pulmonary hypertension. Mol Med (2012) 18:215–23. doi: 10.2119/molmed.2011.00094
136. Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Skinner JT, Champion HC, et al. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol (2009) 296:L582–93. doi: 10.1152/ajplung.90526.2008
137. Johns RA, Takimoto E, Meuchel LW, Elsaigh E, Zhang A, Heller NM, et al. Hypoxia-inducible factor 1α is a critical downstream mediator for hypoxia-induced mitogenic factor (FIZZ1/RELMα)-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol (2016) 36:134–44. doi: 10.1161/ATVBAHA.115.306710
138. Tian H, Liu L, Wu Y, Wang R, Jiang Y, Hu R, et al. Resistin-like molecule β acts as a mitogenic factor in hypoxic pulmonary hypertension via the Ca(2+)-dependent PI3K/Akt/mTOR and PKC/MAPK signaling pathways. Respir Res (2021) 22:8. doi: 10.1186/s12931-020-01598-4
139. Li J, Zhang J, Xie F, Peng J, Wu X. Macrophage migration inhibitory factor promotes warburg effect via activation of the NF−κB/HIF−1α pathway in lung cancer. Int J Mol Med (2018) 41:1062–8. doi: 10.3892/ijmm.2017.3277.
140. Teng X, Li D, Champion HC, Johns RA. FIZZ1/RELMalpha, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circ Res (2003) 92:1065–7. doi: 10.1161/01.RES.0000073999.07698.33
141. Liu C, Sun H, Tang M, Li J, Zhang X, Cao G. Ethyl pyruvate alleviates pulmonary hypertension through the suppression of pulmonary artery smooth muscle cell proliferation via the high mobility group protein B1/Receptor for advanced glycation end-products axis. Ann Thorac Cardiovasc Surg (2021) 27:380–8. doi: 10.5761/atcs.oa.21-00027
142. Li WJ, Hu K, Yang JP, Xu XY, Li N, Wen ZP, et al. HMGB1 affects the development of pulmonary arterial hypertension via RAGE. Eur Rev Med Pharmacol Sci (2017) 21:3950–8.
143. Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol (2011) 187:2711–22. doi: 10.4049/jimmunol.1100479
144. Yu YR, Mao L, Piantadosi CA, Gunn MD. CCR2 deficiency, dysregulation of notch signaling, and spontaneous pulmonary arterial hypertension. Am J Respir Cell Mol Biol (2013) 48:647–54. doi: 10.1165/rcmb.2012-0182OC
145. Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai YY, Tang Y, et al. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J Immunol (2018) 200:3612–25. doi: 10.4049/jimmunol.1701287
146. Amsellem V, Lipskaia L, Abid S, Poupel L, Houssaini A, Quarck R, et al. CCR5 as a treatment target in pulmonary arterial hypertension. Circulation (2014) 130:880–91. doi: 10.1161/CIRCULATIONAHA.114.010757
147. Nie X, Tan J, Dai Y, Liu Y, Zou J, Sun J, et al. CCL5 deficiency rescues pulmonary vascular dysfunction, and reverses pulmonary hypertension via caveolin-1-dependent BMPR2 activation. J Mol Cell Cardiol (2018) 116:41–56. doi: 10.1016/j.yjmcc.2018.01.016
148. Umar S, Cunningham CM, Itoh Y, Moazeni S, Vaillancourt M, Sarji S, et al. The y chromosome plays a protective role in experimental hypoxic pulmonary hypertension. Am J Respir Crit Care Med (2018) 197:952–5. doi: 10.1164/rccm.201707-1345LE
149. Cunningham CM, Li M, Ruffenach G, Doshi M, Aryan L, Hong J, et al. Y-chromosome gene, uty, protects against pulmonary hypertension by reducing proinflammatory chemokines. Am J Respir Crit Care Med (2022) 206:186–96. doi: 10.1164/rccm.202110-2309OC
150. Gambaryan N, Perros F, Montani D, Cohen-Kaminsky S, Mazmanian M, Renaud JF, et al. Targeting of c-kit+ haematopoietic progenitor cells prevents hypoxic pulmonary hypertension. Eur Respir J (2011) 37:1392–9. doi: 10.1183/09031936.00045710
151. Wei L, Zhang B, Cao W, Xing H, Yu X, Zhu D. Inhibition of CXCL12/CXCR4 suppresses pulmonary arterial smooth muscle cell proliferation and cell cycle progression via PI3K/Akt pathway under hypoxia. J Recept Signal Transduct Res (2015) 35:329–39. doi: 10.3109/10799893.2014.984308
152. Sartina E, Suguihara C, Ramchandran S, Nwajei P, Rodriguez M, Torres E, et al. Antagonism of CXCR7 attenuates chronic hypoxia-induced pulmonary hypertension. Pediatr Res (2012) 71:682–8. doi: 10.1038/pr.2012.30
153. Ghosh MC, Zhang DL, Ollivierre WH, Noguchi A, Springer DA, Linehan WM, et al. Therapeutic inhibition of HIF-2α reverses polycythemia and pulmonary hypertension in murine models of human diseases. Blood (2021) 137:2509–19. doi: 10.1182/blood.2020009138
154. Xiang Y, Zheng F, Zhang Q, Zhang R, Pan H, Pang Z, et al. Tanreqing injection regulates cell function of hypoxia-induced human pulmonary artery smooth muscle cells (HPASMCs) through TRPC1/CX3CL1 signaling pathway. Oxid Med Cell Longev (2022) 2022:3235102. doi: 10.1155/2022/3235102
155. Cooper AL, Beasley D. Hypoxia stimulates proliferation and interleukin-1alpha production in human vascular smooth muscle cells. Am J Physiol (1999) 277:H1326–37. doi: 10.1152/ajpheart.1999.277.4.H1326
156. Joppa P, Petrasova D, Stancak Bb, Tkacova R. Systemic inflammation in patients with COPD and pulmonary hypertension. Chest (2006) 130:326–33. doi: 10.1378/chest.130.2.326
157. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
158. Kornberg MD. The immunologic warburg effect: evidence and therapeutic opportunities in autoimmunity. Wiley Interdiscip Rev Syst Biol Med (2020) 12:e1486. doi: 10.1002/wsbm.1486
159. Chan SY, Rubin LJ. Metabolic dysfunction in pulmonary hypertension: from basic science to clinical practice. Eur Respir Rev (2017) 26(146):170094. doi: 10.1183/16000617.0094-2017
160. Young JM, Williams DR, Thompson AAR. Thin air, thick vessels: historical and current perspectives on hypoxic pulmonary hypertension. Front Med (Lausanne) (2019) 6:93. doi: 10.3389/fmed.2019.00093
161. Hoffmann F, Limper U, Zaha VG, Reuter H, Zange L, Schulz-Menger J, et al. Evolution of pulmonary hypertension during severe sustained hypoxia. Circulation (2020) 141:1504–6. doi: 10.1161/CIRCULATIONAHA.119.045192
162. Barnes LA, Mesarwi OA, Sanchez-Azofra A. The cardiovascular and metabolic effects of chronic hypoxia in animal models: a mini-review. Front Physiol (2022) 13:873522. doi: 10.3389/fphys.2022.873522
163. Porteous MK, Fritz JS. Hypoxemia in a patient with pulmonary arterial hypertension: getting to the heart of the matter. Ann Am Thorac Soc (2014) 11:836–40. doi: 10.1513/AnnalsATS.201401-006CC
164. Sztuka K, Jasińska-Stroschein M. Animal models of pulmonary arterial hypertension: a systematic review and meta-analysis of data from 6126 animals. Pharmacol Res (2017) 125:201–14. doi: 10.1016/j.phrs.2017.08.003
165. Hoshikawa Y, Nana-Sinkam P, Moore MD, Sotto-Santiago S, Phang T, Keith RL, et al. Hypoxia induces different genes in the lungs of rats compared with mice. Physiol Genomics (2003) 12:209–19. doi: 10.1152/physiolgenomics.00081.2001
166. Sforza E, Roche F. Chronic intermittent hypoxia and obstructive sleep apnea: an experimental and clinical approach. Hypoxia (Auckl) (2016) 4:99–108. doi: 10.2147/HP.S103091
167. Prieto-Lloret J, Olea E, Gordillo-Cano A, Docio I, Obeso A, Gomez-Niño A, et al. Maladaptive pulmonary vascular responses to chronic sustained and chronic intermittent hypoxia in rat. Antioxidants (Basel) (2021) 11(1):54. doi: 10.3390/antiox11010054
168. Nisbet RE, Graves AS, Kleinhenz DJ, Rupnow HL, Reed AL, Fan TH, et al. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol (2009) 40:601–9. doi: 10.1165/2008-0145OC
169. Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J (2001) 15:427–38. doi: 10.1096/fj.00-0343com
170. Sakao S, Tatsumi K. The effects of antiangiogenic compound SU5416 in a rat model of pulmonary arterial hypertension. Respiration (2011) 81:253–61. doi: 10.1159/000322011
Keywords: hypoxia, pulmonary hypertension, inflammation, immunity, metabolism, HIF
Citation: Ye Y, Xu Q and Wuren T (2023) Inflammation and immunity in the pathogenesis of hypoxic pulmonary hypertension. Front. Immunol. 14:1162556. doi: 10.3389/fimmu.2023.1162556
Received: 10 February 2023; Accepted: 25 April 2023;
Published: 05 May 2023.
Edited by:
Haiyang Tang, University of Arizona, United StatesCopyright © 2023 Ye, Xu and Wuren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tana Wuren, dGFubmEubWdsQGdtYWlsLmNvbQ==