 Yanling Zhao
Yanling Zhao Shizhang Wei
Shizhang Wei Lisheng Chen
Lisheng Chen Xuelin Zhou
Xuelin Zhou Xiao Ma
Xiao Ma- 1Department of Pharmacy, Chinese People's Liberation Army (PLA) General Hospital, Beijing, China
- 2Department of Anatomy, Histology and Embryology, School of Basic Medical Sciences, Peking University, Beijing, China
- 3College of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 4Department of Pharmacology, School of Basic Medical Sciences, Capital Medical University, Beijing, China
Primary biliary cirrhosis (PBC) is a chronic cholestatic immune liver disease characterized by persistent cholestasis, interlobular bile duct damage, portal inflammation, liver fibrosis, eventual cirrhosis, and death. Existing clinical and animal studies have made a good progress in bile acid metabolism, intestinal flora disorder inflammatory response, bile duct cell damage, and autoimmune response mechanisms. However, the pathogenesis of PBC has not been clearly elucidated. We focus on the pathological mechanism and new drug research and development of PBC in clinical and laboratory in the recent 20 years, to discuss the latest understanding of the pathological mechanism, treatment options, and drug discovery of PBC. Current clinical treatment mode and symptomatic drug support obviously cannot meet the urgent demand of patients with PBC, especially for the patients who do not respond to the current treatment drugs. New treatment methods are urgently needed. Drug candidates targeting reported targets or signals of PBC are emerging, albeit with some success and some failure. Single-target drugs cannot achieve ideal clinical efficacy. Multitarget drugs are the trend of future research and development of PBC drugs.
1 Introduction
Primary biliary cirrhosis (PBC) is clinically a type of autoimmune cholestatic liver disease characterized by serum autoantibody antimitochondrial antibodies (AMAs) (1, 2), chronic progressive cholestasis, interlobular bile duct damage, liver inflammation, eventual cirrhosis, and death (3, 4). Studies have found that the incidence and prevalence of PBC have increased globally in the past few decades (5, 6), especially among women (7). About 90% of patients with PBC have developed AMA against PDC-E2 (8). Importantly, AMA is still not detected in the serum of 5% of patients with PBC. However, liver tissues of patients with AMA-negative PBC show anti-nuclear antibodies (9, 10). Many patients with PBC suffer from complications, such as sicca complex (34%) (11), osteoporosis (20%–40%) (12), and hyperlipidemia (75%–95%) (13).
At present, the research on serum markers in PBC has made great breakthroughs. Some new highly sensitive PBC autoantigens (14–16) and PBC biomarkers with extremely high accuracy (17, 18) have been discovered. With the introduction of genome-wide association analysis (GWAS), some key gene loci have been discovered (19–21). It gives us deeper insight into the pathogenesis of PBC. However, the key pathological mechanism of PBC has not yet been clarified. Moreover, effective medicines for PBC still cannot meet clinical needs. Ursodesoxycholic acid (UDCA) can improve approximately two of the three of patients with PBC, but there are still approximately 30% of patients with PBC who are not responsive to UDCA (22). Encouragingly, obeticholic acid (OCA) can treat patients with UDCA-unresponsive PBC. However, it can aggravate the itching symptoms of patients with PBC (23). The only option for advanced PBC is liver transplantation (24). Although the pathogenesis of PBC has a great relationship with the immune disorder, the classic immunosuppressive agents are not ideal for PBC (25–27). Therefore, we hope that, by searching for the PBC pathogenesis, the key pathological mechanism of PBC can be dug out and provide evidence for targeted therapy of PBC in the future.
2 “Bile acid–intestinal flora–bile acid” signal axis in the progress of PBC
There are 1013–1014 microbial cells in the human intestine and more than 1,000 species of bacteria (28–30). The microbiota composition changes may cause increased intestinal permeability and bacterial translocation, eventually leading to chronic liver inflammation and fibrosis (31). Bile acids (BAs; 5%) in the colon are transformed into secondary BAs by intestinal bacteria (32). BAs can regulate the composition of intestinal microbes (32). In the early stage of PBC, the reduction of several potentially beneficial microbiota and the enrichment of opportunistic pathogens were observed (30, 33). Some flora can accurately distinguish patients with PBC from normal people (34).
The synthesis of BAs involves many reaction steps. The classic BA synthesis pathway is initiated by the 7a-hydroxylation of cholesterol catalyzed by cholesterol 7a-hydroxylase (CYP7A1), which produces about 75% of BA production (mainly including CDCA and CA) (35). The alternative synthesis pathway for BAs is initiated by sterol-27-hydroxylase (CYP27A1), which mainly synthesizes CDCA (32). The proportion of CDCA and CA is mainly regulated by sterol 12a-hydroxylase (CYP8B1) and is not regulated by microbial. However, intestinal flora can regulate the levels of CYP7A1, CYP7B1, and CYP27A1 in the liver (36). Studies have proved that, in the absence of bacteria, the BA pool consists of mainly primary conjugated BAs (37). Specifically, BA deconjugation is carried out by bacteria with bile salt hydrolase (BSH) activity (38), preventing the reabsorption of BAs into the enterohepatic circulation. The deconjugated primary BAs enter the colon and are metabolized by gut microbial 7-dehydroxylation into secondary BAs [Lithocholic acid (LCA) and Deoxycholic acid (DCA)] (32, 39, 40). Gut microbial dysbiosis reduces the activity of BSH and 7α-dehydroxylase, followed by increasing the ratio of conjugated/unconjugated BAs and primary/secondary BAs (41). BAs can affect the composition of intestinal flora through direct antibacterial effects and can also induce antibacterial factors (42) and intestinal protection–related genes (32). Moreover, Amphiregulin (AREG) inhibits the activity of CYP7A1 by targeting the activation of the EGFR signal, preventing toxic BA-induced hepatotoxicity (43). A recent study reported that taking a probiotic (Lactobacillus reuteri) can increase circulating BAs more than two-fold (44). Studies have found the potential role of gut microbes in PBC. However, because of a small number of patients, the difference between gut microbiota and patients with different disease stages and different serum immunological substances has not been studied Figure 1 (45).
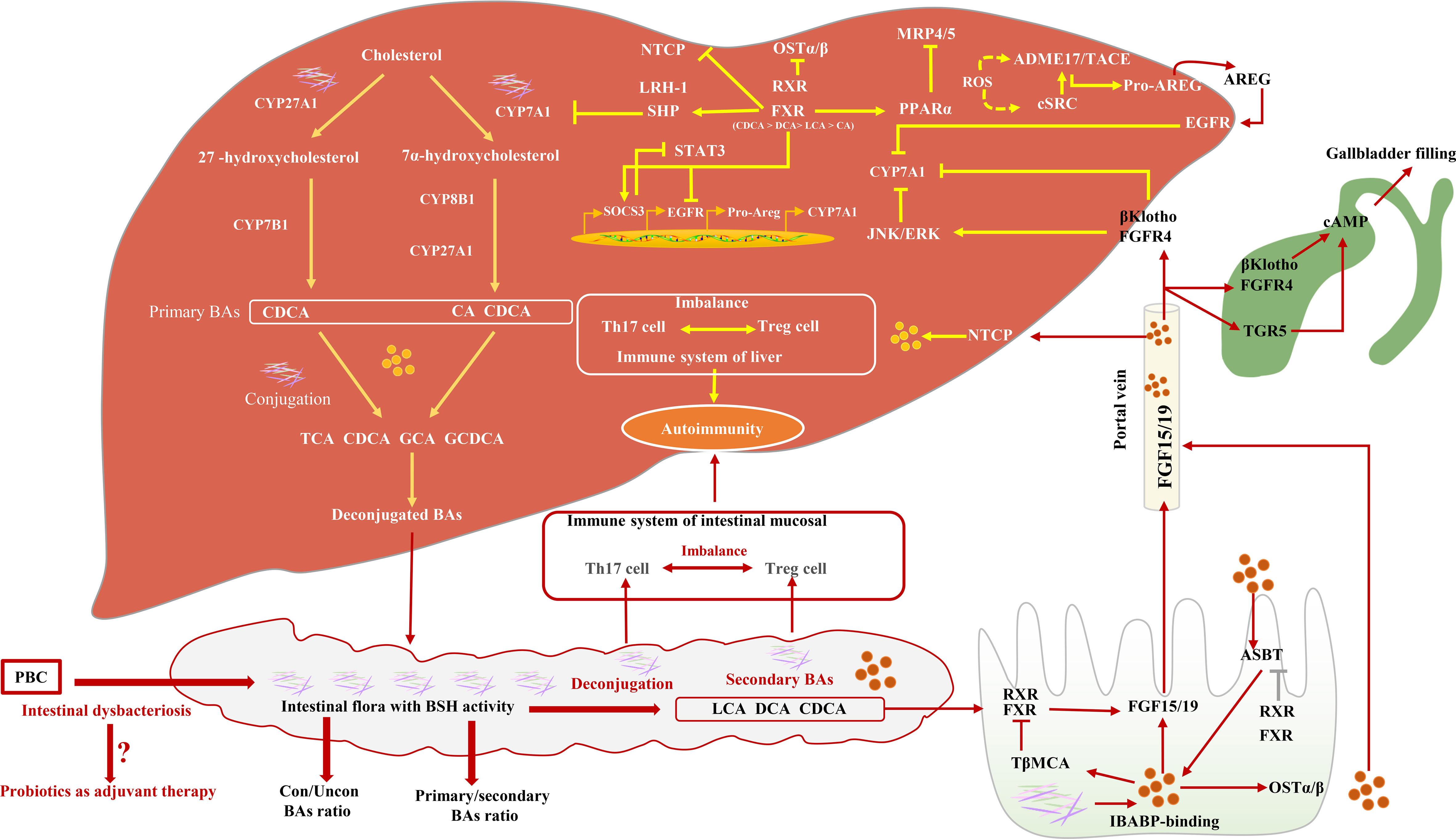
Figure 1 The relationship between intestinal flora and BA synthesis and their possible role in PBC. Intestinal microbial dysbiosis leads to a decrease in BSH and 7α-dehydroxylase activity, subsequently inducing BA pool size and composition changes. Farnesoid X receptor (FXR) activity induces the inhibition of CYP7A1 via activating SHP, the inhibition of Multidrug resistance-associated protein 4/5 (MRP 4/5) via activating Peroxisome proliferator activated receptor alpha (PPARα), and the inhibition of OSTα/β. CYP7A1 is also inhibited by Epidermal growth factor receptor (EGFR) signaling pathways. In the small intestine, BAs, FXR, Apical sodium-dependent bile salt transporter (ASBT), and Ileal bile acid binding protein (IBABP) work together to regulate Fibroblast growth factor 15/19 (FGF15/19), which binds to Fibroblast growth factor receptor 4 (FGFR4) through the portal vein and directly or indirectly inhibits CYP7A1 activity. FGF15/19 induces gallbladder filling through TGR5 or FGFR4 signaling pathways.
3 Small intrahepatic bile duct injury is the main pathological feature of PBC
Biliary epithelial cells (BECs) are the targets of most chronic cholestatic diseases (46). In PBC, continuous damage and senescence of BECs have been observed (47, 48). BECs secrete chemotactic cytokines and inflammatory cytokines during senescence (47, 49, 50) and participate in the induction and recruitment of CD4+ T helper (Th) cells. Toxic BAs can induce mitochondrial dysfunction through oxidative stress in PBC (51) and induce the senescence of BECs through a p38 mitogen-activated protein kinase (p38MAPK)-dependent pathway (52–57). Moreover, the activity of Cyclin-dependent kinase inhibitor p21 (p21WAF1/Cip1) and Cyclin dependent kinase inhibitor 2A (p16INK4) and ataxia telangiectasia-mutated (ATM)/p53/p21WAF1/Cip1 signals in the liver may also induce the senescence of bile duct cells of PBC (48, 58–60). Increasing pieces of evidence suggest that BEC apoptosis may be one of the main mechanisms of the pathogenesis of PBC (61–63). In PBC, emperipolesis of lymphocytes (mainly T lymphocytes) in BECs often be found to be related to inflammation and can reduce the repair of fibrosis (64, 65).
Cholestasis in PBC is related to damage to biliary bicarbonate and a defective biliary bicarbonate “umbrella” on the outer membrane of BECs (46). BECs participate in up to 40% of the bile flow by inducing the active secretion of bicarbonate in the bile (66). By virtue of the favorable Cl− gradient across the plasma membrane of BECs, the activation of Na+-independent anion exchanger 2 (AE2) causes the secretion of bicarbonate in the bile (67). This umbrella is heavily dependent on the AE2 function (68) and prevents hydrophobic BAs from damaging cells (69, 70). The lack of AE2 in the bile duct cells in patients with PBC will cause the cells to be more sensitive to apoptosis induced by cytotoxic hydrophobic Bas (68). There are two separate pathways for bile duct bicarbonate secretion, including cAMP/cystic fibrosis transmembrane conductance regulator (CFTR) signaling and InsP3/Ca2+ signaling (71). Type III inositol 1,4,5-trisphosphate receptor (InsP3R3) promotes the secretion of biliary bicarbonate (71, 72). In BECs, pro-inflammatory cytokines enhance the microRNA-506 (miR-506) expression, which can cause overexpression and mislocalization of PDC-E2 (26, 73, 74) in PBC (26, 75–79). There are pieces of evidence that PAMPs that exist in bile can induce the release of chemokines to stimulate the innate immune response and damages the bile ducts (80, 81). Interestingly, mitochondria are present in all cells. However, only BECs are destroyed in PBC (82), which is an important cause inducing autoimmune reactions.
Multiple BA receptors, including nuclear receptors FXR (83), plasma membrane–bound G protein–coupled receptors (GPCRs) (84), the sphingosine-1-phosphate receptor 2 (S1PR2) (85), and the Takeda G protein–coupled receptor 5 (TGR5) (86), are expressed in BECs and show a variety of biological activities. S1PR2 is only activated by conjugated BAs (86). TGR5 is activated by BAs by increasing reactive oxygen species (ROS) and subsequent EGFR-dependent signals (87) to promote the proliferation of BECs. Ligand binding to TGR5 through coupling to a Gα(s) protein activates adenylate cyclase, increases intracellular cAMP concentrations, and triggers chloride secretion via the CFTR (36, 88–91). BAs can activate FXR and induce the expression of FGF15/19, thereby activating p38 in adjacent BECs and inhibiting the activity of CYP27 (92). Interestingly, in a similar inflammatory environment, compared with other chronic cholestatic liver diseases, the expression of WAF1 and p53 in the BECs in patients with PBC is more significant, which is related to bile duct epithelial cell apoptosis (93) Figure 2.
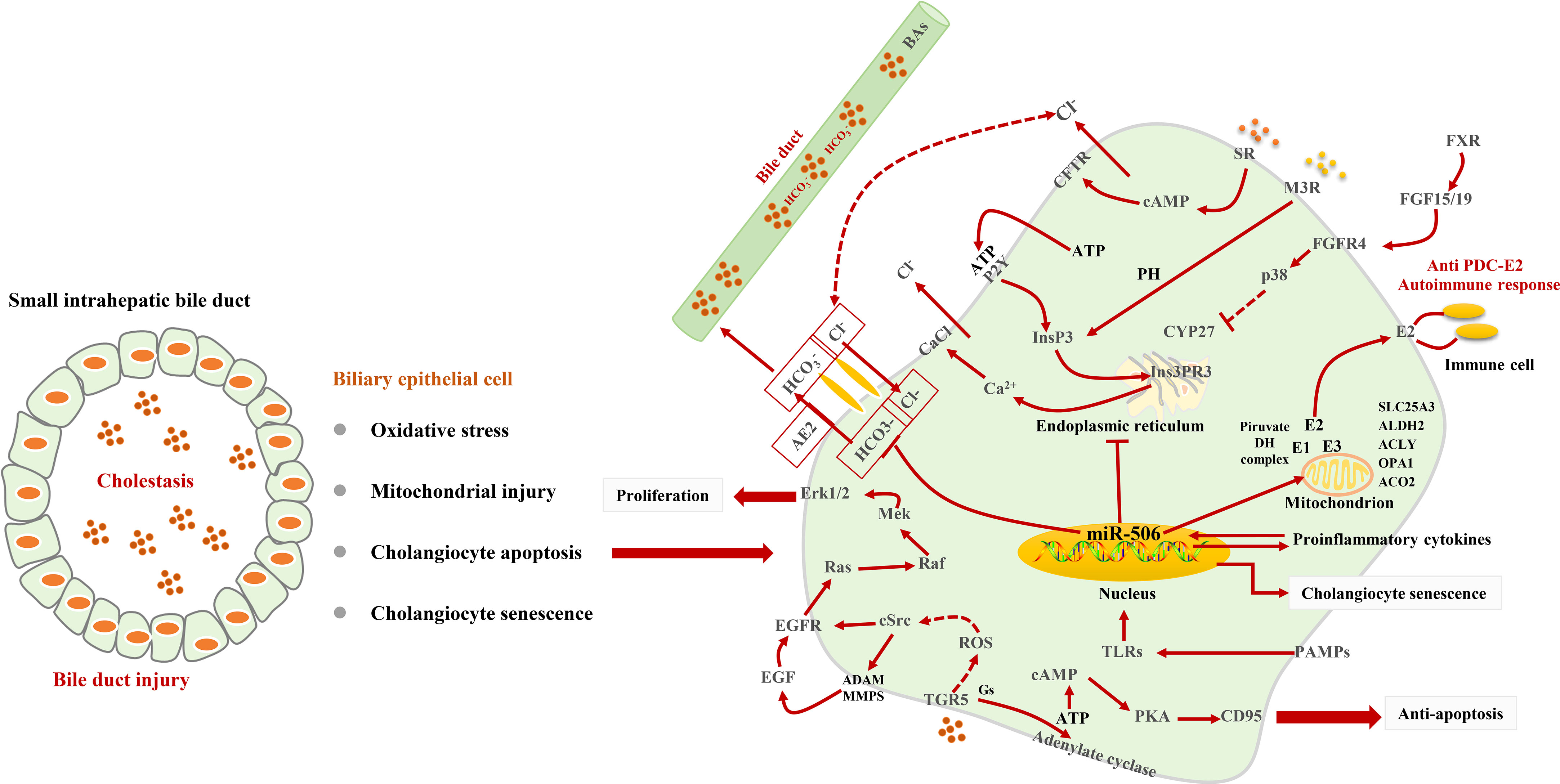
Figure 2 Signaling pathways for BEC injury. Cholestasis induces oxidative stress and mitochondrial injury and leads to BEC senescent and apoptosis. In BECs, TGR5-dependent signaling triggers Cl− secretion, EGFR transactivation, and CD95 to play a role in anti-apoptosis and promote proliferation. The stimulation of M3 muscarinic receptors and P2Y nucleotide receptors induces the release of Ca2+ from InsP3Rs. miR-506 targets AE2 to impair biliary bicarbonate secretion and leads to cholestasis. miR-506 can promote the release of PDC-E2.
4 “Bile acid–inflammation–bile acid” signal axis in PBC
Studies have shown that the pro-inflammatory effects of hydrophobic BAs mediate liver inflammation through various signal pathways (94–100). Normally, 95% of the BAs in the serum of patients with cholestasis are in conjugated form and are rapidly secreted into the gallbladder once they have been transported into the liver cells by Na(+)/taurocholate cotransporter (Ntcp). In the state of cholestasis, BA efflux transporters are restricted, leading to cholestasis in liver cells (101). NTCP only exists in hepatocytes; therefore, BA-induced inflammatory cytokine production is specific to hepatocytes (102). Excessive BAs in hepatocytes can cause the abnormal expression of cytochrome c and Grp78, leading to the release of mitochondrial DNA, which initiates innate immune response by activating Toll-like receptor 9 (TLR9)-dependent and TLR9-independent signals (103). In hepatocytes, FXR can inhibit NF-κB signaling activity by binding between Nuclear Factor Kappa-beta (NF-κB) and DNA sequences. In addition, the NF-κB p50/p65 heterodimer inhibits FXR-mediated gene [Organic solute transporter alpha/beta (OSTα/β), Bile salt export pump (BSEP), Multidrug resistance-associated protein homologs 2 (MRP2), Multidrug resistance protein-2/3 (MDR2/3), Small heterodimer partner (SHP)]) expression by binding to the FXR promoter in turn (104–107). BAs with high concentration can cause the ubiquitination and phosphorylation of NLR family pyrin domain containing 3 (NLRP3) (108) through the TGR5-cAMP-Protein kinase A (PKA) signal axis and inhibit its activity (109, 110). However, because of the expression restriction of FXR and TGR5 in cholestasis, the activity of NLRP3 cannot be restricted to cause liver inflammation (111). Nuclear factor of activated T cells (NFAT) plays an important role in the inflammatory response in PBC (112–114). Studies suggest that the Ca2+–calmodulin–calcineurin–NFAT signaling pathway is involved in BA-induced expression of chemokines in hepatocytes (115), which subsequently recruits neutrophils to mediate the inflammatory response, leading to hepatocyte necrosis (103). Previous studies have confirmed that NFAT signals can interact with innate immune responses through TLR signals (116). The activated TLR9 stimulates Burton’s tyrosine kinase (BTK) and causes phosphorylation and activation of phospholipase Cγ, which, in turn, induces Ca2+/NFAT signal (117). However, whether the Ca2+/NFAT axis is a downstream signal of TLR9 remains to be discussed (103). NLRP6 inflammasomes in intestinal epithelial cells can inhibit the destruction of the intestinal barrier by inducing Interleukin 18 (IL-18) synthesis and promote the production of antimicrobial peptides and mucus secretion by goblet cells Figure 3.
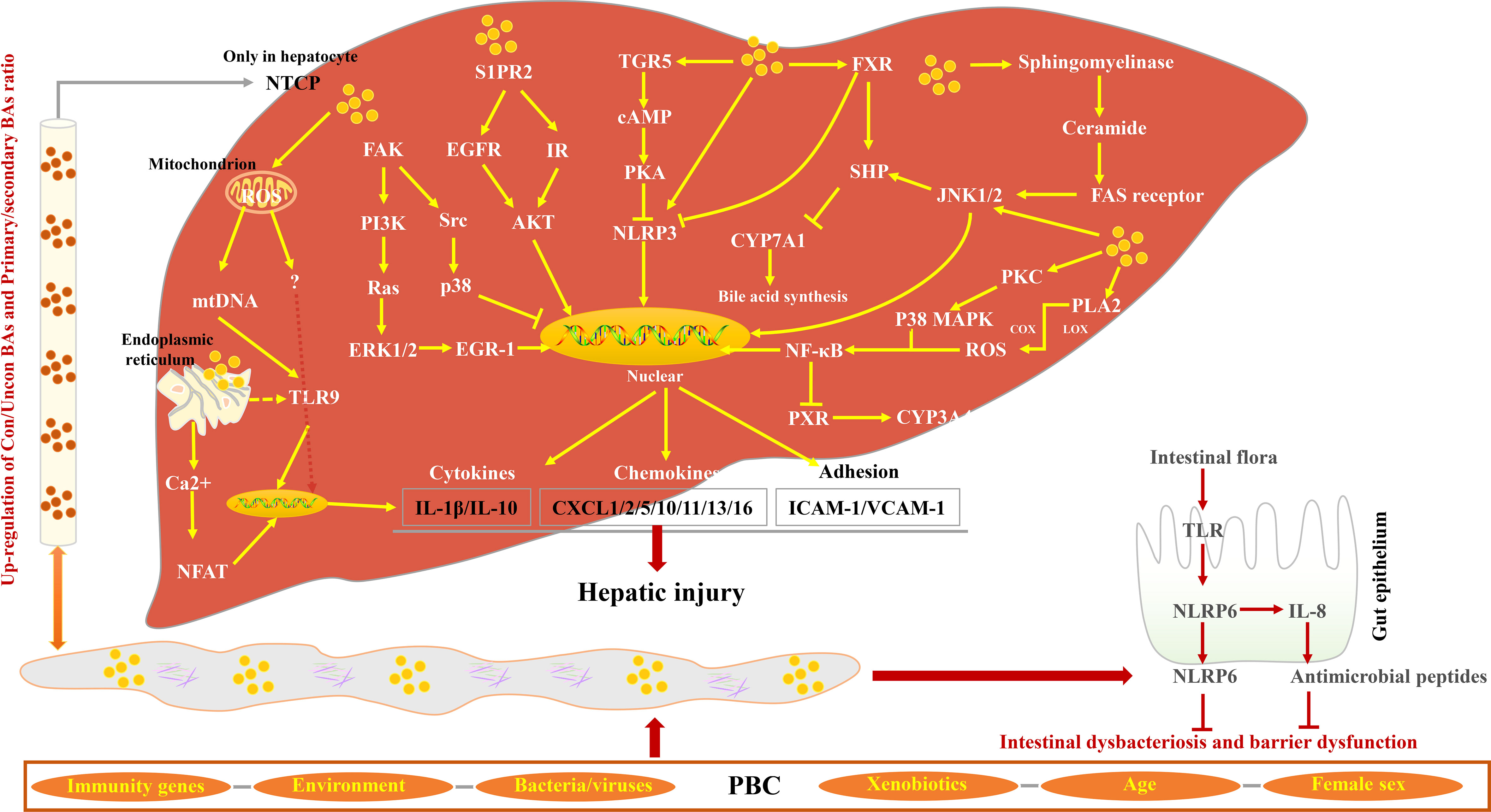
Figure 3 Characteristics of BA-induced inflammatory response in PBC. BAs from enterohepatic circulation activate Focal adhesion kinase (FAK)/EGR-1, FAK/p38, S1PR2/AKT, and sphingomyelinase/Fas cell surface death receptor (FAS) receptor signaling pathways, inducing the release of cytokines, chemokines, and adhesions and subsequently promoting hepatic injury. BAs play an inflammatory effect by promoting the production of ROS and Ca2+. In contrast, the ligands of FXR and TGR5 inhibit NLRP3 activity via different signaling pathways. In the small intestine, intestine flora activates TLR, promotes NLRP6 activity, and inhibits intestinal dysbacteriosis and dysfunction.
5 “Cholestasis–inflammation–liver tissue-specific autoimmunity” signals in PBC
T-cell dysfunction is an important mechanism for PBC (118). The CD4+ cell ratio in the portal area is relatively high. It can develop into two subtypes including T helper type I (Th1) and Th2 cells, respectively, producing corresponding cytokines. Th1 is mainly responsible for cellular immune response (119). The distribution of Th1 is consistent with CD8+ cells in the small bile ducts. CD8+ cells are the main type of lymphocytes that invade BECs in early-stage PBC. However, in the later stage of PBC, the number, proportion, and distribution of immune cells have changed greatly (64, 120). T lymphocytes mediate BEC damage through three pathways: Fas/FasL, T cell receptor (TCR)/major histocompatibility complex (MHC) , and Fas/Fas receptor signaling pathways (63, 64). Targeting nudt1 to inhibit the number and function of CD 103+ TRM cells in the liver has the potential to alleviate immune bile duct injury in patients with PBC (121). Some studies have also found that humoral immune response may also be involved in the inflammatory response in PBC (122, 123), and the CD5–B-cell population may enhance the process of T cells invading BECs (124). The serum and liver tissue cytokine profiles of patients with PBC showed activation and liver recruitment of Th1 and Th17 cells (50).
It has been shown that pro-inflammatory cytokines are involved in the immune response in PBC (125). IL-12 is mainly responsible for Th0 differentiation into Th1 cells. IL-12 can also stimulate the growth and function of T cells. The presence of significant hepatic autoantibodies in IL-12Rβ1–deficient patients suggests that IL-12Rβ1 signaling is closely related to hepatic autoimmunity (126). IL-23 can mediate Th17 differentiation from CD4 T cells and produce IL-6, IL-17, and Transforming growth factor beta (TGF-β) (50, 54). The IL-23– and IL-17–positive monocytes in the portal area in PBC were significantly higher. IL-23 promotes Th0 differentiation into Th17 and induces it to secretion IL-17 (27, 127). Previous studies have proved that IL-6 is essential for Th17 polarization (52, 54). Pro-inflammatory cytokines can activate NK cells and induce the activation of dendritic cells (DCs). DC can further activate T lymphocytes, leading to their differentiation toward the Th1 and Th17 phenotypes. Moreover, it can attack B cells to produce AMA autoantibodies. AMA can recognize the PDC-E2 antigen produced by apoptotic BECs, leading to the formation of antigen–antibody complexes and promoting cell damage (128). IL-17 in PBC not only has a pro-inflammatory effect but may also promote the activation of stellate cells, thereby promoting the occurrence of liver cirrhosis (125, 129). This is also an important factor that PBC progresses from liver fibrosis to cirrhosis. Regulatory T (Treg) cells, as “immune suppressors”, play a protective role in controlling the inflammatory response. Tregs in the damaged bile duct area in the PBC liver tissue are reduced (130). There is increasing evidence that Treg level increases significantly near the inflamed portal area (131). Th17 and Treg cells seem to maintain a delicate balance in liver immune homeostasis (132). It has been found that the main effect of the absence of TGFβ pathway signaling is to downregulate immune regulatory processes and, consequently, upregulate inflammatory processes. In the mouse PBC model, there are extensive differences between the dominant-negative transforming growth factor β receptor II (dnTGFβRII) and normal, wild-type Tregs. Key transcription factors in dnTGFβRII Tregs are downregulated and express an activated pro-inflammatory phenotype (133).
Studies have found that follicular helper T (Tfh) cells, which are differentiated from CD4+ T cells, can promote B-cell activation, proliferation, affinity maturation, and differentiation (134, 135). They contribute to the B-cell germinal center (GC) response, which is then involved in the development of PBC (132, 136, 137). Tfh cells also express a series of transcription factors, such as Signal transducer and activator of transcription 3/4 (STAT3/4), Transcription factor B-cell lymphoma 6 (Bcl-6), Interferon regulatory factor 4 (IRF4), c-musculoaponeurotic fibrosarcoma (cMaf), and show a similar relationship with Th effector subsets (138). Another study also found that the survival, proliferation, and differentiation of GC B cells and Tfh cells are reciprocally dependent on each other (138). Tfr cells can express Foxp3 and suppress the same function as Treg cells (139, 140). Intrahepatic Treg cells and Tfr cells have inadequate inhibition of inflammatory and autoimmune responses (141). In GWAS studies, genetic factors have been found to play a key role in the development of primary cholangitis (PBC). Relatives of patients with PBC had a significantly higher risk of developing the disease, and, even among second - and third-degree relatives, the risk was significantly increased (19, 142). Because of the insufficient sample size of patients with PBC, GWAS has not revealed the key pathological mechanism of PBC. A new CD103+ CD69+ CD8+ T cell has recently been found to invade BEC, and the E-cadherin expressed by this type of cell accelerates its invasive ability Figure 4.
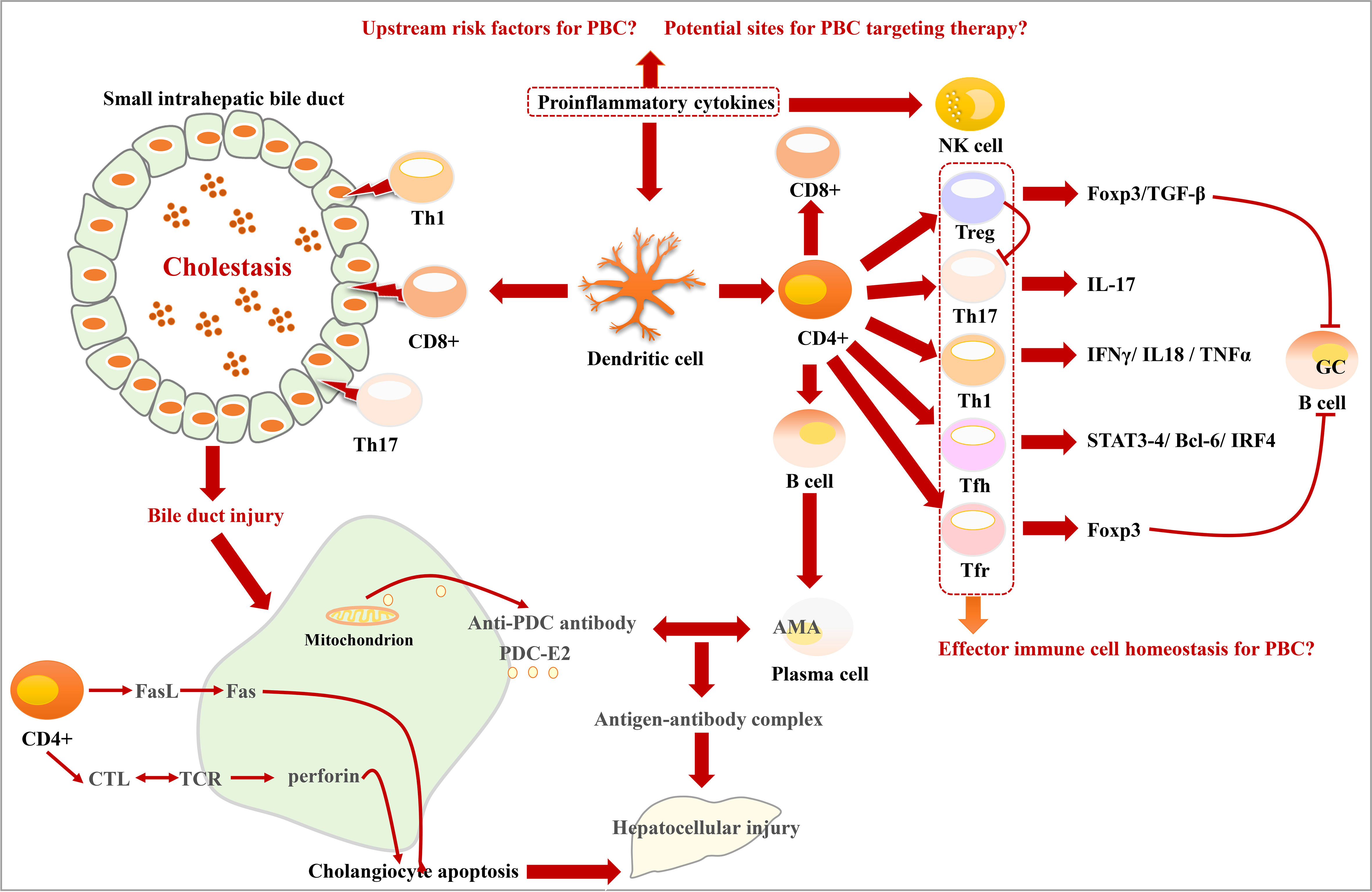
Figure 4 Characteristics of bile duct injury and immune disorder in PBC. DCs activated by proinflammatory cytokines activate T lymphocytes and prompt them to differentiate into Treg, Th17, Th1, Tfh, and Tfr phenotypes. Th1, CD8+, and Th17 cells promote bile duct injury. CD4+ cells induce cholangiocyte apoptosis through cytotoxic T lymphocytes (CTL)/TCR and Fasl/Fas signaling pathways. The mitochondria in damaged cholangiocytes produce PDC-E2 antigen, which can be recognized by AMA, leading to the formation of antigen–antibody complexes.
6 Natural small molecules have great potential in the treatment of PBC
Currently, there are a limited number of drugs that have been approved for clinical use in the treatment of PBC. Natural small molecules are an essential area of research for candidate drugs for PBC treatment. FXR agonists have been found to be a promising direction for PBC-targeted drug research. The approved FXR agonist OCA has shown outstanding therapeutic efficacy in the clinical treatment of PBC (143). It is noteworthy that multi-acid receptor agonists have more tremendous therapeutic potential for PBC (144). On this basis, we reviewed the research progress of laboratory and clinical research on FXR agonists (Table 1).

Table 1 Natural small molecules that activate nuclear receptors.
7 Conclusion
By summarizing the mechanism of PBC in BA cytotoxicity, the correlation characteristics of inflammation with BAs and immune response, and the main targets of hepatocyte necrosis and senescence and apoptosis of BEC, we have a generally in-depth understanding of the pathogenesis of PBC. Immune pathogenesis has a special relationship with the development of PBC. The immune response involves both innate immune response and humoral immune response that directly damage liver cells and BECs. However, the clinical effects of existing classic immunosuppressants and clinical trials of new biological agents modifying the immune system have been disappointing (158–160). Therefore, it is necessary to rethink the role of the immune mechanism in the pathogenesis of PBC. Limiting immune response to “downstream pathogenic factors” cannot achieve the desired efficacy in the treatment of PBC. Therefore, it is necessary to improve the “upstream pathogenic factors” of PBC, such as BA or inflammation, while suppressing immunity. This requires the new functions of new potential therapeutic drugs for PBC. It is worth noting that most patients with PBC have AMA autoantibodies. However, there are still a few patients with PBC who cannot be detected with AMA autoantibodies, but they do have other anti-nuclear antibodies. This suggests that the current research on the immune mechanism is flawed, and the key immune pathogenesis still needs further research and discovery.
As a possible “upstream factor” in the pathogenesis of PBC, BAs also show metabolic abnormalities in viral hepatitis, alcoholic or non-alcoholic steatohepatitis, drug-induced liver injury, and intrahepatic cholestasis of pregnancy, but no AMA autoantibodies appear as in PBC. This distinction suggests whether the relationship between BA metabolism disorder and autoimmune response is mediated by a “third party” in PBC, which requires further study. BAs are synthesized in hepatocytes, excreted into the small intestine through the gallbladder, and absorbed by the liver through blood circulation. Various cells throughout the enterohepatic circulation are exposed to BAs. However, in PBC, only the mitochondria in BECs are damaged. The destruction of mitochondria in BECs is also an important cause of autoimmune reactions. Therefore, we point out that BA-mediated damage to mitochondria in BECs is likely to be mediated by specific components of BECs. This is also an essential step in finding a PBC-targeted therapy strategy. On the basis of the existing research methods and technologies, some new high-throughput and high-sensitivity bioinformatics analysis technologies, such as GWAS, need to be used to reveal the targeting mechanism of PBC.
Author contributions
YZ generated the idea for the review and wrote the manuscript. SW, LC, XZ, and XM designed the figures corrected the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Major Program of the National Natural Science Foundation of China (No. 82192915) and the National Natural Science Foundation of China (81874365).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
PBC, primary biliary cirrhosis; AMAs, antimitochondrial antibodies; PDC-E2, pyruvate dehydrogenase E2 component; GWAS, genome-wide association analysis; UDCA, ursodesoxycholic acid; OCA, obeticholic acid; BAs, bile acids; CYP7A1, cholesterol 7a-hydroxylase; CYP27A1, sterol-27-hydroxylase; CYP8B1, sterol 12a-hydroxylase; BSH, bile salt hydrolase; BECs, biliary epithelial cells; CFTR, cystic fibrosis transmembrane conductance regulator; InsP3R3, type III inositol 1,4,5-trisphosphate receptor; miR-506, microRNA-506; GPCRs, G protein–coupled receptors; S1PR2, sphingosine-1-phosphate receptor 2; TGR5, Takeda G protein–coupled receptor 5; ROS, reactive oxygen species; NFAT, nuclear factor of activated T cells; BTK, Burton’s tyrosine kinase; DC, dendritic cells; Tfh, follicular helper T; GC, germinal center.
References
1. Terziroli Beretta-Piccoli B, Stirnimann G, Mertens J, Semela D, Zen Y, Mazzucchelli L, et al. Primary biliary cholangitis with normal alkaline phosphatase: a neglected clinical entity challenging current guidelines. J Autoimmun (2020) 102578. doi: 10.1016/j.jaut.2020.102578
2. Kilanczyk E, Banales JM, Wunsch E, Barbier O, Avila MA, Mato JM, et al. S-adenosyl-L-methionine (SAMe) halts the autoimmune response in patients with primary biliary cholangitis (PBC) via antioxidant and s-glutathionylation processes in cholangiocytes. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165895. doi: 10.1016/j.bbadis.2020.165895
3. Pariente A, Chazouilleres O, Causse X, Hanslik B, Arpurt JP, Henrion J, et al. Management of primary biliary cholangitis: results from a large real-life observational study in France and Belgium. Eur J Gastroenterol Hepatol (2020). 33(1S Suppl 1): e197–e205 doi: 10.1097/MEG.0000000000002011
4. Gerussi A, Cristoferi L, Carbone M, Asselta R, Invernizzi P. The immunobiology of female predominance in primary biliary cholangitis. J Autoimmun (2018) 95:124–32. doi: 10.1016/j.jaut.2018.10.015
5. Baldursdottir TR, Bergmann OM, Jonasson JG, Ludviksson BR, Axelsson TA, Bjornsson. The epidemiology ES. And natural history of primary biliary cirrhosis: a nationwide population-based study. Eur J Gastroenterol Hepatol (2012) 24:824–30. doi: 10.1097/MEG.0b013e328353753d
6. Myers RP, Shaheen AA, Fong A, Burak KW, Wan A, Swain MG, et al. Epidemiology and natural history of primary biliary cirrhosis in a Canadian health region: a population-based study. Hepatology (2009) 50:1884–92. doi: 10.1002/hep.23210
7. Gatselis NK, Zachou K, Lygoura V, Azariadis K, Arvaniti P, Spyrou E, et al. Geoepidemiology, clinical manifestations and outcome of primary biliary cholangitis in Greece. Eur J Intern Med (2017) 42:81–8. doi: 10.1016/j.ejim.2017.05.006
8. Sun C, Xiao X, Yan L, Sheng L, Wang Q, Jiang P, et al. Histologically proven AMA positive primary biliary cholangitis but normal serum alkaline phosphatase: is alkaline phosphatase truly a surrogate marker? J Autoimmun (2019) 99:33–8. doi: 10.1016/j.jaut.2019.01.005
9. Mendes F, Lindor KD. Antimitochondrial antibody-negative primary biliary cirrhosis. Gastroenterol Clin North Am (2008) 37:479–84. doi: 10.1016/j.gtc.2008.02.006
10. Kouroumalis E, Samonakis D, Voumvouraki A. Biomarkers for primary biliary cholangitis: current perspectives. Hepat Med (2018) 10:43–53. doi: 10.2147/HMER.S135337
11. Floreani A, Franceschet I, Cazzagon N, Spinazze A, Buja A, Furlan P, et al. Extrahepatic autoimmune conditions associated with primary biliary cirrhosis. Clin Rev Allergy Immunol (2015) 48:192–7. doi: 10.1007/s12016-014-8427-x
12. Raszeja-Wyszomirska J, Miazgowski T. Osteoporosis in primary biliary cirrhosis of the liver. Prz Gastroenterol (2014) 9:82–7. doi: 10.5114/pg.2014.42502
13. Sorokin A, Brown JL, Thompson. Primary biliary cirrhosis PD. Hyperlipidemia, and atherosclerotic risk: a systematic review. Atherosclerosis (2007) 194:293–9. doi: 10.1016/j.atherosclerosis.2006.11.036
14. Ninomiya M, Kondo Y, Funayama R, Nagashima T, Kogure T, Kakazu E, et al. Distinct microRNAs expression profile in primary biliary cirrhosis and evaluation of miR 505-3p and miR197-3p as novel biomarkers. PloS One (2013) 8:e66086. doi: 10.1371/journal.pone.0066086
15. Deng C, Hu C, Wang L, Zhang S, Li P, Wu Z, et al. Serological comparative proteomics analysis of mitochondrial autoantibody-negative and -positive primary biliary cirrhosis. Electrophoresis (2015) 36:1588–95. doi: 10.1002/elps.201400342
16. Joshita S, Umemura T, Usami Y, Yamashita Y, Norman GL, Sugiura A, et al. Serum autotaxin is a useful disease progression marker in patients with primary biliary cholangitis. Sci Rep (2018) 8:8159. doi: 10.1038/s41598-018-26531-0
17. Tan Y, Pan T, Ye Y, Ge G, Chen L, Wen D, et al. Serum microRNAs as potential biomarkers of primary biliary cirrhosis. PloS One (2014) 9:e111424. doi: 10.1371/journal.pone.0111424
18. Wang X, Wen X, Zhou J, Qi Y, Wu R, Wang Y, et al. MicroRNA-223 and microRNA-21 in peripheral blood b cells associated with progression of primary biliary cholangitis patients. PloS One (2017) 12:e0184292. doi: 10.1371/journal.pone.0184292
19. Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet (2011) 43:329–32. doi: 10.1038/ng.789
20. Umemura T, Joshita S, Yamazaki T, Komatsu M, Katsuyama Y, Yoshizawa K, et al. Genetic association of PTPN22 polymorphisms with autoimmune hepatitis and primary biliary cholangitis in Japan. Sci Rep (2016) 6:29770. doi: 10.1038/srep29770
21. Paziewska A, Habior A, Rogowska A, Zych W, Goryca K, Karczmarski J, et al. A novel approach to genome-wide association analysis identifies genetic associations with primary biliary cholangitis and primary sclerosing cholangitis in polish patients. BMC Med Genomics (2017) 10:2. doi: 10.1186/s12920-016-0239-9
22. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med (2005) 353:1261–73. doi: 10.1056/NEJMra043898
23. Cristoferi L, Nardi A, Ronca V, Invernizzi P, Mells G, Carbone M. Prognostic models in primary biliary cholangitis. J Autoimmun (2018) 95:171–8. doi: 10.1016/j.jaut.2018.10.024
24. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ, et al. Primary biliary cirrhosis. Hepatology (2009) 50:291–308. doi: 10.1002/hep.22906
25. Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from 'cirrhosis' to 'cholangitis'. Hepatology (2015) 62:1620–2. doi: 10.1002/hep.28140
26. Erice O, Munoz-Garrido P, Vaquero J, Perugorria MJ, Fernandez-Barrena MG, Saez E, et al. MicroRNA-506 promotes primary biliary cholangitis-like features in cholangiocytes and immune activation. Hepatology (2018) 67:1420–40. doi: 10.1002/hep.29533
27. Dyson JK, Hirschfield GM, Adams DH, Beuers U, Mann DA, Lindor KD, et al. Novel therapeutic targets in primary biliary cirrhosis. Nat Rev Gastroenterol Hepatol (2015) 12:147–58. doi: 10.1038/nrgastro.2015.12
28. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight. Diversity R. Stability and resilience of the human gut microbiota. Nature (2012) 489:220–30. doi: 10.1038/nature11550
29. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature (2010) 464:59–65. doi: 10.1038/nature08821
30. Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut (2018) 67:534–41. doi: 10.1136/gutjnl-2016-313332
31. Ma HD, Wang YH, Chang C, Gershwin ME, Lian. The intestinal microbiota ZX. And microenvironment in liver. Autoimmun Rev (2015) 14:183–91. doi: 10.1016/j.autrev.2014.10.013
32. Wahlstrom A, Sayin SI, Marschall HU, Backhed. Intestinal Crosstalk between Bile Acids F. And microbiota and its impact on host metabolism. Cell Metab (2016) 24:41–50. doi: 10.1016/j.cmet.2016.05.005
33. Lv LX, Fang DQ, Shi D, Chen DY, Yan R, Zhu YX, et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ Microbiol (2016) 18:2272–86. doi: 10.1111/1462-2920.13401
34. de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab (2013) 17:657–69. doi: 10.1016/j.cmet.2013.03.013
35. Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov (2008) 7:678–93. doi: 10.1038/nrd2619
36. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab (2013) 17:225–35. doi: 10.1016/j.cmet.2013.01.003
37. Selwyn FP, Csanaky IL, Zhang Y, Klaassen CD. Importance of Large intestine in regulating bile acids and glucagon-like peptide-1 in germ-free mice. Drug Metab Dispos (2015) 43:1544–56. doi: 10.1124/dmd.115.065276
38. Song Z, Cai Y, Lao X, Wang X, Lin X, Cui Y, et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome (2019) 7:9. doi: 10.1186/s40168-019-0628-3
39. Jones BV, Begley M, Hill C, Gahan CG, Marchesi. Functional JR. And comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA (2008) 105:13580–5. doi: 10.1073/pnas.0804437105
40. Ridlon JM, Bajaj JS. The human gut sterolbiome: bile acid-microbiome endocrine aspects and therapeutics. Acta Pharm Sin B (2015) 5:99–105. doi: 10.1016/j.apsb.2015.01.006
41. Chen W, Wei Y, Xiong A, Li Y, Guan H, Wang Q, et al. Comprehensive analysis of serum and fecal bile acid profiles and interaction with gut microbiota in primary biliary cholangitis. Clin Rev Allergy Immunol (2020) 58:25–38. doi: 10.1007/s12016-019-08731-2
42. Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA (2006) 103:3920–5. doi: 10.1073/pnas.0509592103
43. Santamaria E, Rodriguez-Ortigosa CM, Uriarte I, Latasa MU, Urtasun R, Alvarez-Sola G, et al. The epidermal growth factor receptor ligand amphiregulin protects from cholestatic liver injury and regulates bile acids synthesis. Hepatology (2019) 69:1632–47. doi: 10.1002/hep.30348
44. Martoni CJ, Labbe A, Ganopolsky JG, Prakash S, Jones ML. Changes in bile acids, FGF-19 and sterol absorption in response to bile salt hydrolase active l. reuteri NCIMB 30242. Gut Microbes (2015) 6:57–65. doi: 10.1080/19490976.2015.1005474
45. Tang R, Chen H, Miao Q, Bian Z, Ma W, Feng X, et al. The cumulative effects of known susceptibility variants to predict primary biliary cirrhosis risk. Genes Immun (2015) 16:193–8. doi: 10.1038/gene.2014.76
46. Glaser S, Francis H, Demorrow S, Lesage G, Fava G, Marzioni M, et al. Heterogeneity of the intrahepatic biliary epithelium. World J Gastroenterol (2006) 12:3523–36. doi: 10.3748/wjg.v12.i22.3523
47. Brain JG, Robertson H, Thompson E, Humphreys EH, Gardner A, Booth TA, et al. Biliary epithelial senescence and plasticity in acute cellular rejection. Am J Transplant (2013) 13:1688–702. doi: 10.1111/ajt.12271
48. Sasaki M, Ikeda H, Nakanuma Y. Activation of ATM signaling pathway is involved in oxidative stress-induced expression of mito-inhibitory p21WAF1/Cip1 in chronic non-suppurative destructive cholangitis in primary biliary cirrhosis: an immunohistochemical study. J Autoimmun (2008) 31:73–8. doi: 10.1016/j.jaut.2008.03.005
49. Sasaki M, Ikeda H, Sato Y, Nakanuma Y. Proinflammatory cytokine-induced cellular senescence of biliary epithelial cells is mediated via oxidative stress and activation of ATM pathway: a culture study. Free Radic Res (2008) 42:625–32. doi: 10.1080/10715760802244768
50. Yang CY, Ma X, Tsuneyama K, Huang S, Takahashi T, Chalasani NP, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology (2014) 59:1944–53. doi: 10.1002/hep.26979
51. Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol (2009) 15:1677–89. doi: 10.3748/wjg.15.1677
52. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441:231–4. doi: 10.1038/nature04754
53. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
54. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
55. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol (2009) 10:314–24. doi: 10.1038/ni.1698
56. Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol (2009) 182:5904–8. doi: 10.4049/jimmunol.0900732
57. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421:744–8. doi: 10.1038/nature01355
58. Sasaki M, Ikeda H, Haga H, Manabe T, Nakanuma Y. Frequent cellular senescence in small bile ducts in primary biliary cirrhosis: a possible role in bile duct loss. J Pathol (2005) 205:451–9. doi: 10.1002/path.1729
59. Hammond EM, Giaccia AJ. The role of ATM and ATR in the cellular response to hypoxia and re-oxygenation. DNA Repair (Amst) (2004) 3:1117–22. doi: 10.1016/j.dnarep.2004.03.035
60. Sasaki M, Ikeda H, Sato Y, Nakanuma Y. Decreased expression of Bmi1 is closely associated with cellular senescence in small bile ducts in primary biliary cirrhosis. Am J Pathol (2006) 169:831–45. doi: 10.2353/ajpath.2006.051237
61. Tinmouth J, Lee M, Wanless IR, Tsui FW, Inman R, Heathcote EJ. Apoptosis of biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. Liver (2002) 22:228–34. doi: 10.1046/j.0106-9543.2002.01595.x
62. Chen Y, Guo G, Guo S, Shimoda S, Shroyer KR, Tang Y, et al. Intracellular B7-H4 suppresses bile duct epithelial cell apoptosis in human primary biliary cirrhosis. Inflammation (2011) 34:688–97. doi: 10.1007/s10753-010-9280-6
63. Harada K, Furubo S, Ozaki S, Hiramatsu K, Sudo Y, Nakanuma Y. Increased expression of WAF1 in intrahepatic bile ducts in primary biliary cirrhosis relates to apoptosis. J Hepatol (2001) 34:500–6. doi: 10.1016/S0168-8278(00)00075-1
64. Zhao SX, Li WC, Fu N, Zhou GD, Liu SH, Jiang LN, et al. Emperipolesis mediated by CD8(+) T cells correlates with biliary epithelia cell injury in primary biliary cholangitis. J Cell Mol Med (2020) 24:1268–75. doi: 10.1111/jcmm.14752
65. Ichiki Y, Selmi C, Shimoda S, Ishibashi H, Gordon SC, Gershwin ME. Mitochondrial antigens as targets of cellular and humoral auto-immunity in primary biliary cirrhosis. Clin Rev Allergy Immunol (2005) 28:83–91. doi: 10.1385/CRIAI:28:2:083
66. Beuers U, Hohenester S, de Buy Wenniger LJ, Kremer AE, Jansen PL, Elferink RP. The biliary HCO(3)(-) umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology (2010) 52:1489–96. doi: 10.1002/hep.23810
67. Banales JM, Arenas F, Rodriguez-Ortigosa CM, Saez E, Uriarte I, Doctor RB, et al. Bicarbonate-rich choleresis induced by secretin in normal rat is taurocholate-dependent and involves AE2 anion exchanger. Hepatology (2006) 43:266–75. doi: 10.1002/hep.21042
68. Hohenester S, Wenniger LM, Paulusma CC, van Vliet SJ, Jefferson DM, Elferink RP, et al. A biliary HCO3- umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology (2012) 55:173–83. doi: 10.1002/hep.24691
69. Salas JT, Banales JM, Sarvide S, Recalde S, Ferrer A, Uriarte I, et al. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology (2008) 134:1482–93. doi: 10.1053/j.gastro.2008.02.020
70. Arenas F, Hervias I, Uriz M, Joplin R, Prieto J, Medina JF. Combination of ursodeoxycholic acid and glucocorticoids upregulates the AE2 alternate promoter in human liver cells. J Clin Invest (2008) 118:695–709. doi: 10.1172/JCI33156
71. Minagawa N, Nagata J, Shibao K, Masyuk AI, Gomes DA, Rodrigues MA, et al. Cyclic AMP regulates bicarbonate secretion in cholangiocytes through release of ATP into bile. Gastroenterology (2007) 133:1592–602. doi: 10.1053/j.gastro.2007.08.020
72. Hirata K, Dufour JF, Shibao K, Knickelbein R, O'Neill AF, Bode HP, et al. Regulation of Ca(2+) signaling in rat bile duct epithelia by inositol 1,4,5-trisphosphate receptor isoforms. Hepatology (2002) 36:284–96. doi: 10.1053/jhep.2002.34432
73. Banales JM, Saez E, Uriz M, Sarvide S, Urribarri AD, Splinter P, et al. Up-regulation of microRNA 506 leads to decreased cl-/HCO3- anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology (2012) 56:687–97. doi: 10.1002/hep.25691
74. Ananthanarayanan M, Banales JM, Guerra MT, Spirli C, Munoz-Garrido P, Mitchell-Richards K, et al. Post-translational regulation of the type III inositol 1,4,5-trisphosphate receptor by miRNA-506. J Biol Chem (2015) 290:184–96. doi: 10.1074/jbc.M114.587030
75. Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab (2017) 25:1374–1389 e1376. doi: 10.1016/j.cmet.2017.04.021
76. Ni R, Zheng D, Xiong S, Hill DJ, Sun T, Gardiner RB, et al. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy. Diabetes (2016) 65:255–68. doi: 10.2337/db15-0963
77. Houstek J, Pickova A, Vojtiskova A, Mracek T, Pecina P, Jesina. Mitochondrial diseases P. And genetic defects of ATP synthase. Biochim Biophys Acta (2006) 1757:1400–5. doi: 10.1016/j.bbabio.2006.04.006
78. Sasaki M, Ikeda H, Yamaguchi J, Nakada S, Nakanuma Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatology (2008) 48:186–95. doi: 10.1002/hep.22348
79. Sasaki M, Yoshimura-Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J Gastroenterol (2015) 50:984–95. doi: 10.1007/s00535-014-1033-0
80. Lu BR, Mack CL. Inflammation and biliary tract injury. Curr Opin Gastroenterol (2009) 25:260–4. doi: 10.1097/MOG.0b013e328325aa10
81. Chen XM, O'Hara SP, Nelson JB, Splinter PL, Small AJ, Tietz PS, et al. Multiple TLRs are expressed in human cholangiocytes and mediate host epithelial defense responses to cryptosporidium parvum via activation of NF-kappaB. J Immunol (2005) 175:7447–56. doi: 10.4049/jimmunol.175.11.7447
82. Lleo A, Leung PSC, Hirschfield GM, Gershwin EM. The pathogenesis of primary biliary cholangitis: a comprehensive review. Semin Liver Dis (2020) 40:34–48. doi: 10.1055/s-0039-1697617
83. Chan J, Vandeberg JL. Hepatobiliary transport in health and disease. Clin Lipidol (2012) 7:189–202. doi: 10.2217/clp.12.12
84. Sheikh Abdul Kadir SH, Miragoli M, Abu-Hayyeh S, Moshkov AV, Xie Q, Keitel V, et al. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PloS One (2010) 5:e9689. doi: 10.1371/journal.pone.0009689
85. Wang Y, Aoki H, Yang J, Peng K, Liu R, Li X, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology (2017) 65:2005–18. doi: 10.1002/hep.29076
86. Deutschmann K, Reich M, Klindt C, Droge C, Spomer L, Haussinger D, et al. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim Biophys Acta Mol Basis Dis (2018) 1864:1319–25. doi: 10.1016/j.bbadis.2017.08.021
87. Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut (2016) 65:487–501. doi: 10.1136/gutjnl-2015-309458
88. Keitel V, Haussinger D. TGR5 in cholangiocytes. Curr Opin Gastroenterol (2013) 29:299–304. doi: 10.1097/MOG.0b013e32835f3f14
89. Pellicciari R, Gioiello A, Macchiarulo A, Thomas C, Rosatelli E, Natalini B, et al. Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J Med Chem (2009) 52:7958–61. doi: 10.1021/jm901390p
90. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem (2003) 278:9435–40. doi: 10.1074/jbc.M209706200
91. Pean N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology (2013) 58:1451–60. doi: 10.1002/hep.26463
92. Jung D, York JP, Wang L, Yang C, Zhang A, Francis HL, et al. FXR-induced secretion of FGF15/19 inhibits CYP27 expression in cholangiocytes through p38 kinase pathway. Pflugers Arch (2014) 466:1011–9. doi: 10.1007/s00424-013-1364-3
93. Takeda K, Kojima Y, Ikejima K, Harada K, Yamashina S, Okumura K, et al. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc Natl Acad Sci USA (2008) 105:10895–900. doi: 10.1073/pnas.0802702105
94. Terziroli Beretta-Piccoli B, Mieli-Vergani G, Vergani D, Vierling JM, Adams D, Alpini G, et al. The challenges of primary biliary cholangitis: what is new and what needs to be done. J Autoimmun (2019) 105:102328. doi: 10.1016/j.jaut.2019.102328
95. Jansen PL, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology (2017) 65:722–38. doi: 10.1002/hep.28965
96. Gupta S, Natarajan R, Payne SG, Studer EJ, Spiegel S, Dent P, et al. Deoxycholic acid activates the c-jun n-terminal kinase pathway via FAS receptor activation in primary hepatocytes. role of acidic sphingomyelinase-mediated ceramide generation in FAS receptor activation. J Biol Chem (2004) 279:5821–8. doi: 10.1074/jbc.M310979200
97. O'Brien KM, Allen KM, Rockwell CE, Towery K, Luyendyk JP, Copple BL. IL-17A synergistically enhances bile acid-induced inflammation during obstructive cholestasis. Am J Pathol (2013) 183:1498–507. doi: 10.1016/j.ajpath.2013.07.019
98. Copple BL, Li T. Pharmacology of bile acid receptors: evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res (2016) 104:9–21. doi: 10.1016/j.phrs.2015.12.007
99. Anwer MS. Intracellular signaling by bile acids. J Biosci (Rajshari) (2012) 20:1–23. doi: 10.3329/jbs.v20i0.17647
100. Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol (2009) 15:3329–40. doi: 10.3748/wjg.15.3329
101. Woolbright BL, Jaeschke H. Therapeutic targets for cholestatic liver injury. Expert Opin Ther Targets (2016) 20:463–75. doi: 10.1517/14728222.2016.1103735
102. Slijepcevic D, Kaufman C, Wichers CG, Gilglioni EH, Lempp FA, Duijst S, et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+) -taurocholate cotransporting polypeptide knockout mice. Hepatology (2015) 62:207–19. doi: 10.1002/hep.27694
103. Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight (2017) 2:e90780. doi: 10.1172/jci.insight.90780
104. Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut (2011) 60:463–72. doi: 10.1136/gut.2010.212159
105. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology (2008) 48:1632–43. doi: 10.1002/hep.22519
106. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res (2006) 47:241–59. doi: 10.1194/jlr.R500013-JLR200
107. Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol (2013) 58:155–68. doi: 10.1016/j.jhep.2012.08.002
108. Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab (2017) 25:856–867 e855. doi: 10.1016/j.cmet.2017.03.007
109. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity (2016) 45:802–16. doi: 10.1016/j.immuni.2016.09.008
110. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol (2014) 11:55–67. doi: 10.1038/nrgastro.2013.151
111. Wang J, Dong R, Zheng S. Roles of the inflammasome in the gutliver axis (Review). Mol Med Rep (2019) 19:3–14. doi: 10.1007/s00894-018-3886-2
112. Ulrich JD, Kim MS, Houlihan PR, Shutov LP, Mohapatra DP, Strack S, et al. Distinct activation properties of the nuclear factor of activated T-cells (NFAT) isoforms NFATc3 and NFATc4 in neurons. J Biol Chem (2012) 287:37594–609. doi: 10.1074/jbc.M112.365197
113. Kar P, Mirams GR, Christian HC, Parekh AB. Control of NFAT isoform activation and NFAT-dependent gene expression through two coincident and spatially segregated intracellular Ca(2+) signals. Mol Cell (2016) 64:746–59. doi: 10.1016/j.molcel.2016.11.011
114. Cai SY, Yu D, Soroka CJ, Wang J, Boyer JL. Hepatic NFAT signaling regulates the expression of inflammatory cytokines in cholestasis. J Hepatol (2020). 74(3):550–9 doi: 10.1101/2020.03.25.007831
115. Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity (2015) 42:265–78. doi: 10.1016/j.immuni.2015.01.006
116. Ma B, Yu J, Xie C, Sun L, Lin S, Ding J, et al. Toll-like receptors promote mitochondrial translocation of nuclear transcription factor nuclear factor of activated T-cells in prolonged microglial activation. J Neurosci (2015) 35:10799–814. doi: 10.1523/JNEUROSCI.2455-14.2015
117. Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to aspergillus fumigatus. EMBO Mol Med (2015) 7:240–58. doi: 10.15252/emmm.201404556
118. Kita H, Imawari M, Gershwin ME. Cellular immune response in primary biliary cirrhosis. Hepatol Res (2004) 28:12–7. doi: 10.1016/j.hepres.2003.09.003
119. Jones DE, Palmer JM, Burt AD, Walker C, Robe AJ, Kirby JA. Bacterial motif DNA as an adjuvant for the breakdown of immune self-tolerance to pyruvate dehydrogenase complex. Hepatology (2002) 36:679–86. doi: 10.1053/jhep.2002.35067
120. Miao Q, Bian Z, Tang R, Zhang H, Wang Q, Huang S, et al. Emperipolesis mediated by CD8 T cells is a characteristic histopathologic feature of autoimmune hepatitis. Clin Rev Allergy Immunol (2015) 48:226–35. doi: 10.1007/s12016-014-8432-0
121. Huang B, Lyu Z, Qian Q, Chen Y, Zhang J, Li B, et al. NUDT1 promotes the accumulation and longevity of CD103(+) T(RM) cells in primary biliary cholangitis. J Hepatol (2022) 77:1311–24. doi: 10.1016/j.jhep.2022.06.014
122. Ronca V, Mancuso C, Milani C, Carbone M, Oo YH, Invernizzi. Immune system P. And cholangiocytes: a puzzling affair in primary biliary cholangitis. J Leukoc Biol (2020) 108:659–71. doi: 10.1002/JLB.5MR0320-200R
123. Carbone M, Milani C, Gerussi A, Ronca V, Cristoferi L, Invernizzi P. Primary biliary cholangitis: a multifaceted pathogenesis with potential therapeutic targets. J Hepatol (2020) 73:965–6. doi: 10.1016/j.jhep.2020.05.041
124. Jin Q, Moritoki Y, Lleo A, Tsuneyama K, Invernizzi P, Moritoki H, et al. Comparative analysis of portal cell infiltrates in antimitochondrial autoantibody-positive versus antimitochondrial autoantibody-negative primary biliary cirrhosis. Hepatology (2012) 55:1495–506. doi: 10.1002/hep.25511
125. Qian C, Jiang T, Zhang W, Ren C, Wang Q, Qin Q, et al. Increased IL-23 and IL-17 expression by peripheral blood cells of patients with primary biliary cirrhosis. Cytokine (2013) 64:172–80. doi: 10.1016/j.cyto.2013.07.005
126. Ronca V, Chen QB, Lygoura V, Ben-Mustapha I, Shums Z, Trifa M, et al. Autoantibodies in patients with interleukin 12 receptor beta 1 deficiency. J Dig Dis (2019) 20:363–70. doi: 10.1111/1751-2980.12790
127. Kikly K, Liu L, Na S, Sedgwick JD. The IL-23/Th(17) axis: therapeutic targets for autoimmune inflammation. Curr Opin Immunol (2006) 18:670–5. doi: 10.1016/j.coi.2006.09.008
128. Arsenijevic A, Stojanovic B, Milovanovic J, Arsenijevic D, Arsenijevic N, Milovanovic M. Galectin-3 in inflammasome activation and primary biliary cholangitis development. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21145097
129. Sun HQ, Zhang JY, Zhang H, Zou ZS, Wang FS, Jia JH. Increased Th17 cells contribute to disease progression in patients with HBV-associated liver cirrhosis. J Viral Hepat (2012) 19:396–403. doi: 10.1111/j.1365-2893.2011.01561.x
130. Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology (2006) 43:729–37. doi: 10.1002/hep.21123
131. Garetto S, Trovato AE, Lleo A, Sala F, Martini E, Betz AG, et al. Peak inflammation in atherosclerosis, primary biliary cirrhosis and autoimmune arthritis is counter-intuitively associated with regulatory T cell enrichment. Immunobiology (2015) 220:1025–9. doi: 10.1016/j.imbio.2015.02.006
132. Zhang H, Jiang Z, Zhang L. Dual effect of T helper cell 17 (Th17) and regulatory T cell (Treg) in liver pathological process: from occurrence to end stage of disease. Int Immunopharmacol (2019) 69:50–9. doi: 10.1016/j.intimp.2019.01.005
133. Wang YH, Yang W, Yang JB, Jia YJ, Tang W, Gershwin ME, et al. Systems biologic analysis of T regulatory cells genetic pathways in murine primary biliary cirrhosis. J Autoimmun (2015) 59:26–37. doi: 10.1016/j.jaut.2015.01.011
134. Zhang Y, Garcia-Ibanez L, Toellner KM. Regulation of germinal center b-cell differentiation. Immunol Rev (2016) 270:8–19. doi: 10.1111/imr.12396
135. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
136. Zhou ZQ, Tong DN, Guan J, Li MF, Feng QM, Zhou MJ, et al. Circulating follicular helper T cells presented distinctively different responses toward bacterial antigens in primary biliary cholangitis. Int Immunopharmacol (2017) 51:76–81. doi: 10.1016/j.intimp.2017.08.004
137. Chevalier N, Jarrossay D, Ho E, Avery DT, Ma CS, Yu D, et al. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol (2011) 186:5556–68. doi: 10.4049/jimmunol.1002828
138. Cannons JL, Lu KT, Schwartzberg PL. T Follicular helper cell diversity and plasticity. Trends Immunol (2013) 34:200–7. doi: 10.1016/j.it.2013.01.001
139. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and bcl-6 suppress germinal center reactions. Nat Med (2011) 17:983–8. doi: 10.1038/nm.2426
140. Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (2011) 187:4553–60. doi: 10.4049/jimmunol.1101328
141. Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol (2020) 17:93–110. doi: 10.1038/s41575-019-0226-7
142. Ornolfsson KT, Olafsson S, Bergmann OM, Gershwin ME, Bjornsson ES. Using the icelandic genealogical database to define the familial risk of primary biliary cholangitis. Hepatology (2018) 68:166–71. doi: 10.1002/hep.29675
143. Gomez E, Garcia Buey L, Molina E, Casado M, Conde I, Berenguer M, et al. Effectiveness and safety of obeticholic acid in a southern European multicentre cohort of patients with primary biliary cholangitis and suboptimal response to ursodeoxycholic acid. Aliment Pharmacol Ther (2021) 53:519–30. doi: 10.1111/apt.16181
144. Fiorucci S, Baldelli F. Farnesoid X receptor agonists in biliary tract disease. Curr Opin Gastroenterol (2009) 25:252–9. doi: 10.1097/MOG.0b013e328324f87e
145. Chen H, Li J, Hu L, Zhao W, Yu H, Liu HZ, et al. [Effect of geniposidic acid on hepato-enteric circulation in cholestasis rats through Sirt1-FXR signaling pathway]. Zhongguo Zhong Yao Za Zhi (2019) 44:787–95. doi: 10.19540/j.cnki.cjcmm.20181204.013
146. Ding L, Zhang B, Li J, Yang L, Wang Z. Beneficial effect of resveratrol on alphanaphthyl isothiocyanateinduced cholestasis via regulation of the FXR pathway. Mol Med Rep (2018) 17:1863–72. doi: 10.3892/mmr.2017.8051
147. Meng Q, Chen XL, Wang CY, Liu Q, Sun HJ, Sun PY, et al. Alisol b 23-acetate protects against ANIT-induced hepatotoxity and cholestasis, due to FXR-mediated regulation of transporters and enzymes involved in bile acid homeostasis. Toxicol Appl Pharmacol (2015) 283:178–86. doi: 10.1016/j.taap.2015.01.020
148. Meng Q, Chen X, Wang C, Liu Q, Sun H, Sun P, et al. Protective effects of alisol b 23-acetate via farnesoid X receptor-mediated regulation of transporters and enzymes in estrogen-induced cholestatic liver injury in mice. Pharm Res (2015) 32:3688–98. doi: 10.1007/s11095-015-1727-x
149. Kanno Y, Yatsu T, Yamashita N, Zhao S, Li W, Imai M, et al. Alisol b 23-acetate from the rhizomes of alisma orientale is a natural agonist of the human pregnane X receptor. Phytomedicine (2017) 26:22–7. doi: 10.1016/j.phymed.2017.01.003
150. Dong R, Wang J, Gao X, Wang C, Liu K, Wu J, et al. Yangonin protects against estrogen-induced cholestasis in a farnesoid X receptor-dependent manner. Eur J Pharmacol (2019) 857:172461. doi: 10.1016/j.ejphar.2019.172461
151. Kong Y, Gao X, Wang C, Ning C, Liu K, Liu Z, et al. Protective effects of yangonin from an edible botanical kava against lithocholic acid-induced cholestasis and hepatotoxicity. Eur J Pharmacol (2018) 824:64–71. doi: 10.1016/j.ejphar.2018.02.002
152. Chen X, Meng Q, Wang C, Liu Q, Sun H, Huo X, et al. Protective effects of calycosin against CCl4-induced liver injury with activation of FXR and STAT3 in mice. Pharm Res (2015) 32:538–48. doi: 10.1007/s11095-014-1483-3
153. Yang F, Tang X, Ding L, Zhou Y, Yang Q, Gong J, et al. Curcumin protects ANIT-induced cholestasis through signaling pathway of FXR-regulated bile acid and inflammation. Sci Rep (2016) 6:33052. doi: 10.1038/srep33052
154. Li T, Xu L, Zheng R, Wang X, Li L, Ji H, et al. Picroside II protects against cholestatic liver injury possibly through activation of farnesoid X receptor. Phytomedicine (2020) 68:153153. doi: 10.1016/j.phymed.2019.153153
155. Xiong XL, Ding Y, Chen ZL, Wang Y, Liu P, Qin H, et al. Emodin rescues intrahepatic cholestasis via stimulating FXR/BSEP pathway in promoting the canalicular export of accumulated bile. Front Pharmacol (2019) 10:522. doi: 10.3389/fphar.2019.00522
156. Wang J, Fu T, Dong R, Wang C, Liu K, Sun H, et al. Hepatoprotection of auraptene from the peels of citrus fruits against 17alpha-ethinylestradiol-induced cholestasis in mice by activating farnesoid X receptor. Food Funct (2019) 10:3839–50. doi: 10.1039/C9FO00318E
157. Yang F, Wang Y, Li G, Xue J, Chen ZL, Jin F, et al. Effects of corilagin on alleviating cholestasis via farnesoid X receptor-associated pathways in vitro and in vivo. Br J Pharmacol (2018) 175:810–29. doi: 10.1111/bph.14126
158. Bowlus CL, Yang GX, Liu CH, Johnson CR, Dhaliwal SS, Frank D, et al. Therapeutic trials of biologics in primary biliary cholangitis: an open label study of abatacept and review of the literature. J Autoimmun (2019) 101:26–34. doi: 10.1016/j.jaut.2019.04.005
159. Hirschfield GM, Gershwin ME, Strauss R, Mayo MJ, Levy C, Zou B, et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: a proof-of-concept study. Hepatology (2016) 64:189–99. doi: 10.1002/hep.28359
Keywords: PBC, pathological mechanism, treatment options, drug discovery, natural products
Citation: Zhao Y, Wei S, Chen L, Zhou X and Ma X (2023) Primary biliary cholangitis: molecular pathogenesis perspectives and therapeutic potential of natural products. Front. Immunol. 14:1164202. doi: 10.3389/fimmu.2023.1164202
Received: 12 February 2023; Accepted: 05 June 2023;
Published: 30 June 2023.
Edited by:
Lu Liangjing, Shanghai Jiao Tong University, ChinaReviewed by:
Pranav Shivakumar, Cincinnati Children’s Hospital Medical Center, United StatesVincenzo Ronca, University of Birmingham, United Kingdom
Patrick Leung, University of California, Davis, United States
Copyright © 2023 Zhao, Wei, Chen, Zhou and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanling Zhao, emhhb3lsMjg1NUAxMjYuY29t