 Hui Yang
Hui Yang Jiaxian Chen
Jiaxian Chen Jun Li
Jun Li- State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, National Medical Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Acute liver failure (ALF) is a high-mortality syndrome for which liver transplantation is considered the only effective treatment option. A shortage of donor organs, high costs and surgical complications associated with immune rejection constrain the therapeutic effects of liver transplantation. Recently, mesenchymal stem cell (MSC) therapy was recognized as an alternative strategy for liver transplantation. Bone marrow mesenchymal stem cells (BMSCs) have been used in clinical trials of several liver diseases due to their ease of acquisition, strong proliferation ability, multipotent differentiation, homing to the lesion site, low immunogenicity and anti-inflammatory and antifibrotic effects. In this review, we comprehensively summarized the harvest and culture expansion strategies for BMSCs, the development of animal models of ALF of different aetiologies, the critical mechanisms of BMSC therapy for ALF and the challenge of clinical application.
1 Introduction
Acute liver failure (ALF) is defined as a sudden onset of fulminant liver dysfunction in patients without underlying liver disease and is characterized by multiple organ failure with hepatic jaundice, coagulopathy [INR ≥ 1.5] and encephalopathy (1, 2). The interval time from the onset of jaundice to the development of encephalopathy is divided into three classifications: hyperacute, acute and subacute (3). Without therapeutic intervention, ALF can rapidly progress to multiorgan failure, severe systemic inflammation and even death, with a mortality rate often exceeding 90% (4). Liver transplantation, the only curative treatment for acute liver failure, is limited by the high cost, shortage of donor organs and long-term immune rejection (5). Hepatocyte transplantation has been considered an alternative to organ transplantation but has been hampered by the lack of large cell quantities, expansion difficulties ex vivo, rejection of allografts and xenotransplantation, and the rapid loss of liver properties in vitro (6–8). Therefore, other alternatives need to be studied for the constraint of hepatocyte and liver transplantation. Stem cells, including foetal biliary tree stem cells, foetal liver stem cells, haematopoietic stem cells, endothelial progenitor cells, MSCs, induced pluripotent stem cells and others, can transdifferentiate into hepatocyte-like cells to restore the damaged liver and response to stimulation (9). MSC therapy has been extensively studied and shows great clinical promise due to its ease of acquisition, strong proliferation ability, multipotential differentiation, homing to the lesion site, low immunogenicity and anti-inflammatory and antifibrotic effects.
At present, MSCs are utilized to improve liver function while waiting for liver transplantation and can also be used as a potential alternative therapy to organ or hepatocyte transplantation (10). Some recent clinical trials have reported that infusion of MSCs could induce tolerance after liver transplantation to reduce immune rejection due to the low immunogenicity and immunosuppression of MSCs (11–13). MSCs have been isolated from multiple biological tissues, including adult bone marrow, adipose tissue and neonatal tissues, such as the umbilical cord and the placenta. Bone marrow-derived mesenchymal stem cells (BMSCs) were the first multipotential stromal progenitor cells isolated and identified. BMSCs was recognized as the most promising cell sources due to their easy access and well-characterized biological features in clinical trials and preclinical studies (14, 15). However, only a small percentage of BMSCs, ranging from 0.01% to 0.001%, are present in bone marrow tissue, and these cells require substantial expansion ex vivo before they can be used for clinical treatment. Isolating BMSCs through conventional differential adherence and density gradient centrifugation is effective, but the approaches do not yield a relatively homogeneous cell population, and the cells may be contaminated by other cells from the bone marrow (16), which results in differential proliferation, transdifferentiation and therapeutic efficiency of BMSCs. Therefore, cell sorting based on BMSC-specific markers is an attractive technique for homogeneous subsets, and glucose, hypoxia and serum-free conditions are vital to facilitate the proliferation of MSCs and reduce cell senescence. Although BMSCs have been used in numerous clinical therapies and animal investigations (17, 18), the mechanism is unclear. In this review, we summarize the critical mechanism as shown in Figure 1. Under the stimulation of liver failure signals, BMSCs homed to lesion sites through the endothelium and immediately modulated the immune microenvironment, which is beneficial for tissue repair. Furthermore, several animal models of ALF have been described to clarify the mechanisms of BMSCs for the treatment of ALF with different aetiologies.
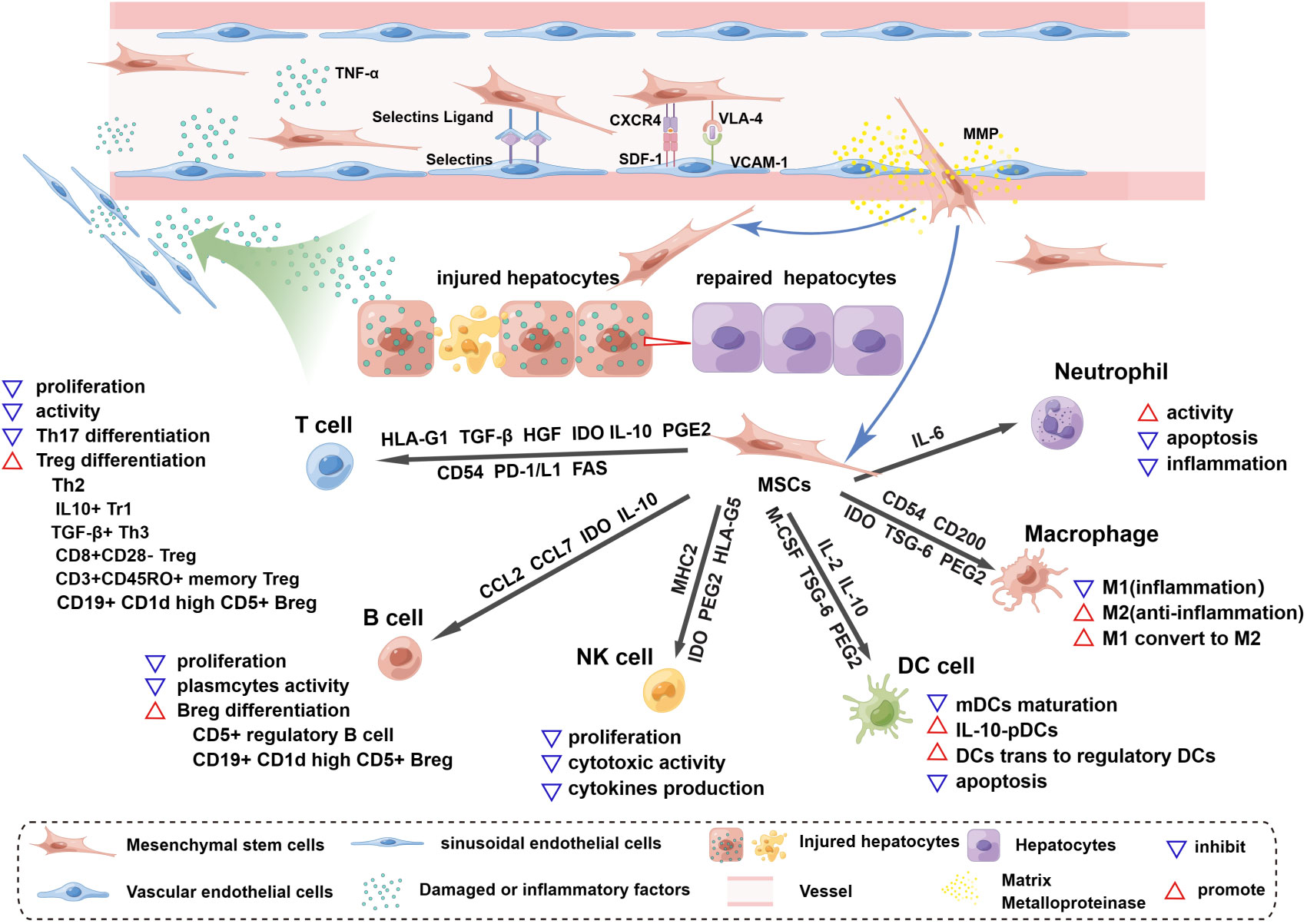
Figure 1 Homing and immunoregulation of MSCs triggered by the injured liver. The injured liver releases a variety of damaged or inflammatory factors, such as TNF-α and histamine, which enter vessels through the hepatic blood sinusoids and activate blood vascular endothelial cells (ECs), upregulating the expression of selectin and VCAM1. Once MSCs roll into the vessel wall, the expression of CD44 (HCAM), CXCR4, and VLA-4 on the surface of MSCs is triggered. MSCs then adhere to ECs through the interaction of selectin ligands and selectins, and an activation phase occurs through the interaction of SDF-1 and CXCR4, which enhances the affinity for integrins. The binding of VLA-4 and VCAM-1 promotes MSC extravasation; this process starts after the activation of matrix metalloproteinases (MMPs) and disrupts type IV collagen in the basement membrane. MSCs cross the basement membrane and are released into the hepatic interstitium, where they perform the functions of liver regeneration and immunoregulation and achieve therapeutic efficacy. MSCs release soluble factors and express surface molecules to regulate adaptive (T cells, B cells) and innate (NK cells, DC cells, macrophages and neutrophils) immune cells. HLA-G1, TGF-β, HGF and IDO secreted by MSCs can inhibit T-cell proliferation and activation, and similarly, the Fas and PD-1/L1 pathways trigger apoptosis of T cells. MSCs expressing CD54, PD-1, IL-10 and PEG2 can inhibit Th17 differentiation. In the presence of MSCs, T cells can differentiate into Tregs, such as Th2 cells, CD3+CD45RO+ memory Treg cells, CD8+CD28- Tregs, IL10+ Tr1 cells and TGFβ+ Th3 cells. MSCs inhibit B-cell proliferation, interfere with the formation of plasma cells, and release CCL2 and CCL7 to inhibit antibody production. IDO from MSCs is involved in the proliferation of CD5+ regulatory B cells, and IL-10 promotes the differentiation of CD19+CD24+CD38+ Bregs. MSCs can inhibit the proliferation, cytotoxic activity and cytokine production of quiescent NK cells by releasing IDO, PGE2 and HLA-G5 and by expressing MHCI. The maturation of myeloid dendritic cells (mDCs) is inhibited by MSC-derived IL-6, macrophage colony-stimulating factor (M-CSF), TSG-6, and PGE2. IL-10-plasmacytoid dendritic cells (pDCs) can be induced by PGE2 release from MSCs. In addition, MSCs can induce the transformation of mature dendritic cells into immunosuppressive regulatory dendritic cells through IL-10 and evasion of apoptosis. Inflammatory macrophages (M1) are converted to anti-inflammatory macrophages (M2) by IDO, TSG-6 and PGE2 secreted by MSCs, and CD54 and CD200 enhance their immunosuppressive effects. MSCs maintain neutrophil activity, and IL-6 delays apoptosis and inflammation.
2 Definition and source of mesenchymal stem cells
MSCs are a heterogeneous population that can adhere to plastic and proliferate ex vivo, forming colonies with a fibroblast-like morphology. They can differentiate into osteocytes, chondrocytes, adipocytes and other mesodermal lineages and have endodermic (19) and ectodermic (20) differentiation potential (Figure 2). Several studies have shown that MSCs can differentiate into functional hepatocytes and cholangiocytes after growth factor induction ex vivo (22). Intrasplenic transplantation of human-derived BMSCs into mice with fulminant liver failure developed a dual humanized mouse model with hepatocytes and immune cells (23). A recent investigation reported that MSCs can also self-assemble a three-dimensional (3D) human liver bud ex vivo by transdifferentiating into hepatocytes, sinusoidal endothelial cells (LECs) and hepatic stellate cells (HSCs) (24). Minimal criteria for human MSCs in basic scientific investigations and preclinical studies were proposed in 2006 by the International Society for Cellular Therapy (ISCT), which included adherence to plastic, potential for differentiation into osteoblasts, adipocytes, and chondroblasts under standard in ex vivo differentiation conditions, and expression (≥95% positive) of CD105, CD73 and CD90, as measured by flow cytometry. Additionally, these cells must lack expression (≤2% positive) of CD45, CD34, CD14 or CD11b, CD79a, CD19 and HLA class II, which are haematopoietic stem cell surface antigens (21).
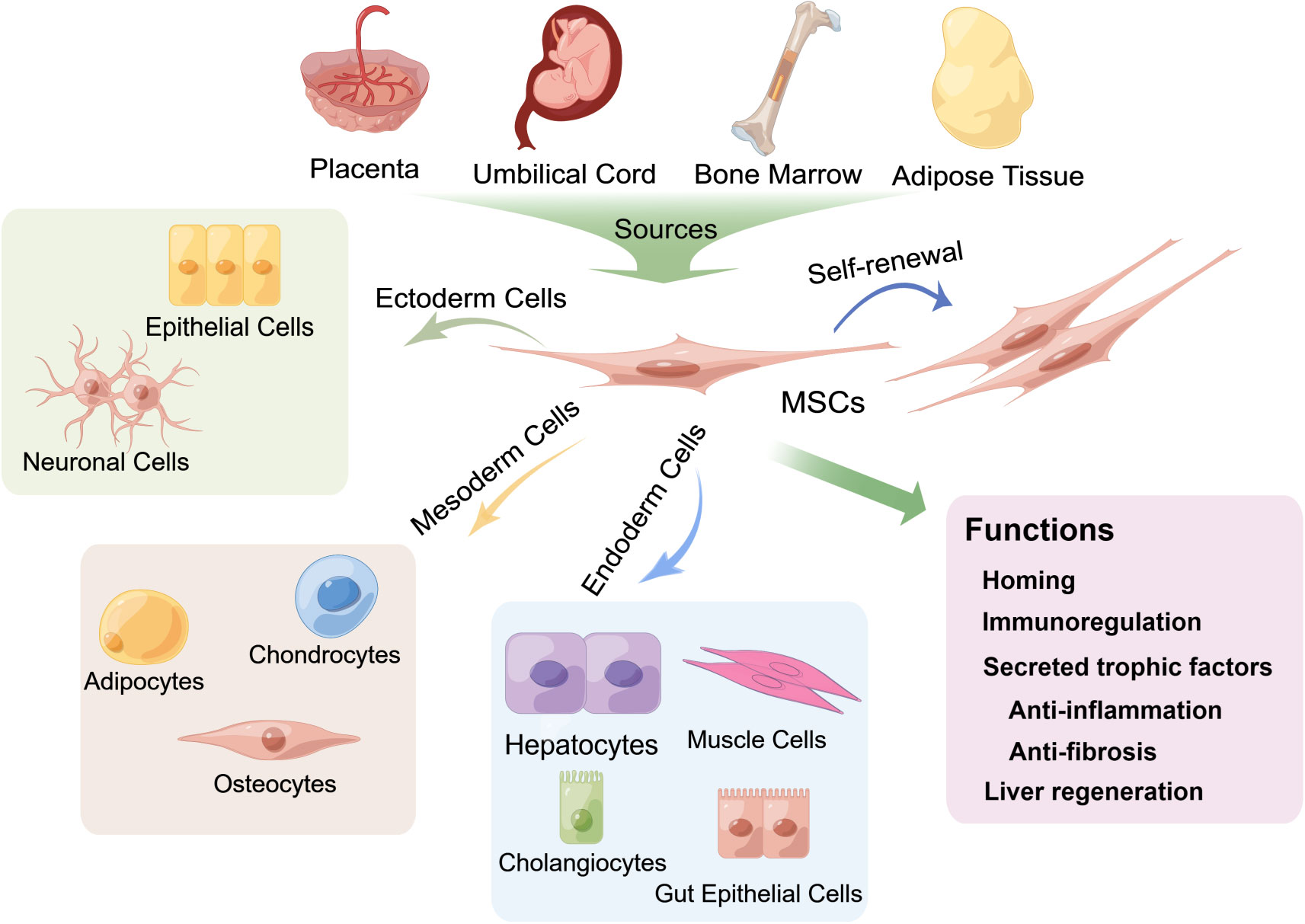
Figure 2 Multipotent differentiation and functions of mesenchymal stem cells (MSCs) derived from various tissues. MSCs can be isolated from multiple biological tissues, including adult bone marrow, adipose tissue and neonatal tissues, such as the umbilical cord and the placenta, and can differentiate into osteocytes, chondrocytes, adipocytes and other mesodermal lineages (21) and exhibit endodermic (such as hepatocytes, muscle cells, gut epithelial cells, and cholangiocytes) (20, 22–28) and ectodermic (epithelial cells and neuronal cells) (29–33) differentiation potential. MSCs also have strong self-renewal properties. The fundamental biological functions of MSCs include homing to sites of damage, immunoregulation of the immune microenvironment and secretion of trophic factors that exert anti-inflammatory and antifibrotic effects, as well as differentiation into hepatocytes to promote liver regeneration.
MSCs are located in multiple adult and neonatal tissues with perivascular niches (34), such as adult bone marrow and adipose tissue, and neonatal tissues, such as the umbilical cord and the placenta. In addition, the fundamental biological functions of MSCs involved in the treatment of liver diseases are mainly homing/migration to sites of damage and the secretion of trophic factors that mediate liver regeneration and regulation of immune responses (17, 35–37) (Figure 2).
3 Culture strategies of BMSCs in vitro
3.1 Isolation of BMSCs
The effect of BMSCs in clinical treatment is highly dependent on their quality. However, the lack of standardized culture procedures and unique markers limits the consistency of BMSC characteristics. The initial step for BMSC standardization is the isolation process (38). Human bone marrow is mainly obtained from the iliac crest via aspiration in the presence of some anticoagulants, such as heparin sodium (23). There are several methods for isolating BMSCs from bone marrow. Traditional differential adhesion is based on the typical capacity of MSCs, such as adherence to plastic and sensitivity to enzyme digestion, and the culture medium is changed every 3-4 days to gradually achieve purification. Although this method is convenient and economical, it does not yield a homogeneous population of cells that contain other bone marrow subpopulations, such as endothelial cells, pericytes, leukocytes, and haematopoietic stem cells (39, 40). Another technique, called density gradient centrifugation, has also been proposed to precipitate different cells in the bone marrow according to size and density using gradient centrifugation solutions with a density of approximately 1.077 g/mL, low viscosity and low permeability, such as Ficoll, Ficoll-Paque, Percoll, and Lymphogre. After centrifugation and stratification, the greyish-white cloudy layer above the separation fluid was purified through differential adhesion. However, the lack of specific subsets and contamination of cell populations have limited its application (38, 41, 42). To improve the homogeneity of BMSC populations, advanced isolation methods, such as fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS), have been used for high-throughput screening of BMSCs via specific surface markers. FACS and MACS employ electric and magnetic fields, respectively, that exert external forces to separate BMSCs. However, there is no evidence assessing their influence on MSC functions and therapeutic effects (43, 44). Therefore, the crucial aspect of this method is specific surface markers. ISCT published minimal guidelines for the isolation of human BMSCs based on the positive markers CD105, CD73, and CD90 and the negative markers CD45, CD34, HLA-DR, CD79a, CD19, CD11b, and CD14 (21), and we summarized a variety of unique markers for human BMSCs in Table 1 (45–58).
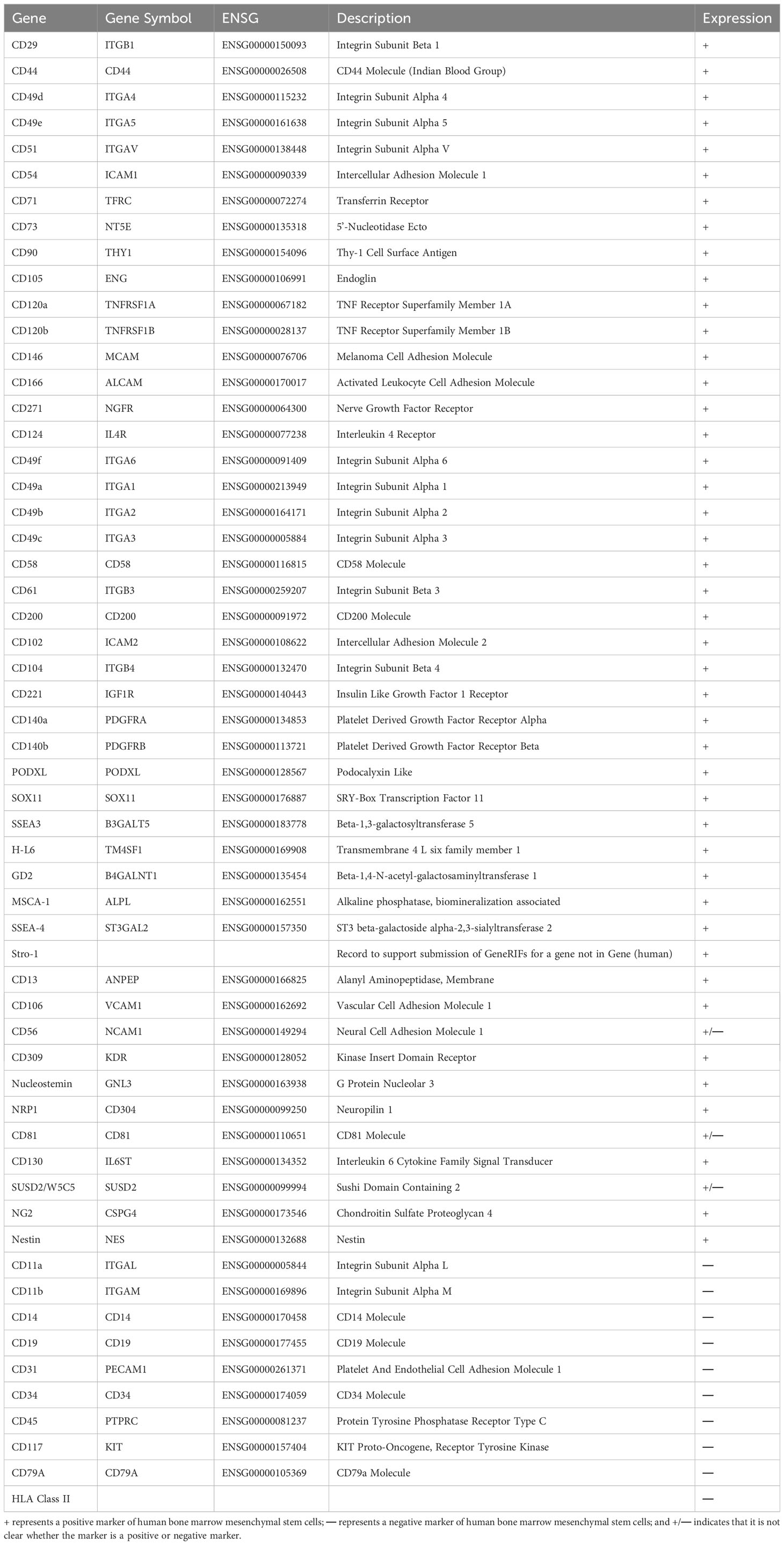
Table 1 Positive and negative markers of human bone marrow mesenchymal stem cells.
3.2 Culture expansion of BMSCs
The scientific studies and clinical applications of BMSCs require substantial expansion ex vivo to obtain sufficient numbers of cells because MSCs are rare in the bone marrow (0.001-0.01% of total nucleated cells and 0.42% of plastic adherent cells) (19). To maintain the functions and activities of BMSCs in vitro, a variety of approaches have been used to optimize the culture conditions, including medium composition, cell seeding density, passages and parameters related to the external environment, such as oxygen tension, pH and temperature.
3.2.1 Serum
Foetal bovine serum (FBS), the classic nutritional supplement for cell culture ex vivo, has been commonly used at a concentration of 5-20% (v/v) for the expansion of BMSCs, predominantly at 10% (59). FBS can provide macromolecules, proteins, adhesion, growth factors, nutrients, hormones and other essential biomolecules for the growth of BMSCs (60). However, the treatment of BMSCs cultured with FBS is controversial due to high batch-to-batch variations, xenoimmune effects and contamination with pathogens (38, 61, 62). Therefore, materials from autologous or allogeneic human blood sources have been explored, and the results showed that human serum (63), platelet lysate (63, 64) and umbilical cord serum (65) significantly increased cell proliferation, but an in-depth study of their efficacy is lacking. To overcome the uncertainties associated with serum, commercial serum-free media (SFMs) showed good performance ex vivo culture of human-derived BMSCs (63, 66–68). Van T Hoang et al. adapted a standardized process to assess the functional characteristics of serum-free cultured MSCs and showed that the MSCs satisfied the criteria (including basic MSC characteristics, normal karyotype, stronger proliferation, clinical-scale production and quality control requirements) (69, 70).
3.2.2 Glucose
Glucose is another critical source of energy for the growth and development of most cell types in vivo (71). Under physiological conditions, the serum glucose concentration of an organism is maintained at approximately 100 mg/dL, suggesting that MSCs should be exposed to the same glucose concentration in the bone marrow niche or ex vivo culture (72). Nevertheless, there is still controversy regarding the culture of BMSCs in terms of high vs. low glycaemic levels. Al-Qarakhli et al. assessed the effects of different glucose concentrations on the proliferation, senescence and multidirectional differentiation ability of MSCs, and the results demonstrated that high concentrations of glucose (450 mg/dL) inhibited osteogenic/adipogenic differentiation and had a limited negative effect on the proliferation and stemness of MSCs (73). Similar studies have shown that low glucose concentrations (100 mg/dL or 350 mg/dL) during culture can promote cell proliferation, colony formation, and multidirectional differentiation and reduce apoptosis and senescence (74–78). Overall, low-glycaemic culture may better maintain the properties of MSCs, which may facilitate the homing and tissue repair of MSCs in ALF.
3.2.3 Oxygen tension
The oxygen tension (pO2) of MSCs exposed to the bone marrow microenvironment typically ranges from 1% to 8% (also referred to hypoxia) (79), whereas ex vivo culture was at an atmospheric oxygen tension (21%), which may lead to cell proliferation cessation after multiple passages as well as cellular senescence. This does not occur in hypoxic conditions (1% pO2), which is possibly related to downregulation of the gene expression of p16 and extracellular signal-regulated kinase (ERK) (80). Several previous studies have revealed that hypoxic culture can promote cell proliferation (81), inhibit differentiation (82, 83) and reduce BMSC senescence (80). Recently, Ben Antebi et al. evaluated the function of human and porcine bone marrow-derived MSCs following long-term (10 days) and short-term (48 hours) hypoxic (1% pO2) culture, and the results demonstrated that short-term culture under hypoxia significantly increased cell proliferation upregulated VEGF expression and downregulated the expression of HMGB1 and the apoptotic genes BCL-2 and CASP3. Additionally, in short-term hypoxic culture at 2% and 5% pO2, BMSCs showed inhibition of the proinflammatory cytokine IL-8 and promotion of the anti-inflammatory agents IL-1Ra and GM-CSF, especially in short-term hypoxic culture at 2% pO2 (84). Yu et al. found that CXCR4 expression was upregulated in the presence of short-term hypoxic culture (24 h, 2% pO2) and low-dose inflammatory stimuli (1 ng/mL TNF-α and 0.5 ng/mL IL-1β), enhancing the homing/migration of BMSCs (85).
3.2.4 Cell seeding density
Cell seeding density is a key factor to be considered when BMSCs are expanded ex vivo. During the primary culture of bone marrow cell suspensions, the seeding density is typically 1 × 106 to 2 × 106 cells/cm2 for differential adhesion and approximately 1 × 104 cells/cm2 for density gradient centrifugation (86). The inoculation density of BMSCs is usually in the range of 2,000 to 5,000 cells/cm2 during the passaging process (38). It has been shown that low-density inoculation may facilitate cell proliferation by reducing contact inhibition and less affect the cell surface antigen phenotype and cell differentiation.
3.2.5 BMSC passage
Passaging is helpful to expanding the number of cells in culture and avoids mass mortality arising from cells entering the plateau stay or even decay. Typical digestion with 0.25% trypsin/EDTA was performed on cells at a confluence of 80-90%. BMSCs face replicative senescence, exhibiting progressive shortening of chromosomal telomeres, reduced stemness and a heightened risk of mutation (87, 88). Although no comprehensive studies have reported which generation will undergo cell senescence, the loss of typical fibroblast-like morphology and the decreased rate of fibroblast colony-forming units (CFU) are representations of ex vivo cellular ageing (88–90). An ex vivo study also revealed a dramatic decrease in the potential for hepatic differentiation at later passages (passage 8, p8) (91), suggesting that early passage of BMSCs may have superior therapeutic benefits for liver failure.
4 Homing functions of MSCs in ALF treatment
Homing to damaged tissue sites is a key property of BMSCs in treating liver failure. Regardless of the method of local or systemic administration, BMSCs are always found at sites of damaged tissue (92). Proinflammatory chemokines, such as TNF-α and histamine secreted by the injured liver, activate blood vascular endothelial cells (ECs), as indicated by the upregulated expression of selectin and VCAM1 (93). Once MSCs rolled to the vascular wall, a few significant ligands related to MSC extravasation, such as CD44 (HCAM), CXCR4, and VLA-4, were triggered. The adherence process occurs when the adhesion molecule selectin ligand CD44 (HCAM) expressed by MSCs interacts with selectins located on ECs (94, 95). Notably, CD44 is a significant target for modifying MSCs. During the activated phase, stromal cell-derived factor (SDF)-1 naturally expressed by ECs binds to the chemokine receptor CXCR4 on MSCs or CXCR7 and other chemokines, such as MCP-1 and MCP-3 (96–99), increasing the affinity for integrins (100). Several preclinical studies indicated that MSCs with enhanced CXCR4 expression after genetic engineering, treatment with ALF rat serum or stimulation with inflammatory cytokines such as TNF-α and IL-1β/3 showed better homing in vivo (101–106). The entrapment phase involves the VLA-4-VCAM-1 interaction, which is the key mediator of the MSC adherence process to ECs (107, 108). During the extravasation stage, the transmigration of MSCs begins with the activation of matrix metalloproteinases (MMPs), which break down type IV collagen in the basement membrane and then cross the endothelial cell layer and basement membrane into the vasculature to migrate to the liver lesion (109, 110), where they can then exert their therapeutic, regeneration and immunomodulatory effects.
5 Immunomodulatory functions of MSCs in ALF treatment
MSCs can improve and repair injured tissue by regulating immune responses by secreting soluble factors and direct cell-to-cell interactions (111). When MSCs migrate to damaged sites, they interact closely with numerous proinflammatory cytokines, such as TNF-α, IL-1β and IL-6, causing the conversion of MSCs to an immunosuppressive phenotype to modulate innate and adaptive immune responses (112). In this section, we mainly focus on soluble factors and membrane-bound molecules involved in MSC immune modulation (Figure 1).
5.1 Adaptive immune cells
MCSs can inhibit T‐cell proliferation and activation and induce the differentiation of Tregs. Several soluble immunosuppressive factors secreted by MSCs are involved in T-cell immunoregulation; the release of HLA-G1, TGF-β and HGF induces cell cycle arrest in G1 phase by downregulating phosphoretinoblastoma (pRb), cyclin D and cyclin A as well as upregulating cyclin-dependent kinase inhibitor 1B (p27Kip1), rendering T-cell activation ineffective (113, 114), and the production of IDO after IFN‐γ stimulation promotes tryptophan metabolism, resulting in the depletion of tryptophan, which inhibits proliferation and induces apoptosis of T cells (115). The direct interaction between MSCs and T cells may trigger T-cell apoptosis through the Fas ligand (FasL)-dependent pathway (116) as well as the PD-L1 pathway (117); of note, Fas ligand-associated T-cell apoptosis can induce macrophages to produce TGF-β, thereby increasing the abundance of Tregs (116). Additionally, several studies revealed that MSCs could inhibit the differentiation of Th17 cells through different mechanisms; the expression of CD54 recruited Th17 cells to MSCs and then upregulated PD‐1, IL‐10 and PGE2, blocking differentiation (118, 119). Some reports have also shown that activated T cells can differentiate into Th2 (120), CD3+CD45RO+ memory Treg cells (121), CD8+CD28‐ Treg (122), IL10+ Tr1 and TGFβ+ Th3 (123) cells in the presence of MSCs, which suppress immune responses and accelerate tissue repair. Although the detailed mechanism of the interaction between MSCs and B cells is controversial, it is known that the inhibition of B-cell proliferation by MSCs seems to be associated with cell cycle disruption at specific stages rather than the induction of apoptosis (124, 125). Activated MSCs can interfere with the formation of plasmacytes and promote the differentiation of Bregs; for example, the soluble molecule IDO secreted by MSCs is involved in the survival and proliferation of CD5+ regulatory B cells (126), and when MSCs express IL-10, they promote the production of CD19+CD24+CD38+ Bregs in humans and CD19+CD1d high CD5+ Bregs in mice (126, 127). In addition, MSCs can promote the formation of naive, transitional and memory B-cell subsets, and these nonactivated B cells can induce Treg differentiation. Notably, the MSC-derived CC chemokine ligands CCL2 and CCL7 can suppress immunoglobulin (such as IgA and IgM IgG) production and release by plasmacytes (128).
5.2 Innate immune cells
Natural killer cells (NK cells) are important effector cells of innate immunity (129). MSCs can inhibit the proliferation, cytotoxic activity and cytokine production of resting NK cells (130). IL-2-mediated proliferation of resting NK cells is inhibited by coculture with MSCs. The mechanism may involve the release of IDO, PGE2 and HLA-G5 (131–133), and upregulating the expression of HLA class I molecules (MHCI) inhibits cytokine-mediated induced NK-cell cytotoxicity and decreases the secretion of cytokines (130). MSCs also maintain the activity of neutrophils for a long period to promote the elimination of invading bacteria (134), and the MSC-derived soluble factor IL-6 can delay apoptosis of neutrophils and inhibit the neutrophil-mediated fulminant inflammatory response, which is called the respiratory burst (135). The immunoregulatory effect of MSCs on macrophages mainly converts polarized inflammatory macrophages (M1) to anti-inflammatory macrophages (M2). The mechanism may involve IDO, TSG-6 and PGE2 (134, 136–138). Furthermore, proinflammatory factors (IFN‐γ, TNF‐α and LPS) can enhance the M2 macrophage polarization of MSCs. To date, the membrane-bound molecules CD54 and CD200 have been found to increase the immunosuppressive function of MSCs (139, 140). Regarding myeloid dendritic cells (mDCs), MSCs can inhibit the development and maturation of mesenchymal/dermal DCs and the conversion of umbilical cord blood and CD34+ haematopoietic progenitors as well as monocytes into DCs (141) (142, 143). Several recent studies suggested that MSC-derived IL-6, macrophage colony-stimulating factor (M-CSF), TSG-6 and PGE2 could be responsible for the immunoregulatory interaction between MSCs and immature dendritic cells (141, 142, 144). Furthermore, MSCs can induce the transformation of mature dendritic cells into immunosuppressive regulatory dendritic cells via jagged-2 and IL-10-activated SOCS3 pathways while escaping from their apoptotic fate (145, 146), and IL-10-plasmacytoid dendritic cells (pDCs) can be induced by PGE2 (124).
These soluble and membrane molecules play important roles in MSC immunomodulation, and researchers can evaluate the effectiveness of pretreatment in vitro for improving the immunomodulation of MSCs. Recently, the concept of immune training of MSCs has been proposed (147–149), where MSCs are stimulated in vitro by proinflammatory factors or cocultured with activated immune cells, and when MSCs are stimulated again with the same stimuli, detection of the immunosuppressive molecules we mentioned can be used to assess whether MSCs can achieve “memory” to rapidly and efficiently suppress inflammatory signaling.
6 Developing animal models of liver failure and the mechanism of MSC therapy
6.1 Devascularization-induced ischaemia-reperfusion model
Devascularization models mainly imitate hepatic ischaemia-reperfusion injury (IRI) caused by liver transplantation, hepatectomy and haemorrhagic shock (150, 151), and such models have also been used to investigate liver regeneration and the therapeutic potential of artificial liver support systems (ALSSs) (152, 153). Hepatocytes and endothelial cells experienced hypoxic insult during a brief period of ischaemia. Subsequently, dysfunctional mitochondrial respiratory chain-activating degradative enzymes cause a range of disruptions in intracellular proteins, lipids and DNA. Reperfusion produces reactive oxygen species (ROS) and hydroxyl radicals that activate Kupffer cell amplification cascades and inflammatory responses, recruit neutrophils (154), and trigger different types of cell death, including apoptosis, autophagy-associated cell death and necrosis (155).
As many investigations have shown, ischaemia-reperfusion models in rats (156), mice (157) (C57BL/6 mice were described as the most popular model) and pigs (158) (in combination with hepatectomy) have been developed via 70% hepatic segmental thermal ischaemia. The critical protocol is a noninvasive vascular clip on the upper left side of the portal triad structure (bile duct, portal vein and hepatic artery) for 45 (159), 60 or 90 (160) minutes to block the blood supply to the left and median lobe of the liver, and reperfusion is initiated by removal of the clamp (161). A previous study revealed reproducible hepatic injury at 60 min of ischaemia and is therefore extensively employed in ischaemia-reperfusion models (157, 161–164), allowing decompression of the portal vein through the right lobe and caudate lobe. To prevent mesenteric vein congestion, all surgical procedures were performed at a constant temperature of 37°C.
Tail vein (165, 166), hepatic vein (167) and peripheral vein (168) injection of MSCs in an ischaemia-reperfusion injury animal model showed that MSC infusion can reduce liver damage and cell death (165, 167), improve the levels of ALT and AST (166–168) and mainly decrease oxidative stress caused by liver excision and ischaemia-reperfusion injury (169). MSCs also inhibit the production of proinflammatory cytokines (TNF-α, IL-1β and iNOS), macrophage activation and neutrophil recruitment and promote anti-inflammatory cytokine secretion (IL-10), which is beneficial for recovery from liver injury and inflammatory responses (167). The potential mechanism of MSCs in the treatment of liver IRI may increase CD47 expression in the liver, and then the CD47-SIRPα interaction activates HEDGEHOG/SMO/Gli1 signaling and further inhibits NEK7/NLRP3 activity to protect the integrity of the liver (166). Zheng et al. found that MSCs upregulated PINK1-dependent mitophagy and exerted a protective effect in liver IRI, which might be associated with the modulation of AMPKα activation (168).
6.2 Acetaminophen-induced drug-induced liver injury models
Acetaminophen-induced drug-induced liver injury (DILI) is the most frequent cause of ALF in many Western countries, such as the United States and the United Kingdom. The animal models caused by acetaminophen are more similar to the pathophysiological characteristics of liver failure in humans (170). The toxicity mechanism of excess acetaminophen-induced oncotic necrosis begins with the accumulation of a toxic metabolite, N-acetyl-benzoquinone imine (NAPQI), catalysed by the cytochrome P450 enzyme system (171). Subsequently, a large amount of reactive oxygen species are formed, which initiates severe mitochondrial oxidative stress (172), mainly through activation of MAP kinase and translocation of phospho-c Jun N-terminal kinase (p-JNK) to mitochondria (173); the mitochondria eventually undergo swelling of the cytoplasm and rupture of the outer membranes (174), with the release of endonucleases to degrade nuclear DNA (175, 176). Several species, such as mice, rats, rabbits, dogs and pigs, have been used to develop acetaminophen models, and mice are widely used because they develop liver failure very close to those reported in humans in pathophysiology structure and acetaminophen doses (177).
Furthermore, the acetaminophen dose, the diluent, the administration route and the mouse strain are critical factors that need to be considered for model development. Many studies have noted that because glutathione can relieve the toxicity associated with NAPQI (178), fasting before acetaminophen administration maintains baseline levels of glutathione in all animals, which increases the consistency of experimental results and the success of acetaminophen-induced liver failure. In previous studies, typical hepatotoxicity was observed in fasted mice at doses of 200-300 mg/kg and in nonfasted mice at doses of 500-600 mg/kg or higher, and acetaminophen was diluted in normal saline (NS) or phosphate-buffered saline (PBS) and administered intraperitoneally to mice, while intravenous or subcutaneous administration is more suitable for large animals.
Some studies have reported that tail vein transplantation of MSCs significantly improves the survival rate of mice with liver failure induced by APAP and ameliorates liver function by reducing intense centrilobular necrosis and inflammatory infiltration (179–183). A recent study showed that MSC therapy can efficiently improve APAP-induced mitochondrial dysfunction and liver injury by inhibiting c-Jun N-terminal kinase (JNK)-mediated mitochondrial retrograde pathways (179). Another study reported that MSC-mediated immunoregulation is associated with the activation of the Notch2/COX2/AMPK/SIRT1 pathway (183). More interestingly, MSCs can enhance antioxidant activity to attenuate liver damage by inhibiting cytochrome P450 activity (by reducing NAPQI production) to reduce the depletion of GSH and oxidative stress. These results might be related to the downregulation of MAPK signalling and the decreased inflammatory responses (180).
6.3 Carbon tetrachloride-induced drug-induced liver injury model
Carbon tetrachloride (CCL4), a classical hepatotoxin, induces DILI after single high-dose administration and can progress to chronic liver disease (CLD) (184) such as nonalcoholic steatohepatitis (NASH) (185), hepatocellular carcinoma (HCC) (186), acute-on-chronic liver failure or other alcohol-related liver disease and fatty liver disease after multiple low-dose administrations (187). After entering the body, CCL4 depends on the cytochrome P450 enzyme system metabolism for conversion into reactive trichloromethyl radicals with high activity (188). These metabolites can cause lipid peroxidation and hepatocyte membrane rupture, as well as DNA strand breakage. In addition, it has been revealed that such damage further affects the transcriptional and replication activity of hepatocytes, resulting in portacaval zone necrosis (189).
In general, CCL4 administration is performed in mice, rats and rabbits (BALB/c mice have been described as the most appropriate model) via intragastric administration, intraperitoneal injection, subcutaneous injection or inhalation to induce acute or chronic liver failure. In some investigations, 6- to 8-week-old male mice (weighing approximately 25-30 g) were used to develop acute mouse models of CCL4 induction (190, 191), in which olive or corn oil served as diluents to solubilize CCL4 at ratios ranging from 10% (v/v) to 50% (v/v), and CCL4 doses of 2 mL/kg or higher can induce acute liver failure in mice (191–194).
MSC therapy efficiently prolonged the survival time of CCL4 induced acute liver failure mice from day 2 to day 7 after transplantations of second trimester amniotic fluid (AF-MSCs), and the ALT and AST levels significantly decreased by 35.36% and 64.72%, respectively (195). In addition, Milosavljevic et al. found that MSCs can modulate the IL17 signaling to treat the immune-mediated liver failure via altering NKT17/NKTreg ratio and suppressing hepatotoxicity of NKT cells in an IDO-dependent manner (196). A previous study reported that the treatment effect of adipose tissue-derived mesenchymal stem cells (AT-MSCs) may relate to the secretion of interleukin 1 receptor (IL-1R), IL-6, IL-8, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), monocyte chemotactic protein 1, nerve growth factor, and hepatocyte growth factor (197).
6.4 D-gel/LPS-induced TNF-α-mediated liver failure model
Lipopolysaccharide (LPS) is a molecule that is present in the outer membrane of gram-negative bacteria and can activate Kupffer cells (198–200), triggering the secretion of multiple inflammatory mediators (200, 201), and the coadministration of 300 mg/kg D-Gal dramatically increased rodent susceptibility to LPS, resulting in extensive liver injury and cell death (202). D-Gal is a hepatocellular phosphate uracil nucleotide interference agent that is metabolized via the galactose pathway and can cause diffuse necrosis and inflammation rather than zonal necrosis, similar to most hepatotoxic drugs (153). The administration of D-Gal/LPS in mice induced liver necrosis and inflammation similar to human hepatitis (203, 204). Previous studies demonstrated that coadministration of LPS at doses of 10-100 µg/kg and D-Gal at doses of 100-1000 mg/kg was performed to establish an acute liver failure mouse model (205–209).
The pathophysiological mechanism of D-Gal/LPS-induced ALF involves the binding of LPS to Toll-like receptor 4 (TLR4) on Kupffer cells, which triggers transcriptional and translational activation of cytokines such as TNF-α, IL-1β and IL-6 (210, 211). In particular, TNF-α has been recognized as a key regulator of hepatitis, as it recruits many neutrophils into the liver sinusoids and induces the expression of various adhesion molecules, including intercellular adhesion molecule 1 (ICAM1), vascular adhesion molecule 1 (VCAM1), selectin, and chemokines on endothelial cells and hepatocytes, after LPS treatment (212, 213). Some of these adhesion molecules are critical for neutrophil extravasation and cytotoxicity. In addition, by binding to its receptor 1 (TNFR1) on hepatocytes, TNF-α activates the nuclear factor kappa beta (NF-κB) pathway, resulting in the expression of proinflammatory and antiapoptotic genes (214, 215). Although high doses of D-gal inhibite the synthesis of antiapoptotic genes by depleting uridine triphosphate in hepatocytes, they promote apoptosis signaling via activation of the caspase cascade and DNA damage. Thus, TNF-α-mediated apoptotic signaling and inflammation are commonly considered pathophysiological mechanisms of D-Gal/LPS-induced ALF. Notably, the interaction between these mechanisms remains uncertain and should be explored in the future.
BMSC transplantation rescued the D-gal-induced liver failure model. In rodents, the 4-week survival rate significantly increased by 80% (216). Numerous hepatocytes were repaired, with only a few necrotic areas. In D-gal-induced liver failure in large animals, BMSC therapy significantly prolonged the survival time from 3.22 days to more than 14 days by suppressing the life-threatening cytokine storm. BMSC-derived hepatocytes were widely distributed in injured livers within 10 weeks, with liver function returning to normal levels (217). During recovery, serum levels of proinflammatory molecules, including IFN-γ, IL-1β, and IL-6, were reduced, while serum levels of the anti-inflammatory cytokine IL-10 were significantly increased through paracrine effects, referring to regulation by the STAT3 signaling pathway (216) and notch-DLL4 signaling pathway (218).
6.5 JO2-induced Fas/FasL-mediated liver failure model
Fas receptor (CD95), a member of the TNF-receptor superfamily with a death domain, mediates the assembly of a death-inducing signaling complex. Inducing caspase activation and cell apoptosis (219) has been considered the critical mechanism of fulminant liver failure (220), ischaemia-reperfusion-associated liver diseases (221), nonalcoholic fatty liver disorders (222) and other acute and chronic hepatic disorders. The liver constitutively and abundantly expresses the Fas receptor and activated caspase (casp) 8 upon binding of FasL or other receptor agonists (220, 223), such as the Fas receptor antibody JO2 and soluble FasL in the hexameric form (MegaFasL) (198). Then, it triggers the caspase cascade accompanied by excessive hepatocyte apoptosis (224), which can quickly progress to secondary necrosis (225). This mechanism may rely on activation of Fas-induced inflammatory signaling via the nonclassical interleukin-1β pathway (225).
Several researchers have reported that JO2 at concentrations such as 0.15, 0.2, 0.23, 0.35, 0.4, 0.42, and 0.5 mg/kg can induce liver failure, and the severity of liver injury is dependent on the JO2 dose (220, 221, 225–231). Thus, the critical element for developing this animal model is the concentration of JO2. Shao et al. (220) observed severe liver damage, including destruction of the hepatic lobules, hepatocyte necrosis and haemorrhage after treatment with 0.5 mg/kg JO2 (dissolved in normal saline (NS)) in BALB/c mice via intraperitoneal injection. However, after treatment of C57BL/6 mice with the same doses and methods, all mice died within 12 h (231). Although this difference could be caused by differences across researchers or other environmental factors, strain differences. In addition, the administration route can affect the pharmacodynamics and pharmacokinetics of the drug.
In recent works, BMSC transplantation rescued mice with JO2-induced liver failure and prolonged the survival time by improving liver function and decreasing extensive hepatic necrosis and haemorrhage. BMSCs can colonize injured mice and transdifferentiate into hepatocytes and cholangiocytes, and many KRT7- and KRT19-positive human cholangiocytes form tubular structures around the portal area (22). Meanwhile, transplanted BMSCs differentiate into immune cell lineages, including T cells, B cells, natural killer (NK) cells, macrophages and dendritic cells, and play a paracrine role by regulating inflammatory cytokine levels (23). Another study confirmed this finding and further identified the two transdifferentiation phases by transcriptomics; hepatic metabolism and liver regeneration were characterized in the first 5 days after BMSC transplantation, and immune cell growth and extracellular matrix (ECM) regulation were observed from day 5 to day 14 (25).
7 BMSC transplantation routes in animal and clinical trials
In clinical trials, MSC transplantation routes involving intravenous injection, followed by intrahepatic injection (through the portal vein and hepatic artery), and intrasplenic injection are minimally used (232). Notably, different routes could affect the number of MSCs homing to sites of damage. Next, we discuss which routes resulted in optimal therapeutic benefits.
Peripheral intravenous injection, such as caudal or jugular venous injection, is the most common administration route in clinical trials and animal models owing to the simplicity of the technique and the success rate rather than promising therapeutic results. In our previous study, we transplanted human-derived BMSCs (hBMSCs) into pigs with D-gal-induced fulminant hepatic failure (FHF) via peripheral and intraportal veins. The results revealed that all animals died of FHF within 96 hours after peripheral intravenous injection of hBMSCs, while most animals were rescued and survived for up to 6 months after intraportal vein injection (217), which suggested that the intraportal route has a better therapeutic effect than peripheral intravenous injection. E. Eggenhofer et al. radiolabelled MSCs with Cr-51 and found that within the first 24 hours after tail vein infusion, most viable MSCs accumulated in the lungs and that beyond 24 hours, MSCs disappeared in the lungs and were probably cleared by immune cells, with less than 10% of the cellular debris transferred to the injured liver (233). Similarly, Mami Higashimoto also found that a large proportion of MSCs resided in the lungs after caudal vein infusion, with only a small number of cells homing to the hepatic site of conA injury (234). This finding indicates that after the intravenous injection of MSCs, the cells first moved into the lungs and subsequently moved towards the liver, where they may be phagocytosed by reticuloendothelial cells in the capillary tissue, diminishing their therapeutic potential.
In contrast to intravenous injection, the intrahepatic portal vein is an important structure in the hepatic portal system that allows MSCs to rapidly home and colonize the liver after grafting and avoids cellular off-target effects. A comprehensive preclinical study compared four different transplantation routes: intraportal injection, intrahepatic artery injection, intravenous injection and intrahepatic injection. The results indicated that compared to other routes, intraportal injection of MSCs efficiently improved liver function, inhibited apoptosis and prolonged survival in ALF swine (235). An additional study confirmed that portal vein grafts can reduce the inflammatory response, inhibit cellular necrosis and promote liver regeneration in pigs with ALF (236). This preclinical evidence can ultimately guide the choice of graft route for the treatment of MSCs in the clinic. Regarding whether portal injection performs better in clinical trials, there was a trial comparing the therapeutic efficacy of MSCs after portal vein and intrasplenic injection in patients with end-stage liver diseases. According to the Fatigue Impact Scale and the MELD score, portal injection was found to be more effective than intrasplenic injection only in the first month, and this difference disappeared in the following months. The results demonstrated that the portal vein is more beneficial for the migration of MSCs. In particular, splenic injection could be the most promising route of transplantation in the future because of its simplicity (237). Recently, Ogasawara et al. found that the limited transplantable space in the spleen resulted in many cells clustered together experiencing high pressure, which may inhibit graft function (238). However, another animal study showed that transplantation of BMSCs via intrasplenic injection rescued a large proportion of 84.6% of FHF mice (23).
It is clear that intraportal injection can be chosen as the optimal administration route for MSCs to treat liver failure. Another possible reason for the excellent performance of portal vein transplantation is that there is an adequate graft area, and the graft can be widely distributed within the hepatic sinusoids and be maintained in good condition (238).
8 Future challenges and perspectives
MSCs are recognized as a promising cell therapy for the treatment of complications of liver transplantation, liver cancer, cirrhosis and liver failure caused by HBV, HCV, alcohol, primary biliary cholangitis and other infections (17, 18). Autologous bone marrow MSCs are the predominant source of cells, but the aspiration of bone marrow from patients themselves is still an invasive procedure, and the therapeutic efficiency of bone marrow MSCs can be limited by cellular senescence and differential proliferation and differentiation capacity (239). Adipose-derived MSCs may be an alternative source of cells in the future with the improvement of complex isolation strategies, and umbilical cord-derived MSCs would be a more desirable source of cells without the limitations mentioned above (240). While embryonic stem cell-derived extracellular vesicles have been verified to rejuvenate senescent MSCs and enhance their therapeutic effects, the antisenescence mechanism may be associated with the IGF1/PI3K/AKT pathway (241). MSC-derived extracellular vesicles have also been explored as a cell-free therapy that can effectively treat liver failure and avoid cell rejection (242–245).
The persistence time of MSCs for continued remission and maintenance of liver function have no consistent conclusion in various studies. Some clinical trials for acute-on-chronic liver failure showed that the MSC group can significantly improve liver function at 24 weeks or 48 weeks of follow-up, as shown in Table 2, alleviate TBIL levels and MELD scores, and decrease the mortality rate (247, 248, 251–253, 255). However, some studies have found that MSC transplantation has no significant effect (249, 256, 257). These results may be caused by the source of MSCs, quantity of MSCs, cell dosage, treatment frequency, endpoints and small number of cohorts.
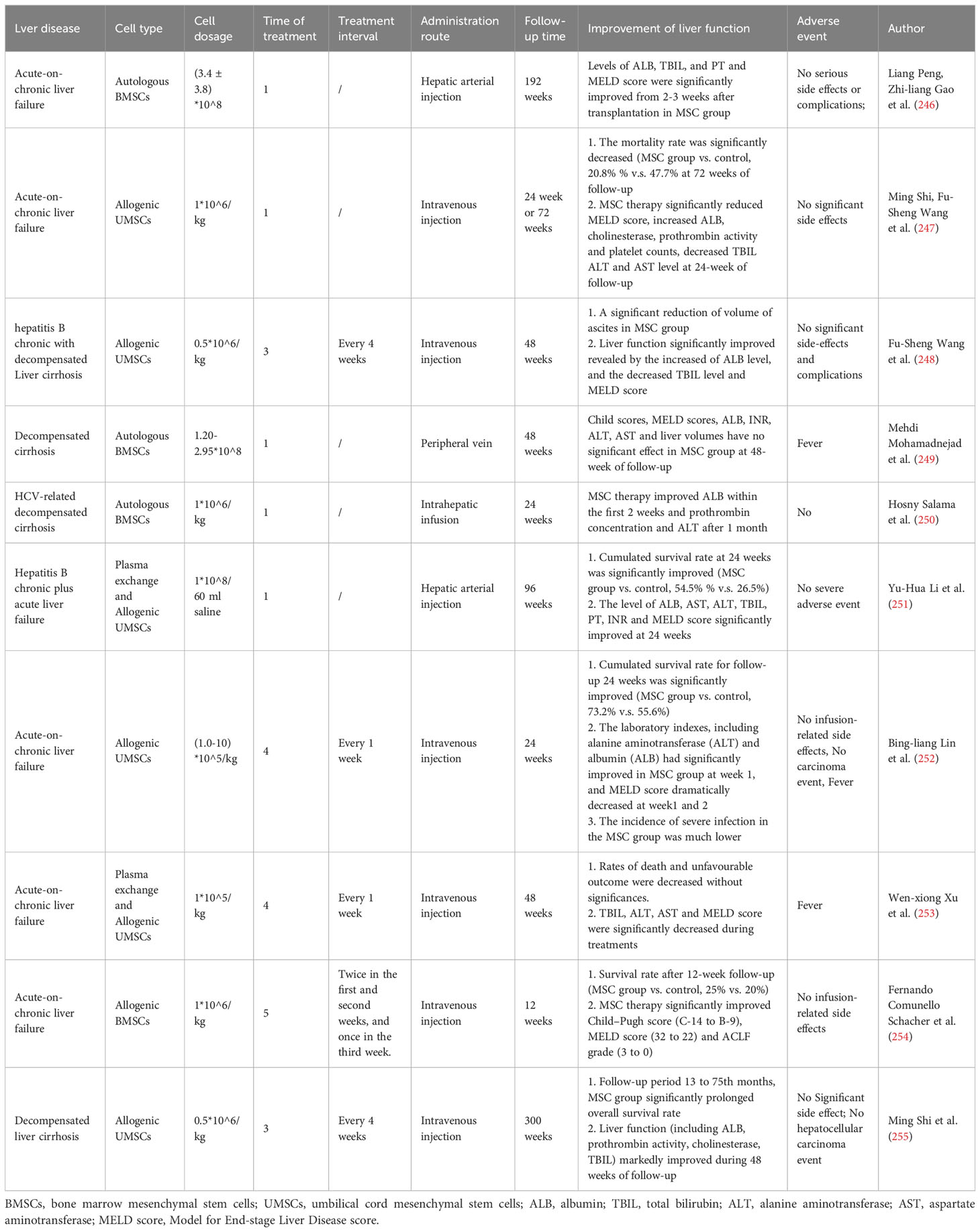
Table 2 Clinical trials of MSC therapy in acute liver disease.
The quality of MSCs is a critical factor that needs to be considered, as in many clinical trials, the characteristics of autologous BMSCs from patients of different ages and disease states vary significantly; therefore, there is an urgent need to establish uniform criteria for evaluating the quality of MSCs to support autologous or allogeneic transplantation.
Although MSCs can improve liver function and effectively treat liver failure in the short term and can be used as a cell source to modulate cellular properties and improve the effectiveness of bioartificial liver systems, their long-term efficacy in patients with decompensated end-stage liver disease remains poor (246). Zhang Z et al. first reported 45 patients with chronic hepatitis B decompensation who received MSC transfusions at 0.5 x 106 cells/kg three times at 4-week intervals. Clinical parameters were measured at 40 weekly follow-ups, and the results demonstrated that MSC treatment markedly reduced ascites and improved liver function in patients with decompensated liver cirrhosis (248). Other similar research also reported the effectiveness of multiple injections. Peripheral intravenous infusion of MSCs at a dose of 0.5 x 106 cells/kg 3 times, 4 weeks apart for chronic hepatic failure and chronic hepatitis B liver failure, effectively prolonged the overall survival time and improved the biochemical liver index (247, 255, 258). A total of 0.5 x 107 cells were injected via the hepatic artery twice in weeks 4 and 8 for alcoholic cirrhosis disease, which improved the patients’ liver histological features (259, 260). Multiple injections of MSCs may achieve long-term therapy. The treatment interval of MSCs that can maintain liver function is shown in Table 2, including every week for 4 weeks, twice in the first and second weeks and once in the third week for a total of 5 times, every four weeks for a total of three times and only one infusion (247, 252–255). However, no uniform guideline has been defined, and there is an urgent need to address this issue through extensive animal and clinical trials.
Current studies on MSC therapy have some limitations. MSCs are a heterogeneous population that limits their consistent treatment effects. Although Table 1 lists the special markers of MSCs, cell subsets with specific biological functions have not been identified. The MSC atlas is an urgent acquirement for screening special cell subpopulations aimed at different diseases. The guidelines for isolating and cultivating high-quality MSCs have not been uniformed. The mechanisms of MSC therapy for acute liver failure are still poorly understood. How does MSC migrate to the site of injury from spatial distribution, and in what form does it treat hepatocyte failure and regulate the immune microenvironment. Therefore, multi-omics combinations, including spatial transcriptomics, single-cell transcriptomics, proteomics, metabolomics, and bulk transcriptomics, are an instant demand to generally clarify the mechanisms of MSC therapy. Highly simulated mouse models of human acute liver failure need to be constructed to better evaluate the efficacy of MSCs in preclinical studies and provide more evidence-based medical evidence for clinical trials. MSCs have been used in perioperative care for liver transplantation and to improve immune rejection after liver transplantation (10, 11). However, the persistence, frequency and early initiation time of MSC treatment have no consistent conclusion, which requires further validation in multi-central, large sample, non-random cohorts. The comparison and combination of MSC therapy and other strategies, such as xenotransplantation, is an important direction for future studies.
Author contributions
HY and JC contributed equally. HY drafted the work. JL conceived and revised the work. JC drew and revised the figure. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (81830073, 81901901), the State’s Key Project of Research and Development Plan of China (2022YFA1104100), the Natural Science Foundation of Zhejiang Province (LY21H030007, LGF21H200006), the National Special support program for high-level personnel recruitment (Ten-thousand Talents Program).
Acknowledgments
We would like to thank Figdraw (www.figdraw.com) for its help in creating the figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee WM, Stravitz RT, Larson AM. Introduction to the revised American Association for the Study of Liver Diseases Position Paper on acute liver failure 2011. Hepatology (2012) 55(3):965–7. doi: 10.1002/hep.25551
2. Bernal W, Wendon J. Acute liver failure. N Engl J Med (2013) 369(26):2525–34. doi: 10.1056/NEJMra1208937
3. O'Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet (1993) 342(8866):273–5. doi: 10.1016/0140-6736(93)91818-7
4. Ezquer F, Huang YL, Ezquer M. New perspectives to improve mesenchymal stem cell therapies for drug-induced liver injury. Int J Mol Sci (2022) 23(5):2669. doi: 10.3390/ijms23052669
5. Londoño MC, Rimola A, O'Grady J, Sanchez-Fueyo A. Immunosuppression minimization vs. complete drug withdrawal in liver transplantation. J Hepatol (2013) 59(4):872–9. doi: 10.1016/j.jhep.2013.04.003
6. Chen Y, Wong PP, Sjeklocha L, Steer CJ, Sahin MB. Mature hepatocytes exhibit unexpected plasticity by direct dedifferentiation into liver progenitor cells in culture. Hepatology (2012) 55(2):563–74. doi: 10.1002/hep.24712
7. Huebert RC, Rakela J. Cellular therapy for liver disease. Mayo Clin Proc (2014) 89(3):414–24. doi: 10.1016/j.mayocp.2013.10.023
8. Lee CW, Chen YF, Wu HH, Lee OK. Historical perspectives and advances in mesenchymal stem cell research for the treatment of liver diseases. Gastroenterology (2018) 154(1):46–56. doi: 10.1053/j.gastro.2017.09.049
9. Li Y, Lu L, Cai X. Liver regeneration and cell transplantation for end-stage liver disease. Biomolecules (2021) 11(12):1907. doi: 10.3390/biom11121907
10. Hu XH, Chen L, Wu H, Tang YB, Zheng QM, Wei XY, et al. Cell therapy in end-stage liver disease: replace and remodel. Stem Cell Res Ther (2023) 14(1):141. doi: 10.1186/s13287-023-03370-z
11. Detry O, Vandermeulen M, Delbouille MH, Somja J, Bletard N, Briquet A, et al. Infusion of mesenchymal stromal cells after deceased liver transplantation: A phase I-II, open-label, clinical study. J Hepatol (2017) 67(1):47–55. doi: 10.1016/j.jhep.2017.03.001
12. Vandermeulen M, Mohamed-Wais M, Erpicum P, Delbouille MH, Lechanteur C, Briquet A, et al. Infusion of allogeneic mesenchymal stromal cells after liver transplantation: A 5-year follow-up. Liver Transpl (2022) 28(4):636–46. doi: 10.1002/lt.26323
13. Casiraghi F, Perico N, Remuzzi G. Mesenchymal stromal cells to promote solid organ transplantation tolerance. Curr Opin Organ Transpl (2013) 18(1):51–8. doi: 10.1097/MOT.0b013e32835c5016
14. Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of Guinea-pig bone marrow and spleen cells. Cell Tissue Kinet (1970) 3(4):393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x
15. Naji A, Eitoku M, Favier B, Deschaseaux F, Rouas-Freiss N, Suganuma N, et al. Biological functions of mesenchymal stem cells and clinical implications. Cell Mol Life Sci (2019) 76(17):3323–48. doi: 10.1007/s00018-019-03125-1
16. Mosna F, Sensebé L, Krampera M. Human bone marrow and adipose tissue mesenchymal stem cells: a user's guide. Stem Cells Dev (2010) 19(10):1449–70. doi: 10.1089/scd.2010.0140
17. Eom YW, Yoon Y, Baik SK. Mesenchymal stem cell therapy for liver disease: current status and future perspectives. Curr Opin Gastroenterol (2021) 37(3):216–23. doi: 10.1097/MOG.0000000000000724
18. Zhang S, Yang Y, Fan L, Zhang F, Li L. The clinical application of mesenchymal stem cells in liver disease: the current situation and potential future. Ann Transl Med (2020) 8(8):565. doi: 10.21037/atm.2020.03.218
19. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science (1999) 284(5411):143–7. doi: 10.1126/science.284.5411.143
20. Petersen BE, Bowen WC, Patrene KD, Mars WM, Sullivan AK, Murase N, et al. Bone marrow as a potential source of hepatic oval cells. Science (1999) 284(5417):1168–70. doi: 10.1126/science.284.5417.1168
21. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy (2006) 8(4):315–7. doi: 10.1080/14653240600855905
22. Sun S, Yuan L, An Z, Shi D, Xin J, Jiang J, et al. DLL4 restores damaged liver by enhancing hBMSC differentiation into cholangiocytes. Stem Cell Res (2020) 47:101900. doi: 10.1016/j.scr.2020.101900
23. Yuan L, Jiang J, Liu X, Zhang Y, Zhang L, Xin J, et al. HBV infection-induced liver cirrhosis development in dual-humanised mice with human bone mesenchymal stem cell transplantation. Gut (2019) 68(11):2044–56. doi: 10.1136/gutjnl-2018-316091
24. Li J, Xing F, Chen F, He L, So KF, Liu Y, et al. Functional 3D human liver bud assembled from MSC-derived multiple liver cell lineages. Cell Transplant (2019) 28(5):510–21. doi: 10.1177/0963689718780332
25. Sun S, Yang H, Xin J, Yao H, Yuan L, Ren K, et al. Transcriptomics confirm the establishment of a liver-immune dual-humanized mouse model after transplantation of a single type of human bone marrow mesenchymal stem cell. Liver Int (2023) 43(6):1345–56. doi: 10.1111/liv.15546
26. Su J, Guo L, Wu C. A mechanoresponsive PINCH-1-Notch2 interaction regulates smooth muscle differentiation of human placental mesenchymal stem cells. Stem Cells (2021) 39(5):650–68. doi: 10.1002/stem.3347
27. Wang F, Zachar V, Pennisi CP, Fink T, Maeda Y, Emmersen J, et al. Hypoxia enhances differentiation of adipose tissue-derived stem cells toward the smooth muscle phenotype. Int J Mol Sci (2018) 19(2):517. doi: 10.3390/ijms19020517
28. Motta LCB, Pereira VM, Pinto PAF, Mançanares CAF, Pieri NCG, de Oliveira VC, et al. 3D culture of mesenchymal stem cells from the yolk sac to generate intestinal organoid. Theriogenology (2023) 209:98–106. doi: 10.1016/j.theriogenology.2023.06.003
29. Kopen GC, Prockop DJ, Phinney DG. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc Natl Acad Sci USA (1999) 96(19):10711–6. doi: 10.1073/pnas.96.19.10711
30. Deng J, Petersen BE, Steindler DA, Jorgensen ML, Laywell ED. Mesenchymal stem cells spontaneously express neural proteins in culture and are neurogenic after transplantation. Stem Cells (2006) 24(4):1054–64. doi: 10.1634/stemcells.2005-0370
31. Tomita M, Mori T, Maruyama K, Zahir T, Ward M, Umezawa A, et al. A comparison of neural differentiation and retinal transplantation with bone marrow-derived cells and retinal progenitor cells. Stem Cells (2006) 24(10):2270–8. doi: 10.1634/stemcells.2005-0507
32. Wislet-Gendebien S, Hans G, Leprince P, Rigo JM, Moonen G, Rogister B. Plasticity of cultured mesenchymal stem cells: switch from nestin-positive to excitable neuron-like phenotype. Stem Cells (2005) 23(3):392–402. doi: 10.1634/stemcells.2004-0149
33. Chaubey S, Wolfe JH. Transplantation of CD15-enriched murine neural stem cells increases total engraftment and shifts differentiation toward the oligodendrocyte lineage. Stem Cells Transl Med (2013) 2(6):444–54. doi: 10.5966/sctm.2012-0105
34. Corselli M, Chen CW, Crisan M, Lazzari L, Péault B. Perivascular ancestors of adult multipotent stem cells. Arterioscler Thromb Vasc Biol (2010) 30(6):1104–9. doi: 10.1161/ATVBAHA.109.191643
35. Wei X, Yang X, Han ZP, Qu FF, Shao L, Shi YF. Mesenchymal stem cells: a new trend for cell therapy. Acta Pharmacol Sin (2013) 34(6):747–54. doi: 10.1038/aps.2013.50
36. Zhang Z, Wang FS. Stem cell therapies for liver failure and cirrhosis. J Hepatol (2013) 59(1):183–5. doi: 10.1016/j.jhep.2013.01.018
37. Alvites R, Branquinho M, Sousa AC, Lopes B, Sousa P, Maurício AC. Mesenchymal stem/stromal cells and their paracrine activity-immunomodulation mechanisms and how to influence the therapeutic potential. Pharmaceutics (2022) 14(2):381. doi: 10.3390/pharmaceutics14020381
38. Mushahary D, Spittler A, Kasper C, Weber V, Charwat V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytometry A (2018) 93(1):19–31. doi: 10.1002/cyto.a.23242
39. McDaniel JS, Antebi B, Pilia M, Hurtgen BJ, Belenkiy S, Necsoiu C, et al. Quantitative assessment of optimal bone marrow site for the isolation of porcine mesenchymal stem cells. Stem Cells Int (2017) 2017:1836960. doi: 10.1155/2017/1836960
40. Schachtele S, Clouser C, Aho J. Markers and Methods to Verify Mesenchymal Stem Cell Identity, Potency, And Quality. (2020) Bio-techne.
41. Yusop N, Battersby P, Alraies A, Sloan AJ, Moseley R, Waddington RJ. Isolation and characterisation of mesenchymal stem cells from rat bone marrow and the endosteal niche: A comparative study. Stem Cells Int (2018) 2018:6869128. doi: 10.1155/2018/6869128
42. Ferrin I, Beloqui I, Zabaleta L, Salcedo JM, Trigueros C, Martin AG. Isolation, culture, and expansion of mesenchymal stem cells. Methods Mol Biol (2017) 1590:177–90. doi: 10.1007/978-1-4939-6921-0_13
43. Ghazanfari R, Zacharaki D, Li H, Ching Lim H, Soneji S, Scheding S. Human primary bone marrow mesenchymal stromal cells and their in vitro progenies display distinct transcriptional profile signatures. Sci Rep (2017) 7(1):10338. doi: 10.1038/s41598-017-09449-x
44. Huang S, Xu L, Sun Y, Wu T, Wang K, Li G. An improved protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. J Orthop Translat (2015) 3(1):26–33. doi: 10.1016/j.jot.2014.07.005
45. Lv FJ, Tuan RS, Cheung KM, Leung VY. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells (2014) 32(6):1408–19. doi: 10.1002/stem.1681
46. Samsonraj RM, Raghunath M, Nurcombe V, Hui JH, van Wijnen AJ, Cool SM. Concise review: multifaceted characterization of human mesenchymal stem cells for use in regenerative medicine. Stem Cells Transl Med (2017) 6(12):2173–85. doi: 10.1002/sctm.17-0129
47. Bourin P, Bunnell BA, Casteilla L, Dominici M, Katz AJ, March KL, et al. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: a joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT). Cytotherapy (2013) 15(6):641–8. doi: 10.1016/j.jcyt.2013.02.006
48. Battula VL, Treml S, Bareiss PM, Gieseke F, Roelofs H, de Zwart P, et al. Isolation of functionally distinct mesenchymal stem cell subsets using antibodies against CD56, CD271, and mesenchymal stem cell antigen-1. Haematologica (2009) 94(2):173–84. doi: 10.3324/haematol.13740
49. Keith MC, Bolli R. "String theory" of c-kit(pos) cardiac cells: a new paradigm regarding the nature of these cells that may reconcile apparently discrepant results. Circ Res (2015) 116(7):1216–30. doi: 10.1161/CIRCRESAHA.116.30555
50. Xu M, Shaw G, Murphy M, Barry F. Induced pluripotent stem cell-derived mesenchymal stromal cells are functionally and genetically different from bone marrow-derived mesenchymal stromal cells. Stem Cells (2019) 37(6):754–65. doi: 10.1002/stem.2993
51. Shen YS, Chen XJ, Wuri SN, Yang F, Pang FX, Xu LL, et al. Polydatin improves osteogenic differentiation of human bone mesenchymal stem cells by stimulating TAZ expression via BMP2-Wnt/β-catenin signaling pathway. Stem Cell Res Ther (2020) 11(1):204. doi: 10.1186/s13287-020-01705-8
52. Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science (2005) 309(5737):1074–8. doi: 10.1126/science.1110955
53. Markiewicz A, Topa J, Nagel A, Skokowski J, Seroczynska B, Stokowy T, et al. Spectrum of epithelial-mesenchymal transition phenotypes in circulating tumour cells from early breast cancer patients. Cancers (Basel) (2019) 11(1):59. doi: 10.3390/cancers11010059
54. Kafienah W, Mistry S, Williams C, Hollander AP. Nucleostemin is a marker of proliferating stromal stem cells in adult human bone marrow. Stem Cells (2006) 24(4):1113–20. doi: 10.1634/stemcells.2005-0416
55. Munshi A, Mehic J, Creskey M, Gobin J, Gao J, Rigg E, et al. A comprehensive proteomics profiling identifies NRP1 as a novel identity marker of human bone marrow mesenchymal stromal cell-derived small extracellular vesicles. Stem Cell Res Ther (2019) 10(1):401. doi: 10.1186/s13287-019-1516-2
56. Rameshwar P. IFNgamma and B7-H1 in the immunology of mesenchymal stem cells. Cell Res (2008) 18(8):805–6. doi: 10.1038/cr.2008.90
57. Barilani M, Peli V, Cherubini A, Dossena M, Dolo V, Lazzari L. NG2 as an identity and quality marker of mesenchymal stem cell extracellular vesicles. Cells (2019) 8(12):1524. doi: 10.3390/cells8121524
58. Lu D, Liao Y, Zhu SH, Chen QC, Xie DM, Liao JJ, et al. Bone-derived Nestin-positive mesenchymal stem cells improve cardiac function via recruiting cardiac endothelial cells after myocardial infarction. Stem Cell Res Ther (2019) 10(1):127. doi: 10.1186/s13287-019-1217-x
59. Heiskanen A, Satomaa T, Tiitinen S, Laitinen A, Mannelin S, Impola U, et al. N-glycolylneuraminic acid xenoantigen contamination of human embryonic and mesenchymal stem cells is substantially reversible. Stem Cells (2007) 25(1):197–202. doi: 10.1634/stemcells.2006-0444
60. Dimarakis I, Levicar N. Cell culture medium composition and translational adult bone marrow-derived stem cell research. Stem Cells (2006) 24(5):1407–8. doi: 10.1634/stemcells.2005-0577
61. Sundin M, Ringdén O, Sundberg B, Nava S, Götherström C, Le Blanc K. No alloantibodies against mesenchymal stromal cells, but presence of anti-fetal calf serum antibodies, after transplantation in allogeneic hematopoietic stem cell recipients. Haematologica (2007) 92(9):1208–15. doi: 10.3324/haematol.11446
62. Tekkatte C, Gunasingh GP, Cherian KM, Sankaranarayanan K. "Humanized" stem cell culture techniques: the animal serum controversy. Stem Cells Int (2011) 2011:504723. doi: 10.4061/2011/504723
63. Gottipamula S, Muttigi MS, Chaansa S, Ashwin KM, Priya N, Kolkundkar U, et al. Large-scale expansion of pre-isolated bone marrow mesenchymal stromal cells in serum-free conditions. J Tissue Eng Regener Med (2016) 10(2):108–19. doi: 10.1002/term.1713
64. Doucet C, Ernou I, Zhang Y, Llense JR, Begot L, Holy X, et al. Platelet lysates promote mesenchymal stem cell expansion: a safety substitute for animal serum in cell-based therapy applications. J Cell Physiol (2005) 205(2):228–36. doi: 10.1002/jcp.20391
65. Jung J, Moon N, Ahn JY, Oh EJ, Kim M, Cho CS, et al. Mesenchymal stromal cells expanded in human allogenic cord blood serum display higher self-renewal and enhanced osteogenic potential. Stem Cells Dev (2009) 18(4):559–71. doi: 10.1089/scd.2008.0105
66. Hoang DH, Nguyen TD, Nguyen HP, Nguyen XH, Do PTX, Dang VD, et al. Differential wound healing capacity of mesenchymal stem cell-derived exosomes originated from bone marrow, adipose tissue and umbilical cord under serum- and xeno-free condition. Front Mol Biosci (2020) 7:119. doi: 10.3389/fmolb.2020.00119
67. Gottipamula S, Ashwin KM, Muttigi MS, Kannan S, Kolkundkar U, Seetharam RN. Isolation, expansion and characterization of bone marrow-derived mesenchymal stromal cells in serum-free conditions. Cell Tissue Res (2014) 356(1):123–35. doi: 10.1007/s00441-013-1783-7
68. Bui HTH, Nguyen LT, Than UTT. Influences of xeno-free media on mesenchymal stem cell expansion for clinical application. Tissue Eng Regener Med (2021) 18(1):15–23. doi: 10.1007/s13770-020-00306-z
69. Mendicino M, Bailey AM, Wonnacott K, Puri RK, Bauer SR. MSC-based product characterization for clinical trials: an FDA perspective. Cell Stem Cell (2014) 14(2):141–5. doi: 10.1016/j.stem.2014.01.013
70. Hoang VT, Trinh QM, Phuong DTM, Bui HTH, Hang LM, Ngan NTH, et al. Standardized xeno- and serum-free culture platform enables large-scale expansion of high-quality mesenchymal stem/stromal cells from perinatal and adult tissue sources. Cytotherapy (2021) 23(1):88–99. doi: 10.1016/j.jcyt.2020.09.004
71. Palm W, Thompson CB. Nutrient acquisition strategies of mamMalian cells. Nature (2017) 546(7657):234–42. doi: 10.1038/nature22379
72. Zhou Y, Tsai TL, Li WJ. Strategies to retain properties of bone marrow-derived mesenchymal stem cells ex vivo. Ann N Y Acad Sci (2017) 1409(1):3–17. doi: 10.1111/nyas.13451
73. Al-Qarakhli AMA, Yusop N, Waddington RJ, Moseley R. Effects of high glucose conditions on the expansion and differentiation capabilities of mesenchymal stromal cells derived from rat endosteal niche. BMC Mol Cell Biol (2019) 20(1):51. doi: 10.1186/s12860-019-0235-y
74. Stolzing A, Coleman N, Scutt A. Glucose-induced replicative senescence in mesenchymal stem cells. Rejuv Res (2006) 9(1):31–5. doi: 10.1089/rej.2006.9.31
75. Chang TC, Hsu MF, Wu KK. High glucose induces bone marrow-derived mesenchymal stem cell senescence by upregulating autophagy. PloS One (2015) 10(5):e0126537. doi: 10.1371/journal.pone.0126537
76. Li YM, Schilling T, Benisch P, Zeck S, Meissner-Weigl J, Schneider D, et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem Biophys Res Commun (2007) 363(1):209–15. doi: 10.1016/j.bbrc.2007.08.161
77. Choudhery MS. Strategies to improve regenerative potential of mesenchymal stem cells. World J Stem Cells (2021) 13(12):1845–62. doi: 10.4252/wjsc.v13.i12.1845
78. Liu Y, Li Y, Nan LP, Wang F, Zhou SF, Wang JC, et al. The effect of high glucose on the biological characteristics of nucleus pulposus-derived mesenchymal stem cells. Cell Biochem Funct (2020) 38(2):130–40. doi: 10.1002/cbf.3441
79. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature (2014) 508(7495):269–73. doi: 10.1038/nature13034
80. Jin Y, Kato T, Furu M, Nasu A, Kajita Y, Mitsui H, et al. Mesenchymal stem cells cultured under hypoxia escape from senescence via down-regulation of p16 and extracellular signal regulated kinase. Biochem Biophys Res Commun (2010) 391(3):1471–6. doi: 10.1016/j.bbrc.2009.12.096
81. Dos Santos F, Andrade PZ, Boura JS, Abecasis MM, da Silva CL, Cabral JM. Ex vivo expansion of human mesenchymal stem cells: a more effective cell proliferation kinetics and metabolism under hypoxia. J Cell Physiol (2010) 223(1):27–35. doi: 10.1002/jcp.21987
82. Fehrer C, Brunauer R, Laschober G, Unterluggauer H, Reitinger S, Kloss F, et al. Reduced oxygen tension attenuates differentiation capacity of human mesenchymal stem cells and prolongs their lifespan. Aging Cell (2007) 6(6):745–57. doi: 10.1111/j.1474-9726.2007.00336.x
83. Boyette LB, Creasey OA, Guzik L, Lozito T, Tuan RS. Human bone marrow-derived mesenchymal stem cells display enhanced clonogenicity but impaired differentiation with hypoxic preconditioning. Stem Cells Transl Med (2014) 3(2):241–54. doi: 10.5966/sctm.2013-0079
84. Antebi B, Rodriguez LA, Walker KP, Asher AM, Kamucheka RM, Alvarado L. Short-term physiological hypoxia potentiates the therapeutic function of mesenchymal stem cells. Stem Cell Res Ther (2018) 9(1):265. doi: 10.1186/s13287-018-1007-x
85. Yu Y, Yin Y, Wu RX, He XT, Zhang XY, Chen FM. Hypoxia and low-dose inflammatory stimulus synergistically enhance bone marrow mesenchymal stem cell migration. Cell Prolif (2017) 50(1):e12309. doi: 10.1111/cpr.12309
86. Li H, Ghazanfari R, Zacharaki D, Lim HC, Scheding S. Isolation and characterization of primary bone marrow mesenchymal stromal cells. Ann N Y Acad Sci (2016) 1370(1):109–18. doi: 10.1111/nyas.13102
87. Zhai W, Yong D, El-Jawhari JJ, Cuthbert R, McGonagle D, Win Naing M, et al. Identification of senescent cells in multipotent mesenchymal stromal cell cultures: Current methods and future directions. Cytotherapy (2019) 21(8):803–19. doi: 10.1016/j.jcyt.2019.05.001
88. Yang YK, Ogando CR, Wang See C, Chang TY, Barabino GA. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res Ther (2018) 9(1):131. doi: 10.1186/s13287-018-0876-3
89. Bertolo A, Mehr M, Janner-Jametti T, Graumann U, Aebli N, Baur M, et al. An in vitro expansion score for tissue-engineering applications with human bone marrow-derived mesenchymal stem cells. J Tissue Eng Regener Med (2016) 10(2):149–61. doi: 10.1002/term.1734
90. Wang YH, Tao YC, Wu DB, Wang ML, Tang H, Chen EQ. Cell heterogeneity, rather than the cell storage solution, affects the behavior of mesenchymal stem cells in vitro and in vivo. Stem Cell Res Ther (2021) 12(1):391. doi: 10.1186/s13287-021-02450-2
91. Luo S, Xiao S, Ai Y, Wang B, Wang Y. Changes in the hepatic differentiation potential of human mesenchymal stem cells aged in vitro. Ann Transl Med (2021) 9(21):1628. doi: 10.21037/atm-21-4918
92. Liesveld JL, Sharma N, Aljitawi OS. Stem cell homing: From physiology to therapeutics. Stem Cells (2020) 38(10):1241–53. doi: 10.1002/stem.3242
93. Teo GS, Ankrum JA, Martinelli R, Boetto SE, Simms K, Sciuto TE, et al. Mesenchymal stem cells transmigrate between and directly through tumor necrosis factor-α-activated endothelial cells via both leukocyte-like and novel mechanisms. Stem Cells (2012) 30(11):2472–86. doi: 10.1002/stem.1198
94. Sackstein R, Merzaban JS, Cain DW, Dagia NM, Spencer JA, Lin CP, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med (2008) 14(2):181–7. doi: 10.1038/nm1703
95. Maric DM, Velikic G, Maric DL, Supic G, Vojvodic D, Petric V, et al. Stem cell homing in intrathecal applications and inspirations for improvement paths. Int J Mol Sci (2022) 23(8):4290. doi: 10.3390/ijms23084290
96. Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells (2009) 27(4):857–65. doi: 10.1002/stem.23
97. Shao Y, Zhou F, He D, Zhang L, Shen J. Overexpression of CXCR7 promotes mesenchymal stem cells to repair phosgene-induced acute lung injury in rats. BioMed Pharmacother (2019) 109:1233–9. doi: 10.1016/j.biopha.2018.10.108
98. Belema-Bedada F, Uchida S, Martire A, Kostin S, Braun T. Efficient homing of multipotent adult mesenchymal stem cells depends on FROUNT-mediated clustering of CCR2. Cell Stem Cell (2008) 2(6):566–75. doi: 10.1016/j.stem.2008.03.003
99. Schenk S, Mal N, Finan A, Zhang M, Kiedrowski M, Popovic Z, et al. Monocyte chemotactic protein-3 is a myocardial mesenchymal stem cell homing factor. Stem Cells (2007) 25(1):245–51. doi: 10.1634/stemcells.2006-0293
100. Ullah M, Liu DD, Thakor AS. Mesenchymal stromal cell homing: mechanisms and strategies for improvement. iScience (2019) 15:421–38. doi: 10.1016/j.isci.2019.05.004
101. Yang H, Feng R, Fu Q, Xu S, Hao X, Qiu Y, et al. Human induced pluripotent stem cell-derived mesenchymal stem cells promote healing via TNF-α-stimulated gene-6 in inflammatory bowel disease models. Cell Death Dis (2019) 10(10):718. doi: 10.1038/s41419-019-1957-7
102. Fan H, Zhao G, Liu L, Liu F, Gong W, Liu X, et al. Pre-treatment with IL-1β enhances the efficacy of MSC transplantation in DSS-induced colitis. Cell Mol Immunol (2012) 9(6):473–81. doi: 10.1038/cmi.2012.40
103. Yu Y, Yoo SM, Park HH, Baek SY, Kim YJ, Lee S, et al. Preconditioning with interleukin-1 beta and interferon-gamma enhances the efficacy of human umbilical cord blood-derived mesenchymal stem cells-based therapy via enhancing prostaglandin E2 secretion and indoleamine 2,3-dioxygenase activity in dextran sulfate sodium-induced colitis. J Tissue Eng Regener Med (2019) 13(10):1792–804. doi: 10.1002/term.2930
104. Wang S, Gao S, Li Y, Qian X, Luan J, Lv X. Emerging importance of chemokine receptor CXCR4 and its ligand in liver disease. Front Cell Dev Biol (2021) 9:716842. doi: 10.3389/fcell.2021.716842
105. Barhanpurkar-Naik A, Mhaske ST, Pote ST, Singh K, Wani MR. Interleukin-3 enhances the migration of human mesenchymal stem cells by regulating expression of CXCR4. Stem Cell Res Ther (2017) 8(1):168. doi: 10.1186/s13287-017-0618-y
106. Zhang C, Zhang W, Zhu D, Li Z, Wang Z, Li J, et al. Nanoparticles functionalized with stem cell secretome and CXCR4-overexpressing endothelial membrane for targeted osteoporosis therapy. J Nanobiotechnol (2022) 20(1):35. doi: 10.1186/s12951-021-01231-6
107. Steingen C, Brenig F, Baumgartner L, Schmidt J, Schmidt J, Bloch W. Characterization of key mechanisms in transmigration and invasion of mesenchymal stem cells. J Mol Cell Cardiol (2008) 44(6):1072–84. doi: 10.1016/j.yjmcc.2008.03.010
108. Aldridge V, Garg A, Davies N, Bartlett DC, Youster J, Beard H, et al. Human mesenchymal stem cells are recruited to injured liver in a β1-integrin and CD44 dependent manner. Hepatology (2012) 56(3):1063–73. doi: 10.1002/hep.25716
109. Nitzsche F, Müller C, Lukomska B, Jolkkonen J, Deten A, Boltze J. Concise review: MSC adhesion cascade-insights into homing and transendothelial migration. Stem Cells (2017) 35(6):1446–60. doi: 10.1002/stem.2614
110. Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P. MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Blood (2007) 109(9):4055–63. doi: 10.1182/blood-2006-10-051060
111. Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell (2013) 13(4):392–402. doi: 10.1016/j.stem.2013.09.006
112. Yang X, Liang L, Zong C, Lai F, Zhu P, Liu Y, et al. Kupffer cells-dependent inflammation in the injured liver increases recruitment of mesenchymal stem cells in aging mice. Oncotarget (2016) 7(2):1084–95. doi: 10.18632/oncotarget.6744
113. Giuliani M, Fleury M, Vernochet A, Ketroussi F, Clay D, Azzarone B, et al. Long-lasting inhibitory effects of fetal liver mesenchymal stem cells on T-lymphocyte proliferation. PloS One (2011) 6(5):e19988. doi: 10.1371/journal.pone.0019988
114. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood (2002) 99(10):3838–43. doi: 10.1182/blood.V99.10.3838
115. Böttcher M, Hofmann AD, Bruns H, Haibach M, Loschinski R, Saul D, et al. Mesenchymal stromal cells disrupt mTOR-signaling and aerobic glycolysis during T-cell activation. Stem Cells (2016) 34(2):516–21. doi: 10.1002/stem.2234
116. Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T, et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell (2012) 10(5):544–55. doi: 10.1016/j.stem.2012.03.007
117. Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells (2017) 35(3):766–76. doi: 10.1002/stem.2509
118. Ghannam S, Pène J, Moquet-Torcy G, Jorgensen C, Yssel H. Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J Immunol (2010) 185(1):302–12. doi: 10.4049/jimmunol.0902007
119. Luz-Crawford P, Noël D, Fernandez X, Khoury M, Figueroa F, Carrión F, et al. Mesenchymal stem cells repress Th17 molecular program through the PD-1 pathway. PloS One (2012) 7(9):e45272. doi: 10.1371/journal.pone.0045272
120. Lin R, Ma H, Ding Z, Shi W, Qian W, Song J, et al. Bone marrow-derived mesenchymal stem cells favor the immunosuppressive T cells skewing in a Helicobacter pylori model of gastric cancer. Stem Cells Dev (2013) 22(21):2836–48. doi: 10.1089/scd.2013.0166
121. Di Ianni M, Del Papa B, De Ioanni M, Moretti L, Bonifacio E, Cecchini D, et al. Mesenchymal cells recruit and regulate T regulatory cells. Exp Hematol (2008) 36(3):309–18. doi: 10.1016/j.exphem.2007.11.007
122. Liu Q, Zheng H, Chen X, Peng Y, Huang W, Li X, et al. Human mesenchymal stromal cells enhance the immunomodulatory function of CD8(+)CD28(-) regulatory T cells. Cell Mol Immunol (2015) 12(6):708–18. doi: 10.1038/cmi.2014.118
123. Mougiakakos D, Jitschin R, Johansson CC, Okita R, Kiessling R, Le Blanc K. The impact of inflammatory licensing on heme oxygenase-1-mediated induction of regulatory T cells by human mesenchymal stem cells. Blood (2011) 117(18):4826–35. doi: 10.1182/blood-2010-12-324038
124. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood (2005) 105(4):1815–22. doi: 10.1182/blood-2004-04-1559
125. Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F. Human mesenchymal stem cells modulate B-cell functions. Blood (2006) 107(1):367–72. doi: 10.1182/blood-2005-07-2657
126. Peng Y, Chen X, Liu Q, Zhang X, Huang K, Liu L, et al. Mesenchymal stromal cells infusions improve refractory chronic graft versus host disease through an increase of CD5+ regulatory B cells producing interleukin 10. Leukemia (2015) 29(3):636–46. doi: 10.1038/leu.2014.225
127. Cho KA, Lee JK, Kim YH, Park M, Woo SY, Ryu KH, et al. Mesenchymal stem cells ameliorate B-cell-mediated immune responses and increase IL-10-expressing regulatory B cells in an EBI3-dependent manner. Cell Mol Immunol (2017) 14(11):895–908. doi: 10.1038/cmi.2016.59
128. Rafei M, Hsieh J, Fortier S, Li M, Yuan S, Birman E, et al. Mesenchymal stromal cell-derived CCL2 suppresses plasma cell immunoglobulin production via STAT3 inactivation and PAX5 induction. Blood (2008) 112(13):4991–8. doi: 10.1182/blood-2008-07-166892
129. Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol (2002) 2(12):957–64. doi: 10.1038/nri956
130. Spaggiari GM, Capobianco A, Becchetti S, Mingari MC, Moretta L. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood (2006) 107(4):1484–90. doi: 10.1182/blood-2005-07-2775
131. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood (2008) 111(3):1327–33. doi: 10.1182/blood-2007-02-074997
132. Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells (2006) 24(1):74–85. doi: 10.1634/stemcells.2004-0359
133. Chatterjee D, Marquardt N, Tufa DM, Beauclair G, Low HZ, Hatlapatka T, et al. Role of gamma-secretase in human umbilical-cord derived mesenchymal stem cell mediated suppression of NK cell cytotoxicity. Cell Commun Signal (2014) 12:63. doi: 10.1186/s12964-014-0063-9
134. Alcayaga-MIranda F, Cuenca J, Khoury M. Antimicrobial activity of mesenchymal stem cells: current status and new perspectives of antimicrobial peptide-based therapies. Front Immunol (2017) 8:339. doi: 10.3389/fimmu.2017.00339
135. Raffaghello L, Bianchi G, Bertolotto M, Montecucco F, Busca A, Dallegri F, et al. Human mesenchymal stem cells inhibit neutrophil apoptosis: a model for neutrophil preservation in the bone marrow niche. Stem Cells (2008) 26(1):151–62. doi: 10.1634/stemcells.2007-0416
136. Németh K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med (2009) 15(1):42–9. doi: 10.1038/nm.1905
137. Chiossone L, Conte R, Spaggiari GM, Serra M, Romei C, Bellora F, et al. Mesenchymal stromal cells induce peculiar alternatively activated macrophages capable of dampening both innate and adaptive immune responses. Stem Cells (2016) 34(7):1909–21. doi: 10.1002/stem.2369
138. Choi H, Lee RH, Bazhanov N, Oh JY, Prockop DJ. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-κB signaling in resident macrophages. Blood (2011) 118(2):330–8. doi: 10.1182/blood-2010-12-327353
139. Espagnolle N, Balguerie A, Arnaud E, Sensebé L, Varin A. CD54-mediated interaction with pro-inflammatory macrophages increases the immunosuppressive function of human mesenchymal stromal cells. Stem Cell Rep (2017) 8(4):961–76. doi: 10.1016/j.stemcr.2017.02.008
140. Li Y, Zhang D, Xu L, Dong L, Zheng J, Lin Y, et al. Cell-cell contact with proinflammatory macrophages enhances the immunotherapeutic effect of mesenchymal stem cells in two abortion models. Cell Mol Immunol (2019) 16(12):908–20. doi: 10.1038/s41423-019-0204-6
141. Nauta AJ, Kruisselbrink AB, Lurvink E, Willemze R, Fibbe WE. Mesenchymal stem cells inhibit generation and function of both CD34+-derived and monocyte-derived dendritic cells. J Immunol (2006) 177(4):2080–7. doi: 10.4049/jimmunol.177.4.2080
142. Spaggiari GM, Abdelrazik H, Becchetti F, Moretta L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: central role of MSC-derived prostaglandin E2. Blood (2009) 113(26):6576–83. doi: 10.1182/blood-2009-02-203943
143. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol (2008) 8(9):726–36. doi: 10.1038/nri2395
144. Liu Y, Yin Z, Zhang R, Yan K, Chen L, Chen F, et al. MSCs inhibit bone marrow-derived DC maturation and function through the release of TSG-6. Biochem Biophys Res Commun (2014) 450(4):1409–15. doi: 10.1016/j.bbrc.2014.07.001
145. Zhang B, Liu R, Shi D, Liu X, Chen Y, Dou X, et al. Mesenchymal stem cells induce mature dendritic cells into a novel Jagged-2-dependent regulatory dendritic cell population. Blood (2009) 113(1):46–57. doi: 10.1182/blood-2008-04-154138
146. Liu X, Qu X, Chen Y, Liao L, Cheng K, Shao C, et al. Mesenchymal stem/stromal cells induce the generation of novel IL-10-dependent regulatory dendritic cells by SOCS3 activation. J Immunol (2012) 189(3):1182–92. doi: 10.4049/jimmunol.1102996
147. Drummer C. 4th, Saaoud F, Shao Y, Sun Y, Xu K, Lu Y, et al. Trained immunity and reactivity of macrophages and endothelial cells. Arterioscler Thromb Vasc Biol (2021) 41(3):1032–46. doi: 10.1161/ATVBAHA.120.315452
148. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
149. Lin T, Pajarinen J, Kohno Y, Huang JF, Maruyama M, Romero-Lopez M, et al. Trained murine mesenchymal stem cells have anti-inflammatory effect on macrophages, but defective regulation on T-cell proliferation. FASEB J (2019) 33(3):4203–11. doi: 10.1096/fj.201801845R
150. Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation–from bench to bedside. Nat Rev Gastroenterol Hepatol (2013) 10(2):79–89. doi: 10.1038/nrgastro.2012.225
151. Anger F, Camara M, Ellinger E, Germer CT, Schlegel N, Otto C, et al. Human mesenchymal stromal cell-derived extracellular vesicles improve liver regeneration after ischemia reperfusion injury in mice. Stem Cells Dev (2019) 28(21):1451–62. doi: 10.1089/scd.2019.0085
152. Wei W, Dirsch O, Mclean AL, Zafarnia S, Schwier M, Dahmen U. Rodent models and imaging techniques to study liver regeneration. Eur Surg Res (2015) 54(3-4):97–113. doi: 10.1159/000368573
153. Tuñón MJ, Alvarez M, Culebras JM, González-Gallego J. An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World J Gastroenterol (2009) 15(25):3086–98. doi: 10.3748/wjg.15.3086
154. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med (2011) 17(11):1391–401. doi: 10.1038/nm.2507
155. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med (2009) 361(16):1570–83. doi: 10.1056/NEJMra0901217
156. Li S, Zheng X, Li H, Zheng J, Chen X, Liu W, et al. Mesenchymal stem cells ameliorate hepatic ischemia/reperfusion injury via inhibition of neutrophil recruitment. J Immunol Res (2018) 2018:7283703. doi: 10.1155/2018/7283703
157. Zhang M, Ueki S, Kimura S, Yoshida O, Castellaneta A, Ozaki KS, et al. Roles of dendritic cells in murine hepatic warm and liver transplantation-induced cold ischemia/reperfusion injury. Hepatology (2013) 57(4):1585–96. doi: 10.1002/hep.26129
158. Jiao Z, Ma Y, Zhang Q, Wang Y, Liu T, Liu X, et al. The adipose-derived mesenchymal stem cell secretome promotes hepatic regeneration in miniature pigs after liver ischaemia-reperfusion combined with partial resection. Stem Cell Res Ther (2021) 12(1):218. doi: 10.1186/s13287-021-02284-y
159. Lee SC, Kim JO, Kim SJ. Secretome from human adipose-derived stem cells protects mouse liver from hepatic ischemia-reperfusion injury. Surgery (2015) 157(5):934–43. doi: 10.1016/j.surg.2014.12.016
160. Haga H, Yan IK, Borrelli DA, Matsuda A, Parasramka M, Shukla N, et al. Extracellular vesicles from bone marrow-derived mesenchymal stem cells protect against murine hepatic ischemia/reperfusion injury. Liver Transpl (2017) 23(6):791–803. doi: 10.1002/lt.24770
161. Tsung A, Stang MT, Ikeda A, Critchlow ND, Izuishi K, Nakao A, et al. The transcription factor interferon regulatory factor-1 mediates liver damage during ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol (2006) 290(6):G1261–8. doi: 10.1152/ajpgi.00460.2005
162. Castellaneta A, Yoshida O, Kimura S, Yokota S, Geller DA, Murase N, et al. Plasmacytoid dendritic cell-derived IFN-α promotes murine liver ischemia/reperfusion injury by induction of hepatocyte IRF-1. Hepatology (2014) 60(1):267–77. doi: 10.1002/hep.27037
163. Yi Z, Deng M, Scott MJ, Fu G, Loughran PA, Lei Z, et al. Immune-responsive gene 1/itaconate activates nuclear factor erythroid 2-related factor 2 in hepatocytes to protect against liver ischemia-reperfusion injury. Hepatology (2020) 72(4):1394–411. doi: 10.1002/hep.31147
164. Sun P, Lu YX, Cheng D, Zhang K, Zheng J, Liu Y, et al. Monocyte chemoattractant protein-induced protein 1 targets hypoxia-inducible factor 1α to protect against hepatic ischemia/reperfusion injury. Hepatology (2018) 68(6):2359–75. doi: 10.1002/hep.30086
165. Owen A, Patten D, Vigneswara V, Frampton J, Newsome PN. PDGFRα/sca-1 sorted mesenchymal stromal cells reduce liver injury in murine models of hepatic ischemia-reperfusion injury. Stem Cells (2022) 40(11):1056–70. doi: 10.1093/stmcls/sxac059
166. Sheng M, Lin Y, Xu D, Tian Y, Zhan Y, Li C, et al. CD47-mediated hedgehog/SMO/GLI1 signaling promotes mesenchymal stem cell immunomodulation in mouse liver inflammation. Hepatology (2021) 74(3):1560–77. doi: 10.1002/hep.31831
167. Sahu A, Jeon J, Lee MS, Yang HS, Tae G. Nanozyme impregnated mesenchymal stem cells for hepatic ischemia-reperfusion injury alleviation. ACS Appl Mater Interfaces (2021) 13(22):25649–62. doi: 10.1021/acsami.1c03027
168. Zheng J, Chen L, Lu T, Zhang Y, Sui X, Li Y, et al. MSCs ameliorate hepatocellular apoptosis mediated by PINK1-dependent mitophagy in liver ischemia/reperfusion injury through AMPKα activation. Cell Death Dis (2020) 11(4):256. doi: 10.1038/s41419-020-2424-1
169. Ge Y, Zhang Q, Jiao Z, Li H, Bai G, Wang H, et al. Adipose-derived stem cells reduce liver oxidative stress and autophagy induced by ischemia-reperfusion and hepatectomy injury in swine. Life Sci (2018) 214:62–9. doi: 10.1016/j.lfs.2018.10.054
170. Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, et al. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol (2014) 279(3):266–74. doi: 10.1016/j.taap.2014.05.010
171. Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, et al. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol (2015) 289(2):213–22. doi: 10.1016/j.taap.2015.09.022
172. Du K, Farhood A, Jaeschke H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch Toxicol (2017) 91(2):761–73. doi: 10.1007/s00204-016-1692-0
173. Xie Y, Ramachandran A, Breckenridge DG, Liles JT, Lebofsky M, Farhood A, et al. Inhibitor of apoptosis signal-regulating kinase 1 protects against acetaminophen-induced liver injury. Toxicol Appl Pharmacol (2015) 286(1):1–9. doi: 10.1016/j.taap.2015.03.019165
174. Win S, Than TA, Fernandez-Checa JC, Kaplowitz N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis (2014) 5(1):e989. doi: 10.1038/cddis.2013.522
175. Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci (2006) 94(1):217–25. doi: 10.1093/toxsci/kfl077
176. Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther (2008) 324(1):8–14. doi: 10.1124/jpet.107.129445
177. McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H. HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology (2011) 53(3):974–82. doi: 10.1002/hep.24132
178. Rosen GM, Rauckman EJ, Ellington SP, Dahlin DC, Christie JL, Nelson SD. Reduction and glutathione conjugation reactions of N-acetyl-p-benzoquinone imine and two dimethylated analogues. Mol Pharmacol (1984) 25(1):151–7.
179. Cen Y, Lou G, Qi J, Li M, Zheng M, Liu Y. Adipose-Derived Mesenchymal Stem Cells Inhibit JNK-Mediated Mitochondrial Retrograde Pathway to Alleviate Acetaminophen-Induced Liver Injury. Antioxidants (Basel) (2023) 12(1):158. doi: 10.3390/antiox12010158
180. Huang YJ, Chen P, Lee CY, Yang SY, Lin MT, Lee HS, et al. Protection against acetaminophen-induced acute liver failure by omentum adipose tissue derived stem cells through the mediation of Nrf2 and cytochrome P450 expression. J BioMed Sci (2016) 23:5. doi: 10.1186/s12929-016-0231-x
181. Liu Z, Meng F, Li C, Zhou X, Zeng X, He Y, et al. Human umbilical cord mesenchymal stromal cells rescue mice from acetaminophen-induced acute liver failure. Cytotherapy (2014) 16(9):1207–19. doi: 10.1016/j.jcyt.2014.05.018
182. Wang P, Cui Y, Wang J, Liu D, Tian Y, Liu K, et al. Mesenchymal stem cells protect against acetaminophen hepatotoxicity by secreting regenerative cytokine hepatocyte growth factor. Stem Cell Res Ther (2022) 13(1):94. doi: 10.1186/s13287-022-02754-x
183. Yu M, Zhou M, Li J, Zong R, Yan Y, Kong L, et al. Notch-activated mesenchymal stromal/stem cells enhance the protective effect against acetaminophen-induced acute liver injury by activating AMPK/SIRT1 pathway. Stem Cell Res Ther (2022) 13(1):318. doi: 10.1186/s13287-022-02999-6
184. Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H. "Second hit" models of alcoholic liver disease. Semin Liver Dis (2009) 29(2):178–87. doi: 10.1055/s-0029-1214373
185. Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol (2018) 69(2):385–95. doi: 10.1016/j.jhep.2018.03.011
186. Kubota N, Kado S, Kano M, Masuoka N, Nagata Y, Kobayashi T, et al. A high-fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non-alcoholic steatohepatitis in mice. Clin Exp Pharmacol Physiol (2013) 40(7):422–30. doi: 10.1111/1440-1681.12102
187. Chiang DJ, Roychowdhury S, Bush K, McMullen MR, Pisano S, Niese K, et al. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PloS One (2013) 8(7):e69114. doi: 10.1371/journal.pone.0069114
188. Slater TF. Necrogenic action of carbon tetrachloride in the rat: a speculative mechanism based on activation. Nature (1966) 209(5018):36–40. doi: 10.1038/209036a0
189. Knockaert L, Berson A, Ribault C, Prost PE, Fautrel A, Pajaud J, et al. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab Invest (2012) 92(3):396–410. doi: 10.1038/labinvest.2011.193
190. Perugorria MJ, Esparza-Baquer A, Oakley F, Labiano I, Korosec A, Jais A, et al. Non-parenchymal TREM-2 protects the liver from immune-mediated hepatocellular damage. Gut (2019) 68(3):533–46. doi: 10.1136/gutjnl-2017-314107
191. Ramavath NN, Gadipudi LL, Provera A, Gigliotti LC, Boggio E, Bozzola C, et al. Inducible T-cell costimulator mediates lymphocyte/macrophage interactions during liver repair. Front Immunol (2021) 12:786680. doi: 10.3389/fimmu.2021.786680
192. Nagamoto Y, Takayama K, Ohashi K, Okamoto R, Sakurai F, Tachibana M, et al. Transplantation of a human iPSC-derived hepatocyte sheet increases survival in mice with acute liver failure. J Hepatol (2016) 64(5):1068–75. doi: 10.1016/j.jhep.2016.01.004
193. Liang H, Huang K, Su T, Li Z, Hu S, Dinh PU, et al. Mesenchymal stem cell/red blood cell-inspired nanoparticle therapy in mice with carbon tetrachloride-induced acute liver failure. ACS Nano (2018) 12(7):6536–44. doi: 10.1021/acsnano.8b00553
194. Kopec AK, Joshi N, Luyendyk JP. Role of hemostatic factors in hepatic injury and disease: animal models de-liver. J Thromb Haemost (2016) 14(7):1337–49. doi: 10.1111/jth.13327
195. Zagoura DS, Roubelakis MG, Bitsika V, Trohatou O, Pappa KI, Kapelouzou A, et al. Therapeutic potential of a distinct population of human amniotic fluid mesenchymal stem cells and their secreted molecules in mice with acute hepatic failure. Gut (2012) 61(6):894–906. doi: 10.1136/gutjnl-2011-300908
196. Milosavljevic N, Gazdic M, Simovic Markovic B, Arsenijevic A, Nurkovic J, Dolicanin Z, et al. Mesenchymal stem cells attenuate acute liver injury by altering ratio between interleukin 17 producing and regulatory natural killer T cells. Liver Transpl (2017) 23(8):1040–50. doi: 10.1002/lt.24784
197. Banas A, Teratani T, Yamamoto Y, Tokuhara M, Takeshita F, Osaki M, et al. IFATS collection: in vivo therapeutic potential of human adipose tissue mesenchymal stem cells after transplantation into mice with liver injury. Stem Cells (2008) 26(10):2705–12. doi: 10.1634/stemcells.2008-0034
198. Newsome PN, Plevris JN, Nelson LJ, Hayes PC. Animal models of fulminant hepatic failure: a critical evaluation. Liver Transpl (2000) 6(1):21–31. doi: 10.1002/lt.500060110
199. Hefler J, Marfil-Garza BA, Pawlick RL, Freed DH, Karvellas CJ, Bigam DL, et al. Preclinical models of acute liver failure: a comprehensive review. PeerJ (2021) 9:e12579. doi: 10.7717/peerj.12579
200. Maes M, Vinken M, Jaeschke H. Experimental models of hepatotoxicity related to acute liver failure. Toxicol Appl Pharmacol (2016) 290:86–97. doi: 10.1016/j.taap.2015.11.016
201. Hamesch K, Borkham-Kamphorst E, Strnad P, Weiskirchen R. Lipopolysaccharide-induced inflammatory liver injury in mice. Lab Anim (2015) 49(1 Suppl):37–46. doi: 10.1177/0023677215570087
202. Ferluga J, Allison AC. Role of mononuclear infiltrating cells in pathogenesis of hepatitis. Lancet (1978) 2(8090):610–1. doi: 10.1016/S0140-6736(78)92828-3
203. Yethon JA, Whitfield C. Lipopolysaccharide as a target for the development of novel therapeutics in gram-negative bacteria. Curr Drug Targets Infect Disord (2001) 1(2):91–106. doi: 10.2174/1568005014606143
204. Silverstein R. D-galactosamine lethality model: scope and limitations. J Endotoxin Res (2004) 10(3):147–62. doi: 10.1179/096805104225004879
205. Ahmedy OA, Salem HH, Sayed NH, Ibrahim SM. Naringenin affords protection against lipopolysaccharide/D-galactosamine-induced acute liver failure: Role of autophagy. Arch Biochem Biophys (2022) 717:109121. doi: 10.1016/j.abb.2022.109121
206. Zhao J, Liu L, Xin L, Lu Y, Yang X, Hou Y. The protective effects of a modified xiaohua funing decoction against acute liver failure in mice induced by D-gal and LPS. Evid Based Complement Alternat Med (2022) 2022:6611563. doi: 10.1155/2022/6611563
207. Wang X, Zhong Y, Zhang R, Chen Y, Wang M, Lv C, et al. Wenyang Huazhuo Tuihuang formula inhibits the th17/treg cell imbalance and protects against acute-on-chronic liver failure. Evid Based Complement Alternat Med (2022) 2022:5652172. doi: 10.1155/2022/5652172
208. Kong D, Xu H, Chen M, Yu Y, Qian Y, Qin T, et al. Co-encapsulation of HNF4α overexpressing UMSCs and human primary hepatocytes ameliorates mouse acute liver failure. Stem Cell Res Ther (2020) 11(1):449. doi: 10.1186/s13287-020-01962-7
209. Liu Y, Lou G, Li A, Zhang T, Qi J, Ye D, et al. AMSC-derived exosomes alleviate lipopolysaccharide/d-galactosamine-induced acute liver failure by miR-17-mediated reduction of TXNIP/NLRP3 inflammasome activation in macrophages. EBioMedicine (2018) 36:140–50. doi: 10.1016/j.ebiom.2018.08.054
210. Zhong W, Qian K, Xiong J, Ma K, Wang A, Zou Y. Curcumin alleviates lipopolysaccharide induced sepsis and liver failure by suppression of oxidative stress-related inflammation via PI3K/AKT and NF-κB related signaling. BioMed Pharmacother (2016) 83:302–13. doi: 10.1016/j.biopha.2016.06.036
211. Yang P, Zhou W, Li C, Zhang M, Jiang Y, Jiang R, et al. Kupffer-cell-expressed transmembrane TNF-α is a major contributor to lipopolysaccharide and D-galactosamine-induced liver injury. Cell Tissue Res (2016) 363(2):371–83. doi: 10.1007/s00441-015-2252-2
212. Jaeschke H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol (2006) 290(6):G1083–8. doi: 10.1152/ajpgi.00568.2005
213. Jing ZT, Liu W, Xue CR, Wu SX, Chen WN, Lin XJ, et al. AKT activator SC79 protects hepatocytes from TNF-α-mediated apoptosis and alleviates d-Gal/LPS-induced liver injury. Am J Physiol Gastrointest Liver Physiol (2019) 316(3):G387–g396. doi: 10.1152/ajpgi.00350.2018
214. Barnabei L, Laplantine E, Mbongo W, Rieux-Laucat F, Weil R. NF-κB: at the borders of autoimmunity and inflammation. Front Immunol (2021) 12:716469. doi: 10.3389/fimmu.2021.716469
215. Liedtke C, Trautwein C. The role of TNF and Fas dependent signaling in animal models of inflammatory liver injury and liver cancer. Eur J Cell Biol (2012) 91(6-7):582–9. doi: 10.1016/j.ejcb.2011.10.001
216. Ma HC, Wang X, Wu MN, Zhao X, Yuan XW, Shi XL. Interleukin-10 contributes to therapeutic effect of mesenchymal stem cells for acute liver failure via signal transducer and activator of transcription 3 signaling pathway. Chin Med J (Engl) (2016) 129(8):967–75. doi: 10.4103/0366-6999.179794
217. Li J, Zhang L, Xin J, Jiang L, Li J, Zhang T, et al. Immediate intraportal transplantation of human bone marrow mesenchymal stem cells prevents death from fulminant hepatic failure in pigs. Hepatology (2012) 56(3):1044–52. doi: 10.1002/hep.25722
218. Shi D, Zhang J, Zhou Q, Xin J, Jiang J, Jiang L, et al. Quantitative evaluation of human bone mesenchymal stem cells rescuing fulminant hepatic failure in pigs. Gut (2017) 66(5):955–64. doi: 10.1136/gutjnl-2015-311146
219. Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity (2009) 30(2):180–92. doi: 10.1016/j.immuni.2009.01.001
220. Shao R, Yang Y, Fan K, Wu X, Jiang R, Tang L, et al. REV-ERBα Agonist GSK4112 attenuates fas-induced acute hepatic damage in mice. Int J Med Sci (2021) 18(16):3831–8. doi: 10.7150/ijms.52011
221. Al-Saeedi M, Steinebrunner N, Kudsi H, Halama N, Mogler C, Büchler MW, et al. Neutralization of CD95 ligand protects the liver against ischemia-reperfusion injury and prevents acute liver failure. Cell Death Dis (2018) 9(2):132. doi: 10.1038/s41419-017-0150-0
222. Alkhouri N, Alisi A, Okwu V, Matloob A, Ferrari F, Crudele A, et al. Circulating soluble fas and fas ligand levels are elevated in children with nonalcoholic steatohepatitis. Dig Dis Sci (2015) 60(8):2353–9. doi: 10.1007/s10620-015-3614-z
223. Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ (2012) 19(1):36–41. doi: 10.1038/cdd.2011.155
224. Schüngel S, Buitrago-Molina LE, Nalapareddy PD, Lebofsky M, Manns MP, Jaeschke H, et al. The strength of the Fas ligand signal determines whether hepatocytes act as type 1 or type 2 cells in murine livers. Hepatology (2009) 50(5):1558–66. doi: 10.1002/hep.23176
225. Lazic M, Eguchi A, Berk MP, Povero D, Papouchado B, Mulya A, et al. Differential regulation of inflammation and apoptosis in Fas-resistant hepatocyte-specific Bid-deficient mice. J Hepatol (2014) 61(1):107–15. doi: 10.1016/j.jhep.2014.03.028
226. Li M, Qin XY, Furutani Y, Inoue I, Sekihara S, Kagechika H, et al. Prevention of acute liver injury by suppressing plasma kallikrein-dependent activation of latent TGF-β. Biochem Biophys Res Commun (2018) 504(4):857–64. doi: 10.1016/j.bbrc.2018.09.026
227. Liu W, Jing ZT, Wu SX, He Y, Lin YT, Chen WN, et al. A novel AKT activator, SC79, prevents acute hepatic failure induced by fas-mediated apoptosis of hepatocytes. Am J Pathol (2018) 188(5):1171–82. doi: 10.1016/j.ajpath.2018.01.013
228. Motoi S, Toyoda H, Obara T, Ohta E, Arita Y, Negishi K, et al. Anti-apoptotic effects of recombinant human hepatocyte growth factor on hepatocytes were associated with intrahepatic hemorrhage suppression indicated by the preservation of prothrombin time. Int J Mol Sci (2019) 20(8):1821. doi: 10.3390/ijms20081821
229. Sharma AD, Narain N, Händel EM, Iken M, Singhal N, Cathomen T, et al. MicroRNA-221 regulates FAS-induced fulminant liver failure. Hepatology (2011) 53(5):1651–61. doi: 10.1002/hep.24243
230. Utaipan T, Otto AC, Gan-Schreier H, Chunglok W, Pathil A, Stremmel W, et al. Ursodeoxycholyl lysophosphatidylethanolamide protects against CD95/FAS-induced fulminant hepatitis. Shock (2017) 48(2):251–9. doi: 10.1097/SHK.0000000000000831
231. Jing ZT, Liu W, Wu SX, He Y, Lin YT, Chen WN, et al. Hepatitis B virus surface antigen enhances the sensitivity of hepatocytes to fas-mediated apoptosis via suppression of AKT phosphorylation. J Immunol (2018) 201(8):2303–14. doi: 10.4049/jimmunol.1800732
232. Yang X, Meng Y, Han Z, Ye F, Wei L, Zong C. Mesenchymal stem cell therapy for liver disease: full of chances and challenges. Cell Biosci (2020) 10:123. doi: 10.1186/s13578-020-00480-6
233. Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, et al. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol (2012) 3:297. doi: 10.3389/fimmu.2012.00297
234. Higashimoto M, Sakai Y, Takamura M, Usui S, Nasti A, Yoshida K, et al. Adipose tissue derived stromal stem cell therapy in murine ConA-derived hepatitis is dependent on myeloid-lineage and CD4+ T-cell suppression. Eur J Immunol (2013) 43(11):2956–68. doi: 10.1002/eji.201343531
235. Sang JF, Shi XL, Han B, Huang T, Huang X, Ren HZ, et al. Intraportal mesenchymal stem cell transplantation prevents acute liver failure through promoting cell proliferation and inhibiting apoptosis. Hepatobil Pancreat Dis Int (2016) 15(6):602–11. doi: 10.1016/S1499-3872(16)60141-8
236. Cao H, Yang J, Yu J, Pan Q, Li J, Zhou P, et al. Therapeutic potential of transplanted placental mesenchymal stem cells in treating Chinese miniature pigs with acute liver failure. BMC Med (2012) 10:56. doi: 10.1186/1741-7015-10-56
237. Amer ME, El-Sayed SZ, El-Kheir WA, Gabr H, Gomaa AA, El-Noomani N, et al. Clinical and laboratory evaluation of patients with end-stage liver cell failure injected with bone marrow-derived hepatocyte-like cells. Eur J Gastroenterol Hepatol (2011) 23(10):936–41. doi: 10.1097/MEG.0b013e3283488b00
238. Ogasawara H, Inagaki A, Fathi I, Imura T, Yamana H, Saitoh Y, et al. Preferable transplant site for hepatocyte transplantation in a rat model. Cell Transplant (2021) 30:9636897211040012. doi: 10.1177/09636897211040012
239. Schubert T, Xhema D, Vériter S, Schubert M, Behets C, Delloye C, et al. The enhanced performance of bone allografts using osteogenic-differentiated adipose-derived mesenchymal stem cells. Biomaterials (2011) 32(34):8880–91. doi: 10.1016/j.biomaterials.2011.08.009
240. Heo JS, Choi Y, Kim HS, Kim HO. Comparison of molecular profiles of human mesenchymal stem cells derived from bone marrow, umbilical cord blood, placenta and adipose tissue. Int J Mol Med (2016) 37(1):115–25. doi: 10.3892/ijmm.2015.2413
241. Zhang Y, Liu S, Lim M, Zhao S, Cui K, et al. Embryonic stem cell-derived extracellular vesicles enhance the therapeutic effect of mesenchymal stem cells. Theranostics (2019) 9(23):6976–90. doi: 10.7150/thno.35305
242. Wu R, Fan X, Wang Y, Shen M, Zheng Y, Zhao S, et al. Mesenchymal stem cell-derived extracellular vesicles in liver immunity and therapy. Front Immunol (2022) 13:833878. doi: 10.3389/fimmu.2022.833878
243. Psaraki A, Ntari L, Karakostas C, Korrou-Karava D, Roubelakis MG. Extracellular vesicles derived from mesenchymal stem/stromal cells: The regenerative impact in liver diseases. Hepatology (2022) 75(6):1590–603. doi: 10.1002/hep.32129
244. Rostom DM, Attia N, Khalifa HM, Abou Nazel MW, El Sabaawy EA. The therapeutic potential of extracellular vesicles versus mesenchymal stem cells in liver damage. Tissue Eng Regener Med (2020) 17(4):537–52. doi: 10.1007/s13770-020-00267-3
245. Qiu X, Liu J, Zheng C, Su Y, Bao L, Zhu B, et al. Exosomes released from educated mesenchymal stem cells accelerate cutaneous wound healing via promoting angiogenesis. Cell Prolif (2020) 53(8):e12830. doi: 10.1111/cpr.12830
246. Peng L, Xie DY, Lin BL, Liu J, Zhu HP, Xie C, et al. Autologous bone marrow mesenchymal stem cell transplantation in liver failure patients caused by hepatitis B: short-term and long-term outcomes. Hepatology (2011) 54(3):820–8. doi: 10.1002/hep.24434
247. Shi M, Zhang Z, Xu R, Lin H, Fu J, Zou Z, et al. Human mesenchymal stem cell transfusion is safe and improves liver function in acute-on-chronic liver failure patients. Stem Cells Transl Med (2012) 1(10):725–31. doi: 10.5966/sctm.2012-0034
248. Zhang Z, Lin H, Shi M, Xu R, Fu J, Lv J, et al. Human umbilical cord mesenchymal stem cells improve liver function and ascites in decompensated liver cirrhosis patients. J Gastroenterol Hepatol (2012) 27 Suppl 2:112–20. doi: 10.1111/j.1440-1746.2011.07024.x
249. Mohamadnejad M, Alimoghaddam K, Bagheri M, Ashrafi M, Abdollahzadeh L, Akhlaghpoor S, et al. Randomized placebo-controlled trial of mesenchymal stem cell transplantation in decompensated cirrhosis. Liver Int (2013) 33(10):1490–6. doi: 10.1111/liv.12228
250. Salama H, Zekri AR, Medhat E, Al Alim SA, Ahmed OS, Bahnassy AA, et al. Peripheral vein infusion of autologous mesenchymal stem cells in Egyptian HCV-positive patients with end-stage liver disease. Stem Cell Res Ther (2014) 5(3):70. doi: 10.1186/scrt459
251. Li YH, Xu Y, Wu HM, Yang J, Yang LH, Yue-Meng W. Umbilical cord-derived mesenchymal stem cell transplantation in hepatitis B virus related acute-on-chronic liver failure treated with plasma exchange and entecavir: a 24-month prospective study. Stem Cell Rev Rep (2016) 12(6):645–53. doi: 10.1007/s12015-016-9683-3
252. Lin BL, Chen JF, Qiu WH, Wang KW, Xie DY, Chen XY, et al. Allogeneic bone marrow-derived mesenchymal stromal cells for hepatitis B virus-related acute-on-chronic liver failure: A randomized controlled trial. Hepatology (2017) 66(1):209–19. doi: 10.1002/hep.29189
253. Xu WX, He HL, Pan SW, Chen YL, Zhang ML, Zhu S, et al. Combination treatments of plasma exchange and umbilical cord-derived mesenchymal stem cell transplantation for patients with hepatitis B virus-related acute-on-chronic liver failure: A clinical trial in China. Stem Cells Int (2019) 2019:4130757. doi: 10.1155/2019/4130757
254. Schacher FC, Martins Pezzi da Silva A, Silla LMDR, Álvares-da-Silva MR. Bone marrow mesenchymal stem cells in acute-on-chronic liver failure grades 2 and 3: A phase I-II randomized clinical trial. Can J Gastroenterol Hepatol (2021) 2021:3662776. doi: 10.1155/2021/3662776
255. Shi M, Li YY, Xu RN, Meng FP, Yu SJ, Fu JL, et al. Mesenchymal stem cell therapy in decompensated liver cirrhosis: a long-term follow-up analysis of the randomized controlled clinical trial. Hepatol Int (2021) 15(6):1431–41. doi: 10.1007/s12072-021-10199-2
256. Yang L, Chang N, Liu X, Han Z, Zhu T, Li C, et al. Bone marrow-derived mesenchymal stem cells differentiate to hepatic myofibroblasts by transforming growth factor-β1 via sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis. Am J Pathol (2012) 181(1):85–97. doi: 10.1016/j.ajpath.2012.03.014
257. Lanthier N, Lin-Marq N, Rubbia-Brandt L, Clément S, Goossens N, Spahr L. Autologous bone marrow-derived cell transplantation in decompensated alcoholic liver disease: what is the impact on liver histology and gene expression patterns? Stem Cell Res Ther (2017) 8(1):88. doi: 10.1186/s13287-017-0541-2
258. Wang L, Li J, Liu H, Li Y, Fu J, Sun Y, et al. Pilot study of umbilical cord-derived mesenchymal stem cell transfusion in patients with primary biliary cirrhosis. J Gastroenterol Hepatol (2013) 28 Suppl 1:85–92. doi: 10.1111/jgh.12029
259. Jang YO, Kim YJ, Baik SK, Kim MY, Eom YW, Cho MY, et al. Histological improvement following administration of autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: a pilot study. Liver Int (2014) 34(1):33–41. doi: 10.1111/liv.12218
Keywords: mesenchymal stem cells (MSCs), acute liver failure (ALF), animal models, culture strategy, surface markers, cell therapy
Citation: Yang H, Chen J and Li J (2023) Isolation, culture, and delivery considerations for the use of mesenchymal stem cells in potential therapies for acute liver failure. Front. Immunol. 14:1243220. doi: 10.3389/fimmu.2023.1243220
Received: 20 June 2023; Accepted: 18 August 2023;
Published: 07 September 2023.
Edited by:
Veronica Veschi, University of Palermo, ItalyReviewed by:
Ashwin Ajith, Augusta University, United StatesRobert Wade Siggins, Louisiana State University, United States
Copyright © 2023 Yang, Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Li, bGlqdW4yMDA5QHpqdS5lZHUuY24=
†These authors have contributed equally to this work