 Cheng-Yen Chang
Cheng-Yen Chang Dominique Armstrong
Dominique Armstrong David B. Corry
David B. Corry Farrah Kheradmand
Farrah Kheradmand- 1Department of Medicine, Baylor College of Medicine, Houston, TX, United States
- 2Department of Pathology and Immunology, Baylor College of Medicine, Houston, TX, United States
- 3Biology of Inflammation Center, Baylor College of Medicine, Houston, TX, United States
- 4Center for Translational Research on Inflammatory Diseases, Michael E. DeBakey Department of Veterans Affairs Medical Center, Houston, TX, United States
Alveolar macrophages (AMs) are critical components of the innate defense mechanism in the lung. Nestled tightly within the alveoli, AMs, derived from the yolk-sac or bone marrow, can phagocytose foreign particles, defend the host against pathogens, recycle surfactant, and promptly respond to inhaled noxious stimuli. The behavior of AMs is tightly dependent on the environmental cues whereby infection, chronic inflammation, and associated metabolic changes can repolarize their effector functions in the lungs. Several factors within the tumor microenvironment can re-educate AMs, resulting in tumor growth, and reducing immune checkpoint inhibitors (ICIs) efficacy in patients treated for non-small cell lung cancer (NSCLC). The plasticity of AMs and their critical function in altering tumor responses to ICIs make them a desirable target in lung cancer treatment. New strategies have been developed to target AMs in solid tumors reprograming their suppressive function and boosting the efficacy of ICIs. Here, we review the phenotypic and functional changes in AMs in response to sterile inflammation and in NSCLC that could be critical in tumor growth and metastasis. Opportunities in altering AMs’ function include harnessing their potential function in trained immunity, a concept borrowed from memory response to infections, which could be explored therapeutically in managing lung cancer treatment.
1 Introduction
Alveolar macrophages (AMs) are critical in maintaining lung homeostasis under normal and diseased conditions. In response to minimal irritants, AMs initiate low-grade inflammation followed by a resolution phase inducing immune tolerance, thus limiting the extent of lung injury. Under a static state, AMs express critical regulatory factors such as interleukin (IL)-10, the canonical anti-inflammatory cytokine (1), and express cell surface receptors including program cell death-ligand 1 (PD-L1) to optimize phagocytosis and prevent excessive inflammation in the lung (2). Severe tissue-damage disrupts the AM pool, primarily composed of tissue-resident AMs (TRAMs), which can be replenished by monocyte-derived AMs (MoAMs) (3, 4). In response to infection, AMs phagocytose infectious agents, which triggers secretion of pro-inflammatory cytokines and chemokines, activating the downstream adaptive immune response (5). Sterile inflammation, which occurs in the absence of microorganisms, can trigger a similar response (5). Chronic inflammation, infectious or sterile, leads to aberrant tissue repair signals and immune responses. The development of lung cancer is initiated and perpetuated by both extrinsic chronic inflammation and intrinsic DNA damage in the lungs related to cigarette smoke. Impaired AMs have been found in heavy smokers, showing deficiencies in antigen recognition and phagocytosis (6).
Antitumor functions of AMs have been described in non-small cell lung cancer (NSCLC) (7), but once integrated within solid tumors, they become a component of a heterogenous class of tumor-associated macrophages (TAMs). TAMs can induce exhausted T cells, promote immune suppression, and reduce the efficacy of ICIs in NSCLC (8). Therefore, several strategies have been developed to target TAMs, aiming to resolve their function in inducing a suppressive microenvironment. These efforts aim to increase T cell trafficking in solid tumors, thus improving the efficacy of ICIs.
NSCLCs account for around 85% of cancer cases and include adeno (40%), squamous (25%), and large cell (10%) carcinoma subtypes (9). Despite differences in histology, early-stage NSCLC is amenable to resection and carries a favorable prognosis (10). However, over 70% of cancer cases are diagnosed at advanced stages (11), and carry a poor prognosis (12). In the past decade, the incorporation of ICIs, targeting cytotoxic T lymphocyte-associated protein 4 (CTLA4), program cell death 1 (PD-1), and PD-L1 have significantly advanced the treatment paradigm in NSCLC. For patients with tumor PD-L1 expression of > 50%, ICIs are the first line treatment choice showing a promising overall survival rate (12). However, the overall response rate remains low at approximately 20% in non-selected NSCLC (13), indicating the need to improve reactivating the exhausted tumor-specific T cells.
The tumor microenvironment (TME) shapes immune cells’ fate and governs treatment outcomes (14). Specifically, tumors have evolved to sustain energy, compete for local resources, evade immune cell surveillance, and thrive under extreme conditions making them highly adaptive to changes in TME (15). A critical mechanism responsible for tumor evasion includes altering macrophage function, which are abundant in the lung TME (16), by disabling their antitumor function while licensing them to increase tolerance. Consistently, increase in macrophage density within TME correlates negatively with patient survival in several types of solid tumors, including NSCLC (17). Tumor cells can further recruit and polarize monocytes accelerating growth/angiogenic factor production, increasing proteinases, and facilitating tumor invasion, suggesting co-evolution between these two cell types (18).
TAMs promote tumor progression and metastasis in several murine models of NSCLC (19). TAMs represent heterogenous populations which exist on a spectrum of phenotypes from inflammatory to anti-inflammatory macrophages (Figure 1) (20). Markers of inflammatory macrophages, major histocompatibility complex (MHC) class II co-stimulatory molecules (CD40, CD80, and CD86), and markers of anti-inflammatory macrophages (CD163 and CD206) have been detected in TAMs in early-stage NSCLC (21). TAMs are often considered anti-inflammatory-skewed macrophages because of their immunosuppressive phenotypes (22); however, the exact expression of inflammatory/anti-inflammatory macrophage phenotype in humans is less clear. Chronic inflammation associated with TAMs is linked to malignant cell transformation (23). For instance, inflammatory TAMs accelerate genome instability, whereas anti-inflammatory TAMs express immunosuppressive molecules facilitating tumor progression (24). Anti-inflammatory TAMs directly inhibit T and natural killer (NK) functions by engaging between checkpoint ligands and receptors. Alternatively, the secretion of immunosuppressive cytokines, exosomes, metabolites, and immunosuppressive enzymes also inhibits anti-tumor immunity and fosters tumor growth (25).
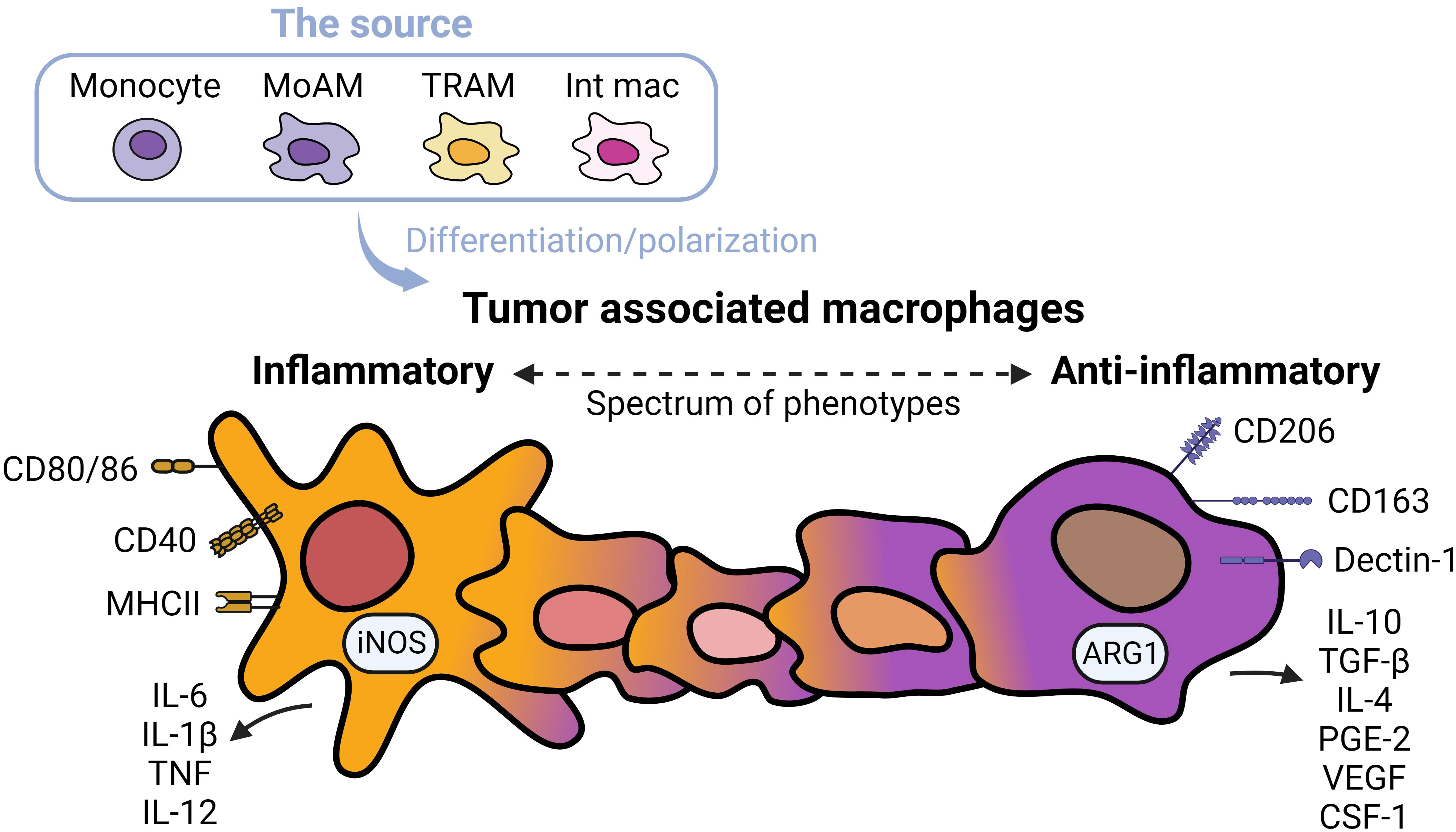
Figure 1 Spectrums of tumor associated macrophages. Tumor associated macrophages (TAMs) can develop from monocytes, monocyte-derived alveolar macrophages (MoAMs), tissue-resident alveolar macrophages (TRAMs), or interstitial macrophages (IMs) in the lung. TAMs exist as a spectrum of phenotypes from inflammatory to anti-inflammatory cells. Inflammatory macrophages are defined by higher expression of antigen-presenting and co-stimulatory molecules including CD80/CD86, CD40, MHCII, and secretion of inflammatory cytokines, whereas anti-inflammatory macrophages are immunosuppressive, expressing CD163 and Dectin-1, higher levels of CD206, and secretion of immunosuppressive cytokines and angiogenic factors. Typically, anti-inflammatory TAMs are more abundant in NSCLC.
The critical roles of AMs in lung cancer have prompted focused studies on identifying their ontogeny and lineages. Although the ontogeny of AMs in humans is undefined, murine TRAMs can self-renew, whereas monocyte-derived macrophages primarily repopulate from hematopoietic stem cells and accumulate in inflamed regions (26). AMs detect foreign substances in the airways, defend the host from pathogens, and recycle lipids (e.g., surfactant). AMs also interpret lung signals from the environmental, leading to rapid and reversible changes in their function (27). However, chronic inflammation polarizes and impairs AMs’ function (28, 29). Here we review relevant discoveries related to AMs in chronic inflammation and in NSCLC. We discuss the regulation of PD-1/PD-L1 in AMs and their potential reprogramming as a novel therapeutic option for NSCLC.
2 Origin and function of alveolar macrophages
2.1 Development and origin of AMs
AMs encompass both TRAMs and MoAMs. Advanced technology and lineage tracing has increasingly enabled the ability to distinguish these populations in mice, but less is known in humans. In mice, TRAMs are derived from yoke sac precursors of fetal monocytes that populate the alveoli shortly after birth and persist over the lifespan. The precursors are a self-renewing, embryo-derived population and are independent of bone marrow contribution (30). TRAMs rely on the granulocyte-macrophage colony-stimulating factor (GM-CSF) for their function (31). TRAM precursors in humans are not well-defined but the process may be similar across species because defects in GM-CSF production or auto-antibodies against GM-CSF disrupts TRAMs function causing alveolar proteinosis in mice and humans (32). GM-CSF is also critical for peroxisome proliferator-activated receptor-γ (PPARγ) expression, a ligand-activated nuclear receptor, required for TRAM differentiation (33). PPARγ is constitutively expressed in TRAMs under steady states and is required for the alveolar lipid homeostasis (34). The autocrine activation of TGF-β can also trigger PPARγ expression (35). Notably, in mice TRAMs can locally restore a moderately depleted pool without the need for recruiting MoAMs. However, in response to severe lung injury or an overwhelming infection, TRAM repletion requires MoAMs to engraft the AM pool (Figure 2) (3, 4). There is some evidence that over the course of natural aging, monocyte infiltrate and differentiate into AMs in the lungs (30). Although in early stages of inflammation, MoAMs can be readily separated from TRAMs, recruited monocytes progressively acquire similar functions, but retain specific genes linked to the circulatory origin (36). The functional differences between these two origins of cells, and their effect on tissue responses to injury, remain less clear.
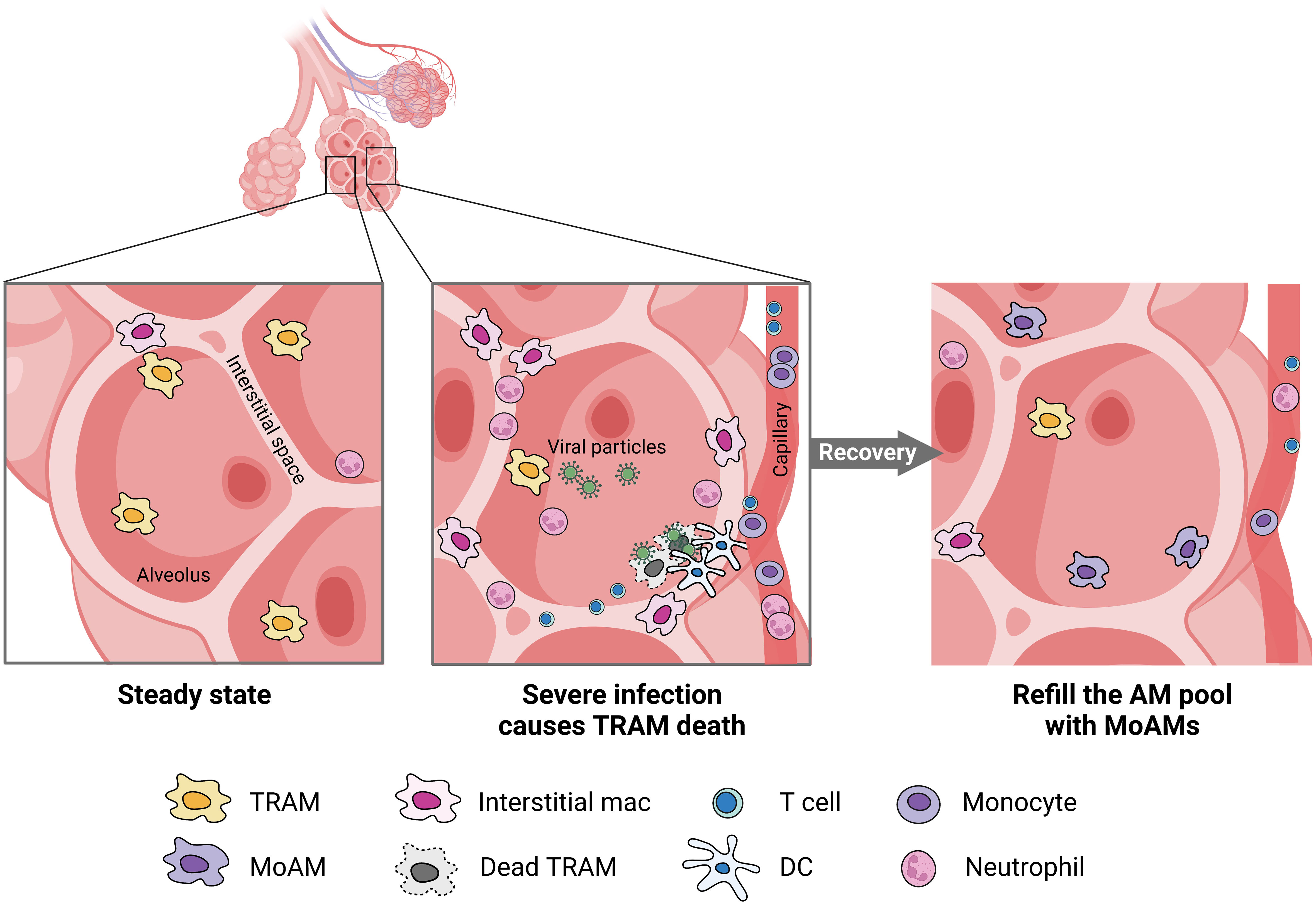
Figure 2 MoAMs replace TRAMs after severe lung infection. Severe infection leads to the death of TRAMs but attracts monocytes, neutrophils, DC, and T cells from the blood stream to the alveolar space. Increased interstitial macrophages can be found in the lungs. After recovery, MoAMs replace the loss of TRAMs in the alveoli.
Lineage tracing including the membrane-spanning 4-domains subfamily A member 3 (Ms4a3), found in granulocyte-monocyte and common monocyte progenitors has been used to study macrophages (37). Ms4a3 reporter and Cre fate-mapping models track monocyte-derived macrophages and granulocytes but not dendritic cells (DCs) or embryonic tissue-resident macrophages in the tissue (38), thereby distinguishing MoAMs from mature TRAMs. Another fate-mapping model used to specify TRAMs from the hematopoietic origin of macrophages is membrane-associated protein 17 (MAP17) reporter, which marks adult hematopoietic stem cells (39). However, a critical caveat is that monocyte fate-mapping models using Cx3cr1Cre or Cx3cr1CreERT2 also label DC, and LyzMCre labels other myeloid cells, restricting their use in understanding TRAMs source (37). Future research is needed to determine whether TRAMs and MoAMs are functionally redundant or whether they have distinct roles in tissue repair, infection, and cancer.
3.2 Function of AMs in the lungs under steady state
AMs are capable of presenting antigens to T lymphocytes; however, under steady state, their low-level expression of co-stimulatory and MHC class II molecules reduces their ability to activate adaptive immunity (27). In contrast, in response to a high expression of damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) such as viral particles, AMs are poised to immediately respond to the threat (40). These responses include secreting pro-inflammatory cytokines IL-6, IL-1β, and tumor necrotic factor (TNF), propagating the innate immune responses (41). AMs are also essential in phagocytosing apoptotic cells (e.g., neutrophils) and toxic particles to prevent further tissue injury and leakage of immunogenic molecules from dead or dying cells, serving as a critical function in resolving inflammation (40). Phagocytosing apoptotic cells, known as efferocytosis, reprograms AMs to increase the expression of transforming growth factor-β (TGF-β), IL-10, and prostaglandin E2 (PGE2), abrogating the inflammatory responses (42). TGF-β further inhibits T lymphocyte activation and promotes regulatory T cells (Treg) differentiation (43), thus restraining activation of adaptive immune responses (Figure 3).
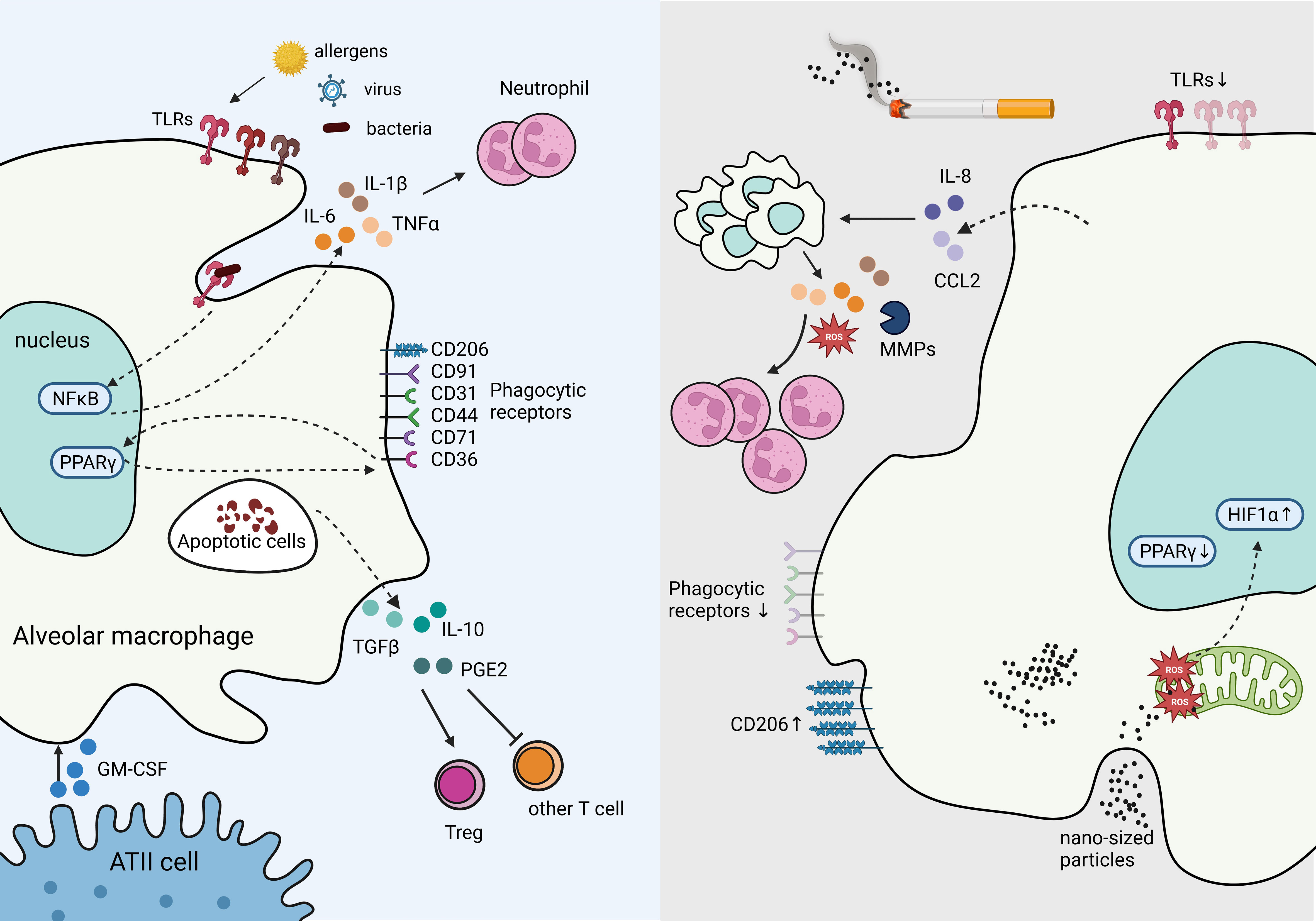
Figure 3 Tissue Resident Alveolar Macrophages in homeostasis and immune suppression in response to chronic inflammation. The production of GM-CSF from ATII cells supports AM survival, proliferation, and differentiation. Under homeostasis, AMs express high levels of pathogen recognition receptors (PRRs), such as toll-like receptors (TLRs) to rapid response to bacterial, and viral insults. Engagement of TLRs induces NFκB activation which stimulates inflammatory cytokines IL-6 and IL-1β production, and recruit neutrophils. Phagocytosing apoptotic cells stimulates suppressive signals IL-10, TGF-β, and PGE2 secretion mitigating tissue damage and inflammation. In contrast, cigarette smoke inhibits TLRs and phagocytic receptors dampening responses to viral and bacterial threats in AMs, while increasing CD206. Cigarette smoke stimulates IL-8 and CCL2 production, which recruits additional macrophages primed to secrete excessive ROS, MMPs, and other inflammatory cytokines, resulting in excessive neutrophils and causing tissue damage. Cigarette smoke generates nano-sized carbon which accumulates in AMs and penetrates mitochondria, inducing ROS and HIF-1α while decreasing PPARγ signaling.
3 Chronic cigarette smoke and sterile inflammation impairs AM function
3.1 Smoking increases trafficking but impairs AM function
Chronic exposure to particulate matter, generated from incomplete combustion of organic matter (e.g., cigarette smoke, forest fires, environmental pollutants, etc.), causes sterile inflammation in the lungs and plays a key role in the activation of adaptive immunity in the lung (44). Cellular profiling of bronchial alveolar lavage (BAL) fluid in cigarette smokers show increased number of AMs, but their immune function is dampened as evidenced by downregulation of interferon-γ (IFN-γ) signaling and increased risk of lung infection (45). The paracrine and/or autocrine effects of IL-8 and CCL2 (also known as CXCL8 and MCP-1, respectively), induced by cigarette smoke are responsible for increased AM trafficking in the lungs (46). AMs secrete pro-inflammatory cytokines, matrix metalloproteinases (MMPs), and reactive oxygen species (ROS), leading to a degradation of extracellular matrix, oxidative stress, excessive recruitment of neutrophils and T helper (Th)17 cells, and tissue damage (28).
Smoking also reduces the expression of many cell surface recognition molecules in AM, including CD31, CD91, CD44, and CD71 and dampens their phagocytic activity (6). Toll-like receptor (TLR) downregulation has been shown in AMs after exposure to cigarette smoke, further diminishing their antimicrobial function (47). Consistently, smoking can reduce AM’s ability to phagocytose fungal pathogen, Candida albicans, and gram-negative bacteria, Klebsiella pneumoniae, in murine models (48). Smoking can reduce the expression of inflammatory phenotypic markers and polarize AMs to an anti-inflammatory-like phenotype (49). Further, dual polarization of AMs has been reported in smokers with Chronic obstructive pulmonary disease (COPD), a chronic inflammatory lung condition that blocks pulmonary airflow, whereby inflammatory and anti-inflammatory macrophage markers are increased at the same time and correlate with disease severity (50). Interestingly, CD206 is an anti-inflammatory marker that is highly expressed in resting AMs (51) but is amplified in smokers (50) and after exposure to particulates (28), indicating a feedback mechanism that may be critical in resolving inflammation. Though increased expression of anti-inflammatory markers suggest an immune suppressive response, cytokine profile and functional assessments are required to determine the exact role of AMs in specific contexts.
3.2 Changes in metabolism alters AM phenotype and function
AMs are capable of intrinsic metabolic switching. Specifically, under steady-state or resting conditions, AMs preferentially use mitochondrial oxidative phosphorylation (OXPHOS), fueled by fatty acid and glucose oxidation to generate ATP (52). Once activated, AMs rapidly shift their metabolism and increase glycolysis to provide a large supply of ATP that further polarizes them toward a pro-inflammatory macrophage. Notably, in response to influenza infection, and in contrast to monocyte-derived macrophages, TRAMs stabilize hypoxia-inducible factor 1 α (HIF-1α), a process that is required for altering their metabolic responses (53). The accessory functions of AMs in the lung include lipid metabolism and homeostasis of surfactant, a complex of phospholipids and proteins that maintains pulmonary compliance and requires constitutive expression of PPARγ (34). The nuclear receptor transcription factor, PPARγ in AMs inhibits the expression of pro-inflammatory cytokines while enhancing phagocytic function (54). Other transcriptional genetic control of AMs includes mitochondrial transcription factor A (TFAM), whereby its deficiency results in impaired OXPHOS, leading to reduced numbers of mature AM, accumulated surfactants, and increased susceptibility to infection (55).
Chronic exposure to cigarette smoke dysregulates AM metabolism (56). Smokers show disrupted redox homeostasis, reduced PPARγ, and increased nitric oxide and ROS production in mitochondria, with reduced antioxidant glutathione, exacerbating lung inflammation (57). Increased ROS is also found in AMs after phagocytosing nanotubes and nano-particulate carbon black, a byproduct of smoking (28). In contrast, PPARγ plays a protective role against oxidative stress by transcriptional repression of pro-inflammatory factors and enzymes, such as NF-κB, induced nitric oxide synthase (iNOS), and the activation of antioxidant genes, such as heme oxygenase-1 (HO-1) and superoxide dismutase (SOD) (58). PPARγ also participates in glucose and lipid metabolism (54), and regulates the scavenger receptor, CD36, to control the surfactant lipid catabolism (59). Current smokers show decreased CD36 in AMs compared to former smokers (60), indicating that smoking may impair surfactant lipid catabolism and increase NF-κB activation (61) through deregulated surfactant recycling. Notably, smoke-induced reduction of lipid metabolism and PPARγ signaling in AMs is independent of COPD phenotype (62). Furthermore, smoking-associated AM clusters show upregulated genes involved in detoxification, inflammation, and oxidative stress that strengthen COPD progression (62). Additionally, transferrin receptor, CD71, is downregulated in smokers, permitting inappropriate pulmonary bacterial growth and pulmonary fibrosis (47, 63). CD71 expression in mature AMs can limit free iron available to be used by pathogenic microorganisms. AMs lacking CD71 exhibit reduced maturation, phagocytic function, and increased profibrotic genes (64). Advanced COPD lungs are enriched with high metallothionein-expressing AMs, indicating disrupted heavy metal metabolism (60). Although metallothionein has protective roles in acute lung injury (65), there is limited mechanistic information about elevated metallothionein in advanced COPD.
4 The role of AMs in NSCLC
4.1 AM polarization
Both the promotion and inhibition of lung tumor have been attributed to AMs. AMs isolated from lung cancer patients when stimulated with IFN-γ or GM-CSF, secrete TNF-α, IL-6, and IL-1β, that can enhance tumor killing (66). However, mounting evidence suggest that many antitumor function of AMs, such as phagocytosis and the expression of cytokines that activate adaptive immunity, wane with the progression of NSCLC (67), pointing to their role in promoting immunosuppressive microenvironment and dysfunctional immunity (Figure 4).
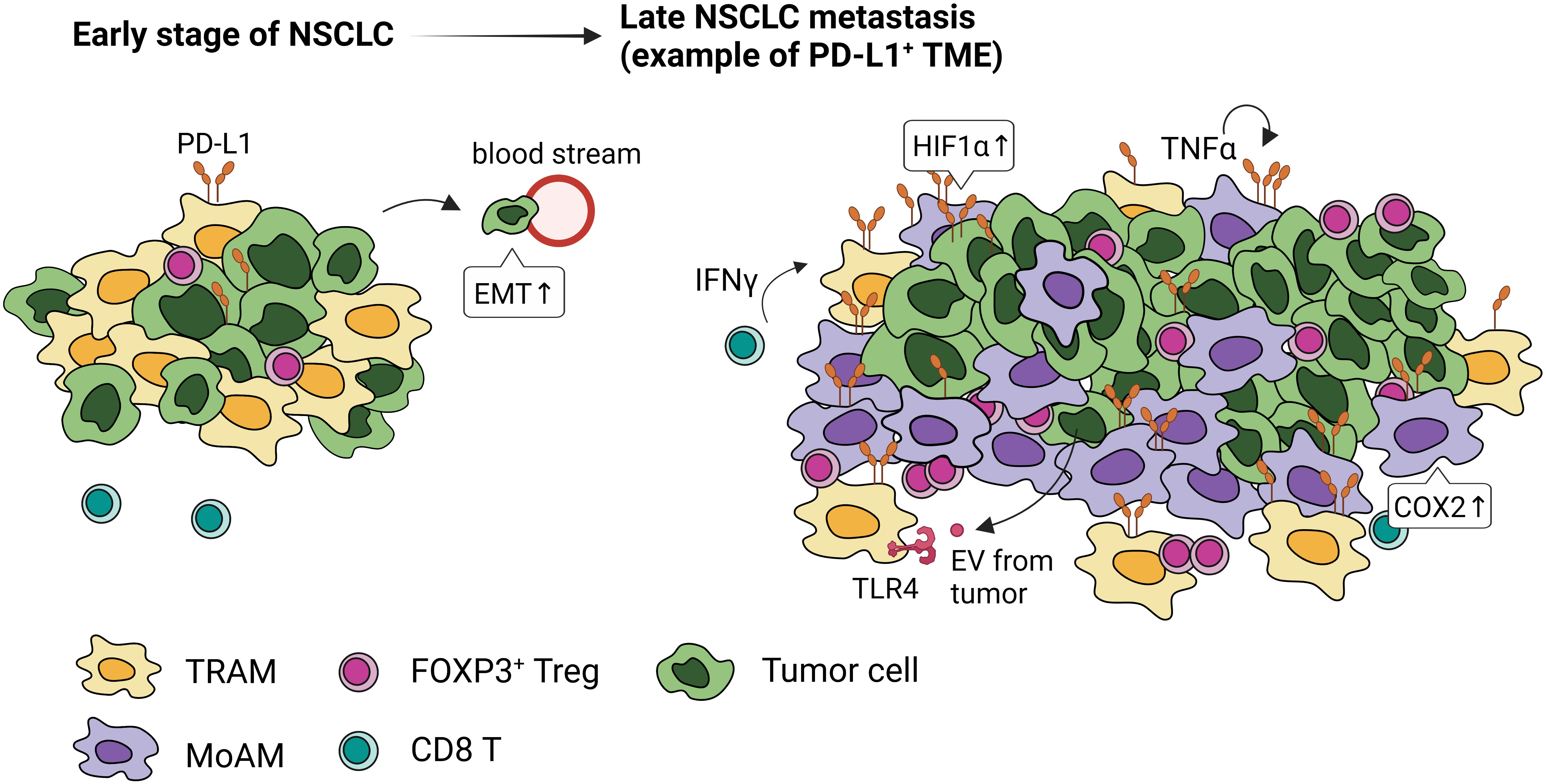
Figure 4 PD-L1 expression and localization of macrophages in early and late stages of NSCLC. Tumor-associated macrophages express high levels of PD-L1 through activation of HIF1α, COX2 signaling, paracrine IFN-γ, autocrine TNFα, and extracellular vesicles (EVs) from tumor cells. In the early stage of NSCLC, embryonic-origin TRAMs are adjacent to the tumor cells. They surround the tumor mass preventing CD8 T cell infiltration, promoting FOXP3+Tregs, and inducing epithelial-mesenchymal transition of tumor cells. In the late stage of NSCLC, these TRAMs are in the periphery and MoAMs dominate the tumor.
Acute inflammation induces DC maturation, required for antigen presentation, and initiation of anti-tumor responses from effector T cells. In contrast, chronic inflammation, a hallmark of advanced NSCLC (68), promotes suppressive environment hindering the efficacy of immunotherapy, exacerbating cancer development (69). Chronic inflammatory mediators recruit TAM to tumors sites inhibiting T cell function and facilitating tumor invasion. While some bone marrow-derived Ly6C+ monocytes express CCR2 in lung tumors other immunosuppressive monocytic myeloid-derived suppressor cells (M-MDSC) can also differentiate into TAM upon exposure to hypoxia (70). TAMs are highly heterogenous in nature and in addition to hematopoietic origin (37), include polarized AMs (71). Although MoAMs and TRAMs make up TAMs population in human NSCLC, their functional differences, surface marker expression, gene regulation, and cell fate, remain largely unknown.
4.2 TRAMs foster lung cancer
TRAMs are highly represented within TAMs in NSCLC (39, 72). Single cell (sc)RNA sequencing and lineage tracing studies have identified distinct temporal and spatial distribution of TAMs in mouse and human lung (39). In the early-stage NSCLC, TRAMs are highly proximal to tumors; however, MoAMs gradually replace TRAMs and infiltrate the advanced NSCLC lesions (39) (Figure 4). TRAMs upregulate genes involved in antigen presentation and tissue remodeling, promoting epithelial mesenchymal transition and tumor invasion (39). Depletion of TRAMs before tumor engraftment can lead to lower tumor burden, reduced FOXP3+ Tregs, and increased IFN-γ+ CD8 T cells, indicating TRAMs support the early stage of NSCLC development (39).
Targeted molecular therapy for epidermal growth factor receptor (EGFR) mutation has improved patients’ responses; however, nearly all tumors develop resistance after treatment (73). Animal models of EGFR-driven NSCLC have shown that AMs are critical in the development of primary lung adenocarcinoma. Using an inducible knock-in model that mimics the development of mutant EGFR lung lepidic adenocarcinoma in humans, the oncogenic signaling led to a massive expansion of AMs with an immunosuppressive phenotype (8, 74). Similar results have been reported in the Kras; p53 mouse model of NSCLC (75). AMs upregulate IL-1R and phagocytosis while downregulating MHCII and co-stimulatory molecules, consistent with an anti-inflammatory phenotype (76). Specific elimination of AMs but not interstitial macrophages (IMs) drastically reduced tumor burden (74, 76) and increased cytotoxic CD8 T cell infiltration in the lung (74). When compared to early and late invasive lung adenocarcinoma, studies have shown that AMs (defined as CD11c+ F4/80+ CD64+ Siglec-F+) are replaced by TAMs (CD11b+ F4/80+ CD64+ Siglec-F-) as cancer progresses (8). Why and how AMs are progressively replaced by monocyte-derived macrophages in the TAM pool during tumor expansion remains to be determined.
TRAMs play a pivotal role in promoting immunosuppression in the metastatic niches. In a model of metastatic breast cancer, TRAMs accumulate in the premetastatic lung area, a process that was dependent on complement C5a receptor and local production of C5 (72). TRAMs inhibited lung DC maturation by promoting Th2 cell generation and reducing tumoricidal activity (72). Depleting TRAMs reversed the immune suppression in the lungs, strengthened Th1 responses, and reduced metastatic burden (72). Notably, bone marrow transplant experiments showed that the transplanted cells did not become AMs in the lung; instead, most of them become MDSCs, expressing CD11b and Gr-1, excluding the contribution of bone marrow to the TRAM pool (72). In the metastatic lung tumor, foamy TRAMs were shown to express the lipid metabolic receptors Lgals3 and Trem2 and inhibit T cell effector function (77). Similarly, in a metastatic hepatocellular carcinoma model, TRAMs exhibited a high level of the inflammatory eicosanoid derivative leukotriene B4 (LTB4), by expressing 5-lipoxygenases (5-LOX), enabling cancer cell proliferation (63). CCR2+ MoAMs are recruited by CCL2+ interstitial macrophages in the metastatic foci, indicating a systemic signal repletes AM pool from bone marrow (63). TRAM depletion decreased metastatic foci numbers, demonstrating a direct role of TRAMs in lung metastasis (63). These studies show that distinct signals from the primary tumor can manipulate TRAMs to promote metastasis support the pool by a variety of mechanisms; however, the actual signal molecules or mediators that come from the primary tumor are still largely unknown.
4.3 Regulation of PD-L1 expression in AMs
The interaction between PD-1 on tumor-infiltrating lymphocytes (TILs) and PD-L1 on tumor cells is a critical mechanism for immune escape and most widely targeted with ICI. Although many solid tumors express PD-L1, immune cells including CD68+ TAMs, exhibit high levels of PD-L1 (78) (Figure 4) and some cases, PD-1 (8). Notably, ICI-treated NSCLC patients with elevated PD-L1+ macrophages have a more favorable survival than those with high PD-L1 in tumor cells (78). Further, a high level of PD-L1+ macrophages positively correlate with PD-L1/PD-1 expression in tumor cells and TILs, respectively (78). The single-cell analysis combined with spatial quantification identified clusters of PD-L1+ macrophages at the tumor-invasive margin, posing a physical barrier that can block T cell entry in the tumor lesion (79). The efficacy of ICI-mediated antitumor responses depends on whether TILs can recognize and kill tumor cells; therefore, increased TIL infiltration indicates “hot tumors” and carries favorable treatment outcome (80). PD-L1+ TAMs that hinder effector T cell function are commonly described in syngenetic tumor transplant and advanced models of solid tumors (81, 82). However, in humans PD-L1+ TAMs in early-stage of NSCLC do not directly inhibit T cell responses (83), highlighting the complex biology of human tumor development. Some of the differences between animal models and human tumors include tumor latency with selective pressures and delayed immune infiltration occurring in the later stages of cancer (84). TRAMs are a critical innate immune player, and their phenotypes are dictated by the surrounding TME in NSCLC. Under static conditions, they constitutively express PD-L1 to increase their phagocytosis and repress the effector T cell activation (2). However, whether and how TRAMs alter PD-L1 expression at different stages of tumor development in humans could provide critical information in predicting those who may benefit from PD-L1/PD-1-based immunotherapy.
Tissue hypoxia, a microenvironmental factor that induces polarization and PD-L1 expression in TAMs, has been shown to promote NSCLC progression (85). HIF-1α upregulates PD-L1 in TAMs, thereby supporting an immunosuppressive TME (86). Combination therapies based on HIF-1α inhibition and PD-1/PD-L1 checkpoint blockade have been shown to induce tumor regression, alleviate immune suppression, and increase survival in a murine model of NSCLC (87). Although specific changes in TAMs were not examined, small-molecule inhibition of HIF-1α promoted antitumor immunity, indicating HIF1α inhibition may be a promising adjuvant with ICI treatment (87).
Cytokines and soluble factors within the TME, including IFN-γ, TNF, COX2, and TLRs, can also induce PD-L1 expression in TAMs. Evidence has shown that the inflammatory cytokines IFN-γ from tumor-specific T cells can drive PD-L1 expression in tumors and surrounding stroma that expresses interferon receptors (88). While endogenous IFN-γ is dispensable for AMs’ PD-L1 expression, intrinsic TNF is required for its maintenance and upregulation (89). The inflammatory COX2/mPGES1/PGE2 pathway contributes to PD-L1 expression in tumor-infiltrating myeloid cells (81). The anti-inflammatory cytokine IL-10 has been reported to increase TAM PD-L1 in several cancers (90). Notably, tumor-derived extracellular vesicles captured by TLR4 in macrophages activate STAT3-dependent PD-L1 transcription, which polarizes them to anti-inflammatory phenotype (91). Together, PD-L1 expression by TAM through paracrine and autocrine signaling emphasizes the multifaceted and complex interactions that occur in the TME. Thus, further studies are needed to more comprehensively delineate the upstream stimulations that induce PD-L1+ TAM to advance the development of therapeutic intervention and treatment regimes.
4.4 Factors Responsible for Trained Immunity in AMs
Metabolic reprogramming and epigenetic imprinting drive innate immune memory responses, also known as trained immunity. AMs responding to pathogens, can protect the host from subsequent related or unrelated microbial exposure (92). Whether TRAMs or MoAMs develop trained immunity is context dependent. TRAMs can increase MHC-II, defense-related genes, glycolysis, and chemokine expression after adenovirus infection. This priming requires contact with IFN-γ+ CD8 T cells (93). In contrast, TRAMs depleted by influenza infection are replaced by MoAMs, which confer protection post-influenza infection against other microbial pathogens (94). These recruited MoAMs show similar surface markers of TRAMs but with a transcriptional profile that resembles CCR2+ monocyte precursors, and open chromatin at loci controlling the expression of inflammatory genes (94). However, MoAM production of IL-6 wanes 2 months post-influenza and protection against bacteria is lost. Interestingly, TRAMs become unresponsive, with poor phagocytic ability after the resolution of systemic inflammation, a process that is dependent on signal-regulatory protein α (SIRPα) (95). In pneumoniae, LPS-mediated induction of trained AMs adoptively transferred to naïve mice and challenged with Streptococcus pneumoniae, failed to protect mice against the infection, indicating niche environments are critical for triggering effective trained immunity (96). These findings suggest that infection severity and the microenvironment can determine whether trained or tolerant AMs are generated.
Less is known about the role of trained macrophages in tumor immunity. Trained macrophages can develop from systemic hematopoietic progenitors and circulating monocytes or TRAMs (97, 98). Antitumor responses in lungs have been described in mice recovered from acute influenza infection, indicating that trained AMs may play a pivotal role in determining the fate of tumor development. Trained AMs can increase phagocytosis, promote cytotoxicity, and resist tumor-induced immune suppression, showing distinct transcriptomic and epigenetic profiles (99). Notably, this trained immunity is dependent on IFN-γ and NK cells (99). Different routes of antigen exposure can train independent pools of macrophages. For example, in the metastatic setting, beta-glucan particles via i.p. has been shown to result in trained interstitial macrophages but not AMs (100). Future research should specify which populations of macrophages are affected by stimulation and what environmental signals are involved in the trained immunity.
4.5 Targeting TAMs to treat NSCLC
A recent trend to control tumor growth is to deplete, block, or reactivate TAMs and synergistically activate T cells in the TME. This is accomplished by depleting or stimulating TAMs to become inflammatory macrophages, concurrently with ICI treatment to awaken tumor-specific T cell responses. Several reviews have discussed this topic in solid tumors (101, 102), therefore, here we provide an update of this information, focusing on targeting TAMs in NSCLC (Figure 5).
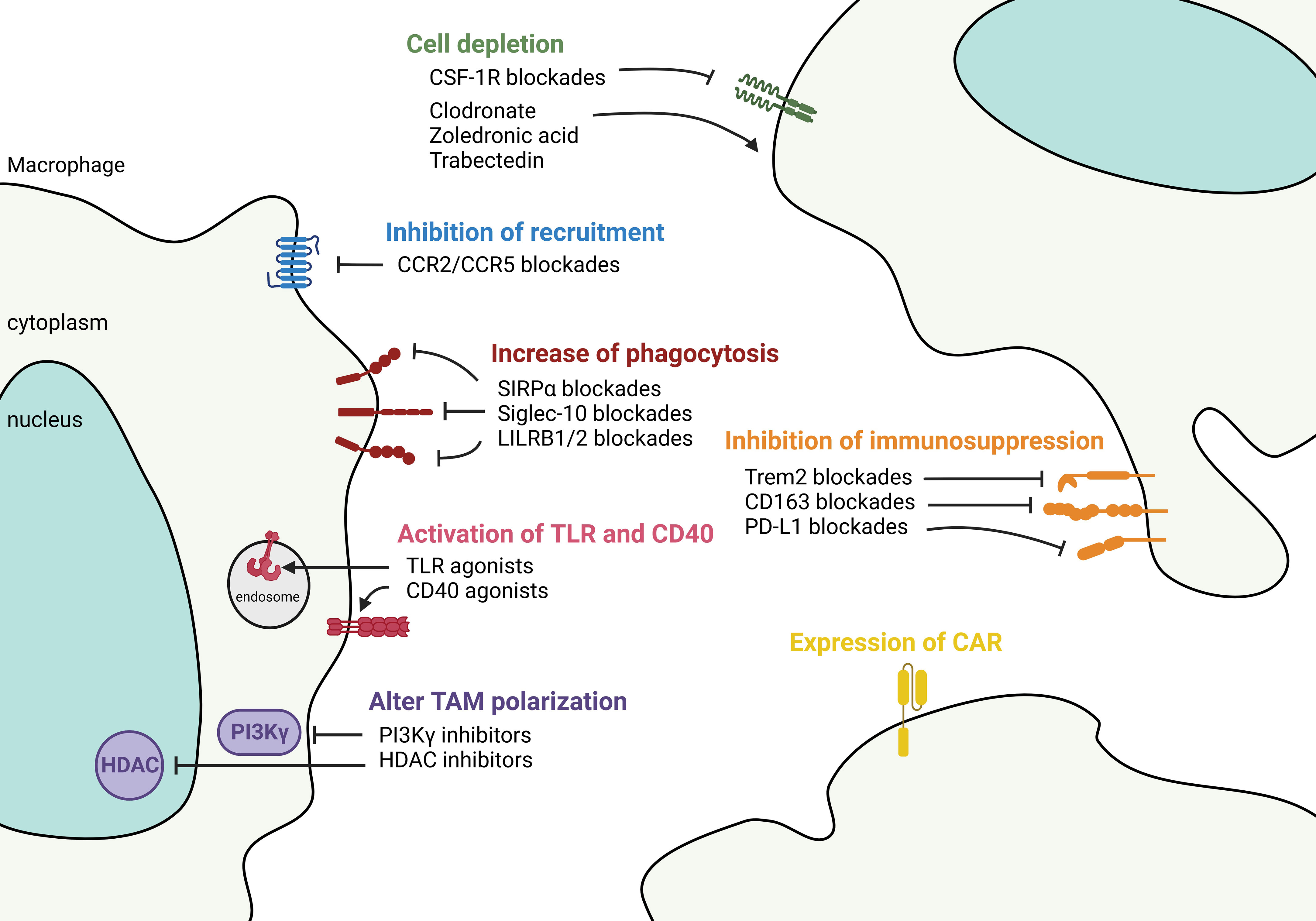
Figure 5 Targeting macrophages to control tumor microenvironment. Therapeutic strategies aimed at controlling TAM in the TME fall into 1) controlling the number of TAMs either by anti-CSF-1R and chemicals such as clodronate, zoledronic acid, and Trabectedin to deplete the cells, or by chemokine blockades to decrease the recruitment of TAMs. 2) Functional-based reprogramming that targets the surface receptors/ligands. For example, increase phagocytosis by SIRPα, Siglec-10, and LILRB1/2 blockades; activate TLR and CD40 signaling by agonists; inhibit immunosuppression by targeting Trem2, CD163, and PD-L1. 3) Pan reprogramming that inhibits the intracellular key regulators such as PI3Kγ and HDAC to prevent TAM polarization. 4) Expression of engineered CAR in TAMs to recognize and phagocytose targeted tumor cells.
Depletion of TAM by inducing cell death can inhibit tumor growth. Both MoAMs and TRAMs express CSF-1R (103). Targeting the CSF-1/CSF-1R pathway can deplete monocytes/macrophages in animal models (104). However, antibody-based CSF-1R blockade alone has only a modest antitumor effect in humans (105). Combining the anti-CSF-1R with ICI in murine models led to significant tumor reduction (106), however, systemic depletion of macrophages either causes undesired side effects (107) or insufficient antitumor response in NSCLC patients (108, 109). A small study that compared the spatial transcriptomic profiles in NSCLC tumors suggested that combined anti-CSF-1R with ICI may not be clinically beneficial because ICI responders showed higher expression of CSF-1R (110). This finding underlines the heterogeneity of TAMs and beneficial roles of macrophage subsets in the TME. Similarly, another study showed that anti-CSF-1R can deplete macrophages but have a limited effect on antitumor responses in mice bearing established tumors (111). Despite limited clinical activities, CSF-1R kinase inhibitors combing with other drugs are currently in phase I trials for advanced solid tumors. Small molecules that mimic bone matrix pyrophosphatases, including clodronate and zoledronic acid, and Trabectedin that induces DNA damage can deplete macrophages (112). More studies are needed to decipher whether TAM depletion combined with other agents could provide clinical applications.
A combination of blacking monocyte recruitment in patients with ICI therapy represents a potential new treatment strategy in NSCLC. Several small molecule inhibitors targeting chemokine receptors can decrease monocyte-derived macrophage recruitment to NSCLC. Targeting CCL2/CCR2 and CCL5/CCR5 pathways have shown promising pre-clinical results in NSCLC (113), and have been expanded to clinical trials in adult solid tumors (NCT04504942 and NCT04123379). The CCL2/CCR2 axis attracts TAMs through PD-1 signaling in esophageal carcinogenesis (114). CCL5 from macrophages facilitates colorectal cancer growth via PD-L1 stabilization (115). A caveat is that once bone marrow-derived macrophages enter the tumor, the expression of chemokine receptors may be downregulated, as has been shown in ovarian cancer, where CCR2 expression is regulated by tumor-derived TNF (116). Also, TRAMs play a pivotal role in cancer initiation similarly express a lower level of chemokine receptors; therefore, targeting chemokine pathways may not be able to remove these cells.
Reprogramming TAMs at the tumor site is a newly emerging strategy for cancer treatment. “Don’t eat me” axis, e.g., CD47/SIRPα and CD24/Siglec-10 between tumor cells and myeloid cells presents an exciting paradigm for using tumor-induced inhibitory pathways to escape immune surveillance. Specifically, CD47 is commonly overexpressed in solid tumors where it is recognized by macrophages’ SIRPα, preventing phagocytosis (117). NSCLCs co-expressing CD47 and PD-L1 are associated with worse clinical outcomes (118). Blocking the interaction between CD47 and macrophage SIRPα increases cytosolic DNA sensing and activates the STING pathway activation that enables detection of tumor mitochondrial DNA and subsequent antigen presentation to T cells (119). More research is needed to better understand the mechanistic effects of blocking CD47/SIRPα in the TME. The first human anti-CD47 phase I trial showed that the drug is well tolerated in advanced tumors (120). Several CD47 blockades used as a monotherapy or combined with ICI or other agents are currently in clinical trials. Another “don’t eat me” signal, the recently discovered CD24/Siglec-10 axis facilitates macrophage infiltration into the tumor by Siglec-10-mediated sensing of CD24 and is complementary to CD47 signaling. Targeting both axes showed an additive phagocytosis response (121). Similarly, the disruption of the MHCI/LILRB1/2 axis potentiates phagocytosis by TAMs (122).
Several new approaches have shown that reprograming TAMs’ intracellular signaling can effectively alter their function. For instance, phosphoinositide 3 kinase gamma (PI3Kγ), a family member of PI3K, controls the switch between immune stimulating or suppressive macrophages (123). PI3Kγ is highly expressed on myeloid cells but not on cancer cells and mediates myeloid cell trafficking in cancer (123). PI3Kγ blockade stimulates inflammatory macrophages, enhances effector CD8 T cell function, and increases ICI responses in tumors (123). The PI3K/AKT/mTOR pathway is critical in NSCLC development. Studies have shown that inhibition of the epigenetic regulator, histone deacetylase (HDAC), can transcriptionally modify TAMs and activate their antitumor activity (124). The class IIa HDAC inhibitor (TMP195) has been shown in a metastatic breast cancer model to induce recruitment and differentiation of phagocytic macrophages, reducing primary tumor burden and incidence of lung metastasis (125). Combination with ICI further enhances the anti-tumor effect and durability of this HDAC IIa inhibition (125). Adjuvant therapy targeting epigenetic modulators (DNA methyltransferase and HDAC) after surgical removal of the tumor also showed inhibition of MDSC accumulation in the lung premetastatic niche and longer disease-free survival (126).
Activation through TLR signaling and CD40 can polarize TAMs to a pro-inflammatory state. This signaling converts tumors from cold to hot, activates the immune system, and overcomes ICI resistance. TLRs regulate TAM phenotypes, DC maturation, and increase effector T cell response (127). The anti-tumor property has led to the expansion of TLR agonists for immunotherapy. The use of TLR agonists in cancer is controversial because TLRs are also expressed on cancer cells and have pro- as well as anti-tumorigenic activities. However, several TLR agonists targeting TLR 3, 7, 8, and 9 are currently in trials for solid tumors (127). TLR agonists paired with ICI have shown synergistic effects on therapeutic efficacy and survival rate in patients (128). Future work is needed to understand the mechanistic regulation between TLR agonists and immune checkpoint expression in the TME. Similarly, CD40 is a potent agent to promote a TAM-mediated antitumor response. Combining CD40 agonist, anti-CSF-1R, and anti-PD-L1 in trio treatment has been evaluated in the MC38 syngeneic model and showed 90% complete response (129). The results of the phase I trial of this trio regimen in advanced tumors, which included NSCLC, showed no overt safety issue (130). Given the safety profile and antitumor activity, more clinical trials regarding these agonists with other therapeutic approaches in diverse solid tumors are warranted and anticipated.
Counteracting the immunosuppressive TME by targeting cell-surface suppressive markers on TAM to potentiate the efficacy of ICI is of growing interest. Triggering receptor expressed on myeloid cells 2 (TREM2) has been found on tumor cells, TAMs, and myeloid cells. In addition to its role in oncogenic signaling (131), TREM2+ TAM derived from monocytes strongly suppress NK cell function and promoting antitumor immunity in NSCLC (132). TREM2+ monocytes-derived macrophages have been found exclusively induced by the TME (133), and inhibit production of IL-18 and IL-15, which are necessary for NK activity (132). Trem2 deficiency or anti-Trem2 treatment improved ICI responses and reduces tumor burden (134). Interestingly, two types of APOE+ TAMs are found, expressing either TREM2 or FOLR2 in human breast cancers. According to spatial analyses, TREM2+ macrophages are inside the tumor nest and close to the invasive margin, whereas FOLR2+ macrophages reside away from the tumor nest and remain in the perivascular area (135). FOLR2+ macrophage-CD8 T cell clusters correlate with a better clinical outcome (135). However, an in-depth characterization of macrophages and determination of their role in NSCLC is needed for their prognostic utilization. Another suppressive target is the scavenger receptor CD163 that positively correlates with tumor progression (136). PD-1+ TAMs in human NSCLC express CD163 and are associated with reduced survival (137). Interestingly, CD163+ C33+ PD-L1+ macrophages were retrospectively found to be higher prior to ICI treatment in those NSCLC patients with hyper progressive disease (138). Other suppressive targets, such as macrophage receptor with collagenous structure (MARCO), have also been described in TAM, and targeting them is expected to reverse the phenotype and curb cancers (139).
Chimeric antigen receptor macrophages (CAR-M) have drawn tremendous attention since they were introduced in 2020 (140). Given the tumor-homing ability of macrophages, CAR-M are expected to enter the solid tumor and exhibit antigen-specific-phagocytosis. They have been demonstrated to promote pro-inflammation, attract T cells, and prevent immunosuppression in TME (141). Similar to the evolution of CAR-T cell therapy, the cell sources of CAR-M have transitioned from using peripheral blood monocytes (140) to induced pluripotent stem cells (iPSC) (142), to now modifying in situ tumor macrophages to bypass cell isolation and tissue rejection issues (141). Additionally, CAR-M may potentially serve as a cargo to deliver drugs (143). CAR-M has shown an extraordinary ability to clear tumor cells in preclinical models as mentioned; however, how they interact with tissue-resident macrophages, whether CAR-M develop an innate memory response, whether trogocytosis occurs between CAR-M and other cells in the TME, and many other questions remain to be resolved.
5 Discussion
AMs are highly plastic immune cells that respond to environmental signals. NSCLCs associated with chronic inflammation and extrinsic stimuli, such as cigarette smoke and environmental pollutants, can transform TAMs phenotypes incapacitating their effector functions. Increasing evidence suggests that TRAMs are transcriptionally and epigenetically distinct from MoAMs, which can contribute to TAMs but exert different functions. These differences highlight the intricate relationships between myeloid cells and their environment. Discovering key regulatory factors that determine these disparities will provide depth to our understanding of the heterogeneity of macrophages and inform us on new lung cancer treatment options. Several tools will be beneficial in this pursuit. Genetic tracing can inform the origin of macrophages. Multi-omics approaches combined with pseudo-timing algorithms or temporal fluorescent labeling techniques can provide insights into the localization, interactions, and evolution between various cell types and dynamics in the disease states. In vivo murine tumor studies combining different therapies with targeting specific macrophage populations or regulators will inform basic biology as well as precision oncology. Given the abundance, tumor-homing ability, and plasticity of macrophages, targeting macrophages and macrophage cell therapy is desirable. Future treatments should be tailored based on the TME structure in each patient and logistically combine ICI to enhance efficacy.
Author contributions
CC: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. DA: Visualization, Writing – review & editing. DC: Writing – review & editing. FK: Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was in part funded by The Nancy Chang Endowment Funds, US National Institutes of Health (NIH) grants R01 ES029442-01, R01 AI135803-01, and the US Veterans Administration (VA) Merit grant CX000104, DOD W81XWH-20-1-0607 to F.K.; HL117181, HL140398, R01 AI135803, and R41 AI124997 and VA Office of Research and Development grant I01BX004828 to D.C. NIH T32GM088129 and Baylor Research Advocates for Student Scientists (BRASS) scholarship to D.A. VA I50CU000161 to F.K.
Acknowledgments
The figures were created with Biorender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Steen EH, Wang X, Balaji S, Butte MJ, Bollyky PL, Keswani SG. The role of the anti-inflammatory cytokine interleukin-10 in tissue fibrosis. Adv Wound Care (New Rochelle) (2020) 9:184–98. doi: 10.1089/wound.2019.1032
2. Sun F, Li L, Xiao Y, Gregory AD, Shapiro SD, Xiao G, et al. Alveolar macrophages inherently express programmed death-1 ligand 1 for optimal protective immunity and tolerance. J Immunol (2021) 207:110–4. doi: 10.4049/jimmunol.2100046
3. Hou F, Xiao K, Tang L, Xie L. Diversity of macrophages in lung homeostasis and diseases. Front Immunol (2021) 12:753940. doi: 10.3389/fimmu.2021.753940
4. Li F, Piattini F, Pohlmeier L, Feng Q, Rehrauer H, Kopf M. Monocyte-derived alveolar macrophages autonomously determine severe outcome of respiratory viral infection. Sci Immunol (2022) 7:eabj5761. doi: 10.1126/sciimmunol.abj5761
5. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10:826–37. doi: 10.1038/nri2873
6. Dewhurst JA, Lea S, Hardaker E, Dungwa JV, Ravi AK, Singh D. Characterisation of lung macrophage subpopulations in COPD patients and controls. Sci Rep (2017) 7:7143. doi: 10.1038/s41598-017-07101-2
7. Kielbassa K, Vegna S, Ramirez C, Akkari L. Understanding the origin and diversity of macrophages to tailor their targeting in solid cancers. Front Immunol (2019) 10:2215. doi: 10.3389/fimmu.2019.02215
8. Jang HJ, Lee HS, Yu W, Ramineni M, Truong CY, Ramos D, et al. Therapeutic targeting of macrophage plasticity remodels the tumor-immune microenvironment. Cancer Res (2022) 82:2593–609. doi: 10.1158/0008-5472.CAN-21-3506
9. Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res (2016) 5:288–300. doi: 10.21037/tlcr.2016.06.07
10. Provencio M, Calvo V, Romero A, Spicer JD, Cruz-Bermudez A. Treatment sequencing in resectable lung cancer: the good and the bad of adjuvant versus neoadjuvant therapy. Am Soc Clin Oncol Educ Book (2022) 42:1–18. doi: 10.1200/EDBK_358995
11. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
12. Shields MD, Marin-Acevedo JA, Pellini B. Immunotherapy for advanced non-small cell lung cancer: A decade of progress. Am Soc Clin Oncol Educ Book (2021) 41:1–23. doi: 10.1200/EDBK_321483
13. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol (2019) 30:1321–8. doi: 10.1093/annonc/mdz167
14. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
15. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30:R921–5. doi: 10.1016/j.cub.2020.06.081
16. Xu F, Wei Y, Tang Z, Liu B, Dong J. Tumor−associated macrophages in lung cancer: Friend or foe? (Review). Mol Med Rep (2020) 22:4107–15. doi: 10.3892/mmr.2020.11518
17. Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang CL. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp Ther Med (2019) 18:4490–8. doi: 10.3892/etm.2019.8068
18. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci (2021) 22(13):6995. doi: 10.3390/ijms22136995
19. Sedighzadeh SS, Khoshbin AP, Razi S, Keshavarz-Fathi M, Rezaei N. A narrative review of tumor-associated macrophages in lung cancer: regulation of macrophage polarization and therapeutic implications. Transl Lung Cancer Res (2021) 10:1889–916. doi: 10.21037/tlcr-20-1241
20. Cassetta L, Pollard JW. A timeline of tumour-associated macrophage biology. Nat Rev Cancer (2023) 23:238–57. doi: 10.1038/s41568-022-00547-1
21. Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther (2021) 6:127. doi: 10.1038/s41392-021-00506-6
22. Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood (2006) 107:2112–22. doi: 10.1182/blood-2005-01-0428
23. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
24. Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch’ng ES. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front Oncol (2019) 9:1512. doi: 10.3389/fonc.2019.01512
25. Xie QH, Zheng JQ, Ding JY, Wu YF, Liu L, Yu ZL, et al. Exosome-mediated immunosuppression in tumor microenvironments. Cells (2022) 11(12):1946. doi: 10.3390/cells11121946
26. Jenkins SJ, Allen JE. The expanding world of tissue-resident macrophages. Eur J Immunol (2021) 51:1882–96. doi: 10.1002/eji.202048881
27. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol (2014) 14:81–93. doi: 10.1038/nri3600
28. Chang CY, You R, Armstrong D, Bandi A, Cheng YT, Burkhardt PM, et al. Chronic exposure to carbon black ultrafine particles reprograms macrophage metabolism and accelerates lung cancer. Sci Adv (2022) 8:eabq0615. doi: 10.1126/sciadv.abq0615
29. Hill W, Lim EL, Weeden CE, Lee C, Augustine M, Chen K, et al. Lung adenocarcinoma promotion by air pollutants. Nature (2023) 616:159–67. doi: 10.1038/s41586-023-05874-3
30. Park MD, Silvin A, Ginhoux F, Merad M. Macrophages in health and disease. Cell (2022) 185:4259–79. doi: 10.1016/j.cell.2022.10.007
31. Gschwend J, Sherman SPM, Ridder F, Feng X, Liang HE, Locksley RM, et al. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J Exp Med (2021) 218(10):e20210745. doi: 10.1084/jem.20210745
32. Ataya A, Knight V, Carey BC, Lee E, Tarling EJ, Wang T. The role of GM-CSF autoantibodies in infection and autoimmune pulmonary alveolar proteinosis: A concise review. Front Immunol (2021) 12:752856. doi: 10.3389/fimmu.2021.752856
33. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol (2014) 15:1026–37. doi: 10.1038/ni.3005
34. Baker AD, Malur A, Barna BP, Ghosh S, Kavuru MS, Malur AG, et al. Targeted PPARgamma deficiency in alveolar macrophages disrupts surfactant catabolism. J Lipid Res (2010) 51:1325–31. doi: 10.1194/jlr.M001651
35. Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, et al. The cytokine TGF-beta promotes the development and homeostasis of alveolar macrophages. Immunity (2017) 47:903–912 e4. doi: 10.1016/j.immuni.2017.10.007
36. Gibbings SL, Goyal R, Desch AN, Leach SM, Prabagar M, Atif SM, et al. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood (2015) 126:1357–66. doi: 10.1182/blood-2015-01-624809
37. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate mapping via ms4a3-expression history traces monocyte-derived cells. Cell (2019) 178:1509–1525 e19. doi: 10.1016/j.cell.2019.08.009
38. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell (2019) 178:686–698 e14. doi: 10.1016/j.cell.2019.05.054
39. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature (2021) 595:578–84. doi: 10.1038/s41586-021-03651-8
40. Allard B, Panariti A, Martin JG. Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front Immunol (2018) 9:1777. doi: 10.3389/fimmu.2018.01777
41. Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol (2011) 2:65. doi: 10.3389/fimmu.2011.00065
42. Byrne A, Reen DJ. Lipopolysaccharide induces rapid production of IL-10 by monocytes in the presence of apoptotic neutrophils. J Immunol (2002) 168:1968–77. doi: 10.4049/jimmunol.168.4.1968
43. Bain CC, MacDonald AS. The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol (2022) 15:223–34. doi: 10.1038/s41385-021-00480-w
44. Kheradmand F, Zhang Y, Corry DB. Contribution of adaptive immunity to human COPD and experimental models of emphysema. Physiol Rev (2023) 103:1059–93. doi: 10.1152/physrev.00036.2021
45. Hong MJ, Gu BH, Madison MC, Landers C, Tung HY, Kim M, et al. Protective role of gammadelta T cells in cigarette smoke and influenza infection. Mucosal Immunol (2018) 11:894–908. doi: 10.1038/mi.2017.93
46. Burgoyne RA, Fisher AJ, Borthwick LA. The role of epithelial damage in the pulmonary immune response. Cells (2021) 10(10):2763. doi: 10.3390/cells10102763
47. Sarir H, Mortaz E, Karimi K, Kraneveld AD, Rahman I, Caldenhoven E, et al. Cigarette smoke regulates the expression of TLR4 and IL-8 production by human macrophages. J Inflammation (Lond) (2009) 6:12. doi: 10.1186/1476-9255-6-12
48. Broug-Holub E, Toews GB, van Iwaarden JF, Strieter RM, Kunkel SL, Paine 3RDR, et al. Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect Immun (1997) 65:1139–46. doi: 10.1128/iai.65.4.1139-1146.1997
49. Yuan F, Fu X, Shi H, Chen G, Dong P, Zhang W. Induction of murine macrophage M2 polarization by cigarette smoke extract via the JAK2/STAT3 pathway. PloS One (2014) 9:e107063. doi: 10.1371/journal.pone.0107063
50. Bazzan E, Turato G, Tine M, Radu CM, Balestro E, Rigobello C, et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir Res (2017) 18:40. doi: 10.1186/s12931-017-0522-0
51. Aggarwal NR, King LS, D’Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol (2014) 306:L709–25. doi: 10.1152/ajplung.00341.2013
52. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
53. Woods PS, Kimmig LM, Meliton AY, Sun KA, Tian Y, O’Leary EM, et al. Tissue-resident alveolar macrophages do not rely on glycolysis for LPS-induced inflammation. Am J Respir Cell Mol Biol (2020) 62:243–55. doi: 10.1165/rcmb.2019-0244OC
54. Asada K, Sasaki S, Suda T, Chida K, Nakamura H. Antiinflammatory roles of peroxisome proliferator-activated receptor gamma in human alveolar macrophages. Am J Respir Crit Care Med (2004) 169:195–200. doi: 10.1164/rccm.200207-740OC
55. Gao X, Zhu B, Wu Y, Li C, Zhou X, Tang J, et al. TFAM-dependent mitochondrial metabolism is required for alveolar macrophage maintenance and homeostasis. J Immunol (2022) 208:1456–66. doi: 10.4049/jimmunol.2100741
56. Skold CM, Lundahl J, Hallden G, Hallgren M, Eklund A. Chronic smoke exposure alters the phenotype pattern and the metabolic response in human alveolar macrophages. Clin Exp Immunol (1996) 106:108–13. doi: 10.1046/j.1365-2249.1996.d01-805.x
57. Lugg ST, Scott A, Parekh D, Naidu B, Thickett DR. Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease. Thorax (2022) 77:94–101. doi: 10.1136/thoraxjnl-2020-216296
58. Lee C. Collaborative power of nrf2 and PPARgamma activators against metabolic and drug-induced oxidative injury. Oxid Med Cell Longev (2017) 2017:1378175. doi: 10.1155/2017/1378175
59. Dodd CE, Pyle CJ, Glowinski R, Rajaram MV, Schlesinger LS. CD36-mediated uptake of surfactant lipids by human macrophages promotes intracellular growth of mycobacterium tuberculosis. J Immunol (2016) 197:4727–35. doi: 10.4049/jimmunol.1600856
60. Sauler M, McDonough JE, Adams TS, Kothapalli N, Barnthaler T, Werder RB, et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing. Nat Commun (2022) 13:494. doi: 10.1038/s41467-022-28062-9
61. Nguyen HA, Rajaram MV, Meyer DA, Schlesinger LS. Pulmonary surfactant protein A and surfactant lipids upregulate IRAK-M, a negative regulator of TLR-mediated inflammation in human macrophages. Am J Physiol Lung Cell Mol Physiol (2012) 303:L608–16. doi: 10.1152/ajplung.00067.2012
62. Liegeois M, Bai Q, Fievez L, Pirottin D, Legrand C, Guiot J, et al. Airway macrophages encompass transcriptionally and functionally distinct subsets altered by smoking. Am J Respir Cell Mol Biol (2022) 67:241–52. doi: 10.1165/rcmb.2021-0563OC
63. Nosaka T, Baba T, Tanabe Y, Sasaki S, Nishimura T, Imamura Y, et al. Alveolar macrophages drive hepatocellular carcinoma lung metastasis by generating leukotriene B(4). J Immunol (2018) 200:1839–52. doi: 10.4049/jimmunol.1700544
64. Allden SJ, Ogger PP, Ghai P, McErlean P, Hewitt R, Toshner R, et al. The transferrin receptor CD71 delineates functionally distinct airway macrophage subsets during idiopathic pulmonary fibrosis. Am J Respir Crit Care Med (2019) 200:209–19. doi: 10.1164/rccm.201809-1775OC
65. Wesselkamper SC, McDowell SA, Medvedovic M, Dalton TP, Deshmukh HS, Sartor MA, et al. The role of metallothionein in the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol (2006) 34:73–82. doi: 10.1165/rcmb.2005-0248OC
66. Almatroodi SA, McDonald CF, Pouniotis DS. Alveolar macrophage polarisation in lung cancer. Lung Cancer Int (2014) 2014:721087. doi: 10.1155/2014/721087
67. Pouniotis DS, Plebanski M, Apostolopoulos V, McDonald CF. Alveolar macrophage function is altered in patients with lung cancer. Clin Exp Immunol (2006) 143:363–72. doi: 10.1111/j.1365-2249.2006.02998.x
68. Tan Z, Xue H, Sun Y, Zhang C, Song Y, Qi Y. The role of tumor inflammatory microenvironment in lung cancer. Front Pharmacol (2021) 12:688625. doi: 10.3389/fphar.2021.688625
69. Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res (2015) 75:3479–91. doi: 10.1158/0008-5472.CAN-14-3587
70. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
71. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity (2019) 50:1317–1334 e10. doi: 10.1016/j.immuni.2019.03.009
72. Sharma SK, Chintala NK, Vadrevu SK, Patel J, Karbowniczek M, Markiewski MM. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J Immunol (2015) 194:5529–38. doi: 10.4049/jimmunol.1403215
73. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3 (2011) 8(2):181–7. doi: 10.1126/scitranslmed.3002003
74. Alikhanyan K, Chen Y, Kraut S, Sotillo R. Targeting alveolar macrophages shows better treatment response than deletion of interstitial macrophages in EGFR mutant lung adenocarcinoma. Immun Inflammation Dis (2020) 8:181–7. doi: 10.1002/iid3.293
75. Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene (2006) 25:2105–12. doi: 10.1038/sj.onc.1209237
76. Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology (2018) 7:e1412909. doi: 10.1080/2162402X.2017.1412909
77. Huggins DN, LaRue RS, Wang Y, Knutson TP, Xu Y, Williams JW, et al. Characterizing macrophage diversity in metastasis-bearing lungs reveals a lipid-associated macrophage subset. Cancer Res (2021) 81:5284–95. doi: 10.1158/0008-5472.CAN-21-0101
78. Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, et al. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res (2020) 26:970–7. doi: 10.1158/1078-0432.CCR-19-1040
79. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell (2017) 169:750–765 e17. doi: 10.1016/j.cell.2017.04.014
80. Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics (2021) 11:5365–86. doi: 10.7150/thno.58390
81. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U.S.A. (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
82. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545:495–9. doi: 10.1038/nature22396
83. Singhal S, Stadanlick J, Annunziata MJ, Rao AS, Bhojnagarwala PS, O’Brien S, et al. Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Sci Transl Med 11 (2019) 11(479):eaat1500. doi: 10.1126/scitranslmed.aat1500
84. Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer (2006) 6:924–35. doi: 10.1038/nrc2013
85. Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest (2016) 126:3672–9. doi: 10.1172/JCI84427
86. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211:781–90. doi: 10.1084/jem.20131916
87. Luo F, Lu FT, Cao JX, Ma WJ, Xia ZF, Zhan JH, et al. HIF-1alpha inhibition promotes the efficacy of immune checkpoint blockade in the treatment of non-small cell lung cancer. Cancer Lett (2022) 531:39–56. doi: 10.1016/j.canlet.2022.01.027
88. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi: 10.1038/nature13954
89. Hartley G, Regan D, Guth A, Dow S. Regulation of PD-L1 expression on murine tumor-associated monocytes and macrophages by locally produced TNF-alpha. Cancer Immunol Immunother (2017) 66:523–35. doi: 10.1007/s00262-017-1955-5
90. Zhang H, Liu L, Liu J, Dang P, Hu S, Yuan W, et al. Roles of tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer (2023) 22:58. doi: 10.1186/s12943-023-01725-x
91. Wen ZF, Liu H, Gao R, Zhou M, Ma J, Zhang Y, et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J Immunother Cancer (2018) 6:151. doi: 10.1186/s40425-018-0452-5
92. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
93. Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, et al. Induction of autonomous memory alveolar macrophages requires T cell help and is critical to trained immunity. Cell (2018) 175:1634–1650 e17. doi: 10.1016/j.cell.2018.09.042
94. Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol (2020) 21:145–57. doi: 10.1038/s41590-019-0568-x
95. Roquilly A, Jacqueline C, Davieau M, Molle A, Sadek A, Fourgeux C, et al. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol (2020) 21:636–48. doi: 10.1038/s41590-020-0673-x
96. Zahalka S, Starkl P, Watzenboeck ML, Farhat A, Radhouani M, Deckert F, et al. Trained immunity of alveolar macrophages requires metabolic rewiring and type 1 interferon signaling. Mucosal Immunol (2022) 15:896–907. doi: 10.1038/s41385-022-00528-5
97. Netea MG, Joosten LAB. Trained immunity and local innate immune memory in the lung. Cell (2018) 175:1463–5. doi: 10.1016/j.cell.2018.11.007
98. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell (2018) 172:176–190 e19. doi: 10.1016/j.cell.2017.12.031
99. Wang T, Zhang J, Wang Y, Li Y, Wang L, Yu Y, et al. Influenza-trained mucosal-resident alveolar macrophages confer long-term antitumor immunity in the lungs. Nat Immunol (2023) 24:423–38. doi: 10.1038/s41590-023-01428-x
100. Ding C, Shrestha R, Zhu X, Geller AE, Wu S, Woeste MR, et al. Inducing trained immunity in pro-metastatic macrophages to control tumor metastasis. Nat Immunol (2023) 24:239–54. doi: 10.1038/s41590-022-01388-8
101. Kumari N, Choi SH. Tumor-associated macrophages in cancer: recent advancements in cancer nanoimmunotherapies. J Exp Clin Cancer Res (2022) 41:68. doi: 10.1186/s13046-022-02272-x
102. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discovery (2022) 21:799–820. doi: 10.1038/s41573-022-00520-5
103. MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood (2010) 116:3955–63. doi: 10.1182/blood-2010-02-266296
104. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
105. Papadopoulos KP, Gluck L, Martin LP, Olszanski AJ, Tolcher AW, Ngarmchamnanrith G, et al. First-in-human study of AMG 820, a monoclonal anti-colony-stimulating factor 1 receptor antibody, in patients with advanced solid tumors. Clin Cancer Res (2017) 23:5703–10. doi: 10.1158/1078-0432.CCR-16-3261
106. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
107. Gomez-Roca C, Cassier P, Zamarin D, Machiels JP, Perez Gracia JL, Stephen Hodi F, et al. Anti-CSF-1R emactuzumab in combination with anti-PD-L1 atezolizumab in advanced solid tumor patients naive or experienced for immune checkpoint blockade. J Immunother Cancer (2022) 10(5):e004076. doi: 10.1136/jitc-2021-004076
108. Razak AR, Cleary JM, Moreno V, Boyer M, Calvo Aller E, Edenfield W, et al. Safety and efficacy of AMG 820, an anti-colony-stimulating factor 1 receptor antibody, in combination with pembrolizumab in adults with advanced solid tumors. J Immunother Cancer (2020) 8(2):e001006. doi: 10.1136/jitc-2020-001006
109. Gomez-Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D’Angelo SP, et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann Oncol (2019) 30:1381–92. doi: 10.1093/annonc/mdz163
110. Larroquette M, Guegan JP, Besse B, Cousin S, Brunet M, Le Moulec S, et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J Immunother Cancer (2022) 10(5):e003890. doi: 10.1136/jitc-2021-003890
111. O’Brien SA, Orf J, Skrzypczynska KM, Tan H, Kim J, DeVoss J, et al. Activity of tumor-associated macrophage depletion by CSF1R blockade is highly dependent on the tumor model and timing of treatment. Cancer Immunol Immunother (2021) 70:2401–10. doi: 10.1007/s00262-021-02861-3
112. Martori C, Sanchez-Moral L, Paul T, Pardo JC, Font A, Ruiz de Porras V, et al. Macrophages as a therapeutic target in metastatic prostate cancer: A way to overcome immunotherapy resistance? Cancers (Basel) (2022) 14(2):440. doi: 10.3390/cancers14020440
113. Xia L, Zhu X, Zhang L, Xu Y, Chen G, Luo J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol Appl Biochem (2020) 67:1011–9. doi: 10.1002/bab.1875
114. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer (2020) 19:41. doi: 10.1186/s12943-020-01165-x
115. Liu C, Yao Z, Wang J, Zhang W, Yang Y, Zhang Y, et al. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ (2020) 27:1765–81. doi: 10.1038/s41418-019-0460-0
116. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood (2004) 104:2224–34. doi: 10.1182/blood-2004-03-1109
117. Takimoto CH, Chao MP, Gibbs C, McCamish MA, Liu J, Chen JY, et al. The Macrophage ‘Do not eat me’ signal, CD47, is a clinically validated cancer immunotherapy target. Ann Oncol (2019) 30:486–9. doi: 10.1093/annonc/mdz006
118. Yang Z, Peng Y, Guo W, Xu J, Li L, Tian H, et al. PD-L1 and CD47 co-expression predicts survival and enlightens future dual-targeting immunotherapy in non-small cell lung cancer. Thorac Cancer (2021) 12:1743–51. doi: 10.1111/1759-7714.13989
119. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity (2017) 47:363–373 e5. doi: 10.1016/j.immuni.2017.07.016
120. Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody hu5F9-G4 in patients with advanced cancers. J Clin Oncol (2019) 37:946–53. doi: 10.1200/JCO.18.02018
121. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
122. Umiker B, Hashambhoy-Ramsay Y, Smith J, Rahman T, Mueller A, Davidson R, et al. Inhibition of LILRB2 by a novel blocking antibody designed to reprogram immunosuppressive macrophages to drive T-cell activation in tumors. Mol Cancer Ther (2023) 22:471–84. doi: 10.1158/1535-7163.MCT-22-0351
123. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
124. Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol (2013) 9:319–25. doi: 10.1038/nchembio.1223
125. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, SChad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature (2017) 543:428–32. doi: 10.1038/nature21409
126. Lu Z, Zou J, Li S, Topper MJ, Tao Y, Zhang H, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature (2020) 579:284–90. doi: 10.1038/s41586-020-2054-x
127. Hoden B, DeRubeis D, Martinez-Moczygemba M, Ramos KS, Zhang D. Understanding the role of Toll-like receptors in lung cancer immunity and immunotherapy. Front Immunol (2022) 13:1033483. doi: 10.3389/fimmu.2022.1033483
128. Koh J, Kim S, Lee SN, Kim SY, Kim JE, Lee KY, et al. Therapeutic efficacy of cancer vaccine adjuvanted with nanoemulsion loaded with TLR7/8 agonist in lung cancer model. Nanomedicine (2021) 37:102415. doi: 10.1016/j.nano.2021.102415
129. Xiong H, Mittman S, Rodriguez R, Moskalenko M, Pacheco-Sanchez P, Yang Y, et al. Anti-PD-L1 treatment results in functional remodeling of the macrophage compartment. Cancer Res (2019) 79:1493–506. doi: 10.1158/0008-5472.CAN-18-3208
130. Weiss SA, Djureinovic D, Jessel S, Krykbaeva I, Zhang L, Jilaveanu L, et al. A phase I study of APX005M and cabiralizumab with or without nivolumab in patients with melanoma, kidney cancer, or non-small cell lung cancer resistant to anti-PD-1/PD-L1. Clin Cancer Res (2021) 27:4757–67. doi: 10.1158/1078-0432.CCR-21-0903
131. Wolf EM, Fingleton B, Hasty AH. The therapeutic potential of TREM2 in cancer. Front Oncol (2022) 12:984193. doi: 10.3389/fonc.2022.984193
132. Park MD, Reyes-Torres I, LeBerichel J, Hamon P, LaMarche NM, Hegde S, et al. TREM2 macrophages drive NK cell paucity and dysfunction in lung cancer. Nat Immunol (2023) 24:792–801. doi: 10.1038/s41590-023-01475-4
133. Zhang H, Liu Z, Wen H, Guo Y, Xu F, Zhu Q, et al. Immunosuppressive TREM2(+) macrophages are associated with undesirable prognosis and responses to anti-PD-1 immunotherapy in non-small cell lung cancer. Cancer Immunol Immunother (2022) 71:2511–22. doi: 10.1007/s00262-022-03173-w
134. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell (2020) 182:886–900 e17. doi: 10.1016/j.cell.2020.07.013
135. Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Nunez NG, et al. Tissue-resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell (2022) 185:1189–1207 e25. doi: 10.1016/j.cell.2022.02.021
136. Skytthe MK, Graversen JH, Moestrup SK. Targeting of CD163(+) macrophages in inflammatory and Malignant diseases. Int J Mol Sci (2020) 21(15):5497. doi: 10.3390/ijms21155497
137. Chen L, Cao MF, Xiao JF, Ma QH, Zhang H, Cai RL, et al. Stromal PD-1(+) tumor-associated macrophages predict poor prognosis in lung adenocarcinoma. Hum Pathol (2020) 97:68–79. doi: 10.1016/j.humpath.2019.12.007
138. Lo Russo G, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, et al. Antibody-fc/fcR interaction on macrophages as a mechanism for hyperprogressive disease in non-small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin Cancer Res (2019) 25:989–99. doi: 10.1158/1078-0432.CCR-18-1390
139. La Fleur L, Botling J, He F, Pelicano C, Zhou C, He C, et al. and IL37R on immunosuppressive macrophages in lung cancer blocks regulatory T cells and supports cytotoxic lymphocyte function. Cancer Res (2021) 81:956–67. doi: 10.1158/0008-5472.CAN-20-1885
140. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
141. Chen C, Jing W, Chen Y, Wang G, Abdalla M, Gao L, et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci Transl Med (2022) 14:eabn1128. doi: 10.1126/scitranslmed.abn1128
142. Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol (2020) 13:153. doi: 10.1186/s13045-020-00983-2
Keywords: tissue-resident alveolar macrophages, smoking, lung cancer, immune checkpoint inhibitors, tumor-associated macrophages, tumor microenvironment
Citation: Chang C-Y, Armstrong D, Corry DB and Kheradmand F (2023) Alveolar macrophages in lung cancer: opportunities and challenges. Front. Immunol. 14:1268939. doi: 10.3389/fimmu.2023.1268939
Received: 28 July 2023; Accepted: 12 September 2023;
Published: 26 September 2023.
Edited by:
Guochang Hu, University of Illinois Chicago, United StatesReviewed by:
Jian Liu, Zhejiang University-University of Edinburgh Institute, ChinaPeter Chen, Department of Medicine, Cedars Sinai Medical Center, United States
Copyright © 2023 Chang, Armstrong, Corry and Kheradmand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farrah Kheradmand, ZmFycmFoa0BiY20uZWR1