 Zhengmin Wang
Zhengmin Wang Nan Liu2†
Nan Liu2† Zhengkun Tu
Zhengkun Tu- 1Department of Hepatology, The First Hospital of Jilin University, Changchun, Jilin, China
- 2Institute of Epigenetic Medicine, First Hospital of Jilin University, Changchun, China
- 3Institute of Liver Diseases, The First Hospital of Jilin University, Changchun, Jilin, China
Hepatitis B Virus (HBV) infections pose a global public health challenge. Despite extensive research on this disease, the intricate mechanisms underlying persistent HBV infection require further in-depth elucidation. Recent studies have revealed the pivotal roles of immunometabolism and epigenetic reprogramming in chronic HBV infection. Immunometabolism have identified as the process, which link cell metabolic status with innate immunity functions in response to HBV infection, ultimately contributing to the immune system’s inability to resolve Chronic Hepatitis B (CHB). Within hepatocytes, HBV replication leads to a stable viral covalently closed circular DNA (cccDNA) minichromosome located in the nucleus, and epigenetic modifications in cccDNA enable persistence of infection. Additionally, the accumulation or depletion of metabolites not only directly affects the function and homeostasis of immune cells but also serves as a substrate for regulating epigenetic modifications, subsequently influencing the expression of antiviral immune genes and facilitating the occurrence of sustained HBV infection. The interaction between immunometabolism and epigenetic modifications has led to a new research field, known as metabolic epigenomics, which may form a mutually reinforcing relationship with CHB. Herein, we review the recent studies on immunometabolism and epigenetic reprogramming in CHB infection and discuss the potential mechanisms of persistent HBV infection. A deeper understanding of these mechanisms will offer novel insights and targets for intervention strategies against chronic HBV infection, thereby providing new hope for the treatment of related diseases.
1 Introduction
1.1 Hepatitis B virus and CHB infection
Chronic hepatitis B (CHB) infection poses a significant global health challenge affecting the health and well-being of millions of people (1). Hepatitis B Virus (HBV) is a partially double-stranded DNA virus with a sophisticated life cycle that can persist by forming covalently closed circular DNA (cccDNA) within the host cell nucleus. The structure of HBV comprises a lipid envelope, with its surface characterized by three types of surface proteins, namely small (S), medium (M), and large (L), collectively referred to as HBsAg in serology. Additionally, there is an internal core-shell enveloping the core particles, known as the hepatitis B core antigen (HBcAg) in serology. The outer membrane forms an icosahedral shell composed of 120 core protein dimers (2–5). cccDNA plays a crucial role in the subgenomic RNA mediated by host RNA polymerase II, encompassing the surface protein HBx and two RNA templates exceeding the genomic length, namely pregenomic (pg) RNA and precore RNA. These two types of RNA exert significant regulatory functions, leading to the generation of the precore protein precursor, ultimately resulting in the secretion of hepatitis B e antigen (HBeAg) (6). Sustained HBV infection has the potential to induce diverse levels of liver impairment, culminating in progression to chronic hepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (7, 8). According to the World Health Organization (WHO), approximately 300 million individuals worldwide are afflicted with chronic HBV infection, and nearly one million succumb annually to complications directly linked to HBV infection (9).
The risk of developing CHB infection depends on the age at exposure, with infants and children being mostly symptom-free. Approximately 95% of newborns, 20–30% of children (1-5 years old), and < 5% of adults fail to clear the virus after six months (10). During CHB infection, the virus remains in the body for a long period. This can lead to severe liver damage, such as cirrhosis and liver cancer, with significant repercussions for affected individuals, their families, and society at large (11–13). Although antiviral drugs can reduce the viral load in patients with hepatitis B, a complete cure remains difficult to achieve (6). Therefore, scientists are attempting to understand the mechanisms underlying this disease more thoroughly and promptly.
HBV infection can cause liver damage and chronic diseases; however, the virus is not cytotoxic. The immune system plays a key role in controlling viral replication and liver injury (14–16). Patients who achieve HBeAg seroconversion spontaneously or after antiviral therapy show improved immune function (17, 18). This implies that combining immunotherapy with antiviral treatment may increase the likelihood of viral elimination (19–22). The immune response to HBV infection is complex and variable. Some studies have reported that immune activation leads to reduced HBV DNA levels and HBeAg seroconversion, indicating effective clearance of infected hepatocytes (23). Other studies have reported that the reduction in HBV DNA levels is only temporary, suggesting ineffective clearance (24). Therefore, the host immune response is a critical factor in determining the outcome of HBV infection (25). The mechanisms by which HBV modulates the hepatic immune microenvironment to promote chronic infection are not fully understood.
1.2 Hepatic immune microenvironment
Within the liver, there is a complex interplay among parenchymal (primarily hepatocytes), non-parenchymal, and recruited immune cells, collectively forming the hepatic immune microenvironment, which includes cellular components, secreted cytokines, and extracellular matrix components (26). One of the most intriguing features of the liver as a remarkable immune organ is its propensity to induce tolerance rather than immunity upon encountering foreign antigens (27). Specialized antigen-presenting cells in the liver, such as Kupffer cells and liver sinusoidal endothelial cells, are characterized by well-induced tolerance owing to their insufficient co-stimulatory signal transmission and tendency to produce immunosuppressive molecules, resulting in the establishment of an inherent hepatic tolerogenic microenvironment in a steady state (28). The nature of immune responses within the liver is intricately shaped by the level of inflammation. In cases of chronic or low-grade inflammation, where an immunosuppressive microenvironment prevails, the liver can serve as a “graveyard” for effector cells (29) or as a “school” for educating regulatory cells (30). These processes can lead to clonal deletion (31, 32) or suppression of antigen-specific T cells (33–35), which constitute the primary mechanisms underlying liver-induced antigen-specific tolerance. During CHB infection, HBV replication within hepatocytes goes unnoticed by the intracellular innate immune system, resulting in a tolerogenic hepatic microenvironment. This gradually leads to the alteration and deletion of the functional capacities of HBV-specific B and T cells, enabling the establishment of intractable persistent (chronic) HBV infection (36). However, how these mechanisms coordinate to sustain the tolerance of the hepatic microenvironment while the virus persists in the liver remains largely unknown (37). According to recent research in the relevant field, immunometabolism and epigenetic reprogramming have emerged as pivotal factors linking hepatocytes and immune cells within the hepatic immune microenvironment, as elaborated in the subsequent sections.
1.3 Immunometabolism and epigenetic modification
The immune system is intricately linked to metabolic functions in a manner that has not been previously recognized. This unconventional mode of thinking has been described in a novel field known as immunometabolism (38, 39). Immunometabolism encompasses a series of processes involving the energy metabolism of immune cells in response to pathogens, accumulation of metabolic products, and regulatory activity of metabolic enzymes (40). Reports of cellular metabolism being hijacked by viruses have a history spanning over half a century; however, the mechanisms and consequences of virus-induced metabolic reprogramming have only begun to be extensively studied over the past decade (41). Following internalization by glucose transporters, glucose molecules enter distinct metabolic pathways, including glycolysis and the pentose phosphate pathway (42, 43), each of which serves specific functions and purposes. To some extent, these changes influence the functionality of immune cells, thereby affecting the effectiveness of antiviral immune responses.
In addition to immunometabolism, epigenetic modifications play crucial roles in regulating gene expression (44, 45). Simultaneously, there is a vital interplay between metabolism and the epigenetic modification in immune regulation that disrupts the functionality and response patterns of immune cells (46, 47). The interaction between metabolism and epigenetics led to a new research field known as metabolic epigenomics (48). Recently emphasized metabolites such as lactate (49, 50), and a newly discovered epigenetic modification, lactylation (51), serve as noteworthy representatives. This combination also includes acetyl-CoA and histone acetylation (52, 53), S-adenosylmethionine and histone methylation (54, 55), as well as NAD+ and the Sirtuin family (56). These intricate interactive relationships play a pivotal role in the dynamic balance between immune responses and sustained viral infections.
During CHB infection, significant alterations occur in host immune metabolism, including glucose and lipid metabolism (57, 58). For instance, during HBV infection, cells involved in pathological responses, such as immune cells and hepatocytes, enhance utilization and glucose uptake to meet their energy and biosynthetic demands (50, 59). Concurrently, during CHB infection, epigenetic regulatory mechanisms such as DNA methylation and histone modification also undergo changes, subsequently influencing the expression patterns of specific genes (60, 61). Here, our aim is to systematically review the current state of research on immunometabolism and epigenetic modifications in CHB infection, as well as to anticipate their potential applications in future clinical treatments. By delving into these aspects of research, we hope to offer new perspectives and strategies for the treatment and prevention of HBV infection, making a positive contribution to the well-being of individuals affected by HBV infection worldwide.
2 Immunometabolism and epigenetic modification of HBV-infected hepatocytes
HBV affects various metabolic pathways within the liver to the extent that it is referred to as a metabolic virus (62). The liver assumes a pivotal function in the daily immune surveillance of the body (63). Its unique blood supply mechanism leads to continuous exposure to a variety of pathogens originating from both the bloodstream and gastrointestinal tract (64). Hepatocytes constitute the vast majority of liver masses (approximately 4/5) and play a role in various biochemical and metabolic functions (65). HBV infection leads to significant alterations in the host hepatocyte metabolic pathways and epigenetic modifications. These changes not only affect the bioenergetic supply of hepatocytes but also impact the immune response and viral replication in liver cells(as discussed subsequently).
2.1 Immunometabolism and antiviral immunity
The regulation of hepatocyte metabolism in CHB is an emerging and significant research area in the development of this disease. Hepatocytes not only act as active drivers of liver inflammation and fibrosis through intercellular communication but also engage in antiviral immunity by secreting type I interferons (IFNs) (66).
IFN serves as a critical initial defense mechanism that exerts control over viral gene replication and expression (67), with its expression directly influencing viral replication. Hepatocytes can trigger innate immune responses through pattern recognition receptors such as Retinoic Acid-Inducible Gene I (RIG-I)-like receptors (RLRs) (68, 69), which recruit the mitochondrial antiviral signaling (MAVS) adaptor (also known as VISA, Cardif, and IPS-1) (70, 71). Upon activation, MAVS molecules aggregate into virus-like supramolecular structures in the mitochondria (72, 73), initiating the activation of transcription factors including IRF3. This activation results in the production of IFN to execute antiviral activities (74, 75).
Recent pivotal studies have elucidated a novel mechanism by which HBV influences immune escape by regulating the immune metabolism of liver cells, thereby affecting their antiviral immune functions (57, 58, 76).
2.1.1 Hepatocellular glycolysis
HBV can enhance the activity of the crucial glycolytic enzyme hexokinase 2 (HK2) in liver cells, promoting glycolysis. Furthermore, it stimulates lactate dehydrogenase A (LDHA), leading to increased lactate production; thus, facilitating the transition toward aerobic glycolysis (58), also known as the Warburg effect (77).
During this metabolic transition, HBV modulates the interplay between RIG-I and MAVS by orchestrating the activities of HK2 and lactate in a concerted manner. On one hand, HBV facilitates the assembly of a trimeric complex comprising HK2, MAVS, and voltage-dependent anion channel protein 1 (VDAC1) within the mitochondria, isolating MAVS from RIG-I and impeding their functional interaction. On the other hand, HBV elevates lactate expression, reinforcing its physical association with MAVS. This, in turn, impedes the interaction between RIG-I and MAVS, MAVS aggregation, and the subsequent signal transduction cascade, ultimately suppressing the production of INFs (58). Similarly, another recent study on metabolic immunity discovered that HBV can interact with the metabolic switch pyruvate kinase M2 isoform (PKM2) through the utilization of HBsAg, especially the large viral surface antigen (LHBS). This interaction inhibits the oligomerization of PKM2 within liver cells, leading to increased glucose consumption and lactate production, thereby promoting viral replication (57). The direct relationship between metabolic shifts and immune function can be explained by the lactate-MAVS-RIGI-IFN theory.
In summary, HBV regulates lactate production within liver cells through two mechanisms, thereby influencing antiviral immunity and promoting CHB infection. On one hand, HBV induces the formation of the HK2-MAVS-VDAC1 trimeric complex and promotes the interaction between lactate, a product of glycolysis, and MAVS. This impedes the interaction between MAVS and RIG-I, suppressing downstream signal transduction and ultimately inhibiting the production of INFs (Figure 1.1). On the other hand, HBV employs HBsAg to interact with PKM2, thereby promoting the expression of the downstream metabolic product lactate and subsequently facilitating HBV virus replication (Figure 1.2).
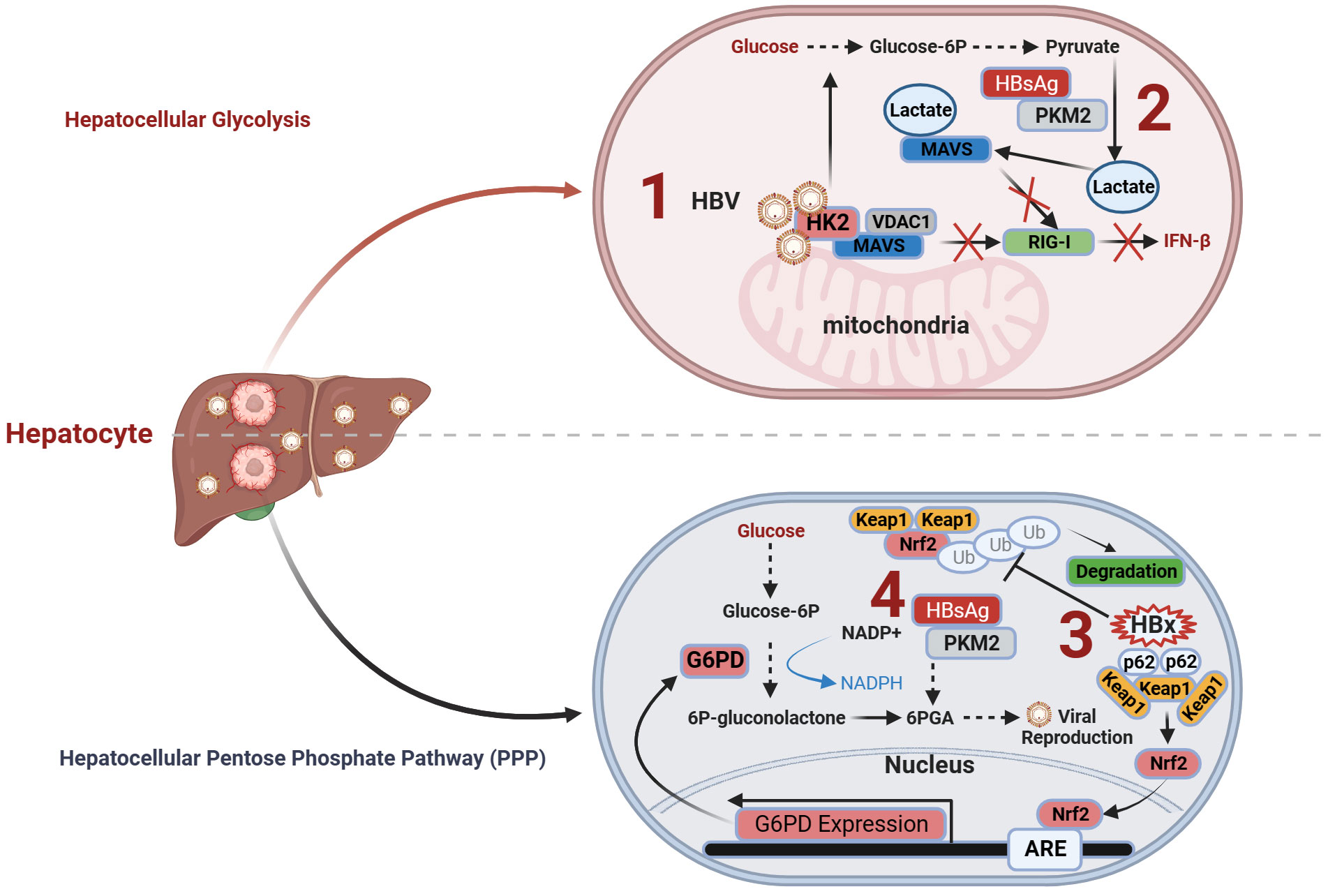
Figure 1 Immunometabolism and antiviral immunity of hepatocytes in CHB. (1) HBV induces the formation of the HK2-MAVS-VDAC1 trimeric complex and promotes the interaction between lactate and MAVS. This impedes the interaction between MAVS and RIG-I, suppressing downstream signal transduction and ultimately inhibiting the production of interferons; (2) HBV employs HBsAg to interact with PKM2, thereby promoting the expression of downstream metabolic product lactate and subsequently facilitating HBV virus replication; (3) The formation of HBx-p62-Keap1 aggregates sequesters Keap1 from Nrf2 and facilitates Nrf2 activation, forming the basis of HBV-induced Nrf2-mediated G6PD expression, promoting PPP and HBV viral replication; (4) HBV employs HBsAg to promote the PPP through the interaction of HBsAg with PKM2, thereby providing an ample supply of precursors for nucleotide and amino acid synthesis required for viral replication, facilitating sustained HBV replication. G6PD, glucose-6-phosphate dehydrogenase; HK2, hexokinase 2; Keap1, Kelch-like ECH-associated protein 1; MACS, mitochondrial antiviral signaling; Nrf2, NF-E2-related factor 2; PKM2, pyruvate kinase M2; RIG-I, Retinoic Acid-Inducible Gene I; VDAC1, voltage-dependent anion channel protein 1.
2.1.2 Hepatocellular Pentose Phosphate Pathway (PPP)
The PPP is the primary route of glucose catabolic metabolism, encompassing the initial phases of glucose metabolism, along with two distinct metabolic stages. PPP directs glucose flux to its oxidative branch through the catalytic action of glucose-6-phosphate dehydrogenase (G6PD), which serves as the rate-limiting enzyme in this pathway. This process generates the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) (78).
HBV infection exerts multilevel effects on the PPP in liver cells, including alterations in glycolytic pathway activity (57) and the regulation of key enzyme activities (76). Seizing control of the host macromolecular metabolic supply is a common strategy employed by viruses for replication (41, 79). Different proteins encoded during HBV infection can affect the PPP within liver cells through distinct mechanisms. HBV enhances the expression of G6PD by activating Nrf2 (76), thereby promoting PPP. Mechanistically, the formation of HBx-p62-Keap1 aggregates sequesters Kelch-like ECH-associated protein 1 (Keap1) from NF-E2-related factor 2 (Nrf2) and facilitates Nrf2 activation, forming the basis for HBV-induced Nrf2-mediated G6PD expression. Nrf2 is a master transcription factor that regulates the expression of numerous antioxidant and cell-protective genes.
Under normal circumstances, Nrf2 associates with its inhibitor Keap1 and undergoes proteasomal degradation (80). Elevated Nrf2 activity amplifies the expression of enzymes within the PPP, including G6PD, thereby accelerating cancer cell proliferation (81). Intriguingly, investigations propose that the accumulation of p62, an autophagic adaptor protein, fosters sustained activation of Nrf2, thereby facilitating the progression of HCC (82, 83). In HBV infection, the HBV X protein (HBx) plays a pivotal role in forming a complex with p62 and Keap1, sequestering Keap1 within the aggregate, thereby preventing Keap1-mediated inhibition of Nrf2, promoting Nrf2 nuclear translocation, and enhancing the expression of the target gene G6PD, the rate-limiting enzyme of PPP (76). Additionally, the interaction between HBV and LHBS with PKM2 promotes aerobic glycolysis and activates PPP. A study found that inhibiting the formation of PKM2 dimers (increasing PKM2 activity) led to the reduced expression of 6-PGA, a component of PPP (57) (Figure 1.4). PPP upregulation provides precursors for nucleotide and amino acid biosynthesis, making it an ideal model to promote viral replication (57).
Altogether, HBV employs HBx to activate G6PD and LHBS-PKM2 to promote the PPP, thereby providing an ample supply of precursors for amino acid and nucleotide synthesis required for viral replication, facilitating sustained HBV replication (Figure 1.3). Additionally, HBV-induced intermediate metabolites of PPP may potentially alter the accessibility of the antiviral immune gene chromatin through epigenetic modification pathways. For further details, please refer to section 4.1.
2.2 Epigenetic modifications and antiviral immunity
Research on epigenetic modifications in CHB has focused primarily on cccDNA domains. Within the nucleus, the HBV DNA template adopts a minichromosomal structure that interacts with both histone and nonhistone proteins (84). Consequently, epigenetic regulatory mechanisms, including DNA methylation and histone modifications, can influence cccDNA activity, potentially offering new therapeutic concepts for treating chronic hepatitis B (CHB) (85, 86) (Figure 2.1). Conversely, HBV may induce alterations in the host genome at both the genetic and epigenetic levels (86). This dysregulation in gene expression may contribute to the development of CHB infection.
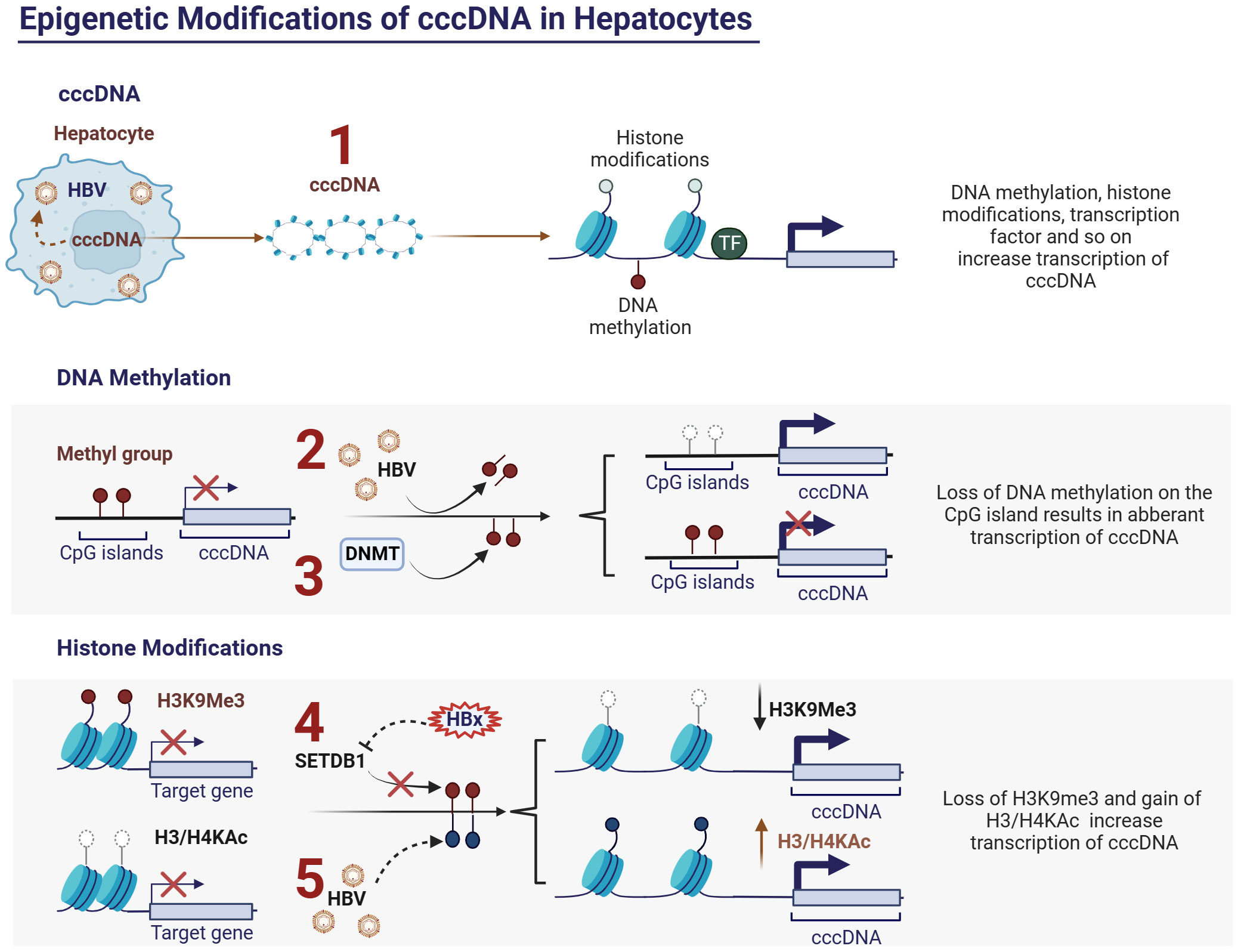
Figure 2 Epigenetic modifications of cccDNA in hepatocytes and HBV. (1) Activity of cccDNA regulated by epigenetic modifications, such as DNA methylation, histone modifications and so on; (2) HBV inhibits DNA methylation at CpG islands of cccDNA and thus promotes cccDNA transcription; (3) Upregulating the expression of DNA methyltransferases (DNMTs) can induce methylation of the viral genome, thereby inhibiting HBV replication; (4) HBx protein can relieve transcriptional silencing of cccDNA by slowing down the histone methylation (H3K9me3) upregulated by histone methyltransferase SETDB1, thereby promoting viral replication. (5) HBV can promote viral replication by modulating the acetylation levels of histones H3 and H4, which are associated with cccDNA. Ac, acetylation; cccDNA, covalently closed circular DNA; DNMTs, DNA methyltransferases; H3, histone 3; H4, histone 4.
2.2.1 DNA methylation modification
DNA methylation occurs within specific CpG islands and is typically mediated by DNA methyltransferases (DNMTs), which is closely associated with transcriptional silencing (87). The life cycle of HBV has been extensively described in numerous articles (85). Briefly, HBV cccDNA is a single minichromosome with a chromatin-like structure containing four open reading frames (ORFs) encoding HBsAg, HBeAg, HBx protein, and viral polymerase (pol) (88–90). Depending on the genotype, cccDNA contains 2-3 CpG islands (91, 92) located at 1) the initiation site of the S gene; 2) the X gene and genome pre-RNA promoter; and 3) the pS1 and polymerase gene promoters (84). Among these, methylation at position 1 occurs infrequently and varies with genotype, whereas positions 2 and 3 are relatively conserved and more prone to methylation. Studies have demonstrated a clear negative correlation between the methylation level at position 2 and transcription and replication levels of cccDNA (93). Additionally, evidence suggests a significant elevation in the methylation levels at CpG island 2 in HBeAg-negative patients relative to infected and cirrhotic tissues. Moreover, the methylation levels of HBV DNA at CpG islands 2 and 3 in HCC tissues are higher than that in infected and cirrhotic tissues (91, 94, 95).
DNA methylation of the viral genome has been identified as a host defense mechanism (96). HBV infection can induce changes in host genome DNA methylation, leading to fluctuations in the methylation levels of specific genes, subsequently affecting their expression (97, 98) (Figure 2.2). This opens up the possibility of suppressing HBV replication by modulating the methylation levels of the host DNA. Upregulation of DNA methyltransferases (DNMTs) can induce methylation of the viral genome in vitro, thereby inhibiting HBV replication (99) (Figure 2.3). However, the host DNA methylation is also a major mechanism leading to the inactivation of tumor suppressor genes in HCC (100). Therefore, the precise regulation of the methylation levels of specific genes is a crucial topic for future research in the field of viral epigenetics.
2.2.2 Histone modifications
Histone modifications, including methylation, acetylation, phosphorylation, and ubiquitination, regulate the accessibility of chromatin and consequently affect the expression levels of associated genes (101). Histone modifications play crucial regulatory roles in the course of CHB infection. HBV infection induces alterations in the histone modification patterns of specific genes, thus influencing the intensity and direction of the host immune response. Experimental evidence has demonstrated that HBV can promote viral replication by modulating the acetylation levels of histones H3 and H4, which are associated with cccDNA (102) (Figure 2.5). Additionally, there is evidence suggesting that the HBx protein can relieve transcriptional silencing of cccDNA by slowing down histone methylation (H3K9me3) upregulated by the histone methyltransferase SETDB1, thereby promoting viral replication (103) (Figure 2.4). Furthermore, specific histone modifications, such as acetylation and methylation, affect the transcriptional activity of immune cells, thereby accelerating immune escape (45). While epigenetic therapies show promise for the treatment of CHB, achieving precise control of epigenetic modifications of specific genes remains a crucial issue that urgently needs to be addressed.
3 Immunometabolism and epigenetic modification of immune cells during CHB
Viral infection disrupts the balance between sustained activation and inhibition of the host immune system, typically through the modulation of the metabolic state of immune cells (104). Similar adaptive changes in immune cell metabolism are also evident during CHB infection, subsequently influencing the effectiveness of host immune responses, involving monocytes/macrophages, kupffer cells, dendritic cells, natural killer cells (NK cells), T cells, and B cells (105–108).
3.1 Monocytes/macrophages
Monocytes/Macrophages, as the primary line of defense against infections, are almost ubiquitously present in various tissues under steady-state physiological conditions. Monocytes/Macrophages exhibit high plasticity and heterogeneity (109). For instance, hepatic macrophages are highly sensitive to the hepatic microenvironment and can rapidly adapt their phenotype (110). HBV infection can polarize macrophages toward an immunosuppressive M2 phenotype, inhibiting the secretion of inflammatory cytokines and stimulating the secretion of the anti-inflammatory cytokine interleukin-10 (IL-10) (111), thereby limiting innate immunity and promoting HBV immune evasion. Consistent with our previous studies, we observed that HBV affects the cytokine secretion functionality of monocytes/macrophages and attenuates their antiviral innate immune responses (112–114).
Metabolism governs the differentiation and function of macrophages (115), whereas epigenetic modifications can directly impact immune function by regulating the activity of metabolic enzymes, thereby influencing energy metabolism (116, 117). Recent studies have demonstrated that HBV-induced hyperacetylation of citrate synthase (CS) and the pyruvate dehydrogenase complex (PDHC) negatively regulates innate immune responses by impairing the TCA cycle in macrophages (105). Mechanistically, HBV activates the TLR2-NF-κB-PGC-1α axis in macrophages, leading to downregulation of the deacetylase SIRT3, thereby increasing acetylation of CS/PDHC and inhibiting their activity, ultimately disrupting the TCA cycle and promoting M2-like polarization of macrophages (105). Additionally, activation of signaling pathways is closely associated with macrophage polarization. AKT signaling activation promotes polarization toward the M2 phenotype, while NF-κB signaling activation suppresses M2 phenotype (118, 119). A recent study on chronic HBV infection suggested that HBV facilitates M2 macrophage polarization through SIRT1-mediated NICD deacetylation, resulting in reduced pro-inflammatory tumor necrosis factor α (TNF-α) and enhanced anti-inflammatory IL-10 expression (120). Mechanistically, HBsAg and HBeAg upregulate the expression of SIRT1 deacetylase, promoting NICD deacetylation, thereby enhancing Akt phosphorylation and inhibiting NF-kB nuclear translocation, ultimately driving M2 macrophage polarization (120).
In summary, HBsAg and HBeAg can, on one hand, upregulate the deacetylase SIRT1, facilitating NICD deacetylation, enhancing Akt phosphorylation, and inhibiting NF-kB nuclear translocation, thereby inducing the formation of immunosuppressive macrophages. On the other hand, HBV can also downregulate the expression of the deacetylase SIRT3 through the TLR2-NF-κB-PGC-1α axis, thereby increasing the acetylation of metabolic enzymes CS/PDHC and inhibiting their activity. This leads to impairment of the TCA cycle and promotion of the M2 phenotype in macrophages (Figure 3.1).
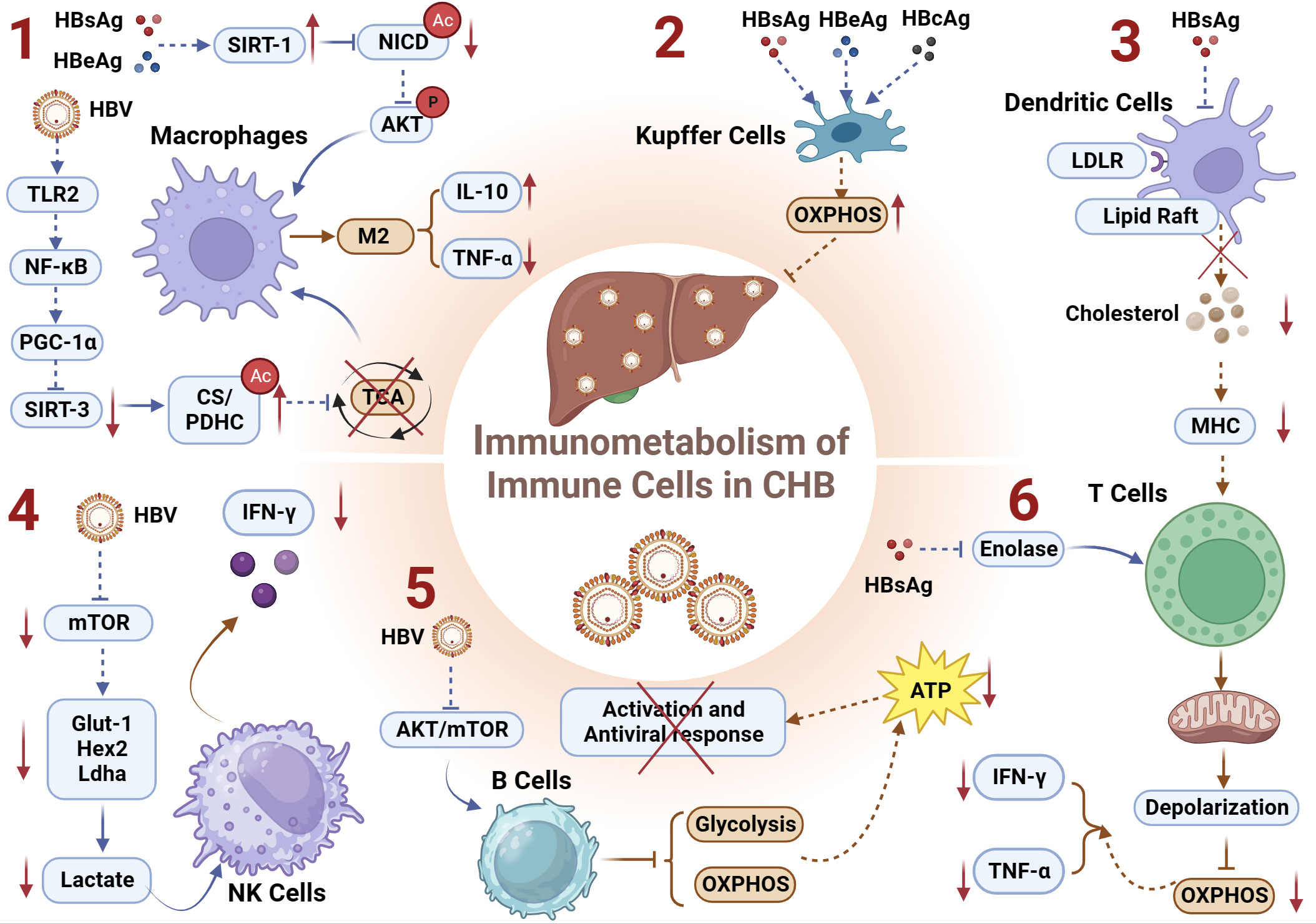
Figure 3 Immunometabolism of Immune Cells and Chronic HBV infection. (1) HBsAg and HBeAg upregulates the deacetylase SIRT1, facilitating NICD deacetylation, enhancing Akt phosphorylation, and inhibiting NF-kB nuclear translocation, thereby inducing the formation of immunosuppressive macrophages; HBV downregulates deacetylase SIRT3 through the TLR2-NF-κB-PGC-1α axis, thereby increasing the acetylation of metabolic enzymes CS/PDHC and inhibiting their activity. This leads to impairment of the TCA cycle and promotion of the M2 macrophages manifested by increased IL-10 and decreased TNF-α; (2) HBsAg, HBcAg, and HBeAg can induce immunometabolic reprogramming in KCs, upregulating OXPHOS and thereby inhibiting the host’s antiviral response; (3) HBsAg inhibits lipid raft formation and/or LDLR-mediated endocytosis on DCs, resulting in reduced levels of free cholesterol during CHB. This inhibition of antigen presentation capacity in DCs induces exhausted HBV-specific CD8+ T cells; (4) HBV regulates the Glut1-Hex2-Ldha-lactate axis-mediated metabolic reprogramming by inhibiting the metabolic regulatory factor mTOR pathway, thereby suppressing the expression of IFN-γ in NK cells; (5) HBV virus impairs the glycolytic and OXPHOS metabolic functions of B cells by inhibiting the AKT/mTOR pathway, and leads to a reduction in ATP production attenuating the activation and the antiviral immune response of B cells; (6) HBsAg can inhibit the key metabolic enzyme enolase, leading to depolarization and impaired OXPHOS function of exhausted T cell mitochondria, manifested as decreased secretion of IFN-γ and TNF-α. CS, citrate synthase; DCs, Dendritic Cells; GLUT-1, glucose transporter-1; IL-10, interleukin-10; KCs, Kupffer Cells; LDLR, low-density lipoprotein receptor; OXPHOS, oxidative phosphorylation; PDHC, pyruvate dehydrogenase complex; TNF-α, tumor necrosis factor α.
3.2 Kupffer cells
The liver is host to the largest population of tissue-resident macrophages, recognized as Kupffer cells (KCs). During CHB, the HBV employs diverse strategies to manipulate KCs, facilitating infection establishment. This includes inducing KCs to attenuate B cell antibody production (121), inhibiting CD8+ T cell cytotoxicity (122), and preferentially producing profibrogenic/anti-inflammatory cytokine TGF-β1 (123) to foster hepatic immune tolerance. A murine study investigating maternally transmitted HBV and its role in vertical virus transmission revealed that HBV can induce an M2-like anti-inflammatory polarization in KCs (124). Subsequent research within this study demonstrated that both M1- and M2-like KCs stimulated by HBV have the capacity to directly inhibit HBV replication in hepatic cells. Mechanistically, the study identified HBV-associated antigens, including HBsAg, HBcAg, and HBeAg, as capable of reprogramming KCs’ metabolism to enhance their OXPHOS activity (Figure 3.2). This reprogramming minimizes host antiviral responses (125). However, the precise mechanisms by which HBV orchestrates metabolic reprogramming in KCs and the impact of immunometabolic alterations on their epigenetic modifications remain elusive. Further investigations in this field hold the potential to unveil novel insights and contribute to a deeper understanding of the intricate interplay between HBV and KCs (125).
3.3 Dendritic cells
As proficient antigen-presenting cells (APCs) integral to the immune system, dendritic cells (DCs) play a pivotal role in bridging the innate and adaptive immune responses (126). The intricate regulation of immunometabolism is recognized as a key determinant influencing the functionality and viability of immune cells, with DCs relying on finely tuned immunometabolic processes to either initiate immune responses or foster immunological tolerance (127). Contrasting the characteristics observed in healthy donors (HD), DCs derived from individuals with CHB manifest impaired antigen presentation capabilities and diminished migratory proficiency (128). A recent investigation within the realm of DCs’ immunometabolism has revealed that HBsAg hampers lipid raft formation and/or low-density lipoprotein receptor (LDLR)-mediated endocytosis on DCs, culminating in diminished levels of free cholesterol during the course of CHB. This impediment in the antigen presentation capacity of DCs precipitates the exhaustion of HBV-specific CD8+ T cells, thereby fostering the protracted presence of HBV (129) (Figure 3.3). The findings underscore the intricate interplay between immunometabolism and viral persistence, shedding light on a mechanistic link between HBsAg-mediated alterations in DC function and the sustained evasion of host immune surveillance by HBV.
Cholesterol stands as an essential lipid molecule intricately involved in pivotal biological processes, encompassing lipid rafts and major histocompatibility complex (MHC) molecules (130). Strategies aimed at increasing cholesterol accumulation on DCs can enhance antigen presentation, thus improving the efficacy of therapeutic vaccines and rescuing the exhausted HBV-specific CD8+ T cells (129).
3.4 NK cells
Natural killer (NK) cells, as another integral component of the innate immune system (131), play a crucial role in defending against pathogenic infections (132). Our previous research has revealed that HBV utilizes monocytes to modulate NK cells, thereby affecting their regulatory functions and antiviral immunity (112, 113). NK cells secrete TNF-α and IFN-γ, which may contribute to the cytotoxicity involved in HBV clearance (133). In patients with acute HBV infection, NK cells are activated during the acute phase before the adaptive immune response kicks in (134–138). However, during the chronic phase of the disease, NK cells exhibit impaired functionality, which may facilitate viral persistence. Indeed, NK cells from CHB patients show reduced capacity to produce cytokines such as TNF-α and IFN-γ, despite maintaining or even augmenting their cytotoxic capabilities (139–142), a phenomenon referred to as functional dichotomy (139). Additionally, NK cells play crucial roles in shaping and influencing adaptive immune responses by exerting immunomodulatory effects (143).
However, the immune capacity of NK cells at different stages of infection is closely linked to their metabolic phenotypes (144–146). Functional deficiency of peripheral NK cells in patients with CHB is associated with defects in a key metabolic regulatory factor, the mTOR pathway (106). This is primarily manifested by a significant inhibition of cytokine secretion, particularly IFN-γ, while the cytotoxic function remains unaffected in the same environment. The mTOR-mediated metabolic reprogramming has been reported to be closely associated with the function of NK cells (147, 148). Inhibiting mTOR signaling can reduce the rate of glucose uptake and glycolysis in NK cells by inhibiting the key rate-limiting enzymes involved in glycolysis, such as Glut1, Hex2, and Ldha, thereby suppressing lactate production. Furthermore, this pathway is closely related to the expression of IFN-γ in NK cells (149).
Recent studies indicate that epigenetic reprogramming may play a central role in the persistent phenotypic and functional changes in the effector and memory NK cell populations (150). During human cytomegalovirus (HCMV) infection or reactivation, the expanded and host-protective regulated by memory-like NK cell subset demonstrates features of epigenetic reprogramming at genes involved in effector functions and intracellular signaling (151–153). For instance, the enhanced IFN-γ response in these memory-like cells is associated with decreased DNA methylation at the IFNG locus, while the reduced expression of signaling molecules FcϵRIγ and EAT-2 correlates with increased DNA methylation at gene loci (152, 153). Recent ATAC-seq and ChIP-seq studies reveal extensive and highly dynamic epigenetic reprogramming in Ly49H NK cells during the progression of effector, contraction, and memory phases in response to murine cytomegalovirus (MCMV) infection (154). Changes in NK cell chromatin accessibility, particularly at promoter regions, have been shown to directly correlate with transcript abundance changes at early time points after MCMV attack (154), notably for genes related to type I and II interferon signaling, such as Ifng and various ISGs (154). Although there is currently no specific literature on the epigenetic modifications of NK cells under CHB infection, this presents a new avenue for investigating the specific mechanisms underlying NK cell functional defects during CHB infection.
Taken together, HBV regulates the Glut1-Hex2-Ldha-lactate axis-mediated metabolic reprogramming by inhibiting the metabolic regulatory factor mTOR pathway, thereby suppressing the expression of IFN-γ in NK cells (Figure 3.4). Exploring whether HBV regulates epigenetic reprogramming of interferon genes (e.g., methylation, lactylation modifications) by inhibiting the metabolic regulator mTOR pathway and thereby influencing the antiviral function of NK cells would be an intriguing research direction.
3.5 T cells
T cell exhaustion is a prominent feature of CHB infection (155–158). Energy metabolism in T-cells is closely associated with the function of effector T cells. When T cells experience nutrient deficiency or the inhibition of key metabolic enzymes, their activation and proliferation are suppressed (159–161). Extensive research has highlighted the significant role of glycolytic enzymes in T cell function and activation, such as the crucial glycolytic metabolite phosphoenolpyruvate (PEP) (162) and others (163–165). During HBV infection, despite significantly elevated glucose uptake in severely exhausted virus-specific CD8+ T cells, there is impaired oxidative phosphorylation, subsequently affecting their ability to secrete IFN-γ and TNF-α, which correlates with HBsAg levels (107).
In terms of mechanism, HBsAg may inhibit the expression of enolase in exhausted T cells, thereby restricting glycolytic flux. This effect can be enhanced by supplementing downstream product pyruvate (PEP) (107). Additionally,during HBV infection, functional and non-exhausted T cells can utilize oxidative phosphorylation (OXPHOS) to meet their energy demands. In contrast, exhausted T cells exhibit dysfunctional depolarized mitochondria despite increased expression of glucose transporter-1 (GLUT-1), indicating impaired OXPHOS function, which subsequently affects the antiviral immune function of T cells (166). Consequently, the compromised functionality of HBV-specific CD8+ T cells does not stem from a deficiency in glucose uptake but rather from their diminished ability to engage OXPHOS to meet their energy demands. This deficiency hampers the capacity of T cells for metabolic reprogramming, transitioning from glycolysis to OXPHOS, which is essential for fulfilling the bioenergetic requisites crucial for the establishment of protective T cell memory (167). Recent investigations have further validated this underlying mechanism, with studies indicating that addressing mitochondrial dysfunction can effectively restore the antiviral activity of exhausted HBV-specific CD8+ T cells in CHB (168).
A recent authoritative review on the metabolic and epigenetic regulation of T cell exhaustion proposed that changes in mitochondrial adaptability and dynamics may drive epigenetic alterations that enhance T cell exhaustion, further impeding T cell immune function by influencing chromatin accessibility (169). This review highlights acetyl-CoA as a plausible candidate with potential supporting evidence. Under aerobic conditions, pyruvate generated from glycolysis enters the tricarboxylic acid (TCA) cycle in the mitochondria to produce acetyl-CoA [31], and the levels of acetyl-CoA are highly dependent on glucose availability and mitochondrial respiratory function (170). Acetyl-CoA forms the basis for the functions of histone acetyltransferases (HATs) (53). Evidence has long established that the availability of acetyl-CoA regulates many protein acetylation modifications (52, 171, 172). A mouse study demonstrated that reducing the levels of acetyl-CoA led to decreased acetylation of lysine 9 residues on histone H3 at the IFNG gene locus in CD4+ T cells, thereby suppressing IFNG expression (59). Additionally, promoting histone acetylation and chromatin accessibility can restore IFN-γ secretion in CD8+ T cells under glucose-restricted conditions (173).
Together, HBsAg can inhibit the key metabolic enzyme enolase, leading to the depolarization of exhausted T-cell mitochondria, manifested as impaired OXPHOS function (Figure 3.6). This specific alteration in metabolic reprogramming may lower acetyl-CoA levels, subsequently inhibiting the acetylation levels of target genes (IFN-γ and TNF-α), hindered the antiviral immune function of T cells.
3.6 B cells
Functioning as essential components of the humoral immune system, B cells play a pivotal role in generating antibodies against HBV (174). Notably, recent studies have focused on investigating deficiencies in both intrahepatic and circulating antiviral B cell responses in individuals with hepatitis B (175–177). Persistent viral infections can compromise the various antiviral functions of B cells, including antigen presentation, humoral immunity, and cytokine production (178–180). The activation status of B cells is closely related to their metabolic pathways. Unlike T cells, activated B cells lead to a wide-ranging increase in metabolism, including enhanced oxygen consumption and extracellular acidification rates (181).
During CHB infection, the virus inhibits the metabolic capacities of B cells, including OXPHOS and glycolysis. This, in turn, affects the energy supply and effector secretion of B cells, leading to a decrease in their proliferation and differentiation abilities, thereby facilitating the sustained presence of HBV (108). Mechanistically, HBV suppresses the AKT/mTOR pathway in B cells, reducing their glucose uptake and causing mitochondrial dysfunction, specifically by impairing OXPHOS function (108). Because OXPHOS provides 85% of the total ATP in activated PBMCs (182), HBV virus impairs the production of ATP in B cells by compromising both glycolysis and OXPHOS functions, thereby attenuating the activation of B cells.
Additionally, epidemiological studies have shown an increased risk of Non-Hodgkin’s lymphoma (NHL) in individuals with CHB infection, indicating a significant correlation (183, 184). Furthermore, recent clinical cohort studies on HBV and B cells have identified a higher occurrence of mutations in the key epigenetic regulator, ARID1A, in HBV cases (185, 186). Notably, HBV-related HCC also exhibits a high frequency of ARID1A mutations (187). ARID1A encodes a subunit of the Switch/Sucrose-Nonfermentable (SWI/SNF) chromatin remodeling complex, which primarily regulates chromatin accessibility to allow for the activation of target gene transcription. Importantly, its function in chromatin remodeling is ATP-dependent (188). Subunits of the SWI/SNF complex are crucial for the expression of B lineage-specific genes and the development of B cells (189).
Collectively, HBV impairs the glycolytic and OXPHOS metabolic functions of B cells by inhibiting the AKT/mTOR pathway (Figure 3.5). This inhibition leads to a reduction in ATP production, and the expression of the ATP-dependent epigenetic modification complex (SWI/SNF) responsible for promoting chromatin accessibility, ultimately affecting the antiviral immune response of B cells.
4 Discussion
This review aimed to examine how HBV affects the immunometabolism and epigenetic reprogramming of crucial cells, such as hepatocytes and immune cells, in the hepatic immune microenvironment. By altering the immunometabolism and epigenetic modifications in these cells, HBV impairs antiviral immune responses and facilitates the chronic persistence of HBV infection. However, some intriguing aspects of this process warrant further attention as they may offer novel insights into the pathogenic mechanisms of HBV.
4.1 Potential connections between immunometabolism, epigenetic modifications, and antiviral immunity in HBV-infected hepatocytes?
In HBV-infected hepatocytes, the virus can modulate the epigenetic modifications of downstream target genes (such as IFN) by regulating immunometabolism, thereby exerting an impact on antiviral immunity. HBV influences the antiviral immune response and viral replication in hepatocytes by intervening in the glycolytic and PPP metabolic pathway (Figure 1). Specifically, the primary outcomes of the impact of HBV on hepatocyte glycolysis are an increase in the metabolic product lactate and suppression of IFN production (58). Coincidentally, lactate derived from glycolysis acts as a threshold inhibitor of IFN production when RLR is activated, providing strong evidence of a negative regulatory relationship between lactate and IFN (190). This underscores the importance of lactate as a crucial metabolic product in innate immune responses in hepatocytes during CHB infection. Notably, a new epigenetic modification, lactylation, was discovered four years ago, elevating our understanding of lactate to unprecedented levels (51). Lactate-driven lactylation modifications play a vital role in suppressing inflammation and immune processes, making them a popular topic in recent research (191, 192). Therefore, exploring whether lactate, through its induced lactylation modifications, reduces the accessibility of IFN gene chromatin, thereby inhibiting the antiviral immune response in hepatocytes, would be an intriguing research direction. Lactate-induced lactylation may serve as a bridge between immunometabolism and antiviral effects.
Concomitantly, HBV promotes the hepatocellular PPP metabolism and sustains viral replication (57, 76). One of the main functions of the PPP is the generation of NADPH (193), which is an indispensable reducing agent in synthetic metabolic processes. The upregulation of HBx in hepatocytes increases the production of intracellular ATP and NADPH (194). Recent research revealed that NADPH can interact with histone deacetylase 3 (HDAC3), inhibiting its activity by disrupting its binding with co-factors (195). Simultaneously, in studies on hepatocytes, overexpression of the deacetylase HDAC3 can inhibit HBV replication (196). Recent studies have found a direct correlation between deacetylase and the production of type I interferons in innate immunity. In U3OS cells infected with Sendai virus (SeV), acetyl groups are present at specific DNA-binding domain (DBD) sites of IRF3/IRF7, thereby eliminating the liquid-liquid phase separation (LLPS) and IFN-I induction of IRF3/IRF7 (197). In other words, inhibiting deacetylases can block IFN signal transduction, thereby promoting viral replication. This further supports the conclusion that HBV protein expression interferes with the transmission of type I IFN signaling in infected hepatocytes (198). In summary, HBV increases the accumulation of NADPH, a product of PPP metabolism, in hepatocytes. NADPH may potentially interact with HDAC3, inhibiting deacetylase activity, which promotes the acetylation of the DNA-binding domain (DBD) of IRF3/IRF7. This, in turn, suppresses IFN expression and facilitates viral replication.
4.2 Could there be a connection between the immunometabolism, epigenetic modifications, and immune function of immune cells during CHB?
From a metabolism-epigenomics perspective, metabolites can serve as substrates or cofactors for epigenetic modifications, thereby altering the structure and function of chromatin and genomic regions associated with diseases (55). Epigenetic modifications can also directly impact energy metabolism and thus influence immune function by either regulating the transcription levels of metabolic enzymes or affecting the activity of metabolic transcription factors (117, 199). This intricate interplay creates a complex network between immunometabolism and epigenetics in CHB infection. In our recent study, we found that HBV can induce immunometabolism and epigenetic reprogramming in monocytes/macrophages, subsequently influencing their functionality and fate (to be published). Therefore, in-depth research into immunometabolism and epigenetic reprogramming will provide new perspectives for understanding the mechanistic origins of CHB.
Furthermore, as described above, exploring the relationships between immunometabolism and epigenetic modifications in various immune cells will be a captivating and highly meaningful research direction. Firstly, it remains an intriguing question whether HBV modulates the epigenetic reprogramming of interferon genes, such as methylation and lactylation, by inhibiting the metabolic regulatory factor mTOR pathway, consequently impacting the antiviral function of NK cells. Secondly, despite HBV inducing specific T cell exhaustion, leading to mitochondrial dysfunction and affecting the secretion of IFN-γ and TNF-α (107), the intricate mechanisms underlying the metabolic reprogramming and altered antiviral activity in T cells remain elusive. The potential reduction of acetyl-CoA levels in T cells through HBV-induced OXPHOS metabolic reprogramming may inhibit the acetylation levels of target genes, such as IFN-γ and TNF-α, representing a profound mechanism underlying the immune deficiency in exhausted T cells. Lastly, HBV, by suppressing the glycolysis of B cells, not only impairs the activation of B cells but also inhibits ATP levels within B cells and the expression of the ATP-dependent epigenetic modification complex SWI/SNF, both of which may synergistically further compromise the antiviral immune response of B cells.
4.3 The dialogue at the immunometabolic and epigenetic levels between hepatocytes and immune cells within the hepatic immune microenvironment
HBV affects metabolic reprogramming of hepatocytes through various pathways, leading to increased lactate production (57, 58). Lactate is a metabolic intermediary that shapes the fate and function of immune cells (200). Lactate plays a key role in oxidation, gluconeogenesis, and cellular signal transduction (201–203). Endogenous lactate is primarily produced through aerobic or anaerobic glycolysis, whereas exogenous lactate primarily enters the cells via monocarboxylate transporters (MCTs) (204) and G protein-coupled receptor 81 (GPR81) (GPR81) (205). MCT-1 mediates the effects of lactate on macrophages and CD8+ T cells (50, 206), potentially providing lactate as a messenger from hepatocytes to regulate immune cells. A decade ago, research indicated that exogenous lactate could induce the polarization of macrophages toward the M2 phenotype, manifested by increased expression of VEGF and ARG1 (50). Similarly, recent findings suggested that lactylation modifications specifically induced by lactate promote the expression of VEGF and ARG1 in macrophages (51). Furthermore, in the field of lactate research, predominantly focused on tumors, physiologically relevant concentrations of lactate can inhibit the activation of CD8+ T and NK cells, resulting in reduced production of IFN-γ (207). Tregs can absorb and metabolize lactate, thereby sustaining their suppressive function in environments characterized by elevated lactate levels, as observed in contexts such as the tumor microenvironment (208). Whether lactate-mediated inhibition of IFN-γ secretion in NK and T cells is facilitated through lactylation modifications presents an intriguing avenue for exploration.
5 Concluding remarks and future perspectives
In this review, we comprehensively explored the interplay and pivotal roles of immunometabolism and epigenetic modifications within the hepatic immune microenvironment across various cell types in CHB infection research. Immunometabolism is considered the link between cellular metabolic status and innate immune function, which is crucial for responding to HBV infection and contributing to the inability of the immune system to resolve CHB. HBV replication forms a stable viral cccDNA minichromosome within the nucleus of liver cells, and epigenetic modifications in cccDNA facilitate persistent infection. Furthermore, the accumulation or depletion of metabolic products not only directly affects the functionality and dynamic balance of immune cells, but also serves as a substrate that regulates epigenetic modifications. This, in turn, influences the expression of antiviral immune genes, thereby promoting sustained HBV infection. By delving into the regulation of host immune metabolic pathways and the impact of epigenetic modifications in response to HBV infection, we not only gain a better understanding of the mechanisms underlying the development of CHB infection but also provide robust theoretical support for the development of novel therapeutic strategies.
Based on research findings on immunometabolism and epigenetic regulation, future developments may lead to personalized therapeutic approaches (209). Further investigations into the functions and regulatory mechanisms of key metabolic enzymes in immune metabolic pathways will contribute to the discovery of novel therapeutic targets for CHB infection. Additionally, a deeper understanding of the dynamic changes in epigenetic modifications during HBV infection and their association with disease progression may lead to the development of therapeutic strategies targeting specific gene-modifying enzymes. Precise analysis of patient metabolic and epigenetic characteristics can facilitate the design of targeted treatment plans, thereby enhancing treatment efficacy and alleviating patient suffering.
The field of immunometabolism and epigenetic modifications in CHB infection has opened new avenues for understanding and treating this disease. We hope that more rigorous and comprehensive research in this area will yield novel insights and strategies to combat the serious health challenges posed by CHB infections.
Author contributions
ZW: Formal analysis, Resources, Writing – original draft, Writing – review & editing. NL: Conceptualization, Methodology, Supervision, Writing – review & editing. YY: Formal analysis, Resources, Supervision, Writing – review & editing. ZT: Conceptualization, Investigation, Methodology, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Program for JLU Science and Technology Innovative Research Team (2017TD-08) and the Fundamental Research Funds for the Central Universities, and Jilin Province Science and Technology Development Plan project, Natural Science Foundation (20190201245JC).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing. In addition, we are very grateful to the biorender website for facilitating our drawing. Imagines created with BioRender.com with permission. Figure 1 adapted from “Cancer Metabolism” by BioRender.com; Figure 2 created with BioRender.com; Figure 3 adapted from “Epigenetic Deregulation in Cancer” by BioRender.com. Retrieved from https://app.biorender.com/biorender-templates.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jeng WJ, Papatheodoridis GV, Lok ASF. Hepatitis B. Lancet (2023) 401:1039–52. doi: 10.1016/s0140-6736(22)01468-4
2. Kenney JM, von Bonsdorff CH, Nassal M, Fuller SD. Evolutionary conservation in the hepatitis B virus core structure: comparison of human and duck cores. Structure (1995) 3:1009–19. doi: 10.1016/s0969-2126(01)00237-4
3. Böttcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature (1997) 386:88–91. doi: 10.1038/386088a0
4. Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature (1997) 386:91–4. doi: 10.1038/386091a0
5. Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell (1999) 3:771–80. doi: 10.1016/s1097-2765(01)80009-5
6. Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut (2015) 64:1972–84. doi: 10.1136/gutjnl-2015-309809
7. McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology (2021) 73 Suppl 1:4–13. doi: 10.1002/hep.31288
8. Wang D, Zheng X, Fu B, Nian Z, Qian Y, Sun R, et al. Hepatectomy promotes recurrence of liver cancer by enhancing IL-11-STAT3 signaling. EBioMedicine (2019) 46:119–32. doi: 10.1016/j.ebiom.2019.07.058
9. Hsu YC, Huang DQ, Nguyen MH. Global burden of hepatitis B virus: current status, missed opportunities and a call for action. Nat Rev Gastroenterol Hepatol (2023) 20:524–37. doi: 10.1038/s41575-023-00760-9
10. Beasley RP. Rocks along the road to the control of HBV and HCC. Ann Epidemiol (2009) 19:231–4. doi: 10.1016/j.annepidem.2009.01.017
11. Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet (2015) 386:1546–55. doi: 10.1016/s0140-6736(15)61412-x
12. Ohsaki E, Ueda K. Interplay between KSHV and the host DNA damage response. Front Cell Infect Microbiol (2020) 10:604351. doi: 10.3389/fcimb.2020.604351
13. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (2012) 380:2095–128. doi: 10.1016/s0140-6736(12)61728-0
14. Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut (2012) 61:1754–64. doi: 10.1136/gutjnl-2011-301073
15. Bertoletti A, Maini MK, Ferrari C. The host-pathogen interaction during HBV infection: immunological controversies. Antivir Ther (2010) 15 Suppl 3:15–24. doi: 10.3851/imp1620
16. Dandri M, Locarnini S. New insight in the pathobiology of hepatitis B virus infection. Gut (2012) 61 Suppl 1:i6–17. doi: 10.1136/gutjnl-2012-302056
17. Boni C, Laccabue D, Lampertico P, Giuberti T, Viganò M, Schivazappa S, et al. Restored function of HBV-specific T cells after long-term effective therapy with nucleos(t)ide analogues. Gastroenterology (2012) 143:963–73.e9. doi: 10.1053/j.gastro.2012.07.014
18. Rehermann B, Lau D, Hoofnagle JH, Chisari FV. Cytotoxic T lymphocyte responsiveness after resolution of chronic hepatitis B virus infection. J Clin Invest (1996) 97:1655–65. doi: 10.1172/jci118592
19. Michel ML, Deng Q, Mancini-Bourgine M. Therapeutic vaccines and immune-based therapies for the treatment of chronic hepatitis B: perspectives and challenges. J Hepatol (2011) 54:1286–96. doi: 10.1016/j.jhep.2010.12.031
20. Zoulim F, Luangsay S, Durantel D. Targeting innate immunity: a new step in the development of combination therapy for chronic hepatitis B. Gastroenterology (2013) 144:1342–4. doi: 10.1053/j.gastro.2013.04.028
21. Wang D, Fu B, Shen X, Guo C, Liu Y, Zhang J, et al. Restoration of HBV-specific CD8(+) T-cell responses by sequential low-dose IL-2 treatment in non-responder patients after IFN-α therapy. Signal Transduct Target Ther (2021) 6:376. doi: 10.1038/s41392-021-00776-0
22. Sun J, Wu G, Pastor F, Rahman N, Wang WH, Zhang Z, et al. RNA helicase DDX5 enables STAT1 mRNA translation and interferon signalling in hepatitis B virus replicating hepatocytes. Gut (2022) 71:991–1005. doi: 10.1136/gutjnl-2020-323126
23. Liaw YF, Chu CM, Su IJ, Huang MJ, Lin DY, Chang-Chien CS. Clinical and histological events preceding hepatitis B e antigen seroconversion in chronic type B hepatitis. Gastroenterology (1983) 84:216–9. doi: 10.1016/S0016-5085(83)80114-0
24. Lok AS, Lai CL. Acute exacerbations in Chinese patients with chronic hepatitis B virus (HBV) infection. Incidence, predisposing factors and etiology. J Hepatol (1990) 10:29–34. doi: 10.1016/0168-8278(90)90069-4
25. Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med (2013) 19:859–68. doi: 10.1038/nm.3251
26. Jin B, Zhang YY, Pan JX. The role and significance of hepatic environmental cells in tumor metastatic colonization to liver. Sichuan Da Xue Xue Bao Yi Xue Ban (2023) 54:469–74. doi: 10.12182/20230560301
27. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
28. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol (2010) 10:753–66. doi: 10.1038/nri2858
29. Crispe IN, Dao T, Klugewitz K, Mehal WZ, Metz DP. The liver as a site of T-cell apoptosis: graveyard, or killing field? Immunol Rev (2000) 174:47–62. doi: 10.1034/j.1600-0528.2002.017412.x
30. Li F, Tian Z. The liver works as a school to educate regulatory immune cells. Cell Mol Immunol (2013) 10:292–302. doi: 10.1038/cmi.2013.7
31. Dobrzynski E, Mingozzi F, Liu YL, Bendo E, Cao O, Wang L, et al. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood (2004) 104:969–77. doi: 10.1182/blood-2004-03-0847
32. Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity (2004) 20:327–36. doi: 10.1016/s1074-7613(04)00050-0
33. Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood (2007) 110:1132–40. doi: 10.1182/blood-2007-02-073304
34. Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology (2009) 50:612–21. doi: 10.1002/hep.23043
35. Xu L, Yin W, Sun R, Wei H, Tian Z. Liver type I regulatory T cells suppress germinal center formation in HBV-tolerant mice. Proc Natl Acad Sci USA (2013) 110:16993–8. doi: 10.1073/pnas.1306437110
36. Fanning GC, Zoulim F, Hou J, Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov (2019) 18:827–44. doi: 10.1038/s41573-019-0037-0
37. Zeng Z, Li L, Chen Y, Wei H, Sun R, Tian Z. Interferon-γ facilitates hepatic antiviral T cell retention for the maintenance of liver-induced systemic tolerance. J Exp Med (2016) 213:1079–93. doi: 10.1084/jem.20151218
38. Lercher A, Baazim H, Bergthaler A. Systemic immunometabolism: challenges and opportunities. Immunity (2020) 53:496–509. doi: 10.1016/j.immuni.2020.08.012
39. Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity (2017) 47:406–20. doi: 10.1016/j.immuni.2017.08.009
40. O'Neill LA. A broken krebs cycle in macrophages. Immunity (2015) 42:393–4. doi: 10.1016/j.immuni.2015.02.017
41. Thaker SK, Ch'ng J, Christofk HR. Viral hijacking of cellular metabolism. BMC Biol (2019) 17:59. doi: 10.1186/s12915-019-0678-9
42. DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell (2012) 148:1132–44. doi: 10.1016/j.cell.2012.02.032
43. Noch E, Khalili K. Oncogenic viruses and tumor glucose metabolism: like kids in a candy store. Mol Cancer Ther (2012) 11:14–23. doi: 10.1158/1535-7163.Mct-11-0517
44. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet (2016) 17:487–500. doi: 10.1038/nrg.2016.59
45. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol (2011) 32:335–43. doi: 10.1016/j.it.2011.04.001
46. O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature (2013) 493:346–55. doi: 10.1038/nature11862
47. Werlen G, Jain R, Jacinto E. MTOR signaling and metabolism in early T cell development. Genes (Basel) (2021) 12:728. doi: 10.3390/genes12050728
48. Fanucchi S, Domínguez-Andrés J, Joosten LAB, Netea MG, Mhlanga MM. The intersection of epigenetics and metabolism in trained immunity. Immunity (2021) 54:32–43. doi: 10.1016/j.immuni.2020.10.011
49. Ratter JM, Rooijackers HMM, Hooiveld GJ, Hijmans AGM, de Galan BE, Tack CJ, et al. In vitro and in vivo Effects of Lactate on Metabolism and Cytokine Production of Human Primary PBMCs and Monocytes. Front Immunol (2018) 9:2564. doi: 10.3389/fimmu.2018.02564
50. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513:559–63. doi: 10.1038/nature13490
51. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
52. Christ A, Latz E. The Western lifestyle has lasting effects on metaflammation. Nat Rev Immunol (2019) 19:267–8. doi: 10.1038/s41577-019-0156-1
53. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (2009) 324:1076–80. doi: 10.1126/science.1164097
54. Ding W, Smulan LJ, Hou NS, Taubert S, Watts JL, Walker AK. s-adenosylmethionine levels govern innate immunity through distinct methylation-dependent pathways. Cell Metab (2015) 22:633–45. doi: 10.1016/j.cmet.2015.07.013
55. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab (2015) 22:861–73. doi: 10.1016/j.cmet.2015.08.024
56. Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem (2011) 286:9856–64. doi: 10.1074/jbc.M110.196790
57. Wu YH, Yang Y, Chen CH, Hsiao CJ, Li TN, Liao KJ, et al. Aerobic glycolysis supports hepatitis B virus protein synthesis through interaction between viral surface antigen and pyruvate kinase isoform M2. PloS Pathog (2021) 17:e1008866. doi: 10.1371/journal.ppat.1008866
58. Zhou L, He R, Fang P, Li M, Yu H, Wang Q, et al. Hepatitis B virus rigs the cellular metabolome to avoid innate immune recognition. Nat Commun (2021) 12:98. doi: 10.1038/s41467-020-20316-8
59. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (2016) 354:481–4. doi: 10.1126/science.aaf6284
60. Jin J, Xu H, Wu R, Niu J, Li S. Aberrant DNA methylation profile of hepatitis B virus infection. J Med Virol (2019) 91:81–92. doi: 10.1002/jmv.25284
61. Mak LY, Wong DK, Pollicino T, Raimondo G, Hollinger FB, Yuen MF. Occult hepatitis B infection and hepatocellular carcinoma: Epidemiology, virology, hepatocarcinogenesis and clinical significance. J Hepatol (2020) 73:952–64. doi: 10.1016/j.jhep.2020.05.042
62. Zhang J, Ling N, Lei Y, Peng M, Hu P, Chen M. Multifaceted interaction between hepatitis B virus infection and lipid metabolism in hepatocytes: A potential target of antiviral therapy for chronic hepatitis B. Front Microbiol (2021) 12:636897. doi: 10.3389/fmicb.2021.636897
63. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol (2017) 17:306–21. doi: 10.1038/nri.2017.11
64. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol (2017) 14:55–66. doi: 10.1038/nrgastro.2016.168
65. Tu T, Calabro SR, Lee A, Maczurek AE, Budzinska MA, Warner FJ, et al. Hepatocytes in liver injury: Victim, bystander, or accomplice in progressive fibrosis? J Gastroenterol Hepatol (2015) 30:1696–704. doi: 10.1111/jgh.13065
66. Gong J, Tu W, Liu J, Tian D. Hepatocytes: A key role in liver inflammation. Front Immunol (2022) 13:1083780. doi: 10.3389/fimmu.2022.1083780
67. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207:2053–63. doi: 10.1084/jem.20101664
68. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
69. Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity (2016) 44:739–54. doi: 10.1016/j.immuni.2016.04.002
70. Schlee M, Hartmann G. Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol (2016) 16:566–80. doi: 10.1038/nri.2016.78
71. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
72. You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, et al. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol (2009) 10:1300–8. doi: 10.1038/ni.1815
73. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
74. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi: 10.1038/nri3581
75. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi: 10.1016/j.immuni.2012.03.013
76. Liu B, Fang M, He Z, Cui D, Jia S, Lin X, et al. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis (2015) 6:e1980. doi: 10.1038/cddis.2015.322
77. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
78. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell (2014) 5:592–602. doi: 10.1007/s13238-014-0082-8
79. Mayer KA, Stöckl J, Zlabinger GJ, Gualdoni GA. Hijacking the supplies: metabolism as a novel facet of virus-host interaction. Front Immunol (2019) 10:1533. doi: 10.3389/fimmu.2019.01533
80. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev (2013) 27:2179–91. doi: 10.1101/gad.225680.113
81. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell (2012) 22:66–79. doi: 10.1016/j.ccr.2012.05.016
82. Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol (2011) 193:275–84. doi: 10.1083/jcb.201102031
83. Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell (2013) 51:618–31. doi: 10.1016/j.molcel.2013.08.003
84. Dandri M. Epigenetic modulation in chronic hepatitis B virus infection. Semin Immunopathol (2020) 42:173–85. doi: 10.1007/s00281-020-00780-6
85. Hong X, Kim ES, Guo H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis B. Hepatology (2017) 66:2066–77. doi: 10.1002/hep.29479
86. Moreau P, Cournac A, Palumbo GA, Marbouty M, Mortaza S, Thierry A, et al. Tridimensional infiltration of DNA viruses into the host genome shows preferential contact with active chromatin. Nat Commun (2018) 9:4268. doi: 10.1038/s41467-018-06739-4
87. Schübeler D. Function and information content of DNA methylation. Nature (2015) 517:321–6. doi: 10.1038/nature14192
88. Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology (2015) 479-480:672–86. doi: 10.1016/j.virol.2015.02.031
89. Bock CT, Schranz P, Schröder CH, Zentgraf H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes (1994) 8:215–29. doi: 10.1007/bf01703079
90. Bock CT, Schwinn S, Locarnini S, Fyfe J, Manns MP, Trautwein C, et al. Structural organization of the hepatitis B virus minichromosome. J Mol Biol (2001) 307:183–96. doi: 10.1006/jmbi.2000.4481
91. Vivekanandan P, Thomas D, Torbenson M. Hepatitis B viral DNA is methylated in liver tissues. J Viral Hepat (2008) 15:103–7. doi: 10.1111/j.1365-2893.2007.00905.x
92. Zhang Y, Li C, Zhang Y, Zhu H, Kang Y, Liu H, et al. Comparative analysis of CpG islands among HBV genotypes. PloS One (2013) 8:e56711. doi: 10.1371/journal.pone.0056711
93. Zhang Y, Mao R, Yan R, Cai D, Zhang Y, Zhu H, et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PloS One (2014) 9:e110442. doi: 10.1371/journal.pone.0110442
94. Guo Y, Li Y, Mu S, Zhang J, Yan Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J Med Virol (2009) 81:1177–83. doi: 10.1002/jmv.21525
95. Jain S, Chang TT, Chen S, Boldbaatar B, Clemens A, Lin SY, et al. Comprehensive DNA methylation analysis of hepatitis B virus genome in infected liver tissues. Sci Rep (2015) 5:10478. doi: 10.1038/srep10478
96. Barlow DP. Methylation and imprinting: from host defense to gene regulation? Science (1993) 260:309–10. doi: 10.1126/science.8469984
97. Ye C, Tao R, Cao Q, Zhu D, Wang Y, Wang J, et al. Whole-genome DNA methylation and hydroxymethylation profiling for HBV-related hepatocellular carcinoma. Int J Oncol (2016) 49:589–602. doi: 10.3892/ijo.2016.3535
98. Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell (2019) 179:561–577.e22. doi: 10.1016/j.cell.2019.08.052
99. Vivekanandan P, Daniel HD, Kannangai R, Martinez-Murillo F, Torbenson M. Hepatitis B virus replication induces methylation of both host and viral DNA. J Virol (2010) 84:4321–9. doi: 10.1128/jvi.02280-09
100. Shim YH, Yoon GS, Choi HJ, Chung YH, Yu E. p16 Hypermethylation in the early stage of hepatitis B virus-associated hepatocarcinogenesis. Cancer Lett (2003) 190:213–9. doi: 10.1016/s0304-3835(02)00613-4
101. Ueda M, Seki M. Histone modifications form epigenetic regulatory networks to regulate abiotic stress response. Plant Physiol (2020) 182:15–26. doi: 10.1104/pp.19.00988
102. Pollicino T, Belloni L, Raffa G, Pediconi N, Squadrito G, Raimondo G, et al. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology (2006) 130:823–37. doi: 10.1053/j.gastro.2006.01.001
103. Rivière L, Gerossier L, Ducroux A, Dion S, Deng Q, Michel ML, et al. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J Hepatol (2015) 63:1093–102. doi: 10.1016/j.jhep.2015.06.023
104. Chiramel AI, Brady NR, Bartenschlager R. Divergent roles of autophagy in virus infection. Cells (2013) 2:83–104. doi: 10.3390/cells2010083
105. Bei J, Chen Y, Zhang Q, Wang X, Lin L, Huang J, et al. HBV suppresses macrophage immune responses by impairing the TCA cycle through the induction of CS/PDHC hyperacetylation. Hepatol Commun (2023) 7:e0294. doi: 10.1097/hc9.0000000000000294
106. Marotel M, Villard M, Drouillard A, Tout I, Besson L, Allatif O, et al. Peripheral natural killer cells in chronic hepatitis B patients display multiple molecular features of T cell exhaustion. Elife (2021) 10:e60095. doi: 10.7554/eLife.60095
107. Winkler F, Hipp AV, Ramirez C, Martin B, Villa M, Neuwirt E, et al. Enolase represents a metabolic checkpoint controlling the differential exhaustion programmes of hepatitis virus-specific CD8(+) T cells. Gut (2023) 72:1971–84. doi: 10.1136/gutjnl-2022-328734
108. Huang C, Shao J, Lou C, Wu F, Ge T, Gao H, et al. Reduced energy metabolism impairs T cell-dependent B cell responses in patients with advanced HBV-related cirrhosis. Front Immunol (2021) 12:660312. doi: 10.3389/fimmu.2021.660312
109. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3:23–35. doi: 10.1038/nri978
110. Possamai LA, Thursz MR, Wendon JA, Antoniades CG. Modulation of monocyte/macrophage function: a therapeutic strategy in the treatment of acute liver failure. J Hepatol (2014) 61:439–45. doi: 10.1016/j.jhep.2014.03.031
111. Faure-Dupuy S, Delphin M, Aillot L, Dimier L, Lebossé F, Fresquet J, et al. Hepatitis B virus-induced modulation of liver macrophage function promotes hepatocyte infection. J Hepatol (2019) 71:1086–98. doi: 10.1016/j.jhep.2019.06.032
112. Li H, Zhai N, Wang Z, Song H, Yang Y, Cui A, et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut (2018) 67:2035–44. doi: 10.1136/gutjnl-2017-314098
113. Li Y, Yang Y, Li T, Wang Z, Gao C, Deng R, et al. Activation of AIM2 by hepatitis B virus results in antiviral immunity that suppresses hepatitis C virus during coinfection. J Virol (2023) 97:e0109023. doi: 10.1128/jvi.01090-23
114. Song H, Tan G, Yang Y, Cui A, Li H, Li T, et al. Hepatitis B virus-induced imbalance of inflammatory and antiviral signaling by differential phosphorylation of STAT1 in human monocytes. J Immunol (2019) 202:2266–75. doi: 10.4049/jimmunol.1800848
115. Angajala A, Lim S, Phillips JB, Kim JH, Yates C, You Z, et al. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol (2018) 9:1605. doi: 10.3389/fimmu.2018.01605
116. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA (2008) 105:14447–52. doi: 10.1073/pnas.0803790105
117. Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol (2015) 33:125–31. doi: 10.1016/j.ceb.2015.02.003
118. Xu Y, Liao C, Liu R, Liu J, Chen Z, Zhao H, et al. IRGM promotes glioma M2 macrophage polarization through p62/TRAF6/NF-κB pathway mediated IL-8 production. Cell Biol Int (2019) 43:125–35. doi: 10.1002/cbin.11061
119. Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci USA (2015) 112:2853–8. doi: 10.1073/pnas.1501441112
120. Li J, Yu M, Zong R, Fan C, Ren F, Wu W, et al. Deacetylation of Notch1 by SIRT1 contributes to HBsAg- and HBeAg-mediated M2 macrophage polarization. Am J Physiol Gastrointest Liver Physiol (2022) 322:G459–g471. doi: 10.1152/ajpgi.00338.2021
121. Xu L, Yin W, Sun R, Wei H, Tian Z. Kupffer cell-derived IL-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice. Hepatology (2014) 59:443–52. doi: 10.1002/hep.26668
122. Li M, Sun R, Xu L, Yin W, Chen Y, Zheng X, et al. Kupffer cells support hepatitis B virus-mediated CD8+ T cell exhaustion via hepatitis B core antigen-TLR2 interactions in mice. J Immunol (2015) 195:3100–9. doi: 10.4049/jimmunol.1500839
123. Li H, Zheng HW, Chen H, Xing ZZ, You H, Cong M, et al. Hepatitis B virus particles preferably induce Kupffer cells to produce TGF-β1 over pro-inflammatory cytokines. Dig Liver Dis (2012) 44:328–33. doi: 10.1016/j.dld.2011.11.005
124. Tian Y, Kuo CF, Akbari O, Ou JH. Maternal-Derived Hepatitis B Virus e Antigen Alters Macrophage Function in Offspring to Drive Viral Persistence after Vertical Transmission. Immunity (2016) 44:1204–14. doi: 10.1016/j.immuni.2016.04.008
125. Li Y, Zhu Y, Feng S, Ishida Y, Chiu TP, Saito T, et al. Macrophages activated by hepatitis B virus have distinct metabolic profiles and suppress the virus via IL-1β to downregulate PPARα and FOXO3. Cell Rep (2022) 38:110284. doi: 10.1016/j.celrep.2021.110284
126. Novak N, Koch S, Allam JP, Bieber T. Dendritic cells: bridging innate and adaptive immunity in atopic dermatitis. J Allergy Clin Immunol (2010) 125:50–9. doi: 10.1016/j.jaci.2009.11.019
127. Møller SH, Wang L, Ho PC. Metabolic programming in dendritic cells tailors immune responses and homeostasis. Cell Mol Immunol (2022) 19:370–83. doi: 10.1038/s41423-021-00753-1
128. Yonejima A, Mizukoshi E, Tamai T, Nakagawa H, Kitahara M, Yamashita T, et al. Characteristics of impaired dendritic cell function in patients with hepatitis B virus infection. Hepatology (2019) 70:25–39. doi: 10.1002/hep.30637
129. Zhao H, Yu Y, Wang Y, Zhao L, Yang A, Hu Y, et al. Cholesterol accumulation on dendritic cells reverses chronic hepatitis B virus infection-induced dysfunction. Cell Mol Immunol (2022) 19:1347–60. doi: 10.1038/s41423-022-00939-1
130. You Z, Chi H. Lipid metabolism in dendritic cell biology. Immunol Rev (2023) 317:137–51. doi: 10.1111/imr.13215
131. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Science (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
132. Freeman BE, Raué HP, Hill AB, Slifka MK. Cytokine-mediated activation of NK cells during viral infection. J Virol (2015) 89:7922–31. doi: 10.1128/jvi.00199-15
133. Fisicaro P, Rossi M, Vecchi A, Acerbi G, Barili V, Laccabue D, et al. The good and the bad of natural killer cells in virus control: perspective for anti-HBV therapy. Int J Mol Sci (2019) 20:5080. doi: 10.3390/ijms20205080
134. Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology (2009) 137:1289–300. doi: 10.1053/j.gastro.2009.06.054
135. Fisicaro P, Valdatta C, Boni C, Massari M, Mori C, Zerbini A, et al. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut (2009) 58:974–82. doi: 10.1136/gut.2008.163600
136. Zhao J, Li Y, Jin L, Zhang S, Fan R, Sun Y, et al. Natural killer cells are characterized by the concomitantly increased interferon-γ and cytotoxicity in acute resolved hepatitis B patients. PloS One (2012) 7:e49135. doi: 10.1371/journal.pone.0049135
137. Lunemann S, Malone DF, Hengst J, Port K, Grabowski J, Deterding K, et al. Compromised function of natural killer cells in acute and chronic viral hepatitis. J Infect Dis (2014) 209:1362–73. doi: 10.1093/infdis/jit561
138. Yu WH, Cosgrove C, Berger CT, Cheney PC, Krykbaeva M, Kim AY, et al. ADCC-mediated CD56(DIM) NK cell responses are associated with early HBsAg clearance in acute HBV infection. Pathog Immun (2018) 3:2–18. doi: 10.20411/pai.v3i1.228
139. Oliviero B, Varchetta S, Paudice E, Michelone G, Zaramella M, Mavilio D, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology (2009) 137:1151–60, 1160.e1-7. doi: 10.1053/j.gastro.2009.05.047
140. Peppa D, Micco L, Javaid A, Kennedy PT, Schurich A, Dunn C, et al. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PloS Pathog (2010) 6:e1001227. doi: 10.1371/journal.ppat.1001227
141. Sun C, Fu B, Gao Y, Liao X, Sun R, Tian Z, et al. TGF-β1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PloS Pathog (2012) 8:e1002594. doi: 10.1371/journal.ppat.1002594
142. Tjwa ET, van Oord GW, Hegmans JP, Janssen HL, Woltman AM. Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis B. J Hepatol (2011) 54:209–18. doi: 10.1016/j.jhep.2010.07.009
143. Sepulveda FE, Maschalidi S, Vosshenrich CA, Garrigue A, Kurowska M, Ménasche G, et al. A novel immunoregulatory role for NK-cell cytotoxicity in protection from HLH-like immunopathology in mice. Blood (2015) 125:1427–34. doi: 10.1182/blood-2014-09-602946
144. Cong J. Metabolism of natural killer cells and other innate lymphoid cells. Front Immunol (2020) 11:1989. doi: 10.3389/fimmu.2020.01989
145. Zhao K, Huang J, Dai D, Feng Y, Liu L, Nie S. Serum iron level as a potential predictor of coronavirus disease 2019 severity and mortality: A retrospective study. Open Forum Infect Dis (2020) 7:ofaa250. doi: 10.1093/ofid/ofaa250
146. Osuna-Espinoza KY, Rosas-Taraco AG. Metabolism of NK cells during viral infections. Front Immunol (2023) 14:1064101. doi: 10.3389/fimmu.2023.1064101
147. Yang C, Malarkannan S. Transcriptional regulation of NK cell development by mTOR complexes. Front Cell Dev Biol (2020) 8:566090. doi: 10.3389/fcell.2020.566090
148. Wang D, Sun Z, Zhu X, Zheng X, Zhou Y, Lu Y, et al. GARP-mediated active TGF-β1 induces bone marrow NK cell dysfunction in AML patients with early relapse post-allo-HSCT. Blood (2022) 140:2788–804. doi: 10.1182/blood.2022015474
149. Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol (2014) 193:4477–84. doi: 10.4049/jimmunol.1401558
150. Beaulieu AM. Transcriptional and epigenetic regulation of memory NK cell responses. Immunol Rev (2021) 300:125–33. doi: 10.1111/imr.12947
151. Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity (2015) 42:431–42. doi: 10.1016/j.immuni.2015.02.013
152. Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD, et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity (2015) 42:443–56. doi: 10.1016/j.immuni.2015.02.008
153. Luetke-Eversloh M, Hammer Q, Durek P, Nordström K, Gasparoni G, Pink M, et al. Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PloS Pathog (2014) 10:e1004441. doi: 10.1371/journal.ppat.1004441
154. Lau CM, Adams NM, Geary CD, Weizman OE, Rapp M, Pritykin Y, et al. Epigenetic control of innate and adaptive immune memory. Nat Immunol (2018) 19:963–72. doi: 10.1038/s41590-018-0176-1
155. Chisari FV, Isogawa M, Wieland SF. Pathogenesis of hepatitis B virus infection. Pathol Biol (Paris) (2010) 58:258–66. doi: 10.1016/j.patbio.2009.11.001
156. Hoogeveen RC, Robidoux MP, Schwarz T, Heydmann L, Cheney JA, Kvistad D, et al. Phenotype and function of HBV-specific T cells is determined by the targeted epitope in addition to the stage of infection. Gut (2019) 68:893–904. doi: 10.1136/gutjnl-2018-316644
157. Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol (2007) 81:4215–25. doi: 10.1128/jvi.02844-06
158. Park JJ, Wong DK, Wahed AS, Lee WM, Feld JJ, Terrault N, et al. Hepatitis B virus–specific and global T-cell dysfunction in chronic hepatitis B. Gastroenterology (2016) 150:684–695.e5. doi: 10.1053/j.gastro.2015.11.050
159. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
160. Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol (2005) 174:4670–7. doi: 10.4049/jimmunol.174.8.4670
161. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi: 10.1084/jem.20110278
162. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
163. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep (2018) 22:1509–21. doi: 10.1016/j.celrep.2018.01.040
164. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
165. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14:1064–72. doi: 10.1038/ni.2687
166. Schurich A, Pallett LJ, Jajbhay D, Wijngaarden J, Otano I, Gill US, et al. Distinct metabolic requirements of exhausted and functional virus-specific CD8 T cells in the same host. Cell Rep (2016) 16:1243–52. doi: 10.1016/j.celrep.2016.06.078
167. Barili V, Boni C, Rossi M, Vecchi A, Zecca A, Penna A, et al. Metabolic regulation of the HBV-specific T cell function. Antiviral Res (2021) 185:104989. doi: 10.1016/j.antiviral.2020.104989
168. Fisicaro P, Barili V, Montanini B, Acerbi G, Ferracin M, Guerrieri F, et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med (2017) 23:327–36. doi: 10.1038/nm.4275
169. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab (2020) 2:1001–12. doi: 10.1038/s42255-020-00280-9
170. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun (2020) 11:102. doi: 10.1038/s41467-019-13668-3
171. Shi L, Tu BP. Protein acetylation as a means to regulate protein function in tune with metabolic state. Biochem Soc Trans (2014) 42:1037–42. doi: 10.1042/bst20140135
172. Cai L, Tu BP. On acetyl-CoA as a gauge of cellular metabolic state. Cold Spring Harb Symp Quant Biol (2011) 76:195–202. doi: 10.1101/sqb.2011.76.010769
173. Qiu J, Villa M, Sanin DE, Buck MD, O'Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep (2019) 27:2063–2074.e5. doi: 10.1016/j.celrep.2019.04.022
174. Cai Y, Yin W. The multiple functions of B cells in chronic HBV infection. Front Immunol (2020) 11:582292. doi: 10.3389/fimmu.2020.582292
175. Burton AR, Pallett LJ, McCoy LE, Suveizdyte K, Amin OE, Swadling L, et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J Clin Invest (2018) 128:4588–603. doi: 10.1172/jci121960
176. Ma Z, Zhang E, Gao S, Xiong Y, Lu M. Toward a functional cure for hepatitis B: the rationale and challenges for therapeutic targeting of the B cell immune response. Front Immunol (2019) 10:2308. doi: 10.3389/fimmu.2019.02308
177. Neumann-Haefelin C, Thimme R. Entering the spotlight: hepatitis B surface antigen-specific B cells. J Clin Invest (2018) 128:4257–9. doi: 10.1172/jci124098
178. Barnaba V, Franco A, Alberti A, Benvenuto R, Balsano F. Selective killing of hepatitis B envelope antigen-specific B cells by class I-restricted, exogenous antigen-specific T lymphocytes. Nature (1990) 345:258–60. doi: 10.1038/345258a0
179. Milich DR, Chen M, Schödel F, Peterson DL, Jones JE, Hughes JL. Role of B cells in antigen presentation of the hepatitis B core. Proc Natl Acad Sci USA (1997) 94:14648–53. doi: 10.1073/pnas.94.26.14648
180. Shen P, Fillatreau S. Antibody-independent functions of B cells: a focus on cytokines. Nat Rev Immunol (2015) 15:441–51. doi: 10.1038/nri3857
181. Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol (2014) 192:3626–36. doi: 10.4049/jimmunol.1302062
182. Roos D, Loos JA. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res (1973) 77:127–35. doi: 10.1016/0014-4827(73)90561-2
183. Sinha M, Sundar K, Premalata CS, Asati V, Murali A, Bajpai AK, et al. Pro-oncogenic, intra host viral quasispecies in Diffuse large B cell lymphoma patients with occult Hepatitis B Virus infection. Sci Rep (2019) 9:14516. doi: 10.1038/s41598-019-51157-1
184. Li M, Shen Y, Chen Y, Gao H, Zhou J, Wang Q, et al. Characterization of hepatitis B virus infection and viral DNA integration in non-Hodgkin lymphoma. Int J Cancer (2020) 147:2199–209. doi: 10.1002/ijc.33027
185. Fernández-Rodríguez C, Rodríguez-Sevilla JJ, Fernández-Ibarrondo L, Sánchez-González B, Gibert J, Bento L, et al. Worse outcome and distinct mutational pattern in follicular lymphoma with anti-HBc positivity. Blood Adv (2022) 6:82–6. doi: 10.1182/bloodadvances.2021005316
186. Ren W, Ye X, Su H, Li W, Liu D, Pirmoradian M, et al. Genetic landscape of hepatitis B virus-associated diffuse large B-cell lymphoma. Blood (2018) 131:2670–81. doi: 10.1182/blood-2017-11-817601
187. An P, Xu J, Yu Y, Winkler CA. Host and viral genetic variation in HBV-related hepatocellular carcinoma. Front Genet (2018) 9:261. doi: 10.3389/fgene.2018.00261
188. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv (2015) 1:e1500447. doi: 10.1126/sciadv.1500447
189. Choi J, Ko M, Jeon S, Jeon Y, Park K, Lee C, et al. The SWI/SNF-like BAF complex is essential for early B cell development. J Immunol (2012) 188:3791–803. doi: 10.4049/jimmunol.1103390
190. Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell (2019) 178:176–189.e15. doi: 10.1016/j.cell.2019.05.003
191. Rho H, Terry AR, Chronis C, Hay N. Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell Metab (2023) 35:1406–1423.e8. doi: 10.1016/j.cmet.2023.06.013
192. Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res (2022) 131:893–908. doi: 10.1161/circresaha.122.320488
193. Liu X, Olszewski K, Zhang Y, Lim EW, Shi J, Zhang X, et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol (2020) 22:476–86. doi: 10.1038/s41556-020-0496-x
194. Wang MD, Wu H, Huang S, Zhang HL, Qin CJ, Zhao LH, et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget (2016) 7:6711–26. doi: 10.18632/oncotarget.6817
195. Li W, Kou J, Qin J, Li L, Zhang Z, Pan Y, et al. NADPH levels affect cellular epigenetic state by inhibiting HDAC3-Ncor complex. Nat Metab (2021) 3:75–89. doi: 10.1038/s42255-020-00330-2
196. Wei F, Meng D. Study on the role of histone epigenetic modification in replication of hepatitis B virus. Biochem Biophys Res Commun (2023) 669:1–9. doi: 10.1016/j.bbrc.2023.05.045
197. Qin Z, Fang X, Sun W, Ma Z, Dai T, Wang S, et al. Deactylation by SIRT1 enables liquid-liquid phase separation of IRF3/IRF7 in innate antiviral immunity. Nat Immunol (2022) 23:1193–207. doi: 10.1038/s41590-022-01269-0
198. Lütgehetmann M, Bornscheuer T, Volz T, Allweiss L, Bockmann JH, Pollok JM, et al. Hepatitis B virus limits response of human hepatocytes to interferon-α in chimeric mice. Gastroenterology (2011) 140:2074–83, 2083.e1-2. doi: 10.1053/j.gastro.2011.02.057
199. Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol (2012) 13:270–6. doi: 10.1038/nrm3305
200. Caslin HL, Abebayehu D, Pinette JA, Ryan JJ. Lactate is a metabolic mediator that shapes immune cell fate and function. Front Physiol (2021) 12:688485. doi: 10.3389/fphys.2021.688485
201. Brooks GA. Cell-cell and intracellular lactate shuttles. J Physiol (2009) 587:5591–600. doi: 10.1113/jphysiol.2009.178350
202. Daw CC, Ramachandran K, Enslow BT, Maity S, Bursic B, Novello MJ, et al. Lactate elicits ER-mitochondrial mg(2+) dynamics to integrate cellular metabolism. Cell (2020) 183:474–489.e17. doi: 10.1016/j.cell.2020.08.049
203. Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z, et al. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep (2020) 33:108487. doi: 10.1016/j.celrep.2020.108487
204. Felmlee MA, Jones RS, Rodriguez-Cruz V, Follman KE, Morris ME. Monocarboxylate transporters (SLC16): function, regulation, and role in health and disease. Pharmacol Rev (2020) 72:466–85. doi: 10.1124/pr.119.018762
205. Brown TP, Ganapathy V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol Ther (2020) 206:107451. doi: 10.1016/j.pharmthera.2019.107451
206. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PloS Biol (2015) 13:e1002202. doi: 10.1371/journal.pbio.1002202
207. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
208. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature (2021) 591:645–51. doi: 10.1038/s41586-020-03045-2
Keywords: chronic hepatitis B infection, immunometabolism reprogramming, epigenetic modification, antiviral immunity, hepatic immune microenvironment
Citation: Wang Z, Liu N, Yang Y and Tu Z (2024) The novel mechanism facilitating chronic hepatitis B infection: immunometabolism and epigenetic modification reprogramming. Front. Immunol. 15:1349867. doi: 10.3389/fimmu.2024.1349867
Received: 05 December 2023; Accepted: 02 January 2024;
Published: 15 January 2024.
Edited by:
Yong Liu, Nanjing Drum Tower Hospital, ChinaReviewed by:
Hua-Jun Zhao, Shandong University, ChinaDongyao Wang, University of Science and Technology of China, China
Copyright © 2024 Wang, Liu, Yang and Tu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengkun Tu, dHV6aGVuZ2t1bkBqbHUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship