 Maximilian Schmid1,2,3,4†
Maximilian Schmid1,2,3,4† Patrick Fischer1,2,3,4†
Patrick Fischer1,2,3,4† Magdalena Engl1,2,4,5Joachim Widder1,2
Magdalena Engl1,2,4,5Joachim Widder1,2 Sylvia Kerschbaum-Gruber1,2,3
Sylvia Kerschbaum-Gruber1,2,3 Dea Slade1,2,3,4*
Dea Slade1,2,3,4*- 1Department of Radiation Oncology, Medical University of Vienna, Vienna, Austria
- 2Comprehensive Cancer Center, Medical University of Vienna, Vienna, Austria
- 3MedAustron Ion Therapy Center, Wiener Neustadt, Austria
- 4Department of Medical Biochemistry, Medical University of Vienna, Max Perutz Labs, Vienna Biocenter, Vienna, Austria
- 5Vienna Biocenter PhD Program, a Doctoral School of the University of Vienna and Medical University of Vienna, Vienna, Austria
Autophagy is an intracellular process that targets various cargos for degradation, including members of the cGAS-STING signaling cascade. cGAS-STING senses cytosolic double-stranded DNA and triggers an innate immune response through type I interferons. Emerging evidence suggests that autophagy plays a crucial role in regulating and fine-tuning cGAS-STING signaling. Reciprocally, cGAS-STING pathway members can actively induce canonical as well as various non-canonical forms of autophagy, establishing a regulatory network of feedback mechanisms that alter both the cGAS-STING and the autophagic pathway. The crosstalk between autophagy and the cGAS-STING pathway impacts a wide variety of cellular processes such as protection against pathogenic infections as well as signaling in neurodegenerative disease, autoinflammatory disease and cancer. Here we provide a comprehensive overview of the mechanisms involved in autophagy and cGAS-STING signaling, with a specific focus on the interactions between the two pathways and their importance for cancer.
1 Autophagy
Autophagy is an adaptive, highly conserved cellular process that acts as a recycling mechanism to maintain cellular homeostasis. It has long been thought of as an unspecific degradation system to regenerate nutrients. However, in recent years autophagy emerged as a selective process that degrades potentially dangerous cellular materials ranging from misfolded proteins and protein aggregates to damaged mitochondria and intracellular bacteria (1–3). Autophagy can also be used to secrete cytosolic proteins (termed secretory autophagy) and can regulate immune responses by secreting damage-associated molecular patterns (DAMPs) or cytokines (4–7). There are three main types of digestive autophagy in mammalian cells: macroautophagy, microautophagy and chaperon-mediated autophagy. They employ different mechanisms to converge at the same final step of cargo delivery to lysosomes followed by its proteolytic degradation. In this review we will only focus on macroautophagy and will, for simplicity, refer to it as “autophagy”. In brief, the process of macroautophagy involves the formation of double membrane vesicles called autophagosomes that form around cellular cargo and travel to lysosomes where they subsequently fuse with them to induce cargo degradation (1, 8, 9). Autophagy is induced by different forms of cellular stress and is thereby connected to the main stress signaling pathways of the cell: nutrient availability [mTOR (10, 11)], energy status [AMPK (12, 13)], hypoxia [HIF (14)] and infection [inflammation (15)]. As a consequence, autophagy is also implicated in major pathologies like neurodegenerative disease, pathogenic infections and cancer, where it can either act in a cytoprotective or cytotoxic fashion and can even accelerate the course of the disease (1, 2, 16–19). Targeting autophagy therefore represents a promising strategy against these pathologies.
1.1 The mechanism of autophagy
Generally, autophagy consists of four distinct phases: (1) initiation, (2) nucleation, (3) elongation, (4) fusion and degradation, which involve many protein complexes required for autophagosome biogenesis, trafficking, fusion with lysosomes, and cargo degradation (1, 20–22).
1.1.1 Initiation
During the initiation of autophagy, the Unc-51-like kinase (ULK) complex, consisting of either ULK1 or ULK2 (a serine/threonine kinase), RB1-inducible coiled-coil protein 1 (FIP200), autophagy related protein 13 (ATG13) and autophagy related protein 101 (ATG101), induces the formation of autophagosomes by acting as a scaffold to recruit autophagic machinery (1, 20, 22). ULK1 directly interacts with the mammalian target of rapamycin complex I (mTORC1) and AMP-activated protein kinase (AMPK) via phosphorylation (23, 24). While autophagy inhibition by mTORC1 and induction by AMPK are the most common routes, there are many more pathways that can activate autophagy. These range from endoplasmic reticulum (ER) stress to hypoxia, microbial infection, DNA damage and mechanical stress (14, 25–32). Some of these will be mentioned in more detail in later parts of this review.
Under high nutrient availability, mTORC1 is activated and negatively regulates the ULK complex by directly binding and inactivating ULK1 and ATG13 by phosphorylation. Upon starvation, however, mTORC1 is deactivated and ULK1 is dephosphorylated, causing it to dissociate from mTORC1 (Figure 1, panel 1). Simultaneously, ULK1 is activated by autophosphorylation, which triggers phosphorylation of its substrates ATG13 and FIP200 (24, 33, 34). ULK1 then forms multimeric complexes that act as a recruitment platform for downstream autophagy proteins such as the phosphoinositol 3-kinase (PI3K) complex I as well as multiple adaptor proteins that are essential for autophagosome biogenesis (35–37).
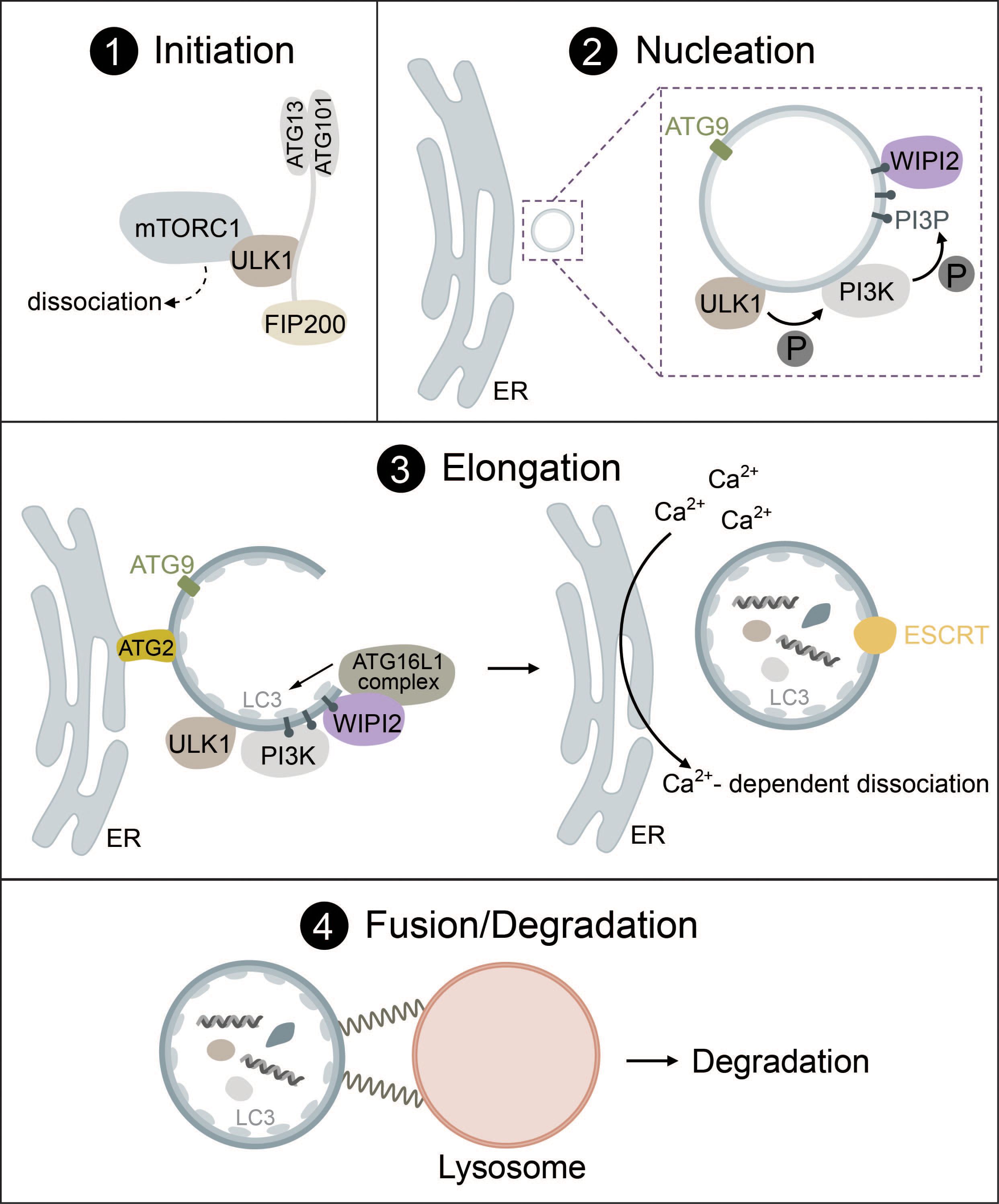
Figure 1 The mechanism of autophagosome biogenesis. 1) Upon starvation, mTORC1 dissociates from ULK1, which is then activated via autophosphorylation and phosphorylates ATG13 and FIP200. 2) ULK1 is recruited to the phagophore assembly site together with ATG9-positive vesicles. ULK1 phosphorylates the PI3K complex, inducing the production of PI3P. PI3P leads to the recruitment of WIPI2. 3) The ATG16L1 complex is recruited to the growing autophagosome via WIPI2 and conjugates ATG8-family proteins such as LC3 to PE. Lipid influx is controlled via ATG2, which acts as a channel funneling lipids from donor compartments. Autophagosome closure is mediated by the ESCRT complex. Membrane contacts are dissolved via VMP1-mediated changes in local Ca2+ concentration. 4) Fully formed autophagosomes travel to the lysosome and undergo fusion mediated by SNARE proteins, tethering factors and RABs. Cargo is then degraded by lysosomal hydrolases.
Another regulator of autophagy is the energy-sensing protein kinase AMPK. Cellular starvation caused by, for example, low levels of glucose results in an increased AMP/ATP ratio, which activates AMPK. Activated AMPK can initiate autophagy indirectly by inactivating mTORC1 or directly by phosphorylating ULK1 (24, 38, 39).
Autophagy regulation by both mTORC1 and AMPK is, however, generally very unspecific and only responds to certain nutrient conditions such as glucose or amino acid starvation (24). Selective autophagy follows a similar initiation procedure that relies on the assembly of multiple ULK complexes but is mostly independent of mTORC1 (40, 41). These ULK1 assemblies form on cargo marked by specific autophagic cargo receptors, such as sequestosome-1 (SQSTM1/p62) and nuclear domain 10 protein 52 (NDP52) (42–44).
1.1.2 Nucleation
The nucleation phase of autophagy has proven to be exceedingly complicated and hard to study, which is why it is still not fully understood. In the following section we will outline the main steps based on the current state of knowledge.
While various organelles have been shown to contribute to autophagosome biogenesis, it has become clear that the ER serves as a platform for this process in most cases (45–47). During nucleation, the ULK1 complex is recruited to the phagophore assembly site (PAS) at ER regions, which are enriched in vacuole membrane protein 1 (VMP1) (48–50). ER transmembrane proteins VAPA/B facilitate the recruitment of the ULK1 complex through direct interaction with FIP200 and ULK1 (51). ATG9, a transmembrane protein found in vesicles that originate from the trans-Golgi, endosomes or even the plasma membrane, is recruited to the PAS together with ULK1 and has been implicated as a seed for the initial establishment of membrane contact sites in yeast (52) and mammals (53, 54) (Figure 1, panel 2).
In the next step, ULK1 initiates the synthesis of phosphatidylinositol 3-phosphate (PI3P) via the PI3K complex I that consists of vacuolar sorting protein 34 (VPS34), 150kDa protein (p150), Bcl-2 interacting protein (Beclin-1), ATG14L and nuclear receptor-binding factor 2 (NRBF2). ULK1 directly phosphorylates Beclin-1, which leads to the activation of VPS34 kinase activity that produces PI3P (55–59). PI3P then regulates the recruitment of several downstream autophagic factors such as WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) or the PI3P-binding protein zinc-finger FYVE domain-containing protein 1 (DFCP1) (49, 60, 61). These PI3P-rich, DFCP1-positive regions are termed the omegasome and are distinct from the autophagosome or pre-autophagosome. Omegasomes act as a platform for the transformation of phagophores into fully-fledged autophagosomes as well as for membrane expansion and budding of fully-formed autophagosomes (49, 62–64). WIPI proteins serve as PI3P effectors and function as an adaptor for the lipidation machinery (ATG16L1 complex) driving membrane expansion in a PI3P-dependent manner (60, 65) (Figure 1, panel 2).
1.1.3 Elongation
Elongation of the phagophore requires the interplay of two complexes that function as ubiquitin-like systems: the ATG16L1 complex and the ATG8 lipidation machinery. The formation of the ATG16L1 complex is initiated by activation of the ubiquitin-like protein ATG12 by ATG7 (E1-like enzyme) in an ATP-dependent manner, resulting in the formation of a covalent bond between ATG12 and ATG7. ATG3 then acts as an E2 facilitating the binding of ATG12 to ATG5. In the last step, ATG16 is attached forming ATG12-ATG5-ATG16, also known as the ATG16L1 complex (66–68). WIPI2b, a WIPI2 isoform, directly interacts with ATG16L1 and recruits the complex to the phagophore, where ATG16L1 acts similar to E3 enzymes to promote the lipidation of ATG8 family proteins (65).
The ATG8 lipidation machinery contains six ATG8 family proteins, which can be divided into two subfamilies: the microtubule-associated protein light chain 3 (LC3A, LC3B, LC3C) and gamma-aminobutyric acid receptor-associated protein (GABARAP, GABARAPL1, GABARAPL2) (69–71). LC3 is one of the most well-known markers of autophagosomes (72, 73). Analogous to the conjugation of ubiquitin to target proteins, lipidation occurs through the conjugation of phosphatidylethanolamine (PE) to ATG8 proteins such as LC3. ATG4, a cysteine protease, cleaves the C-terminal tail of LC3 exposing a glycine residue. This residue then interacts with the E1-like ATG7 and is conjugated to PE by the E2-like ATG3. To fulfill its function, ATG3 requires stimulation by the ATG16L1 complex acting in an E3-like manner. Lipidation transforms LC3-I (nascent and cytosolic) to LC3-II (conjugated to PE and attached to the autophagosome) (22, 65, 68) (Figure 1, panel 3).
ATG4 can also induce deconjugation of ATG8 proteins from PE and must be tightly regulated to prevent autophagosome biogenesis from stalling. This process might be regulated by reactive oxygen species (ROS) generated in mitochondria (74) or by phosphorylation as shown in yeast (75).
Lipids required for ATG8 lipidation and membrane extension are delivered to the phagophore via ATG2 and ATG9, where ATG9 acts as a seed to initiate lipid transfer by ATG2 that acts as a hose funneling lipids from relevant compartments (e.g., the ER) to the phagophore (52–54, 76–78) (Figure 1, panel 3). ATG9 may also promote redistribution of the lipids to the inner layer through lipid scramblases (52, 79). ATG8 lipidation facilitates further membrane expansion by recruiting the rest of the autophagic machinery as detailed below or by linking cargo receptors to phagophores during selective autophagy via LC3 interacting regions (LIRs) (40).
After the phagophore has reached its final size, VMP1 promotes the dissociation of contacts between the ER and the phagophore via local changes in Ca2+ concentrations until the final scission event is then mediated by the endosomal sorting complexes required for transport (ESCRT) machinery (80–83) (Figure 1, panel 3).
1.1.4 Fusion and degradation
For an autophagosome to successfully complete its life cycle, it needs to recruit proteins that facilitate travel from the PAS to the perinuclear region, where autophagosomes fuse with lysosomes. ATG8 family proteins link autophagosomes to kinesins via kinesin adaptors, such as FYVE and coiled-coil domain containing protein 1 (FYCO1) (84). Kinesins, but also dynein motor proteins then mediate travel to the lysosome via microtubules. ATG8 proteins are also responsible for the recruitment of the homotypic fusion and protein sorting (HOPS) complex, a tethering factor required for autophagosome-lysosome fusion (85). The fusion between autophagosomes and lysosomes is driven by the coordinated action of two distinct soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes (STX17-SNAP29-VAMP8 and YKT6-SNAP29-STX7), tethering factors (e.g., the HOPS complex) and Ras-associated binding proteins (RABs) (e.g., Rab7) (81, 86–89) (Figure 1, panel 4). The exact mechanism of fusion between autophagosomes and lysosomes is heavily regulated and has been thoroughly reviewed (81, 86, 90).
Finally, after the fusion of mature autophagosomes with lysosomes, autophagic cargo is degraded by lysosomal hydrolases. This process allows recycling of essential nutrients such as amino acids but also elimination of potentially pathogenic substances (91).
1.2 Selective autophagy
During the process of selective autophagy, the autophagic machinery is targeted to specific cargos via selective autophagy receptors independent of the energy sensing pathways mTORC1 and AMPK. Selective autophagy can be classified based on different types of cargo into mitophagy (damaged mitochondria), aggrephagy (protein aggregates), xenophagy (intracellular pathogens), and many more (3).
Cargo selectivity is conferred by selective autophagy receptors. The majority of selective autophagy receptors are soluble proteins referred to as sequestosome-like cargo receptors (SLRs). Among them the most prominent ones are SQSTM1/p62, NDP52, next to BRCA1 gene 1 protein (NBR1), Tax1-binding protein 1 (TAX1BP1) or optineurin (OPTN). Selective autophagy and its receptors have been reviewed expertly elsewhere (3, 40, 92–95). In general, their primary function is to create a direct link between the cargo and the autophagic machinery allowing their selective degradation in the lysosome.
These receptor proteins are characterized by the presence of LIR motifs for the interaction with ATG8 family proteins such as LC3 on the surface of phagophores, and many have ubiquitin-binding domains capable of recognizing ubiquitinated cargo, thus establishing a bridge between the cargo and the autophagic machinery (3, 40, 92, 93). Selective autophagy receptors such as NDP52, TAX1BP1 and p62 are also capable of directly recruiting the ULK1/2 complex through binding to FIP200 (subunit of the ULK1 complex) to initiate de novo autophagosome biogenesis at the site of the cargo (40, 42, 43). Moreover, OPTN can directly recruit ATG9A or TANK-binding kinase 1 (TBK1), while NIX can recruit WIPI2, resulting in mitophagy induction (96–98).
A special role during the process of selective autophagy is allocated to TBK1. Initially it was thought that the main role of TBK1 during selective autophagy was facilitating the interaction between ATG8 family protein members and the selective cargo via selective autophagy receptors. We know now that the role of TBK1 in regulating selective autophagy is much broader than previously thought with recent studies implicating TBK1 in the direct recruitment of cargo receptors as well as the early autophagic machinery to intracellular bacteria and mitochondria, which will be described in more detail below (40, 99).
2 cGAS-STING signaling
One of the main defense systems against pathogenic bacteria or viruses is the innate immune system. It utilizes pattern recognition receptors (PRRs) to detect and respond to various pathogen- or damage-associated molecular patterns (PAMPs or DAMPs) (100). One such innate immune pathway that was discovered over the last two decades is the cGAS-STING signaling cascade, which recognizes cytosolic double-stranded DNA (dsDNA) and triggers the expression of interferons (IFN) and interferon-stimulated genes (ISGs) (101). ISGs boost the body’s defense against disease by inducing the antiviral state. Cyclic GMP-AMP synthase (cGAS) acts as a PRR for cytosolic dsDNA and generates the second messenger 2´3´-cyclic GMP-AMP (cGAMP). cGAMP binds to stimulator of interferon genes (STING), which further activates interferon regulatory factors 3 and 7 (IRF3, IRF7) that drive type I IFN expression or nuclear factor kappa-light-chain-enhancer of activated B-cell (NF-κB) that induces the expression of proinflammatory genes (101–104).
Chronic or heightened activation of cGAS-STING signaling has been associated with autoimmune diseases such as Aicardi-Goutières syndrome and other pathologies. Additionally, it is also a prominent target for cancer research due to its interplay with many chemotherapeutic treatments, radiation therapy and immune checkpoint inhibitors, which could lead to abscopal responses and improve patient outcome (105, 106).
Recent reports have linked the cGAS-STING pathway with autophagy, revealing multiple regulatory mechanisms and interactions that highlight the potential of exploiting these pathways for cancer therapy.
2.1 The mechanism of cGAS-STING signaling
Under physiological conditions, DNA is exclusively found in the nucleus and in mitochondria. However, certain types of cellular stress can lead to the accumulation of DNA in the cytosol, where it is then recognized by cGAS. Cytosolic DNA may originate from DNA viruses, intracellular bacteria or RNA viruses causing the release of mitochondrial DNA (mtDNA) (101, 107–111). In addition, cGAS can recognize not only foreign DNA but also endogenous DNA that is released into the cytosol from mitochondria or micronuclei resulting from mitotic defects (Figure 2). Extracellular DNA originating from dead cells can also be taken up by cells and stimulate the cGAS-STING pathway (111–116). cGAS is also found in the nucleus but is kept inactive due to nucleosome binding and chromatin tethering (117). However, nuclear cGAS can be activated by human immunodeficiency virus (HIV) in dendritic cells and macrophages (118).
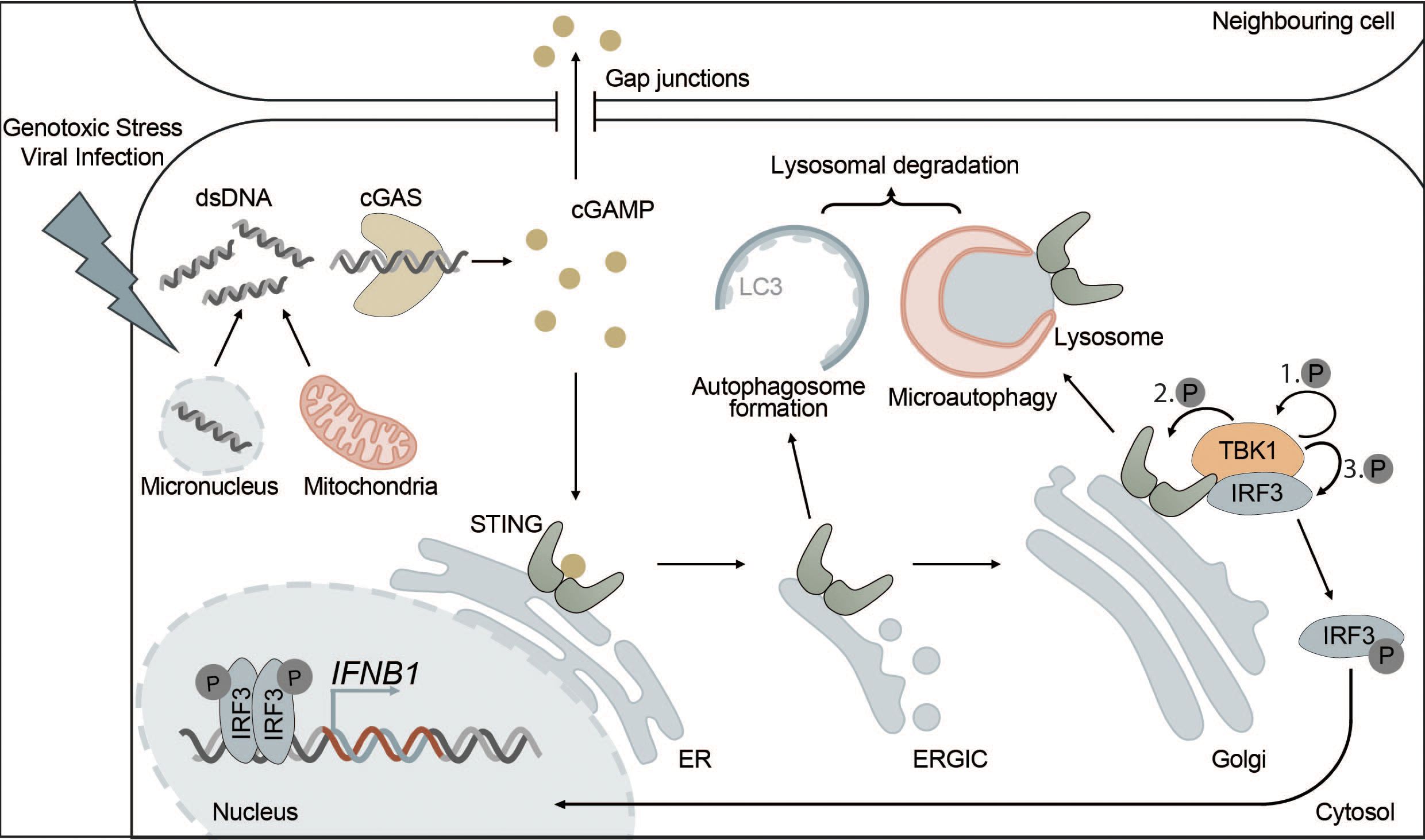
Figure 2 The cGAS-STING pathway is directly connected to autophagy. cGAS-STING signaling is an innate immune pathway, responsible for the production of type I IFNs. Extracellular stressors such as genotoxic stress or viral infection as well as genomic instability can lead to the appearance of dsDNA in the cytosol. cGAS is activated upon binding dsDNA, which leads to the production of the secondary messenger cGAMP. cGAMP then activates STING, located at the ER, inducing its oligomerization and translocation to the Golgi via the ERGIC. Alternatively, cGAMP can also trigger the activation of STING in neighbouring cells via intercellular transport mediated by gap junctions. At the Golgi, STING induces the phosphorylation of TBK1 and ultimately IRF3 leading to its translocation to the nucleus where phosphorylated IRF3 drives the transcription of type I IFNs. Each distinct step of the cGAS-STING pathway is interconnected with autophagy. For example, STING, upon activation, leads to the formation of autophagosomes at the ERGIC in a WIPI2-dependent manner. This acts as a negative feedback loop by degrading dsDNA and other members of the pathway, such as cGAS or IRF3. Additionally, microautophagy is responsible for the final termination of STING signaling in an ESCRT-dependent manner.
Structurally, cGAS can bind double-stranded DNA as well as single-stranded DNA with secondary structures and has even been shown to bind RNA-DNA hybrids (119–122). Even though both small and large DNA fragments can be bound, cGAS activation requires a certain minimal fragment size and has been shown to improve with increasing DNA length (120–124). A very recent publication proposes that mechanical flexibility of DNA, which is increased in the case of DNA damage and higher AT content, promotes the binding and activation of cGAS (125).
Upon DNA binding, cGAS undergoes a conformational change, leading to activation and dimerization with two DNA molecules sandwiched in between (124, 126–128). Those dimers can undergo liquid-liquid phase separation in a DNA length-dependent manner, forming condensates that act as “minireactors” and concentrate cGAS as well as substrates to increase the catalytic activity of cGAS. This could also explain why longer DNA fragments activate cGAS more efficiently (129).
Once activated, cGAS uses GTP and ATP as substrates to produce the secondary messenger cGAMP, which can then be recognized by the ER transmembrane protein STING (130–132). Additionally, cGAMP can diffuse into neighboring cells via gap junctions or be released into the local microenvironment to activate STING signaling in surrounding cells (133–137).
STING resides at the ER membrane as a dimer, where two C-terminal ligand-binding domains form a binding pocket for cGAMP. Binding of cGAMP induces a conformational change that facilitates oligomerization of STING dimers and STING activation (138–140).
Active STING is translocated from the ER to the ER-Golgi intermediate compartment (ERGIC) and Golgi via COPII vesicles, which requires secretion-associated and Ras-related GTPase 1A (SAR1), SEC24 homolog C (SEC24C) and ADP-ribosylation factor (ARF) family members (141).
At the Golgi and in post-Golgi compartments, TBK1 is recruited and then activated by STING (142). Activated TBK1 phosphorylates the C-terminal tail (CTT) of STING at several residues including Ser366, which is part of a highly conserved motif (pLXIS) (143, 144). This motif recruits IRF3, which is then phosphorylated by TBK1, inducing its dimerization. Phosphorylated IRF3 dimers can translocate into the nucleus where they act as transcription factors for type I interferons and other immune-related proteins (143–145) (Figure 2). After activation of IRF3, cGAMP-bound STING oligomers are transported through the trans-Golgi and sorted into RAB7-positive late endosomes that travel to lysosomes for degradation via multivesicular bodies (141).
In addition to its function as an activator of type I IFN synthesis and autophagy, STING can also activate the NF-κB pathway in a TBK1-IKKϵ-dependent or TBK1-independent manner that involves DNA damage response factors ataxia telangiectasia mutated (ATM) and poly(ADP-ribose) polymerase 1 (PARP1) (146–148).
3 Interactions of autophagy with the cGAS-STING pathway
3.1 cGAS
The interplay between autophagy and cGAS occurs through (i) cGAS-Beclin-1 interaction that promotes autophagic degradation of cytosolic dsDNA, (ii) cGAS-LC3 interaction that promotes micronucleophagy, and (iii) degradation of cGAS through p62-mediated selective autophagy (Figure 3).
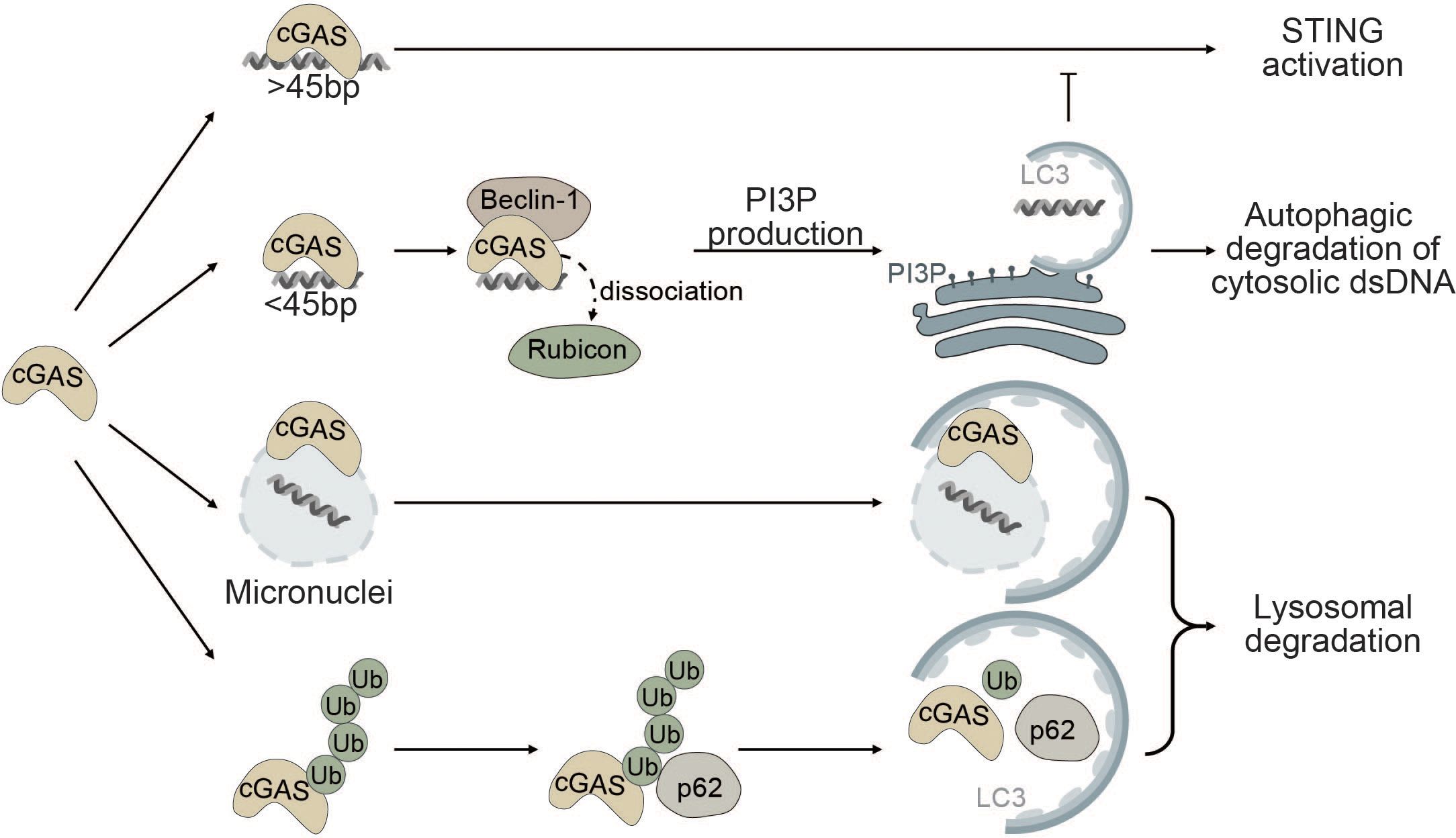
Figure 3 Autophagy as a negative regulator of cGAS. cGAS is a cytosolic sensor for long cytosolic dsDNA fragments. Binding to long dsDNA fragments leads to productive cGAS-STING signaling culminating in the production of type I IFNs. Conversely, binding of small cytosolic dsDNA (<45bp) promotes binding of cGAS to Beclin-1, thereby reducing the interaction of Beclin-1 with Rubicon, a negative regulator of autophagy. This gives rise to the production of PI3P and the formation of autophagosomes allowing degradation of cytosolic dsDNA and negatively impacting cGAS activation. cGAS can also act as a selective autophagy receptor for micronuclei causing their ultimate degradation and thereby prohibiting them from releasing DNA into the cytosol. Finally, K48-linked ubiquitination of cGAS promotes selective p62-dependent autophagic degradation of cGAS, limiting the available amount of activatable cGAS.
Robust activation of the enzymatic function of cGAS is dependent on the length of dsDNA fragments and longer fragments (>45bp) promote robust activation of cGAS (123, 149). Conversely, small cytosolic dsDNA fragments decrease cGAS-STING signaling by competing with long dsDNA fragments for cGAS binding. Binding of small dsDNA fragments to cGAS promotes the binding of cGAS to Beclin-1. By binding to Beclin-1, cGAS outcompetes run domain Beclin-1 interacting and cysteine-rich containing protein (Rubicon) (150). Rubicon negatively regulates maturation and fusion of the autophagosome with the lysosome and endocytic trafficking by suppressing VPS34 kinase activity (151–153). Binding of cGAS to Beclin-1 leads to the release of Rubicon, thereby enabling autophagic degradation of cytosolic dsDNA and resulting in decreased cGAS-STING signaling (Figure 3). Moreover, Beclin-1 also suppresses the NTase activity of cGAS to decrease the production of the secondary messenger cGAMP and type I IFN production (150, 154).
Furthermore, cGAS itself has been proposed as a potential selective autophagy receptor for autophagic degradation of micronuclei also known as micronucleophagy (155). Overexpression of cGAS significantly reduces the number of micronuclei in an autophagy-dependent manner independent of its role in cGAS-STING signaling. cGAS was shown to directly interact with the ATG8 family protein LC3 and colocalize at micronuclei, thereby facilitating the recruitment of the autophagic machinery to enable lysosomal degradation of micronuclei (Figure 3). Moreover, cGAS-driven micronucleophagy also dampens cGAMP production and altogether exerts a negative effect on cGAS-STING signaling and innate immunity (155).
In another report, it was shown that K48-linked ubiquitinated cGAS is directly sequestered into the lysosome for degradation by interacting with the selective autophagy receptor p62 (Figure 3). Upon viral infection, the ISG tripartite motif 14 (TRIM14) directly binds to the C-terminus of cGAS and recruits ubiquitin-specific peptidase 14 (USP14) to prevent K48-linked ubiquitination and degradation of cGAS. Consequently, cGAS evades p62-mediated autophagic degradation and can stimulate type I IFN signaling after viral infection (156).
3.2 STING
STING and autophagy are connected on four levels: (i) STING facilitates non-canonical induction of autophagy, (ii) microautophagy and selective macroautophagy can induce STING degradation, (iii) ULK1 negatively regulates STING-IRF3 signaling by phosphorylating STING, and (iv) non-canonical RAB22-mediated autophagy can promote intercellular spreading of activated STING (Figure 4).
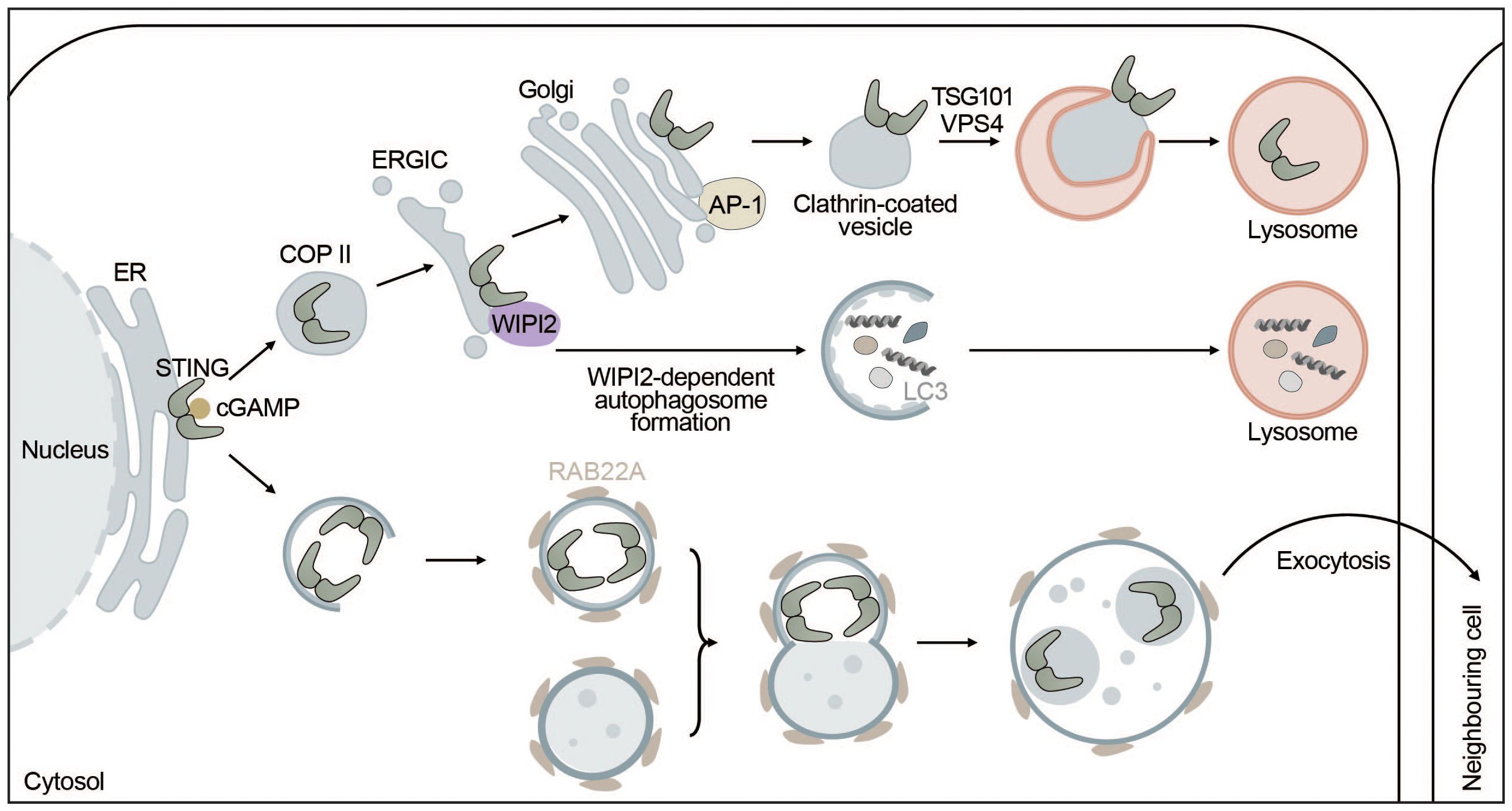
Figure 4 STING signaling is linked to autophagy. STING directly induces the formation of autophagosomes at the ERGIC in a WIPI2-dependent manner utilizing the ERGIC as a membrane source. This process is independent of “canonical” upstream autophagic machinery such as ULK1 or VPS34. However, STING itself is not degraded this way but rather by microautophagy where it is packaged into clathrin-coated vesicles from the Golgi by AP-1. These vesicles are directly taken up by lysosomes thereby terminating STING signaling. Alternatively, upon activation by cGAMP, STING can be transported to neighbouring cells inside non-canonical RAB22A-dependent autophagosomes that fuse with endosomes. The emerging new vesicle can inactivate Rab7, thereby escaping lysosomal degradation and allowing STING pathway activation in surrounding tissues.
Autophagy induction has been proposed to be the primordial function of cGAS-STING signaling before interferon induction evolved. STING can induce autophagy independent of TBK1 and IRF3 via an LR motif at the C-terminus of STING (141, 157). This motif can also be found in other organisms such as Nematostella vectensis that are lacking the C-terminal activation domain required for interferon production. Upon cGAMP binding, STING moves to the ERGIC in a COPII- and ARF GTPase-dependent manner. The ERGIC can then serve as a membrane source for LC3 lipidation, which is dependent on the adaptor protein WIPI2 and ATG5 but does not require ULK1 or VPS34, indicating that this non-canonical STING-dependent induction of autophagy can “skip” early steps of the canonical autophagy mechanism (141). STING directly recruits WIPI2 to STING-positive vesicles and competes with PI3P (canonical bulk autophagy) for binding to WIPI2, adding another layer of complexity to the feedback mechanism between autophagy and cGAS-STING signaling (158) (Figure 4). STING-induced non-canonical autophagy was shown to inhibit herpes simplex virus (HSV-1) (159).
Myristic acid has recently emerged as a regulator of STING degradation via N-myristoylation of ARF1, which is involved in directing the membrane trafficking and autophagic degradation of STING, thereby limiting cGAS-STING-induced interferon response. Upon viral infection the production of myristic acid is reduced due to a disruption in the cellular lipid metabolism, which alleviates autophagy-dependent STING degradation (160).
Another recent report postulated that STING-induced LC3 lipidation happens independent of WIPI2 at perinuclear single membrane vesicles, rather than double-membrane autophagosomes, and requires both the V-ATPase as well as the WD40 domain of ATG16L1 required for interaction with V-ATPase (161, 162). Autophagy-independent LC3 lipidation occurs during influenza A virus infection and mouse models lacking the WD40 domain of ATG16L1 show a reduction in major histocompatibility complex class II (MHC-II)-dependent antigen presentation in dendritic cells (162). The V-ATPase-ATG16L1 axis was also shown to initiate intracellular clearance of bacteria (163). In line with this, STING was recently shown to have proton channel activity that is essential for both non-canonical LC3 lipidation and inflammasome activation. This would allow for mechanistic decoupling of IFN induction via phosphorylated STING and proton channel-dependent LC3 lipidation and inflammasome activation (164). In conclusion, STING can induce non-canonical autophagy in a WIPI2-dependent manner at the ERGIC and autophagy-independent LC3 lipidation at perinuclear single membrane vesicles facilitating defense against viruses and bacteria.
While STING can induce autophagy, autophagy can degrade STING through multiple mechanisms including microautophagy and selective macroautophagy. In microautophagy, cytoplasmic cargo is directly engulfed by the lysosome. STING is transported by clathrin-coated vesicles and recycling endosomes (141) and directly encapsulated in lysosomal associated membrane protein 1 (LAMP-1) positive compartments through the ESCRT components tumor susceptibility gene 101 (TSG101) and vacuolar sorting-associated protein 4 (VPS4) (165). STING packaging from the trans-Golgi into clathrin-coated vesicles is mediated by the cargo adaptor complex 1 (AP-1), which also controls the termination of STING-dependent immune activation by an as of yet unknown mechanism (166) (Figure 4). In addition, basal STING levels seem to be kept under control by the ER-associated degradation (ERAD) system. The ERAD protein complex SEL1L-HRD1 directly interacts with and mediates targeted degradation of basal inactive STING thereby limiting the activatable STING pool and preventing overactivation of the immune response (167). Alternatively, STING may be degraded through selective macroautophagy, whereby TBK1-mediated phosphorylation of p62 at S403 increases its affinity for K63-ubiquitinated STING (168) and ubiquitously expressed transcript (UXT) may facilitate the interaction between p62 and STING (169). Taken together, microautophagy and p62-mediated selective autophagy regulate STING degradation and thereby keep the interferon response in check.
Autophagy has also been shown to have a more direct function in negatively regulating STING-dependent signaling. ULK1 phosphorylates STING at S366, thereby inhibiting IRF3- but not NF-κB-dependent interferon production upon stimulation with dsDNA (170).
A non-canonical autophagy pathway that relies on RAB22A was shown to facilitate the spreading of activated STING to neighboring cells. RAB22A activates phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit alpha (PI3K2A) to produce phosphatidylinositol 4-phosphate (PI4P) that recruits ATG12-ATG5-ATG16L1. The ATG16L1 complex then induces the formation of ER-derived non-canonical autophagosomes containing activated STING. RAB22A-induced autophagosomes fuse with early endosomes, while inhibiting RAB7-mediated fusion with lysosomes. Secretion of the RAB22A-induced extracellular vesicles enables intercellular transfer of activated STING and the spreading of interferon induction to neighboring cells similarly to cGAMP (171) (Figure 4).
In summary, STING can induce non-canonical formation of autophagosomes via WIPI2 using the ERGIC as a source for autophagosome lipidation and promotes autophagy-independent LC3 lipidation. However, none of these pathways seem to mediate the degradation of STING. Instead, STING is degraded by direct microautophagic uptake by lysosomes after being packaged into clathrin-coated vesicles, which is mediated by AP-1. In addition to microautophagic degradation, p62-dependent degradation of STING might also contribute to this phenomenon. Lastly, STING can be taken up by non-canonical RAB22A-induced autophagosomes that do not fuse with lysosomes but are secreted to surrounding cells, spreading active STING. Overall, STING positively regulates and induces different types of autophagy, while autophagy negatively impacts STING signaling (Figure 4).
3.3 TBK1
The kinase TBK1 is known to fulfill a broad range of functions in our cells ranging from IFN signaling, innate immunity and inflammation to autophagy. It is likely that distinct functions of TBK1 in different cellular pathways are due to its cellular localization and association with different adaptor proteins (99, 172). Moreover, TBK1 may simultaneously regulate autophagy and IFN signaling by controlling starvation-induced autophagy and different forms of selective autophagy (e.g., mitophagy or xenophagy). TBK1 may regulate the clearance of dsDNA in the cytosol and thereby modulate cGAS-STING activation and IFN production. TBK1 can regulate autophagy by: (i) activating or inhibiting mTORC1, (ii) inhibiting AMPK, (iii) promoting mitophagy, and (iv) promoting xenophagy (Figure 5).
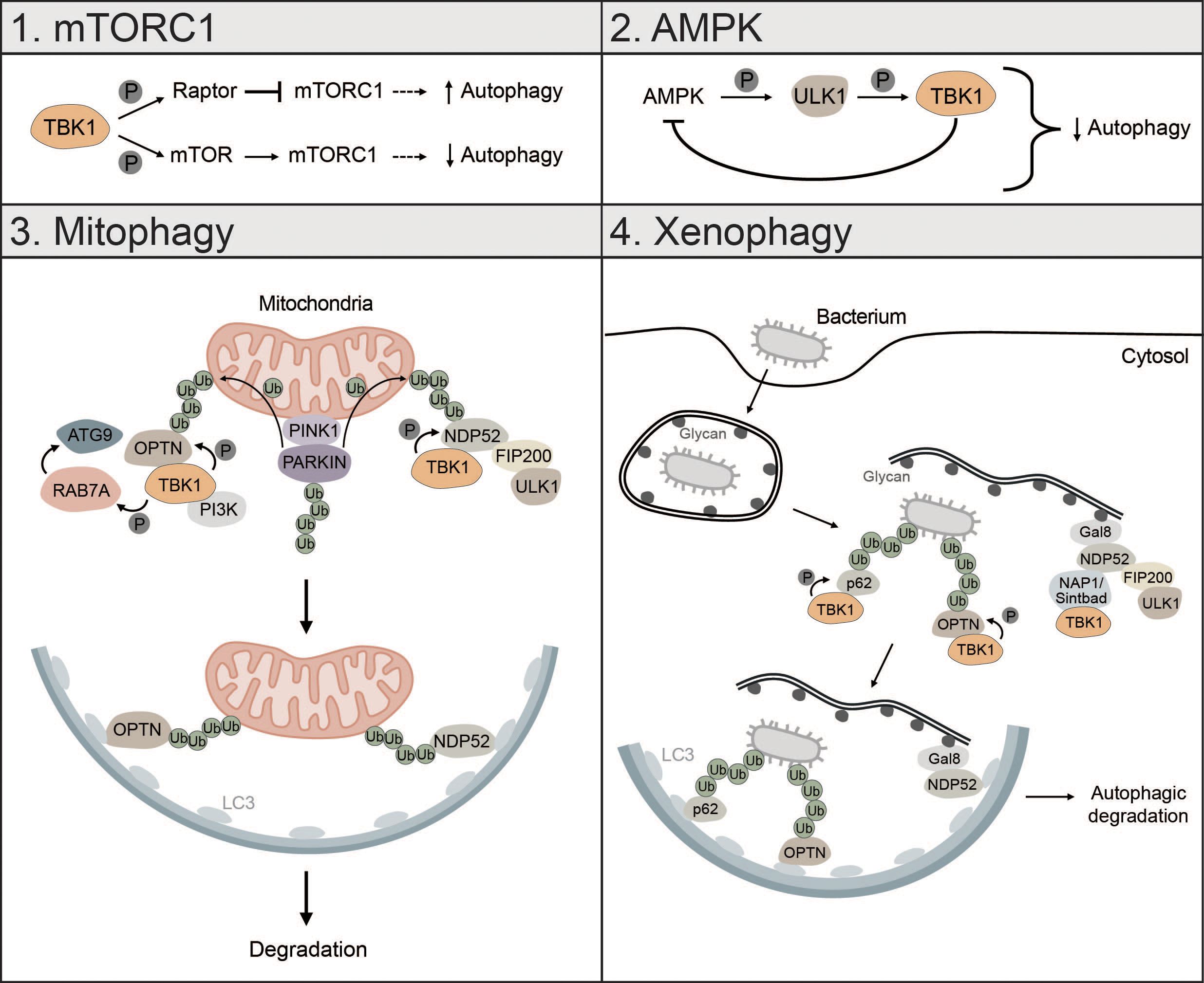
Figure 5 TBK1 regulates different types of autophagy. 1) mTORC1: TBK1 can positively and negatively affect autophagic flux via direct or indirect regulation of mTORC1. Phosphorylation of RAPTOR by TBK1 decreases the activity of mTORC1 and promotes autophagy. In addition, TBK1 can directly phosphorylate mTOR, which increases mTORC1 activity and decreases autophagic flux. 2) AMPK: AMPK can enhance TBK1 activity via ULK1-mediated phosphorylation of TBK1. High levels of active TBK1, however, negatively regulate AMPK, thus creating a negative feedback loop that can negatively affect autophagic flux. 3) Mitophagy: During PINK1-PARKIN-mediated mitophagy, OPTN and NDP52 redundantly activate TBK1, which in turn phosphorylates OPTN and NDP52 to ensure their retention at damaged mitochondria and facilitate the recruitment of ATG8 family proteins. In addition, TBK1-mediated phosphorylation enables spatial proximity to the autophagic machinery by facilitating the binding of NDP52 to the FIP200/ULK1 complex. During OPTN-induced autophagy, TBK1 can take over the functions of the ULK1 complex and directly interact with the PI3K complex to drive mitophagy. Moreover, by phosphorylating RAB7A, TBK1 enables recruitment of ATG9-positive vesicles to damaged mitochondria. 4) Xenophagy: Gal8 recognizes exposed glycans and recruits NDP52. Subsequently, TBK1 is recruited via the two adapter proteins NAP1 and SINTBAD leading to the formation of a NDP52-ULK1-TBK1 super complex, which is essential for phagophore formation. In addition, TBK1 directly phosphorylates the selective autophagy receptors p62 and OPTN, which are crucial for autophagic clearance of ubiquitin-coated bacteria.
During starvation-induced autophagy, TBK1 can positively and negatively regulate autophagic flux by inhibiting or activating mTORC1 respectively (173, 174). TBK1 can phosphorylate and activate regulatory-associated protein of mTOR (RAPTOR), which decreases activity of mTORC1 and promotes autophagy (173). Besides inhibition of mTORC1, TBK1 can directly activate mTORC1 by phosphorylation of mTOR on S2159 to control innate immunity (174) (Figure 5). Whether TBK1 activates or inhibits mTORC1 seems to be cell-context dependent. TBK1 can also initiate autophagy under starvation conditions by phosphorylating and activating the autophagosome-lysosomal fusion protein syntaxin 17 (STX17). Phosphorylated STX17 translocates from the Golgi to the mammalian PAS and controls the formation and assembly of the ULK1 complex, thereby promoting initiation of the autophagic process (175).
The energy sensor AMPK is also regulated by TBK1. Mice with high fat diet have high protein levels of TBK1, which negatively regulates AMPK activity. At the same time, active AMPK indirectly enhances TBK1 activity by promoting ULK1-mediated phosphorylation of TBK1. This creates a negative feedback loop whereby AMPK activates TBK1, which downregulates AMPK, possibly explaining diminished obesity in TBK1 KO mice (176). Since AMPK is known to promote autophagy via direct activation of ULK1 (24), this negative feedback loop between TBK1 and AMPK can negatively affect autophagic flux (Figure 5).
TBK1 is also involved in the regulation of mitophagy, especially in the early steps, where it is instrumental for initiating autophagosome biogenesis (43, 97, 177, 178). Mitophagy is activated by PTEN-induced kinase I (PINK1), which upon mitochondrial depolarization accumulates at the outer mitochondrial membrane (OMM) followed by the recruitment of PARKIN and ubiquitination of mitochondrial proteins (40, 179). During PINK1-PARKIN-induced mitophagy, the selective autophagy receptors OPTN and NDP52 redundantly activate TBK1, which in turn phosphorylates the mitophagy receptors OPTN, NDP52 and p62 to ensure their retention at damaged mitochondria and to facilitate the recruitment of ATG8 family proteins (180, 181). TBK1-mediated phosphorylation of NDP52 facilitates direct interaction with the FIP200/ULK1 complex thus enabling spatial proximity to ubiquitinated cargo to drive autophagosome biogenesis (43). OPTN-induced mitophagy is not necessarily dependent on the binding to the FIP200/ULK1 complex as shown for NDP52. In fact, TBK1 takes over the functions of the ULK1 complex and directly interacts with the PI3K complex I to initiate mitophagy (97). TBK1-mediated phosphorylation of OPTN supports mitophagy by stabilizing the binding of OPTN to ubiquitinated cargo (181). Reciprocally, OPTN-mediated TBK1 relocalization to mitochondria is crucial for proper TBK1 activation (182, 183). TBK1 also phosphorylates RAB7A, which promotes the recruitment of ATG9-positive vesicles to damaged mitochondria enabling efficient mitophagy (184) (Figure 5). Recently, it was shown that mitochondria can be cleared upon PINK1-PARKIN mediated mitophagy in the absence of the mATG8-conjugation machinery and independent of lysosomal degradation via a process called autophagic secretion of mitochondria (ASM). ASM promotes cGAS-STING signaling in recipient cells. The exact mechanism of how mtDNA-containing extracellular vesicles get in contact with cytosolic cGAS of recipient cells remains to be elucidated (185).
Xenophagy is another form of selective autophagy responsible for elimination of pathogens or clearance of invading bacteria which, like mitophagy, relies on TBK1 (3). During early xenophagy, Galectin-8 (GAL8) recognizes exposed glycans on ruptured vacuoles and subsequently recruits the selective autophagy receptor NDP52. Binding of NDP52 to the two adaptor proteins NAP1 and SINTBAD enables recruitment of TBK1 and the formation of an NDP52-ULK1-TBK1 super complex, which is essential for WIPI2-positive phagophore formation and for the recruitment of LC3 (42) (Figure 5). In addition, the ubiquitin ligase RNF213 directly ubiquitylates lipopolysaccharides (LPS) upon vacuole rupture, followed by the recruitment of linear (M1) Ub assembly complex (LUBAC), which is responsible for the assembly of M1 ubiquitin chains. The recruitment of the selective autophagy receptor OPTN and the immune adaptor NF-κB essential modulator (NEMO) is dependent on LUBAC function, while the recruitment of the selective autophagy receptors p62 and NDP52 requires only RNF213 (186). TBK1 directly phosphorylates S403 of p62, which is required for the autophagic function of p62 in driving autophagosome maturation (187). OPTN is also directly phosphorylated by TBK1, thereby enhancing LC3 binding and autophagic clearance of ubiquitin-coated bacteria (188) (Figure 5).
TBK1 is known to be tightly regulated by multiple post-translational modifications to prevent sustained and prolonged immune activation (99, 189–191). The E3 ubiquitin ligase neural precursor cell-expressed developmentally downregulated gene 4 (NEDD4) controls the stability of TBK1 through K27-linked polyubiquitination. The selective autophagy receptor NDP52 binds polyubiquitinated TBK1 and elicits autophagic degradation, resulting in decreased type I IFN signaling (189) (Figure 6). Lysosomal degradation of TBK1 is also promoted by ubiquitin specific peptidase 19 (USP19), which interacts with the TBK1-HSPA8 complex via the chaperone-mediated autophagy (CMA) motif of TBK1 (190) (Figure 6). Another way in which autophagy negatively regulates TBK1 stability is through the autophagy protein ATG4B, which serves as an adaptor for the recruitment of TBK1 to gamma-aminobutyric acid receptor-associated protein (GABARAP) via its LIR motif. Subsequently, TBK1 is sequestered into the lysosome and degraded (Figure 6). Pharmacological inhibition of ATG4B-dependent autophagic degradation of TBK1 was shown to increase the overall antiviral response (192). Taken together, TBK1 stability is regulated by autophagy to prevent sustained type I IFN production and prolonged immune reaction.
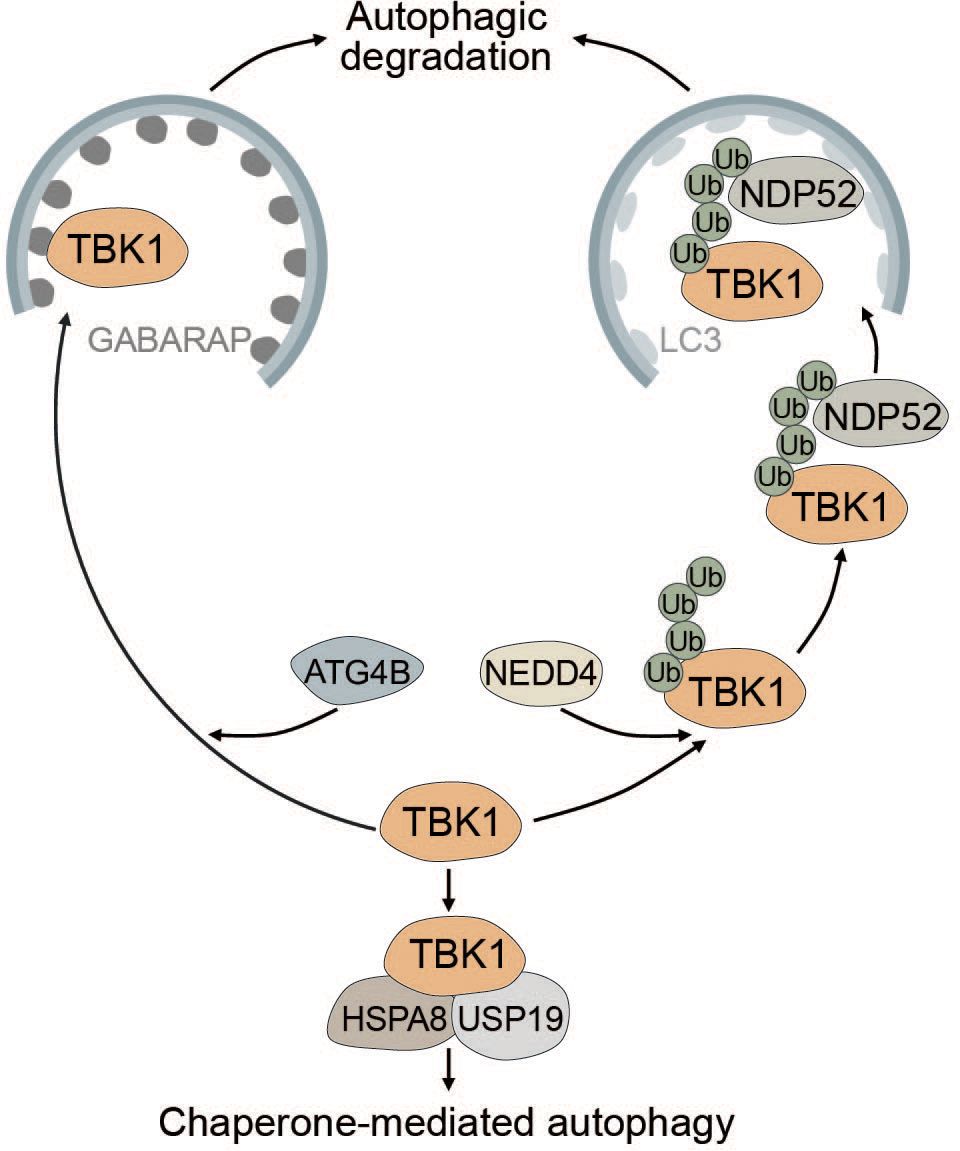
Figure 6 Multiple modes of autophagy negatively regulate TBK1. ATG4B directly associates with TBK1 and acts as an adapter mediating the direct LIR-dependent interaction between the ATG8 family protein GABARAP and TBK1 leading to TBK1 degradation via the autophagosome. The E3 ubiquitin ligase NEDD4 is responsible for the deposition of K27-linked polyubiquitin chains on TBK1, which can be recognized by NDP52 causing selective degradation of TBK1. Conversely, TBK1 can also be degraded by chaperone-mediated autophagy via TBK1 CMA motif that is recognized by the CMA adapter proteins HSPA8 and USP19.
3.4 IRF3
Autophagic degradation of IRF3 is another mechanism of keeping immune activation in check. NDP52 mediates the selective degradation of IRF3 upon viral infection (Figure 7). Under homeostatic conditions, the deubiquitinase PSMD14 cleaves K27-linked polyubiquitin chains at K313 of IRF3, thereby preventing autophagic recognition and degradation (193). Furthermore, OUT deubiquitinase 7B (OTUD7B) promotes autophagic degradation of IRF3 upon viral infection by enhancing the association between IRF3 and p62, thereby negatively regulating the immune response. OTUD7B directly interacts with IRF3 and activates p62 by removing K63 polyubiquitin chains leading to enhanced p62 oligomerization. This allows more efficient degradation of IRF3 in a p62-dependent manner (194) (Figure 7). The histone deacetylase HDAC10 can bind to IRF3 in a deacetylase-independent manner and inhibit its TBK1-dependent phosphorylation at S396. Viral infection induces targeted autophagic degradation of HDAC10 resulting in increased phosphorylation of IRF3 and enhanced interferon response (195).
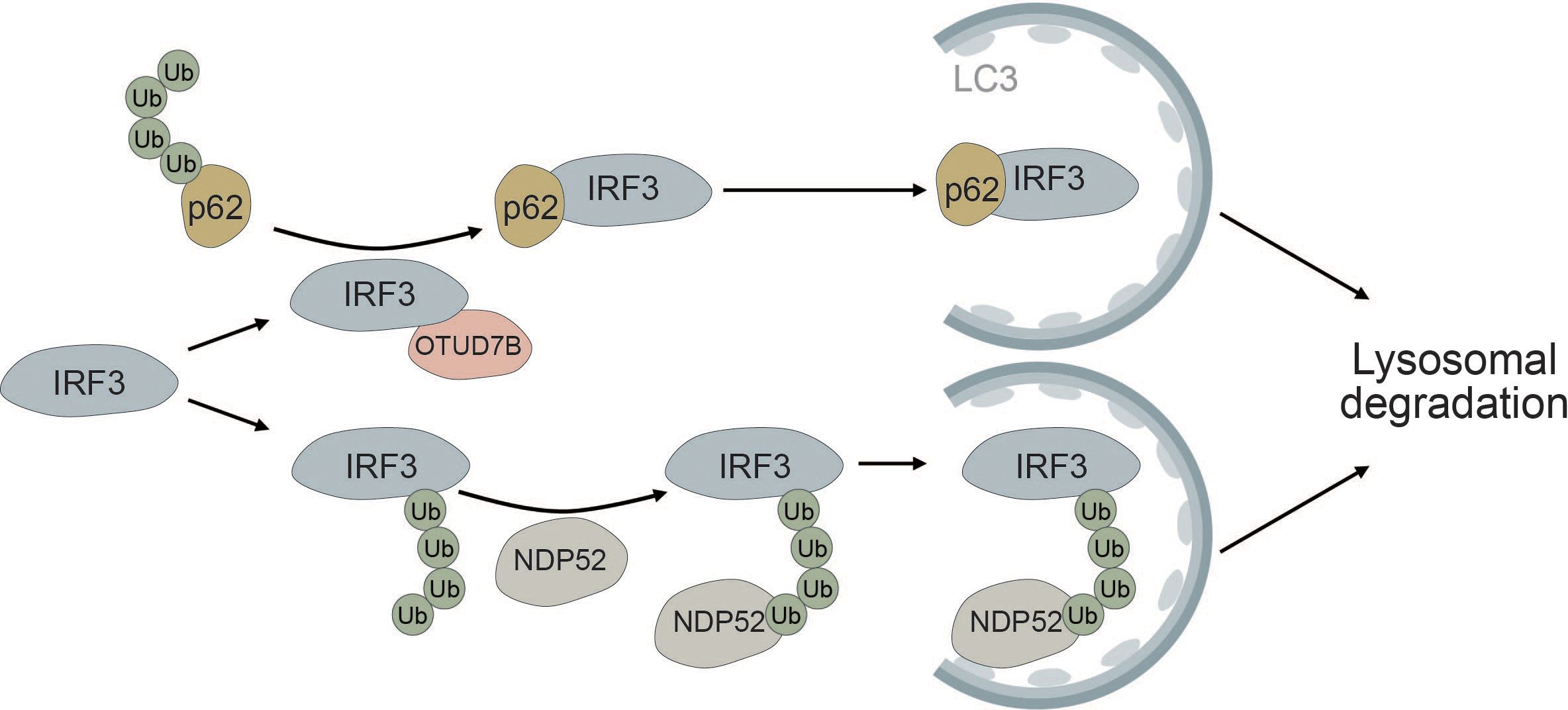
Figure 7 IRF3 is selectively degraded via the autophagic cargo receptors p62 and NDP52. The deubiquitinase OTUD7B removes ubiquitin chains from p62, thereby allowing it to more efficiently oligomerize. This allows for tighter association between p62 and IRF3, causing selective autophagic degradation of IRF3. Autophagic degradation of IRF3 is further promoted by the selective cargo receptor NDP52, which recognizes K27-linked polyubiquitin chains on IRF3, thereby inducing its autophagic degradation.
4 Autophagy and cGAS-STING signaling in cancer
One of the major functions of the immune system is the prevention of malignant transformation, cancer progression as well as metastasis (196, 197). Modern cancer therapies utilize the immune system to target and destroy cancer cells and to provide lasting recovery. One of the main pathways involved in response to radiotherapy and chemotherapies is cGAS-STING signaling. In recent years, many therapies specifically targeting the immune signaling pathway have been tested (198).
Since autophagy is one of the main players in the degradation of damaged organelles and protein aggregates to maintain cellular homeostasis and is also tightly connected to other essential cellular signaling pathways like nutrient sensing or cell death, deregulation of this pathway can have severe consequences in cancer development and progression (19, 199).
Both cGAS-STING signaling and autophagy were shown to form multiple signaling complexes with different cellular effectors that may exert pro- or anti-tumorigenic functions at different stages of the disease (1, 200). We will provide an overview of the involvement of cGAS-STING signaling and autophagy in cancer progression and treatment strategies individually, as well as their interactions in the context of cancer and how this might affect potential therapeutic strategies.
4.1 cGAS-STING and cancer
Genomic instability resulting from defects in DNA damage response, chromosomal instability or defects in chromosome segregation can lead to the formation of micronuclei and generation of aberrant cytosolic dsDNA fragments (113, 201, 202). Micronuclei are small nuclei-like structures containing chromatin encapsulated by the nuclear envelope, which arise from chromosomal instability (203). Micronuclei can rupture and release genomic dsDNA into the cytosol, which is directly engaged by cGAS (113, 204). In addition to genomic dsDNA, dsDNA released from mitochondria can also directly activate the cGAS-STING pathway (205). Another way to activate cGAS is through dying tumor cells, which release DNA into their surroundings. This DNA can be taken up by dendritic cells and activate the cGAS-STING pathway (206).
One of the major concepts in cancer biology is the cancer-immunity cycle, a cyclic self-propagating process that leads to the initiation and amplification of T-cell responses against cancer cells (197). Functional STING signaling affects multiple stages of the cancer-immunity cycle and can enhance tumor-specific T-cell responses (197, 198, 207). Type I IFNs produced by activation of the cGAS-STING pathway instigate the differentiation and maturation of antigen-presenting cells (APCs) and the expression of MHC molecules, thereby facilitating enhanced tumor antigen presentation (208–211). Consequently, cGAS-STING signaling is important for priming, activation and infiltration of tumor-specific cytotoxic T cells (212–214). Active cGAS-STING signaling has also been linked with the production of certain chemokines that recruit T cells to the tumor site such as CXCL10, as well as induction of T-cell stemness (197, 215). Generally, the induction of the type I IFN response targets different immune cells from antigen-presenting dendritic cells, T cells to natural killer (NK) cells (200). In addition to the production of type I IFNs, STING activation can induce the NF-κB pathway and the expression of various proinflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-a (TNF-α) (200). Apart from its intrinsic function in tumor and immune cells, cGAS-STING can signal intercellularly by cGAMP spreading to neighboring cells via gap junctions (134, 216, 217). This process is an active field of study and has recently been reviewed (217). Conventional cancer therapies including radio- and chemotherapy can induce cGAS-STING and anti-tumor immunity alone or in combination with STING agonists and immune checkpoint blockade (198, 218, 219).
In contrast to its anti-tumorigenic function, cGAS-STING signaling has also been implicated in oncogenesis (200, 220–222). The major culprits seem to be chronic type I IFN production and inflammation, which promote cancer cell invasion, metastasis and cancer cell stemness (200, 223–225). Recently, active STING signaling has been linked to the survival of chromosomally unstable cancers in an IL-6 dependent manner involving both NF-κB and signal transducer and activator of transcription 3 (STAT3) activation (226). Additionally, chromosomal instability can promote metastasis via STING-dependent NF-κB activation (227).
Overall, the relationship between pro- and antitumorigenic functions of the cGAS-STING pathway are complex and highly dependent on the genetic makeup of the tumor as well as the tumor microenvironment. Recent therapeutic approaches have mostly focused on the induction of the pathway with some promising results that seem to outweigh the negative effects. However, preventing chronic induction of type I interferons by using short, focused bursts of interferon induction might prove valuable in the future in preventing possible side effects.
4.2 Autophagy and cancer
Similar to cGAS-STING signaling, the function of autophagy in cancer development and therapy is complex. The effect of autophagy on cancer is strongly dependent on the stage of the disease. Initially, autophagy is involved in the control and prevention of malignant transformation. Since autophagy mostly acts in a cytoprotective fashion, this also applies to cells that have already turned to malignancy, providing them with multiple tools to avoid detection and destruction (1, 19, 228).
4.2.1 Tumor suppression
In early stages of tumorigenesis autophagy may act as a tumor suppressor by exhibiting various cytoprotective effects such as degradation of damaged organelles or proteins and maintenance of cellular homeostasis. Indeed, several reports have shown that depletion of certain autophagy-related genes frequently occurs in a variety of cancers to enable proper tumorigenesis (229). The first step in tumorigenesis is the transformation of a pre-malignant cell into a neoplastic precursor, which can be inhibited by autophagy via cellular senescence. Autophagy can remove damaged mitochondria that would otherwise release reactive oxygen species (ROS) or micronuclei that act as a source of genomic instability. Autophagy can also maintain healthy stem cell niches; however, upon completion of malignant transformation, autophagy may promote tumor growth by maintaining cancer stem cells, especially in combination with tumor hypoxia (230).
4.2.2 Tumor promotion
In advanced late-stage tumors, autophagy was shown to promote tumor growth. At this stage, the tumor is exposed to various types of stress such as nutrient deprivation or hypoxia. Due to high proliferation rates of the tumor, the availability of high amounts of nutrients is indispensable. Because of the recycling capacity of autophagy, nutrients can be made available for the tumor to fulfill its high energetic demands. Therefore, the tumor can hijack the autophagic machinery resulting in enhanced stress tolerance and cancer cell survival (229, 231)
RAS-driven cancers are especially dependent on autophagy for tumor progression and malignancy (232). Autophagy inhibition in those tumors affects multiple aspects of cancer metabolism such as lipid metabolism (233), amino acid recycling (234) and nucleotide homeostasis (235), making it a promising therapeutic strategy for RAS-driven cancers.
Furthermore, autophagy has been implicated in chemotherapy resistance. Treatment with chemotherapeutics like 5-fluorouracil or cisplatin was shown to induce protective autophagy in a variety of different cancer types, thereby restricting the efficacy of those drugs and promoting resistance. Combining autophagy inhibitors with chemotherapeutic drugs may provide a promising strategy for overcoming cancer drug resistance (236–239).
A lot of effort has been poured into finding suitable biomarkers associated with autophagy that could guide available treatments options. LC3B has been identified as a potential biomarker and high LC3B rates can predict poor prognosis in hepatocellular carcinoma as well as colon cancer and TNBC, while p62 has also been used as a biomarker for colon cancer (240–242). In TNBCs with high LC3B rates, autophagy inhibition showed promising results as proof of concept (243).
Since many pancreatic cancers seem to rely on high autophagy rates for tumor growth, autophagy inhibitors may prove beneficial for the treatment of pancreatic tumors (244, 245). Autophagy inhibition may sensitize pancreatic cancer to radiotherapy, as radiation was shown to induce autophagy (246). In pancreatic cancer, autophagy has been linked with the degradation of MHC-I, thereby limiting antigen presentation and promoting immune evasion (247).
Currently the only two Food and Drug Agency (FDA)-approved autophagy inhibitor drugs are chloroquine (CQ) and hydroxychloroquine (HCQ). In the context of cancer, multiple clinical trials using CQ or HCQ either in combination or as monotherapy are currently in progress showing partly promising results in terms of anticancer effects and toxicity in early phase studies. Since the majority of clinical trials are either in phase I or II, further evaluation and testing is needed regarding long term toxicity and potential adverse effects associated with those drugs (248–252). Unfortunately, one of the main downsides of using CQ and HCQ is the high degree of off target effects (253). An effort to produce highly specific small molecule inhibitors that might be better suited as targeted treatments in patients is under way (18, 254).
In conclusion, targeting autophagy might be a promising strategy for cancer therapy. However, the lack of clinically suitable autophagy inhibitors and the accompanying effects on the tumor, which are dependent on the genetic makeup and stage of the tumor, must be considered and further improved to ensure favorable outcomes for patients.
4.3 Autophagy and hypoxia
Cancer cells are part of the tumor microenvironment (TME), which includes non-cancerous cells present in the tumor as well as surrounding stroma and various cytokines and chemokines that are secreted. The TME generally tends to promote cancer progression as well as therapy resistance (255, 256). One of the players shaping the TME and its tumorigenic effects is hypoxia. Due to their fast proliferation, tumors can rapidly outgrow their vasculature leading to low oxygen levels. To compensate for their lack of oxygen, cells utilize hypoxic signaling mediated mainly by HIF1 (257). HIF1 is a transcription factor that can bind to hypoxia response elements in promoters, which leads to the transcription of genes involved in cell survival, proliferation and autophagy (258–260). HIF1 is a heterodimer that consists of HIF1α and HIF1β. Both subunits are constitutively expressed, however, during normoxia HIF1α is polyubiquitinated by the VHL complex mediating its degradation by the proteasome. Upon induction of hypoxia, HIF1α is no longer polyubiquitinated and can form a functional heterodimer with HIF1β, allowing it to translocate to the nucleus and induce transcription (14, 261).
BCL2-interacting protein 3 (BINP3) and BCL2-interacting protein 3-like (BINP3L) are induced by HIF1. Under normoxia, B-cell lymphoma 2 (Bcl-2) forms low-affinity complexes with Beclin-1, thereby decreasing the overall rate of autophagy. Under low oxygen, BINP3 and BNIP3L are upregulated, bind to Bcl-2 in place of Beclin-1, freeing it from the complex and leading to higher rates of autophagy (262).
Another way by which hypoxia can lead to the induction of autophagy is via ER stress followed by the unfolded protein response (UPR). Prolonged hypoxia leads to ER stress, which can be sensed by protein kinase R-like endoplasmatic reticulum kinase (PERK) (263). PERK induces the translation of ATF4, which induces the expression of the transcription factor C/EBP homologous protein (CHOP) (264). CHOP regulates the expression of autophagy genes and the switching between autophagy and apoptosis (265).
Since severe hypoxia also goes hand in hand with lack of nutrients, AMPK, which is one of the main regulators of metabolic stress, is often activated under hypoxia and can regulate autophagy directly by phosphorylation of ULK1 as well as indirectly by inactivation of mTOR (266).
4.4 Interaction of cGAS-STING signaling with autophagy in cancer
Given that cGAS-STING signaling is intricately connected with autophagy, the following section will provide insights into how autophagy affects cGAS-STING signaling in the context of cancer.
In breast cancer cell lines, irradiation-induced Bcl-2-associated X (BAX)-dependent permeabilization of the mitochondrial membranes results in the release of mtDNA into the cytosol, thus increasing cGAS-STING dependent type I IFN production. Autophagy negatively regulates this effect by clearing cytosolic dsDNA and thereby promoting radio-resistance. Yamazaki et al. further identified cytosolic DNA originating from mitochondria as the main driver of the abscopal response, a phenomenon whereby distant tumor sites are affected by irradiation of the main malignant lesion. In autophagy-deficient mice, irradiation shows a strong induction of type I interferons, which are responsible for the regression of abscopal tumors. The authors linked this to clinical outcomes in breast cancer patients; autophagy rates inversely correlate with the amount of mtDNA and type I IFN response and patients with higher amounts of mtDNA show improved disease-free survival and overall survival (205).
In esophageal squamous cell carcinoma (ESCC), however, disruption of mitochondrial biogenesis through transcription factor A, mitochondrial (TFAM) downregulation strongly induces autophagy and promotes ESCC cancer growth, in line with the poor overall survival rate of ESCC patients with low expression of TFAM. Mechanistically, TFAM downregulation promotes the release of mtDNA, thereby promoting non-canonical autophagy through the activation of the cGAS-STING pathway. In TFAM-deficient ESCC cells, ISGs are significantly downregulated supporting the idea that STING degradation through autophagy attenuates the expression of type I IFN genes, thereby promoting cancer cell progression (267). Blockage of autophagy using the late-stage autophagy inhibitor bafilomycin A1 was shown to reduce STING degradation leading to enhanced downstream signaling and improved anti-tumor response (268).
Further building on the inverse relationship between autophagy and cGAS-STING signaling, inhibition of autophagy has been shown to increase the cGAS-STING signaling activity by promoting the accumulation of irradiation-induced dsDNA in the cytosol. Moreover, inhibition of autophagy combined with irradiation induces a potent antitumor effect in PD-L1-deficient lung cancer mouse models via cGAS-STING-mediated T-cell activation, suggesting a possible combination therapy of irradiation, anti-PD-L1 immune checkpoint blockade and autophagy inhibition in patients with lung cancer (218).
Furthermore, inhibition of the deubiquitinase TRAF-binding domain-containing protein (TRABID) that regulates cell division leads to accumulation of micronuclei through defects in cell division and autophagy. Moreover, cGAS is no longer autophagically degraded and can recognize micronuclei generated by mitotic defects, thereby inducing cGAS-STING signaling (269).
In addition to the induction of type I IFNs via phosphorylated IRF3, the activated cGAS-STING signaling cascade also induces the NF-κB signaling pathway, leading to the production of inflammatory cytokines like IL-6 or TNF-α, chemokines such as CXCL10 and cell cycle regulators (104, 270–272). cGAS-STING-dependent IL-6 production contributes to the survival of chromosomally unstable cancers in an IL-6-STAT3 dependent manner, at the same time inhibiting STAT1 signaling, preventing apoptosis and promoting cell survival and growth (226). In addition to cell survival and proliferation, STAT3 has been linked to various other protumorigenic functions such as angiogenesis and immune escape and is often categorized as immunosuppressive (273, 274). Inhibition of hypoxia-induced autophagy was shown to disrupt pSTAT3 signaling through accumulation of p62, which selectively induces pSTAT3 degradation by the proteasome system and thereby restores cytotoxic T-cell mediated killing of tumor cells (273). In triple negative breast cancer cell lines, myc overexpression was shown to downregulate STING signaling and its downstream effectors in a STAT3-dependent manner (274).
In colorectal cancer, IL-6 induces Beclin-1 phosphorylation at Y333 by the JAK2 kinase, which increases Beclin-1 affinity for VPS34. This enhances autophagic rates and promotes chemoresistance in a STAT3-independent manner. Patients with Beclin-1 Y333 phosphorylation also display increased chemotherapy resistance and poor prognosis (275). Recently, it was shown that IL-6-induced autophagy can also contribute to cancer cachexia, a condition characterized by severe loss of body weight and increased muscle wasting and is associated with an increased risk of cancer treatment failure and poor overall survival. Mechanistically, cancer cells capable of inducing cancer cachexia are secreting IL-6, which subsequently induces autophagy in differentiated muscle cells via IL-6 signaling and therefore might be relevant for the characteristic muscle and weight loss during cancer cachexia (276).
4.5 Clinical implications
4.5.1 Sting agonists
Given the complex involvement of STING signaling in the cancer immunity cycle and its capability to induce an anti-tumor immune response, efforts have been made to develop new STING agonists. In a pre-clinical setting, numerous STING agonists such as DMXAA or ADU-S100 have shown promising results by inducing a potent anti-tumor immune response either alone or in combination with other conventional cancer therapies including radio- and chemotherapy (277–281). Therefore, it was only a matter of time until STING agonists entered clinical trials, with DMXAA being the first. Contrary to pre-clinical studies where DMXAA was shown to induce a robust anti-tumor immune response, clinical trials did not show any beneficial effects (282, 283). Later studies, however, revealed that DMXAA binds to mouse STING, but not to its human counterpart, explaining the lack of response in clinical trials (284). The new STING agonist ADU-S100 capable of activating human STING showed modest efficacy with low overall response rates (285, 286). Nevertheless, multiple clinical trials testing different cyclic dinucleotides [e.g., BMS-986301 (NCT03956680) or BI 1387446 (NCT04147234)] either alone or in combination with other cancer therapies are currently ongoing and will ultimately determine the future relevance of STING agonists in a clinical setting (www.clinicaltrials.gov). One of the major constraints is the delivery of these drugs, which significantly limits their clinical application. This led to the development of new delivery systems such as extracellular vesicles loaded with cyclic dinucleotides (referred to as exoSTING) or SYNB1891, an engineered E. coli strain capable of producing cyclic dinucleotides when injected intratumorally (287–289).
While first-generation STING agonists showed only modest activities, new potent STING agonists in combination with other cancer therapies and improved delivery methods might improve treatment options for cancer patients.
4.5.2 Autophagy modulators
Chloroquine and hydroxychloroquine remain the most clinically tested autophagy inhibitors to date, due to their previous FDA approval as anti-malaria drugs. While they appear to be relatively safe, the outcomes of clinical studies are inconsistent, largely due to their lack of specificity (290, 291). This instigated the development of specific small molecule inhibitors of autophagy such as SAR405 or repurposing of FDA approved drugs (254, 292–297). The chemoproteomic platform has also been used to screen for novel small molecule inhibitors targeting autophagy and could provide an invaluable tool in the future (298–300). Despite promising preclinical results with new inhibitors, CQ or HCQ are still predominantly used in clinical trials in combination with histone deacetylase inhibitors, antiangiogenic kinase inhibitors or MEK inhibitors in different tumors (301–304).
In recent years, activation of autophagy has also been considered for cancer therapy (296, 305). For example, fasting cycles, especially in combination with chemotherapeutic agents, led to increased long-term survival in mice (306). Starvation, as induced by caloric restriction, downregulates mTOR activity and activates autophagy (307, 308). Diets or drugs mimicking caloric restriction such as hydroxycitrate could effectively delay progression in breast cancer, melanoma and lung cancer and led to the autophagy-dependent depletion of regulatory T cells in mice (307, 309).
Considering the heterogenicity of the tumor and its microenvironment, modulation of autophagy should be assessed on a case-by-case basis depending on specific tumor types and biomarkers to avoid adverse effects (19, 291, 310–312). For example, chronic inhibition of autophagy is associated with an irreversible increase in cancer risk (310). In breast cancer, samples positive for both LC3B and nuclear HMGB1 positively correlate with prolonged metastasis-free survival (313, 314). Conversely, LC3-positive extracellular vesicles (EVs) can promote lung metastasis in breast cancer patients and the number of LC3-positive EVs positively correlates with disease progression (315). In comparison, high autophagy rates correlate with poor survival, lymph node metastasis and hepatic metastasis in gastric cancer (316). Low LACTB expression and high LC3 levels are associated with poor patient responses to neoadjuvant chemotherapy (317). However, in multiple myeloma, patients with strong immunoreactivity to Beclin-1 or LC3 show significantly improved overall survival compared to those with medium or low levels of Beclin-1 or LC3 (318). This heterogenicity of tumors and the resulting convoluted nature of biomarkers makes personalized cancer treatments challenging, especially considering that combinations of multiple markers correlate with different outcomes compared to single markers.
4.5.3 Radiotherapy
Radiotherapy is a commonly used treatment approach for many different types of cancer with approximately 50% of all cancer patients receiving radiotherapy during the course of their illness. It is used to treat both benign and malignant cancers, alone or in combination with other therapeutical approaches such as chemotherapy or surgery (319).
Radiotherapy can activate the immune system through the cGAS-STING signaling pathway and thereby potentiate immunotherapy with immune checkpoint inhibitors (ICIs) (209, 320, 321). Irradiated tumor cells secrete type I interferons, which activate several immune cell types, including dendritic cells and T cells (320). Indeed, in several preclinical studies with immunocompetent mouse models radiotherapy was shown to synergize with ICIs (322–325). Clinical evidence is, however, conflicting. A combinatorial treatment of radiotherapy with ICIs was effective in lung, prostate cancer and melanoma but failed to show superior effects over radiotherapy or immunotherapy alone in glioblastoma and head and neck cancer (326–331).
Conventionally, the main goal of radiotherapy has been to kill cancer cells, notwithstanding the local immunosuppression through various pathways such as vascular disruption and hypoxia, which might result in diminished recruitment of certain immune effector cells, exhaustion of effector T cells, and stimulation of regulatory T cells (321, 332–334). Furthermore, standard radiotherapeutic approaches sometimes target the local draining lymph nodes to reduce the risk of tumor recurrence. Draining lymph nodes, however, are essential for T-cell priming, immunostimulatory effects and successful checkpoint blockade (321, 335). Therefore, efforts must be made to adapt radiotherapy to work in a more immunostimulatory way and thereby enable a clinically superior radiotherapy-immunotherapy combinatorial approach. For example, hypofractionated radiation was shown to be more immunogenic compared to a single high dose in breast cancer and was shown to sensitize mice with breast cancer, melanoma, colon carcinoma or pancreatic cancer to ICI (336–340). Synergistic effects of hypofractionated radiation and ICI were confirmed in patients with lung cancer or melanoma (328, 330). Spacing fractions apart in personalized ultrafractionated stereotactic adaptive radiotherapy (PULSAR) has shown promising results in combination with ICI in colon and lung cancer mouse models, which is currently being evaluated in clinical studies (341). Moreover, particle therapy with protons or carbon ions was shown to induce a stronger immunogenic response compared to conventional photon therapy in mouse melanoma models (342).
5 Conclusion and outlook
Autophagy and cGAS-STING signaling are two highly connected pathways. Both are crucial for defense against pathogen invasion and different diseases such as cancer or neurodegeneration. Here we provided a comprehensive overview of the current state of knowledge on how these two pathways interact. cGAS-STING signaling is tightly regulated by autophagy at almost all levels. In the context of cancer therapy, targeting cGAS-STING signaling is a promising strategy to activate the innate immune system to fight and ultimately kill tumor cells. In most cases, autophagy negatively regulates cGAS-STING signaling and may therefore negatively impact cancer therapy. Preclinical data indicates that boosting cGAS-STING signaling while inhibiting autophagy might be a promising treatment option to improve patients’ outcomes. Enhancing the immunostimulatory effects of radiotherapy, improving delivery methods for STING agonists and developing specific autophagy modulators will allow further investigation of their clinical potential as monotherapy or combinatorial therapy. Moreover, approaches such as proteogenomics (343–346) that provide information about tumor subtypes and tumor microenvironment may reveal new biomarkers to guide personalized therapy options. Finally, further efforts in understanding cGAS-STING signaling and autophagy pathways individually as well as their interactions will be instrumental for their targeting in cancer.
Author contributions
MS: Writing – original draft, Writing – review & editing. PF: Writing – original draft, Writing – review & editing. ME: Writing – review & editing, Visualization. JW: Writing – review & editing. SK-G: Writing – review & editing. DS: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank Sascha Martens and Christian Zierhut for critical feedback on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
2. Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. (2018) 19:579–93. doi: 10.1038/s41580-018-0033-y
3. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. (2022) 24:167–85. doi: 10.1038/s41580-022-00542-2
4. Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V, et al. Secretory autophagy. Curr Opin Cell Biol. (2015) 35:106–16. doi: 10.1016/j.ceb.2015.04.016
5. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. (2011) 30:4701–11. doi: 10.1038/emboj.2011.398
6. Iula L, Keitelman IA, Sabbione F, Fuentes F, Guzman M, Galletti JG, et al. Autophagy mediates interleukin-1β Secretion in human neutrophils. Front Immunol. (2018) 9:269. doi: 10.3389/fimmu.2018.00269
7. Kim YH, Kwak MS, Lee B, Shin JM, Aum S, Park IH, et al. Secretory autophagy machinery and vesicular trafficking are involved in HMGB1 secretion. Autophagy. (2021) 17:2345–62. doi: 10.1080/15548627.2020.1826690
8. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. (2010) 221:3–12. doi: 10.1002/path.2697
9. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. (2014) 20:460–73. doi: 10.1089/ars.2013.5371
10. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. (2009) 122:3589–94. doi: 10.1242/jcs.051011
11. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
12. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. (2011) 13:1016–23. doi: 10.1038/ncb2329
13. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
14. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. (2020) 21:268–83. doi: 10.1038/s41580-020-0227-y
15. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. (2011) 469:323–35. doi: 10.1038/nature09782
16. Saha S, Panigrahi DP, Patil S, Bhutia SK. Autophagy in health and disease: A comprehensive review. Biomed Pharmacother. (2018) 104:485–95. doi: 10.1016/j.biopha.2018.05.007
17. Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O, et al. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci. (2019) 134:116–37. doi: 10.1016/j.ejps.2019.04.011
18. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. (2019) 176(1-2):11–42. doi: 10.1016/j.cell.2018.09.048
19. Yamazaki T, Bravo-San Pedro JM, Galluzzi L, Kroemer G, Pietrocola F. Autophagy in the cancer-immunity dialogue. Adv Drug Deliv Rev. (2021) 169:40–50. doi: 10.1016/j.addr.2020.12.003
20. Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. (2020) 21:439–58. doi: 10.1038/s41580-020-0241-0
21. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. (2013) 14:759–74. doi: 10.1038/nrm3696
22. Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy. (2011) 7:1273–94. doi: 10.4161/auto.7.11.17661
23. Lee JW, Park S, Takahashi Y, Wang H-G. The association of AMPK with ULK1 regulates autophagy. PloS One. (2010) 5:e15394. doi: 10.1371/journal.pone.0015394
24. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
25. Bhardwaj M, Leli NM, Koumenis C, Amaravadi RK. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin Cancer Biol. (2020) 66:116–28. doi: 10.1016/j.semcancer.2019.11.007
26. Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. (2015) 40:141–8. doi: 10.1016/j.tibs.2015.01.002
27. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. (2015) 22:377–88. doi: 10.1038/cdd.2014.150
28. Hu W, Chan H, Lu L, Wong KT, Wong SH, Li MX, et al. Autophagy in intracellular bacterial infection. Semin Cell Dev Biol. (2020) 101:41–50. doi: 10.1016/j.semcdb.2019.07.014
29. Rosenberg G, Riquelme S, Prince A, Avraham R. Immunometabolic crosstalk during bacterial infection. Nat Microbiol. (2022) 7:497–507. doi: 10.1038/s41564-022-01080-5
30. Juretschke T, Beli P. Causes and consequences of DNA damage-induced autophagy. Matrix Biol. (2021) 100–101:39–53. doi: 10.1016/j.matbio.2021.02.004
31. Zhang D, Tang B, Xie X, Xiao Y-F, Yang S-M, Zhang J-W, et al. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol Ther. (2015) 16:1005–13. doi: 10.1080/15384047.2015.1046022
32. King JS, Veltman DM, Insall RH. The induction of autophagy by mechanical stress. Autophagy. (2011) 7:1490–9. doi: 10.4161/auto.7.12.17924
33. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1–atg13–FIP200 complex required for autophagy. Mol Biol Cell. (2009) 20:1981–91. doi: 10.1091/mbc.e08-12-1248
34. Jung CH, Jun CB, Ro S-H, Kim Y-M, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. (2009) 20:1992–2003. doi: 10.1091/mbc.e08-12-1249
35. Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. (2011) 440:283–91. doi: 10.1042/BJ20101894
36. Yamamoto H, Fujioka Y, Suzuki SW, Noshiro D, Suzuki H, Kondo-Kakuta C, et al. The intrinsically disordered protein atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev Cell. (2016) 38:86–99. doi: 10.1016/j.devcel.2016.06.015
37. Kamber RA, Shoemaker CJ, Denic V. Receptor-bound targets of selective autophagy use a scaffold protein to activate the atg1 kinase. Mol Cell. (2015) 59:372–81. doi: 10.1016/j.molcel.2015.06.009
38. Inoki K, Zhu T, Guan K-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. (2003) 115:577–90. doi: 10.1016/S0092-8674(03)00929-2
39. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
40. Adriaenssens E, Ferrari L, Martens S. Orchestration of selective autophagy by cargo receptors. Curr Biol. (2022) 32:R1357–71. doi: 10.1016/j.cub.2022.11.002
41. Goodall EA, Kraus F, Harper JW. Mechanisms underlying ubiquitin-driven selective mitochondrial and bacterial autophagy. Mol Cell. (2022) 82:1501–13. doi: 10.1016/j.molcel.2022.03.012
42. Ravenhill BJ, Boyle KB, Muhlinen von Ellison N CJ, Masson GR, Otten EG, et al. The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol-invading bacteria. Mol Cell. (2019) 74:320–329.e6. doi: 10.1016/j.molcel.2019.01.041
43. Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, et al. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell. (2019) 74:347–362.e6. doi: 10.1016/j.molcel.2019.02.010
44. Turco E, Witt M, Abert C, Bock-Bierbaum T, Su M-Y, Trapannone R, et al. FIP200 Claw Domain Binding to p62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol Cell. (2019) 74:330–46.e11. doi: 10.1016/j.molcel.2019.01.035
45. Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. (2009) 11:1433–7. doi: 10.1038/ncb1991
46. Takahashi S, Saito C, Koyama-Honda I, Mizushima N. Quantitative 3D correlative light and electron microscopy of organelle association during autophagy. Cell Struct Funct. (2022) 47:89–99. doi: 10.1247/csf.22071
47. Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen E-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. (2009) 5:1180–5. doi: 10.4161/auto.5.8.10274
48. Koyama-Honda I, Itakura E, Fujiwara TK, Mizushima N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy. (2013) 9:1491–9. doi: 10.4161/auto.25529
49. Karanasios E, Stapleton E, Manifava M, Kaizuka T, Mizushima N, Walker SA, et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci. (2013) 126:5224–38. doi: 10.1242/jcs.132415
50. Tábara L-C, Escalante R. VMP1 establishes ER-microdomains that regulate membrane contact sites and autophagy. PloS One. (2016) 11:e0166499. doi: 10.1371/journal.pone.0166499
51. Zhao YG, Liu N, Miao G, Chen Y, Zhao H, Zhang H. The ER contact proteins VAPA/B interact with multiple autophagy proteins to modulate autophagosome biogenesis. Curr Biol. (2018) 28:1234–1245.e4. doi: 10.1016/j.cub.2018.03.002
52. Sawa-Makarska J, Baumann V, Coudevylle N, von Bülow S, Nogellova V, Abert C, et al. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science. (2020) 369:eaaz7714. doi: 10.1126/science.aaz7714
53. Olivas TJ, Wu Y, Yu S, Luan L, Choi P, Guinn ED, et al. ATG9 vesicles comprise the seed membrane of mammalian autophagosomes. J Cell Biol. (2023) 222:e202208088. doi: 10.1083/jcb.202208088
54. Broadbent DG, Barnaba C, Perez GI, Schmidt JC. Quantitative analysis of autophagy reveals the role of ATG9 and ATG2 in autophagosome formation. J Cell Biol. (2023) 222:e202210078. doi: 10.1083/jcb.202210078
55. Russell RC, Tian Y, Yuan H, Park HW, Chang Y-Y, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating Vps34 lipid kinase. Nat Cell Biol. (2013) 15:741–50. doi: 10.1038/ncb2757
56. Young LN, Goerdeler F, Hurley JH. Structural pathway for allosteric activation of the autophagic PI 3-kinase complex I. Proc Natl Acad Sci USA. (2019) 116:21508–13. doi: 10.1073/pnas.1911612116
57. Tremel S, Ohashi Y, Morado DR, Bertram J, Perisic O, Brandt LTL, et al. Structural basis for VPS34 kinase activation by Rab1 and Rab5 on membranes. Nat Commun. (2021) 12:1564. doi: 10.1038/s41467-021-21695-2
58. Stjepanovic G, Baskaran S, Lin MG, Hurley JH. Vps34 kinase domain dynamics regulate the autophagic PI 3-kinase complex. Mol Cell. (2017) 67:528–34.e3. doi: 10.1016/j.molcel.2017.07.003
59. Mercer TJ, Ohashi Y, Boeing S, Jefferies HBJ, De Tito S, Flynn H, et al. Phosphoproteomic identification of ULK substrates reveals VPS15-dependent ULK/VPS34 interplay in the regulation of autophagy. EMBO J. (2021) 40:e105985. doi: 10.15252/embj.2020105985
60. Polson HEJ, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. (2010) 6:506–22. doi: 10.4161/auto.6.4.11863
61. Proikas-Cezanne T, Takacs Z, Dönnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. (2015) 128:207–17. doi: 10.1242/jcs.146258
62. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. (2008) 182:685–701. doi: 10.1083/jcb.200803137
63. Walker SA, Ktistakis NT. Autophagosome biogenesis machinery. J Mol Biol. (2020) 432:2449–61. doi: 10.1016/j.jmb.2019.10.027
64. Roberts R, Ktistakis NT. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem. (2013) 55:17–27. doi: 10.1042/bse0550017
65. Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze SA, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting atg12–5-16L1. Mol Cell. (2014) 55:238–52. doi: 10.1016/j.molcel.2014.05.021
66. Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem. (2002) 277:18619–25. doi: 10.1074/jbc.M111889200
67. Fujioka Y, Noda NN, Nakatogawa H, Ohsumi Y, Inagaki F. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem. (2010) 285:1508–15. doi: 10.1074/jbc.M109.053520
68. Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. (2008) 9:859–64. doi: 10.1038/embor.2008.163
69. Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. (2020) 432:80–103. doi: 10.1016/j.jmb.2019.07.016
70. Shpilka T, Weidberg H, Pietrokovski S, Elazar Z. Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol. (2011) 12:226. doi: 10.1186/gb-2011-12-7-226
71. Jatana N, Ascher DB, Pires DEV, Gokhale RS, Thukral L. Human LC3 and GABARAP subfamily members achieve functional specificity via specific structural modulations. Autophagy. (2020) 16:239–55. doi: 10.1080/15548627.2019.1606636
72. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) 1. Autophagy. (2021) 17:1–382. doi: 10.1080/15548627.2020.1797280
73. Slobodkin MR, Elazar Z. The Atg8 family: multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. (2013) 55:51–64. doi: 10.1042/bse0550051
74. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. (2007) 26:1749–60. doi: 10.1038/sj.emboj.7601623
75. Sánchez-Wandelmer J, Kriegenburg F, Rohringer S, Schuschnig M, Gómez-Sánchez R, Zens B, et al. Atg4 proteolytic activity can be inhibited by Atg1 phosphorylation. Nat Commun. (2017) 8:295. doi: 10.1038/s41467-017-00302-3
76. Prinz WA, Hurley JH. A firehose for phospholipids. J Cell Biol. (2020) 219:e202003132. doi: 10.1083/jcb.202003132
77. Li P, Lees JA, Lusk CP, Reinisch KM. Cryo-EM reconstruction of a VPS13 fragment reveals a long groove to channel lipids between membranes. J Cell Biol. (2020) 219:e202001161. doi: 10.1083/jcb.202001161
78. Chang C, Jensen LE, Hurley JH. Autophagosome biogenesis comes out of the black box. Nat Cell Biol. (2021) 23:450–6. doi: 10.1038/s41556-021-00669-y
79. Matoba K, Kotani T, Tsutsumi A, Tsuji T, Mori T, Noshiro D, et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol. (2020) 27:1185–93. doi: 10.1038/s41594-020-00518-w
80. Zhao YG, Chen Y, Miao G, Zhao H, Qu W, Li D, et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol Cell. (2017) 67:974–989.e6. doi: 10.1016/j.molcel.2017.08.005
81. Yu S, Melia TJ. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr Opin Cell Biol. (2017) 47:92–8. doi: 10.1016/j.ceb.2017.03.010
82. Takahashi Y, He H, Tang Z, Hattori T, Liu Y, Young MM, et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun. (2018) 9:2855. doi: 10.1038/s41467-018-05254-w
83. Schmidt O, Teis D. The ESCRT machinery. Curr Biol. (2012) 22:R116–20. doi: 10.1016/j.cub.2012.01.028
84. Pankiv S, Alemu EA, Brech A, Bruun J-A, Lamark T, Overvatn A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. (2010) 188:253–69. doi: 10.1083/jcb.200907015
85. Takáts S, Pircs K, Nagy P, Varga Á, Kárpáti M, Hegedűs K, et al. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell. (2014) 25:1338–54. doi: 10.1091/mbc.e13-08-0449
86. Zhao YG, Codogno P, Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol. (2021) 22:733–50. doi: 10.1038/s41580-021-00392-4
87. Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. (2012) 151:1256–69. doi: 10.1016/j.cell.2012.11.001
88. Langemeyer L, Fröhlich F, Ungermann C. Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol. (2018) 28:957–70. doi: 10.1016/j.tcb.2018.06.007
89. Matsui T, Jiang P, Nakano S, Sakamaki Y, Yamamoto H, Mizushima N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol. (2018) 217:2633–45. doi: 10.1083/jcb.201712058
90. Zhao YG, Zhang H. Autophagosome maturation: An epic journey from the ER to lysosomes. J Cell Biol. (2019) 218:757–70. doi: 10.1083/jcb.201810099
91. Shen H-M, Mizushima N. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci. (2014) 39:61–71. doi: 10.1016/j.tibs.2013.12.001
92. Lamark T, Johansen T. Mechanisms of selective autophagy. Annu Rev Cell Dev Biol. (2021) 37:143–69. doi: 10.1146/annurev-cellbio-120219-035530
93. Zaffagnini G, Martens S. Mechanisms of selective autophagy. J Mol Biol. (2016) 428:1714–24. doi: 10.1016/j.jmb.2016.02.004
94. Sarraf SA, Shah HV, Kanfer G, Pickrell AM, Holtzclaw LA, Ward ME, et al. Loss of TAX1BP1-directed autophagy results in protein aggregate accumulation in the brain. Mol Cell. (2020) 80:779–95.e10. doi: 10.1016/j.molcel.2020.10.041
95. Turco E, Savova A, Gere F, Ferrari L, Romanov J, Schuschnig M, et al. Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat Commun. (2021) 12:5212. doi: 10.1038/s41467-021-25572-w
96. Yamano K, Kikuchi R, Kojima W, Hayashida R, Koyano F, Kawawaki J, et al. Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Biol. (2020) 219:e201912144. doi: 10.1083/jcb.201912144
97. Nguyen TN, Sawa-Makarska J, Khuu G, Lam WK, Adriaenssens E, Fracchiolla D, et al. Unconventional initiation of PINK1/Parkin mitophagy by Optineurin. Mol Cell. (2023) 83:1693–1709.e9. doi: 10.1016/j.molcel.2023.04.021
98. Bunker EN, Le Guerroué F, Wang C, Strub M-P, Werner A, Tjandra N, et al. Nix interacts with WIPI2 to induce mitophagy. EMBO J. (2023) 42:e113491. doi: 10.15252/embj.2023113491
99. Herhaus L. TBK1 (TANK-binding kinase 1)-mediated regulation of autophagy in health and disease. Matrix Biol. (2021) 100–101:84–98. doi: 10.1016/j.matbio.2021.01.004
100. Remick BC, Gaidt MM, Vance RE. Effector-triggered immunity. Annu Rev Immunol. (2023) 41:453–81. doi: 10.1146/annurev-immunol-101721-031732
101. Collins AC, Cai H, Li T, Franco LH, Li X-D, Nair VR, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for mycobacterium tuberculosis. Cell Host Microbe. (2015) 17:820–8. doi: 10.1016/j.chom.2015.05.005
102. Hopfner K-P, Hornung V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol. (2020) 21:501–21. doi: 10.1038/s41580-020-0244-x
103. Zhang X, Bai X, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. (2020) 53:43–53. doi: 10.1016/j.immuni.2020.05.013
104. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
105. Kwon J, Bakhoum SF. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. (2020) 10:26–39. doi: 10.1158/2159-8290.CD-19-0761
106. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS–STING pathway in health and disease. Nat Rev Genet. (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
107. Tan X, Sun L, Chen J, Chen ZJ. Detection of microbial infections through innate immune sensing of nucleic acids. Annu Rev Microbiol. (2018) 72:447–78. doi: 10.1146/annurev-micro-102215-095605
108. Sun B, Sundström KB, Chew JJ, Bist P, Gan ES, Tan HC, et al. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci Rep. (2017) 7:3594. doi: 10.1038/s41598-017-03932-1
109. Franz KM, Neidermyer WJ, Tan Y-J, Whelan SPJ, Kagan JC. STING-dependent translation inhibition restricts RNA virus replication. Proc Natl Acad Sci USA. (2018) 115:E2058–67. doi: 10.1073/pnas.1716937115
110. Li X-D, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. (2013) 341:1390–4. doi: 10.1126/science.1244040
111. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
112. Motwani M, Fitzgerald KA. cGAS micro-manages genotoxic stress. Immunity. (2017) 47:616–7. doi: 10.1016/j.immuni.2017.09.020
113. Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. (2017) 548:461–5. doi: 10.1038/nature23449
114. Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. (2017) 19:1061–70. doi: 10.1038/ncb3586
115. He B, Yu H, Liu S, Wan H, Fu S, Liu S, et al. Mitochondrial cristae architecture protects against mtDNA release and inflammation. Cell Rep. (2022) 41:111774. doi: 10.1016/j.celrep.2022.111774
116. Jm D, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, et al. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol Res. (2018) 6:910–20. doi: 10.1158/2326-6066.CIR-17-0581
117. Bai J, Liu F. Nuclear cGAS: sequestration and beyond. Protein Cell. (2022) 13:90–101. doi: 10.1007/s13238-021-00869-0
118. Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, et al. NONO detects the nuclear HIV capsid to promote cGAS-mediated innate immune activation. Cell. (2018) 175:488–501.e22. doi: 10.1016/j.cell.2018.08.062
119. Crossley MP, Song C, Bocek MJ, Choi J-H, Kousouros JN, Sathirachinda A, et al. R-loop-derived cytoplasmic RNA-DNA hybrids activate an immune response. Nature. (2023) 613:187–94. doi: 10.1038/s41586-022-05545-9
120. Herzner A-M, Hagmann CA, Goldeck M, Wolter S, Kübler K, Wittmann S, et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat Immunol. (2015) 16:1025–33. doi: 10.1038/ni.3267
121. Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. (2014) 33:2937–46. doi: 10.15252/embj.201488726
122. Luecke S, Holleufer A, Christensen MH, Jønsson KL, Boni GA, Sørensen LK, et al. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. (2017) 18:1707–15. doi: 10.15252/embr.201744017
123. Andreeva L, Hiller B, Kostrewa D, Lässig C, de Oliveira Mann CC, Jan Drexler D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. (2017) 549:394–8. doi: 10.1038/nature23890
124. Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. (2013) 39:1019–31. doi: 10.1016/j.immuni.2013.10.019
125. Wang L, Li S, Wang K, Wang N, Liu Q, Sun Z, et al. DNA mechanical flexibility controls DNA potential to activate cGAS-mediated immune surveillance. Nat Commun. (2022) 13:7107. doi: 10.1038/s41467-022-34858-6
126. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. (2013) 498:332–7. doi: 10.1038/nature12305
127. Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. (2014) 6:421–30. doi: 10.1016/j.celrep.2014.01.003
128. Hooy RM, Sohn J. The allosteric activation of cGAS underpins its dynamic signaling landscape. Elife. (2018) 7:e39984. doi: 10.7554/eLife.39984
129. Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. (2018) 361:704–9. doi: 10.1126/science.aat1022
130. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. (2013) 498:380–4. doi: 10.1038/nature12306
131. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
132. Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. (2013) 3:1355–61. doi: 10.1016/j.celrep.2013.05.009
133. SChadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. (2019) 29:1236–1248.e7. doi: 10.1016/j.celrep.2019.09.065
134. Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. (2013) 503:530–4. doi: 10.1038/nature12640
135. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. (2016) 533:493–8. doi: 10.1038/nature18268
136. Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature. (2019) 573:434–8. doi: 10.1038/s41586-019-1553-0
137. Ritchie C, Cordova AF, Hess GT, Bassik MC, Li L. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol Cell. (2019) 75:372–381.e5. doi: 10.1016/j.molcel.2019.05.006
138. Shang G, Zhu D, Li N, Zhang J, Zhu C, Lu D, et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol. (2012) 19:725–7. doi: 10.1038/nsmb.2332
139. Shang G, Zhang C, Chen ZJ, Bai X-C, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature. (2019) 567:389–93. doi: 10.1038/s41586-019-0998-5
140. Ergun Sl, Fernandez D, Weiss TM, Li L. STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell. (2019) 178:290–301. doi: 10.1101/552166
141. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
142. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
143. Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. (2019) 569:718–22. doi: 10.1038/s41586-019-1228-x
144. Zhang C, Shang G, Gui X, Zhang X, Bai X-C, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
145. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
146. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell. (2018) 71:745–760.e5. doi: 10.1016/j.molcel.2018.07.034
147. Fang R, Wang C, Jiang Q, Lv M, Gao P, Yu X, et al. NEMO-IKKβ Are essential for IRF3 and NF-κB activation in the cGAS-STING pathway. J Immunol. (2017) 199:3222–33. doi: 10.4049/jimmunol.1700699
148. Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol. (2014) 88:5328–41. doi: 10.1128/JVI.00037-14
149. Zhou W, Whiteley AT, Mann de CC O, Morehouse BR, Nowak RP, Fischer ES, et al. Structure of the human cGAS–DNA complex reveals enhanced control of immune surveillance. Cell. (2018) 174:300–11.e11. doi: 10.1016/j.cell.2018.06.026
150. Liu Y, Chen X, Zhao Y, Wang X-Y, Luo Y-W, Chen L, et al. Small cytosolic double-stranded DNA represses cyclic GMP-AMP synthase activation and induces autophagy. Cell Rep. (2023) 42:112852. doi: 10.1016/j.celrep.2023.112852
151. Nakamura S, Oba M, Suzuki M, Takahashi A, Yamamuro T, Fujiwara M, et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat Commun. (2019) 10:847. doi: 10.1038/s41467-019-08729-6
152. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. (2009) 11:385–96. doi: 10.1038/ncb1846
153. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. (2009) 11:468–76. doi: 10.1038/ncb1854
154. Liang Q, Seo GJ, Choi YJ, Kwak M-J, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. (2014) 15:228–38. doi: 10.1016/j.chom.2014.01.009
155. Zhao M, Wang F, Wu J, Cheng Y, Cao Y, Wu X, et al. CGAS is a micronucleophagy receptor for the clearance of micronuclei. Autophagy. (2021) 17:3976–91. doi: 10.1080/15548627.2021.1899440
156. Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol Cell. (2016) 64:105–19. doi: 10.1016/j.molcel.2016.08.025
157. Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. (2019) 26:1735–49. doi: 10.1038/s41418-018-0251-z
158. Wan W, Qian C, Wang Q, Li J, Zhang H, Wang L, et al. STING directly recruits WIPI2 for autophagosome formation during STING-induced autophagy. EMBO J. (2023) 42:e112387. doi: 10.15252/embj.2022112387
159. Zheng Z, Zhao M, Shan H, Fang D, Jin Z, Tang J, et al. Noncanonical autophagy is a new strategy to inhibit HSV-1 through STING1 activation. Autophagy. (2023) 0:3096–112. doi: 10.1080/15548627.2023.2237794
160. Jia M, Wang Y, Wang J, Qin D, Wang M, Chai L, et al. Myristic acid as a checkpoint to regulate STING-dependent autophagy and interferon responses by promoting N-myristoylation. Nat Commun. (2023) 14:660. doi: 10.1038/s41467-023-36332-3
161. Fischer TD, Wang C, Padman BS, Lazarou M, Youle RJ. STING induces LC3B lipidation onto single-membrane vesicles via the V-ATPase and ATG16L1-WD40 domain. J Cell Biol. (2020) 219:e202009128. doi: 10.1083/jcb.202009128
162. Fletcher K, Ulferts R, Jacquin E, Veith T, Gammoh N, Arasteh JM, et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. (2018) 37:e97840. doi: 10.15252/embj.201797840
163. Xu Y, Zhou P, Cheng S, Lu Q, Nowak K, Hopp A-K, et al. A bacterial effector reveals the V-ATPase-ATG16L1 axis that initiates xenophagy. Cell. (2019) 178:552–566.e20. doi: 10.1016/j.cell.2019.06.007
164. Liu B, Carlson RJ, Pires IS, Gentili M, Feng E, Hellier Q, et al. Human STING is a proton channel. Science. (2023) 381:508–14. doi: 10.1126/science.adf8974
165. Kuchitsu Y, Mukai K, Uematsu R, Takaada Y, Shinojima A, Shindo R, et al. STING signalling is terminated through ESCRT-dependent microautophagy of vesicles originating from recycling endosomes. Nat Cell Biol. (2023) 25:453–66. doi: 10.1038/s41556-023-01098-9
166. Liu Y, Xu P, Rivara S, Liu C, Ricci J, Ren X, et al. Clathrin-associated AP-1 controls termination of STING signalling. Nature. (2022) 610:761–7. doi: 10.1038/s41586-022-05354-0
167. Ji Y, Luo Y, Wu Y, Sun Y, Zhao L, Xue Z, et al. SEL1L–HRD1 endoplasmic reticulum-associated degradation controls STING-mediated innate immunity by limiting the size of the activable STING pool. Nat Cell Biol. (2023) 25:726–39. doi: 10.1038/s41556-023-01138-4
168. Prabakaran T, Bodda C, Krapp C, Zhang B, Christensen MH, Sun C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. (2018) 37:e97858. doi: 10.15252/embj.201797858
169. Pan M, Yin Y, Hu T, Wang X, Jia T, Sun J, et al. UXT attenuates the CGAS-STING1 signaling by targeting STING1 for autophagic degradation. Autophagy. (2023) 19:440–56. doi: 10.1080/15548627.2022.2076192
170. Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. (2013) 155:688–98. doi: 10.1016/j.cell.2013.09.049
171. Gao Y, Zheng X, Chang B, Lin Y, Huang X, Wang W, et al. Intercellular transfer of activated STING triggered by RAB22A-mediated non-canonical autophagy promotes antitumor immunity. Cell Res. (2022) 32:1086–104. doi: 10.1038/s41422-022-00731-w
172. Helgason E, Phung QT, Dueber EC. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: The emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. (2013) 587:1230–7. doi: 10.1016/j.febslet.2013.01.059
173. Antonia RJ, Castillo J, Herring LE, Serafin DS, Liu P, Graves LM, et al. TBK1 limits mTORC1 by promoting phosphorylation of raptor ser877. Sci Rep. (2019) 9:13470. doi: 10.1038/s41598-019-49707-8
174. Bodur C, Kazyken D, Huang K, Ekim Ustunel B, Siroky KA, Tooley AS, et al. The IKK-related kinase TBK1 activates mTORC1 directly in response to growth factors and innate immune agonists. EMBO J. (2018) 37:19–38. doi: 10.15252/embj.201696164
175. Kumar S, Gu Y, Abudu YP, Bruun J-A, Jain A, Farzam F, et al. Phosphorylation of syntaxin 17 by TBK1 controls autophagy initiation. Dev Cell. (2019) 49:130–144.e6. doi: 10.1016/j.devcel.2019.01.027
176. Zhao P, Wong K, Sun X, Reilly SM, Uhm M, Liao Z, et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. (2018) 172:731–43.e12. doi: 10.1016/j.cell.2018.01.007
177. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. (2015) 524:309–14. doi: 10.1038/nature14893
178. Moore AS, Holzbaur ELF. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci. (2016) 113:E3349–58. doi: 10.1073/pnas.1523810113
179. Eiyama A, Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol. (2015) 33:95–101. doi: 10.1016/j.ceb.2015.01.002
180. Heo J-M, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. (2015) 60:7–20. doi: 10.1016/j.molcel.2015.08.016
181. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci. (2016) 113:4039–44. doi: 10.1073/pnas.1523926113
182. Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. (2015) 24:4429–42. doi: 10.1093/hmg/ddv179
183. Yamano K, Sawada M, Kikuchi R, Nagataki K, Kojima W, Sugihara A, et al. Optineurin provides a mitophagy contact site for TBK1 activation. EMBO J (2024) 2023.02:24. doi: 10.1038/s44318-024-00036-1
184. Heo J-M, Ordureau A, Swarup S, Paulo JA, Shen K, Sabatini DM, et al. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv. (2018) 4:eaav0443. doi: 10.1126/sciadv.aav0443
185. Tan HWS, Lu G, Dong H, Cho Y-L, Natalia A, Wang L, et al. A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat Commun. (2022) 13:3720. doi: 10.1038/s41467-022-31213-7
186. Otten EG, Werner E, Crespillo-Casado A, Boyle KB, Dharamdasani V, Pathe C, et al. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature. (2021) 594:111–6. doi: 10.1038/s41586-021-03566-4
187. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. (2012) 37:223–34. doi: 10.1016/j.immuni.2012.04.015
188. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts salmonella growth. Science. (2011) 333:228–33. doi: 10.1126/science.1205405
189. Xie W, Jin S, Zhang C, Yang S, Wu Y, Zhao Y, et al. Selective autophagy controls the stability of TBK1 via NEDD4 to balance host defense. Cell Death Differ. (2022) 29:40–53. doi: 10.1038/s41418-021-00833-9
190. Zhao X, Di Q, Yu J, Quan J, Xiao Y, Zhu H, et al. USP19 (ubiquitin specific peptidase 19) promotes TBK1 (TANK-binding kinase 1) degradation via chaperone-mediated autophagy. Autophagy. (2022) 18:891–908. doi: 10.1080/15548627.2021.1963155
191. Guo Y, Li R, Tan Z, Shi J, Fu Y, Song Y, et al. E3 ubiquitin ligase ASB8 negatively regulates interferon via regulating TBK1/IKKi homeostasis. Mol Immunol. (2020) 121:195–203. doi: 10.1016/j.molimm.2020.03.011
192. Xie W, Zhang C, Wang Z, Chen H, Gu T, Zhou T, et al. ATG4B antagonizes antiviral immunity by GABARAP-directed autophagic degradation of TBK1. Autophagy. (2023) 0:1–16. doi: 10.1080/15548627.2023.2233846
193. Wu Y, Jin S, Liu Q, Zhang Y, Ma L, Zhao Z, et al. Selective autophagy controls the stability of transcription factor IRF3 to balance type I interferon production and immune suppression. Autophagy. (2021) 17:1379–92. doi: 10.1080/15548627.2020.1761653
194. Xie W, Tian S, Yang J, Cai S, Jin S, Zhou T, et al. OTUD7B deubiquitinates SQSTM1/p62 and promotes IRF3 degradation to regulate antiviral immunity. Autophagy. (2022) 18:2288–302. doi: 10.1080/15548627.2022.2026098
195. Zhou W, Wang J, Wang X, Wang B, Zhao Z, Fu J, et al. Degradation of HDAC10 by autophagy promotes IRF3-mediated antiviral innate immune responses. Sci Signal. (2022) 15:eabo4356. doi: 10.1126/scisignal.abo4356
196. Miller JFAP, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell. (2015) 27:439–49. doi: 10.1016/j.ccell.2015.03.007
197. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
198. Garland KM, Sheehy TL, Wilson JT. Chemical and biomolecular strategies for STING pathway activation in cancer immunotherapy. Chem Rev. (2022) 122:5977–6039. doi: 10.1021/acs.chemrev.1c00750
199. Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. (2021) 54:437–53. doi: 10.1016/j.immuni.2021.01.018
200. Samson N, Ablasser A. The cGAS–STING pathway and cancer. Nat Cancer. (2022) 3:1452–63. doi: 10.1038/s43018-022-00468-w
201. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA, et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. (2017) 548:466–70. doi: 10.1038/nature23470
202. Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. (2015) 42:332–43. doi: 10.1016/j.immuni.2015.01.012
203. Krupina K, Goginashvili A, Cleveland DW. Causes and consequences of micronuclei. Curr Opin Cell Biol. (2021) 70:91–9. doi: 10.1016/j.ceb.2021.01.004
204. Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, et al. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet. (2017) 26:3960–72. doi: 10.1093/hmg/ddx283
205. Yamazaki T, Kirchmair A, Sato A, Buqué A, Rybstein M, Petroni G, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. (2020) 21:1160–71. doi: 10.1038/s41590-020-0751-0
206. Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen EM. STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol. (2014) 193:6124–34. doi: 10.4049/jimmunol.1401869
207. Zhu Y, An X, Zhang X, Qiao Y, Zheng T, Li X. STING: a master regulator in the cancer-immunity cycle. Mol Cancer. (2019) 18:152. doi: 10.1186/s12943-019-1087-y
208. Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci. (2017) 114:1637–42. doi: 10.1073/pnas.1621363114
209. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
210. Wang X, Zhang H, Wang Y, Bramasole L, Guo K, Mourtada F, et al. DNA sensing via the cGAS/STING pathway activates the immunoproteasome and adaptive T-cell immunity. EMBO J. (2023) 42:e110597. doi: 10.15252/embj.2022110597
211. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. (2015) 15:760–70. doi: 10.1038/nri3921
212. Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
213. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci. (2015) 112:15408–13. doi: 10.1073/pnas.1512832112
214. Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Trans Med. (2015) 7:283ra52–283ra52. doi: 10.1126/scitranslmed.aaa4306
215. Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING–mediated DNA sensing maintains CD8+ T cell stemness and promotes antitumor T cell therapy. Sci Trans Med. (2020) 12:eaay9013. doi: 10.1126/scitranslmed.aay9013
216. Wu Q, Leng X, Xu P. Intercellular transmission of cGAS-STING signaling in cancer. Cancer Biol Med. (2023) 20:93–7. doi: 10.20892/j.issn.2095-3941.2022.0750
217. Xie W, Patel DJ. Structure-based mechanisms of 2′3′-cGAMP intercellular transport in the cGAS–STING immune pathway. Trends Immunol. (2023) 44:450–67. doi: 10.1016/j.it.2023.04.006
218. Zhao X, Hu S, Zeng L, Liu X, Song Y, Zhang Y, et al. Irradiation combined with PD-L1–/– and autophagy inhibition enhances the antitumor effect of lung cancer via cGAS-STING-mediated T cell activation. iScience. (2022) 25:104690. doi: 10.1016/j.isci.2022.104690
219. Zhang P, Rashidi A, Zhao J, Silvers C, Wang H, Castro B, et al. STING agonist-loaded, CD47/PD-L1-targeting nanoparticles potentiate antitumor immunity and radiotherapy for glioblastoma. Nat Commun. (2023) 14:1610. doi: 10.1038/s41467-023-37328-9
220. Hoong BYD, Gan YH, Liu H, Chen ES. cGAS-STING pathway in oncogenesis and cancer therapeutics. Oncotarget. (2020) 11:2930–55. doi: 10.18632/oncotarget.v11i30
221. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature. (2018) 563:131–6. doi: 10.1038/s41586-018-0629-6
222. Xia T, Konno H, Ahn J, Barber GN. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. (2016) 14:282–97. doi: 10.1016/j.celrep.2015.12.029
223. Musella M, Guarracino A, Manduca N, Galassi C, Ruggiero E, Potenza A, et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nat Immunol. (2022) 23:1379–92. doi: 10.1038/s41590-022-01290-3
224. Qiu J, Xu B, Ye D, Ren D, Wang S, Benci JL, et al. Cancer cells resistant to immune checkpoint blockade acquire interferon-associated epigenetic memory to sustain T cell dysfunction. Nat Cancer. (2023) 4:43–61. doi: 10.1038/s43018-022-00490-y
225. Li J, Duran MA, Dhanota N, Chatila WK, Bettigole SE, Kwon J, et al. Metastasis and immune evasion from extracellular cGAMP hydrolysis. Cancer Discov. (2021) 11:1212–27. doi: 10.1158/2159-8290.CD-20-0387
226. Hong C, Schubert M, Tijhuis AE, Requesens M, Roorda M, van den Brink A, et al. cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature. (2022) 607:366–73. doi: 10.1038/s41586-022-04847-2
227. Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. (2018) 553:467–72. doi: 10.1038/nature25432
228. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. (2017) 17:528–42. doi: 10.1038/nrc.2017.53
229. Yun C, Lee S. The roles of autophagy in cancer. IJMS. (2018) 19:3466. doi: 10.3390/ijms19113466
230. Rybstein MD, Bravo-San Pedro JM, Kroemer G, Galluzzi L. The autophagic network and cancer. Nat Cell Biol. (2018) 20:243–51. doi: 10.1038/s41556-018-0042-2
231. Anderson CM, Macleod KF. Autophagy and cancer cell metabolism. Int Rev Cell Mol Biol. (2019) 347:145–90. doi: 10.1016/bs.ircmb.2019.06.002
232. Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. (2019) 25:620–7. doi: 10.1038/s41591-019-0367-9
233. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. (2013) 27:1447–61. doi: 10.1101/gad.219642.113
234. Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature. (2015) 524:361–5. doi: 10.1038/nature14587
235. Guo JY, Teng X, Laddha SV, Ma S, Nostrand SCV, Yang Y, et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. (2016) 30:1704–17. doi: 10.1101/gad.283416.116
236. Liang X, Tang J, Liang Y, Jin R, Cai X. Suppression of autophagy by chloroquine sensitizes 5-fluorouracil-mediated cell death in gallbladder carcinoma cells. Cell Biosci. (2014) 4:10. doi: 10.1186/2045-3701-4-10
237. Myung Park J, Huang S, Wu T-T, Foster NR, Sinicrope FA. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol Ther. (2013) 14:100–7. doi: 10.4161/cbt.22954
238. Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. (2014) 289:17163–73. doi: 10.1074/jbc.M114.558288
239. Bao L, Jaramillo MC, Zhang Z, Zheng Y, Yao M, Zhang DD, et al. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol Med Rep. (2015) 11:91–8. doi: 10.3892/mmr.2014.2671
240. Niklaus M, Adams O, Berezowska S, Zlobec I, Graber F, Slotta-Huspenina J, et al. Expression analysis of LC3B and p62 indicates intact activated autophagy is associated with an unfavorable prognosis in colon cancer. Oncotarget. (2017) 8:54604–15. doi: 10.18632/oncotarget.v8i33
241. Lee YJ, Ha YJ, Na Kang Y, Kang KJ, Hwang JS, Chung WJ, et al. The autophagy-related marker LC3 can predict prognosis in human hepatocellular carcinoma. PloS One. (2013) 8:e81540. doi: 10.1371/journal.pone.0081540
242. Zhao H, Yang M, Zhao J, Wang J, Zhang Y, Zhang Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol. (2013) 30:475. doi: 10.1007/s12032-013-0475-1
243. Lefort S, Joffre C, Kieffer Y, Givel A-M, Bourachot B, Zago G, et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy. (2015) 10:2122–42. doi: 10.4161/15548627.2014.981788
244. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. (2011) 25:717–29. doi: 10.1101/gad.2016111
245. Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. (2014) 4:905–13. doi: 10.1158/2159-8290.CD-14-0362
246. Yazal T, Bailleul J, Ruan Y, Sung D, Chu F-I, Palomera D, et al. Radiosensitizing pancreatic cancer via effective autophagy inhibition. Mol Cancer Ther. (2022) 21:79–88. doi: 10.1158/1535-7163.MCT-20-1103
247. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. (2020) 581:100–5. doi: 10.1038/s41586-020-2229-5
248. Mohsen S, Sobash PT, Algwaiz GF, Nasef N, Al-Zeidaneen SA, Karim NA. Autophagy agents in clinical trials for cancer therapy: A brief review. Curr Oncol. (2022) 29:1695–708. doi: 10.3390/curroncol29030141
249. Espina VA, Liotta L, Rassulova S, Gallimore H, Grant-Wisdom T, Menezes G, et al. Abstract CT140: PINC trial: Preventing invasive breast neoplasia with chloroquine. Cancer Res. (2017) 77:CT140. doi: 10.1158/1538-7445.AM2017-CT140
250. Malhotra J, Jabbour S, Orlick M, Riedlinger G, Guo Y, White E, et al. Phase Ib/II study of hydroxychloroquine in combination with chemotherapy in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Treat Res Commun. (2019) 21:100158. doi: 10.1016/j.ctarc.2019.100158
251. Malhotra J, Jabbour S, Orlick M, Riedlinger G, Joshi S, Guo JY, et al. Modulation of autophagy with hydroxychloroquine in patients with advanced non-small cell lung cancer (NSCLC): A phase Ib study. JCO. (2018) 36:e21138–8. doi: 10.1200/JCO.2018.36.15_suppl.e21138
252. Goldberg SB, Supko JG, Neal JW, Muzikansky A, Digumarthy S, Fidias P, et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J Thorac Oncol. (2012) 7:1602–8. doi: 10.1097/JTO.0b013e318262de4a
253. Liang DH, Choi DS, Ensor JE, Kaipparettu BA, Bass BL, Chang JC. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. (2016) 376:249–58. doi: 10.1016/j.canlet.2016.04.002
254. He S, Li Q, Jiang X, Lu X, Feng F, Qu W, et al. Design of small molecule autophagy modulators: A promising druggable strategy. J Med Chem. (2018) 61:4656–87. doi: 10.1021/acs.jmedchem.7b01019
255. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. (2019) 79:4557–66. doi: 10.1158/0008-5472.CAN-18-3962
256. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
257. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci. (1995) 92:5510–4. doi: 10.1073/pnas.92.12.5510
258. Xia X, Kung AL. Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. (2009) 10:R113. doi: 10.1186/gb-2009-10-10-r113
259. Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. (2011) 117:e207–17. doi: 10.1182/blood-2010-10-314427
260. Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts *. J Biol Chem. (2009) 284:16767–75. doi: 10.1074/jbc.M901790200
261. Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. (2000) 19:4298–309. doi: 10.1093/emboj/19.16.4298
262. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol Cell Biol. (2009) 29:2570–81. doi: 10.1128/MCB.00166-09
263. Feldman DE, Chauhan V, Koong AC. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol Cancer Res. (2005) 3:597–605. doi: 10.1158/1541-7786.MCR-05-0221
264. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. (2000) 6:1099–108. doi: 10.1016/S1097-2765(00)00108-8
265. B’chir W, Chaveroux C, Carraro V, Averous J, Maurin A-C, Jousse C, et al. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal. (2014) 26:1385–91. doi: 10.1016/j.cellsig.2014.03.009
266. Hardie DG. AMPK and autophagy get connected. EMBO J. (2011) 30:634–5. doi: 10.1038/emboj.2011.12
267. Li Y, Yang Q, Chen H, Yang X, Han J, Yao X, et al. TFAM downregulation promotes autophagy and ESCC survival through mtDNA stress-mediated STING pathway. Oncogene. (2022) 41:3735–46. doi: 10.1038/s41388-022-02365-z
268. Gonugunta VK, Sakai T, Pokatayev V, Yang K, Wu J, Dobbs N, et al. Trafficking-mediated STING degradation requires sorting to acidified endolysosomes and can be targeted to enhance anti-tumor response. Cell Rep. (2017) 21:3234–42. doi: 10.1016/j.celrep.2017.11.061
269. Chen Y-H, Chen H-H, Wang W-J, Chen H-Y, Huang W-S, Kao C-H, et al. TRABID inhibition activates cGAS/STING-mediated anti-tumor immunity through mitosis and autophagy dysregulation. Nat Commun. (2023) 14:3050. doi: 10.1038/s41467-023-38784-z
270. Zhang L, Wei X, Wang Z, Liu P, Hou Y, Xu Y, et al. NF-κB activation enhances STING signaling by altering microtubule-mediated STING trafficking. Cell Rep. (2023) 42:112185. doi: 10.1016/j.celrep.2023.112185
271. Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D’Silva DB, et al. TBK1 and IKKϵ Act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep. (2020) 31:107492. doi: 10.1016/j.celrep.2020.03.056
272. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Sig Transduct Target Ther. (2017) 2:1–9. doi: 10.1038/sigtrans.2017.23
273. Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. (2011) 71:5976–86. doi: 10.1158/0008-5472.CAN-11-1094
274. Zimmerli D, Brambillasca CS, Talens F, Bhin J, Linstra R, Romanens L, et al. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat Commun. (2022) 13:6579. doi: 10.1038/s41467-022-34000-6
275. Hu F, Song D, Yan Y, Huang C, Shen C, Lan J, et al. IL-6 regulates autophagy and chemotherapy resistance by promoting BECN1 phosphorylation. Nat Commun. (2021) 12:3651. doi: 10.1038/s41467-021-23923-1
276. Pettersen K, Andersen S, Degen S, Tadini V, Grosjean J, Hatakeyama S, et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci Rep. (2017) 7:2046. doi: 10.1038/s41598-017-02088-2
277. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. (2015) 11:1018–30. doi: 10.1016/j.celrep.2015.04.031
278. Vonderhaar EP, Barnekow NS, McAllister D, McOlash L, Eid MA, Riese MJ, et al. STING activated tumor-intrinsic type I interferon signaling promotes CXCR3 dependent antitumor immunity in pancreatic cancer. Cell Mol Gastroenterol Hepatol. (2021) 12:41–58. doi: 10.1016/j.jcmgh.2021.01.018
279. Shi F, Su J, Wang J, Liu Z, Wang T. Activation of STING inhibits cervical cancer tumor growth through enhancing the anti-tumor immune response. Mol Cell Biochem. (2021) 476:1015–24. doi: 10.1007/s11010-020-03967-5
280. Zaidi AH, Kelly RJ, Gorbunova A, Omstead AN, Salvitti MS, Zheng P, et al. Intratumoral immunotherapy with STING agonist, ADU-S100, induces CD8+ T-cell mediated anti-tumor immunity in an esophageal adenocarcinoma model. Oncotarget. (2021) 12:292–303. doi: 10.18632/oncotarget.v12i4
281. Ghaffari A, Peterson N, Khalaj K, Vitkin N, Robinson A, Francis J-A, et al. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br J Cancer. (2018) 119:440–9. doi: 10.1038/s41416-018-0188-5
282. Lara PN, Douillard J-Y, Nakagawa K, von Pawel J, McKeage MJ, Albert I, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non–small-cell lung cancer. JCO. (2011) 29:2965–71. doi: 10.1200/JCO.2011.35.0660
283. Carboplatin and Paclitaxel and ASA404 in Non-Small Cell Lung Cancer. Clinical Trials Registry - ICH GCP. Available at: https://clinicaltrials.gov/study/NCT00662597?cond=Carboplatin%20and%20Paclitaxel%20and%20ASA404%20in%20Non-Small%20Cell%20Lung%20Cancer&rank=1.
284. Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, et al. Mouse, but not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. J Immunol. (2013) 190:5216–25. doi: 10.4049/jimmunol.1300097
285. Meric-Bernstam F, Sweis RF, Kasper S, Hamid O, Bhatia S, Dummer R, et al. Combination of the STING agonist MIW815 (ADU-S100) and PD-1 inhibitor spartalizumab in advanced/metastatic solid tumors or lymphomas: an open-label, multicenter, phase ib study. Clin Cancer Res. (2023) 29:110–21. doi: 10.1158/1078-0432.CCR-22-2235
286. Meric-Bernstam F, Sweis RF, Hodi FS, Messersmith WA, Andtbacka RHI, Ingham M, et al. Phase I dose-escalation trial of MIW815 (ADU-S100), an intratumoral STING agonist, in patients with advanced/metastatic solid tumors or lymphomas. Clin Cancer Res. (2022) 28:677–88. doi: 10.1158/1078-0432.CCR-21-1963
287. Leventhal DS, Sokolovska A, Li N, Plescia C, Kolodziej SA, Gallant CW, et al. Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat Commun. (2020) 11:2739. doi: 10.1038/s41467-020-16602-0
288. Jang SC, Economides KD, Moniz RJ, Sia CL, Lewis N, McCoy C, et al. ExoSTING, an extracellular vesicle loaded with STING agonists, promotes tumor immune surveillance. Commun Biol. (2021) 4:1–17. doi: 10.1038/s42003-021-02004-5
289. Luke JJ, Piha-Paul SA, Medina T, Verschraegen CF, Varterasian M, Brennan AM, et al. Phase I study of SYNB1891, an engineered E. coli nissle strain expressing STING agonist, with and without atezolizumab in advanced malignancies. Clin Cancer Res. (2023) 29(13):2435–44. doi: 10.1158/1078-0432.CCR-23-0118
290. Shi T-T, Yu X-X, Yan L-J, Xiao H-T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol. (2017) 79:287–94. doi: 10.1007/s00280-016-3197-1
291. Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell. (2014) 26:190–206. doi: 10.1016/j.ccr.2014.06.025
292. Lin Y, Shi Q, Yang G, Shi F, Zhou Y, Wang T, et al. A small-molecule drug inhibits autophagy gene expression through the central regulator TFEB. Proc Natl Acad Sci. (2023) 120:e2213670120. doi: 10.1073/pnas.2213670120
293. Noman MZ, Parpal S, Van Moer K, Xiao M, Yu Y, Viklund J, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv. (2020) 6:eaax7881. doi: 10.1126/sciadv.aax7881
294. Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol. (2014) 10:1013–9. doi: 10.1038/nchembio.1681
295. Vakifahmetoglu-Norberg H, Xia H, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest. (2015) 125:5–13. doi: 10.1172/JCI73937
296. Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. (2017) 16:487–511. doi: 10.1038/nrd.2017.22
297. Filippakis H, Belaid A, Siroky B, Wu C, Alesi N, Hougard T, et al. Vps34-mediated macropinocytosis in Tuberous Sclerosis Complex 2-deficient cells supports tumorigenesis. Sci Rep. (2018) 8:14161. doi: 10.1038/s41598-018-32256-x
298. Salvi A, Young AN, Huntsman AC, Pergande MR, Korkmaz MA, Rathnayake RA, et al. PHY34 inhibits autophagy through V-ATPase V0A2 subunit inhibition and CAS/CSE1L nuclear cargo trafficking in high grade serous ovarian cancer. Cell Death Dis. (2022) 13:45. doi: 10.1038/s41419-021-04495-w
299. Yik-Sham Chung C, Shin HR, Berdan CA, Ford B, Ward CC, Olzmann JA, et al. Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat Chem Biol. (2019) 15:776–85. doi: 10.1038/s41589-019-0308-4
300. Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat Rev Drug Discovery. (2022) 21:881–98. doi: 10.1038/s41573-022-00542-z
301. Karasic TB, Brown TJ, Schneider C, Teitelbaum UR, Reiss KA, Mitchell TC, et al. Phase I trial of regorafenib, hydroxychloroquine, and entinostat in metastatic colorectal cancer. Oncologist. (2022) 27:716–e689. doi: 10.1093/oncolo/oyac078
302. Arora SP, Tenner L, Sarantopoulos J, Morris J, Liu Q, Mendez JA, et al. Modulation of autophagy: a Phase II study of vorinostat plus hydroxychloroquine versus regorafenib in chemotherapy-refractory metastatic colorectal cancer (mCRC). Br J Cancer. (2022) 127:1153–61. doi: 10.1038/s41416-022-01892-6
303. Aggarwal C, Maity AP, Bauml JM, Long Q, Aleman T, Ciunci C, et al. A phase II open-label trial of binimetinib and hydroxychloroquine in patients with advanced KRAS-mutant non-small cell lung cancer. Oncologist. (2023) 28:644–e564. doi: 10.1093/oncolo/oyad106
304. Mehnert JM, Mitchell TC, Huang AC, Aleman TS, Kim BJ, Schuchter LM, et al. BAMM (BRAF autophagy and MEK inhibition in melanoma): A phase I/II trial of dabrafenib, trametinib, and hydroxychloroquine in advanced BRAFV600-mutant melanoma. Clin Cancer Res. (2022) 28:1098–106. doi: 10.1158/1078-0432.CCR-21-3382
305. Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. (2017) 14:247–58. doi: 10.1038/nrclinonc.2016.183
306. Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Trans Med. (2012) 4:124ra27–124ra27. doi: 10.1126/scitranslmed.3003293
307. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. (2016) 30:147–60. doi: 10.1016/j.ccell.2016.05.016
308. Saleh A, Simone B, Palazzo J, Savage JE, Sano Y, Dan T, et al. Caloric restriction augments radiation efficacy in breast cancer. Cell Cycle. (2013) 12:1955–63. doi: 10.4161/cc.25016
309. Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng C-W, et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell. (2016) 30:136–46. doi: 10.1016/j.ccell.2016.06.005
310. Cassidy LD, Young ARJ, Young CNJ, Soilleux EJ, Fielder E, Weigand BM, et al. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat Commun. (2020) 11:307. doi: 10.1038/s41467-019-14187-x
311. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. (2011) 334:1573–7. doi: 10.1126/science.1208347
312. Ko A, Kanehisa A, Martins I, Senovilla L, Chargari C, Dugue D, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. (2014) 21:92–9. doi: 10.1038/cdd.2013.124
313. Ladoire S, Penault-Llorca F, Senovilla L, Dalban C, Enot D, Locher C, et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy. (2015) 11:1878–90. doi: 10.1080/15548627.2015.1082022
314. Ladoire S, Enot D, Senovilla L, Ghiringhelli F, Poirier-Colame V, Chaba K, et al. The presence of LC3B puncta and HMGB1 expression in Malignant cells correlate with the immune infiltrate in breast cancer. Autophagy. (2016) 12:864–75. doi: 10.1080/15548627.2016.1154244
315. Sun X, Wang X, Yan C, Zheng S, Gao R, Huang F, et al. Tumor cell-released LC3-positive EVs promote lung metastasis of breast cancer through enhancing premetastatic niche formation. Cancer Sci. (2022) 113:3405–16. doi: 10.1111/cas.15507
316. Masuda GO, Yashiro M, Kitayama K, Miki Y, Kasashima H, Kinoshita H, et al. Clinicopathological correlations of autophagy-related proteins LC3, beclin 1 and p62 in gastric cancer. Anticancer Res. (2016) 36:129–36.
317. Yang F, Yan Z, Nie W, Liu Z, Cheng X, Wang W, et al. LACTB and LC3 could serve as potential biomarkers of gastric cancer to neoadjuvant chemotherapy with oxaliplatin plus S-1. Oncol Lett. (2021) 21:470. doi: 10.3892/ol
318. Jung G, Roh J, Lee H, Gil M, Yoon DH, Suh C, et al. Autophagic markers BECLIN 1 and LC3 are associated with prognosis of multiple myeloma. Acta Haematol. (2015) 134:17–24. doi: 10.1159/000368848
319. Baskar R, Lee KA, Yeo R, Yeoh K-W. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. (2012) 9:193–9. doi: 10.7150/ijms.3635
320. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The efficacy of radiotherapy relies upon induction of type I interferon–dependent innate and adaptive immunity. Cancer Res. (2011) 71:2488–96. doi: 10.1158/0008-5472.CAN-10-2820
321. Galluzzi L, Aryankalayil MJ, Coleman CN, Formenti SC. Emerging evidence for adapting radiotherapy to immunotherapy. Nat Rev Clin Oncol. (2023) 20:543–57. doi: 10.1038/s41571-023-00782-x
322. Rodríguez-Ruiz ME, Rodríguez I, Mayorga L, Labiano T, Barbes B, Etxeberria I, et al. TGFβ Blockade enhances radiotherapy abscopal efficacy effects in combination with anti-PD1 and anti-CD137 immunostimulatory monoclonal antibodies. Mol Cancer Ther. (2019) 18:621–31. doi: 10.1158/1535-7163.MCT-18-0558
323. Rodriguez-Ruiz ME, Rodriguez I, Garasa S, Barbes B, Solorzano JL, Perez-Gracia JL, et al. Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mAbs and are dependent on CD8 T cells and crosspriming. Cancer Res. (2016) 76:5994–6005. doi: 10.1158/0008-5472.CAN-16-0549
324. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. (2005) 11:728–34. doi: 10.1158/1078-0432.728.11.2
325. Laurent P-A, Morel D, Meziani L, Depil S, Deutsch E. Radiotherapy as a means to increase the efficacy of T-cell therapy in solid tumors. OncoImmunology. (2023) 12:2158013. doi: 10.1080/2162402X.2022.2158013
326. Lee NY, Ferris RL, Psyrri A, Haddad RI, Tahara M, Bourhis J, et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. (2021) 22:450–62. doi: 10.1016/S1470-2045(20)30737-3
327. Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. (2023) 25:123–34. doi: 10.1093/neuonc/noac099
328. Formenti SC, Rudqvist N-P, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. (2018) 24:1845–51. doi: 10.1038/s41591-018-0232-2
329. Zhou Q, Chen M, Jiang O, Pan Y, Hu D, Lin Q, et al. Sugemalimab versus placebo after concurrent or sequential chemoradiotherapy in patients with locally advanced, unresectable, stage III non-small-cell lung cancer in China (GEMSTONE-301): interim results of a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. (2022) 23:209–19. doi: 10.1016/S1470-2045(21)00630-6
330. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. (2012) 366:925–31. doi: 10.1056/NEJMoa1112824
331. Fizazi K, Drake CG, Beer TM, Kwon ED, Scher HI, Gerritsen WR, et al. Final analysis of the ipilimumab versus placebo following radiotherapy phase III trial in postdocetaxel metastatic castration-resistant prostate cancer identifies an excess of long-term survivors. Eur Urol. (2020) 78:822–30. doi: 10.1016/j.eururo.2020.07.032
332. Vignali PDA, DePeaux K, Watson MJ, Ye C, Ford BR, Lontos K, et al. Hypoxia drives CD39-dependent suppressor function in exhausted T cells to limit antitumor immunity. Nat Immunol. (2023) 24:267–79. doi: 10.1038/s41590-022-01379-9
333. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. (2021) 22:205–15. doi: 10.1038/s41590-020-00834-9
334. Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
335. Prokhnevska N, Cardenas MA, Valanparambil RM, Sobierajska E, Barwick BG, Jansen C, et al. CD8+ T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity. (2023) 56:107–124.e5. doi: 10.1016/j.immuni.2022.12.002
336. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. (2014) 74:5458–68. doi: 10.1158/0008-5472.CAN-14-1258
337. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. (2009) 15:5379–88. doi: 10.1158/1078-0432.CCR-09-0265
338. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. (2017) 8:15618. doi: 10.1038/ncomms15618
339. Fujiwara K, Saung MT, Jing H, Herbst B, Zarecki M, Muth S, et al. Interrogating the immune-modulating roles of radiation therapy for a rational combination with immune-checkpoint inhibitors in treating pancreatic cancer. J Immunother Cancer. (2020) 8:e000351. doi: 10.1136/jitc-2019-000351
340. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. (2015) 520:373–7. doi: 10.1038/nature14292
341. Moore C, Hsu C-C, Chen W-M, Chen BPC, Han C, Story M, et al. Personalized ultrafractionated stereotactic adaptive radiotherapy (PULSAR) in preclinical models enhances single-agent immune checkpoint blockade. Int J Radiat Oncol Biol Phys. (2021) 110:1306–16. doi: 10.1016/j.ijrobp.2021.03.047
342. Zhou H, Tu C, Yang P, Li J, Kepp O, Li H, et al. Carbon ion radiotherapy triggers immunogenic cell death and sensitizes melanoma to anti-PD-1 therapy in mice. Oncoimmunology. (2022) 11:2057892. doi: 10.1080/2162402X.2022.2057892
343. Petralia F, Ma W, Yaron TM, Caruso FP, Tignor N, Wang JM, et al. Pan-cancer proteogenomics characterization of tumor immunity. Cell. (2024) 187:1255–77.e27. doi: 10.1016/j.cell.2024.01.027
344. Johansson HJ, Socciarelli F, Vacanti NM, Haugen MH, Zhu Y, Siavelis I, et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat Commun. (2019) 10:1600. doi: 10.1038/s41467-019-09018-y
345. Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, et al. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell. (2020) 183:1436–56.e31. doi: 10.1016/j.cell.2020.10.036
Keywords: autophagy, cGAS/STING signaling, innate immunity, cancer, radiotherapy
Citation: Schmid M, Fischer P, Engl M, Widder J, Kerschbaum-Gruber S and Slade D (2024) The interplay between autophagy and cGAS-STING signaling and its implications for cancer. Front. Immunol. 15:1356369. doi: 10.3389/fimmu.2024.1356369
Received: 15 December 2023; Accepted: 26 March 2024;
Published: 10 April 2024.
Edited by:
Shawn Iadonato, Kineta, United StatesReviewed by:
Wenzheng Guo, University of Kentucky, United StatesPinglong Xu, Zhejiang University, China
Copyright © 2024 Schmid, Fischer, Engl, Widder, Kerschbaum-Gruber and Slade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dea Slade, ZGVhLnNsYWRlQG1lZHVuaXdpZW4uYWMuYXQ=
†These authors contributed equally to this work