 Xiao-Xin Wang1,2
Xiao-Xin Wang1,2 Ze-Hui Li1,2Hua-Yong Du1,2Wu-Bo Liu1,2,3
Ze-Hui Li1,2Hua-Yong Du1,2Wu-Bo Liu1,2,3 Chun-Jia Zhang1,2
Chun-Jia Zhang1,2 Xin Xu1,2
Xin Xu1,2 Han Ke1,2,3
Han Ke1,2,3 Run Peng1,2De-Gang Yang1,2
Run Peng1,2De-Gang Yang1,2 Jian-Jun Li1,2,3,4*
Jian-Jun Li1,2,3,4* Feng Gao1,2*
Feng Gao1,2*- 1School of Rehabilitation, Capital Medical University, Beijing, China
- 2Department of Spinal and Neural Functional Reconstruction, China Rehabilitation Research Center, Beijing, China
- 3Cheeloo College of Medicine, Shandong University, Jinan, Shandong, China
- 4Institute of Rehabilitation Medicine, China Rehabilitation Research Center, Beijing, China
Spinal cord injury (SCI) results in a large amount of tissue cell debris in the lesion site, which interacts with various cytokines, including inflammatory factors, and the intrinsic glial environment of the central nervous system (CNS) to form an inhibitory microenvironment that impedes nerve regeneration. The efficient clearance of tissue debris is crucial for the resolution of the inhibitory microenvironment after SCI. Macrophages are the main cells responsible for tissue debris removal after SCI. However, the high lipid content in tissue debris and the dysregulation of lipid metabolism within macrophages lead to their transformation into foamy macrophages during the phagocytic process. This phenotypic shift is associated with a further pro-inflammatory polarization that may aggravate neurological deterioration and hamper nerve repair. In this review, we summarize the phenotype and metabolism of macrophages under inflammatory conditions, as well as the mechanisms and consequences of foam cell formation after SCI. Moreover, we discuss two strategies for foam cell modulation and several potential therapeutic targets that may enhance the treatment of SCI.
1 Introduction
Spinal cord injury (SCI) induces primary damage to neurons and glia due to mechanical trauma, followed by secondary injury characterized by a cascade of inflammatory processes that cause further cell death of oligodendrocytes, astrocytes, and especially neurons (1). A key feature of the inflammatory response in SCI is the infiltration of monocyte-derived macrophages (MDMs) into the lesion site and the activation of resident microglia in the central nervous system (CNS). Both cell types act as major phagocytes that remove tissue debris after injury, which is crucial for creating a regenerative environment. It is well established that tissue debris from peripheral injuries is cleared by MDMs within weeks after injury. However, after SCI, the persistent demyelination produces myelin debris and other lipid-rich histiocyte debris that induce the transformation of macrophages into foamy macrophages, resembling those found in atherosclerotic plaques. These foam cells persist in the lesion site and mediate chronic inflammation, which hampers nerve repair (2, 3). T The molecular mechanisms of foam cell formation and function are poorly understood, but their phagocytic and migratory abilities have been reported to be impaired (2–6). Thus, foam cells after SCI represent a potential target for novel SCI research and intervention.
In this review, we provide a brief overview of the lipid metabolism of macrophages in the inflammatory milieu, and how it influences their function and phenotype. This will help to elucidate the mechanisms and consequences of macrophage phenotypic alterations after SCI. Furthermore, we summarise and discuss the factors that induce foam cell formation after SCI and the potential targets for modulating them, aiming to identify the strategies for foam cell regulation after SCI.
2 Macrophage metabolic regulation in inflammation
Different types of stimuli induce different macrophage phenotypes and metabolic profiles, and immune activation can be divided into four main components: inducers (signals that trigger an inflammatory response), sensors (proteins that detect the inducers), mediators (molecules that transmit signals to activate an effector response), and effectors (downstream metabolic programmes that facilitate the maturation of the desired effector phenotype) (7, 8). Based on this general classification, the macrophage response to stimulation can be described more clearly. The phenotypes that emerge from macrophage activation are commonly categorised as the M1 phenotype and the M2 phenotype. The M1 phenotype is mainly associated with acute bacterial infections, and the typical inducers are pathogen-associated molecular patterns (PAMPs), which are sensed by cell surface pattern recognition receptors (PRRs), leading to the production of the intermediate mediator, HIF-1α, which connects glycolytic metabolism to the inflammatory and microbicidal programmes of the macrophage (9). The M2 phenotype is driven by IL-4 and IL-13 produced by innate and adaptive immune cells (e.g., mast cells, basophils, and T cells) during processes such as helminthic and parasitic infections (10, 11). After IL-4 and IL-13 bind to their respective receptors (IL-4Rα vs. IL-13Rα), the subsequent activation of the M2 cells largely depends on STAT6, which is transcriptionally activated to upregulate fatty acid β-oxidation, OXPHOS and metabolic genes essential for mitochondrial biogenesis (12). Additionally, STAT6 induces the expression of transcriptional regulatory proteins and co-activator proteins, such as PPARγ, PPARδ and PGC-1β, which cooperate with STAT6 to sustain the switch of the cellular metabolic profile to oxidative metabolism (12–15). Different metabolic profiles of the M1 and M2 phenotypes stem from this, with the M1 macrophage using Warburg metabolism and the M2 macrophage using OXPHOS, which has been extensively discussed by others in the literature (16–19). The metabolism of M1 and M2 macrophages is bi-directionally linked to their functional phenotypes. Glycolysis in M1 macrophages enhances PPP flux, which provides intermediates for the biosynthesis of ribose, fatty acids and non-essential amino acids to sustain vital metabolic pathways. In contrast, the activation programme in M2 macrophages requires high levels of both duration and intensity, making the TCA cycle more prominent than glycolysis. Fatty acid oxidation is especially important in these cells to support the TCA cycle. One source of fatty acids is triglycerides, which are internalised by M2 macrophages via CD36 and then hydrolysed by lysosomal acid lipase (20). Besides uptake from external sources, macrophages also have the ability for lipid synthesis (21, 22). Microbial stimulation can augment macrophage de novo lipogenesis (DNL). Microbial stimulation also increases glucose utilisation, redirects intracellular glucose metabolism, and supplies a reaction substrate for intracellular lipid regeneration (DNL) in macrophages (23, 24). On the other hand, it can also activate the signalling pathway triggered by the transcription factor NF-κB, which directly binds to the response element in the promoter of SREBP1a (an isoform expressed in macrophages), thereby enhancing the expression of SREBP1a, which in turn promotes the oxidation of fatty acids in the DNL, providing nutrients for the TCA cycle (25). The inhibition of lipolysis reduces M2 macrophage-mediated resistance to parasite infection (26). These metabolic changes are not irreversible. Unlike metabolic shifts in cancer (genetically driven aerobic glycolytic transitions) or slow exercise-induced myofibre type conversions in skeletal muscle, many immune cells can swiftly switch between glycolysis and OXPHOS in response to external signals.
It is noteworthy that some vitro studies have proposed that the key determinant of the two phenotypes is a difference in the metabolic pathways used by the cells, i.e. the use of metabolic substrates controls macrophage polarization. However, this conclusion is disputed, with Chang HR et al. demonstrating that cellular lipid metabolism does not correlate with macrophage phenotype, and that the tissue environment is the main factor in determining how subpopulations of macrophages respond to changes in lipid synthesis and metabolic capacity (27). Furthermore, although the M1 and M2 phenotypes of macrophage activation facilitate the understanding of macrophage responses to stimuli, the in vivo phenotype of macrophages is more complex, especially in spinal cord injury (28).
3 Mechanisms and consequences of foam cell formation after spinal cord injury
As discussed above, macrophages display various functional states that influence tissue repair, and understanding the basic macrophage metabolism in inflammation will help to elucidate the complex changes that occur in macrophages after SCI. It is well known that the surrounding microenvironment is a key determinant of macrophage function (29). The spinal cord microenvironment undergoes drastic changes shortly after SCI. The early microenvironmental changes in SCI are characterised by an inflammatory response that induces monocytes to migrate to the injury site and differentiate into macrophages, which further amplify the inflammation. Inflammation after SCI is complicated and involves multiple cell types and a variety of inflammatory cytokines, such as tumour necrosis factor α (TNFα), interleukin-1β (IL-1β) and interleukin-6 (IL-6). Despite some beneficial effects of inflammation after SCI, excessive infiltration of immune cells is the main cause of neurodegeneration. After the acute inflammation resolves, a large number of cells undergo apoptosis, generating large amounts of cellular debris, particularly myelin debris. The accumulation of myelin and cellular debris at the injury site, combined with the pre-existing CNS glial environment, forms an inhibitory environment in the centre of injury, which impairs nerve repair processes, such as oligodendrocyte precursor cells (OPCs) differentiation and synapse regeneration (30, 31). By removing myelin debris, lipid peroxidation of myelin-derived lipids in the injury centre is prevented and the inhibitory microenvironment is partially deregulated. Previous studies have demonstrated that the function of macrophages after SCI is significantly influenced by myelin debris, depending on the type and timing of stimulation, but this issue remains highly contentious (2, 32, 33).. Membrane lipids are classified into three major groups: cholesterol, phospholipids (e.g., plasminogen, lecithin, sphingomyelin), and glycolipids (e.g., galactose ceramide). Unlike most biological membranes (25%:65%:10%), myelin has a distinctive lipid composition, with a high proportion of cholesterol and glycolipids in a 40%:40%:20% ratio (cholesterol, phospholipids, and glycolipids) (34), and a total lipid dry weight of 70-75% (35, 36). This creates a unique lipid-rich environment. Dysfunctional lipid metabolism in macrophages, coupled with the lipid-rich environment, leads to the accumulation of intracellular lipids in macrophages, resulting in the formation of cell types resembling the specialised macrophages called foam cells in atherosclerotic plaques (Figure 1), which are also referred to as foam cells after spinal cord injury. Foam cell is a term widely used in the study of diseases such as atherosclerosis and tuberculosis. Foam cell formation is induced by various factors, such as uncontrolled uptake of modified low-density lipoprotein (LDL), increased cholesterol esterification, and impaired cholesterol release mechanisms (37). Macrophages internalize modified lipoproteins via surface scavenger receptors (SRs) like CD36 and SR-A. These SRs are macrophage PRRs that recognize and bind oxidized LDL, thereby promoting foam cell formation by internalizing these lipoproteins. Coated-pit endocytosis, phagocytosis and pinocytosis also mediate lipoprotein internalization. After internalization, cleared lipoproteins are transported to endosomes or lysosomes for degradation, where cholesteryl esters (CE) are hydrolyzed by lysosomal acid lipase (LAL) to unesterified free cholesterol (FC). Free cholesterol is transported to the endoplasmic reticulum where it is re-esterified by acyl-CoA: cholesterol acyltransferase 1 (ACAT1) and stored as cytoplasmic lipid droplets. These lipid droplets look like bubbles within macrophages, hence the name foam cells. The foam cell formation process above has been detailed in articles (38, 39). It is noteworthy that while most studies have focused on macrophages as the main or only source of foam cells, the role of smooth muscle cell-derived foam cells in atherosclerosis has gained more attention (40). This article focuses on macrophage-derived foam cells, but the reader should be aware that foam cells are defined by their morphology and not by their precursor cells.
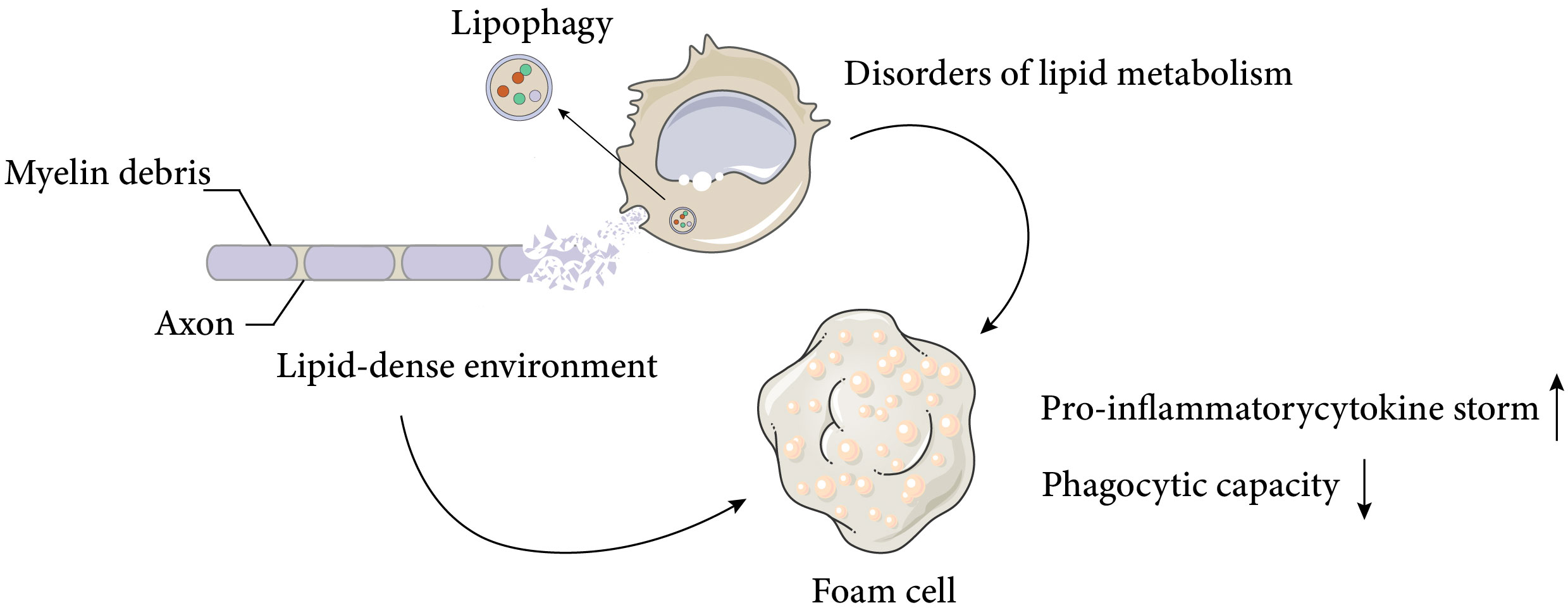
Figure 1 Causes and negative effects of foam cell formation after spinal cord injury. Spinal cord injury results in massive cell death and the accumulation of cellular and myelin debris at the lesion site, creating a lipid-rich environment. Macrophages can phagocytose these debris and promote a regenerative milieu that stimulates axonal myelination and repair. However, chronic exposure to the lipid-rich environment and impaired cellular lipid metabolism lead to the formation of foam cells, which produce pro-inflammatory cytokines and exhibit reduced phagocytic activity. These factors adversely affect neural recovery after spinal cord injury. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
These foam cells are typically observed in the injury area 1 week after SCI in mice and persist for at least 4 weeks (2).. Early studies suggested that the uptake of myelin residues biased macrophage polarisation towards a beneficial anti-inflammatory phenotype (41), but more recent studies have shown that foamy macrophages in SCI, which lose their ability to phagocytose apoptotic cells and tissue debris, activate NF-κ B signalling, which promotes inflammation and may contribute to further neurodegeneration (2, 3). The direction and modes of intervention of foam cells will be discussed later.
4 Possible strategies for reducing foam cell formation
Pathway analysis of genes involved in macrophage lipolysis metabolism revealed that lipolysis metabolic processes were a major function of macrophages seven days after spinal cord injury (compared to three days after spinal cord injury), with hubs including TNF, CD36, LPL, PPARγ, and ABCA1, among which the LXR/RXR pathways (CPs) were considered to be one of the most enriched pathways (42). Except for TNF expression, which was decreased, all gene-enriched pathways were increased, and the above receptors and pathways involved in phagocytosis of tissue debris and lipid efflux might be a key direction in dealing with foam cells (Figure 2). To discuss the direction of foam cell intervention, we first need to clarify the functional differences between MDMs and microglia after spinal cord injury. Recent lineage tracing studies have indicated that almost all tissue-resident macrophages in adult mice originated from embryonic yolk sac progenitor cells and that their numbers were maintained by local proliferation throughout the organism’s life cycle (43–45). It is worth noting that there was confusion about the distinction between MDMs and microglia in previous studies due to the difficulty in distinguishing them from each other. The two types of cells showed similarities in morphology as well as gene and cell surface protein expression after activation, but the two types of cells differed in the timing of activation, location, and efficiency of processing of myelin debris after SCI. After SCI, as a result of primary mechanical injury and secondary death due to apoptosis, the number of microglia at and near the site of injury was significantly reduced (46). However, surviving microglia responded rapidly to ATP released by damaged cells. They also responded to danger and pathogen-associated molecular patterns (D/PAMPs) by releasing inflammatory molecules and were in turn affected by inflammatory molecules. Once activated, microglia could proliferate, migrate, and perform various effector functions including phagocytosis and proliferation of neuroinflammation by secreting additional chemokines and cytokines, and had better myelin debris processing efficiency compared to MDMs (46, 47). However, MDMs that subsequently infiltrated into the injury area inhibited microglia phagocytosis and gradually replaced microglia as the primary phagocytes after injury (48). Although the reason why microglia were less susceptible to foam cell formation remained to be explored, their potential as a target for intervention in the regulation of lipid metabolism after spinal cord injury had been well demonstrated. Since MDMs were the main phagocytes that formed foam cells, the potential interventions discussed subsequently would mainly target MDMs cells.
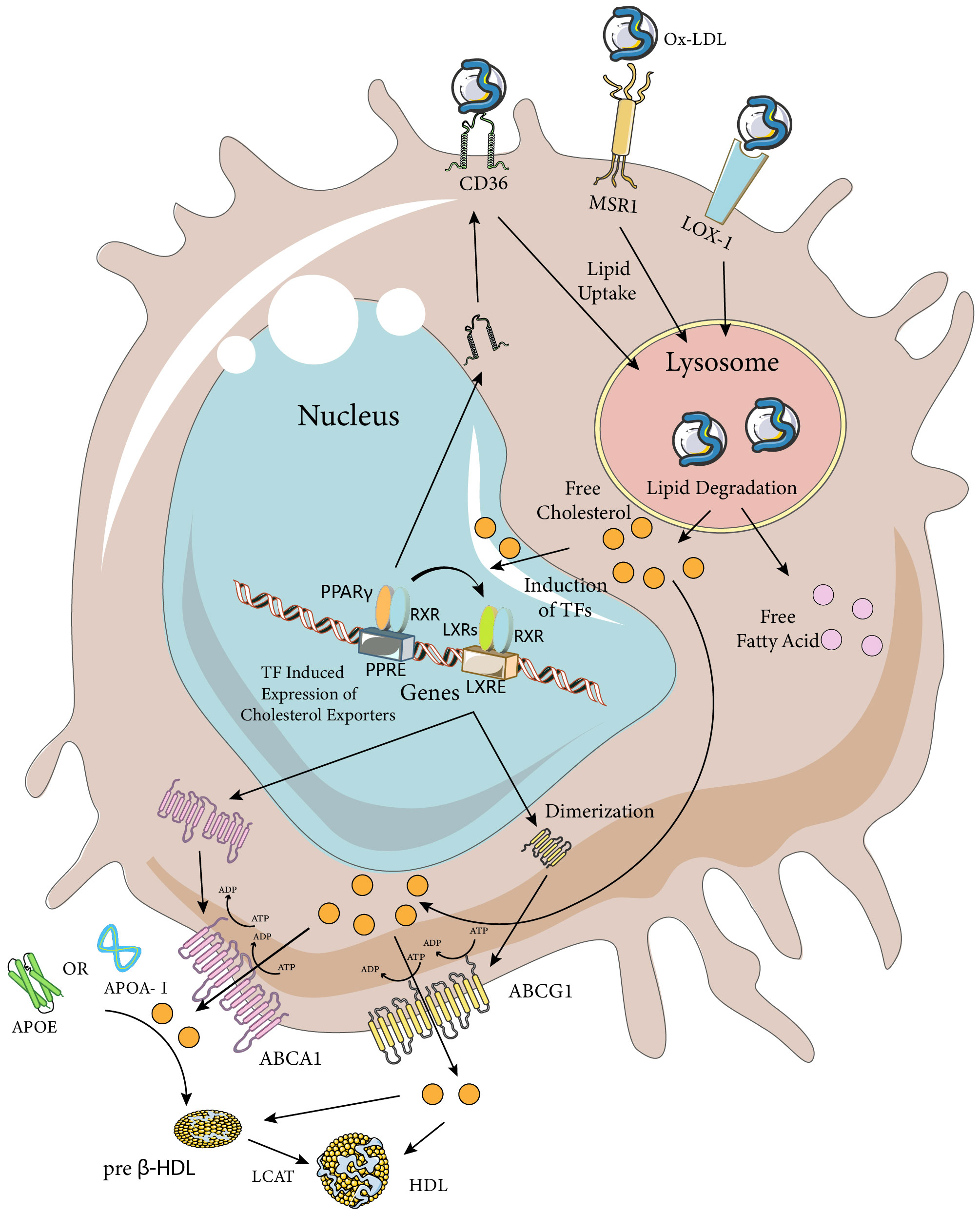
Figure 2 Macrophage Ox-LDL uptake and cholesterol efflux pathways. Macrophages internalize modified LDL (mainly Ox-LDL) via MSR1, CD36, and LOX-1 receptors. Ox-LDL is degraded in lysosomes to release lipids such as free cholesterol and fatty acids. Excess cellular cholesterol activates several transcription factors, including PPARγ, LXRs, and RXRs. Activated PPARγ and LXRs upregulate the expression of their target genes by heterodimerizing with RXRs and binding to the PPARE and LXRE response elements, respectively. The PPARγ transcriptome further enhances the expression of LXRs and CD36, while the LXR transcriptome induces the expression of the cholesterol efflux transporters ABCA1 and ABCG1. ABCA1 is a full-length membrane transporter, while ABCG1 functions as a dimer. Both proteins have two nucleotide-binding and two transmembrane domains and use ATP hydrolysis to transport substrates. ABCA1 mediates the efflux of cholesterol (chol) and phosphatidylcholine (PC) to ApoA-I or ApoE, forming preb-HDL, and ABCG1 mediates the efflux of cholesterol, sphingomyelin (SM), and phosphatidylcholine to preb-HDL or HDL (only the cholesterol transporter portion is highlighted in the figure). Cholesterol in preb-HDL and HDL is esterified by lecithin-cholesterol acyltransferase (LCAT) to form mature HDL. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license (https://creativecommons.org/licenses/by/3.0/).
4.1 Decreased lipid phagocytosis by macrophages
As mentioned earlier, a major cause of foam cell formation after SCI is the high lipid content of the tissue debris and cells phagocytosed by macrophages. One study found that about half of the neutral lipids in foamy macrophages originated from non-myelin sources by culturing macrophages with spinal cord homogenates, suggesting that the conclusions of in vitro experiments on myelin-induced foamy cells might be controversial and heterogeneous (49). Therefore, modulating macrophage phagocytosis to reduce myelin and other lipids could be a way to reduce foam cell formation. Among the various subtypes of the scavenger receptor (SR) family, SR-A type I and II, CD-36 and LOX-1 have been found to be involved in foam cell formation through uptake of modified LDL (Figure 2) (50). In vitro studies have shown that SR-A and CD36 on macrophages account for up to 75% to 90% of oxidized low-density lipoprotein (Ox-LDL) or acetylated low-density lipoprotein (Ac-LDL) uptake, and both have been extensively studied in spinal cord injury (51). LOX-1 has been less studied in relation to spinal cord injury, but its role in foam cell formation has been systematically investigated in vitro and in diseases such as atherosclerosis. Therefore, a detailed discussion of the phagocytosis and regulation of foam cells mediated by these three scavenger receptor subtypes in spinal cord injury will follow. It is worth noting that myelin sheaths and associated tissue debris after spinal cord injury have an inhibitory role in neural repair, and the formation of foam cells after phagocytosis of tissue debris by macrophages can also have a negative role in neural repair, implying that simply decreasing or increasing macrophage phagocytosis may not be a perfect and reasonable intervention. Nevertheless, several studies manipulating macrophage phagocytosis have obtained positive results. This may be attributed to various factors, such as the phagocytic capacity of different cell types within the CNS. Macrophages, as “specialized” phagocytes, mediate the phagocytosis of most tissue debris after spinal cord injury. However, some “non-specialized” phagocytes (e.g., fibroblasts, astrocytes, neurons, and oligodendrocytes) also contribute to this process. In addition, macrophage phagocytosis-associated receptors may have multiple functions that require further exploration.
4.1.1 CD36
CD36 is a type 2 cell surface scavenger receptor widely expressed on many immune and non-immune cells. It acts as both a signalling receptor in response to DAMPs and PAMPs and a long-chain free fatty acid transporter. Recent studies have indicated that CD36 can integrate cell signalling pathways and metabolic pathways through its dual functions, thereby influencing immune cell differentiation and activation, and ultimately cell fate. CD36 recognises and binds with high affinity to Ox-LDL and to specific oxidised phospholipids fractions on the surface of some apoptotic cells (52–54). Binding and uptake of ox-LDL by human macrophages lacking CD36 is reduced by 40% (42). Knockout of CD36 in spinal cord injured mice resulted in reduced lipid droplet content in macrophages, reduced foamy macrophage formation, reduced area of injury, and better functional recovery compared to wild-type mice (42, 55). CD36 deletion can also lead to a reduction in vascular ERSR, which improves functional recovery after spinal cord injury (55). A recent study showed that CD36 is expressed in fibroblasts, a major cellular component of glial scar formation in the spinal cord. Jun is a key factor in the formation of pathologic cutaneous scarring, and CD36 is a downstream effector (56). Increased expression of Jun in fibroblasts after SCI and induced expression of CD36 activated fibroblasts to form in the mouse scarring, implying that CD36 is one of the regulators of the glial scarring response after SCI (57). CD36 knockout improves the area of injury at 1 and 3 days post-injury in terms of vascular supply, it is noteworthy that the angiogenic response after spinal cord injury in wild-type mice does not begin until 3 dpi (58), so this early vascular improvement may be due to increased vascular retention or it may be related to an increase in vascular perfusion within the injured area as a result of the deregulation of the negative regulatory effect of CD36 on endothelial cell nitric oxide synthase and cyclic guanosine monophosphate (cGMP) production (59, 60). Notably, the role of CD36 for endothelial cells and angiogenesis and vascular repair remains unclear, with studies showing that blockade of the CD36 signalling pathway increases proliferation of endothelial cells from the CNS (61), with an improved angiogenic response consistent with the known antiangiogenic function of CD36, and studies showing that endothelial cell CD36 deficiency prevents normal angiogenesis and vascular repair (62). CD36 antagonists have been extensively explored. For example, salvianolic acid B, curcumin, and sulfosuccinimidyl oleate have been found to act as CD36 antagonists (63). In addition to these well-defined molecules, herbal extracts such as shibaosaponin A, epimedium glycosides, andrographolide, resveratrol, and quercetin have also been shown to modulate CD36 (57, 64, 65).
In summary, blocking the CD36 signalling pathway may reduce spinal cord injury lesion size, attenuate inflammation, decrease vascular ERSR, enhance acute heterotopic vascular proliferation and microvascular perfusion, improve scarring, and diminish foamy macrophage formation. These effects suggest that CD36 may be a promising target for SCI intervention.
4.1.2 CD204-MSR1 (scavenger receptor A - SR-A)
Macrophage scavenger receptor 1 (MSR1) belongs to the scavenger receptor (SR) family and has three isoforms (1, 2, and 3) that are generated by alternative splicing of the MSR1 gene. These isoforms have distinct functions, but they share a common structure as macrophage-specific trimeric integral membrane glycoproteins. To avoid confounding effects, the study of MSR1 as a whole will be addressed in subsequent studies, but researchers should consider the functional differences among the isoforms when investigating MSR1. MSR1 is a receptor that is mainly expressed on immune cells and vascular endothelial cells, and it recognises and phagocytoses modified LDL. When LDL undergoes modifications, such as oxidation or glycosylation, within the vascular wall, it is recognised by MSR1 and taken up by macrophages, resulting in foam cell formation due to the accumulation of intracellular cholesterol. This is considered a key step in the pathogenesis of atherosclerosis, as it triggers the secretion of chemokines and pro-inflammatory cytokines (66, 67). Similar to other SRs, MSR1 binds, internalises and degrades various ligands, including heat shock proteins, surface molecules of bacteria and viruses (68, 69). These ligands are foreign substances that are recognised by the innate immune system, thus MSR1 acts as an innate pattern recognition receptor that mediates cellular processes such as host defence, phagocytosis and apoptosis. Besides its role in the immune response, MSR1 also participates in cellular functions such as lipid metabolism and cell adhesion, thereby influencing the functional regulation of several organs and diseases. For instance, MSR1 has been implicated in Alzheimer’s disease, tumours, myocardial infarction, coronary artery disease, etc., with either beneficial or detrimental effects. MSR1 is a receptor of broad biological significance, as it plays an important role in the development and progression of various diseases (70–74). Reichert et al. reported that MSR1 facilitates the phagocytosis of peripheral nerve myelin fragments in vitro (75). In spinal cord injury mice, the knockout of the MSR1 gene did not affect the macrophage infiltration in the injury area compared to wild-type mice, but MSR1 WT mice had a large number of myelin debris patches in macrophages. This suggests that the formation of foamy macrophages is restricted in mice lacking MSR1, and that MSR1 promotes foamy macrophage formation and exacerbates the detrimental effects of spinal cord injury. Since the phagocytosis of myelin debris by macrophages is closely related to lipid metabolism, further experiments are required to verify whether MSR1 influences the metabolism and efflux of myelin lipids (76).
4.1.3 LOX-1
Lectin-like oxidised low-density lipoprotein receptor-1 (LOX-1) is also known as the Ox-LDL receptor. Ox-LDL interacts with the transmembrane glycoprotein LOX-1 and affects various cell types, such as endothelial cells, platelets, macrophages, fibroblasts and smooth muscle cells (77). Under normal conditions, LOX-1 expressed in macrophages accounts for 5-10% of Ox-LDL uptake. However, in pro-inflammatory states, LOX-1 expression is increased and contributes to approximately 40% of Ox-LDL turnover in macrophages (78, 79). Abnormal conditions, such as activation of pro-inflammatory pathways, hyperglycemia and hypertension, enhance LOX-1 expression and Ox-LDL uptake, leading to lipid accumulation and foam cell formation (80, 81). Several studies have shown that inhibition or down-regulation of macrophage LOX-1 expression reduces foam cell formation and lipid accumulation (82–84), suggesting its potential role in regulating foam cell formation and inflammatory factor expression in SCI. LOX-1 receptor expression is elevated at 14 days after spinal cord injury (85), but the expression pattern of LOX-1 at different time points after spinal cord injury is not well defined, and there is a lack of studies on the therapeutic effects of LOX-1 receptor intervention on SCI. This may be because the two receptors, CD36 and CD204, have received more attention, while the role of LOX-1 receptor has been overlooked. In conclusion, the observation and study of the expression and regulation of LOX-1 receptors may be important for understanding the formation of foam cells in the injury centre after spinal cord injury.
4.2 Enhancement of reverse cholesterol transport mechanisms in macrophages
Foam cells are formed when macrophages ingest excessive amounts of lipids after spinal cord injury. The prolonged persistence of foam cells after spinal cord injury indicates that the lipids accumulated in foam cells are not properly cleared (2). The persistence of foam cells impairs the subsequent phagocytic activity of macrophages and mediates chronic inflammation and poor neurological recovery. Therefore, besides regulating macrophage phagocytosis, enhancing cholesterol efflux from macrophages in the spinal cord injury site may be a strategy to address foam cell formation and accumulation. Generally, there are four cholesterol efflux pathways from cells: simple diffusion, scavenger receptor B1 (SR-BI, which belongs to the same family as CD36 but has different functions) mediated facilitated diffusion, and ATP-binding cassette transporters A1 (ABCA1) and G1 (ABCG1) in cooperation with extracellular lipid-poor apolipoproteins or more mature high-density lipoprotein (HDL) mediate cholesterol efflux (86). The role of SR-B1 in macrophage cholesterol efflux remains unresolved and is beyond the scope of this review. Here, we focus on ABCA1 and ABCG1, as well as apolipoprotein A-I (ApoA-I) and apolipoprotein E (ApoE), which are key mediators of cellular cholesterol efflux (Figure 2). We also discuss their relevance to spinal cord injury, where some studies have investigated their roles. It is noteworthy that two key issues should be considered when investigating cellular cholesterol efflux: the first is the existence of species variability in the cholesterol efflux pathway, which implies that the same receptor mediating cholesterol efflux may have different roles in different species; and the second is that the blood-brain barrier separates the CNS from the periphery, which allows for a different mechanism of cholesterol metabolism and cycling in the CNS compared to that of the peripheral tissues, such as the different structure and apolipoprotein content of HDL particles. The differences between the two should be taken into account when choosing and performing interventions.
4.2.1 ABCA1 and ABCG
ABCA1 is an intact membrane transporter protein, ABCG1 is a half-transporter and needs to function as a dimer biologically. At the cellular level, macrophage-like cell lines from mouse or human origin consistently express ABCA1 and ABCG1 proteins, which are both important for maintaining cellular cholesterol homeostasis (87). The basal levels of ABCA1 and ABCG1 proteins are low in macrophages, but they are induced by cholesterol loading and modulated by HDL-mediated cholesterol efflux (88–90). Reverse cholesterol transport (RCT) is one of the major pathways for the removal of excess cholesterol from tissues (91). In this process, ABCA1 mediates the initial transport of cellular cholesterol to ApoA-I to form nascent HDL particles, and ABCG1 facilitates the subsequent sustained transport of cholesterol to HDL for further maturation (92). When mouse peritoneal macrophages are fully loaded with cholesterol and treated with diluted human serum, ABCA1 mediates approximately half of the cholesterol output and ABCG1 mediates 20% of the cholesterol output (93). Considering the critical role of ABCA1 and ABCG1 in mediating intracellular cholesterol export, combined up-regulation of the expression of these two transporter proteins may be more effective in inhibiting foam cell formation. LXR, a member of the nuclear receptor superfamily, plays a key role in the stimulation of ABCA1 transcription. LXR binds to RXR to form a specialized heterodimer. The LXR/RXR complex then binds to the promoter region of the ABCA1 gene, inducing its expression and playing a similar role to ABCA1 during ABCG1 transcription, a good way to regulate the expression of both proteins. It is noteworthy that the expression of ABCA1 in macrophages at the lesion site was significantly reduced after 7 days of spinal cord injury, suggesting a decrease in the capacity of macrophage RCT (2), and that this down-regulation is associated with the formation of foam cells, whereas the expression of the ABCG1 protein after spinal cord injury is still unknown. Similarly, several studies have observed and regulated the expression of ABCA1 protein after spinal cord injury: biosignature analysis showed that ABCA1 is a key gene closely associated with immune cell infiltration and may contribute to the pathogenesis of ischemic or hypoxic SCI by regulating vascular injury, inflammation, and immune infiltration (94). Atorvastatin indirectly upregulates ABCA1 through activation of PPAR-γ and promotes functional recovery (95). Allicin activates peroxisome proliferator-activated factor receptor (PPAR-γ), an inducer of ABCA1, stimulating macrophage lipid efflux and reducing foam cell formation (96). Whereas little research has been done on ABCG1 protein after spinal cord injury compared to ABCA1 protein, which may be related to the species-specific variability of its cholesterol efflux action, the important role of ABCG1 protein in RCT implies that it is likely to be one of the key targets for foam cell intervention after spinal cord injury.
4.2.2 Apolipoprotein A-I
As discussed above, ApoA-I is a protein component of HDL that mediates reverse cholesterol transport. Although it is present in cerebrospinal fluid (CSF), its mRNA is not detected in the CNS, suggesting that ApoA-I is not synthesised in the brain and that it can enter the CNS with HDL via SR-BI (belonging to the CD36 family) mediated uptake (97). In contrast to ApoJ, ApoD, ApoA-II and ApoA-IV, which are present in the CNS in low abundance, ApoA-I (0.3.7 ± 0.0.8 mg/dl) and ApoE (0.3 ± 0.2.1 mg/dl) are the most abundant apolipoproteins in the CSF (98–100). ApoA-I has been shown to have a role in CNS inflammation and myelin regeneration (101–104). After spinal cord injury, ApoA-I expression is upregulated in injured spinal cord tissues (105). PPI network analysis showed it to be one of the 10 most altered core proteins 1 week after SCI (106), and changes in ApoA-I in the cerebrospinal fluid correlate with the severity of SCI injury, implying that ApoA-I may be a potential target for intervention after spinal cord injury (107). Monocytes and macrophages have the ability to synthesise and secrete endogenous ApoA-I (108). Two lipid-poor ApoA-I molecules bind ABCA1 dimers, allowing the transport of lipids in macrophages as high-density lipoproteins (HDL) (109). Recombinant exogenous human ApoA-I in mouse macrophages increased cholesterol efflux from macrophages and reduced the development of atherosclerosis, suggesting that macrophage-specific ApoA-I expression plays an important role in the prevention of atherosclerotic disease (110–112). Similarly, ApoA-I may be important for processing intracellular lipids in macrophages after spinal cord injury. Currently, there is still no systematic summary of ApoA-I expression levels after spinal cord injury, and there are few relevant interventions. An ApoA-I peptide mimetic made from D-amino acids, D-4F, improved HDL-mediated cholesterol efflux from macrophages and reduced foam cell formation in spinal cord injury (113). Notably, given the synergistic role that ABCA1 and ABCG1 proteins play with ApoA-I and, as subsequently described, ApoE during lipid efflux, their simultaneous intervention may have a better effect on foam cell formation and accumulation.
4.2.3 Apolipoprotein E
ApoE is a well-characterised lipoprotein that is expressed in several organs of the body, with the highest levels of expression in the liver, followed by the CNS, and it can also be synthesised by other tissues including monocytes (such as macrophages). The main cell type expressing ApoE in the CNS is astrocytes, while microglia also express it to some extent (114, 115). ApoE is involved in various activities including lipid transport, synaptic growth and neuroplasticity, and has been shown to play an important role in a variety of neurological diseases (116–120). Previous studies have shown that APOE4 is associated with poorer neurological recovery and longer rehabilitation time in patients with traumatic cervical spinal cord injury (121), s suggesting a potential role for ApoE in spinal cord injury. ApoE expression was significantly increased during the subacute phase of traumatic spinal cord injury in mice (122, 123). Single-cell RNA sequencing showed that ApoE was the most up-regulated gene expressed in macrophages and microglia during the subacute and chronic phases, and also ApoE was a hub gene for macrophages and microglia during the subacute and chronic phases of SCI (124). These results suggest that ApoE plays an important role in macrophages and microglia in the subacute and chronic phases of SCI. Deficiency of ApoE exacerbates the inflammatory response and mediates poor functional recovery after spinal cord injury (122, 124, 125), which is related to the leakage of macromolecules, such as inflammatory cells and immune proteins, from the blood-spinal cord barrier due to the increased permeability caused by the ApoE deficiency, and the direct effect of ApoE on the immune response. ApoE is the main cholesterol carrier involved in lipid efflux from CNS cells (126). Under normal conditions, the ABCA1 transporter protein interacts with lipid-poor/free ApoA-I, which acquires cellular PL and FC to form nascent HDL and initiate the RCT (127). imilarly, in the CNS, ABCA1 interacts with lipid-poor/free ApoE to form HDL that is structurally distinct from ApoA-I-HDL (128), and it is recognised that HDL formed by different ApoE isoforms (ϵ2, ϵ3 and ϵ4) have different roles in the CNS, which has been particularly evident in studies related to Alzheimer’s disease (129). Transmission electron microscopy observations revealed an increase in the number of lipid droplets and dense lysosomal material in macrophages and microglia in ApoE-/- mice (124), suggesting that ApoE may be a therapeutic target to promote the restoration of lipid metabolism and to reduce foam cell formation after spinal cord injury. Notably, the molecular size and structure of ApoE dictate that it is difficult to cross the blood-brain barrier, and thus exogenous ApoE mimetic peptides have been developed for use as alternative therapies, which retain the anti-inflammatory and neuroprotective effects of natural ApoE proteins but can still cross the blood-brain barrier (120, 130). The neuroprotective effects of exogenous apoE mimetic peptides have now also been demonstrated in several models of neurological disease including spinal cord injury (116, 120, 124, 131–133). In conclusion, endogenous ApoE has important neuroprotective effects after spinal cord injury and exogenous apoE mimetic peptides may be a new promising neuroprotective agent with the potential to inhibit foam cell formation after spinal cord injury.
5 Discussion
After spinal cord injury, the clearance of tissue debris is important for the inflammatory process and the nerve repair process. However, the clearance of tissue debris is hindered by the formation of foam cells. Therefore, the effective regulation of the phagocytosis and metabolic profiles of MDMs for the smooth clearance and metabolism of tissue debris is a key issue for promoting nerve repair after spinal cord injury. We propose two intervention strategies targeting foam cells: (I) reducing the phagocytosis of lipids by macrophages: the phagocytosis of lipid-rich tissue debris (including but not limited to myelin debris) leads to lipid accumulation in macrophages, and this can be improved by regulating or knocking down the relevant lipid uptake receptors (e.g., CD36, CD204, LOX-1); (II) regulating the RCT of macrophages: this process mainly involves ApoA-I, ApoE, and ABCA1 and ABCG1. Accelerating the lipid efflux from macrophages can reduce the excessive accumulation of intracellular lipids and thus prevent the transformation of macrophages into foam cells. It should be noted that the intervention strategies for foam cells are not limited to the ones proposed here; for example, since CE can only be transported to the extracellular compartment after being hydrolysed to FC, the hydrolysis of CE in macrophages limits the cholesterol efflux to some extent, and thus the intervention of related enzymes may also have a positive effect; moreover, since FC available for efflux is mainly transported to the autophagosome via CE in the autophagosome translocation to lysosomes to be supplied by hydrolysis by acid lipase rather than by hydrolysis by neutral cholesterol esterase, the induction of macrophage autophagy or the modulation of lysosomal function may also be effective intervention strategies. Traditionally, foam cell formation has been mainly linked to cholesterol uptake via natural and modified LDL, and the high cholesterol levels in the microenvironment due to the unique lipid composition of myelin sheaths after spinal cord injury have led to the article’s focus on cholesterol efflux. However, other lipids, such as nonesterified fatty acids and triacylglycerol (TAG)-rich lipoproteins (very low-density lipoproteins and chymotrypsin), can also interact with macrophages. Moreover, the intracellular composition of lipid droplets in foam cells after spinal cord injury and the proportion of lipid components phagocytosed and accumulated by macrophages are unclear, as there is a lack of analysis and studies on these aspects. Therefore, besides the foam cell treatment methods summarized in this article based on cholesterol efflux, we encourage researchers to explore more diverse approaches to foam cells after spinal cord injury. In conclusion, although many studies have obtained positive results, further studies are required to understand the differential effects of foam cell intervention and its potential as a pharmacological target for SCI repair.
Author contributions
XW: Conceptualization, Validation, Visualization, Writing – original draft, Writing – review & editing. ZL: Investigation, Software, Writing – review & editing. HD: Conceptualization, Formal Analysis, Software, Writing – review & editing. WL: Conceptualization, Visualization, Writing – review & editing. CZ: Formal Analysis, Project administration, Writing – review & editing. XX: Supervision, Visualization, Writing – review & editing. HK: Writing – review & editing, Resources. RP: Supervision, Writing – review & editing. D–GY: Supervision, Writing – review & editing. JL: Funding acquisition, Supervision, Writing – review & editing. FG: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Fundamental Research Funds for Central Public Welfare Research Institutes (2023CZ-2) and the Fundamental Research Funds for Central Public Welfare Research Institutes (2019CZ-13).
Acknowledgments
We thank SMART - Servier Medical ART for the support of the diagram drawing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alizadeh A, Dyck SM, Karimi-Abdolrezaee S. Traumatic spinal cord injury: an overview of pathophysiology, models and acute injury mechanisms. Front Neurol. (2019) 10:282. doi: 10.3389/fneur.2019.00282
2. Wang X, Cao K, Sun X, Chen Y, Duan Z, Sun L, et al. Macrophages in spinal cord injury: phenotypic and functional change from exposure to myelin debris. Glia. (2015) 63:635–51. doi: 10.1002/glia.22774
3. Kong FQ, Zhao SJ, Sun P, Liu H, Jie J, Xu T, et al. Macrophage MSR1 promotes the formation of foamy macrophage and neuronal apoptosis after spinal cord injury. J Neuroinflammation. (2020) 17:62. doi: 10.1186/s12974-020-01735-2
4. Nguyen MA, Karunakaran D, Geoffrion M, Cheng HS, Tandoc K, Perisic Matic L, et al. Extracellular vesicles secreted by atherogenic macrophages transfer microRNA to inhibit cell migration. Arterioscler Thromb Vasc Biol. (2018) 38:49–63. doi: 10.1161/ATVBAHA.117.309795
5. Sun X, Wang X, Chen T, Li T, Cao K, Lu A, et al. Myelin activates FAK/Akt/NF-kappaB pathways and provokes CR3-dependent inflammatory response in murine system. PloS One. (2010) 5:e9380. doi: 10.1371/journal.pone.0009380
6. Williams K, Ulvestad E, Waage A, Antel JP, McLaurin J. Activation of adult human derived microglia by myelin phagocytosis in vitro. J Neurosci Res. (1994) 38:433–43. doi: 10.1002/jnr.490380409
7. Medzhitov R. Origin and physiological roles of inflammation. Nature. (2008) 454:428–35. doi: 10.1038/nature07201
8. Odegaard JI, Chawla A. The immune system as a sensor of the metabolic state. Immunity. (2013) 38:644–54. doi: 10.1016/j.immuni.2013.04.001
9. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. (2003) 112:645–57. doi: 10.1016/s0092-8674(03)00154-5
10. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. (1992) 176:287–92. doi: 10.1084/jem.176.1.287
11. Doyle AG, Herbein G, Montaner LJ, Minty AJ, Caput D, Ferrara P, et al. Interleukin-13 alters the activation state of murine macrophages in vitro: comparison with interleukin-4 and interferon-gamma. Eur J Immunol. (1994) 24:1441–5. doi: 10.1002/eji.1830240630
12. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
13. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. (2008) 7:496–507. doi: 10.1016/j.cmet.2008.04.003
14. Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. (2008) 7:485–95. doi: 10.1016/j.cmet.2008.04.002
15. Lee YJ, Kim BM, Ahn YH, Choi JH, Choi YH, Kang JL. STAT6 signaling mediates PPARγ Activation and resolution of acute sterile inflammation in mice. Cells. (2021) 10(3):501. doi: 10.3390/cells10030501
16. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. (2011) 6:275–97. doi: 10.1146/annurev-pathol-011110-130138
17. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
18. Mehla K, Singh PK. Metabolic regulation of macrophage polarization in cancer. Trends Cancer. (2019) 5:822–34. doi: 10.1016/j.trecan.2019.10.007
19. Devanney NA, Stewart AN, Gensel JC. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp Neurol. (2020) 329:113310. doi: 10.1016/j.expneurol.2020.113310
20. Chen Y, Zhang J, Cui W, Silverstein RL. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J Exp Med. (2022) 219(6):e20211314. doi: 10.1084/jem.20211314
21. Iverson NM, Plourde NM, Sparks SM, Wang J, Patel EN, Shah PS, et al. Dual use of amphiphilic macromolecules as cholesterol efflux triggers and inhibitors of macrophage athero-inflammation. Biomaterials. (2011) 32(32):8319–27. doi: 10.1016/j.biomaterials.2011.07.039
22. Bidault G, Virtue S, Petkevicius K, Jolin HE, Dugourd A, Guénantin AC, et al. SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defences to promote their alternative activation. Nat Metab. (2021) 3:1150–62. doi: 10.1038/s42255-021-00440-5
23. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. (2014) 15:323–32. doi: 10.1038/ni.2833
24. Cader MZ, Boroviak K, Zhang Q, Assadi G, Kempster SL, Sewell GW, et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat Immunol. (2016) 17:1046–56. doi: 10.1038/ni.3532
25. Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. (2011) 13:540–9. doi: 10.1016/j.cmet.2011.04.001
26. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
27. Chang HR, Josefs T, Scerbo D, Gumaste N, Hu Y, Huggins LA, et al. Role of lpL (Lipoprotein lipase) in macrophage polarization in vitro and in vivo. Arterioscler Thromb Vasc Biol. (2019) 39:1967–85. doi: 10.1161/ATVBAHA.119.312389
28. Brennan FH, Li Y, Wang C, Ma A, Guo Q, Li Y, et al. Microglia coordinate cellular interactions during spinal cord repair in mice. Nat Commun. (2022) 13:4096. doi: 10.1038/s41467-022-31797-0
29. Amit I, Winter DR, Jung S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat Immunol. (2016) 17:18–25. doi: 10.1038/ni.3325
30. McKerracher L, Rosen KM. MAG. myelin and overcoming growth inhibition in the CNS. Front Mol Neurosci. (2015) 8:51. doi: 10.3389/fnmol.2015.00051
31. Baer AS, Syed YA, Kang SU, Mitteregger D, Vig R, Ffrench-Constant C, et al. Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling. Brain. (2009) 132:465–81. doi: 10.1093/brain/awn334
32. Greenhalgh AD, David S. Differences in the phagocytic response of microglia and peripheral macrophages after spinal cord injury and its effects on cell death. J Neurosci. (2014) 34:6316–22. doi: 10.1523/JNEUROSCI.4912-13.2014
33. Kopper TJ, Zhang B, Bailey WM, Bethel KE, Gensel JC. The effects of myelin on macrophage activation are phenotypic specific via cPLA(2) in the context of spinal cord injury inflammation. Sci Rep. (2021) 11:6341. doi: 10.1038/s41598-021-85863-6
34. O’Brien JS. STABILITY OF THE MYELIN MEMBRANE. Science. (1965) 147:1099–107. doi: 10.1126/science.147.3662.1099
35. Dimas P, Montani L, Pereira JA, Moreno D, Trötzmüller M, Gerber J, et al. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife. (2019) 8:e44702. doi: 10.7554/eLife.44702
36. Nave KA, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol. (2014) 30:503–33. doi: 10.1146/annurev-cellbio-100913-013101
37. Hutchins PM, Heinecke JW. Cholesterol efflux capacity, macrophage reverse cholesterol transport and cardioprotective HDL. Curr Opin Lipidol. (2015) 26:388–93. doi: 10.1097/mol.0000000000000209
38. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (Berl). (2017) 95:1153–65. doi: 10.1007/s00109-017-1575-8
39. Chistiakov DA, Bobryshev YV, Orekhov AN. Macrophage-mediated cholesterol handling in atherosclerosis. J Cell Mol Med. (2016) 20:17–28. doi: 10.1111/jcmm.12689
40. Pi S, Mao L, Chen J, Shi H, Liu Y, Guo X, et al. The P2RY12 receptor promotes VSMC-derived foam cell formation by inhibiting autophagy in advanced atherosclerosis. Autophagy. (2021) 17:980–1000. doi: 10.1080/15548627.2020.1741202
41. Bogie JF, Jorissen W, Mailleux J, Nijland PG, Zelcer N, Vanmierlo T, et al. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol Commun. (2013) 1:43. doi: 10.1186/2051-5960-1-43
42. Zhu Y, Lyapichev K, Lee DH, Motti D, Ferraro NM, Zhang Y, et al. Macrophage transcriptional profile identifies lipid catabolic pathways that can be therapeutically targeted after spinal cord injury. J Neurosci. (2017) 37:2362–76. doi: 10.1523/JNEUROSCI.2751-16.2017
43. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
44. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
45. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
46. Bellver-Landete V, Bretheau F, Mailhot B, Vallieres N, Lessard M, Janelle ME, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun. (2019) 10:518. doi: 10.1038/s41467-019-08446-0
47. Zhou X, Wahane S, Friedl MS, Kluge M, Friedel CC, Avrampou K, et al. Microglia and macrophages promote corralling, wound compaction and recovery after spinal cord injury via Plexin-B2. Nat Neurosci. (2020) 23:337–50. doi: 10.1038/s41593-020-0597-7
48. Greenhalgh AD, Zarruk JG, Healy LM, Baskar Jesudasan SJ, Jhelum P, Salmon CK, et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PloS Biol. (2018) 16:e2005264. doi: 10.1371/journal.pbio.2005264
49. Ryan CB, Choi JS, Al-Ali H, Lee JK. Myelin and non-myelin debris contribute to foamy macrophage formation after spinal cord injury. Neurobiol Dis. (2022) 163:105608. doi: 10.1016/j.nbd.2021.105608
50. de Beer MC, Zhao Z, Webb NR, van der Westhuyzen DR, de Villiers WJ. Lack of a direct role for macrosialin in oxidized LDL metabolism. J Lipid Res. (2003) 44:674–85. doi: 10.1194/jlr.M200444-JLR200
51. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. (2002) 277:49982–8. doi: 10.1074/jbc.M209649200
52. Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. (2006) 203:2613–25. doi: 10.1084/jem.20060370
53. Nicholson AC, Frieda S, Pearce A, Silverstein RL. Oxidized LDL binds to CD36 on human monocyte-derived macrophages and transfected cell lines. Evidence implicating the lipid moiety of the lipoprotein as the binding site. Arterioscler Thromb Vasc Biol. (1995) 15:269–75. doi: 10.1161/01.atv.15.2.269
54. Podrez EA, Febbraio M, Sheibani N, Schmitt D, Silverstein RL, Hajjar DP, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. (2000) 105:1095–108. doi: 10.1172/jci8574
55. Myers SA, Andres KR, Hagg T, Whittemore SR. CD36 deletion improves recovery from spinal cord injury. Exp Neurol. (2014) 256:25–38. doi: 10.1016/j.expneurol.2014.03.016
56. Griffin MF, Borrelli MR, Garcia JT, Januszyk M, King M, Lerbs T, et al. JUN promotes hypertrophic skin scarring via CD36 in preclinical in vitro and in vivo models. Sci Transl Med. (2021) 13:eabb3312. doi: 10.1126/scitranslmed.abb3312
57. Gong L, Gu Y, Han X, Luan C, Liu C, Wang X, et al. Spatiotemporal dynamics of the molecular expression pattern and intercellular interactions in the glial scar response to spinal cord injury. Neurosci Bull. (2023) 39:213–44. doi: 10.1007/s12264-022-00897-8
58. Benton RL, Maddie MA, Minnillo DR, Hagg T, Whittemore SR. Griffonia simplicifolia isolectin B4 identifies a specific subpopulation of angiogenic blood vessels following contusive spinal cord injury in the adult mouse. J Comp Neurol. (2008) 507:1031–52. doi: 10.1002/cne.21570
59. Jiménez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. (2000) 6:41–8. doi: 10.1038/71517
60. Isenberg JS, Frazier WA, Roberts DD. Thrombospondin-1: a physiological regulator of nitric oxide signaling. Cell Mol Life Sci. (2008) 65:728–42. doi: 10.1007/s00018-007-7488-x
61. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. (2002) 30:207–10. doi: 10.1093/nar/30.1.207
62. Bou Khzam L, Son NH, Mullick AE, Abumrad NA, Goldberg IJ. Endothelial cell CD36 deficiency prevents normal angiogenesis and vascular repair. Am J Transl Res. (2020) 12:7737–61.
63. Jian X, Liu Y, Zhao Z, Zhao L, Wang D, Liu Q. The role of traditional Chinese medicine in the treatment of atherosclerosis through the regulation of macrophage activity. BioMed Pharmacother. (2019) 118:109375. doi: 10.1016/j.biopha.2019.109375
64. Yang J, Park KW, Cho S. Inhibition of the CD36 receptor reduces visceral fat accumulation and improves insulin resistance in obese mice carrying the BDNF-Val66Met variant. J Biol Chem. (2018) 293:13338–48. doi: 10.1074/jbc.RA118.002405
65. Jiang J, Shi Y, Cao J, Lu Y, Sun G, Yang J. Role of ASM/Cer/TXNIP signaling module in the NLRP3 inflammasome activation. Lipids Health Dis. (2021) 20:19. doi: 10.1186/s12944-021-01446-4
66. Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. (1997) 386:292–6. doi: 10.1038/386292a0
67. Ouyang Z, Zhong J, Shen J, Zeng Y. The cell origins of foam cell and lipid metabolism regulated by mechanical stress in atherosclerosis. Front Physiol. (2023) 14:1179828. doi: 10.3389/fphys.2023.1179828
68. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. (2010) 115:e10–9. doi: 10.1182/blood-2009-07-235028
69. Prodjinotho UF, Lema J, Lacorcia M, Schmidt V, Vejzagic N, Sikasunge C, et al. Host immune responses during Taenia solium Neurocysticercosis infection and treatment. PloS Negl Trop Dis. (2020) 14:e0008005. doi: 10.1371/journal.pntd.0008005
70. Cornejo F, Vruwink M, Metz C, Munoz P, Salgado N, Poblete J, et al. Scavenger Receptor-A deficiency impairs immune response of microglia and astrocytes potentiating Alzheimer’s disease pathophysiology. Brain Behav Immun. (2018) 69:336–50. doi: 10.1016/j.bbi.2017.12.007
71. Ben J, Zhu X, Zhang H, Chen Q. Class A1 scavenger receptors in cardiovascular diseases. Br J Pharmacol. (2015) 172:5523–30. doi: 10.1111/bph.13105
72. Wu X, Cheng B, Guo X, Wu Q, Sun S, He P. PPARalpha/gamma signaling pathways are involved in Chlamydia pneumoniae-induced foam cell formation via upregulation of SR-A1 and ACAT1 and downregulation of ABCA1/G1. Microb Pathog. (2021) 161:105284. doi: 10.1016/j.micpath.2021.105284
73. Zong G, Zhu Y, Zhang Y, Wang Y, Bai H, Yang Q, et al. SR-A1 suppresses colon inflammation and tumorigenesis through negative regulation of NF-kappaB signaling. Biochem Pharmacol. (2018) 154:335–43. doi: 10.1016/j.bcp.2018.05.017
74. Huang J, Jiang Y, Ji R, Jia Y, Wang S, Zhou Z, et al. Macrophage scavenger receptor A1 antagonizes abdominal aortic aneurysm via upregulating IRG1. Biochem Pharmacol. (2023) 213:115631. doi: 10.1016/j.bcp.2023.115631
75. Reichert F, Rotshenker S. Complement-receptor-3 and scavenger-receptor-AI/II mediated myelin phagocytosis in microglia and macrophages. Neurobiol Dis. (2003) 12:65–72. doi: 10.1016/s0969-9961(02)00008-6
76. Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol. (2018) 330:27–42. doi: 10.1016/j.cellimm.2018.01.020
77. Akhmedov A, Sawamura T, Chen CH, Kraler S, Vdovenko D, Lüscher TF. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1): a crucial driver of atherosclerotic cardiovascular disease. Eur Heart J. (2021) 42:1797–807. doi: 10.1093/eurheartj/ehaa770
78. Pirillo A, Norata GD, Catapano AL. LOX-1, oxLDL, and atherosclerosis. Mediators Inflamm. (2013) 2013:152786. doi: 10.1155/2013/152786
79. Schaeffer DF, Riazy M, Parhar KS, Chen JH, Duronio V, Sawamura T, et al. LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J Lipid Res. (2009) 50:1676–84. doi: 10.1194/jlr.M900167-JLR200
80. Yang HY, Bian YF, Zhang HP, Gao F, Xiao CS, Liang B, et al. LOX−1 is implicated in oxidized low−density lipoprotein−induced oxidative stress of macrophages in atherosclerosis. Mol Med Rep. (2015) 12:5335–41. doi: 10.3892/mmr.2015.4066
81. Kattoor AJ, Goel A, Mehta JL. LOX-1: regulation, signaling and its role in atherosclerosis. Antioxidants (Basel). (2019) 8(7):218. doi: 10.3390/antiox8070218
82. Wang X, Ding Z, Lin J, Guo Z, Mehta JL. LOX-1 in macrophage migration in response to ox-LDL and the involvement of calpains. Biochem Biophys Res Commun. (2015) 467:135–9. doi: 10.1016/j.bbrc.2015.09.100
83. Dai Y, Wu X, Dai D, Li J, Mehta JL. MicroRNA-98 regulates foam cell formation and lipid accumulation through repression of LOX-1. Redox Biol. (2018) 16:255–62. doi: 10.1016/j.redox.2018.03.003
84. Lu J, Chen X, Xu X, Liu J, Zhang Z, Wang M, et al. Active polypeptides from Hirudo inhibit endothelial cell inflammation and macrophage foam cell formation by regulating the LOX-1/LXR-alpha/ABCA1 pathway. BioMed Pharmacother. (2019) 115:108840. doi: 10.1016/j.biopha.2019.108840
85. An Y, Li J, Liu Y, Fan M, Tian W. Protective effect of D-pinitol on the experimental spinal cord injury in rats. Metab Brain Dis. (2020) 35:473–82. doi: 10.1007/s11011-020-00537-y
86. Yu XH, Tang CK. ABCA1, ABCG1, and cholesterol homeostasis. Adv Exp Med Biol. (2022) 1377:95–107. doi: 10.1007/978-981-19-1592-5_7
87. Zarubica A, Trompier D, Chimini G. ABCA1, from pathology to membrane function. Pflugers Arch. (2007) 453:569–79. doi: 10.1007/s00424-006-0108-z
88. Langmann T, Klucken J, Reil M, Liebisch G, Luciani MF, Chimini G, et al. Molecular cloning of the human ATP-binding cassette transporter 1 (hABC1): evidence for sterol-dependent regulation in macrophages. Biochem Biophys Res Commun. (1999) 257:29–33. doi: 10.1006/bbrc.1999.0406
89. Wang D, Yan X, Xia M, Yang Y, Li D, Li X, et al. Coenzyme Q10 promotes macrophage cholesterol efflux by regulation of the activator protein-1/miR-378/ATP-binding cassette transporter G1-signaling pathway. Arterioscler Thromb Vasc Biol. (2014) 34:1860–70. doi: 10.1161/ATVBAHA.113.302879
90. Mo ZC, Xiao J, Tang SL, Ouyang XP, He PP, Lv YC, et al. Advanced oxidation protein products exacerbates lipid accumulation and atherosclerosis through downregulation of ATP-binding cassette transporter A1 and G1 expression in apolipoprotein E knockout mice. Circ J. (2014) 78:2760–70. doi: 10.1253/circj.cj-14-0193
91. Yu X-H, Zhang D-W, Zheng X-L, Tang C-K. Cholesterol transport system: An integrated cholesterol transport model involved in atherosclerosis. Prog Lipid Res. (2019) 73:65–91. doi: 10.1016/j.plipres.2018.12.002
92. Gutierrez PS. Foam cells in atherosclerosis. Arq Bras Cardiol. (2022) 119:542–3. doi: 10.36660/abc.20220659
93. Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, et al. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. (2007) 48:2453–62. doi: 10.1194/jlr.M700274-JLR200
94. Zhu L, Gao N, Zhu Z, Zhang S, Li X, Zhu J. Bioinformatics analysis of differentially expressed genes related to ischemia and hypoxia in spinal cord injury and construction of miRNA-mRNA or mRNA-transcription factor interaction network. Toxicol Mech Methods. (2023) 21:1–27. doi: 10.1080/15376516.2023.2286363
95. Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. (2008) 454:470–7. doi: 10.1038/nature07202
96. Lin XL, Hu HJ, Liu YB, Hu XM, Fan XJ, Zou WW, et al. Allicin induces the upregulation of ABCA1 expression via PPARgamma/LXRalpha signaling in THP-1 macrophage-derived foam cells. Int J Mol Med. (2017) 39:1452–60. doi: 10.3892/ijmm.2017.2949
97. Elliott DA, Weickert CS, Garner B. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol. (2010) 51:555–73. doi: 10.2217/clp.10.37
98. Sengupta MB, Basu M, Iswarari S, Mukhopadhyay KK, Sardar KP, Acharyya B, et al. CSF proteomics of secondary phase spinal cord injury in human subjects: perturbed molecular pathways post injury. PloS One. (2014) 9:e110885. doi: 10.1371/journal.pone.0110885
99. Chakrabarti A, Chatterjee A, Sengupta MB, Chattopadhyay P, Mukhopadhyay D. Altered levels of amyloid precursor protein intracellular domain-interacting proteins in Alzheimer disease. Alzheimer Dis Assoc Disord. (2014) 28:283–90. doi: 10.1097/wad.0000000000000011
100. Borghini I, Barja F, Pometta D, James RW. Characterization of subpopulations of lipoprotein particles isolated from human cerebrospinal fluid. Biochim Biophys Acta. (1995) 1255:192–200. doi: 10.1016/0005-2760(94)00232-N
101. Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. (1995) 96:2758–67. doi: 10.1172/jci118345
102. Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SP, et al. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol. (2008) 28:2071–7. doi: 10.1161/ATVBAHA.108.168690
103. Vanherle S, Jorissen W, Dierckx T, Loix M, Grajchen E, Mingneau F, et al. The ApoA-I mimetic peptide 5A enhances remyelination by promoting clearance and degradation of myelin debris. Cell Rep. (2022) 41:111591. doi: 10.1016/j.celrep.2022.111591
104. McComb M, Krikheli M, Uher T, Browne RW, Srpova B, Oechtering J, et al. Neuroprotective associations of apolipoproteins A-I and A-II with neurofilament levels in early multiple sclerosis. J Clin Lipidol. (2020) 14:675–84. doi: 10.1016/j.jacl.2020.07.001
105. Kang SK, So HH, Moon YS, Kim CH. Proteomic analysis of injured spinal cord tissue proteins using 2-DE and MALDI-TOF MS. Proteomics. (2006) 6:2797–812. doi: 10.1002/pmic.200500621
106. Liu S, Kang Y, Zhang C, Lou Y, Li X, Lu L, et al. Isobaric tagging for relative and absolute protein quantification (iTRAQ)-based quantitative proteomics analysis of differentially expressed proteins 1 week after spinal cord injury in a rat model. Med Sci Monit. (2020) 26:e924266. doi: 10.12659/MSM.924266
107. Sengupta MB, Mukhopadhyay D. Possible role of apolipoprotein A1 in healing and cell death after neuronal injury. Front Biosci (Elite Ed). (2016) 8:460–77. doi: 10.2741/e780
108. Shavva VS, Mogilenko DA, Nekrasova EV, Trulioff AS, Kudriavtsev IV, Larionova EE, et al. Tumor necrosis factor α stimulates endogenous apolipoprotein A-I expression and secretion by human monocytes and macrophages: role of MAP-kinases, NF-κB, and nuclear receptors PPARα and LXRs. Mol Cell Biochem. (2018) 448:211–23. doi: 10.1007/s11010-018-3327-7
109. Phillips MC. Is ABCA1 a lipid transfer protein? J Lipid Res. (2018) 59:749–63. doi: 10.1194/jlr.R082313
110. Alexander ET, Weibel GL, Joshi MR, Vedhachalam C, de la Llera-Moya M, Rothblat GH, et al. Macrophage reverse cholesterol transport in mice expressing ApoA-I Milano. Arterioscler Thromb Vasc Biol. (2009) 29:1496–501. doi: 10.1161/ATVBAHA.109.191379
111. Ishiguro H, Yoshida H, Major AS, Zhu T, Babaev VR, Linton MF, et al. Retrovirus-mediated expression of apolipoprotein A-I in the macrophage protects against atherosclerosis in vivo. J Biol Chem. (2001) 276:36742–8. doi: 10.1074/jbc.M106027200
112. Tavori H, Su YR, Yancey PG, Giunzioni I, Wilhelm AJ, Blakemore JL, et al. Macrophage apoAI protects against dyslipidemia-induced dermatitis and atherosclerosis without affecting HDL. J Lipid Res. (2015) 56:635–43. doi: 10.1194/jlr.M056408
113. Li J, Zhu Z, Li Y, Chen Y, Hu X, Liu Y, et al. D-4F, an apolipoprotein A-I mimetic, promotes the clearance of myelin debris and the reduction of foamy macrophages after spinal cord injury. Bioengineered. (2022) 13:11794–809. doi: 10.1080/21655979.2022.2073063
114. Grehan S, Tse E, Taylor JM. Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J Neurosci. (2001) 21:812–22. doi: 10.1523/jneurosci.21-03-00812.2001
115. Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. (1987) 262:14352–60. doi: 10.1016/s0021-9258(18)47945-8
116. Tu TM, Kolls BJ, Soderblom EJ, Cantillana V, Ferrell PD, Moseley MA, et al. Apolipoprotein E mimetic peptide, CN-105, improves outcomes in ischemic stroke. Ann Clin Transl Neurol. (2017) 4:246–65. doi: 10.1002/acn3.399
117. Raulin AC, Doss SV, Trottier ZA, Ikezu TC, Bu G, Liu CC. ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies. Mol Neurodegener. (2022) 17:72. doi: 10.1186/s13024-022-00574-4
118. Laskowitz DT, Song P, Wang H, Mace B, Sullivan PM, Vitek MP, et al. Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J Neurotrauma. (2010) 27:1983–95. doi: 10.1089/neu.2010.1396
119. Kaufman NA, Beare JE, Tan AA, Vitek MP, McKenna SE, Hoane MR. COG1410, an apolipoprotein E-based peptide, improves cognitive performance and reduces cortical loss following moderate fluid percussion injury in the rat. Behav Brain Res. (2010) 214:395–401. doi: 10.1016/j.bbr.2010.06.017
120. Laskowitz DT, Wang H, Chen T, Lubkin DT, Cantillana V, Tu TM, et al. Neuroprotective pentapeptide CN-105 is associated with reduced sterile inflammation and improved functional outcomes in a traumatic brain injury murine model. Sci Rep. (2017) 7:46461. doi: 10.1038/srep46461
121. Jha A, Lammertse DP, Coll JR, Charlifue S, Coughlin CT, Whiteneck GG, et al. Apolipoprotein E epsilon4 allele and outcomes of traumatic spinal cord injury. J Spinal Cord Med. (2008) 31:171–6. doi: 10.1080/10790268.2008.11760708
122. Yang X, Chen S, Shao Z, Li Y, Wu H, Li X, et al. Apolipoprotein E deficiency exacerbates spinal cord injury in mice: inflammatory response and oxidative stress mediated by NF-kappaB signaling pathway. Front Cell Neurosci. (2018) 12:142. doi: 10.3389/fncel.2018.00142
123. Seitz A, Kragol M, Aglow E, Showe L, Heber-Katz E. Apolipoprotein E expression after spinal cord injury in the mouse. J Neurosci Res. (2003) 71:417–26. doi: 10.1002/jnr.10482
124. Yao XQ, Chen JY, Yu ZH, Huang ZC, Hamel R, Zeng YQ, et al. Bioinformatics analysis identified apolipoprotein E as a hub gene regulating neuroinflammation in macrophages and microglia following spinal cord injury. Front Immunol. (2022) 13:964138. doi: 10.3389/fimmu.2022.964138
125. Cheng X, Zheng Y, Bu P, Qi X, Fan C, Li F, et al. Apolipoprotein E as a novel therapeutic neuroprotection target after traumatic spinal cord injury. Exp Neurol. (2018) 299:97–108. doi: 10.1016/j.expneurol.2017.10.014
126. Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil MT, Su M, Sen P, et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science. (2018) 359:684–8. doi: 10.1126/science.aan4183
127. Favari E, Chroni A, Tietge UJ, Zanotti I, Escolà-Gil JC, Bernini F. Cholesterol efflux and reverse cholesterol transport. Handb Exp Pharmacol. (2015) 224:181–206. doi: 10.1007/978-3-319-09665-0_4
128. Van Valkenburgh J, Meuret C, Martinez AE, Kodancha V, Solomon V, Chen K, et al. Understanding the exchange of systemic HDL particles into the brain and vascular cells has diagnostic and therapeutic implications for neurodegenerative diseases. Front Physiol. (2021) 12:700847. doi: 10.3389/fphys.2021.700847
129. Jin Y, Chifodya K, Han G, Jiang W, Chen Y, Shi Y, et al. High-density lipoprotein in Alzheimer’s disease: From potential biomarkers to therapeutics. J Control Release. (2021) 338:56–70. doi: 10.1016/j.jconrel.2021.08.018
130. Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, et al. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. (2003) 278:48529–33. doi: 10.1074/jbc.M306923200
131. Cao F, Jiang Y, Wu Y, Zhong J, Liu J, Qin X, et al. Apolipoprotein E-mimetic COG1410 reduces acute vasogenic edema following traumatic brain injury. J Neurotrauma. (2016) 33:175–82. doi: 10.1089/neu.2015.3887
132. Lei B, James ML, Liu J, Zhou G, Venkatraman TN, Lascola CD, et al. Neuroprotective pentapeptide CN-105 improves functional and histological outcomes in a murine model of intracerebral hemorrhage. Sci Rep. (2016) 6:34834. doi: 10.1038/srep34834
Keywords: spinal cord injury, macrophage, foam cell, scavenger receptor, cholesterol reverse transcription
Citation: Wang X-X, Li Z-H, Du H-Y, Liu W-B, Zhang C-J, Xu X, Ke H, Peng R, Yang D-G, Li J-J and Gao F (2024) The role of foam cells in spinal cord injury: challenges and opportunities for intervention. Front. Immunol. 15:1368203. doi: 10.3389/fimmu.2024.1368203
Received: 10 January 2024; Accepted: 22 February 2024;
Published: 13 March 2024.
Edited by:
Jeffrey K. Huang, Georgetown University, United StatesReviewed by:
Caroline Perner, Hannover Medical School, GermanyMichael Davin Kornberg, Johns Hopkins Medicine, United States
Copyright © 2024 Wang, Li, Du, Liu, Zhang, Xu, Ke, Peng, Yang, Li and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-Jun Li, Y3JyY2xpampAMTYzLmNvbQ==; Feng Gao, Z2FvZmVuZzU5NjBAMTI2LmNvbQ==