 Ulrike Schulze-Späte1*
Ulrike Schulze-Späte1* Ludwig Wurschi1
Ludwig Wurschi1 Emiel P. C. van der Vorst2,3,4,5Frank Hölzle6
Emiel P. C. van der Vorst2,3,4,5Frank Hölzle6 Rogerio B. Craveiro7
Rogerio B. Craveiro7 Michael Wolf7
Michael Wolf7 Heidi Noels2,3,8*
Heidi Noels2,3,8*- 1Section of Geriodontics, Department of Conservative Dentistry and Periodontics, University Hospital Jena, Jena, Germany
- 2Institute for Molecular Cardiovascular Research (IMCAR), Uniklinik RWTH Aachen, RWTH Aachen University, Aachen, Germany
- 3Aachen-Maastricht Institute for Cardiorenal Research (AMICARE), Uniklinik RWTH Aachen, RWTH Aachen University, Aachen, Germany
- 4Interdisciplinary Center for Clinical Research (IZKF), RWTH Aachen University, Aachen, Germany
- 5Institute for Cardiovascular Prevention (IPEK), Ludwig-Maximilians-University Munich, Munich, Germany
- 6Department of Oral and Maxillofacial Surgery, School of Medicine, Uniklinik RWTH Aachen, Aachen, Germany
- 7Department of Orthodontics, Dental Clinic, Uniklinik RWTH Aachen, Aachen, Germany
- 8Biochemistry Department, Cardiovascular Research Institute Maastricht (CARIM), Maastricht University, Maastricht, Netherlands
Recent demographic developments resulted in an aged society with a rising disease burden of systemic and non-communicable diseases (NCDs). In cardiovascular disease (CVD), a NCD with high morbidity and mortality, recent preventive strategies include the investigation of comorbidities to reduce its significant economic burden. Periodontal disease, an oral bacterial-induced inflammatory disease of tooth-supporting tissue, is regulated in its prevalence and severity by the individual host response to a dysbiotic oral microbiota. Clinically, both NCDs are highly associated; however, shared risk factors such as smoking, obesity, type II diabetes mellitus and chronic stress represent only an insufficient explanation for the multifaceted interactions of both disease entities. Specifically, the crosstalk between both diseases is not yet fully understood. This review summarizes current knowledge on the clinical association of periodontitis and CVD, and elaborates on how periodontitis-induced pathophysiological mechanisms in patients may contribute to increased cardiovascular risk with focus on atherosclerosis. Clinical implications as well as current and future therapy considerations are discussed. Overall, this review supports novel scientific endeavors aiming at improving the quality of life with a comprehensive and integrated approach to improve well-being of the aging populations worldwide.
Introduction
Within the ageing global population, the disease burden of systemic and non-communicable diseases (NCDs) is increasing (1, 2). Cardiovascular disease (CVD) represents most cases in this context and it is globally the leading cause of death, being responsible for 32% of all global deaths in 2019 (3). Hence, defining effective ways for prevention and treatment of CVD is of great importance. This often involves detailed analysis of local and systemic tissue pathogenesis in association with the disease. Among the diseases associated with CVD, periodontitis (PD) is particularly common. PD is a chronic inflammatory disease affecting the tooth-supporting tissues (4) and severe PD is present in approximately 19% of the global population older than 15 years, representing more than 1 billion cases, which makes it one of the most common NCDs (5). By itself, PD has been associated with all-cause mortality as well as with mortality following cardiovascular events (6). By researching influencing factors, the noticeable association of both diseases has been continually established and will be described in detail in this review. Specific focus will be on the missing links of this multifaceted crosstalk that potentially could affect outcome, address therapeutic resistance and open novel avenues for preventive measures in the aging population.
Periodontitis: pathophysiology
General pathophysiology
PD is a disease of the tissues attaching the teeth, where dental plaque accumulation and the propagation of bacterial key pathogens within an oral dysbiosis provoke an escalated inflammatory host response and impaired resolution of inflammation, leading to periodontal tissue damage and subsequent loss of alveolar bone and teeth (7–10) (Figure 1). It is part of the spectrum classifying inflammatory diseases of the periodontium categorized in a collaborative work by the American Academy of Periodontology (AAP) and European Federation of Periodontology (EFP) (11, 12). Its characteristic clinical symptoms comprise gingival bleeding, attachment loss, dental hypermobility, halitosis and subsequent loss of alveolar bone and teeth (11). Besides these local symptoms, PD leads to systemic consequences (10) and its connection to conditions such as type II diabetes mellitus (T2DM), rheumatoid arthritis, obesity and CVD has been of rising interest in the past decades CVD (13–15). Overall, PD has become a global health problem with an economic burden due to the global demographic development with a growth in the aging population and subsequent increasing prevalence in the elderly (16, 17).
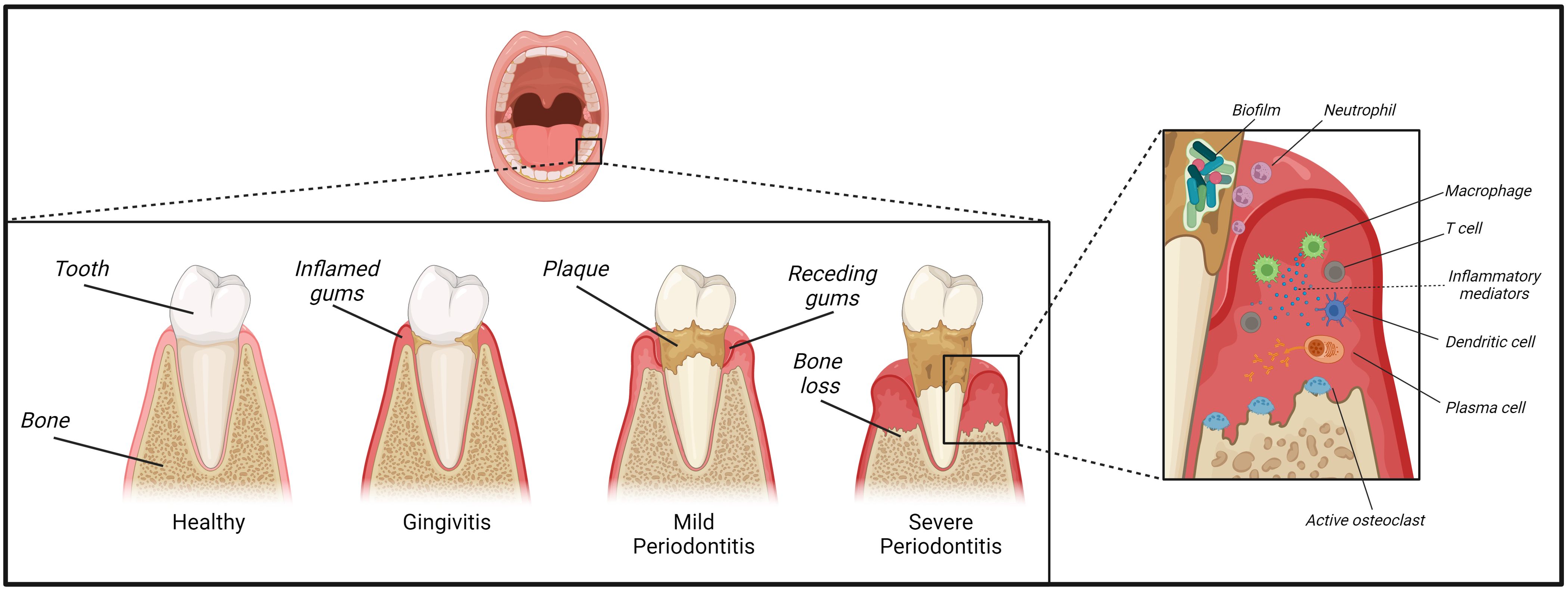
Figure 1. Development and pathophysiology of periodontitis. Plaque deposited on our teeth provides a substrate for a variety of bacterial species. A lack in oral hygiene and insufficient plaque removal from the tooth surfaces and their surrounding junctional gingival tissue can trigger the development of gingivitis. Inflammation can then further progress towards the development of mild to severe periodontitis paralleled by establishment of a dysbiotic biofilm of inflammophilic bacteria. Initially, the periodontal tissue is infiltrated by neutrophils, T-lymphocytes and mononuclear cells such as macrophages, followed by mainly plasma cells in a later stage. These plaques can potentially expand into a destructive advanced lesion inducing loss of periodontal ligament and alveolar bone and thus causing the characteristic clinical symptoms of periodontitis.
Local pathophysiology
The tissue destructive process of PD is associated with the establishment of a plaque-induced dysbiosis (18), subgingival microbial communities, and the presence of responsive immuno-inflammatory infiltrates (14). Opposite to PD as a tissue destructive disease of tooth-supporting tissue, gingivitis is a reversible inflammatory condition of the marginal gingival tissue attachment, which results from plaque accumulation or manifestation of an underlying systemic disease (19). In gingivitis, the inflammatory response to bacterial plaque is contained within epithelium and connective tissue without affecting deeper bony compartments. The prevalent aetiological path is a lack in oral hygiene and insufficient plaque removal from the tooth surfaces and their surrounding junctional gingival tissue (20). This plaque provides a substrate for a variety of bacterial species (21, 22) that are abundant in a planktonic form within the saliva to colonize these dental surfaces and form a matrix that provides protection against elution, phagocytosis, mechanical stress and antimicrobial salivary factors (23). The oral cavity is home to approximately 700 predominant bacterial taxa that only partly have been cultivated in vitro (24).
Gingivitis can, however, be both the precursor, risk factor and accompanying symptom of a later PD (14). Within the maturing biofilm, potentially pathogenic species establish themselves and elucidate a destructive inflammatory response by the immune system. Bacterial presence is required for PD pathogenesis in the susceptible host, however, predominantly an ineffective and dysregulated host response to the microbial challenge drives periodontal tissue damage (24). Non-resolving inflammation and the selective support of dysbiotic inflammophilic communities result in a vicious cycle that is the hallmark of chronic PD and its associated tissue loss (14). In contrast, a tissue-protective immune response fosters balanced host-microbe interactions resulting in tissue homeostasis within a healthy periodontium. Compared to gingivitis or PD, lower numbers of neutrophils as the early responder cells are present together with a small population of γδ T cells and innate lymphoid cells (25, 26).
To describe the developmental process of PD and its associated cellular cascades, Page and Schroeder first established a progressive model, which subsequent studies used to expand on (27, 28). Thereby, PD correlates with an increasingly complex cellular infiltrate- with neutrophils dominating an initial lesion characterized by beginning degradation of perivascular collagen and an exudative vasculitis. Neutrophils arrive from the circulation to the afflicted site and complex molecular interactions with the endothelium involve distinct chemokine- and selectin-induced signaling and adhesive β2 integrins cascades (14). Furthermore, pro-inflammatory mediators such as arachidonic acid derivatives [e.g. Prostaglandins and leukotrienes (29)], and complement activation products, e.g. anaphylatoxins C3a and C5a, can influence neutrophil extravasation and drive pathogenesis of PD (30). Subsequently, inflammation progresses into an early lesion, where periodontal tissue is infiltrated by T-lymphocytes and antigen-presenting cells such as macrophages and dendritic cells (DCs). Ultimately, B and plasma cells dominate the periodontal infiltrate in a fully established lesion, although bone loss does not commence yet (14). This established lesion may remain stable for a prolonged period, but it potentially expands into a destructive advanced lesion inducing loss of periodontal ligament and alveolar bone - thus causing the characteristic clinical symptoms of PD (11, 31).
The oral microbiome
Previous analysis of the oral microbiome in PD patients involved systematization of the bacterial communities based on adaptation to aerobic versus anaerobic conditions and bacterial virulence factors during different stages of disease progression (32). With mineralization of the plaque and its progression into the sulcus, the inflammatory focus moves into the deepening gingival crevice, which corresponds to establishment of an increasingly anaerobic milieu. Thus, presence of Streptococcus species such as S. mitis, S. oralis, S. intermedius and S. gordonii, followed by Actinomyces naeslundii and odontolyticus, Veillonella parvula and Fusobacterium nucleatum and subsequently Porphyromonas gingivalis (P. gingivalis), Treponema denticola and Tannerella forsythia together with Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans) were described in decreasing order. However, recent findings focused on the identification of specific key pathogens (e.g. P. gingivalis) within the polymicrobial community and the establishment of dysbiosis, an alteration in abundance and influence of individual species within the biofilm (14). Keystone pathogens, assisted by accessory pathogens in the provision of nutrients and colonization, can initially escape host immunity, which allows them to establish a dysbiotic microbiota that also includes commensal-turned pathobionts. Ultimately, polymicrobial dysbiotic communities over-activate the inflammatory host response and periodontal tissue loss progresses.
From local to systemic inflammation
Additionally, generalized spread of causative bacteria, bacterial products and mediators of non-resolving inflammatory pathways could compromise systemic tissue homeostasis. Hence, locally produced pro-inflammatory cytokines such as tumor-necrosis factor alpha (TNFα), interleukins (IL)-1 or IL-6 and prostaglandin E2 could move via systemic circulation to distant organs (33) and contribute to cellular disbalance far away from the side of infection. In general, this supports PD`s crosstalk and association with other chronic systemic diseases such as diabetes, obesity, cardiovascular and musculoskeletal diseases (7, 34). The immune reaction shows great individual variability (35) and it is influenced by multiple factors such as genetic, epigenetic, environmental (e.g. smoking, stress, diet) factors, aging and underlying diseases such as T2DM (36), Papillon-Lefèvre syndrome (37), osteoporosis, obesity or specifically atherosclerosis and CVD (13–15).
Cellular and molecular players of the involved immune response
Certain cellular signaling cascades play a special role in the immune reaction. At first, a reaction of the innate immune system is occurring, where bacteria are identified by certain pathogen-associated molecular patterns via pattern-recognition receptors such as toll-like receptors (TLR) of DCs or macrophages. These antigen-presenting cells release a variety of pro-inflammatory cytokines and chemokines and activate surrounding periodontal ligament fibroblasts, thrombocytes and polymorphonuclear leukocytes. The developing cytokine cascade, comprising factors like TNF-α, IL-1β, IL-6, IL-8, IL-12, IL-17 or Prostaglandin E2, amplifies the immunological response (38). TNF-α and IL-1β cause a disinhibition of nuclear factor kappa B (NF-κB), a transcription factor promoting the transcription of pro-inflammatory genes. Subsequently this leads to the expression of matrix metalloproteinases like Collagenase 1 (MMP-1) by macrophages and fibroblasts, Collagenase 2 (MMP-8) by macrophages, macrophage elastase (MMP-12) by T-cells, neutrophils, macrophages and fibroblasts, Gelatinase A (MMP-2) by fibroblasts and Gelatinase B (MMP-9) by neutrophils (39). These MMPs are responsible for the degradation of the extracellular matrix when the resolution of inflammation fails and a chronic condition develops. The IL-17-stimulated expression of the receptor activator of NF-κB ligand (RANKL) by osteoblasts and periodontal ligament cells leads together with macrophage colony-stimulating factor to the activation, fusion and differentiation of osteoclast progenitor cells (40). RANKL is also produced by activated T and B lymphocytes in the inflamed periodontal tissue. Consequently, the balance of osteolysis and osteogenesis is displaced and results in alveolar bone resorption (41). Of note, neutrophils not only support acute but drive chronic inflammation processes by recruiting and stimulating Interleukin-17 release via Th17, CD4+ T helper cells (42). The inflammatory response is counteracted by the network activation of the inflammation-resolution response that is promoted by lipid-metabolic factors such as lipoxins, protectins, maresins, 4-hydroxydocosahexaenoic acid (4-HDHA), 17-HDHA or resolvin E1 (43, 44). For instance, the application of resolvin E1 has been shown to reduce C-reactive protein (CRP), intima-media ratio, leukocyte infiltration, formation of arteriosclerotic plaques, and periodontal inflammation in general (45). Furthermore, immunomodulatory interventions support direct involvement of additional lipid-metabolic factors such as fatty acids and associated ceramides (9), diacylglycerols, triglycerides and stress-reducing lipokines (1,2-dioleoyl-sn-glycero-3-phospho-(1’-myo-inositol) [PI(18:1/18:1)]) (46) in periodontal cellular cascades impacting tissue homeostasis and alveolar bone microarchitecture (9).
In summary, periodontitis involves a chronic oral microbial dysbiosis and overactivation of the host immune response, which ultimately result into a locally destructive lesion triggering a progressive loss of periodontal tissue as well as chronic systemic inflammation.
Cardiovascular disease and atherosclerosis: pathophysiology
General pathophysiology
Atherosclerosis is an important underlying pathology of coronary artery syndrome and refers to the development and progression of lipid-rich, inflammatory lesions in the arterial wall (Figure 2). By severely reducing the vessel lumen or by inducing thrombosis through atherosclerotic plaque rupture or erosion, atherosclerosis can ultimately trigger acute coronary syndrome, myocardial infarction and sudden cardiac death. Mechanistically, atherosclerosis is driven by a combination of hyperlipidemia [with high levels of low-density lipoprotein-cholesterol (LDL-C) and triglycerides but reduced levels of high-density lipoprotein-cholesterol (HDL-C)] and systemic inflammation. Triggered by initial endothelial dysfunction - characterized by endothelial inflammation and increased endothelial permeability -, LDL and inflammatory leukocytes gradually accumulate into the subintimal space of the arterial wall, where they drive vascular inflammation (47). The pro-inflammatory milieu in the vasculature triggers the oxidation of LDL to oxidized LDL (oxLDL), which drives macrophage foam cell formation by oxLDL uptake and pro-inflammatory responses in both macrophages and endothelial cells (47). Also, polymorphonuclear granulocytes or neutrophils infiltrate the vascular wall, where they elucidate inflammatory responses by secreting pro-inflammatory mediators as myeloperoxidase (MPO), produce reactive oxygen species (ROS) and contribute to the recruitment and activation of monocytes/macrophages by secreting chemotactic granule proteins as well as the production of neutrophil extracellular traps (NETs) (48). Lipid overload triggers macrophage death and the formation of necrotic cores with high-inflammatory potential. Smooth muscle cells (SMCs) can migrate from the media into the atherosclerotic lesion to form a protective fibrous cap. However, as for macrophages, lipid loading can trigger a phenotyping switching of SMCs, which increases their inflammatory potential and can induce SMC apoptosis and necrosis (48).
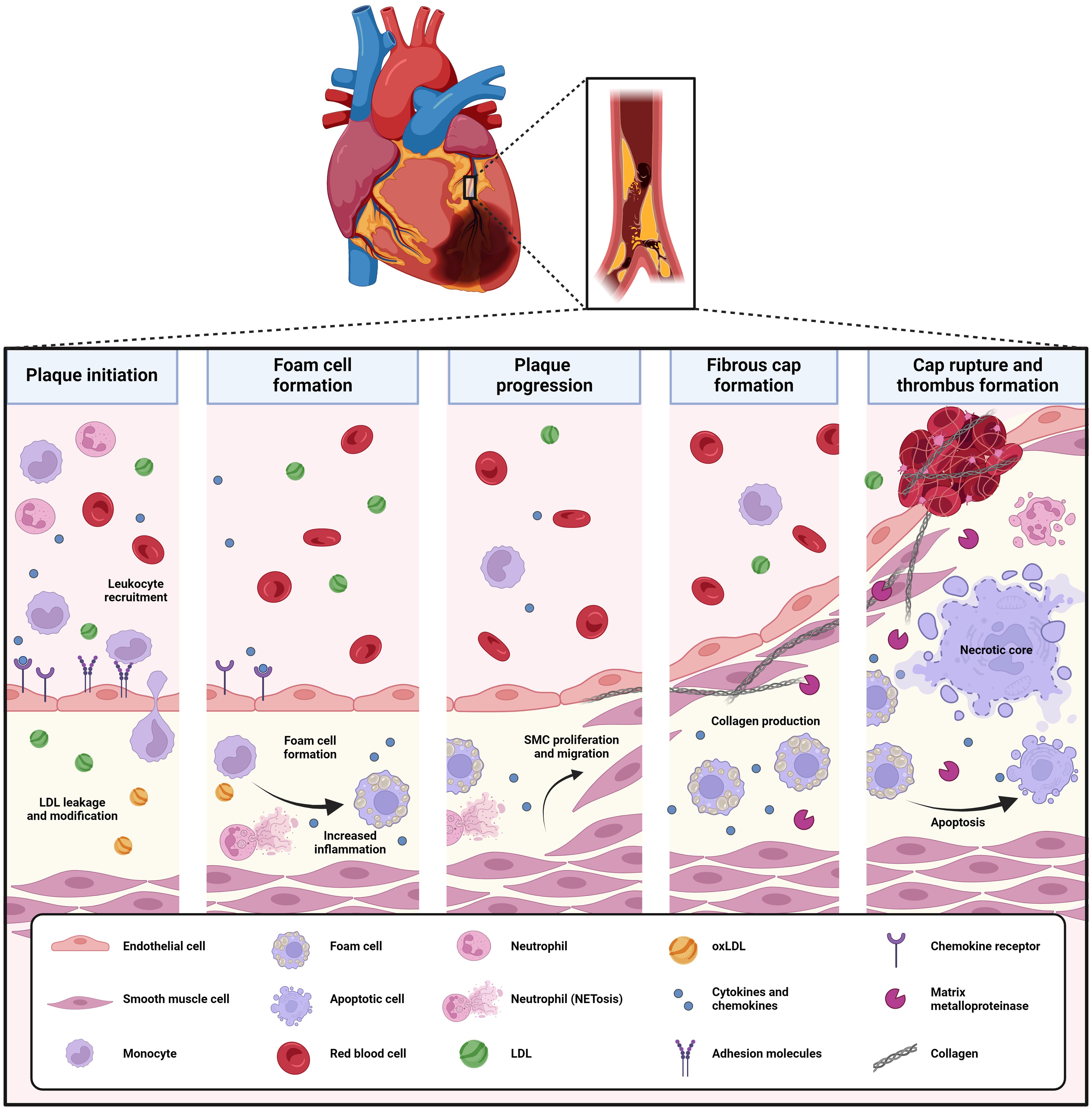
Figure 2. Development and pathophysiology of atherosclerosis. Atherosclerosis is a progressive disease driven by lipid deposition and inflammation into the arterial wall. Initial endothelial dysfunction triggers LDL leakage in the vessel, where it is oxidized to oxLDL. Also, it induces the recruitment and adhesion of leukocytes (neutrophils, monocytes, T-lymphocytes) and their migration into the vascular intima. Here, oxLDL drives monocyte-derived macrophages to lipid uptake and foam cell formation, triggering a pro-inflammatory profile and ultimately, lipid overload and cell death. Neutrophils elucidate inflammatory responses by secreting pro-inflammatory mediators as MPO, produce ROS and contribute to the recruitment and activation of monocytes/macrophages by secreting chemotactic granule proteins as well as the production of NETs. Vascular SMCs migrate from the vessel wall into the developing atherosclerotic lesion, where they form a protective fibrous cap. Upon plaque progression and increasing inflammation, the fibrous cap thins, which makes the atherosclerotic plaque prone to rupture and trigger thrombus formation. LDL, low-density lipoprotein; MPO, myeloperoxidase; NETs, neutrophil extracellular traps; oxLDL, oxidized LDL; ROS, reactive oxygen species; SMCs, smooth muscle cells.
Beyond neutrophils and monocytes/macrophages, also DCs and T-lymphocytes influence atherosclerosis. Among T-cells, Th1 and Th17 cells drive atheroprogression, whereas regulatory T-cells (Tregs) are atheroprotective (47, 49). Also, DCs are found in atherosclerotic lesions, where they contribute to pro-inflammatory cytokine production and Th1 cell polarization (47).
Upon atherosclerotic plaque progression, the production of collagenases and other enzymes degrading extracellular matrix proteins - for example neutrophil elastase produced by neutrophils - triggers the degradation of the fibrous cap, making the plaque increasingly prone to rupture. In such case, the content of the atherosclerotic lesion is exposed to the blood stream, which triggers instant thrombosis (47). On the other hand, plaque disruption with subsequent thrombosis can also be caused by superficial erosion of atherosclerotic lesions, which - relatively to plaque rupture - is increasing in importance due to the improved success of controlling LDL levels in CVD patients (50). Clinical data suggest that plaque erosion may account for around one third of acute coronary syndrome cases (51, 52). Mechanistically, plaque erosion is triggered by a disturbed blood flow triggering TLR2-mediated activation, death and desquamation of the endothelium, followed by neutrophil adhesion and NET formation (53). Plaque erosion occurs also in plaques that have a relatively low lipid-content and inflammatory profile (52).
Endothelial dysfunction, systemic inflammation and innate immune activation as link between CVD and its comorbidities
All combined, this highlights the crucial contribution of endothelial dysfunction and systemic inflammation to atherosclerosis and CVD risk, and explains why comorbidities that impact on each of these processes, also enhance cardiovascular risk. This has for example been shown for chronic kidney disease (CKD), which elicits endothelial dysfunction and a chronic, low-grade systemic inflammation along with increased cardiovascular risk (54–57). Similarly, T2DM as important risk factor for CVD triggers endothelial dysfunction, systemic inflammation and oxidative stress (58) and also caloric intake and obesity induce an increased mobilization of monocytes (59, 60) and neutrophils (59) into the periphery, where these myeloid cells contribute to increased inflammatory and cardiovascular risk. In recent years, an increasing number of studies revealed that inflammatory triggers can prime hematopoietic stem and progenitor cells (HSPCs) in the bone marrow for adaptation towards increased myelopoiesis, a pathological mechanism that has now been recognized to crucially contribute to increased cardiovascular risk induced by cardiovascular risk factors like obesity, hyperlipidemia, chronic stress or sleep interruption, amongst others (61). For example, in obesity, adipose tissue-derived S100A8/A9 triggers IL-1β production by adipose tissue macrophages, which then signals to IL-1 receptor-expressing bone marrow myeloid progenitor cells to increase neutrophil and monocyte production (59). Chronic stress triggers sympathetic nervous system activation and noradrenaline production, which signals to bone marrow niche cells over the β3 adrenergic receptor to reduce CXCL12 production in the bone marrow and increase HSPC proliferation, resulting in a higher production of monocytes (monocytosis) and neutrophils (neutrophilia) (62). Of note, HSPCs can retain this capacity for increased myelopoiesis for a prolonged time even after resolution of the initial trigger. This process - referred to as ‘trained myelopoiesis’ - has now been acknowledged as a very important aspect of maladaptive innate immune adaptation. Mechanistically, it can be induced by epigenetic changes in HSPC triggering bone marrow memory. For example, hyperlipidemia was shown to trigger long-lasting NLRP3-dependent transcriptional and epigenetic reprogramming of granulocyte-monocyte progenitor cells with increased responsiveness to inflammatory triggers (63).
In summary, hyperlipidemia, endothelial dysfunction, systemic inflammation and innate immune activation are important drivers of atherosclerosis.
Clinical association of periodontitis and atherosclerosis
The association between cardiovascular and periodontal disease is supported by an increasing evidence reviewed by Sanz et al. in a consensus report based on a collaborative work of AAP and EFP (64). When PD is present, the risk to develop CVD is generally increased, being possibly raised by 25%, and the likelihood to suffer particularly from a myocardial infarction (MI) or to develop a first MI is elevated too (65–67).
Furthermore, edentulism as a severe symptom of chronic PD is associated with CVD as well (6, 68, 69), however other causes of edentulism besides PD exist. On the contrary, a comparatively lesser degree of tooth loss correlates favorably with systolic blood pressure, glomerular filtration rate, levels of blood glucose or LDL-C, waist circumference and high-sensitivity C-reactive protein (hs-CRP), risk factors and parameters of CVD and metabolic syndrome (70). The degree of severity, represented by the stage of PD, also associates with the risk for CVD (71) and older patients with CVD reversely show signs of periodontal infection (72). Furthermore, patients suffering heart failure, a pro-inflammatory syndrome with multi-organ involvement caused by the inability of the heart to meet the metabolic demands of the body, exhibit more severe periodontal disease associated with increased bone turnover markers when compared with control patients (73).
As a limitation of these clinical studies, the co-existence of periodontitis and CVD may be triggered by shared risk factors, such as smoking, obesity, T2DM and chronic stress. These factors influence the progression of both PD (14) and atherosclerotic CVD (74), and may thus be potential confounders in the association of both diseases.
From shared risk factors to PD-induced pathophysiological effects contributing to atherosclerotic risk
However, in addition to a simultaneous development due to shared risk factors, PD pathogenesis could also by itself be responsible for the detected associations since the oral dysbiosis could influence cardiovascular health and existing atherosclerotic lesions directly and indirectly, as discussed below and visualized in Figure 3.
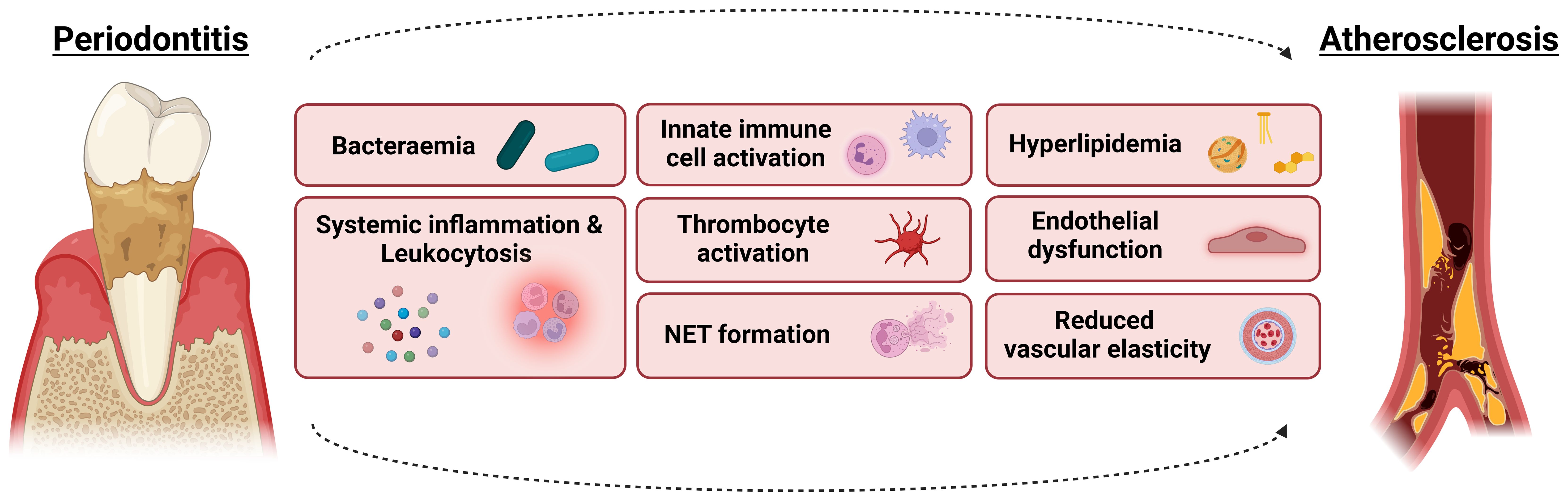
Figure 3. From shared risk factors to periodontitis-induced pathophysiological effects contributing to atherosclerotic risk. The co-existence of periodontitis and atherosclerosis may be triggered by shared risk factors such as smoking, obesity, T2DM and chronic stress. However, the pathogenesis of periodontitis could also by itself be responsible for the detected associations since the oral dysbiosis could influence cardiovascular health and existing atherosclerotic lesions directly and indirectly. Involved mechanisms include bacteraemia, systemic inflammation and leukocytosis, innate immune cell activation, hyperlipidemia, endothelial dysfunction and reduced vascular elasticity, thrombocyte activation as well as the formation of neutrophil extracellular traps (NETs). For more details, see text.
Bacteraemia
Periodontal tissues are intensely perfused and the hyperaemia in case of inflammation could cause both clinically visible bleeding on probing and bacteraemia, where microbes originating in the dental biofilm enter the bloodstream through vascular lesions. In patients with PD, nearly half of them showed bacteraemia after therapeutic periodontal treatment (75). In this way, periodontal bacteria can spread throughout the body, where they can promote inflammatory processes by disrupting immune surveillance mechanisms of the host, as discussed in detail elsewhere (4, 76, 77).
For example, the prominent parodontopathogens A. actinomycetemcomitans and P. gingivalis could be detected within7nbsp;atherosclerotic plaques and viable specimens of A. actinomycetemcomitans and P. gingivalis have been isolated from arteriosclerotic tissue (78–80). CVD patients showed a comparatively more extensive and diverse microbial infection of atheromatous plaques in case of comorbidity with PD (81). Also, PD altered the gut microbiota in an atherosclerosis mouse model and in parallel increased intestinal permeability and atherosclerosis severity, suggesting a role for altered gut microbiota in the link between PD and atherosclerosis (82).
Systemic inflammation
Furthermore, the quality of a systemic inflammatory condition becomes clear when leukocyte numbers and levels of pro-inflammatory markers are determined. Patients with PD displayed increased leukocyte numbers in blood (83) and a large prospective study with a follow-up of 11 years revealed that white blood cell count was longitudinally associated with periodontal disease severity (84). Furthermore, serum concentrations of CRP were elevated in patients affected by PD (85), but also in cases of PD and acute coronary syndrome (86). Moreover, interleukin 6 (IL-6), being a pro-inflammatory cytokine inducing CRP production and mediator of the acute phase response to infection, has been described to increase in case of PD, which might influence comorbidities (87–89). Levels of hs-CRP are used in the clinic to quantify cardiovascular risk that is remaining after intensive lipid-lowering therapy, being referred to as ‘residual inflammatory risk’ (90), and high hs-CRP levels in patients without prevalent CVD were shown to be associated with future adverse vascular events (91). Also, in the CANTOS trial, MI patients that showed high levels of IL-6 despite successful IL-1β blocking suffered from a significant residual inflammatory risk (92), suggesting an important contribution of IL-6 to future cardiovascular events.
Besides CRP, serum amyloid A (SAA) is an important acute phase protein. It supports the cellular inflammatory reaction by its chemotactic function for neutrophils and monocytes, it promotes production of pro-inflammatory cytokines via TLR2 signaling in human gingival fibroblasts and inhibits neutrophil apoptosis (93, 94). Thus, as its involvement in periodontal inflammation has been described (95, 96), increased levels of SAA in serum and gingival crevicular fluid are highly associated with chronic PD (97, 98).
Results from an in vitro study on human aortic endothelial cells suggest the SAA-induced expression of endothelial adhesion molecules through TLR2 receptors as a possible mechanism linking PD and atherosclerosis (99). In patients suffering from comorbid CVD and PD, SAA levels have been found to be significantly increased, along with an accumulation of SAA and CRP in samples from carotid atheroma (100). The furthermore noted interactions between SAA and HDL indicate that SAA is involved in lipoprotein regulation, which has been identified as an important element in the pathogenesis of atheromata (101–103).
Innate immune cell activation
Peripheral blood mononuclear cells from PD patients showed increased expression of pro-inflammatory molecules (such as TNF-α and CCR2), as revealed by single-cell RNAseq (104). Monocytes also demonstrated long-term activation upon exposure to P. gingivalis, with a prior microbial infection triggering increased production of IL-6 and TNF-α upon a secondary inflammatory stimulus six days later (83). Furthermore, neutrophils from PD patients showed a hyperreactivity upon stimulation with either LPS or P. gingivalis, with increased secretion of IL-8, IL-6, TNF-α and IL-1β (105), increased ROS production in basal conditions (106) as well as upon stimulation of the FcγR (107) and an increased interaction with endothelium (106). Such increased FcγR-triggered ROS production could also be induced upon priming healthy neutrophils with type I interferon (IFN). In this context, PD patients revealed enhanced circulating levels of IFN-α and an increased expression signature of type I IFN-induced genes in peripheral blood neutrophils. Furthermore, periodontitis treatment could reduce IFN-α plasma levels, and FcγR-stimulated neutrophil ROS production to levels observed in controls (108).
PD patients also show an increased frequency of myeloid dendritic cells (myeloid DCs) in blood, and P. gingivalis could stimulate monocyte differentiation to immature mDCs in vitro (109). Myeloid DCs from patients with chronic PD as well as P. gingivalis-infected DCs in vitro display an upregulated expression of the homeostatic chemokine receptor CXCR4 but a downregulation of CCR7. Whereas CCR7 confers homing capacity of DCs to secondary lymphoid organs via CCL19, CXCR4-positive cells migrate towards the CXCR4 ligand CXCL12, which was shown to be increasingly expressed by P. gingivalis-infected endothelium in vitro (110). Also, P. gingivalis-infected DCs showed increased expression of the chemokine receptor CCR2 (109), an enhanced inflammatory profile (e.g. with increased expression of C1q, CXCL16 and HSP60/70), a higher secretion of atherosclerotic plaque-destabilizing MMP-9 (109) as well as a downregulated apoptosis and autophagy (111). Combined, this may contribute to enhanced microbial dissemination, atherosclerotic plaque recruitment and inflammatory capacity. Myeloid DCs infected by P. gingivalis have also been detected in atherosclerotic lesions within coronary artery biopsies of PD patients with coronary artery disease (109).
Hyperlipidemia
Patients with PD display increased levels of LDL, oxLDL and triglycerides but reduced levels of HDL, as concluded from a prior meta-analysis study (112). In addition, also structural modifications of both HDL and LDL/VLDL indicative of more pro-inflammatory forms of these lipoprotein particles have been identified in PD patients (113). Post-translational modifications of lipoprotein particles have been increasingly detected in different patient cohorts, including in patients with severe CVD (114) and patients with chronic kidney disease (115). Both in CVD and CKD, these modifications were able to turn LDL into an even more atherogenic lipoprotein particle and modify HDL from a cardioprotective into a detrimental molecule, which contributed to increased cardiovascular risk (114–116).
Reduced vascular elasticity and endothelial dysfunction
There are different possible parameters to assess cardiovascular health and investigating how those are potentially affected by PD allows to discern further connections, examples being an observed higher systolic blood pressure in cases of tooth loss or an augmented likelihood for hypertension in general (117, 118). Considering the function of blood vessels, an association of PD and dysfunction of the vascular endothelium as well as a sclerotization and increased thickness of the intima and media of the carotid artery has been described (119, 120). Further evidence exists for an increase in pulse wave velocity accompanying PD, indicating a loss in vascular elasticity and thus an increased risk of CVD (121, 122). The regenerative capacity of the endothelium seems to be impaired by the systemic inflammation too: The number of endothelial progenitor cells negatively correlates with clinical parameters of periodontal health like number of teeth, probing depth, bleeding on probing and clinical attachment loss (123). Pathologically elevated serum levels of homocysteine are described as a possible causal cofactor of endothelial damage and atherosclerosis (124, 125). Hyperhomocysteinaemia has been found in cases of PD, adding another possible path of cardiovascular damage linked to periodontitis (126).
Thrombocyte activation and neutrophil extracellular traps
The expression of CD40-ligand (CD40L) on the surface of thrombocytes is promoted by pathogenic species, especially an infection with P. gingivalis is associated with elevated levels of CD40L through TLR 2 and 4 (127). The ligand of CD40 plays a significant role in the development of atherosclerosis and the disintegration of atherosclerotic plaques (128, 129). It enhances haemostasis by stimulating the expression of tissue factor on endothelial cells and macrophages. In addition, increased levels of soluble CD40L associate with myocardial infarction and worsened clinical outcome after acute ischemic stroke (130), making it a potential biomarker for acute cardiovascular events (131).
Additionally, P. gingivalis infection promotes thrombocyte aggregation by increasing platelet expression of P-selectin and induces the formation of NETs (132). NETs emerge when neutrophil granulocytes release chromatin and granule proteins and they serve as an innate defense mechanism to catch bacteria, hindering their sprawl and facilitating their exposure to antimicrobial factors (133). Recent findings support a direct interaction of parodontopathogenic species and their products with NETs, possibly evading or neutralizing the antimicrobial potential of the latter. Several species including the potent triad of P. gingivalis, Tannerella forsythia and Treponema denticola within the red complex as well as A. actinomycetemcomitans and Prevotella intermedia have been identified to produce DNAses which degrade NETs (134–136). P. gingivalis, in particular, is able to citrullinate NET proteins via a peptidylarginine deiminase, neutralizing their positive charge and thereby their pathogen-binding property (137). Despite their primary anti-infective purpose, NETs show possibly detrimental effects too, for instance by promoting activation of factor XII and thrombocytes and formation of atherosclerotic plaques, specifically, if their presence is prolonged and they remain in lesions for an extended period (138, 139). Patients with PD showed increased concentrations of extracellular DNA, nucleosomes and NET-associated myeloperoxidase (MPO) and neutrophil elastase in saliva (140). Moreover, the decomposition of NETs is impaired in patients with untreated PD (141) and, conversely, is amplified after periodontal therapy (142). Nevertheless, detailed information about their specific role in PD and its systemic impact, also in relation to PD-associated CVD, is still missing and requires further investigation.
All combined, these studies reveal an important impact of PD on pathophysiological processes that drive the development and progression of atherosclerotic CVD.
Animal studies support a contribution of periodontal disease to atherosclerosis
Combined PD-atherosclerosis model
Given the many confounding factors that can jointly influence PD and atherosclerotic CVD, animal models are essential to demonstrate a causal role of PD to atherosclerosis and to reveal underlying pathophysiological mechanisms. In such animal models, atherosclerosis models are combined with PD induced either by a ligature around the posterior teeth (ligature-induced PD) or by oral gavage with human periodontal pathogens, mostly P. gingivalis. Although these models cannot reproduce the natural progression of periodontitis in humans, they enable to investigate the contribution of pathophysiological mediators to pathophysiological mechanisms underlying periodontitis development and/or progression. Overall, PD promoted atherosclerosis in experimental models with mice (80, 143, 144), rabbits (145) as well as pigs (146). In mice, the most applied atherosclerosis models were Apolipoprotein E-deficient (ApoE-/-) mice on high-fat diet, although also C57BL/6J wild-type mice on long-term high-fat diet as model of metabolic disturbance have been studied.
Dyslipidemia
Mechanistically, experimental PD enhanced circulatory levels of triglycerides (143, 147, 148), total cholesterol (147, 148) and more specifically VLDL in hyperlipidemic ApoE-/- mice (143), as well as of oxidized LDL (80, 143). Others observed also a reduction of HDL levels upon infection of mice with P. gingivalis (149). However, such effects on HDL or lipid levels in general were not observed in all studies, with for example another study not observing lipid changes in either wild-type or hyperlipidemic ApoE-/- mice subjected to ligature-induced PD (144), or also HDL remaining unaltered in other studies of experimental PD (147). In relation to PD-induced changes in lipid metabolism (and more specifically increased cholesterol and triglyceride levels), it could be shown that ligature-induced PD in hyperlipidemic ApoE-/- mice triggered the colonization of the oral cavity and the liver with F. nucleatum, which subsequently promoted glycolysis and thereby lipogenesis in hepatocytes (148).
Vascular inflammation and endothelial dysfunction
Experimental PD induced vascular inflammation with increased expression of IL-6, TNF-α and the endothelial activation marker VCAM-1, as well as with increased activation of the prototypical pro-inflammatory transcription factor NF-κB (150). Experimental PD also triggered endothelial dysfunction in rats, evidenced by a reduced endothelial-dependent vasodilatation (151).
In vitro, P. gingivalis triggered endothelial inflammation and a dysregulation of the circadian clock genes via TLR-NF-κB signaling, resulting in increased oxidative stress and inflammatory signaling in endothelial cells (152). P. gingivalis also induced the shedding of microvesicles by endothelial cells, with a subsequent pro-inflammatory effect towards the endothelium (153). Furthermore, P. gingivalis induced endothelial mitochondrial dysfunction, evidenced by an increase in mitochondrial fragmentation and ROS production vs. reduced mitochondrial DNA copy numbers and ATP levels (154). P. gingivalis also increased endothelial permeability in vitro and in zebrafish larvae by reducing the expression of PECAM1 and VE-Cadherin (76), both important gatekeepers of endothelial cell junctional integrity.
On the other hand, endothelial inflammation in C57BL/6J mice on high-fat diet and infected by P. gingivalis was only observed after 28 weeks along with the onset of lipid deposition in aorta and early-stage atherosclerosis, without bacterial infection found in the aortic endothelium itself (155). Instead, P. gingivalis did colonize perivascular adipose tissue (PVAT) and triggered PVAT inflammation, increasing the inflammatory milieu with increased Th1 but fewer Treg T-cells, increased levels of pro-inflammatory adipokines (IFNγ, leptin, resistin) but reduced levels of the anti-inflammatory adipokine adiponectin. PVAT inflammation preceded endothelial inflammation and early atherosclerosis in this mouse model, suggesting a potential contribution to atherosclerosis development by increasing systemic inflammation (155).
Treatment with an anti-inflammatory peptide (156) or with rosuvastatin (157) - which not only lowers LDL cholesterol but is also anti-inflammatory - counteracted both PD-induced vascular inflammation and lipid deposition in hyperlipidemic ApoE-/- mice. Also, topical application of resolvin E1 - a specialized proresolving lipid – prevented PD and reduced systemic inflammation (CRP levels in blood) as well as atherosclerosis upon P. gingivalis infection of rabbits on high-cholesterol diet (45).
Increased systemic inflammation, innate immune activation and trained innate immunity
(Transiently) increased systemic levels of the pro-inflammatory markers CRP, IL-6 and IL-1β have been observed upon ligation-induced PD (144, 151), with stronger effects in hyperlipidemic ApoE-/- mice compared to wild-type mice on high-fat diet (144). Instead, the atheroprotective factor nitric oxide - which induces vasodilatation and confers endothelial protective properties - was significantly reduced in blood in experimental PD (80, 143).
That experimental PD increased systemic inflammation was also evident from the raised activation status of innate immune cells. PD activated monocytes/macrophages, with increased expression of TNF-α and IL-6 in circulating mononuclear cells of rats subjected to PD (150). In these PD rats, monocytes were also more prone to adhere to endothelium both in vitro as in vivo, with PD-derived monocytes able to induce pro-inflammatory NF-κB activation in endothelial cells (150). Also, P. gingivalis triggered the expression of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in both endothelial cells and monocytes (158), with LOX-1 known to contribute to atherosclerosis by inducing endothelial inflammation and monocyte-endothelial interaction (159). Furthermore, P. gingivalis enhanced macrophage foam cell formation by increasing the uptake of oxLDL (160) and blocking lipid efflux (161).
Beyond increased innate immune cell activation, PD was recently shown to trigger inflammation-induced trained myelopoiesis: Ligature-induced PD triggered myelopoiesis in the bone marrow along with transcriptional changes in HSPC in the bone marrow. After resolution of PD and local periodontal inflammation, a secondary systemic challenge with LPS triggered increased myelopoiesis in bone marrow and myeloid cell recruitment to the periphery compared to mice that were previously not subjected to PD (162). Also, ex vivo, neutrophils and monocytes from PD-trained mice produced more IL-6 and TNF-α upon a secondary LPS challenge compared to mice that were not previously subjected to PD (162). Mechanistically, long-lasting epigenetic rewiring was identified in HSPC of PD-trained mice (162, 163), with global hypomethylation (163) - generally associated with increased gene transcription - and increased chromatin accessibility of genes suggestive of a biased differentiation towards myeloid cells upon a secondary stimulus as well as an increased pro-inflammatory profile in granulocyte-myeloid progenitor cells (162). Such PD-induced maladaptive training of myelopoiesis was observed along with increased IL-1β levels in the bone marrow and could be shown to be dependent on IL1R-signaling in HSPC. Of note, PD-induced maladaptive innate immune training triggered the susceptibility of the development of other inflammatory comorbidities, as shown for experimental arthritis upon bone marrow transfer of PD-trained bone marrow (162).
Alterations in adaptive immune cells
Besides effects on myeloid cells, P. gingivalis infection increased Th17 cells but downregulated Treg cells in ApoE-/- mice on high-fat diet, along with increasing atherosclerosis. Whereas Tregs are atheroprotective, Th17 lymphocytes contribute to inflammation and plaque formation by the production of IL-17A (49).
Altered gut homeostasis
P. gingivalis-enhanced atherosclerosis in hyperlipidemic ApoE-/- mice was associated with changes in the intestinal microflora (82, 164), increased intestinal permeability (82), as well as with raised systemic levels of LPS and TMAO (164) or altered metabolites indicating changes in lipid metabolism and primary bile acid synthesis (82). TMAO is an oxidation product of the gut-derived metabolite TMA, and is associated with higher cardiovascular risk by increasing inflammation (165).
All combined, animal models support a mechanistic contribution of PD to atherosclerosis by driving pathophysiological mechanisms that are known to contribute to the development and progression of atherosclerotic lesions, including endothelial dysfunction, systemic inflammation and innate immune cell activation.
Beyond atherosclerosis, PD also increases the risk of cardiovascular risk factors as diabetes and chronic kidney disease
Beyond aggravating atherosclerosis, PD also enhances the progression of chronic kidney disease through inflammatory stimuli and oxidative stress (166). Experimental PD led to kidney inflammation with induction of IL-1β and TNF-α, and of tubular and glomerular injury, as shown in ApoE-/- mice injected with P. gingivalis-LPS (147). Furthermore, the combination of PD and hyperlipidaemia aggravated circulating cholesterol and triglyceride levels as well as kidney inflammation in a synergistic way, with a strong increase in neutrophil infiltration, IL-1β and TNF-α expression in kidney tissue beyond levels reached by each disease alone (147).
Also, experimental PD induced in C57BL/6J mice by infection with the periodontal bacterial species Aggregatibacter actinomycetemcomitans - a low-abundance gram-negative periodontal pathobiont - triggered the development of glucose tolerance impairment and insulin resistance along with alterations in the gut microbiota, and - under high-fat diet - induced stronger hepatic steatosis (167). These observations may contribute to the known association of PD and diabetes (168).
However, additional experimental studies are required to pinpoint in more detail the key mediators and mechanisms underlying the association of PD with the aforementioned morbidities.
Clinical implications & current and future therapy considerations
The effect of PD treatment on cardiovascular health has been assessed in a current S3 guideline (169). In patients without systemic co-morbidities, therapy of PD may reduce the overall cardiometabolic risk and systemic inflammatory biomarkers. For patients affected by a co-morbid NCD, periodontal treatment is suggested for a favorable influence on cardiovascular risk, metabolic control and systemic inflammation based on moderate evidence. However, in case of a co-morbid NCD, it is unclear whether periodontal therapy improves the outcome in severe cases of NCD, as there is no evidence supporting a significant effect on cardiovascular events (169).
As connections in the pathophysiology of CVD and PD have been established, it suggests itself that the therapy of one ailment might prove beneficial for the other. Evidence on the influence of periodontal therapy on vascular function is diverging. An improvement in endothelial function after therapy of PD in patients affected by ST-elevation myocardial infarction has been described (170–173), while other authors found no significant correlation (174, 175). In addition, even after successful therapy the risk for an adverse cardiovascular event remains elevated (176). Periodontal treatment attenuates systemic inflammation, which becomes visible through an improvement of particular inflammatory markers. Accordingly, a lowered concentration of CRP following treatment of PD could be shown (170, 177–182). Blood pressure, IL-6, IL-8 and IFN-γ correlate in a similar way (171, 177, 181, 183). The described amelioration of cardiovascular risk factors by periodontal therapy corroborates the previously suggested correlations between both diseases.
Overall, due to manifold influencing factors, both diseases cannot be completely controlled. On the other hand, the identified disease connections discussed here open various therapeutic ways by targeting inflammation and innate immune activation as link between PD and CVD risk. In these areas, the cardiovascular field is currently investing strong efforts towards clinical translation opportunities. This has been stimulated by the beneficial, lipid-independent effects of IL-1β blockade on cardiovascular outcome in patients with a previous myocardial infarction and high hs-CRP levels in the CANTOS trial (184). These findings boosted an increased focus on inflammation and innate immune activation as targets of therapy in CVD, as recently discussed in detail elsewhere (57, 185). For periodontitis, novel treatments based on the connection between periodontal inflammation, immune-regulatory signaling cascades and the polymicrobial biofilm in the periodontal microenvironment will complement existing anti-infectious therapies and thereby improve clinical outcome (186–188). These current emerging immunomodulatory therapies address inflammation-resolution, relieve inflammation, reduce platelet aggregation and establish a viable local immune microenvironment with subsequent tissue regeneration. Ultimately, this will also be of benefit to address the pathophysiological interaction between PD and atherosclerosis. Overall, unraveling key molecular mediators underlying PD-induced atherosclerosis in further detail may thereby further expand therapeutic options in the future and help to resolve the huge burden of increased cardiovascular risk in PD.
Conclusions
Taken together, mechanistic and animal studies support a pathophysiological interaction between PD and atherosclerosis beyond sharing of the same risk factors, with PD driving pathophysiological processes that support the development and progression of atherosclerosis. Inflammation and innate immune activation are a central hub in connecting both diseases. Furthermore, they may also represent a key connector of PD and other inflammation-associated diseases such as chronic kidney diseases and diabetes. It is expected that future developments in these fields – as currently highly being investigated in the area of CVD – can also contribute to alleviating comorbid disease presentation. By providing an overview of the current understanding of PD, atherosclerosis and their crosstalk, this review aims to support future research efforts that can uncover novel mechanistic insights into comorbidity development in PD as important step towards advancing clinical translation opportunities.
Author contributions
US-S: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. LW: Writing – original draft. EV: Visualization, Writing – review & editing. FH: Writing – review & editing. RB: Writing – review & editing. MW: Writing – review & editing. HN: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the German Research Foundation (DFG) Project-ID 322900939 – SFB/TRR219 (to HN and EV), Project-ID 403224013 – SFB 1382 (to HN); the “Else Kröner-Fresenius-Stiftung” (Project 2020_EKEA.60 to HN), a grant from the Interdisciplinary Centre for Clinical Research within the faculty of Medicine at the RWTH Aachen University (PTD 1-12 to HN and EV) as well as by the DZHK (German Centre for Cardiovascular Research) (funding code: B23Ex to HN). The project was supported by the Interdisciplinary Center of Clinical Research of the Medical Faculty Jena (IZKF UKJ FF02), the German Federal Ministry of Education and Research (BMBF: 01EC1901B, Project 2), Foundation „Else Kröner Fresenius Stiftung” within the Graduate Program „Else Kröner Promotionskolleg JSAM”, Interdisciplinary Center of Clinical Research of the Medical Faculty Jena (Doctoral Program). Open access funding provided by the Open Access Publishing Fund of RWTH Aachen University.
Conflict of interest
H. Noels is founding shareholder of AMICARE Development GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. (2020) 396:1204–22. doi: 10.1016/s0140-6736(20)30925-9
2. United Nations Department of Economic and Social Affairs, P.D.. World Population Prospects 2022: Summary of Results. (2022) UN DESA/POP/2022/TR/NO. 3.
3. World health organization. Cardiovascular diseases (CVDs) (2021). Available online at: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
4. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. (2015) 15:30–44. doi: 10.1038/nri3785
5. Geneva World Health Organization. Global oral health status report: towards universal health coverage for oral health by 2023. (2022).
6. Romandini M, Baima G, Antonoglou G, Bueno J, Figuero E, Sanz M. Periodontitis, edentulism, and risk of mortality: A systematic review with meta-analyses. J Dent Res. (2021) 100:37–49. doi: 10.1177/0022034520952401
7. Martinez-Garcia M, Hernandez-Lemus E. Periodontal inflammation and systemic diseases: an overview. Front Physiol. (2021) 12:709438. doi: 10.3389/fphys.2021.709438
8. Gasmi Benahmed A, Kumar Mujawdiya P, Noor S, Gasmi A. Porphyromonas gingivalis in the development of periodontitis: impact on dysbiosis and inflammation. Arch Razi Inst. (2022) 77:1539–51. doi: 10.22092/ari.2021.356596.1875
9. Döding A, Hüfner M, Nachtsheim F, Iffarth V, Bölter A, Bastian A, et al. Mediterranean diet component oleic acid increases protective lipid mediators and improves trabecular bone in a Porphyromonas gingivalis inoculation model. J Clin Periodontol. (2023) 50:380–95. doi: 10.1111/jcpe.13751
10. Döding A, Zimmermann S, Maghames A, Reimann M, Symmank J, Thürmer M, et al. Immunometabolic capacities of nutritional fatty acids in regulation of inflammatory bone cell interaction and systemic impact of periodontal infection. Front Immunol. (2023) 14:1213026. doi: 10.3389/fimmu.2023.1213026
11. Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol. (2018) 89 Suppl 1:S173–s182. doi: 10.1002/jper.17-0721
12. Sanz M, Herrera D, Kebschull M, Chapple I, Jepsen S, Berglundh T, et al. Treatment of stage I–III periodontitis—The EFP S3 level clinical practice guideline. J Clin Periodontology. (2020) 47:4–60. doi: 10.1111/jcpe.13290
13. Graves DT, Correa JD, Silva TA. The oral microbiota is modified by systemic diseases. J Dent Res. (2019) 98:148–56. doi: 10.1177/0022034518805739
14. Hajishengallis G, Chavakis T, Lambris JD. Current understanding of periodontal disease pathogenesis and targets for host-modulation therapy. Periodontol 2000. (2020) 84:14–34. doi: 10.1111/prd.12331
15. Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. (2021) 21:426–40. doi: 10.1038/s41577-020-00488-6
16. Cui Y, Tian G, Li R, Shi Y, Zhou T, Yan Y. Epidemiological and sociodemographic transitions of severe periodontitis incidence, prevalence, and disability-adjusted life years for 21 world regions and globally from 1990 to 2019: An age-period-cohort analysis. J Periodontol. (2023) 94:193–203. doi: 10.1002/JPER.22-0241
17. Trindade D, Carvalho R, MaChado V, Chambrone L, Mendes JJ, Botelho J. Prevalence of periodontitis in dentate people between 2011 and 2020: A systematic review and meta-analysis of epidemiological studies. J Clin Periodontol. (2023) 50:604–26. doi: 10.1111/jcpe.13769
18. Curtis MA, Diaz PI, Van Dyke TE. The role of the microbiota in periodontal disease. Periodontol 2000. (2020) 83:14–25. doi: 10.1111/prd.12296
19. Chapple ILC, Mealey BL, Van Dyke TE, Bartold PM, Dommisch H, Eickholz P, et al. Periodontal health and gingival diseases and conditions on an intact and a reduced periodontium: Consensus report of workgroup 1 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Periodontol. (2018) 89 Suppl 1:S74–s84. doi: 10.1002/jper.17-0719
20. Kinane DF. Causation and pathogenesis of periodontal disease. Periodontol 2000. (2001) 25:8–20. doi: 10.1034/j.1600-0757.2001.22250102.x
21. Abusleme L, Hoare A, Hong BY, Diaz PI. Microbial signatures of health, gingivitis, and periodontitis. Periodontol 2000. (2021) 86:57–78. doi: 10.1111/prd.12362
22. Iniesta M, Chamorro C, Ambrosio N, Marín MJ, Sanz M, Herrera D. Subgingival microbiome in periodontal health, gingivitis and different stages of periodontitis. J Clin Periodontol. (2023) 50:905–20. doi: 10.1111/jcpe.13793
23. Jakubovics NS, Goodman SD, Mashburn-Warren L, Stafford GP, Cieplik F. The dental plaque biofilm matrix. Periodontol 2000. (2021) 86:32–56. doi: 10.1111/prd.12361
24. Krishnan K, Chen T, Paster BJ. A practical guide to the oral microbiome and its relation to health and disease. Oral Dis. (2017) 23:276–86. doi: 10.1111/odi.12509
25. Dutzan N, Konkel JE, Greenwell-Wild T, Moutsopoulos NM. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. (2016) 9:1163–72. doi: 10.1038/mi.2015.136
26. Moutsopoulos NM, Konkel JE. Tissue-specific immunity at the oral mucosal barrier. Trends Immunol. (2018) 39:276–87. doi: 10.1016/j.it.2017.08.005
27. Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest. (1976) 34:235–49.
28. Hajishengallis G, Korostoff JM. Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontol 2000. (2017) 75:116–51. doi: 10.1111/prd.12181
29. Capra V, Rovati GE, Mangano P, Buccellati C, Murphy RC, Sala A. Transcellular biosynthesis of eicosanoid lipid mediators. Biochim Biophys Acta. (2015) 1851:377–82. doi: 10.1016/j.bbalip.2014.09.002
30. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
31. Page RC, Eke PI. Case definitions for use in population-based surveillance of periodontitis. J Periodontol. (2007) 78:1387–99. doi: 10.1902/jop.2007.060264
32. Belibasakis GN, Belstrom D, Eick S, Gursoy UK, Johansson A, Kononen E. Periodontal microbiology and microbial etiology of periodontal diseases: Historical concepts and contemporary perspectives. Periodontol 2000. (2023). doi: 10.1111/prd.12473
33. Cecoro G, Annunziata M, Iuorio MT, Nastri L, Guida L. Periodontitis, low-grade inflammation and systemic health: A scoping review. Medicina (Kaunas). (2020) 56. doi: 10.3390/medicina56060272
34. Wang CJ, McCauley LK. Osteoporosis and periodontitis. Curr Osteoporos Rep. (2016) 14:284–91. doi: 10.1007/s11914-016-0330-3
35. Bamashmous S, Kotsakis GA, Kerns KA, Leroux BG, Zenobia C, Chen D, et al. Human variation in gingival inflammation. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2012578118
36. Preshaw PM, Alba AL, Herrera D, Jepsen S, Konstantinidis A, Makrilakis K, et al. Periodontitis and diabetes: a two-way relationship. Diabetologia. (2012) 55:21–31. doi: 10.1007/s00125-011-2342-y
37. Giannetti L, Apponi R, Dello Diago AM, Jafferany M, Goldust M, Sadoughifar R. Papillon-Lefevre syndrome: Oral aspects and treatment. Dermatol Ther. (2020) 33:e13336. doi: 10.1111/dth.13336
38. Pan W, Wang Q, Chen Q. The cytokine network involved in the host immune response to periodontitis. Int J Oral Sci. (2019) 11:30. doi: 10.1038/s41368-019-0064-z
39. Checchi V, Maravic T, Bellini P, Generali L, Consolo U, Breschi L, et al. The role of matrix metalloproteinases in periodontal disease. Int J Environ Res Public Health. (2020) 17. doi: 10.3390/ijerph17144923
40. Tsukasaki M. RANKL and osteoimmunology in periodontitis. J Bone Miner Metab. (2021) 39:82–90. doi: 10.1007/s00774-020-01165-3
41. Hienz SA, Paliwal S, Ivanovski S. Mechanisms of bone resorption in periodontitis. J Immunol Res 2015. (2015) 615486. doi: 10.1155/2015/615486
42. Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood. (2010) 115:335–43. doi: 10.1182/blood-2009-04-216085
43. Ferguson B, Bokka NR, Maddipati KR, Ayilavarapu S, Weltman R, Zhu L, et al. Distinct profiles of specialized pro-resolving lipid mediators and corresponding receptor gene expression in periodontal inflammation. Front Immunol. (2020) 11:1307. doi: 10.3389/fimmu.2020.01307
44. Van Dyke TE. Shifting the paradigm from inhibitors of inflammation to resolvers of inflammation in periodontitis. J Periodontol 91 Suppl 1(Suppl. (2020) 1):S19–25. doi: 10.1002/JPER.20-0088
45. Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol. (2015) 35:1123–33. doi: 10.1161/atvbaha.115.305324
46. Thürmer M, Gollowitzer A, Pein H, Neukirch K, Gelmez E, Waltl L, et al. PI(18:1/18:1) is a SCD1-derived lipokine that limits stress signaling. Nat Commun. (2022) 13:2982. doi: 10.1038/s41467-022-30374-9
47. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. (2011) 17:1410–22. doi: 10.1038/nm.2538
48. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis - from experimental insights to the clinic. Nat Rev Drug Discovery. (2021) 20:589–610. doi: 10.1038/s41573-021-00198-1
49. Wang Q, Wang Y, Xu D. Research progress on Th17 and T regulatory cells and their cytokines in regulating atherosclerosis. Front Cardiovasc Med. (2022) 9:929078. doi: 10.3389/fcvm.2022.929078
50. Quillard T, Franck G, Mawson T, Folco E, Libby P. Mechanisms of erosion of atherosclerotic plaques. Curr Opin Lipidol. (2017) 28:434–41. doi: 10.1097/mol.0000000000000440
51. Pasterkamp G, den Ruijter HM, Libby P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat Rev Cardiol. (2017) 14:21–9. doi: 10.1038/nrcardio.2016.166
52. Kolte D, Libby P, Jang IK. New insights into plaque erosion as a mechanism of acute coronary syndromes. Jama. (2021) 325:1043–4. doi: 10.1001/jama.2021.0069
53. Franck G, Mawson T, Sausen G, Salinas M, Masson GS, Cole A, et al. Flow perturbation mediates neutrophil recruitment and potentiates endothelial injury via TLR2 in mice: implications for superficial erosion. Circ Res. (2017) 121:31–42. doi: 10.1161/circresaha.117.310694
54. Baaten C, Vondenhoff S, Noels H. Endothelial cell dysfunction and increased cardiovascular risk in patients with chronic kidney disease. Circ Res. (2023) 132:970–92. doi: 10.1161/circresaha.123.321752
55. Junho CVC, Frisch J, Soppert J, Wollenhaupt J, Noels H. Cardiomyopathy in chronic kidney disease: clinical features, biomarkers and the contribution of murine models in understanding pathophysiology. Clin Kidney J. (2023) 16:1786–803. doi: 10.1093/ckj/sfad085
56. Noels H, Jankowski J. Increased risk of cardiovascular complications in chronic kidney disease: introduction to a compendium. Circ Res. (2023) 132:899–901. doi: 10.1161/circresaha.123.322806
57. Zoccali C, Mallamaci F. Innate immunity system in patients with cardiovascular and kidney disease. Circ Res. (2023) 132:915–32. doi: 10.1161/circresaha.122.321749
58. An Y, Xu BT, Wan SR, Ma XM, Long Y, Xu Y, et al. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc Diabetol. (2023) 22:237. doi: 10.1186/s12933-023-01965-7
59. Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. (2014) 19:821–35. doi: 10.1016/j.cmet.2014.03.029
60. Jordan S, Tung N, Casanova-Acebes M, Chang C, Cantoni C, Zhang D, et al. Dietary intake regulates the circulating inflammatory monocyte pool. Cell. (2019) 178:1102–1114.e1117. doi: 10.1016/j.cell.2019.07.050
61. Schloss MJ, Swirski FK, Nahrendorf M. Modifiable cardiovascular risk, hematopoiesis, and innate immunity. Circ Res. (2020) 126:1242–59. doi: 10.1161/circresaha.120.315936
62. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. (2014) 20:754–8. doi: 10.1038/nm.3589
63. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–175.e114. doi: 10.1016/j.cell.2017.12.013
64. Sanz M, Marco Del Castillo A, Jepsen S, Gonzalez-Juanatey JR, D'Aiuto F, Bouchard P, et al. Periodontitis and cardiovascular diseases: Consensus report. J Clin Periodontol. (2020) 47:268–88. doi: 10.1111/jcpe.13189
65. Rydén L, Buhlin K, Ekstrand E, de Faire U, Gustafsson A, Holmer J, et al. Periodontitis increases the risk of a first myocardial infarction: A report from the PAROKRANK study. Circulation. (2016) 133:576–83. doi: 10.1161/circulationaha.115.020324
66. Larvin H, Kang J, Aggarwal VR, Pavitt S, Wu J. Risk of incident cardiovascular disease in people with periodontal disease: A systematic review and meta-analysis. Clin Exp Dent Res. (2021) 7:109–22. doi: 10.1002/cre2.336
67. Guo X, Li X, Liao C, Feng X, He T. Periodontal disease and subsequent risk of cardiovascular outcome and all-cause mortality: A meta-analysis of prospective studies. PloS One. (2023) 18:e0290545. doi: 10.1371/journal.pone.0290545
68. Gao S, Tian J, Li Y, Liu T, Li R, Yang L, et al. Periodontitis and number of teeth in the risk of coronary heart disease: an updated meta-analysis. Med Sci Monit. (2021) 27:e930112. doi: 10.12659/msm.930112
69. Pejcic A, Kostic M, Marko I, Obradovic R, Minic I, Bradic-Vasic M, et al. Tooth loss and periodontal status in patients with cardiovascular disease in the Serbian population: A randomized prospective study. Int J Dent Hyg. (2023) 21:317–27. doi: 10.1111/idh.12663
70. Vedin O, Hagström E, Gallup D, Neely ML, Stewart R, Koenig W, et al. Periodontal disease in patients with chronic coronary heart disease: Prevalence and association with cardiovascular risk factors. Eur J Prev Cardiol. (2015) 22:771–8. doi: 10.1177/2047487314530660
71. Sumayin Ngamdu K, Mallawaarachchi I, Dunipace EA, Chuang LH, Jafri SH, Shah NR, et al. Association between periodontal disease and cardiovascular disease (from the NHANES). Am J Cardiol. (2022) 178:163–8. doi: 10.1016/j.amjcard.2022.05.028
72. Aoyama N, Kobayashi N, Hanatani T, Ashigaki N, Yoshida A, Shiheido Y, et al. Periodontal condition in Japanese coronary heart disease patients: A comparison between coronary and non-coronary heart diseases. J Periodontal Res. (2019) 54:259–65. doi: 10.1111/jre.12626
73. Schulze-Späte U, Mizani I, Salaverry KR, Chang J, Wu C, Jones M, et al. Periodontitis and bone metabolism in patients with advanced heart failure and after heart transplantation. ESC Heart Fail. (2017) 4:169–77. doi: 10.1002/ehf2.12126
74. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
75. Horliana AC, Chambrone L, Foz AM, Artese HP, Rabelo Mde S, Pannuti CM, et al. Dissemination of periodontal pathogens in the bloodstream after periodontal procedures: a systematic review. PloS One. (2014) 9:e98271. doi: 10.1371/journal.pone.0098271
76. Farrugia C, Stafford GP, Potempa J, Wilkinson RN, Chen Y, Murdoch C, et al. Mechanisms of vascular damage by systemic dissemination of the oral pathogen Porphyromonas gingivalis. FEBS J. (2021) 288:1479–95. doi: 10.1111/febs.15486
77. Martinez-Garcia M, Hernandez-Lemus E. Periodontal inflammation and systemic diseases: an overview. Front Physiol. (2021) 12:709438. doi: 10.3389/fphys.2021.709438
78. Kozarov EV, Dorn BR, Shelburne CE, Dunn WA Jr., Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. (2005) 25:e17–18. doi: 10.1161/01.ATV.0000155018.67835.1a
79. Rafferty B, Jönsson D, Kalachikov S, Demmer RT, Nowygrod R, Elkind MS, et al. Impact of monocytic cells on recovery of uncultivable bacteria from atherosclerotic lesions. J Intern Med. (2011) 270:273–80. doi: 10.1111/j.1365-2796.2011.02373.x
80. Velsko IM, Chukkapalli SS, Rivera MF, Lee JY, Chen H, Zheng D, et al. Active invasion of oral and aortic tissues by Porphyromonas gingivalis in mice causally links periodontitis and atherosclerosis. PloS One. (2014) 9:e97811. doi: 10.1371/journal.pone.0097811
81. Armingohar Z, Jørgensen JJ, Kristoffersen AK, Abesha-Belay E, Olsen I. Bacteria and bacterial DNA in atherosclerotic plaque and aneurysmal wall biopsies from patients with and without periodontitis. J Oral Microbiol. (2014) 6. doi: 10.3402/jom.v6.23408
82. Gan G, Lin S, Luo Y, Zeng Y, Lu B, Zhang R, et al. Unveiling the oral-gut connection: chronic apical periodontitis accelerates atherosclerosis via gut microbiota dysbiosis and altered metabolites in apoE(-/-) Mice on a high-fat diet. Int J Oral Sci. (2024) 16:39. doi: 10.1038/s41368-024-00301-3
83. Noz MP, Plachokova AS, Smeets EMM, Aarntzen E, Bekkering S, Vart P, et al. An explorative study on monocyte reprogramming in the context of periodontitis in vitro and in vivo. Front Immunol. (2021) 12:695227. doi: 10.3389/fimmu.2021.695227
84. Pink C, Kocher T, Meisel P, Dorr M, Markus MR, Jablonowski L, et al. Longitudinal effects of systemic inflammation markers on periodontitis. J Clin Periodontol. (2015) 42:988–97. doi: 10.1111/jcpe.12473
85. Sharma A, Astekar M, Metgud R, Soni A, Verma M, Patel S. A study of C-reactive protein, lipid metabolism and peripheral blood to identify a link between periodontitis and cardiovascular disease. Biotech Histochem. (2014) 89:577–82. doi: 10.3109/10520295.2014.918280
86. Gu Y, Golub LM, Lee HM, Hou W, Ryan ME. Periodontal disease in acute coronary syndrome patients. Am J Dent. (2021) 34:97–100.
87. Radvar M, Tavakkol-Afshari J, Bajestan MN, Naseh MR, Arab HR. The effect of periodontal treatment on IL-6 production of peripheral blood monocytes in aggressive periodontitis and chronic periodontitis patients. Iran J Immunol. (2008) 5:100–6.
88. Costa PP, Trevisan GL, Macedo GO, Palioto DB, Souza SL, Grisi MF, et al. Salivary interleukin-6, matrix metalloproteinase-8, and osteoprotegerin in patients with periodontitis and diabetes. J Periodontol. (2010) 81:384–91. doi: 10.1902/jop.2009.090510
89. Nibali L, Fedele S, D'Aiuto F, Donos N. Interleukin-6 in oral diseases: a review. Oral Dis. (2012) 18:236–43. doi: 10.1111/j.1601-0825.2011.01867.x
90. Ridker PM. Residual inflammatory risk: addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J. (2016) 37:1720–2. doi: 10.1093/eurheartj/ehw024
91. Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. (2010) 375:132–40. doi: 10.1016/s0140-6736(09)61717-7
92. Ridker PM, MacFadyen JG, Thuren T, Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1β inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. (2020) 41:2153–63. doi: 10.1093/eurheartj/ehz542
93. Basheer S, Peeran S, Elhassan A, Zameer M, Mustafa M, Thiruneervannan M. Role of serum amyloid A (SAA) protein in various diseases with special reference to periodontal and periapical inflammation-A review. J Clin Diagn Res. (2020) 14:ZE01–5. doi: 10.7860/JCDR/2020/46072.14296
94. Song L-T, Lai W, Li J-S, Mu Y-Z, Li C-Y, Jiang S-Y. The interaction between serum amyloid A and Toll-like receptor 2 pathway regulates inflammatory cytokine secretion in human gingival fibroblasts. J Periodontology. (2020) 91:129–37. doi: 10.1002/JPER.19-0050
95. Ardila Medina CM, Bedoya García J, Guzmán Zuluaga IC. Asociación entre niveles patológicos de amiloide A sérico y periodontitis. Rev Archivo Médico Camagüey. (2016) 20:470–6.
96. Lu X, Li P, Li J, Hu J, Tian R. Clinical diagnostic value of IL-14, 1L-16 and SAA in periodontitis. Clin Oral Investigations. (2023) 27:6627–35. doi: 10.1007/s00784-023-05269-8
97. Ardila CM, Guzmán IC. Comparison of serum amyloid A protein and C-reactive protein levels as inflammatory markers in periodontitis. J Periodontal Implant Sci. (2015) 45:14–22. doi: 10.5051/jpis.2015.45.1.14
98. Türer Ç C, Ballı U, Güven B. Fetuin-A, serum amyloid A and tumor necrosis factor alpha levels in periodontal health and disease. Oral Dis. (2017) 23:379–86. doi: 10.1111/odi.12625
99. Nishida E, Aino M, Kobayashi SI, Okada K, Ohno T, Kikuchi T, et al. Serum amyloid A promotes E-Selectin expression via toll-Like receptor 2 in human aortic endothelial cells. Mediators Inflammation. (2016) 2016:7150509. doi: 10.1155/2016/7150509
100. Glurich I, Grossi S, Albini B, Ho A, Shah R, Zeid M, et al. Systemic inflammation in cardiovascular and periodontal disease: comparative study. Clin Vaccine Immunol. (2002) 9:425–32. doi: 10.1128/CDLI.9.2.425-432.2002
101. Getz GS, Krishack PA, Reardon CA. Serum amyloid A and atherosclerosis. Curr Opin Lipidol. (2016) 27:531–5. doi: 10.1097/mol.0000000000000331
102. Shridas P, Tannock LR. Role of serum amyloid A in atherosclerosis. Curr Opin Lipidol. (2019) 30:320–5. doi: 10.1097/mol.0000000000000616
103. Webb NR. High-density lipoproteins and serum amyloid A (SAA). Curr Atheroscler Rep. (2021) 23:7. doi: 10.1007/s11883-020-00901-4
104. Lee H, Joo JY, Song JM, Kim HJ, Kim YH, Park HR. Immunological link between periodontitis and type 2 diabetes deciphered by single-cell RNA analysis. Clin Transl Med. (2023) 13:e1503. doi: 10.1002/ctm2.1503
105. Ling MR, Chapple IL, Matthews JB. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun. (2015) 21:714–25. doi: 10.1177/1753425915589387
106. Martinez-Herrera M, Lopez-Domenech S, Silvestre FJ, Silvestre-Rangil J, Banuls C, Victor VM, et al. Chronic periodontitis impairs polymorphonuclear leucocyte-endothelium cell interactions and oxidative stress in humans. J Clin Periodontol. (2018) 45:1429–39. doi: 10.1111/jcpe.13027
107. Fredriksson MI, Gustafsson AK, Bergstrom KG, Asman BE. Constitutionally hyperreactive neutrophils in periodontitis. J Periodontol. (2003) 74:219–24. doi: 10.1902/jop.2003.74.2.219
108. Wright HJ, Matthews JB, Chapple IL, Ling-Mountford N, Cooper PR. Periodontitis associates with a type 1 IFN signature in peripheral blood neutrophils. J Immunol. (2008) 181:5775–84. doi: 10.4049/jimmunol.181.8.5775
109. Carrion J, Scisci E, Miles B, Sabino GJ, Zeituni AE, Gu Y, et al. Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J Immunol. (2012) 189:3178–87. doi: 10.4049/jimmunol.1201053
110. Miles B, Zakhary I, El-Awady A, Scisci E, Carrion J, O'Neill JC, et al. Secondary lymphoid organ homing phenotype of human myeloid dendritic cells disrupted by an intracellular oral pathogen. Infect Immun. (2014) 82:101–11. doi: 10.1128/iai.01157-13
111. Meghil MM, Tawfik OK, Elashiry M, Rajendran M, Arce RM, Fulton DJ, et al. Disruption of Immune Homeostasis in Human Dendritic Cells via Regulation of Autophagy and Apoptosis by Porphyromonas gingivalis. Front Immunol. (2019) 10:2286. doi: 10.3389/fimmu.2019.02286
112. Nepomuceno R, Pigossi SC, Finoti LS, Orrico SRP, Cirelli JA, Barros SP, et al. Serum lipid levels in patients with periodontal disease: A meta-analysis and meta-regression. J Clin Periodontol. (2017) 44:1192–207. doi: 10.1111/jcpe.12792
113. Ljunggren S, Bengtsson T, Karlsson H, Starkhammar Johansson C, Palm E, Nayeri F, et al. Modified lipoproteins in periodontitis: a link to cardiovascular disease? Biosci Rep. (2019) 39. doi: 10.1042/bsr20181665
114. Soppert J, Lehrke M, Marx N, Jankowski J, Noels H. Lipoproteins and lipids in cardiovascular disease: from mechanistic insights to therapeutic targeting. Adv Drug Delivery Rev. (2020) 159:4–33. doi: 10.1016/j.addr.2020.07.019
115. Noels H, Lehrke M, Vanholder R, Jankowski J. Lipoproteins and fatty acids in chronic kidney disease: molecular and metabolic alterations. Nat Rev Nephrol. (2021) 17:528–42. doi: 10.1038/s41581-021-00423-5
116. Noels H, Jankowski V, Schunk SJ, Vanholder R, Kalim S, Jankowski J. Post-translational modifications in kidney diseases and associated cardiovascular risk. Nat Rev Nephrol. (2024) 20:495–512. doi: 10.1038/s41581-024-00837-x
117. Muñoz Aguilera E, Suvan J, Buti J, Czesnikiewicz-Guzik M, Barbosa Ribeiro A, Orlandi M, et al. Periodontitis is associated with hypertension: a systematic review and meta-analysis. Cardiovasc Res. (2020) 116:28–39. doi: 10.1093/cvr/cvz201
118. Tada A, Tano R, Miura H. The relationship between tooth loss and hypertension: a systematic review and meta-analysis. Sci Rep. (2022) 12:13311. doi: 10.1038/s41598-022-17363-0
119. Moura MF, Navarro TP, Silva TA, Cota LOM, Soares Dutra Oliveira AM, Costa FO. Periodontitis and endothelial dysfunction: periodontal clinical parameters and levels of salivary markers interleukin-1β, tumor necrosis factor-α, matrix metalloproteinase-2, tissue inhibitor of metalloproteinases-2 complex, and nitric oxide. J Periodontol. (2017) 88:778–87. doi: 10.1902/jop.2017.170023
120. Wang W, Yang Z, Wang Y, Gao H, Wang Y, Zhang Q. Association between periodontitis and carotid artery calcification: A systematic review and meta-analysis. BioMed Res Int. (2021) 2021:3278351. doi: 10.1155/2021/3278351
121. Schmitt A, Carra MC, Boutouyrie P, Bouchard P. Periodontitis and arterial stiffness: a systematic review and meta-analysis. J Clin Periodontol. (2015) 42:977–87. doi: 10.1111/jcpe.12467
122. Darnaud C, Courtet A, Schmitt A, Boutouyrie P, Bouchard P, Carra MC. Association between periodontitis and pulse wave velocity: a systematic review and meta-analysis. Clin Oral Investig. (2021) 25:393–405. doi: 10.1007/s00784-020-03718-2
123. Isola G, Giudice AL, Polizzi A, Alibrandi A, Patini R, Ferlito S. Periodontitis and tooth loss have negative systemic impact on circulating progenitor cell levels: A clinical study. Genes (Basel). (2019) 10. doi: 10.3390/genes10121022
124. Lai WK, Kan MY. Homocysteine-induced endothelial dysfunction. Ann Nutr Metab. (2015) 67:1–12. doi: 10.1159/000437098
125. Djuric D, Jakovljevic V, Zivkovic V, Srejovic I. Homocysteine and homocysteine-related compounds: an overview of the roles in the pathology of the cardiovascular and nervous systems. Can J Physiol Pharmacol. (2018) 96:991–1003. doi: 10.1139/cjpp-2018-0112
126. Botelho J, MaChado V, Leira Y, Proença L, Mendes JJ. Periodontal inflamed surface area mediates the link between homocysteine and blood pressure. Biomolecules. (2021) 11. doi: 10.3390/biom11060875
127. Assinger A, Laky M, Badrnya S, Esfandeyari A, Volf I. Periodontopathogens induce expression of CD40L on human platelets via TLR2 and TLR4. Thromb Res. (2012) 130:e73–78. doi: 10.1016/j.thromres.2012.04.017
128. Heeschen C, Dimmeler S, Hamm CW, van den Brand MJ, Boersma E, Zeiher AM, et al. Soluble CD40 ligand in acute coronary syndromes. N Engl J Med. (2003) 348:1104–11. doi: 10.1056/NEJMoa022600
129. Lacy M, Burger C, Shami A, Ahmadsei M, Winkels H, Nitz K, et al. Cell-specific and divergent roles of the CD40L-CD40 axis in atherosclerotic vascular disease. Nat Commun. (2021) 12:3754. doi: 10.1038/s41467-021-23909-z
130. Tony AA, Tony EA, Mohammed WS, Kholef EF. Evaluation of plasma levels of neopterin and soluble CD40 ligand in patients with acute ischemic stroke in upper Egypt: can they surrogate the severity and functional outcome? Neuropsychiatr Dis Treat. (2019) 15:575–86. doi: 10.2147/ndt.S177726
131. Lowenstern A, Storey RF, Neely M, Sun JL, Angiolillo DJ, Cannon CP, et al. Platelet-related biomarkers and their response to inhibition with aspirin and p2y(12)-receptor antagonists in patients with acute coronary syndrome. J Thromb Thrombolysis. (2017) 44:145–53. doi: 10.1007/s11239-017-1516-y
132. Chen WA, Fletcher HM, Payne KJ, Aka S, Thornburg MB, Gheorghe JD, et al. Platelet and neutrophil responses to Porphyromonas gingivalis in human whole blood. Mol Oral Microbiol. (2021) 36:202–13. doi: 10.1111/omi.12336
133. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
134. Palmer LJ, Chapple IL, Wright HJ, Roberts A, Cooper PR. Extracellular deoxyribonuclease production by periodontal bacteria. J Periodontal Res. (2012) 47:439–45. doi: 10.1111/j.1600-0765.2011.01451.x
135. Morita C, Sumioka R, Nakata M, Okahashi N, Wada S, Yamashiro T, et al. Cell wall-anchored nuclease of Streptococcus sanguinis contributes to escape from neutrophil extracellular trap-mediated bacteriocidal activity. PloS One. (2014) 9:e103125. doi: 10.1371/journal.pone.0103125
136. Doke M, Fukamachi H, Morisaki H, Arimoto T, Kataoka H, Kuwata H. Nucleases from Prevotella intermedia can degrade neutrophil extracellular traps. Mol Oral Microbiol. (2017) 32:288–300. doi: 10.1111/omi.12171
137. Stobernack T, du Teil Espina M, Mulder LM, Palma Medina LM, Piebenga DR, Gabarrini G, et al. A secreted bacterial peptidylarginine deiminase can neutralize human innate immune defenses. mBio. (2018) 9. doi: 10.1128/mBio.01704-18
138. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
139. Klopf J, Brostjan C, Eilenberg W, Neumayer C. Neutrophil extracellular traps and their implications in cardiovascular and inflammatory disease. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22020559
140. Kovalcikova AG, Novak B, Roshko O, Kovalova E, Pastorek M, Vlkova B, et al. Extracellular DNA and markers of neutrophil extracellular traps in saliva from patients with periodontitis-A case-control study. J Clin Med. (2024) 13. doi: 10.3390/jcm13020468
141. White P, Sakellari D, Roberts H, Risafi I, Ling M, Cooper P, et al. Peripheral blood neutrophil extracellular trap production and degradation in chronic periodontitis. J Clin Periodontol. (2016) 43:1041–9. doi: 10.1111/jcpe.12628
142. Moonen CG, Buurma KG, Faruque MR, Balta MG, Liefferink E, Bizzarro S, et al. Periodontal therapy increases neutrophil extracellular trap degradation. Innate Immun. (2020) 26:331–40. doi: 10.1177/1753425919889392
143. Chukkapalli SS, Velsko IM, Rivera-Kweh MF, Zheng D, Lucas AR, Kesavalu L. Polymicrobial oral infection with four periodontal bacteria orchestrates a distinct inflammatory response and atherosclerosis in apoE null mice. PloS One. (2015) 10:e0143291. doi: 10.1371/journal.pone.0143291
144. Suh JS, Kim SYJ, Lee SH, Kim RH, Park NH. Hyperlipidemia is necessary for the initiation and progression of atherosclerosis by severe periodontitis in mice. Mol Med Rep. (2022) 26. doi: 10.3892/mmr.2022.12789
145. Jain A, Batista EL Jr., Serhan C, Stahl GL, Van Dyke TE. Role for periodontitis in the progression of lipid deposition in an animal model. Infect Immun. (2003) 71:6012–8. doi: 10.1128/iai.71.10.6012-6018.2003
146. Brodala N, Merricks EP, Bellinger DA, Damrongsri D, Offenbacher S, Beck J, et al. Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. (2005) 25:1446–51. doi: 10.1161/01.ATV.0000167525.69400.9c
147. Fan T, Guo K, Cao F, Deng Z, Liu B, Shi M, et al. Study on the effect of periodontitis on renal tissue in atherosclerotic mice. J Periodontal Res. (2023) 58:655–67. doi: 10.1111/jre.13128
148. Zhou LJ, Lin WZ, Meng XQ, Zhu H, Liu T, Du LJ, et al. Periodontitis exacerbates atherosclerosis through Fusobacterium nucleatum-promoted hepatic glycolysis and lipogenesis. Cardiovasc Res. (2023) 119:1706–17. doi: 10.1093/cvr/cvad045
149. Maekawa T, Takahashi N, Tabeta K, Aoki Y, Miyashita H, Miyauchi S, et al. Chronic oral infection with Porphyromonas gingivalis accelerates atheroma formation by shifting the lipid profile. PloS One. (2011) 6:e20240. doi: 10.1371/journal.pone.0020240
150. Miyajima S, Naruse K, Kobayashi Y, Nakamura N, Nishikawa T, Adachi K, et al. Periodontitis-activated monocytes/macrophages cause aortic inflammation. Sci Rep. (2014) 4:5171. doi: 10.1038/srep05171
151. Brito LC, DalBó S, Striechen TM, Farias JM, Olchanheski LR Jr., Mendes RT, et al. Experimental periodontitis promotes transient vascular inflammation and endothelial dysfunction. Arch Oral Biol. (2013) 58:1187–98. doi: 10.1016/j.archoralbio.2013.03.009
152. Xie M, Tang Q, Nie J, Zhang C, Zhou X, Yu S, et al. BMAL1-downregulation aggravates porphyromonas gingivalis-induced atherosclerosis by encouraging oxidative stress. Circ Res. (2020) 126:e15–29. doi: 10.1161/circresaha.119.315502
153. Bugueno IM, Zobairi El-Ghazouani F, Batool F, El Itawi H, Anglès-Cano E, Benkirane-Jessel N, et al. Porphyromonas gingivalis triggers the shedding of inflammatory endothelial microvesicles that act as autocrine effectors of endothelial dysfunction. Sci Rep. (2020) 10:1778. doi: 10.1038/s41598-020-58374-z
154. Dong Q, Luo Y, Yin Y, Ma Y, Yu Y, Wang L, et al. RhoA/ROCK1 regulates the mitochondrial dysfunction through Drp1 induced by Porphyromonas gingivalis in endothelial cells. J Cell Mol Med. (2023) 27:2123–35. doi: 10.1111/jcmm.17796
155. Du F, Zhu Z, Lai Z, Li K, Chen J, Zhang E, et al. Imbalance of helper T cell subtypes and adipokine secretion in perivascular adipose tissue as a trigger of atherosclerosis in chronic Porphyromonas gingivalis W83 infection. Microbes Infect. (2023) 25:105181. doi: 10.1016/j.micinf.2023.105181
156. Kim SY, Kim YJ, Kim S, Momeni M, Lee A, Treanor A, et al. GV1001 inhibits the severity of the ligature-induced periodontitis and the vascular lipid deposition associated with the periodontitis in mice. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241612566
157. Suh JS, Lee SH, Fouladian Z, Lee JY, Kim T, Kang MK, et al. Rosuvastatin prevents the exacerbation of atherosclerosis in ligature-induced periodontal disease mouse model. Sci Rep. (2020) 10:6383. doi: 10.1038/s41598-020-63350-8
158. Li Q, Liu J, Liu W, Chu Y, Zhong J, Xie Y, et al. LOX-1 regulates P. gingivalis-induced monocyte migration and adhesion to human umbilical vein endothelial cells. Front Cell Dev Biol. (2020) 8:596. doi: 10.3389/fcell.2020.00596
159. Lu J, Mitra S, Wang X, Khaidakov M, Mehta JL. Oxidative stress and lectin-like ox-LDL-receptor LOX-1 in atherogenesis and tumorigenesis. Antioxid Redox Signal. (2011) 15:2301–33. doi: 10.1089/ars.2010.3792
160. Afzoon S, Amiri MA, Mohebbi M, Hamedani S, Farshidfar N. A systematic review of the impact of Porphyromonas gingivalis on foam cell formation: Implications for the role of periodontitis in atherosclerosis. BMC Oral Health. (2023) 23:481. doi: 10.1186/s12903-023-03183-9
161. Rho JH, Kim HJ, Joo JY, Lee JY, Lee JH, Park HR. Periodontal pathogens promote foam cell formation by blocking lipid efflux. J Dent Res. (2021) 100:1367–77. doi: 10.1177/00220345211008811
162. Li X, Wang H, Yu X, Saha G, Kalafati L, Ioannidis C, et al. Maladaptive innate immune training of myelopoiesis links inflammatory comorbidities. Cell. (2022) 185:1709–1727.e1718. doi: 10.1016/j.cell.2022.03.043
163. Omar M, Alexiou M, Rekhi UR, Lehmann K, Bhardwaj A, Delyea C, et al. DNA methylation changes underlie the long-term association between periodontitis and atherosclerotic cardiovascular disease. Front Cardiovasc Med. (2023) 10:1164499. doi: 10.3389/fcvm.2023.1164499
164. Xiao L, Huang L, Zhou X, Zhao D, Wang Y, Min H, et al. Experimental periodontitis deteriorated atherosclerosis associated with trimethylamine N-Oxide metabolism in mice. Front Cell Infect Microbiol. (2021) 11:820535. doi: 10.3389/fcimb.2021.820535
165. Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. (2014) 124:4204–11. doi: 10.1172/jci72331
166. Li L, Zhang YL, Liu XY, Meng X, Zhao RQ, Ou LL, et al. Periodontitis exacerbates and promotes the progression of chronic kidney disease through oral flora, cytokines, and oxidative stress. Front Microbiol. (2021) 12:656372. doi: 10.3389/fmicb.2021.656372
167. Komazaki R, Katagiri S, Takahashi H, Maekawa S, Shiba T, Takeuchi Y, et al. Periodontal pathogenic bacteria, Aggregatibacter actinomycetemcomitans affect non-alcoholic fatty liver disease by altering gut microbiota and glucose metabolism. Sci Rep. (2017) 7:13950. doi: 10.1038/s41598-017-14260-9
168. Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. (2011) 7:738–48. doi: 10.1038/nrendo.2011.106
169. Herrera D, Sanz M, Kebschull M, Jepsen S, Sculean A, Berglundh T, et al. Treatment of stage IV periodontitis: The EFP S3 level clinical practice guideline. J Clin Periodontol. (2022) 49 Suppl 24:4–71. doi: 10.1111/jcpe.13639
170. Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, et al. Treatment of periodontitis and endothelial function. N Engl J Med. (2007) 356:911–20. doi: 10.1056/NEJMoa063186
171. Czesnikiewicz-Guzik M, Osmenda G, Siedlinski M, Nosalski R, Pelka P, Nowakowski D, et al. Causal association between periodontitis and hypertension: evidence from Mendelian randomization and a randomized controlled trial of non-surgical periodontal therapy. Eur Heart J. (2019) 40:3459–70. doi: 10.1093/eurheartj/ehz646
172. Lobo MG, Schmidt MM, Lopes RD, Dipp T, Feijó IP, Schmidt KES, et al. Treating periodontal disease in patients with myocardial infarction: A randomized clinical trial. Eur J Intern Med. (2020) 71:76–80. doi: 10.1016/j.ejim.2019.08.012
173. Gugnani N, Gugnani S. Can treatment of severe periodontitis in patients with ST-segment elevation myocardial infarction improve endothelial function? Evid Based Dent. (2021) 22:5–7. doi: 10.1038/s41432-021-0157-3
174. Saffi MAL, Rabelo-Silva ER, Polanczyk CA, Furtado MV, Montenegro MM, Ribeiro IWJ, et al. Periodontal therapy and endothelial function in coronary artery disease: A randomized controlled trial. Oral Dis. (2018) 24:1349–57. doi: 10.1111/odi.12909
175. Okada A, Murata T, Matin K, Ariyoshi M, Otsuka R, Yamashita M, et al. Effect of advanced periodontal self-care in patients with early-stage periodontal diseases on endothelial function: An open-label, randomized controlled trial. PloS One. (2021) 16:e0257247. doi: 10.1371/journal.pone.0257247
176. Chou SH, Tung YC, Lin YS, Wu LS, Lin CP, Liou EJ, et al. Major adverse cardiovascular events in treated periodontitis: A population-based follow-up study from Taiwan. PloS One. (2015) 10:e0130807. doi: 10.1371/journal.pone.0130807
177. Shimada Y, Komatsu Y, Ikezawa-Suzuki I, Tai H, Sugita N, Yoshie H. The effect of periodontal treatment on serum leptin, interleukin-6, and C-reactive protein. J Periodontol. (2010) 81:1118–23. doi: 10.1902/jop.2010.090741
178. Bokhari SA, Khan AA, Butt AK, Azhar M, Hanif M, Izhar M, et al. Non-surgical periodontal therapy reduces coronary heart disease risk markers: a randomized controlled trial. J Clin Periodontol. (2012) 39:1065–74. doi: 10.1111/j.1600-051X.2012.01942.x
179. López NJ, Quintero A, Casanova PA, Ibieta CI, Baelum V, López R. Effects of periodontal therapy on systemic markers of inflammation in patients with metabolic syndrome: a controlled clinical trial. J Periodontol. (2012) 83:267–78. doi: 10.1902/jop.2011.110227
180. Caúla AL, Lira-Junior R, Tinoco EM, Fischer RG. The effect of periodontal therapy on cardiovascular risk markers: a 6-month randomized clinical trial. J Clin Periodontol. (2014) 41:875–82. doi: 10.1111/jcpe.12290
181. Montenegro MM, Ribeiro IWJ, Kampits C, Saffi MAL, Furtado MV, Polanczyk CA, et al. Randomized controlled trial of the effect of periodontal treatment on cardiovascular risk biomarkers in patients with stable coronary artery disease: Preliminary findings of 3 months. J Clin Periodontol. (2019) 46:321–31. doi: 10.1111/jcpe.13085
182. Montero E, López M, Vidal H, Martínez M, Virto L, Marrero J, et al. Impact of periodontal therapy on systemic markers of inflammation in patients with metabolic syndrome: A randomized clinical trial. Diabetes Obes Metab. (2020) 22:2120–32. doi: 10.1111/dom.14131
183. Zhou QB, Xia WH, Ren J, Yu BB, Tong XZ, Chen YB, et al. Effect of intensive periodontal therapy on blood pressure and endothelial microparticles in patients with prehypertension and periodontitis: A randomized controlled trial. J Periodontol. (2017) 88:711–22. doi: 10.1902/jop.2017.160447
184. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
185. Speer T, Dimmeler S, Schunk SJ, Fliser D, Ridker PM. Targeting innate immunity-driven inflammation in CKD and cardiovascular disease. Nat Rev Nephrol. (2022) 18:762–78. doi: 10.1038/s41581-022-00621-9
186. Van Dyke TE, Bartold PM, Reynolds EC. The nexus between periodontal inflammation and dysbiosis. Front Immunol 11. (2020) 511:511. doi: 10.3389/fimmu.2020.00511
187. Yang B, Pang X, Li Z, Chen Z, Wang Y. Immunomodulation in the treatment of periodontitis: progress and perspectives. Front Immunol. (2021) 12:781378. doi: 10.3389/fimmu.2021.781378
Keywords: periodontitis, cardiovascular disease, innate immunity, inflammation, dysbiosis
Citation: Schulze-Späte U, Wurschi L, van der Vorst EPC, Hölzle F, Craveiro RB, Wolf M and Noels H (2024) Crosstalk between periodontitis and cardiovascular risk. Front. Immunol. 15:1469077. doi: 10.3389/fimmu.2024.1469077
Received: 23 July 2024; Accepted: 06 November 2024;
Published: 09 December 2024.
Edited by:
Dunfang Zhang, Sichuan University, ChinaReviewed by:
Sweta Ghosh, University of Louisville, United StatesLiliana C. Patiño Molano, National Institute of Dental and Craniofacial Research (NIH), United States
Copyright © 2024 Schulze-Späte, Wurschi, van der Vorst, Hölzle, Craveiro, Wolf and Noels. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heidi Noels, aG5vZWxzQHVrYWFjaGVuLmRl; Ulrike Schulze-Späte, dWxyaWtlLnNjaHVsemUtc3BhZXRlQG1lZC51bmktamVuYS5kZQ==