 Sophia Trombello1,2
Sophia Trombello1,2 Andrea Jarisch1Andre Willasch1Eva Rettinger1Julia Fekadu-Siebald1
Andrea Jarisch1Andre Willasch1Eva Rettinger1Julia Fekadu-Siebald1 Dirk Holzinger3,4Roland Adelmann5
Dirk Holzinger3,4Roland Adelmann5 Peter Bader1
Peter Bader1 Shahrzad Bakhtiar1*
Shahrzad Bakhtiar1*- 1Division for Stem Cell Transplantation and Immunology, Department for Pediatrics, Goethe University Hospital Frankfurt, Frankfurt am Main, Germany
- 2Children’s Hospital, Heidelberg University Hospital, Heidelberg, Germany
- 3Department of Pediatric Hematology-Oncology, University of Duisburg-Essen, Essen, Germany
- 4Department of Applied Health Sciences, University of Applied Sciences Bochum, Bochum, Germany
- 5Department for Children and Adolescents Medicine, Hospital Oberberg, Gummersbach, Germany
Dedicator of cytokinesis 8 (DOCK8) deficiency is a combined immunodeficiency (CID) due to biallelic mutations in the gene encoding DOCK8. Major clinical phenomena are recurrent severe infections of the lungs and skin, atopic eczema, and predisposition to malignancy leading to a poor prognosis. Typical findings include highly elevated IgE and eosinophilia. Allogeneic hematopoietic stem cell transplantation (alloHSCT) is indicated as the only curative treatment option. We present a patient with advanced disease undergoing alloHSCT at the age of 11 years after individualized pre-treatment using dupilumab and rituximab resulting in a decrease in IgE levels and clinical improvement of the skin condition. Additionally, in a review of the literature, we summarize morbidity and outcome in DOCK8-deficient patients older than 8 years of age receiving alloHSCT. Life-threatening infections, malignancy, and disease-related complications with organ damage pre-transplant are challenging in older DOCK8-deficient patients. The therapeutic role of dupilumab in DOCK8 deficiency should be evaluated in larger studies.
1 Introduction
Dedicator of cytokinesis 8 deficiency (DOCK8, OMIM 611432) is a combined immunodeficiency (1–3) with clinical presentation of severe susceptibility to infections, immune dysregulation as atopic disease, autoimmunity, and elevated IgE, as well as predisposition for cancer (4, 5). Atopic disease most commonly manifests as eczema and food allergies (5, 6). Most patients display severe viral infections of the skin due to herpes simplex virus (HSV), human papilloma virus (HPV), and molluscum contagiosum (MC), followed by bacterial skin abscesses and mucocutaneous candidiasis (5, 7). Rare features include vasculopathy in part associated with cerebral events and sclerosing cholangitis due to chronic infection with cryptosporidium (4, 5, 7, 8). Nearly all patients display highly elevated IgE. Increased levels of IgG and IgA, along with low levels of IgM, are seen in many cases (5). Chronic Epstein-Barr virus (EBV) viremia likely results in higher risk for malignancies (5, 9). DOCK8 protein (190 kDa, 1,701 amino acids, cytogenic localization 9p24.3) (10) is part of the actin remodeling process (11). Biallelic homozygous or compound heterozygous mutations or deletions in the DOCK8 gene impact the persistence and activation of CD8+ T and NK cells, dysfunctional regulatory T cells (Treg) (12), and cause an imbalanced differentiation and cytokine production of T helper cells (5, 8, 13–15). The differentiation of B cells and their capacity of immunoglobulin production are impaired as well (4, 13, 16–19). AlloHSCT is the only life-saving treatment in DOCK8-deficient patients (13, 20–22). The importance of an early alloHSCT and adequate control of disease complications pre-transplant has been shown (23, 24).
Aydin et al., on behalf of the EBMT Inborn Errors Working Party (IEWP), studied the natural course of the disease in DOCK8 deficiency. In their manuscript published in 2015, they showed that both overall survival and event-free survival rapidly decreased beyond the age of 10 years (4).
In 2019, a study was published on the outcome of alloHSCT in patients with DOCK8 deficiency, including data analysis by age at transplantation. Although the analysis did not reach significance level, there was a tendency toward inferior outcome for patients above the age of 8 years (22). Based on these two large EBMT IEWP studies, we decided to conduct a literature review and focus on patients over 8 years of age.
Literature search for alloHSCT in DOCK8 deficiency in patients older than 8 years included information on comorbidities, immune modulating treatment, post-transplantation complications, survival, and outcome. This retrospective study was performed in the Division for Stem Cell Transplantation and Immunology in Frankfurt/Main, Germany (EBMT Centre Code 138). Written informed consent was obtained from the parents. The study was approved by the local ethics committee of the Frankfurt Goethe University (IRB approval no. 167/16). The results of this study add valuable information to the literature, especially when clinicians counsel patients with a delayed diagnosis of DOCK8 deficiency.
2 Case description
An 11-year-old boy of Syrian descent was referred for further treatment after a diagnosis of homozygous biallelic frameshift mutation in the DOCK8 gene (c.3339delT) was established. The patient suffered from inflammatory bowel disease, eczema, and transfusion-dependent autoimmune-hemolytic anemia since his early childhood. Skin diseases included verrucae vulgares due to HPV infection, tinea capitis, xerosis cutis, candida albicans, and superinfection with methicillin-resistant Staphylococcus aureus (MRSA) resulting in therapy-resistant ulcerations (Figure 1). Furthermore, he suffered from recurrent sinopulmonary infections and one episode of severe catheter-related sepsis due to MRSA. Ophthalmology work up revealed chronic conjunctivitis and uveitis, possibly due to the underlying CMV infection, resulting in corneal scars and loss of sight on both eyes. Evolving symptoms are displayed in Figure 2. He showed significant growth delay (Figure 1).
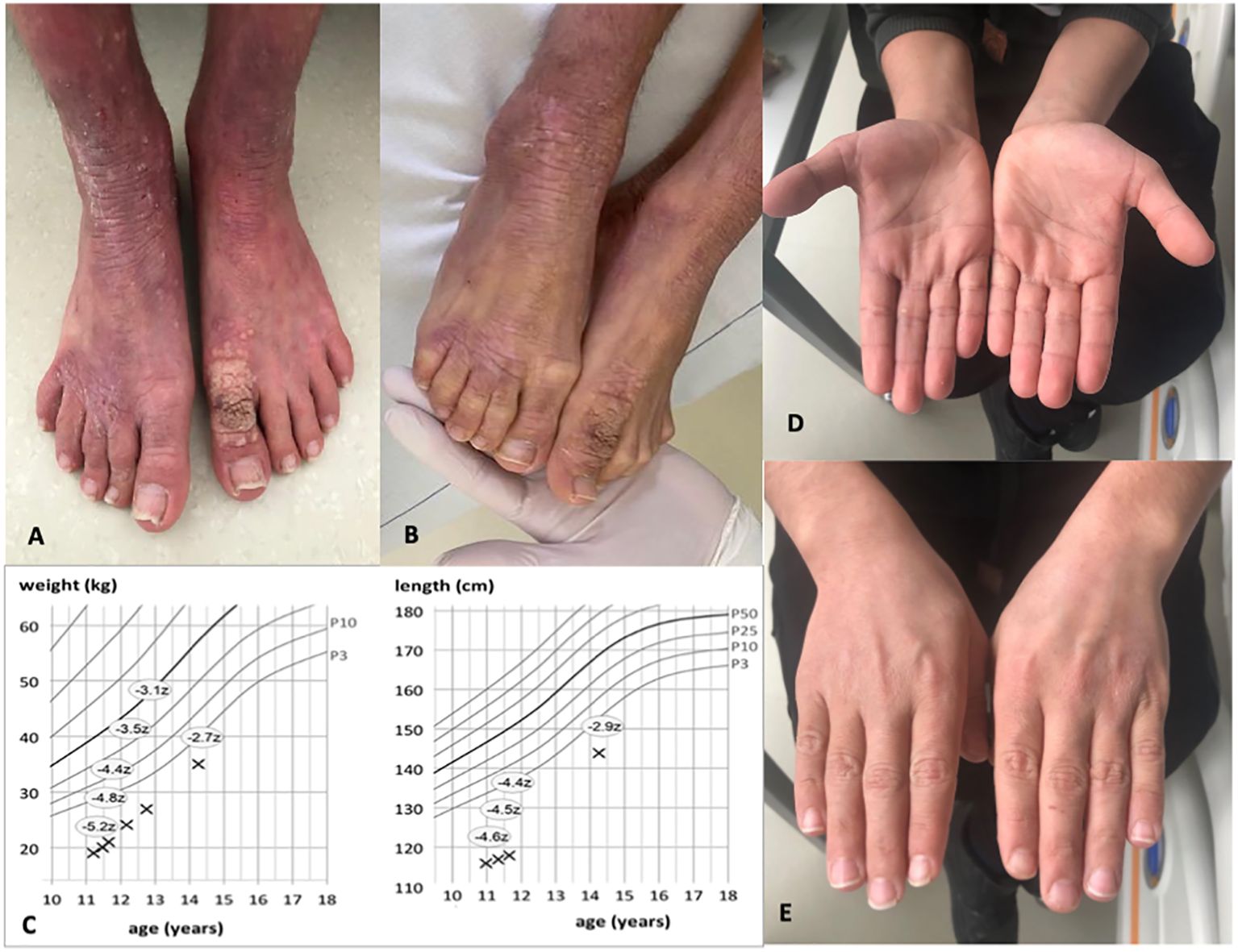
Figure 1. Images of the patient before and 1 year after alloHSCT. (A, B) Show patient’s feet with xerosis cutis, lichenification, and warts pre-alloHSCT. (C) indicates weight and length gain after alloHSCT. Z-scores measure the distance from the 50th percentile via standard deviation. (D, E) Show healthy skin of both hands with resolution of eczema post-alloHSCT.
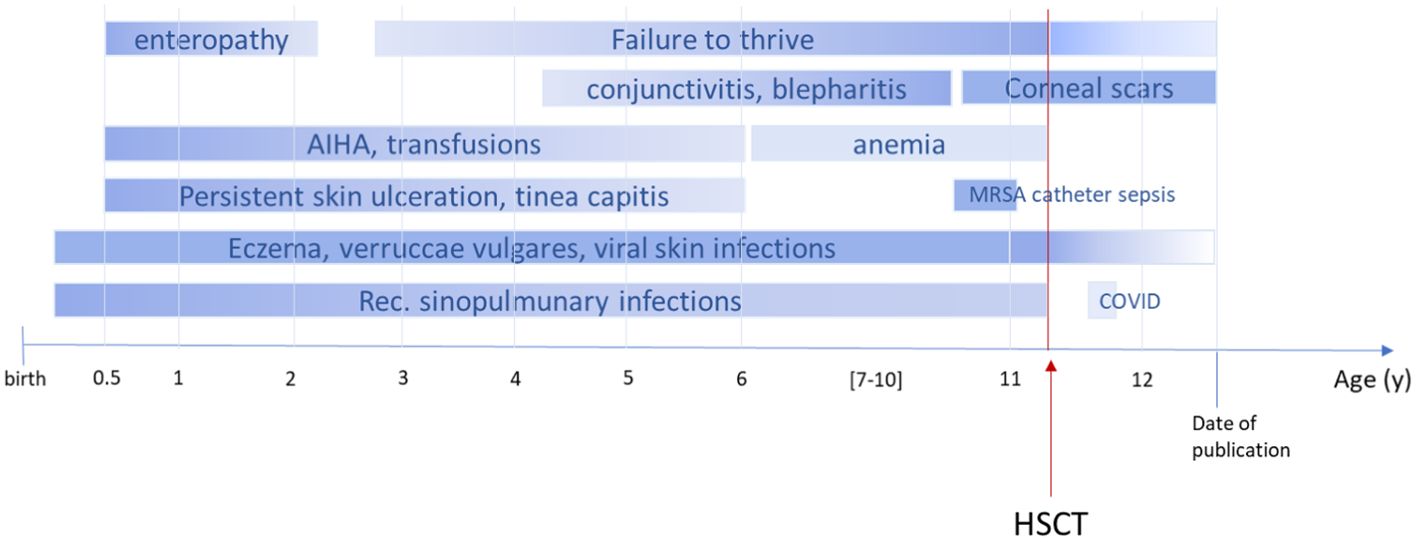
Figure 2. Timeline course of disease and symptoms pre- and post-alloHSCT. The figure illustrates the onset of symptoms in our patient. AIHA, autoimmune hemolytic anemia; MRSA, methicillin-resistant Staphylococcus aureus; COVID, coronavirus disease; shading indicates progress or regression; dark color refers to severity.
2.1 Immunological work up
Naïve CD4 (CD4+CD45RA+CD62L+) and CD8 (CD8+CD45RA+CD62L+) cells as well as terminally differentiated effector memory T cells re-expressing CD45RA (TEMRA) counts were reduced, while activated CD4+ and CD8+ (HLA-DR+) cells were increased. Regulatory T cells were within the normal range. CD19+ B-cell maturation was impaired with higher naïve B cells and decreased number of non-switched und switched memory B cells. Further findings included high levels of immunoglobulins (Ig): IgG of 2167 mg/dl (reference 700–1550 mg/dl), IgA of 582 mg/dl (reference 58–290 mg/dl), IgE of 34000 U/ml (reference <200 U/ml), and low IgM of 8 mg/dl (reference 49–180 mg/dl). Eosinophils were elevated at 2,000/µl (reference 20–700/µl) (detailed laboratory work up is shown in Supplementary Table S1).
2.2 Pre-treatment
To reduce pre-existing inflammation and minimize the risk for inflammatory complications post-transplant, a treatment with dupilumab and rituximab was started. Dupilumab is a human monoclonal antibody (immunoglobulin G4 subclass) that suppresses the response to the cytokines IL-4 and IL-13 (25, 26) by blocking the shared subunit of IL-4 and IL-13 receptor (27). Dupilumab was administered subcutaneously bi-weekly with an initial loading dose of 2 × 300 mg and following dose of 300 mg. We observed a rapid decrease in IgE (Figure 3). Rituximab was administered twice to eliminate B cells as reservoir for EBV. The effect of rituximab is shown through the course of IgG levels (Figure 3). During alloHSCT and post-transplant, all investigations of EBV viremia remained negative.
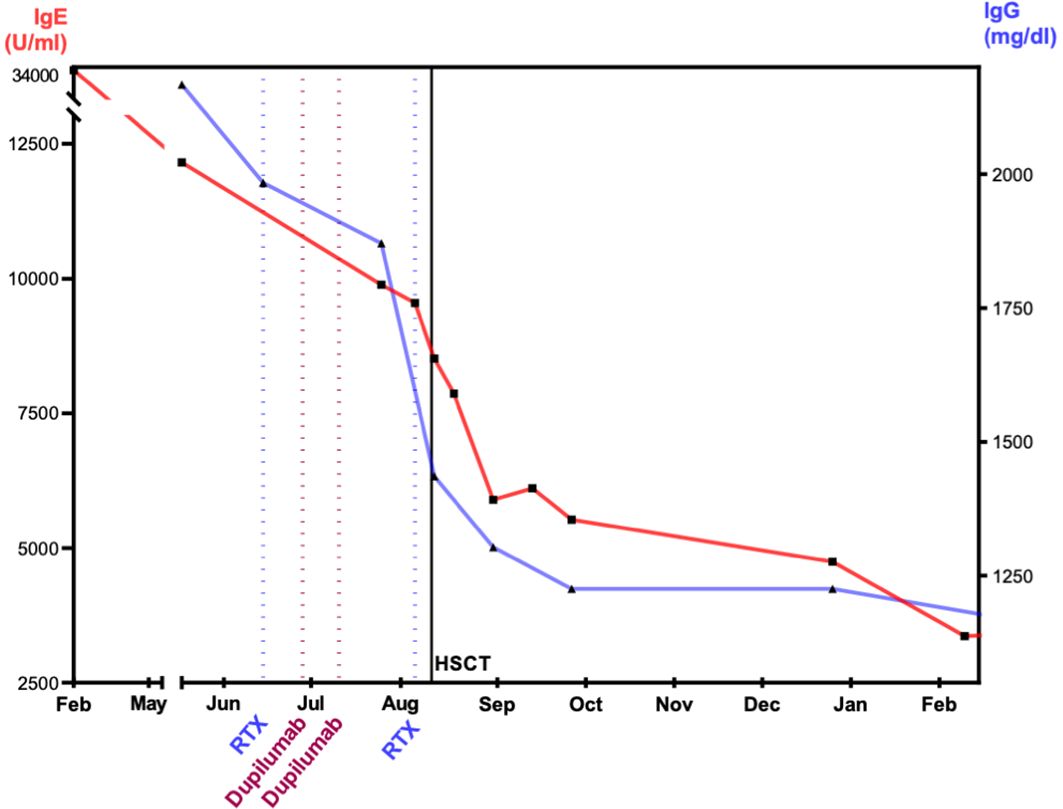
Figure 3. Effect of dupilumab, rituximab, and alloHSCT on IgE and IgG levels. The figure visualizes the level of immunoglobulins after immune-modulating therapy with dupilumab and rituximab. IgE, immunoglobulin E; IgG, immunoglobulin G; RTX, rituximab; alloHSCT, hematopoietic stem cell transplantation. IgE and IgG display decrease prior and after alloHSCT. At 1 year after alloHSCT, IgG shows normal levels. IgE decreased at 1 year and within the normal range at 2 years post-alloHSCT.
2.3 AlloHSCT and outcome
The conditioning regimen consisted of treosulfan (12 g/m2/day), fludarabine (40 mg/m2), and thiotepa (5 mg/kg). For prophylaxis of graft-versus-host disease (GvHD) anti-thymocyte globulin (ATG) (20 mg/kg) and methotrexate (10 mg/m2 on days +2, +4, and +6) were used. A 10/10 HLA-matched unrelated donor (MUD) was available, from whom unmanipulated bone marrow was harvested. Cell dose was 6.19 × 106 cells/kg of CD34+ and 75.3 × 106/kg of CD3+ cells. Antiviral and antimycotic prophylaxis included acyclovir and fluconazole, followed by liposomal amphotericin B. The pre-treatment and the conditioning regimen were tolerated well without any signs of relevant organ toxicity. One episode of fever during aplasia required antibiotic treatment. Leukocyte and neutrophile recovery (cell count >500/µl) was achieved at day +17, thrombocyte recovery (cell count >50/nl) at day +25, and full donor chimerism was detected at day +28 and in all following assessments (bone marrow and peripheral blood). Starting with pre-transplant high count, T cells never fell below 1,000/µl. After the administration of rituximab, B cell reconstitution was delayed at day +90, which required transient intravenous substitution of immunoglobulins. No signs of acute or chronic GvHD appeared. Four months post-transplant, the patient presented with mild upper respiratory infection due to SARS-CoV-2 resulting in positive PCR through nasopharyngeal swab. We observed a full recovery from SARS-CoV-2 infection without additional antiviral or antibody treatment.
At the last follow up 2 years post-alloHSCT, the patient is alive and well without immunosuppression. The skin pathology is in complete remission, without evidence for eczema, warts, and/or GvHD. IgE level is within the normal range (150 U/ml) as well as eosinophile count, IgG, and IgA. We observed a rapid catch-up growth as shown in the course of growth and weight percentiles. He caught up weight from −5.2 to −2.7 standard deviations from the 50th percentile and length from −4.6 to −2.9 (Figure 1).
3 Review of the literature
The review of literature included a systemic research of Medline database from the National Library of Medicine (NLM) via PubMed using terms such as DOCK8, HSCT, dupilumab, and rituximab. Results were filtered for DOCK8-deficient patients who underwent alloHSCT at the age of 8 years or older. Duplications were excluded (13, 20, 28). Diagnosis in the screened references was determined through either genetic mutation detection and/or flow cytometrical detection of the loss of DOCK8 protein. Donors are considered matched [either matched sibling donors (MSD), or matched unknown donors (MUD)] if displaying an HLA-match of at least 9 out of 10.
The literature review revealed 173 patients with DOCK8 deficiency who underwent alloHSCT (Figure 4). Eighty-one out of 173 were reported and analyzed in a large study of Aydin et al. and will be referred to separately (22). Out of the remaining 92 patients, 53 were transplanted at the age of 8 years or older (53/92, 57.6%) (13, 20, 21, 23, 24, 28–37). Within this cohort of older patients, there were more female patients (64%; 34 females, 18 males). The age ranged from 8 to 27 years. Pre-transplant morbidity ranged from recurrent respiratory tract and skin infections with eczema in nearly all patients to less frequent complications as liver disease due to chronic cryptosporidium infection, vasculopathy, and malignancy. IgE levels ranged from 8.3 to 36.000 U/ml, while five cases were over 15.000 U/ml (12.5%) and two over 34.000 U/ml (5%). Immune-modulating pre-treatment was initiated in EBV-positive patients using rituximab (n = 6), and one patient was treated with mycophenolate mofetil (MMF) due to aortitis (35) (Table 1).
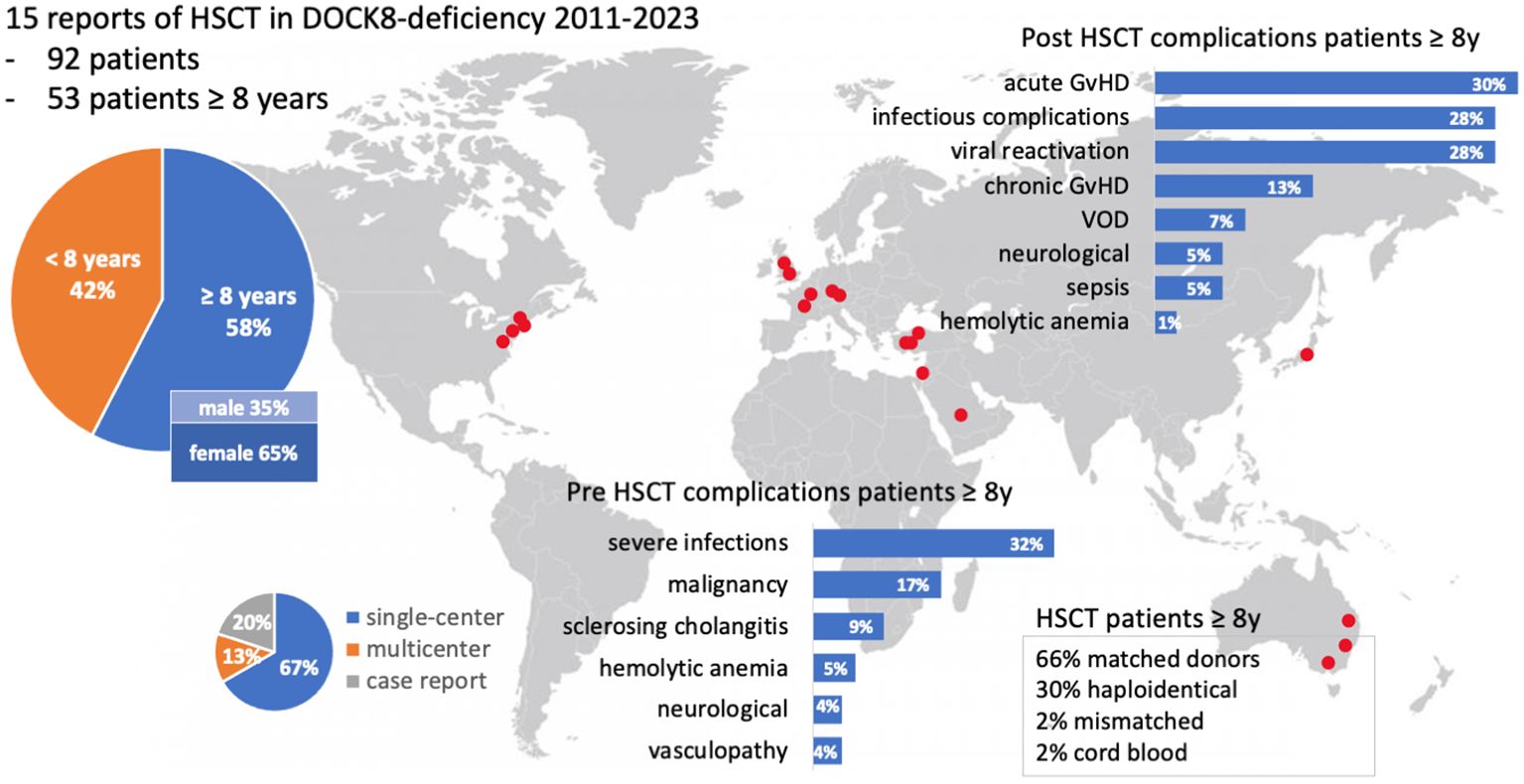
Figure 4. Visual abstract of the review of the literature. Fifteen publications on DOCK8 deficiency treated with alloHSCT were available, including single- and multicenter studies. Red dots indicate the locations of participating centers. A focus lies on patients with at least 8 years of age. Two bar charts illustrate pre- and post-HSCT complications (%). GvHD, graft-versus-host-disease; HSCT, hematopoietic stem cell transplantation; VOD, veno-occlusive disease; y, year.
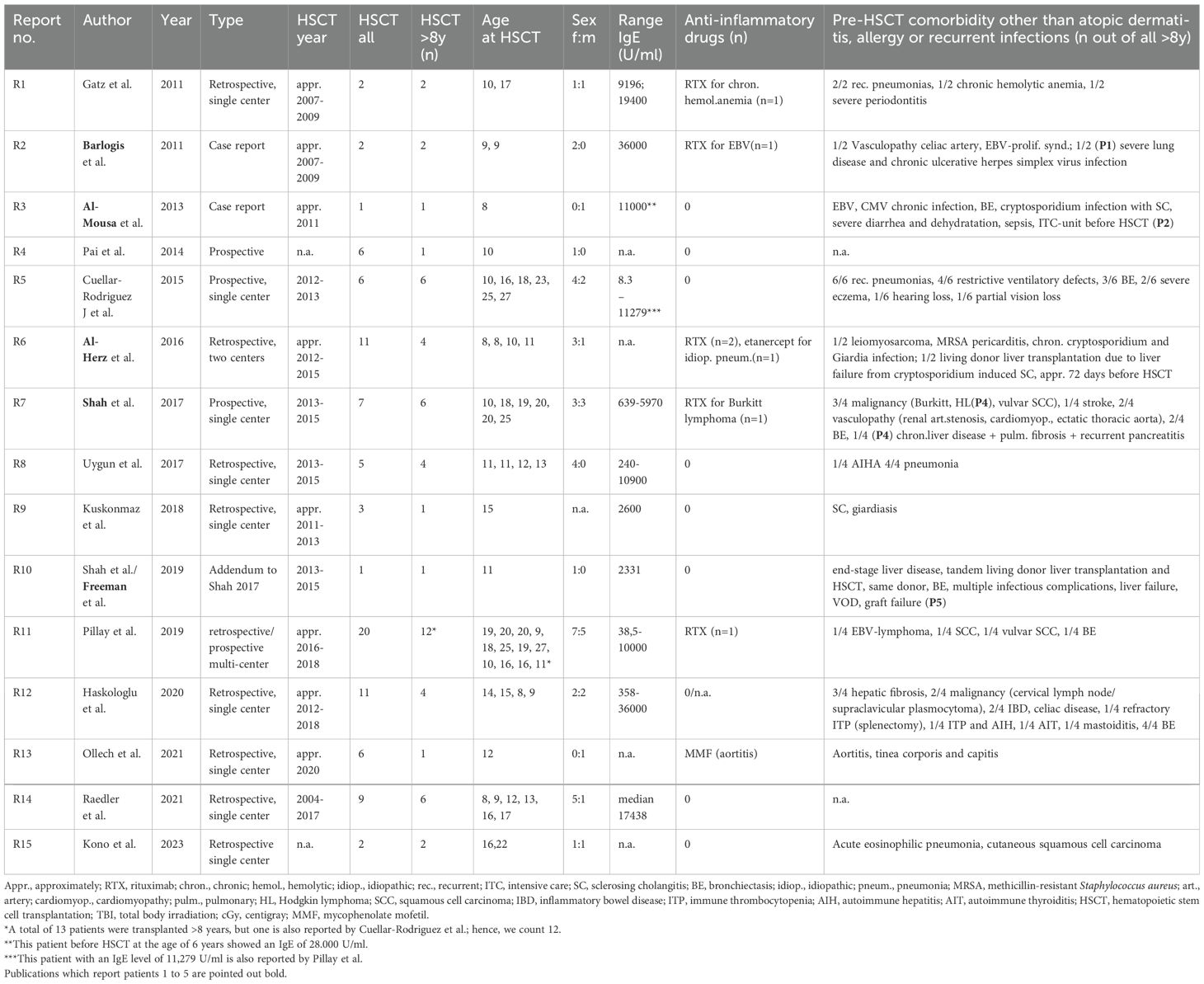
Table 1. Review of the literature part 1 (authors, publication year, type of study, transplantation year, number of patients with HSCT, number of patients ≥8 years at transplantation, sex distribution, concentration of IgE, anti-inflammatory pre-treatment, complications, and comorbidity prior to HSCT).
3.1 AlloHSCT and outcome in patients >8 years of age
The conditioning regimens were busulfan-based (n = 36), treosulfan-based (n = 11), and fludarabine/melphalan-based (n = 3). In one case (P2), unconditioned matched-sibling donor transplantation was conducted presumably as a rescue strategy due to severe intensive care-dependent sepsis (31). Thirty-five patients (66%) received their graft from an MUD, while 34% underwent either mismatched unrelated or haploidentical alloHSCT.
Within the cohort by Aydin et al., 6/81 (7%) had a haploidentical donor, and 62/81 (70%) received their graft from a matched donor. Bone marrow was the major source of stem cells, 37/53 (69%), while 9/53 (17%) received peripheral blood stem cells (PBSC), and two patients received cord blood. Full chimerism was achieved in 45 of 53 patients (84%), very similar to the larger cohort reported by Aydin et al. (22). Acute GvHD [skin grades I–III (n = 10), intestinal grades I–III (n = 7), lung grade II (n = 1); no GvHD data available (n = 2)] occurred in 14 out of 53 patients (26%). Five out of 53 patients (9%) displayed chronic GvHD [gut (n = 1), skin (n = 2), myositis (n = 1), oral lichen planus (n = 1), mucosa (n = 1), liver (n = 1)] and one with suspected bronchiolitis obliterans syndrome (BOS). Acute GvHD occurred in 33% (27% grades II–IV and 11% grades III–IV) and chronic GvHD in 10% (three mild, two moderate, two severe) of patients.
Five patients (9%, P1-5) died within a range of +40 days (P5) to 1 year (P2) post-transplant (23, 28, 30, 31, 34). P1 died due to “transplant-related” cause after haploidentical alloHSCT, approximately in 2007, without further detailed information available (30). One patient (P2) underwent unconditioned alloHSCT of an MSD following treatment in an intensive care unit with sepsis and severe diarrhea, and dehydration due to cryptosporidium infection. Transplantation led to mixed chimerism. Death, 1 year after transplantation, was attributed to uncontrolled chronic GvHD (skin, gut) and sepsis (31). P3, despite MSD and full chimerism, deceased on day +58 due to an invasive Klebsiella species infection at the age of 8.9 years (23). A multifactorial reason for death was reported in P4 (at 25 years), including a history of Hodgkin’s lymphoma, subsequent bleomycin-associated pulmonary fibrosis, recurrent pneumonias, recurrent pancreatitis, and chronic cholestatic liver disease, aggravated by non-compliance, e.g., tobacco abuse (28). One patient (P5) died on day +40 after haploidentical alloHSCT, which had been conducted on day +70 after a living-donor liver transplantation from the same donor. Severe infections—including candida sepsis, acyclovir-resistant herpes simplex viremia, and resistant pseudomonas infection—complicated the course of alloHSCT. Further deterioration of the clinical condition occurred due to veno-occlusive disease (VOD), graft failure, and ultimately multi-organ failure (28, 34) (Table 2). Furthermore, 10 patients were reported to have suffered from a malignancy, 9 of whom survived and recovered fully by their last follow-up.
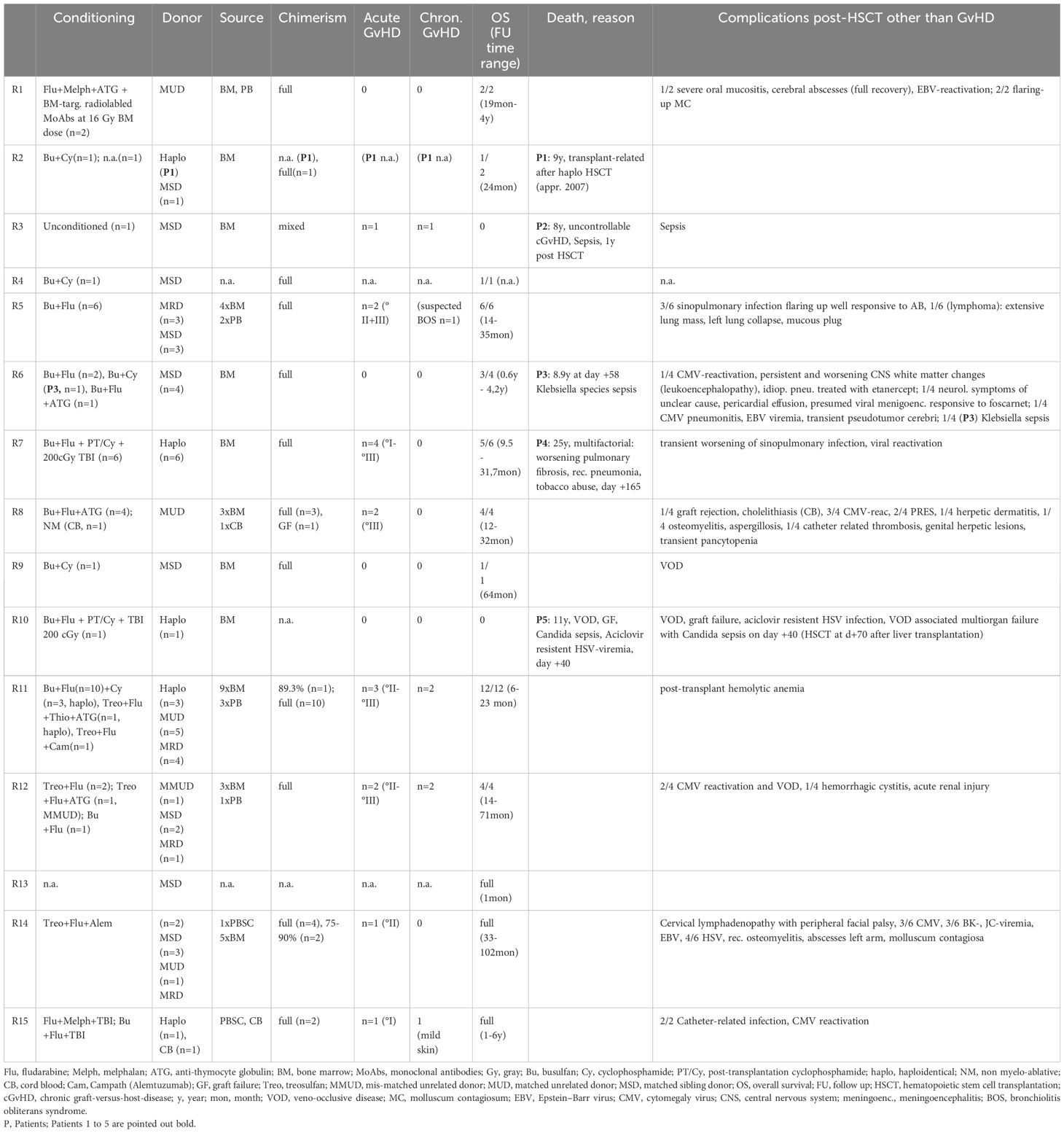
Table 2. Review of the literature part 2 (conditioning regimen, donor characteristics, chimerism, graft-versus-host-disease (GvHD), overall survival, death, reason for death, and post-HSCT complications other than GvHD out of all patients over 8 years of age at transplantation).
4 Discussion
In this work we present an 11-year-old DOCK8-deficient patient with a severe inflammatory disease manifestation and very high IgE levels. To reduce the inflammatory disease prior to alloHSCT, dupilumab was used in combination with rituximab. A substantial decrease in IgE levels and amelioration of skin eczema were achieved. Treatment was well tolerated without adverse events.
Dupilumab, an IL-4/IL-13 receptor inhibitor, suppresses the overwhelming T-helper type 2 inflammatory response (13, 38–40). There is growing evidence for the use of dupilumab either bi-weekly or once every 4 weeks in patients with immunodeficiency syndromes (35, 41–45). The bi-weekly administration seems to be more effective (40). To our knowledge, there is no larger study available on pre-transplantation use of dupilumab in pediatric patients. Nevertheless, there is some evidence for the efficacy of dupilumab in DOCK8 deficiency (35, 45, 46). Ollech et al. described two patients (2 and 10 years of age) receiving dupilumab and discussed the use of dupilumab as a bridge to alloHSCT. However, at the time of publication, none of their patients had received an alloHSCT (35). Additionally, we could find one recent case report on dupilumab and DOCK8 deficiency with high IgE levels in a 6-year-old girl resulting in a successful remission of disseminated eczema herpeticum (46).
Our case is the first reported DOCK8-deficient patient who underwent alloHSCT shortly after treatment with dupilumab. One year post-transplant, the patient’s IgE levels remained slightly elevated. Prolonged elevation of IgE post-transplant in DOCK8 deficiency has been reported in the literature and presumably is due to long lasting chemo-evading plasma B cells, which decline with variable delay (22, 23, 47). The eosinophile count increased after initiation of treatment and eventually decreased over time. This phenomenon is also reported in the literature (25). The patient’s eczema improved significantly before alloHSCT and eventually disappeared within a few weeks after transplantation. No skin toxicity or skin GvHD occurred in our patient. This might be due to the effective control of the patient’s skin disease by pre-transplant use of dupilumab and should be investigated in a larger cohort of patients with DOCK8 deficiency undergoing alloHSCT.
In our patient omalizumab, which is an anti-IgE antibody, was discussed at the time of treatment initiation (2020). Since there is an IgE level and body weight-dependent dosing strategy recommended for this antibody, we decided not to utilize omlizumab for our patient.
The current dosing recommendation is given only for IgE levels below 1,300 IU/ml in the US and 1,500 IU/ml in EU (48). In a study by Menzella et al. published in 2023, the authors conducted a detailed literature review on the efficacy and safety of omalizumab in patients with severe allergic asthma and other allergic diseases focusing on data of patients with higher IgE levels (49). This literature review included a small number of patients with IgE levels above 1,500 IU/ml. Also, in these patients, a beneficial effect of omalizumab could be shown, while there were no severe adverse events reported. Therefore, the authors suggested further analyzing the optimal dose in patients with (very) high IgE levels and eventually extending the dosing recommendation for this patient group (49). Comparative data for the efficacy of dupilumab versus omalizumab for patients with (very) high IgE levels are not available; nevertheless, we postulate that dupilumab might be more effective in this setting as it targets the hyper-IgE disease at the level of cell signaling.
Patients affected by DOCK8 deficiency suffer from combined immune deficiency with severe immune dysregulation. Clinical presentation and laboratory findings might overlap with other IEI, including “treg-o-pathies” (16, 50, 51), Wiskott–Aldrich syndrome (WAS) (4, 52) and hyper-IgE syndromes (autosomal dominant and recessive HIES) (15, 53). Unlike HIES, there are usually no syndromic features in DOCK8 deficiency (4, 54). Patients’ long-term outcome is poor due to an accumulation of life-threatening infections, especially by the age of 20 years, and a predisposition to malignancies (4). To date, alloHSCT is the only curative treatment option (13, 20, 22, 24, 28, 52). Several cohorts have shown promising results also for patients receiving a haploidentical transplantation (28, 37, 55). In almost all surviving patients, the skin disease resolved after successful alloHSCT (13, 22, 24, 28). In a few cases, other comorbidities, including vasculopathy (28, 30, 35), stroke (28), and malignancy (23, 24, 28), were also reported as being in remission. In some cases, remaining food allergies were described (23, 47). There is common agreement on offering alloHSCT to the affected individuals at an early stage of the disease, prior to the accumulation of organ damage, to achieve an optimal transplantation outcome (20, 24, 28, 30, 36). Fatal outcome was observed in five patients (P1, P2, P3, P4, P5), including three patients receiving a haploidentical graft (P1, P4, P 5). For P1, data was incomplete. In P2, P4, and P5, there was a high disease activity pre-transplant. P3 suffered from fulminant bacterial infection at +d58 post-alloHSCT. With regard to malignancy in DOCK8 deficiency, the predisposition to virus-driven malignancy after an alloHSCT remains to be investigated. A successful alloHSCT results in an excellent CD3+-chimerism (13, 20, 24) and recovery of CD8+ cytotoxic T-cell response after alloHSCT (13), which should be preventive of viral-driven malignancy development; nevertheless, long-term data focusing on malignancy and late-onset post-transplant complications are indicated.
In this review of the literature, we focused on DOCK8-deficient patients over 8 years of age receiving alloHSCT and pointed out severe comorbidities and complications related to the accumulation of organ damage by the underlying disease. Especially, for patients with high disease activity, an anti-inflammatory pre-treatment prior to alloHSCT is indicated. A short course of treatment by dupilumab was beneficial in our patient. Further studies in larger IEI cohorts with elevated IgE levels are indicated to confirm a possible improvement of the transplant outcome in these patients.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by IRB Goethe University Hospital Frankfurt. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
ST: Data curation, Formal analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. AW: Data curation, Investigation, Writing – review & editing. AJ: Data curation, Writing – review & editing. ER: Data curation, Writing – review & editing. JF: Data curation, Investigation, Writing – review & editing. DH: Data curation, Investigation, Writing – review & editing. RA: Data curation, Investigation, Writing – review & editing. PB: Supervision, Data curation, Investigation, Writing – review & editing. SB: Supervision, Data curation, Investigation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. SB received funding from the Clinical Scientist Program of the Goethe University Frankfurt.
Acknowledgments
We want to thank the patient and his family, the referring colleagues, and the nursing staff.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1507494/full#supplementary-material
Supplementary Table 1 | Laboratory work up of the patient with dedicator of cytokinesis 8 deficiency. WBC, white blood cell count; Hb, hemoglobin; NK, natural killer cells; DPT, double positive T cells; DNT, double-negative T cells; T4, CD4+ T helper cells; T8, CD8+ cytotoxic T cells; Ig, immunoglobulin; TSH, thyroid stimulation hormone; fT4, free thyroxine; fT3, free triiodothyronine; Ab, antibody; TG, thyroglobulin; TPO, thyroid peroxidase, TSHR, thyroid-stimulating hormone receptor; hGH, human growth hormone, IGFBP-3, insulin-like growth factor binding protein 3; IGF-1, insulin-like growth factor 1; LH, luteinizing hormone; FSH, follicle-stimulating hormone.
References
1. Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. (2014) 133(5):1400–9. doi: 10.1016/j.jaci.2014.02.013
2. Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving DOCK8 in the autosomal recessive form of the hyper-IgE syndrome. J Allergy Clin Immunol. (2009) 124(6):1289. doi: 10.1016/j.jaci.2009.10.038
3. Randall KL, Chan SSY, Ma CS, Fung I, Mei Y, Yabas M, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med. (2011) 208(11):2305–20. doi: 10.1084/jem.20110345
4. Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options - a review of 136 patients. J Clin Immunol. (2015) 35(2):189–98. doi: 10.1007/s10875-014-0126-0
5. Engelhardt KR, Gertz ME, Keles S, Schäffer AA, Sigmund EC, Glocker C, et al. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. (2015) 136(2):402–12. doi: 10.1016/j.jaci.2014.12.1945
6. Boos AC, Hagl B, Schlesinger A, Halm BE, Ballenberger N, Pinarci M, et al. Atopic dermatitis, STAT3- and DOCK8-hyper-IgE syndromes differ in IgE-based sensitization pattern. Allergy. (2014) 69(7):943–53. doi: 10.1111/all.12416
7. Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Arch Dermatol. (2012) 148(1):79–84. doi: 10.1001/archdermatol.2011.262
8. Su HC, Jing H, Angelus P, Freeman AF. Insights into immunity from clinical and basic science studies of DOCK8 immunodeficiency syndrome. Immunol Rev. (2019) 287(1):9–19. doi: 10.1111/imr.12723
9. Biggs CM, Keles S, Chatila TA. DOCK8 deficiency: Insights into pathophysiology, clinical features and management. Clin Immunol. (2017) 181:75–82. doi: 10.1016/j.clim.2017.06.003
10. Ruusala A, Aspenström P. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS Lett. (2004) 572(1–3):159–66. doi: 10.1016/j.febslet.2004.06.095
11. McGhee SA, Chatila TA. DOCK8 immune deficiency as a model for primary cytoskeletal dysfunction. Dis Markers. (2010) 29(3–4):151–6. doi: 10.1155/2010/397291
12. Janssen E, Morbach H, Ullas S, Bannock JM, Massad C, Menard L, et al. DOCK8 deficient patients have a breakdown in peripheral B cell tolerance and defective regulatory T cells. J Allergy Clin Immunol. (2014) 134(6):1365–74. doi: 10.1016/j.jaci.2014.07.042
13. Pillay BA, Avery DT, Smart JM, Cole T, Choo S, Chan D, et al. Hematopoietic stem cell transplant effectively rescues lymphocyte differentiation and function in DOCK8-deficient patients. JCI Insight. (2019) 5:e127527. doi: 10.1172/jci.insight.127527
14. Tangye SG, Pillay B, Randall KL, Avery DT, Phan TG, Gray P, et al. Dedicator of cytokinesis 8-deficient CD4+ T cells are biased to a TH2 effector fate at the expense of TH1 and TH17 cells. J Allergy Clin Immunol. (2017) 139(3):933–49. doi: 10.1016/j.jaci.2016.07.016
15. Keles S, Charbonnier LM, Kabaleeswaran V, Reisli I, Genel F, Gulez N, et al. DOCK8 regulates STAT3 activation and promotes Th17 cell differentiation. J Allergy Clin Immunol. (2016) 138(5):1384–1394.e2. doi: 10.1016/j.jaci.2016.04.023
16. Alroqi FJ, Charbonnier LM, Keles S, Ghandour F, Mouawad P, Sabouneh R, et al. DOCK8 deficiency presenting as an IPEX-like disorder. J Clin Immunol. (2017) 37(8):811–9. doi: 10.1007/s10875-017-0451-1
17. Randall KL, Lambe T, Johnson AL, Johnson A, Treanor B, Kucharska E, et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat Immunol. (2009) 10(12):1283–91. doi: 10.1038/ni.1820
18. Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links toll-like receptor–MyD88 signaling to B cell activation. Nat Immunol. (2012) 13(6):612–20. doi: 10.1038/ni.2305
19. Mizesko MC, Banerjee PP, Monaco-Shawver L, Mace EM, Bernal WE, Sawalle-Belohradsky J, et al. Defective actin accumulation impairs human natural killer cell function in patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. (2013) 131(3):840–8. doi: 10.1016/j.jaci.2012.12.1568
20. Cuellar-Rodriguez J, Freeman AF, Grossman J, Su H, Parta M, Murdock H, et al. Matched related and unrelated donor hematopoietic stem cell transplantation for DOCK8 deficiency. Biol Blood Marrow Transplant.. (2015) 21(6):1037–45. doi: 10.1016/j.bbmt.2015.01.022
21. Kuşkonmaz B, Ayvaz D, Tezcan İ, Yüce A, Sanal Ö, Çetinkaya DU. Successful hematopoietic stem cell transplantation after myeloablative conditioning in three patients with dedicator of cytokinesis 8 deficiency (DOCK8) related hyper IgE syndrome. Bone Marrow Transplant.. (2018) 53(3):339–43. doi: 10.1038/s41409-017-0040-1
22. Aydin SE, Freeman AF, Al-Herz W, Al-Mousa HA, Arnaout RK, Aydin RC, et al. Hematopoietic stem cell transplantation as treatment for patients with DOCK8 deficiency. J Allergy Clin Immunol Pract. (2019) 7(3):848–55. doi: 10.1016/j.jaip.2018.10.035
23. Al-Herz W, Chu JI, van der Spek J, Raghupathy R, Massaad MJ, Keles S, et al. Hematopoietic stem cell transplantation outcomes for 11 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. (2016) 138(3):852–859.e3. doi: 10.1016/j.jaci.2016.02.022
24. Haskologlu S, Kostel Bal S, Islamoglu C, Aytekin C, Guner S, Sevinc S, et al. Clinical, immunological features and follow up of 20 patients with dedicator of cytokinesis 8 (DOCK8) deficiency. Pediatr Allergy Immunol. (2020) 31(5):515–27. doi: 10.1111/pai.13236
25. Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, et al. Dupilumab efficacy and safety in moderate-to-Severe uncontrolled asthma. N Engl J Med. (2018) 378(26):2486–96. doi: 10.1056/NEJMoa1804092
26. Marone G, Granata F, Pucino V, Pecoraro A, Heffler E, Loffredo S, et al. The intriguing role of interleukin 13 in the pathophysiology of asthma. Front Pharmacol. (2019) 10:1387. doi: 10.3389/fphar.2019.01387
27. Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. (2014) 371(2):130–9. doi: 10.1056/NEJMoa1314768
28. Shah NN, Freeman AF, Su H, Cole K, Parta M, Moutsopoulos NM, et al. Haploidentical related donor hematopoietic stem cell transplantation for dedicator-of-Cytokinesis 8 deficiency using post-transplantation cyclophosphamide. Biol Blood Marrow Transplant.. (2017) 23(6):980–90. doi: 10.1016/j.bbmt.2017.03.016
29. Gatz SA, Benninghoff U, Schütz C, Schulz A, Hönig M, Pannicke U, et al. Curative treatment of autosomal-recessive hyper-IgE syndrome by hematopoietic cell transplantation. Bone Marrow Transplant. (2011) 46(4):552–6.
30. Barlogis V, Galambrun C, Chambost H, Lamoureux-Toth S, Petit P, Stephan JL, et al. Successful allogeneic hematopoietic stem cell transplantation for DOCK8 deficiency. J Allergy Clin Immunol. (2011) 128(2):420–422.e2. doi: 10.1016/j.jaci.2011.03.025
31. Al-Mousa H, Hawwari A, Alsum Z. In DOCK8 deficiency donor cell engraftment post-genoidentical hematopoietic stem cell transplantation is possible without conditioning. J Allergy Clin Immunol. (2013) 131(4):1244–5.
32. Pai SY. Treatment of primary immunodeficiency with allogeneic transplant and gene therapy. Hematology. (2019) 2019(1):457–65.
33. Uygun DFK, Uygun V, Reisli İ, Keleş S, Özen A, Yılmaz M, et al. Hematopoietic stem cell transplantation from unrelated donors in children with DOCK8 deficiency. Pediatr Transplant. (2017) 21(7).
34. Shah NN, Freeman AF, Hickstein DD. Addendum to: Haploidentical Related Donor Hematopoietic Stem Cell Transplantation for DOCK8 Deficiency Using Post-Transplantation Cyclophosphamide. Biol Blood Marrow Transplant. (2019) 25(2):e65–7.
35. Ollech A, Mashiah J, Lev A, Simon AJ, Somech R, Adam E, et al. Treatment options for DOCK8 deficiency-related severe dermatitis. J Dermatol. (2021) 48(9):1386–93.
36. Raedler J, Magg T, Rohlfs M, Klein C, Vallee T, Hauck F, et al. Lineage-specific chimerism and outcome after hematopoietic stem cell transplantation for DOCK8 deficiency. J Clin Immunol. (2021) 41(7):1536–48. doi: 10.1007/s10875-021-01069-5
37. Kono A, Wakamatsu M, Umezawa Y, Muramatsu H, Fujiwara H, Tomomasa D, et al. Successful treatment of DOCK8 deficiency by allogeneic hematopoietic cell transplantation from alternative donors. Int J Hematol. (2023) 118(4):519–25. doi: 10.1007/s12185-023-03613-y
38. LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM, et al. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. (2008) 132(2):259–72. doi: 10.1016/j.cell.2007.12.030
39. Gandhi NA, Pirozzi G, Graham NMH. Commonality of the IL-4/IL-13 pathway in atopic diseases. Expert Rev Clin Immunol. (2017) 13(5):425–37. doi: 10.1080/1744666X.2017.1298443
40. Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. (2016) 375(24):2335–48. doi: 10.1056/NEJMoa1610020
41. Lévy R, Béziat V, Barbieux C, Puel A, Bourrat E, Casanova JL, et al. Efficacy of dupilumab for controlling severe atopic dermatitis in a patient with hyper-IgE syndrome. J Clin Immunol. (2020) 40(2):418–20. doi: 10.1007/s10875-020-00751-4
42. Sogkas G, Hirsch S, Jablonka A, Witte T, Schmidt RE, Atschekzei F. Dupilumab to treat severe atopic dermatitis in autosomal dominant hyper-IgE syndrome. Clin Immunol. (2020) 215:108452. doi: 10.1016/j.clim.2020.108452
43. Votquenne N, Dupire G, Michel O, Ben Said B. Dupilumab for severe generalized eczematous eruption complicating common variable immunodeficiency. Eur J Dermatol. (2021) 31(1):93–4. doi: 10.1684/ejd.2020.3954
44. Charvet E, Bourrat E, Hickman G, Donadieu J, Bellanné-Chantelot C, Jachiet M, et al. Efficacy of dupilumab for controlling severe atopic dermatitis with dominant-negative CARD11 variant. Clin Exp Dermatol. (2021) 46(7):1334–5. doi: 10.1111/ced.14686
45. Guo T, Wei L, Karki S, Wen S, Li Q, Lin Y. Omalizumab and dupilumab for the treatment of autosomal-recessive DOCK8 hyper-IgE syndrome. Indian J Dermatol Venereol Leprol (2024) 1–3. doi: 10.25259/IJDVL_348_2023
46. Johar RA, Hasanain A, Khouqeer Y. Efficacy of dupilumab in treating atopic dermatitis with recurrent eczema herpeticum in a patient with DOCK8-deficiency hyper-IgE syndrome: A case report. Cureus 15(8):e43360. doi: 10.7759/cureus.43360
47. Happel CS, Stone KD, Freeman AF, Shah NN, Wang A, Lyons JJ, et al. Food allergies can persist after myeloablative hematopoietic stem cell transplantation in DOCK8-deficient patients. J Allergy Clin Immunol. (2016) 137(6):1895–1898.e5. doi: 10.1016/j.jaci.2015.11.017
48. Xolair | european medicines agency (EMA) [Internet]. (2009). Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/xolair (Accessed 2024 Nov 27).
49. Menzella F, Just J, Sauerbeck IS, Mailaender C, Saccheri F, Thonnelier C, et al. Omalizumab for the treatment of patients with severe allergic asthma with immunoglobulin e levels above >1500 IU/mL. World Allergy Organ J. (2023) 16(6):100787. doi: 10.1016/j.waojou.2023.100787
50. Gambineri E, Ciullini Mannurita S, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, x-linked (IPEX) syndrome. Front Immunol [Internet] 9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6223101/. doi: 10.3389/fimmu.2018.02411
51. Singh AK, Eken A, Hagin D, Komal K, Bhise G, Shaji A, et al. DOCK8 regulates fitness and function of regulatory T cells through modulation of IL-2 signaling. JCI Insight. (2017) 2(19):e94275, 94275. doi: 10.1172/jci.insight.94275
52. Albert MH, Freeman AF. Wiskott-aldrich syndrome (WAS) and dedicator of cytokinesis 8- (DOCK8) deficiency. Front Pediatr. (2019) 7:451. doi: 10.3389/fped.2019.00451
53. Béziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. (2018) 3(24):eaat4956. doi: 10.3389/fped.2019.00451
54. Ma CS, Tangye SG. Flow cytometric-based analysis of defects in lymphocyte differentiation and function due to inborn errors of immunity. Front Immunol [Internet] 10:2108/full. doi: 10.3389/fimmu.2019.02108/full
55. Ghosh S, Schuster FR, Adams O, Babor F, Borkhardt A, Comoli P, et al. Haploidentical stem cell transplantation in DOCK8 deficiency - successful control of pre-existing severe viremia with a TCRaß/CD19-depleted graft and antiviral treatment. Clin Immunol. (2014) 152(1–2):111–4. doi: 10.1016/j.clim.2014.03.006
Keywords: DOCK8-deficiency, alloHSCT, dupilumab, combined immunodeficiency, omalizumab
Citation: Trombello S, Jarisch A, Willasch A, Rettinger E, Fekadu-Siebald J, Holzinger D, Adelmann R, Bader P and Bakhtiar S (2025) Case report: Advanced age at transplantation and pre-emptive treatment with dupilumab in DOCK8 deficiency. Front. Immunol. 15:1507494. doi: 10.3389/fimmu.2024.1507494
Received: 07 October 2024; Accepted: 02 December 2024;
Published: 28 January 2025.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Victoria R. Dimitriades, University of California, Davis, United StatesElizabeth Secord, Wayne State University, United States
Copyright © 2025 Trombello, Jarisch, Willasch, Rettinger, Fekadu-Siebald, Holzinger, Adelmann, Bader and Bakhtiar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shahrzad Bakhtiar, YmFraHRpYXJAbWVkLnVuaS1mcmFua2Z1cnQuZGU=