 Zixin Zhang
Zixin Zhang Jiahui Wang2
Jiahui Wang2 Zijian Zeng
Zijian Zeng Haijian Dong
Haijian Dong- 1Central Laboratory, Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2School of Clinical Medicine, Chengdu University of Traditional Chinese Medicine, Chengdu, China
Liver fibrosis represents a wound-healing response to chronic liver injury caused by viral infections, alcohol, and chemicals agents. It is a critical step in the progression from chronic liver disease to cirrhosis and hepatocellular carcinoma. No chemical or biological drugs have been approved for the treatment of liver fibrosis. Relevant studies have demonstrated that effective inhibition of hepatitis B virus (HBV) replication by nucleoside (acid) analogs or polyethylene glycol alpha-interferon can lead to recovery in some patients with hepatitis B liver fibrosis, However, some patients with liver fibrosis do not show improvement, even after achieving a complete serologic and virologic response. A similar situation occurs in patients with hepatitis C-related liver fibrosis. The liver, with its unique anatomical and immunological structure, is the largest immune organ and produces a large number of cytokines in response to external stimuli, which are crucial for the progression of liver fibrosis. cytokines can act either by directly affecting hepatic stellate cells (HSCs) or by indirectly regulating immune target cells. Among these, the interleukin family activates a complex cascade of responses, including cytokines, chemokines, adhesion molecules, and lipid mediators, playing a key role in the initiation and regulation of inflammation, as well as innate and adaptive immunity. In this paper, we systematically summarize recent literature to elucidate the pathogenesis of interleukin-mediated liver fibrosis and explore potential therapeutic targets for liver fibrosis treatment.
Introduction
Liver fibrosis is a pathological change that occurs during the course of most chronic liver diseases. It is caused by chronic inflammation due to the persistent effects of various etiologies such as viral hepatitis infections, non-alcoholic fatty liver disease, autoimmune liver disease, and drug-induced liver injury. This inflammation leads to liver damage and, consequently, to liver fibrosis (1, 2). Currently, no chemical or biological drugs have been approved for the treatment of liver fibrosis (3). In modern medicine, the treatment of liver fibrosis is primarily etiological. Although studies have demonstrated that liver fibrosis is a reversible pathological process (4), without early intervention, it can progress to cirrhosis or even hepatocellular carcinoma. Approximately more than 2 million people die each year from chronic liver disease (5).
The primary pathological feature of liver fibrosis is the excessive pathological proliferation and deposition of extracellular matrix (ECM) (2). HSCs, the main fibrotic cell type, reside in the perisinusoidal space between hepatic sinusoidal endothelial cells and hepatocytes (6). These cells are non-substantial hepatocytes, accounting for about 15% of the total number of resident cells (7). In the normal liver, HSCs maintain a nonproliferative, quiescent phenotype. However, during liver injury, hepatocytes undergo apoptosis or necrosis, releasing proinflammatory and profibrotic cytokines. These cytokines stimulate the recruitment and activation of inflammatory cells in the liver. At this point, HSCs became activated and transform from vitamin A-storing cells into myofibroblasts with proliferative, contractile, and chemotactic functions. These activated HSCs produce excessive ECM components that accumulate in liver parenchymal cells, disrupting liver structure and forming characteristic scar tissue (8). Activation of HSCs is central to the pathogenesis of liver fibrosis, and HSCs are the primary cellular source of ECM production (7). A variety of cytokines act as “messenger” proteins involved in regulating immunity, cell growth, and tissue repair through paracrine and autocrine effects on target cell-specific receptors. Several cytokines regulate liver fibrosis, particularly through the regulation of collagen metabolism in HSCs and intercellular matrix.
Cytokines include interleukins, interferons, growth factors, chemokines. Among them, the role of the interleukin family in liver fibrosis has gained increasing attention. There are 33 known interleukins, along with numerous derivatives such as Interleukin-1 beta (IL-1β), Interleukin-36 alpha (IL-36α), and Interleukin-36 gamma (IL-36γ). Interleukins are primarily secreted by various of immune cells (e.g., macrophages, neutrophils), and are glycoproteins that act on a variety of cells throughout the body. They regulate immune cell activation, proliferation, secretion, and other processes,playing an essential role in the progression of the inflammatory response. Numerous studies have shown that the interleukin family plays a critical role in the development of liver fibrosis. On one hand, pro-inflammatory interleukins such as Interleukin-1 (IL-1), Interleukin-6 (IL-6), Interleukin-17 (IL-17), Interleukin-18 (IL-18), Interleukin-33 (IL-33), and Interleukin-36 (IL-36) enhance tissue damage and inflammation. On the other hand, anti-inflammatory interleukins such as Interleukin-10 (IL-10), Interleukin-35 (IL-35), and Interleukin-37 (IL-37) promote tissue regeneration and play a protective role in the liver. Additionally, Interleukin-4 (IL-4), Interleukin-22 (IL-22) exerts both anti-inflammatory and pro-inflammatory roles in liver fibrosis.
Currently, interleukins are widely recognized for their regulatory roles in the development of liver fibrosis. However, the overall regulatory mechanisms remain unclear, and the specific signaling pathways and transduction links involved need further investigated. Therefore, in this article, we systematically summarize recent literature to elucidate the pathogenesis of interleukin-family-mediated liver fibrosis and review fibrosis drugs currently targeting interleukins as therapeutic candidates. We aim to provide new insights into the treatment of liver fibrosis.
Interleukins exerting pro-liver fibrosis effects
Interleukin-1
IL-1 is one of the earliest cytokines discovered.It is mainly produced by monocytes, macrophages, T lymphocytes (T cells), B lymphocytes (B cells) and Natural Killer cells (NK cells) (9). Additionally, almost all nucleated cells can produce IL-1. In the liver, it is primarily expressed by Kupffer cells (KCs), hepatic endothelial cells, and HSCs, mainly in the form of Interleukin-1 alpha (IL-1α) and IL-1β, both forms recognize a common receptor, Interleukin-1 Receptor (IL-1R), which consists of the subunits IL-1R1 and Interleukin-1 Receptor Accessory Protein (IL-1RacP). These receptors mediate IL-1’s involvement in immune responses, inflammation, and fibrogenesis (10). IL-1 is synthesized as a precursor, pro-IL-1, which is converted to its active form. The active cytokines promotes fibrosis by being released extracellularly, thereby initiating a cascade of inflammatory responses in target cells, such as HSCs (11). Among the two isoforms, IL-1α is a bifunctional cytokine. Extracellularly, IL-1α binds to IL-1R1 on the cell surface and recruits its co-receptor IL-1R3, initiating pro-inflammatory signaling similar to IL-1β. Intracellularly, IL-1α can shuttle rapidly between the nucleus and cytoplasm, enabling distinct biological functions in different cellular compartments. Since there is limited research on the role of IL-1α in liver fibrosis, this review primarily focuses on IL-1β, an inducible and highly inflammatory cytokine.IL-1β activates a complex signaling cascade via IL-1R1, which subsequently triggers transcription factors such as nuclear factor-kappa B (NF-κB) and induces the production of inflammatory cytokines (12). It has been shown that IL-1β contributes to fibrosis through the MyD88-IRAK-NF-κB signaling pathway by forming a tripartite complex with IL-1R1 and IL-1RacP (13–15).
In patients with chronic liver disease, serum expression of IL-1β is elevated, and IL-1β has been shown implicated of hepatic steatosis to steatohepatitis and hepatic fibrosis (16, 17).Hepatic tissue expression of IL-1α and IL-β is significantly increased in diet-induced nonalcoholic steatohepatitis (NASH) models, indicating that these cytokines are involved in the regulation of steatosis and steatohepatitis. Despite a reduced inflammatory response, both liver cholesterol and serum cholesterol levels were elevated in IL-1α-deficient mice, suggesting that IL-1α may influence hepatic fat accumulation and inflammation through distinct pathways. However, studies using IL-1α and IL- 1β knockout mice showed improved diet-induced steatosis, indicating that both IL-1α and IL- 1β contribute to NASH development (18). Furthermore, liver and non-bone marrow-derived IL-1α/β deficiency ameliorated diet-induced liver inflammation and fibrosis, suggesting a critical role for liver-derived IL-1 in this context (18). High expression of the immune checkpoint T-cell Immunoglobulin and Mucin-domain containing-3 (Tim-3) on hepatic macrophages has been shown to attenuates inflammation-related hepatic injury in a NASH mouse model. In vitro assays demonstrated that Tim-3 negatively regulates reactive oxygen species production and secretion of pro-inflammatory cytokines, such as IL-1β, in macrophages, thereby reducing the severity of inflammatory injury in NASH (19). In mice with high-fat diet-induced nonalcoholic fatty liver disease (NAFLD), upregulation of IL-1β in hepatocytes contributes to increased fat aggregation, liver inflammation, insulin resistance and liver fibrosis (20). KCs play an important role in liver injury in alcohol-fed models. Acute and chronic alcohol feeding activates KCs via the “lipopolysaccharide-toll-like receptor 4” signaling axis (21), leading to the production of pro-inflammatory mediators such as IL-1β and Tumor Necrosis Factor alpha (TNF-α). This cascade ultimately results in hepatocellular dysfunction, apoptosis, necrosis, and the ECM production by HSCs, contributes to liver fibrosis and cirrhosis (22). In alcoholic liver injury models using IL-1β-deficient mice, liver damage was significantly reduced, highlighting the regulator role of the IL-1β signaling pathway is steatosis, inflammation, and liver fibrosis (22). In a rat model of chronic alcoholic liver disease, elevated IL-1β exacerbated hepatocyte injury, promoted the release of pro-fibrotic factors such as Transforming Growth Factor beta (TGF-β) and platelet-derived growth factor, and activated HSCs. These findings underscore the crucial role of IL-1β in alcoholic liver injury and its progression fibrosis, suggesting that targeting the IL-1β signaling pathway could offer therapeutic potential (18).
Caspase-1-mediated pyroptosis is a classical mechanism that induces the maturation of inflammatory cytokines, such as IL-1β, triggering cell lysis and death, promoting pyroptosis, activating inflammasomes, and driving fibrosis development (23). Experimental studies have shown that KCs activation by Lipopolysaccharide releases pro-inflammatory cytokines and fibrogenic factors, inducing hepatocyte pyroptosis, HSCs activation, and ECM production (24); The NOD-like Receptor Pyrin domain-containing 3 (NLRP3) inflammasome has been found to induced IL-1β secretion in primary HSCs or HSCs lines(e.g., LX-2 or HSC-T6) treated with exogenous stimulants such as MSU or bacterial RNA (25, 26). IL-1β interacts with receptors on HSCs membranes to activate NF-kB, leading to the production of Alpha-Smooth Muscle Actin (α-SMA) and type I collagen, both markers of fibrosis (25, 26).
Currently, IL-1β is being explored as a potential therapeutic target for liver fibrosis. The regulation of the Peroxisome Proliferator-Activated Receptor (PPAR) nuclear receptor family plays an important role in NASH treatment. Peroxisome Proliferator-Activated Receptor gamma (PPARγ) antagonists have demonstrated therapeutic efficacy in NASH, while Peroxisome Proliferator-Activated Receptor Delta (PPARδ) activation improved fatty acid oxidation and inhibited hepatic liposynthesis and gluconeogenesis (27). PPARδ antagonists also alleviated liver inflammation and fibrosis by inhibiting the production of pro-inflammatory factors such as IL-1β. MCC950 an inhibitor of NLRP3 inflammasome activation, downregulated IL-1β expression and significantly reduced hepatic fibrosis (28). Additionally, chuanxiongzine has been shown to protect against hepatic injury by modulating the NLRP3 inflammasome pathway, lowering IL-1β levels, and reducing fibrosis-related inflammation (29). Polysaccharides extracted from Angelica sinensis root attenuated hepatic fibrosis by inhibiting IL-1β secretion and HSCs activation (30, 31). In vivo and in vivo studies revealed that Aspergillus extracts ameliorated carbon tetrachloride (CCl4)-induced liver fibrosis and TGF-β-induced HSCs activation, likely through the Nrf2-mediated inhibition of the Reactive Oxygen Species/NOD-like Receptor Pyrin domain-containing 3/Interleukin-1 alpha (ROS/NLRP3/IL-1β) signaling pathway (32). Collectively, these studies highlight the therapeutic potential of targeting IL-1β in liver fibrosis.
Interleukin-6
IL-6, initially identified as B-cell growth/stimulating factor II (33), is produced by various cells types, including fibroblasts, hepatocytes, monocytes/macrophages, T-cells, and endothelial cells (34). In the liver, IL-6 is predominantly expressed in hepatocytes, KCs, and HSCs. IL-6 is a pro-inflammatory cytokines with pleiotropic biological activities. Numerous studies have demonstrated that, in addition to promoting inflammatory responses, IL-6 induces lymphocyte differentiation and proliferation, facilitates HSCs activation, and contributes to the development of liver fibrosis (35).
As a pro-inflammatory cytokines, IL-6 participates in liver inflammatory responses and plays a critical role in liver fibrosis progression by regulating the secretion of pro-fibrotic factors, activating signaling pathway, and promoting the proliferation and differentiation of fibroblasts. In the transformation of chronic hepatitis B (CHB)-associated liver fibrosis to cirrhosis, significantly elevated levels of IL-6 mRNA have been detected in liver tissues, peripheral blood mononuclear cells, and serum of patients with cirrhosis. Correspondingly, IL-6 protein levels are markedly higher in cirrhosis patients compared to those with liver fibrosis (36). A positive correlation between serum IL-6 levels and the degree of fibrosis in NASH has also been established (37). Additionally, IL-6 levels were observed to increase progressively in patients with varying severities of NAFLD. These levels were significantly associated with liver enzymes, steatosis, and fibrosis, indicating that elevated peripheral serum IL-6 may promote the progression of liver fibrosis to cirrhosis (38).
Studies have demonstrated that serum IL-6 expression increases before hepatocyte necrosis occurs. Additionally, cytokines such as TGF-β, IL-6, and Interleukin-8 (IL-8) are significantly elevated in the perihepatic sinusoidal wall and interlobular septa of the liver, correlating with the degree of inflammation and liver fibrosis (39, 40). In one study (41), IL-6 gene knockout mice subjected to acute and chronic liver injury models induced by CCl4 exhibited increased α-SMA levels in HSCs, indicating the role of IL-6 in fibrosis. Glycoprotein 130 (gp130), the signal transduction receptor subunit for IL-6, plays a critical role in this pathway. In high-fat diet-induced fatty liver disease models, IL-6 or gp130 gene knockout resulted in significant improvement in liver inflammation and steatosis, as well as varying degrees of inhibition of tissue remodeling and fibrosis (42). However, in a CCl4-induced chronic liver injury model, selective gp130 gene knockout in non-parenchymal liver cells aggravated liver fibrosis, highlighting the complex role of gp130 signaling in fibrosis (43).
Mice deficient in acetaldehyde dehydrogenase and fed ethanol were more susceptible to liver inflammation and fibrosis, likely due to higher levels of malondialdehyde-acetaldehyde adducts. These adducts activate the Interleukin-6/Signal Transducer and Activator of Transcription 3 (IL-6/STAT3) pathway in the liver (44). The Extracellular Signal-Regulated Kinase (ERK) pathway also plays a significant role. ERK, a serine/threonine protein kinase with two subtypes (ERK1 and ERK2), is activated by IL-6. IL-6 synergizes with acetaldehyde or malondialdehyde to activate the Mitogen-Activated Protein Kinase (MAPK) cascade through gp130, thereby phosphorylating and initiating the ERK1/2 signaling pathway. This activation promotes HSCs activation and liver fibrosis formation in mice (45, 46). Additionally, IL-6 induces HSCs activation and liver fibrosis via the Janus Kinase/Signal Transducer and Activator of Transcription 3 (JAK/STAT3) signaling pathway (47).
Interestingly, studies have reported conflicting results regarding IL-6’s role in liver fibrosis. For instance, in high-fat diet-fed mice with myeloid-specific IL-6 receptor A knockout (IL-6RA-KO), liver fibrosis was more severe compared to wild-type mice. This phenomenon was linked to decreased levels of anti-fibrotic microRNA-223 (miR-223). IL-6 treatment was shown to prompt macrophages to release miR-223-rich exosomes, which subsequently reduced the expression of the fibrosis-promoting transcriptional coactivator with PDZ-binding motif (TAZ) in hepatocytes, thereby ameliorating liver fibrosis (48). Moreover, myeloid-specific IL-6Rα knockout mice (Il6raMye-/-) and IL-6-silenced mice subjected to high-fat diets exhibited reduced inflammation but increased liver fibrosis, further highlighting the complex and sometimes contradictory role of IL-6 in fibrosis (48). These discrepancies may stem from differences in experimental models, the broad expression of transmembrane IL-6Rα and its signaling chain gp130, and the involvement of soluble IL-6R and soluble gp130 (49).
IL-6 plays an important role in liver fibrosis caused by various etiologies. As a key pathway in fibrosis development, the IL-6-related signaling cascade holds promise as a novel serum marker for evaluating the severity of liver fibrosis. Furthermore, antibodies-based therapies targeting the IL-6 pathway represent a potential new approach for treating liver fibrosis.
Interleukin-17
IL-17 is a characteristic cytokines of T helper 17 cells (Th17 cells), predominantly produced by Th17 cells and primarily expressed in Th17 cells, hepatocytes, and KCs in the liver (50). Interleukin-17A (IL-17A), a member of the IL-17 family, is commonly regarded as an important inflammatory mediator. It promotes the progression of liver fibrosis by acting on HSCs and contributing to ECM remodeling (51).
IL-17A has been shown to stimulate the activation of HSCs and contribute to liver fibrosis by increasing the expression of pro-inflammatory cytokines, such as IL-6, IL-1β, and TNF-α, as well as pro-fibrotic factors like TGF-β and α-SMA. The synergistic effects of IL-17A and TGF-β cytokines further activate HSCs, leading to increased collagen production and exacerbating liver fibrosis (52, 53). In a CCl4-induced mouse model of liver fibrosis, exosome-mediated activation of Toll-like Receptor 3 in HSCs during the early stages of liver injury promoted the progression of liver fibrosis by enhancing the production of IL-17A from Gamma Delta T cells (54). KCs, expressing IL-17RA and IL-17RC are activated by IL-17A, further promoting liver inflammation and fibrosis by secreting pro-inflammatory mediators and pro-fibrotic cytokines such as TGF-β, which induce HSCs activation (53, 55). In vitro studies have demonstrated that IL-17A induces the expression of Matrix Metalloproteinase-2 and Matrix Metalloproteinase-9 (56), and another study found that liver fibrosis was significantly reduced in a model of IL-17A receptor-deficient mice infected with Schistosoma haematobium (57). An analysis of 22 human liver samples at different fibrosis stages (F0 ~ F4) suggested that IL-17A promotes fibrosis by inducing the expression of IL-6 and other pro-fibrotic markers such as IL-22 and TGF-β1 (58). These findings confirm that IL-17A contributes to liver fibrosis through two primary pathways: (1) IL-17A expressed in the liver interstitium directly activates HSCs to produce large amounts of collagen; (2) IL-17A stimulates endothelial cells and fibroblasts to secrete various cytokines, chemokines, and cell adhesion factors, inhibited ECM degradation, and promotes fibroblasts proliferation. IL-17A-mediated immune responses significantly affect the hepatic microenvironment, advancing liver fibrosis and correlating positively with fibrosis severity (53).
IL-17A plays a key role in the inflammatory pathways associated with liver injury and may serve as a potential adjunctive diagnostic marker for liver fibrosis (59). Since Cirrhosis can progress to Hepatocellular Carcinoma, studies have suggested that the combined measurement of Alpha-Fetoprotein and IL-17 levels in peripheral serum can predict the prognosis of cirrhotic patients (60). In Hepatitis C Virus (HCV)-associated liver fibrosis, elevated serum IL-17A levels are positively correlated with aminotransferases levels, alpha-fetoprotein concentrations, and fibrosis staging scores, indicating that IL-17A could serve as a biomarker for inflammation and fibrosis progression in chronic HCV infection (61). Therapeutically, IL-17A inhibitors have shown promise in preclinical and clinical studies. These inhibitors suppress HSCs activation and collagen production (51). In a mouse model of Bile Duct Ligation (BDL)-induced liver fibrosis, IL-17A antibody therapy reduced hepatocyte necrosis, decreased pro-inflammatory cytokines, and mitigated neutrophil and macrophage infiltration (62). Furthermore, IL-17A antibody therapy ameliorated hepatic fibrosis in a NASH mouse model (63). Interestingly, IL-17A antibody treatment improved liver fibrosis in 10 psoriasis patients (64), highlighting its potential as a therapeutic target for anti-fibrotic therapies.
Interleukin-18
IL-18, a member of the IL-1 superfamily originally identified as an Interferon-gamma (IFN-γ) inducer, plays a critical role in regulating both innate and adaptive immune responses (65). IL-18 exerts its immunomodulatory effects primarily binding to the Interleukin-18 Receptor (IL-18R) on the cell membrane (66). It is predominantly synthesized and secreted by KCs and monocytes (67).
In mice with NASH-associated liver fibrosis induced by a high-fat diet, Studies have shown that IL-18 promotes the progression of liver fibrosis. Single-cell RNA sequencing data revealed high expression levels of IL-18 and IL-18R1 on mouse HSCs. Treatment of primary mouse HSCs with recombinant IL-18 accelerated their differentiation into myofibroblasts. Furthermore, the activation of HSCs triggered by NLRP3 inflammasome activation was inhibited when IL-18 signaling was blocked by its natural antagonist, Interleukin-18 Binding Protein (68).
A recent study found that, in a mouse model of CCl4-induced liver fibrosis, the Citrus aurantium extract astragalin inhibited the expression of TNF-α, IL-18, and IL-1β mRNAs in the livers of fibrotic mice through the NF-κB/NLRP3 inflammasome pathway, significantly ameliorating liver fibrosis (69). In the mouse model of liver fibrosis induced by thioacetamide, ginsenoside was shown to inhibit the entry of IL-1β and IL-18 into the extracellular matrix by regulating ERRα-P2X7r signaling pathway, thereby reducing inflammatory responses, improving hepatocyte damage, and suppressing liver fibrosis (70). In clinical studies, the expression level of IL-18 in the peripheral serum of patients with hepatitis B-associated cirrhosis complicated by hepatorenal syndrome (HRS) was significantly higher than in patients without this complication. The sensitivity and specificity of IL-18 for predicting HRS were 90.32% and 71.70%, respectively, suggesting that IL-18 could predict the prognosis of patients with hepatitis B-related cirrhosis (71). Additionally, another study suggested a potential association between the IL-18 -137G/C gene variant and the risk of cirrhosis susceptibility (72). These studies indicate that traditional Chinese medicine monomer compounds can improve liver fibrosis by inhibiting IL-18. Furthermore, IL-18 could serve as a therapeutic target and potential biomarker for the prognosis of liver fibrosis. However, extensive clinical and basic research is still required to confirm these findings.
Interleukin-33
IL-33, a member of the IL-1 cytokine superfamily, is a key regulator in pathological inflammation, immune homeostasis, and fibrosis. IL-33 plays a key role in innate and adaptive immunity, contributes to tissue homeostasis, and responds to environmental stress. It is abundantly expressed in the endothelial and epithelial cells during both homeostasis and inflammation (73). In the liver, IL-33 is primarily expressed in HSCs and KCs (74). Its reporter, Suppression of Tumorigenicity 2 (ST2), also known as Interleukin-1 Receptor-Like 1, is predominantly expressed in tissue-resident immune cells (75). Upon cellular damage, IL-33 binds to ST2, stimulating immune cell activity (76). The IL-33/ST2 signaling axis is pivotal in liver fibrosis, balancing inflammation with tissue regeneration, wound healing, and tissue repair.
IL-33 has been found to correlate positively with collagen expression, with HSCs being the primary source of IL-33 in fibrotic liver (77). Protein and mRNA levels of IL-33 and ST2 are elevated in fibrotic livers of both mice and humans, with these levels significantly increasing as fibrosis progresses (78, 79). In a mouse model of hepatic fibrosis, IL-33 knockdown led to a marked improvement in fibrosis (80). However, another study reported that IL-33 deficiency did not attenuate liver fibrosis in a high-fat diet-induced steatohepatitis model (81). In a schistosome mouse model, both IL-33 and ST2 levels were elevated, suggesting that the IL-33/ST2 axis might serve as a therapeutic target for liver fibrosis (82). However, while IL-33-targeted therapy mitigates high-fat diet-induced hepatic steatosis, it can exacerbate liver fibrosis through ST2 signaling (79).
Numerous studies have confirmed that the Interleukin-33/T helper 2 cells (IL-33/Th2) axis contributes to liver fibrosis (83). Group 2 innate lymphoid cells (ILC2s), a novel type of innate immune cell in the lymphocyte lineage, are important for liver immune homeostasis and secrete pro-fibrotic cytokines that mediate fibrogenesis. IL-33 attracts ILC2s and activates HSCs by inducing cytokines such as IL-13. Animal studies have shown that ST2 deletion reduces liver injury, inflammatory cell infiltration, and fibrosis. IL-33 activates and aggregates ILC2 cells via ST2 in the liver, and the activated ILC2s secrete IL-13, which in turn activates HSCs through the IL-4Rα-STAT6 transcription factor pathway (80). Furthermore, IL-33 recruits and activates Th2-like CD4+ T cells, which enhance HSCs activation in an Interleukin-13 (IL-13)-dependent manner (84). The IL-33-mediated Th2 immune response promotes HSCs proliferation, TGF-β synthesis, and collagen deposition, with overexpression of IL-33 inducing liver fibrosis. This suggests that IL-33 exerts its pro-fibrotic effects primarily through IL-13 (77).
Regulatory T cells (Tregs), which provide negative feedback regulation of immune responses, are also influenced by IL-33. Studies have shown that Tregs activation by IL-33 is dependent on MyD88. IL-33 binding to ST2 on Tregs activates MyD88, resulting in the expansion of Foxp3+ Tregs in vivo (85). In liver fibrosis, IL-33 may play a dual regulatory role through ST2+Treg. On one hand, activated Tregs can promote fibrogenesis. In chronic hepatitis C, Tregs accumulate at fibrotic sites and secrete interleukin-8, which acts on HSCs to upregulation fibrosis-related factors and collagen (86). Additionally, Tregs may adopt a Th2-like role, with IL-33 inducing ST2+ Foxp3+ Tregs to promote fibrosis (87). On the other hand, Tregs exert anti-fibrotic effects via IL-10 secretion. In a BDL-induced liver fibrosis model, Treg inhibition resulted in reduced IL-10 expression, increased fibrosis, and greater inflammatory cell infiltration (88).
Genetic studies have identified associations between IL-33 gene variants and susceptibility to HBV-related cirrhosis. IL33-rs4742170C, rs1048274G, and rs10975519C variants may serve as potential biomarkers for diagnosing HBV-associated cirrhosis (89). In HCV-associated fibrosis, serum IL-33 levels correlate with fibrosis stage and viral load, suggesting that IL-33 could be a biomarker for disease progression (90). Recent research (91) has identified a new chemokine, PSMP (PC3-secreted microprotein), whose receptor is C-C Chemokine Receptor 2 (CCR2). PSMP promotes hepatic fibrosis by polarizing macrophages and directly activating HSCs via CCR2. IL-33, as a damage-associated molecular pattern (DAMP), enhances PSMP production, highlighting its critical role in hepatic fibrosis progression (92). In BDL-induced hepatic fibrosis models, injury-induced release of endogenous IL-33 triggers inflammation and fibrosis. However, acute massive liver injury sees IL-33 activating tissue-protective mechanisms, whereas in chronic injury, it promotes fibrosis (77). Melatonin has demonstrated protective effects in hepatic ischemia-reperfusion injury by inhibiting oxidative stress and apoptosis through the IL-33 signaling pathway (93). The role of IL-33 in liver fibrosis warrants further exploration and holds potential as a therapeutic target for fibrosis treatment.
Interleukin-36
IL-36 is a member of the IL-1 superfamily. It Regulates immune cell responses, and activates fibroblasts. IL-36 consists of three receptor agonists- IL-36α, IL-36β, and IL-36γ-as well as the IL-36 receptor antagonist (IL-36Ra). Initially, IL-36 was also referred to as IL-1F6, IL-1F8, among another names. The receptors for IL-36 are primarily IL-36 receptor alpha (IL-36Rα) and IL-1 receptor accessory protein IL-1RAcP, which together form a complex consisting of two subunits (94). IL-36 cytokines are expressed in various cell types, including keratinocytes, monocytes, and dendritic cells (DCs). In the liver, IL-36 is predominantly expressed in macrophages, DCs, and endothelial cells. IL-36α is expressed during embryonic development and is highly enriched in epithelial cells, monocytes, B cells, and T cells (95, 96). IL-36β is also expressed in epithelial cells and is regulated by epidermal growth factor (97). IL-36γ is expressed in stimulated esophageal keratinocytes and squamous epithelium cells (96, 98). IL-36 influences inflammatory responses through multiple mechanisms, including inducing the secretion of inflammatory mediators and chemokines and modulating immune cell function.
Research indicates that IL-36 participates in early tissue inflammatory responses via multiple immune pathways (99). In an animal model of liver injury, IL-36 was implicated in the early inflammatory response, as high levels of C-C Motif Chemokine Ligand 20 (CCL20) expression were detected in liver tissues. CCL20 activation promotes liver fibrosis development (100). In a mouse model of acetaminophen-induced liver injury, IL-36γ and CCL20 were both highly expressed. Treatment with IL-36Ra reduced CCL20 expression at both the mRNA and protein levels and alleviated liver injury. This suggests that blocking IL-36 signaling can mitigate liver inflammation by decreasing CCL20 expression and, consequently, may intervene in the progression of liver fibrosis (101).
IL-36 plays an important role in liver fibrosis. Studies have shown that IL-36 exerts pro-fibrotic effects by binding to its receptor. However, no drugs have been developed specifically targeting IL-36 for liver fibrosis treatment. The use of recombinant IL-36Ra or IL-36R blockers represents a potential therapeutic strategy and warrants further investigation.
The role of interleukins in promoting liver fibrosis is shown in Table 1. Table 2 summarizes the signaling pathways involved in the pro-fibrotic effects of interleukins, and Figure 1 illustrates the mechanisms through which these pro-fibrotic interleukins act in liver fibrosis.
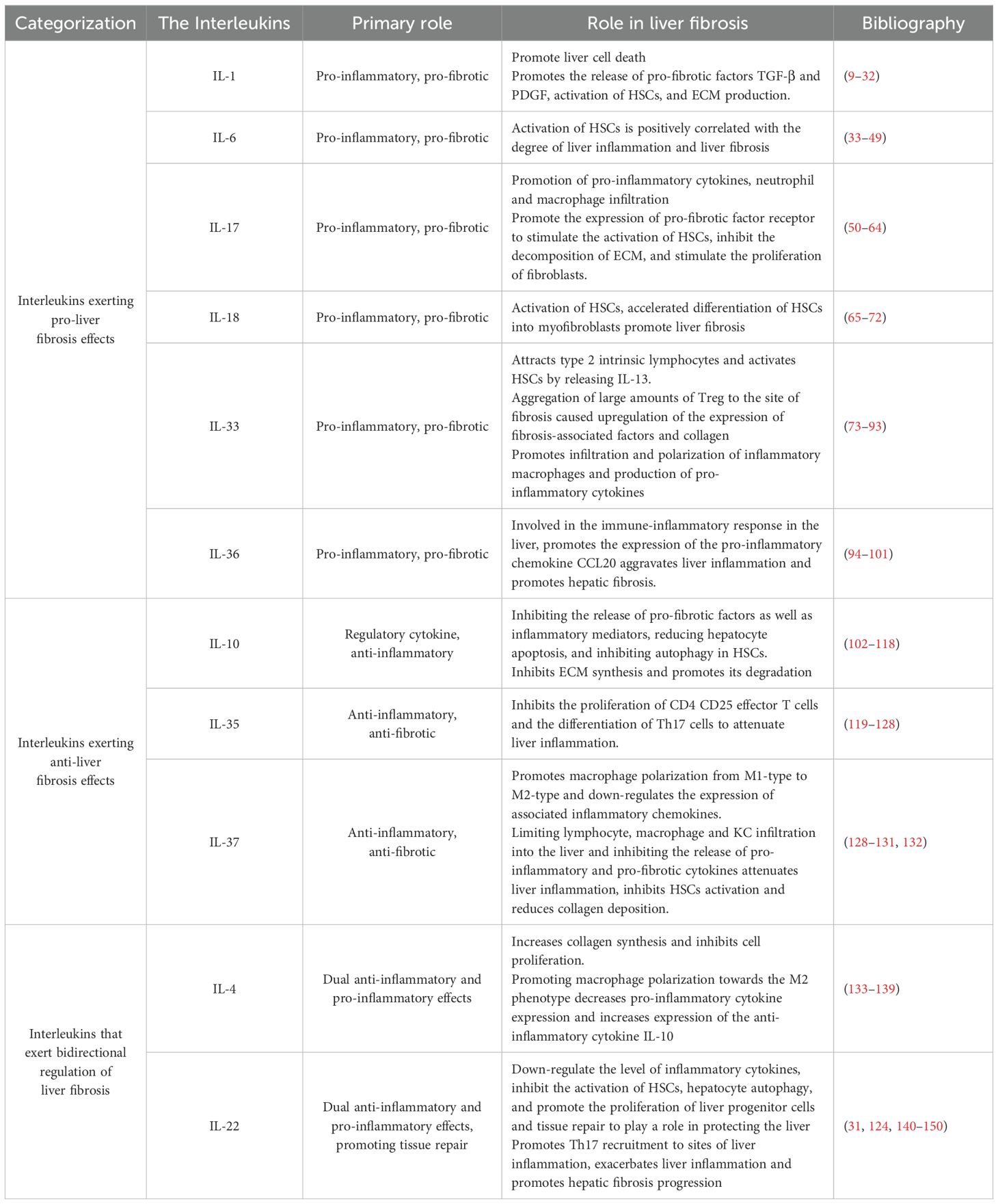
Table 1. Role of interleukins associated with liver fibrosis.
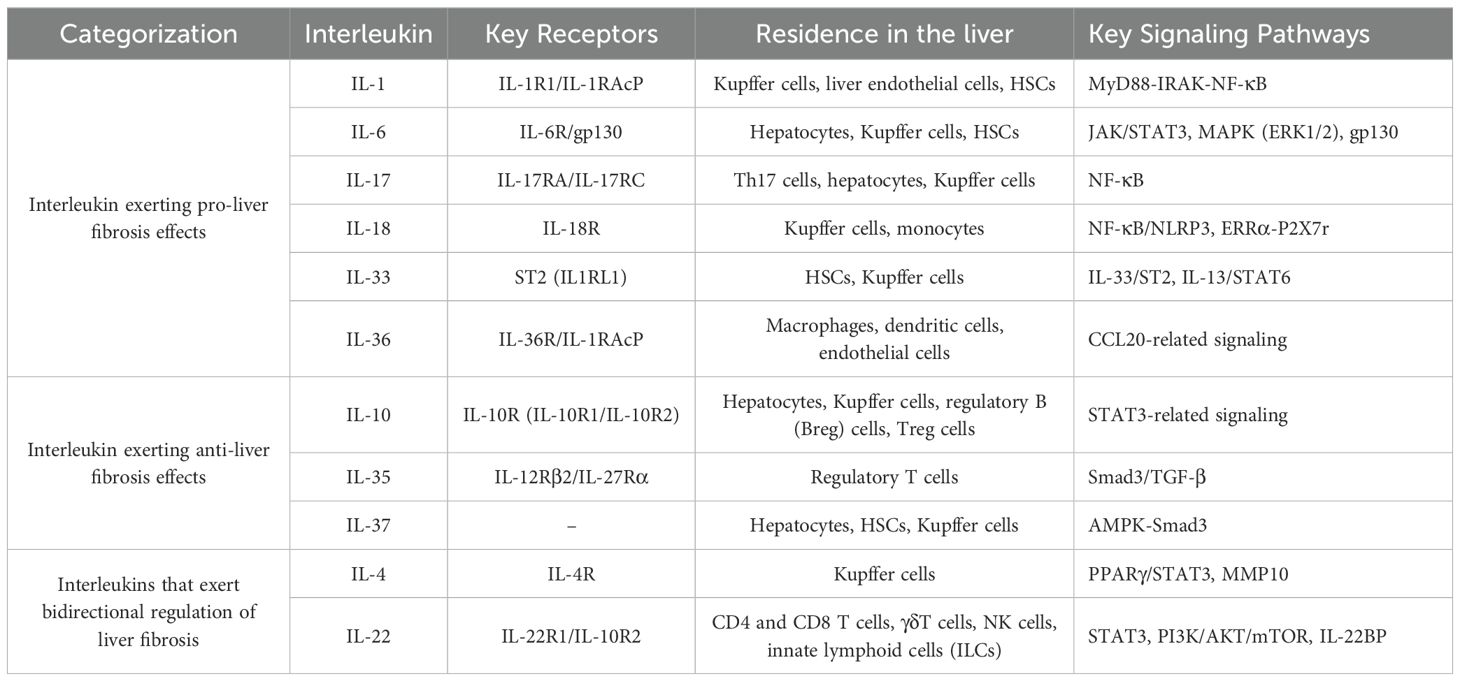
Table 2. Signaling pathways involved in interleukins in liver fibrosis.
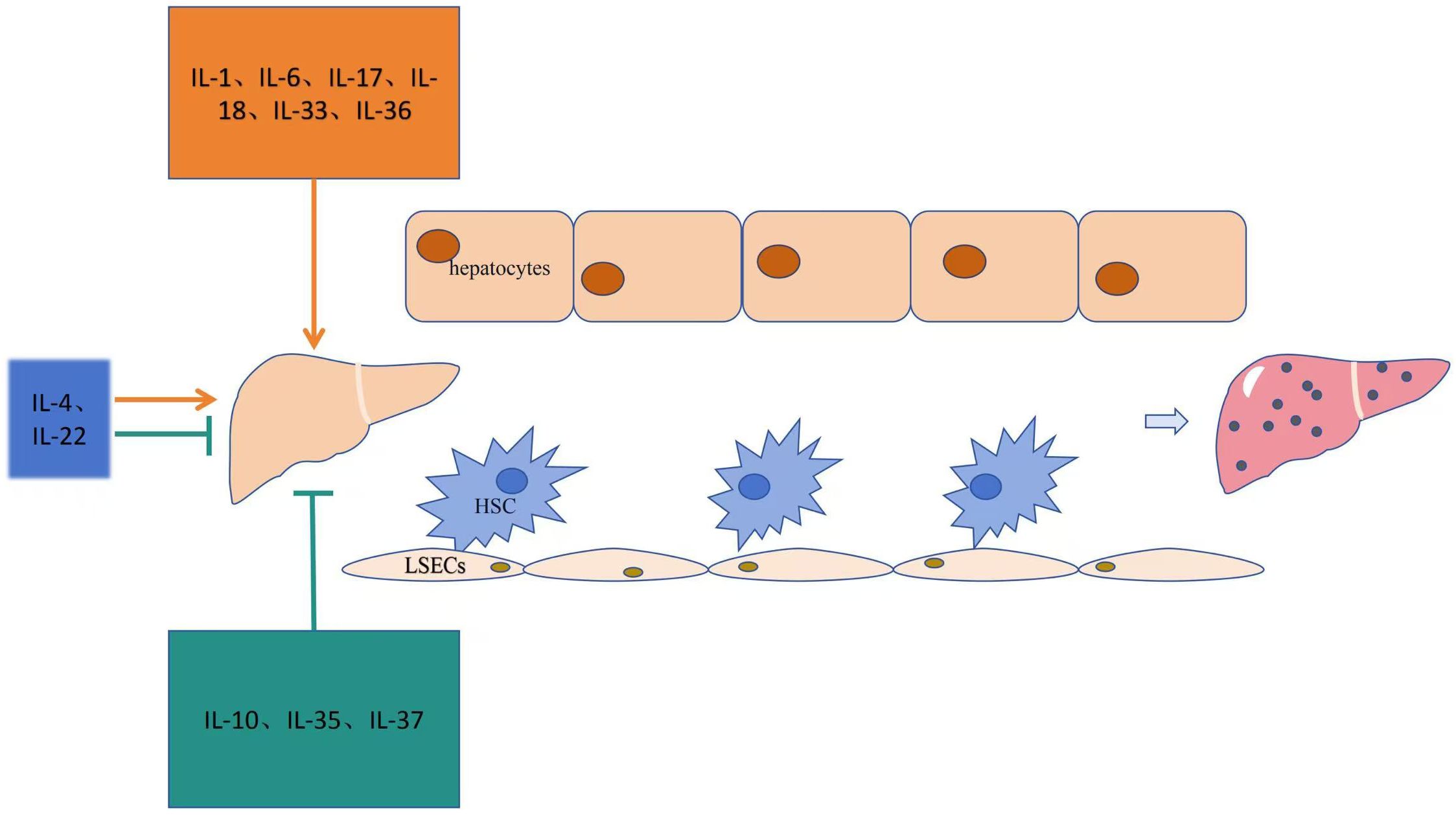
Figure 1. Mechanism of action of the interleukin family in liver fibrosis.
Interleukins exerting anti-liver fibrosis effects
Interleukin-10
IL-10 is a cytokines with anti-inflammatory and immunosuppressive effects. It reduces immune cell infiltration and inhibits the release of inflammatory factors. The IL-10 receptor (IL-10R) is primarily composed of two subunits: IL-10R1 and IL-10R2 (102). In the liver, IL-10 can be secreted by hepatocytes, KCs, regulatory B (Breg) cells and Treg cells, among other cell types (103). IL-10 promotes the translocation of phosphorylated STAT3 from the cytoplasm to the nucleus by enhancing phosphorylation signal transduction and STAT3 expression, thereby activating STAT3 and inhibiting autophagy (104). It also regulates autophagy in HSC-T6 cells through the STAT3-mTOR-p70S6K axis (105) and inhibits autophagosome formation in HSCs induced by oxidative stress by activating the mTOR-STAT3 pathway (106). This process suppresses the release of inflammatory factors, inhibits immune cell activation, and alleviates liver fibrosis.
IL-10 inhibits immune cell activation, attenuates hepatic inflammatory, and exerts protective effects on liver fibrosis progression (107). It effectively reduces the production of the pro-fibrotic factor TGF-β1, inhibits macrophages synthesis and secretion of TNF-α, downregulate the downstream effector NF-κB, and remodels the ECM (108). During liver fibrosis, IL-10 mitigates fibrosis by suppressing inflammatory cellular immune responses and the production of TGF-β1, TNF-α, and tissue inhibitors of metalloproteinases (107, 109). Additionally, IL-10 inhibits ECM synthesis and promotes its degradation (110, 111).
In a mouse model of liver fibrosis induced by BDL, IL-10 was highly expressed in HSCs (112). Elevated IL-10 expression in human liver tissues reduces hepatocyte apoptosis and reverses liver fibrosis (113, 114). HSCs may secrete autocrine IL-10 to inhibit collagen synthesis, suppress liver inflammatory responses, and slow the progression of liver fibrosis (115). A study reported that using IL-10-modified bone marrow-derived DCs, where the DCs were immature and infused into CCL4-induced hepatic fibrotic mice, significantly alleviated liver fibrosis. This treatment increased Treg cell expression in the liver while reducing inflammatory cytokines such as Interleukin-12 (IL-12), IL-22, and TNF-α (116). Furthermore, in vitro studies showed that IL-10 secretion by immature dendritic cells (imDCs) increased apoptosis and inhibited the proliferation of LX-2 cells. It also downregulated α-SMA mRNA expression and decreased TGF-β1 and Smad3 proteins levels, suggesting that IL-10 secretion by imDCs inhibit LX-2 activation via suppressing of the TGF-β1/Smad3 pathway (117). Recent studies indicate that IL-10 gene intervention enhances the accumulation of NK cells in the liver by improving their immune functions, including activation, cytotoxicity, development, and migration. It also increases the expression of NKG2D, IFN-γ, and CD107a in the liver tissue, thereby alleviating liver fibrosis (118).
The finding provide new theoretical support for the anti-fibrotic effects of IL-10. As a key negative feedback regulator, IL-10 prevents the onset and progression of liver fibrosis by inhibition the release of inflammatory mediator, suppressing NF-κB activity, and acting through other mechanisms to protect the liver. Although the anti-fibrotic properties of IL-10 are well-recognized, its precise mechanisms and clinical application prospects require further in-depth research and exploration.
Interleukin-35
IL-35 is a newly identified cytokines and a member of the IL-12 cytokine superfamily. It is a heterodimer composed of EBV-inducible gene 3 and IL-12p35, primarily secreted by Treg cells. IL-35 enhances Treg function through positive feedback and plays a critical role in maintaining immune homeostasis within the hepatic microenvironment. The IL-35 receptor (IL-35R) is primarily composed of two subunits: IL-12 receptor beta 2 (IL-12Rβ2) and IL-27 receptor alpha (IL-27Rα) (119). IL-35 inhibits the proliferation of CD4+ CD25+ effector T cells and the differentiation of Th17 cells, thus contributing to immune balance in the liver (120–122).
Serum IL-35 levels are significantly elevated in patients with HBV-associated cirrhosis compared to healthy controls and show a positive correlation with IL-17, IL-22, and IL-33 (123, 124). In the early stage of HBV-associated cirrhosis, the number of Th17 cells increases, and HSCs become activated, secreting pro-inflammatory cytokines such as IL-17 and TGF-β. To mitigate inflammatory damage to hepatocytes, Treg cells secrete IL-35 and IL-10, effectively suppressing Th17 differentiation and IL-17 production in HBV-associated cirrhosis (125). Furthermore, IL-35 knockdown increase the expression levels of IL-17 and IL-22 (126). IL-35 inhibits the binding of TGF-β to its receptor and suppresses the phosphorylation of Smad3, a downstream effector of the TGF-β receptor. This mechanism reduces Th17 differentiation and IL-17 synthesis in patients with HBV-associated cirrhosis (126). Additionally, the expression of IL-35 correlates with the histological grade and severity of PBC. serum IL-35 levels are higher in patients with stage III and IV disease compared to those in stage II. Moreover, IL-35 mRNA and protein levels negatively correlate with the Child-Pugh score for cirrhosis severity (127, 128).
IL-35 shows potential to improve liver fibrosis by inhibiting Th17 differentiation and IL-17 synthesis. However, its precise mechanisms remain unclear, and no anti-fibrotic drugs targeting IL-35 have been developed. Further research is needed to elucidate the specific roles and mechanisms of IL-35 in liver fibrosis.
Interleukin-37
IIL-37 is an important anti-inflammatory cytokines within the IL-1 family. It plays a crucial role in the prevention and treatment of liver fibrosis. IL-37 is expressed in various immune cells, including NK cells, B cells, T cells, and macrophages (129). In liver tissues, IL-37 is predominantly expressed in hepatocytes and can also be detected in intrahepatic cholangiocytes, HSCs and KCs (130).
IL-37 mitigates liver inflammation and alleviates liver fibrosis by inhibiting the expression of pro-inflammatory cytokines and chemokines in hepatocytes and KCs, reducing neutrophil activity, and acting directly on hepatocytes. Serum IL-37 levels have been found to be higher in patients with cirrhosis compared to healthy controls and positively correlated with cirrhosis stage score. In a BDL-induced mouse model of liver fibrosis, IL-37 overexpression reduced the inflammatory response and HSCs activation (131). In IL-37 transgenic mice with BDL-induced liver fibrosis, reduced expression levels of early liver fibrosis markers, such as C-X-C Motif Chemokine Ligand 2, were observed, along with decreased collagen deposition and liver fibrosis. In vitro experiments further demonstrated that IL-37 inhibits IL-1-induced activation of HSCs (131). Additionally, another study found that IL-37 promotes macrophages polarization from the pro-inflammatory M1 type to the anti-inflammatory M2 type and downregulates the expression of inflammatory chemokines, thereby inhibiting liver fibrosis. This mechanism may be associated with AMP-activated Protein Kinase (AMPK) pathway activation induced by Smad3 interaction (132). Recent studies have reported that IL-37 inhibits the activation of KCs and HSCs and interferes with TGF-β signaling, thereby reducing liver fibrosis and inflammation levels (131). Furthermore, IL-37 exerts protective effect against hepatic ischemia/reperfusion injury by decreasing pro-inflammatory cytokines and chemokines produced by hepatocytes and KCs, directly protecting hepatocytes from damage. As a novel anti-inflammatory factor, IL-37 holds promise as a potential therapeutic target for liver fibrosis.
Table 1 summarizes the role of interleukins in anti-liver fibrosis. Table 2 outlines the signaling pathways involved in the anti-fibrotic effects of interleukins, and Figure 1 illustrates the mechanisms through which these interleukins exert their anti-fibrotic effects.
Interleukins that exert bi-directional regulation of liver fibrosis
Interleukin-4
IL-4 is a Th2 cytokines that regulates the immune response, including eosinophil recruitment, parasite clearance, and IgE class switching, which can lead to hypersensitivity reaction. Additionally, IL-4 inhibits the activity of Th1 cells, thereby exerting some anti-inflammatory effects (94). It is mainly expressed in KCs in the liver. Its receptor is IL-4R (133). In the liver, IL-4 plays a dual role in liver fibrosis by inducing KCs to transform into multinucleated giant cells, stimulating the proliferation of HSCs, upregulating PPARγ, and regulating macrophage polarization (134).
Studies have shown that IL-4 induces hepatic KCs to transform into multinucleated giant cells and stimulates HSCs proliferation (135). Moreover, IL-4 can act directly on HSCs to promote their proliferation, increase collagen production, and accelerate the progression of liver fibrosis (136, 137). In addition, IL-4 can stimulate macrophage polarization toward the M2 phenotype. During this process, PPARγ binds to the matrix metallopeptidase 10 (MMP10) promoter, upregulating MMP10 expression. This mechanism further activates the downstream STAT3 signaling pathway, inducing M2 macrophage polarization. As a result, pro-inflammatory cytokines such as IL-1β and TNF-α are downregulated, while the anti-inflammatory cytokines IL-10 is upregulated. These changes contribute to the amelioration of hepatic steatosis and fibrosis (138). Furthermore, melatonin has been shown to attenuate thioacetamide-induced liver fibrosis in male rats by modulating IL-6, IL-4, apoptosis, and urokinase-type plasminogen activator receptor-related protein/Endo180 expression (139). These findings suggest that IL-4 could serve as a potential therapeutic target for liver fibrosis.
Interleukin-22
IL-22,also known as IL-10-related T cell-derived inducer (IL-T1F), belongs to the IL-10 family. IL-22 is produced by a variety of immune cells, including CD4+ and CD8+ T cells, γδT cells, NK cells, and ILCs (140). It exerts its biological function by binding to the heterodimeric membrane receptor complex IL-22R1/IL-10R2, which is specifically expressed on the surface of tissues such as the skin, kidney, and liver. The IL-22/IL-22R1/IL-22R2 complex primarily activates the downstream JAK-STAT signaling pathway, mainly involving STAT3. IL-22 can also activate the p38 kinase, c-jun N-terminal kinase (JNK), and ERK1/2 pathways (141).
IL-22 has been found to have anti-liver fibrosis effects, IL-22 overexpression attenuates hepatic fibrosis by inhibiting the activation of HSCs and downregulating inflammatory cytokines (142). In a CCl4-induced mouse model of liver fibrosis, IL-22 attenuated fibrosis by regulating cell polarization, inhibiting the STAT3/Erk/Akt pathway, and increasing the M2/M1-KCs ratio of KCs (143). In addition, in the CCL4 mouse model of liver fibrosis, it was found that HSCs could expressed IL-22 receptor 1 in large quantities. IL-22 activated the STAT3 signaling pathway by binding to its receptor, which in turn induced senescence in HSCs and attenuated liver fibrosis (144). In the BDL-induced mouse model of liver fibrosis, IL-22 was shown to increase collagen type I expression while significantly reducing α-SMA mRNA expression, suggesting its antifibrotic effect (53). In the schistosome-induced mouse liver fibrosis model, liposomal IL-22 improved fibrosis via the miR-let7a/STAT3 signaling pathway (145). In another study, inflammatory cells in CHB patients, which promoted hepatic progenitor cell proliferation and tissue repair through the STAT3 signaling pathway, suggesting that IL-22 plays a protective role in liver repair (146). It has also been found that IL-22 binding protein (IL-22BP), an inhibitor of IL-22, can aggravate fibrosis and cirrhosis in chronic HCV-infected patients (147). In an alcoholic mouse model of liver fibrosis, IL-22 ameliorated fibrosis partly by inhibiting hepatocyte autophagy and the PI3K/AKT/mTOR pathway (148).
However, IL-22 has also been reported to exert pro-liver fibrosis effects. In HBV-infected cirrhotic patients and an HBV transgenic mouse model of chronic liver inflammation and fibrosis, IL-22 induced the recruitment of Th17 cells to the sites of liver inflammation. It promoted the production of more IL-22, creating a positive feedback loop that exacerbated chronic inflammation and fibrosis. In a study involving 74 CHB patients, 36 hepatitis B cirrhosis patients, and 10 healthy controls, IL-22 levels positively correlated with the degree of liver fibrosis. When IL-22 was applied to stimulate HSCs in vitro, it produced chemokines that attracted Th17 cells, accelerating liver inflammation and fibrosis (124). IL-22 may lose its protective effect in the presence of IL-17 and even have pathogenicity (149). Studies have reported that an increase in IL-22-positive cells in the liver of HCV-infected patients correlated with fibrosis staging scores and clinical progression from chronic hepatitis to cirrhosis. In vitro experiments showed that IL-22 increased α-SMA expression and collagen production by inhibiting apoptosis and promoting proliferation of LX-2 cells (150).
Angelica sinensis polysaccharide (ASP) has hepatoprotective effects. In a CCl4-induced mouse model of liver fibrosis, ASP promoted IL-22 secretion, inhibited HSCs activation, and effectively attenuates fibrosis through the IL-22/STAT3 pathway (31). The balance between anti-inflammatory and pro-inflammatory effects of IL-22 may determine its role in liver fibrosis, and its exact mechanisms require further investigation. The relationship between IL-22 and liver fibrosis remains controversial. These seemingly contradictory results may be due to differences in disease models, degrees of liver injury, immune microenvironment, and cytokines interactions, The dual anti-inflammatory and pro-inflammatory roles of IL-22 in fibrosis caused by HBV and HCV infections warrant in-depth exploration to better understand its role in liver fibrosis.
Table 1 illustrates the bidirectional regulatory effects of interleukins on liver fibrosis. Table 2 shows the signaling pathways related to interleukins involved in liver fibrosis, and Figure 1 explains the mechanisms through which these interleukins act in liver fibrosis.
Conclusions and perspectives
Liver fibrosis is a dynamic pathological process involving multiple factors and pathways, resulting from intricate cellular crosstalk. In recent years, significant progress has been made in understanding the pathogenesis and exploring treatments for liver fibrosis. However, effective anti-fibrotic therapies are still lacking in clinical practice. The interleukin family of cytokines plays a pivotal role in the initiation and regulation of inflammation, as well as in innate and adaptive immunity, by activating complex cascades involving cytokines, chemokines, adhesion molecules, and lipid mediators. In the context of liver fibrosis, many interleukins, such as IL-1, IL-6, IL-17, IL-18, IL-33, and IL-36, exhibit pro-fibrotic effects. These cytokines exacerbate fibrosis by inducing the infiltration monocytes/macrophages infiltration into liver tissues, upregulating pro-inflammatory and pro-fibrotic cytokines, and promoting the proliferation and activation of HSCs. Conversely, certain interleukins, including IL-10, IL-35 and IL-37 have protective effects against liver fibrosis. These cytokines suppress the release of pro-inflammatory mediators, inhibit fibrosis-related pathways and ECM synthesis, and promote ECM degradation.
Moreover, interleukins such as IL-4 and IL-22 exhibit dual roles, functioning as both anti-inflammatory and pro-inflammatory mediators in liver fibrosis, warranting further investigation. Developing interleukin-targeting therapies, either inhibitors or agonists, holds promise as a potential treatment strategy. However, current research is predominantly at the cellular and animal model levels. Further studies are needed to elucidate the underlying mechanisms and evaluate the clinical applicability of these therapies. Emerging technologies and advancements in interleukin-based treatments are anticipated to play a positive role in managing liver fibrosis in the future.
Author contributions
ZXZ: Writing – review & editing, Conceptualization, Writing – original draft. JW: Writing – review & editing. HL: Writing – review & editing. QN: Writing – review & editing. YT: Writing – review & editing. XZ: Writing – review & editing. ZJZ: Writing – review & editing. HD: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Natural Science Foundation of China (No.82274323); Sichuan Science and Technology Program (No. 2024YFFK0150).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: Concept to treatment. J Hepatol. (2015) 62:S15–24. doi: 10.1016/j.jhep.2015.02.039
2. Aydın MM, Akçalı KC. Liver fibrosis. Turk J Gastroenterol. (2018) 29:14–21. doi: 10.5152/tjg.2018.17330
3. Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. (2013) 381:468–75. doi: 10.1016/S0140-6736(12)61425-1
4. Atta HM. Reversibility and heritability of liver fibrosis: Implications for research and therapy. World J Gastroenterol. (2015) 21:5138–48. doi: 10.3748/wjg.v21.i17.5138
5. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. (2019) 70:151–71. doi: 10.1016/j.jhep.2018.09.014
6. Luo N, Li J, Wei Y, Lu J, Dong R. Hepatic stellate cell: A double-edged sword in the liver. Physiol Res. (2021) 70:821–9. doi: 10.33549/physiolres.934755
7. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Delivery Rev. (2017) 121:27–42. doi: 10.1016/j.addr.2017.05.007
8. Yazdani S, Bansal R, Prakash J. Drug targeting to myofibroblasts: Implications for fibrosis and cancer. Adv Drug Delivery Rev. (2017) 121:101–16. doi: 10.1016/j.addr.2017.07.010
9. Ge Y, Huang M, Yao YM. Recent advances in the biology of IL-1 family cytokines and their potential roles in development of sepsis. Cytokine Growth Factor Rev. (2019) 45:24–34. doi: 10.1016/j.cytogfr.2018.12.004
10. Martin P, Goldstein JD, Mermoud L, Diaz-Barreiro A, Palmer G. IL-1 family antagonists in mouse and human skin inflammation. Front Immunol. (2021) 12:652846. doi: 10.3389/fimmu.2021.652846
11. Lee HJ, Yeon JE, Ko EJ, Yoon EL, Suh SJ, Kang K, et al. Peroxisome proliferator-activated receptor-delta agonist ameliorated inflammasome activation in nonalcoholic fatty liver disease. World J Gastroenterol. (2015) 21:12787–99. doi: 10.3748/wjg.v21.i45.12787
12. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. (2010) 3:cm1. doi: 10.1126/scisignal.3105cm1
13. Wang D, Zhang S, Li L, Liu X, Mei K, Wang X. Structural insights into the assembly and activation of IL-1β with its receptors. Nat Immunol. (2010) 11:905–11. doi: 10.1038/ni.1925
14. Mantovani A, Barajon I, Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev. (2018) 281:57–61. doi: 10.1111/imr.12614
15. Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity. (2019) 50:778–95. doi: 10.1016/j.immuni.2019.03.012
16. Praktiknjo M, Schierwagen R, Monteiro S, Ortiz C, Uschner FE, Jansen C, et al. Hepatic inflammasome activation as origin of Interleukin-1α and Interleukin-1β in liver cirrhosis. Gut. (2021) 70:1799–800. doi: 10.1136/gutjnl-2020-322621
17. Gieling RG, Wallace K, Han YP. Interleukin-1 participates in the progression from liver injury to fibrosis. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G1324–31. doi: 10.1152/ajpgi.90564.2008
18. Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. (2011) 55:1086–94. doi: 10.1016/j.jhep.2011.01.048
19. Du X, Wu Z, Xu Y, Liu Y, Liu W, Wang T, et al. Increased Tim-3 expression alleviates liver injury by regulating macrophage activation in MCD-induced NASH mice. Cell Mol Immunol. (2019) 16:878–86. doi: 10.1038/s41423-018-0032-0
20. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. (2010) 139:323–34.e7. doi: 10.1053/j.gastro.2010.03.052
21. Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. (2008) 48:1224–31. doi: 10.1002/hep.22470
22. Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat Commun. (2016) 7:10791. doi: 10.1038/ncomms10791
23. Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Müller DJ. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. (2018) 37:e98321. doi: 10.15252/embj.201798321
24. Palacios-Macapagal D, Connor J, Mustelin T, Ramalingam TR, Wynn TA, Davidson TS. Cutting edge: eosinophils undergo caspase-1-mediated pyroptosis in response to necrotic liver cells. J Immunol. (2017) 199:847–53. doi: 10.4049/jimmunol.1601162
25. Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, et al. Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G1248–57. doi: 10.1152/ajpgi.90223.2008
26. Wang H, Liu S, Wang Y, Chang B, Wang B. Nod-like receptor protein 3 inflammasome activation by Escherichia coli RNA induces transforming growth factor beta 1 secretion in hepatic stellate cells. Bosn J Basic Med Sci. (2016) 16:126–31. doi: 10.17305/bjbms.2016.699
27. Chachay VS, Macdonald GA, Martin JH, Whitehead JP, O’Moore-Sullivan TM, Lee P, et al. Resveratrol does not benefit patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2014) 12:2092–103.e1-6. doi: 10.1016/j.cgh.2014.02.024
28. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. (2015) 21:248–55. doi: 10.1038/nm.3806
29. Wu X, Zhang F, Xiong X, Lu C, Lian N, Lu Y, et al. Tetramethylpyrazine reduces inflammation in liver fibrosis and inhibits inflammatory cytokine expression in hepatic stellate cells by modulating NLRP3 inflammasome pathway. IUBMB Life. (2015) 67:312–21. doi: 10.1002/iub.1348
30. Wei WL, Zeng R, Gu CM, Qu Y, Huang LF. Angelica sinensis in China-A review of botanical profile, ethnopharmacology, phytochemistry and chemical analysis. J Ethnopharmacol. (2016) 190:116–41. doi: 10.1016/j.jep.2016.05.023
31. Wang K, Wang J, Song M, Wang H, Xia N, Zhang Y. Angelica sinensis polysaccharide attenuates CCl4-induced liver fibrosis via the IL-22/STAT3 pathway. Int J Biol Macromol. (2020) 162:273–83. doi: 10.1016/j.ijbiomac.2020.06.166
32. Shi YS, Li XX, Li HT, Zhang Y. Pelargonidin ameliorates CCl4-induced liver fibrosis by suppressing the ROS-NLRP3-IL-1β axis via activating the Nrf2 pathway. Food Funct. (2020) 11:5156–65. doi: 10.1039/d0fo00660b
33. Widjaja AA, Chothani SP, Cook SA. Different roles of interleukin 6 and interleukin 11 in the liver: implications for therapy. Hum Vaccin Immunother. (2020) 16:2357–62. doi: 10.1080/21645515.2020.1761203
34. O’Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem. (2014) 289:9952–60. doi: 10.1074/jbc.M113.545822
35. Yu Y, Cui X, Zhao J, Jia T, Ren B, Zhang X. Effect of entecavir combined with adefovir dipivoxil on clinical efficacy and TNF-α and IL-6 levels in patients with hepatitis B cirrhosis. J Oncol. (2021) 2021:9162346. doi: 10.1155/2021/9162346
36. Yang Z, Peng Y, Yang S. MicroRNA-146a regulates the transformation from liver fibrosis to cirrhosis in patients with hepatitis B via interleukin-6. Exp Ther Med. (2019) 17:4670–6. doi: 10.3892/etm.2019.7490
37. Kar S, Paglialunga S, Jaycox SH, Islam R, Paredes AH. Assay validation and clinical performance of chronic inflammatory and chemokine biomarkers of NASH fibrosis. PloS One. (2019) 14:e0217263. doi: 10.1371/journal.pone.0217263
38. Sohrabi M, Gholami A, Amirkalali B, Taherizadeh M, Kolahdoz M, SafarnezhadTameshkel F, et al. Is melatonin associated with pro-inflammatory cytokine activity and liver fibrosis in non-alcoholic fatty liver disease (NAFLD) patients? Gastroenterol Hepatol Bed Bench. (2021) 14:229–36.
39. Tan H, He Q, Li R, Lei F, Lei X. Trillin reduces liver chronic inflammation and fibrosis in carbon tetrachloride (CCl4) induced liver injury in mice. Immunol Invest. (2016) 45:371–82. doi: 10.3109/08820139.2015.1137935
40. Elnfarawy AA, Nashy AE, Abozaid AM, Komber IF, Elweshahy RH, Abdelrahman RS. Vinpocetine attenuates thioacetamide-induced liver fibrosis in rats. Hum Exp Toxicol. (2021) 40:355–68. doi: 10.1177/0960327120947453
41. Kovalovich K, DeAngelis RA, Li W, Furth EE, Ciliberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6-deficient mice. Hepatology. (2000) 31:149–59. doi: 10.1002/hep.510310123
42. Kroy DC, Beraza N, Tschaharganeh DF, Sander LE, Erschfeld S, Giebeler A, et al. Lack of interleukin-6/glycoprotein 130/signal transducers and activators of transcription-3 signaling in hepatocytes predisposes to liver steatosis and injury in mice. Hepatology. (2010) 51:463–73. doi: 10.1002/hep.23322
43. Streetz KL, Tacke F, Leifeld L, Wüstefeld T, Graw A, Klein C, et al. Interleukin 6/gp130-dependent pathways are protective during chronic liver diseases. Hepatology. (2003) 38:218–29. doi: 10.1053/jhep.2003.50268
44. Kwon HJ, Won YS, Park O, Chang B, Duryee MJ, Thiele GE, et al. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology. (2014) 60:146–57. doi: 10.1002/hep.27036
45. Masjedi A, Hajizadeh F, Beigi Dargani F, Beyzai B, Aksoun M, Hojjat-Farsangi M, et al. Oncostatin M: A mysterious cytokine in cancers. Int Immunopharmacol. (2021) 90:107158. doi: 10.1016/j.intimp.2020.107158
46. Fujii H, Kawada N. Fibrogenesis in alcoholic liver disease. World J Gastroenterol. (2014) 20:8048–54. doi: 10.3748/wjg.v20.i25.8048
47. Kagan P, Sultan M, Tachlytski I, Safran M, Ben-Ari Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PloS One. (2017) 12:e0176173. doi: 10.1371/journal.pone.0176173
48. Hou X, Yin S, Ren R, Liu S, Yong L, Liu Y, et al. Myeloid-cell-specific IL-6 signaling promotes microRNA-223-enriched exosome production to attenuate NAFLD-associated fibrosis. Hepatology. (2021) 74:116–32. doi: 10.1002/hep.31658
49. He Y, Hwang S, Ahmed YA, Feng D, Li N, Ribeiro M, et al. Immunopathobiology and therapeutic targets related to cytokines in liver diseases. Cell Mol Immunol. (2021) 18:18–37. doi: 10.1038/s41423-020-00580-w
50. Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol. (2011) 2011:345803. doi: 10.1155/2011/345803.
51. Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C, et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol. (2013) 191:1835–44. doi: 10.4049/jimmunol.1203013
52. Sun XF, Gu L, Deng WS, Xu Q. Impaired balance of T helper 17/T regulatory cells in carbon tetrachloride-induced liver fibrosis in mice. World J Gastroenterol. (2014) 20:2062–70. doi: 10.3748/wjg.v20.i8.2062
53. Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. (2012) 143:765–776.e3. doi: 10.1053/j.gastro.2012.05.049
54. Seo W, Eun HS, Kim SY, Yi HS, Lee YS, Park SH, et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by γδ T cells in liver fibrosis. Hepatology. (2016) 64:616–31. doi: 10.1002/hep.28644
55. Bunney PE, Zink AN, Holm AA, Billington CJ, Kotz CM. Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high-fat diet. Physiol Behav. (2017) 176:139–48. doi: 10.1016/j.physbeh.2017.03.040
56. Wu Z, He D, Zhao S, Wang H. IL-17A/IL-17RA promotes invasion and activates MMP-2 and MMP-9 expression via p38 MAPK signaling pathway in non-small cell lung cancer. Mol Cell Biochem. (2019) 455:195–206. doi: 10.1007/s11010-018-3483-9
57. Zhang Y, Huang D, Gao W, Yan J, Zhou W, Hou X, et al. Lack of IL-17 signaling decreases liver fibrosis in murine schistosomiasis japonica. Int Immunol. (2015) 27:317–25. doi: 10.1093/intimm/dxv017
58. Kartasheva-Ebertz D, Gaston J, Lair-Mehiri L, Mottez E, Buivan TP, Massault PP, et al. IL-17A in human liver: significant source of inflammation and trigger of liver fibrosis initiation. Int J Mol Sci. (2022) 23:9773. doi: 10.3390/ijms23179773
59. Yang X, Liao L, Liang Z, Yu S, Guo Z. Correlation analysis of IL-17, IL-21, IL-23 with non-alcoholic liver fibrosis and cirrhosis. J Inflammation Res. (2024) 17:2327–35. doi: 10.2147/JIR.S452061
60. Liang KH, Lai MW, Lin YH, Chu YD, Lin CL, Lin WR, et al. Plasma interleukin-17 and alpha-fetoprotein combination effectively predicts imminent hepatocellular carcinoma occurrence in liver cirrhotic patients. BMC Gastroenterol. (2021) 21:177. doi: 10.1186/s12876-021-01761-1
61. Elbanan WK, Fathy SA, Ibrahim RA, Hegazy MGA. Assessment of interleukin 17 and transforming growth factor-beta 1 in hepatitis C patients with disease progression. Trop Biomed. (2020) 37:1093–104. doi: 10.47665/tb.37.4.1093
62. Zhang S, Huang D, Weng J, Huang Y, Liu S, Zhang Q, et al. Neutralization of interleukin-17 attenuates cholestatic liver fibrosis in mice. Scand J Immunol. (2016) 83:102–8. doi: 10.1111/sji.12395
63. Yamato M, Sakai Y, Mochida H, Kawaguchi K, Takamura M, Usui S, et al. Adipose tissue-derived stem cells prevent fibrosis in murine steatohepatitis by suppressing IL-17-mediated inflammation. J Gastroenterol Hepatol. (2019) 34:1432–40. doi: 10.1111/jgh.14647
64. Magdaleno-Tapial J, López-Martí C, Ortiz-Salvador JM, Hernández-Bel P, Tamarit-García JJ, Diago-Madrid M, et al. Can secukinumab improve liver fibrosis? A pilot prospective study of 10 psoriatic patients. Dermatol Ther. (2021) 34:e15065. doi: 10.1111/dth.15065
65. Tapia VS, Daniels MJD, Palazón-Riquelme P, Dewhurst M, Luheshi NM, Rivers-Auty J, et al. The three cytokines IL-1β, IL-18, and IL-1α share related but distinct secretory routes. J Biol Chem. (2019) 294:8325–35. doi: 10.1074/jbc.RA119.008009
66. Li Z, Yu X, Werner J, Bazhin AV, D’Haese JG. The role of interleukin-18 in pancreatitis and pancreatic cancer. Cytokine Growth Factor Rev. (2019) 50:1–12. doi: 10.1016/j.cytogfr.2019.11.001
67. Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in health and disease. Int J Mol Sci. (2019) 20:649. doi: 10.3390/ijms20030649
68. Knorr J, Kaufmann B, Inzaugarat ME, Holtmann TM, Geisler L, Hundertmark J, et al. Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis. Hepatology. (2023) 77:1968–82. doi: 10.1002/hep.32776
69. Wang SW, Lan T, Zheng F, Lei MK, Zhang F. Effect of extract of Quzhou Aurantii Fructus on hepatic inflammation and NF-κB/NLRP3 inflammasome pathway in CCl_4-induced liver fibrosis mice. Zhongguo Zhong Yao Za Zhi. (2021) 46:1474–9. doi: 10.19540/j.cnki.cjcmm.20201201.401
70. Cui L, Tan YJ, Xu SQ, Qin BF, Xiu MX, Zhang X, et al. Ginsenoside Rd, a natural production for attenuating fibrogenesis and inflammation in hepatic fibrosis by regulating the ERRα-mediated P2X7r pathway. Food Funct. (2023) 14:5606–19. doi: 10.1039/d3fo01315d
71. Wang L, Qiao Q, Hou L. Changes in the IL-18, IL-22, and T lymphocyte subset levels in patients with hepatitis B-related liver cirrhosis and their predictive values for hepatorenal syndrome. Am J Transl Res. (2023) 15:3976–91.
72. Xiao S, Pan X, Huang X, Liu Y, Wen SW, Liu A. Gene polymorphisms of inflammatory factors in liver cirrhosis. Front Genet. (2023) 14:1140427. doi: 10.3389/fgene.2023.1140427
73. Alam A, Levanduski E, Denz P, Villavicencio HS, Bhatta M, Alhorebi L, et al. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell. (2022) 40:153–167.e11. doi: 10.1016/j.ccell.2022.01.003
74. Kotsiou OS, Gourgoulianis KI, Zarogiannis SG. IL-33/ST2 axis in organ fibrosis. Front Immunol. (2018) 9:2432. doi: 10.3389/fimmu.2018.02432
75. Hung LY, Tanaka Y, Herbine K, Pastore C, Singh B, Ferguson A, et al. Cellular context of IL-33 expression dictates impact on anti-helminth immunity. Sci Immunol. (2020) 5:eabc6259. doi: 10.1126/sciimmunol.abc6259
76. Chen WY, Wu YH, Tsai TH, Li RF, Lai AC, Li LC, et al. Group 2 innate lymphoid cells contribute to IL-33-mediated alleviation of cardiac fibrosis. Theranostics. (2021) 11:2594–611. doi: 10.7150/thno.51648
77. Marvie P, Lisbonne M, L’helgoualc’h A, Rauch M, Turlin B, Preisser L, et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. (2010) 14:1726–39. doi: 10.1111/j.1582-4934.2009.00801.x
78. Sun Z, Chang B, Gao M, Zhang J, Zou Z. IL-33-ST2 axis in liver disease: progression and challenge. Mediators Inflamm. (2017) 2017:5314213. doi: 10.1155/2017/5314213
79. McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. (2013) 39:357–71. doi: 10.1016/j.immuni.2013.07.018
80. Vasseur P, Dion S, Filliol A, Genet V, Lucas-Clerc C, Jean-Philippe G, et al. Endogenous IL-33 has no effect on the progression of fibrosis during experimental steatohepatitis. Oncotarget. (2017) 8:48563–74. doi: 10.18632/oncotarget.18335
81. Li ZY, Xiao L, Lin G, Tang J, Chen Y, Chen L, et al. Contribution of tissue transglutaminase to the severity of hepatic fibrosis resulting from Schistosoma japonicum infection through the regulation of IL-33/ST2 expression. Parasit Vectors. (2019) 12:302. doi: 10.1186/s13071-019-3542-4
82. Gao Y, Liu Y, Yang M, Guo X, Zhang M, Li H, et al. IL-33 treatment attenuated diet-induced hepatic steatosis but aggravated hepatic fibrosis. Oncotarget. (2016) 7:33649–61. doi: 10.18632/oncotarget.9259
83. Lurje I, Tacke F. The interleukin 33-T helper 2 cell axis promotes human liver fibrosis. Cell Mol Gastroenterol Hepatol. (2024) 17:657–9. doi: 10.1016/j.jcmgh.2024.01.004
84. Reißing J, Berres M, Strnad P, Wree A, Inzaugarat ME, Trautwein C, et al. Th2 cell activation in chronic liver disease is driven by local IL33 and contributes to IL13-dependent fibrogenesis. Cell Mol Gastroenterol Hepatol. (2024) 17:517–38. doi: 10.1016/j.jcmgh.2023.12.011
85. Xu L, Li W, Wang X, Zhang L, Qi Q, Dong L, et al. The IL-33-ST2-MyD88 axis promotes regulatory T cell proliferation in the murine liver. Eur J Immunol. (2018) 48:1302–7. doi: 10.1002/eji.201747402
86. Langhans B, Krämer B, Louis M, Nischalke HD, Hüneburg R, Staratschek-Jox A, et al. Intrahepatic IL-8 producing Foxp3+CD4+ regulatory T cells and fibrogenesis in chronic hepatitis C. J Hepatol. (2013) 59:229–35. doi: 10.1016/j.jhep.2013.04.011
87. MacDonald KG, Dawson NAJ, Huang Q, Dunne JV, Levings MK, Broady R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J Allergy Clin Immunol. (2015) 135:946–955.e9. doi: 10.1016/j.jaci.2014.12.1932
88. Katz SC, Ryan K, Ahmed N, Plitas G, Chaudhry UI, Kingham TP, et al. Obstructive jaundice expands intrahepatic regulatory T cells, which impair liver T lymphocyte function but modulate liver cholestasis and fibrosis. J Immunol. (2011) 187:1150–6. doi: 10.4049/jimmunol.1004077
89. Ma N, Xu M, Dong Y, Yu F, Zhang X, Gao X, et al. Genetic variants in IL33 and IL1RL1 genes confer susceptibility to HBV-related liver cirrhosis in Chinese Han population. Infect Genet Evol. (2021) 94:104983. doi: 10.1016/j.meegid.2021.104983
90. Askoura M, Abbas HA, Al Sadoun H, Abdulaal WH, Abu Lila AS, Almansour K, et al. Elevated levels of IL-33, IL-17 and IL-25 indicate the progression from chronicity to hepatocellular carcinoma in hepatitis C virus patients. Pathogens. (2022) 11:57. doi: 10.3390/pathogens11010057
91. She S, Wu X, Zheng D, Pei X, Ma J, Sun Y, et al. PSMP/MSMP promotes hepatic fibrosis through CCR2 and represents a novel therapeutic target. J Hepatol. (2020) 72:506–18. doi: 10.1016/j.jhep.2019.09.033
92. Tan Z, Liu Q, Jiang R, Lv L, Shoto SS, Maillet I, et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell Mol Immunol. (2018) 15:388–98. doi: 10.1038/cmi.2016.63
93. Kim SH, Lee SM. Cytoprotective effects of melatonin against necrosis and apoptosis induced by ischemia/reperfusion injury in rat liver. J Pineal Res. (2008) 44:165–71. doi: 10.1111/j.1600-079X.2007.00504.x
94. Andoh A, Nishida A. Pro- and anti-inflammatory roles of interleukin (IL)-33, IL-36, and IL-38 in inflammatory bowel disease. J Gastroenterol. (2023) 58:69–78. doi: 10.1007/s00535-022-01936-x
95. Smith DE, Renshaw BR, Ketchem RR, Kubin M, Garka KE, Sims JE. Four new members expand the interleukin-1 superfamily. J Biol Chem. (2000) 275:1169–75. doi: 10.1074/jbc.275.2.1169
96. Debets R, Timans JC, Homey B, Zurawski S, Sana TR, Lo S, et al. Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J Immunol. (2001) 167:1440–6. doi: 10.4049/jimmunol.167.3.1440
97. Franzke CW, Cobzaru C, Triantafyllopoulou A, Löffek S, Horiuchi K, Threadgill DW, et al. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J Exp Med. (2012) 209:1105–19. doi: 10.1084/jem.20112258
98. Kumar S, Hanning CR, Brigham-Burke MR, Rieman DJ, Lehr R, Khandekar S, et al. Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine. (2002) 18:61–71. doi: 10.1006/cyto.2002.0873
99. Iznardo H, Puig L. IL-1 family cytokines in inflammatory dermatoses: pathogenetic role and potential therapeutic implications. Int J Mol Sci. (2022) 23:9479. doi: 10.3390/ijms23169479
100. Hanson A, Piras IS, Wilhelmsen D, Still CD, Chu X, Petrick A, et al. Chemokine ligand 20 (CCL20) expression increases with NAFLD stage and hepatic stellate cell activation and is regulated by miR-590-5p. Cytokine. (2019) 123:154789. doi: 10.1016/j.cyto.2019.154789
101. Scheiermann P, Bachmann M, Härdle L, Pleli T, Piiper A, Zwissler B, et al. Application of IL-36 receptor antagonist weakens CCL20 expression and impairs recovery in the late phase of murine acetaminophen-induced liver injury. Sci Rep. (2015) 5:8521. doi: 10.1038/srep08521
102. Zhou YC, Chen S, Cao JJ, Chen SY, Xie YF, Niu QX. Adenovirus-mediated viral interleukin-10 gene transfer prevents concanavalin A-induced liver injury. Dig Liver Dis. (2012) 44:398–405. doi: 10.1016/j.dld.2011.11.013
103. Hammerich L, Tacke F. Interleukins in chronic liver disease: lessons learned from experimental mouse models. Clin Exp Gastroenterol. (2014) 7:297–306. doi: 10.2147/CEG.S43737
104. Zhang XW, Mi S, Li Z, Zhou JC, Xie J, Hua F, et al. Antagonism of Interleukin-17A ameliorates experimental hepatic fibrosis by restoring the IL-10/STAT3-suppressed autophagy in hepatocytes. Oncotarget. (2017) 8:9922–34. doi: 10.18632/oncotarget.14266
105. Chen D, Chen J, Chen Y, Chen F, Wang X, Huang Y. Interleukin-10 regulates starvation-induced autophagy through the STAT3-mTOR-p70s6k axis in hepatic stellate cells. Exp Biol Med (Maywood). (2022) 247:832–41. doi: 10.1177/15353702221080435
106. Chen J, Guo Q, Chen Q, Chen Y, Chen D, Chen Z, et al. Interleukin 10 inhibits oxidative stress-induced autophagosome formation in hepatic stellate cells by activating the mTOR-STAT3 pathway. Exp Cell Res. (2022) 411:113001. doi: 10.1016/j.yexcr.2021.113001
107. Gou Y, Weng Y, Chen Q, Wu J, Wang H, Zhong J, et al. Carboxymethyl chitosan prolongs adenovirus-mediated expression of IL-10 and ameliorates hepatic fibrosis in a mouse model. Bioeng Transl Med. (2022) 7:e10306. doi: 10.1002/btm2.10306
108. Chen YX, Huang YH, Zheng WD, Chen ZX, Zhang LJ, Wang XZ. Interleukin-10 gene modification attenuates hepatocyte activation of rat hepatic stellate cells in vitro. Mol Med Rep. (2013) 7:371–8. doi: 10.3892/mmr.2012.1228
109. Franco KGS, de Amorim FJR, Santos MA, Rollemberg CVV, de Oliveira FA, França AVC, et al. Association of IL-9, IL-10, and IL-17 cytokines with hepatic fibrosis in human schistosoma mansoni infection. Front Immunol. (2021) 12:779534. doi: 10.3389/fimmu.2021.779534
110. Caligiuri A, Gentilini A, Pastore M, Gitto S, Marra F. Cellular and molecular mechanisms underlying liver fibrosis regression. Cells. (2021) 10:2759. doi: 10.3390/cells10102759
111. Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis. J Hepatobiliary Pancreat Sci. (2015) 22:512–8. doi: 10.1002/jhbp.245
112. Chen W, Yan X, Xu A, Sun Y, Wang B, Huang T, et al. Dynamics of elastin in liver fibrosis: Accumulates late during progression and degrades slowly in regression. J Cell Physiol. (2019) 234:22613–22. doi: 10.1002/jcp.28827
113. Chou WY, Lu CN, Lee TH, Wu CL, Hung KS, Concejero AM, et al. Electroporative interleukin-10 gene transfer ameliorates carbon tetrachloride-induced murine liver fibrosis by MMP and TIMP modulation. Acta Pharmacol Sin. (2006) 27:469–76. doi: 10.1111/j.1745-7254.2006.00304.x
114. Hung KS, Lee TH, Chou WY, Wu CL, Cho CL, Lu CN, et al. Interleukin-10 gene therapy reverses thioacetamide-induced liver fibrosis in mice. Biochem Biophys Res Commun. (2005) 336:324–31. doi: 10.1016/j.bbrc.2005.08.085
115. Thompson KC, Trowern A, Fowell A, Marathe M, Haycock C, Arthur MJ, et al. Primary rat and mouse hepatic stellate cells express the macrophage inhibitor cytokine interleukin-10 during the course of activation in vitro. Hepatology. (1998) 28:1518–24. doi: 10.1002/hep.510280611
116. Xu Y, Tang X, Yang M, Zhang S, Li S, Chen Y, et al. Interleukin 10 gene-modified bone marrow-derived dendritic cells attenuate liver fibrosis in mice by inducing regulatory T cells and inhibiting the TGF-β/smad signaling pathway. Mediators Inflamm. (2019) 2019:4652596. doi: 10.1155/2019/4652596
117. Xu Y, Liu F, He D, Han L, Zheng X, Hu M, et al. Monocyte-derived immature dendritic cells negatively regulate hepatic stellate cells in vitro by secreting IL-10. Immunobiology. (2023) 228:152315. doi: 10.1016/j.imbio.2022.152315
118. Chen Y, Huang Y, Huang R, Chen Z, Wang X, Chen F, et al. Interleukin-10 gene intervention ameliorates liver fibrosis by enhancing the immune function of natural killer cells in liver tissue. Int Immunopharmacol. (2024) 127:111341. doi: 10.1016/j.intimp.2023.111341
119. Hu S, Lian PP, Hu Y, Zhu XY, Jiang SW, Ma Q, et al. The role of IL-35 in the pathophysiological processes of liver disease. Front Pharmacol. (2021) 11:569575. doi: 10.3389/fphar.2020.569575
120. Huang Y, Hu H, Liu L, Ye J, Wang Z, Que B, et al. Interleukin-12p35 deficiency reverses the Th1/Th2 imbalance, aggravates the Th17/Treg imbalance, and ameliorates atherosclerosis in ApoE-/- mice. Mediators Inflamm. (2019) 2019:3152040. doi: 10.1155/2019/3152040
121. Luo M, Peng H, Chen P, Zhou Y. The immunomodulatory role of interleukin-35 in fibrotic diseases. Expert Rev Clin Immunol. (2019) 15:431–9. doi: 10.1080/1744666X.2019.1564041
122. Zhao N, Liu X, Guo H, Zhao X, Qiu Y, Wang W. Interleukin-35: An emerging player in the progression of liver diseases. Clin Res Hepatol Gastroenterol. (2021) 45:101518. doi: 10.1016/j.clinre.2020.07.023
123. Wang J, Zhao P, Guo H, Sun X, Jiang Z, Xu L, et al. Serum IL-33 levels are associated with liver damage in patients with chronic hepatitis C. Mediators Inflamm. (2012) 2012:819636. doi: 10.1155/2012/819636
124. Zhao J, Zhang Z, Luan Y, Zou Z, Sun Y, Li Y, et al. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology. (2014) 59:1331–42. doi: 10.1002/hep.26916
125. Shi M, Wei J, Dong J, Meng W, Ma J, Wang T, et al. Function of interleukin-17 and -35 in the blood of patients with hepatitis B-related liver cirrhosis. Mol Med Rep. (2015) 11:121–6. doi: 10.3892/mmr.2014.2681
126. Ming D, Yu X, Guo R, Deng Y, Li J, Lin C, et al. Elevated TGF-β1/IL-31 pathway is associated with the disease severity of hepatitis B virus-related liver cirrhosis. Viral Immunol. (2015) 28:209–16. doi: 10.1089/vim.2014.0142
127. Ma Y, Liu X, Wei Z, Wang X, Xu D, Dai S, et al. The expression of a novel anti-inflammatory cytokine IL-35 and its possible significance in childhood asthma. Immunol Lett. (2014) 162:11–7. doi: 10.1016/j.imlet.2014.06.002
128. Li T, Huang Y, Liu P, Liu Y, Guo J, Zhang W, et al. Lower plasma levels of IL-35 in patients with primary biliary cirrhosis. Tohoku J Exp Med. (2018) 244:123–31. doi: 10.1620/tjem.244.123
129. Jiang B, Zhou Y, Liu Y, He S, Liao B, Peng T, et al. Research progress on the role and mechanism of IL-37 in liver diseases. Semin Liver Dis. (2023) 43:336–50. doi: 10.1055/a-2153-8836
130. Griessmair L, Pirringer L, Mountford S, Sendelhofert A, Makeschin MC, Koletzko S, et al. Expression of IL-37 correlates with immune cell infiltrate and fibrosis in pediatric autoimmune liver diseases. J Pediatr Gastroenterol Nutr. (2022) 74:742–9. doi: 10.1097/MPG.0000000000003443
131. Mountford S, Effenberger M, Noll-Puchta H, Griessmair L, Ringleb A, Haas S, et al. Modulation of liver inflammation and fibrosis by interleukin-37. Front Immunol. (2021) 12:603649. doi: 10.3389/fimmu.2021.603649
132. Ren C, Liu F, Xing C, Zhao R, Tang X, Liu M, et al. IL-37 alleviates liver granuloma caused by Schistosoma japonicum infection by inducing alternative macrophage activation. Parasit Vectors. (2022) 15:300. doi: 10.1186/s13071-022-05420-6
133. Zhu J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine. (2015) 75:14–24. doi: 10.1016/j.cyto.2015.05.010
134. Weng SY, Wang X, Vijayan S, Tang Y, Kim YO, Padberg K, et al. IL-4 receptor alpha signaling through macrophages differentially regulates liver fibrosis progression and reversal. EBioMedicine. (2018) 29:92–103. doi: 10.1016/j.ebiom.2018.01.028
135. Atsukawa K, Saito H, Tsukada N, Akiba Y, Toda K, Kumagai N, et al. Th1 and Th2 cytokines differentially regulate the transformation of Kupffer cells into multinucleated giant cells but similarly enhance the Kupffer cell-induced hepatic stellate cell proliferation. Hepatol Res. (2001) 20:193–206. doi: 10.1016/s1386-6346(00)00133-9
136. Bhatt S, Shen GQ, Li Y, Qian S, Ragni MV, Lu L. Hepatic stellate cell-conditioned myeloid cells provide a novel therapy for prevention of factor VIII antibody formation in mice. Exp Hematol. (2015) 43:277–85. doi: 10.1016/j.exphem.2014.12.001
137. Wu TJ, Wang YC, Wu TH, Lee CF, Chan KM, Lee WC. Inhibition of allogenic T-cell cytotoxicity by hepatic stellate cell via CD4+ CD25+ Foxp3+ regulatory T cells in vitro. Transplant Proc. (2012) 44:1055–9. doi: 10.1016/j.transproceed.2012.03.029
138. Chang L, Gao J, Yu Y, Liao B, Zhou Y, Zhang J, et al. MMP10 alleviates non-alcoholic steatohepatitis by regulating macrophage M2 polarization. Int Immunopharmacol. (2023) 124:111045. doi: 10.1016/j.intimp.2023.111045
139. Elhadidy MG, Elmasry AI, El Nashar EM, Alghamdi MA, Al-Khater KM, Alasmari WA, et al. Melatonin attenuates thioacetamide-induced liver fibrosis in male rats through modulation of interleukin-6, interleukin-4, apoptosis and urokinase plasminogen activator receptor-associated protein/Endo180. J Physiol Pharmacol. (2022) 73:613–24. doi: 10.26402/jpp.2022.5.05
140. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discovery. (2014) 13:21–38. doi: 10.1038/nrd4176
141. Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. (2010) 32:17–31. doi: 10.1007/s00281-009-0188-x
142. Wu Y, Min J, Ge C, Shu J, Tian D, Yuan Y, et al. Interleukin 22 in liver injury, inflammation and cancer. Int J Biol Sci. (2020) 16:2405–13. doi: 10.7150/ijbs.38925
143. Su SB, Qin SY, Xian XL, Huang FF, Huang QL, ZhangDi HJ, et al. Interleukin-22 regulating Kupffer cell polarization through STAT3/Erk/Akt crosstalk pathways to extenuate liver fibrosis. Life Sci. (2021) 264:118677. doi: 10.1016/j.lfs.2020.118677
144. Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. (2012) 56:1150–9. doi: 10.1002/hep.25744
145. El-Shorbagy AA, Shafaa MW, Salah Elbeltagy R, El-Hennamy RE, Nady S. Liposomal IL-22 ameliorates liver fibrosis through miR-let7a/STAT3 signaling in mice. Int Immunopharmacol. (2023) 124:111015. doi: 10.1016/j.intimp.2023.111015
146. Feng D, Kong X, Weng H, Park O, Wang H, Dooley S, et al. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology. (2012) 143:188–98.e7. doi: 10.1053/j.gastro.2012.03.044
147. Sertorio M, Hou X, Carmo RF, Dessein H, Cabantous S, Abdelwahed M, et al. IL-22 and IL-22 binding protein (IL-22BP) regulate fibrosis and cirrhosis in hepatitis C virus and schistosome infections. Hepatology. (2015) 61:1321–31. doi: 10.1002/hep.27629
148. Meng YX, Zhao R, Huo LJ. Interleukin-22 alleviates alcohol-associated hepatic fibrosis, inhibits autophagy, and suppresses the PI3K/AKT/mTOR pathway in mice. Alcohol Clin Exp Res (Hoboken). (2023) 47:448–58. doi: 10.1111/acer.15021
149. Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P, et al. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci (Lond). (2016) 130:193–203. doi: 10.1042/CS20150405
Keywords: cytokines, interleukins, interleukin receptors, inflammation, liver fibrosis
Citation: Zhang Z, Wang J, Li H, Niu Q, Tao Y, Zhao X, Zeng Z and Dong H (2025) The role of the interleukin family in liver fibrosis. Front. Immunol. 16:1497095. doi: 10.3389/fimmu.2025.1497095
Received: 16 September 2024; Accepted: 22 January 2025;
Published: 10 February 2025.
Edited by:
Thiago Almeida Pereira, Stanford University, United StatesReviewed by:
Ping Zhou, University of California, Los Angeles, United StatesNatalia A Osna, University of Nebraska Medical Center, United States
Copyright © 2025 Zhang, Wang, Li, Niu, Tao, Zhao, Zeng and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Li, bGlodWlAY2R1dGNtLmVkdS5jbg==
†ORCID: Hui Li, orcid.org/0000-0002-5919-1396