 Ran Liu
Ran Liu Xinyan Jiang
Xinyan Jiang Ruijuan Dong
Ruijuan Dong Yuting Zhang
Yuting Zhang Cong Gai
Cong Gai Peng Wei
Peng Wei- School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing, China
Host immune responses to antigens are tightly regulated through the activation and inhibition of synergistic signaling networks that maintain homeostasis. Stimulatory checkpoint molecules initiate attacks on infected or tumor cells, while inhibitory molecules halt the immune response to prevent overreaction and self-injury. Multiple immune checkpoint proteins are grouped into families based on common structural domains or origins, yet the variability within and between these families remains largely unexplored. In this review, we discuss the current understanding of the mechanisms underlying the co-suppressive functions of CTLA-4, PD-1, and other prominent immune checkpoint pathways. Additionally, we examine the IgSF, PVR, TIM, SIRP, and TNF families, including key members such as TIGIT, LAG-3, VISTA, TIM-3, SIRPα, and OX40. We also highlight the unique dual role of VISTA and SIRPα in modulating immune responses under specific conditions, and explore potential immunotherapeutic pathways tailored to the distinct characteristics of different immune checkpoint proteins. These insights into the unique advantages of checkpoint proteins provide new directions for drug discovery, emphasizing that emerging immune checkpoint molecules could serve as targets for novel therapies in cancer, autoimmune diseases, infectious diseases, and transplant rejection.
Introduction
Cancer is a leading cause of premature death worldwide, with its high mortality rate necessitating innovative therapeutic approaches (1). On April 4, 2024, A Cancer Journal for Clinicians published the most recent global cancer burden data for 2022, which revealed that lung cancer has overtaken breast cancer, once again becoming the most prevalent cancer worldwide (2).However, in many cases, durable remission is not achieved using treatments such as radiotherapy and chemotherapy. Therefore, the development of new therapies for the treatment of cancer is essential.
The tumor microenvironment is a highly heterogeneous ecosystem composed of tumor cells, immune cells, and other stromal cells. Immunotherapy is a promising emerging therapeutic modality for the treatment of many types of cancer (3). Recent advances in immunotherapy have demonstrated the potential of leveraging the immune system to combat cancer (4). Specifically, the immune biomarkers associated with checkpoint immunotherapy responses offer valuable insights into patients’ reactions to treatment (5). The immune microenvironment is a complex network comprising various immune cells, fibroblasts, cytokines, chemokines, and extracellular matrix proteins (6). These components interact extensively with each other and with tumor cells, thereby regulating cancer growth and progression. In certain cases, the immune system is capable of recognizing and attacking cancer cells, leading to tumor regression (7).
The idea that the immune system can recognize and control tumor growth dates back to 1893 when William Coley, a surgeon, used live bacteria as a form of immunotherapy to treat cancer. This early work laid the foundation for the modern understanding of cancer immunology (8). PD-1 was first discovered in 1991 by Yasuya Ishida in cDNA libraries of unstimulated and stimulated mouse T cells. It was subsequently named programmed cell death 1 (PD-1) due to its association with T cell apoptosis induced by specific stimuli.” (9).However, the effectiveness of cancer immunotherapy has been moderate due to its limited clinical efficacy. This limitation arises from the ability of tumor cells to evade recognition and elimination by the immune system, resulting in a tumor escape mechanism (10). Over the past few decades, significant progress has been made in understanding how cancer evades the immune system. This understanding has led to the development of new strategies aimed at blocking cancer’s immune escape, thereby enhancing the elimination of tumor cells (11). In some cases, the immune system fails to recognize and respond to cancer cells, allowing the tumor to evade detection and grow unchecked. Increasing evidence indicates that immune escape plays a crucial role in the survival and progression of tumors (12). Within the tumor microenvironment, tumor cells can recruit immunosuppressive cells, such as CD4+ T cells, which compromise the cytotoxic function of CD8+ T cells (13).
Currently, extensive biological and medical research has categorized immune checkpoint proteins into distinct families based on their conserved domains, expanding the scope of immunotherapy research. (Figure 1) By reviewing the literature on immune checkpoints across various immune protein families, this paper aims to summarize the current research status of key immune checkpoints and offer new perspectives on cancer immunotherapy.
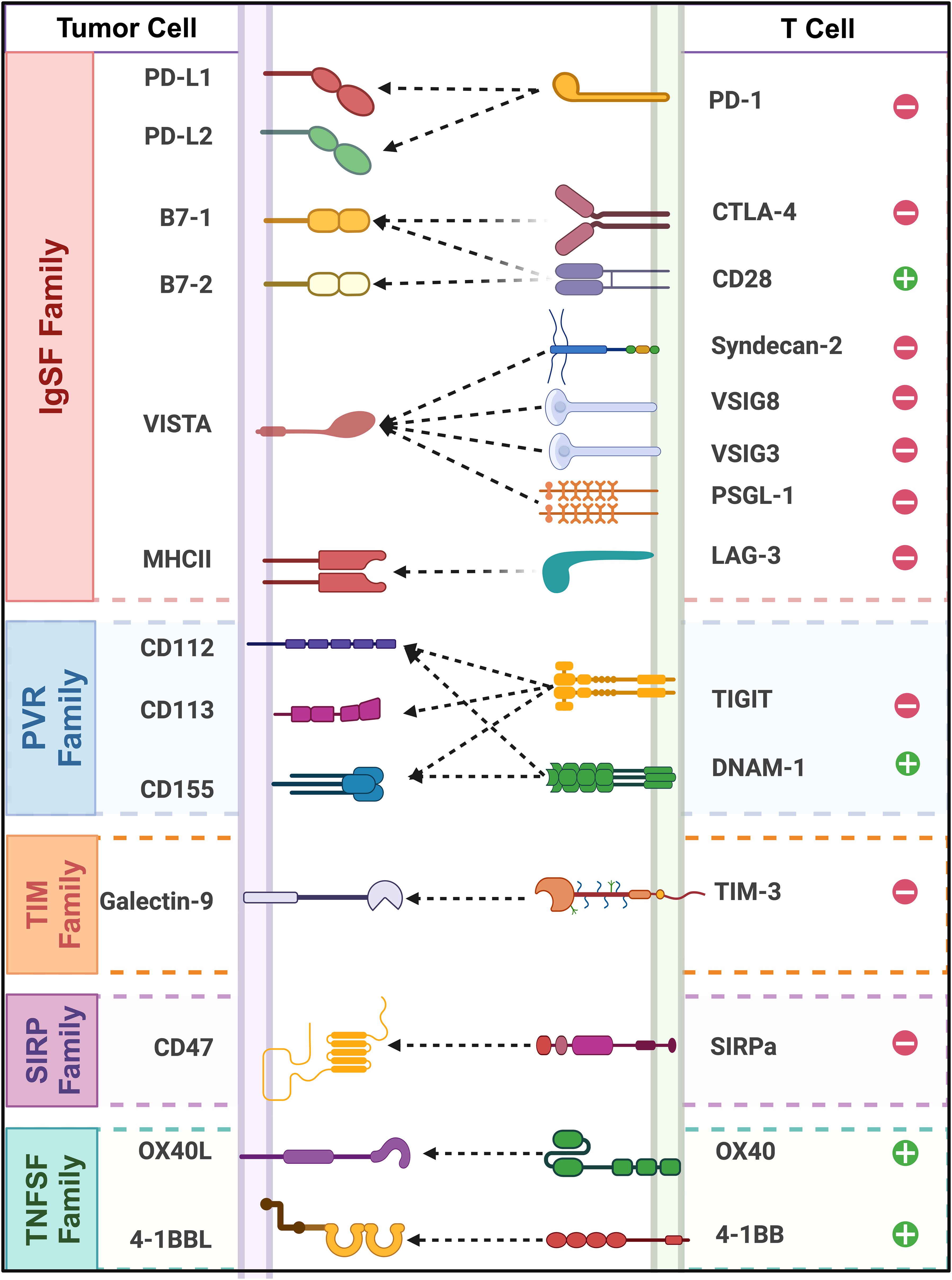
Figure 1. Classification and interactions of immune checkpoint receptors and ligands across different protein families. (This schematic illustrates key immune checkpoint molecules and their interactions between tumor cells and T cells. Tumor cell ligands (left) and their corresponding TCRs (right).”+” and “−” symbols represent stimulatory and inhibitory functions.).
IgSF family
The immunoglobulin superfamily (IgSF) is one of the largest and most versatile families of structural domains in animal genomes (14). IgSF protein genes account for more than 2% of human genes, making them the largest gene family in the human genome (15). Although the amino acid sequences of different family members vary considerably, the structural characteristics of the IgSF are traditionally defined by a few key site-specific residues critical for proper protein folding (16). During ontogeny, IgSF recognition molecules play essential roles in neuronal processes such as cell survival, migration, axonal guidance, and synaptic targeting (17). Many immune checkpoint proteins contain Ig structural domains or exhibit high homology with the V and C regions of immunoglobulins. PD-1, PD-L1, CTLA-4, BTLA, and VISTA are all part of the IgSF (18). All IgSF members contain 1-7 Ig-like structures, with each structure comprising approximately 70-110 amino acid residues (19). The secondary structure is a β-sheet formed by two anti-parallel β-strands, each composed of 3-5 amino acid residues, with 5-10 residues per strand. The hydrophobic amino acids within the β-sheet stabilize the folds (20). Techniques such as X-ray diffraction analysis and DNA sequence analysis have revealed that many cell membrane surface molecules and some protein molecules in the body share a similar peptide folding pattern with Ig structures (21). These molecules exhibit high homology with the variable (V) and conserved (C) regions of immunoglobulins, suggesting they may have evolved from a common ancestor (22). The genes encoding these polypeptide chains are referred to as the immunoglobulin gene superfamily, and their products are known as the IgSF (23).
We have summarized the structure and function of representative immune checkpoint proteins from different immune protein families, including the number of amino acids and Ig structural domains they contain (Table 1).
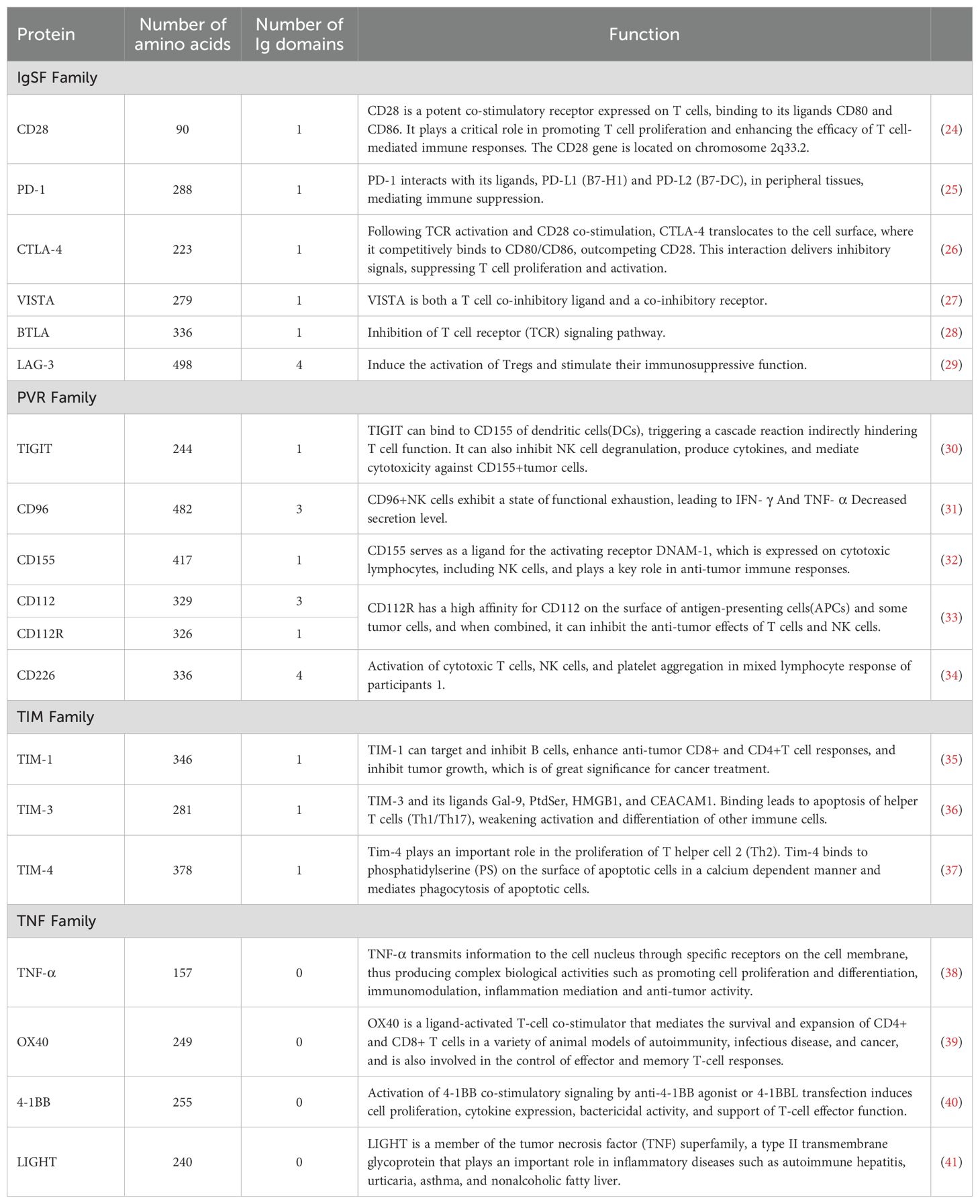
Table 1. Summary of receptor structures of representative immune checkpoints from different families.
Most IgSF members are membrane proteins located on the surface of lymphocytes, playing a crucial role in various immune activities (42). The discovery of the Ig structure in invertebrate cellular adhesion molecules, which lack an immune system, suggests that Ig proteins originally functioned as adhesion molecules during early evolution, and later adapted to serve immune functions (43). The identification of Ig proteins as intermediaries in the evolution of cellular slime molds in invertebrates, followed by the discovery of their immune functions in vertebrates, indicates that the multifunctional nature of IgSF was likely created through gene duplication and subsequent divergence. Japanese scientist Susumu Tonegawa was awarded the Nobel Prize in Physiology or Medicine in 1987 for his groundbreaking research on the structure of immunoglobulin genes (44).
PD-1
Programmed death-1 (PD-1) is a crucial immunoregulatory receptor expressed by activated T cells. PD-1 is a type I transmembrane protein composed of 288 amino acids and is a member of the CD28/CTLA-4 family of T cell regulators. The protein structure includes an extracellular IgV domain, a transmembrane domain, and an intracellular tail (45). The intracellular tail contains two phosphorylation sites within the immune receptor tyrosine-based inhibitory motif (ITIM) and the immune receptor tyrosine-based switch motif (ITSM), indicating that PD-1 negatively regulates TCR signaling (46). PD-1 primarily binds to its ligands, PD-L1 (B7-H1) and PD-L2 (B7-DC), to mediate immunosuppression. PD-L1 and PD-L2 are expressed by tumor cells, stromal cells, or both (25). The discovery and application of PD-1 indicates that the research of tumor therapy has entered a new stage (47).
In the presence of PD-L1, PD-1 and CD28 colocalize at the center of TCR-enriched regions. PD-1, upon activation, recruits the protein tyrosine phosphatase SHP-2, which reduces CD28 phosphorylation and suppresses TCR signaling intensity (48). The PD-1-mediated dephosphorylation of CD28 significantly disrupts PI3K recruitment to the TCR signalosome, leading to decreased activation of the PI3K/AKT pathway and reduced expression of its transcriptional targets, such as Bcl-xL. Furthermore, SHP-2 not only blocks CD28 co-stimulatory signaling but also inhibits TCR-mediated phosphorylation of ZAP70, impairing ERK activation and subsequent IL-2 production and amplification (25) (Figure 2).
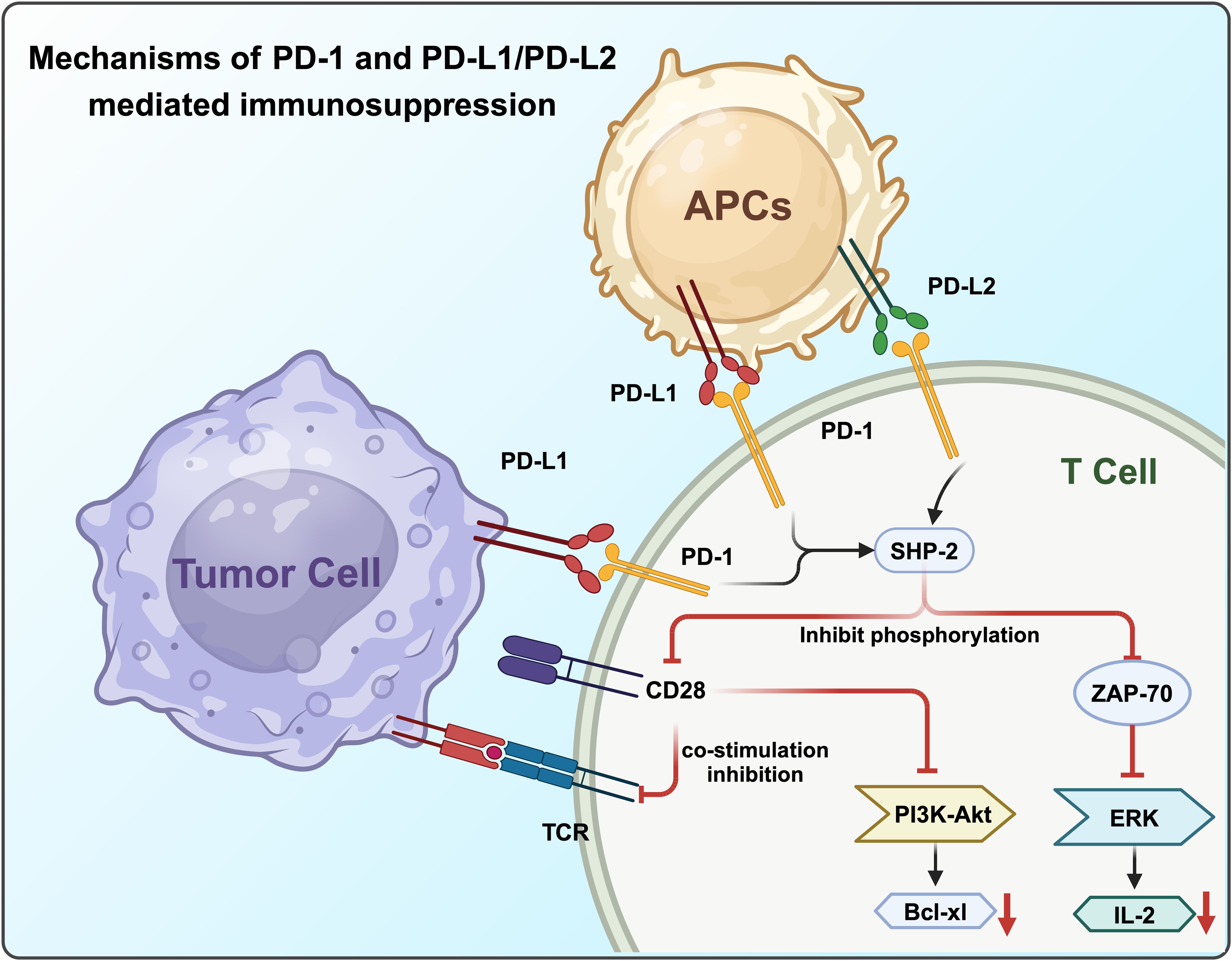
Figure 2. Mechanisms of PD-1 and PD-L1/PD-L2 mediated immunosuppression.
PD-L1 is expressed by APCs, including human peripheral blood interferon-stimulated monocytes that activate human and mouse DCs. It is also expressed in non-lymphoid tissues such as heart and lung (49). Monoclonal antibodies can restore the anti-tumor activity of CD8+ T cells by blocking the inhibitory signaling pathways (50). However, targeting a single immunosuppressive pathway may not completely eliminate tumors. Another ligand of PD-1, PD-L2, acts as a T cell inhibitory receptor (51).
Although much research has focused on the PD-1/PD-L1 interaction, PD-L2 (B7-DC), a member of the B7 family, was identified in DCs in 2001 (52). Binding of PD-L2 to PD-1 significantly inhibited TCR-mediated CD4+ T cell proliferation and cytokine production, leading to the discovery of the overlapping functions of PD-L1 and PD-L2 (53).While initially thought to be expressed primarily in macrophages in the presence of interleukins, recent studies have shown that PD-L2 is expressed in various tumor cells depending on the tumor microenvironment (54). The activation of the PD-1 signaling pathway can lead to T cell apoptosis and exhaustion, resulting in immunosuppression due to T cell dysfunction. Immune checkpoint blockade against PD-1 inhibits its interaction with both PD-L1 and PD-L2 (55).
Compared with PD-L1, the expression of PD-L2 is relatively limited, mainly found on APCs such as activated macrophages and DC (56). Although the interaction affinity between PD-L2 and PD-1 is several times higher than that of PD-L1, PD-L2 is usually expressed at lower levels, making PD-L1 the primary ligand. Consequently, the PD-1/PD-L1 signaling pathway remains a major focus of research (57).
To date, five anti-PD-1/PD-L1 drugs have received approval from the US Food and Drug Administration (FDA). These include anti-PD-1 drugs such as pembrolizumab (Keytruda; Merck & Co., Inc., Kenilworth, NJ, USA) and nivolumab (Opdivo; Bristol-Myers Squibb Company, New York, NY, USA), as well as anti-PD-L1 drugs like atezolizumab (Tecentriq; Genentech, Inc., South San Francisco, CA, USA), avelumab (Bavencio; EMD Serono, Inc., Merck KGaA, Darmstadt, Germany), and durvalumab (Imfinzi; AstraZeneca UK Limited, Cambridge, UK).Of these, pembrolizumab and nivolumab have been used with good efficacy in a variety of diseases (Table 2).

Table 2. Summary of selected anti-PD-1 and anti-PD-L1 drugs approved for marketing by the FDA.
There is growing evidence that drugs targeting immune checkpoints can provide significant clinical benefits, including prolonged response and survival. Monoclonal antibodies targeting the programmed death-1/programmed death ligand-1 (PD-1/PD-L1) immune checkpoint pathway—such as Nivolumab, Pembrolizumab, Atezolizumab, Avelumab, and Durvalumab—have demonstrated considerable efficacy and offer new therapeutic opportunities for many cancer patients. However, reports indicate that the effectiveness of these monoclonal antibodies is often limited due to the emergence of intrinsic or acquired resistance mechanisms and a lack of durable responses in some patients with melanoma (72).
CTLA-4
CTLA-4, or cytotoxic T lymphocyte-associated antigen-4 (also known as CD152), is located on band 33 (2q33) of the long arm of chromosome 2 (73). It exhibits high homology with the costimulatory receptor CD28 found on T cells (74). CTLA-4 is a membrane protein with a relatively short intracellular domain consisting of only 36 amino acids. This domain contains an immune tyrosine inhibitory motif (ITIM), which contrasts with the immune tyrosine activating motif (ITAM) present in CD28 (75).
Both CTLA-4 and CD28 are expressed on the surface of activated CD4+ and CD8+ T cells and are members of the IgSF. They share the same ligands, CD86 (B7-2) and CD80 (B7-1). The binding of CD28 to B7-1/2 generates stimulatory signals that promote cytokine IL-2 mRNA production, cell cycle entry, T cell activation, helper T cell differentiation, and immunoglobulin isotype switching (76). In contrast, CTLA-4 inhibits T cell activation by competitively binding to B7-1 and B7-2, which are normally bound by CD28. This competitive binding downregulates the TCR signaling pathway, reduces IL-2 secretion, and serves as a negative regulator of T cell responses (77).
Regulatory Tregs further inhibit T cell activation by down-regulating CD80/CD86 expression via CTLA-4, thereby disrupting the CD28 signaling pathway. CTLA-4 inhibitors exert anti-tumor effects by preventing Tregs from down-regulating CD80/86 expression and depleting Tregs through antibody-dependent cell-mediated cytotoxicity (ADCC) and phagocytosis (ADCP) (78). This increases the infiltration of CD4+/CD8+ T cells into tumor tissues and enhances the clonality of memory T cells (79).
Compared with PD-1/PD-L1 monoclonal antibodies, CTLA-4 monoclonal antibody drugs, despite being introduced and clinically applied earlier, are relatively limited in variety and are primarily approved for use in combination with other monoclonal antibodies. Currently, the only CTLA-4 inhibitors approved by the US FDA are ipilimumab and tremelimumab. Of these, only ipilimumab is approved by the FDA for the treatment of melanoma, kidney cancer and advanced metastatic colorectal cancer (80–82).
The goal of cancer immunotherapy should remain the complete and safe eradication of cancer from the patient’s body (83). Achieving this goal requires a unique immunotherapy regimen based on the biology present in a given patient’s body, and some patients may require only a single therapy, while others may require a combination of therapies (84). The introduction of CTLA-4 inhibitors has deepened the understanding of immunotherapy among clinicians and increased interest in dual immunotherapy (85). A Phase II clinical trial (CheckMate-069) demonstrated that the combination therapy of nivolumab (a PD-1 monoclonal antibody) and ipilimumab (a CTLA-4 monoclonal antibody), also known as “O+Y,” resulted in a higher objective response rate (ORR) and complete response rate in BRAF wild-type patients compared to ipilimumab monotherapy (61% vs. 11% and 22% vs. 0%, respectively) (86). Additionally, in BRAF-mutant patients, combination therapy significantly prolonged median progression-free survival (mPFS) (8.5 months vs. 2.7 months). Another combination therapy, “D+T” (Durvalumab, a PD-L1 monoclonal antibody, and Tremelimumab, a CTLA-4 monoclonal antibody), has been applied in the first-line treatment of advanced hepatocellular carcinoma. We have summarized the approved combination therapies and their effects across different diseases (87) (Table 3).
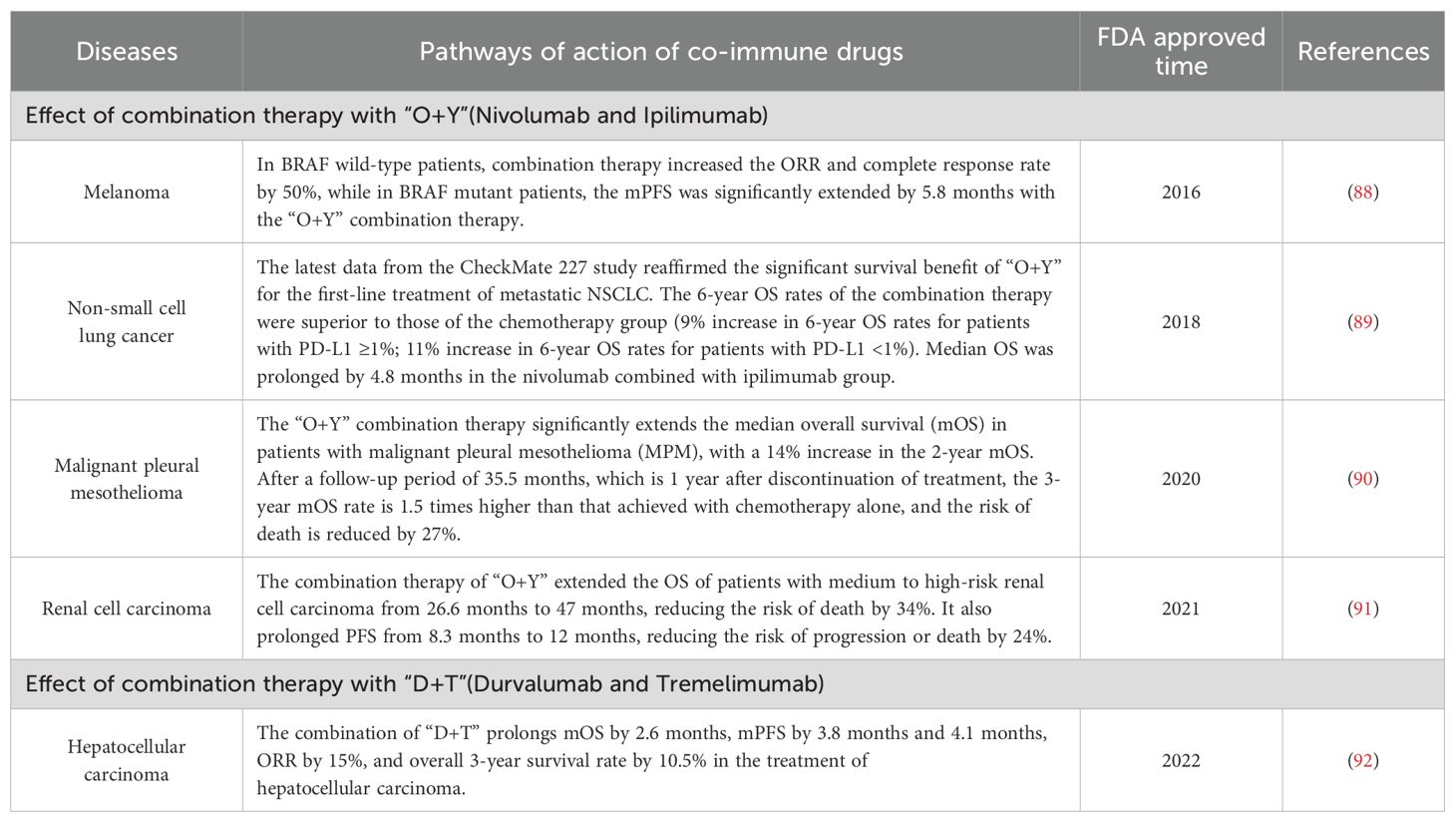
Table 3. Summary of selected anti-PD-1 combined with anti-CTLA-4 drugs approved for marketing by the FDA.
CTLA-4 inhibitors, such as ipilimumab, have been available for several years but have not achieved significant breakthroughs in monotherapy for various solid tumors. This may be due to an incomplete understanding of CTLA-4’s mechanism of action and its relationship with the PD-1/PD-L1 signaling pathway. Additional factors, including variations in IgG antibody types, pH-dependent antibodies, and antigenic epitopes, complicate achieving the expected clinical efficacy of these drugs. However, the development of PD-1/CTLA-4 combination therapies may address these challenges.
Given the complexity, uncertainty, and associated risks of immunotherapy, along with the notable variability in immune checkpoint therapy effectiveness among patients with different clinical profiles, there is a need for more comprehensive evidence-based medicine. Precise biomarkers are required to identify patient populations that are most likely to benefit from immunotherapy, thereby mitigating risks. The use of CTLA-4 inhibitors across various tumor types and treatment stages should be guided by evidence-based medicine and relevant clinical guidelines.
VISTA
VISTA, also known as V-type immunoglobulin domain-containing suppressor of T cell activation or PD-1H, is an immune checkpoint protein that plays a critical role in suppressing T cell-mediated anti-cancer responses (93). The VISTA protein spans 279 amino acids, including a 162-aa extracellular domain, a 21-aa transmembrane domain, and a 96-aa cytoplasmic domain (94). The cytoplasmic domain contains multiple phosphorylation sites for casein kinase 2 and protein kinase C. Similar to PD-1 and CTLA-4, VISTA inhibitors have the potential to enhance the immune system’s ability to eliminate tumors. The immunoglobulin variable (IgV)-like folding in VISTA’s extracellular domain includes two additional disulfide bonds and an extended loop with additional helices, forming a clinically relevant continuous binding epitope for antiviral antibodies. This antibody-binding region is closely related to the Ig domain (VSIG3), a significant ligand for VISTA (95).
Compared to peripheral lymph nodes, VISTA is more abundantly expressed in MDSCs within the tumor microenvironment (TME). Under the hypoxic conditions of the TME, VISTA expression is significantly upregulated, leading to the suppression of TLR signaling and inhibition of cell migration (96). By reprogramming myeloid cells, VISTA reduces the production of pro-inflammatory cytokines such as TNF-α while increasing anti-inflammatory mediators like IL-10, thereby enhancing the immunosuppressive function of myeloid cells. Additionally, VISTA promotes peripheral immune tolerance by facilitating activation-induced T cell death (27).
VISTA may also be crucial in regulating inflammation and autoimmune diseases, such as graft-versus-host disease (GVHD), acute hepatitis, encephalitis, and lupus (27). Additionally, VISTA acts as a co-inhibitory receptor on T cells, significantly modulating antigen-specific CD4+ T cell responses and protecting mice from GVHD, acute hepatitis, and asthma (97).VISTA is primarily expressed on CD45+ cells located near tumors and is also present in the hematopoietic system, with notable expression in myeloid cell compartments (98). It is most abundantly expressed on myeloid cells and DCs, and less so on T cells. The extracellular domain of VISTA contains numerous histidine residues, which confer pH-dependent functionality. Specifically, histidine residues interact with ligands when the extracellular pH decreases from 7.4 to 6.0, a condition found in the tumor microenvironment, lymph node regions, or healing wounds (99). Five ligands of VISTA - PSGL-1, Syndecan-2, LRIG-1, VSIG8, and VSIG3 - were found to bind differently at pH values of 6.0 and 7.4 (100).
Antibodies that selectively bind to and block interactions in acidic environments can potentially reverse VISTA-mediated immunosuppression in vivo. PSGL-1, expressed on T and B cells, myeloid cells, and DCs, can inhibit T cell proliferation and promote a depletion phenotype, although the precise mechanism by which VISTA mediates this effect remains unclear (101). This selective interaction and inhibition of T cells at acidic pH values are mediated by histidine residues along the periphery of the VISTA extracellular domain, which facilitate binding to the adhesion and co-inhibitory receptor PSGL-1 (102). To illustrate the structural and functional diversity of immune checkpoint molecules, we have depicted the complex mechanisms by which these molecules regulate immune responses, particularly in the context of cancer and autoimmune diseases (Figure 3).
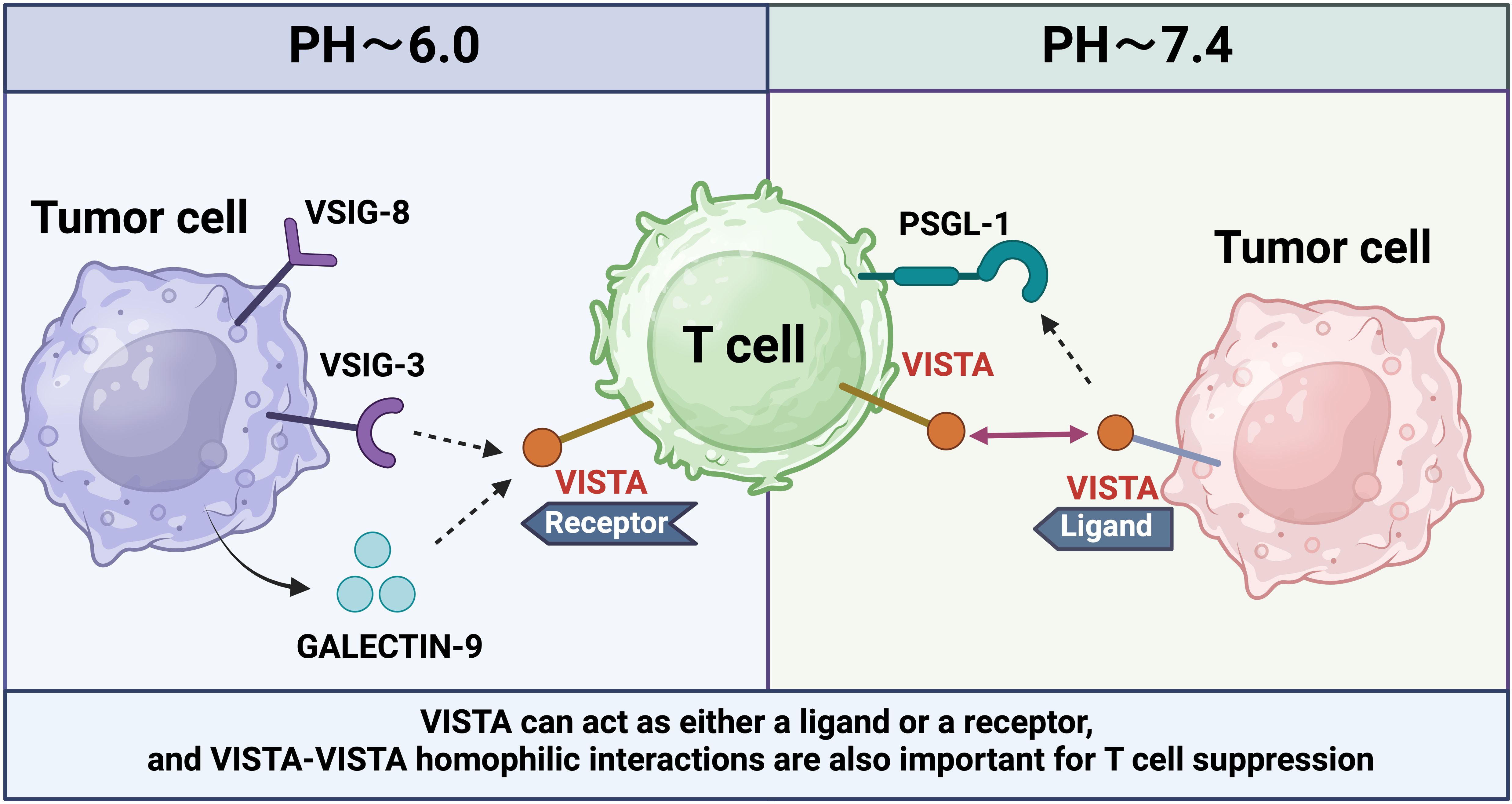
Figure 3. VISTA is expressed on T cells or tumor cells in different PH environments to regulate different immune responses.
Additionally, the interactions between VISTA and its ligands VSIG3 and VSIG8 inhibit T cell activation and effector functions. VISTA also induces the formation of regulatory Tregs from human CD4+ T cells (103). Furthermore, VISTA promotes the inhibition of myeloid cells and tolerogenic DCs by interfering with the MAPK and NF-kB pathways within the TLR signaling cascade (104). Early studies utilizing rat anti-mouse viral antibodies in combination with anti-PD-1 or anti-PD-L1 antibodies have demonstrated efficacy across various mouse tumor models (105). In these models, selective blockade of the interaction with PSGL-1 at pH 6.0, rather than at pH 7.4, offers additional therapeutic benefits against PD-1. These pH-selective antibodies accumulate in the acidic tumor microenvironment rather than in major viral expression sites like the spleen. Compared to non-pH-selective antibodies, pH-selective antibodies have shown improved safety and efficacy in non-human primates (106). The development of pH-selective VISTA antibodies represents a promising new strategy for cancer therapy.
The dual role of VISTA as both a receptor and a ligand has been demonstrated through its ability to engage in homologous interactions. Homologous VISTA-VISTA binding facilitates the phagocytosis of apoptotic cells by macrophages, thereby contributing to the clearance of apoptotic cells from the internal environment. A prior study utilizing VISTA-Ig fusion protein to treat wild-type (WT) T cells and VISTA knockout (KO) T cells in vitro revealed that VISTA KO T cell proliferation was less affected by the VISTA-Ig protein compared to WT T cells (107).
B7-H3
B7 homologous protein 3 (B7-H3, also known as CD276), a newly discovered member of the B7 family, is an immunomodulatory protein with co-stimulatory/co-inhibitory effects and is an attractive and promising target for cancer immunotherapy, playing a dual role in the immune system (108).
B7-H3 is a type I transmembrane protein containing 316 amino acids with a molecular weight of ~45-66 kDa, which was first discovered in 2001 from a cDNA library derived from human DCs (109).The human B7-H3 gene is located on chromosome 15 and the mouse B7-H3 gene is localized on chromosome 9 (110).Upper B7-H3 shares 20-27% amino acid homology with other B7 family members (111).B7-H3 is abundantly expressed on the surface of tumor cells, with limited expression in normal cells, and is also involved in the formation of the tumor microenvironment (TME) (112).
TREM-like transcript 2 (TLT-2) was identified as a potential receptor for B7-H3 (113). However, TLT-2 may not be the only receptor for B7-H3. In contrast to other immune checkpoints, B7-H3 also regulates cancer cell invasiveness through various non-immune pathways (114).A study in 2019, using a new interactome platform with high-throughput data, identified interleukin-20 receptor subunit alpha (IL20RA) as the first target for B7-H3 binding (115).The significance of IL20RA as a cancer biomarker has been investigated and overexpression of IL20RA promotes cancer stemness through the transcription factor SOX2 and suppresses immunity through increased PD-L1 expression (116).In addition, a 2021 study detected phospholipase A2 receptor 1 (PLA2R1) as another high-level binding protein among all single-channel transmembrane proteins and their exogenous sources based on the leaflet vesicle interactions group platform (117).
B7-H3 was initially found to be an immune co-stimulant (118), in which B7-H3-Ig induced the proliferation of CD4+ and CD8+ T cells and increased the secretion of interferon γ thereby enhancing T cell activity.B7-H3 also enhances T cell activity by promoting the production of IL-10, TGF-β1.In addition, the positive correlation between the expression of FOXP3+ tregs and B7-H3 favoring the immune system to suppress the tumor microenvironment (119, 120). On the other hand, B7-H3 inhibited the secretion of IFN-γ, IL-2, perforin, and granzyme B, thereby suppressing the activity of CD4+ T cells, CD8+ T cells, γδ T cells, CAR-T cells, Vδ2 T cells, T17 cells, CD3+ T cells, NK cells, macrophages, neutrophils, and DCs (121–124), while B7-H3 regulated the differentiation of tumor-associated macrophages, promotes polarization of type 2 macrophages, and converts the M1 phenotype to the M2 phenotype (125). B7-H3 triggers different signaling cascades to activate downstream molecules that contribute to the malignant behavior of cancer cells, e.g., B7-H3 activates signaling pathways such as ERK, PI3K, and Stat3 in cancer cells, leading to accelerated cell proliferation and tumor growth (126).
Studies have shown that B7-H3 is abundantly expressed in mouse and human adipose tissue and preferentially expressed in adipocyte progenitor cells (APs), and knockdown of the gene leads to spontaneous obesity in mice, demonstrating a role for B7-H3 in adipocyte progenitor cell differentiation, lipid oxidation, and obesity, in addition to its immunomodulatory function (127). In addition, this study revealed a plausible link between diabetes mellitus (DM) and B7-H3. B7-H3 knockout mice exhibited an increased propensity for obesity and related metabolic syndrome. In another study, patients with type 1 diabetes had significantly higher serum B7-H3 levels than healthy controls. Given this evidence, the role of B7-H3 in the pathologic process of diabetes needs to be further explored (128).
However, the multifaceted role of B7-H3 in the tumor microenvironment has been extensively studied, and B7-H3 has been found to induce malignant behaviors and promote tumor progression through complex pathways. Role of B7-H3 in Tumor Cells, T Cells, DCs, NK Cells, CAFs, Neutrophils, and Endothelial CellsB7-H3 is a key regulator of the tumor microenvironment, and a valuable immunotherapeutic target (129).
LAG-3
Lymphocyte activation gene 3 (LAG-3) is a cell surface inhibitory receptor that regulates T cell activation and effector functions (130). LAG-3, a member of the IgSF, is encoded on human chromosome 12, recognized as a third-generation inhibitory receptor, it is considered a promising therapeutic target following PD-1 and CTLA-4. First identified by Triebel et al. in 1990 on activated human NK and T cells, LAG-3 has gained attention as an immune checkpoint molecule and a key target in cancer immunotherapy (131).
LAG-3 is a type I transmembrane protein weighs approximately 70 kDa and comprises 498 amino acids, spanning extracellular, transmembrane, and cytoplasmic regions. Its expression correlates with tumor prognosis and is found on effector T cells and regulatory Tregs, influencing T lymphocyte and APC signaling (132). The LAG-3 gene is located near the CD4 gene and shares structural similarities, suggesting both evolved from a common ancestral IgSF-encoding gene (133).
The cytoplasmic tail of LAG-3 is crucial for its negative signal transduction function within the cell, its loss completely abolishes this function. The cytoplasmic region of LAG-3 contains three conserved motifs. The first region includes serine phosphorylation sites, the second contains a single lysine residue within the unique “KIEELE” motif, and the third includes glutamate-proline (EP) repeat sequences (134). The absence of the KIEELE motif completely disrupts LAG-3 function on CD4 T cells, underscoring its critical role in inhibiting signal transduction (135).
LAG-3 is expressed in NK cells, B cells, and plasmacytoid DCs. Its expression is induced by TCR activation or cytokines such as IL-12, IL-27, IL-15, IL-2, and IL-7 (136). LAG-3 may serve as a depletion marker similar to PD-1 in CD8+ T cells, particularly in response to repeated antigen stimulation during chronic viral infections or cancer (137). Evidence suggests that LAG-3 interferes with common pathways involved in CD4 and CD8 activation and regulates the activation and expansion of memory T cells (138).
LAG-3 is associated with the TCR: CD3 complex on the T cell membrane, where it negatively regulates TCR signaling, leading to the suppression of cell proliferation and cytokine secretion (139). The co-participation of LAG-3 and CD3 in the immune synapse is essential for attenuating TCR signaling (140). Additionally, the simultaneous engagement of LAG-3/TCR with their respective ligands inhibits TCR: CD3-dependent intracellular calcium flux, further dampening TCR-dependent signaling cascades and suppressing T cell responses (131).
MHC class II (MHC-II) molecules are recognized as typical ligands for LAG-3. These molecules, which are abnormally expressed by APCs or melanoma cells, stably interact with LAG-3 through its D1 domain, exhibiting significantly higher affinity than with CD4 (141). This interaction negatively regulates T cell activation, cytotoxicity, and cytokine production. In fact, the LAG-3-Ig fusion protein competes for binding in CD4/MHC-II-dependent cell adhesion assays. Once LAG-3 binds to MHC-II, it transmits inhibitory signals through its cytoplasmic domain, thereby inhibiting the activation of CD4+ T cells (142).
The second identified ligand of LAG-3 is Galectin-3 (Gal-3), a soluble lectin that binds to galactosides and has a molecular weight of approximately 31 kDa. Gal-3 regulates T cell activation and is highly expressed in various tumor cells and activated T lymphocytes (83). The interaction between Gal-3 and LAG-3 is essential for optimal inhibition of CD8+ T cell cytotoxicity (143). Within the tumor microenvironment, Gal-3, via LAG-3 expression, inhibits the activation of antigen-specific CD8+ T cells and suppresses the expansion of plasmacytoid DCs, thereby impeding the formation of an effective anti-tumor immune response (144).
Fibrinogen-like protein 1 (FGL1), secreted by the liver, has recently been identified as a functional ligand for LAG-3 (145). FGL1 binds to the D1 and D2 domains of LAG-3, and while a single point mutation (Y73F) in the D1 domain disrupts MHC-II binding, it does not affect FGL1-Ig binding. This suggests that FGL1 and LAG-3 interact independently of MHC-II (146). FGL1 expression is induced by IL-6 and is present at low levels in the liver but highly upregulated in certain human cancers, such as lung cancer, melanoma, anterior adenocarcinoma, and colorectal cancer in the United States. FGL1 exhibits high affinity for LAG-3, and their interaction facilitates tumor immune escape. Blocking the FGL1-LAG-3 pathway has been shown to enhance the anti-tumor activity of CD8+ T cells (147).
In addition to FGL1, several other ligands for LAG-3 have been identified. One such ligand is LSECtin (liver sinusoidal endothelial cell lectin), a member of the C-type lectin receptor superfamily and a type II transmembrane protein. It is highly expressed in the liver and melanoma cells, where it inhibits the immune responses of CD8+ T cells and NK cells through its interaction with LAG-3. Another ligand, α-Synuclein, like MHC-II, binds to the LAG-3 D1 region and relies on the D2, D3, or intracellular domains (148).
PVR family
The poliovirus receptor (PVR) family, a group of proteins associated with immune regulation, belongs to the IgSF (149). Initially referred to as the PVR-related Ig domain (PVRIG) due to its inclusion of an Ig domain, this family comprises multiple members, including T cell immunoglobulin and immune receptor tyrosine inhibitory motif domains (TIGIT), CD96, CD226, as well as their ligands CD155 and CD112 (150).
Members of the PVR family share structural homology and exert synergistic or inhibitory effects through highly interactive interactions, forming a complex immune regulatory network (151). These proteins are of significant importance in immunotherapy, particularly in the treatment of hematological malignancies, making them a focal point of research.
PVR/nectin family members are expressed on various lymphocytes, including NK cells, CD8+, CD4+, and Tregs. TIGIT, DNAM-1 (CD226), CD96, and CD112R are expressed on T cells and natural killer (NK) cells, while their ligands—CD155, CD112, CD113, and CD111—are expressed on APCs or tumor cells (152). NK cells play a crucial role in eliminating and preventing metastasis during the early stages of cancer. As cytolytic effector cells, NK cells are involved in the release of tumor antigens, and the regulation of NK cell function by TIGIT significantly impacts the initial phase of the cancer immune cycle (153).
TIGIT, CD155 (PVR), CD96, CD226, and other related proteins share structural similarities and are collectively known as the CD155 family (154). Unlike typical immune checkpoint-ligand interactions, which generally follow a one-to-one or one-to-many relationship, TIGIT maintains a “many-to-many” relationship with CD226, CD96, CD112, and CD155 (155). This positions TIGIT within a complex regulatory network that includes multiple receptors (such as CD96 and CD112R), a competitive co-stimulatory receptor (CD226), and multiple ligands (such as CD155 and CD112) (156). This network is somewhat analogous to the CD28/CTLA-4/CD80/CD86 pathway, where inhibitory and co-stimulatory receptors compete for binding to the same ligands (157).
TIGIT
TIGIT (T-cell immunoreceptor with Ig and ITIM domains) (also known as WUCAM, Vstm3, VSIG9) is a member of the PVR/adhesin family, which belongs to the IgSF (158). It consists of an extracellular immunoglobulin variable region (IgV) domain, a type 1 transmembrane domain, and an intracellular domain with a classical immune receptor tyrosine inhibitory motif (ITIM) and immunoglobulin tyrosine tail (ITT) motif (159). TIGIT was initially discovered in a gene study on T cell specific expression by Genentech’s research team. The TIGIT gene is located on chromosome 3q13.31 and encodes a protein with 244 amino acids (160, 161).
TIGIT has been reported as a marker of CD8+T cell failure and a characteristic marker of Tregs in the tumor microenvironment (162). Another notable feature of TIGIT is that it is N-linked glycosylation, which often occurs on the asparagine residue in the N-X-S/T glycosylation sequence. N is asparagine, X is any amino acid except proline, S is serine, and T is threonine. N-glycosylation involves many aspects of cell biology, such as intercellular information transmission, ligand/receptor interactions, and cellular signal transduction. A study on PD-1 suggests that the interaction between immunosuppressive ligands/receptors is also widely dependent on n-glycosylation (163). In order to investigate whether the n-glycosylation of TIGIT is crucial for its ligand binding activity, a study combined TIGIT deglycosylation with in vitro PVR/TIGIT binding experiments. It was found that eliminating n-glycans from TIGIT inhibited the binding of TIGIT to PVR, indicating that the n-glycosylation of TIGIT is crucial for the involvement of PVR/TIGIT (160).
TIGIT is thought to compete with co-stimulatory receptors CD226 (also known as DNAM-1) and CD96 on T cells for binding to ligands such as CD155, CD112, and CD113 (164). The primary ligand for TIGIT is CD155, though immunoprecipitation experiments have demonstrated that CD112 and CD113 can also weakly interact with TIGIT. The IgV domain of TIGIT contains unique motifs, including (V/I)(S/T)Q, AX6G, and T(F/Y)PX1G subunits, which are involved in mediating trans interactions with PVR family cis dimers (165). These conserved motifs are characteristic of the PVR/nectin family, which includes TIGIT, CD226, CD96, CD112R, PVR, CD112, and CD113 (also known as PVRL3/nectin3) (166).
In mice, phosphorylation of the ITIM (Y227) or ITT-like motif residue (Y233) can trigger TIGIT-mediated inhibitory signaling (30). However, in the human NK cell line YTS, TIGIT/CD155 interaction predominantly initiates inhibitory signaling through the ITT-like motif. Upon TIGIT/CD155 engagement, phosphorylation of Tyr225 within the ITT-like motif occurs, facilitating the recruitment of cytoplasmic signaling molecules Grb2 and β-arrestin 2, which subsequently recruit the inositol-containing SH2 phosphatase-1 (SHIP-1). SHIP-1 inhibits the activation of PI3K and MAPK pathways while also suppressing TRAF6 and NF-κB signaling, leading to reduced IFN-γ production by NK cells (167). Moreover, TIGIT binding to DCs induces CD155 phosphorylation and activates a signaling cascade that promotes the formation of tolerogenic DCs, characterized by decreased IL-12 production and increased IL-10 secretion (168).
Recently, Nectin-4 has been identified as a novel ligand for TIGIT. Nectin-4 binds to TIGIT with an affinity similar to that of CD155 but uniquely does not interact with CD226, CD96, or CD112 (169). TIGIT, DNAM-1, CD96, and CD112R are expressed on T cells and natural killer (NK) cells, while their respective ligands—CD155, CD112, CD113, and CD111—are expressed on APCs or tumor cells. CD155 is predominantly expressed on DCs, T cells, B cells, and macrophages, whereas CD112 is broadly expressed in both hematopoietic and non-hematopoietic tissues, including bone marrow, lungs, pancreas, and kidneys. In contrast, CD113 expression is restricted to non-hematopoietic tissues, such as the lungs, liver, testes, kidneys, and fetal tissues (170) (Figure 4).
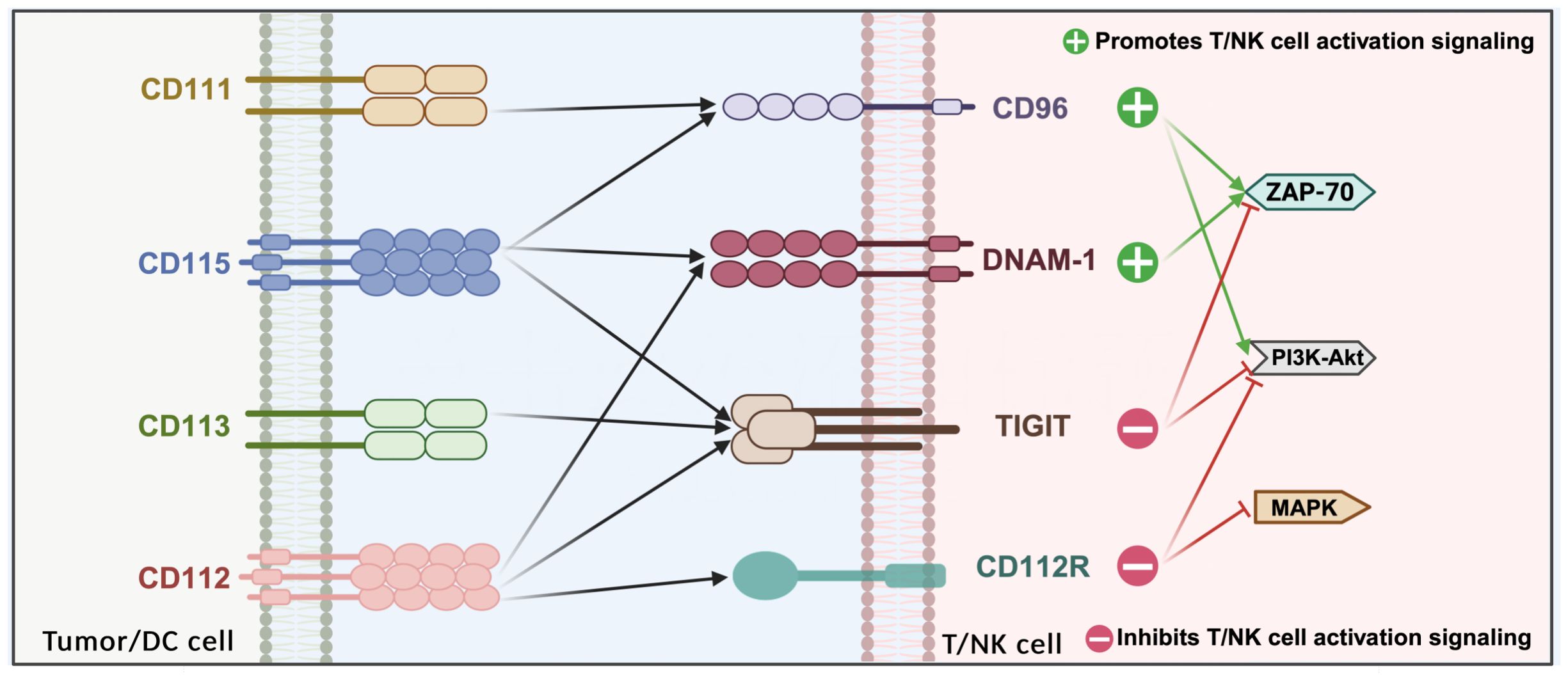
Figure 4. Interactions between the various members of the PVR family.
TIGIT, CD112R, and CD155 transmit inhibitory signals to cells through their cytoplasmic tails, whereas DNAM-1 continues to transmit activation signals. The crystal structure of TIGIT bound to CD155 reveals that two TIGIT/CD155 dimers assemble into a heterotetramer with a core TIGIT/TIGIT cis homodimer, where each TIGIT molecule binds to a CD155 molecule. This cis-trans receptor aggregation mediates cell adhesion and signal transduction (171). TIGIT effectively inhibits both innate and adaptive immunity through various mechanisms. Antibodies that competitively bind to TIGIT can directly inhibit T cell proliferation and function by attenuating TCR-driven activation signals. Moreover, TIGIT binding induces the phosphorylation of CD155 in DCs, triggering a signaling cascade that reduces the expression of interleukin-12 and interleukin-10 in tolerogenic DCs, thereby indirectly impairing T cell function. Concurrently, TIGIT inhibits NK cell degranulation, cytokine production, and the cytotoxicity of NK cells against tumor cells expressing CD155. By competing with CD155 with high affinity, TIGIT hinders CD155-mediated activation of CD226. In CD226-deficient mouse models, CD8+ T cells and NK cells exhibit defects in immune synapse formation, which impairs their anti-tumor immune functions (172, 173).
TIGIT also presents a safety advantage in therapeutic applications. The interaction of TIGIT on Tregs disrupts cytokine balance, inhibits Th1 or Th17 phenotypes, and induces Th2 phenotypes. However, unlike CTLA-4 and PD-1, TIGIT knockout in mice does not result in a severe spontaneous autoimmune phenotype, suggesting that TIGIT moderates the immune response without triggering severe autoimmunity (174).
Currently, targeting the TIGIT-PVR pathway is gaining importance, with several biotechnology and pharmaceutical companies developing antibodies or dual antibodies against TIGIT that are at various stages of clinical development. Globally, major pharmaceutical companies such as Roche, Bristol-Myers Squibb, and MSD are leading the way, with Roche and MSD having made the most progress, both being in Phase III clinical trials. Meanwhile, additional immune checkpoint inhibitors have exhibited promising efficacy across a diverse spectrum of cancers, with ongoing research into novel checkpoint molecules and combination therapies advancing at a rapid pace. Nevertheless, challenges such as drug resistance and immune-related adverse effects remain significant barriers in the development process. Future studies are therefore expected to focus on refining drug efficacy and safety profiles to facilitate broader and more effective clinical applications (Table 4).
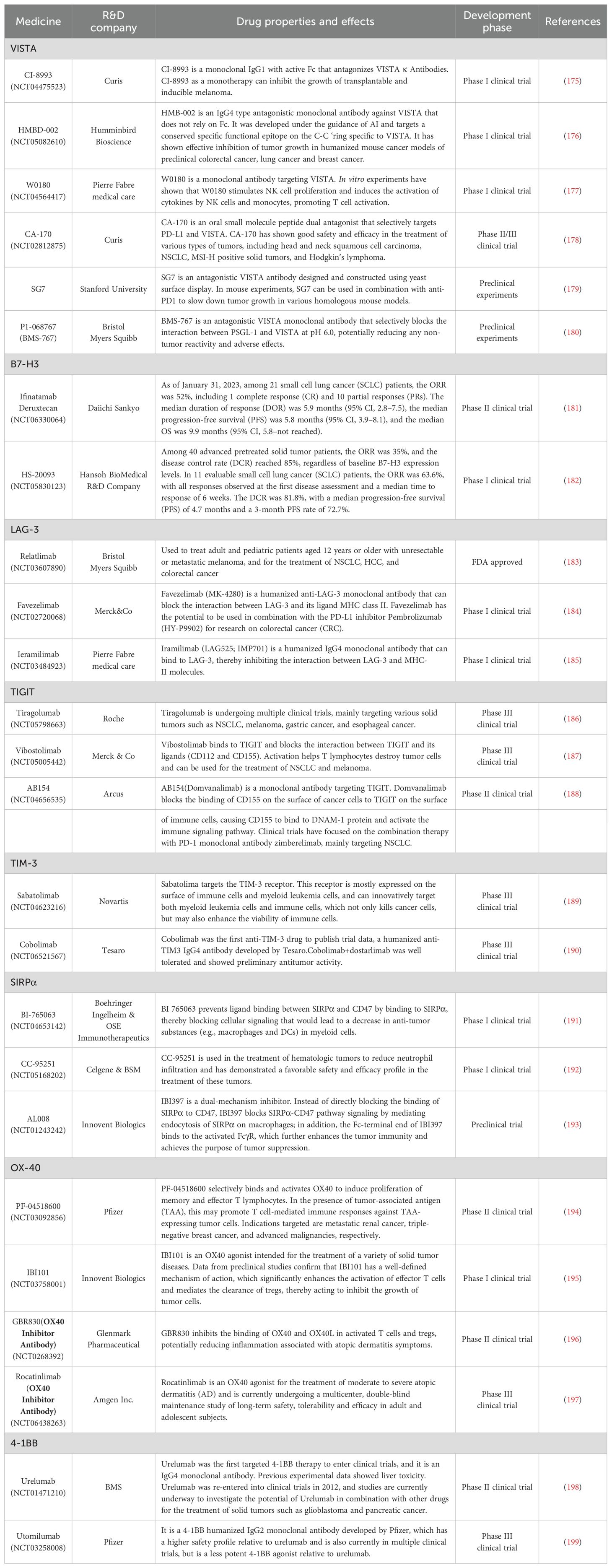
Table 4. Summary of immune checkpoint monoclonal antibody drugs and their pathways of action.
TIM family
In humans, the TIM family includes TIM1, TIM3, and TIM4, located on chromosome 5q33.2. In mice, the TIM family includes TIM1 to TIM8, located on chromosome 11B1.1. TIM proteins are a class of transmembrane glycoproteins characterized by a common motif. Their structure comprises five regions: signal peptide, immunoglobulin, mucin, transmembrane, and intracellular tail (200). Except for TIM-4, the intracellular regions of TIM-1, TIM-2, and TIM-3 contain tyrosine phosphorylation motifs that participate in transmembrane signal transduction.
The TIM (T cell/transmembrane, immunoglobulin, and mucin) gene family proteins first garnered attention in virology due to their phosphatidylserine (PtdSer) receptor epitopes, which play a crucial role in enhancing viral entry (201). Subsequently, substantial data has accumulated indicating that this gene family is pivotal in regulating immune responses, including transplant immune tolerance, autoimmunity, allergies, and asthma (202).
The TIM proteins may function as a novel receptor family for phosphatidylserine (PtdSer), binding to this key “Eat me” signaling molecule, mediating the phagocytic clearance of apoptotic cells, and playing a crucial role in regulating immune tolerance in vivo while maintaining internal homeostasis (203). The unique structure of the TIM immunoglobulin variable domain enables highly specific recognition of PtdSer exposed on the surface of apoptotic cells. The crystal structures of Tim-1, Tim-2, Tim-3, and Tim-4 in rodents reveal a characteristic FG-CC’ motif (204). While TIM-1, TIM-3, and TIM-4 can recognize PtdSer, their expression on different cells suggests distinct functions in immune regulation. Consequently, the TIM gene family is essential for immune response and tolerance. Research has demonstrated that the PS receptor TIM-4 regulates adaptive immune responses in vivo by mediating the antigen-specific clearance of apoptotic T cells (205).
TIM-1 is a significant susceptibility gene for asthma and allergy, preferentially expressed on T helper cell 2 (Th2) cells, and serves as an effective co-stimulatory molecule for T cell activation. TIM-3, expressed on the surface of Th1 cells, binds to its ligand galectin-9. Through the TIM-3-galectin-9 binding pathway, it generates inhibitory signals, induces Th1 cell death, and negatively regulates the Th1 immune response (206). It has been found that TIM-4 expressed by APCs is a ligand for TIM-1. In vivo injection of either soluble TIM-1 immunoglobulin (TIM-1-Ig) fusion proteins or TIM-4-Ig fusion proteins resulted in T-cell over proliferation, and TIM-4-Ig stimulated CD3- and CD28-mediated T-cell proliferation in vitro. These data suggest that TIM-1-TIM-4 interaction is involved in the regulation of T cell proliferation (207).
TIM-1
The T cell immunoglobulin and mucin (TIM) family plays a critical role in regulating T cell-mediated immune responses. Among its members, TIM-1 is notably involved in modulating Th1/Th2 cell differentiation (208). The TIM-1 gene, identified on mouse chromosome 11, has been shown to confer protection against Th2-mediated airway hyperresponsiveness, making it a valuable focus of asthma research. Beyond its association with airway hyperresponsiveness, TIM-1 is predominantly expressed by Th2 cells, further underscoring its significance in Th2-driven immune processes (209). Additionally, TIM-1 signaling was found to influence antibody production both in vitro and in vivo, with higher levels of IgG2b and IgG3 detected in the culture supernatants of anti-TIM-1-stimulated B cells. When immunized with the T-independent antigen TNP-Ficoll, TNP-specific IgG1, IgG2b, and IgG3 antibodies were slightly increased in anti-TIM-1-treated mice (210).
In 2023, a team from Harvard Medical School identified TIM-1 as a critical immune checkpoint in B cells and investigated strategies to bypass this checkpoint to enhance the anti-tumor potential of T cells. Targeting TIM-1 to inhibit B cells can amplify anti-tumor CD8+ and CD4+ T-cell responses and suppress tumor growth. This study identifies TIM-1 as a pivotal immune checkpoint for B-cell activation. TIM-1 modulates the type 1 interferon (IFN-1) response in B cells, thereby limiting B-cell activation, antigen presentation, and co-stimulation, which underscores TIM-1 as a potential target for enhancing B-cell-mediated anti-tumor immunity (35).Given that TIM-4 is a homologous ligand of TIM-1, it is insightful to consider the role of TIM-1 in promoting T-cell expansion and survival via its interaction with TIM-4, suggesting that the TIM-1 pathway serves as a natural stimulator of T-cell function (211).
TIM-3
T-cell immunoglobulin mucin 3 (TIM-3), also known as HAVCR2, is a critical tumor immune checkpoint that was first identified in 2002,TIM-3 functions as a negatively regulated immune checkpoint. The TIM-3 gene is located on chromosome 5q33.2, encodes a protein comprising 281 amino acids, and consists of an extracellular region, a single transmembrane structural domain, and a C-terminal cytoplasmic tail (212). TIM-3 is a class of inhibitory molecules found on the surface of T cells, which contribute to T-cell exhaustion in the context of cancer and chronic viral infections. Similar to PD-1 and CTLA-4, TIM-3 is one of the most extensively studied targets for immunotherapy. It has been observed that patients treated with anti-PD-1 or anti-PD-L1 monoclonal antibodies often develop resistance, and TIM-3 expression is upregulated in response to adaptive resistance to anti-PD-1 therapy (213).
TIM-3 is selectively expressed on IFN-γ-secreting helper T cells (Th1 and Th17), Tregs, mast cells, DCs, NK cells, tumor-infiltrating lymphocytes (TILs), monocytes, as well as on tumor cells such as melanoma, gastric cancer, and B-cell lymphoma (214).
The mechanism by which TIM-3 functions as a crucial immune checkpoint is primarily due to its identification of the most dysfunctional subpopulation of tumor-infiltrating CD8+ PD-1+ T cells (215). Antibodies that simultaneously block the TIM-3 and PD-1 pathways exhibit a synergistic effect, enhancing tumor growth inhibition and improving the response of tumor antigen-specific CD8+ T cells (216).
Transcriptomic analysis revealed a significant enrichment of the PI3K-AKT and MAPK signaling pathways in TIM-3 knockout (KO) tumor cells compared to TIM-3+ tumor cells. Furthermore, evaluation of an anti-TIM-3 monoclonal antibody demonstrated its efficacy in significantly prolonging the survival of DIPG mice (217).This chromosomal region has been consistently associated with asthma, allergies, and autoimmune diseases. TIM proteins are a class of transmembrane glycoproteins characterized by a common motif, with a structure comprising five regions: a signal peptide, an immunoglobulin region, a mucin region, a transmembrane region, and an intracellular tail (Figure 5).
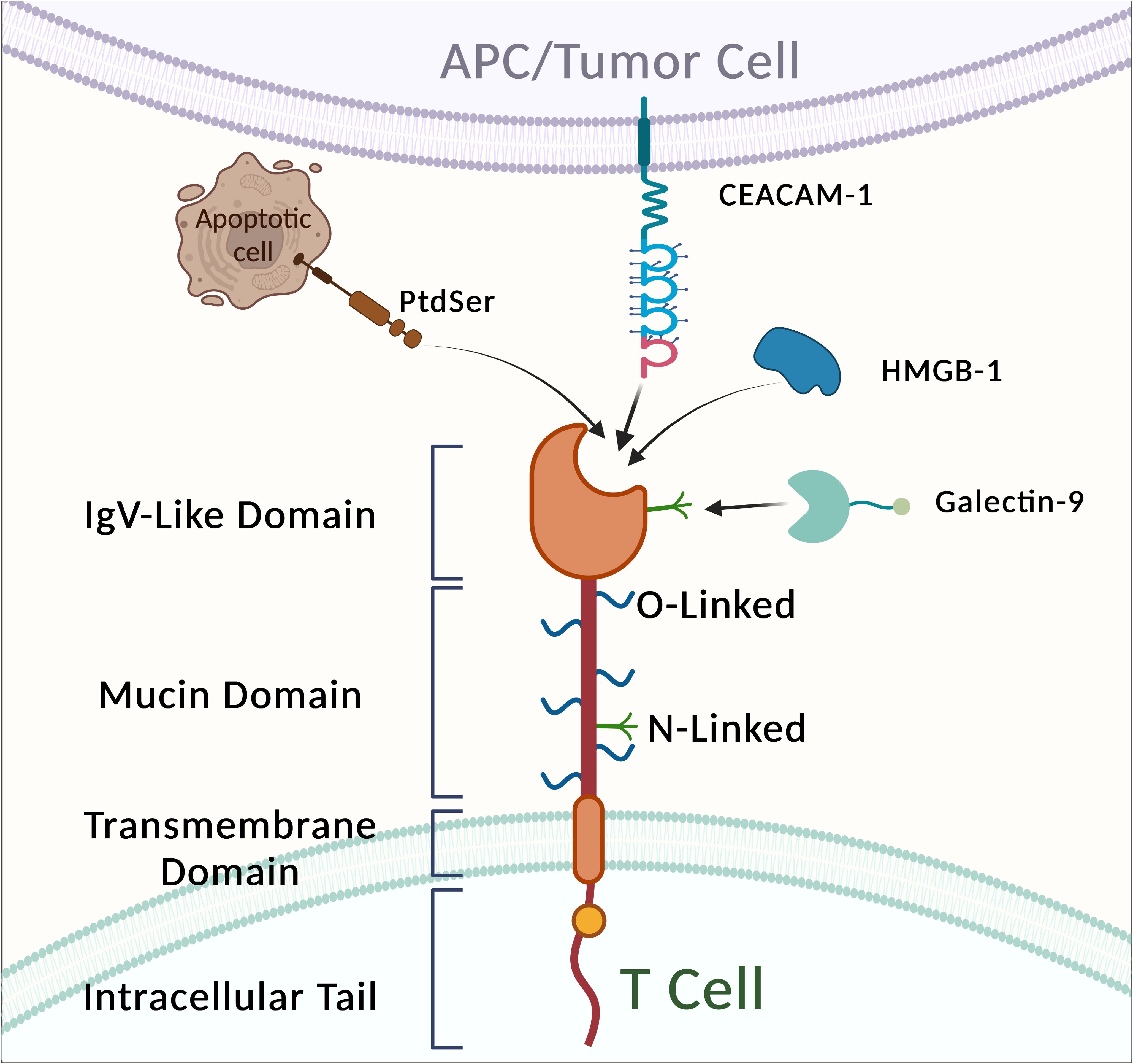
Figure 5. Structure and ligands of TIM-3.
While TIM-3-expressing fibroblasts and APCs are involved in the phagocytosis of apoptotic cells, TIM-3-expressing T cells bind to but do not phagocytose these cells. These observations suggest that TIM-3-expressing DCs, macrophages, and T cells are capable of detecting apoptotic cells (218). TIM-3 has four known ligands: galectin-9 (Gal-9), carcinoembryonic antigen cell adhesion molecule-1 (CEACAM-1), high mobility group protein B1 (HMGB1), and phosphatidylserine (PS) (219). Gal-9, the first ligand identified, is a carbohydrate-binding protein that recognizes N-linked glycans in the TIM-3 IgV domain. The interaction between TIM-3 and Gal-9 inhibits tumor immunity by suppressing T-cell activity, effectively halting Th1 immune responses through binding to the TIM-3 IgV domain (220).
Recent findings indicate that elevated TIM-3 expression is observed on CD4+ and CD8+ T cells in the peripheral blood of patients with acute hepatitis B (AHB) and chronic hepatitis B (CHB) (221). Furthermore, an increase in TIM-3+ T cells correlates positively with conventional liver injury markers, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin (TB), and the international normalized ratio (INR). Conversely, TIM-3 expression is negatively correlated with T-bet mRNA expression and plasma interferon-gamma (IFN-γ) levels. These results suggest that TIM-3 overexpression is involved in CHB disease progression and may contribute to the skewed Th1/Tc1 response that leads to persistent HBV infection.
HCV(hepatitis C virus) evades host immune attack and apoptosis through various mechanisms, including the production of quasispecies, viral-specific and general immunosuppression, Tregs, and induction of PD-1/TIM-3-mediated exhaustion in effector T cells (Teff) (222). TIM-3 may play a significant role in the natural immune response by interacting with the negative regulators Programmed Death-1 (PD-1) and Suppressor of Cytokine Signaling-1 (SOCS-1) (223). This interaction inhibits STAT-1 phosphorylation and negatively regulates the production of interleukin-12 (IL-12), suggesting that TIM-3 may serve as a crucial target for HCV treatment (224).
TIM-3 is among the most extensively researched targets in immunotherapy. However, no TIM-3-targeted drugs are currently approved or marketed globally. Novartis and GSK are advancing TIM-3 inhibitors through Phase III clinical trials, while Roche and Bajaj Shenzhou are conducting Phase II trials. In China, Hengrui and Zhikang Hongyi are in Phase I clinical trials. Additionally, Fuhong Hanklin, Vannes, Zhao Derivatives, Zhiren Meibao, and Lizumab are at the preclinical stage. TIM-3 remains a prominent focus in immunotherapy research, with no TIM-3-targeted drugs yet listed. Novartis and GSK are in Phase III trials, Roche and Bajaj Shenzhou are in Phase II, and AZD7789, a key PD-1/TIM-3 bispecific monoclonal antibody developed by AstraZeneca, is set to enter clinical trials in the U.S. in 2021. This antibody targets advanced solid tumors and hematological malignancies. This led us to summarize multiple immunotherapeutic agents with immune checkpoints that have similar bidirectional specificity to AZD7789 (Table 5).
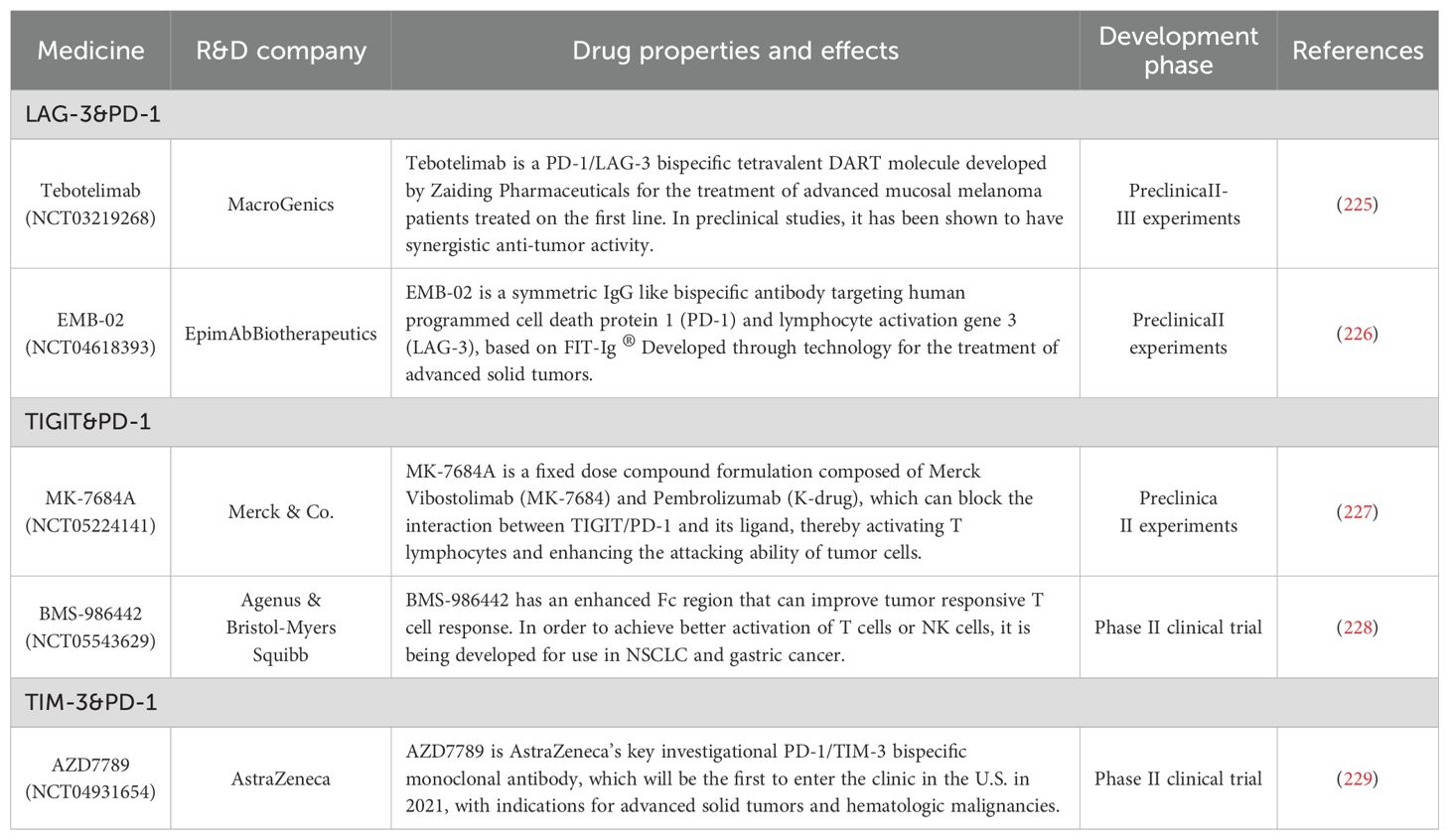
Table 5. Summary of immune checkpoint bispecific antibody drugs and their pathways of action.
SIRP family
Signal regulatory proteins (SIRPs) are a family of cell surface signaling receptors, consisting of five members: SIRPα, SIRPβ1, SIRPγ, SIRPβ2, and SIRPδ (230). These receptors are differentially expressed in leukocytes and the central nervous system, with predominant expression on the surface of myeloid cells, such as monocytes, macrophages, granulocytes, and myeloid DCs in humans (230). SIRPs are also expressed in certain cancer cells and neuronal cells of the nervous system, of all the members. SIRPα is notable for being the immune checkpoint protein with the strongest binding affinity to CD47 (231).
Structurally, SIRPs belong to the IgSF, characterized by an N-terminal extracellular domain containing three cysteine-binding Ig-like loops, a single transmembrane domain, and a C-terminal intracellular domain (232). The C-terminal intracellular domain of the SIRPα subfamily contains a relatively long amino acid sequence (110 amino acids in SIRPα) that includes four tyrosine residues, which form two immunoreceptor tyrosine-based inhibitory motifs (ITIMs) (233).
SIRPa
SIRPα (also known as PTPNS1, SHPS-1, CD172a, and P84) is known for binding to CD47. Signal regulatory protein alpha (SIRPα) is a transmembrane protein whose extracellular region consists of three Ig-like structural domains and a cytoplasmic region containing immunoreceptor tyrosine-based inhibitory motifs (ITIMs) that mediate binding of the protein tyrosine phosphatases SHP-1 and SHP-2 (234). SIRPα is particularly abundant in myeloid cells such as macrophages and DCs (235), with lower expression levels in T cells, B cells, NK cells, and NKT cells. Polymorphic allelic variants in the ligand-binding domain have been reported in African, Japanese, Chinese, and Caucasian populations, with three of them (SIRPαV1, SIRPαV2, and SIRPαV8) being the most prominent haplotypes, covering about 90% of the population (236).
SIRPα inhibits macrophage phagocytosis by interacting with its ligand, CD47, a key immunosuppressive signaling molecule involved in the immune escape of tumor cells. CD47 is typically upregulated on the surface of malignant cells, sending a “don’t-eat-me” signal to immune cells, helping to maintain immune tolerance in non-malignant cells under physiological conditions (237). However, this mechanism can also enable cancer cells to survive in various types of cancer. In many cancer types, CD47, which binds to signal-regulatory protein alpha (SIRPα), initiates inhibitory signaling pathways that prevent malignant cells from being phagocytosed by macrophages (238).
A 2022 study from Texas MD Anderson Cancer Center highlighted the dual role of SIRPα in cancer treatment. Analysis of 60 immuno-oncology genes in melanoma patients revealed that higher SIRPα expression in tumor cells correlated with better responses to anti-PD-1 therapy and improved patient outcomes, contrasting with its traditional immunosuppressive role in macrophages. Single-cell proteomics confirmed that elevated SIRPα expression originated from melanoma cells rather than macrophages and enhanced T cell-mediated tumor killing. These findings suggest tumor cell-expressed SIRPα enhances sensitivity to immunotherapy, while macrophage-expressed SIRPα maintains its inhibitory role. Additionally, SIRPα-targeting antibodies show promise as safer immunotherapy agents, requiring low doses to block CD47-SIRPα interactions without significant hematological side effects (239).
The above studies have shown that the same target in different cell types can have different effects on immunotherapy, thus positioning SIRPα as a promising target with dual immune effects (Figure 6).

Figure 6. Immune responses regulated by SIRPα expression on different cells.
SIRPα-targeting antibodies are considered safer because SIRPα is primarily expressed on myeloid cells. A small dose of SIRPα antibody is sufficient to block the CD47-SIRPα pathway in tumor cells without leading to erythrocyte destruction or other hematological adverse effects. This distinction makes SIRPα-targeting antibodies a potentially safer alternative in cancer immunotherapy (240).
TNFSF family
The Tumor Necrosis Factor Superfamily (TNFSF) consists of proteins that share TNF homology domains at the C-terminus and form a trimeric structure (241). TNFSF ligands can bind to members of the Tumor Necrosis Factor Receptor Superfamily (TNFRSF), thereby regulating a variety of cellular processes, including immune responses, inflammation, and cell proliferation, differentiation, and apoptosis (242). The TNFSF/TNFRSF system includes 19 ligands and 29 receptors, with some ligands capable of binding to multiple receptors and some receptors interacting with more than one ligand. This ligand-receptor sharing creates an extensive communication network that facilitates the regulation of complex cellular responses (243).
When TNFRSF binds to its ligands, the resulting interaction can regulate cell survival and function through activation of the NF-κB or MAPK pathways via TNFR-associated factors (TRAFs) (244). Conversely, binding of TNFRSF to ligands containing death domains can ultimately lead to the activation of caspases and programmed cell death (245). Another subgroup within TNFRSF, such as CD137, glucocorticoid-induced TNF receptor (GITR), and OX40, activate NF-κB, promoting cell survival (246).
In a study on rheumatoid arthritis by Michael Croft and colleagues, interactions between TNFSF ligands and TNFRSF receptors were observed among APCs, B cells, and T cells of the immune system (247).
Upon antigen stimulation, T cells receive signals through TNFRSF members such as OX40, GITR, DR3, CD27, and 4-1BB, which promote follicular helper T (TFH) cell differentiation, regulating antibody responses and cytokine expression linked to histopathology. APCs, DCs and macrophages, enhance T cell responses by upregulating MHC molecules, co-stimulatory ligands, and inflammatory cytokines via CD40 signaling. Additionally, reverse signaling through membrane-bound TNFSF ligands on DCs, macrophages, and B cells enhances inflammatory cytokine production and supports B cell differentiation (247).
OX40
OX40, also known as TNFRSF4 (tumor necrosis factor receptor superfamily, member 4), is predominantly expressed on the surface of activated CD4+ and CD8+ T cells. Binding of OX40 to its ligand, OX40L, stimulates the activation of CD8+ T cells and enhances various T cell functions, including cytokine production, proliferation, and survival. OX40 antibody activators (agonists) have been shown to reduce intratumoral Tregs and improve anti-tumor activity. Structurally, OX40 is a type 1 transmembrane glycoprotein, primarily expressed by tregs and, upon activation, also expressed by effector T cells (248).
OX40L, the ligand for OX40, was initially identified on HTLV-1-transformed T cells and is also known as pg34. It is predominantly expressed on APCs but can also be found on NK cells, mast cells, and activated T cells. The interaction between OX40 and OX40L facilitates the migration of activated T cells into tissues in response to inflammatory signals.
The OX40/OX40L interaction recruits TNFR-associated factors (TRAFs) within the intracellular region of OX40, forming a signaling complex that includes IKKα, IKKβ, PI3K, and PKB (Akt) (249). OX40 synergizes with TCR signaling, enhancing NFAT entry into the nucleus by increasing intracellular Ca2+ levels (250). OX40 signaling activates both the classical NF-κB1 pathway and the non-classical PI3K/PKB, NFAT pathway, and NF-κB2 pathway (251). This regulation controls genes involved in T-cell division and survival, promotes cytokine gene transcription, and increases cytokine receptor expression, which is crucial for cell survival (252). Additionally, OX40 signaling leads to the downregulation of CTLA-4 and Foxp3 and induces the expression of anti-apoptotic proteins (Bcl-2, Bcl-xL, and Bfl-1) and cell cycle progression proteins (Survivin) (253).IL-33, released by barrier-disrupted epidermal keratinocytes, stimulates type 2 innate lymphoid cells (ILC2s) and DCs to express OX40L. Moreover, the OX40-OX40L signaling pathway also plays a role in regulating IL-22 production in T cells (197).
Studies have analyzed tumor tissues from mouse models of B-cell lymphomas and human cases of condylomatous and follicular lymphomas, revealing high expression of OX40 and CTLA-4 on the surface of tumor-specific Tregs (CD4+, Foxp3+) (254). OX40 has emerged as a specific biomarker in various cancers. For example, high expression of OX40 in primary ovarian immune cells and recurrent tumor cells is associated with increased chemotherapy sensitivity, while patients lacking OX40 expression are more prone to relapse (255). In patients with cutaneous melanoma, OX40 expression in T cells from sentinel lymph nodes negatively correlates with poor prognostic features such as tumor size, ulceration, and lymph node involvement (256).
Given its role in enhancing the immune response to tumors, several therapeutic strategies have been developed to stimulate the OX40 signaling pathway. These include OX40-specific agonistic antibodies, OX40L-Fc fusion proteins, transfection of DCs with OX40L mRNA, and the use of surface-engineered OX40L-expressing tumor cells (257).
4-1BB
4-1BB (CD137) is a co-stimulatory immune checkpoint molecule belonging to the TNF receptor superfamily (TNFRSF) and plays a crucial role in regulating the immune response. The CD137 gene, located on chromosome 1p36, is situated near other co-stimulatory TNFRSF members (258). Identified in 1989, 4-1BB is expressed on antigen-activated T cells but not on resting T cells (259). It is also found on DCs, NKs, activated CD4+ and CD8+ T lymphocytes, eosinophils, natural killer T-cells (NKTs), and mast cells (260) though myeloid-derived suppressor cells (MDSCs) do not express this molecule. Additionally, 4-1BB is present on various tumor cells, including human leukemia cells and several lung tumor cell lines. Its ligand, 4-1BBL, is expressed on some APCs such as B lymphocytes, macrophages, DCs, and activated T cells (261). Anti-4-1BB antibodies have shown the ability to activate cytotoxic T cells and enhance γ-interferon (IFN-γ) production. Both dual and multi-specific antibodies targeting 4-1BB are demonstrating significant potential in cancer therapy (40).
4-1BB recruits TNFR-associated factors TRAF1 and TRAF2, forming a heterotrimeric complex that activates the c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) pathways, while also enhancing signaling through the β-catenin and AKT pathways. Additionally, 4-1BB signaling is regulated by the master transcription factor NF-κB, which promotes cytokine production and secretion. NF-κB activation further enhances CD8+ T lymphocyte survival by upregulating the expression of anti-apoptotic genes Bcl-xL and Bfl-1 (262).
Dual and multi-specific antibodies targeting 4-1BB have shown significant potential in cancer therapy. The human-derived 4-1BB is a type I transmembrane receptor characterized by four extracellular cysteine-rich domains, a short transmembrane domain, and a C-terminal cytoplasmic domain essential for binding adaptor proteins and facilitating signaling. Its ligand, 4-1BBL, is a type II transmembrane protein presented in a soluble form. It consists of a short N-terminal cytoplasmic region, a transmembrane domain, and an extracellular domain that binds 4-1BB (263). The 4-1BB monomer is elongated, with four cysteine-rich domains arranged linearly. Binding of 4-1BBL to 4-1BB induces signaling through TRAF1 and TRAF2, activating the NF-κB, AKT, p38 MAPK, and ERK pathways (264).
CD137 and/or CD137L agonists stimulate the production of several inflammatory cytokines, such as IL-6, TNF-α, and MCP-1, in adipocytes and macrophages (265). Cross-linking CD137 on B cells enhances immune signaling and induces B cell proliferation (266).
Depletion of DCs in vivo significantly diminishes the level of cytotoxic T lymphocyte (CTL) stimulation, thereby impairing the overall efficacy of 4-1BB antibodies. These antibodies activate various immune cells through 4-1BB signaling, modulating T cell activity, inducing cytokine production, and preventing activation-induced cell death (AICD), ultimately enhancing CTL activity. 4-1BB is considered a highly promising target in immuno-oncology and remains one of the most attractive T-cell co-stimulatory receptors within the TNF receptor superfamily (TNFRSF). Phase I trials for next-generation 4-1BB targeting agents are currently focusing on mitigating hepatotoxicity while maintaining therapeutic efficacy (267).
Outstanding questions and concluding remarks
In summary, the co-inhibitory and co-stimulatory pathways of immune checkpoint proteins are crucial for maintaining immune homeostasis, preventing infections, and avoiding autoimmunity. These pathways regulate not only the activation of naïve T cells but also the immune responses of memory cells and Tregs. Although significant progress has been made in understanding the immunoregulatory roles of these pathways, challenges remain, such as adverse effects associated with immune checkpoint inhibition during antibody drug development, including hepatotoxicity observed with 4-1BB agonists (198).
Currently, combination therapies targeting immune checkpoints have been widely adopted for treating various diseases. Additionally, it has been observed that immune checkpoint expression can be modulated by the tumor microenvironment—for instance, pH levels influence VISTA expression (99). While previous research has largely concentrated on T-cell responses, emerging data on TIM-1’s stimulatory effects on B cells offer new biological insights and strategies (210). This evolving knowledge enhances our understanding of the efficacy of current immunotherapies and opens avenues for developing novel therapeutic approaches. The FDA’s approval of CTLA-4, PD-1, and PD-L1 antibodies underscores the therapeutic potential of a deeper understanding of co-inhibitory pathways, with agonistic antibodies for autoimmune diseases showing promise. Continued research will refine our grasp of these pathways in health and disease, leading to more effective and safe treatments for various immune-mediated conditions.
Immune checkpoint combination therapy represents a pivotal advancement in tumor immunotherapy, offering significant clinical potential. By simultaneously targeting multiple immune checkpoints, such as PD-1/PD-L1 and CTLA-4, this approach overcomes the limitations of single-target therapies and amplifies anti-tumor immune responses. For instance, PD-1/PD-L1 inhibitors restore effector TCR functionality, while CTLA-4 inhibitors promote the activation of naïve T cells (46, 47). The synergistic effects of these pathways have demonstrated substantial improvements in therapeutic efficacy. Combination therapies have achieved high ORRs and durable efficacy in solid tumors, such as melanoma and NSCLC, leading to significant improvements in long-term OS (88, 89).
Furthermore, combining emerging immune checkpoint molecules, such as LAG-3, TIGIT, and TIM-3, with classical checkpoint inhibitors has opened new avenues for immunotherapy. For example, the combination of LAG-3 and PD-1 inhibition has shown notable efficacy across various tumor models (225, 226). Similarly, strategies targeting TIGIT in combination with PD-L1 inhibitors have demonstrated promising potential in both solid tumors and hematologic malignancies (227, 228).
The primary advantage of immune checkpoint combination therapies lies in their ability to enhance therapeutic efficacy through multi-targeted interventions while addressing the resistance often encountered in monotherapies. However, this approach also presents challenges, including increased toxicity and the complexity of designing individualized treatment regimens for patients. Future research will prioritize optimizing combination strategies, selecting precise checkpoint combinations, and integrating biomarkers to predict treatment responses and patient outcomes.
In conclusion, immune checkpoint combination therapy is a transformative innovation in tumor immunotherapy. It not only provides novel therapeutic options for various malignancies but also lays a solid foundation for the development of precision medicine. This approach highlights its vast potential in advancing anti-tumor therapy and improving patient outcomes.
Checkpoint-blocking immunotherapies have demonstrated efficacy across a broad range of cancers and have significantly impacted clinical practice in oncology. Among the next-generation immune checkpoint targets—such as LAG-3, the Ig domain-containing VISTA, TIM-3, TIGIT, B7-H3, and SIRPα—each shows promising therapeutic potential, though it remains uncertain which will become the next major breakthrough like PD-1.
Author contributions
RL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. XJ: Conceptualization, Data curation, Funding acquisition, Methodology, Supervision, Writing – review & editing. YZ: Conceptualization, Data curation, Funding acquisition, Writing – review & editing. RD: Formal Analysis, Funding acquisition, Project administration, Resources, Validation, Visualization, Writing – review & editing. CG: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. PW: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the National Natural Science Foundation of China (Grant No. 31800652 and 82104644), the Key Project of Beijing University of Chinese Medicine: Leader Selection and Appointment (2024-JYB-JBZD-063), the National Administration of Traditional Chinese Medicine High-level Key Discipline construction Project (Grant No. zyyzdxk-2023262), and the QiHuang YingCai Program of Beijing University of Chinese Medicine (Grant No. BUCM-2023KYBJRC-008).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
IgSF, Immunoglobulin superfamily; APCs, Antigen presenting cells; TCR, T cell receptor; DCs, Dendritic cells; ADCC, Antibody-dependent cell-mediated cytotoxicity; ADCP, Antibody-dependent cell-mediated phagocytosis; ORR, Objective response rate; OS, Overall survival; PFS, Progression-free survival; Tregs, Regulatory T cells; PVR, Poliovirus receptor; NKs, Natural killer cells; ITT, Immunoglobulin tyrosine tail; NSCLC, non-small cell lung cancer; IgV, Immunoglobulin variable region; NTCs, Non-targeted controls.
References
1. Bray F, Laversanne M, Weiderpass E, Soerjomataram I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer. (2021) 127:3029–30. doi: 10.1002/cncr.33587
2. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74(3):229–63. doi: 10.3322/caac.21834
3. Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. (2016) 375(19):1845–55. doi: 10.1056/NEJMoa1611299
4. Dagher OK, Schwab RD, Brookens SK, Posey AD Jr. Advances in cancer immunotherapies. Cell. (2023) 186:1814–1814.e1. doi: 10.1016/j.cell.2023.02.039
5. Peterson C, Denlinger N, Yang Y. Recent advances and challenges in cancer immunotherapy. Cancers (Basel). (2022) 14:3972. doi: 10.3390/cancers14163972
6. Xu L, Tang L, Zhang L. Proteoglycans as miscommunication biomarkers for cancer diagnosis. Prog Mol Biol Transl Sci. (2019) 162:59–92. doi: 10.1016/bs.pmbts.2018.12.003
7. Assia H, Carlotta A, Chiara C, Lebbé C, Ferraresi V, Smylie M, et al. Light on life: immunoscore immune-checkpoint, a predictor of immunotherapy response. Oncoimmunology. (2023) 12(1):2243169–2243169. doi: 10.1080/2162402X.2023.2243169
8. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
9. Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. (2006) 90:51–81. doi: 10.1016/S0065-2776(06)90002-9
10. Galassi C, Chan TA, Vitale I, Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. (2024) 42:1825–63. doi: 10.1016/j.ccell.2024.09.010
11. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. (2015) 125:3335–7. doi: 10.1172/JCI83871
12. George JT, Levine H. Implications of tumor-immune coevolution on cancer evasion and optimized immunotherapy. Trends Cancer. (2021) 7:373–83. doi: 10.1016/j.trecan.2020.12.005
13. Mitra A, Thompson B, Strange A, Amato CM, Vassallo M, Dolgalev I, et al. A population of tumor-infiltrating CD4+ T cells co-expressing CD38 and CD39 is associated with checkpoint inhibitor resistance. Clin Cancer Res. (2023) 29(20):4242–55. doi: 10.1158/1078-0432.CCR-23-0653
14. Huene AL, Sanders SM, Ma Z, Nguyen AD, Koren S, Michaca MH, et al. A family of unusual immunoglobulin superfamily genes in an invertebrate histocompatibility complex. Proc Natl Acad Sci U S A. (2022) 119(40):e2207374119. doi: 10.1073/pnas.2207374119
15. Srinivasan M, Roeske RW. Immunomodulatory peptides from IgSF proteins: a review. Curr Protein Pept Sci. (2005) 6:185–96. doi: 10.2174/1389203053545426
16. Halaby DM, Mornon JP. The immunoglobulin superfamily: an insight on its tissular, species, and functional diversity. J Mol Evol. (1998) 46:389–400. doi: 10.1007/PL00006318
17. Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration (published correction appears in Nat Neurosci. 2007 Feb;10(2):263). Nat Neurosci. (2007) 10:19–26. doi: 10.1038/nn1827
18. Le Mercier I, Lines JL, Noelle RJ. Beyond CTLA-4 and PD-1, the generation Z of negative checkpoint regulators. Front Immunol. (2015) 6:418. doi: 10.3389/fimmu.2015.00418
19. Barclay AN. Membrane proteins with immunoglobulin-like domains–a master superfamily of interaction molecules. Semin Immunol. (2003) 15:215–23. doi: 10.1016/S1044-5323(03)00047-2
20. Berisio R, Ciccarelli L, Squeglia F, De Simone A, Vitagliano L. Structural and dynamic properties of incomplete immunoglobulin-like fold domains. Protein Pept Lett. (2012) 19:1045–53. doi: 10.2174/092986612802762732
21. Wang JH. The sequence signature of an Ig-fold. Protein Cell. (2013) 4:569–72. doi: 10.1007/s13238-013-3903-2
22. Oreste U, Ametrano A, Coscia MR. On origin and evolution of the antibody molecule. Biol (Basel). (2021) 10:140. doi: 10.3390/biology10020140
23. Yum JI, Hong YK. Terminating Cancer by Blocking VISTA as a Novel Immunotherapy: Hasta la vista, baby. Front Oncol. (2021) 11:658488. doi: 10.3389/fonc.2021.658488
24. Bharati K. Human genetic polymorphism and Leishmaniasis. Infect Genet Evol. (2022) 98:105203. doi: 10.1016/j.meegid.2021.105203
25. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
26. Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer. (2018) 4:418–28. doi: 10.1016/j.trecan.2018.04.001
27. Zheng M, Zhang Z, Yu L, Wang Z, Dong Y, Tong A, et al. Immune-checkpoint protein VISTA in allergic, autoimmune disease and transplant rejection. Front Immunol. (2023) 14:1194421. doi: 10.3389/fimmu.2023.1194421
28. Zhang J, Zhang H, Wang Z, Yang H, Chen H, Cheng H, et al. BTLA suppress acute rejection via regulating TCR downstream signals and cytokines production in kidney transplantation and prolonged allografts survival. Sci Rep. (2019) 9:12154. doi: 10.1038/s41598-022-24559-x
29. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in tregs. Immunity. (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
30. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. (2020) 8:e000957. doi: 10.1136/jitc-2020-000957
31. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. (2019) 70(1):168–83. doi: 10.1002/hep.30347
32. Molfetta R, Zitti B, Lecce M, Milito ND, Stabile H, Fionda C, et al. CD155: A multi-functional molecule in tumor progression. Int J Mol Sci. (2020) 21(3):922. doi: 10.3390/ijms21030922
33. Zeng T, Cao Y, Jin T, Tian Y, Dai C, Xu F. The CD112R/CD112 axis: a breakthrough in cancer immunotherapy. J Exp Clin Cancer Res. (2021) 40:285. doi: 10.1186/s13046-021-02053-y
34. Shibuya K, Shirakawa J, Kameyama T, Honda S, Tahara-Hanaoka S, Miyamoto A, et al. CD226 (DNAM-1) is involved in lymphocyte function-associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J Exp Med. (2003) 198(12):1829–39. doi: 10.1084/jem.20030958
35. Bod L, Kye YC, Shi J, Torlai Triglia E, Schnell A, Fessler J, et al. B-cell-specific checkpoint molecules that regulate anti-tumor immunity. Nature. (2023) 619(7969):348–56. doi: 10.1038/s41586-023-06231-0
36. Kandel S, Adhikary P, Li G, Cheng K. The TIM3/Gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett. (2021) 510:67–78. doi: 10.1016/j.canlet.2021.04.011
37. Min C, Park J, Kim G, Moon H, Lee SA, Kim D, et al. Tim-4 functions as a scavenger receptor for phagocytosis of exogenous particles. Cell Death Dis. (2020) 11(7):561. doi: 10.1038/s41419-020-02773-7
38. Tian T, Wang M, Ma D. TNF-α, a good or bad factor in hematological diseases? Stem Cell Investig. (2014) 1:12. doi: 10.3978/j.issn.2306-9759.2014.04.02
39. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev. (2009) 229:173–91. doi: 10.1111/j.1600-065X.2009.00766.x
40. Bartkowiak T, Curran MA. 4-1BB agonists: multi-potent potentiators of tumor immunity. Front Oncol. (2015) 5:117. doi: 10.3389/fonc.2015.00117
41. Anand S, Wang P, Yoshimura K, Choi IH, Hilliard A, Chen YH, et al. Essential role of TNF family molecule LIGHT as a cytokine in the pathogenesis of hepatitis. J Clin Invest. (2006) 116(4):1045–51. doi: 10.1172/JCI27083
42. Wang J, Springer TA. Structural specializations of immunoglobulin superfamily members for adhesion to integrins and viruses. Immunol Rev. (1998) 163:197–215. doi: 10.1111/j.1600-065X.1998.tb01198.x
43. Müller WE, Blumbach B, Müller IM. Evolution of the innate and adaptive immune systems: relationships between potential immune molecules in the lowest metazoan phylum (Porifera) and those in vertebrates. Transplantation. (1999) 68:1215–27. doi: 10.1097/00007890-199911150-00001
44. MLA style: The Nobel Prize in Physiology or Medicine 1987. NobelPrize.org. Nobel Prize Outreach 2025 (2025). Available online at: https://www.nobelprize.org/prizes/medicine/1987/summary/.
45. Wartewig T, Ruland J. PD-1 tumor suppressor signaling in T cell lymphomas. Trends Immunol. (2019) 40:403–14. doi: 10.1016/j.it.2019.03.005
46. Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, et al. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure. (2015) 23(12):2341–8. doi: 10.1016/j.str.2015.09.010
47. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
48. Hui E, Cheung J, Zhu J, Fouser L, Carter L, Ling V, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
49. Ai L, Xu A, Xu J. Roles of PD-1/PD-L1 pathway: signaling, cancer, and beyond. Adv Exp Med Biol. (2020) 1248:33–59. doi: 10.1007/978-981-15-3266-5_3
50. Jiang M, Liu M, Liu G, Ma J, Zhang L, Wang S. Advances in the structural characterization of complexes of therapeutic antibodies with PD-1 or PD-L1. MAbs. (2023) 15:2236740. doi: 10.1080/19420862.2023.2236740
51. Zhang J, Guo Y, Fang H, Guo X, Zhao L. Oncolytic virus oHSV2 combined with PD-1/PD-L1 inhibitors exert antitumor activity by mediating CD4 + T and CD8 + T cell infiltration in the lymphoma tumor microenvironment. Autoimmunity. (2023) 56:2259126. doi: 10.1080/08916934.2023.2259126
52. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2(3):261–8. doi: 10.1038/85330
53. Guo PD, Sun ZW, Lai HJ, Yang J, Wu PP, Guo YD, et al. Clinicopathological analysis of PD-L2 expression in colorectal cancer. Onco Targets Ther. (2018) 11:7635–42. doi: 10.2147/OTT.S177329
54. Yang Y, Yan X, Bai X, Yang J, Song J. Programmed cell death-ligand 2: new insights in cancer. Front Immunol. (2024) 15:1359532. doi: 10.3389/fimmu.2024.1359532
55. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. (2001) 193(7):839–46. doi: 10.1084/jem.193.7.839
56. Philips EA, Garcia-España A, Tocheva AS, Ahearn IM, Adam KR, Pan R, et al. The structural features that distinguish PD-L2 from PD-L1 emerged in placental mammals. J Biol Chem. (2020) 295(14):4372–80. doi: 10.1074/jbc.AC119.011747
57. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
58. Zhang LN, Chen JY, Liu YX, Zhang Y, Hong LL, Li XX, et al. Identification of lncRNA dual targeting PD-L1 and PD-L2 as a novel prognostic predictor for gastric cancer. Frontiers in Oncology. (2024) 14:1341056. doi: 10.3389/fonc.2024.1341056
59. Gomar M, Najafi M, Aghili M, Cozzi S, Jahanbakhshi A. Durable complete response to pembrolizumab in microsatellite stable colorectal cancer. Daru. (2021) 29:501–6. doi: 10.1007/s40199-021-00404-w
60. Cybulska-Stopa B, Piejko K, Ostaszewski K, Dziura R, Galus Ł, Ziółkowska B, et al. Long-term clinical evidence of comparable efficacy and toxicity of nivolumab and pembrolizumab in advanced melanoma treatment. Melanoma Res. (2023) 33(3):208–17. doi: 10.1097/CMR.0000000000000885
61. Jones L, Rittberg R, Leung B, Shokoohi A, Pender A, Wong S, et al. Alternate pembrolizumab dosing interval in advanced NSCLC with PD-L1 TPS ≥ 50%: 3 weekly compared to 6 weekly dosing. Curr Oncol. (2022) 29(11):8686–92. doi: 10.3390/curroncol29110685
62. Al Hadidi SA, Lee HJ. Pembrolizumab for the treatment of hodgkin lymphoma. Expert Opin Biol Ther. (2020) 20:1275–82. doi: 10.1080/14712598.2020.1830056
63. Specenier P. Nivolumab in melanoma. Expert Rev Anticancer Ther. (2016) 16:1247–61. doi: 10.1080/14737140.2016.1249856
64. Cortinovis DL, Canova S, Abbate M, Colonese F, Bidoli P. Focus on nivolumab in NSCLC. Front Med (Lausanne). (2016) 3:67. doi: 10.3389/fmed.2016.00067
65. Li S, Manochakian R, Chen R, Patel J, Inampudi JV, Hiren KR, et al. Clinical outcomes of atezolizumab versus standard-of-care docetaxel with and without ramucirumab in patients with advanced non-small-cell lung cancer who received prior immunotherapy. Front Oncol. (2024) 14:1306311. doi: 10.3389/fonc.2024.1306311
66. Kwapisz D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol Immunother. (2021) 70:607–17. doi: 10.1007/s00262-020-02736-z
67. Bellmunt J, Hussain M, Gschwend JE, Albers P, Oudard S, Castellano D, et al. Adjuvant atezolizumab versus observation in muscle-invasive urothelial carcinoma (IMvigor010): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. (2021) 22(4):525–37. doi: 10.1016/S1470-2045(21)00004-8
68. Heymach JV, Harpole D, Mitsudomi T, Taube JM, Galffy G, Hochmair M, et al. Perioperative durvalumab for resectable non-small-cell lung cancer. N Engl J Med. (2023) 389(18):1672–84. doi: 10.1056/NEJMoa2304875
69. Powles T, van der Heijden MS, Castellano D, Galsky MD, Loriot Y, Petrylak DP, et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. (2021) 22:e5.
70. Shirley M. Avelumab: A review in metastatic merkel cell carcinoma. Target Oncol. (2018) 13:409–16. doi: 10.1007/s11523-018-0571-4
71. Aldin A, Besiroglu B, Adams A, Monsef I, Piechotta V, Tomlinson E, et al. First-line therapy for adults with advanced renal cell carcinoma: a systematic review and network meta-analysis. Cochrane Database Syst Rev. (2023) 5(5):CD013798. doi: 10.1002/14651858.CD013798.pub2
72. Teo MY, Rosenberg JE. Nivolumab for the treatment of urothelial cancers. Expert Rev Anticancer Ther. (2018) 18:215–21. doi: 10.1080/14737140.2018.1432357
73. Nisticò L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E, et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet. (1996) 5(7):1075–80. doi: 10.1093/hmg/5.7.1075
74. Harper K, Balzano C, Rouvier E, Mattéi MG, Luciani MF, Golstein P. CTLA-4 and CD28 activated lymphocyte molecules are closely related in both mouse and human as to sequence, message expression, gene structure, and chromosomal location. J Immunol. (1991) 147:1037–44.
75. Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL, et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. (1993) 366:76–9. doi: 10.1038/366076a0
76. Slavik JM, Hutchcroft JE, Bierer BE. CD28/CTLA-4 and CD80/CD86 families: signaling and function. Immunol Res. (1999) 19:1–24. doi: 10.1007/BF02786473
77. Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. (1996) 183:2533–40. doi: 10.1084/jem.183.6.2533
78. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U S A. (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
79. Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. (2015) 36:63–70. doi: 10.1016/j.it.2014.12.001
80. Gattinoni L, Ranganathan A, Surman DR, Palmer DC, Antony PA, Theoret MR, et al. CTLA-4 dysregulation of self/tumor-reactive CD8+ T-cell function is CD4+ T-cell dependent. Blood. (2006) 108(12):3818–23. doi: 10.1182/blood-2006-07-034066
81. Wolchok JD, Saenger Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation, Oncologist. (2008) 13:2–9. doi: 10.1634/theoncologist.13-S4-2
82. Weber J. Overcoming immunologic tolerance to melanoma: targeting CTLA-4 with ipilimumab (MDX-010), Oncologist. (2008) 13:16–25. doi: 10.1634/theoncologist.13-S4-16
83. Chretien KC, Swenson R, Yoon B, Julian R, Keenan J, Croffoot J, et al. Tell me your story: A pilot narrative medicine curriculum during the medicine clerkship. J Gen Intern Med. (2015) 30:1025–8. doi: 10.1007/s11606-015-3211-z
84. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
85. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
86. Zhang X, Wu T, Cai X, Dong J, Xia C, Zhou Y, et al. Neoadjuvant immunotherapy for MSI-H/dMMR locally advanced colorectal cancer: new strategies and unveiled opportunities. Front Immunol. (2022) 13:795972. doi: 10.3389/fimmu.2022.795972
87. Johnson ML, Cho BC, Luft A, Alatorre-Alexander J, Geater SL, Laktionov K, et al. Durvalumab with or without tremelimumab in combination with chemotherapy as first-line therapy for metastatic non-small-cell lung cancer: the phase III POSEIDON study. J Clin Oncol. (2023) 41(6):1213–27. doi: 10.1200/JCO.22.00975
88. Willsmore ZN, Coumbe BGT, Crescioli S, Reci S, Gupta A, Harris RJ, et al. Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: Treatment of melanoma and immune mechanisms of action. Eur J Immunol. (2021) 51(3):544–56. doi: 10.1002/eji.202048747
89. John T, Sakai H, Ikeda S, Cheng Y, Kasahara K, Sato Y, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in advanced NSCLC: a subanalysis of Asian patients in CheckMate 9LA. Int J Clin Oncol. (2022) 27(4):695–706. doi: 10.1007/s10147-022-02120-0
90. Baas P, Scherpereel A, Nowak AK, Fujimoto N, Peters S, Tsao AS, et al. First-line nivolumab plus ipilimumab in unresectable Malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. (2021) 397(10272):670. doi: 10.1016/S0140-6736(20)32714-8
91. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. (2018) 378(14):1277–90. doi: 10.1056/NEJMoa1712126
92. Kudo M. Durvalumab plus tremelimumab: A novel combination immunotherapy for unresectable hepatocellular carcinoma. Liver Cancer. (2022) 11:87–93. doi: 10.1159/000523702
93. Muller S, Victoria Lai W, Adusumilli PS, Desmeules P, Frosina D, Jungbluth A, et al. V-domain Ig-containing suppressor of T-cell activation (VISTA), a potentially targetable immune checkpoint molecule, is highly expressed in epithelioid Malignant pleural mesothelioma. Mod Pathol. (2020) 33(2):303–11. doi: 10.1038/s41379-019-0364-z
94. ElTanbouly MA, Schaafsma E, Noelle RJ, Lines JL. VISTA: Coming of age as a multi-lineage immune checkpoint. Clin Exp Immunol. (2020) 200:120–30. doi: 10.1111/cei.13415
95. Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory functions of VISTA. Immunol Rev. (2017) 276(1):66–79. doi: 10.1111/imr.2017.276.issue-1
96. Deng J, Li J, Sarde A, Lines JL, Lee YC, Qian DC, et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol Res. (2019) 7(7):1079–90. doi: 10.1158/2326-6066.CIR-18-0507
97. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. (2011) 208(3):577–92. doi: 10.1084/jem.20100619
98. Hong S, Yuan Q, Xia H, Zhu G, Feng Y, Wang Q, et al. Analysis of VISTA expression and function in renal cell carcinoma highlights VISTA as a potential target for immunotherapy. Protein Cell. (2019) 10(11):840–5. doi: 10.1007/s13238-019-0642-z
99. Yuan L, Tatineni J, Mahoney KM, Freeman GJ. VISTA: A mediator of quiescence and a promising target in cancer immunotherapy. Trends Immunol. (2021) 42:209–27. doi: 10.1016/j.it.2020.12.008
100. Luk SJ, Schoppmeyer R, Ijsselsteijn ME, Somarakis A, Acem I, Remst DFG, et al. VISTA expression on cancer-associated endothelium selectively prevents T-cell extravasation. Cancer Immunol Res. (2023) 11(11):1480–92. doi: 10.1158/2326-6066.CIR-22-0759
101. Díaz-García E, García-Sánchez A, Alfaro E, López-Fernández C, Mañas E, Cano-Pumarega I, et al. PSGL-1: a novel immune checkpoint driving T-cell dysfunction in obstructive sleep apnea. Front Immunol. (2023) 14:1277551. doi: 10.3389/fimmu.2023.1277551
102. Lines JL, Sempere LF, Broughton T, Wang L, Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. (2014) 2:510–7. doi: 10.1158/2326-6066.CIR-14-0072
103. Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature. (2019) 574:565–70. doi: 10.1038/s41586-019-1674-5
104. Xu W, Dong J, Zheng Y, Zhou J, Yuan Y, Ta HM, et al. Immune-checkpoint protein VISTA regulates antitumor immunity by controlling myeloid cell-mediated inflammation and immunosuppression. Cancer Immunol Res. (2019) 7:1497–510. doi: 10.1158/2326-6066.CIR-18-0489
105. Guldner IH, Wang Q, Yang L, Golomb SM, Zhao Z, Lopez JA, et al. CNS-native myeloid cells drive immune suppression in the brain metastatic niche through cxcl10. Cell. (2020) 183:1234–1248.e25. doi: 10.1016/j.cell.2020.09.064
106. Smith FD, Thisted T, Feng F, Kleschenko Y, Mukherjee A, Biesova Z, et al. SNS-101, a clinical-stage pH-dependent and TME-selective anti-VISTA antibody with favorable pharmacokinetic and safety characteristics, promotes anti tumoral M1 macrophage polarization in PD-1 refractory settings. Milan, Italy: European Society for Medical Oncology (2023).
107. Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. (2015) 349:1261669. doi: 10.1126/science.1261669
108. Hofmeyer KA, Ray A, Zang X. The contrasting role of B7-H3. Proc Natl Acad Sci USA. (2008) 105:10277–8. doi: 10.1073/pnas.0805458105
109. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. (2001) 2:269–74. doi: 10.1038/85339
110. Li G, Quan Y, Che F, Wang L. B7-H3 in tumors: friend or foe for tumor immunity? Cancer Chemother Pharmacol. (2018) 81:245–53. doi: 10.1007/s00280-017-3508-1
111. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7–H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. (2001) 2:269–74. doi: 10.1038/85339
112. Zhou WT, Jin WL. B7-H3/CD276: an emerging cancer immunotherapy. Front Immunol. (2021) 12:701006. doi: 10.3389/fimmu.2021.701006
113. Hashiguchi M, Kobori H, Ritprajak P, Kamimura Y, Kozono H, Azuma M. Triggering receptor expressed on myeloid cell-like transcript 2 (TLT-2) is a counter-receptor for B7-H3 and enhances T cell responses. P Natl Acad Sci USA. (2008) 105:14744–4. doi: 10.1073/pnas.0807700105
114. Yang S, Wei W, Zhao Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int J Biol Sci. (2020) 16:1767–73. doi: 10.7150/ijbs.41105
115. Husain B, Ramani SR, Chiang E, Lehoux I, Paduchuri S, Arena TA, et al. A platform for extracellular interactome discovery identifes novel functional binding partners for the immune receptors B7-H3/CD276 and PVR/CD155. Mol Cell Proteom. (2019) 18:2310–23. doi: 10.1074/mcp.TIR119.001433
116. Gao W, Wen H, Liang L, Dong X, Du R, Zhou W, et al. IL20RA signaling enhances stemness and promotes the formation of an immunosuppressive microenvironment in breast cancer. Theranostics. (2021) 11:2564–80. doi: 10.7150/thno.45280
117. Cao S, Peterson SM, Muller S, Reichelt M, McRoberts Amador C, Martinez-Martin N. A membrane protein display platform for receptor interactome discovery. Proc Natl Acad Sci. (2021) 118:e2025451118. doi: 10.1073/pnas.2025451118
118. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-γ production. Nat Immunol. (2001) 2:269–74. doi: 10.1038/85339
119. Han S, Wang Y, Shi X, Zong L, Liu L, Zhang J, et al. Negative roles of B7– H3 and B7–H4 in the microenvironment of cervical cancer. Exp Cell Res. (2018) 371:222–30. doi: 10.1016/j.yexcr.2018.08.014
120. Inamura K, Amori G, Yuasa T, Yamamoto S, Yonese J, Ishikawa Y. Relationship of B7–H3 expression in tumor cells and tumor vasculature with FOXP3+ tregs in renal cell carcinoma. Cancer Manag Res. (2019) 11:7021–30. doi: 10.2147/CMAR.S209205
121. Lu H, Shi T, Wang M, Li X, Gu Y, Zhang X, et al. B7–H3 inhibits the IFNgamma-dependent cytotoxicity of Vgamma9Vdelta2 T cells against colon cancer cells. Oncoimmunology. (2020) 9:1748991. doi: 10.1080/2162402X.2020.1748991
122. Si S, Wang L, Cao H, Xu Y, Zhan Q. Co-defciency of B7–H3 and B7–H4 identifes high CD8 + T cell infltration and better prognosis in pancreatic cancer. BMC Cancer. (2022) 22:211. doi: 10.1186/s12885-022-09294-w
123. Lee CC, Ho KH, Huang TW, Shih CM, Hsu SY, Liu AJ, et al. A regulatory loop among CD276, miR-29c-3p, and Myc exists in cancer cells against natural killer cell cytotoxicity. Life Sci. (2021) 277:119438. doi: 10.1016/j.lfs.2021.119438
124. Long C, Li G, Zhang C, Jiang T, Li Y, Duan X, et al. B7–H3 as a target for CAR-T cell therapy in skull base chordoma. Front Oncol. (2021) 11:659662. doi: 10.3389/fonc.2021.659662
125. Mao Y, Chen L, Wang F, Zhu D, Ge X, Hua D, et al. Cancer cellexpressed B7–H3 regulates the differentiation of tumor-associated macrophages in human colorectal carcinoma. Oncol Lett. (2017) 14:6177–83. doi: 10.3892/ol.2017.6935
126. Getu AA, Tigabu A, Zhou M, Lu J, Fodstad Ø, Tan M, et al. New frontiers in immune checkpoint B7-H3 (CD276) research and drug development. Mol Cancer. (2023) 22:43. doi: 10.1186/s12943-023-01751-9
127. Picarda E, Galbo PM Jr, Zong H, Rajan MR, Wallenius V, Zheng D, et al. The immune checkpoint B7-H3 (CD276) regulates adipocyte progenitor metabolism and obesity development. Sci Adv. (2022) 8:eabm7012. doi: 10.1126/sciadv.abm7012
128. Fang C, Li S, Xu R, Guo H, Jiang R, Ding S, et al. Soluble B7–H3 (sB7-H3) is over-expressed in the serum of type 1 diabetes patients. Diabetes Res Clin Pract. (2018) 143:332–6. doi: 10.1016/j.diabres.2018.08.004
129. Zhao B, Li H, Xia Y, Wang Y, Wang Y, Shi Y, et al. Immune checkpoint of B7-H3 in cancer: from immunology to clinical immunotherapy. J Hematol Oncol. (2022) 15:153. doi: 10.1186/s13045-022-01364-7
130. Mariuzza RA, Shahid S, Karade SS. The immune checkpoint receptor LAG3: Structure, function, and target for cancer immunotherapy. J Biol Chem. (2024) 300:107241. doi: 10.1016/j.jbc.2024.107241
131. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
132. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. (2007) 117:3383–92. doi: 10.1172/JCI31184
133. Chocarro L, Blanco E, Zuazo M, Arasanz H, Bocanegra A, Fernández-Rubio L, et al. Understanding LAG-3 signaling. Int J Mol Sci. (2021) 22:5282. doi: 10.3390/ijms22105282
134. Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. (2002) 169:5392–5. doi: 10.4049/jimmunol.169.10.5392
135. Annunziato F, Manetti R, Tomasévic I, Guidizi MG, Biagiotti R, Giannò V, et al. Expression and release of LAG-3-encoded protein by human CD4+ T cells are associated with IFN-gamma production. FASEB J. (1996) 10:769–76. doi: 10.1096/fasebj.10.7.8635694
136. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. (2018) 558:454–9. doi: 10.1038/s41586-018-0206-z
137. Maçon-Lemaître L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. (2005) 115:170–8. doi: 10.1111/j.1365-2567.2005.02145.x
138. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol. (1995) 25:2718–21. doi: 10.1002/eji.1830250949
139. Guy C, Mitrea DM, Chou PC, Temirov J, Vignali KM, Liu X, et al. LAG3 associates with TCR-CD3 complexes and suppresses signaling by driving co-receptor-Lck dissociation. Nat Immunol. (2022) 23:757–67. doi: 10.1038/s41590-022-01176-4
140. Hashimoto-Tane A, Bowman EP, Sakuma M, Yoneda N, Yugi K, de Waal Malefyt R, et al. Dissociation of LAG-3 inhibitory cluster from TCR microcluster by immune checkpoint blockade. Front Immunol. (2024) 15:1444424. doi: 10.3389/fimmu.2024.1444424
141. Lui Y, Davis SJ. LAG-3: a very singular immune checkpoint. Nat Immunol. (2018) 19:1278–9. doi: 10.1038/s41590-018-0257-1
142. Chung LY, Tang SJ, Wu YC, Sun GH, Liu HY, Sun KH. Galectin-3 augments tumor initiating property and tumorigenicity of lung cancer through interaction with β-catenin. Oncotarget. (2015) 6:4936–52. doi: 10.18632/oncotarget.3210
143. Srejovic I, Selakovic D, Jovicic N, Jakovljević V, Lukic ML, Rosic G. Galectin-3: roles in neurodevelopment, neuroinflammation, and behavior. Biomolecules. (2020) 10:798. doi: 10.3390/biom10050798
144. Belli C, Antonarelli G, Repetto M, Boscolo Bielo L, Crimini E, Curigliano G. Targeting cellular components of the tumor microenvironment in solid Malignancies. Cancers (Basel). (2022) 14:4278. doi: 10.3390/cancers14174278
145. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. (2019) 176:334–347.e12. doi: 10.1016/j.cell.2018.11.010
146. Hua N, Chen A, Yang C, Dong H, He X, Ru G, et al. The correlation of fibrinogen-like protein-1 expression with the progression and prognosis of hepatocellular carcinoma. Mol Biol Rep. (2022) 49:7911–9. doi: 10.1007/s11033-022-07624-6
147. Luo Y, Cai X, Yang B, Lu F, Yi C, Wu G. Advances in understanding the role of immune checkpoint LAG-3 in tumor immunity: a comprehensive review. Front Oncol. (2024) 14:1402837. doi: 10.3389/fonc.2024.1402837
148. Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. (2016) 353:aah3374. doi: 10.1126/science.aah3374
149. Yang J, Wang L, Byrnes JR, Kirkemo LL, Driks H, Belair CD, et al. PVRL2 suppresses antitumor immunity through PVRIG- and TIGIT-independent pathways. Cancer Immunol Res. (2024) 12:575–91. doi: 10.1158/2326-6066.CIR-23-0722
150. Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers (Basel). (2019) 11:877. doi: 10.3390/cancers11060877
151. Murakami K, Ganguly S. The Nectin family ligands, PVRL2 and PVR, in cancer immunology and immunotherapy. Front Immunol. (2024) 15:1441730. doi: 10.3389/fimmu.2024.1441730
152. Li XY, Das I, Lepletier A, Addala V, Bald T, Stannard K, et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J Clin Invest. (2018) 128:2613–25. doi: 10.1172/JCI98769
153. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
154. Jin HS, Park Y. Hitting the complexity of the TIGIT-CD96-CD112R-CD226 axis for next-generation cancer immunotherapy. BMB Rep. (2021) 54:2–11. doi: 10.5483/BMBRep.2021.54.1.229
155. Chiang EY, Mellman I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J Immunother Cancer. (2022) 10:e004711. doi: 10.1136/jitc-2022-004711
156. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via tregs [retraction of: J Clin Invest. 2015 Nov 2;125(11):4053-62. J Clin Invest. (2024) 134:e183278. doi: 10.1172/JCI183278
157. Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, et al. Structure of TIGIT immunoreceptor bound to PVR reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc Natl Acad Sci U S A. (2012) 109:5399–404. doi: 10.1073/pnas.1120606109
158. Sanjabi S, Oh SA, Li MO. Regulation of the immune response by TGF-β: from conception to autoimmunity and infection. Cold Spring Harb Perspect Biol. (2017) 9:a022236. doi: 10.1101/cshperspect.a022236
159. Gorvel L, Olive D. Targeting the “PVR-TIGIT axis” with immune checkpoint therapies. F1000Res. (2020) 9:F1000. doi: 10.12688/f1000research.22877.1
160. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory DCs. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
161. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol. (2009) 39:695–703. doi: 10.1002/eji.200839116
162. Chew GM, Fujita T, Webb GM, Burwitz BJ, Wu HL, Reed JS, et al. TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV infection. PloS Pathog. (2016) 12:e1005349. doi: 10.1371/journal.ppat.1005349
163. Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. (2018) 33:187–201.e10. doi: 10.1016/j.ccell.2018.01.009
164. Conner M, Hance KW, Yadavilli S, Smothers J, Waight JD. Emergence of the CD226 axis in cancer immunotherapy. Front Immunol. (2022) 13:914406. doi: 10.3389/fimmu.2022.914406
165. Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, et al. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc Natl Acad Sci U S A. (2012) 109:5399–404. doi: 10.1073/pnas.1120606109
166. Lin YX, Hung MC, Hsu JL, Hsu JM. The N-linked glycosylations of TIGIT Asn32 and Asn101 facilitate PVR/TIGIT interaction. Biochem Biophys Res Commun. (2021) 562:9–14. doi: 10.1016/j.bbrc.2021.05.034
167. Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J Biol Chem. 289(25):17647–57. doi: 10.1074/jbc.M114.572420
168. Rotte A, Sahasranaman S, Budha N. Targeting TIGIT for immunotherapy of cancer: update on clinical development. Biomedicines. (2021) 9:1277. doi: 10.3390/biomedicines9091277
169. Harjunpää H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol. (2020) 200:108–19. doi: 10.1111/cei.13407
170. Eberlé F, Dubreuil P, Mattei MG, Devilard E, Lopez M. The human PRR2 gene, related to the human poliovirus receptor gene (PVR), is the true homolog of the murine MPH gene. Gene. (1995) 159:267–72. doi: 10.1016/0378-1119(95)00180-e
171. Lopez M, Aoubala M, Jordier F, Isnardon D, Gomez S, Dubreuil P. The human poliovirus receptor related 2 protein is a new hematopoietic/endothelial homophilic adhesion molecule. Blood. (1998) 92:4602–11.
172. Anderson AC, Joller N, Kuchroo VK. Lag-3, tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
173. Zhang B, Zhao W, Li H, Chen Y, Tian H, Li L, et al. Immunoreceptor TIGIT inhibits the cytotoxicity of human cytokine-induced killer cells by interacting with CD155. Cancer Immunol Immunother. (2016) 65:305–14. doi: 10.1007/s00262-016-1799-4
174. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via tregs. J Clin Invest. (2015) 125:4053–62. doi: 10.1172/JCI81187
175. Scott F, Wichmann C, Burvenich I, Sokol L, Sahin D, McCarter JD, et al. 324 Preclinical evaluation of anti-VISTA antibody CI-8993 in a syngeneic huVISTA-KI model. J Immuno Ther Cancer. (2021) 9(Suppl 2):A354. doi: 10.1136/jitc-2021-SITC2021.324
176. Fitzpatrick E, Filippi C, Jagadisan B, Shivapatham D, Anand H, Lyne M, et al. Intraperitoneal transplant of Hepatocytes co-Encapsulated with mesenchymal stromal cells in modified alginate microbeads for the treatment of acute Liver failure in Pediatric patients (HELP)-An open-label, single-arm Simon’s two stage phase 1 study protocol. PloS One. (2023) 18:e0288185. doi: 10.1371/journal.pone.0288185
177. Melero IJ, Gomez-Roca CA, Ferre P, Chetaille E, Petain A, Thuilliez C, et al. 315 W0180 novel anti-VISTA antibody: Rationale for target patient population and first-in-human trial design in monotherapy and in combination with anti-PD1 antibody. J ImmunoTher Cancer. (2020) 8. doi: 10.1136/jitc-2020-SITC2020.0315
178. Sasikumar PG, Sudarshan NS, Adurthi S, Ramachandra RK, Samiulla DS, Lakshminarasimhan A, et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun Biol. (2021) 4:699. doi: 10.1038/s42003-021-02191-1
179. Mehta N, Maddineni S, Kelly RL, Lee RB, Hunter SA, Silberstein JL, et al. An engineered antibody binds a distinct epitope and is a potent inhibitor of murine and human VISTA. Sci Rep. (2020) 10:15171. doi: 10.1038/s41598-020-71519-4
180. Mahoney KM, Freeman GJ. Acidity changes immunology: a new VISTA pathway. Nat Immunol. (2020) 21:13–6. doi: 10.1038/s41590-019-0563-2
181. Johnson M, Awad M, Koyama T, Gutierrez M, Falchook GS, Piha-Paul SA, et al. OA05.05 ifinatamab deruxtecan (I-DXd; DS-7300) in patients with refractory SCLC: A subgroup analysis of a phase 1/2 study. J Thorac Oncol. (2023) 18:54–5. doi: 10.1016/j.jtho.2023.09.042
182. Wang J, Xing L, Sun Y, Guo W, Wang H, Chen J, et al. ARTEMIS-001: phase 1 study of HS-20093, a B7-H3–targeting antibody-drug conjugate, in patients with advanced solid tumor. J Clin Oncol. (2023) 41(16_suppl):3017. doi: 10.1200/JCO.2023.41.16_suppl.3017
183. McMinn ME, Brister KA, Orr WS, Sheehan NT, Christopher WO. Complete remission of metastatic melanoma of the scalp following treatment with nivolumab plus relatlimab. Am Surg. (2024) 90:2078–9. doi: 10.1177/00031348241241642
184. Garralda E, Sukari A, Lakhani NJ, Patnaik A, Lou Y, Im SA, et al. A first-in-human study of the anti-LAG-3 antibody favezelimab plus pembrolizumab in previously treated, advanced microsatellite stable colorectal cancer. ESMO Open. (2022) 7:100639. doi: 10.1016/j.esmoop.2022.100639
185. Schöffski P, Tan DSW, Martín M, Ochoa-de-Olza M, Sarantopoulos J, Carvajal RD, et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced Malignancies. J Immunother Cancer. (2022) 10:e003776. doi: 10.1136/jitc-2021-003776
186. Tiragolumab impresses in multiple trials. Cancer Discov. (2020) 10:1086–7. doi: 10.1158/2159-8290.CD-NB2020-063
187. Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer. Ann Oncol. (2022) 33:169–80. doi: 10.1016/j.annonc.2021.11.002
188. Chaudhry A, Johnson M, Colburn D, Gilbert H, Garofalo A, Paoloni M, et al. 1419TiP ARC-7: A phase II study to evaluate the safety and efficacy of zimberelimab alone, AB154 in combination with zimberelimab, and AB154 in combination with zimberelimab and AB928 in front-line, PD-L1 expressing, NSCLC (NSCLC). Ann Oncol. (2020) 31, Supplement 4. doi: 10.1016/j.annonc.2020.08.1733
189. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res. (2021) 27:3620–9. doi: 10.1158/1078-0432.CCR-20-4746
190. Acoba JD, Rho Y, Fukaya E. Phase II study of cobolimab in combination with dostarlimab for the treatment of advanced hepatocellular carcinoma. J Clin Oncol. (2023) 41:580–0. doi: 10.1200/JCO.2023.41.4_suppl.580
191. Doi T, Aogi K, Tamiya M, Machida N, Noguchi M, Arai Y, et al. MO11-1 Phase I dose-finding study of the SIRPα inhibitor BI 765063 as monotherapy in Japanese patients with advanced cancer. Ann Oncol. (2023) 34, Supplement 3:S1403. doi: 10.1016/j.annonc.2023.09.182
192. Chan H, Trout C, Mikolon D, Adams P, Guzman R, Fenalti G, et al. Discovery and preclinical characterization of CC-95251, an anti-SIRPα Antibody that enhances macrophage-mediated phagocytosis of non-hodgkin lymphoma (NHL) cells when combined with rituximab. Blood. (2021) 138:2271. doi: 10.1182/blood-2021-147262
193. Yang J, Deresa I, Ho WH, Long H, Maslyar D, Rosenthal A, et al. AL008 enhances myeloid antitumor function by inhibiting SIRPα Signaling and activating fc receptors. J Immunol. (2023) 210:204–15. doi: 10.4049/jimmunol.2200157
194. Knisely A, Ahmed J, Stephen B, Piha-Paul SA, Karp D, Zarifa A, et al. Phase 1/2 trial of avelumab combined with utomilumab (4-1BB agonist), PF-04518600 (OX40 agonist), or radiotherapy in patients with advanced gynecologic Malignancies. Cancer. (2024) 130:400–9. doi: 10.1002/cncr.v130.3
195. Zhang J, Jiang X, Gao H, Zhang F, Zhang X, Zhou A, et al. Development and characterization of a novel anti-OX40 antibody for potent immune activation. Cancer Immunol Immunother. (2020) 69:939–50. doi: 10.1007/s00262-020-02501-2
196. Guttman-Yassky E, Lee JS, Pavel A, Salhi Y, Wolff G. Evidence for activity of GBR830 (anti-OX40) in extrinsic and intrinsic atopic dermatitis (AD) in a phase 2a Study. J Allergy Clin Immunol. (2019) 143:Ab127. doi: 10.1016/j.jaci.2018.12.385
197. Lé AM, Torres T. OX40-OX40L inhibition for the treatment of atopic dermatitis-focus on rocatinlimab and amlitelimab. Pharmaceutics. (2022) 14:2753. doi: 10.3390/pharmaceutics14122753
198. Khushalani NI, Ott PA, Ferris RL, Cascone T, Schadendorf D, Le DT, et al. Final results of urelumab, an anti-CD137 agonist monoclonal antibody, in combination with cetuximab or nivolumab in patients with advanced solid tumors. J Immunother Cancer. (2024) 12:e007364. doi: 10.1136/jitc-2023-007364
199. Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, et al. Phase I study of single-agent utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin Cancer Res. (2018) 24:1816–23. doi: 10.1158/1078-0432.CCR-17-1922
200. Liu Y, Chen H, Chen Z, Qiu J, Pang H, Zhou Z. Novel roles of the tim family in immune regulation and autoimmune diseases. Front Immunol. (2021) 12:748787. doi: 10.3389/fimmu.2021.748787
201. Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, Monahan S, et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PloS Pathog. (2013) 9:e1003232. doi: 10.1371/journal.ppat.1003232
202. Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol Med. (2005) 11:362–9. doi: 10.1016/j.molmed.2005.06.008
203. Nurtanio N, Yang PC. Role of TIM-4 in innate or adaptive immune response. N Am J Med Sci. (2011) 3:217–21. doi: 10.4297/najms.2011.3217
204. Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. (2010) 235:172–89. doi: 10.1111/j.0105-2896.2010.00903.x
205. Albacker LA, Karisola P, Chang YJ, Umetsu SE, Zhou M, Akbari O, et al. TIM-4, a receptor for phosphatidylserine, controls adaptive immunity by regulating the removal of antigen-specific T cells. J Immunol. (2010) 185:6839–49. doi: 10.4049/jimmunol.1001360
206. Rodriguez-Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT. The costimulatory role of TIM molecules. Immunol Rev. (2009) 229:259–70. doi: 10.1111/j.1600-065X.2009.00772.x
207. Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, Umetsu SE, et al. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol. (2005) 6:455–64. doi: 10.1038/ni1185
208. McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, et al. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat Immunol. (2001) 2:1109. doi: 10.1038/ni739
209. Umetsu SE, Lee WL, McIntire JJ, Downey L, Sanjanwala B, Akbari O, et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol. (2005) 6:447. doi: 10.1038/ni1186
210. Ma J, Usui Y, Takeda K, Harada N, Yagita H, Okumura K, et al. TIM-1 signaling in B cells regulates antibody production. Biochem Biophys Res Commun. (2011) 406:223–8. doi: 10.1016/j.bbrc.2011.02.021
211. Rodriguez-Manzanet R, Meyers JH, Balasubramanian S, Slavik J, Kassam N, Dardalhon V, et al. TIM-4 expressed on APCs induces T cell expansion and survival. J Immunol. (2008) 180:4706–13. doi: 10.4049/jimmunol.180.7.4706
212. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
213. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
214. Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. (2009) 39:2492–501. doi: 10.1002/eji.200939274
215. Granier C, Dariane C, Combe P, Verkarre V, Urien S, Badoual C, et al. Tim-3 expression on tumor-infiltrating PD-1+CD8+ T cells correlates with poor clinical outcome in renal cell carcinoma. Cancer Res. (2017) 77:1075–82. doi: 10.1158/0008-5472.CAN-16-0274
216. Solinas C, De Silva P, Bron D, Willard-Gallo K, Sangiolo D. Significance of TIM3 expression in cancer: From biology to the clinic. Semin Oncol. (2019) 46:372–9. doi: 10.1053/j.seminoncol.2019.08.005
217. Ausejo-Mauleon I, Labiano S, de la Nava D, Laspidea V, Zalacain M, Marrodán L, et al. TIM-3 blockade in diffuse intrinsic pontine glioma models promotes tumor regression and antitumor immune memory. Cancer Cell. (2023) 41:1911–1926.e8. doi: 10.1016/j.ccell.2023.09.001
218. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. (2010) 184:1918–30. doi: 10.4049/jimmunol.0903059
219. Du W, Yang M, Turner A, Xu C, Ferris RL, Huang J, et al. TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int J Mol Sci. (2017) 18:645. doi: 10.3390/ijms18030645
220. He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. (2018) 11:7005–9. doi: 10.2147/OTT.S170385
221. Jiang D, Chen C, Yan D, Zhang X, Liu X, Yan D, et al. Exhausted phenotype of circulating CD8+ T cell subsets in hepatitis B virus carriers. BMC Immunol. (2022) 23:18. doi: 10.1186/s12865-022-00488-2
222. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. (2011) 117:4501–10. doi: 10.1182/blood-2010-10-310425
223. Moorman JP, Wang JM, Zhang Y, Ji XJ, Ma CJ, Wu XY, et al. Tim-3 pathway controls regulatory and effector T cell balance during hepatitis C virus infection. J Immunol. (2012) 189:755–66. doi: 10.4049/jimmunol.1200162
224. Wu W, Shi Y, Li J, Chen F, Chen Z, Zheng M. Tim-3 expression on peripheral T cell subsets correlates with disease progression in hepatitis B infection. Virol J. (2011) 8:113. doi: 10.1186/1743-422X-8-113
225. Luke JJ, Patel MR, Blumenschein GR, et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat Med. (2023) 29:2814–24. doi: 10.1038/s41591-023-02593-0
226. Day D, Ganju V, Chung K, Si L, Mao L, Aghmesheh M, et al. 1028P Preliminary phase I results from a first-in-human study of EMB-02, a PD-1xLAG-3 bispecific antibody, in patients (pts) with advanced solid tumors. Ann Oncol. (2023) 34, Supplement 2:S625. doi: 10.1016/j.annonc.2023.09.2167
227. Jabbour S, Lu B, Fu X, Goksel T, Sugawara S, Song A, et al. 969TiP Randomized, phase III study of MK-7684A plus concurrent chemoradiotherapy (cCRT) followed by MK-7684A vs cCRT followed by durvalumab for unresectable, locally advanced, stage III NSCLC (NSCLC): KEYVIBE-006. Ann Oncol. (2022) 33, Supplement 7:S990. doi: 10.1016/j.annonc.2022.07.1095
228. Ward R, Ramsay N, Campbell S, Patel H, Bushell M, Morin B, et al. Abstract 3915: BMS-986442 (AGEN1777), a novel TIGIT/CD96 bispecific antibody, demonstrates superior monotherapy and combination activity versus conventional anti-TIGIT antibodies in preclinical models. Cancer Res. (2024) 84(16_Supplement):3915. doi: 10.1158/1538-7445.AM2024-3915
229. Clancy-Thompson E, Perry T, Pryts S, Jaiswal A, Oganesyan V, van Dyk N, et al. 461 Generation of AZD7789, a novel PD-1 and TIM-3 targeting bispecific antibody, which binds to a differentiated epitope of TIM-3. J ImmunoTher Cancer. (2022) 10:461. doi: 10.1136/jitc-2022-SITC2022.0461
230. van Beek EM, Cochrane F, Barclay AN, van den Berg TK. Signal regulatory proteins in the immune system. J Immunol. (2005) 175:7781–7. doi: 10.4049/jimmunol.175.12.7781
231. Liu Y, Soto I, Tong Q, Chin A, Bühring HJ, Wu T, et al. SIRPbeta1 is expressed as a disulfide-linked homodimer in leukocytes and positively regulates neutrophil transepithelial migration. J Biol Chem. (2005) 280:36132–40. doi: 10.1074/jbc.M506419200
232. Veillette A, Thibaudeau E, Latour S. High expression of inhibitory receptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 in macrophages. J Biol Chem. (1998) 273:22719–28. doi: 10.1074/jbc.273.35.22719
233. Taniguchi H, Okumura N, Hamada J, Inagaki M, Nakahata Y, Sano S, et al. Cold exposure induces tyrosine phosphorylation of BIT through NMDA receptors in the rat hypothalamus. Biochem Biophys Res Commun. (2004) 319:178–84. doi: 10.1016/j.bbrc.2004.04.173
234. Murata Y, Saito Y, Kotani T, Matozaki T. CD47-signal regulatory protein α signaling system and its application to cancer immunotherapy. Cancer Sci. (2018) 109:2349–57. doi: 10.1111/cas.13663
235. Sugimura-Nagata A, Koshino A, Inoue S, Matsuo-Nagano A, Komura M, Riku M, et al. Expression and prognostic significance of CD47-SIRPA macrophage checkpoint molecules in colorectal cancer. Int J Mol Sci. (2021) 22:2690. doi: 10.3390/ijms22052690
236. Adams S, van der Laan LJ, Vernon-Wilson E, Renardel de Lavalette C, Döpp EA, Dijkstra CD, et al. Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. J Immunol. (1998) 161:1853–9.
237. Voets E, Paradé M, Lutje Hulsik D, Spijkers S, Janssen W, Rens J, et al. Functional characterization of the selective pan-allele anti-SIRPα antibody ADU-1805 that blocks the SIRPα-CD47 innate immune checkpoint. J Immunother Cancer. (2019) 7:340. doi: 10.1186/s40425-019-0772-0
238. Huang B, Bai Z, Ye X, Zhou C, Xie X, Zhong Y, et al. Structural analysis and binding sites of inhibitors targeting the CD47/SIRPα interaction in anticancer therapy. Comput Struct Biotechnol J. (2021) 19:5494–503. doi: 10.1016/j.csbj.2021.09.036
239. Zhou Z, Chen MM, Luo Y, Mojumdar K, Peng X, Chen H, et al. Tumor-intrinsic SIRPA promotes sensitivity to checkpoint inhibition immunotherapy in melanoma. Cancer Cell. (2022) 40:1324–1340.e8. doi: 10.1016/j.ccell.2022.10.012
240. Behrens LM, van den Berg TK, van Egmond M. Targeting the CD47-SIRPα Innate immune checkpoint to potentiate antibody therapy in cancer by neutrophils. Cancers (Basel). (2022) 14:3366. doi: 10.3390/cancers14143366
241. Eck MJ, Sprang SR. The structure of tumor necrosis factor-alpha at 2.6 A resolution. Implications for receptor binding. J Biol Chem. (1989) 264:17595–605. doi: 10.2210/pdb1tnf/pdb
242. Dostert C, Grusdat M, Letellier E, Brenner D. The TNF family of ligands and receptors: communication modules in the immune system and beyond. Physiol Rev. (2019) 99:115–60. doi: 10.1152/physrev.00045.2017
243. Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. (2012) 119:651–65. doi: 10.1182/blood-2011-04-325225
244. Shi JH, Sun SC. Tumor necrosis factor receptor-associated factor regulation of nuclear factor κB and mitogen-activated protein kinase pathways. Front Immunol. (2018) 9:1849. doi: 10.3389/fimmu.2018.01849
245. Chinnaiyan AM, Tepper CG, Seldin MF, O’Rourke K, Kischkel FC, Hellbardt S, et al. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. (1996) 271:4961–5. doi: 10.1074/jbc.271.9.4961
246. Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity. (2016) 44:1005–19. doi: 10.1016/j.immuni.2016.04.019
247. Croft M, Siegel RM. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol. (2017) 13:217–33. doi: 10.1038/nrrheum.2017.22
248. Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood. (2018) 131:39–48. doi: 10.1182/blood-2017-07-741025
249. Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem. (1998) 273:5808–14. doi: 10.1074/jbc.273.10.5808
250. Fu N, Xie F, Sun Z, Wang Q. The OX40/OX40L axis regulates T follicular helper cell differentiation: implications for autoimmune diseases. Front Immunol. (2021) 12:670637. doi: 10.3389/fimmu.2021.670637
251. Song J, So T, Croft M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J Immunol. (2008) 180:7240–8. doi: 10.4049/jimmunol.180.11.7240
252. Chao JL, Savage PA. Unlocking the complexities of tumor-associated regulatory T cells. J Immunol. (2018) 200:415–21. doi: 10.4049/jimmunol.1701188
253. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. (2001) 15:445–55. doi: 10.1016/S1074-7613(01)00191-1
254. Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. (2013) 123:2447–63. doi: 10.1172/JCI64859
255. Ramser M, Eichelberger S, Däster S, Weixler B, Kraljević M, Mechera R, et al. High OX40 expression in recurrent ovarian carcinoma is indicative for response to repeated chemotherapy. BMC Cancer. (2018) 18:425. doi: 10.1186/s12885-018-4339-0
256. Sarff M, Edwards D, Dhungel B, Wegmann KW, Corless C, Weinberg AD, et al. OX40 (CD134) expression in sentinel lymph nodes correlates with prognostic features of primary melanomas. Am J Surg. (2008) 195:621–5. doi: 10.1016/j.amjsurg.2007.12.036
257. Alves Costa Silva C, Facchinetti F, Routy B, Derosa L. New pathways in immune stimulation: targeting OX40. ESMO Open. (2020) 5:e000573. doi: 10.1136/esmoopen-2019-000573
258. Schwarz H, Arden K, Lotz M. CD137, a member of the tumor necrosis factor receptor family, is located on chromosome 1p36, in a cluster of related genes, and colocalizes with several Malignancies. Biochem Biophys Res Commun. (1997) 235:699–703. doi: 10.1006/bbrc.1997.6870
259. Kwon BS, Weissman SM. cDNA sequences of two inducible T-cell genes. Proc Natl Acad Sci U S A. (1989) 86:1963–7. doi: 10.1073/pnas.86.6.1963
260. Vinay DS, Kwon BS. 4-1BB signaling beyond T cells. Cell Mol Immunol. (2011) 8:281–4. doi: 10.1038/cmi.2010.82
261. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
262. Cheuk AT, Mufti GJ, Guinn BA. Role of 4-1BB:4-1BB ligand in cancer immunotherapy. Cancer Gene Ther. (2004) 11:215–26. doi: 10.1038/sj.cgt.7700670
263. Chin SM, Kimberlin CR, Roe-Zurz Z, Zhang P, Xu A, Liao-Chan S, et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat Commun. (2018) 9:4679. doi: 10.1038/s41467-018-07136-7
264. Cannons JL, Choi Y, Watts TH. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol. (2000) 165:6193–204. doi: 10.4049/jimmunol.165.11.6193
265. Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. (2012) 481:463–8. doi: 10.1074/jbc.271.9.4961
266. Pollok KE, Kim YJ, Hurtado J, Zhou Z, Kim KK, Kwon BS. 4-1BB T-cell antigen binds to mature B cells and macrophages, and costimulates anti-mu-primed splenic B cells. Eur J Immunol. (1994) 24:367–74. doi: 10.1002/eji.1830240215
Keywords: immunotherapy, immune checkpoint proteins, tumor microenvironment specificity, co-suppressive pathways, protein families
Citation: Liu R, Jiang X, Dong R, Zhang Y, Gai C and Wei P (2025) Revealing the mechanisms and therapeutic potential of immune checkpoint proteins across diverse protein families. Front. Immunol. 16:1499663. doi: 10.3389/fimmu.2025.1499663
Received: 21 September 2024; Accepted: 28 March 2025;
Published: 28 April 2025.
Edited by:
Lily Wang, Cleveland Clinic, United StatesReviewed by:
Mohammad Asad, Albert Einstein College of Medicine, United StatesLiangxue Zhou, Sichuan University, China
Kenji Morimoto, Kyoto Prefectural University of Medicine, Japan
Copyright © 2025 Liu, Jiang, Dong, Zhang, Gai and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Wei, d2VpcGVuZ0BidWNtLmVkdS5jbg==; Cong Gai, Z2FpY29uZ0BidWNtLmVkdS5jbg==