 Yuting Zhuo
Yuting Zhuo Shangui Fu
Shangui Fu Yue Qiu
Yue Qiu- 1Department of Endocrinology and Metabolism, Jiujiang Hospital of Traditional Chinese Medicine, Jiujiang, Jiangxi, China
- 2The Second School of Clinical Medicine, Jiangxi Medical College, Nanchang University, Nanchang, Jiangxi, China
Post-translational modifications such as SUMOylation are crucial for the functionality and signal transduction of a diverse array of proteins. Analogous to ubiquitination, SUMOylation has garnered significant attention from researchers and has been implicated in the pathogenesis of various human diseases in recent years, such as cancer, neurological lesions, cardiovascular diseases, diabetes mellitus, and so on. The pathogenesis of diabetes, particularly type 1 and type 2 diabetes, has been closely associated with immune dysfunction, which constitutes the primary focus of this review. This review will elucidate the process of SUMOylation and its impact on diabetes mellitus development and associated complications, focusing on its regulatory effects on the immune microenvironment. This article summarizes various signaling pathways at both cellular and molecular levels that are implicated in these processes. Furthermore, it proposes potential new targets for drug development aimed at the prevention and treatment of diabetes mellitus based on insights gained from the SUMOylation process.
1 Introduction
In the context of globalization, diabetes mellitus (DM) has emerged as a critical public health issue and a substantial economic burden on healthcare systems worldwide. The estimated prevalence of DM in the adult population was 10.5% in 2021, with projections indicating a continued rise in prevalence, as reported in the survey (1). From 2007 to 2017, there was a notable increase in the prevalence of DM among Chinese adults, and the number of diabetes-related risk factors remained uncontrolled (2). Therefore, addressing the challenge posed by DM has emerged as an urgent priority for the public health protection system. DM is a group of metabolic disorders that are characterized by hyperglycemia. Based on the diagnostic and classification criteria set forth by the American Diabetes Association (ADA), DM is classified into four categories: type 1 diabetes mellitus (T1DM), type 2 diabetes mellitus(T2DM), gestational diabetes mellitus (GDM), and other types of DM. T1DM is regarded as an autoimmune disease, which is characterized by the autoimmune destruction of pancreatic β-cells. T2DM constitutes over 90% of all DM types, with a more intricate underlying mechanism than that of T1DM. This condition is marked by insulin resistance and a relative insulin deficiency (3, 4). DM is frequently a multifactorial disease that can present in various forms, including immune dysregulation, glucose metabolism disorders, insulin resistance, and β-cell destruction (5). In these cases, immune dysfunction exerts an influence on the development of DM, which is caused by various factors and contributes to the onset of DM and its associated complications (6, 7).
Since the initial identification of small ubiquitin-like modifier (SUMO) proteins in 1996, these proteins have increasingly been recognized as versatile and important post-translation modifications (PTMs) (8). SUMO proteins constitute a family of proteins that modulate their function through the processes of associating with other proteins (SUMOylation) and dissociating from them (deSUMOylation) (9). The modification of the substrate protein is reversible. Similar to ubiquitination, SUMOylation plays a critical role in regulating various cellular processes by modulating distinct substrates. This modulation influences the stability and subcellular localization of proteins, as well as impacting essential processes such as cell cycle regulation (10), DNA synthesis and repair (11), signal transduction, and cellular immunity (12).
2 SUMOylation
2.1 Overview of SUMOylation
The attachment of SUMO proteins to a cysteine residue within a protein is termed SUMOylation. In contrast, the removal of SUMO proteins from the substrate is referred to as deSUMOylation. In vivo, the SUMOylation and deSUMOylation processes reciprocally regulate the SUMOylated state of proteins.
The three-dimensional structure of SUMO proteins exhibits a similarity to that of ubiquitin, as both proteins belong to the conserved ubiquitin-like protein (UBL) family. SUMO proteins covalently bind to the lysine residues of target proteins through a cascade reaction facilitated by a series of enzymes. SUMO1, SUMO2, SUMO3, SUMO4 (13), and SUMO5 (14) are the five isoforms of SUMO proteins that have been described. Of these, there is widespread expression of SUMO1, SUMO2, and SUMO3 in vivo. The amino acid sequences of SUMO2 and SUMO3 are essentially identical but have only 46% homogeneity with SUMO1 (15–17). The expression of SUMO4 is predominantly observed in immune tissues, including the kidneys, spleen, and lymph nodes (18). Numerous studies have demonstrated a strong association between SUMO4 and the development of T1DM and T2DM (19–22). Additionally, SUMO5 is tissue-specific, highly conserved in primates, and is also involved in the formation and destruction of promyelocytic leukemia nuclear body (PML-NB) (14).
2.2 Mechanism of SUMOylation
The process of SUMOylation is analogous to ubiquitination, as the SUMO protein binds to the substrate protein through a series of enzymatic reactions. (Table 1) The principal enzymes involved in this process are, in order: the activation enzyme 1 (E1, Aos1/Uba2 heterodimer), conjugation enzyme 2 (E2, Ubc9), and SUMO ligation enzyme 3 (E3, such as PIAS, ZNF451, and RanBP2) (23–28). The PIAS family is the major SUMO E3 ligase and comprises PIAS1, PIAS2 (PIASx), PIAS3, and PIAS4 (PIASy) (29, 30). Subsequently, SUMO-specific proteases (SENPs) execute the deSUMOylation process.
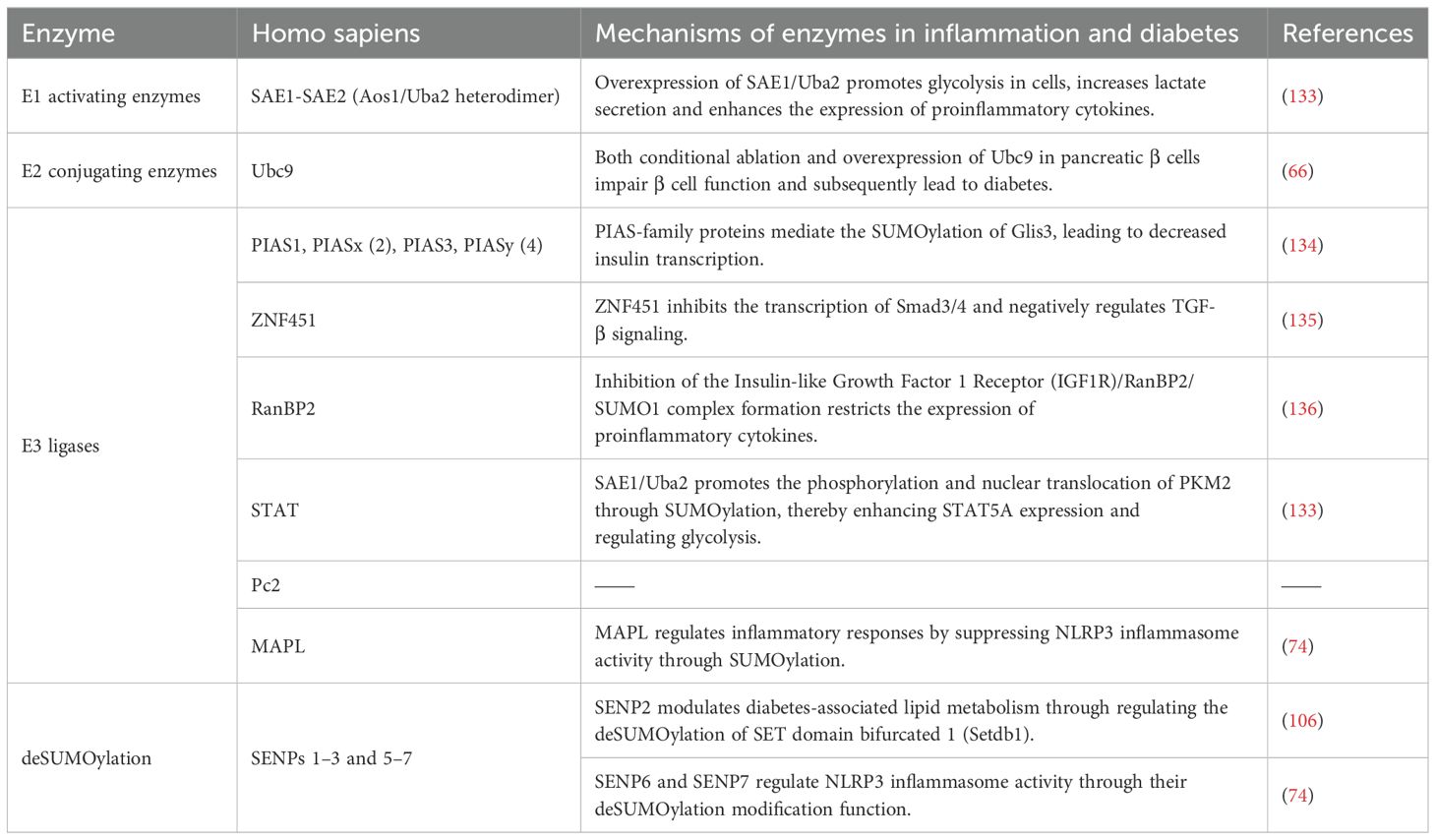
Table 1. The enzymes of SUMOylation in mammal cells.
The process of SUMOylation can be categorized into four principal stages: the maturation of SUMO, activation of SUMO by the E1 enzyme, the activated SUMO binds to the E2 enzyme and promotion of the conjugation of SUMO to the substrate by the E3 enzyme. (Figure 1) (і) The first step in SUMO maturation begins with SENPs degrading several amino acids at the C-terminus of SUMOs to expose the diglycine residues. (ii) Next, in the presence of ATP, the diglycine residues of mature SUMOs interact with the cysteine residue of the E1 enzyme, forming high-energy thioester bonds. E1s are constituted by two subunits: the SUMO-activating enzyme subunit 1 (SAE1 or Aos1) and the SUMO-activating enzyme subunit 2 (SAE2 or Uba2). Typically, these two subunits assemble into ATP-dependent heterodimers consisting of SAE1 and SAE2 (31, 32). The process of SUMO activation is accomplished through the mechanism above (33). (iii) Subsequently, SUMOs are transferred from the E1 to the E2, resulting in the formation of a SUMO-E2 complex. The only E2 binding enzyme identified is ubiquitin conjugating enzyme 9 (Ubc9) (34–36). (iv) Ultimately, under the catalytic influence of E3 ligases, Ubc9 directly recognizes the conserved sequence Ψ-Kx-D/E (Ψ represents a hydrophobic group, K denotes lysine conjugated to SUMO, x signifies any amino acid, and D/E indicates an acidic amino acid, either aspartic acid or glutamic acid) within the substrate. This recognition facilitates the conjugation of SUMO proteins to lysine residues of the substrate, forming isopeptide bonds and thereby completing the transfer of SUMO from the E2 enzyme to the substrate (37). The process of deSUMOylation is defined as the dissociation of SUMO from the substrate protein. The SENPs encompass various species, with seven members of the SENP family identified in the human genome, specifically SENPs 1–3 and 5–7 (38). In summary, SUMOylation is characterized by two major features: specificity and reversibility. The specificity is determined by the combined action of Ubc9 and E3 ligases, while the reversibility is achieved through the deSUMOylation process mediated by SENPs. This ensures the dynamic regulation of SUMOylation.
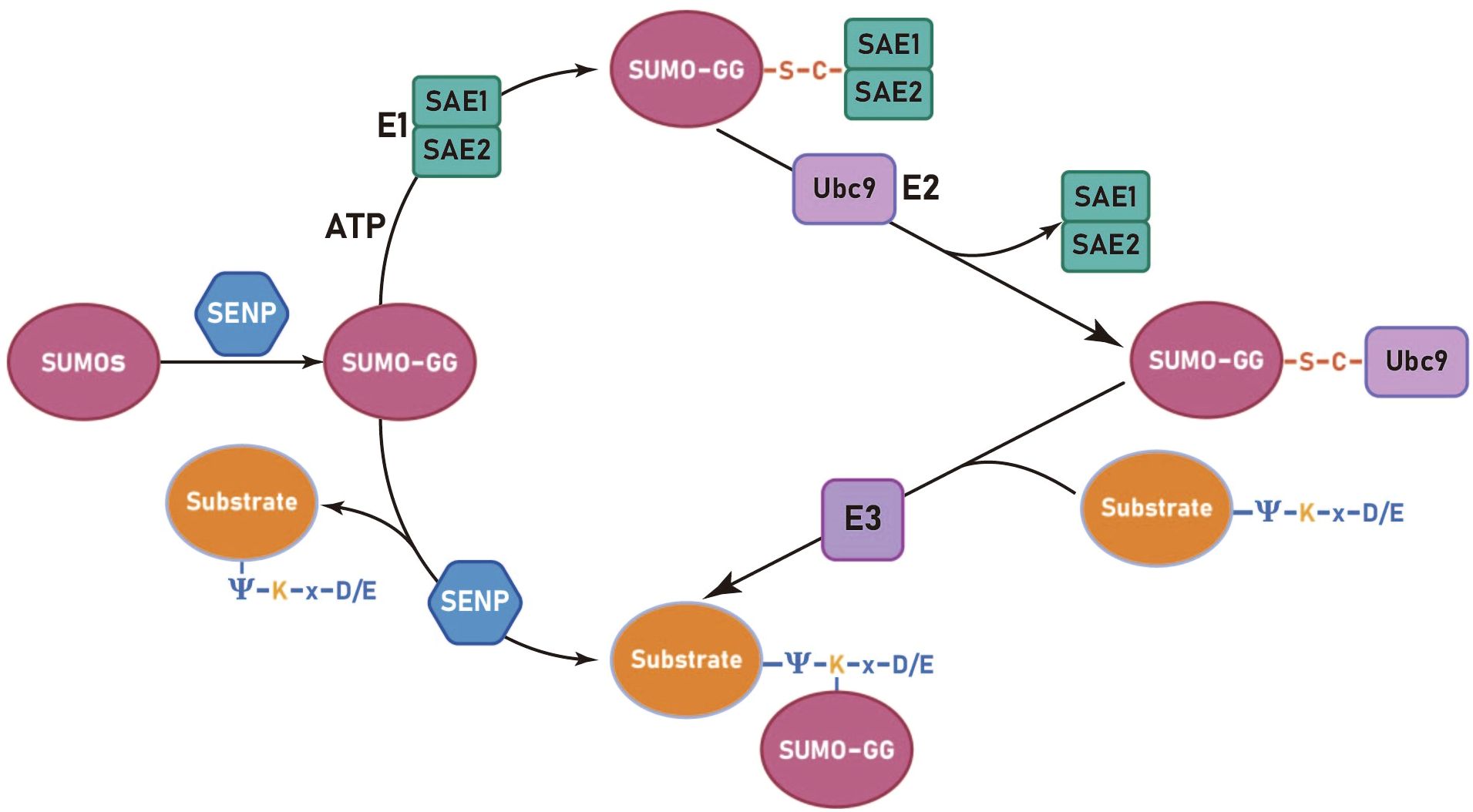
Figure 1. The SUMOylation cycle in mammalian cells involves several sequential steps. Initially, SUMO paralogues are cleaved by a SUMO-specific protease (SENP) to expose carboxy-terminal diglycine residues (GG). Subsequently, an ATP-dependent activation step is carried out by the activation enzyme 1 (SAE1 and SAE2), resulting in the formation of a high-energy thioester bond. The activated SUMO is then transferred from the E1 to the E2, forming a SUMO-E2 complex through a thioester linkage. Finally, the E2 enzyme, Ubc9, recognizes the conserved sequence Ψ-K-x-D/E (Ψ represents a hydrophobic group, K denotes lysine conjugated to SUMO, x signifies any amino acid, and D/E indicates an acidic amino acid, either aspartic acid or glutamic acid) on the substrate protein and facilitates the formation of an isopeptide bond between the SUMO and substrate protein, a reaction catalyzed by the E3 ligase.
SUMOylation, a crucial post-translational modification of proteins, regulates key cellular processes through diverse molecular mechanisms. Notably, the fragile replication fork structure becomes particularly susceptible to chromosomal rearrangements and mutations during DNA replication. Studies have revealed that Pli1, an E3 SUMO ligase, mediates SUMOylation to perform dual functions: it not only maintains replication fork integrity but also promotes their translocation to nuclear pore complexes (NPCs) via poly-SUMOylation-induced SUMO chain formation. This regulatory mechanism orchestrates the precise repair of replication forks and is essential for preserving genomic stability (11).
Emerging evidence has highlighted the pivotal role of SUMOylation regulation in cell cycle control. Mechanistically, lactate has been shown to inhibit the deSUMOylation activity of SENP1 through zinc ion chelation at its active site, leading to enhanced APC4 SUMOylation. This post-translational modification triggers structural remodeling of the anaphase-promoting complex (APC/C), facilitating its interaction with UBE2C and thereby orchestrating proper cell cycle progression and mitotic regulation (10). Capitalizing on the essential role of SUMOylation in cell cycle regulation, scientists have developed TAK-981, a novel SUMO E1 inhibitor. This small molecule exhibits dual anti-tumor mechanisms: it not only arrests the cancer cell cycle through SUMOylation inhibition but also potentiates anti-tumor immunity by stimulating immune activation, with a specific emphasis on cellular immune responses. These pharmacological properties underscore its promising clinical applications in oncology (12).
3 Regulation of immune microenvironment by SUMOylation
3.1 Regulation of immune cells by SUMOylation
3.1.1 SUMOylation and regulatory T cells
Regulation of immune homeostasis is well known to be the function of regulatory T (Treg) cells, which can exert immunosuppressive and inflammatory control effects by promoting the formation of anti-inflammatory (M2) macrophages (39), inhibiting T helper (Th) cell populations (40), and secreting inhibitory cytokines, such as interleukin-10 (IL-10) and IL-35 (41). SUMOylation is involved in maintaining peripheral T cell homeostasis and immune tolerance. Stimulation of the T-cell receptor (TCR) and cluster of differentiation 28 (CD28) can induce the production of reactive oxygen species (ROS), resulting in the accumulation of SENP3. SENP3 promotes the nuclear localization of BACH2 by mediating the deSUMOylation of the transcriptional repressor BACH2, thereby preserving the stability of Treg cells and their immunosuppressive function (42).
3.1.2 SUMOylation and CD8+ T cells
TCR-induced ROS also prompts rapid translocation of SENP7 to the cytoplasm, thereby mediating the deSUMOylation, ubiquitination, and subsequent degradation of phosphatase and tensin homolog (PTEN) protein. In CD8 T cells, SENP7-dependent reduction of PTEN maintains metabolic fitness and effector function. Moreover, SENP7 enhances the activation of PI3K/mTOR signal transduction and is involved in the maintenance of glycolysis and oxidative phosphorylation (OXPHOS) in CD8 T cells (43). Several studies have indicated that SUMOylation can impede the presentation of major histocompatibility complex class I (MHC I) antigens, thereby promoting immune evasion by tumor cells. Conversely, the inhibition of SUMOylation has been shown to repair the MHC I antigen presentation mechanism, activate CD8+ T cells, and augment their cytotoxic efficacy against target cells (44).
SUMOylation is crucial for the growth of hepatocellular carcinoma (HCC) cells, and SUMOylation inhibitors TAK-981 and ML-792 effectively reduce SUMOylation in HCC cells. Furthermore, they enhance anti-tumor immunity by restoring the killing ability of T cells, promoting the activity of natural killer (NK) cells and inflammatory (M1) macrophages, activating innate immune cells, and modulating the intestinal microbiota (45). Moreover, the inhibitory effect of TAK-981 on SUMOylation in T cells has been demonstrated. The interferon (IFN)-like response mediated by TAK-981 inhibits the differentiation of Treg cells while enhancing various cytotoxic features of primary chronic lymphocytic leukemia (CLL)-derived CD8+ T cells, including degranulation (CD107a), and upregulated perforin, Granzyme B, and IFN-γ expression activity (46).
3.1.3 SUMOylation and Th17 cell differentiation
Th cell subsets are important players in chronic inflammation and insulin resistance in DM, with the Th17 cell population garnering significant interest from researchers due to its heterogeneity (47, 48). Th17 cells represent a critical component of the pathogenic T cell population. A variety of parenteral autoimmune diseases and tissue inflammation have been linked to Th17 cells48. In the absence of SENP2, the co-transcriptional factor Smad4 serves as the modification site of SUMO1 to up-regulate the expression level and nuclear localization of Smad4 and promote the differentiation of T cells to pathogenic Th17 cells (49). PIAS4 catalyzes SUMO3 transcription factor retinoic acid-related orphan receptor gamma t (RORγt) to SUMOylation at the lysine residue 31 (K31), promoting the binding of two RORγt binding proteins (KAT2A and SRC1) to RORγt, which drives the differentiation and development of Th17 cells (50). Under the catalysis of two SUMO E3 ligases, PIASxβ and PIAS3, SUMO1 modifies the K54 of phospholipase C-γ1 (PLC-γ1) to regulate T cell activation and induce IL-2 production by facilitating the assembly of PLC-γ1 microclusters (51).
3.1.4 SUMOylation and macrophage polarization
Macrophages are of significant importance in the regulation of the body’s immune response and metabolism. After polarization, macrophages form different subtypes: Th17 cells represent a critical component of the pathogenic T cell population. A variety of parenteral autoimmune diseases and tissue inflammation have been linked to Th17 cells, while M2 macrophages play an anti-inflammatory role (52). Studies have demonstrated that mice with conditional knockout of SENP3 exhibit diminished polarization of M1 macrophages and reduced production of pro-inflammatory cytokines under lipopolysaccharide (LPS) induction, thereby indicating that SENP3 plays an important pro-inflammatory role in LPS-induced lung injury and regulates M1 macrophage polarization by activating pyruvate kinase M2 (PKM2) in a hypoxia-inducible factor-1α(HIF-1α)dependent manner (53).
3.1.5 Summary
SUMOylation exerts distinct effects on various types of immune cells, with regulatory mechanisms that differ across cell types and involve multiple aspects such as activation, differentiation and functional activities, affecting the secretion of downstream cytokines (such as IL-2, IL-6, IL-8, IL-10, etc.) or direct killing effects. While maintaining or breaking immune tolerance, SUMOylation also regulates the production and quantity of various inflammatory mediators, affecting the inflammatory response.
The above analysis reveals an antagonistic relationship between Treg cells and Th17 cells. SUMOylation has been demonstrated to regulate both of these cell types, suggesting that the modulation of Treg/Th17 cells under SUMOylation conditions may prove beneficial for the advancement and prognosis of autoimmune diseases. Additionally, the opposite effects on macrophage polarization and activated cells indicate that it is feasible to create an environment conducive to M2 macrophage polarization rather than M1 macrophage polarization by influencing SUMOylation, thereby controlling chronic inflammation or excessively amplified inflammatory responses.
3.2 SUMOylation and inflammatory response
3.2.1 SUMOylation and the NF-KB signaling pathway
Post-translational modifications serve as critical regulators of immune signaling-related proteins. SUMOylated nuclear factor kappa B (NF-κB) p65 inhibits NF-κB activation and its downstream pathways in HCC cells by assembling with mesencephalic astrocyte-derived neurotrophic factor (MANF). As a transcription factor, NF-κB regulates gene expression activity, and its activation can result in the production of numerous inflammatory factors. The NF-κB signaling pathway is a classic pathway for regulating immune and inflammatory responses and is also key to the pathogenesis of DM and its complications. SUMO1 overexpression can also SUMOylate MANF and promote the nuclear import of MANF (54, 55).
Further study revealed that SUMOylated annexin-A1 (ANXA1) promotes inhibitory kappa B (IκB) kinase α (IKKα) degradation through selective autophagy, while numerous studies have indicated that IKKα/β-mediated phosphorylation-dependent degradation of IκB is the most critical step in canonical NF-κB signaling pathway activation, thereby inhibiting its overall activation (56).
Additionally, SENP6 could stabilize IKKα by deSUMOylating ANXA1, leading to inflammation (57). Under the regulation of the telomere-associated protein SLX4IP, the SUMO E3 ligase PIAS1 SUMOylates the telomere-binding protein RAP1, facilitating its interaction with IKK and subsequently leading to the activation of the transcription factor NF-κB (58). NF-κB essential molecule (NEMO) is a critical regulator in NF-κB. Intermittent hypoxia (IH) conditions enhance the SUMOylation of NEMO, resulting in NF-κB activation and upregulating the expression levels of tumor necrosis factor-alpha (TNF-α) and IL-6. SENP1-mediated deSUMOylation of NEMO can reduce the inflammatory response of microglia (59, 60). SENP2 can also inhibit the activation of the NF-κB signaling pathway through NEMO deSUMOylation, a mechanism that plays a crucial role in reducing doxorubicin resistance in breast cancer (61). The SUMO E3 ligase tripartite motifcontaining protein 60 (TRIM60) mediates the SUMOylation of TAK1 binding protein TAB2 at K329 and K562, and the SUMOylation of TAB2 interferes with the formation of TRAF6/TAB2/TAK1 complex to inhibit the MAPK/NF-κB pathway. This negatively regulates the innate immune response to toll-like receptor (TLR) signaling (62) (Figure 2).
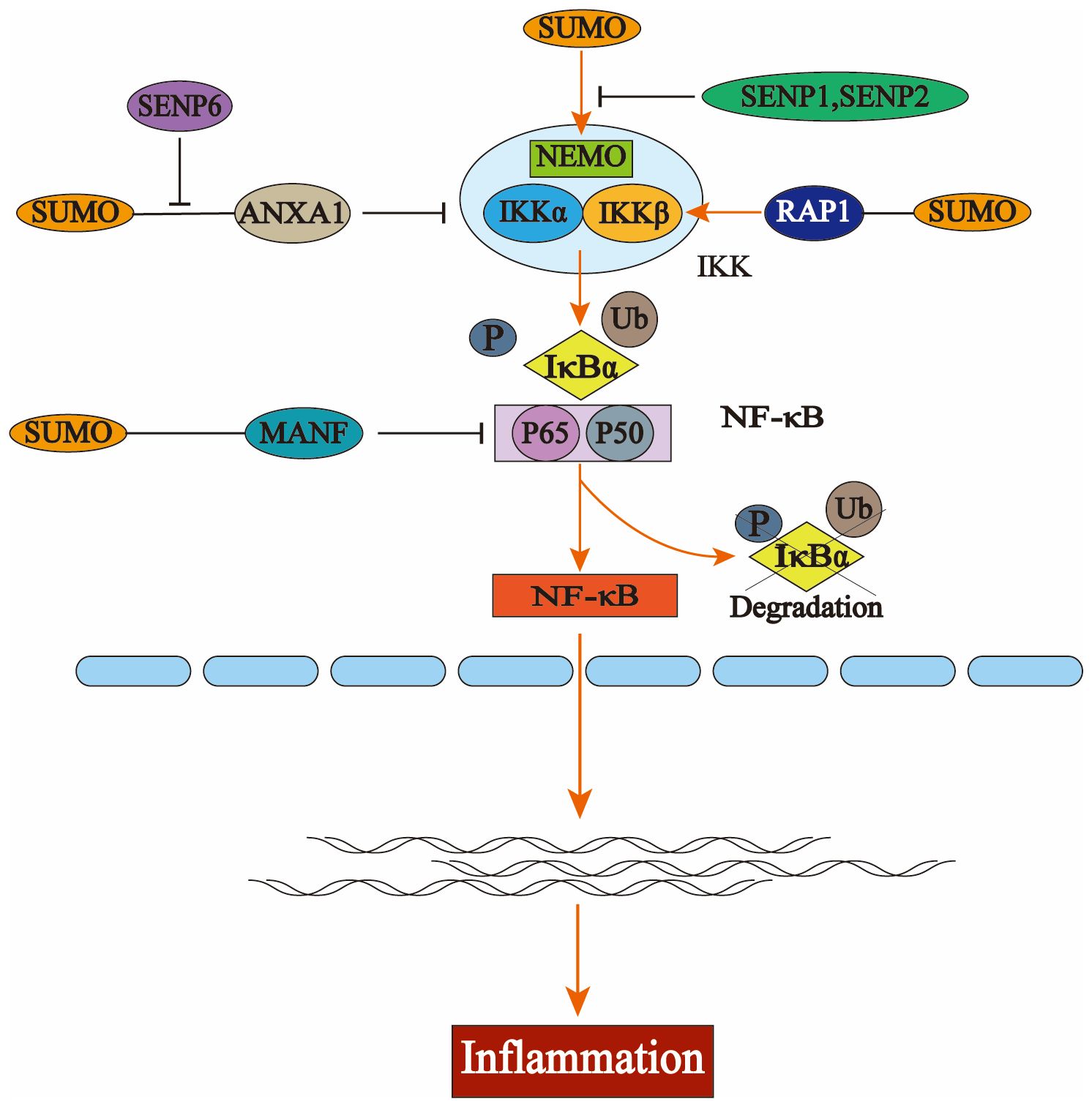
Figure 2. The activation process of nuclear factor kappa B (NF-κB) and the regulation of the NFκB signaling pathway by the small ubiquitin-like modifier (SUMO). In the resting state, NF-κB binds to the inhibitory protein IκBα to form an inactive trimer in the cytoplasm, thereby preventing its translocation into the nucleus. Upon stimulation by extracellular stressors, the inhibitory kappa Bn(IκB) kinase complex (IKK) is activated, resulting in phosphorylation and subsequent degradation of IκBα via the ubiquitin-proteasome system. This leads to dissociation of the trimer, allowing for nuclear translocation of the p50-p65 heterodimer that exhibits NF-κB activity and engages in gene transcription and protein synthesis. Several key components within the NF-κB pathway—including IκBα, NF-κB essential molecule (NEMO), P65, IKKα, and IKKβ—are subject to SUMO modification. The interaction between NEMO and SUMO facilitates the activation of IKK; similarly, SUMO-modified RAP1 enhances this activation process which ultimately promotes the degradation of IκBα and triggers NF-κB pathway activation. Additionally, the binding of annexin-A1 (ANXA1) or mesencephalic astrocyte-derived neurotrophic factor (MANF) to SUMO inhibits this pathway’s activation by interfering with key signaling molecules involved in NF-κB regulation.
3.2.2 SUMOylation and the JNK signaling pathway
C-Jun-N-Terminal kinase (JNK) is a member of the mitogen-activated protein kinase (MAPK) family and serves as a critical component of the inflammatory signaling system. Under conditions of elevated H2O2, JNK can be activated as a substrate for SUMO1, with both molecules participating in the apoptotic death pathway caused by oxidative stress. Curcumin acts as an antiinflammatory antioxidant molecule to counteract this effect, which has important implications in retinal layer associated diseases (63, 64).
3.2.3 SUMOylation and the NRF2 signaling pathway
Nuclear factor erythroid 2-related factor 2 (NRF2) has been demonstrated to be a key regulatory factor in maintaining redox homeostasis, typically existing in a low-activity state due to the negative regulation by kelch-like ECH-associated protein 1 (KEAP1), the substrate recognition component of the ubiquitin E3 ligase (65). Researchers have found that SUMOylation preserves and enhances the survival and function of β-cells while reducing the risk of DM. This process largely depends on the SUMOylation process of NRF2, a critical antioxidant factor, which plays a protective role in β-cells (66). SUMO2 has been demonstrated to modify NRF2 at K110 and K533, thereby influencing its transcriptional activity and effective nucleocytoplasmic localization (67). In addition, studies have shown that amyloid-β (aβ) can inhibit the binding of NRF2 and its activation signaling pathway by reducing the SUMOylation of both NRF2 and small musculoaponeurotic fibrosarcoma (sMaf) protein, which is essential for NRF2 activation. This process plays an important role in the onset and progression of Alzheimer’s disease (AD) (68). ROS could induce abnormal nucleolar activation of NRF2 through deSUMOylation of NRF2 mediated by SENP3 (69). Serine starvation diminishes SENP1 expression in HCC cells, facilitates SUMO1 modification of NRF2 at the conserved K110, enhances de novo serine synthesis, and perpetuates HCC tumorigenesis (70).
3.2.4 SUMOylation and the inflammasome
The assembly of inflammasome is a critical factor in immune response, and SUMOylation is also involved in the activation of inflammasome as a post-translational modification mechanism. The non-obese diabetic (NOD)-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) is essential for the activation of the NLRP3 inflammasome (71). The SUMO E3 ligase tripartite motifcontaining protein 28 (TRIM28) interacts with NLRP3, facilitating the SUMO1- and SUMO2/3catalyzed SUMOylation of NLRP3, thereby inhibiting its ubiquitination and subsequent proteasomal degradation. Consequently, TRIM28 stabilizes NLRP3 protein expression and enhances subsequent inflammasome activation (72). Other studies have similarly demonstrated that SUMO1 can mediate the SUMOylation of NLRP3 (73). In addition, previous studies have demonstrated that SUMOylation exerts an inhibitory effect on the activation of the NLRP3 inflammasome. For instance, the SUMOylation of NLRP3 mediated by mitochondrial‐anchored protein ligase (MAPL), identified as the first mitochondrial‐anchored SUMO E3 ligase, negatively regulates inflammasome formation, while SUMO-specific proteases SENP6 and SENP7 reverse this phenomenon (74). Additional experiments have also shown that the knockdown of SENP7 inhibits the activation of the NLRP3 inflammasome pathway (75). The nuclear zinc-finger and BTB domaincontaining protein 16 (ZBTB16) serves a positive regulatory role in the assembly of inflammasomes. The underlying mechanism involves ZBTB16 facilitating inflammasome assembly through the regulation of the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) by SUMO1 modification at K21 and/or K109 (76) (Table 2).
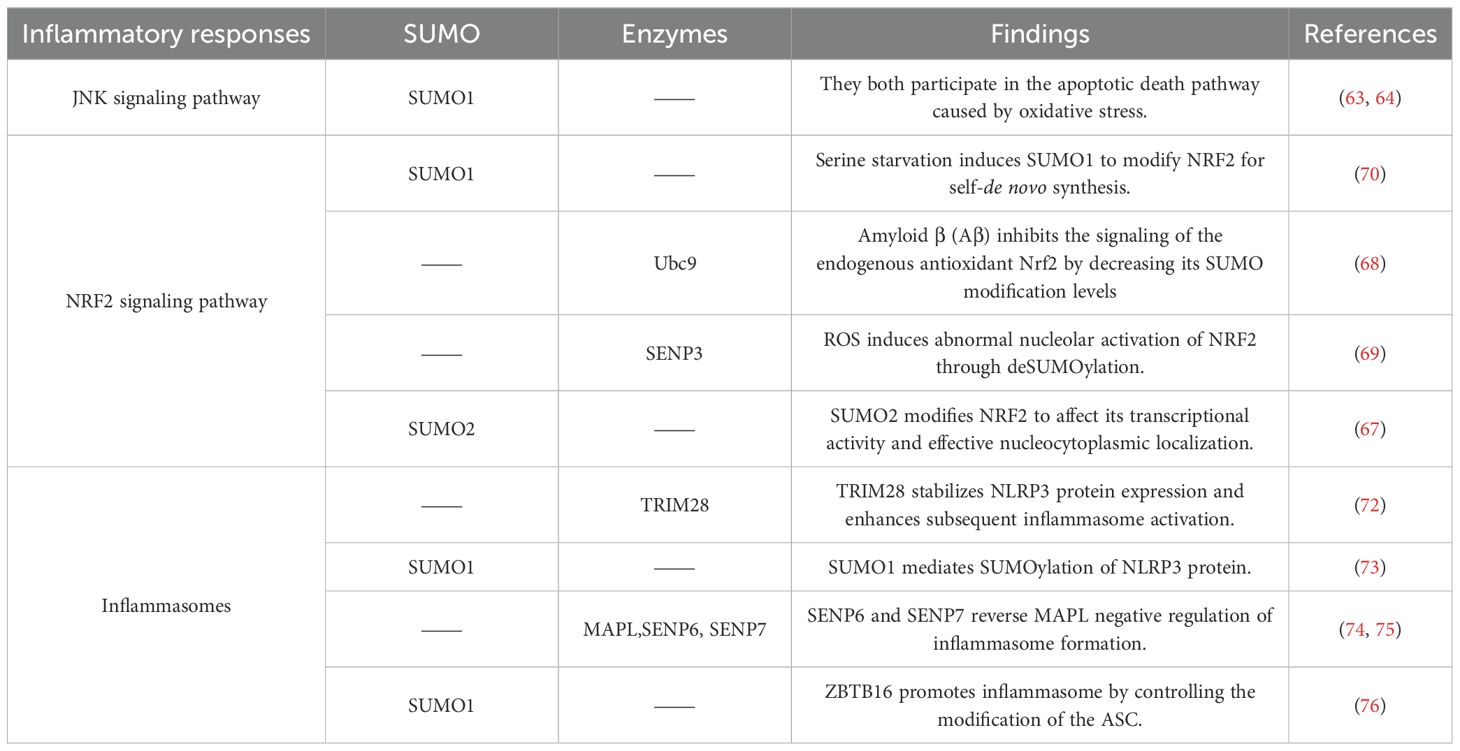
Table 2. SUMOylation and inflammatory response.
4 Diabetes and immune system interactions
4.1 Overview of diabetes
Diabetes mellitus represents a group of metabolic disorders characterized primarily by chronic hyperglycemia, which serves as a key diagnostic criterion. In addition, patients with diabetes have typical clinical manifestations including polydipsia, polyphagia and polyuria. Diabetes-associated complications, categorized into macrovascular (primarily cardiovascular disease) and microvascular complications (including diabetic nephropathy, retinopathy, and neuropathy), can lead to multi-organ damage. These complications significantly impair patients’ quality of life while increasing morbidity and mortality rates (77). Diabetes is generally divided into four clinical categories, namely type 1 diabetes(T1DM), type 2 diabetes(T2DM), gestational diabetes (GDM) and other specific types of diabetes, and the vast majority of them are type II diabetes, accounting for 90%-95% of all diabetes (3, 78). This review primarily focuses on type 1 and type 2 diabetes, which represent the most clinically significant forms of the disease.
T1DM and T2DM exhibit distinct heterogeneity, with significant differences in their pathogenesis, clinical manifestations, and disease progression. T1DM predominantly affects younger populations, particularly children and adolescents, although its prevalence is highest in adults due to patient longevity (79). Autoimmune mediated beta cell destruction leading to insufficient insulin secretion is a recognized mechanism, symptoms include polydipsia, polyuria, weight loss, and in severe cases, diabetic ketoacidosis. Currently, the main treatment is insulin replacement therapy (80, 81).
In contrast, T2DM typically manifests later in life and is frequently associated with obesity. Its pathogenesis involves progressive insulin secretory dysfunction coupled with insulin resistance. Notably, patients with T2DM exhibit a significantly higher risk of cardiovascular events compared to their T1DM counterparts (82).
4.2 Interaction between diabetes and the immune system
4.2.1 Interaction between T1DM and the immune system
It is well established that T1DM is an autoimmune disorder characterized by the destruction of β-cells mediated by T lymphocytes. T cells recognize islet antigens, drive islet autoimmunity and islet inflammatory infiltration, while activated autoreactive CD8+ T cells induce β-cell apoptosis through various mechanisms (83, 84), resulting in impaired insulin secretion. In addition to T cells, other immune components including innate immune cells (B cells, NK cells, macrophages, and neutrophils) and pro-inflammatory cytokines (such as TNF-α, IL-6, IFN-γ, and CXCL10) significantly contribute to disease progression by amplifying the autoimmune cascade (85–88).
Recent studies have demonstrated that β-cells are not merely passive ‘victims’ of autoimmunity, rather, they can also exacerbate the condition (89). Cellular alterations in β-cells, including enhanced immunogenicity, cellular senescence, oxidative stress, and endoplasmic reticulum dysfunction, can trigger the formation of novel antigenic epitopes, thereby amplifying immune responses. This self-perpetuating process represents a crucial mechanism through which β-cells contribute to T1DM pathogenesis (90). These results indicated that the islet - immune cell interaction promoted the progression of T1DM to a great extent, and the occurrence of T1DM could not be attributed to the immune system, and the involvement and metabolic changes of β cells were also worthy of consideration.
4.2.2 Interaction between T2DM and the immune system
T2DM is acknowledged as a metabolic disorder characterized by insulin resistance and progressive insulin insufficiency, with obesity identified as the primary risk factor predisposing individuals to T2DM (91). Adipose tissue hyperplasia results in elevated free fatty acid (FFA) levels, triggering the release of monocyte chemokines and subsequent macrophage activation (92), while also facilitating macrophage polarization (93) that exacerbates the inflammatory response. Research has demonstrated that cytokines secreted by activated macrophages not only reduce adipocyte insulin sensitivity but also promote further macrophage infiltration and inflammatory signaling amplification. This cascade ultimately disrupts insulin receptor signaling pathways in adipocytes, resulting in systemic insulin resistance (94). Macrophages can also directly or indirectly cause β-cell dysfunction, leading to impaired insulin secretion (95, 96). In addition, other immune cells (B cells and T cells) and their secreted cytokines have also been shown to contribute to the development of insulin resistance and T2DM (97, 98).
T2DM is characterized by complex metabolic disorders, and in addition to obesity, the gut microbiome has also been shown to be involved in the development of T2DM (99). Disruption of the intestinal barrier leads to bacterial penetration, and recognition of bacterial components [mainly lipopolysaccharide (LPS)] by Toll-like receptors (TLRS) and their adaptor molecules leads to the activation of inflammasomes and inflammatory signaling pathways, resulting in insulin resistance and chronic inflammation (100). Furthermore, studies have demonstrated that IL-17 receptor deficiency under high-fat diet (HFD) conditions disrupts neutrophil migration in the intestinal mucosa. This impairment results in gut microbiota dysbiosis and subsequent LPS translocation to adipose tissue, ultimately contributing to insulin resistance and metabolic dysfunction. Additionally, IL-17 receptor deficiency impairs the phosphorylation and activation of key kinases in insulin signaling pathways, leading to compensatory hyperinsulinemia and exacerbation of insulin resistance (101).
4.3 Summary of the role of SUMOylation regulating the immune microenvironment in the development of diabetes mellitus
4.3.1 The impact of SUMOylation on lipid metabolism
The function of adipose tissue is intricately linked to the pathogenesis of DM, with peripancreatic adipocytes (PAT) secreting pro-inflammatory cytokines that recruit immune cells, resulting in damage to pancreatic islet β-cells and exacerbating the progression of T1DM. The elevated SUMOylation of the component NEMO at the lysine-277/309 sites is pivotal in this process, as it induces NF-κB activation and cytokine expression in SENP1-deficient adipocytes. Moreover, NEMO SUMOylation and NF-κB activation in PAT exhibit greater sensitivity to variations in SENP1 expression and activity compared to other adipose depots (54). Relevant studies have also elaborated and summarized the role of SENP1-mediated protein SUMOylation in the pancreatic immune response, β-cell damage, and the progression of DM, while also proposing the use of NEMO inhibitory peptides or NF-κB inhibitors as potential therapeutic strategies for T1DM (102).
It is well established that inflammation plays a significant role in the pathogenesis of T2DM. In particular, inflammation within adipose tissue, resulting from both malnutrition and obesity, can impair insulin receptor function and diminish insulin sensitivity through the action of inflammatory mediators and immune cell infiltration (103). Recent evidence indicates that ubiquitin-conjugating enzyme E2 I (Ube2i) plays a crucial role in the maintenance of growth and normal function of white adipose tissue (WAT). Impaired WAT expansion, inflammatory responses, and adipocyte apoptosis were observed in Ube2i-deficient mice with adipocyte-specific deletion. These alterations led to metabolic abnormalities such as ectopic lipid deposition and insulin resistance in the liver, mirroring the insulin resistance seen in patients with adipose dystrophy. Notably, a sex difference was evident, with effects being more pronounced in females. This underscores the critical role of Ube2i in maintaining functional stability and normal expansion of adipose tissue, however, its specific downstream transcription factors and mechanisms of action require further investigation (104).
Another study demonstrated that adipocyte Ubc9 enhances the SUMOylation of Endoplasmic reticulum protein44 (ERp44) and promotes its covalent binding to endoplasmic reticulum oxidoreductase 1 (Ero1α), thereby exacerbating cell-to-cell transmission of endoplasmic reticulum stress (ER) signals and retention of Ero1α, which ultimately leads to disorders associated with ER stress. ER stress and impaired ER function disrupt systemic glucose homeostasis, resulting in abnormalities in lipid metabolism and insulin resistance. Ubc9 knockdown in adipocytes demonstrated that inhibition of SUMO modification attenuated high-fat-induced ER stress, facilitated lipolysis and energy metabolism, and restored ER function, ultimately leading to the alleviation of insulin resistance and obesity. These findings suggest that modulation of ERp44’s SUMOylation may represent a viable strategy for ameliorating obesity and insulin resistance in clinical settings (105). SUMOylation of SET structural domain bifurcation 1 (Setdb1) promotes Setdb1 to occupy the promoter site of the Pparg and Cebpa genes. Setdb1 functions as a histone methyltransferase responsible for H3K9 trimethylation, which increases H3K9 trimethylation in adipocytes and represses the expression of Pparg and Cebpa, ultimately leading to a reduction in lipid storage capacity. This process was validated in adipocyte-specific SENP2 knockout mice subjected to a diet rich in fats, which exhibited ectopic fat accumulation and insulin resistance pathology (106).
SUMOylation exerts diverse effects on lipid metabolism through its regulation of various target proteins. The primary mechanisms involve activation of adipose tissue inflammatory signaling pathways, induction of endoplasmic reticulum stress, and dysregulation of lipid homeostasis. Both adipose tissue inflammation and lipid metabolism dysregulation contribute to insulin receptor impairment, disrupted insulin signaling pathways, and subsequent development of insulin resistance and T2DM. Furthermore, adipose-derived inflammatory infiltration can induce pancreatic β-cell dysfunction, potentially exacerbating T1DM progression. These findings suggest that modulation of protein SUMOylation in adipocytes may represent a promising therapeutic strategy for mitigating insulin resistance and protecting β-cell function.
4.3.2 The impact of SUMOylation on transcription factors and their downstream effectors
It has been reported that under high glucose conditions, retinal microvascular endothelial cells (mRMEC) exhibit significantly elevated levels of SUMO1 and SUMO2/3 proteins, along with enhanced stabilization of RUNX family transcription factor 1 (RUNX1) protein through SUMO2/3dependent SUMOylation, a process that is heavily reliant on the K182 and K144 residues of RUNX1. RUNX1 protein enhances the proliferation, migration, and angiogenesis of mRMECs, contributing to the exacerbation of diabetic retinopathy (DR) symptoms. This process can be alleviated by SENP1 overexpression, suggesting a potential correlation between SUMOylation and the severity of DR (107, 108). Similarly, high glucose induction can elevate the expression levels of SUMO1 and silent information regulator 1 (SIRT1) proteins in human lens epithelial cells (HLECs), moreover, SUMO1 overexpression enhances the SUMOylation of IκBα, thereby stabilizing this protein and facilitating its binding to NF-κB p65, which inhibits NF-κB p65 activation and oxidative stress. This mechanism protects the lens from high glucose-induced oxidative damage. Collectively, these findings suggest that SUMO1 may represent a novel therapeutic target for the treatment of diabetic cataracts (DC) (109).
NF-κB functions as a transcription factor that regulates gene expression activity, and its activation can result in the production of substantial amounts of inflammatory factors. It has been previously mentioned that several signaling molecules within the NF-κB pathway, including IKKα, NEMO, and p65, are subject to modification by SUMOylation. Contrary to previous reports indicating that high glucose and high osmolality diminish the interaction between IκBα and SUMO1, reduced SUMOylation of IκBα leads to decreased expression levels of IκBα, resulting in the activation of NF-κB signaling, which is implicated in the pathogenesis of diabetic nephropathy (DN). Therefore, novel therapeutic strategies targeting specific regulators of the NF-κB pathway may prove effective in treating DN (110).
Previous studies have also investigated the complex role of mouse SUMO2 (mSUMO2) in the development of autoimmune diabetes by establishing genetically modified mice with a enhanced repressor of NF-κB (IκBαΔN) and overexpression of mSUMO2, which demonstrated that Th1 cytokines were suppressed in the IκBαΔN T cells. Conversely, T cells with elevated levels of mSUMO2 showed a reduction in both Th1 and Th2 cytokine production. Furthermore, IκBαΔN completely prevented diabetes regardless of mSUMO2 overexpression, however, mice expressing only mSUMO2 exhibited susceptibility to diabetes comparable to that of wild-type mice. It is suggested that SUMO2 may differentially inhibit NF-κB expression at least in T cells, but the specific regulatory mechanism and the signaling molecules involved remain to be clearly elucidated (111). Similarly, the further development of DM and its complications through activation of the NF-κB pathway can also be observed through the enhanced phosphorylation and SUMOylation of IκB kinase γ (IKKγ) (30).
The NF-κB pathway plays a vital role in the development of DM through various mechanisms. For instance, deficiency of thioredoxin 2 (Trx2) in adipose tissue results in the stimulation of NFκB, which mediates the accumulation of the autophagy receptor p62, leading to mitochondrial autophagy and subsequently promotes insulin resistance and lipid metabolism disorders in T2DM, thereby linking NF-κB to metabolic disorders associated with insulin resistance (112). NF-κB also upregulates the expression of complement C3, thereby promoting the development of DM (113), C3 is a crucial component in the activation process of all three complement pathways and is not only associated with insulin resistance (114), reflecting the progression of metabolic derangement but also serves as a biomarker for T1DM and T2DM (115, 116). Epidemiological studies have demonstrated that elevated plasma levels of complement C3 can increase the risk of diabetic retinopathy, diabetic nephropathy, and neuropathy (113) in the general population (117). Furthermore, the pathogenesis of diabetes-related periodontitis mediated by C3 in patients with T2DM is supported by existing studies (118). Induced by hyperglycemia, the NF-κB signaling pathway mediates the activation of valvular interstitial cells (VICs), resulting in valvular calcification and dysfunction, which significantly contributes to the exacerbation of valvular calcification in patients with T2DM suffering from aortic stenosis (AS) (119).
Moreover, the NF-κB signaling pathway, along with its related inflammatory mediators, could represent a critical nexus in microRNA-92a-mediated diabetic cardiovascular disease (120). The aforementioned findings suggest that inhibition of the NF-κB signaling pathway may serve as an effective therapeutic target for DM. The mechanisms underlying NF-κB pathway activation are intricate and involve a diverse array of signaling molecules. It is anticipated that further investigation into potential sumo-chemical modifications regulating the NF-κB pathway will yield significant insights, particularly regarding their critical roles in the pathogenesis of TM and its associated complications.
The age-related decrease in c-Maf SUMOylation within CD4 T cells negatively correlates with IL-21 expression, which facilitates the differentiation of T follicular helper (Tfh) cells and the proliferation of granzyme B-producing effector/memory CD8+ T cells, thereby contributing to the development of autoimmune diabetes in NOD mice. Moreover, the findings indicate that SUMOdeficient c-Maf inhibits Death domain-associated protein 6 (Daxx)/Histone Deacetylase (HDAC) recruitment to the IL-21 promoter (IL-21p) and enhances histone acetylation mediated by CREBbinding protein (CBP) and p300. It is hypothesized that inhibitors of CBP/p300 may mitigate several effects induced by IL-21 (121). This study elucidates a potential link between the SUMO status of a single transcription factor and the pathogenesis of autoimmune diabetes, detailing the downstream signaling molecules involved (e.g., IL-21, Daxx, etc.) and their pathogenic effects supported by experimental data. It suggests that the low SUMOylation levels of c-Maf may serve as a precursor to autoimmune diabetes, thereby facilitating early preventive and therapeutic interventions. Additionally, the role and downstream effects of IL-21 offer novel insights for the treatment of T1DM.
The role of transcription factor c-Maf in the pathogenesis of autoimmune diabetes is further corroborated by reports indicating that c-Maf directly influences the expression of glucose transporter genes Sglt2 and Glut2. Additionally, c-Maf deficiency has been shown to ameliorate diabetic nephropathy associated with hyperglycemia and oxidative stress by downregulating Sglt2 and Glut2 expression (122).
IL-21, a cytokine primarily produced by Tfh, is an essential element of the immune system that modulates various immune subpopulations, including B cells and CD8+ T cells. It is instrumental in the pathogenesis of multiple autoimmune diseases, including DM (123). IL-21 has been demonstrated to exhibit pro-diabetic activity, contributing to β-cell destruction and the onset of spontaneous T1DM (124). Additionally, it is implicated in Tfh cell-mediated chronic vascular inflammatory responses and the progression of diabetic retinopathy (125). Clinical trials have demonstrated that the combination of anti-IL-21 antibodies and liraglutide preserves β-cell activity and enhances islet function, offering a promising approach to addressing the treatment of T1DM (126).
Besides IL-21, other cytokines, including members of the IL-12 family members (IL-12, IL23, IL-27 and IL-35) (127), along with IL-6 (128), TNF-α (129), have been closely associated with DM and its complications. Identifying these cytokines in relation to the development of diabetes mellitus also provides targeted therapeutic avenues for managing diabetes and its related conditions.
Similarly in NOD mouse CD4+ T cells, SUMO1 reduces IL-4 production by enhancing its interaction with c-Maf, thereby inhibiting c-Maf from binding to the IL-4p half-MARE site. This reduction is detrimental to the normal development of protective Th2 cells. Numerous studies have demonstrated that T1DM is associated with an attenuated Th2 response, and thus this pathway may be involved in the immune bias of T1DM (130).
SENP1 has been demonstrated to enhance cell death in the insulin secreting cell line (INS-1 832/13) and human islet cells, leading to islet secretory dysfunction that correlates with decreased expression of inducible nitric oxide synthase (iNOS) and nuclear translocation of NF-κB. In contrast, up-regulation of SUMO1 or knockdown of SENP1 can reduce the nuclear translocation of NF-κB and its target gene expression, suggesting that preservation of islet activity and restoration of islet survival can be achieved through the processes of SUMOylation and de-SUMOylation (131). Glycolipotoxicity and the pro-inflammatory cytokine TNF-α convert extracellular stress into pattern recognition receptor (PKR) activation via the second messenger ceramide and activated PKR interacts with the conjugating enzyme Ubc9 to promote SUMOylation-dependent stabilization of p53, whose enhanced transcriptional activity leads to cell cycle arrest and disrupts β-cell proliferation, thereby contributing to the progression of T2DM. These findings indicate that targeting the ceramide/PKR/Ubc9/p53 signaling pathway may represent an effective strategy for the treatment of T2DM (132) (Table 3).

Table 3. Molecular mechanism and pathogenesis of SUMOylation on diabetes mellitus and its complications.
5 Conclusions and future perspectives
Recently, significant advancements in SUMOylation research has led to a deeper insight into the molecular mechanisms and enzymatic systems that are involved. More importantly, SUMOylation, as a novel reversible protein regulation process, plays a crucial role in the development of various physiopathological states within the body, offering new insights into the pathogenesis of multiple diseases. Building on this foundation, the present paper offers a comprehensive overview of how SUMO chemical modifications influence downstream signaling molecules by regulating the functions of proteins within the immune environment, thereby potentially impacting the pathogenesis of DM.
This review comprehensively examines the molecular mechanisms of SUMOylation and its regulatory effects on the immune system, with particular emphasis on their pivotal roles in the pathogenesis of both T1DM and T2DM. By exploring the intricate relationships among these components, we aim to clarify the critical function of SUMOylation in modulating the immune microenvironment and its implications for diabetes progression and associated complications.
Additionally, it summarizes several intricate molecular mechanisms, the majority of which are classical signaling pathways linked to immune homeostasis and inflammation, including adipose tissue inflammatory factors, NF-κB pathway activation, Tfh cytokines, and other cellular signaling pathways. This underscores the significant potential of SUMO in diabetes-related therapies. SUMOylation influences the activity of key protein kinases involved in the pathogenesis by disrupting downstream signaling pathways. Although several studies have supported this notion, most are based on similar animal models, and the detailed mechanisms of SUMO modification on the regulation of various physiological and pathological states of the organism remain inadequately elucidated. Furthermore, the proteins subject to this modification warrant further investigation. In future research, it is anticipated that a more comprehensive understanding of how SUMO-regulated key proteins induce islet inflammation and autoimmunity will be achieved, leading to the identification of new therapeutic targets for diabetes based on these key proteins and existing knowledge.
Author contributions
YZ: Writing – original draft, Writing – review & editing. SF: Writing – original draft, Writing – review & editing. YQ: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ong KL, Stafford LK, McLaughlin SA, Boyko EJ, Vollset SE, Smith AE, et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the global burden of disease study 2021. Lancet (London England). (2023) 402:203–34. doi: 10.1016/s0140-6736(23)01301-6
2. Jin C, Lai Y, Li Y, Teng D, Yang W, Teng W, et al. Changes in the prevalence of diabetes and control of risk factors for diabetes among chinese adults from 2007 to 2017: an analysis of repeated national cross-sectional surveys. J Diabetes. (2024) 16:e13492. doi: 10.1111/1753-0407.13492
3. ElSayed NA, Aleppo G, Bannuru RR, Bruemmer D, Collins BS, Ekhlaspour L, et al. Diagnosis and classification of diabetes: standards of care in diabetes-2024. Diabetes Care. (2024) 47:S20–s42. doi: 10.2337/dc24-S002
4. American Diabetes Association. Classification and diagnosis of diabetes: standards of medical care in diabetes-2019. Diabetes Care. (2019) 42:S13–s28. doi: 10.2337/dc19-S002
5. Guo H, Wu H, Li Z. The pathogenesis of diabetes. Int J Mol Sci. (2023) 24(8):6978. doi: 10.3390/ijms24086978
6. Ruze R, Liu T, Zou X, Song J, Chen Y, Xu R, et al. Obesity and type 2 diabetes mellitus: connections in epidemiology, pathogenesis, and treatments. Front Endocrinol (Lausanne). (2023) 14:1161521. doi: 10.3389/fendo.2023.1161521
7. Sticht J, Álvaro-Benito M, Konigorski S. Type 1 diabetes and the hla region: genetic association besides classical hla class ii genes. Front Genet. (2021) 12:683946. doi: 10.3389/fgene.2021.683946
8. Okura T, Gong L, Kamitani T, Wada T, Okura I, Wei CF, et al. Protection against fas/apo-1- and tumor necrosis factor-mediated cell death by a novel protein, sentrin. J Immunol (Baltimore Md: 1950). (1996) 157:4277–81. doi: 10.4049/jimmunol.157.10.4277
9. Su HL, Li SS. Molecular features of human ubiquitin-like sumo genes and their encoded proteins. Gene. (2002) 296:65–73. doi: 10.1016/s0378-1119(02)00843-0
10. Liu W, Wang Y, Bozi LHM, Fischer PD, Jedrychowski MP, Xiao H, et al. Lactate regulates cell cycle by remodelling the anaphase promoting complex. Nature. (2023) 616:790–7. doi: 10.1038/s41586-023-05939-3
11. Kramarz K, Schirmeisen K, Boucherit V, Ait Saada A, Lovo C, Palancade B, et al. The nuclear pore primes recombination-dependent DNA synthesis at arrested forks by promoting sumo removal. Nat Commun. (2020) 11:5643. doi: 10.1038/s41467-020-19516-z
12. Kumar S, Schoonderwoerd MJA, Kroonen JS, de Graaf IJ, Sluijter M, Ruano D, et al. Targeting pancreatic cancer by tak-981: A sumoylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut. (2022) 71:2266–83. doi: 10.1136/gutjnl-2021-324834
13. Guo D, Li M, Zhang Y, Yang P, Eckenrode S, Hopkins D, et al. A functional variant of sumo4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat Genet. (2004) 36:837–41. doi: 10.1038/ng1391
14. Liang YC, Lee CC, Yao YL, Lai CC, Schmitz ML, Yang WM. Sumo5, a novel poly-sumo isoform, regulates pml nuclear bodies. Sci Rep. (2016) 6:26509. doi: 10.1038/srep26509
15. Tatham MH, Jaffray E, Vaughan OA, Desterro JM, Botting CH, Naismith JH, et al. Polymeric chains of sumo-2 and sumo-3 are conjugated to protein substrates by sae1/sae2 and ubc9. J Biol Chem. (2001) 276:35368–74. doi: 10.1074/jbc.M104214200
16. Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers sumo-1 versus sumo-2/3. J Biol Chem. (2000) 275:6252–8. doi: 10.1074/jbc.275.9.6252
17. Kamitani T, Kito K, Nguyen HP, Fukuda-Kamitani T, Yeh ET. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J Biol Chem. (1998) 273:11349–53. doi: 10.1074/jbc.273.18.11349
18. Bohren KM, Nadkarni V, Song JH, Gabbay KH, Owerbach D. A M55v polymorphism in a novel sumo gene (Sumo-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J Biol Chem. (2004) 279:27233–8. doi: 10.1074/jbc.M402273200
19. Li H, Ning M, Li Q, Wang T, Li W, Xiao J, et al. The association of five polymorphisms with diabetic retinopathy in a chinese population. Ophthalmic Genet. (2023) 44:346–51. doi: 10.1080/13816810.2023.2194494
20. Tong Z, Qi J, Ma W, Wang D, Hu B, Li Y, et al. Sumo4 gene snp rs237025 and the synergistic effect with weight management: A study of risk factors and interventions for mets. Front Genet. (2021) 12:786393. doi: 10.3389/fgene.2021.786393
21. Li YY, Wang H, Yang XX, Geng HY, Gong G, Kim HJ, et al. Small ubiquitin-like modifier 4 (Sumo4) gene M55v polymorphism and type 2 diabetes mellitus: A meta-analysis including 6,823 subjects. Front Endocrinol. (2017) 8:303. doi: 10.3389/fendo.2017.00303
22. Coppola A, Tomasello L, Pizzolanti G, Pucci-Minafra I, Albanese N, Di Cara G, et al. In vitro phenotypic, genomic and proteomic characterization of a cytokine-resistant murine B-tc3 cell line. PloS One. (2012) 7:e32109. doi: 10.1371/journal.pone.0032109
23. Schmidt D, Müller S. Members of the pias family act as sumo ligases for C-jun and P53 and repress P53 activity. Proc Natl Acad Sci U.S.A. (2002) 99:2872–7. doi: 10.1073/pnas.052559499
24. Karvonen U, Jääskeläinen T, Rytinki M, Kaikkonen S, Palvimo JJ. Znf451 is a novel pml body- and sumo-associated transcriptional coregulator. J Mol Biol. (2008) 382:585–600. doi: 10.1016/j.jmb.2008.07.016
25. Zeng W, Gu S, Yu Y, Feng Y, Xiao M, Feng XH. Znf451 stabilizes twist2 through sumoylation and promotes epithelial-mesenchymal transition. Am J Cancer Res. (2021) 11:898–915.
26. Moreno-Oñate M, Herrero-Ruiz AM, García-Dominguez M, Cortés-Ledesma F, Ruiz JF. Ranbp2-mediated sumoylation promotes human DNA polymerase lambda nuclear localization and DNA repair. J Mol Biol. (2020) 432:3965–79. doi: 10.1016/j.jmb.2020.03.020
27. Liu X, Liu J, Xiao W, Zeng Q, Bo H, Zhu Y, et al. Sirt1 regulates N(6) -methyladenosine rna modification in hepatocarcinogenesis by inducing ranbp2-dependent fto sumoylation. Hepatol (Baltimore Md). (2020) 72:2029–50. doi: 10.1002/hep.31222
28. Werner A, Flotho A, Melchior F. The ranbp2/rangap1*Sumo1/ubc9 complex is a multisubunit sumo E3 ligase. Mol Cell. (2012) 46:287–98. doi: 10.1016/j.molcel.2012.02.017
29. Wu R, Fang J, Liu M, A J, Liu J, Chen W, et al. Sumoylation of the transcription factor zfhx3 at lys-2806 requires sae1, ubc9, and pias2 and enhances its stability and function in cell proliferation. J Biol Chem. (2020) 295:6741–53. doi: 10.1074/jbc.RA119.012338
30. Huang W, Liang Y, Dong J, Zhou L, Gao C, Jiang C, et al. Sumo E3 ligase piasy mediates high glucose-induced activation of nf-Kb inflammatory signaling in rat mesangial cells. Mediators Inflammation. (2017) 2017:1685194. doi: 10.1155/2017/1685194
31. Desterro JM, Rodriguez MS, Kemp GD, Hay RT. Identification of the enzyme required for activation of the small ubiquitin-like protein sumo-1. J Biol Chem. (1999) 274:10618–24. doi: 10.1074/jbc.274.15.10618
32. Gong L, Li B, Millas S, Yeh ET. Molecular cloning and characterization of human aos1 and uba2, components of the sentrin-activating enzyme complex. FEBS Lett. (1999) 448:185–9. doi: 10.1016/s0014-5793(99)00367-1
33. Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin-like protein smt3p is activated for conjugation to other proteins by an aos1p/uba2p heterodimer. EMBO J. (1997) 16:5509–19. doi: 10.1093/emboj/16.18.5509
34. Lin D, Tatham MH, Yu B, Kim S, Hay RT, Chen Y. Identification of a substrate recognition site on ubc9. J Biol Chem. (2002) 277:21740–8. doi: 10.1074/jbc.M108418200
35. Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated sumo conjugation revealed by a complex between ubiquitin-conjugating enzyme ubc9 and rangap1. Cell. (2002) 108:345–56. doi: 10.1016/s0092-8674(02)00630-x
36. Desterro JM, Thomson J, Hay RT. Ubch9 conjugates sumo but not ubiquitin. FEBS Lett. (1997) 417:297–300. doi: 10.1016/s0014-5793(97)01305-7
37. Li O, Ma Q, Li F, Cai GY, Chen XM, Hong Q. Progress of small ubiquitin-related modifiers in kidney diseases. Chin Med J (Engl). (2019) 132:466–73. doi: 10.1097/cm9.0000000000000094
38. Nayak A, Müller S. Sumo-specific proteases/isopeptidases: senps and beyond. Genome Biol. (2014) 15:422. doi: 10.1186/s13059-014-0422-2
39. Song J, Gong YH, Yan X, Liu Y, Zhang M, Luo J, et al. Regulatory T cells accelerate the repair process of renal fibrosis by regulating mononuclear macrophages. Am J Med Sci. (2021) 361:776–85. doi: 10.1016/j.amjms.2021.01.022
40. Jiao WE, Xu S, Qiao YL, Kong YG, Sun L, Deng YQ, et al. Notch2-dependent gata3+ Treg cells alleviate allergic rhinitis by suppressing the th2 cell response. Int Immunopharmacol. (2022) 112:109261. doi: 10.1016/j.intimp.2022.109261
41. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of il-10(+) and il-35(+) T(Reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. (2019) 20:724–35. doi: 10.1038/s41590-019-0346-9
42. Yu X, Lao Y, Teng XL, Li S, Zhou Y, Wang F, et al. Senp3 maintains the stability and function of regulatory T cells via bach2 desumoylation. Nat Commun. (2018) 9:3157. doi: 10.1038/s41467-018-05676-6
43. Wu Z, Huang H, Han Q, Hu Z, Teng XL, Ding R, et al. Senp7 senses oxidative stress to sustain metabolic fitness and antitumor functions of cd8+ T cells. J Clin Invest. (2022) 132(7):e155224. doi: 10.1172/jci155224
44. Demel UM, Böger M, Yousefian S, Grunert C, Zhang L, Hotz PW, et al. Activated sumoylation restricts mhc class I antigen presentation to confer immune evasion in cancer. J Clin Invest. (2022) 132(9):e152383. doi: 10.1172/jci152383
45. Wang Z, Pan B, Su L, Yu H, Wu X, Yao Y, et al. Sumoylation inhibitors activate anti-tumor immunity by reshaping the immune microenvironment in a preclinical model of hepatocellular carcinoma. Cell Oncol (Dordr). (2023) 47(2):513–32. doi: 10.1007/s13402-023-00880-z
46. Lam V, Roleder C, Liu T, Bruss N, Best S, Wang X, et al. T cell-intrinsic immunomodulatory effects of tak-981 (Subasumstat), a sumo-activating enzyme inhibitor, in chronic lymphocytic leukemia. Mol Cancer Ther. (2023) 22:1040–51. doi: 10.1158/1535-7163.Mct-22-0762
47. Schnell A, Huang L, Singer M, Singaraju A, Barilla RM, Regan BML, et al. Stem-like intestinal th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell. (2021) 184:6281–98.e23. doi: 10.1016/j.cell.2021.11.018
48. Schnell A, Littman DR, Kuchroo VK. T(H)17 cell heterogeneity and its role in tissue inflammation. Nat Immunol. (2023) 24:19–29. doi: 10.1038/s41590-022-01387-9
49. Yang TT, Chiang MF, Chang CC, Yang SY, Huang SW, Liao NS, et al. Senp2 restrains the generation of pathogenic th17 cells in mouse models of colitis. Commun Biol. (2023) 6:629. doi: 10.1038/s42003-023-05009-4
50. He Z, Zhang J, Huang Z, Du Q, Li N, Zhang Q, et al. Sumoylation of rorγt regulates T(H)17 differentiation and thymocyte development. Nat Commun. (2018) 9:4870. doi: 10.1038/s41467-018-07203-z
51. Wang QL, Liang JQ, Gong BN, Xie JJ, Yi YT, Lan X, et al. T cell receptor (Tcr)-induced plc-Γ1 sumoylation via piasxβ and pias3 sumo E3 ligases regulates the microcluster assembly and physiological function of plc-Γ1. Front Immunol. (2019) 10:314. doi: 10.3389/fimmu.2019.00314
52. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
53. He S, Fan C, Ji Y, Su Q, Zhao F, Xie C, et al. Senp3 facilitates M1 macrophage polarization via the hif-1α/pkm2 axis in lipopolysaccharide-induced acute lung injury. Innate Immun. (2023) 29:25–34. doi: 10.1177/17534259231166212
54. Shao L, Zhou HJ, Zhang H, Qin L, Hwa J, Yun Z, et al. Senp1-mediated nemo desumoylation in adipocytes limits inflammatory responses and type-1 diabetes progression. Nat Commun. (2015) 6:8917. doi: 10.1038/ncomms9917
55. Liu J, Wu Z, Han D, Wei C, Liang Y, Jiang T, et al. Mesencephalic astrocyte-derived neurotrophic factor inhibits liver cancer through small ubiquitin-related modifier (Sumo)Ylation-related suppression of nf-Kb/snail signaling pathway and epithelial-mesenchymal transition. Hepatol (Baltimore Md). (2020) 71:1262–78. doi: 10.1002/hep.30917
56. Li X, Xia Q, Mao M, Zhou H, Zheng L, Wang Y, et al. Annexin-A1 sumoylation regulates microglial polarization after cerebral ischemia by modulating ikkα Stability via selective autophagy. Sci Adv. (2021) 7(4):eabc5539. doi: 10.1126/sciadv.abc5539
57. Mao M, Xia Q, Zhan GF, Chu QJ, Li X, Lian HK. Senp6 Induces Microglial Polarization and Neuroinflammation through De-Sumoylation of Annexin-A1 after Cerebral Ischaemia-Reperfusion Injury. Cell bioscience. (2022) 12:113. doi: 10.1186/s13578-022-00850-2
58. Robinson NJ, Miyagi M, Scarborough JA, Scott JG, Taylor DJ, Schiemann WP. Slx4ip promotes rap1 sumoylation by pias1 to coordinate telomere maintenance through nf-Kb and notch signaling. Sci Signaling. (2021) 14(689):eabe9613. doi: 10.1126/scisignal.abe9613
59. Yang T, Sun J, Wei B, Liu S. Senp1-mediated nemo de-sumoylation inhibits intermittent hypoxia induced inflammatory response of microglia in vitro. J Cell Physiol. (2020) 235:3529–38. doi: 10.1002/jcp.29241
60. Wang H, Yang T, Sun J, Zhang S, Liu S. Senp1 Modulates Microglia-Mediated Neuroinflammation toward Intermittent Hypoxia-Induced Cognitive Decline through the De-Sumoylation of Nemo. J Cell Mol Med. (2021) 25:6841–54. doi: 10.1111/jcmm.16689
61. Gao X, Wu Y, Qiao L, Feng X. Senp2 suppresses nf-Kb activation and sensitizes breast cancer cells to doxorubicin. Eur J Pharmacol. (2019) 854:179–86. doi: 10.1016/j.ejphar.2019.03.051
62. Gu Z, Chen X, Yang W, Qi Y, Yu H, Wang X, et al. The sumoylation of tab2 mediated by trim60 inhibits mapk/nf-Kb activation and the innate immune response. Cell Mol Immunol. (2021) 18:1981–94. doi: 10.1038/s41423-020-00564-w
63. Buccarello L, Dragotto J, Iorio F, Hassanzadeh K, Corbo M, Feligioni M. The pivotal role of sumo-1-jnk-tau axis in an in vitro model of oxidative stress counteracted by the protective effect of curcumin. Biochem Pharmacol. (2020) 178:114066. doi: 10.1016/j.bcp.2020.114066
64. Buccarello L, Dragotto J, Hassanzadeh K, Maccarone R, Corbo M, Feligioni M. Retinal ganglion cell loss in an ex vivo mouse model of optic nerve cut is prevented by curcumin treatment. Cell Death Discovery. (2021) 7:394. doi: 10.1038/s41420-021-00760-1
65. Yamamoto M, Kensler TW, Motohashi H. The keap1-nrf2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. (2018) 98:1169–203. doi: 10.1152/physrev.00023.2017
66. He X, Lai Q, Chen C, Li N, Sun F, Huang W, et al. Both conditional ablation and overexpression of E2 sumo-conjugating enzyme (Ubc9) in mouse pancreatic beta cells result in impaired beta cell function. Diabetologia. (2018) 61:881–95. doi: 10.1007/s00125-017-4523-9
67. Walters TS, McIntosh DJ, Ingram SM, Tillery L, Motley ED, Arinze IJ, et al. Sumo-modification of human nrf2 at K(110) and K(533) regulates its nucleocytoplasmic localization, stability and transcriptional activity. Cell Physiol biochemistry: Int J Exp Cell physiology biochemistry Pharmacol. (2021) 55:141–59. doi: 10.33594/000000351
68. Wang P, Wang X, Qiao K, Zhang Y, Nie Q, Cui J, et al. Reduced sumoylation of nrf2 signaling contributes to its inhibition induced by amyloid-B. Neurosci Lett. (2023) 799:137118. doi: 10.1016/j.neulet.2023.137118
69. Zhou Z, Xu J, Bao X, Shi J, Liu B, Chen Y, et al. Nuclear nrf2 activity in laryngeal carcinoma is regulated by senp3 after cisplatin-induced reactive oxygen species stress. J Cancer. (2019) 10:3427–34. doi: 10.7150/jca.30318
70. Guo H, Xu J, Zheng Q, He J, Zhou W, Wang K, et al. Nrf2 sumoylation promotes de novo serine synthesis and maintains hcc tumorigenesis. Cancer Lett. (2019) 466:39–48. doi: 10.1016/j.canlet.2019.09.010
71. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. Nalp3 forms an il-1beta-processing inflammasome with increased activity in muckle-wells autoinflammatory disorder. Immunity. (2004) 20:319–25. doi: 10.1016/s1074-7613(04)00046-9
72. Qin Y, Li Q, Liang W, Yan R, Tong L, Jia M, et al. Trim28 sumoylates and stabilizes nlrp3 to facilitate inflammasome activation. Nat Commun. (2021) 12:4794. doi: 10.1038/s41467-021-25033-4
73. Shao L, Liu Y, Wang W, Li A, Wan P, Liu W, et al. Sumo1 sumoylates and senp3 desumoylates nlrp3 to orchestrate the inflammasome activation. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2020) 34:1497–515. doi: 10.1096/fj.201901653R
74. Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH, et al. Sumo-mediated regulation of nlrp3 modulates inflammasome activity. Nat Commun. (2018) 9:3001. doi: 10.1038/s41467-018-05321-2
75. Li X, Jiao F, Hong J, Yang F, Wang L, Gong Z. Senp7 knockdown inhibited pyroptosis and nf-Kb/nlrp3 inflammasome pathway activation in raw 264.7 cells. Sci Rep. (2020) 10:16265. doi: 10.1038/s41598-020-73400-w
76. Dong D, Du Y, Fei X, Yang H, Li X, Yang X, et al. Inflammasome activity is controlled by zbtb16-dependent sumoylation of asc. Nat Commun. (2023) 14:8465. doi: 10.1038/s41467-023-43945-1
77. Cole JB, Florez JC. Genetics of diabetes mellitus and diabetes complications. Nat Rev Nephrol. (2020) 16:377–90. doi: 10.1038/s41581-020-0278-5
78. Harreiter J, Roden M. Diabetes mellitus: definition, classification, diagnosis, screening and prevention (Update 2023). Wiener klinische Wochenschrift. (2023) 135:7–17. doi: 10.1007/s00508-022-02122-y
79. Vanderniet JA, Jenkins AJ, Donaghue KC. Epidemiology of type 1 diabetes. Curr Cardiol Rep. (2022) 24:1455–65. doi: 10.1007/s11886-022-01762-w
80. Zajec A, Trebušak Podkrajšek K, Tesovnik T, Šket R, Čugalj Kern B, Jenko Bizjan B, et al. Pathogenesis of type 1 diabetes: established facts and new insights. Genes. (2022) 13(4):706. doi: 10.3390/genes13040706
81. Stene LC, Lernmark A. Epidemiology and pathogenesis of type 1 diabetes. In: Gruessner RWG, Gruessner AC, editors. Transplantation of the Pancreas., vol. . p . Springer International Publishing, Cham (2023). p. 13–39.
82. Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci. (2020) 21(17):6275. doi: 10.3390/ijms21176275
83. Knight RR, Kronenberg D, Zhao M, Huang GC, Eichmann M, Bulek A, et al. Human B-cell killing by autoreactive preproinsulin-specific cd8 T cells is predominantly granule-mediated with the potency dependent upon T-cell receptor avidity. Diabetes. (2013) 62:205–13. doi: 10.2337/db12-0315
84. Trivedi P, Graham KL, Krishnamurthy B, Fynch S, Slattery RM, Kay TW, et al. Perforin facilitates beta cell killing and regulates autoreactive cd8+ T-cell responses to antigen in mouse models of type 1 diabetes. Immunol Cell Biol. (2016) 94:334–41. doi: 10.1038/icb.2015.89
85. Kendall PL, Woodward EJ, Hulbert C, Thomas JW. Peritoneal B cells govern the outcome of diabetes in non-obese diabetic mice. Eur J Immunol. (2004) 34:2387–95. doi: 10.1002/eji.200324744
86. Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “Speed congenic” Stock of nod.Ig mu null mice. J Exp Med. (1996) 184:2049–53. doi: 10.1084/jem.184.5.2049
87. Bruggeman Y, Martens PJ, Sassi G, Viaene M, Wasserfall CH, Mathieu C, et al. Footprint of pancreas infiltrating and circulating immune cells throughout type 1 diabetes development. Front Endocrinol. (2023) 14:1275316. doi: 10.3389/fendo.2023.1275316
88. Bender C, Rajendran S, von Herrath MG. New insights into the role of autoreactive cd8 T cells and cytokines in human type 1 diabetes. Front Endocrinol. (2020) 11:606434. doi: 10.3389/fendo.2020.606434
89. Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A. Type 1 diabetes mellitus as a disease of the B-cell (Do not blame the immune system)? Nat Rev Endocrinol. (2021) 17:150–61. doi: 10.1038/s41574-020-00443-4
90. Atkinson MA, Mirmira RG. The pathogenic “Symphony” in type 1 diabetes: A disorder of the immune system, B Cells, and exocrine pancreas. Cell Metab. (2023) 35:1500–18. doi: 10.1016/j.cmet.2023.06.018
91. Okamura T, Hashimoto Y, Hamaguchi M, Obora A, Kojima T, Fukui M. Ectopic fat obesity presents the greatest risk for incident type 2 diabetes: A population-based longitudinal study. Int J Obes (2005). (2019) 43:139–48. doi: 10.1038/s41366-018-0076-3
92. Jiao P, Chen Q, Shah S, Du J, Tao B, Tzameli I, et al. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: involvement of nuclear factor-kappab and C-jun nh2-terminal kinase pathways. Diabetes. (2009) 58:104–15. doi: 10.2337/db07-1344
93. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/jci29881
94. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. (2003) 112:1821–30. doi: 10.1172/jci19451
95. Ying W, Fu W, Lee YS, Olefsky JM. The role of macrophages in obesity-associated islet inflammation and B-cell abnormalities. Nat Rev Endocrinol. (2020) 16:81–90. doi: 10.1038/s41574-019-0286-3
96. Wentworth JM, Fourlanos S, Harrison LC. Reappraising the stereotypes of diabetes in the modern diabetogenic environment. Nat Rev Endocrinol. (2009) 5:483–9. doi: 10.1038/nrendo.2009.149
97. DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U.S.A. (2013) 110:5133–8. doi: 10.1073/pnas.1215840110
98. Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, et al. T-lymphocyte infiltration in visceral adipose tissue: A primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arteriosclerosis thrombosis Vasc Biol. (2008) 28:1304–10. doi: 10.1161/atvbaha.108.165100
99. Zhong H, Ren H, Lu Y, Fang C, Hou G, Yang Z, et al. Distinct gut metagenomics and metaproteomics signatures in prediabetics and treatment-naïve type 2 diabetics. EBioMedicine. (2019) 47:373–83. doi: 10.1016/j.ebiom.2019.08.048
100. Scheithauer TPM, Rampanelli E, Nieuwdorp M, Vallance BA, Verchere CB, van Raalte DH, et al. Gut microbiota as a trigger for metabolic inflammation in obesity and type 2 diabetes. Front Immunol. (2020) 11:571731. doi: 10.3389/fimmu.2020.571731
101. Pérez MM, Martins LMS, Dias MS, Pereira CA, Leite JA, Gonçalves ECS, et al. Interleukin-17/interleukin-17 receptor axis elicits intestinal neutrophil migration, restrains gut dysbiosis and lipopolysaccharide translocation in high-fat diet-induced metabolic syndrome model. Immunology. (2019) 156:339–55. doi: 10.1111/imm.13028
102. Shao L, Feng B, Zhang Y, Zhou H, Ji W, Min W. The role of adipose-derived inflammatory cytokines in type 1 diabetes. Adipocyte. (2016) 5:270–4. doi: 10.1080/21623945.2016.1162358
103. Kahn CR, Wang G, Lee KY. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest. (2019) 129:3990–4000. doi: 10.1172/jci129187
104. Cox AR, Chernis N, Kim KH, Masschelin PM, Saha PK, Briley SM, et al. Ube2i deletion in adipocytes causes lipoatrophy in mice. Mol Metab. (2021) 48:101221. doi: 10.1016/j.molmet.2021.101221
105. Xie H, Wang YH, Liu X, Gao J, Yang C, Huang T, et al. Sumoylation of erp44 enhances ero1α Er retention contributing to the pathogenesis of obesity and insulin resistance. Metabolism. (2023) 139:155351. doi: 10.1016/j.metabol.2022.155351
106. Zheng Q, Cao Y, Chen Y, Wang J, Fan Q, Huang X, et al. Senp2 regulates adipose lipid storage by de-sumoylation of setdb1. J Mol Cell Biol. (2018) 10:258–66. doi: 10.1093/jmcb/mjx055
107. Zhang W, Li F, Hou J, Cheng Y, Zhang W, Liang X, et al. Aberrant sumo2/3 modification of runx1 upon senp1 inhibition is linked to the development of diabetic retinopathy in mice. Exp Eye Res. (2023) 237:109695. doi: 10.1016/j.exer.2023.109695
108. Zhang W, Zhang D, Cheng Y, Liang X, Wang J. Runx1 regulates tff1 expression to expedite viability of retinal microvascular endothelial cells in mice with diabetic retinopathy. Exp Eye Res. (2022) 217:108969. doi: 10.1016/j.exer.2022.108969
109. Han X, Dong XX, Shi MY, Feng L, Wang XL, Zhang JS, et al. Sumoylation and deacetylation affect nf-Kb P65 activity induced by high glucose in human lens epithelial cells. Int J Ophthalmol. (2019) 12:1371–9. doi: 10.18240/ijo.2019.09.01
110. Huang W, Xu L, Zhou X, Gao C, Yang M, Chen G, et al. High glucose induces activation of nf-Kb inflammatory signaling through Iκbα Sumoylation in rat mesangial cells. Biochem Biophys Res Commun. (2013) 438:568–74. doi: 10.1016/j.bbrc.2013.07.065
111. Hwang KW, Won TJ, Kim H, Chun HJ, Chun T, Park Y. Erratum to ‘‘Characterization of the regulatory roles of the sumo. Diabetes Metab Res Rev. (2012) 28:196–202. doi: 10.1002/dmrr.2273
112. He F, Huang Y, Song Z, Zhou HJ, Zhang H, Perry RJ, et al. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J Exp Med. (2021) 218(3):e20201416. doi: 10.1084/jem.20201416
113. Jiang T, Li Y, He S, Huang N, Du M, Zhai Q, et al. Reprogramming astrocytic ndrg2/nf-Kb/C3 signaling restores the diabetes-associated cognitive dysfunction. EBioMedicine. (2023) 93:104653. doi: 10.1016/j.ebiom.2023.104653
114. Wlazlo N, van Greevenbroek MM, Ferreira I, Feskens EJ, van der Kallen CJ, Schalkwijk CG, et al. Complement factor 3 is associated with insulin resistance and with incident type 2 diabetes over a 7-year follow-up period: the codam study. Diabetes Care. (2014) 37:1900–9. doi: 10.2337/dc13-2804
115. Sarkar S, Elliott EC, Henry HR, Ludovico ID, Melchior JT, Frazer-Abel A, et al. Systematic review of type 1 diabetes biomarkers reveals regulation in circulating proteins related to complement, lipid metabolism, and immune response. Clin Proteomics. (2023) 20:38. doi: 10.1186/s12014-023-09429-6
116. Jiang J, Wang H, Liu K, He S, Li Z, Yuan Y, et al. Association of complement C3 with incident type 2 diabetes and the mediating role of bmi: A 10-year follow-up study. J Clin Endocrinol Metab. (2023) 108:736–44. doi: 10.1210/clinem/dgac586
117. Rasmussen KL, Nordestgaard BG, Nielsen SF. Complement C3 and risk of diabetic microvascular disease: A cohort study of 95202 individuals from the general population. Clin Chem. (2018) 64:1113–24. doi: 10.1373/clinchem.2018.287581
118. Li Y, Wang X, Wang S, Zhu C, Guo J, Li K, et al. Complement 3 mediates periodontal destruction in patients with type 2 diabetes by regulating macrophage polarization in periodontal tissues. Cell proliferation. (2020) 53:e12886. doi: 10.1111/cpr.12886
119. Kopytek M, Mazur P, Ząbczyk M, Undas A, Natorska J. Diabetes concomitant to aortic stenosis is associated with increased expression of nf-Kb and more pronounced valve calcification. Diabetologia. (2021) 64:2562–74. doi: 10.1007/s00125-021-05545-w
120. Wang WY, Zheng YS, Li ZG, Cui YM, Jiang JC. Mir-92a contributes to the cardiovascular disease development in diabetes mellitus through nf-Kb and downstream inflammatory pathways. Eur Rev Med Pharmacol Sci. (2019) 23:3070–9. doi: 10.26355/eurrev_201904_17589
121. Hsu CY, Yeh LT, Fu SH, Chien MW, Liu YW, Miaw SC, et al. Sumo-defective C-maf preferentially transactivates il21 to exacerbate autoimmune diabetes. J Clin Invest. (2018) 128:3779–93. doi: 10.1172/jci98786
122. Fujino M, Morito N, Hayashi T, Ojima M, Ishibashi S, Kuno A, et al. Transcription factor C-maf deletion improves streptozotocin-induced diabetic nephropathy by directly regulating sglt2 and glut2. JCI Insight. (2023) 8(6):e163306. doi: 10.1172/jci.insight.163306
123. Ren HM, Lukacher AE, Rahman ZSM, Olsen NJ. New developments implicating il-21 in autoimmune disease. J Autoimmun. (2021) 122:102689. doi: 10.1016/j.jaut.2021.102689
124. Sutherland AP, Van Belle T, Wurster AL, Suto A, Michaud M, Zhang D, et al. Interleukin-21 is required for the development of type 1 diabetes in nod mice. Diabetes. (2009) 58:1144–55. doi: 10.2337/db08-0882
125. Liu Y, Yang Z, Lai P, Huang Z, Sun X, Zhou T, et al. Bcl-6-directed follicular helper T cells promote vascular inflammatory injury in diabetic retinopathy. Theranostics. (2020) 10:4250–64. doi: 10.7150/thno.43731
126. von Herrath M, Bain SC, Bode B, Clausen JO, Coppieters K, Gaysina L, et al. Anti-interleukin-21 antibody and liraglutide for the preservation of B-cell function in adults with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. (2021) 9:212–24. doi: 10.1016/s2213-8587(21)00019-x
127. Luo J, Ning T, Li X, Jiang T, Tan S, Ma D. Targeting il-12 family cytokines: A potential strategy for type 1 and type 2 diabetes mellitus. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2024) 170:115958. doi: 10.1016/j.biopha.2023.115958
128. Gunes A, Schmitt C, Bilodeau L, Huet C, Belblidia A, Baldwin C, et al. Il-6 trans-signaling is increased in diabetes, impacted by glucolipotoxicity, and associated with liver stiffness and fibrosis in fatty liver disease. Diabetes. (2023) 72:1820–34. doi: 10.2337/db23-0171
129. Roca-Rivada A, Marín-Cañas S, Colli ML, Vinci C, Sawatani T, Marselli L, et al. Inhibition of the type 1 diabetes candidate gene ptpn2 aggravates tnf-A-induced human beta cell dysfunction and death. Diabetologia. (2023) 66:1544–56. doi: 10.1007/s00125-023-05908-5
130. Leavenworth JW, Ma X, Mo YY, Pauza ME. Sumo conjugation contributes to immune deviation in nonobese diabetic mice by suppressing C-maf transactivation of il-4. J Immunol. (2009) 183:1110–9. doi: 10.4049/jimmunol.0803671
131. Hajmrle C, Ferdaoussi M, Plummer G, Spigelman AF, Lai K, Manning Fox JE, et al. Sumoylation protects against il-1β-induced apoptosis in ins-1 832/13 cells and human islets. Am J Physiol Endocrinol Metab. (2014) 307:E664–73. doi: 10.1152/ajpendo.00168.2014
132. Song Y, Wan X, Gao L, Pan Y, Xie W, Wang H, et al. Activated pkr inhibits pancreatic B-cell proliferation through sumoylation-dependent stabilization of P53. Mol Immunol. (2015) 68:341–9. doi: 10.1016/j.molimm.2015.09.007
133. Wang C, Xiao Y, Lao M, Wang J, Xu S, Li R, et al. Increased sumo-activating enzyme sae1/uba2 promotes glycolysis and pathogenic behavior of rheumatoid fibroblast-like synoviocytes. JCI Insight. (2020) 5(18):e135935. doi: 10.1172/jci.insight.135935
134. Hoard TM, Yang XP, Jetten AM, ZeRuth GT. Pias-family proteins negatively regulate glis3 transactivation function through sumo modification in pancreatic B Cells. Heliyon. (2018) 4:e00709. doi: 10.1016/j.heliyon.2018.e00709
135. Feng Y, Wu H, Xu Y, Zhang Z, Liu T, Lin X, et al. Zinc finger protein 451 is a novel smad corepressor in transforming growth factor-B Signaling. J Biol Chem. (2014) 289:2072–83. doi: 10.1074/jbc.M113.526905
Keywords: immune, SUMOylation, diabetes mellitus, inflammation, posttranslation modification, signaling pathway
Citation: Zhuo Y, Fu S and Qiu Y (2025) Regulation of the immune microenvironment by SUMO in diabetes mellitus. Front. Immunol. 16:1506500. doi: 10.3389/fimmu.2025.1506500
Received: 05 October 2024; Accepted: 12 February 2025;
Published: 26 February 2025.
Edited by:
Uzma Saqib, Indian Institute of Technology Indore, IndiaReviewed by:
Sutripta Sarkar, Barrackpore Rastraguru Surendranath College, IndiaXinxing Wan, Central South University, China
Copyright © 2025 Zhuo, Fu and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue Qiu, MTUxNzkyODA4MDZAMTYzLmNvbQ==
†These authors have contributed equally to this work