 Camila de Freitas Oliveira-Tore1
Camila de Freitas Oliveira-Tore1 Amarilis Giaretta de Moraes2
Amarilis Giaretta de Moraes2 Helena Musetti B. S. Plácido1
Helena Musetti B. S. Plácido1 Nathalia M. D. L. Signorini1Pamela Dias Fontana1Tatiane da Piedade Batista Godoy1
Nathalia M. D. L. Signorini1Pamela Dias Fontana1Tatiane da Piedade Batista Godoy1 Angelica Beate Winter Boldt2†
Angelica Beate Winter Boldt2† Iara de Messias1*†
Iara de Messias1*†- 1Laboratory Molecular Immunopathology, Postgraduate Program in Internal Medicine and Health Sciences, Federal University of Paraná (UFPR), Curitiba, PR, Brazil
- 2Laboratory Human Molecular Genetics, Postgraduate Program in Genetics, Department of Genetics, Federal University of Paraná (UFPR), Curitiba, PR, Brazil
The Complement System (CS) comprises three catalytic pathways that can be activated by specific immune triggers. However, within the tumor microenvironment (TME), CS intracellular components, recently named as complosome, play roles that extend beyond the activation and regulation of its pathways. The interaction between TME elements and tumor cells alters the local immune response, leading to inflammation, cell proliferation, and tumor invasion. Our focus is on understanding the significance of complosome and non-canonical pathways in cancer. In this scoping review, we analyzed 45 articles that discussed the various roles of CS components in carcinogenesis. Many CS components, including C1q, C3a-C3aR, C5a-C5aR, factor H, and properdin, some of them at the intracellular level, may play a dual role in tumor progression, demonstrating either anti-tumor or pro-tumor activity independent of complement pathway activation. The specific function of each component can influence both the type and stage of tumor cells. There is a notable lack of studies on the role of the lectin pathway in tumor development, and this knowledge gap must be addressed to fully understand the role of complosome in cancer. Nevertheless, the activation of CS and the roles of its components in complosome pathways are crucial steps in tumor development.
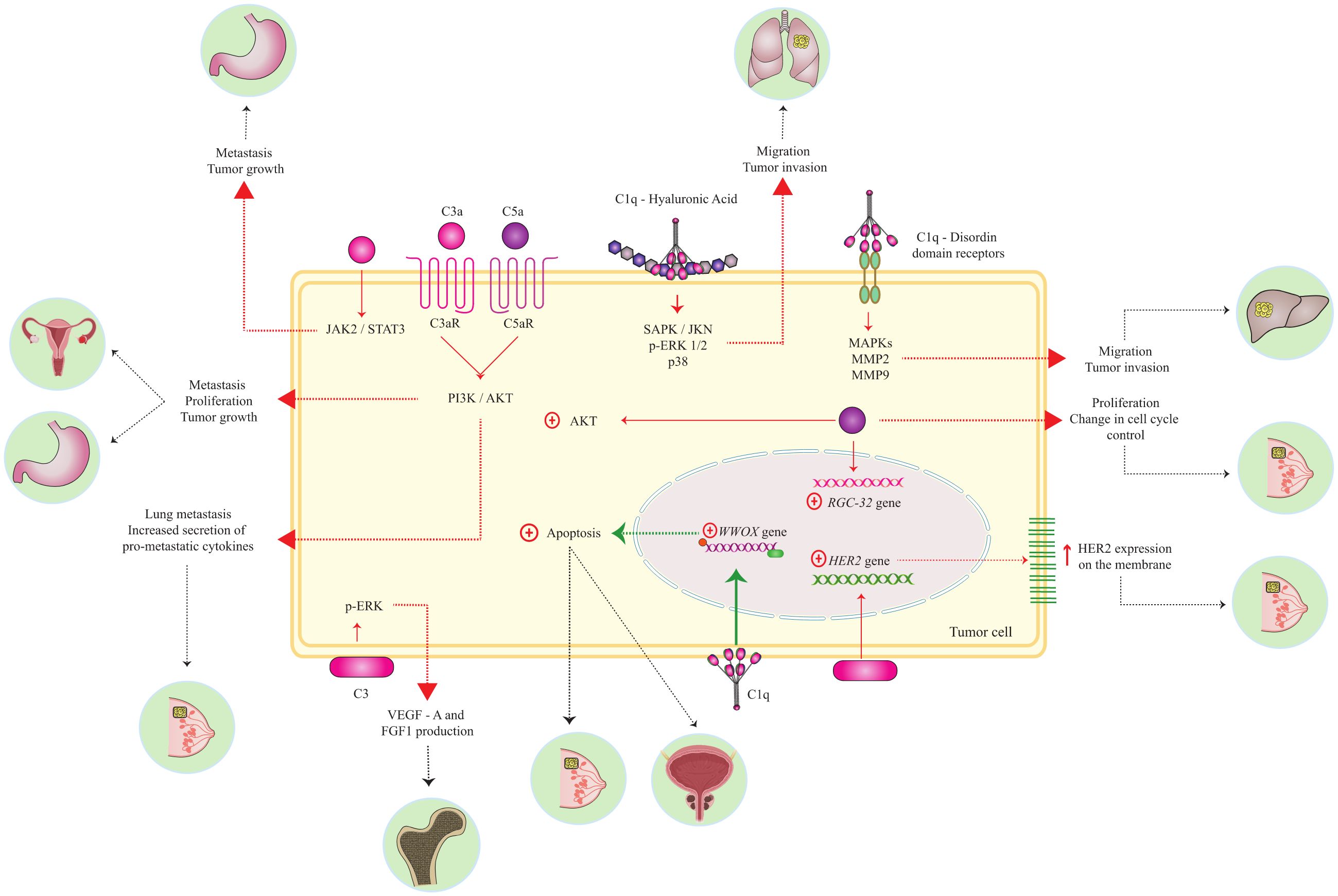
Graphical Abstract.
Highlights
● C1q has an anti-tumor role through the WWOX oncogene in breast and colon cancers.
● The C3a-C3aR and C5a-C5aR axis activate the PI3K/AKT and ERK/MEK1-2 pathways.
● Activation of C3 plays a negative regulatory role for the oncogene Her2.
● C5a promotes overexpression of the RGCC gene, which controls cell cycle progression.
● Properdin showed an antitumor function by regulating pro-apoptotic factors.
1 Introduction
The complement system (CS) comprises a set of soluble, membrane-bound, and regulatory proteins that are involved in homeostasis processes. It serves as a central component of innate immunity while also providing a critical link to adaptive immunity. CS is known as a key mediator of inflammation, coagulation cascade, in the elimination of immune complexes and apoptotic cells (1–3).
Canonical complement activation induces proteolytic cleavage of key molecules through three pathways: the classical (CP), lectin (LP), and alternative (AP) pathways. CP activation occurs through antibody binding to the C1q-C1r/C1s complex (4, 5). LP is activated through the recognition of pathogen-associated, damage-associated, and altered cell-associated molecular patterns (PAMPs, DAMPs, and ACAMPs) by pattern recognition receptors (PRRs, as mannose-binding lectin -MBL, ficolins, and collectins), thereby activating the MBL-associated serine proteases MASP-1 or MASP-2 (mannan-binding lectin-associated serine proteases -1 or -2). Activation of the CP and LP pathways generate a crucial C3 convertase - C2aC4b. Yet the AP is continuously activated at low levels by the spontaneous hydrolysis of native C3, also circulating as C3(H2O), generating the C3bBb convertase (Figure 1). C3 convertases (C2aC4b or C3bBb) cleave C3 molecules into C3a and C3b fragments (6). The C3a fragment is an anaphylatoxin that may bind to its cell receptor, C3aR, influencing several immunological pathways (as discussed in this review), whereas C3b continues the cascade, leading to the formation of C5 convertase, which cleaves C5 into C5a and C5b. C5a is an effective chemotactic factor and proinflammatory mediator and like C3a, acts as an anaphylatoxin with several biological roles. C5b binds to C6, C7, C8, and C9 and forms the membrane attack complex (MAC), which may disrupt cell membranes by causing lytic pores (Figure 1) (5).
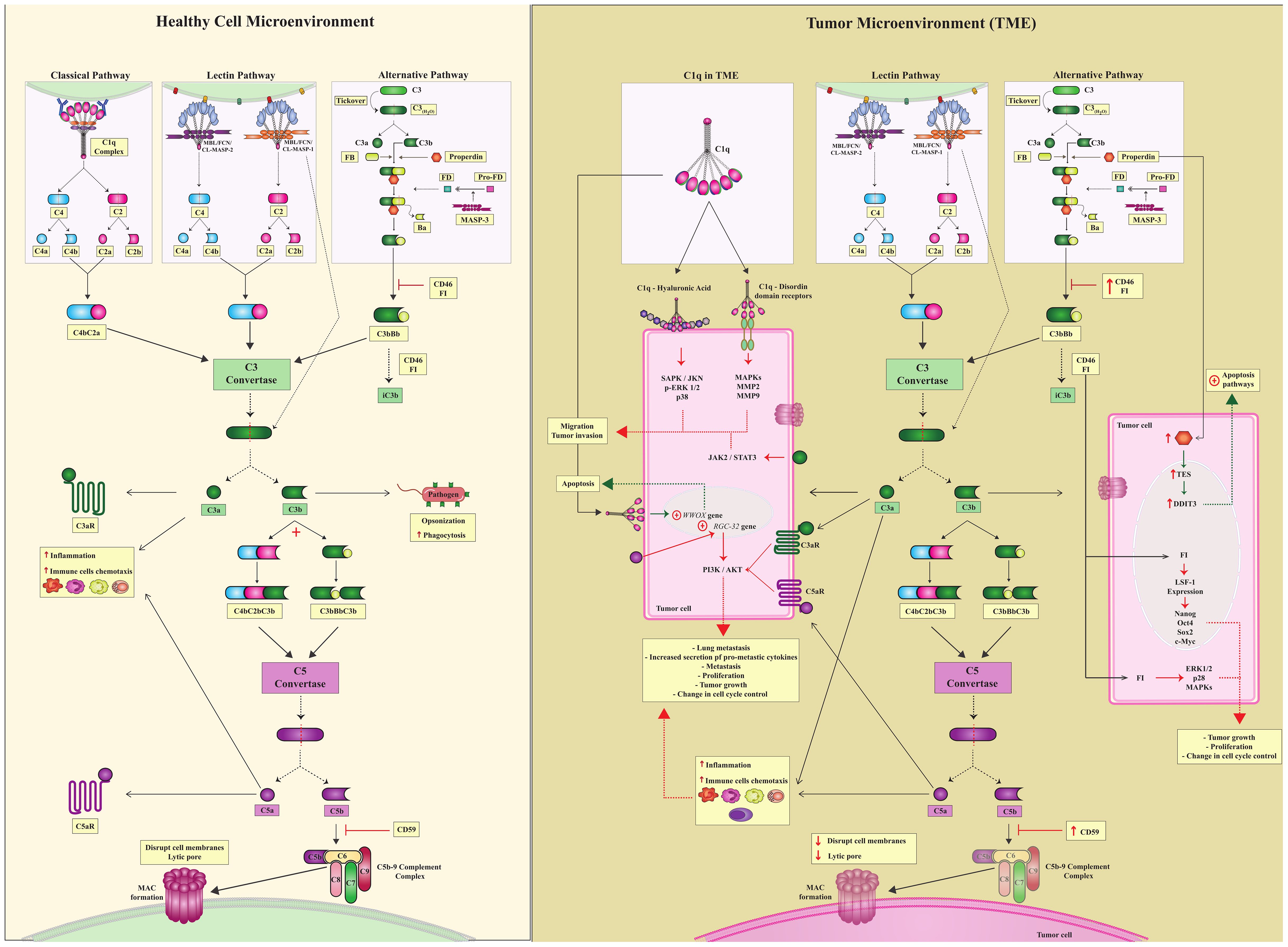
Figure 1. Canonical complement system and complosome pathways. (Healthy Cell Microenvironment) There are three canonical CS pathways in the microenvironment of healthy cells: classical, lectin, and alternative pathways. Different triggers activate all of them; however, these three pathways converge to form C3 convertase, which is a central complement enzyme. C3 converts cleaves C3 into C3a and C3b. C3a is a potent anaphylatoxin capable of attracting immune cells and contributing to inflammation. C3b continues in the complement cascade and forms another essential enzyme, C5 convertases. C5 convertase cleaves C5 into C5a and C5b.C5a, just as C3a is an anaphylatoxin, and C3b follows the cascade and leads to the formation of the C5bC9 complement complex, which culminates in the formation of MAC. To ensure controlled complement activation, complement regulators must act efficiently. (TME) In the tumor microenvironment, in addition to the canonical action of the complement system, there is also a complosome. Complosome proteins play a role in different intracellular pathways, leading to pro- or anti-tumor profiles in these cells. Tumor cells are pink. Green dotted arrows indicate the anti-tumor profile, and red dotted arrows indicate a pro-tumor profile.
Recent evidence suggests that the role of the CS extends well beyond the three traditional catalytic pathways activated in the extracellular environment (7–10). To recognize this, two new terms have been introduced by complement researchers in the past decade: complosome and non-canonical pathways (NCP). They encompass the roles of complement proteins, regulators, and receptors that do not depend on activation of the canonical pathways CP, LP and AP. Although these concepts remain under discussion (9, 11), they will be used in this scoping review to emphasize the rather unexpected involvement of complement components in cancer development. Specifically, the term NCP will refer to complement activation within the extracellular environment, without the direct involvement of any of the three canonical pathways. In contrast, the term complosome will be used to describe the activity of intracellular complement components (Figure 1).
NCPs are less well defined but have gained increasing recognition for their roles in immune regulation and the modulation of inflammation. CS components that do not directly involve the activation of the CP, LP or AP in the extracellular space include serine proteases as MASPs, which interact with other proteins and receptors, such as protease-activated receptor 4 (PAR4) in endothelial cells (12–14), leading to pro-inflammatory and/or pro-thrombotic responses (15). They involve complex mechanisms, including activation via PRRs, tissue-specific factors, or indirect modulation through cytokine signaling, and contribute to the complement system’s broad regulatory functions in immunity, particularly in inflammation, tissue injury, and pathogen defense.
The complosome comprises all CS proteins able to act in intracellular processes independently of the activation of the canonical pathways. It represents a recent and evolving perspective, still requiring new study designs to achieve a more comprehensive definition (9, 11, 16, 17). Despite ongoing discussion regarding which pathways and functions of CS proteins can be specifically attributed to the complosome, it is evident that these proteins have the potential to regulate intracellular pathways through endosomes (18, 19), mitochondrial membranes (20, 21) and direct gene regulation (22, 23). The core complosome components are C3 and C5, as well as their respective membrane receptors. Furthermore, the complosome can be found in immune and non-immune cells, as well as in different tissues (11). Although still poorly investigated in the TME, the complosome may hold an additional key for a pro- or anti-tumor outcome (16).
Cancer is one of the most common health problems worldwide. In 2020, the International Agency for Research on Cancer reported 19.3 million new cases and almost 10 million deaths from cancer worldwide (24–26). The mechanisms underlying cancer development and progression are not completely understood. The TME is considered to be a key promoter of carcinogenesis, along with genetic and epigenetic factors (27–31). Crosstalk between TME elements and tumor cells modifies the local immune response, inducing inflammation and suppressing the anti-tumor response, thereby facilitating tumor proliferation and invasion (32–35). The hypothesis that chronic inflammation is closely related to malignant tumor transformation and progression has been substantiated by human epidemiological data and genetic experiments (36). However, the interplay between non-canonical complement pathways, the complosome and the TME has been largely out of the spotlight in cancer research. In this scoping review, we discuss the available data published till 2020 at the light of recent evidence, focusing on understanding the implications of activating these poorly known arms of the CS, within tumoral cells and the TME.
2 Materials and methods
2.1 Data sources and literature search strategy
For this scoping review, we followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses extension for Scoping Reviews Checklist (37). We performed a systematic search of the role of the complement system in TME on two main databases: PubMed and Web of Science. For the descriptors, we selected MeSH Terms in PubMed (“neoplasms”[MeSH Terms]) AND (“complement system proteins”[MeSH Terms]) and topics (Ts) in Web of Science (cancer AND (Complement system)) AND (OPEN ACCESS) AND (ARTICLE). We retrieved a total of 550 articles from PubMed and 69 from the Web of Science, and excluded duplicate articles using Excel (n=32).
2.2 Study eligibility – inclusion and exclusion criteria, and limitations
Independently of each other, two of us (CFO-T and AGM) screened the titles and abstracts of 619 articles for the following inclusion criteria: articles in English, with available abstract, open access, in humans or animal models, in vivo, in vitro, or in silico, and published from November 26th, 2010 to November 26th, 2020 (Figure 2). Exclusion criteria were: articles whose subjects were not components of the CS and cancer; studies classified either as review, commentary, editorial, letter, systematic review and/or meta-analysis, news, guidelines, clinical trials/controlled trials, and patents. We further excluded articles without an abstract and those with an exclusive focus on treatment strategies/drugs that inhibit CS components, retrieving a total of 135 articles, grouped according to the investigated topic. Next, five of us (CFO-T, HMBSP, NMDLS, PDF, and TPB) distributed the 135 articles among themselves according to their area of expertise and assessed their full content. We discussed and resolved any doubts by consensus, retrieving 45 studies for the final analysis (Figure 2, Supplementary Table 1). We extracted the following information from the selected studies: first author’s name, year of publication, title, experimental model, cancer type, complement components, anti/pro-tumoral role of complement components in the cancer context, and experimental methods. This review is limited by the inclusion of articles published up to 2020 and accessible through open access. As a result, relevant studies published after this period or behind paywalls may not have been considered. Additionally, as with any scoping review that relies on keyword searches in databases, some articles may have been unintentionally excluded due to variations in search terms or database indexing. Nevertheless, we made every effort to incorporate all relevant studies within the defined scope of this review.
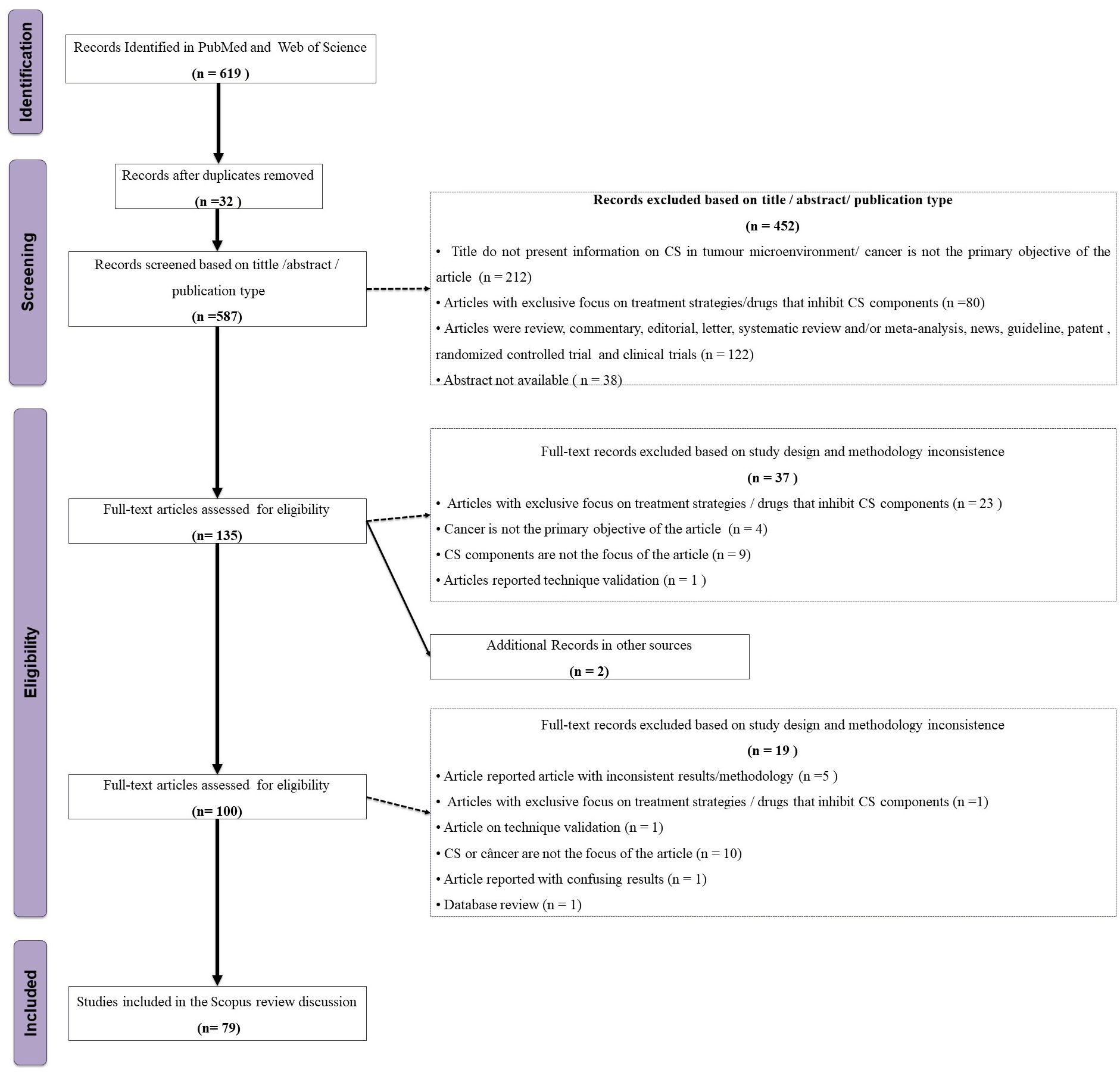
Figure 2. Flowchart of the study eligibility based on the PRISMA-ScR.
3 Results
3.1 C1q complex
The C1 complex is composed of three proteins, C1q, C1r, and C1s. C1q is the recognition unit which is bound to two molecules each of the serine proteases C1r and C1s (C1q-C1r2-C1s2) (38, 39). C1q has a structural protein configuration consisting of a globular region and collagen-like domain. The globular region comprises three different subunits, A, B, and C, which form a trimer that is capable of recognizing a wide range of ligands. Apart from its role in CP activation, C1q interacts with complement receptor 1 (CR1), promoting phagocytosis of opsonized elements by mononuclear phagocytes and regulating their cytokine profile. C1q-stimulated macrophages upregulate pro-phagocytic genes, enhancing their phagocytic activity and altering cytokine expression, thereby suppressing the expression of IL-1 (interleukins 1 alpha and beta) and increasing secretion of interleukin-10 (IL-10), IL-1 receptor antagonist, monocyte chemoattractant protein-1, and interleukin-6 (IL-6) (40–42). It also plays an important anti-inflammatory role in maintaining homeostasis by aiding immune cells such as monocytes/macrophages and red blood cells in clearing immune complexes and apoptotic cells. Below we present our principal results, regarding C1q’s CP-independent role in cancer.
3.1.1 C1q and C1r/C1s gene expression and protein levels in cancer
C1q-deficient mice lacking the C1qa gene exhibited reduced B16 melanoma tumor mass and vascular density, as well as prolonged survival. C1q further induces proliferation, migration and murine melanoma cell adhesion, corroborating the hypothesis that C1q promotes melanoma progression in vivo independent of the CP activation (43). In line with this, tumor-associated macrophages (TAMs), resembling M2-like macrophages, as well as monocytoid B cells, contribute to elevated C1q levels in the TME. C1q-producing TAMs were associated with an immunosuppressed TME in clear cell renal cell carcinoma (ccRCC), characterized by high expression of immune-checkpoint molecules (PD1, LAG3, PDL1 and PDL2) (44). High C1q concentrations also occur in the TME of both human low-grade gliomas (LGGs) and glioblastoma multiforme (GBMs). In GBM, C1q was specifically associated with vascular endothelial cells, and its source could again be attributed to TAMs (45). C1q protein was diffusely present in the TME and expressed by TAMs in basal-like breast cancer and lung adenocarcinoma, whereas in clear cell renal cell carcinoma it was found in the small vessels and on the cell membrane of tumor cells - besides the TME. Expression of all three genes encoding C1q chains was associated with tumor growth and a poor prognosis in these types of lung and kidney cancer, despite representing a favorable prognosis in Her-2 positive and basal-type breast cancer (Figure 3) (46). Notably, there was no evidence of C3d nor C4d immunoreactivity and thus for complement activation in gliomas (45). In fact, deposition of other CS components seems to be infrequent in most tumors, suggesting that C1q operates in the TME independently of CP activation (43).
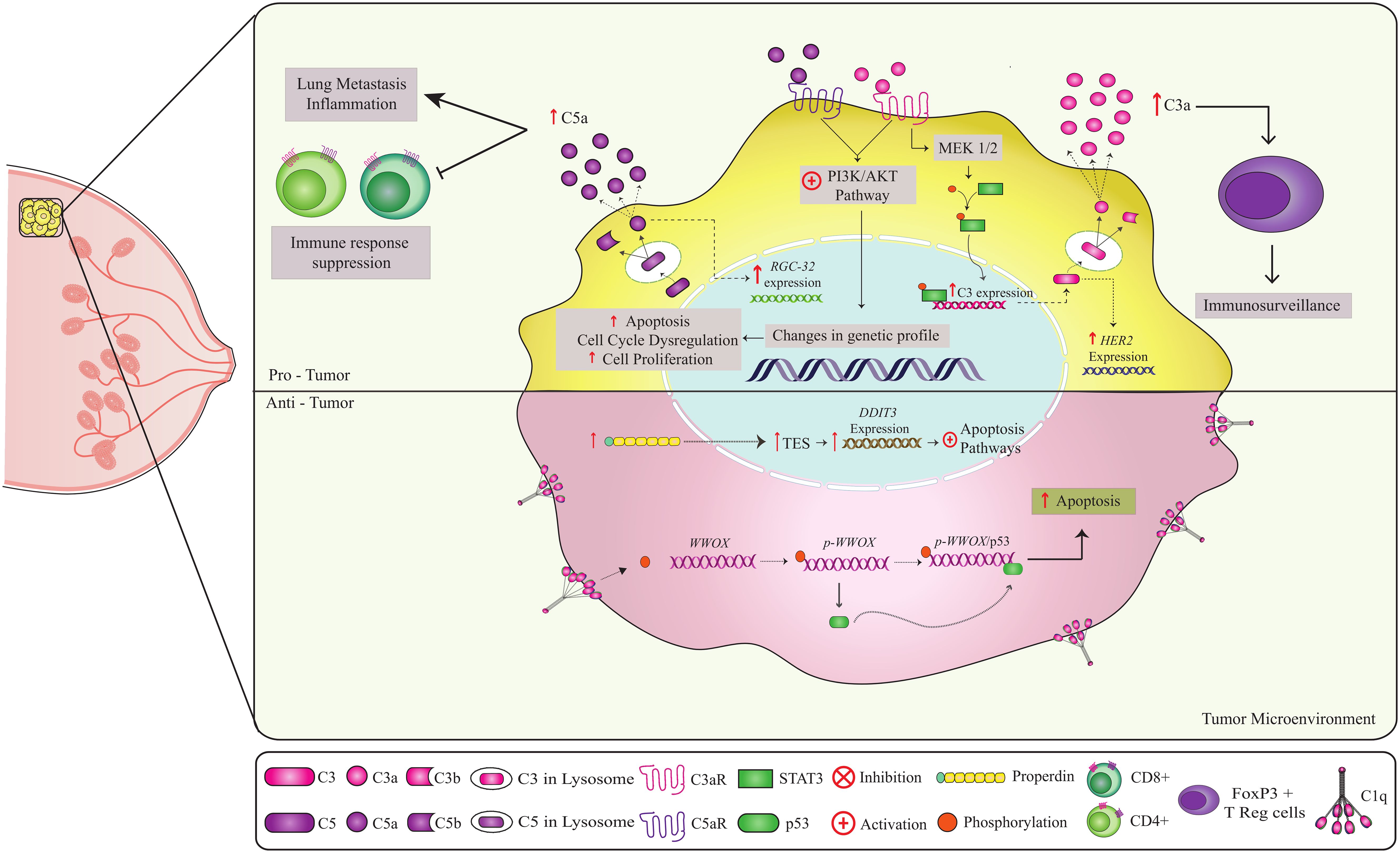
Figure 3. Non-canonical pathways involving complement components in breast cancer. Complement components play a role in both anti-tumor and pro-tumor responses. C1q and properdin have anti-tumor effects in breast cancer (51, 53, 91, 92). Binding of C1q to the cell at the breast tumor membrane leads to Tyr33 site phosphorylation of the WWOX protein, enabling the binding of p53 to WWOX gene. The p-WWOx-p53 complex is carried to the mitochondria and nucleus, where it activates apoptotic pathways (51). The high expression of intracellular properdin in tumor cells induces stress on the endoplasmic reticulum, leading to the expression of molecules that act in the increase of TES transcription, causing an increase in the expression of the DDIT3 gene that activates apoptotic pathways. Thus, both C1q and properdin contribute to the elimination of tumor cells. In contrast, on the C3a-C3aR and C5a-C5aR axes, CD46 and CD59 may have a pro-tumor role. Activation of the C3a-C3aR axis leads to PI3K/AKT and MEK1/2 pathway activation (63, 64). The PI3K/AKT pathway is a key regulator of the cellular processes involved in cell growth, metabolism, motility, survival, and apoptosis. Uncontrolled activation of this pathway promotes tumor cell survival and proliferation, leading to tumor progression and resistance to cancer therapy. Intracellular production of C5a leads to the C5a-C5aR axis, which can activate the PI3K/AKT pathway. C5a also promotes overexpression of the RGC-32 gene (complement response gene 32), which controls cell cycle progression. High expression of the regulators CD46 and CD55 can potentially worsen prognosis and inhibit apoptosis, respectively. In addition, C3 has been shown to be involved in tumor immunosurveillance, probably through interactions with FOXP3+ T (Tregs), which are essential for the maintenance of immune tolerance and immune homeostasis. Thus, the combined action of these components can have a crucial effect on clinical outcomes.
3.1.2 C1q or C1r/C1s NCP signaling in cancer
In vitro studies have shown that C1q binds to high molecular weight hyaluronan (HMW-HA) in the extracellular matrix, triggering adhesion and proliferation of mesothelioma cells. This occurs through an increased phosphorylation of ERK1/ERK2 (extracellular signal-regulated kinases), SAPK/JNK, and p38, regardless of CP activation in a NCP manner (47). In a similar way, C1q has been shown to bind in vitro to discoidin domain tyrosine kinase receptors (DDRs) at the membrane of hepatocellular carcinoma cell lines. The C1q-DDR complex interacts with MMP2 and MMP9, two zinc-dependent matrix metalloproteinases that degrade extracellular matrix, promoting tumoral cell migration and tissue invasion (48). These data support the idea that C1q may increase the aggressiveness of liver cancer, as well as of mesothelioma, though through different mechanisms. Yet in breast and prostate cancer cell lines, extracellular C1q was involved in initiating a signaling pathway that activates the expression of the WWOX (WW domain-containing oxidoreductase) tumor-suppressor gene. In these particular tumor types, diminished C1q deposition was associated with an enhanced cell proliferation and less apoptosis (Figure 3) (22, 23).
In contrast to previous examples, cutaneous squamous cell (cSCC) lines exhibit high expression levels of C1r and C1s mRNA and protein, while lacking expression of genes producing C1q proteins. These cell lines secrete C1s and C1r, which leads to their activation in a C1q-independent manner. Consequently, activation of latent MMP9 occurs, accompanied by inhibition of extracellular signal-related kinase 1/2 and Akt activation (49). These findings suggest a potential role for these serine proteases in promoting cSCC tumor growth, angiogenesis, and metastasis.
3.2 C3 component
The canonical AP can contribute to up to 80% of the overall complement response and remains active even after activation of the CP and LP. These pathways converge to form and activate C3 convertase, which is a central step of the cascade that leads to the cleavage of C3, producing C3a and C3b. Moreover, AP C3 convertase amplifies C3b deposition on cell membranes, facilitating immune surveillance. C3a acts as a potent anaphylatoxin and induces chemotaxis, cell activation, and inflammation by binding to the C3aR receptor. The conformational change from C3 to C3b exposes previously hidden binding sites, enabling distinct interactions and efficient immune surveillance. Interactions between C3b and complement receptors/regulators, such as CR1/CD35 and CD46, was shown to lead to its degradation into iC3b and/or C3dg fragments, allowing immune adhesion, phagocytosis, and adaptive stimulation (Figure 1) (6).
3.2.1 C3 gene expression and protein levels in cancer
In the neuT mouse model of breast cancer, C3 accumulates specifically in the vessels and stroma undergoing tumorigenesis in the mammary gland, while remaining absent in the surrounding tissue (Figure 3). These cancer cells express high levels of Her2 and of the complement regulator CD55, preventing C3 cleavage by the C3 convertase. In the neuT C3-/- knockout mice, autochthonous mammary carcinomas started earlier and grew faster, reaching larger volumes in less time and presenting greater multiplicity, sooner seeding lung metastases. Tumors in these C3−/− mice showed 6- to 8-fold higher levels of Her2/neu expression. Thus, C3 may limit Her2 expression and, consequently, cell proliferation. The número of regulatory T cells (Tregs) did also increase in tumors from neuT-C3−/− mice, contributing to a more immunosuppressive environment and favoring tumor growth. Although the number of tumor vessels was similar between neuT and neuT-C3−/− mice, the vessels in C3-deficient tumors were wider and more permeable, resulting in better nutrient and oxygen supply to the tumor (Figure 3) (50).
3.2.2 C3 and C3aR NCP signaling in cancer
In lung cancer, CD4+ T cells present intracellular C3 cleavage by cathepsin and C3a signaling inhibits the production of multiple cytokines by CD4+ T cells, independently of FOXP3+ Tregs (51) (Figure 4). In vitro studies revealed that iC3b is inactivated by prostate-specific antigen (PSA) in prostatic fluid, generating a novel 37 kDa fragment independently of factors H and I, smaller than that produced by the canonical pathways. Notably, the new iC3b fragments appear to promote an immunosuppressive prostatic tumor profile, although their biological function is still unclear (52).
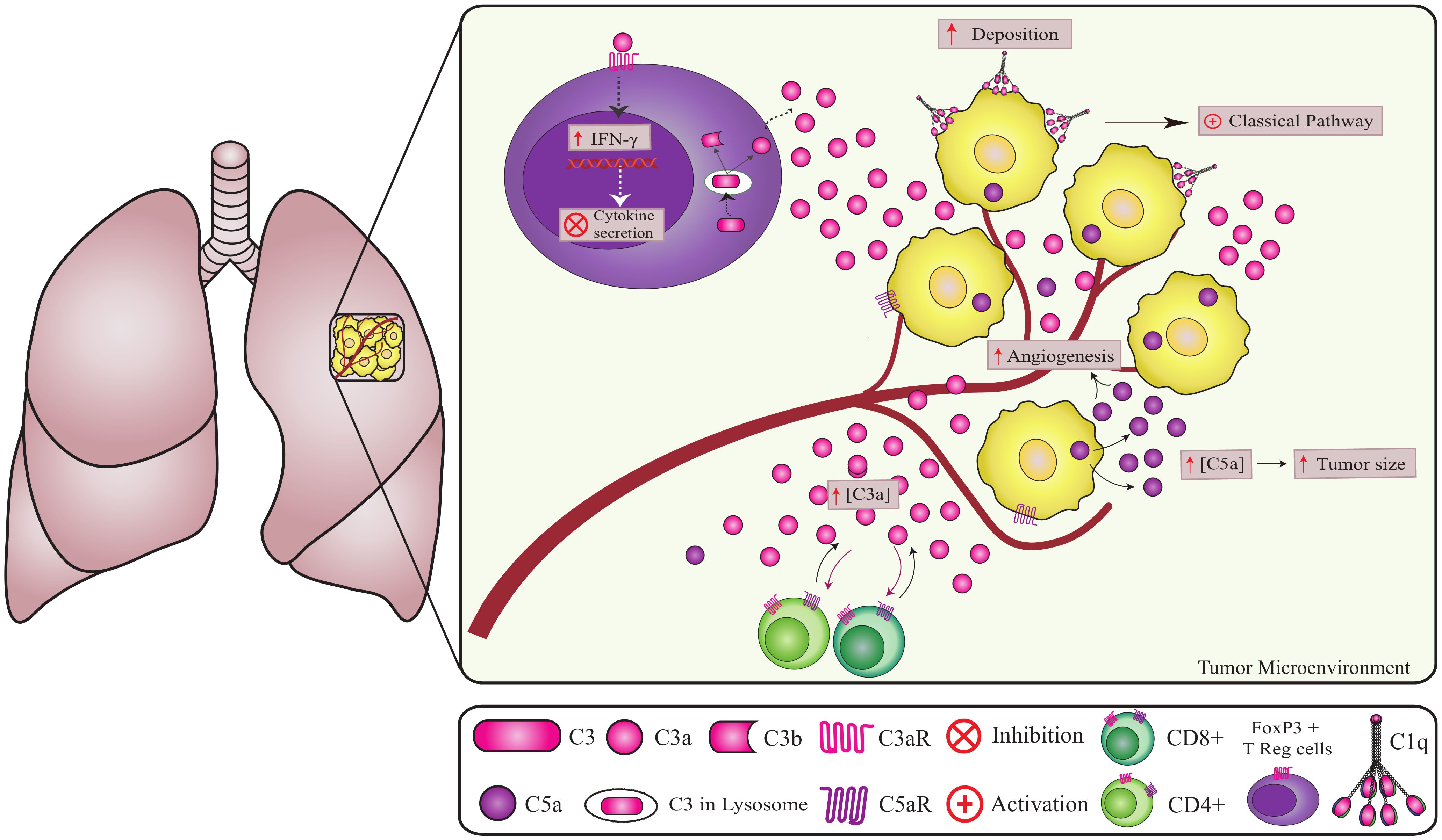
Figure 4. Non-canonical pathways involving complement components in lung cancer. Complement components play a crucial role in lung cancer progression. C1q promotes angiogenesis independent of CS activation (2). However, the origin of intracellular C5a in cancer cells remains unknown. Increased extracellular C5a levels lead to tumor growth and angiogenesis (72, 74). The high deposition of factor H on the tumor cell membrane contributes to tumor progression and can be considered a marker for lung adenocarcinomas. A high concentration of C3 in the TME reduces CD4 T cells, promoting metastasis and tumor growth. In addition, CD4+ T cells can produce and cleave C3 intracellularly via cathepsin leading to the regulation of IFNγ (interferon-gamma) secretion (60, 61).
C3a has the potential to activate ERK/MEK1-2 kinases promoting cell proliferation and apoptosis resistance, regulating synaptic plasticity and p53 phosphorylation, e.g. human bone osteosarcoma epithelial cells (U2-OS) exposed to normal serum exhibited increased ERK phosphorylation compared to U2-OS treated with heat-inactivated serum (53). In gastric cancer, deposition of C3 fragments on tumor cells activates the JAK2/STAT3 pathway, increasing levels of phosphorylated STAT3 (promoting cell proliferation and migration) and IL-6 (54). Ovarian cancer cells secrete high amounts of C3, whose non-canonical cleavage (independent of CP, AP and LP) leads to C3a production in the TME. C3a binding to C3aRs on the tumoral cell membrane activate the PI3K/AKT/mTOR pathway, boosting protein synthesis, cell proliferation, survival, and motility by increasing AKT mRNA levels and phosphorylating p85, a PI3K regulatory subunit (55). Similarly, in ovarian cancer mice models (C57BL/6), the C3a-C3aR interaction triggers PI3K/AKT pathway activation, enhancing C3 secretion, tumor growth, and proliferation via a C3-dependent autocrine loop (55). Finally, C3a-C3aR signaling through PI3K-AKT promote lung metastases of breast cancer in a murine model, modulating carcinoma-associated fibroblasts to increase secretion of pro-metastatic cytokines and expression of extracellular matrix components (Figure 3) (56).
3.3 C5 component in cancer
The final phase of the CS cascade starts with the formation of C5 convertase, which catalyzes the cleavage of the C5 component into C5a and C5b (Figure 1). C5a plays a role as a potent anaphylatoxin and chemoattractant for neutrophils, monocytes, and macrophages (57). Additionally, intracellular C5a modulates cytokine expression in various cell types, playing a role in activating coagulation pathways and inducing the formation of neutrophil extracellular traps (NETs). The C5a receptor (C5aR), a member of the rhodopsin family of G protein-coupled receptors, is predominantly expressed by myeloid cells in the lung and liver. Activation of C5aR triggers diverse local responses depending on the cell type, with implications in cancer, particularly in the generation and modulation of anti-apoptotic responses, among other functions (Figure 1) (58).
3.3.1 C5 and C5aR gene expression and protein levels in cancer
In lung (59), colon (60), ovarian (36), and bile duct cancers (60), C5a production does not appear to be associated with the activation of the CP or AP, a matter that still needs to be clarified (59, 60). In vitro, lung cancer cells produced C5a independently of the expression of factor B, properdin, C1q, C1r, C1s, C1, and C4 (Figure 4) (59). However, C5a production was reduced by inhibitors of trypsin-like serine proteases. MASP-1, a serine protease from the LP with trypsin-like activity, might play a role in producing tumoral C5a. However, further experiments using inhibitors of LP serine proteases are required to confirm this possibility (59).
3.3.2 C5a and C5aR NCP signaling in cancer
Similarly to C3, C5 activation plays a pro-tumoral role in different types of cancer. Certain types of cancer cells, such as those found in lung, colon, ovarian, and bile duct cancers, secrete C5a into the TME, independently of CP and AP, initiating an autocrine loop that enhances cell proliferation and fosters metastasis. Mice with lung cancer, if treated with C5aR antagonist, slow the growth of the tumor and decrease angiogenesis (59, 60).
In lung cancer tissue and cell lines, C5a contributes to an immunosuppressive microenvironment by recruiting myeloid-derived suppressor cells and inducing the expression of several immunomodulators, including arginase 1 (ARG1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), IL-6, IL-10, lymphocyte-activation gene 3 (LAG3), and programmed cell death 1 ligand 1 (PD-L1 or CD274) (59). Similarly, in ovarian cancer, elevated levels of C5a impact the levels of immunoregulators in the TME, such as arginase, inducible nitric oxide synthase (iNOS), vascular endothelial growth factor (VEGF), and tumor necrosis factor alpha (TNF-α), thereby reducing the infiltration of CD4+ and CD8+ T cells and promoting tumor growth (36). High C5a levels stimulate linfoma with decreased infiltration of CD4+ and CD8+ T cells, while low levels of C5a in the TME decrease tumor progression, thus, the local concentration of C5a, independent of complement activation, seem to be critical to determine its role in tumor progression (61).
In human breast cancer cell lines, C5a induces the overexpression of the RGCC gene (response gene to complement 32 protein, involved in cell cycle progression), by activating the Akt pathway (Figure 3) (62). Additionally, C5a-C5aR signaling facilitates lung metastasis, a common occurrence in breast cancer, by suppressing the responses of CD8+ and CD4+ T cells and promoting the generation of regulatory T cells (Tregs) in situ (Figure 3) (63). The mechanism of this C5aR-mediated T cell suppression in the metastatic target involves the recruitment of immature myeloid cells and increased production of transforming growth factor β (TGF-β) and IL-10, favoring the generation of Treg and Th2-oriented responses that render CD8+ T cells, dysfunctional (Figure 3) (63).
In gastric cancer, C5a-C5aR recognition regulates p21 expression through the PI3K/AKT axis. Moreover, increased C5aR expression by the MKN1 and MKN7 gastric cancer cells enhances their invasiveness and promotes liver metastasis, which is associated with a poor prognosis (64). In these cells, C5a-C5aR signaling promotes the conversion of RhoA-GDP (RhoA-guanosine diphosphate) to RhoA-GTP (RhoA-guanosine triphosphate) in the cytosol, leading to cytoskeletal rearrangement and increased invasive capacity (65). In fact, in renal cell carcinoma the activation of the C5a-C5aR axis was indicated as a prognostic marker, with C5a stimulating ERK and PI3K-dependent invasion in renal cell carcinoma cells expressing C5aR (66, 67).
In metastatic pancreatic invasive ductal adenocarcinoma, podocalyxin-like protein 1 (PODXL1) activates C5aR on the cell membrane, increasing cellular motility. This activation confers invasive and metastatic properties to PDAC cells (68). Furthermore, C5aR1 signaling induces the secretion of chemokine C-C motif ligand 2 (CCL2) by melanoma cells. This attracts immunosuppressive populations of myeloid cells, supporting tumoral growth and facilitating the infiltration of leukocytes with an immunosuppressive profile in the TME. Conversely, C5aR2 plays a more limited yet beneficial role in restraining tumor growth (69).
In summary, C5a/C5aR signaling plays a significant pro-tumoral activity in several tumor types. Binding of C5a to C5aR in tumor cells correlates with reduced overall survival and recurrence-free survival, while also elevating the incidence of microvascular invasion and metastasis of gastric cancer and renal cell carcinoma (Figures 1, 3, 4) (66, 67).
3.4 The final complement lytic pathway in cancer
The final complement lytic pathway serves as the common intersection for the three canonical complement pathways, which culminate with the disruption of plasma membranes of target cells or microorganisms (Figure 1) (70). Complement regulators restrict CS activation on most tumors. However, sublytic MAC deposition on cancer cells raises intracellular Ca2+ levels, altering cellular functionality and causing cellular damage (70). MAC activity is crucial for maintaining cellular and tissue homeostasis and promoting protection against infections. It may also maintain tumor-associated inflammatory signaling, assuming a dual role in cancer (71, 72).
3.4.1 The role of MAC and C7 in cancer
Exposure of cancer cells to sublytic levels of MAC alters the expression of G protein and Ca2+ signal transduction (ITPRIP, RGS16), transcription factors (EGR1, EGR2), inflammatory response genes (Interferon Regulatory Factor 1 - IRF1), as well as four other extracellular protein genes (Amphiregulin - AREG, C-X-C Motif Chemokine Ligand 1 - CXCL1, Matrix Metalloproteinase 3 - MMP3, and MMP13), impacting cell proliferation and survival (73). In this way, sublytic MAC deposition on cancer cells emerges as a potent stimulator of tumor progression.
C7 is an important protein in the MAC formation. Increased amounts of C7 protein in the nuclei of hepatocellular carcinoma cell lines promotes in vivo cell growth, by upregulating late SV40 factor (LSF-1) protein levels and the expression of stemness factors such as POU Class 5 Homeobox 1, SRY-Box Transcription Factor 2, and MYC Proto-Oncogene Transcription Factor (encoded by OCT4, SOX2, and MYC, respectively) (74). These alterations in gene expression sustain the replication capacity of liver tumor-initiating cells, while suppression of C7 inhibited the establishment of human tumors in NOD/SCID (non obese diabetic/severe combined immunodeficiency) mice (74).
On the other hand, in non-small cell lung cancer (NSCLC), C7 overexpression suppresses colony formation in vitro and lower C7 expression was associated with worse outcome, advanced clinical stage and grade, increased likelihood of relapse and death (75). Therefore, the anti- or pro-tumorigenic activity of C7 is influenced by the different cancer types. While these components act as independent drivers of tumor immune response and surveillance, further investigation is needed to clarify their in vivo effects on carcinogenesis and tumor progression.
3.5 Complement regulators in cancer
Complement activation leads to various biological processes extending beyond its protective function and can, in some cases, pose harm. Therefore, safeguarding self-tissues from complement-mediated damage is crucial, achieved through the actions of several soluble regulators or membrane-bound counterparts. Different cell types rely on a combination of complement regulatory proteins (CRPs) to modulate the cascade at various points and in diverse manners. Noteworthy membrane-bound CRPs in tumors include CD46, CD55, and CD59, while soluble ones encompass factor I, factor H, and properdin (76, 77).Although CD46, CD55, and CD59 are pivotal complement regulators expressed across most cell types and tumor cells, they exhibit a double-edged sword role. While they avert complement-mediated autologous lysis in normal cells, their aberrant overexpression impedes complement-mediated lysis, thereby fostering tumor cell survival and progression. Consequently, membrane-bound CRPs may serve as biomarkers of malignant transformation (78–81).
CD59, a prevalent regulator expressed in most tumor cells, is a glycosylphosphatidylinositol (GPI)-anchored membrane protein that impedes C9 polymerization and the attachment of C9 units to the C5b-8 complex and MAC through its physical incorporation into the complex (79), thereby preventing membrane disruption and cell lysis. CD55, referred to as a complement decay-accelerating factor (DAF), expedites the decay of C3 and C5 convertases by swiftly dissociating the catalytic subunit C2a or Bb from the cell surface, thus preventing the generation of anaphylatoxins, opsonins, and MAC. Additionally, CD55 can recognize C4b and C3b fragments produced during C4 or C3 activation (78). These CRPs exert significant modulation over complement activity and may profoundly influence tumor progression.
The factor H (FH) is a plasma glycoprotein that acts as a soluble inhibitor of complement, where its binding to self markers such as glycan structures prevents complement activation and amplification on cell surfaces (82, 83). FH ensures the complement system spares host tissues from damage, by destabilizing the AP C3 convertase complex (C3bBb). Its dysfunction in age-related macular degeneration and atypical hemolytic uremic syndrome highlights its importance in maintaining the balance between effective immunity and self-damage. In contrast, properdin stabilizes C3bBb, amplifying pathogen opsonization, inflammation, and MAC formation. Properdin is unique as one of the few complement components that act as a positive regulator in the system (84). As a plasma glycoprotein, the complement factor properdin (CFP) is the only known positive regulator of the CS. It plays a crucial role in stabilizing surface-bound alternative pathway (AP) C3 and C5 convertases (64, 85).
3.5.1 CD59, CD55 and CD46 gene expression and protein levels in cancer
Head and neck cancer cells exhibit high expression of CD46, CD55, and CD59 (86). CD59 is highly expressed on tumoral cells, protecting them against complement lysis, and aiding in immune evasion. CD59 expression correlated with TAM infiltration in pancreatic cancer tissues. Pancreatic cancer-educated macrophages upregulate CD59 expression on pancreatic cancer cell lines through STAT3 phosphorylation via the IL-6R/STAT3 signaling pathway and protect them from complement-dependent cytotoxicity (81). Similarly, in human ovarian A2780 cancer cells, IL-6 stimulates CD59 expression at low concentrations, but at high concentrations or with IL-8, it presents a post-transcriptional inhibitory effect (87). Indeed, CD59 immunoreactivity is detected in up to 50% of ovarian tumors and at the border areas between normal and malignant tissue (87). Similarly, increased CD55 and CD59 expression was observed on cell membranes and in the TME of ovarian and uterine cancers (80, 88). Additionally, CD55 overexpression predicts poorer clinical outcomes in colorectal cancer, with stromal CD55 overexpression correlating with unfavorable prognostic markers (78). Beyond its role in inhibiting canonical complement action within the TME, CD59 was shown to impede apoptosis of breast cancer cells, thereby contributing to tumor development (79).
High CD46 expression is an unfavorable prognostic factor associated with lower relapse-free survival in breast cancer (89). Notably, the CD46 gene promoter has two binding sites for STAT3. If induced by STAT3 and IL-6, CD46 expression protects cancer cells against complement lysis and fosters a pro-tumoral profile in breast and prostate cancers (89). On the other hand, low CD46 expression rates in cervical cancer correlate with increased patient´s survival (90). CD46 shifts its expression profile in cervical cancer cells, compared to normal female reproductive tissue, suggesting its involvement in tumorigenesis.
3.5.2 Complosomes with FH and properdin in cancer
As is the case with complosome involving C7, intranuclear FH promotes via LSF-1, the expression of NANOG, OCT4, SOX2, and MYC genes. On their turn, these stemness factors promote liver tumor formation and growth in vivo (Figure 1). Notably, increased FH expression, particularly within the nucleus, suggests enrichment in tumor cells (74). Similarly, FH and its isoform FHL-1 expression is specifically induced during cutaneous carcinogenesis (cSCC) and exert effects on intracellular pathways, being negatively regulated by the inhibition of ERK1/2, p38, and MAPK pathways, while being upregulated by IFNγ, IL-1b, TGF-α, TGF-β, and TNF-α. FH/FHL-1 silencing inhibited tumoral cell proliferation and migration, pointing to a critical protumorigenic role (91).
In the complosome network, properdin presented an anti-tumoral role through a novel tumor suppressor pathway identified in breast cancer (Figures 1, 3). High expression of intracellular properdin induces the transcription of the Testin LIM domain protein (TES), subsequently leading to increased expression of DDIT3 (DNA-Damage-Inducible Transcript 3), a pro-apoptotic transcription factor associated with the endoplasmic reticulum stress response. Consequently, increased properdin expression modulates cell apoptosis, playing an anti-tumoral role in breast cancer (Figure 3) (92). Furthermore, in vitro studies have demonstrated that bone marrow-derived macrophages (BMDM) from properdin-deficient mice can stimulate B16F10 melanoma cell lines. This properdin deficiency results in low levels of IL-1b mRNA and higher levels of arginase-1, monocyte chemotactic protein-1 (MCP-1), and IL-10 mRNAs, suggesting that a properdin-deficient tumor microenvironment may induce a profile of M2 macrophages with pro-tumoral activity (92).
Although there is currently no significant association between properdin expression and cancer prognosis, this regulator plays an important anti-tumoral role in complosome pathways in breast cancer³ (93). In summary, FH acts as a pro-tumoral factor in hepatocellular carcinoma, while properdin serves as an anti-tumoral factor suppressing breast cancer/melanoma. These two CRPs likely play a role in complosome pathways by modulating tumor formation and growth.
4 Discussion
Research over the past decade has firmly established that CS components play a pivotal role in driving hallmark features of cancer. These elements contribute to critical tumor processes such as angiogenesis, invasion, metastasis, immunomodulation, metabolic reprogramming, microenvironmental remodeling, unchecked proliferation, and therapy resistance. While canonical complement activation pathways are implicated in many of these mechanisms, emerging evidence highlights the substantial involvement of CS components through activation-independent pathways. Its role in cancer is context-dependent, shaped by its cellular source, interaction partners, and downstream signaling pathways.
We found little evidence for complosome activity in cancer, but rather for NCP-mediated activation of intracellular signaling pathways, suppressing tumors in certain settings (e.g., for C1q, by activating WWOX in breast and prostate cancer cells (22, 23), and more commonly driving malignancy by stromal interactions, receptor signaling, and matrix remodeling, (e.g. C1q by engaging receptors as DDR1 and HA to activate MAPK signaling or modulate MMPs, fostering tumor aggression) (47, 48). A recent study published after the timeframe of our scoping review described a pro-tumorigenic C1q-driven mechanism in pleural mesothelioma (PM). The researchers identified a functional interplay between hyaluronidase-2 (HYAL2)—a prognostic marker associated with poor PM outcomes—and the hyaluronic acid-binding protein gC1qR (globular C1q receptor/HABP1/p32). C1q binding to hyaluronic acid (HA) via gC1qR activates intracellular signaling pathways that up-regulate the expression of both HYAL2 and hyaluronan synthase HAS3 mRNA and protein expression. Through this NCP, C1q both increases HA synthesis and stimulates its degradation into low-molecular-weight fragments, creating a feedback loop that supports tumor growth and maintenance (94, 95).
Beside complement regulators and receptors, components normally produced in the liver, as C1q, C1r, C1s, C3 and C5 are locally synthesized and deposited in different tumor types and in the stroma regardless of complement activation, being predominantly tumorigenic in the majority of cancer types. C3a-C3aR and C5a-C5aR binding activate the PI3K/AKT/mTOR pathways, which promote cell proliferation, protein synthesis, survival and motility. Concurrently, C3 cleavage fragments stimulate the JAK2/STAT3 and ERK pathways, driving cellular reprogramming that promotes tumor progression. These pathways collectively orchestrate pro-tumorigenic changes such as enhanced survival, proliferation, and immune evasion (56).
Thus, tumor fate depends on all the dynamic interactions between complement components, immune cells, and signaling pathways in the TME. A tumor-friendly TME would probably present one or more of the following features: PI3K/AKT and MEK1/2 signaling via C3a-C3aR and/or C5a-C5aR axes, JAK2/STAT3 activation driven by tumor-deposited C3 fragments, promoting growth/metastasis, anti-apoptotic effects via CD59-mediated inhibition of terminal complement complexes, C5a-induced Akt-dependent oncogenic RGC-32 expression, overexpression of CD55, CD46, and CD59 on tumor cells, Factor H overexpression, enhancing stemness (via LSF-1) and angiogenesis. On the contrary, TME-driven tumor suppression would include C1q-mediated phosphorylation of the WWOX tumor suppressor, Properdin-dependent activation of the TES-DDIT3 tumor suppressor pathway, CD4+/CD8+ T-cell infiltration due to high intratumoral C3 expression and C3-mediated inhibition of HER2 oncogene expression.
While no studies meeting our inclusion criteria (Section 2.2) directly demonstrated LP components influencing carcinogenesis via complosomes or NCPs, two references are worth mentioning. Ficolin-2 was found to inhibit epithelial–mesenchymal transition in HCC, both in vivo and in vitro, by decreasing TGF beta levels and phosphorylation of Smad2 and Smad3 (96). FCN-2 was also shown to interact with Toll-like receptor 4 (TLR4) and drive macrophage polarization toward the M1 phenotype. This activation enhanced antigen presentation to CD8+ T lymphocytes, ultimately reducing tumor growth (CT26 colon carcinoma, Lewis lung carcinoma, and Hca-f hepatocarcinoma) across three distinct murine models (BALB/c, C57BL/6, and C3H/He). Notably, FCN-A knockout mice exhibited accelerated CT26 colon and Lewis lung carcinoma progression – a phenomenon reversed by FCN-2 or FCN-A supplementation (97). In fact, LP components seem very relevant in oncogenic processes (98, 99). Their absence from currently characterized carcinogenic pathways reflects the paucity of mechanistic studies, underscoring the need for systematic functional characterization of these molecules in oncogenic processes.
A central unresolved question persists: do CS components drive tumorigenesis, or are they merely bystanders in the TME? While some studies suggest individual CS elements may foster tumor-friendly niches, this perspective conflicts with cancer’s multifactorial etiology. Instead, we propose that CS components act as dynamic regulators of the TME, through direct involvement in complement activation cascades, in NCP and complosomes leading to receptor signaling, and crosstalk with stromal cells. Critically, the TME itself dictates whether CS components promote or suppress tumor progression. For example, tumor cells and TME-associated immune cells (e.g., macrophages, monocytes) can locally produce CS proteins. These elements may then paradoxically support or antagonize tumor growth depending on contextual factors like tumor type, stage, and immune landscape (44, 45). This complexity underscores the need for cancer-specific investigations to unravel the precise role and therapeutic potential of each complement component.
Author contributions
CdFO-T: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AdM: Conceptualization, Data curation, Formal analysis, Investigation, Project administration, Supervision, Visualization, Writing – review & editing. HP: Data curation, Investigation, Methodology, Validation, Writing – review & editing. NS: Conceptualization, Investigation, Methodology, Validation, Writing – review & editing. PF: Conceptualization, Investigation, Methodology, Validation, Writing – review & editing. TdPBG: Conceptualization, Investigation, Methodology, Validation, Writing – review & editing. AB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. IdM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. We thank the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES/PROAP - Finance Code 001) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for financial support and for the scholarships provided to AB (research productivity CNPq-313741/2021-2) CO-T (CAPES 40001016012P1), NS (CAPES 40001016012P1), PF (CAPES 40001016012P1), TG (CAPES 40001016012P1), HP (CAPES 134403/2020-7) and IdM (CNPq - Bolsa de Produtividade em Pesquisa).
Acknowledgments
To the Postgraduate Program in Internal Medicine and Health Sciences - UFPR and to the Laboratory of Molecular Immunopathology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1519465/full#supplementary-material
References
1. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. (2010) 20:34–50. doi: 10.1038/cr.2009.139
2. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
3. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
4. Andrade FA, Lidani KCF, Catarino SJ, Messias-Reason IJ. Serine proteases in the lectin pathway of the complement system. In: Chakraborti S, Dhalla N, editors. Proteases Physiol Pathol. Springer, SG (2017). p. 397–420. doi: 10.1007/978-981-10-2513-6_18
5. Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol. (2018) 18:5–18. doi: 10.1038/nri.2017.97
6. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 – The “swiss army knife” of innate immunity and host defense. Immunol Rev. (2016) 274:33–58. doi: 10.1111/imr.12500
7. Arbore G, Kemper C, Kolev M. Intracellular complement – the complosome – in immune cell regulation. Mol Immunol. (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
8. West EE, Kunz N, Kemper C. Complement and human T cell metabolism: Location, location, location. Immunol Rev. (2020) 295:68–81. doi: 10.1111/imr.12852
9. Singh P, Kemper C. Complement, complosome, and complotype: A perspective. Eur J Immunol. (2023) 53:e2250042. doi: 10.1002/eji.202250042
10. Kolev M, Le Friec G, Kemper C. Complement–tapping into new sites and effector systems. Nat Rev Immunol. (2014) 14:811–20. doi: 10.1038/nri3761
11. King BC, Blom AM. Intracellular complement: evidence, definitions, controversies, and solutions. Immunol Rev. (2023) 313:104–19. doi: 10.1111/imr.13135
12. Dobó J, Schroeder V, Jenny L, Cervenak L, Závodszky P, Gál P. Multiple roles of complement MASP-1 at the interface of innate immune response and coagulation. Mol Immunol. (2014) 61:69–78. doi: 10.1016/j.molimm.2014.05.013
13. Hess K, Ajjan R, Phoenix F, Dobó J, Gál P, Schroeder V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PloS One. (2012) 7:e35690. doi: 10.1371/journal.pone.0035690
14. Jenny L, Noser D, Larsen JB, Dobó J, Gál P, Pál G, et al. MASP-1 of the complement system alters fibrinolytic behaviour of blood clots. Mol Immunol. (2019) 114:1–9. doi: 10.1016/j.molimm.2019.07.005
15. Bumiller-Bini V, de-Freitas-Oliveira-Toré C, Carvalho TM, Kretzschmar GC, Gonçalves LB, Alencar NM, et al. MASPs at the crossroad between the complement and the coagulation cascades - the case for COVID-19. Genet Mol Biol. (2021) 17:e20200199. doi: 10.1590/1678-4685-GMB-2020-0199
16. West EE, Kemper C. Complosome - the intracellular complement system. Nat Rev Nephrol. (2023) 19:426–39. doi: 10.1038/s41581-023-00704-1
17. Xiao F, Guo J, Tomlinson S, Yuan G, He S. The role of the complosome in health and disease. Front Immunol. (2023) 14:1146167. doi: 10.3389/fimmu.2023.1146167
18. Ghannam A, Pernollet M, Fauquert JL, Monnier N, Ponard D, Villiers MB, et al. Human C3 deficiency associated with impairments in dendritic cell differentiation, memory B cells, and regulatory T cells. J Immunol. (2008) 181:5158–66. doi: 10.4049/jimmunol.181.7.5158
19. Naslavsky N, Caplan S. The enigmatic endosome - sorting the ins and outs of endocytic trafficking. J Cell Sci. (2018) 131:jcs216499. doi: 10.1242/jcs.216499
20. Daugan MV, Revel M, Thouenon R, Dragon-Durey MA, Robe-Rybkine T, Torset C, et al. Intracellular factor H drives tumor progression independently of the complement cascade. Cancer Immunol Res. (2021) 9:909–25. doi: 10.1158/2326-6066.CIR-20-0787
21. Kenney MC, Chwa M, Atilano SR, Pavlis JM, Falatoonzadeh P, Ramirez C, et al. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: implications for age-related macular degeneration. PloS One. (2013) 8:e54339. doi: 10.1371/journal.pone.0054339
22. Hong Q, Sze CI, Lin SR, Lee MH, He RY, Schultz L, et al. Complement C1q activates tumor suppressor WWOX to induce apoptosis in prostate cancer cells. PloS One. (2009) 4:e5755. doi: 10.1371/journal.pone.0005755
23. Bandini S, Macagno M, Hysi A, Lanzardo S, Conti L, Bello A, et al. The non-inflammatory role of C1q during Her2/neu-driven mammary carcinogenesis. Oncoimmunol. (2016) 5:e1253653. doi: 10.1080/2162402X.2016.1253653
24. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
25. World Health Organization. Cancer(2022). Available online at: https://www.who.int/news-room/fact-sheets/detail/cancer (Accessed March 07, 2025).
26. World Health Organization. International agency for research on cancer(2022). Available online at: https://www.iarc.who.int (Accessed March 07, 2025).
27. Macaluso M, Paggi MG, Giordano A. Genetic and epigenetic alterations as hallmarks of the intricate road to cancer. Oncog. (2003) 22:6472–8. doi: 10.1038/sj.onc.1206955
28. Koumenis C, Hammond E, Giaccia A. Tumor microenvironment and cellular stress: signaling, metabolism, imaging, and therapeutic targets. Adv Exp Med Biol. (2014) 772:111–45. doi: 10.1007/978-1-4614-5915-6
29. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Sci. (2017) 357:eaal12380. doi: 10.1126/science.aal2380
30. Drak Alsibai K, Meseure D. Tumor microenvironment and noncoding RNAs as co-drivers of epithelial-mesenchymal transition and cancer metastasis. Dev Dyn. (2017) 247:405–31. doi: 10.1002/dvdy.24548
31. Wu P, Gao W, Su M, Nice EC, Zhang W, Lin J, et al. Adaptive mechanisms of tumor therapy resistance driven by tumor microenvironment. Front Cell Dev Biol. (2021) 9:641469. doi: 10.3389/fcell.2021.641469
32. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncog. (2008) 27:5904–12. doi: 10.1038/onc.2008.271
33. Barriga V, Kuol N, Nurgali K, Apostolopoulos V. The complex interaction between the tumor micro-environment and immune checkpoints in breast cancer. Cancers. (2019) 11:1205. doi: 10.3390/cancers11081205
34. Giraldo NA, Sanchez-Salas R, Peske JD, Vano Y, Becht E, Petitprez F, et al. The clinical role of the TME in solid cancer. Br J Cancer. (2019) 120:45–53. doi: 10.1038/s41416-018-0327-z
35. Petitprez F, Meylan M, Reyniès A, Sautès-Fridman C, Fridman WH. The tumor microenvironment in the response to immune checkpoint blockade therapies. Front Immunol. (2020) 11:784. doi: 10.3389/fimmu.2020.00784
36. Nunez-Cruz S, Gimotty PA, Guerra MW, Connolly DC, Wu YQ, DeAngelis RA, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplas. (2012) 14:994–1004. doi: 10.1593/neo.121262
37. Tricco AC, Lillie E, Zarin W, O’Brien KK, Colquhoun H, Levac D, et al. PRISMA extension for scoping reviews (PRISMA-ScR): checklist and explanation. Ann Intern Med. (2018) 169:467–73. doi: 10.1084/jem.117.6.983
38. Lepow IH, Naff GB, Todd EW, Pensky J, Hmz F. Chromatographic resolution of the first component of human complement into three activities. J Exp Med. (1963) 117:983–1008. doi: 10.1084/jem.117.6.983
39. Naff GB, Pensky J, Lepow IH. The macromolecular nature of the first component of human complement. J Exp Med. (1964) 119:593–613. doi: 10.1084/jem.119.4.593
40. Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ. C1q: a fresh look upon an old molecule. Mol Immunol. (2017) 89:73–83. doi: 10.1016/j.molimm.2017.05.025
41. Fraser DA, Bohlson SS, Jasinskiene N, Rawal N, Palmarini G, Ruiz S, et al. C1q and MBL, components of the innate immune system, influence monocyte cytokine expression. J Leukoc Biol. (2006) 80:107–16. doi: 10.1189/jlb.1105683
42. Bohlson SS, O’Conner SD, Hulsebus HJ, Ho MM, Fraser DA. Complement, C1Q, and C1q-related molecules regulate macrophage polarization. Front Immunol. (2014) 5:402. doi: 10.3389/fimmu.2014.00402
43. Bulla R, Tripodo C, Rami D, Ling GS, Agostinis C, Guarnotta C, et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat Commun. (2016) 7:10346. doi: 10.1038/ncomms10346
44. Roumenina LT, Daugan MV, Noe R, Petitprez F, Vano YA, Sanchez-Salas R, et al. Tumor cells hijack macrophage-produced complement C1q to promote tumor growth. Cancer Immunol Res. (2019) 7:1091–105. doi: 10.1158/2326-6066.CIR-18-0891
45. Mangogna A, Belmonte B, Agostinis C, Zacchi P, Iacopino DG, Martorana A, et al. Prognostic implications of the complement protein C1q in gliomas. Front Immunol. (2019) 10:2366. doi: 10.3389/fimmu.2019.02366
46. Mangogna A, Agostinis C, Bonazza D, Belmonte B, Zacchi P, Zito G, et al. Is the complement protein C1q a pro- or anti-tumorigenic factor? Bioinformatics analysis involving human carcinomas. Front Immunol. (2019) 10:865. doi: 10.3389/fimmu.2019.00865
47. Agostinis C, Vidergar R, Belmonte B, Mangogna A, Amadio L, Geri P, et al. Complement protein C1q binds to hyaluronic acid in the Malignant pleural mesothelioma microenvironment and promotes tumor growth. Front Immunol. (2017) 8:1559. doi: 10.3389/fimmu.2017.01559
48. Lee JH, Poudel B, Ki HH, Nepali S, Lee YM, Shin JS, et al. Complement C1q stimulates the progression of hepatocellular tumor through the activation of discoidin domain receptor 1. Sci Rep. (2018) 8:4908. doi: 10.1038/s41598-018-23240-6
49. Riihilä P, Viiklepp K, Nissinen L, Farshchian M, Kallajoki M, Kivisaari A, et al. Tumour-cell-derived complement components C1r and C1s promote growth of cutaneous squamous cell carcinoma. Br J Dermatol. (2020) 182:658–70. doi: 10.1111/bjd.18095
50. Bandini S, Curcio C, Macagno M, Quaglino E, Arigoni M, Lanzardo S, et al. Early onset and enhanced growth of autochthonous mammary carcinomas in C3-deficient Her2/neu transgenic mice. Oncoimmunol. (2013) 2:1–14. doi: 10.4161/onci.26137
51. Kwak JW, Laskowski J, Li HY, McSharry MV, Sippel TR, Bullock BL, et al. Complement activation via a C3a receptor pathway alters CD4+ T lymphocytes and mediates lung cancer progression. Cancer Res. (2018) 78:143–56. doi: 10.1158/0008-5472.CAN-17-0240
52. Manning ML, Williams SA, Jelinek CA, Kostova MB, Denmeade SR. Proteolysis of complement factors iC3b and C5 by the serine protease prostate-specific antigen in prostatic fluid and seminal plasma. J Immunol. (2013) 190:2567–74. doi: 10.4049/jimmunol.1200856
53. Jeon H, Han SR, Lee S, Park SJ, Kim JH, Yoo SM, et al. Activation of the complement system in an osteosarcoma cell line promotes angiogenesis through enhanced production of growth factors. Sci Rep. (2018) 8:1–11. doi: 10.1038/s41598-018-23851-z
54. Yuan K, Ye J, Liu Z, Ren Y, He W, Xu J, et al. Complement C3 overexpression activates JAK2/STAT3 pathway and correlates with gastric cancer progression. J Exp Clin Cancer Res. (2020) 3:1–15. doi: 10.1186/s13046-019-1514-3
55. Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine effects of tumor-derived complement. Cell Rep. (2014) 6:1085–95. doi: 10.1016/j.celrep.2014.02.014
56. Shu C, Zha H, Long H, Wang X, Yang F, Gao J, et al. C3a-C3aR signaling promotes breast cancer lung metastasis via modulating carcinoma associated fibroblasts. J Exp Clin Cancer Res. (2020) 39:1–14. doi: 10.1186/s13046-019-1515-2
57. Afshar-Kharghan V. The role of the complement system in cancer. J Clin Investig. (2017) 127:780–9. doi: 10.1172/JCI90962
59. Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. (2012) 189:4674–83. doi: 10.4049/jimmunol.1201654
60. Nitta H, Murakami Y, Wada Y, Eto M, Baba H, Imamura T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. (2014) 32:1715–9. doi: 10.3892/or.2014.3341
61. Gunn L, Ding C, Liu M, Ma Y, Qi C, Cai Y, et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. (2012) 189:2985–94. doi: 10.4049/jimmunol.1200846
62. Lu Y, Hu XB. C5a stimulates the proliferation of breast cancer cells via Akt-dependent RGC-32 gene activation. Oncol Rep. (2014) 32:2817–23. doi: 10.3892/or.2014.3489
63. Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, et al. Complement C5a receptor facilitates cancer metastasis by altering t-cell responses in the metastatic niche. Cancer Res. (2014) 74:3454–65. doi: 10.1158/0008-5472.CAN-14-0157
64. Chen J, Li GQ, Zhang L, Tang M, Cao X, Xu GL, et al. Complement C5a/C5aR pathway potentiates the pathogenesis of gastric cancer by down-regulating p21 expression. Cancer Lett. (2018) 412:30–6. doi: 10.1016/j.canlet.2017.10.003
65. Kaida T, Nitta H, Kitano Y, Yamamura K, Arima K, Izumi D, et al. C5a receptor (CD88) promotes motility and invasiveness of gastric cancer by activating RhoA. Oncotarget. (2016) 7:84798–809. doi: 10.18632/oncotarget.12656
66. Maeda Y, Kawano Y, Wada Y, Yatsuda J, Motoshima T, Murakami Y, et al. C5aR is frequently expressed in metastatic renal cell carcinoma and plays a crucial role in cell invasion via the ERK and PI3 kinase pathways. Oncol Rep. (2015) 33:1844–50. doi: 10.3892/or.2015.3800
67. Xi W, Liu L, Wang J, Xia Y, Bai Q, Xiong Y, et al. Enrichment of C5a-C5aR axis predicts poor postoperative prognosis of patients with clear cell renal cell carcinoma. Oncotarget. (2016) 7:80925–34. doi: 10.18632/oncotarget.13108
68. Saito K, Iioka H, Maruyama S, Sumardika IW, Sakaguchi M, Kondo E. PODXL1 promotes metastasis of the pancreatic ductal adenocarcinoma by activating the C5aR/C5a axis from the tumor microenvironment. Neoplas. (2019) 21:1121–32. doi: 10.1016/j.neo.2019.09.003
69. Nabizadeh JA, Manthey HD, Panagides N, Steyn FJ, Lee JD, Li XX, et al. C5a receptors C5aR1 and C5aR2 mediate opposing pathologies in a mouse model of melanoma. FASEB J. (2019) 33:11060–71. doi: 10.1096/fj.201800980RR
70. Triantafilou K, Hughes TR, Triantafilou M, Morgan PP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. (2013) 126:2903–13. doi: 10.1242/JCS.124388
71. Tegla CA, Cudrici C, Patel S, Trippe R, Rus V, Niculescu F, et al. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res. (2011) 51:45–60. doi: 10.1007/s12026-011-8239-5
72. Wong EKS, Kavanagh D. Diseases of complement dysregulation—an overview. Semin Immunopathol. (2018) 40:49–64. doi: 10.1007/s00281-017-0663-8
73. Towner LD, Wheat RA, Hughes TR, PaulMorgan B. Complement membrane attack and tumorigenesis a systems biology approach. J Biol Chem. (2016) 291:14927–38. doi: 10.1074/jbc.M115.708446
74. Seol HS, Lee SE, Song JS, Rhee JK, Singh SR, Chang S, et al. Complement proteins C7 and CFH control the stemness of liver cancer cells via LSF-1. Cancer Lett. (2016) 372:24–35. doi: 10.1016/j.canlet.2015.12.005
75. Ying L, Zhang F, Pan X, Chen K, Zhang N, Jin J, et al. Complement component 7 (C7), a potential tumor suppressor, is correlated with tumor progression and prognosis. Oncotarget. (2016) 7:86536–46. doi: 10.18632/oncotarget
76. Bao D, Zhang C, Li L, Wang H, Li Q, Ni L, et al. Integrative analysis of complement system to prognosis and immune infiltrating in colon cancer and gastric cancer. Front Oncol. (2021) 10:553297. doi: 10.3389/fonc.2020.553297
77. O’Brien R, Cannon A, Reynolds J, Lysaght J, Lynam-Lennon N. Complement in tumourigenesis and the response to cancer therapy. Cancers. (2021) 13:1209. doi: 10.3390/cancers13061209
78. Baek TH, Kim JH, Park MJ, Lee HK, Son HJ, Soon HK, et al. The stromal overexpression of decay accelerating factor (DAF/CD55) correlates with poor clinical outcome in colorectal cancer patients. Korean J Pathol. (2011) 45:445–54. doi: 10.4132/KoreanJPathol.2011.45.5.445
79. Li B, Chu X, Gao M, Xu Y. The effects of CD59 gene as a target gene on breast cancer cells. Cell Immunol. (2011) 272:61–70. doi: 10.1016/j.cellimm.2011.09.006
80. Kapka-Skrzypczak L, Wolinska E, Szparecki G, Wilczynski GM, Czajka M, Skrzypczak M. CD55, CD59, factor H and factor H-like 1 gene expression analysis in tumors of the ovary and corpus uteri origin. Immunol Lett. (2015) 167:67–71. doi: 10.1016/j.imlet.2015.06.017
81. Zhang R, Liu Q, Peng J, Wang M, Gao X, Liao Q, et al. Pancreatic cancer-educated macrophages protect cancer cells from complement-dependent cytotoxicity by up-regulation of CD59. Cell Death Dis. (2019) 10:836. doi: 10.1038/s41419-019-2065-4
82. Kajander T, Lehtinen MJ, Hyvärinen S, Bhattacharjee A, Leung E, Isenman DE, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci USA. (2011) 108:2897–902. doi: 10.1073/pnas.1017087108
83. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrín D, Stehle T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol. (2015) 11:77–82. doi: 10.1038/nchembio.1696
84. Michels MAHM, Volokhina EB, Van de Kar NCAJ, Van den Heuvel LPWJ. The role of properdin in complement-mediated renal diseases: a new player in complement-inhibiting therapy? Pediatr Nephrol. (2019) 34:1349–67. doi: 10.1007/s00467-018-4042-z
85. Moore SR, Menon SS, Galwankar NS, Khuder SA, Pangburn MK, Ferreira VP. A novel assay that characterizes properdin function shows neutrophil-derived properdin has a distinct oligomeric distribution. Front Immunol. (2023) 13:918856. doi: 10.3389/fimmu.2022.918856
86. Kesselring R, Thiel A, Pries R, Fichtner-, Brunner S, Seidel P, et al. The complement receptors CD46, CD55 and CD59 are regulated by the tumour microenvironment of head and neck cancer to facilitate escape of complement attack. Eur J Cancer. (2014) 50:2152–61. doi: 10.1016/j.ejca.2014.05.005
87. Kapka-Skrzypczak L, Popek S, Sawicki K, Wolińska E, Czajka M, Skrzypczak M. Effect of IL-6 and IL-8 on the expression of the complement activation inhibitors MAC-inhibitory protein and decay-accelerating factor in ovarian cancer A2780 cells. Oncol Lett. (2016) 12:1507–12. doi: 10.3892/ol.2016.4795
88. Kapka-Skrzypczak L, Wolinska E, Szparecki G, Czajka M, Skrzypczak M. The immunohistochemical analysis of membrane-bound CD55, CD59 and fluid-phase FH and FH-like complement inhibitors in cancers of ovary and corpus uteri origin. Cent Eur J Immunol. (2015) 40:349–53. doi: 10.5114/ceji.2015.54598
89. Maciejczyk A, Szelachowska J, Omiej BS, Szulc R, Szulc A, Wysocka T, et al. CD46 expression is an unfavorable prognostic factor in breast cancer cases. Appl Immunohistochem Mol Morphol. (2011) 19:540–6. doi: 10.1097/PAI.0b013e31821a0be9
90. Mo X, Leung TH-Y, Siu MK-Y, Ngan HY-S. Elucidating the potential role of membrane complement regulatory proteins (CD46, CD55, CD59) in tumorigenesis of cervical cancer. Cancer Res. (2020) 80:3816. doi: 10.1158/1538-7445.AM2020-3816
91. Riihilä PM, Nissinen LM, Ala-Aho R, Kallajoki M, Grénman R, Meri S, et al. Complement factor H: a biomarker for progression of cutaneous squamous cell carcinoma. J Investig Dermatol. (2014) 134:498–506. doi: 10.1038/jid.2013.346
92. Block I, Müller C, Sdogati D, Pedersen H, List M, Jaskot AM, et al. CFP suppresses breast cancer cell growth by TES-mediated upregulation of the transcription factor DDIT3. Oncog. (2019) 38:4560–73. doi: 10.1038/s41388-019-0739-0
93. Human Protein Atlas. Expression of CFP in cancer(2025). Available online at: https://www.proteinatlas.org/ENSG00000126759-CFP/cancer (Accessed March 07, 2025).
94. Vidergar R, Balduit A, Zacchi P, Agostinis C, Mangogna A, Belmonte B, et al. C1q-HA matrix regulates the local synthesis of hyaluronan in Malignant pleural mesothelioma by modulating HAS3 expression. Cancers (Basel). (2021) 13:416. doi: 10.3390/cancers13030416
95. Balduit A, Vidergar R, Zacchi P, Mangogna A, Agostinis C, Grandolfo M, et al. Complement protein C1q stimulates hyaluronic acid degradation via gC1qR/HABP1/p32 in Malignant pleural mesothelioma. Front Immunol. (2023) 14:1151194. doi: 10.3389/fimmu.2023.1151194
96. Yang G, Liang Y, Zheng T, Song R, Wang J, Shi H, et al. FCN2 inhibits epithelial-mesenchymal transition-induced metastasis of hepatocellular carcinoma via TGF-β/Smad signaling. Cancer Lett. (2016) 378:80–6. doi: 10.1016/j.canlet.2016.05.007
97. Ding Q, Shen Y, Li D, Yang J, Yu J, Yin Z, et al. Ficolin-2 triggers antitumor effect by activating macrophages and CD8+ T cells. Clin Immunol. (2017) 183:145–57. doi: 10.1016/j.clim.2017.08.012
98. Cedzyński M, Świerzko AS. Components of the lectin pathway of complement in haematologic Malignancies. Cancers (Basel). (2020) 12:1792. doi: 10.3390/cancers12071792
Keywords: C3, C1q, C5, cancer, complement system, non-canonical activation pathway
Citation: de Freitas Oliveira-Tore C, de Moraes AG, Plácido HMBS, Signorini NMDL, Fontana PD, da Piedade Batista Godoy T, Boldt ABW and de Messias I (2025) Non-canonical extracellular complement pathways and the complosome paradigm in cancer: a scoping review. Front. Immunol. 16:1519465. doi: 10.3389/fimmu.2025.1519465
Received: 29 October 2024; Accepted: 13 March 2025;
Published: 30 April 2025.
Edited by:
Anna Swierzko, Polish Academy of Sciences, PolandReviewed by:
Hadida Yasmin, Cooch Behar Panchanan Barma University, IndiaAndrea Balduit, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), Italy
Copyright © 2025 de Freitas Oliveira-Tore, de Moraes, Plácido, Signorini, Fontana, da Piedade Batista Godoy, Boldt and de Messias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iara de Messias, aWFyYS5yZWFzb25AaGMudWZwci5icg==
†These authors have contributed equally to this work