 Pan Li
Pan Li Zhengbo Fan2†
Zhengbo Fan2† Yanlan Huang
Yanlan Huang Xiaoyan Wu
Xiaoyan Wu- 1Department of Environment and Safety Engineering, Taiyuan Institute of Technology, Taiyuan, China
- 2People’s Government of Huangshui Town, Shizhu Tujia Autonomous County, Chongqing, China
- 3College of Veterinary Medicine, Southwest University, Chongqing, China
- 4National Engineering Research Center of Immunological Products, Department of Microbiology and Biochemical Pharmacy, College of Pharmacy and Laboratory Medicine, Third Military Medical University, Chongqing, China
Macrophages are vital sentinels in innate immunity, and their functions cannot be performed without internal metabolic reprogramming. Mitochondrial dynamics, especially mitochondrial fusion and fission, contributes to the maintenance of mitochondrial homeostasis. The link between mitochondrial dynamics and macrophages in the past has focused on the immune function of macrophages. We innovatively summarize and propose a link between mitochondrial dynamics and macrophage metabolism. Among them, fusion-related FAM73b, MTCH2, SLP-2 (Stomatin-like protein 2), and mtSIRT, and fission-related Fis1 and MTP18 may be the link between mitochondrial dynamics and macrophage metabolism association. Furthermore, post-translational modifications (PTMs) of mtSIRT play prominent roles in mitochondrial dynamics-macrophage metabolism connection, such as deacetylates and hypersuccinylation. MicroRNAs such as miR-150, miR-15b, and miR-125b are also possible entry points. The metabolic reprogramming of macrophages through the regulation of mitochondrial dynamics helps improve their adaptability and resistance to adverse environments and provides therapeutic possibilities for various diseases.
1 Introduction
Macrophages are important sentinels in innate immunity. Classically activated macrophages and alternatively activated macrophages play roles predominantly in inflammatory processes and injury repair or parasitic diseases, respectively (1). It has been reported that in vivo M1-like and in vitro classical activated macrophages and in vivo M2-like and in vitro alternatively activated macrophage are not completely equivalent (2). Undoubtedly, the different polarization forms of macrophages affect the immune regulation, which in turn determines the prognosis of host in adverse environments. The fate of macrophages is interfered by a variety of factors. For example: (1) Multiple signaling pathways and factors affect macrophage polarization. For example, PI3K/AKT/Rac-1 and peroxisome proliferator-activated receptor-γ (PPAR-γ) are involved in macrophage anti-inflammatory (3), Janus kinase (JAK)-signal transducer and activator of transcription (STAT) 1, Adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK)/nuclear factor kappa-B (NF-κB) signal pathways are involved in macrophage inflammatory response (4–6). (2) Micro-environmental nutrients and metabolites regulate macrophage metabolism and thus affect its polarization. Such as serine by regulating glutathione (GSH) synthesis (7), and γ-aminobutyric acid (GABA) perturbing macrophage oxidative phosphorylation (OXPHOS) via succinate-flavin adenine dinucleotide (FAD)-lysine specific demethylase1 (LSD1) (8) are involved in the regulation of LPS or LPS+IFN-γ induced macrophages. A creatine-related metabolic pathway is involved in IL-4 induced macrophage polarization (9). Endothelial cells secrete lactate through glycolysis, and lactic acid, after ingestion by macrophages, affects the acetylation of specific molecules through p300/CBP pathway, increases endothelium permeability, and finally affects the course of disease by regulating the polarization of macrophages (10, 11). (3) In recent years, the role of epigenetic modifications in macrophage polarization has been emphasized, which is also significant for trained immunity (12, 13).
As we listed above, macrophages in different microenvironment have different requirements for nutrients and immunity. Mitochondria are important multifunctional organelles in macrophages, including the core energy and metabolism stations (14), cell division (15, 16), signaling platform (17) and other biological functions. Thus, mitochondria have a non-negligible impact on macrophage fate decision. Mitochondrial morphology involves a series of temporal and spatial mitochondrial processes (18, 19), including fission and fusion, as well as adaptive behaviors such as mitochondrial autophagy (termly named as “mitophagy”) and mitochondrial transport, which are closely related to the biological activities and are the adaptive strategies of mitochondria in different cellular microenvironments. Therefore, changes in morphology, quality and location of mitochondria bestow macrophages energy supplementation and/or immunomodulatory assistances.
Mitochondrial dynamics regulates macrophage immunity by affecting macrophage immunity-related signals and pathways, and their entanglement has been extensively reviewed elsewhere (14, 18, 20, 21). LPS-activated macrophages benefit fragmented mitochondria for its increased mitochondrial fission through Stat2 promoting dynamin-related protein 1 (DRP1) Ser616 phosphorylation (22). Subsequent mitochondrial remodeling, such as fragmentation, loose cristae structure, and reduced membrane potential [ΔΨ(m)], promotes their function to ROS production, which further facilitate the pro-inflammatory response of macrophages (22). Inhibition of pyruvate dehydrogenase kinase (PDHK) in macrophages can promote mitochondrial fusion and reduce inflammatory stress (23).
Reparative macrophages show the aggregation of elongated mitochondria and induce mitochondrial fusion (24). Interferon regulator 1 (IRF1), a transcription factor that coordinates the expression of multiple inflammatory genes, it plays a key role in the removal of damaged mitochondria, macrophage autophagy, and inflammation control (25). Studies have also shown that IRF1 plays a bidirectional role in regulating mitochondrial dynamics, responding to TLR-induced macrophage mitochondrial fusion or fission in macrophages via a cascade reaction with CHIP (a HSC70-interacting protein) monoubiquitination translocation or ubiquitination degradation (26). Apart from IRF1, extracellular signal-regulated kinase (ERK1/2) is probably another bond in the mitochondrial morphological network that adapt to the polarization of macrophage. For one part, ERK1/2 is related to LPS induced polarization in RAW264.7 cells and THP-1 macrophages (27, 28), or alternatively activated macrophages in patients with endometriosis or gastric cancer-liver metastasis (29, 30). For another, ERK1/2 probably participates in the inhibition of mitochondrial-dependent apoptosis (31) and facilitates mitochondrial biogenesis in virtue of promoting CREB phosphorylation and finally increasing PGC1α (PPARγ co-activator 1 alpha, a mitochondrial biogenesis regulator) (32). However, ERK1/2 is proved to induce fragmented mitochondria by phosphorylating DRP1 at S616 to induce mitochondrial fission (33, 34), and mediate MFN1 (Mitofusin 1) T562-phosphorylation (35) to decrease mitochondrial fusion. Reviewed by Sabouny R. and Shutt T. E. concluded protein kinase A (PKA), protein kinase B (AKT) and cyclic adenosine monophosphate (cAMP) work effectively in facilitating hyperfused mitochondria, while ERK1/2 and Ca2+ promote fragmented mitochondria (20). Unfortunately, this paper does not correlate these mitochondrial morphology regulating signaling molecules with the polarization state of macrophages, but we predict that mitochondrial dynamics have connection with macrophage immunity. More significantly, Xie et al. reviewed and speculated that mitochondrial dynamic network regulation (e.g., MFN1/2) probably participate in the release of IFN-β (type I interferons) and activation of the inflammasome NLRP3 in macrophages (21). In conclusion, mitochondrial dynamic networks regulating the immune responses of macrophages have been extensively summarized, so we will not dwell on it.
In addition to the immune-related factors and/or pathways mentioned above in geared to mitochondrial dynamics and macrophage polarization, macrophage immunity is closely associated with intracellular metabolism. As all accepted, the high plasticity of macrophages is reflected in OXPHOS and glycolysis dependent metabolic switch, which supports the determination of the polarization fate of macrophages under different stimulations (36). In IL-4 induced macrophages, OXPHOS is the dominant metabolic state, accompanied by a complete tricarboxylic acid cycle (TCA) cycle and enhanced electron transport chain (ETC), while LPS+IFN-γ activated macrophages prefer to use glycolysis (37). Recently, it was uncovered that mitochondrial dynamics can reflect different polarization phenotype of macrophages. Classically activated macrophages show glycolysis and discrete mitochondria, however, OXPHOS metabolically active macrophage has elongated mitochondria (38). Based on this mitochondrial dynamics tether to macrophage metabolism coincident with polarization states, mitochondrial subcellular imaging has potential to monitor metabolic progression of macrophages and ultimately instruct clinical treatment. The conclusion above depends on mitochondrial fluorescence confocal imaging and ImageJ macro tools to analyze its mass (foodprint), branching, network morphology and size and other traits (38). In addition to these intuitive and visual methods, the fusion and fission of mitochondria take place under the coordination of complex life activities in cells. For example, the guanosine triphosphatases (GTPase) proteins MFN1 and MFN2 are responsible for fusion, and DRP1 is responsible for fission (39). In addition, mitochondrial dynamics have been shown to induce metabolic reprogramming in skeletal muscle atrophy or liver tumors (40), suggesting that mitochondrial dynamics have significant impacts on the fate of body health. Whether there are deeper connections between mitochondrial dynamics and macrophage metabolism, we will discuss those in the following part.
2 Mitochondrial fission and macrophage metabolism
2.1 Mitochondrial fission event
As we discussed above, cytosolic protein DRP1 is the primary regulate factor during mitochondrial fission (39). The post-translational modification (PTM) of DRP1 has been extensively summarized and annotated (41), such as ubiquitination, phosphorylation, palmitoylation, O-GlcNAcylation (OGA), etc. Different PTM or the same type of modifications occurs at different sites may have completely opposite effects on the reconciliation of mitochondrial state. Therefore, it is of great significance to study PTM.
Mitochondrial fission is a multifactor derived biological event (Figure 1, right). Endoplasmic reticulum (ER)-Mitochondrial signaling plays a key role in mitochondrial fission, involving special regions of the ER called mitochondria-associated membranes (MAMs) (42). The ER tubule wraps mitochondrial tubule physically to form the earlier fission event (43), constitutes a subsequent assembly site and assists fission in an actin-dependent manner. In recent years, it has been found that acetylation of actin regulates actin polymerization via inverted guanidine protein 2 (INF2) located in the ER, and that mitochondria-localized myosin 19 (Myo19) collaborates with INF2 and spiretype actin nucleation factor 1 (Spire1C) to regulate actin assembly (44–46). Actin-depolymerizing protein cofilin1 acts as a reverse regulator to balance INF2/Spire1C in mitochondrial actin dynamics (47). Mff (the tail-anchored mitochondrial fission factor) controls midzone fission, leading to mitochondrial proliferation, while mitochondrial outer membrane protein fission 1 protein (Fis1) regulates peripheral fission, leading to mitochondrial autophagy (48). MiD49/51 (N-terminally anchored mitochondrial dynamics proteins, 49 kDa and 51 kDa) is responsible for Drp1 recruitment to the mitochondrial OM in the form of inactive dimer, and Drp1-MiD is essential for Drp1 assemble and subsequent cytochrome c (cyt c) release and cristae remolding during apoptosis (49). Mechanically, Hidenori et al. demonstrated visually through TEM that WT and Mff-KO cells showed abnormal mitochondria with balloon or lamella-less cristae, while most mitochondria in MiD49/51-KO and Drp1-KO cells retained lamellar cristae structure after apoptosis induction (49). This suggests that induced cristae stabilization in MiD49/51-KO and Drp1-KO cells leads to resistance to cyt c release (49). It was further demonstrated that MiD51’s N-SA, a mutation in the N-terminal OMM targeting signal anchor of MiD51, is necessary to correctly pinpoint the Drp1-MiD51 complex in the proximity of the crista-remodeling system under apoptotic signal, thereby modulating cristae junction disruption to release cyt c (49). In mammals, classical dynamin 2 (DYN2) assists DRP1 in promoting mitochondrial fission (40). In addition, cortactin, Arp2/3 (Actin-related protein 2/3) complex (50) and Myosin II also works in mitochondrial fission (51).
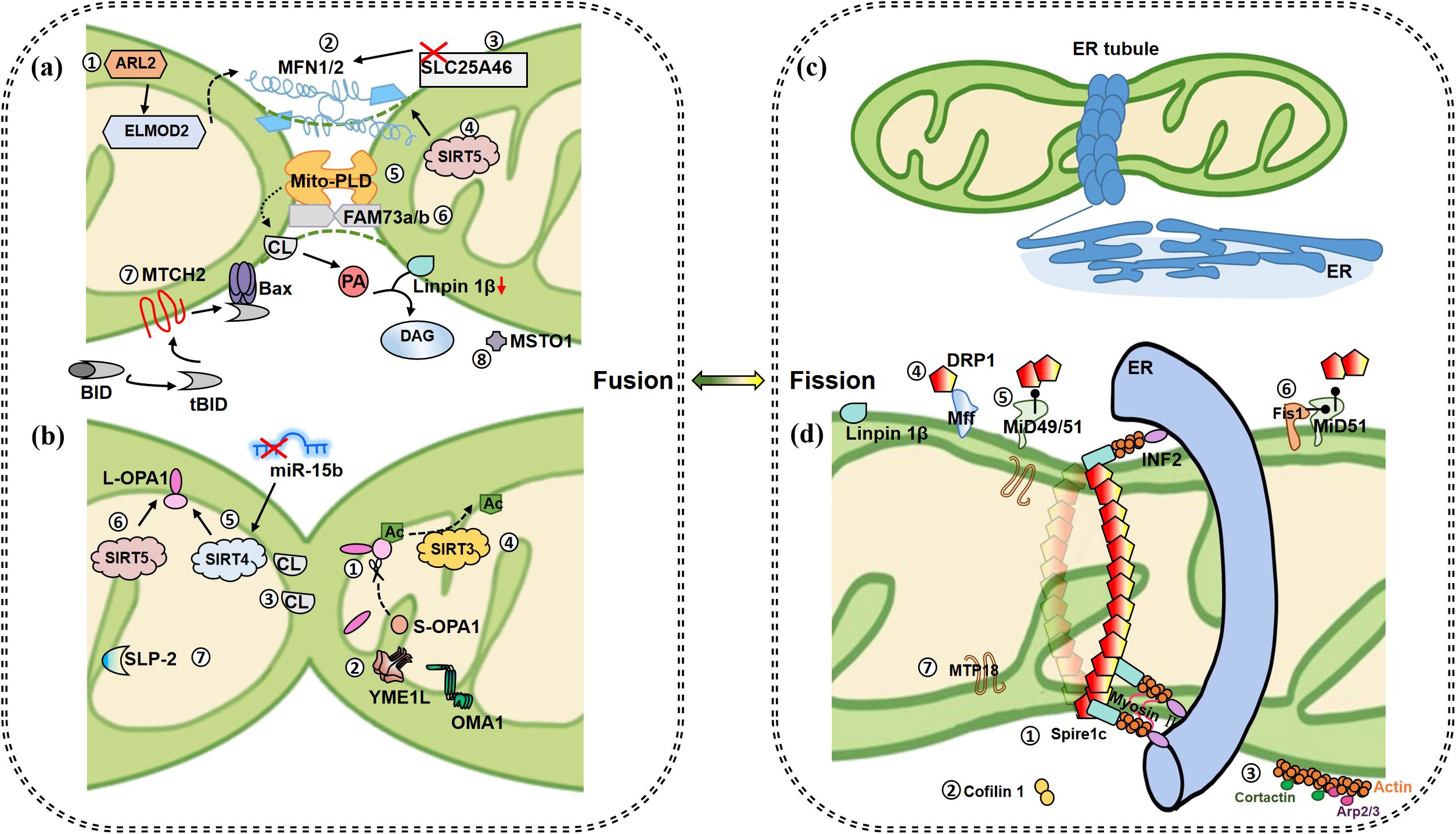
Figure 1. Mitochondrial fusion and fission events. (a) Fusion of OMM. ①IMS protein ARL2 promotes mitochondrial fusion during constitutive activation, and its downstream ELMOD2 is a necessary effector. ARL2 and ELMOD2 work upstream of MFNs. ②When MFN1/2 appear on two opposite membranes of mitochondria, it causes the fusion of OMM. ③Inhibition of OMM protein SLC25A46 promotes the stability of MFN1 and MNF2, which further promotes hyperfusion. ④SIRT5 overexpression promotes the increase of MFN2 and promotes fusion. ⑤At a certain time point when MFNs bind mitochondria together, MitoPLD modifies the lipid surface of the opposite mitochondrial OM in a PA-dependent manner for subsequent mitochondrial fusion. MitoPLD anchors on OM and hydrolyzes CL to produce PA. PA is converted into DAG by absorption of Lipin 1β, which ultimately contributes to mitochondrial fusion. ⑥Integrin protein FAM73A/B promotes the formation of mitoPLD dimer and inhibits its degradation. ⑦MTCH2 is a direct regulator of fusion, by regulating tBID and promoting the interaction between tBID and Bax, thereby inducing MOMP and accelerating apoptosis. ⑧MTSO1 is mainly located in the cytoplasm and is involved in OM fusion, but the specific mechanism is unknown. (b) Fusion of IMM. ①GTPase OPA1 contributes to IM fusion. Specifically, IMM fusion is dependent on L-OPA1 and its hydrolyzed form S-OPA1, and L-OPA1 alone is sufficient to promote mitochondrial fusion. ②OMA1 and YME1L are involved in the hydrolysis of L-OPA1 to produce c-, e- and d-type s-OPA1, respectively. ③IM fusion occurs asymmetrically. As long as OPA1 on one side and a sufficient concentration (15%-20%) of CL on the other side, these two elements bridge to promote fusion. S-OPA1 facilitates the bridging. ④SIRT3 promotes deacetylation of OPA1 at K926/931 to promote fusion. ⑤Up-regulated SIRT4 is accompanied by higher levels of L-OPA1 expression, a phenomenon that could also be repeated by inhibiting miR-15b. ⑥SIRT5 overexpression promotes the increase of OPA1, which contributes to mitochondrial fusion. ⑦SLP-2 cooperates with L-OPA1 to promote hyperfusion. (c) Initiation of mitochondrial fission. ER tubules wrap mitochondria to form early fission events. (d) Subsequent fission events. ①Located on the ER, INF2 recruits actin and forms a complex with Spire1c to promote mitochondrial contraction with the assistance of myosin II. ②The actin depolymerase protein cofilin1 acts as a reverse regulator to balance the effects of INF2/Spire1C. ③Cortactin and ARP2/3 complex regulate actin assembly on OMM and affect Drp1 dynamics and mitochondrial fission. Plentiful receptors on the OMM are involved in fission regulation. ④Drp1-Mff promotes GTPase-dependent shrinkage and breakage of fission helical rings. ⑤Drp1-MiD has been shown to promote MFN2 independent fusion by blocking Drp1. ⑥The activity of MiD51 is low when combined with Fis1, so Fis1 indirectly promotes fission. ⑦MTP18, located in the IMM, induces fission together with Drp1 and Fis1. Whether MTP18 plays a role in IM fission remains unknown. (Arrows represent promotion and dots represent inhibition.).
Little is known about mitochondrial IM fission events. Interestingly, mitochondrial protein 18 kDa (MTP18) induces fission along with Drp1 and Fis1 (52). Because located on IMM (mitochondria inner membrane), MTP18 may drive the IM fission assembly, which is only a speculation and needs experimental proof. In recent years, studies have shown that MTP18 deficiency can reduce mitochondrial division, and two microRNAs negatively regulate MTP18 (miR-652-3p and miR-668) (53).
In the ROS induced cardiac mitochondrial fission, MTP18 interfered with Drp1 accumulation but failed to mediate fission alone when Drp1 expression was scarcely (54). Therefore, MTP18 must collaborate with other factors to promote fission.
Mitochondrial fission is a complex biological process. We have summarized the current knowledge of fission-regulating proteins (Table 1), hoping to describe mitochondrial fission more comprehensively, but the existing content is far from sufficient. More research is still needed on mitochondrial fission, especially IMM.
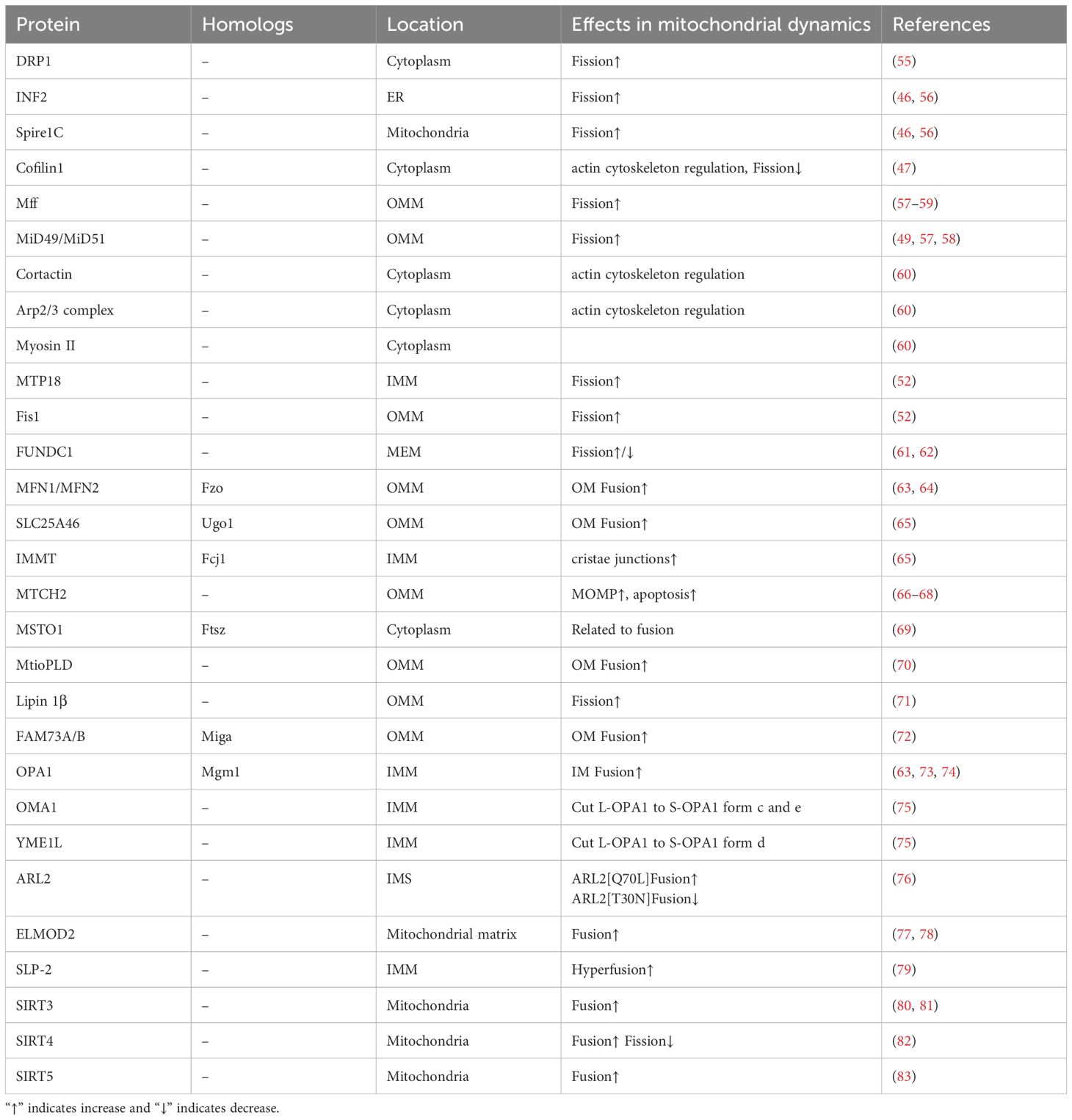
Table 1. Mitochondrial dynamics related proteins and their location.
2.2 The connection between mitochondrial fission and macrophage metabolism
Preliminary studies of mitochondrial dynamics are usually performed in fibroblasts and/or stem cells (66, 84). Nowadays, mitochondrial dynamics have also been found to be crucial to the fate determination of macrophages. The classical cognition of macrophage metabolism is that glycolysis is the metabolic state of LPS-induced macrophages, and OXPHOS is extremely important for resting macrophages (85). The mitochondria of LPS (0.5 µg/mL) -induced BMDMs rapidly displayed small punctate and fragmented forms within 2 h, and remain in a fission state after 12 h of stimulation, and the expressions of Mfn1, Mfn2, Fam73a, and Fam73b are decreased, the dephosphorylation of Drp1 is enhanced (26). Mitochondria in IL-4-induced BMDM are tubular in fusion state, and Fam73b expression is elevated (26). Under the two completely different metabolic states of macrophages (glycolysis and OXPHOS), mitochondria also present extremely different morphology. Whether the dynamic changes of mitochondria can directly represent the metabolism of macrophages, or whether there are some ways to correlate these two phenotypes, we will analyze the argument in the following article.
In the process of mitochondrial fission, excessive activation of DRP1 leads to mitochondrial dysfunction, which is manifested as increased permeability of OM, decreased ATP production, and increased release of ROS and cyt c, thus leading to cell apoptosis (86). Therefore, mitochondrial fission is closely related to cell metabolism (Figure 2).
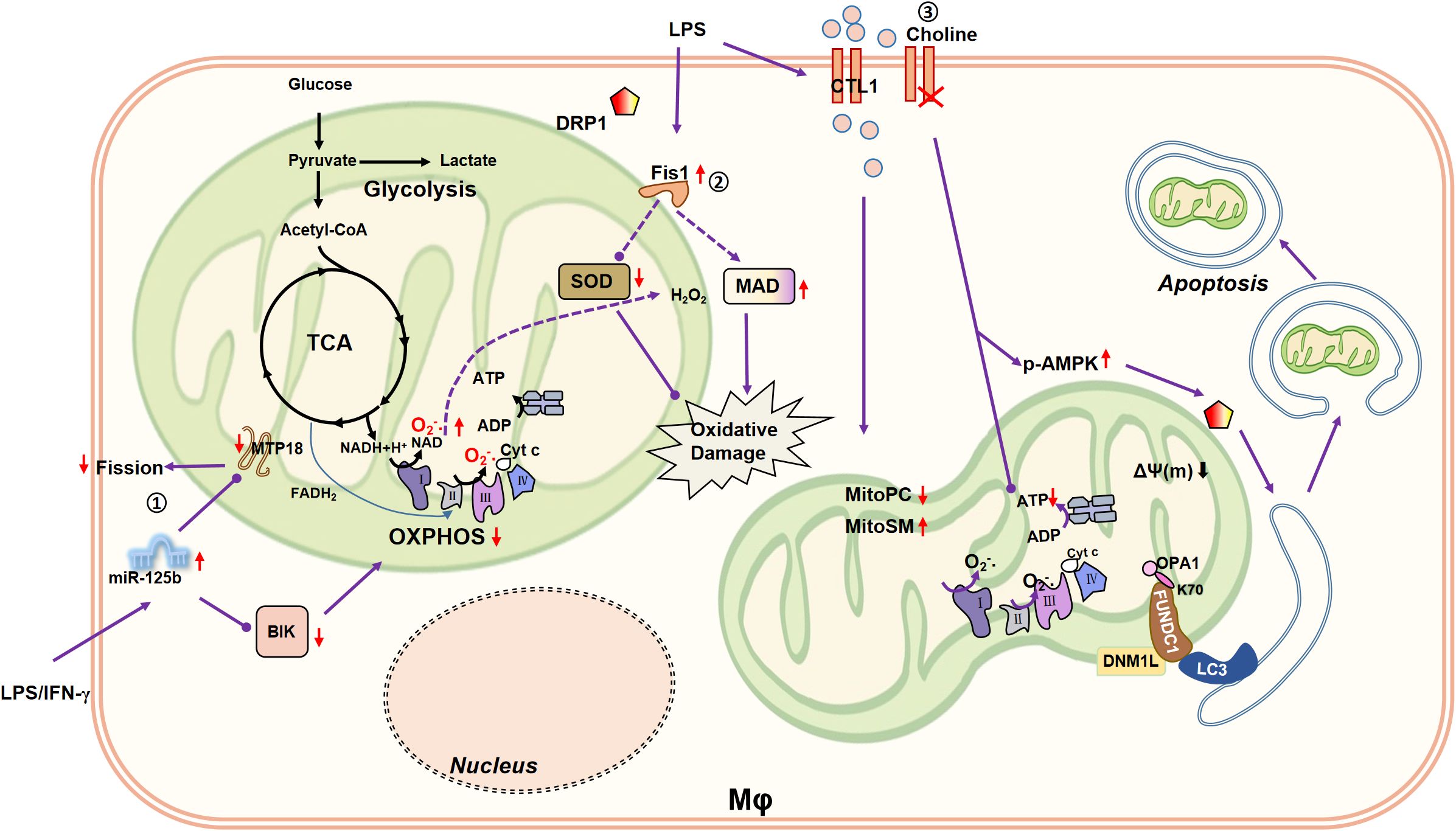
Figure 2. Connection between mitochondrial fission/mitophagy and macrophage metabolism. ①MiR-125b induces mitochondrial fusion by silencing MTP18 and reduces OXPHOS of macrophages by inhibiting BIK, reducing the pro-inflammatory activity of macrophages. ②Mitochondrial fission-related receptor Fis1 is upregulated in LPS (10 µg/mL)-induced rat alveolar macrophages, accompanied by a decrease in cellular SOD and an increase in MDA. SOD is capable to catalyze the disproportionation of O2•- to H2O2. Therefore, Fis1 may be involved in aggravated macrophage oxidative damage. ③LPS (0.2 µg/mL) enhances the expression of CTL1, promotes the choline uptake mediated by CTL1, and remodels macrophage lipid metabolism, such as reducing Mito-PC, and increasing Mito-SM. Impaired choline uptake disrupts mitochondrial ATP synthesis and triggers AMPK activation to promote DRP1-driven mitophagy.
The crystal structure of MiD51, an adaptor protein of mitochondria, shows its ability to bind to ADP (87). MiD51 greatly promotes the hydrolytic activity and oligomerization of Drp1 in the presence of ADP, and then promoted mitochondrial fission, while when ADP absence, it inhibits the hydrolytic activity of Drp1 GTPase, which fully indicated the objective relationship between mitochondrial fission and cell metabolism (88, 89). MiD51 has been shown to regulate basal or glucose-induced oxygen consumption and insulin secretion levels in both mouse and human pancreatic islet cells (90). Unfortunately, there are few studies about MiD51 functions in macrophages at present, and the relationship between MiD51 and macrophage metabolism is also a study of far-reaching significance. Fis1, another key protein associated to mitochondrial fission, is up-regulated in LPS (10 µg/mL)-induced rat alveolar macrophage, and simultaneously causes cellular SOD (superoxide dismutase) decrease and MDA (malondialdehyde) increase, suggesting that Fis1 is involved in the aggravation of oxidative damage in macrophages (91). Whether the changes of SOD and MDA directly related to Fis1 was not explored in this article.
Recently, DRP1 has been confirmed to be involved in the differentiation of osteoclasts and the regulation of bone loss (92). It is well known that osteoclast differentiation involves a series of comprehensive metabolic reprogramming such as OXPHOS, glycolysis and fatty acid synthesis (93). Therefore, Drp1 regulation of osteoclast differentiation is closely related to macrophage metabolism regulation. However, it remains unclear whether there is a causal relationship between metabolic changes and mitochondrial dynamics. A large number of literature research to mitochondrial fission is associated with macrophage inflammatory state, including the mice BMDM and human monocytes THP-1 cells (22, 94, 95), but those research mainly focus on macrophage inflammatory signals, including inflammatory pathways or mediators, however, the metabolic changes of macrophages are worth exploring. In human monocytes, pro-apoptotic protein BIK and mitochondrial fission process 1 protein MTP18 correlate apoptosis with mitochondrial dynamics through the miR-125b (96). In monocyte derived macrophages, pro-inflammatory activity of macrophages are correlated with the decrease of BIK and MTP18 and the increase of miR-125b (96). Moreover, miR-125b induces mitochondrial fusion by silencing MTP18 and reduces the OXPHOS of macrophages (96).
Mitochondrial fission-related proteins have been studied more rarely on cell metabolism, let alone on macrophage metabolism (Table 2, Figure 2). Surprisingly, we find that micro-RNA (e.g., miR-125b) could be used as a breakthrough point to study the effect of mitochondrial fission on the metabolism of macrophages, which provides a good idea for subsequent research.
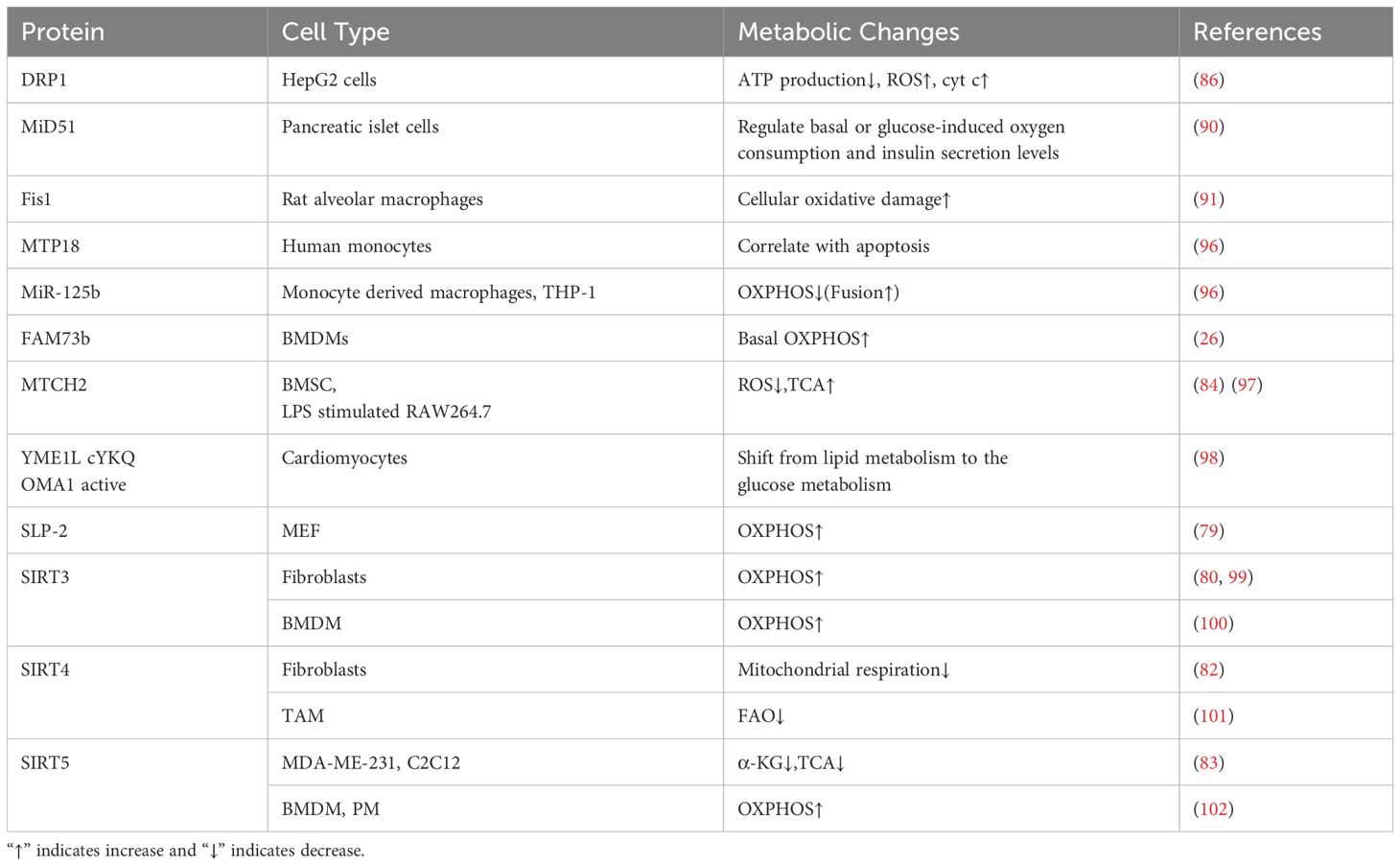
Table 2. The effects of mitochondrial dynamics related protein in cell metabolism.
3 Mitochondrial fusion and macrophage metabolism
3.1 Mitochondrial fusion event
Mitochondrial fusion largely divides into OMM fusion and IMM fusion (21, 103, 104). During mitochondrial fusion, the mixing of contents facilitates the transmission of information [e.g., mtDNA (105)] and leads to the rapidly dilution of cytoplasmic contents (106), which helps to rescue dysfunctional mitochondria. MFN1 and MFN2 are responsible for OMM fusion and optic atrophy 1 (OPA1) is responsible for IMM fusion (63). The fusion of two mitochondria occurs first in OM and then in IM (the inner membrane) (Figure 1, left).
3.1.1 OMM fusion
About mitochondrial fusion and fission-related proteins were first discovered in yeast (107), drosophila (108), etc. Later, homologous proteins were found in humans and/or other mammals. MFN1 and MFN2 involved in OMM fusion in mammals are homologies of fuzzy onions (Fzo) (64). Similarly, Ugo1 is a OMM protein that coordinates the fusion events of OMM and IMM, but no obvious homologues of Ugo1 have been found in higher eukaryotes (109). The discovery of SLC25A46, a member of the mitochondrial solute carrier family 14 (SLC25), which matched Ugo1 in yeast (Schizosaccharomyces japonicus), confirmed that the OMM and IMM can fuse independently in mammalian cells (65). SLC25A46 may be recruited to the OMM as a pre-fission factor and interact with the IMM remodeling protein mitofilin (Fcj1), the IMMT homolog of mammal (65). Knocking down SLC25A46 causes delay in mitochondrial fission or elongation (65), reduced expression of SLC25A46 increases the stability of MFN1 and MFN2 (110), which both leading to mitochondrial hyperfusion. Interestingly, SLC25A46 mutation also results in hyperfusion of mitochondria, but with down-regulated OXPHOS, because excessive fusion seriously damages the subcellular structure of mitochondria, showing that the cristae structure is destroyed or even disappeared (111).
Mitochondrial carrier homolog 2 (MTCH2, SLC25A50), located at OMM, is a direct regulator of mitochondrial fusion/elongation in mouse embryonic fibroblasts (MEFs) and embryonic stem cells (ESCs) (66). As an effective binding partner of BID, MTCH2/MIMP (Met-induced mitochondrial protein) interacts with truncated BID (tBID) at OMM, promoting tBID translocation to mitochondria and ultimately inducing OMM permeabilization (MOMP) to accelerate apoptosis (67, 68).
Similar to Ftsz of prokaryotic (69), MSTO1 (Misato homolog 1), mainly located to the cytoplasm, is a protein involved in the fusion and network formation of OMM (69). Currently, MSTO1 is only known to be related to the fusion of OMM. It is worth exploring how MSTO1 participates in the fusion process, whether there is structural interaction with OMM, or whether MSTO1 promotes the mitochondrial fusion by enhancing the chemotaxis of other factors.
Intriguingly, remolding membrane phospholipid content may affect mitochondrial fusion. For example, mitochondrial phospholipase D (MitoPLD) belongs to Phospholipase D (PLD) signaling enzyme superfamily. A series of metabolic cascades catalyzed by MitoPLD results in mitochondrial tubular morphology, that is, promote mitochondrial fusion. Concretely, due to the special N-terminal domain, MitoPLD is anchored on OMM, which can hydrolyze cardiolipin (CL) on the surface of mitochondria to generate phospholipid acid (PA) (70). At certain points in time when MFNs tethered mitochondria together, MitoPLD modifies the lipid surface of the OM of the opposite mitochondria in a PA-dependent manner to perform the subsequent steps of mitochondrial fusion (70). Later studies found that PA absorption phosphatase (Lipin 1β) produced by MitoPLD hydrolysis converts PA into diacylglycerol (DAG) and terminates the lipid-signaling pathway of browning activated by PA (71). The catalytic domain of Lipin 1β locates at the tip of small mitochondria, or in a spot-like manner on tubular mitochondria, converting long mitochondrial tubules into medium-size fragments (71). Lipin 1β facilitates mitochondrial fission (71), and likewise, skeletal muscle of Lipin-1-deficient patients and extensor digitorum longus muscle of Lipin-1-deficient fatty liver dystrophy mice have been observed with mitochondrial aggregation or large shape (112–114). In conclusion, MitoPLD promotes mitochondrial fusion by lowering Lipin 1β through PA generation. On the other hand, PA generated by the MitoPLD is converted to DAG, which eventually leads to the increase of DAG. An animal study revealed that DAG probably reduces body fat mass by stimulating thermogenesis in brown adipose tissue (BAT) while inducing lipolysis in white adipose tissue (WAT) (115). From a more macroscopic perspective, MitoPLD shows that there is a correlation between cell metabolism and mitochondrial dynamics.
Moreover, mammalian FAM73A (Miga1) and FAM73B (Miga2) were discovered as orthologs of drosophila mitoguardin (miga) gene (72). As membrane integrins on OMM, MIGA1 and MIGA2 promote the formation of MitoPLD dimers and inhibit its degradation (72). Therefore, miga acts downstream of MitoPLD, and both miga and MitoPLD act downstream of MFNs to promote mitochondrial fusion. In addition to the effect on mitochondrial morphology, miga is also involved in the regulation of ΔΨ(m), mitochondrial ATP and ROS production (72). Therefore, miga may be a pleiotropic factor that affects mitochondrial morphological dynamics and cell metabolism.
3.1.2 IMM fusion
Mgm1, the first member of the Dynamin family identified in yeast, is required for mitochondrial fusion of IMM and is also associated with the structure maintenance of crista (116), and the human homolog of Mgm1 is OPA1 (73). OPA1 is a dynamin-related GTPase that plays an important role in promoting fusion of IMM, maintaining cristae structure, promoting IMM structure and integrity (74) and controlling apoptosis (117, 118). OPA1 exists in two forms, namely, membrane-bound Long-OPA1 (L-OPA1) and Short-OPA1 (S-OPA1), the hydrolysate of L-OPA1 limited to the membrane space. Mitochondrial fusion is believed to depend on the presence of both L- and S-OPA1 (119), and various stress conditions destroy these complexes and trigger the complete transformation of L-OPA1 into S-OPA1, thus inhibiting mitochondrial fusion (119, 120). In recent years, it has been found that the L-OPA1 is sufficient to mediate mitochondrial fusion and has anti-apoptotic properties (121). Therefore, the morphology of OPA1 has a great influence on mitochondrial morphology.
The proteolytic sites S1 and S2 of OPA1 are encoded by exons 5 and 5B, respectively (122). The IM peptidase OMA1 and i-AAA protease YME1L cut OPA1 separately at S1 and S2, resulting in the aggregation or transformation of L-/S-OPA1 and interfering with mitochondrial fusion. The OMA1 cleaves L-OPA1 in S1 to generate S-OPA1 forms c and e (75). In Oma1 knockout cells, fusion ability is preserved in the presence of L-OPA1, while the c and e forms of S-OPA1 are blocked, thus maintaining the tubular mitochondrial network and protecting cells from apoptosis (121). L-OPA1 protein in Yme1l knockout cells is equivalent to that in WT cells, but S-OPA1 accumulates due to OMA1 hydrolysis (121). This unbalanced processing of OPA1 results in the mitochondrial fragmentation, which also indicates that S-OPA1 may be involved in mitochondrial fission. According to the current conclusions, L-OPA1 is beneficial for promoting mitochondrial fusion, while S-OPA1 appears to be detrimental to mitochondrial fusion. However, S-OPA1, despite its lack of fusion function, has recently been shown to maintain cristae and mitochondrial energy through its GTPase activity (123), to some extent facilitating cell survival under stress.
Intriguingly, IM fusion has a different pattern from OM fusion. This is mainly reflected in the fact that OM fusion requires MFNs to appear on two opposite membranes, while for IM fusion, OPA1 regulates the fusion in an asymmetric way, which occurs as long as the unilateral IM contains OPA1 (124). Speaking of asymmetric regulation of OPA1, we have to mention a negatively charged acidic phospholipid CL. CL is enriched in IMM, and as long as there is sufficient concentration (15%-20%) on the other side of the mitochondria, L-OPA1 and CL pair to induce membrane-tethering and further induce mitochondrial fusion (125). S-OPA1 promotes the bridging between L-OPA1 and CL (125). However, fusion does not occur when the CL concentrations are low on the opposite or both sides of OPA1 (125). At this time, the interrelationship of homotypic trans-OPA1 mediates tethering of IM, which is conducive to the formation and maintenance of cristae (125). CL is re-assigned from IMM to OMM during the induction of damage signals such as mitochondrial damage and depolarization (126). In aging mitochondria, CL is oxidized by cyt c peroxidase and the content of CL decreases (127). Therefore, this regulatory mode of OPA1- CLhigh bridging contributes to the specificity of mitochondrial fusion and may be involved in preventing the fusion of healthy mitochondria with damaged/senescent mitochondria and improving the efficiency of cells. However, the mechanism by which OPA1 combined with CL to regulate mitochondrial fusion remains unclear and needs to be explored experimentally.
In addition, CL is required for optimal activity of a variety of mitochondrial carrier proteins. For instance, CL endows ADP/ATP carrier (AAC) 2 with optimal activity by promoting the stable association between AAC2 and respiratory supercomplexs, and reduces the travel distance of cyt c between ETC complex III and IV, contributing to the efficiency of OXPHOS (128). AACs are responsible for the exchange of ADP/ATP, specifically the transfer of ATP formed by OXPHOS from IMM to the intermembrane space (IMS) (129). In addition to regulating ATP production (coupled energy conversion) in mitochondria, AAC is shown to mediate the uncoupled energy conversion (130). Therefore, in the process of regulating mitochondrial fusion with OPA1, CL is likely to associate mitochondrial dynamics with cell metabolism by influencing AACs.
3.1.3 IMS proteins participated fusion
ARL2 (ADP-ribosylation factor (ARF) like 2) exists in the IMS and is uniformly distributed along the mitochondria in a spot-like pattern with MFN1/2 (76). The dominant negative mutation of ARL2[T30N] results in a decrease in mitochondrial fusion rate (24h) and a loss of specific mitochondrial motility (30h), and the dominant activating mutant ARL2[Q70L] leads to a significant increase in mitochondrial tubular structure (48h), independent of its effect on microtubules (76). Although there are some differences in the dynamics of cellular effects, as indicated activation time in parentheses, ARL2 generally promotes mitochondrial fusion during constitutively activation, and its reversal of mitochondrial fragmentation in the absence of fusion factors requires the presence of OPA1 and at least one MFN (76).
Richard A. et al. conducted the above systematic studies on ARL2 also found ELMOD2 (ELMO domain containing 2), which is essential for ARL2 to exert its active functions (131). ELMOD2 is an ARL2 GTPase-activating protein (GAP) that acts downstream of ARL2, and its influence on mitochondrial morphology is independent of GAP activity (77). In MEFs with MFN1 or MFN2 deficiency, the effect of ELMOD2 on the reverse of mitochondrial fragmentation is to a lower degree than that of ARL2, and ELMOD2 and ARL2 have no significant effect in MFN1 and MFN2 double-knockout cells (78). This also suggests that ARL2 and ELMOD2 both act upstream of MFNs, and ARL2 activation without ELMOD2 cannot induce mitochondrial elongation, so ELMOD2 is an effector necessary for ARL2 to promote mitochondrial elongation and tubular structure maintenance (77). Interestingly, ELMOD2 and ARL2, as well as MFN1, MFN2, and MitoPLD, are all located in a regular spot-like discrete distribution along the mitochondria forming bands (77), suggesting that they may form some special complexes in the mitochondria to function together, or there are unknown binding domains on the mitochondria, which recruits these factors to alter mitochondrial dynamics.
In the process of mitochondrial fusion, various elements of OM, IM, and IMS work together to change the dynamics of mitochondria, forming a complex and precise regulatory network (Table 1.). Among them, CL and DAG are involved in the metabolic regulation of OXPHOS and lipid metabolism, respectively, suggesting that mitochondrial dynamics is related to cell metabolism.
3.2 The association between mitochondrial fusion and macrophage metabolism
As we can see in Figure 1, a large number of proteins play a role in mitochondrial fusion and together maintain this complex biological process. Among them, no matter the OM proteins FAM73b, MTCH2, or mtROS are found to be involved in macrophage metabolism, while the relationship between IM proteins involved in fusion and macrophage metabolism still needs to be studied (Figure 3).
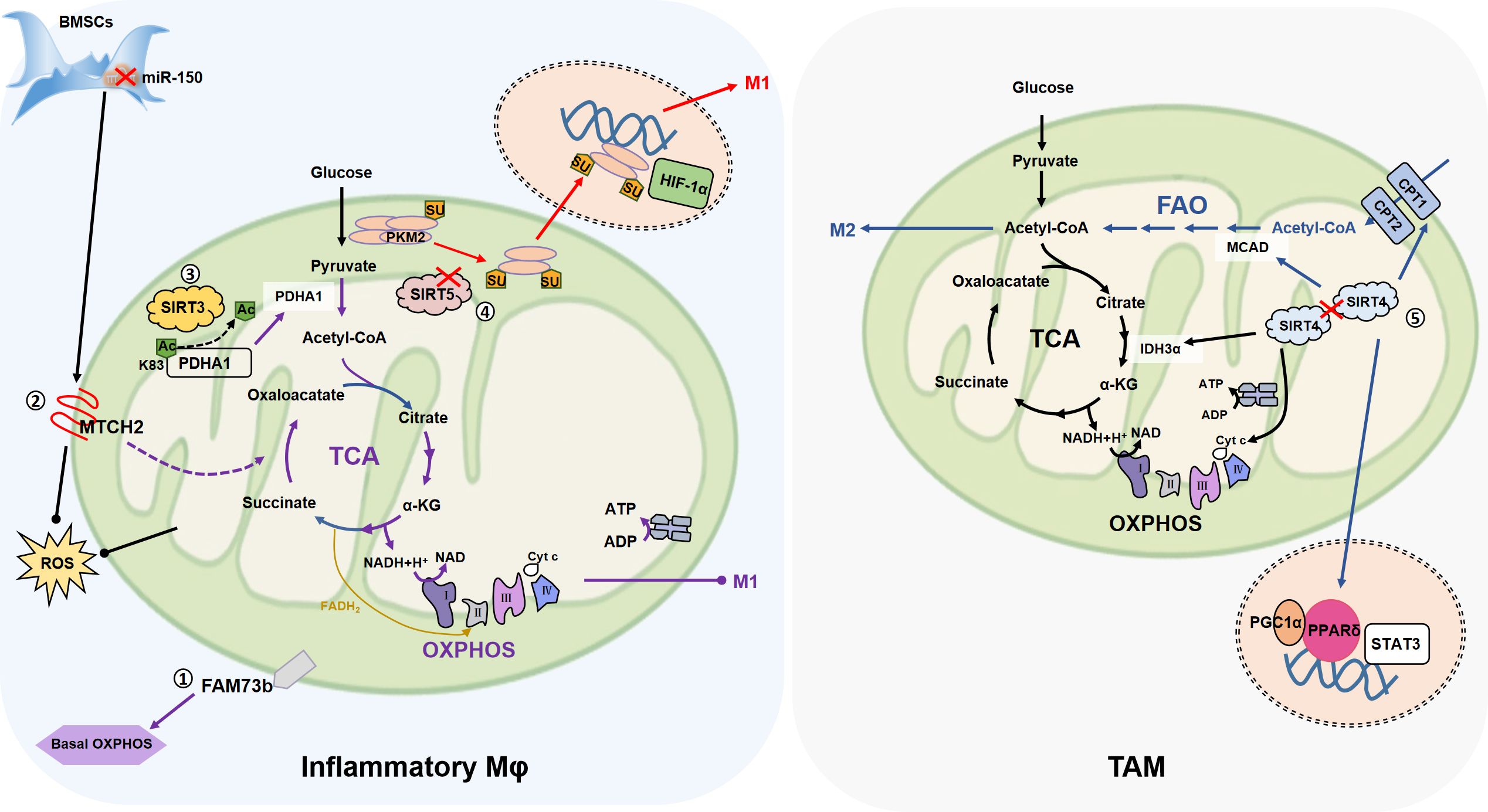
Figure 3. The association of mitochondrial fusion with macrophage metabolism. ①FAM73b which promotes mitochondrial OM fusion, promotes OXPHOS of resting macrophages, but has no effect on glycolysis, and neither OXPHOS nor glycolysis in inflammatory state. ②In the co-culture of BMSCs and macrophages, inhibition of miR-150 increases the expression of OMM fusion-related MTCH2 and promotes the transfer of mitochondria from BMSCs to macrophages (not shown in the figure). MTCH2 may enhance OXPHOS by promoting TCA cycle, thereby alleviating LPS (0.5 μg/mL)-induced apoptosis. ③MtSIRT promotes mitochondrial fusion, in which SIRT3 promotes PDHA1 deacetylation at the K83 site, and activated PDHA1 catalyzes pyruvate production of acetyl-coA to promote OXPHOS, inhibit glycolysis, and reduce macrophage inflammation. ④SIRT5 knockdown promotes PKM2 hypersuccinylation, reduces its pyruvate kinase activity, then induces PKM2 entry into the nucleus, and further persuading macrophage inflammation. ⑤SIRT4 knockdown activates FAO by promoting the expression of lipid catabolism genes such as MCAD and CPT1, and promotes the M2-like transformation of TAM through FAO-PPARδ-STAT3. On the other hand, SIRT4 silencing increases the expression of IDH3α, cyt c and other mitochondrial genes, so SIRT4 silencing contributes to the strengthening of mitochondrial OXPHOS. (Arrows represent promotion and dots represent inhibition.).
3.2.1 FAM73b
Compared with wild type (WT) cells, FAM73b KO BMDMs show a severe decline in the basal OXPHOS level and the mitochondria are in a state of fragmentation (26). However, there is almost no difference in ECAR between WT and FAM73b KO BMDMs after LPS (0.5 µg/mL) stimulation and the difference in OCR are converged (26). These results suggest that FAM73b KO has little effect on the glycolysis ability of macrophages, and affects OXPHOS mainly in the resting state. Therefore, we speculate that mitochondrial dynamics affect macrophage metabolism, but are biased and depend on specific cell state.
3.2.2 MTCH2
In bone marrow mesenchymal stem cells (BMSCs), inhibition of miR (microRNA)-150 mitigates LPS (0.5 µg/mL)-induced apoptosis in RAW264.7 (84), suggesting that the metabolic status of RAW264.7 is changed. The co-culture of BMSC also reduces ROS production of RAW264.7 after LPS stimulation (84). Mitochondrial respiratory chain is the main source of intracellular ROS, and the antioxidant system of mitochondria is responsible for removing ROS (132), so mitochondria play an important role in this process. As expected, the inhibition of miR-150 leads to the expression of MTCH2 (84), which is related to mitochondrial fusion (66). In addition, it is indicated that increased MTCH2 promotes mitochondrial transfer from BMSCs to RAW264.7 (84), so miR-150 may regulate the metabolism of RAW264.7 by affecting the morphology of transferred mitochondria. Unfortunately, this research did not cover studies related to mitochondrial dynamics, so this conclusion needs to be validated experimentally. On the one hand, miR-150 has been shown to negatively regulate brown-like transformation of white adipose tissue in inguinal peritoneum of mice, so regulates metabolism (133). In AML (acute myelocytic leukemia) cells, MTCH2 deletion has been shown to reduce glucose entry into the mitochondrial TCA cycle, thereby inducing glutamine to maintain the TCA cycle in the form of oxaloacetate, as well as reduced mitochondrial pyruvate levels and increased nuclear pyruvate and pyruvate dehydrogenase (PDH) levels (97). Further, in the cell nucleus, PDH converts pyruvate into acetyl-CoA and increases histone acetylation. This series of metabolic changes promotes the differentiation of AML (97). Therefore, we hypothesized that MTCH2, which affected mitochondrial fusion, is transferred from BMSCs to RAW264.7, which may promote TCA circulation by affecting macrophage metabolism, thus enhancing OXPHOS and alleviating LPS-induced apoptosis.
3.2.3 OMA1/YME1L
Cardiomyocytes-specific deletion of Yme1l (cYKO) in mice inhibits the formation of S-OPA1 form d, and activated OMA1 accelerates the proteolysis of OPA1, leading to the accumulation of S-OPA1 form c and e, triggering mitochondrial fragmentation and leading to dilated cardiomyopathy and heart failure (98). The cYKO shifts the cardiomyocytes from lipid metabolism to the glucose metabolism, and this metabolic shift leads to heart failure in cYKO mice (98). However, double knockout mice (additional OMA1-deficient in Yme1l-/- mice) show normal cardiac function by preventing OPA1 cleavage (98). The additional muscle specific-knockout of Yme1l maintains cardiac function without restoring the mitochondrial morphological defects of cYKO cardiomyocytes (98). Interestingly, the loss of Yme1l in skeletal muscle systematically impairs glucose homeostasis, inhibits insulin secretion, and thus reduces cardiac glucose uptake, alters cardiac metabolism, and slows down cellular involuntary metabolism, demonstrating the physiological importance of tissue crossover disturbance (98). Metabolic intervention in cYKO mice fed a high-fat diet replicates the protective effect of hmYKO (heart and muscle-specific deletion of Yme1l) mice (98). In conclusion, coordinated mitochondrial dynamics or balanced mitochondrial fusion and fission plays a key role in cardiac metabolism and cardiac function. YME1L and OMA1 are still lacking in research in macrophages, but they affect mitochondrial dynamics by shearing L-OPA1 to produce different forms of S-OPA1, which may affect macrophage metabolism in this process, which also provides new ideas for studying the macrophage metabolism regulation.
3.2.4 SLP-2
When mitochondria are exposed to selective stress in cells, mitochondrial hyperfusion occurs prior to mitochondrial fission (79). In this process, mitochondrial IM protein SLP-2 cooperates with L-OPA1 and MFN1 to induce hyperfusion, which in turn promotes the production of OXPHOS and ATP (79).
3.2.5 MtSIRT
Mitochondrial Sirtuins (mtSIRT), including SIRT3, SIRT4 and SIRT5, are important parts of the sirtuin family (SIRT1-7) and possess enzyme activity such as NAD+-dependent deacylases and ADP-ribotransferase, which endowing mtSIRT with the function of regulating energy metabolism (134).
3.2.6 SIRT3
On the one hand, SIRT3 promotes mitochondrial function by regulating the activity of metabolic enzymes, such as by binding to NDUFA9 subunits of ETC complexes I and deacetylation to maintain OXPHOS (99). SIRT3, on the other hand, directly binds to OPA1 at its lysine site (K926 and/or K931) for deacetylation, and enhances mitochondrial respiratory complex activity (80). Since mitochondrial morphology depends on the integrity of the coiled-coil domain at C-terminal of OPA1 (81), where these two acetylated lysine residues of OPA1 located, therefore, SIRT3 deacetylation promotes mitochondrial fusion (80). According to the TEM results, in SIRT3-KO heart, mitochondria are mostly clustered together, forming multiple island-like shapes, and the OMM is continuous, but the IMM fusion is defective (80), which also is consist with the function of OPA1 responsible for IMM fusion and reminds us that OMM fusion can occur independently of IMM fusion.
3.2.7 SIRT4
Down-regulation of SIRT4 in aging chondrocytes was accompanied by excessive fragmentation of mitochondrial network, elevated ROS levels, and impaired ΔΨ(m) (135). Overexpression of SIRT4 saved mitochondrial fragmentation and restored mitochondrial homeostasis in aging chondrocytes (135). Therefore, SIRT4 has the potential to promote mitochondrial fusion. In addition to reported dehydrogenase activity (136, 137), in fibroblast models, SIRT4 expression inhibits mitochondrial respiration under basal conditions, resulting in reduced electron transport system capacity (ETS) and ΔΨ(m) (82). The up-regulated SIRT4 is accompanied by a higher level of mitochondrial fusion regulatory factor L-OPA1 expression, resulting in a higher degree of mitochondrial elongation/fusion and inhibiting mitochondrial fission and autophagy (82), and this phenomenon can also be repeated by upregulation of SIRT4 by inhibiting miR-15b (82). Meanwhile, the overexpression of SIRT4 in Muller glial cells increased the L-OPA/S-OPA1 ratio and MFN2 (138); SIRT4 positively regulates OPA1 and MFN1 in skeletal muscle (139); In mammary epithelial cells, SIRT4 deletion reduces MFN1/2 and increases DRP1 and Fis1 (140). Taken together, these evidences support the role of SIRT4 in promoting mitochondrial fusion.
3.2.8 SIRT5
SIRT5 has deacetylase, deglutarylase and desuccinylase activities and participate in a variety of intracellular metabolic activities, such as the TCA, ETC, glycolysis, fatty acid β-oxidation and other processes (141, 142). For tumor cells, SIRT5 supports cell transformation and promotes breast carcinoma proliferation and tumorigenesis (143). Therefore, functional evaluation of SIRT5 should refer to its specific cell environment. Glutamine is converted into glutamate and ammonia in mitochondria by glutaminase (GLS), and glutamate is converted into α-ketoglutarate (α-KG) through glutamate dehydrogenase 1 (GLUD1) to participate in TCA (144). SIRT5 inhibits glutamine metabolism and regulates ammonia production by desuccinylation of GLS at Lys245 and/or Lys32, thus affecting autophagy (83). However, SIRT3 but not SIRT5 is co-immunoprecipitation with GLUD1 (83). After SIRT5 silencing or inhibition (by MC3482), autophagy is promoted, the expressions of MFN2 and OPA1 are decreased, and mitochondria are short-round shapes that cluster around the nucleus (83). In contrast, SIRT5 overexpression leads to the increase of MFN2 or OPA1, which is conducive to mitochondrial fusion, with increased number of mitochondria, elongated morphology, high ΔΨ(m), and reduced lactate production (83). STIR5, which regulates glutamine homeostasis, is also involved in mitochondrial dynamics regulation, suggesting there is a correlation between cellular metabolism and mitochondrial dynamics.
3.2.9 MtSIRT for macrophage metabolism
The relationship between SIRT3/4/5 and macrophage metabolism has been emphasized in recent years. Generally speaking, SIRT3 promotes OXPHOS and FAO (fat acid oxidation), SIRT4 promotes glycolytic anabolism, and SIRT5 changes the metabolic state of macrophages by regulating PTM of key metabolic enzymes (100, 101, 145, 146). For example, in BMDM, SIRT3 deacetylates at Lys83 to activate PDHA1 (pyruvate dehydrogenase E1 α), then PDHA1 catalyzes the decarboxylation of pyruvate into acetyl-CoA and promotes OXPHOS, inhibits glycolysis, which helpful for reducing inflammation of macrophages (100). Interestingly, the ability of SIRT3 to promote OXPHOS and support FAO may lead to immune tolerance in monocytes (147). During the immune tolerance process, the increased SIRT4 controls the expression of PDK1 and SIRT1, thereby transforming the FAO pathway into glucose oxidation, promoting pyruvate to enter glycolysis, which is conducive to restoring immune homeostasis (145). The expression of SIRT4 is reduced in hepatocellular carcinoma (HCC) tumor tissues (101). By increasing the expression of lipid catabolisc genes, such as MCAD (medium chain acyl-CoA dehydrogenase), CPT1 (carnitine palmitoyl transferase 1), etc., SIRT4 knockdown activates the FAO-PPARδ (peroxisome proliferator-activated receptor-δ)-STAT3 signal pathway and promotes the M2 markers (CD206, Arg-1) expression of TAM (101). This has also proved that SIRT4 is not conducive to FAO metabolic pathway from the opposite side. Moreover, SIRT4 silencing increases the expression of mitochondrial genes, including PGC1α, EER-α, CytC, CoxV (mitochondrial cytochrome oxidase V) and IDH3α (isocitrate dehydrogenase 3 alpha) in TAMs (101). PKM2 (M2-type pyruvate kinase), the physiological substrate of SIRT5, is a key determinant of the aerobic glycolytic transition of LPS-activated macrophages (102). In LPS (0.1 µg/mL)-stimulated BMDM, SIRT5-knockdown -regulated hypersuccinylation inhibits the pyruvate kinase activity of PKM2, promotes PKM2 dimerization entering into the nucleus, and ultimately mediates the up-regulation of IL-1β (102). Similarly, SIRT5 promotes inflammation of primary macrophages by promoting p65 acetylation (146). SIRT5 mediates the deglutarylation of GLUD1 in IL-4-polarized BMDM, thereby enhancing GLUD1 enzyme activity and promoting the production of αKG, which has a positive effect on the polarization of M2-like macrophages (148). It has also been shown that after the inhibition of SIRT5 by glutamine, PDH activity was enhanced due to the inhibition of PDHA1 desuccinylation, which promoted the polarization of IL-4 treated BMDM, and the glutamine metabolite α-KG played a role in the inhibition of SIRT5 (149).This seems to be the opposite of the previous sentence in which SIRT5 promotes α-KG production, possible causes include different disease models (eosinophilic chronic rhinosinusitis or burn sepsis) or different amounts of IL-4 stimulation (10 ng/mL or 50 ng/mL), but the specific reasons remain to be further explored (148, 149).
Therefore, mtSIRT is an important link between cellular metabolism and mitochondrial dynamics. SIRT3/4/5 promotes mitochondrial fusion by affecting OPA1 or/and MFN2, which produces different regulation on cell metabolism. SIRT3 promotes mitochondrial respiration, while SIRT4 and SIRT5 inhibit mitochondrial respiration in fibroblasts, MDA-ME-231 or C2C21. As for macrophages, SIRT3/4/5 affect macrophage metabolism in OXPHOS, glycolysis and FAO, illustrating that mtSIRT reconciles the metabolic state of macrophages to meet their demands. Unfortunately, these researches do not cover the change of mitochondrial dynamics under the remolding of macrophage metabolic state. Thence, there are complex regulatory mechanisms for mtSIRT to reconcile cellular metabolism and mitochondrial dynamics, and more researches are urgently needed to unearth their connection.
In conclusion, mitochondrial fusion is driven by multifactor and has undergone a biological process from OM fusion to IM fusion. In addition to mitochondrial fusion-related proteins regulating its morphology, ΔΨ(m), cristae formation, etc., some proteins have been confirmed to be involved in regulating macrophage metabolism (Table 2). However, there are few and scattered articles relating mitochondrial dynamics to macrophage metabolism, but we still conclude with a gratifying possibility. Whether there is a direct connection between mitochondrial fusion and macrophage metabolism and whether there is a logical relationship such as primary and secondary or causality is worthy of in-depth exploration.
4 Mitophagy and macrophage metabolism
Mitochondrial dynamics is involved in the elimination of part of damaged mitochondria and the maintenance of their own structural and functional integrity. When fusion/fission fails to meet the repair requirements, mitochondria are cleared by selective autophagy, also known as mitophagy. As researchers reviewed, the autophagosome mediated mitophagy is mainly induced by the LC3 adaptor protein, with or without ubiquitin-dependent or LC3 directly interacting with its receptor (150). In addition, some studies have pointed out that mitophagy requires Drp1 to break the mitochondrial network (151) or regulate PINK1/Parkin pathways (152, 153). The overexpression of OPA1 in myocardial infarction model induces mitophagy, and then increases the expression of antioxidant items (e.g., GSH, SOD), restores mitochondrial ΔΨ(m), blocks the accumulation of cytoplasmic cyt c, alleviates mitochondrial dysfunction, and ultimately saves cardiomyocytes from further damage (154). Both phenomena remind us that mitochondrial dynamics possibly participates in the regulation of mitophagy. However, the internal relationship between mitochondrial dynamics and mitochondrial autophagy remains unclear. Meaningfully, Fun-14 domain containing protein 1 (FUNDC1), a key receptor associated with mitochondrial fission and mitophagy, was discovered. OPA1 and DNM1L (DRP1 Dynamin 1-like) are core targets of FUNDC1 (155). In the process of hypoxia-induced mitophagy, FUNDC1 first acts on the MAMs and is enriched at the mitochondrial associated membrane (MAM), and then recruits DNM1L/DRP1 and separates from CANX to promote mitochondria fission in response to hypoxic stress (61). When Lys70 (K70) of FUNDC1 is mutated to Ala (A) [not Arg (R)], the interaction of FUNDC1 on OPA1 reduced and that of DNM1L is enhanced, which in turn stimulates mitophagy (155). However, recent studies have found that mitophagy caused by FUNDC1 protects the kidney from ischemic damage by inhibiting DRP1-induced mitochondrial fission (62). Therefore, the effect of FUNDC1 on mitochondrial fission depends on the microenvironment of cells, illustrating the metabolic state of cells may alter mitochondrial dynamics. In addition, other connections between mitochondrial dynamics and mitophagy deserve further investigation. On the other hand, FUNDC1-deficient mice experienced increased liver damage, accelerated adipose tissue macrophage infiltration and M1 polarization, and were accompanied by metabolic changes such as up-regulation of genes associated with adipogenesis (156). Overexpression of FUNDC1 inhibited the production of IL-1β and ROS in BMDMs (157). Therefore, FUNDC1-mediated mitochondrial autophagy has an effect on the reprogramming of macrophage metabolic state. However, the existing body of research predominantly focuses on FUNDC1’s canonical role in mitophagy regulation during disease pathogenesis, wherein macrophage metabolic parameters are occasionally measured as secondary endpoints. This conspicuous gap in direct mechanistic investigations positions the FUNDC1-macrophage metabolism axis as a novel and scientifically significant area of investigation.
Choline transporter 1 (CTL1) -mediated choline uptake affects mitochondrial phosphatidylcholine (PC) and sphingomyelin (SM) (158). Impaired choline uptake disrupts mitochondrial ATP synthesis and triggers AMPK activation to promote DRP1-driven mitophagy (158). Moreover, LPS (0.2 µg/mL) enhances the expression of CTL1 for 4h, and then promotes the choline uptake of BMDM (158). After 24h of LPS treatment, the lipid metabolism of macrophages (mainly occurring in mitochondria) is remodeled, for example, mitochondrial PC (mito-PC) decreased and mito-SM increased (158). In conclusion, it is reasonable to assume that choline acts as a connecting factor between mitophagy and macrophage metabolism. Besides, choline is considered to be a water-soluble vitamin related to the B vitamins, which is involved in the synthesis of acetylcholine and the formation of methyl donors in the methionine cycle in the body (159). Whether the above-mentioned regulation of mitochondrial dynamics and macrophage metabolism by choline is directly regulated or occurs through these biosynthetic or oxidative processes is worth exploring at this point (Figure 2).
5 The connection of macrophage metabolism with “kiss and run”
In addition to the above-mentioned mitochondrial complete fission and fusion, mitochondria have a transient fusion-fission mode called “kiss and run”. This process can be completed in a very short time (as short as 4 seconds), and relative to the longitudinal alignment of fully fused mitochondria, transient fusions tend to occur from oblique or lateral sites, so transient fusions always preserve the original topology (160). During transient fusion, the soluble contents of mitochondria undergo passive diffusion under the MFNs- and OPA1-dependent action (160). Transient fusion occurs in mitochondria on two microtubules, and DRP1 is recruited when transient fusion occurs due to the force of motor proteins and the tension of anchoring to the microtubules, causing transient fission (160). Liu et al. also found that transiently fused mitochondria maintain normal bioenergetics such as ΔΨ(m) and respiration (160). To sum up, the “kiss and run” mode is similar to that of athletes on two tracks. After a brief high-five, they exchange part of the material, and then quickly separate without affecting each other’s motivation.
The instantaneity of mitochondrial kiss and run enables it to exchange or dilute part of its contents in the shortest time without affecting mitochondrial metabolism. Although the role of kiss and run in macrophage metabolism is currently lacking, we believe this will be a meaningful research direction.
6 Metabolism-related diseases of macrophages are linked to mitochondrial dynamics
6.1 Atherosclerosis
Macrophage metabolic reprogramming determines the progression of atherosclerosis. During the transition from early to inflammatory progression of atherosclerosis, the proportion of M[LPS+IFN-γ]-like macrophages gradually increased, accompanied by the increase in glycolysis, and in the regression stage of atherosclerotic plaques, the proportion of M[IL-4]-like macrophages increased with up-regulated FAO, and M(Ox)-like macrophages with lower phagocytic migration ability are also found (161).
In the dynamic regulation of mitochondria, mitophagy removes damaged mitochondria and plays a role in preventing oxidative stress and apoptosis (150), so mitophagy is crucial for cardiovascular-derived cellular homeostasis (162). Studies have linked impaired mitophagy to atherosclerosis (163). For example, treatment with the DRP1 inhibitor mdivi-1 reduces mitochondrial fragmentation and attenuates atherosclerosis in diabetic ApoE-/- mice (164). This is a very interesting point. Although the metabolic state of macrophages in an atherosclerotic environment is variable, we can treat atherosclerosis by regulating mitochondrial dynamic to active mitophagy as an entry point.
6.2 Asthma
Asthma is a chronic inflammatory disease of the respiratory system, which is divided into various phenotypes according to the immune cells enriched in the airways (165). Macrophage function is altered in asthmatic patients, including decreased phagocytic capacity, increased production of inflammatory mediators, and disturbed production of anti-inflammatory factors and so on, moreover, there is substantial evidence that macrophages may serve as therapeutic targets for asthma (165).
Metabolic alterations in human and mouse airway cells under cigarette smoke (CS) suggest a vital role for mitochondria, specifically, the mitochondrial TCA and OXPHOS are converted to FAO, and mtROS increases with increasing damage, and the ATP demand for cellular repair increases at this time (166). In chronic obstructive pulmonary disease (COPD), studies have found increased mitophagy in airway epithelial cells, and unfortunately, no studies have linked mitochondrial dynamics to macrophage therapy for asthma (167). We speculate, however, that the metabolic reprogramming of macrophages by regulating mitochondrial dynamics would be a possible strategy for the treatment of asthma. Emerging mechanistic studies suggest that DRP1 is a key amplifier in multifactorial asthma, establishing its therapeutic potential through pathway regulation (168–170). Notably, in recombinant DEK-induced mouse asthma models, DRP1 expression is significantly upregulated and physically interacts with ATAD3A (169). Mechanistic inquiry revealed that inhibition of ATAD3A mediates inhibition of DRP1, thereby resolving mitochondrial oxidative stress by regulating mitophagy, and achieving attenuation of airway inflammation (169). In ovalbumin-induced asthma, mitochondrial fusion/fission dysregulation promotes ROS production and intensifies the activation of NLRP3 inflammasome (171). After Abscisic acid treatment, NLRP3 activation was down-regulated, mitochondrial fusion/fission markers such as OPA1, MFN2 and DRP1 were decreased, and the expression of PPARγ was further increased, which inhibited airway inflammation (171). Studies have shown that PPARγ regulates FAO metabolism in macrophages (172). Therefore, mitochondrial dynamics and metabolic adaptation are key regulators of macrophage behavior in asthma.
6.3 Other diseases
A recent article summarizes the relationship between mitophagy and macrophage-related chronic inflammatory and autoimmune diseases (173), including inflammatory bowel disease (IBD), systemic lupus erythematosus (SLE), and primary biliary cirrhosis (PBC). Although this article does not mention the role of mitochondrial dynamics in IBD, SLE, and PBC, according to our previous description of mitochondrial dynamics perturbation and mitophagy, we believe that mitochondrial dynamics regulation may be related to macrophages metabolic reprogramming through disturbing mitophagy ultimately serves as a target for the treatment of macrophage-related diseases.
7 Conclusion
Mitochondria are the factories of cell energy metabolism and the key organelles to maintain body homeostasis (174). With the change of cell microenvironment, mitochondria undergo a series of dynamic changes, and form a complex regulatory network through hyperfusion, elongation, fragmentation, etc. (18, 20), to adapt to or meet the needs of cell metabolism and immunity. When the dynamics of mitochondrial fission and fusion is unable to rescue the cell, mitophagy occurs (150), thereby eliminating the damaged mitochondria. Recently, researchers corrected some statements about mitochondrial morphology, that is the hyperfused and fragmented mitochondrial network are caused by the equilibrium between fission and fusion processes, and thus affect mitochondrial functional fitness, rather than described mitochondrial morphology as single fusion and/or fission (20). The up-regulated fusion to fission ratio promotes mitochondrial hyperfusion whether fusion increases or fission decreases, or both, instead, when the ratio of fusion to fission decreased, mitochondria tend to fragmentation. Intrinsically, the equilibrium fluctuations described above are more consistent with natural phenomena, but in order to describe mitochondria in different states more intuitively, fusion and fission are also used to describe the morphology of mitochondria in this paper.
Macrophages are important members of the natural immunity, and their polarization is accompanied by glycolysis and OXPHOS metabolic programming (85). These metabolic changes are inseparable from the regulation of mitochondria. Therefore, previous studies focused on the association between mitochondrial metabolism and macrophage immunity, such as the dual role of ERK1/2 on mitochondrial fission (35, 175, 176), and the role of PKA and AKT on mitochondrial fusion (20).
Based on existing literature, we reviewed the dynamic changes of mitochondrial dynamics during macrophages polarization and the effects of mitochondrial dynamics on the functional diversity of macrophages. In LPS-activated macrophages (M1 type or classically activated), enhanced phosphorylation of DRP1 at Ser616 promotes mitochondrial fission, reduced ΔΨ(m), and morphological changes such as mitochondrial fragmentation and disorganized cristae (22). During this process, the remodeled mitochondria redirect their function towards increased ROS production, thereby driving the transcription of inflammatory cytokines and exacerbating the inflammatory response (22). On the other hand, mitochondrial fission acts as an atypical regulator of macrophage functional plasticity. For example, DRP1-mediated mitochondrial fission is required for the uptake of apoptotic cells by macrophages (177). Mitochondrial fission promotes Ca2+ release from ER, activates vesicular trafficking and cytoskeletal remodeling, enabling macrophages to sequentially engulf multiple apoptotic cells without functional exhaustion (177). Importantly, mitochondrial fission and increased cytoplasmic calcium prompt protein kinase C-θ of macrophages to phosphorylate WIP (Wiskott-Aldrich Syndrome Interacting Protein) during phagocytosis and are necessary for therapeutic antibody-induced phagocytosis of live tumor cells (178). Furthermore, by modulating metabolic pathways through the inhibition of Glutamine-fructose-6-phosphate transaminase 2 (GFPT2) in tumor cells, glutamine metabolism is regulated, thereby restoring mitochondrial dynamics and consequently influencing the phagocytic capacity of macrophages (178). Mdivi-1 inhibits DRP1-dependent mitochondrial fission, reduces mitochondrial reactive oxygen species (mito-ROS) and NLRP3 inflammasome activation, thereby reducing macrophage M1 polarization and down-regulating IL-6 and TNF-α release, which is beneficial for alleviating atherosclerotic diseases (179). Importantly, it was found that following M1 polarization, the mitochondrial ETC complex of macrophages was either partially or completely suppressed, leading to impaired OXPHOS function, and macrophages were unable to transform to M2 phenotype even when exposure to IL-4 stimulation (180). However, macrophages showed a tendency to increase M2 markers (e.g., CD206 and IL-10) after DRP1 inhibition by Mdivi-1 (179), suggesting that mitochondrial dynamics may acts as metabolic rheostats, allowing macrophages to switch between polarized states. Alternatively activated macrophages (M2-like) are predisposed to mitochondrial fusion, produce elongated mitochondria, and have high levels of FAO and OXPHOS (21). As previously discussed, OPA1, MFN1/2, and other proteins participate in the dynamic regulation of mitochondrial fusion (39, 125). Research has shown that restoration of mitochondrial dynamics by up-regulation of MFN1/2, which promotes mitochondrial fusion, successfully inhibits pro-inflammatory macrophage polarization (181). The fused mitochondria facilitate FAO and ATP synthesis, presenting an optimal therapeutic strategy to promote M2 polarization in chronic inflammatory conditions (181). However, the effect of mitochondrial fusion-associated proteins on macrophage function appears to be more complex than a simple M2-to-M1-type conversion. Myeloid specific OPA1 deletion in mice leads to mitochondrial network fragmentation and cristae disorganization (182). Interestingly, macrophages deficient in OPA1 exhibit up-regulated expression of M2 markers such as Arg1, Mrc1 and Retnla, suggesting that OPA1 deletion promotes M2-like phenotype (182). Paradoxically, under M2-polarized conditions, OPA1 deletion also interferes with macrophage metabolism and immune pathways by shifting macrophages to glycolytic metabolism and inability to maintain ATP levels due to mitochondrial uncoupling (182). OPA1 deletion also disrupts TCA cycle, resulting in succinate and citrate accumulation and metabolic perturbation, which then impairs NF-κB/p65 signaling and ultimately suppress the pro-inflammatory response (182).
Regulation of mitochondrial dynamics-associated proteins, including DRP1 and OPA1, reshaped the metabolic profile of macrophages (179, 182). DRP1-mediated mitochondrial fission disrupts cristae morphology and enhances mito-ROS generation (179). Elevated mito-ROS stabilizes hypoxia-inducible factor-1α (HIF-1α), driving a metabolic shift toward glycolysis characterized by increased glycolytic ATP production and enhanced glycolytic flux (183). OPA1 deficiency induces macrophage dependency on glycolysis; however, defective assembly of respiratory chain supercomplexes and impaired ATP production compromise the acquisition of the M1 phenotype, leading instead to the manifestation of classical M2 characteristics (182). Enhancing OPA1 activity or inhibiting its degradation restores OXPHOS capacity (184). Thus, OPA1 functions as a “metabolic switch” in macrophage polarization by orchestrating mitochondrial dynamics and metabolic homeostasis. Consistently, we summarize and induction that mitochondrial dynamics are closely related to macrophage metabolism. A large number of mitochondrial fission/fusion-related proteins have been shown to affect macrophage metabolism. Among them, FAM73b (26), MTCH2 (84), SLP-2 (79), and mtSIRT (80, 82, 83, 100–102) related to mitochondrial fusion, Fis1 and MTP18 related to mitochondrial fission are closely related to macrophage metabolism. Specifically, mitochondrial fusion tends to macrophage OXPHOS, while mitochondrial fission tends to glycolysis and fat acid metabolism. These proteins serve as the vinculum between mitochondrial dynamics and macrophage metabolism. PTM plays an important role in mitochondrial dynamics and macrophage metabolism, especially in DRP1 (22, 41) and mtSIRT (100–102). MiRs are involved in the regulation of macrophage polarization (185) and mediate metabolic shifts (186). Although studies on the regulation of macrophage metabolism by miRs are lacking, we suggest that miRs may be the link through which mitochondrial dynamics regulate macrophage metabolism (82, 84, 96).
In addition, we have conducted a comprehensive search for the relationship between mitophagy (150, 154, 158, 159), mitochondrial “kiss and run” (160) and macrophage metabolism. Although relevant research is insufficient, it still needs to be verified. Mitocytosis, a recently discovered new mitochondrial quality control mechanism, has the ability to maintain mitochondrial homeostasis in macrophages (187). As mitocytosis is a newly discovered mechanism, its impact on macrophage metabolism is currently unknown, but it would be an interesting research direction. Study of Chen et al. revealed that the antimalarial drug chloroquine (CQ), as an anti-tumor immunomodulator, transformed TAM from M2 to tumor killing M1 phenotype (transformed TAM to tumor-killing macrophages) by reprogramming the transformation mechanism of TAM metabolism from OXPHOS to glycolysis (188), which was a strategy to improve the adaptability of macrophages in immunosuppressed environment. This suggests that mitochondrial dynamic regulation has huge potential to be used as a means of metabolic reprogramming of macrophages to enhance the resistance of macrophages in adverse environments such as immunosuppression, thus contributing to host survival. On the other hand, we summarize the relationship between macrophage-related chronic inflammatory diseases and autoimmune diseases (including atherosclerosis, asthma) and mitochondrial dynamics, and further suggest that mitochondrial metabolism regulation may be an effective strategy for the treatment of these diseases. Overall, the relationship between mitochondrial dynamics and macrophage metabolism still requires a large amount of experimental input to verify its logical relationship.
Author contributions
PL: Funding acquisition, Supervision, Visualization, Writing – original draft, Writing – review & editing. ZF: Resources, Writing – original draft, Writing – review & editing. YH: Data curation, Writing – review & editing. LL: Data curation, Writing – review & editing. XW: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by Taiyuan Institute of Technology Scientific Research Initial Funding (2022KJ016), Fundamental Research Program of Shanxi Province (202303021212276), and the Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (No. 2022L545).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
AAAC: ADP/ATP carrier
AKT: Protein kinase B
AML: acute myelocytic leukemia
AMP: Adenosine 5’-monophosphate
AMPK: AMP-activated protein kinase
ARL2: ADP-ribosylation factor (ARF) like 2
Arp2/3: Actin-related protein 2/3
BAT: Brown adipose tissue
BMDM: Bone marrow derived macrophages
BMSCs: Bone marrow mesenchymal stem cells
cAMP: Cyclic adenosine monophosphate
CANX: Calnexin
CL: Cardiolipin
COPD: Chronic obstructive pulmonary disease
CoxV: Mitochondrial cytochrome oxidase V
CPT1: Carnitine palmitoyl transferase 1
CQ: Chloroquine
CS: Cigarette smoke
CTL1: Choline transporter 1
Cyt c: Cytochrome c
DAG: Diacylglycerol
DNM1L: DRP1 Dynamin 1-like
DRP1: Dynamin-related protein 1
ELMOD2: ELMO domain containing 2
ER: Endoplasmic reticulum
ERK: Extracellular signal-regulated kinase
ESCs: Embryonic stem cells
ETC: Electron transport chain
ETS: Electron transport system
FAO: Fat acid oxidation
Fis1: Mitochondrial fission 1 protein
FUNDC1: Fun-14 domain containing protein 1
Fzo: fuzzy onions
GABA: γ-aminobutyric acid
GAP: GTPase-activating protein
GFPT2: Glutamine-fructose-6-phosphate transaminase 2
GLS: Glutaminase
GLUD1: Glutamate dehydrogenase 1
GSH: Glutathione
GTPase: Guanosine triphosphatases
HCC: Hepatocellular carcinoma
IBD: Inflammatory bowel disease
IDH3α: Isocitrate dehydrogenase 3 alpha
IFN-β: Type I interferons
IM: The inner membrane
IMM: Mitochondrial inner membrane
IMS: The intermembrane space
INF2: Inverted formin 2
IRF1: Interferon regulator 1
JAK: Janus kinase
L-OPA1: Long-OPA1
MAM: Mitochondrial associated membrane
MCAD: Medium chain acyl-CoA dehydrogenase
MDA: Malondialdehyde
MEFs: Mouse embryonic fibroblasts
Mff: Mitochondrial fission factor
MFN1/2: Mitofusin 1/2
MiD: N-terminally anchored mitochondrial dynamics proteins
Miga: Mitoguardin
MIMP: Met-induced mitochondrial protein
MiR: MicroRNA
Mito-PC: Mitochondrial PC
MitoPLD: Mitochondrial phospholipase D
Mito-ROS: Mitochondrial reactive oxygen species
Mito-SM: Mitochondrial SM
MOMP: OMM permeabilization
MSTO1: Misato homolog 1
MTCH2: Mitochondrial carrier homolog 2
mtDNA: Mitochondrial DNA
MTP18: Mitochondrial protein 18 kDa
MtSIRT: Mitochondrial sirtuins
NF-κB: nuclear factor kappa-B
OGA: O-GlcNAcylation
OM: The outer membrane
OMM: Mitochondrial outer membrane
OPA1: Optic atrophy 1
OXPHOS: Oxidative phosphorylation
PA: Phospholipid acid
PBC: Primary biliary cirrhosis
PC: Phosphatidylcholine
PDH: Pyruvate dehydrogenase
PDHA1: Pyruvate dehydrogenase E1 α
PGC1α: PPARγ co-activator 1 alpha
PKA: Protein kinase A
PKM2: M2-type pyruvate kinase
PLD: Phospholipase D
PPAR-γ: peroxisome proliferator-activated receptor-γ
PPARδ: Peroxisome proliferator-activated receptor-δ
PTM: Post-translational modification
RIP1: receptor-interacting protein kinase 1
SAM: S-adenosylmethionine
SLE: Systemic lupus erythematosus
SLP-2: Stomatin-like protein 2
SM: Sphingomyelin
SOD: Superoxide dismutase
S-OPA1: Short-OPA1
Spire1C: Spiretype actin nucleation factor 1
STAT: signal transducer and activator of transcription
Stat2: Signal transducers and activators of transcription 2
TAM: Tumor-associated macrophage
tBID: Truncated BID
WAT: White adipose tissue
WT: Wild type
α-KG: α-ketoglutarate
ΔΨ(m): Membrane potential
References
1. Sharifiaghdam M, Shaabani E, Faridi-Majidi R, De Smedt SC, Braeckmans K, Fraire JC. Macrophages as a therapeutic target to promote diabetic wound healing. Mol Ther. (2022) 30:2891–908. doi: 10.1016/j.ymthe.2022.07.016
2. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Corrigendum: Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front Immunol. (2020) 11:234. doi: 10.3389/fimmu.2020.00234
3. Yu T, Gao M, Yang P, Liu D, Wang D, Song F, et al. Insulin promotes macrophage phenotype transition through PI3K/Akt and PPAR-γ signaling during diabetic wound healing. J Cell Physiol. (2019) 234:4217–31. doi: 10.1002/jcp.27185
4. Shan X, Hu P, Ni L, Shen L, Zhang Y, Ji Z, et al. Serine metabolism orchestrates macrophage polarization by regulating the IGF1-p38 axis. Cell Mol Immunol. (2022) 19:1263–78. doi: 10.1038/s41423-022-00925-7
5. Franz S, Ertel A, Engel KM, Simon JC, Saalbach A. Overexpression of S100A9 in obesity impairs macrophage differentiation via TLR4-NFkB-signaling worsening inflammation and wound healing. Theranostics. (2022) 12:1659–82. doi: 10.7150/thno.67174
6. Cui Y, Chen J, Zhang Z, Shi H, Sun W, Yi Q. The role of AMPK in macrophage metabolism, function and polarisation. J Transl Med. (2023) 21:892. doi: 10.1186/s12967-023-04772-6
7. Rodriguez AE, Ducker GS, Billingham LK, Martinez CA, Mainolfi N, Suri V, et al. Serine metabolism supports macrophage IL-1β Production. Cell Metab. (2019) 29:1003–1011.e4. doi: 10.1016/j.cmet.2019.01.014
8. Fu J, Han Z, Wu Z, Xia Y, Yang G, Yin Y, et al. GABA regulates IL-1β production in macrophages. Cell Rep. (2022) 41:111770. doi: 10.1016/j.celrep.2022.111770
9. Yu L, Wang L, Hu G, Ren L, Qiu C, Li S, et al. Reprogramming alternative macrophage polarization by GATM-mediated endogenous creatine synthesis: A potential target for HDM-induced asthma treatment. Front Immunol. (2022) 13:937331. doi: 10.3389/fimmu.2022.937331
10. Zhang J, Muri J, Fitzgerald G, Gorski T, Gianni-Barrera R, Masschelein E, et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. (2020) 31:1136–1153.e7. doi: 10.1016/j.cmet.2020.05.004
11. Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. (2022) 29:133–46. doi: 10.1038/s41418-021-00841-9
12. Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res. (2022) 131:893–908. doi: 10.1161/circresaha.122.320488
13. Komal S, Han SN, Cui LG, Zhai MM, Zhou YJ, Wang P, et al. Epigenetic regulation of macrophage polarization in cardiovascular diseases. Pharm (Basel). (2023) 16:141. doi: 10.3390/ph16020141
14. Dong T, Chen X, Xu H, Song Y, Wang H, Gao Y, et al. Mitochondrial metabolism mediated macrophage polarization in chronic lung diseases. Pharmacol Ther. (2022) 239:108208. doi: 10.1016/j.pharmthera.2022.108208
15. Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. (2014) 15:634–46. doi: 10.1038/nrm3877
16. Umemoto T, Hashimoto M, Matsumura T, Nakamura-Ishizu A, Suda T. Ca(2+)-mitochondria axis drives cell division in hematopoietic stem cells. J Exp Med. (2018) 215:2097–113. doi: 10.1084/jem.20180421
17. Dumont A, Lee M, Barouillet T, Murphy A, Yvan-Charvet L. Mitochondria orchestrate macrophage effector functions in atherosclerosis. Mol Aspects Med. (2021) 77:100922. doi: 10.1016/j.mam.2020.100922
18. Cervantes-Silva MP, Cox SL, Curtis AM. Alterations in mitochondrial morphology as a key driver of immunity and host defence. EMBO Rep. (2021) 22:e53086. doi: 10.15252/embr.202153086
19. Yildirim RM, Seli E. The role of mitochondrial dynamics in oocyte and early embryo development. Semin Cell Dev Biol. (2024) 159-160:52–61. doi: 10.1016/j.semcdb.2024.01.007
20. Sabouny R, Shutt TE. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem Sci. (2020) 45:564–77. doi: 10.1016/j.tibs.2020.03.009
21. Xie JH, Li YY, Jin J. The essential functions of mitochondrial dynamics in immune cells. Cell Mol Immunol. (2020) 17:712–21. doi: 10.1038/s41423-020-0480-1
22. Yu W, Wang X, Zhao J, Liu R, Liu J, Wang Z, et al. Stat2-Drp1 mediated mitochondrial mass increase is necessary for pro-inflammatory differentiation of macrophages. Redox Biol. (2020) 37:101761. doi: 10.1016/j.redox.2020.101761
23. Meyers AK, Wang Z, Han W, Zhao Q, Zabalawi M, Duan L, et al. Pyruvate dehydrogenase kinase supports macrophage NLRP3 inflammasome activation during acute inflammation. Cell Rep. (2023) 42:111941. doi: 10.1016/j.celrep.2022.111941
24. Umezu R, Koga JI, Matoba T, Katsuki S, Wang L, Hasuzawa N, et al. Macrophage (Drp1) dynamin-related protein 1 accelerates intimal thickening after vascular injury. Arterioscler Thromb Vasc Biol. (2020) 40:e214–26. doi: 10.1161/atvbaha.120.314383
25. Liang S, Zhong Z, Kim SY, Uchiyama R, Roh YS, Matsushita H, et al. Murine macrophage autophagy protects against alcohol-induced liver injury by degrading interferon regulatory factor 1 (IRF1) and removing damaged mitochondria. J Biol Chem. (2019) 294:12359–69. doi: 10.1074/jbc.RA119.007409
26. Gao Z, Li Y, Wang F, Huang T, Fan K, Zhang Y, et al. Mitochondrial dynamics controls anti-tumour innate immunity by regulating CHIP-IRF1 axis stability. Nat Commun. (2017) 8:1805. doi: 10.1038/s41467-017-01919-0
27. Lu C, Cheng RJ, Zhang Q, Hu Y, Pu Y, Wen J, et al. Herbal compound cepharanthine attenuates inflammatory arthritis by blocking macrophage M1 polarization. Int Immunopharmacol. (2023) 125:111175. doi: 10.1016/j.intimp.2023.111175
28. Takuathung MN, Potikanond S, Sookkhee S, Mungkornasawakul P, Jearanaikulvanich T, Chinda K, et al. Anti-psoriatic and anti-inflammatory effects of Kaempferia parviflora in keratinocytes and macrophage cells. BioMed Pharmacother. (2021) 143:112229. doi: 10.1016/j.biopha.2021.112229
29. Gou Y, Wang H, Wang T, Wang H, Wang B, Jiao N, et al. Ectopic endometriotic stromal cells-derived lactate induces M2 macrophage polarization via Mettl3/Trib1/ERK/STAT3 signalling pathway in endometriosis. Immunology. (2023) 168:389–402. doi: 10.1111/imm.13574
30. Qiu S, Xie L, Lu C, Gu C, Xia Y, Lv J, et al. Gastric cancer-derived exosomal miR-519a-3p promotes liver metastasis by inducing intrahepatic M2-like macrophage-mediated angiogenesis. J Exp Clin Cancer Res. (2022) 41:296. doi: 10.1186/s13046-022-02499-8
31. Li Y, Zhou Y, Liu D, Wang Z, Qiu J, Zhang J, et al. Glutathione Peroxidase 3 induced mitochondria-mediated apoptosis via AMPK/ERK1/2 pathway and resisted autophagy-related ferroptosis via AMPK/mTOR pathway in hyperplastic prostate. J Transl Med. (2023) 21:575. doi: 10.1186/s12967-023-04432-9
32. Sola-Penna M, Paixão LP, Branco JR, Ochioni AC, Albanese JM, Mundim DM, et al. Serotonin activates glycolysis and mitochondria biogenesis in human breast cancer cells through activation of the Jak1/STAT3/ERK1/2 and adenylate cyclase/PKA, respectively. Br J Cancer. (2020) 122:194–208. doi: 10.1038/s41416-019-0640-1
33. Wu F, Zhang YT, Teng F, Li HH, Guo SB. S100a8/a9 contributes to sepsis-induced cardiomyopathy by activating ERK1/2-Drp1-mediated mitochondrial fission and respiratory dysfunction. Int Immunopharmacol. (2023) 115:109716. doi: 10.1016/j.intimp.2023.109716
34. Lai YS, Chang CC, Chen YY, Nguyen TMH, Xu J, Chen YC, et al. Optogenetically engineered Ca2+ oscillation-mediated DRP1 activation promotes mitochondrial fission and cell death. J Cell Sci. (2023) 136:jcs260819. doi: 10.1242/jcs.260819
35. Pyakurel A, Savoia C, Hess D, Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell. (2015) 58:244–54. doi: 10.1016/j.molcel.2015.02.021
36. Wculek SK, Heras-Murillo I, Mastrangelo A, Mañanes D, Galán M, Miguel V, et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity. (2023) 56:516–530.e9. doi: 10.1016/j.immuni.2023.01.011
37. Lundahl MLE, Mitermite M, Ryan DG, Case S, Williams NC, Yang M, et al. Macrophage innate training induced by IL-4 and IL-13 activation enhances OXPHOS driven anti-mycobacterial responses. Elife. (2022) 11:e74690. doi: 10.7554/eLife.74690
38. Li Y, He Y, Miao K, Zheng Y, Deng C, Liu TM. Imaging of macrophage mitochondria dynamics in vivo reveals cellular activation phenotype for diagnosis. Theranostics. (2020) 10:2897–917. doi: 10.7150/thno.40495
39. Gilkerson R, Kaur H, Carrillo O, Ramos I. OMA1-mediated mitochondrial dynamics balance organellar homeostasis upstream of cellular stress responses. Int J Mol Sci. (2024) 25:4566. doi: 10.3390/ijms25084566
40. Lei Y, Gan M, Qiu Y, Chen Q, Wang X, Liao T, et al. The role of mitochondrial dynamics and mitophagy in skeletal muscle atrophy: from molecular mechanisms to therapeutic insights. Cell Mol Biol Lett. (2024) 29:59. doi: 10.1186/s11658-024-00572-y
41. Jin JY, Wei XX, Zhi XL, Wang XH, Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin. (2021) 42:655–64. doi: 10.1038/s41401-020-00518-y
42. Beaulant A, Dia M, Pillot B, Chauvin MA, Ji-Cao J, Durand C, et al. Endoplasmic reticulum-mitochondria miscommunication is an early and causal trigger of hepatic insulin resistance and steatosis. J Hepatol. (2022) 77:710–22. doi: 10.1016/j.jhep.2022.03.017
43. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. (2011) 334:358–62. doi: 10.1126/science.1207385
44. Mu A, Latario CJ, Pickrell LE, Higgs HN. Lysine acetylation of cytoskeletal proteins: Emergence of an actin code. J Cell Biol. (2020) 219:e202006151. doi: 10.1083/jcb.202006151
45. Coscia SM, Thompson CP, Tang Q, Baltrusaitis EE, Rhodenhiser JA, Quintero-Carmona OA, et al. Myo19 tethers mitochondria to endoplasmic reticulum-associated actin to promote mitochondrial fission. J Cell Sci. (2023) 136:jcs260612. doi: 10.1242/jcs.260612
46. Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, et al. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife. (2015) 4:e08828. doi: 10.7554/eLife.08828
47. Rehklau K, Hoffmann L, Gurniak CB, Ott M, Witke W, Scorrano L, et al. Cofilin1-dependent actin dynamics control DRP1-mediated mitochondrial fission. Cell Death Dis. (2017) 8:e3063. doi: 10.1038/cddis.2017.448
48. Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. (2021) 593:435–9. doi: 10.1038/s41586-021-03510-6
49. Otera H, Miyata N, Kuge O, Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J Cell Biol. (2016) 212:531–44. doi: 10.1083/jcb.201508099
50. Li S, Xu S, Roelofs BA, Boyman L, Lederer WJ, Sesaki H, et al. Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J Cell Biol. (2015) 208:109–23. doi: 10.1083/jcb.201404050
51. Fung TS, Chakrabarti R, Higgs HN. The multiple links between actin and mitochondria. Nat Rev Mol Cell Biol. (2023) 24:651–67. doi: 10.1038/s41580-023-00613-y
52. Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J, Santel A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. (2005) 118:3049–59. doi: 10.1242/jcs.02415
53. Wang K, Gan TY, Li N, Liu CY, Zhou LY, Gao JN, et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. (2017) 24:1111–20. doi: 10.1038/cdd.2017.61
54. Aung LHH, Li YZ, Yu H, Chen X, Yu Z, Gao J, et al. Mitochondrial protein 18 is a positive apoptotic regulator in cardiomyocytes under oxidative stress. Clin Sci (Lond). (2019) 133:1067–84. doi: 10.1042/cs20190125
55. Ding M, Feng N, Tang D, Feng J, Li Z, Jia M, et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J Pineal Res. (2018) 65:e12491. doi: 10.1111/jpi.12491
56. Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. (2013) 339:464–7. doi: 10.1126/science.1228360
57. Lee H, Yoon Y. Mitochondrial fission and fusion. Biochem Soc Trans. (2016) 44:1725–35. doi: 10.1042/bst20160129
58. Liu R, Chan DC. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol Biol Cell. (2015) 26:4466–77. doi: 10.1091/mbc.E15-08-0591
59. Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, et al. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci. (2016) 129:2170–81. doi: 10.1242/jcs.185165
60. Korobova F, Gauvin TJ, Higgs HN. A role for myosin II in mammalian mitochondrial fission. Curr Biol. (2014) 24:409–14. doi: 10.1016/j.cub.2013.12.032
61. Wu W, Li W, Chen H, Jiang L, Zhu R, Feng D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy. (2016) 12:1675–6. doi: 10.1080/15548627.2016.1193656
62. Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. (2020) 30:101415. doi: 10.1016/j.redox.2019.101415
63. Li M, Wang L, Wang Y, Zhang S, Zhou G, Lieshout R, et al. Mitochondrial fusion via OPA1 and MFN1 supports liver tumor cell metabolism and growth. Cells. (2020) 9:121. doi: 10.3390/cells9010121
64. Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. (2001) 114:867–74. doi: 10.1242/jcs.114.5.867
65. Abrams AJ, Hufnagel RB, Rebelo A, Zanna C, Patel N, Gonzalez MA, et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet. (2015) 47:926–32. doi: 10.1038/ng.3354
66. Bahat A, Goldman A, Zaltsman Y, Khan DH, Halperin C, Amzallag E, et al. MTCH2-mediated mitochondrial fusion drives exit from naïve pluripotency in embryonic stem cells. Nat Commun. (2018) 9:5132. doi: 10.1038/s41467-018-07519-w
67. Shamas-Din A, Bindner S, Zhu W, Zaltsman Y, Campbell C, Gross A, et al. tBid undergoes multiple conformational changes at the membrane required for Bax activation. J Biol Chem. (2013) 288:22111–27. doi: 10.1074/jbc.M113.482109
68. Zaltsman Y, Shachnai L, Yivgi-Ohana N, Schwarz M, Maryanovich M, Houtkooper RH, et al. MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nat Cell Biol. (2010) 12:553–62. doi: 10.1038/ncb2057
69. Gal A, Balicza P, Weaver D, Naghdi S, Joseph SK, Várnai P, et al. MSTO1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol Med. (2017) 9:967–84. doi: 10.15252/emmm.201607058
70. Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. (2006) 8:1255–62. doi: 10.1038/ncb1487
71. Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, et al. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell. (2011) 20:376–87. doi: 10.1016/j.devcel.2011.01.004
72. Zhang Y, Liu X, Bai J, Tian X, Zhao X, Liu W, et al. Mitoguardin regulates mitochondrial fusion through mitoPLD and is required for neuronal homeostasis. Mol Cell. (2016) 61:111–24. doi: 10.1016/j.molcel.2015.11.017
73. Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. (2000) 26:211–5. doi: 10.1038/79944
74. Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, et al. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. (2017) 19:2557–71. doi: 10.1016/j.celrep.2017.05.073
75. Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. (2009) 187:959–66. doi: 10.1083/jcb.200906083
76. Newman LE, Schiavon CR, Turn RE, Kahn RA. The ARL2 GTPase regulates mitochondrial fusion from the intermembrane space. Cell Logist. (2017) 7:e1340104. doi: 10.1080/21592799.2017.1340104
77. Newman LE, Schiavon CR, Zhou C, Kahn RA. The abundance of the ARL2 GTPase and its GAP, ELMOD2, at mitochondria are modulated by the fusogenic activity of mitofusins and stressors. PloS One. (2017) 12:e0175164. doi: 10.1371/journal.pone.0175164
78. Schiavon CR, Turn RE, Newman LE, Kahn RA. ELMOD2 regulates mitochondrial fusion in a mitofusin-dependent manner, downstream of ARL2. Mol Biol Cell. (2019) 30:1198–213. doi: 10.1091/mbc.E18-12-0804
79. Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. (2009) 28:1589–600. doi: 10.1038/emboj.2009.89
80. Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. (2014) 34:807–19. doi: 10.1128/mcb.01483-13
81. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. (2004) 101:15927–32. doi: 10.1073/pnas.0407043101
82. Lang A, Anand R, Altinoluk-Hambüchen S, Ezzahoini H, Stefanski A, Iram A, et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging (Albany NY). (2017) 9:2163–89. doi: 10.18632/aging.101307
83. Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy. (2015) 11:253–70. doi: 10.1080/15548627.2015.1009778
84. Zhou X, Zhang K, He Z, Deng Y, Gao Y. Downregulated miR-150 in bone marrow mesenchymal stem cells attenuates the apoptosis of LPS-stimulated RAW264.7 via MTCH2-dependent mitochondria transfer. Biochem Biophys Res Commun. (2020) 526:560–7. doi: 10.1016/j.bbrc.2020.03.098
85. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
86. Zhang C, Huang J, An W. Hepatic stimulator substance resists hepatic ischemia/reperfusion injury by regulating Drp1 translocation and activation. Hepatology. (2017) 66:1989–2001. doi: 10.1002/hep.29326
87. Losón OC, Meng S, Ngo H, Liu R, Kaiser JT, Chan DC. Crystal structure and functional analysis of MiD49, a receptor for the mitochondrial fission protein Drp1. Protein Sci. (2015) 24:386–94. doi: 10.1002/pro.2629
88. Losón OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, et al. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure. (2014) 22:367–77. doi: 10.1016/j.str.2014.01.001
89. Atkins K, Dasgupta A, Chen KH, Mewburn J, Archer SL. The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: implications for human disease. Clin Sci (Lond). (2016) 130:1861–74. doi: 10.1042/cs20160030
90. Schultz J, Warkus J, Wolke C, Waterstradt R, Baltrusch S. MiD51 is important for maintaining mitochondrial health in pancreatic islet and MIN6 cells. Front Endocrinol (Lausanne). (2020) 11:232. doi: 10.3389/fendo.2020.00232
91. Shi J, Yu J, Zhang Y, Li Z, Gong L, Dong S, et al. Phosphatidylinositol 3-kinase-mediated HO-1/CO represses Fis1 levels and alleviates lipopolysaccharide-induced oxidative injury in alveolar macrophages. Exp Ther Med. (2018) 16:2735–42. doi: 10.3892/etm.2018.6448
92. Jeong S, Seong JH, Kang JH, Lee DS, Yim M. Dynamin-related protein 1 positively regulates osteoclast differentiation and bone loss. FEBS Lett. (2021) 595:58–67. doi: 10.1002/1873-3468.13963
93. Park-Min KH. Metabolic reprogramming in osteoclasts. Semin Immunopathol. (2019) 41:565–72. doi: 10.1007/s00281-019-00757-0
94. Kapetanovic R, Afroz SF, Ramnath D, Lawrence GM, Okada T, Curson JE, et al. Lipopolysaccharide promotes Drp1-dependent mitochondrial fission and associated inflammatory responses in macrophages. Immunol Cell Biol. (2020) 98:528–39. doi: 10.1111/imcb.12363
95. Alalaiwe A, Chen CY, Chang ZY, Sung JT, Chuang SY, Fang JY. Psoriasiform inflammation is associated with mitochondrial fission/GDAP1L1 signaling in macrophages. Int J Mol Sci. (2021) 22:10410. doi: 10.3390/ijms221910410
96. Duroux-Richard I, Roubert C, Ammari M, Présumey J, Grün JR, Häupl T, et al. miR-125b controls monocyte adaptation to inflammation through mitochondrial metabolism and dynamics. Blood. (2016) 128:3125–36. doi: 10.1182/blood-2016-02-697003
97. Khan DH, Mullokandov M, Wu Y, Voisin V, Gronda M, Hurren R, et al. Mitochondrial carrier homolog 2 is necessary for AML survival. Blood. (2020) 136:81–92. doi: 10.1182/blood.2019000106
98. Wai T, García-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. (2015) 350:aad0116. doi: 10.1126/science.aad0116
99. Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. (2008) 105:14447–52. doi: 10.1073/pnas.0803790105
100. Wei T, Gao J, Huang C, Song B, Sun M, Shen W. SIRT3 (Sirtuin-3) prevents ang II (Angiotensin II)-induced macrophage metabolic switch improving perivascular adipose tissue function. Arterioscler Thromb Vasc Biol. (2021) 41:714–30. doi: 10.1161/atvbaha.120.315337
101. Li Z, Li H, Zhao ZB, Zhu W, Feng PP, Zhu XW, et al. SIRT4 silencing in tumor-associated macrophages promotes HCC development via PPARδ signalling-mediated alternative activation of macrophages. J Exp Clin Cancer Res. (2019) 38:469. doi: 10.1186/s13046-019-1456-9
102. Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1β Production and to prevent DSS-induced colitis in mice. Cell Rep. (2017) 19:2331–44. doi: 10.1016/j.celrep.2017.05.065
103. Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. (2016) 78:505–31. doi: 10.1146/annurev-physiol-021115-105011
104. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
105. Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. (2010) 141:280–9. doi: 10.1016/j.cell.2010.02.026
106. Meyer JN, Bess AS. Involvement of autophagy and mitochondrial dynamics in determining the fate and effects of irreparable mitochondrial DNA damage. Autophagy. (2012) 8:1822–23. doi: 10.4161/auto.21741
107. Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J. The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol. (2000) 151:341–52. doi: 10.1083/jcb.151.2.341
108. Hales KG, Fuller MT. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. (1997) 90:121–9. doi: 10.1016/s0092-8674(00)80319-0
109. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. (2013) 5:a011072. doi: 10.1101/cshperspect.a011072
110. Steffen J, Vashisht AA, Wan J, Jen JC, Claypool SM, Wohlschlegel JA, et al. Rapid degradation of mutant SLC25A46 by the ubiquitin-proteasome system results in MFN1/2-mediated hyperfusion of mitochondria. Mol Biol Cell. (2017) 28:600–12. doi: 10.1091/mbc.E16-07-0545
111. Zou W, Chen Q, Slone J, Yang L, Lou X, Diao J, et al. Nanoscopic quantification of sub-mitochondrial morphology, mitophagy and mitochondrial dynamics in living cells derived from patients with mitochondrial diseases. J Nanobiotechnol. (2021) 19:136. doi: 10.1186/s12951-021-00882-9
112. Michot C, Hubert L, Romero NB, Gouda A, Mamoune A, Mathew S, et al. Study of LPIN1, LPIN2 and LPIN3 in rhabdomyolysis and exercise-induced myalgia. J Inherit Metab Dis. (2012) 35:1119–28. doi: 10.1007/s10545-012-9461-6
113. Alshudukhi AA, Zhu J, Huang D, Jama A, Smith JD, Wang QJ, et al. Lipin-1 regulates Bnip3-mediated mitophagy in glycolytic muscle. FASEB J. (2018) 32:6796–807. doi: 10.1096/fj.201800374
114. Zhang P, Verity MA, Reue K. Lipin-1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab. (2014) 20:267–79. doi: 10.1016/j.cmet.2014.05.003
115. Kim H, Choe JH, Choi JH, Kim HJ, Park SH, Lee MW, et al. Medium-chain enriched diacylglycerol (MCE-DAG) oil decreases body fat mass in mice by increasing lipolysis and thermogenesis in adipose tissue. Lipids. (2017) 52:665–73. doi: 10.1007/s11745-017-4277-7
116. Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, et al. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. (2006) 127:383–95. doi: 10.1016/j.cell.2006.09.021
117. Anderson CJ, Kahl A, Fruitman H, Qian L, Zhou P, Manfredi G, et al. Prohibitin levels regulate OMA1 activity and turnover in neurons. Cell Death Differ. (2020) 27:1896–906. doi: 10.1038/s41418-019-0469-4
118. Lai Y, Lin P, Chen M, Zhang Y, Chen J, Zheng M, et al. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. (2020) 34:101503. doi: 10.1016/j.redox.2020.101503
119. Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. (2007) 178:749–55. doi: 10.1083/jcb.200704110
120. Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. (2006) 281:37972–9. doi: 10.1074/jbc.M606059200
121. Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. (2014) 204:919–29. doi: 10.1083/jcb.201308006
122. Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. (2006) 25:2966–77. doi: 10.1038/sj.emboj.7601184
123. Lee H, Smith SB, Sheu SS, Yoon Y. The short variant of optic atrophy 1 (OPA1) improves cell survival under oxidative stress. J Biol Chem. (2020) 295:6543–60. doi: 10.1074/jbc.RA119.010983
124. Liu R, Chan DC. OPA1 and cardiolipin team up for mitochondrial fusion. Nat Cell Biol. (2017) 19:760–2. doi: 10.1038/ncb3565
125. Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol. (2017) 19:856–63. doi: 10.1038/ncb3560
126. Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. (2013) 15:1197–205. doi: 10.1038/ncb2837
127. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. (2016) 118:1593–611. doi: 10.1161/circresaha.116.307505
128. Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol. (2008) 182:937–50. doi: 10.1083/jcb.200801152
129. Nury H, Dahout-Gonzalez C, Trézéguet V, Lauquin GJ, Brandolin G, Pebay-Peyroula E. Relations between structure and function of the mitochondrial ADP/ATP carrier. Annu Rev Biochem. (2006) 75:713–41. doi: 10.1146/annurev.biochem.75.103004.142747
130. Bertholet AM, Chouchani ET, Kazak L, Angelin A, Fedorenko A, Long JZ, et al. H(+) transport is an integral function of the mitochondrial ADP/ATP carrier. Nature. (2019) 571:515–20. doi: 10.1038/s41586-019-1400-3
131. Bowzard JB, Cheng D, Peng J, Kahn RA. ELMOD2 is an Arl2 GTPase-activating protein that also acts on Arfs. J Biol Chem. (2007) 282:17568–80. doi: 10.1074/jbc.M701347200
132. Venditti P, Di Stefano L, Di Meo S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion. (2013) 13:71–82. doi: 10.1016/j.mito.2013.01.008
133. Chou CF, Lin YY, Wang HK, Zhu X, Giovarelli M, Briata P, et al. KSRP ablation enhances brown fat gene program in white adipose tissue through reduced miR-150 expression. Diabetes. (2014) 63:2949–61. doi: 10.2337/db13-1901
134. van de Ven RAH, Santos D, Haigis MC. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol Med. (2017) 23:320–31. doi: 10.1016/j.molmed.2017.02.005
135. Lin S, Wu B, Hu X, Lu H. Sirtuin 4 (Sirt4) downregulation contributes to chondrocyte senescence and osteoarthritis via mediating mitochondrial dysfunction. Int J Biol Sci. (2024) 20:1256–78. doi: 10.7150/ijbs.85585
136. Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, et al. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell. (2014) 159:1615–25. doi: 10.1016/j.cell.2014.11.046
137. Betsinger CN, Cristea IM. Mitochondrial function, metabolic regulation, and human disease viewed through the prism of sirtuin 4 (SIRT4) functions. J Proteome Res. (2019) 18:1929–38. doi: 10.1021/acs.jproteome.9b00086
138. Luo H, Jin M, Hu H, Ying Q, Hu P, Sheng W, et al. SIRT4 protects müller glial cells against apoptosis by mediating mitochondrial dynamics and oxidative stress. Mol Neurobiol. (2024). doi: 10.1007/s12035-024-04349-4
139. Noone J, Rochfort KD, O’Sullivan F, O’Gorman DJ. SIRT4 is a regulator of human skeletal muscle fatty acid metabolism influencing inner and outer mitochondrial membrane-mediated fusion. Cell Signal. (2023) 112:110931. doi: 10.1016/j.cellsig.2023.110931
140. Ding Q, Wang Y, Xia SW, Zhao F, Zhong JF, Wang HL, et al. SIRT4 expression ameliorates the detrimental effect of heat stress via AMPK/mTOR signaling pathway in BMECs. Int J Mol Sci. (2022) 23:13307. doi: 10.3390/ijms232113307
141. Kumar S, Lombard DB. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit Rev Biochem Mol Biol. (2018) 53:311–34. doi: 10.1080/10409238.2018.1458071
142. Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. (2013) 18:920–33. doi: 10.1016/j.cmet.2013.11.013
143. Greene KS, Lukey MJ, Wang X, Blank B, Druso JE, Lin MJ, et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc Natl Acad Sci U S A. (2019) 116:26625–32. doi: 10.1073/pnas.1911954116
144. Yu Y, Newman H, Shen L, Sharma D, Hu G, Mirando AJ, et al. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab. (2019) 29:966–978.e4. doi: 10.1016/j.cmet.2019.01.016
145. Tao J, Zhang J, Ling Y, McCall CE, Liu TF. Mitochondrial sirtuin 4 resolves immune tolerance in monocytes by rebalancing glycolysis and glucose oxidation homeostasis. Front Immunol. (2018) 9:419. doi: 10.3389/fimmu.2018.00419
146. Qin K, Han C, Zhang H, Li T, Li N, Cao X. NAD(+) dependent deacetylase Sirtuin 5 rescues the innate inflammatory response of endotoxin tolerant macrophages by promoting acetylation of p65. J Autoimmun. (2017) 81:120–9. doi: 10.1016/j.jaut.2017.04.006
147. Baeza J, Smallegan MJ, Denu JM. Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem Sci. (2016) 41:231–44. doi: 10.1016/j.tibs.2015.12.006
148. Cha XD, Zou QY, Li FZ, Wang TY, Wang SL, Cai BY, et al. SIRT5 exacerbates eosinophilic chronic rhinosinusitis by promoting polarization of M2 macrophage. J Allergy Clin Immunol. (2024) 154:644–56. doi: 10.1016/j.jaci.2024.04.028
149. Zhu Y, Chen X, Lu Y, Xia L, Fan S, Huang Q, et al. Glutamine mitigates murine burn sepsis by supporting macrophage M2 polarization through repressing the SIRT5-mediated desuccinylation of pyruvate dehydrogenase. Burns Trauma. (2022) 10:tkac041. doi: 10.1093/burnst/tkac041
150. Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells. (2018) 41:18–26. doi: 10.14348/molcells.2018.2277
151. Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. (2016) 35:1368–84. doi: 10.15252/embj.201593102
152. Park YS, Choi SE, Koh HC. PGAM5 regulates PINK1/Parkin-mediated mitophagy via DRP1 in CCCP-induced mitochondrial dysfunction. Toxicol Lett. (2018) 284:120–8. doi: 10.1016/j.toxlet.2017.12.004
153. Li S, Lin Q, Shao X, Zhu X, Wu J, Wu B, et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic Biol Med. (2020) 152:632–49. doi: 10.1016/j.freeradbiomed.2019.12.005
154. Xin T, Lu C. Irisin activates Opa1-induced mitophagy to protect cardiomyocytes against apoptosis following myocardial infarction. Aging (Albany NY). (2020) 12:4474–88. doi: 10.18632/aging.102899
155. Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. (2016) 12:689–702. doi: 10.1080/15548627.2016.1151580
156. Wu H, Wang Y, Li W, Chen H, Du L, Liu D, et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy. (2019) 15:1882–98. doi: 10.1080/15548627.2019.1596482
157. Huang J, Zhu T, Rong R, You M, Ji D, Li H. FUN14 domain-containing 1-mediated mitophagy suppresses interleukin-1β production in macrophages. Int Immunopharmacol. (2020) 88:106964. doi: 10.1016/j.intimp.2020.106964
158. Sanchez-Lopez E, Zhong Z, Stubelius A, Sweeney SR, Booshehri LM, Antonucci L, et al. Choline uptake and metabolism modulate macrophage IL-1β and IL-18 production. Cell Metab. (2019) 29:1350–1362.e7. doi: 10.1016/j.cmet.2019.03.011
159. Hollenbeck CB. An introduction to the nutrition and metabolism of choline. Cent Nerv Syst Agents Med Chem. (2012) 12:100–13. doi: 10.2174/187152412800792689
160. Liu X, Weaver D, Shirihai O, Hajnóczky G. Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. (2009) 28:3074–89. doi: 10.1038/emboj.2009.255
161. Koelwyn GJ, Corr EM, Erbay E, Moore KJ. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. (2018) 19:526–37. doi: 10.1038/s41590-018-0113-3
162. Bravo-San-Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. (2017) 120:1812–24. doi: 10.1161/circresaha.117.311082
163. Grootaert MOJ, Roth L, Schrijvers DM, De Meyer GRY, Martinet W. Defective autophagy in atherosclerosis: to die or to senesce? Oxid Med Cell Longev. (2018) 2018:7687083. doi: 10.1155/2018/7687083
164. Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of drp1-mediated mitochondrial fission. Diabetes. (2017) 66:193–205. doi: 10.2337/db16-0915
165. Fricker M, Gibson PG. Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J. (2017) 50:1700196. doi: 10.1183/13993003.00196-2017
166. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest. (2017) 152:618–26. doi: 10.1016/j.chest.2017.03.020
167. Araya J, Tsubouchi K, Sato N, Ito S, Minagawa S, Hara H, et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy. (2019) 15:510–26. doi: 10.1080/15548627.2018.1532259
168. Liu J, Chen Y, Chen H, Wang Y, Li D, Zhang Q, et al. Macrophage migration inhibitory factor exacerbates asthmatic airway remodeling via dynamin-related protein 1-mediated autophagy activation. Respir Res. (2023) 24:216. doi: 10.1186/s12931-023-02526-y
169. Bai Q, Liu R, Quan C, Han X, Wang D, Wang C, et al. DEK deficiency suppresses mitophagy to protect against house dust mite-induced asthma. Front Immunol. (2023) 14:1289774. doi: 10.3389/fimmu.2023.1289774
170. Abbasi Moajani F, Soozangar N, Amani M, Jeddi F, Salimnejad R, Aslani MR. The suppressive effects of crocin from saffron on allergic airway inflammation through Drp1/Nfr1/Mfn2/Pgc1-alpha signaling pathway in mice. J Ethnopharmacol. (2025) 337:118862. doi: 10.1016/j.jep.2024.118862
171. Zhao CC, Xu J, Xie QM, Zhang HY, Fei GH, Wu HM. Abscisic acid suppresses the activation of NLRP3 inflammasome and oxidative stress in murine allergic airway inflammation. Phytother Res. (2021) 35:3298–309. doi: 10.1002/ptr.7051
172. Liu S, Zhang H, Li Y, Zhang Y, Bian Y, Zeng Y, et al. S100A4 enhances protumor macrophage polarization by control of PPAR-γ-dependent induction of fatty acid oxidation. J Immunother Cancer. (2021) 9:e002548. doi: 10.1136/jitc-2021-002548
173. Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. (2020) 16:3–17. doi: 10.1080/15548627.2019.1603547
174. Kramer P, Bressan P. Our (Mother’s) mitochondria and our mind. Perspect Psychol Sci. (2018) 13:88–100. doi: 10.1177/1745691617718356
175. Kim JH, Lee ER, Jeon K, Choi HY, Lim H, Kim SJ, et al. Role of BI-1 (TEGT)-mediated ERK1/2 activation in mitochondria-mediated apoptosis and splenomegaly in BI-1 transgenic mice. Biochim Biophys Acta. (2012) 1823:876–88. doi: 10.1016/j.bbamcr.2012.01.016
176. Prieto J, León M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, et al. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. (2016) 7:11124. doi: 10.1038/ncomms11124
177. Wang Y, Subramanian M, Yurdagul A Jr., Barbosa-Lorenzi VC, Cai B, de-Juan-Sanz J, et al. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell. (2017) 171:331–345.e22. doi: 10.1016/j.cell.2017.08.041
178. Li J, Ye Y, Liu Z, Zhang G, Dai H, Li J, et al. Macrophage mitochondrial fission improves cancer cell phagocytosis induced by therapeutic antibodies and is impaired by glutamine competition. Nat Cancer. (2022) 3:453–70. doi: 10.1038/s43018-022-00354-5
179. Su ZD, Li CQ, Wang HW, Zheng MM, Chen QW. Inhibition of DRP1-dependent mitochondrial fission by Mdivi-1 alleviates atherosclerosis through the modulation of M1 polarization. J Transl Med. (2023) 21:427. doi: 10.1186/s12967-023-04270-9
180. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
181. Li Z, Pan H, Yang J, Chen D, Wang Y, Zhang H, et al. Xuanfei Baidu formula alleviates impaired mitochondrial dynamics and activated NLRP3 inflammasome by repressing NF-κB and MAPK pathways in LPS-induced ALI and inflammation models. Phytomedicine. (2023) 108:154545. doi: 10.1016/j.phymed.2022.154545
182. Sánchez-Rodríguez R, Tezze C, Agnellini AHR, Angioni R, Venegas FC, Cioccarelli C, et al. OPA1 drives macrophage metabolism and functional commitment via p65 signaling. Cell Death Differ. (2023) 30:742–52. doi: 10.1038/s41418-022-01076-y
183. Sun X, Yegambaram M, Lu Q, Garcia Flores AE, Pokharel MD, Soto J, et al. Mitochondrial fission produces a Warburg effect via the oxidative inhibition of prolyl hydroxylase domain-2. Redox Biol. (2025) 81:103529. doi: 10.1016/j.redox.2025.103529
184. Wu Z, Zuo M, Zeng L, Cui K, Liu B, Yan C, et al. OMA1 reprograms metabolism under hypoxia to promote colorectal cancer development. EMBO Rep. (2021) 22:e50827. doi: 10.15252/embr.202050827
185. Qian M, Wang S, Guo X, Wang J, Zhang Z, Qiu W, et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene. (2020) 39:428–42. doi: 10.1038/s41388-019-0996-y
186. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene. (2019) 38:5158–73. doi: 10.1038/s41388-019-0782-x
187. Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. (2021) 184:2896–2910.e13. doi: 10.1016/j.cell.2021.04.027
Keywords: mitochondrial dynamics, fusion, fission, macrophage, metabolism
Citation: Li P, Fan Z, Huang Y, Luo L and Wu X (2025) Mitochondrial dynamics at the intersection of macrophage polarization and metabolism. Front. Immunol. 16:1520814. doi: 10.3389/fimmu.2025.1520814
Received: 31 October 2024; Accepted: 04 March 2025;
Published: 24 March 2025.
Edited by:
Yayun Wang, Air Force Medical University, ChinaReviewed by:
Quanfu Li, Tongji University, ChinaJingjing Ye, Peking University People’s Hospital, China
Copyright © 2025 Li, Fan, Huang, Luo and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Wu, eGlhb3lhbnd1MjAyMEAxMjYuY29t
†These authors have contributed equally to this work