 Iliana Michailidou1
Iliana Michailidou1 Anastasia Patsiarika2
Anastasia Patsiarika2 Evangelia Kesidou1
Evangelia Kesidou1 Marina Kleopatra Boziki1,3
Marina Kleopatra Boziki1,3 Dimitrios Parisis3
Dimitrios Parisis3 Christos Bakirtzis3
Christos Bakirtzis3 Elisabeth Chroni4
Elisabeth Chroni4 Nikolaos Grigoriadis1,3*
Nikolaos Grigoriadis1,3*- 1Laboratory of Experimental Neurology and Neuroimmunology, AHEPA University Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
- 2AstraZeneca, Medical Affairs Department Rare Disease, Athens, Greece
- 32ndDepartment of Neurology, AHEPA University Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
- 4Department of Neurology, Medical School, University of Patras, Patra, Greece
The complement system is a key component of the innate immune system. In antiacetylcholine receptor (AChR) antibody-positive (Ab+) generalized myasthenia gravis (MG), complement activation has long been considered a principal driver of pathology. Understanding the role of complement in AChR-Ab+ generalized MG has gained increasing importance in recent years, as anticomplement drugs have been approved for clinical use or are undergoing phase II/III clinical trials. This review aims to discuss recent and previous findings on the role of complement in AChR-Ab+ MG pathology, including its interaction with pathogenic antibodies and mechanisms beyond the classical pathway activation.
Introduction
Myasthenia gravis (MG) is a rare, chronic neuromuscular autoimmune disease with a median global prevalence of 10 per 100,000 and an in-hospital mortality rate of 1.8%. The hallmark of MG pathology is defective transmission at the neuromuscular junction. MG is characterized by fatigability and fluctuating weakness of the ocular, bulbar, respiratory, and limb muscles. Most people with MG (pwMG) develop ocular symptoms at some point during the disease course. Approximately 80% of pwMG with ocular onset progress to generalized MG within 2 years of disease onset (1–3).
MG is a T-cell-dependent disease in which CD4+ T cells produce cytokines and promote B-cell differentiation into plasma cells (4). The trigger for T-cell activation and MG development remains unknown but has been linked to thymomas (1, 5). Plasma cells produce autoantibodies that target neuromuscular junction proteins, a specialized synapse that transmits nerve impulses to muscles across the synaptic cleft. MG autoantibodies and serum molecules, including complement proteins, can bind to and become activated at the neuromuscular junction, as this structure lacks protection from the blood–nerve barrier (4).
MG autoantibodies vary in IgG subclasses, protein targets, and pathogenic actions, forming the basis for disease subgroup classification. In particular, autoantibodies against the acetylcholine receptor (AChR) belong to the IgG1 and IgG3 subclasses (6) and account for 85% of cases. There are three pathogenic mechanisms by which AChR autoantibodies lead to MG: antibody blocking, antigenic modulation, and complement activation as described below. Another 5% of all pwMG have autoantibodies against muscle-specific kinase (MuSK), which belongs to the IgG4 subclass (7). IgG4 antibodies have a limited capacity to interact with complement proteins and are therefore considered noncomplement-fixing antibodies (8). Additionally, 1%–5% of pwMG have autoantibodies targeting low-density lipoprotein receptor-related protein 4 (LRP4), which belongs to the IgG1 subclass (9). Approximately, 10% of pwMG are diagnosed without detectable autoantibodies and are classified as seronegative.
The complement system is a key effector mechanism in anti-AChR antibody-positive (Ab+) MG (4, 10). Complement activation may occur as an early and crucial event in MG (11–13), while the impact of autoantibodies alone remains poorly understood (14). Therefore, a causal rather than a bystander role for complement in MG pathology should be considered, given its involvement in primarily degenerative processes such as autophagy and synaptic pruning (15, 16). This review discusses the mechanisms of complement-mediated damage at the neuromuscular junction in the presence or absence of autoantibodies.
AChR autoantibody-mediated pathogenic mechanisms in MG
AChRs are heteropentamers consisting of two α-subunits and one β-, δ-, and either γ-subunit (in embryonic muscle) or ϵ-subunit (in adult muscle), organized around a central ion channel. These receptors are located at the postsynaptic membrane of the neuromuscular junction and facilitate the generation of a muscle fiber action potential upon binding of Ach, which is released by the firing nerve ending. In MG, the anti-AChR antibody response is polyclonal; however, at least half of the AChR autoantibodies target the α-subunits, specifically the N-terminal region of AChR-α, which includes the main immunogenic region (MIR). The MIR is a cluster of conformation-dependent epitopes recognized by functionally and structurally heterogenous monoclonal anti-MIR antibodies (17). These anti-MIR antibodies may promote AChR clustering at the neuromuscular junction by binding to the α-subunits of adjacent receptors (18).
MG pathology may result from the direct blockade of AChRs by autoantibodies, which inhibits ACh binding to its receptor. Steric interference with ACh-AChR binding may lead to an activity-dependent reduction in neuromuscular junction efficacy, preventing striated muscle contraction and ultimately causing denervation of the affected muscle (19, 20). Anti-AChR autoantibodies have also been found to slow AChR channel closure, leading to abnormal AChR desensitization and the formation of miniature rather than normal endplate currents (21). In MG, miniature endplate currents are insufficient to reach the threshold level necessary for generating a muscle fiber action potential (22).
However, most MG autoantibodies do not block the binding of ACh to its binding sites. This may—at least in part—explain the lack of a significant relationship between antibody titers and disease severity (23, 24). PwMG in remission may present with the highest antibody abundance (25), whereas severe disease is primarily associated with the loss of AChRs and the destruction of junctional folds (26). Previous studies have categorized pathogenic antibodies as blocking or binding using standard immunoprecipitation assays to quantify binding antibodies and assays to measure antibodies that block α-bungarotoxin binding to receptors (24, 25, 27). Although the degree of AChR blockade was significantly correlated with the generalization of muscle weakness (25), people with severe generalized MG had either both blocking and binding antibodies or binding antibodies alone (28). Whether blocking and binding MG antibodies bind to distinct AChR sites with higher or lower agonist affinity—and whether the two types of antibodies act synergistically to exert a functional action on AChR—remains to be determined.
The second hypothesis for MG pathogenesis is based on the ability of anti-AChR autoantibodies and their divalent F(Ab′)2 fragments to modulate—rather than block—AChRs (29). Antigenic modulation activity in MG has been linked to antibody titers (30). Autoantibodies targeting AChRs are believed to accelerate the natural AChR degradation cycle by binding to the MIR and inducing AChR cross-linking (18). AChR cross-linking is followed by internalization and lysosomal degradation of the autoantibody-bound receptors, which—similar to the first hypothesis—reduces AChR densities at the neuromuscular junction (29). Notably, studies in rodents have indicated that increased AChR internalization is compensated by enhanced AChR synthesis, resulting in similar AChR levels before and after internalization (31, 32). However, other studies have reported contradictory data (33, 34). A key question remains: what is the impact of coexisting pathological mechanisms on the synthesis and insertion of newly synthesized AChRs at the neuromuscular junction? In addition, further research is needed on the kinetics and recycling potential of internalized AChRs to better understand the exact contribution of the modulatory effect of autoantibodies on MG pathogenesis and progression.
Complement-mediated pathogenic mechanisms in MG
The complement system consists of more than 30 proteins found in plasma and on cell surfaces, which enhance the activity of antibodies and phagocytic cells in removing microbes, immunocomplexes, and dead or modified self-cells. It primarily serves the innate immune system while also interacting with components of the adaptive immune system to regulate antibody-mediated responses. Complement proteins, mainly produced by hepatocytes, have both effector and regulatory roles (35).
Activation of complement occurs through three pathways: the classical, lectin, and alternative pathways (Figure 1). The classical pathway is triggered when C1q binds to immune complexes or “eat me” signals displayed by apoptotic cells. Similarly, the lectin pathway is initiated when mannose-binding lectin (MBL) or ficolins bind to carbohydrate patterns on the cell membrane. The alternative pathway is activated by the slow spontaneous hydrolysis (tick-over) of C3 to C3(H2O), allowing a continuous but subtle complement activation that contributes to immune surveillance and can amplify neuroinflammatory responses. All complement pathways ultimately lead to three key outcomes: opsonization of a target for phagocytosis via the covalently bound C3b component, induction or amplification of inflammation through the released C3a and C5a anaphylatoxins, and target cell damage via the membrane attack complex (MAC). MAC is a pore-forming structure that integrates into the lipid bilayer of the plasma membrane, causing lysis (13, 35) or triggering proinflammatory responses when formed at sublytic levels (36).
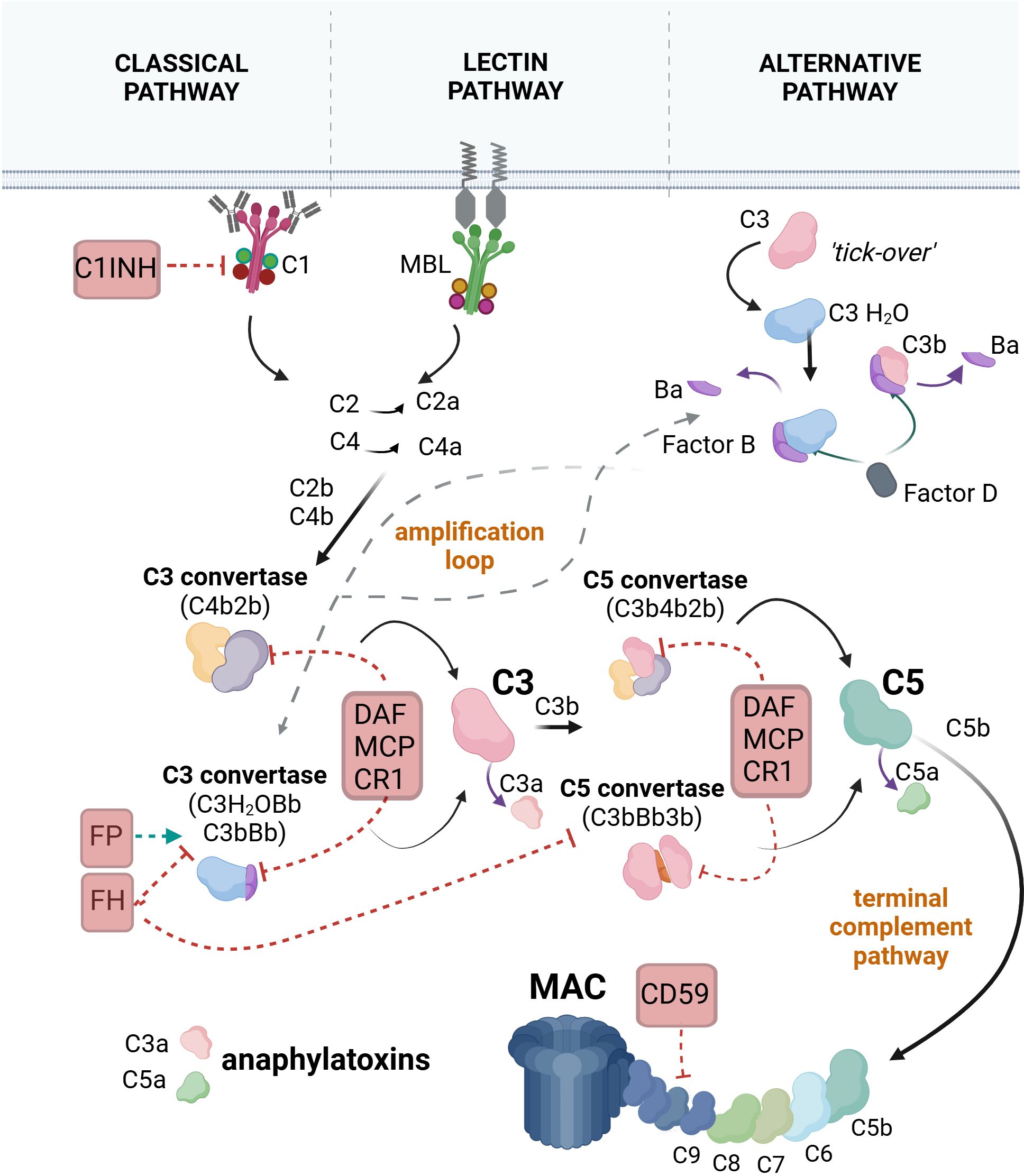
Figure 1. The pathways of complement system activation. In the classical pathway, C1q, a subunit of the C1 complex, binds to an antibody-fixed cell. This binding activates the proteases C1r and C1s (also components of the C1 complex). C1s cleave the C4 protein into C4a and C4b. C4b binds covalently to the target and attracts C2, which is cleaved by C1s into C2a and C2b. C2b binds to C4b to form the C4b2b complex, also known as C3 convertase. C3 convertase cleaves C3 into C3a and C3b. C3b may bind to the C3 convertase, forming the C4b2b3b complex, also known as C5 convertase. C5 convertase cleaves C5 into C5a and C5b. C5b associates with C6, C7, C8, and C9 to form the final product of complement activation, the membrane attack complex (MAC). In the lectin pathway, mannose-binding lectin (MBL) or ficolins bind to carbohydrate patterns on the target cell, leading to the activation of MBL-associated serine proteases (MASP)1 and MASP2, which are complexed with MBL. MASPs cleave C4 and C2, forming C3 convertase. The pathway continues with the generation of the C5 convertase and the formation of MAC. In the alternative pathway, inactive C3 undergoes spontaneous hydrolysis, forming C3(H2O). Factor B binds to C3(H2O), leading to its cleavage by factor D into Ba and Bb, resulting in the formation of the fluid-phase C3 convertase, C3(H2O)Bb. C3(H2O)Bb converts C3 into C3a and C3b, with some C3b molecules binding to the target and associating with factor B, which is subsequently cleaved by factor D to form the C3 convertase C3bBb. The pathway progresses with the formation of the C5 convertase (C3bBb3b), cleavage of C5, and the assembly of the MAC. Selected regulators of the complement cascade that interfere with different steps of the cascade are illustrated. C1 inhibitor (C1INH) promotes the decomposition of the C1 complex. Decay-accelerating factor (DAF), membrane cofactor protein (MCP), and complement receptor 1 (CR1) accelerate the decay of the C3 and C5 convertases. CD59 inhibits the formation of MAC. Factor H (FH) prevents the assembly of the alternative pathway C3 and C5 convertases, while factor P (properdin) stabilizes the alternative pathway C3 convertase, thereby promoting the amplification loop. Created with BioRender.com.
Complement can have the potential to harm self-tissues; therefore, its activation is tightly regulated (Figure 1) to eliminate pathogens or “unwanted” cells without injuring the host. When this balance is disrupted, excessive complement activation can lead to tissue injury and contribute to the pathology of various diseases (35).
Numerous studies assessing the levels of complement activation products and regulators in plasma (37–41) and biopsied material (42–46) from individuals with AChR-Ab+ MG have confirmed that the activated complement system is a key mediator of AChR-Ab+ MG pathology (10, 13, 47). Its physiological role in eliminating antibody-targeted pathogens (35) leads to binding and activation at the autoantibody-fixed AChR-Ab+ MG neuromuscular junction, resulting in tissue damage (42–44). Activation occurs via the classical pathway when the C1q molecule binds to the Fc region of IgG1 and IgG3 autoantibodies attached to AChRs (48). Since optimal C1q binding requires multiple IgG molecules to bind one C1q, the clustering of autoantibody-bound AChRs on the postsynaptic membrane plays an important role in classical pathway activation (49). Activation of the classical pathway leads to the assembly of the pore-forming MAC, which serves as the key effector of complement-mediated damage in AChR-Ab+ MG. MAC induces direct destruction of the postsynaptic membrane, reducing AChRs and voltage-gated channels, and significantly contributing to the widening of the postsynaptic cleft and muscle denervation (50). Next to the role of MAC, activation of the classical pathway results in the covalent binding of C4b and C3b opsonins on the muscle surface. These opsonins be recognized by complement receptor type 1 (CR1)- and type 2 (CR2)-bearing cells, including T and B cells. Furthermore, activation leads to the release of C3a and C5a, which modulate leucocyte responses by binding to C3aR and C5aR (Figure 2). It remains unclear whether, in AChR-Ab+ MG, C1q activates complement in the absence of antibodies by binding to motifs or molecules exposed on muscle endplates. This possibility might explain its deposition on the motor endplates in the intercostal muscle of amyotrophic lateral sclerosis (ALS) donors (51). Alternatively, C1q activation may be linked to activity patterns at the neuromuscular junction, a mechanism previously suggested to underlie its role in the synapse elimination in the central nervous system by microglia (52).
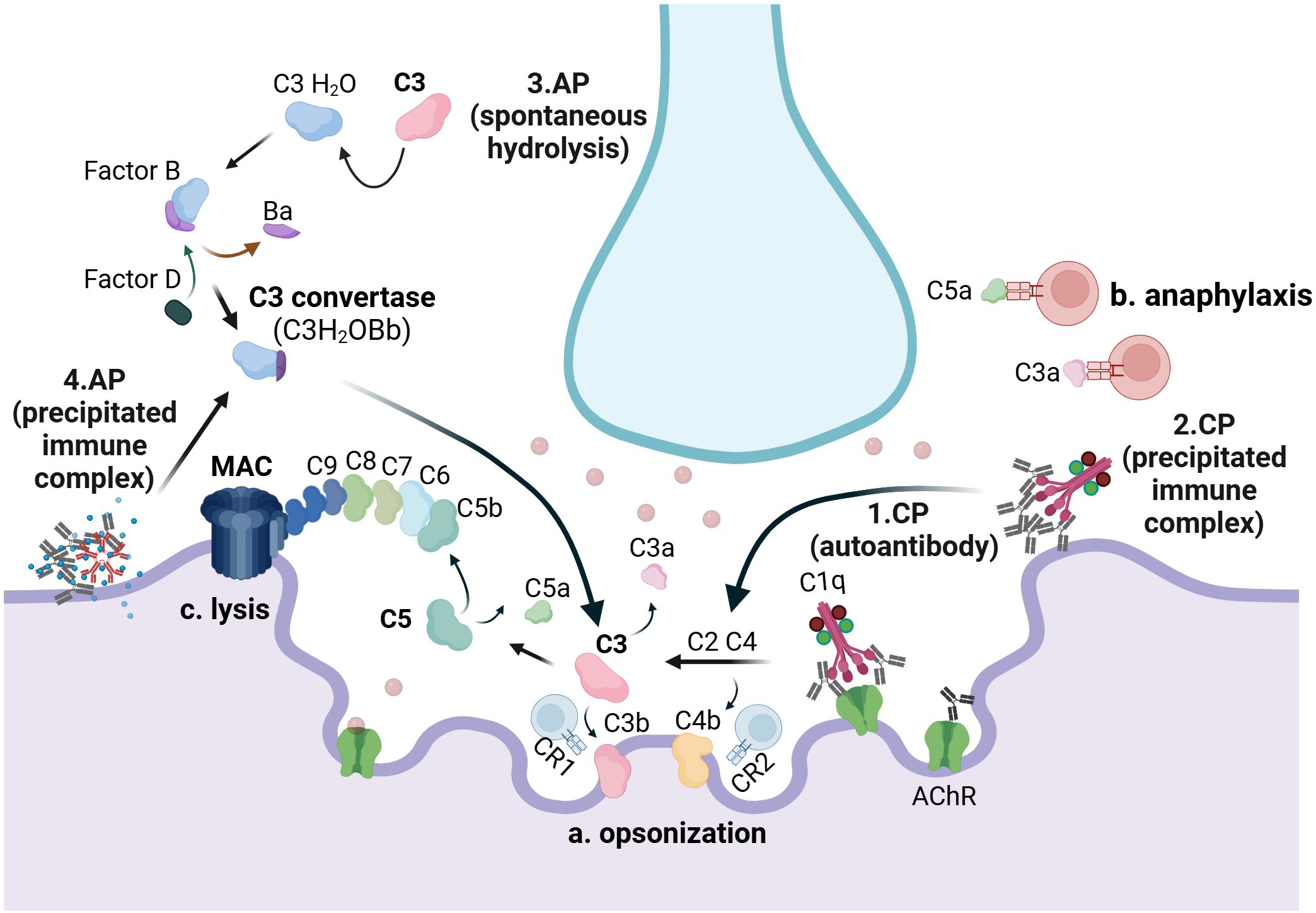
Figure 2. The complement system in AChR-Ab+ generalized myasthenia gravis: C1q binds to autoantibodies attached to AChRs (1) and to precipitated immune complexes (2), when present, at the neuromuscular junction, thereby activating the classical pathway (CP). Hydrolysis of C3, either spontaneously (3) or triggered by precipitated immunocomplexes (4), induces activation of the alternative pathway (AP). CP activation results in the opsonization of the muscle membrane by C4b and C3b, which serve as potent activators of CR1- and CR2-bearing lymphocytes (a). Both the CP and the AP result in the release of C3a, a product of C3 cleavage, and C5a, a product of C5 cleavage, which may interact with immune cells bearing C3a and C5a receptors, respectively (b). Additionally, both pathways contribute to the formation of the membrane attack complex (MAC), composed of C5b, C6, C7, C8, and C9, which serves as the key driver of neuromuscular junction damage (c). CP, classical pathway; AP, alternative pathway. Created with BioRender.com.
The molecular contribution of MAC to MG pathology has been inferred from studies conducted in experimental autoimmune myasthenia gravis (EAMG) models (53, 54). Pharmacological inhibition of MAC in EAMG has been achieved using C5 inhibitors, such as the 4G2 monoclonal antibody (55) and the rEV576 compound (56), both of which block rat C5 and thereby inhibit MAC formation as well as C5a-induced anaphylaxis. Additionally, MAC inhibitors, including the 7E5 monoclonal antibody targeting human C6 (57) and the TPP1820 monoclonal antibody targeting mouse C7 (58), have also been used to suppress MAC activity. The administration of C5, C6, or C7 terminal complement pathway inhibitors in rodents mitigated EAMG.
Moreover, inducing EAMG in mice deficient in C5 (59) or C6 (53), both components of the terminal pathway, resulted in a reduced incidence of severe disease and better preservation of AChRs compared with wildtype littermates. In contrast, animals lacking the CD59a gene, which encodes the CD59 regulator of MAC formation, exhibited high levels of C9 immunoreactivity at the diaphragm neuromuscular junctions, indicating MAC deposition (60). In line with these data, C3 or C4 knockout mice, which are unable to form MAC, exhibited autoantibody deposition at the neuromuscular junctions when immunized with anti-AChR antibodies but remained resistant to EAMG development, with no alterations in AChR densities (61). This demonstrates the role of complement in the induction and progression of EAMG. In contrast, mice deficient in the CD55/decay-accelerating factor (DAF), a regulator of C3 and C5 convertase decay, exhibited excessive MAC formation and extensive damage to the postsynaptic membrane (60). Notably, EAMG rodents deficient in the C3 protein exhibited reduced serum IgG levels, including anti-AChR IgGs, and a decreased number of B cells, impacting the adaptive immune system (61). Collectively, these findings indicate that complement serves as the primary effector mechanism in EAMG (Table 1).
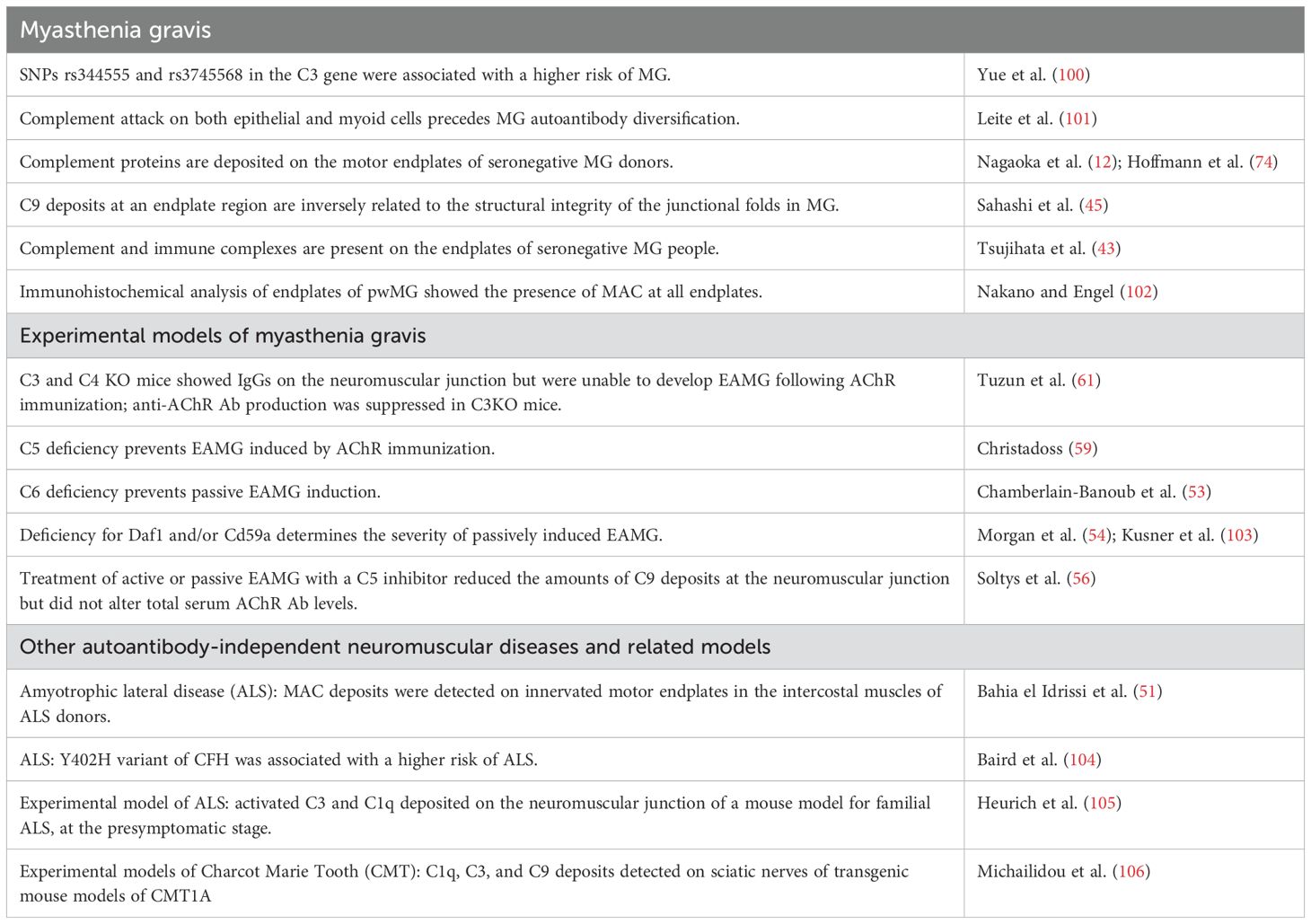
Table 1. Published data support an AChR-Ab-independent role of complement in generalized MG and suggest a causal role in EAMG pathology.
Limiting complement activation vs. lowering AChR autoantibody titers for suppression of MG
Since 2017, terminal complement pathway inhibition has been a therapeutic option for individuals with AChR-Ab+ MG. Eculizumab, a C5 inhibitor, was the first anticomplement drug approved for the treatment of refractory generalized MG in people aged 6 years and older with AChR-Ab+. Four mutations on eculizumab led to the development of ravulizumab, the first long-acting C5 inhibitor, which received regulatory approval for AChR-Ab+ generalized MG in 2022. Ravulizumab is administered via intravenous infusion every 8 weeks, offering a longer dosing interval compared to eculizumab, which is given every 2 weeks (62, 63). In 2023, a year later, zilucoplan, a peptidic C5 inhibitor, was authorized for daily self-administration via subcutaneous injection. Zilucoplan has a favorable safety profile and has been shown to maintain or improve MG symptoms in pwMG switching from intravenous C5 inhibitor therapy, based on data from the interim analysis of a phase C3b study (64). The results of phase III trials (65, 66), along with subsequent market authorizations of the aforementioned C5 inhibitors, the ongoing preclinical testing of the CP010 C6 inhibitor (https://gravitoncorp.com), and the extensive list of complement inhibitors that have been evaluated in clinical phase I/II and III trials or are currently undergoing phase II/III clinical testing—including ALXN1720 (NCT05556096), iptacopan (NCT06517758), DNTH103 (NCT06282159), and forelimb/cemdisiran (NCT05070858) inhibitors—suggest that anticomplement therapeutics may play a central role in the treatment of AChR-Ab+ generalized MG. Several phase III trials were followed by corresponding open-label extension studies (Table 2).
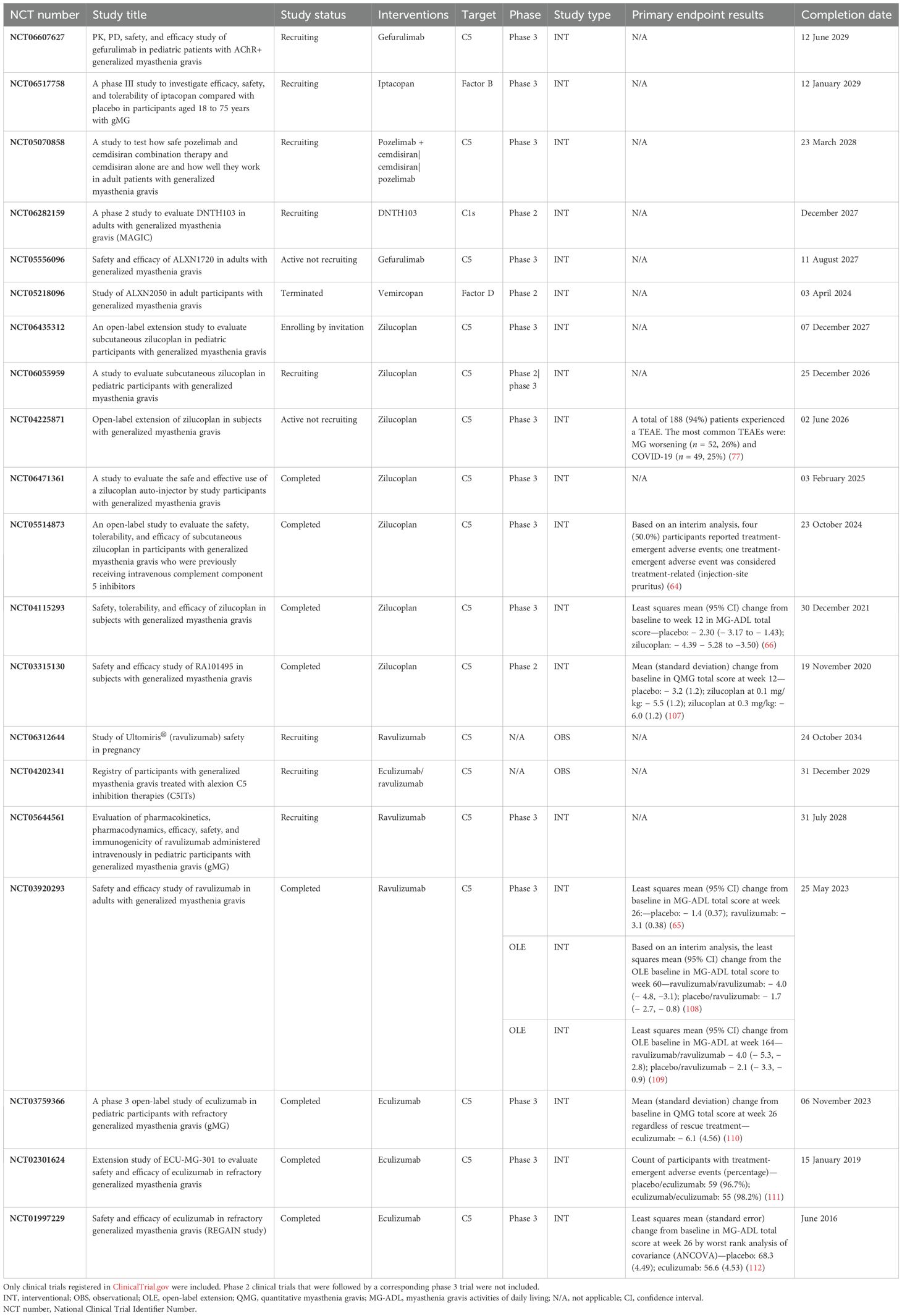
Table 2. Clinical trials of complement inhibitors for the treatment of generalized MG.
Terminal complement pathway inhibitors are promising for AChR-Ab+ MG therapy because they block the destructive MAC while preserving upstream complement mechanisms. This ensures physiological protection against infection and may facilitate immune complex clearance in MG. Activated C3 is known to mediate immune complex clearance via CR1-receptor-bearing cells (67) and aids in their solubilization to prevent precipitation. The solubilization of preformed immune complexes is considered a primary function of the alternative complement pathway (68), though the classical pathway is also involved (69). Precipitated immune complexes, particularly those formed by IgG1 or IgG3 antibodies, can have significant pathological impacts on various tissues (70), as they activate the complement system. Immune complexes and C3 have previously been identified in forelimb muscles in EAMG (45) and within degenerating junctional folds in humans (71, 72). Additionally, IgG, C3, and C9 were detected at the limb muscle endplates of pwMG (43). Interestingly, some pwMG with immune complexes and activated complement exhibited no detectable serum anti-AChR antibodies (43), suggesting that insufficient immune complex clearance, rather than autoantibody production, may contribute to complement-mediated damage in seronegative pwMG (Table 1). However, the possibility cannot be excluded that complement activation at the neuromuscular junction of seronegative pwMG is triggered by an unknown autoantibody targeting the synapse or that the sensitivity of current antibody quantification assays is insufficient to detect low levels of circulating antibodies. Live cell-based assays for detecting anti-AChR antibodies may serve as a valuable tool for diagnosing patients with radioimmunoassay-double seronegative MG (73). Furthermore, the use of intercostal muscle biopsy to identify complement depositions at the neuromuscular junction (74) may need to be considered for diagnosis and/or therapeutic decision-making in patients for whom live cell-based assay has also failed to detect antibodies.
The advantage of selectively blocking only the damaging arm of the complement system, rather than promoting broad immunosuppression, has been inferred by real-world studies demonstrating sustained improvement (65, 75–77) and a reduced need for corticosteroids (78–85) in people with AChR-Ab+ generalized MG receiving C5 inhibitor therapies. In contrast, plasmapheresis, which depletes MG autoantibodies from the serum, is essential for symptom relief during a crisis but provides only a short-lasting (approximately 6 weeks) effect, as symptoms return once autoantibody levels revert to baseline (86). Similarly, intravenous immunoglobulin (IVIg) and other immunosuppressive therapies—such as corticosteroids, azathioprine, and the neonatal Fc receptor (FcRn) inhibitors—regulate autoantibody levels and indirectly modulate complement-dependent damage. However, these drugs cannot block the effects of residual autoantibodies bound to the neuromuscular junction or suppress potential activation of the antibody-independent alternative pathway, meaning they do not specifically block complement-mediated effects (11). Notably, these treatments are ineffective in reducing alternative pathway activation, which implies the effects of initial classical pathway activation. This has significant implications for MG, particularly given data demonstrating changes in serum levels of the alternative pathway regulator properdin in pwMG (87). Remarkably, robust classical pathway activation may bypass the effect of the alternative pathway (88), suggesting that alternative complement pathway inhibitors may be beneficial only when classical pathway activation is modest. This may explain the lack of efficacy of vemircopan, a factor D inhibitor, in a phase II clinical trial. A phase III clinical trial evaluating the effects of iptacopan, a factor B inhibitor, is currently underway, and its results are awaited. The anti-B-cell rituximab is particularly effective in MuSK-Ab+ MG and is often prescribed for individuals with AChR-Ab+ refractory MG based on long-term clinical experience (89, 90). However, rituximab may promote complement activation as an adverse reaction, which could explain the lack of response in some pwMG (89), including those with refractory MG who are double seronegative for anti-AChR and anti-MuSK antibodies (91). It is noteworthy that anti-LRP4 antibodies can—at least partially—activate the complement system (92), but the impact of complement-mediated mechanisms in anti-LRP4 Ab+ MG remains underrecognized in clinical practice.
Targeting the terminal complement pathway in AChR-Ab+ MG may regulate the disease course beyond MAC formation. Zilucoplan has been shown to prevent C5b6 formation through plasmin-mediated noncanonical C5 activation, thereby interfering with red blood cell hemolysis (93). Meanwhile, eculizumab has been found to induce functional pathways related to antioxidant activity, cholesterol transfer, and cellular detoxification, while downregulating pathways associated with antigen binding. In addition, eculizumab reduced leukotriene production, possibly due to the inhibition of C5a-induced anaphylaxis (94).
Studies indicate that despite current therapeutic interventions for AChR-Ab+ generalized MG, a proportion of people, approximately 10% or higher, do not respond adequately to treatment. These pwMG may remain refractory, develop drug dependency, or experience intolerable side effects (95–98). For those receiving C5 inhibitors, vaccination against all meningococcal strains should be administered as close as possible to therapy initiation. When the interval between vaccination and treatment is insufficient, prophylactic antibiotics are strongly recommended (99). This is because activation of upstream complement functions alone is insufficient for protection against Neisseria species, a group of bacteria that requires activation of the terminal pathway and can cause meningococcal sepsis or meningitis. Since no reliable biomarkers of MG pathology are currently available, the present treatment approach relies on trial and error. There is an urgent need for personalized MG treatment, which may be achieved by gaining deeper insights into both the beneficial and adverse effects of existing therapies.
Overall, terminal complement activation is destructive, making its inhibition a primary goal in treating people with AChR-Ab+ generalized MG. Identifying biomarkers to guide therapeutic decisions and understanding inhibitor-induced molecular actions may help improve treatment responses.
Concluding remarks
Complement plays a crucial role in MG pathology, as the formation of MAC at MG endplates can lead to the destruction of the neuromuscular junction. AChR blocking and antigenic modulation trigger complement activation in MG. In addition to the classical pathway, the role of the activated alternative pathway in MG pathology requires further evaluation, as antibody-targeting therapies may not efficiently suppress it.
Author contributions
IM: Writing – original draft, Writing – review & editing. AP: Writing – original draft; Writing – review & editing. EK: Writing – original draft. MB: Writing – review & editing. DP: Writing – review & editing. CB: Writing – review & editing. EC: Writing – review & editing. NG: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors declare this work was supported by an unrestricted grant of the Aristotle University of Thessaloniki Research Committee.
Acknowledgments
We thank Prof. Frank Baas (LUMC, The Netherlands) for his critical review of this manuscript.
Conflict of interest
AP is an employee of AstraZeneca Rare Disease. EC has participated in advisory meetings and satellite symposia organized by Merck, Sanofi Genzyme, Biogen, Genesis Pharma, Teva Pharmaceuticals, UCB Pharma S.A., Lavipharm, ITF Hellas, Medison Pharma-Argenx, Alexion-Astra Zeneca, and Takeda, and has received unrestricted research grants from Genesis Pharma, Merck, Novartis, and Lavipharm. NG has received honoraria, travel support, consultancy, and lecture fees from Biogen Idec, Novartis, TEVA, Bayer, Merck Serono, Genesis Pharma, Sanofi-Genzyme, Celgene, ELPEN, ROCHE, Alexion-Astra Zeneca, UCB, Medison, Lavipharn, and Pharmaserve, as well as research grants from Biogen Idec, Novartis, TEVA, Merck Serono, Genesis Pharma, Sanofi-Genzyme, and ROCHE.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Berrih-Aknin S, Frenkian-Cuvelier M, Eymard B. Diagnostic and clinical classification of autoimmune myasthenia gravis. J Autoimmun. (2014) 48–49:143–8. doi: 10.1016/j.jaut.2014.01.003
2. Oosterhuis HJ. The natural course of myasthenia gravis: a long term follow up study. J Neurol Neurosurg Psychiatry. (1989) 52:1121–7. doi: 10.1136/jnnp.52.10.1121
3. Hong YH, Kwon SB, Kim BJ, Kim BJ, Kim SH, Kim JK, et al. Prognosis of ocular myasthenia in Korea: a retrospective multicenter analysis of 202 patients. J Neurol Sci. (2008) 273:10–4. doi: 10.1016/j.jns.2008.05.023
4. Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. (2006) 116:2843–54. doi: 10.1172/JCI29894
5. Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. Lancet. (2001) 357:2122–8. doi: 10.1016/S0140-6736(00)05186-2
6. Rødgaard A, Nielsen FC, Djurup R, Somnier F, Gammeltoft S. Acetylcholine receptor antibody in myasthenia gravis: predominance of IgG subclasses 1 and 3. Clin Exp Immunol. (1987) 67:82–8.
7. Niks EH, van Leeuwen Y, Leite MI, Dekker FW, Wintzen AR, Wirtz PW, et al. Clinical fluctuations in MuSK myasthenia gravis are related to antigen-specific IgG4 instead of IgG1. J Neuroimmunol. (2008) 195:151–6. doi: 10.1016/j.jneuroim.2008.01.013
8. van der Zee JS, van Swieten P, Aalberse RC. Inhibition of complement activation by IgG4 antibodies. Clin Exp Immunol. (1986) 64:415–22.
9. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol. (2011) 69:418–22. doi: 10.1002/ana.22312
10. Howard JF Jr. Myasthenia gravis: the role of complement at the neuromuscular junction. Ann New York Acad Sci. (2018) 1412:113–28. doi: 10.1111/nyas.13522
11. Stascheit F, Chuquisana O, Keller CW, Ambrose PA, Hoffmann S, Gross CC, et al. Complement activation profiles in anti-acetylcholine receptor positive myasthenia gravis. Eur J Neurol. (2023) 30:1409–16. doi: 10.1111/ene.15730
12. Nagaoka A, Tsujino A, Shiraishi H, Kanamoto T, Shima T, Yoshimura S, et al. Motor end-plate analysis to diagnose immune-mediated myasthenia gravis in seronegative patients. J Neurological Sci. (2022) 443:120494. doi: 10.1016/j.jns.2022.120494
13. Albazli K, Kaminski HJ, Howard JF. Complement inhibitor therapy for myasthenia gravis. Front Immunol. (2020) 11:917/full. doi: 10.3389/fimmu.2020.00917/full
14. Ruff RL, Lennon VA. How myasthenia gravis alters the safety factor for neuromuscular transmission. J Neuroimmunol. (2008) 201:201–202:13–20.
15. Michailidou I, Willems JGP, Kooi EJ, van Eden C, Gold SM, Geurts JJG, et al. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann Neurol. (2015) 77:1007–26. doi: 10.1002/ana.24398
16. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
17. Tzartos SJ, Barkas T, Cung MT, Mamalaki A, Marraud M, Orlewski P, et al. Anatomy of the antigenic structure of a large membrane autoantigen, the muscle-type nicotinic acetylcholine receptor. Immunol Rev. (1998) 163:89–120. doi: 10.1111/j.1600-065X.1998.tb01190.x
18. Koneczny I, Herbst R. Myasthenia gravis: pathogenic effects of autoantibodies on neuromuscular architecture. Cells. (2019) 8:671. doi: 10.3390/cells8070671
19. Almon RR, Andrew CG, Appel SH. Serum globulin in myasthenia gravis: inhibition of alpha-bungarotoxin binding to acetylcholine receptors. Science. (1974) 186:55–7. doi: 10.1126/science.186.4158.55
20. Hara H, Hayashi K, Ohta K, Itoh N, Nishitani H, Ohta M. Detection and characterization of blocking-type anti-acetylcholine receptor antibodies in sera from patients with myasthenia gravis. Clin Chem. (1993) 39:2053–7. doi: 10.1093/clinchem/39.10.2053
21. Wintzen AR, Plomp JJ, Molenaar PC, van Dijk JG, van Kempen GT, Vos RM, et al. Acquired slow-channel syndrome: a form of myasthenia gravis with prolonged open time of the acetylcholine receptor channel. Ann Neurol. (1998) 44:657–64. doi: 10.1002/ana.410440412
22. Maselli RA, Richman DP, Wollmann RL. Inflammation at the neuromuscular junction in myasthenia gravis. Neurology. (1991) 41:1497–504. doi: 10.1212/WNL.41.9.1497
23. Tindall RS. Humoral immunity in myasthenia gravis: clinical correlations of anti-receptor antibody avidity and titer. Ann N Y Acad Sci. (1981) 377:316–31. doi: 10.1111/j.1749-6632.1981.tb33741.x
24. Vernet-der Garabedian B, Morel E, Bach JF. Heterogeneity of antibodies directed against the alpha-bungarotoxin binding site on human acetylcholine receptor and severity of myasthenia gravis. J Neuroimmunol. (1986) 12:65–74. doi: 10.1016/0165-5728(86)90098-6
25. Howard FM, Lennon VA, Finley J, Matsumoto J, Elveback LR. Clinical correlations of antibodies that bind, block, or modulate human acetylcholine receptors in myasthenia gravis. Ann N Y Acad Sci. (1987) 505:526–38. doi: 10.1111/j.1749-6632.1987.tb51321.x
26. Drachman DB, Adams RN, Josifek LF, Self SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med. (1982) 307:769–75. doi: 10.1056/NEJM198209233071301
27. Besinger UA, Toyka KV, Hömberg M, Heininger K, Hohlfeld R, Fateh-Moghadam A. Myasthenia gravis: long-term correlation of binding and bungarotoxin blocking antibodies against acetylcholine receptors with changes in disease severity. Neurology. (1983) 33:1316–21. doi: 10.1212/WNL.33.10.1316
28. Kang SY, Oh JH, Song SK, Lee JS, Choi JC, Kang JH. Both binding and blocking antibodies correlate with disease severity in myasthenia gravis. Neurol Sci. (2015) 36:1167–71. doi: 10.1007/s10072-015-2236-8
29. Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med. (1978) 298:1116–22. doi: 10.1056/NEJM197805182982004
30. Tzartos SJ, Sophianos D, Zimmerman K, Starzinski-Powitz A. Antigenic modulation of human myotube acetylcholine receptor by myasthenic sera. Serum titer determines receptor internalization rate. J Immunol. (1986) 136:3231–8. doi: 10.4049/jimmunol.136.9.3231
31. Fumagalli G, Engel AG, Lindstrom J. Ultrastructural aspects of acetylcholine receptor turnover at the normal end-plate and in autoimmune myasthenia gravis. J Neuropathol Exp Neurol. (1982) 41:567–79. doi: 10.1097/00005072-198211000-00001
32. Wilson S, Vincent A, Newsom-Davis J. Acetylcholine receptor turnover in mice with passively transferred myasthenia gravis. II. Receptor synthesis. J Neurol Neurosurg Psychiatry. (1983) 46:383–7. doi: 10.1136/jnnp.46.5.383
33. Merlie JP, Heinemann S, Einarson B, Lindstrom JM. Degradation of acetylcholine receptor in diaphragms of rats with experimental autoimmune myasthenia gravis. J Biol Chem. (1979) 254:6328–32. doi: 10.1016/S0021-9258(18)50366-5
34. Engel AG, Fumagalli G. Mechanisms of acetylcholine receptor loss from the neuromuscular junction. Ciba Found Symp. (1982) 90):197–224. doi: 10.1002/9780470720721.ch12
35. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
36. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci. (2013) 126:2903–13. doi: 10.1242/jcs.124388
37. Romi F, Kristoffersen EK, Aarli JA, Gilhus NE. The role of complement in myasthenia gravis: serological evidence of complement consumption in vivo. J Neuroimmunol. (2005) 158:191–4. doi: 10.1016/j.jneuroim.2004.08.002
38. Liu A, Lin H, Liu Y, Cao X, Wang X, Li Z. Correlation of C3 level with severity of generalized myasthenia gravis. Muscle Nerve. (2009) 40:801–8. doi: 10.1002/mus.21398
39. Iacomino N, Vanoli F, Frangiamore R, Ballardini M, Scandiffio L, Bortone F, et al. Complement activation profile in myasthenia gravis patients: perspectives for tailoring anti-complement therapy. Biomedicines. (2022) 10:1360. doi: 10.3390/biomedicines10061360
40. Barohn RJ, Brey RL. Soluble terminal complement components in human myasthenia gravis. Clin Neurol Neurosurg. (1993) 95:285–90. doi: 10.1016/0303-8467(93)90103-n
41. Kamolvarin N, Hemachudha T, Ongpipattanakul B, Phanthumchinda K, Sueblinvong T. Plasma C3c in immune-mediated neurological diseases: a preliminary report. Acta Neurol Scand. (1991) 83:382–7. doi: 10.1111/j.1600-0404.1991.tb03968.x
42. Iwasa K, Furukawa Y, Yoshikawa H, Yamada M, Ono K. CD59 expression in skeletal muscles and its role in myasthenia gravis. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200057. doi: 10.1212/NXI.0000000000200057
43. Tsujihata M, Yoshimura T, Satoh A, Kinoshita I, Matsuo H, Mori M, et al. Diagnostic significance of IgG, C3, and C9 at the limb muscle motor end-plate in minimal myasthenia gravis. Neurology. (1989) 39:1359–63. doi: 10.1212/wnl.39.10.1359
44. Engel AG, Arahata K. The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci. (1987) 505:326–32. doi: 10.1111/j.1749-6632.1987.tb51301.x
45. Sahashi K, Engel AG, Lambert EH, Howard FM. Ultrastructural localization of the terminal and lytic ninth complement component (C9) at the motor end-plate in myasthenia gravis. J Neuropathol Exp Neurol. (1980) 39:160–72. doi: 10.1097/00005072-198003000-00005
46. Fazekas A, Komoly S, Bózsik B, Szobor A. Myasthenia gravis: demonstration of membrane attack complex in muscle end-plates. Clin Neuropathol. (1986) 5:78–83.
47. Chamberlain JL, Huda S, Whittam DH, Matiello M, Morgan BP, Jacob A. Role of complement and potential of complement inhibitors in myasthenia gravis and neuromyelitis optica spectrum disorders: a brief review. J Neurol. (2021) 268:1643–64. doi: 10.1007/s00415-019-09498-4
48. Behan WM, Behan PO. Immune complexes in myasthenia gravis. J Neurol Neurosurg Psychiatry. (1979) 42:595–9. doi: 10.1136/jnnp.42.7.595
49. Rose N, Holdermann S, Callegari I, Kim H, Fruh I, Kappos L, et al. Receptor clustering and pathogenic complement activation in myasthenia gravis depend on synergy between antibodies with multiple subunit specificities. Acta Neuropathol. (2022) 144:1005–25. doi: 10.1007/s00401-022-02493-6
50. San PP, Jacob S. Role of complement in myasthenia gravis. Front Neurol. (2023) 14:1277596. doi: 10.3389/fneur.2023.1277596
51. Bahia El Idrissi N, Bosch S, Ramaglia V, Aronica E, Baas F, Troost D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J Neuroinflammation. (2016) 13:72. doi: 10.1186/s12974-016-0538-2
52. Presumey J, Bialas AR, Carroll MC. Complement system in neural synapse elimination in development and disease. Adv Immunol. (2017) 135:53–79. doi: 10.1016/bs.ai.2017.06.004
53. Chamberlain-Banoub J, Neal JW, Mizuno M, Harris CL, Morgan BP. Complement membrane attack is required for endplate damage and clinical disease in passive experimental myasthenia gravis in Lewis rats. Clin Exp Immunol. (2006) 146:278–86. doi: 10.1111/j.1365-2249.2006.03198.x
54. Morgan BP, Chamberlain-Banoub J, Neal JW, Song W, Mizuno M, Harris CL. The membrane attack pathway of complement drives pathology in passively induced experimental autoimmune myasthenia gravis in mice. Clin Exp Immunol. (2006) 146:294–302. doi: 10.1111/j.1365-2249.2006.03205.x
55. Zelek WM, Taylor PR, Morgan BP. Development and characterization of novel anti-C5 monoclonal antibodies capable of inhibiting complement in multiple species. Immunology. (2019) 157:283–95. doi: 10.1111/imm.13083
56. Soltys J, Kusner LL, Young A, Richmonds C, Hatala D, Gong B, et al. Novel complement inhibitor limits severity of experimentally myasthenia gravis. Ann neurology. (2009) 65:67. doi: 10.1002/ana.21536
57. Gytz Olesen H, Michailidou I, Zelek WM, Vreijling J, Ruizendaal P, de Klein F, et al. Development, characterization, and in vivo validation of a humanized C6 monoclonal antibody that inhibits the membrane attack complex. J Innate Immun. (2023) 15:16–36. doi: 10.1159/000524587
58. Zelek WM, Morgan BP. Monoclonal antibodies capable of inhibiting complement downstream of C5 in multiple species. Front Immunol. (2020) 11:612402. doi: 10.3389/fimmu.2020.612402
59. Christadoss P. C5 gene influences the development of murine myasthenia gravis. J Immunol. (1988) 140:2589–92.
60. Kaminski HJ, Kusner LL, Richmonds C, Medof ME, Lin F. Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp Neurol. (2006) 202:287–93. doi: 10.1016/j.expneurol.2006.06.003
61. Tüzün E, Scott BG, Goluszko E, Higgs S, Christadoss P. Genetic evidence for involvement of classical complement pathway in induction of experimental autoimmune myasthenia gravis. J Immunol. (2003) 171:3847–54. doi: 10.4049/jimmunol.171.7.3847
62. Sheridan D, Yu ZX, Zhang Y, Patel R, Sun F, Lasaro MA, et al. Design and preclinical characterization of ALXN1210: A novel anti-C5 antibody with extended duration of action. PloS One. (2018) 13:e0195909. doi: 10.1371/journal.pone.0195909
64. Freimer M, Govindarajan R, Khatri B, Levine T, Boroojerdi B, Delicha EM, et al. Safety, efficacy, and patient preference for subcutaneous zilucoplan in myasthenia gravis after switching from intravenous complement component 5 inhibitors: an interim analysis of a phase 3b study (P10-11.006). Neurology. (2024) 102:3200.
65. Vu T, Meisel A, Mantegazza R, Annane D, Katsuno M, Aguzzi R, et al. Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evid. (2022) 1:EVIDoa2100066. doi: 10.1056/EVIDoa2100066
66. Howard JF, Bresch S, Genge A, Hewamadduma C, Hinton J, Hussain Y, et al. Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Neurol. (2023) 22:395–406. doi: 10.1016/S1474-4422(23)00080-7
67. Yoshida K, Yukiyama Y, Miyamoto T. Interaction between immune complexes and C3b receptors on erythrocytes. Clin Immunol Immunopathol. (1986) 39:213–21. doi: 10.1016/0090-1229(86)90085-1
68. Miller GW, Nussenzweig V. A new complement function: solubilization of antigen-antibody aggregates. Proc Natl Acad Sci U S A. (1975) 72:418–22. doi: 10.1073/pnas.72.2.418
69. Schifferli JA, Woo P, Peters DK. Complement-mediated inhibition of immune precipitation. I. Role of the classical and alternative pathways. Clin Exp Immunol. (1982) 47:555–62.
70. Mohammed I, Holborow EJ, Fry L, Thompson BR, Hoffbrand AV, Stewart JS. Multiple immune complexes and hypocomplementaemia in dermatitis herpetiformis and coeliac disease. Lancet. (1976) 1:487–90. doi: 10.1016/s0140-6736(76)90787-x
71. Engel AG, Lambert EH, Howard FM. Immune complexes (IgG and C3) at the motor end-plate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlations. Mayo Clin Proc. (1977) 52:267–80.
72. Sahashi K, Engel AG, Linstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of immune complexes (IgG and C3) at the end-plate in experimental autoimmune myasthenia gravis. J Neuropathol Exp Neurol. (1978) 37:212–23. doi: 10.1097/00005072-197803000-00008
73. Spagni G, Gastaldi M, Businaro P, Chemkhi Z, Carrozza C, Mascagna G, et al. Comparison of fixed and live cell-based assay for the detection of AChR and muSK antibodies in myasthenia gravis. Neurol Neuroimmunol Neuroinflamm. (2023) 10:e200038. doi: 10.1212/NXI.0000000000200038
74. Hoffmann S, Harms L, Schuelke M, Rückert JC, Goebel HH, Stenzel W, et al. Complement deposition at the neuromuscular junction in seronegative myasthenia gravis. Acta Neuropathol. (2020) 139:1119–22. doi: 10.1007/s00401-020-02147-5
75. Nelke C, Schroeter CB, Stascheit F, Pawlitzki M, Regner-Nelke L, Huntemann N, et al. Eculizumab versus rituximab in generalised myasthenia gravis. J Neurol Neurosurg Psychiatry. (2022) 93:548–54. doi: 10.1136/jnnp-2021-328665
76. Abdulrazaq A, Smith R, Digala LP, Govindarajan R. Minimal manifestations with eculizumab therapy in a patient with refractory generalized seropositive myasthenia gravis. J Clin Neuromuscul Dis. (2022) 23:170–3. doi: 10.1097/CND.0000000000000388
77. Howard JF, Bresch S, Farmakidis C, Freimer M, Genge A, Hewamadduma C, et al. Long-term safety and efficacy of zilucoplan in patients with generalized myasthenia gravis: interim analysis of the RAISE-XT open-label extension study. Ther Adv Neurol Disord. (2024) 17:17562864241243186. doi: 10.1177/17562864241243186
78. Blackowicz M, Weiskopf E, Clark K, Grant J, Scheiner C. Long-term corticosteroid treatment patterns and steroid-sparing effects of approved treatments for generalized myasthenia gravis in the United States. Neurology. (2024) 103:S24–4. doi: 10.1212/01.wnl.0001051040.22359.0b
79. Katyal N, Govindarajan R, Goyal N, Muley S, Muppidi S. Ravulizumab use for acetylcholine receptor-positive generalized myasthenia gravis in clinical practice. Front Neurol. (2024) 15:1378080/full. doi: 10.3389/fneur.2024.1378080/full
80. Katyal N, Narula N, Govindarajan R. Clinical experience with eculizumab in treatment-refractory acetylcholine receptor antibody-positive generalized myasthenia gravis. J Neuromuscular Diseases. (2021) 8:287–94. doi: 10.3233/JND-200584
81. Durmus H, Cakar A, Parman Y. 510P Eculizumab versus rituximab for refractory anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: a single-center experience. Neuromuscular Disord. (2024) 43(Suppl 1):174. doi: 10.1016/j.nmd.2024.07.652
82. Channa H, Angela G, Miriam F, Isabel LM, Raphaëlle BL, Babak B, et al. Corticosteroid dose tapering in patients with generalised myasthenia gravis on zilucoplan: interim analysis of RAISE-XT. J Neurol Neurosurg Psychiatry. (2024) 95:A45–6. doi: 10.1136/jnnp-2024-ABN.148
83. Marchio L, Freimer M, Genge A, Hewamadduma C, Leite MI, Lejdstrom RB, et al. 2997 Corticosteroid dose tapering in patients with generalised myasthenia gravis on zilucoplan: interim analysis of RAISE-XT. BMJ Neurol Open. (2024) 6(Suppl 1):A1–A65. doi: 10.1136/bmjno-2024-ANZAN.35
84. Nicolle MW, Annane D, Meisel A, Vu T, Mantegazza R, Katsuno M, et al. P.053 Concomitant corticosteroid use in ravulizumab-treated adults with anti-AChR antibody-positive gMG: results from the CHAMPION MG open-label extension. Can J Neurological Sci. (2024) 51:S30–0.
85. Nowak RJ, Muppidi S, Beydoun SR, O’Brien FL, Yountz M, Howard JF. Concomitant immunosuppressive therapy use in eculizumab-treated adults with generalized myasthenia gravis during the REGAIN open-label extension study. Front Neurol. (2020) 11:556104. doi: 10.3389/fneur.2020.556104
86. Guptill JT, Juel VC, Massey JM, Anderson AC, Chopra M, Yi JS, et al. Effect of therapeutic plasma exchange on immunoglobulins in myasthenia gravis. Autoimmunity. (2016) 49:472–9. doi: 10.1080/08916934.2016.1214823
87. Ozawa Y, Uzawa A, Yasuda M, Kojima Y, Oda F, Himuro K, et al. Changes in serum complements and their regulators in generalized myasthenia gravis. Eur J Neurol. (2021) 28:314–22. doi: 10.1111/ene.14500
88. de Boer EC, Thielen AJ, Langereis JD, Kamp A, Brouwer MC, Oskam N, et al. The contribution of the alternative pathway in complement activation on cell surfaces depends on the strength of classical pathway initiation. Clin Transl Immunol. (2023) 12:e1436. doi: 10.1002/cti2.1436
89. Narayanaswami P, Sanders DB, Wolfe G, Benatar M, Cea G, Evoli A, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. (2021) 96:114–22. doi: 10.1212/WNL.0000000000011124
90. Kefalopoulou ZM, Veltsista D, Germeni A, Lykouras D, Tsiamaki E, Chroni E. Rituximab as a sole steroid-sparing agent in generalized myasthenia gravis: Long-term outcomes. Neurol Sci. (2024) 45:1233–42. doi: 10.1007/s10072-023-07082-3
91. Veltsista D, Kefalopoulou Z, Tzartos J, Chroni E. Autoantibody profile in myasthenia gravis patients with a refractory phase. Muscle Nerve. (2022) 65:607–11. doi: 10.1002/mus.27521
92. Hoffmann S, Meisel A, Brokamp K, Helmig L, Schülke-Gerstenfeld M, Rückert J, et al. 513P Deposition of MAC and IgG subclasses at the neuromuscular junction in LRP4+ myasthenia gravis. Neuromuscular Disord. (2024) 43:104441.646. doi: 10.1016/j.nmd.2024.07.655
93. Tang GQ, Tang Y, Dhamnaskar K, Hoarty MD, Vyasamneni R, Vadysirisack DD, et al. Zilucoplan, a macrocyclic peptide inhibitor of human complement component 5, uses a dual mode of action to prevent terminal complement pathway activation. Front Immunol. (2023) 14:1213920. doi: 10.3389/fimmu.2023.1213920
94. Nelke C, Schroeter CB, Stascheit F, Huntemann N, Pawlitzki M, Willison A, et al. Eculizumab treatment alters the proteometabolome beyond the inhibition of complement. JCI Insight. (2023) 8:e169135. doi: 10.1172/jci.insight.169135
95. Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther Adv Neurol Disord. (2018) 11:1756285617749134. doi: 10.1177/1756285617749134
96. Silvestri NJ, Wolfe GI. Treatment-refractory myasthenia gravis. J Clin Neuromuscular Disease. (2014) 15:167. doi: 10.1097/CND.0000000000000034
97. Schneider-Gold C, Hagenacker T, Melzer N, Ruck T. Understanding the burden of refractory myasthenia gravis. Ther Adv Neurol Disord. (2019) 12:1756286419832242. doi: 10.1177/1756286419832242
98. Engel-Nitz NM, Boscoe A, Wolbeck R, Johnson J, Silvestri NJ. Burden of illness in patients with treatment refractory myasthenia gravis. Muscle Nerve. (2018) 58:99–105. doi: 10.1002/mus.26114
99. Folaranmi T, Rubin L, Martin SW, Patel M, MacNeil JR, Centers for Disease Control (CDC). Use of serogroup B meningococcal vaccines in persons aged ≥10 years at increased risk for serogroup B meningococcal disease: recommendations of the advisory committee on immunization practices, 2015. MMWR Morb Mortal Wkly Rep. (2015) 64:608–12.
100. Yue YX, Gao X, Tang TP, Xie Y, Gu CK, Hao HJ, et al. Complement C3 polymorphism is associated with the susceptibility of myasthenia gravis in Chinese adult patients. J Neuroimmunol. (2021) 353:577487. doi: 10.1016/j.jneuroim.2021.577487
101. Leite MI, Jones M, Ströbel P, Marx A, Gold R, Niks E, et al. Myasthenia gravis thymus. Am J Pathol. (2007) 171:893–905. doi: 10.2353/ajpath.2007.070240
102. Nakano S, Engel AG. Myasthenia gravis: quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology. (1993) 43:1167–72. doi: 10.1212/WNL.43.6.1167
103. Kusner LL, Halperin JA, Kaminski HJ. Cell surface complement regulators moderate experimental myasthenia gravis pathology. Muscle Nerve. (2013) 47:33–40. doi: 10.1002/mus.23448
104. Baird PN, Islam FMA, Richardson AJ, Cain M, Hunt N, Guymer R. Analysis of the Y402H variant of the complement factor H gene in age-related macular degeneration. Invest Ophthalmol Vis Sci. (2006) 47:4194–8. doi: 10.1167/iovs.05-1285
105. Heurich B, El Idrissi NB, Donev RM, Petri S, Claus P, Neal J, et al. Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J Neuroimmunol. (2011) 235:104–9. doi: 10.1016/j.jneuroim.2011.03.011
106. Michailidou I, Vreijling J, Rumpf M, Loos M, Koopmans B, Vlek N, et al. The systemic inhibition of the terminal complement system reduces neuroinflammation but does not improve motor function in mouse models of CMT1A with overexpressed PMP22. Curr Res Neurobiol. (2023) 4:100077. doi: 10.1016/j.crneur.2023.100077
107. Howard JF Jr, Nowak RJ, Wolfe GI, Freimer ML, Vu TH, Hinton JL, et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurology. (2020) 77:582–92. doi: 10.1001/jamaneurol.2019.5125
108. Meisel A, Annane D, Vu T, Mantegazza R, Katsuno M, Aguzzi R, et al. Long-term efficacy and safety of ravulizumab in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: results from the phase 3 CHAMPION MG open-label extension. J Neurol. (2023) 270:3862–75. doi: 10.1007/s00415-023-11699-x
109. Vu T, Mantegazza R, Annane D, Katsuno M, Meisel A, Nicolle M, et al. Long-term efficacy and safety of ravulizumab, a long-acting terminal complement inhibitor, in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: final results from the phase 3 CHAMPION MG open-label extension (S15.010). Neurology. (2024) 102:2591. doi: 10.1212/WNL.0000000000204620
110. Brandsema JF, Ginsberg M, Hoshino H, Mimaki M, Nagata S, Rao VK, et al. Eculizumab in adolescent patients with refractory generalized myasthenia gravis: A phase 3, open-label, multicenter study. Pediatr Neurol. (2024) 156:198–207. doi: 10.1016/j.pediatrneurol.2024.04.020
111. Muppidi S, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. (2019) 60:14–24. doi: 10.1002/mus.26447
112. Howard JF, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. (2017) 16:976–86. doi: 10.1016/S1474-4422(17)30369-1
Keywords: myasthenia gravis, complement system, anti-acetylcholine receptor antibodies, membrane attack complex, therapy
Citation: Michailidou I, Patsiarika A, Kesidou E, Boziki MK, Parisis D, Bakirtzis C, Chroni E and Grigoriadis N (2025) The role of complement in the immunopathogenesis of acetylcholine receptor antibody-positive generalized myasthenia gravis: bystander or key player? Front. Immunol. 16:1526317. doi: 10.3389/fimmu.2025.1526317
Received: 11 November 2024; Accepted: 24 March 2025;
Published: 15 April 2025.
Edited by:
Paola Cavalcante, IRCCS Carlo Besta Neurological Institute Foundation, ItalyReviewed by:
Nicholas Sanderson, University of Basel, SwitzerlandNiklas Huntemann, University Hospital of Düsseldorf, Germany
Copyright © 2025 Michailidou, Patsiarika, Kesidou, Boziki, Parisis, Bakirtzis, Chroni and Grigoriadis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolaos Grigoriadis, bmdyaWdvcmlhZGlzQGF1dGguZ3I=