 Tanmaya Atre
Tanmaya Atre Gregor S. D. Reid
Gregor S. D. Reid- 1Michael Cuccione Childhood Cancer Research Program, BC Children’s Hospital Research Institute, Vancouver, BC, Canada
- 2Department of Pediatrics, University of British Columbia, Vancouver, BC, Canada
B-cell acute lymphoblastic leukemia (B-ALL) is the most common pediatric malignancy, accounting for 20-25% of all new cancer diagnoses in North American children each year. The leukemia arises, most commonly after a latency of 3–5 years, from a preleukemic B cell precursor population generated in utero. Despite the generally low immunogenicity of B-ALL cells, emerging evidence implicates T cell exhaustion - a state marked by sustained expression of inhibitory receptors and progressive functional decline - as a contributor to disease progression. Expression of inhibitory receptors is frequently detected on T cells from children with B-ALL at diagnosis and during therapy. As T cell exhaustion presents an actionable target for enhancing protective immune activity, in this review we summarize evidence from both clinical and pre-clinical settings for T cell exhaustion during pediatric B-ALL progression and discuss the opportunities and challenges to incorporating immune checkpoint blockade into pediatric B-ALL therapy regimens.
1 Introduction
T cell activation is initiated when T cell receptor (TCR) engagement with peptide-MHC complexes on antigen-presenting cells (APC) (signal 1) is accompanied by co-stimulatory signals (signal 2) and cytokine support (signal 3). This coordinated signaling cascade drives lymphocyte activation, proliferation, and differentiation that mediates antigen clearance (1, 2). Typically, immune checkpoints (IC) then regulate the immune response by engaging with inhibitory receptors on activated T cells (3, 4). The timing and balance of stimulatory and inhibitory signals influences the quality and duration of T cell responses (5). However, in cases where antigen clearance is not achieved, prolonged T cell stimulation can lead to an altered differentiation state, known as ‘exhaustion’ (6). T cell exhaustion is characterized by sustained upregulation and co-expression of multiple inhibitory IC receptors, along with transcriptional (7), metabolic (8), and epigenetic modifications (9), resulting in a progressive loss of effector function in antigen-specific T cells (10). The accumulation of exhausted T cells is observed in many cancers (11).
The genomic alterations that drive tumor formation can lead to the generation of immunogenic neoantigens (12). Through iterations of the tumor-immune cycle, recognition of these altered-self epitopes by the adaptive immune system can initiate and maintain immune responses that exert ongoing immunosurveillance (13, 14). However, if these responses fail to eliminate the nascent tumor, exposure to an increasing burden of neoantigens can lead to T cell exhaustion and downregulation of protective immune activity, leading to tumor progression (15, 16). The development of immune checkpoint blockade (ICB) therapy to overcome inhibitory signaling pathways and re-establish T cell-mediated anti-tumor activity has markedly transformed the therapeutic landscape for several human malignancies (17–19). While outcomes achieved with ICB therapy have been unprecedented, there is a growing recognition from adult cancer studies that properties of the patient’s immune system can significantly influence the outcome of these therapies (20, 21). Whether these same immune variables will modulate responses to ICB in pediatric cancer patients remains largely unknown.
Pediatric cancers generally exhibit a lower tumor mutation burden (TMB) than adult cancers (22–24), likely resulting in reduced neoantigen-driven immune stimulation and infiltration. Notably, ICB treatment responses are strongest for those rare pediatric cancers that possess TMB approaching or exceeding those of adult tumors (25, 26). Furthermore, many pediatric malignancies, such as B cell acute lymphoblastic leukemia (B-ALL), neuroblastoma, Wilm’s tumor, and medulloblastoma, have a prenatal origin and manifest in early childhood following a relatively short latency period (27). Fetal and neonatal development are times of extensive immune tolerance induction (28, 29), suggesting that the early genetic alterations driving pediatric cancers may evade immune detection. Given these potential constraints on the effectiveness of checkpoint inhibition in the setting of childhood cancer, this review examines the evidence supporting the application of ICB therapy to improve outcomes for children with B-ALL, the most common of the prenatally initiated pediatric cancers (Figure 1).
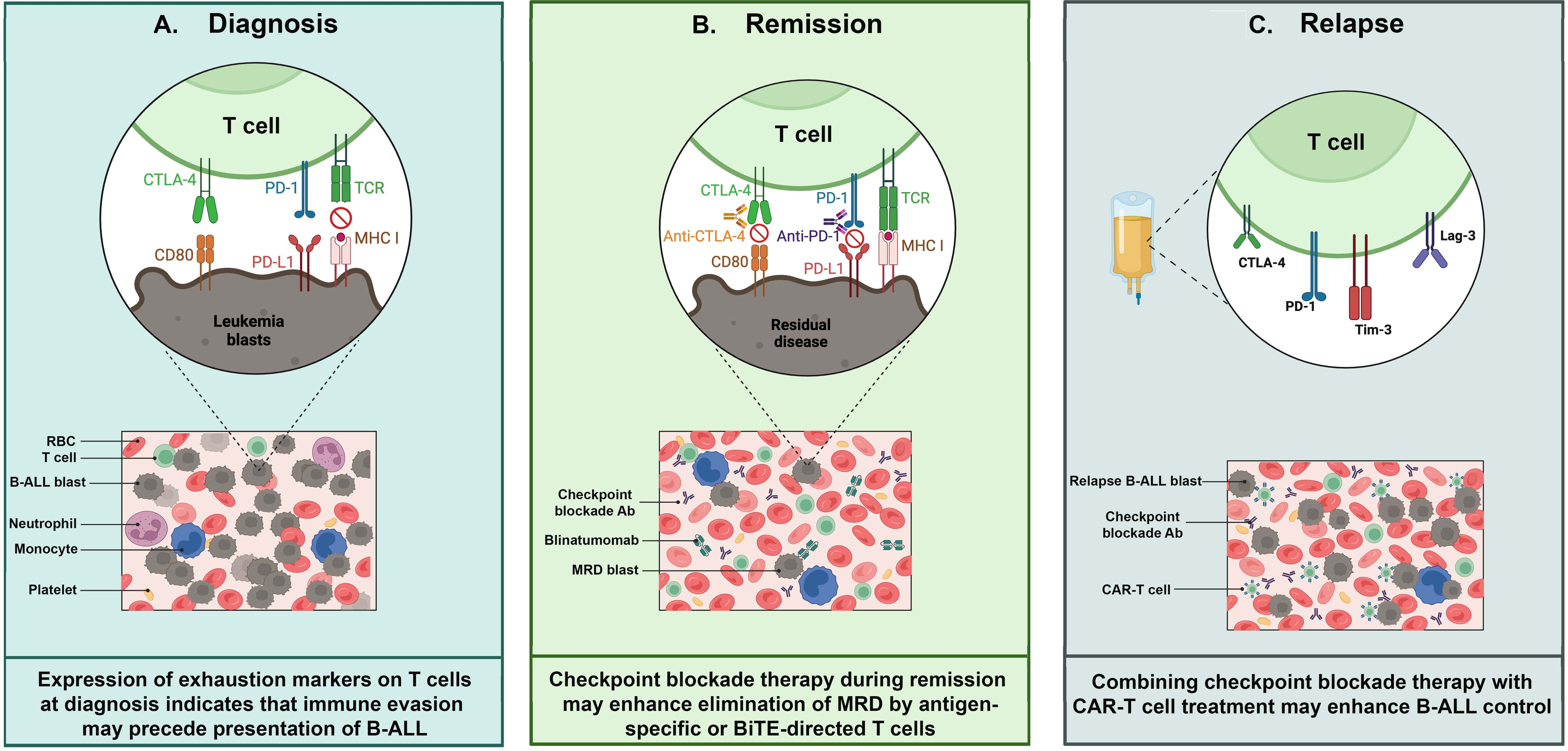
Figure 1. T cell exhaustion in pediatric B-cell acute lymphoblastic leukemia (B-ALL). Exhausted T cells can be detected and targeted at various timepoints during B-ALL progression: (A) Diagnosis: Exhausted T cells in bone marrow aspirates from patients at diagnosis express multiple inhibitory receptors such as PD-1,TIM-3 and CTLA-4, which impair T cell function. These receptors interact with their respective ligands expressed on leukemia blasts, contributing to immune evasion. (B) Remission: When leukemia burden is reduced to minimal residual disease (MRD) levels by induction chemoptherapy, there is an opportunity to restore anti-leukemia immunity. At this stage, immune checkpoint inhibitors - e.g. anti-PD-1 and anti-CTLA-4 antibodies (Abs) - may reinvigorate T cells, especially when combined with approved immunotherapies such as CD3/CD19 bispecific T cell engagers. (C) Relapse: CAR-T cells show an exhausted phenotype, characterized by high expression of inhibitory receptors including PD-1, TIM-3, LAG-3, and CTLA-4, at various times during production and application. ICB may enhance CAR-T functionality, but the optimal timing to achieve this remains to be determined. Abbreviations – Minimal residual disease (MRD); Bispecific T cell engager (BiTE); Chimeric antigen receptor (CAR); red blood cell (RBC); antibody (Ab).
2 B-ALL immunogenicity
B-ALL comprises a heterogeneous group of malignancies characterized by the uncontrolled proliferation of B cell progenitors within the bone marrow (30). It is the most common pediatric malignancy, accounting for almost a quarter of childhood cancer cases worldwide (31, 32). While the incidence of B-ALL peaks between 3 and 5 years of age, the initiating event usually occurs in utero: more than 70% of patients have detectable leukemia-initiating cytogenetic abnormalities at birth (33–37). Full transformation to overt leukemia requires additional genetic lesions to occur within the early arising preleukemic population (38). Treatment advancements have significantly improved outcomes for children with B-ALL, with the current 5-year event-free survival rate exceeding 85% in North America (39, 40). Nevertheless, around 20% of patients will remain unresponsive to initial therapy or experience relapse (41, 42). For these patients, targeted immune therapies, such as chimeric antigen receptor (CAR) T cells and bi-specific T cell engagers (BiTE), offer considerable hope (43, 44). However, achieving more durable responses with these novel treatments has emerged as a clinical imperative (45, 46).
The mutation burden in pediatric B-ALL is relatively low, ranging from 0.15 to 0.66 single nucleotide variants (SNVs) per megabase (22), a frequency predicted to result in relatively few neoantigens (47, 48). Additionally, B-ALL blasts often lack or have limited expression of essential co-stimulatory molecules, including CD80 and CD86 (49, 50). The combination of weak neoantigen expression and poor co-stimulation is predicted to favor induction of T cell anergy over activation. Consistent with this prediction, early studies reported that antigen presentation by B-ALL blasts often induces T cell deletion or anergy (51–54). The low immunogenicity of B-ALL, however, is not absolute, as ALL-specific CD4+ and CD8+ T cell responses can be generated under experimental conditions, indicating that the leukmeic blasts can present immunogenic epitopes (55, 56). Subsequent research demonstrated that 88% of B-ALL cases contain at least one predicted neoepitope (48). Further, leukemia antigen-specific T cells can be generated from pediatric B-ALL patients during maintenance therapy, and these T cells exhibited cytotoxicity against autologous leukemia cells (57). High-throughput studies further support this finding, showing that CD8+ T cells from patients with B-ALL can recognize and respond to neoantigens derived from fusion proteins, such as ETV6::RUNX1 (58)
Mouse models of B-ALL further support the feasibility of T cell-mediated control over leukemia, although these studies rely primarily on leukemia transplant approaches that do not capture the potential impact of neonatal tolerance. In a recent study using a murine Arf−/− Bcr-Abl1 mouse model, BCR::ABL1-specific CD4+ memory T cells played a protective role, with T cell depletion drastically increasing leukemia outgrowth after dasatinib or cytotoxic chemotherapy (59). Similarly, protective T cell responses are generated by toll-like receptor-mediated immune modulation in the Eμ-ret model of hyperdiploid B-ALL (60). In addition, evidence that tolerance mechanisms affect the durability of T cell-mediated protection against Eμ-ret B-ALL outgrowth has been reported, suggesting that secondary, less immunogenic antigens might contribute to anti-leukemia T cell activity (61). However, the contribution of such antigens to ongoing immunosurveillance prior to disease presentation remains unknown.
Collectively, findings support a model of B-ALL progression in which an in utero-generated preleukemic cell population persists due to the undermining of effective immunosurveillance against the driver mutation by early-life tolerance mechanisms. The occurrence of secondary genomic lesions in preleukemic cells drives transformation but may also induce neoantigen-targeted immune responses against the emerging leukemia cells. According to this model, if T cell exhaustion is a pathway that enables immune escape and the emergence of overt leukemia, B-ALL blasts and patient T cells should be characterized by the expression of inhibitory IC molecules.
3 Clinical evidence of T cell exhaustion in pediatric B-ALL
The expression of exhaustion markers at various timepoints during pediatric B-ALL progression has been reported, summarized in Table 1. At diagnosis, there is an upregulation of IC receptors on patient T cells and their corresponding ligands on B-ALL blasts (68, 69, 71–73). Elevated expression of PD-1 and CTLA-4 have been observed on both αβ+ and γδ+ T cells in newly diagnosed ALL patients prior to chemotherapy (62). Notably, higher CTLA-4 levels on γδ+ T cells and CD86 expression on blasts has been linked to poor prognosis in high-risk B-ALL. Similarly, analysis of the bone marrow (BM) immune microenvironment in B-ALL showed increased expression of TIGIT, LAG3, and PD-1 on CD4+ and CD8+ T cells compared to healthy controls (63). In addition, several studies have reported upregulation of multiple immune checkpoint molecules at diagnosis across different compartments, including serum, peripheral blood mononuclear cells (PBMCs), and bone marrow mononuclear cells (BMMCs). These findings include elevated PD-L1 levels in the serum of children with ALL at diagnosis (67); upregulation of inhibitory molecules such as TIM-3, NR4A1, and BATF on CD8+ T cells in bone marrow aspirates from children with PAX5 mutation (64), and significantly higher TIM-3 mRNA expression in peripheral blood and BM of ALL patients (1.7- and 5-fold higher, respectively, compared to controls) (66). A bioinformatic analysis further suggested that increased expression of CD39, CTLA-4, TNFR2, TIGIT, and TIM-3 on Tregs and CD8+ T cells may contribute to disease progression (65).
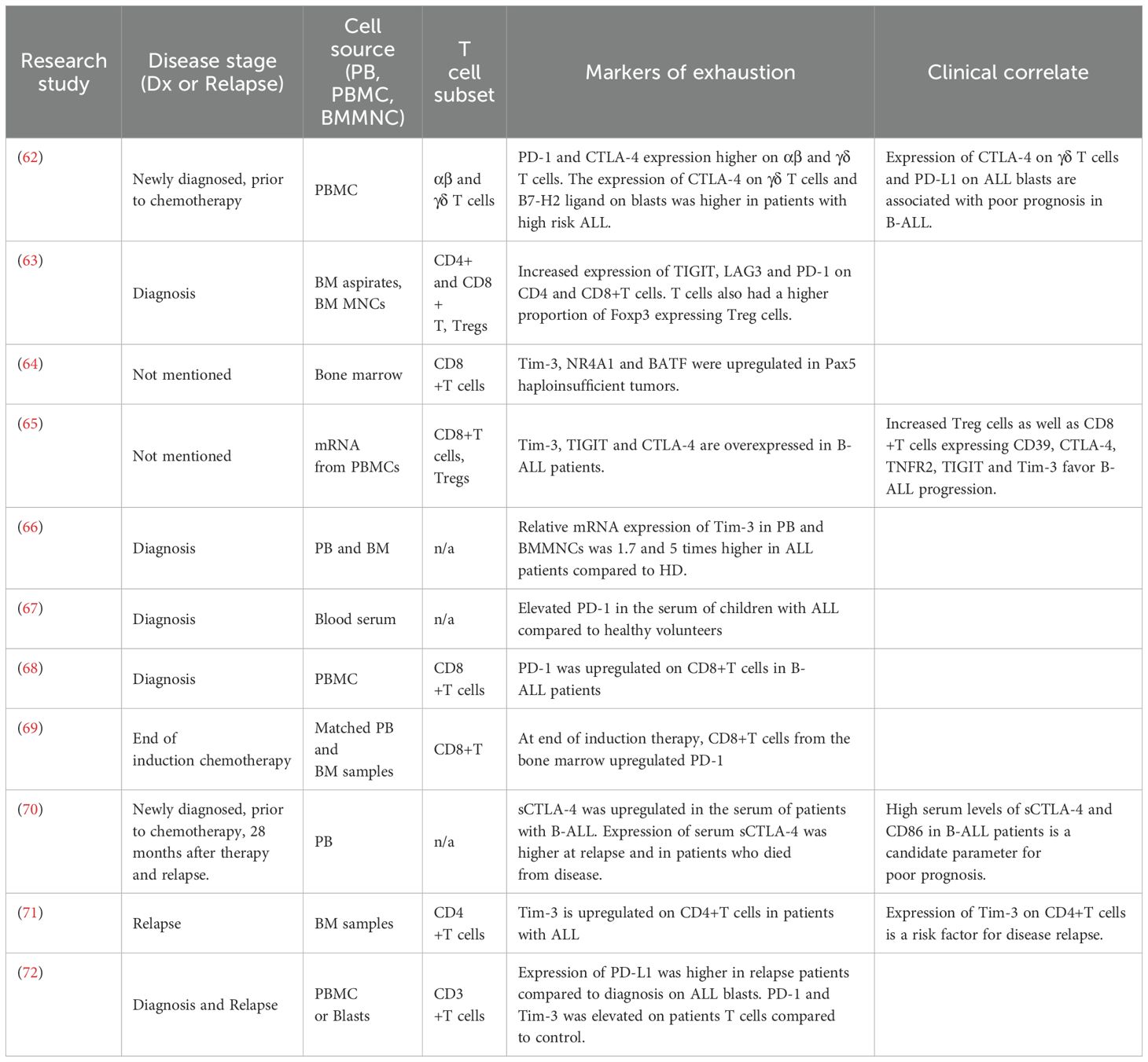
Table 1. Overview of exhaustion markers on T cells from pediatric B-ALL patients at diagnosis, during treatment, and at relapse.
Elevated IC molecule expression continue to be detected after the initiation of B-ALL treatment. Evaluation of matched PB and BM samples from B-ALL patients post induction therapy indicated higher PD-1 expression on BM T cells, with PD-1 and LAG3 levels further upregulated on CD4+ and CD8+ T cells following ex vivo expansion (69). High circulating soluble CTLA-4 (sCTLA-4) levels have been detected in 70% of pediatric B-ALL patients with active disease (74), with elevated sCTLA-4 and CD86 levels associated with poor prognosis (70). In relapsed B-ALL following allogeneic hematopoietic stem cell transplantation (allo-HSCT), increased co-expression of PD-1 and TIM-3 on CD4+ and CD8+ T cells correlated with reduced proliferative capacity, cytokine production, and cytotoxic potential (73). Notably, the frequency of PD-1+TIM-3+ CD8+ T cells was lower in patients who achieved a complete remission. Lastly, while CD8+T cells are undoubtedly important mediators of anti-tumor immunity (75), recent studies suggest that exhausted CD4+ T cells may predict risk of relapse. In a study by Blaeschke et al., B-ALL was associated with a late-stage CD4+ phenotype, with high TIM-3 expression on BM CD4+ T cells correlating with a higher risk of relapse (71). Collectively, these findings suggest a functional relevance of IC expression on both CD4+ and CD8+ T cells during B-ALL development and relapse.
4 Pre-clinical support for functional T cell exhaustion in pediatric B-ALL
Preclinical studies using murine models of B-ALL have shed light on the role of ICs in disease progression. While exhausted CD8+ T cells are characterized by distinct functional, epigenetic and transcriptional features, many of these characteristics remain poorly defined for exhausted CD4+T cells. Recent work using primary patient samples and a murine model of Ph+ B-ALL have shown that phenotypic exhaustion predominantly occurs within a unique subset of CD4+ T cells (76). This subset, defined by its transcriptomic profile, displays hybrid functionality, exhibiting both cytotoxic and helper functions. In a syngeneic murine model of TCF3::PBX1 leukemia, an upregulation of PD-1, TIM-3, and LAG3 on CD4+ and CD8+ T cells was observed in the presence of leukemia (77). The resulting leukemia-induced T cell dysfunction was independent of TCR signaling and led to the generation of suboptimal autologous CAR-T cells, which were less effective in clearing leukemia blasts compared to CAR-T cells generated from naïve mice.
Further research has indicated that inhibiting myeloid–epithelial–reproductive tyrosine kinase (MERTK), a gene linked to the induction of an antiapoptotic gene expression signature in B-ALL cells, decreased PD-1 expression on both CD4+ and CD8+ T cells, leading to enhanced T cell activation and anti-ALL immune activity (78). Similarly, IL-12-mediated leukemia clearance in a syngeneic murine model of B-ALL was dependent on T cell activity. T cells from mice that failed to achieve leukemia clearance exhibited expression of exhaustion-associated genes, including LAG3 and TIGIT (79). Lastly, in a syngeneic model of Eμ−ret B-ALL, mice that failed to control non−immunogenic wild−type ALL blasts exhibited an upregulation of PD−1 and CTLA−4 on both CD4+ and CD8+ splenic T cells, whereas mice receiving B-ALL cells that express GFP/luciferase (which act as a model antigens) did not (80). This elevated checkpoint expression in non−responders was accompanied by higher CD80 on conventional dendritic cells and increased PD−L1 on plasmacytoid DCs. In contrast, T cells from leukemia−responsive mice downregulated these inhibitory receptors, allowing effective DC maturation, IL−12 production, and IFN−γ release by naïve T cells against otherwise non−immunogenic leukemia antigens. These results suggest that PD−1 and CTLA−4 inhibit epitope spreading and support combined checkpoint blockade strategies to broaden anti−ALL immunity.
In summary, increasing evidence from both clinical and preclinical studies indicates that B-ALL progression is accompanied by impaired T cell function, characterized by the overexpression of multiple IC molecules. This finding has significant implications for the application of immune therapies to children with B-ALL.
5 Immune checkpoint blockade in B-ALL
The role for immune checkpoint pathways in cancer progression, and rationale for ICB therapy, was first identified when antibodies targeting CTLA-4 demonstrated efficacy in reducing melanoma tumor size in mice (81). This early discovery led to the development of ipilimumab, an anti-CTLA-4 monoclonal antibody (mAb) that became the first therapy to improve survival in patients with metastatic melanoma (82). PD-1 then emerged as another critical immune checkpoint. Anti-PD-1/PD-L1 therapies, such as pembrolizumab and nivolumab, showed promising efficacy in controlling tumor progression, leading to their approval in 2014 for metastatic melanoma (83, 84). In general, ICB therapy with blocking mAbs has shown most success in solid tumors with high mutation loads, where it can achieve durable clinical responses (85). However, clinical efficacy is largely confined to a subset of patients (86). In the years since their approval, hundreds of clinical trials have explored the impact of ICB mAbs across diverse cancers, with varying degrees of success. In hematologic malignancies like B-ALL, the application of immune checkpoint inhibitors remains under investigation (87–89).
Given the low TMB and minimal neoantigen-specific T cell generation in pediatric B-ALL, ICB alone was predicted to be insufficient to achieve meaningful therapeutic activity. However, the recent findings described above have challenged this notion, prompting a re-examination of anti-ALL T cell activity. Preclinical B-ALL models show early evidence that ICB, alone or combined, can induce remissions. For instance, CTLA-4 blockade in Eμ-ret mice, which are likely tolerized to antigens derived from the leukemia-driving transgene, enhanced immune control and extended survival by 50% (61). In a BCR::ABL+ ALL mouse model, PD-L1 blockade led to clonal expansion of leukemia-specific CD4+T cells with a helper/cytotoxic phenotype, while reducing exhaustion marker expression (76). Additionally, combining PD-L1 mAb with nilotinib, a tyrosine kinase inhibitor (TKI), significantly improved survival of BCR::ABL+ leukemia-bearing mice. Studies with dasatinib, another TKI, in combination with anti-PD-1 eliminated BCR::ABL+ ALL cells, prolonged survival, and induced anti-leukemic immune memory upon rechallenge in syngeneic mice (90). These intriguing findings suggest that the administration of ICB during standard therapy is worthy of thorough preclinical investigation. One key question is whether targeting established or emerging exhaustion pathways (during immune reconstitution following induction chemotherapy) can enhance immune-mediated clearance of residual disease (91). Notably, a Phase 2 study of pembrolizumab for treating minimal residual disease (MRD) in adults with B-ALL found limited clinical benefit from anti-PD-1 therapy in this setting (92). However, given the distinct differences in immune reconstitution and treatment responses between adults and children, investigating this approach in pediatric B-ALL remains warranted. Finally, although B-ALL is characterized by low TMB, levels vary across different B-ALL subtypes. For instance, KMT2A-rearranged (KMT2A-r) ALL typically exhibits a low TMB, whereas iAMP21 ALL tends to have a comparatively higher TMB (93–95). Current data are insufficient to establish a clear association between TMB variations across B-ALL subtypes and their responsiveness to ICB therapy.
6 Integrating immune checkpoint blockade with approved immunotherapies
With the current shortage of empirical evidence supporting single-agent ICB use during pediatric B-ALL treatment, ongoing efforts are focused on exploring ICB in combination with other immunotherapies. Redirected T cell therapies, such as CAR-T and BiTE, facilitate cytotoxicity by directing autologous T cells toward leukemia cell surface antigens (96). Both CARs and BiTEs operate independently of the TCR and MHC molecules and rely on single-chain variable fragment (scFv) to recognize tumor-associated antigens (97). However, similar to peptide-specific T cells, continuous exposure of redirected T cells to tumor antigen can lead to T cell exhaustion (98), posing a significant challenge for both therapies. Emerging evidence supports that integration of ICB into these therapy protocols may achieve superior outcomes (99, 100).
6.1 ICB with CD3/CD19 BiTE
Blinatumomab, a CD3/CD19 BiTE, has notably improved outcomes for pediatric patients with relapsed or refractory (R/R) B-ALL and is now a frontline treatment option (101, 102). As of 2025, it remains the only immunotherapy approved for pediatric B-ALL patients who are minimal residual disease (MRD) positive, showing promise in low burden early-stage disease in combination with standard chemotherapy (103). Despite this, patient responses to blinatumomab can vary considerably. Some patients show little to no response to the treatment (104, 105), while others experience a loss of response after multiple cycles (104). Blinatumomab has been shown to induce an upregulation of inhibitory receptors on T cells and their corresponding ligands on B-ALL blasts, such as PD-1 and PD-L1, respectively. A comparison of IC expression at diagnosis and relapse showed higher PD-L1 expression on blasts obtained at relapse or from patients refractory to the anti-CD19 BiTE blinatumomab. Additionally, relapsed patients who failed to respond to blinatumomab exhibited increased expression of PD-1 and TIM-3 on T cells, alongside elevated PD-L1 on B-ALL blasts (71, 72). Recent data from a study of 11 pediatric patients treated with a continuous 28-day infusion of blinatumomab revealed progressive acquisition of T-cell exhaustion features (106). T cells exhibited phenotypic and transcriptomic upregulation of inhibitory receptors including PD-1, TIM-3, and TIGIT, a shift toward CD8+ T effector memory cells re-expressing CD45RA (TEMRA) subsets, and reduced cytotoxic and proliferative capacity. In a patient-derived xenograft (PDX) model of B-ALL using umbilical cord blood-reconstituted immunodeficient mice, treatment with either blinatumomab or pembrolizumab alone led to partial disease protection (107). Notably, the combination of both treatments resulted in a lower incidence of MRD and improved leukemia-free survival. Lastly, treatment of a single refractory ALL patient with a combination of blinatumomab and anti-PD-1 antibody induced anti-leukemic responses, reducing the disease burden from 45% to 1% (72).
6.2 ICB with CAR-T
CD19-directed CAR-T therapy has achieved over 70% complete remission rates in pediatric R/R B-ALL patients (108–111). However, 30–50% of these children experience relapse within the first year (112–114). Relapse in B−ALL patients following CAR−T therapy is most often driven by loss of leukemia-associated antigen (e.g., CD19 or CD22) or limited CAR−T cell persistence and proliferative capacity due to T cell exhaustion (115). In a syngeneic B−ALL mouse model, DeGolier et al. demonstrated that CD8+ CAR−T cells inherit epigenetic and transcriptional programs from their prior TCR antigen exposure, which dictate their exhaustion susceptibility (116). Although memory−derived CAR−T cells mount superior initial effector responses, they rapidly develop exhaustion phenotypes under low−antigen or low−dose conditions, which are marked by an increased expression of PD-1, TIM-3, Tox and CD39. In contrast, naive−derived CAR−T cells sustain proliferation and resist dysfunction. Complementing these findings, Zebley et al. analyzed CD8+ CD19-CAR T cells from pediatric B-ALL patients and found that ongoing antigen stimulation drives exhaustion-associated DNA methylation reprogramming (117). This includes demethylation at genes such as CX3CR1, BATF, and TOX, alongside repression of memory-associated genes like TCF7 and LEF1. This collectively promotes a progenitor-exhausted phenotype which limits CAR-T cell persistence. In a related study, it was shown that transiently interrupting CAR signaling—using either a drug-regulatable system or dasatinib—can restore functionality in exhausted CAR-T cells through epigenetic remodeling (118). This brief period of rest reprograms CAR-T cells toward a memory-like state, enhancing their cytokine production, proliferative potential, and antitumor efficacy.
As the limited long-term efficacy of CAR-T therapy has been linked to CAR-T cell exhaustion, ICB has been explored as a strategy to enhance persistence (119). In a small cohort study involving 14 pediatric B-ALL patients, the addition of anti-PD-1 mAbs to CD19 CAR-T cell therapy enhanced CAR-T cell persistence. Remarkably, three out of six patients treated with the PD-1 inhibitor at the time of early B cell recovery re-established B cell aplasia, signifying restoration of CAR-T cell activity (120). Other potential strategies for ICB integration with CAR-T therapy include engineering CAR-T cells to secrete soluble PD-1-blocking scFv. This approach has shown anti-leukemic efficacy comparable to combination therapy with CAR-T cells and anti-PD-1 antibodies (121), and has demonstrated improved eradication of CD19+PD-L1+ leukemia cells (122). Another approach involves the development of TIM-3-CD28 fusion proteins, which convert inhibitory TIM-3 signaling into an activating, immunostimulatory signal (123). However, the complex outcomes of ICB require further investigation, as increased CAR-T cell activation can reduce cell survival and exacerbate exhaustion by upregulating TIGIT (124).
A growing body of research emphasizes the significance of patient-derived T cell quality on CAR-T cell performance. Evaluating both the apheresis starting material and the post-infusion CAR-T product is crucial for identifying correlates of CAR-T cell persistence (125). Notably, increased expression of exhaustion markers, such as PD-1 and LAG-3, on CD8+ T cells within the apheresis material has been associated with reduced CAR-T cell efficacy (126). Conversely, higher levels of these markers on CD4+ CAR-T cells during peak expansion in recipients - triggered by cognate antigen-mediated activation - have been associated with prolonged event-free survival (127). These observations highlight that timing of IC assessments is crucial, as elevated expression at different times are associated with very different outcomes. A recent study examined the apheresis materials from pediatric and young adult patients with R/R B-ALL undergoing CD22 CAR-T cell therapy and found that T cells from non-responders had a more differentiated phenotype and overexpressed exhaustion-associated genes (128). These differences in the apheresis material could predict response to therapy, with the exhausted phenotype being a key predictor of poor outcomes. The study indicates that early identification of exhaustion markers in apheresis material could guide targeted manufacturing adjustments to optimize CAR-T cell efficacy.
6.3 Challenges to integration
Despite the promise of ICB, several translational and clinical barriers must be overcome if it is to become an established treatment in the pediatric setting. First, ICB-associated immune-related adverse events (irAEs), such as colitis, endocrinopathies, and hepatitis, represent a significant risk in children, whose immune and endocrine systems are still developing (129–131). The long-term effects of checkpoint inhibition on immune and organ development in children remain unknown. Second, the optimal timing and patient selection for ICB remain open questions. Introducing checkpoint inhibitors during immune reconstitution, such as post-chemotherapy or allo-HSCT, may either enhance anti-leukemic responses or disrupt essential tolerance pathways. Third, empirical evidence supporting the clinical use of ICB for pediatric B-ALL is limited. Most existing data are derived from adult trials or preclinical models, and few clinical trials that include pediatric B-ALL have been initiated (Table 2). These challenges highlight the importance of careful patient stratification, pediatric-specific trial design, and long-term monitoring if the field is to move toward clinical translation of ICB in B-ALL. These goals are made even more challenging to achieve by the diverstiy of clinical trials available for this patient population.
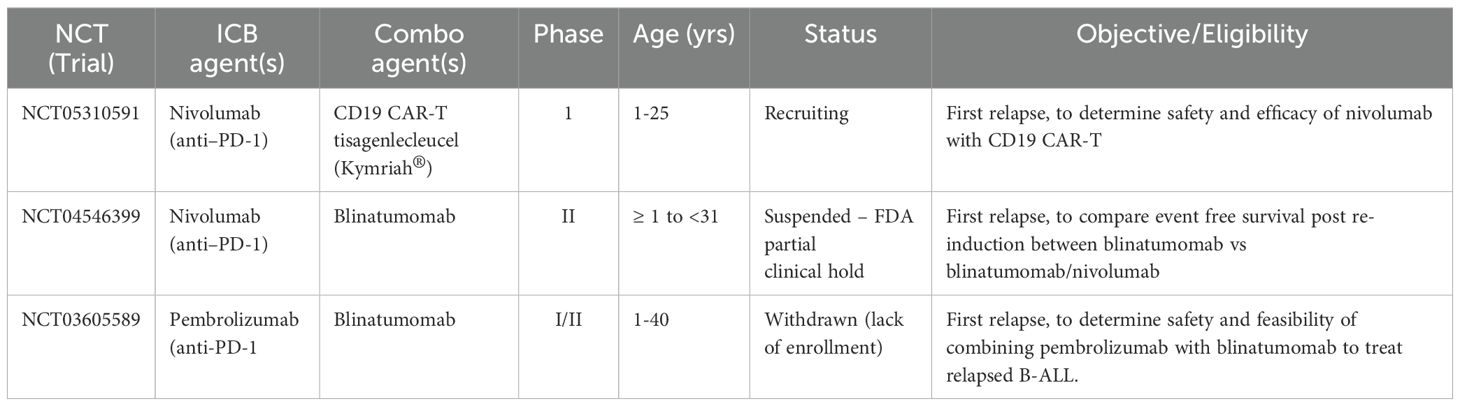
Table 2. Pediatric trials of ICB in B-ALL.
7 Future considerations
Over the past decade, it has become clear that T cell-based immunotherapies can overcome the hurdles of low immunogenicity and tolerance to achieve significant therapeutic activity in children with B-ALL. However, T cell exhaustion has emerged as a characteristic feature of B-ALL progression. This finding identifies ICB therapy as an important consideration for improved treatment of children with progressive disease. Several strategies to integrate ICB into current treatment regimens merit further investigation, including during immune reconstitution following chemotherapy and in combination with redirected T cell therapies. Considerable work will be needed to establish ICB as a central component of B-ALL therapy, but the continued application of both preclinical models and clinical studies to unveil underlying biology, identify the variables determining outcome, and optimize protocols could quickly set new immunotherapeutic standards that ultimately improving long-term outcomes for pediatric patients with B-ALL.
Author contributions
TA: Conceptualization, Funding acquisition, Visualization, Writing – original draft, Writing – review & editing. GR: Conceptualization, Funding acquisition, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. TA is the recipient of a Graduate Scholarship from the Michael Cuccione Foundation.
Acknowledgments
We would like to thank Dr. Ali Farrokhi for critically reviewing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hwang J-R, Byeon Y, Kim D, and Park S-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med. (2020) 52:750–61. doi: 10.1038/s12276-020-0435-8
2. Shah K, Al-Haidari A, Sun J, and Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther. (2021) 6:412. doi: 10.1038/s41392-021-00823-w
3. Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki I, et al. PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front Immunol. (2019) 10:630. doi: 10.3389/fimmu.2019.00630
4. Rieder SA, Wang J, White N, Qadri A, Menard C, Stephens G, et al. B7-H7 (HHLA2) inhibits T-cell activation and proliferation in the presence of TCR and CD28 signaling. Cell Mol Immunol. (2021) 18:1503–11. doi: 10.1038/s41423-020-0361-7
5. Chen L and Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
7. Seo W, Jerin C, and Nishikawa H. Transcriptional regulatory network for the establishment of CD8+ T cell exhaustion. Exp Mol Med. (2021) 53:202–9. doi: 10.1038/s12276-021-00568-0
8. Wu H, Campillo Prados M, and Vaeth M. Metabolic regulation of T cell exhaustion. Immune Discov. (2025) 1:10005–5. doi: 10.70322/immune.2025.10005
9. Belk JA, Daniel B, and Satpathy AT. Epigenetic regulation of T cell exhaustion. Nat Immunol. (2022) 23:848–60. doi: 10.1038/s41590-022-01224-z
10. Wherry EJ and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
11. Chow A, Perica K, Klebanoff CA, and Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol. (2022) 19:775–90. doi: 10.1038/s41571-022-00689-z
12. Mardis ER. Neoantigens and genome instability: impact on immunogenomic phenotypes and immunotherapy response. Genome Med. (2019) 11:71. doi: 10.1186/s13073-019-0684-0
13. Mellman I, Chen DS, Powles T, and Turley SJ. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity. (2023) 56:2188–205. doi: 10.1016/j.immuni.2023.09.011
14. Roerden M and Spranger S. Cancer immune evasion, immunoediting and intratumour heterogeneity. Nat Rev Immunol. (2025) 25:353–69. doi: 10.1038/s41577-024-01111-8
15. Galassi C, Chan TA, Vitale I, and Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. (2024) 42:1825–63. doi: 10.1016/j.ccell.2024.09.010
16. Xie N, Shen G, Gao W, Huang Z, Huang C, and Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. (2023) 8:9. doi: 10.1038/s41392-022-01270-x
17. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, and Zhang Y. T cell dysfunction and exhaustion in cancer. Front Cell Dev Biol. (2020) 8:17. doi: 10.3389/fcell.2020.00017
18. Topalian SL, Forde PM, Emens LA, Yarchoan M, Smith KN, and Pardoll DM. Neoadjuvant immune checkpoint blockade: A window of opportunity to advance cancer immunotherapy. Cancer Cell. (2023) 41:1551–66. doi: 10.1016/j.ccell.2023.07.011
19. Sinicrope FA and Turk MJ. Immune checkpoint blockade: timing is everything. J Immunother Cancer. (2024) 12:e009722. doi: 10.1136/jitc-2024-009722
20. Chen DS and Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
21. Gicobi JK, Barham W, and Dong H. Immune resilience in response to cancer therapy. Cancer Immunol Immunother. (2020) 69:2165–7. doi: 10.1007/s00262-020-02731-4
22. Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. (2022) 54:1376–89. doi: 10.1038/s41588-022-01159-z
23. Sweet-Cordero EA and Biegel JA. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science. (2019) 363:1170–5. doi: 10.1126/science.aaw3535
24. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
25. Wu Y, Xu J, Du C, Wu Y, Xia D, Lv W, et al. The predictive value of tumor mutation burden on efficacy of immune checkpoint inhibitors in cancers: A systematic review and meta-analysis. Front Oncol. (2019) 9:1161. doi: 10.3389/fonc.2019.01161
26. Wan L, Wang Z, Xue J, Yang H, and Zhu Y. Tumor mutation burden predicts response and survival to immune checkpoint inhibitors: a meta-analysis. Transl Cancer Res. (2020) 9:5437–49. doi: 10.21037/tcr-20-1131
27. Marshall GM, Carter DR, Cheung BB, Liu T, Mateos MK, Meyerowitz JG, et al. The prenatal origins of cancer. Nat Rev Cancer. (2014) 14:277–89. doi: 10.1038/nrc3679
28. Guerau-de-Arellano M, Martinic M, Benoist C, and Mathis D. Neonatal tolerance revisited: a perinatal window for Aire control of autoimmunity. J Exp Med. (2009) 206:1245–52. doi: 10.1084/jem.20090300
29. Rackaityte E and Halkias J. Mechanisms of fetal T cell tolerance and immune regulation. Front Immunol. (2020) 11:588. doi: 10.3389/fimmu.2020.00588
30. Roberts KG and Mullighan CG. The biology of B-progenitor acute lymphoblastic leukemia. Cold Spring Harb Perspect Med. (2020) 10. doi: 10.1101/cshperspect.a034835
31. Hunger SP and Mullighan CG. Acute lymphoblastic leukemia in children. New Engl J Med. (2015) 373:1541–52. doi: 10.1056/NEJMra1400972
32. Lee SHR, Antillon-Klussmann F, Pei D, Yang W, Roberts KG, Li Z, et al. Association of genetic ancestry with the molecular subtypes and prognosis of childhood acute lymphoblastic leukemia. JAMA Oncol. (2022) 8:354–63. doi: 10.1001/jamaoncol.2021.6826
33. Wiemels J, Cazzaniga G, Daniotti M, Eden O, Addison G, Masera G, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. (1999) 354:1499–503. doi: 10.1016/S0140-6736(99)09403-9
34. Hein D, Borkhardt A, and Fischer U. Insights into the prenatal origin of childhood acute lymphoblastic leukemia. Cancer Metastasis Rev. (2020) 39:161–71. doi: 10.1007/s10555-019-09841-1
35. Hjalgrim LL, Madsen HO, Melbye M, Jørgensen P, Christiansen M, Andersen MT, et al. Presence of clone-specific markers at birth in children with acute lymphoblastic leukaemia. Br J Cancer. (2002) 87:994–9. doi: 10.1038/sj.bjc.6600601
36. Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci. (2002) 99:8242–7. doi: 10.1073/pnas.112218799
37. Rüchel N, Jepsen VH, Hein D, Fischer U, Borkhardt A, and Gössling KL. In utero development and immunosurveillance of B cell acute lymphoblastic leukemia. Curr Treat Options Oncol. (2022) 23:543–61. doi: 10.1007/s11864-022-00963-3
38. Greaves M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat Rev Cancer. (2018) 18:471–84. doi: 10.1038/s41568-018-0015-6
39. Maloney KW, Devidas M, Wang C, Mattano LA, Friedmann AM, Buckley P, et al. Outcome in children with standard-risk B-cell acute lymphoblastic leukemia: results of children’s oncology group trial AALL0331. J Clin Oncol. (2020) 38:602–12. doi: 10.1200/JCO.19.01086
40. Pui C-H, Yang JJ, Bhakta N, and Rodriguez-Galindo C. Global efforts toward the cure of childhood acute lymphoblastic leukaemia. Lancet Child Adolesc Health. (2018) 2:440–54. doi: 10.1016/S2352-4642(18)30066-X
41. Hunger SP and Raetz EA. How I treat relapsed acute lymphoblastic leukemia in the pediatric population. Blood. (2020) 136:1803–12. doi: 10.1182/blood.2019004043
42. Sun W, Malvar J, Sposto R, Verma A, Wilkes JJ, Dennis R, et al. Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia. (2018) 32:2316–25. doi: 10.1038/s41375-018-0094-0
43. Hodder A, Mishra AK, Enshaei A, Baird S, Bhuller K, Elbeshlawi I, et al. Blinatumomab for first-line treatment of children and young persons with B-ALL. J Clin Oncol. (2024) 42:907–14. doi: 10.1200/JCO.23.01392
44. Gardner RA and Shah NN. CAR T-cells for cure in pediatric B-ALL. J Clin Oncol. (2023) 41:1646–8. doi: 10.1200/JCO.22.02345
45. Jasinski S, De Los Reyes FA, Yametti GC, Pierro J, Raetz E, and Carroll WL. Immunotherapy in pediatric B-cell acute lymphoblastic leukemia: advances and ongoing challenges. Paediatr Drugs. (2020) 22:485–99. doi: 10.1007/s40272-020-00413-3
46. Aldoss I, Bargou RC, Nagorsen D, Friberg GR, Baeuerle PA, and Forman SJ. Redirecting T cells to eradicate B-cell acute lymphoblastic leukemia: bispecific T-cell engagers and chimeric antigen receptors. Leukemia. (2017) 31:777–87. doi: 10.1038/leu.2016.391
47. Zamora AE, Crawford JC, and Thomas PG. Hitting the target: how T cells detect and eliminate tumors. J Immunol. (2018) 200:392–9. doi: 10.4049/jimmunol.1701413
48. Chang T-C, Carter RA, Li Y, Li Y, Wang H, Edmonson MN, et al. The neoepitope landscape in pediatric cancers. Genome Med. (2017) 9:78. doi: 10.1186/s13073-017-0468-3
49. Luczyński W, Stasiak-Barmuta A, Iłendo E, Kovalchuk O, Krawczuk-Rybak M, Malinowska I, et al. Low expression of costimulatory molecules and mRNA for cytokines are important mechanisms of immunosuppression in acute lymphoblastic leukemia in children? Neoplasma. (2006) 53:301–4.
50. Kebelmann-Betzing C, Körner G, Badiali L, Buchwald D, Möricke A, Korte A, et al. Characterization of cytokine, growth factor receptor, costimulatory and adhesion molecule expression patterns of bone marrow blasts in relapsed childhood B cell precursor all. Cytokine. (2001) 13:39–50. doi: 10.1006/cyto.2000.0794
51. D’Amico G, Vulcano M, Bugarin C, Bianchi G, Pirovano G, Bonamino M, et al. CD40 activation of BCP-ALL cells generates IL-10–producing, IL-12–defective APCs that induce allogeneic T-cell anergy. Blood. (2004) 104:744–51. doi: 10.1182/blood-2003-11-3762
52. Cardoso A, Schultze J, Boussiotis V, Freeman G, Seamon M, Laszlo S, et al. Pre-B acute lymphoblastic leukemia cells may induce T-cell anergy to alloantigen. Blood. (1996) 88:41–8. doi: 10.1182/blood.V88.1.41.41
53. Yotnda P, Mintz P, Grigoriadou K, Lemonnier F, Vilmer E, and Langlade-Demoyen P. Analysis of T-cell defects in the specific immune response against acute lymphoblastic leukemia cells. Exp Hematol. (1999) 27:1375–83. doi: 10.1016/s0301-472x(99)00083-1
54. Curran EK, Godfrey J, and Kline J. Mechanisms of immune tolerance in leukemia and lymphoma. Trends Immunol. (2017) 38:513–25. doi: 10.1016/j.it.2017.04.004
55. Hatakeyama N, Tamura Y, Sahara H, Suzuki N, Suzuki K, Hori T, et al. Induction of autologous CD4- and CD8-mediated T-cell responses against acute lymphocytic leukemia cell line using apoptotic tumor cell-loaded dendritic cells. Exp Hematol. (2006) 34:197–207. doi: 10.1016/j.exphem.2005.11.004
56. D’Amico G, Bonamino M, Dander E, Marin V, Basso G, Balduzzi A, et al. T cells stimulated by CD40L positive leukemic blasts-pulsed dendritic cells meet optimal functional requirements for adoptive T-cell therapy. Leukemia. (2006) 20:2015–24. doi: 10.1038/sj.leu.2404390
57. Weber G, Caruana I, Rouce RH, Barrett AJ, Gerdemann U, Leen AM, et al. Generation of tumor antigen-specific T cell lines from pediatric patients with acute lymphoblastic leukemia–implications for immunotherapy. Clin Cancer Res. (2013) 19:5079–91. doi: 10.1158/1078-0432.CCR-13-0955
58. Zamora AE, Crawford JC, Allen EK, Guo X-ZJ, Bakke J, Carter RA, et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8+ T cell responses. Sci Transl Med. (2019) 11. doi: 10.1126/scitranslmed.aat8549
59. Li Y, Yang X, Sun Y, Li Z, Yang W, Ju B, et al. Impact of T-cell immunity on chemotherapy response in childhood acute lymphoblastic leukemia. Blood. (2022) 140:1507–21. doi: 10.1182/blood.2021014495
60. Seif AE, Barrett DM, Milone M, Brown VI, Grupp SA, and Reid GSD. Long-term protection from syngeneic acute lymphoblastic leukemia by CpG ODN-mediated stimulation of innate and adaptive immune responses. Blood. (2009) 114:2459–66. doi: 10.1182/blood-2009-02-203984
61. Jo S, Lee JH, Mattei JJ, Barrett DM, Van Den Elzen P, Grupp SA, et al. Generation of a multi-antigen-directed immune response for durable control of acute lymphoblastic leukemia. Leukemia. (2018) 32:539–72. doi: 10.1038/leu.2017.290
62. Kang SH, Hwang HJ, Yoo JW, Kim H, Choi ES, Hwang S-H, et al. Expression of immune checkpoint receptors on T-cells and their ligands on leukemia blasts in childhood acute leukemia. Anticancer Res. (2019) 39:5531–9. doi: 10.21873/anticanres.13746
63. Bailur JK, McCachren SS, Pendleton K, Vasquez JC, Lim HS, Duffy A, et al. Risk-associated alterations in marrow T cells in pediatric leukemia. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.140179
64. Liang M, Gong D, Wang L, Liang X, Meng J, Huang W, et al. PAX5 haploinsufficiency induced CD8+ T cells dysfunction or exhaustion by high expression of immune inhibitory-related molecules. Cancer Treat Res Commun. (2021) 28:100437. doi: 10.1016/j.ctarc.2021.100437
65. Naghavi Alhosseini M, Palazzo M, Cari L, Ronchetti S, Migliorati G, and Nocentini G. Overexpression of potential markers of regulatory and exhausted CD8+ T cells in the peripheral blood mononuclear cells of patients with B-acute lymphoblastic leukemia. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24054526
66. Balajam NZ, Shabani M, Aghaei M, Haghighi M, and Kompani F. Study of T-cell immunoglobulin and mucin domain-3 expression profile in peripheral blood and bone marrow of human acute lymphoblastic leukemia patients. J Res Med Sci. (2020) 25:69. doi: 10.4103/jrms.JRMS_759_19
67. Ruan Y, Xie L, and Zou A. Association of CDKN2A/B mutations, PD-1, and PD-L1 with the risk of acute lymphoblastic leukemia in children. J Cancer Res Clin Oncol. (2023) 149:10841–50. doi: 10.1007/s00432-023-04974-x
68. Akbar A, Asgarian-Omran H, Valadan R, Dindarloo M-M, Najafi A, Kahrizi A, et al. Expression of Galectin-9-related immune checkpoint receptors in B-cell acute lymphoblastic leukemia. Iran J Basic Med Sci. (2023) 26:1468–74. doi: 10.22038/IJBMS.2023.73159.15901
69. Palen K, Thakar M, Johnson BD, and Gershan JA. Bone marrow-derived CD8+ T cells from pediatric leukemia patients express PD1 and expand ex vivo following induction chemotherapy. J Pediatr Hematol Oncol. (2019) 41:648–52. doi: 10.1097/MPH.0000000000001244
70. Mansour A, Elkhodary T, Darwish A, and Mabed M. Increased expression of costimulatory molecules CD86 and sCTLA-4 in patients with acute lymphoblastic leukemia. Leuk Lymphoma. (2014) 55:2120–4. doi: 10.3109/10428194.2013.869328
71. Blaeschke F, Willier S, Stenger D, Lepenies M, Horstmann MA, Escherich G, et al. Leukemia-induced dysfunctional TIM-3+CD4+ bone marrow T cells increase risk of relapse in pediatric B-precursor ALL patients. Leukemia. (2020) 34:2607–20. doi: 10.1038/s41375-020-0793-1
72. Feucht J, Kayser S, Gorodezki D, Hamieh M, Döring M, Blaeschke F, et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget. (2016) 7:76902–19. doi: 10.18632/oncotarget.12357
73. Liu L, Chang Y-J, Xu L-P, Zhang X-H, Wang Y, Liu K-Y, et al. T cell exhaustion characterized by compromised MHC class I and II restricted cytotoxic activity associates with acute B lymphoblastic leukemia relapse after allogeneic hematopoietic stem cell transplantation. Clin Immunol. (2018) 190:32–40. doi: 10.1016/j.clim.2018.02.009
74. Simone R, Tenca C, Fais F, Luciani M, De Rossi G, Pesce G, et al. A soluble form of CTLA-4 is present in paediatric patients with acute lymphoblastic leukaemia and correlates with CD1d+ expression. PloS One. (2012) 7:e44654. doi: 10.1371/journal.pone.0044654
75. Dolina JS, Van Braeckel-Budimir N, Thomas GD, and Salek-Ardakani S. CD8+ T cell exhaustion in cancer. Front Immunol. (2021) 12:715234. doi: 10.3389/fimmu.2021.715234
76. Tracy SI, Venkatesh H, Hekim C, Heltemes-Harris LM, Knutson TP, Bachanova V, et al. Combining nilotinib and PD-L1 blockade reverses CD4+ T-cell dysfunction and prevents relapse in acute B-cell leukemia. Blood. (2022) 140:335–48. doi: 10.1182/blood.2021015341
77. Qin H, Ishii K, Nguyen S, Su PP, Burk CR, Kim B-H, et al. Murine pre-B-cell ALL induces T-cell dysfunction not fully reversed by introduction of a chimeric antigen receptor. Blood. (2018) 132:1899–910. doi: 10.1182/blood-2017-12-815548
78. Lee-Sherick AB, Jacobsen KM, Henry CJ, Huey MG, Parker RE, Page LS, et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.97941
79. Hunter R, Imbach KJ, Zhou C, Dougan J, Hamilton JAG, Chen KZ, et al. B-cell acute lymphoblastic leukemia promotes an immune suppressive microenvironment that can be overcome by IL-12. Sci Rep. (2022) 12:11870. doi: 10.1038/s41598-022-16152-z
80. Gil-de-Gómez L, Mattei JJ, Lee JH, Grupp SA, Reid GSD, and Seif AE. CD4 + T cells mediate dendritic cell licensing to promote multi-antigen anti-leukemic immune response. Cancer Med. (2025) 14. doi: 10.1002/cam4.70508
81. Leach DR, Krummel MF, and Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
82. Savoia P, Astrua C, and Fava P. Ipilimumab (Anti-Ctla-4 Mab) in the treatment of metastatic melanoma: Effectiveness and toxicity management. Hum Vaccin Immunother. (2016) 12:1092–101. doi: 10.1080/21645515.2015.1129478
83. Simeone E, Grimaldi AM, and Ascierto PA. Anti-PD1 and anti-PD-L1 in the treatment of metastatic melanoma. Melanoma Manag. (2015) 2:41–50. doi: 10.2217/mmt.14.30
84. Vaddepally RK, Kharel P, Pandey R, Garje R, and Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel). (2020) 12:738. doi: 10.3390/cancers12030738
85. Sun Q, Hong Z, Zhang C, Wang L, Han Z, and Ma D. Immune checkpoint therapy for solid tumours: clinical dilemmas and future trends. Signal Transduct Target Ther. (2023) 8:320. doi: 10.1038/s41392-023-01522-4
86. Liu Y, Altreuter J, Bodapati S, Cristea S, Wong CJ, Wu CJ, et al. Predicting patient outcomes after treatment with immune checkpoint blockade: A review of biomarkers derived from diverse data modalities. Cell Genomics. (2024) 4:100444. doi: 10.1016/j.xgen.2023.100444
87. Tsumura A, Levis D, and Tuscano JM. Checkpoint inhibition in hematologic Malignancies. Front Oncol. (2023) 13:1288172. doi: 10.3389/fonc.2023.1288172
88. Pophali P, Varela JC, and Rosenblatt J. Immune checkpoint blockade in hematological Malignancies: current state and future potential. Front Oncol. (2024) 14:1323914. doi: 10.3389/fonc.2024.1323914
89. Ciurej A, Lewis E, Gupte A, and Al-Antary E. Checkpoint immunotherapy in pediatric oncology: will we say checkmate soon? Vaccines (Basel). (2023) 11:1843. doi: 10.3390/vaccines11121843
90. Koller P, Baran N, Harutyunyan K, Cavazos A, Mallampati S, Chin RL, et al. PD-1 blockade in combination with dasatinib potentiates induction of anti-acute lymphocytic leukemia immunity. J Immunother Cancer. (2023) 11. doi: 10.1136/jitc-2022-006619
91. Barrett AJ and Savani BN. Does chemotherapy modify the immune surveillance of hematological Malignancies? Leukemia. (2009) 23:53–8. doi: 10.1038/leu.2008.273
92. Cassaday RD, Garcia K-LA, Fromm JR, Percival M-EM, Turtle CJ, Nghiem PT, et al. Phase 2 study of pembrolizumab for measurable residual disease in adults with acute lymphoblastic leukemia. Blood Adv. (2020) 4:3239–45. doi: 10.1182/bloodadvances.2020002403
93. Gao Q, Ryan SL, Iacobucci I, Ghate PS, Cranston RE, Schwab C, et al. The genomic landscape of acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21. Blood. (2023) 142:711–23. doi: 10.1182/blood.2022019094
94. Pilheden M, Ahlgren L, Hyrenius-Wittsten A, Gonzalez-Pena V, Sturesson H, Hansen Marquart HV, et al. Duplex sequencing uncovers recurrent low-frequency cancer-associated mutations in infant and childhood KMT2A-rearranged acute leukemia. Hemasphere. (2022) 6:e785. doi: 10.1097/HS9.0000000000000785
95. Forgione MO, McClure BJ, Eadie LN, Yeung DT, and White DL. KMT2A rearranged acute lymphoblastic leukaemia: Unravelling the genomic complexity and heterogeneity of this high-risk disease. Cancer Lett. (2020) 469:410–8. doi: 10.1016/j.canlet.2019.11.005
96. Strohl WR and Naso M. Bispecific T-cell redirection versus chimeric antigen receptor (CAR)-T cells as approaches to kill cancer cells. Antibodies (Basel). (2019) 8. doi: 10.3390/antib8030041
97. Goebeler M-E and Bargou RC. T cell-engaging therapies — BiTEs and beyond. Nat Rev Clin Oncol. (2020) 17:418–34. doi: 10.1038/s41571-020-0347-5
98. Lopez de Rodas M and Schalper KA. Tumour antigen-induced T cell exhaustion - the archenemy of immune-hot Malignancies. Nat Rev Clin Oncol. (2021) 18:749–50. doi: 10.1038/s41571-021-00562-5
99. Zhu WM and Middleton MR. Combination therapies for the optimisation of Bispecific T-cell Engagers in cancer treatment. Immunotherapy Adv. (2023) 3:ltad013. doi: 10.1093/immadv/ltad013
100. Wang H, Kaur G, Sankin AI, Chen F, Guan F, and Zang X. Immune checkpoint blockade and CAR-T cell therapy in hematologic Malignancies. J Hematol Oncol. (2019) 12:59. doi: 10.1186/s13045-019-0746-1
101. Hogan LE, Brown PA, Ji L, Xu X, Devidas M, Bhatla T, et al. Children’s oncology group AALL1331: phase III trial of blinatumomab in children, adolescents, and young adults with low-risk B-cell ALL in first relapse. J Clin Oncol. (2023) 41:4118–29. doi: 10.1200/JCO.22.02200
102. von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. (2016) 34:4381–9. doi: 10.1200/JCO.2016.67.3301
103. Gupta S, Rau RE, Kairalla JA, Rabin KR, Wang C, Angiolillo AL, et al. Blinatumomab in standard-risk B-cell acute lymphoblastic leukemia in children. N Engl J Med. (2024). doi: 10.1056/NEJMoa2411680
104. Brown PA, Ji L, Xu X, Devidas M, Hogan LE, Borowitz MJ, et al. Effect of postreinduction therapy consolidation with blinatumomab vs chemotherapy on disease-free survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia. JAMA. (2021) 325:833. doi: 10.1001/jama.2021.0669
105. Locatelli F, Zugmaier G, Rizzari C, Morris JD, Gruhn B, Klingebiel T, et al. Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia. . JAMA. (2021) 325:843. doi: 10.1001/jama.2021.0987
106. Ma J, Luong A, Doan A, Lin TY, Ji L, Wang CP, et al. T-cell dysfunction during blinatumomab therapy in pediatric acute lymphoblastic leukemia. Blood Adv. (2025). doi: 10.1182/bloodadvances.2025015894
107. Wunderlich M, Manning N, Sexton C, O’Brien E, Byerly L, Stillwell C, et al. PD-1 inhibition enhances blinatumomab response in a UCB/PDX model of relapsed pediatric B-cell acute lymphoblastic leukemia. Front Oncol. (2021) 11:642466. doi: 10.3389/fonc.2021.642466
108. Narula G, Keerthivasagam S, Jain H, Punatar S, Chichra A, Dhamne C, et al. Novel humanized CD19-CAR-T (Now talicabtagene autoleucel, Tali-celTM) cells in relapsed/refractory pediatric B-acute lymphoblastic leukemia- an open-label single-arm phase-I/Ib study. Blood Cancer J. (2025) 15:75. doi: 10.1038/s41408-025-01279-9
109. Martínez-Gamboa DA, Hans R, Moreno-Cortes E, Figueroa-Aguirre J, Garcia-Robledo JE, Vargas-Cely F, et al. CAR T-cell therapy landscape in pediatric, adolescent and young adult oncology – A comprehensive analysis of clinical trials. Crit Rev Oncol Hematol. (2025) 209:104648. doi: 10.1016/j.critrevonc.2025.104648
110. Wang Y, Xue Y, Zuo Y, Jia Y, Lu A, Zeng H, et al. CD19-specific CAR-T cell treatment of 115 children and young adults with acute B lymphoblastic leukemia: long-term follow-up. Cancer Res Treat. (2024) 56:945–55. doi: 10.4143/crt.2023.1205
111. Annesley C, Seidel KD, Wu Q, Summers C, Wayne AS, Pulsipher MA, et al. Outcomes of PLAT-02 and PLAT-03: evaluating CD19 CAR T-cell therapy and CD19-expressing T-APC support in pediatric B-ALL. Blood J. (2025). doi: 10.1182/blood.2025028359
112. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
113. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
114. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
115. Dreyzin A, Rankin AW, Luciani K, Gavrilova T, and Shah NN. Overcoming the challenges of primary resistance and relapse after CAR-T cell therapy. Expert Rev Clin Immunol. (2024) 20:745–63. doi: 10.1080/1744666X.2024.2349738
116. DeGolier KR, Danis E, D’Antonio M, Cimons J, Yarnell M, Kedl RM, et al. Antigen experience history directs distinct functional states of CD8+ CAR T cells during the antileukemia response. Nat Immunol. (2025) 26:68–81. doi: 10.1038/s41590-024-02034-1
117. Zebley CC, Brown C, Mi T, Fan Y, Alli S, Boi S, et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. (2021) 37:110079. doi: 10.1016/j.celrep.2021.110079
118. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Sci (1979). (2021) 372. doi: 10.1126/science.aba1786
119. Pietrobon V, Todd LA, Goswami A, Stefanson O, Yang Z, and Marincola F. Improving CAR T-cell persistence. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms221910828
120. Li AM, Hucks GE, Dinofia AM, Seif AE, Teachey DT, Baniewicz D, et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood. (2018) 132:556–6. doi: 10.1182/blood-2018-99-112572
121. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
122. Zhang A, Sun Y, Wang S, Du J, Gao X, Yuan Y, et al. Secretion of human soluble programmed cell death protein 1 by chimeric antigen receptor-modified T cells enhances anti-tumor efficacy. Cytotherapy. (2020) 22:734–43. doi: 10.1016/j.jcyt.2020.05.007
123. Blaeschke F, Ortner E, Stenger D, Mahdawi J, Apfelbeck A, Habjan N, et al. Design and evaluation of TIM-3-CD28 checkpoint fusion proteins to improve anti-CD19 CAR T-cell function. Front Immunol. (2022) 13:845499. doi: 10.3389/fimmu.2022.845499
124. Kalinin RS, Ukrainskaya VM, Chumakov SP, Moysenovich AM, Tereshchuk VM, Volkov DV, et al. Engineered removal of PD-1 from the surface of CD19 CAR-T cells results in increased activation and diminished survival. Front Mol Biosci. (2021) 8:745286. doi: 10.3389/fmolb.2021.745286
125. Anderson ND, Birch J, Accogli T, Criado I, Khabirova E, Parks C, et al. Transcriptional signatures associated with persisting CD19 CAR-T cells in children with leukemia. Nat Med. (2023) 29:1700–9. doi: 10.1038/s41591-023-02415-3
126. Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest. (2019) 129:2123–32. doi: 10.1172/JCI125423
127. García-Calderón CB, Sierro-Martínez B, García-Guerrero E, Sanoja-Flores L, Muñoz-García R, Ruiz-Maldonado V, et al. Monitoring of kinetics and exhaustion markers of circulating CAR-T cells as early predictive factors in patients with B-cell Malignancies. Front Immunol. (2023) 14:1152498. doi: 10.3389/fimmu.2023.1152498
128. Dreyzin A, Shao L, Cai Y, Han KL, Prochazkova M, Gertz M, et al. Immunophenotype of CAR T cells and apheresis products predicts response in CD22 CAR T cell trial for B cell acute lymphoblastic leukemia. Mol Ther. (2025). doi: 10.1016/j.ymthe.2025.03.019
129. Zhao P, Zhao T, Yu L, Ma W, Liu W, and Zhang C. The risk of endocrine immune-related adverse events induced by PD-1 inhibitors in cancer patients: a systematic review and meta-analysis. Front Oncol. (2024) 14:1381250. doi: 10.3389/fonc.2024.1381250
130. Okiyama N and Tanaka R. Immune-related adverse events in various organs caused by immune checkpoint inhibitors. Allergology Int. (2022) 71:169–78. doi: 10.1016/j.alit.2022.01.001
Keywords: pediatric B cell precursor acute lymphoblastic leukemia, T cell exhaustion (Tex), bispecific T cell engager (BiTE), chimeric antigen receptor (CAR T), immune checkpoint inhibition
Citation: Atre T and Reid GSD (2025) T cell exhaustion in pediatric B-ALL: current knowledge and future perspectives. Front. Immunol. 16:1531145. doi: 10.3389/fimmu.2025.1531145
Received: 19 November 2024; Accepted: 12 May 2025;
Published: 28 May 2025.
Edited by:
Shahram Salek-Ardakani, Inhibrx, United StatesReviewed by:
Vera Muench, Ulm University Medical Center, GermanyCopyright © 2025 Atre and Reid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregor S. D. Reid, Z3JlZ29yLnJlaWRAdWJjLmNh
†ORCID: Gregor S.D. Reid, orcid.org/0000-0002-7567-3424