 Robert Sertori1
Robert Sertori1 Billy Truong1Manoj K. Singh1
Billy Truong1Manoj K. Singh1 Susan Shinton1Rachael Price1Andrew Sharo2Paulameena Shultes2†
Susan Shinton1Rachael Price1Andrew Sharo2Paulameena Shultes2† Uma Sunderam3Sadhna Rana3
Uma Sunderam3Sadhna Rana3 Rajgopal Srinivasan3Sutapa Datta3
Rajgopal Srinivasan3Sutapa Datta3 Joan Font-Burgada4Steven E. Brenner2
Joan Font-Burgada4Steven E. Brenner2 Jennifer M. Puck5,6
Jennifer M. Puck5,6 David L. Wiest1*
David L. Wiest1*- 1Nuclear Dynamics and Cancer Program, Fox Chase Cancer Center, Philadelphia, PA, United States
- 2Center for Computational Biology, University of California, Berkeley, Berkeley, CA, United States
- 3Innovation Labs, Tata Consultancy Services, Hyderabad, India
- 4Cancer Signaling and Microenvironment Program, Fox Chase Cancer Center, Philadelphia, PA, United States
- 5Department of Pediatrics, University of California, San Francisco, San Francisco, CA, United States
- 6Department of Pediatrics, University of California, San Francisco (UCSF) and UCSF Benioff Children’s Hospital, San Francisco, CA, United States
Introduction: Newborn screening for immunodeficiency has led to the identification of numerous cases for which the causal etiology is unknown.
Methods: Here we report the diagnosis of T lymphopenia of unknown etiology in a male proband. Whole exome sequencing (WES) was employed to nominate candidate variants, which were then analyzed functionally in zebrafish and in mice bearing orthologous mutations.
Results: WES revealed missense mutations in CHTF18 that were inherited in an autosomal recessive manner. CTF18, encoded by the CHTF18 gene, is a component of a secondary clamp loader, which is primarily thought to function by promoting DNA replication. We determined that the patient’s variants in CHTF18 (CTF18 R751W and E851Q) were damaging to function and severely attenuated the capacity of CTF18 to support hematopoiesis and lymphoid development, strongly suggesting that they were responsible for his T lymphopenia; however, the function of CTF18 appeared to be unrelated to its role as a clamp loader. DNA-damage, expected when replication is impaired, was not evident by expression profiling in murine Chtf18 mutant hematopoietic stem and progenitor cells (HSPC), nor was development of Ctf18-deficient progenitors rescued by p53 loss. Instead, we observed an expression signature suggesting disruption of HSPC positioning and migration. Indeed, the positioning of HSPC in ctf18 morphant zebrafish embryos was perturbed, suggesting that HSPC function was impaired through disrupted positioning in hematopoietic organs.
Discussion: Accordingly, we propose that T lymphopenia in our patient resulted from disturbed cell-cell contacts and migration of HSPC, caused by a non-canonical function of CHTF18 in regulating gene expression.
Introduction
Primary immune deficiencies are rare, with severe combined immunodeficiency (SCID), where T and B lymphocytes are absent or not functional, occurring in approximately 1/58,000 live births in the United States (1). The recently increased frequency of diagnosis is likely a result of newborn screening for SCID and T lymphopenia that is now occurring in all 50 states in the US (2–4). Newborn screening has allowed cases to be diagnosed and treated before maternal immunity has waned and infections occur, which has greatly improved the efficacy of treatment and led to identification of many new genes implicated in the etiology of SCID (3–5). Newborn screening relies on identification of reduced T cell receptor excision circles (TREC) in peripheral blood, which is a surrogate marker for impaired T cell output from the thymus (6, 7). This screening method has also identified many cases of T lymphopenia for which the causal etiology was initially unknown (8–10). Elucidating the causal etiology in such cases not only opens potential new avenues for the diagnosis and treatment of immunodeficiency diseases, but also represents an opportunity to identify novel regulators of lymphocyte development.
Unbiased investigation of the causal etiology of SCID or T lymphopenia cases can lead to the identification of genes of unknown function or identification of new functions for known genes. Candidate variant identification is pursued by whole exome sequencing (WES) followed by functional screening in zebrafish. Zebrafish are an attractive model for functional screening of candidate alleles, as they are amenable to genetic analysis and their hematopoietic processes, including lymphoid development, are highly conserved (11). For example, we previously employed this strategy to reveal a new function for STN1, a component of the CST complex (CTC1-STN1-TEN1), which plays multiple roles in telomere maintenance specifically and genome stability generally (12, 13). Interestingly, the developmental anomalies produced by the STN1 patient variant did not appear to result from impairing telomere maintenance or genome integrity, as the defects were rescued by treatment with Thalidomide, and were not phenocopied by knockdown of another CST complex component, CTC1 (Unpublished data) (14). Finally, we have also identified mutations in genes critical for essential processes, such as actin remodeling (ARPC1B), transcription (MED14) or clamp loader function (CHTF18) (15;10), yet were compatible with live birth of immunodeficient patients.
Clamp loaders facilitate replication by loading onto DNA, a clamp comprising a trimer of proliferating cell nuclear antigen (PCNA), which is required for DNA replication (16, 17). PCNA is typically loaded onto DNA by the primary clamp loading complex, composed of five replication factor C (RFC) components, RFC1-5, which are essential for life in yeast and higher vertebrates (18, 19). In contrast, secondary clamp loader component, Chromosome Transmission Fidelity Factor 18 (CTF18), is not essential for life (20), either because it is a secondary clamp loader and/or because its expression is more tissue restricted than primary clamp loader component RFC1 (biogps/human atlas).
Here we describe our investigation of the causal etiology in an infant whose T lymphopenia was detected by newborn screening. Functional screening in zebrafish revealed that missense mutations in the secondary clamp loader component CHTF18, were potentially responsible for disease in the proband. Despite the prevailing view that the predominant function of CTF18 (encoded by the CHTF18 gene) is facilitating DNA replication as part of a clamp loader, our data suggest its role in supporting hematopoiesis may be unrelated to this activity. Indeed, Ctf18 loss produced no evidence of DNA-damage, and did not impair proliferation; however, it did cause selective changes in the expression of genes in hematopoietic stem and progenitor cells (HSPC) that influence cell-cell interactions and migration, suggesting that CHTF18 mutant HSPC would make suboptimal contacts with supporting stroma and fail to execute the prescribed migration patterns required or successful hematopoiesis. Thus, we propose that CTF18 supports normal hematopoiesis and lymphopoiesis through a “moonlighting function” unrelated to the support of DNA replication, but rather controls proper expression of genes regulating HSPC positioning and migration.
Materials and methods
Patient information
T lymphopenia was identified in the male proband by routine newborn SCID screening for T cell receptor excision circles (TREC) using blood from a Guthrie blood spot filter card, as described (1, 21, 22). Research activities were performed with parental informed consent under protocols approved by the institutional review board (IRB) at the University of California, San Francisco.
Research-based whole exome sequencing (WES) was performed on cells from the proband and parents with bioinformatic analysis and variant calling, as described (9, 23). Briefly, libraries were prepared by appending TruSeq adaptors (Illumina: San Diego, CA) to genomic DNA fragments of 200–300 bp that were enriched with 10 cycles of PCR, pooled and submitted to exon capture using a Roche Nimblegen kit (V3.0). After 10 additional amplification cycles, 100 bp paired end sequence reads were generated (HiSeq2000, llumina) and aligned against GRCh37 (Aug 2009 release) using BWA (v0.6.2). The results were converted to BAM, sorted by coordinate, indexed, and marked for PCR duplicate reads using the Picard toolkit (v 1.81) (http://picard.sourceforge.net). Local realignment was performed around known indel locations, and base quality scores were re-calibrated using GATK (v 2.6.5). Variants were called using GATK UnifiedGenotyper and freebayes (vesion 0.9.10) and variant quality scores were re-calibrated by VQSR (Variant Quality Score Recalibration) using the trio of exomes in this report, as well as 65 others sequenced at our site. HapMap v3.3, 1000 genomes high confidence SNPs (phase1 v3 2010-11-23) and Omni chip array sets were used as training data, and to provided truth sites for SNPs, while the Mills dataset from the 1000 genomes was used for indels, using a truth sensitivity cutoff of 99% (24). Variant annotation (including region, effect, allele frequency, disease phenotype annotation, and conservation) was performed using our custom tool Varant (http://compbio.berkeley.edu/proj/varant/). Particular attention was given to high-confidence, rare, likely-damaging, protein-altering variants in genes associated with primary cellular immunodeficiency (25). WES data from the JPSCID-15 trio were submitted to dbGaP, accession number phs002968.v1.p1.
Zebrafish
Tubingen long fin zebrafish were maintained at 28.5°C under standard aquaculture conditions. Animal housing and handling were all performed in accordance with the approved protocol from the Institutional Animal Care and Use Committee (IACUC).
Analysis of candidate variants in zebrafish
The zebrafish CHTF18 ortholog chtf18 (NM_001110102.3) was previously identified. CTBP2 paralogues ctbp2a (NM_001195491.1 and NM_131715.1), and ctbp2l (NM_001015064.1) were identified by homology and synteny, as described (10, 26). Antisense morpholino oligonucleotides (MO; Gene Tools) were used to block splicing of zebrafish chtf18 (5’ ACTGGAACAAAACACACCTCTTCAT 3’), ctbp2a (5’ TTCGGTCAGAAACAATCATACACAC 3’) and ctbp2l (5’ CCGGAAACAAGTAATGATGCAACTG 3’) upon injection into one-cell embryos using a PM1000 microinjector (Microdata Instrument, Inc.). The efficacy of the MO in disrupting splicing was assessed by reverse-transcriptase (RT)–PCR using the following oligonucleotides (Table 1): 1) chtf18: F-AGGAGGCAAGATGTGGCG and R-CATTTCAGCAGACAGCGATTAG; 2) ctbp2a: F-CGGTGGGTGCGATGATG and R-TGTACCAGGCTGTGTGAGGG; 3) ctbp2l: F-CAGGAGCGGCTCCGAAGACGTTTTCCGGC and R-ACAGAAGGCAACGGTTGCCAGATC; and 4) actb: F-TGGCATCACACCTTCTAC and R-AGACCATCACCAGAGTCC (27). Tp53 rescue experiments were conducted as described in (28). To assess the capacity of wildtype and variant human CHTF18 orthologs to rescue the loss of zebrafish, wildtype and patient variant CHTF18 were cloned into heat shock-inducible expression vector pSGH2 using standard approaches. Sanger sequencing confirmed the R751W, V776L and E851Q variant alleles. Ectopic expression of wild-type and mutant human CHTF18 was achieved by injection of the heat-inducible pSGH2 vector (29) into one-cell stage embryos, which were heated to 37°C for 1 hour at 30 hours post fertilization. GFP+ embryos were selected at 5 dpf for analysis by WISH using a probe for lck as described (9). The impact of chtf18 knockdown on HSPC localization was assessed by injection of chtf18e6 splice blocking MO followed by the performance of WISH at 36 hours post fertilization, as described (28).
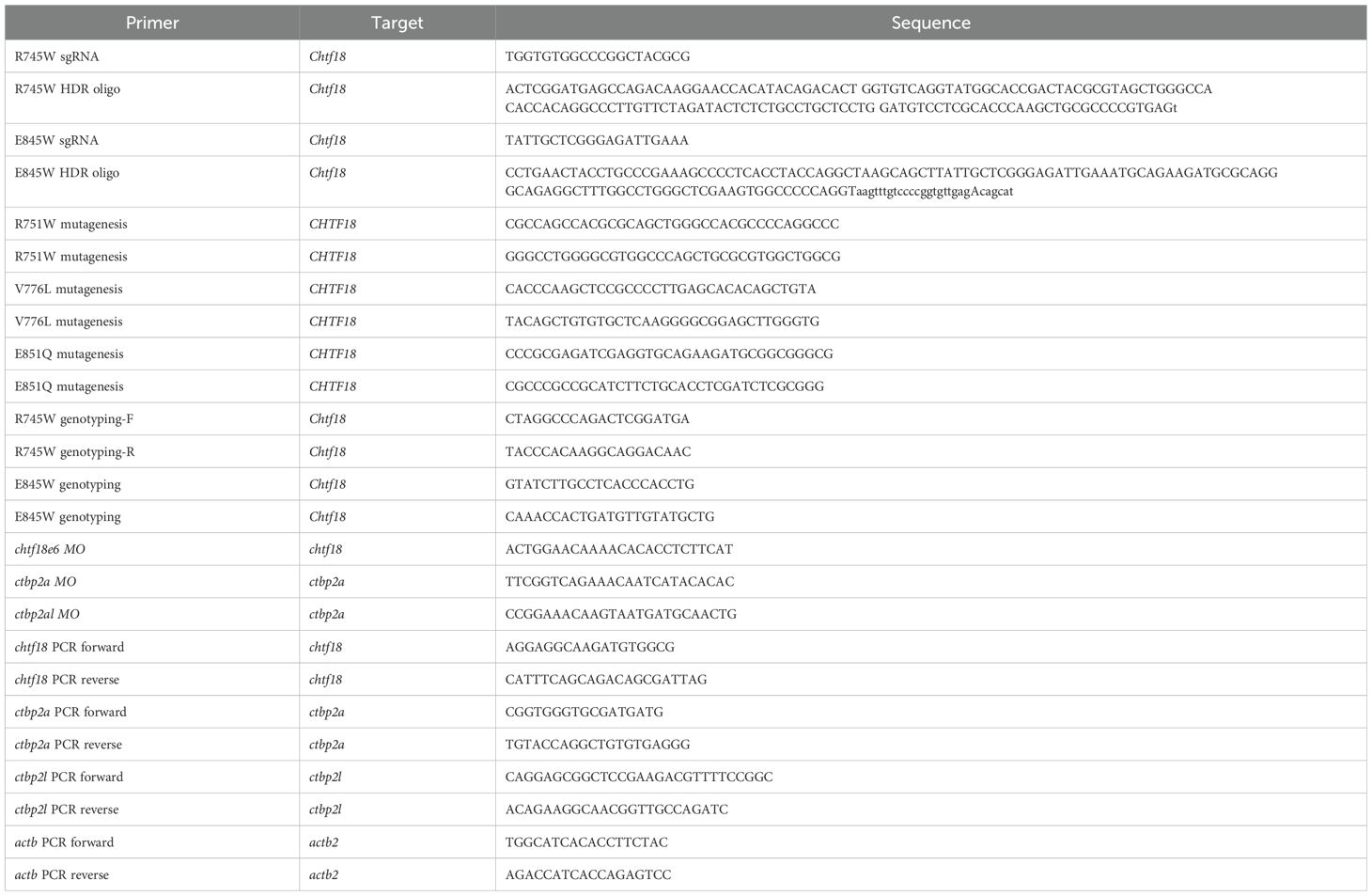
Table 1. Oligonucleotides.
WISH analysis
WISH was performed as described (30) using anti-sense probes for lck, ikaros, tcrd, runx1, and foxn1 (28, 31). The stained embryos were photographed using the Nikon SMZ1500 stereomicroscope equipped with DS-Fi1 digital camera and Nikon Ar imaging software. Image J software was used to measure integrated density of the lck WISH staining of zebrafish thymi.
Structural modeling
Genomic sequences were obtained using the NCBI and ENSEMBL databases. Multiple alignments of human, mouse and zebrafish CTF18 and CTBP2 amino acid sequences were obtained using Clustal X. Structural modeling was performed as described for human CTF18 using UCSF Chimera software and AlphaFold predicted structures for CTF18 wildtype, the arginine 751 to tryptophan (R751W) variant and glutamic acid 851 to glutamine (E851Q) variant, as well as the RFC1 clamp loading structure (PDB code 6VVO) (10, 32). CTF18 has limited homology to the clamp loader paralog RFC1 found in the CryoEM structure of human clamp loader bound to PCNA (32). AlphaFold-Multimer v2.3 in the ColabFold implementation (33) was used to predict the structure of a conserved portion of CTF18 (residues 595 to 865) that contained the 3 missense mutations identified in the proband and their interactions with the nearest neighbor subunit in the clamp loader complex, RFC3. The resulting heterodimeric models were subjected to a coordinate constrained method of protein relaxation using the Rosetta (34) molecular modeling suite to optimize side chain packing (35–37). These relaxed models were aligned by the RFC3 subunit using the UCSF Chimera 1.15 software package (38) and analyzed for contacts between CTF18 variants and RFC3 with its bound ADP. As a test to validate our modeling procedure, we used the same AlphaFold methods to recapitulate the RFC1-RFC3 interaction, and the resulting model superposed with RFC1 subunit of the 6VVO structure with an RMSD < 0.76 Å2 for 200 alpha carbons in this region. Final refined modeling methods utilized AlphaFold3 (AF3) (39), which can predict the combined structure of multicomponent complexes including proteins, nucleic acids, small molecules, ions and modified residues. We retained a focus on the conserved fragment of CTF18 (583-863) bound to RFC3, and added an ADP molecule and magnesium ion that is often bound in the active site. Multiple AF3 runs were used to generate 20 models each of the WT R751, mutant R751W, and the mutant E851Q. In all cases the models scored with high confidence (ipTM range 0.89 to 0.91) and backbone superposition of models yielded RMSD values in the range of 0.4 to 0.8 Å2. The WT R751 rotamer chosen by the AF3 modeling was the same as that found in the PDB structure 8UN0 (state7) for 17 of the top 20 models, albeit with very minor differences in position due to slight backbone variation. In all cases the top ranked models were used to analyze the interface interactions and prepare figures with UCSF Chimerax version 1.7.1. To obtain a general overall interface assessment, we also analyzed the CTF18(583-863)-RFC3 interface with PISA analysis (40).
Mice
All mouse strains were housed in the Laboratory Animal Facility at Fox Chase, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and handled in accordance with an IACUC approved protocol. Ctf18 R745W and E845Q knockin mutant mice were generated by CRISPR genome editing (10). Chf18 R745W mice were created using a 150 bp oligo donor to mutate R745 to W and silent cytosine (C) to thymine (T) to inactivate the PAM and prevent re-cutting of the repaired allele. Similarly, the Ctf18 E845Q mice were created using a 150 bp oligo to mutate E845 to Q and inactivate the PAM to prevent re-cutting of the repaired allele. The knockin mutation created BseYI and HpyCH4V restriction enzyme sites, respectively, to genotype the mutated alleles. All oligonucleotides used in study are listed in Table 1.
Flow cytometry
Single-cell suspensions prepared from mouse thymus, spleen, bone marrow, or peripheral blood were stained with optimal amounts of the following fluorochrome-conjugated antibodies: anti-CD4 (GK1.5), anti-CD8 (53-6.7), anti-CD24 (M1/69), anti-CD25 (PC61), anti-CD44 (IM7), anti-CD62L (MEL-14), anti-CD69 (H1.2F3), anti-CD73 (TY/11.8), anti-CD90.2 (30-H12) anti-B220 (RA3-6B2), anti-NK1.1 (PK136), anti-TCRγδ (GL3), anti-TCRβ (H57-597), anti-CD19 (eBio1D3), anti-CD3 (17A2), anti-Gr1 (RB6-8C5), anti-Sca1 (D7), and anti-cKit (2B8). The antibodies were purchased from BD Biosciences, eBioscience, BioLegend, or Tonbo Biosciences. Dead cells were excluded from analyses using propidium iodide (PI). Data were analyzed on either a BD LSRII or Symphony A5 flow cytometer (BD Pharmingen) using Flowjo 9.96 software (Treestar, Inc).
Single cell RNA-seq
30,000 lineage depleted bone marrow cells from wildtype or mutant mice were loaded with the objective of collecting a single library of 10,000 cells by Chromium controller (V3 Chemistry version, 10X Genomics Inc, San Francisco, USA). Barcoding and DNA purification were performed according to the manufacturer’s protocol. Sequencing was performed using 2 x 150 pair-end configuration on an NovaSeqX Plus platform at a sequencing depth of 375G/lane. Cell-barcode and unique molecular identifiers were extracted and aligned through Cell Ranger and expression analysis was performed using Seurat V4, as described (41). Uniform Manifold Approximation and Projection (UMAP) plots were used to visualize the data based on canonical component analysis of the top 1000 genes with highest dispersion between all sample groups. Gene ontology and Gene Set Enrichment analysis was performed using EnrichR.
Transplant assays
CD45.1 mice were lethally irradiated with a total dose of 11 Gy, split in two doses of 6.5Gy and 4.5Gy, separated by three hours. After 24 hours, mice were injected i.v. in the tail vein with either 100,000 lineage depleted adult bone marrow (BM) CD45.2 hematopoietic progenitors alone or together with 100,000 adult BM CD45.1+ competitor progenitors delivered in 200μl Hank’s Balanced Salt Solution (HBSS), as described (10). Engraftment was monitored by retro-orbital bleeding at the indicated intervals. Mice were sacrificed after 6 weeks of engraftment and bone marrow, thymus and spleen were analyzed by flow cytometry. For fetal liver (FL) transplants, 100,000 embryonic day 14.5 (E14.5) FL cells from CD45.2 mice were transferred as above with 100,000 CD45.1 FL competitor cells as described above. Seeding assays were performed by transplanting adult BM progenitors as above and determining the seeding of the BM of recipient mice 4 days later by performing flow cytometry on explanted bone marrow cells for the allelic marker (CD45.2/1).
HSPC function in situ
To assess the capacity of WT and compound Chtf18 mutant HSPC to respond to repeated entry into cell cycle, mice were treated twice weekly for 60 days with two intraperitoneal doses of pIpC (15mg/kg) as described (42), with weekly monitoring of peripheral blood lymphocyte content by flow cytometry.
In vitro cultures
E14.5 FL progenitors from WT and compound Chtf18 mutant mice were cultured on OP9-DL1 monolayers, as described (43). Developmental progression was monitored by quantitating cell growth of control triplicate cultures or those treated with agents including Interferon-β, Mitomycin-C, Ganetespib, and Aramycin-C.
Graphing and statistics
Graphic representations of data were generated using GraphPad prism V9, following statistical analysis using ANOVA and Student’s t tests. Significance values are indicated.
Results
Clinical history of a proband with T lymphopenia
The male infant (JPSCID-15) was born at term after an uncomplicated pregnancy to nonconsanguineous, healthy parents with no known family history of immune deficiency. Newborn screening revealed low T cell receptor excision circles (TREC; normal >25), with T-cell lymphopenia subsequently documented by flow cytometry (Table 2) (21). The proportion of naïve recent thymic emigrant (i.e., CD45RA+) CD4+ and CD8+ T cells was reduced, with CD8+ cells exhibiting a slightly greater reduction (available data shown in Table 2) (46, 47). B and NK cell numbers were normal. The patient had a normal physical exam with no syndromic features. Proliferation to phytohemagglutinin, immunoglobulin levels and titers to killed vaccines were obtained over the ensuing months and were normal (44, 45). During his first year the patient experienced one episode of pneumonia treated successfully with oral antibiotics, but no other infections. Growth and development remained normal until he was lost to follow-up after his second birthday. Genetic workup included normal fluorescent in situ hybridization for chromosome 22q11.2 deletion and normal alpha-fetoprotein that ruled out DiGeorge syndrome and ataxia telangiectasia, respectively. Sanger sequencing of a panel of SCID genes, including ADA, DCLRE1C, IL2RG, IL7R, JAK3, RAG1, RAG2, RMRP, and ZAP70, revealed no mutations.
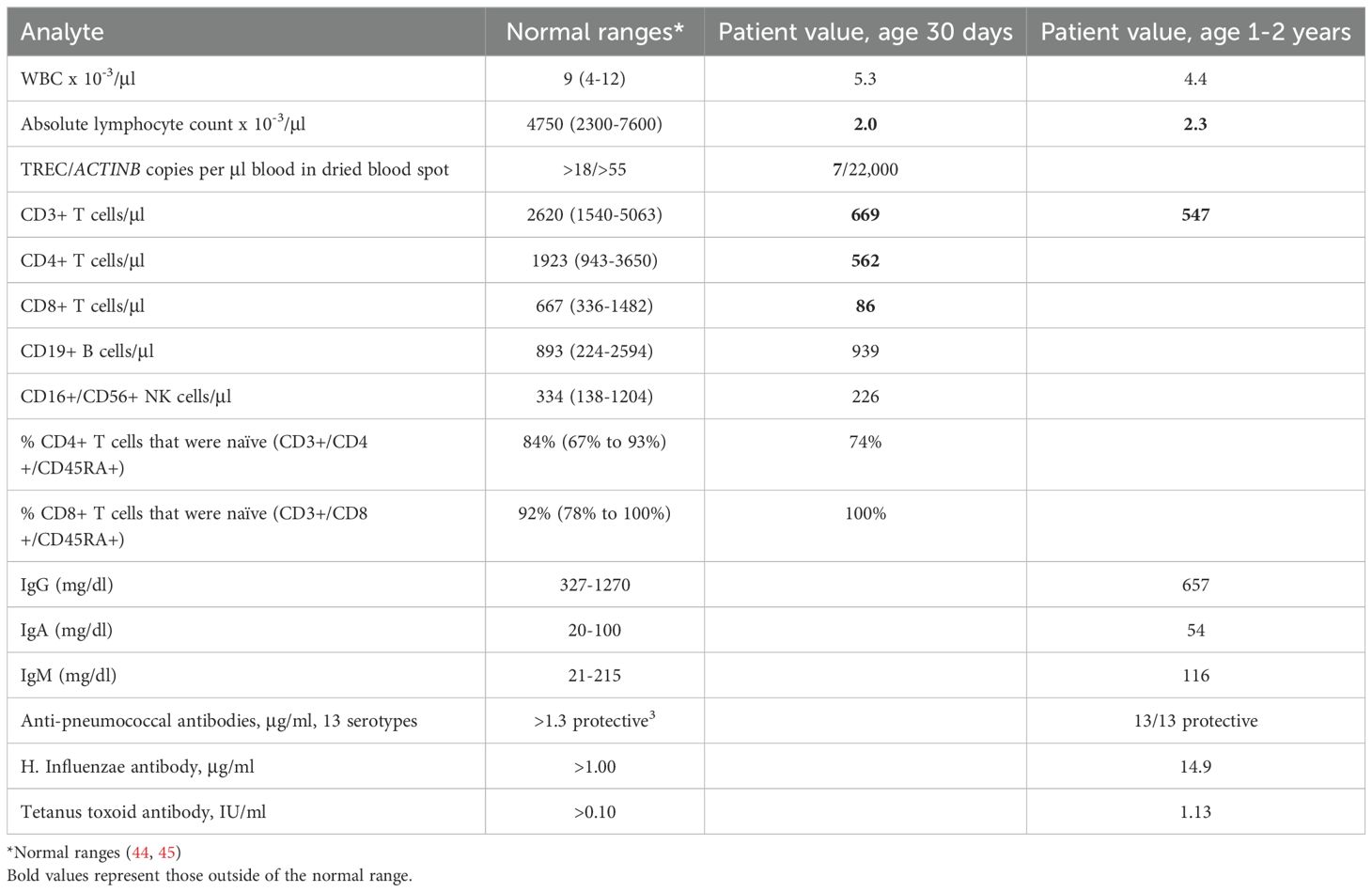
Table 2. Patient Laboratory Test Values.
Identification and vetting of patient variants found upon exome sequencing
While sequencing of known genes implicated in immunodeficiency revealed no mutations, whole exome sequencing (WES) revealed several potential variants, with the most likely variants being mutations in CTBP2 and CHTF18. CTBP2 is a transcriptional corepressor which binds Proline-Isoleucine-Aspartate-Leucine-Serine (PIDLS) to repress target genes, including IL-2 (48, 49). In zebrafish, ctbp2a/ribeye a and ctbp2l/ribeye b are the major proteins in synaptic ribbons and loss of both genes leads to decreased electron density, synaptic localization and recruitment of calcium channels (50). The patient had two variants in CTFBP2: 1) a de novo CTFBP2 D437N missense mutation affecting an aspartic acid (D) residue conserved in mice and zebrafish Ctbp2a isoform 2, but not in Ctbp2a isoform 1 or Ctbp2l; and 2) a 12aa insertion following P389 in a portion of the protein conserved in all isoforms (Supplementary Figure 1A). To investigate the role of CTBP2 in T cell development, we employed morpholino oligonucleotides (MO) to knock down ctbp2 expression in zebrafish embryos and assessed the impact on T cell development by performing whole mouse in situ hybridization (WISH) using a T cell specific lck probe at five days post fertilization (dpf; Supplementary Figure 1B). Splice site MO targeting ctbp2a, ctbp2l, alone or in combination attenuated ctbp2a and ctbp2l expression, but did not arrest in T cell development, suggesting that the CTBP2 patient variants were not responsible for disease (Supplementary Figure 1B).
The only other highly ranked set of variants impacted the CHTF18 gene. The CTF18 protein, encoded by the CHTF18 gene, is a component of a secondary clamp loader complex, which loads PCNA onto DNA during replication (16, 17). CTF18 forms a pentamer with RFC2, RFC3, RFC4 and RFC5 (51). The proband exhibited two maternal (R751W and V776L) and one paternal (E851Q) missense variant in the human CHTF18 gene, in residues that are conserved from human to mouse and zebrafish (Figures 1, 2A; Supplementary Figure 2). To assess the extent to which these variants might disrupt the structure of the CTF18-containing secondary clamp loader, we examined a recently solved cryo-EM structure of the complex (51) and compared it to that of the RFC1-containing primary clamp loader complex using AlphaFold 2 (AF2) and AlphaFold 3 (AF3), with a particular focus on the impact of the CTF18 variants (Figures 2B, C) (32, 33, 54) (39). In the RFC1 complex, the RFC3-R9 residue is a key component of the ADP binding pocket and the ε amino group hydrogen bonds to the 3´ hydroxyl of ADP (Figure 2B). The analogous CTF18 loop, which is in close contact with RFC-R9, revealed a slightly different conformation containing a short alpha helix, as there are two extra amino acids in this CTF18 loop compared to the RFC1 loop (Figure 2C). Nevertheless, the analogous CTF18 residue to RFC1-R986, CTF18-R751, also points toward RFC3-R9 and the ADP binding pocket (Figures 2B, C; arrow), suggesting that it may also contribute to the dynamics of ADP binding and the ATP hydrolysis cycle. Moreover, the CryoEM structure led to speculation that the alternative CTF18 clamp loader complex is a much more mobile multi-subunit assembly compared to the canonical RFC1-based clamp loader. This may lower loading activity, thus limiting the alternative clamp loader complex to leading strand loading (51). Interestingly, the AF3 models of R751W variant forces a downward rotamer change for RFC3-R9 resulting in RFC3-R9 stably making four hydrogen bonds with either RFC3-D6 or the 3’hydroxyl of ADP (see blue dashed lines in Figure 2D, arrows indicate R751W and RFC3 R9). The R751W variant could cause a perturbation of the ATP hydrolysis cycle (i.e. slower ADP release) by rigidly fixing the rotamer of RFC3-R9. In contrast, the CTF18-E851Q variant is located away from the interface with RFC3 in a solvent exposed location and not near any other subunit in the CryoEM structures (Figure 2E, arrow indicates E851Q). This is a mild side chain substitution and it is unclear how this variant could alter function.

Figure 1. Sequence analysis of the parent and proband CHTF18 alleles. NextGen sequence traces of the paternal, maternal, and proband CHTF18 alleles are depicted, with the sequence variants boxed in white.
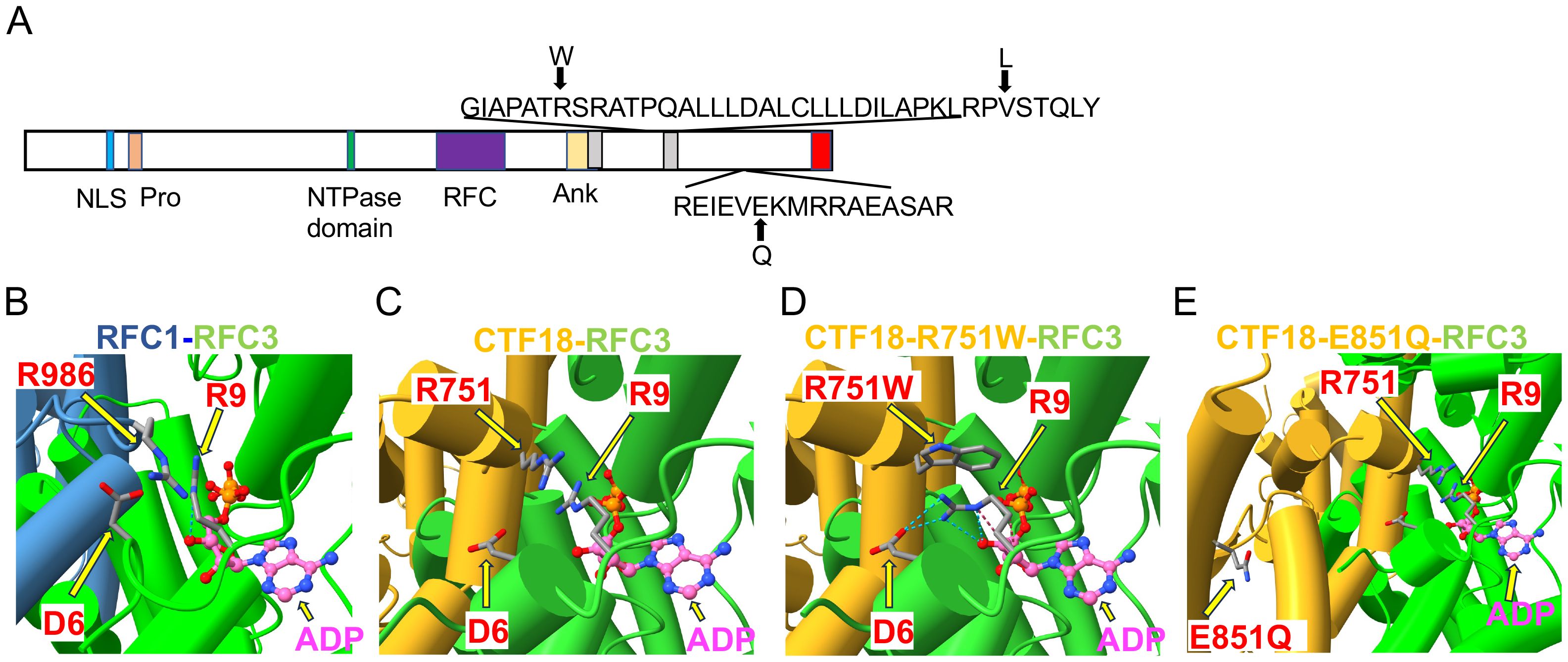
Figure 2. Structural analysis of CHTF18 patient variants. (A) The structure of human CTF18 is schematized with the structural domains and the location of variants inherited from the mother and father indicated above and below the diagram, respectively (52). NLS, nuclear localization signal; Pro, proline-rich domain; Ank, ankyrin repeat domain; TBD, triple barrel domain in red where CTF18 interacts with DSCC1 and CTF8 (53). (B-E) Clamp loader interface of either RFC1 or CTF18 with RFC3. (B) RFC1 structure (PDB #6VVO) shown in blue cylinders for helices and RFC3 in green. The ADP molecule (displayed with pink carbon atoms) bound by RFC3 is shown in ball-and-stick representation, and the key residues RFC1-986 and RFC3-R9 are labeled. (C) AF3 model of CTF18 (gold; residues 583-863) bound to RFC3. The wildtype R751 makes contact with RFC3-R9 in a similar manner compared to RFC1-R986. (D) AF3 model of the R751W variant makes more extensive contacts with RFC3-R9, changing the RFC-R9 rotamer conformation to where it makes 4 hydrogen bonds with RFC-D6 and the 3’ hydroxyl of ADP. (E) AF3 model of the CTF18-E851Q variant in a solvent exposed location distal from the RFC3 interface.
Functional tests of CTF18 variants in zebrafish
To investigate the role of CTF18 protein in supporting T-cell development in vivo, we employed zebrafish, a tractable genetic model in which hematopoietic development is highly conserved (26, 55). Expression of the zebrafish chtf18 ortholog was knocked down using a MO that interferes with chtf18 splicing and induces nonsense mediated decay (Figure 3A). The chtf18 MO attenuated chtf18 expression and markedly impaired T cell development at 5dpf, as indicated by decreased whole mount in situ hybridization (WISH) staining with an RNA probe for the T cell specific kinase, lck (Figure 3A). The blockade in T cell development caused by knockdown of chtf18 indicated that Ctf18 performs an important function in supporting T cell development; however, it did not provide insight into whether the CTF18 patient variants damage CTF18 function. To determine whether the patient variants (R751W, V776L and E851Q) abrogated the function of CTF18 protein, we performed rescue experiments in zebrafish embryos. Endogenous chtf18 was knocked down by MO as in Figure 3A and heat shock induction was employed to re-express the wild type and variant human CHTF18 orthologs to determine if they rescue the block in T cell development caused by loss of zebrafish Ctf18 (Figure 3B) (9, 10). Re-expression of the wild type (WT) human ortholog compensated for the loss of Ctf18 and rescued T cell development, indicating that CTF18 function was conserved from human to zebrafish (Figure 3B). Moreover, the maternally inherited V776L variant also rescued development, indicating that this mutation is not damaging (Figure 3B); however, re-expression of either the R751W (maternal) or the E851Q (paternal) variant failed to rescue T cell development, indicating that each of these variants damage CTF18 function (Figure 3B). Taken together, these data suggest that T cell insufficiency in patient JPSCID-15 may result from compound heterozygosity of two damaging CHTF18 mutations (R751W and E851Q).
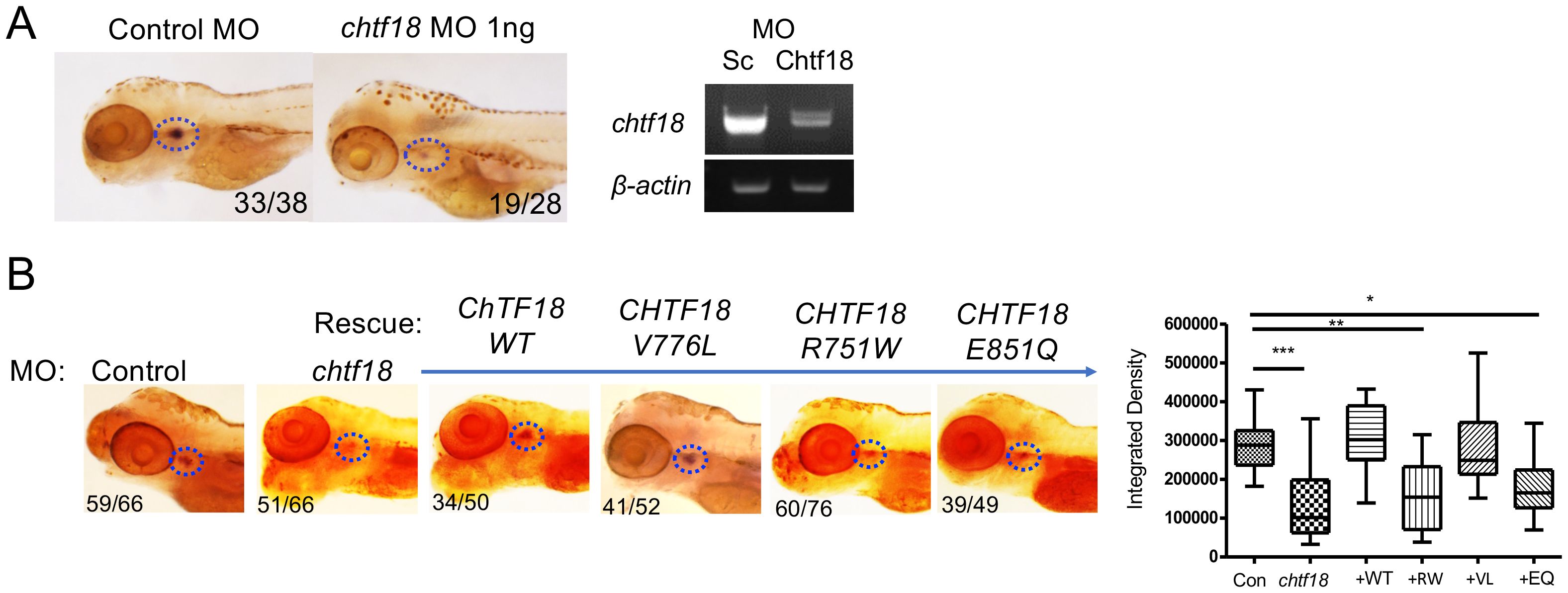
Figure 3. CHTF18 patient variants fail to support T cell development. (A) Effect of chtf18 knockdown on T cell development at 5 days post fertilization (dpf) in zebrafish embryos. T cell development was assessed by whole-mount in situ hybridization (WISH) using a probe for the T cell specific kinase, lck. Numbers on images represent the fractions of embryos with the depicted phenotype. Thymus is indicated by dashed blue ovals. (right panel) The efficacy of chtf18 E2I2 splice-blocking MO (1ng) was assessed by RT-PCR at 1 dpf. (B) Capacity of patient CHTF18 variants to rescue T cell development in zebrafish. Lateral images of zebrafish treated with control or chtf18 MO and subjected to rescue by heat-induced re-expression of the WT or mutant human CHTF18 orthologs. Rescue of T-cell development was evaluated by lck WISH at 5 dpf. Thymus staining is indicated by dashed blue ovals and the fraction of embryos with the depicted phenotype is indicated on each micrograph. The intensity of the lck WISH signal was quantified by ImageJ software and is depicted graphically as box and whisker plots. Statistical significance was evaluated using one way ANOVA. *p < 0.05; **p < 0.01; ***p < 0.001.
Loss of Chtf18 selectively blocks αβ T cell development in the thymus
To gain insight into the breadth and stage of developmental arrest caused by Ctf18 loss, we performed WISH using probes for other T cell subsets and thymic cell types. Chtf18 knockdown did not appear to impair thymic seeding as staining for ikaros, a marker of early thymic progenitors, was not altered (Supplementary Figure 3A). The arrest of T cell development appeared to be restricted to αβ lineage T cells, since the γδ T cell subset identified using WISH with a tcrd probe was not affected by Ctf18 loss (Supplementary Figure 3A). Moreover, the block in αβ T cell development was likely to be cell autonomous, since WISH using a foxn1 probe suggests that thymic stroma was not disrupted by chtf18 knockdown (Supplementary Figure 3A). Finally, because CTF18 functions as a secondary clamp loader during DNA replication (17, 53, 56), we reasoned that chtf18 knockdown might cause DNA damage resulting in a p53-dependent developmental arrest; however, trp53 knockdown failed to rescue development in chtf18 morphants, despite rescuing the p53-dependent arrest caused by knockdown of rpl22 (Supplementary Figure 3B) (28). Together, these findings suggest that ctf18 loss arrests the development of αβ lineage T cells in a cell-autonomous, but p53-independent, manner.
Generation of Chtf18 mutant knockin mice
To understand the mechanistic basis for Ctf18 action, we generated knockin mice bearing patient variants in the orthologous residues of mouse Ctf18, R745W and E845Q (Supplementary Figure 4). The knockin mice were generated using CRISPR-induced cutting and HDR oligos for repair. The resulting compound heterozygous Chtf18R745W/E845Q (Chtf18 mut) mice were fertile and viable, with no outward signs of abnormalities (data not shown). We next examined T cell development by flow cytometry (Figure 4A). We observed no significant difference in any of the thymic subsets defined by CD4, CD8, CD44, and CD25. Moreover, splenic T cell numbers were not diminished; nor did they exhibit any signs of homeostatic proliferation marked by changes in expression of CD62L and CD44 (Figures 4B, C) (57). Developmental defects in some cases are evident only when hematopoietic stem and progenitor cells (HSPC) are stressed (10, 58). To place Chtf18 mut HSPC under stress, we performed mixed bone marrow chimera analysis using allotype marked (CD45.1) competitor cells (Figure 5A). While the wild type Chtf18+/+ (WT) HSPC effectively reconstituted the thymus and spleen of recipient mice, the Chtf18 mut HSPC were unable to reconstitute recipient mice (Figures 5A, B). The failure to reconstitute recipient mice did not result from failure to home to the bone marrow, as a seeding assay revealed no defect in Chtf18 mut HSPC reaching the bone marrow of the recipients (Figure 5C). Together, these data indicate that Chtf18 mut HSPC are able to support hematopoiesis at baseline, but fail to do so under stress during competitive bone marrow transplantation.
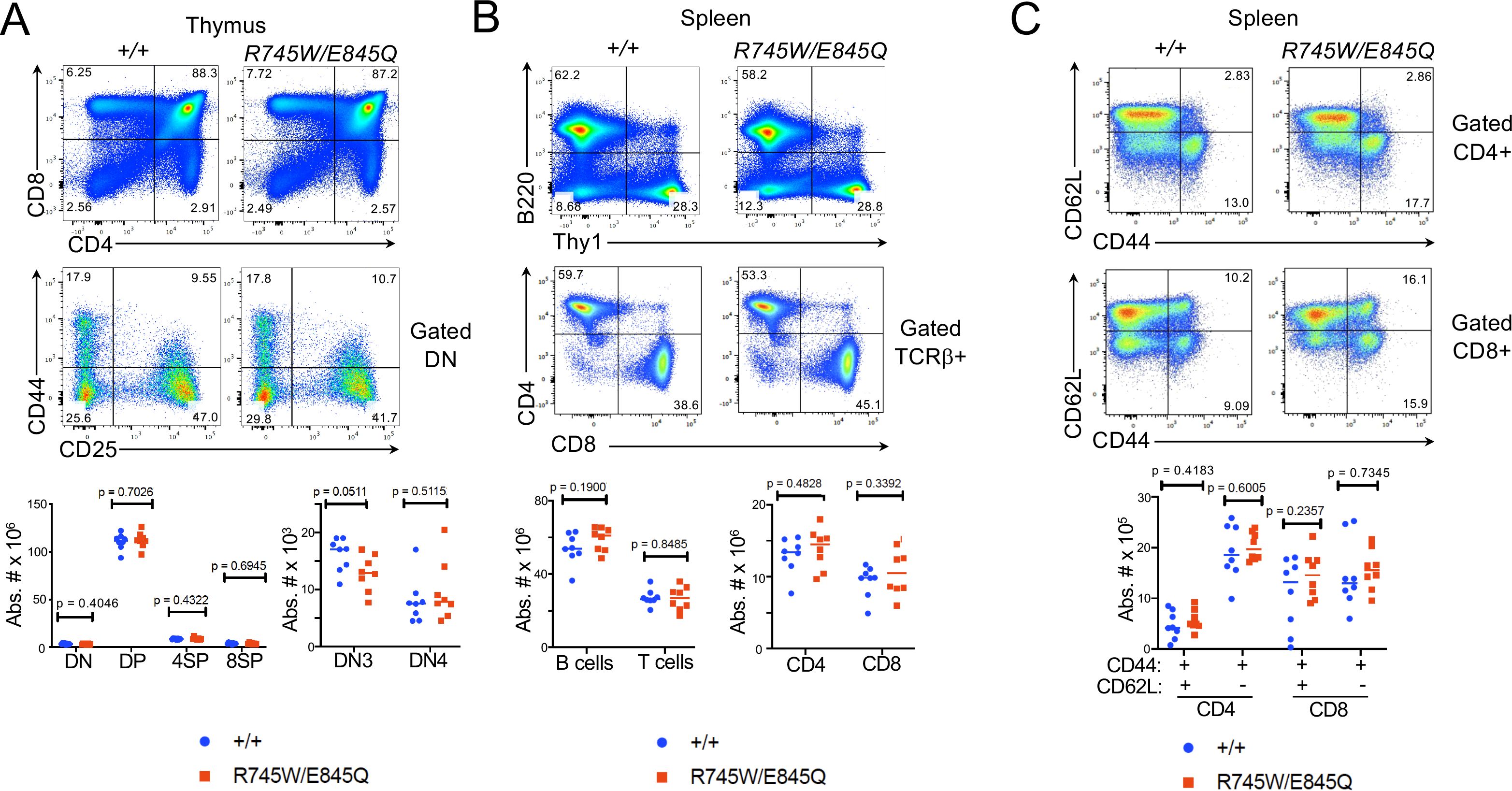
Figure 4. Phenotype of Chtf18 mutant mice. (A-C) Flow cytometric analysis of lymphoid development assessed by flow cytometry on thymus (A) and spleen (B, C). Flow cytometry analysis was performed on explanted thymus and spleen from adult WT (+/+) and Cht18 mut (R745W/E845Q) mice using the indicated antibodies to define the following populations in: (A) Thymus: CD4-CD8- (DN), CD4+CD8+ (DP), CD4+ (4SP), CD8 (8SP); (B) Spleen: B220+ (B cells); Thy1+ (T cells), CD4+, CD8+; (C) CD44+CD62L+ (effector memory); CD44+CD62L- (central memory). Absolute numbers of the indicated populations were depicted graphically as scatter plots with each symbol representing an individual animal. Statistical significance as assessed by multiple unpaired t tests.

Figure 5. Assessment of Chtf18 mut HSPC by competitive transplantation. (A, B) The ability of adult Chtf18 mut bone marrow HSPC to repopulate hematopoiesis was assessed by competitive transplantation using allotype marked (CD45.1) competitor. 100,000 Lineage– HSPC from CD45.2 WT or Chtf18 mut mice were mixed with an equal quantity of CD45.1 competitor cells and transferred into irradiated recipients. After 6 weeks, the hematopoietic reconstitution was assessed on explanted thymus (A) and spleen (B) cell suspensions by flow cytometry using anti-CD45.1 and anti-CD45.2 antibodies. Cell frequencies were displayed graphically as scatter plots. Statistical significance was assessed by multiple unpaired t tests. (C) The capacity of CD45.2 WT and Chtf18 mut HSPC to home to the bone marrow was assessed as above expect that bone marrow seeding was measured by flow cytometry 4 days after transfer. The number of transferred cells found in the recipient bone marrow was exhibited by scatter plot. Statistical significance was assessed by multiple unpaired t tests.
Transcriptomic analysis of Chtf18 mutant HSPC
To elucidate the molecular basis for the impaired capacity of Chtf18 mut HSPC to reconstitute hematopoiesis in competitive transplant experiments, we performed single-cell RNA-seq (scRNA-seq) on lineage negative (Lin-) HSPC. Lin- HSPC were selected because of their functional impairment in competitive transfer experiments, and because publicly available scRNA-Seq analysis revealed that Chtf18 mRNA levels are highest in the stem and progenitor cells in the bone marrow (Supplementary Figures 5A, B). Chtf18 mRNA levels were also found to elevated in CD8 immature single positive cells (ISP), which are transitioning from the CD4-8- double negative (DN) to the CD4+8+ double positive (DP) stage of thymocyte development (Supplementary Figure 5C, D). The scRNA-seq analysis revealed eight clusters defined by lineage specific stem cell markers derived from the Tabula Muris dataset (Supplementary Figure 6A) (https://www.czbiohub.org/sf/tabula-muris/). The clusters showed minimal changes in representation in the Chtf18 mut HSPC, except for the myeloerythroid progenitors (MEP), which were substantially reduced (Figure 6A). Pathway analysis revealed that among the differentially expressed genes (DEG), there was no enrichment for DNA-damage response pathways, but there was marked enrichment in pathways related to interferon signaling and protein folding (Figure 6B; Supplementary Figures 6B, C). Expression of the remaining clamp loader components (RFC2-5) was increased in Chtf18mut myeloid progenitors (Figure 6C). Interestingly, expression of Myc, which has been implicated in controlling HSC quiescence, was markedly elevated in Chtf18 mut HSC/MPP (Figure 6C) (59). Using transcriptomic analysis to infer cell cycle status, we found that long-term HSC (LT-HSC), short term HSC (ST-HSC) and CD34 expressing LT-HSC (LT-34F) all exhibited an increase in cells in cycle (G2M/S) in the Chtf18 mut mice, consistent with the potential loss of quiescence by HSC (Figure 6D).
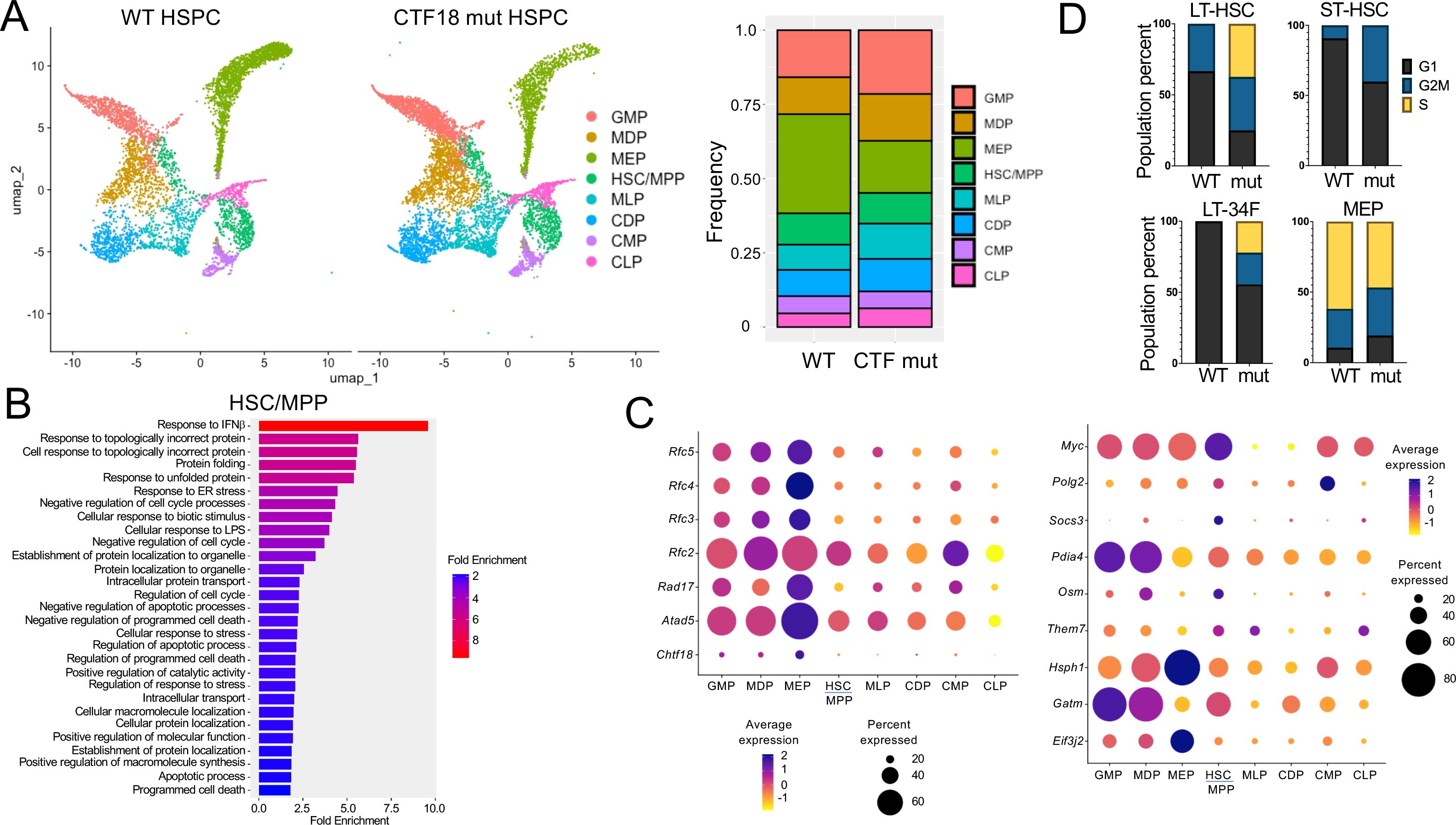
Figure 6. scRNA-Seq analysis of WT and Chtf18 mut HSPC. (A) Uniform Manifold Approximation Projection (UMAP) plots showing eight distinct HSPC clusters and their relative frequencies in Lineage- HSPC from Chtf18 wildtype and mut mice. (B) Gene Set Enrichment analysis (EnrichGo) of the highly variable expressed genes between Chtf18 wildtype and mut HSC/MPP cluster. (C) Bubble plot representation showing expression of DNA clamp loader proteins and top upregulated genes in the Chtf18 mut HSPCs. (D) Percentage of cells in G1, G2/M and S phase in Chtf18 WT and mut HSPCs. Reference cell type abbreviations: LT-34F, CD34+ Long-term reconstituting stem cell; LT-HSC, CD34- Long-term reconstituting stem cell; MEP, Megakaryocyte-Erythroid Progenitor; CMP, Common Myeloid Progenitor; GMP, Granulocyte-Monocyte Progenitor; MLP, Multilineage Progenitor; HSC/MPP, Hematopoietic Stem Cell and Multi-Potent Progenitor; CLP, Common Lymphoid Progenitor; MDP, Monocyte-Dendritic cell Progenitor; CDP, Common Dendritic cell Progenitor.
Analysis of the basis for impaired Chtf18 mut HSPC function
The increased expression of Myc and proliferative transcriptomic signature of Chtf18 mut HSC raised the possibility that their failure to reconstitute hematopoiesis in the competitive setting resulted from exhaustion. To test this possibility, we performed competitive transfers using FL HSC, which are highly proliferative (60). Interestingly, Chtf18 mut FL HSC also failed to support hematopoiesis upon competitive transfer into irradiated hosts (Supplementary Figure 7A). The failure of Cht18 mut HSC to support hematopoiesis was not evident in non-competitive transfers (Supplementary Figure 7B), indicating that Chtf18 mut HSPC had the capacity to support hematopoiesis but were unable to do so when forced to compete with fully competent WT HSPC. The elevated transcriptomic signatures of IFN signaling and protein folding in Cht18 mut HSPC were reminiscent of what is observed in some mouse models of Fanconi Anemia (Fanca-/-) (61, 62), in which defects do not manifest until stressed (42). To test this possibility, Chtf18 mut mice were repeatedly stressed by biweekly pIpC treatments; however, unlike the lethality and hematopoietic defects observed following serial treatment of Fanca-/- mice with pIpC, biweekly treatment of Chtf18 mut mice had no significant impact on their viability or peripheral lymphoid populations (Supplementary Figure 7C). Likewise, Cht18 mut FL HSPC grew normally in vitro and did not exhibit increased sensitivity to interferon-β (IFNβ), heat shock protein inhibitors (Ganetespib), or DNA damage induced by Mitomycin c (MMC) (Supplementary Figures 7D–F), unlike what has been observed for cells with mutations in FANC family proteins (42, 61, 62). Finally, contrary to reports in cell lines lacking CTF18, Chtf18 mutant HSPC did not exhibit increased sensitivity to the chain-terminating chemotherapeutic agent, Cytrabine (AraC) (Supplementary Figure 7G) (63).
Because the defect in Chtf18 mutant HSPC did not phenocopy that seen in Fanca-/- mice, we re-examined the expression signature alterations in Chtf18 mut HSPC. Interestingly, we found that Chtf18 mut common lymphoid progenitors (CLP) exhibited changes in the expression of genes regulating cell-cell interactions, migration, and chemotaxis (Figures 7A; Supplementary Figure 8A). Specifically, the expression of several Rho family GTPases and their regulators (RhoA, Cdc42, Pak2, and Rock1) was diminished in Chtf18 mut CLP (Figures 7A; Supplementary Figure 8A). Rho family GTPases have been shown to regulate HSC migration, and localization, in addition to playing a critical role in the asymmetric division that is required to balance self-renewal with differentiation (64–67). Indeed, loss of these effectors markedly perturbs these processes and so may explain the defective function of Chtf18 mut HSPC in the competitive transplant setting (64–66). To determine if Chtf18 mut HSPC caused the mis-localization of HSPC, we employed the zebrafish model, in which HSPC positioning can be readily evaluated in embryos by WISH using a runx1 probe to identify emerging HSPC (Figure 7B). Indeed, we observed that knockdown of chtf18 using MO resulted in a marked dispersal of HSPC away from the embryo midline, indicating that Ctf18 loss caused the mis-localization of HSPC (Figure 7B). Together, these data suggest that the competitive disadvantage displayed by Chtf18 mut HSPC resulted from alteration of their positioning during development.

Figure 7. Impact of Chtf18 variants on HSPC migration. (A) Gene Set Enrichment analysis (EnrichGo) of the highly variable expressed genes between Chtf18 WT and mut CLP cluster (upper panel). (Bottom Panel) Volcano plot of the top differentially expressed genes between Chtf18 WT and mut CLP cluster. (B) The impact of Ctf18 loss on the localization of HSPC in zebrafish embryos was examined by MO knockdown of chtf18 (chtf18e6), following which the location of HSPC in the caudal hematopoietic tissue was assessed by WISH using a probe for runx1. Statistical significance was assessed by Fisher’s Exact Test, with the p=6.8x10-7 for Con. vs Ctfe6.
Discussion
Here we report our investigation of the etiology of T lymphopenia in a male proband identified through newborn screening, but who was unfortunately lost to follow-up with only limited clinical and laboratory data available. Informatic analysis of WES data from the parents and proband revealed that the highest scoring variant was a component of a secondary clamp loader, CHTF18, a novel candidate in immunodeficiency (16). The proband exhibited compound heterozygosity of the CTF18-R751W maternal and CTF18-E851Q paternal missense variants. Functional analysis in zebrafish revealed that both mutations damaged CTF18 function and impaired their ability to support T cell development, suggesting that the proband’s T lymphopenia resulted from autosomal recessive inheritance of compound-heterozygous, damaging mutations in the CHTF18 gene. Modeling the orthologous, compound-heterozygous mutation in mice produced a distinct phenotype. While there was no baseline deficit in T cell development specifically or more generally in hematopoiesis in the Chtf18 mut mice, the compound heterozygous mutations in Chtf18 abrogated HSPC function in the competitive transplant setting. The defect in function was associated with reduced expression in Rho family GTPases and their regulators, which in turn may disturb cell-cell contacts and migration of HSPC, since they exhibited altered positioning in zebrafish embryos. Taken together, these data indicate that impairing CTF18 function negatively impacts hematopoietic development through a novel mechanism that is unrelated to its canonical role as a secondary clamp loader in supporting DNA replication (16, 17).
The proband was initially identified by TREC-based newborn screening, which has both enhanced treatment efficacy for SCID and led to the identification of a number of novel immunodeficiency genes (1, 6, 9, 10, 22). While initially presenting with neonatal T lymphopenia that did not resolve in over two years, the infant experienced normal growth, and recovered from one episode of pneumonia, before being lost to follow-up. WES implicated two distinct variants with the potential to cause disease, CHTF18 and CTBP2. CTBP2 is a transcriptional corepressor that represses target genes, including IL-2 (48, 49), but its elimination did not impair T cell development in zebrafish. It remains possible that the two putative inactivating CTBP2 variants in the proband could impair CTBP2 function, leading to IL-2 de-repression, which at very high levels can cause severe side effects (68); however, the patient did not manifest symptoms related to excess IL-2 in early life. In contrast, CTF18 loss in zebrafish attenuated T cell development, attesting to the importance of CTF18 function in supporting T cell development. Moreover, replacement of zebrafish Ctf18 with wild type human CTF18 restored development, indicating conservation of CTF18 function from human to zebrafish. Both the maternal (R751W) and paternal (E851Q) variants failed to complement the loss of zebrafish Ctf18 in restoring T cell development, supporting the conclusion that the patient’s compound heterozygous mutations in CHTF18 were responsible for the proband’s T lymphopenia. Moreover, our zebrafish studies suggested that impaired T cell development might be restricted to αβ T cells after thymic seeding, which is consistent with the normal numbers and function of B and NK cells in the patient. Together, these data suggest that CHTF18 mutations like those in this patient are more likely to be associated with T lymphopenia than SCID; however, it remains possible that other variants that more severely attenuate CTF18 function, or alter canonical functions of CTF18, might cause SCID. Finally, the block in zebrafish T cell development caused by loss of Ctf18 function was not dependent upon p53, suggesting that it was not the result of accumulated DNA damage, as might be expected if the developmental block was caused by loss of Ctf18’s support for DNA-replication as part of a clamp loader complex (53, 69).
Mice bearing orthologous mutations exhibited a different phenotype than was observed in zebrafish and our human infant, in that there was no baseline phenotype but the Chtf18 mut HSPC were severely impaired in their capacity to reconstitute hematopoiesis in the competitive transplant setting. This may relate to the observation that MO knockdown in zebrafish can result in mosaic loss of target gene expression, creating a de facto competitive context similar to that seen during competitive transplantation in mice (70). Nevertheless, the murine developmental arrest was far earlier than that observed in zebrafish, in that HSPC were unable to seed the thymus or contribute to the production of any blood lineage.
We have previously found that zebrafish better modeled human disease than mice. Indeed, loss of zebrafish and human ARPC1B caused a block in T cell and thrombocyte development (15); however, Arpc1b-deficient mice do not exhibit a change in T cells or thrombocytes, but do exhibit one aspect of the anomalies seen in patients, increased IgE levels (71). Moreover, the interleukin 7 receptor alpha (IL7Rα) chain plays a critical role in T cell development in humans, mice and zebrafish. However, while IL7Rα-loss in humans and zebrafish selectively reduces T cells (72–74), IL7Rα-deficiency in mice reduces both B and T cells (75). A final example of zebrafish more faithfully modeling human disease relates to the impact of loss-of-function of PLOD2. PLOD2 mutations in humans cause Bruck syndrome, a disease marked by bone fragility and congenital joint contractures (76), which is faithfully modeled by Plod2-deficient zebrafish that exhibit a shortened body axis and severe skeletal abnormalities with evidence of bone fragility (77). In contrast, Plod2-deficient mice are embryonically lethal due to exacerbation of ER stress and apoptosis (78). These examples emphasize how the zebrafish can more faithfully model human disease than mice bearing orthologous mutations.
Nevertheless, mouse models can serve as a useful platforms to gain mechanistic insights into the basis by which a human variant disrupts function. CTF18 is a component of a secondary clamp loader (16, 51, 53). The primary clamp loader complex comprises five components, the large RFC1 subunit and four smaller RFC subunits, RFC2-5 (16, 17). Primary clamp loaders ensure that the trimeric PCNA ring, which acts a clamp, is loaded onto DNA and functions as a landing pad for the DNA polymerase and other processivity factors required for DNA replication (16, 17). Loss of RFC complex components is lethal in yeast and higher vertebrates (18, 19). In contrast, loss of CTF18, the largest subunit of a secondary clamp loader, is not lethal in yeast or mice, though its loss does result in non-Mendelian inheritance of knockout offspring, which are smaller and have reduced testis size (20, 79). The Chtf18 mut mice (R745W/E845Q) were born with Mendelian inheritance ratios and exhibit no defects in fertility (data not shown). Structural modeling of the CHTF18 patient variants based on a recent cryo-EM structure of the CTF18 clamp loader suggested that the stability of the CTF18 interaction with the RFC3 subunit would be altered by R751W, thereby affecting catalysis (51); however, because E851Q was solvent facing, its impact on clamp loader function was unclear, but could affect the interaction of CTF18 and pol ∈. Nevertheless, the structural modeling will not be informative regarding the mechanism of action of the CHTF18 variants, if the role of CTF18 is supporting hematopoiesis is unrelated to its clamp loader function.
The absence of a DNA-damage signature in the scRNA-Seq analysis of Chtf18 mut HSPC and the normal proliferation exhibited by Chtf18 mut FL progenitors suggests that CTF18 may be supporting hematopoiesis through activities unrelated to clamp loader function. Consistent with this idea, both CTF18 and RFC1 have also been shown to have functions independent of their clamp loading function. RFC1 can regulate NFKB activity (80) and inhibit the activity of polymerase η (Polη), while CTF18 stimulates Polη activity (69). Polη has also been shown to be enriched in actively transcribed regions of the yeast genome, so this could be a mechanism by which CTF18 could influence transcription (81). CTF18 has also been implicated in cohesin loading onto chromatin, which could influence gene expression through chromatin looping (82, 83). Finally, CTF18 has been found to associate with the nuclear pore complex, which has been implicated in gene silencing at sub-telomeric regions (84). Consequently, any of these mechanisms could be responsible for the changes in gene expression exhibited by Chtf18 mut HSPC, including signatures for interferon signaling, protein folding, migration and RhoA GTPase signaling. The interferon and protein folding signatures were similar to those found in Fanca-/- mice, which also had no baseline phenotype, but exhibited HSC attrition after repeated inflammatory insults (42); however, Chtf18 mut HSPC were not preferentially susceptible to attrition in response to those treatments. Interestingly, the expression of genes from the Rho family of GTPases, and their regulators, were reduced in Chtf18 mut HSPC. Rho family GTPases have been shown to regulate HSC migration, localization and mobilization in addition to playing a critical role in the asymmetric division that is required to balance self-renewal with differentiation (64–67). Consequently, downregulation of these effectors is likely to explain the defective function of mouse Chtf18 mut HSPC in the competitive transplant setting and the mis-localization of HSPC in zebrafish embryos. Specifically, CXCR4 signaling in response to CXCL12 produced by Leptin Receptor expressing cells in the perivascular niche plays a critical role in the proper positioning required to support HSC function (85–87). Thus, reduced chemokine receptor signaling may impair migration of HSC to these supportive perivascular niches, thereby depriving them of the trophic signals required for development. We have previously found that perturbation of Ccr7 and Ccr9 signaling displaced bcl11b mutant HSPC, although in this case their displacement resulted from increased signaling and so hematopoiesis was restored by repression of Ccr9 (9). Whether restoration of Rho family GTPase function is sufficient to reestablish the hematopoietic potential of Chtf18 mutant HSPC remains to be tested. The impact of the Chtf18 mut on HSPC localization caused different perturbations of hematopoiesis in mice and zebrafish, with Chtf18 mut mouse HSPC showing no activity in the competitive setting, and the zebrafish HSPC being able to seed the thymus but not support T cell development. The basis for this difference is unclear but both defects could be the result of failure to maintain cell-cell contacts or migrate properly, given that intrathymic migration is critical for normal T cell development (88–90).
While the precise mechanism by which the CHTF18 missense mutations could disrupt hematopoiesis and reasons for species differences in the impact of those mutations remain unclear, the analysis in zebrafish appears to align with the defect observed in the proband. Moreover, these studies highlight how the analysis of human genetic variants can uncover novel non-canonical functions for these candidate genes. Indeed, together our data identify CHTF18 as a novel immunodeficiency gene that supports T cell development through its capacity to influence gene expression in a non-canonical manner, distinct from its traditional role as a secondary clamp loader. It remains be determined how the CHTF18 missense mutations interfere with its noncanonical function, but this is being actively investigated.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation. WES data from the JPSCID-15 trio were submitted to dbGaP, accession number phs002968.v1.p1. scRNA-Seq data for Ctf18wt and mutant HSPC have been deposited in GEO, Accession #GSE289457; GSM8791734; GSM8791735; GSM8791736; GSM8791737.
Ethics statement
Research activities were performed with parental informed consent under protocols approved by the institutional review board (IRBs) at the University of California, San Francisco. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. The animal studies were approved by the Fox Chase Cancer Center Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
RoS: Conceptualization, Data curation, Formal analysis, Writing – original draft. BT: Data curation, Formal analysis, Writing – review & editing. MS: Formal analysis, Writing – review & editing. SS: Formal analysis, Writing – review & editing. RP: Formal analysis, Writing – review & editing. AS: Data curation, Formal analysis, Writing – review & editing. PS: Data curation, Formal analysis, Writing – review & editing. US: Data curation, Formal analysis, Writing – review & editing. SR: Data curation, Formal analysis, Writing – review & editing. RaS: Data curation, Formal analysis, Writing – review & editing. SD: Data curation, Formal analysis, Writing – review & editing. JF-B: Methodology, Writing – review & editing. SB: Conceptualization, Data curation, Formal analysis, Writing – review & editing. JP: Conceptualization, Data curation, Formal analysis, Writing – review & editing. DW: Conceptualization, Data curation, Formal analysis, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Institutes of Health (NIH) grants P30CA006927, P01AI138962, and the Bishop Fund. SB received support from TCS to the University of California, Berkley.
Acknowledgments
We thank the following Fox Chase Cancer Center core facilities for their vital service: Cell Culture, Cell Sorting, Imaging, Molecular Modeling, Transgenic Mouse, Laboratory Animal/Zebrafish.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1539848/full#supplementary-material
References
1. Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. Jama. (2014) 312:729–38. doi: 10.1001/jama.2014.9132
2. Currier R, Puck JM. SCID newborn screening: What we’ve learned. J Allergy Clin Immunol. (2021) 147:417–26. doi: 10.1016/j.jaci.2020.1010.1020
3. Dvorak CC, Haddad E, Heimall J, Dunn E, Buckley RH, Kohn DB, et al. The diagnosis of severe combined immunodeficiency (SCID): The Primary Immune Deficiency Treatment Consortium (PIDTC) 2022 Definitions. J Allergy Clin Immunol. (2023) 151:539–46. doi: 10.1016/j.jaci.2022.1010.1022
4. Thakar MS, Logan BR, Puck JM, Dunn EA, Buckley RH, Cowan MJ, et al. Measuring the effect of newborn screening on survival after haematopoietic cell transplantation for severe combined immunodeficiency: a 36-year longitudinal study from the Primary Immune Deficiency Treatment Consortium. Lancet. (2023) 402:129–40. doi: 10.1016/S0140-6736(1023)00731-00736
5. Forlanini F, Chan A, Dara J, Dvorak CC, Cowan MJ, Puck JM, et al. Impact of genetic diagnosis on the outcome of hematopoietic stem cell transplant in primary immunodeficiency disorders. J Clin Immunol. (2023) 43:636–46. doi: 10.1007/s10875-10022-01403-10875
6. Puck JM. Neonatal screening for severe combined immunodeficiency. Curr Opin Pediatr. (2011) 23:667–73. doi: 10.1097/MOP.1090b1013e32834cb32839b32830
7. Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. J Allergy Clin Immunol. (2012) 129:607–16. doi: 10.1016/j.jaci.2012.1001.1032
8. Mallott J, Kwan A, Church J, Gonzalez-Espinosa D, Lorey F, Tang LF, et al. Newborn screening for SCID identifies patients with ataxia telangiectasia. J Clin Immunol. (2013) 33:540–9. doi: 10.1007/s10875-10012-19846-10871
9. Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A, et al. Multisystem anomalies in severe combined immunodeficiency with mutant BCL11B. N Engl J Med. (2016) 375:2165–76. doi: 10.1056/NEJMoa1509164
10. Sertori R, Lin JX, Martinez E, Rana S, Sharo A, Kazemian M, et al. Investigation of the causal etiology in a patient with T-B+NK+ immunodeficiency. Front Immunol. (2022) 13:928252. doi: 10.3389/fimmu.2022.928252
11. Trede NS, Langenau DM, Traver D, Look AT, Zon LI. The use of zebrafish to understand immunity. Immunity. (2004) 20:367–79. doi: 10.1016/S1074-7613(04)00084-6
12. Lyu X, Sang PB, Chai W. CST in maintaining genome stability: Beyond telomeres. DNA Repair (Amst). (2021) 102:103104. doi: 10.1016/j.dnarep.2021.103104
13. He Q, Lim CJ. Models for human telomere C-strand fill-in by CST-Polα-primase. Trends Biochem Sci. (2023) 48:860–72. doi: 10.1016/j.tibs.2023.1007.1008
14. Simon AJ, Lev A, Zhang Y, Weiss B, Rylova A, Eyal E, et al. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J Exp Med. (2016) 213:1429–40. doi: 10.1084/jem.20151618
15. Somech R, Lev A, Lee YN, Simon AJ, Barel O, Schiby G, et al. Disruption of thrombocyte and T lymphocyte development by a mutation in ARPC1B. J Immunol. (2017) 199:4036–45. doi: 10.4049/jimmunol.1700460
16. Lee KY, Park SH. Eukaryotic clamp loaders and unloaders in the maintenance of genome stability. Exp Mol Med. (2020) 52:1948–58. doi: 10.1038/s12276-12020-00533-12273
17. Arbel M, Choudhary K, Tfilin O, Kupiec M. PCNA loaders and unloaders-one ring that rules them all. Genes (Basel). (2021) 12:1812–35. doi: 10.3390/genes12111812
18. Howell EA, Mcalear MA, Rose D, Holm C. CDC44: a putative nucleotide-binding protein required for cell cycle progression that has homology to subunits of replication factor C. Mol Cell Biol. (1994) 14:255–67. doi: 10.1128/mcb.14.1.255-267.1994
19. Cullmann G, Fien K, Kobayashi R, Stillman B. Characterization of the five replication factor C genes of Saccharomyces cerevisiae. Mol Cell Biol. (1995) 15:4661–71. doi: 10.1128/MCB.15.9.4661
20. Mayer ML, Gygi SP, Aebersold R, Hieter P. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol Cell. (2001) 7:959–70. doi: 10.1016/S1097-2765(01)00254-4
21. Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, Kohn DB, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J Allergy Clin Immunol. (2013) 132:140–50. doi: 10.1016/j.jaci.2013.1004.1024
22. Thorsen J, Kolbert K, Joshi A, Baker M, Seroogy CM. Newborn screening for severe combined immunodeficiency: 10-year experience at a single referral cente-2018). J Clin Immunol. (2021) 41:595–602. doi: 10.1007/s10875-10020-00956-10877
23. Patel JP, Puck JM, Srinivasan R, Brown C, Sunderam U, Kundu K, et al. Nijmegen breakage syndrome detected by newborn screening for T cell receptor excision circles (TRECs). J Clin Immunol. (2015) 35:227–33. doi: 10.1007/s10875-10015-10136-10876
24. Mills RE, Pittard WS, Mullaney JM, Farooq U, Creasy TH, Mahurkar AA, et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. (2011) 21:830–9. doi: 10.1101/gr.115907.115110
25. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. (2014) 5:162. doi: 10.3389/fimmu.2014.00162
26. Sertori R, Zhang Y, Wiest DL. Zebrafish: A tractabl model for analysis of T cell development. Methods Mol Biol. (2023) 2580:355–77. doi: 10.1007/1978-1001-0716-2740-1002_1022
27. Draper BW, Morcos PA, Kimmel CB. Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis. (2001) 30:154–6. doi: 10.1002/gene.v30:3
28. Zhang Y, Duc AC, Rao S, Sun XL, Bilbee AN, Rhodes M, et al. Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev Cell. (2013) 24:411–25. doi: 10.1016/j.devcel.2013.01.018
29. Bajoghli B, Aghaallaei N, Heimbucher T, Czerny T. An artificial promoter construct for heat-inducible misexpression during fish embryogenesis. Dev Biol. (2004) 271:416–30. doi: 10.1016/j.ydbio.2004.04.006
30. Bennett CM, Kanki JP, Rhodes J, Liu TX, Paw BH, Kieran MW, et al. Myelopoiesis in the zebrafish, Danio rerio. Blood. (2001) 98:643–51. doi: 10.1182/blood.V98.3.643
31. Schorpp M, Leicht M, Nold E, Hammerschmidt M, Haas-Assenbaum A, Wiest W, et al. A zebrafish orthologue (whnb) of the mouse nude gene is expressed in the epithelial compartment of the embryonic thymic rudiment. Mech Dev. (2002) 118:179–85. doi: 10.1016/S0925-4773(02)00241-1
32. Gaubitz C, Liu X, Magrino J, Stone NP, Landeck J, Hedglin M, et al. Structure of the human clamp loader reveals an autoinhibited conformation of a substrate-bound AAA+ switch. Proc Natl Acad Sci U.S.A. (2020) 117:23571–80. doi: 10.1073/pnas.2007437117
33. Mirdita M, Ovchinnikov S, Steinegger M. ColabFold - Making protein folding accessible to all. bioRxiv. (2021) 2008:2015.456425. doi: 10.1038/s41592-022-01488-1
34. Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. (2011) 487:545–74. doi: 10.1016/B978-0-12-381270-4.00019-6
35. Khatib F, Cooper S, Tyka MD, Xu K, Makedon I, Popovic Z, et al. Algorithm discovery by protein folding game players. Proc Natl Acad Sci U.S.A. (2011) 108:18949–53. doi: 10.1073/pnas.1115898108
36. Alford RF, Leaver-Fay A, Jeliazkov JR, O’meara MJ, Dimaio FP, Park H, et al. The rosetta all-atom energy function for macromolecular modeling and design. J Chem Theory Comput. (2017) 13:3031–48. doi: 10.1021/acs.jctc.7b00125
37. Maguire JB, Haddox HK, Strickland D, Halabiya SF, Coventry B, Griffin JR, et al. Perturbing the energy landscape for improved packing during computational protein design. Proteins. (2021) 89:436–49. doi: 10.1002/prot.26030
38. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. (2004) 25:1605–12. doi: 10.1002/jcc.v25:13
39. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. (2024) 630:493–500. doi: 10.1038/s41586-41024-07487-w
40. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. (2007) 372:774–97. doi: 10.1016/j.jmb.2007.1005.1022
41. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, et al. Comprehensive integration of single-cell data. Cell. (2019) 177:1888–1902.e1821. doi: 10.1016/j.cell.2019.1805.1031
42. Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. (2015) 520:549–52. doi: 10.1038/nature14131
43. Lauritsen JP, Wong GW, Lee SY, Lefebvre JM, Ciofani M, Rhodes M, et al. Marked induction of the helix-loop-helix protein Id3 promotes the gammadelta T cell fate and renders their functional maturation Notch independent. Immunity. (2009) 31:565–75. doi: 10.1016/j.immuni.2009.1007.1010
44. Lockitch G, Halstead AC, Quigley G, Maccallum C. Age- and sex-specific pediatric reference intervals: study design and methods illustrated by measurement of serum proteins with the Behring LN Nephelometer. Clin Chem. (1988) 34:1618–21. doi: 10.1093/clinchem/34.8.1618
45. Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. (2015) 136:1186–1205.e1181-1178. doi: 10.1016/j.jaci.2015.1104.1049
46. Amatuni GS, Currier RJ, Church JA, Bishop T, Grimbacher E, Nguyen AA, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in californi-2017. Pediatrics. (2019) 143:e20182300. doi: 10.20181542/peds.20182018-20182300
47. Amatuni GS, Sciortino S, Currier RJ, Naides SJ, Church JA, Puck JM. Reference intervals for lymphocyte subsets in preterm and term neonates without immune defects. J Allergy Clin Immunol. (2019) 144:1674–83. doi: 10.1016/j.jaci.2019.1605.1038
48. Wang J, Lee S, Teh CE, Bunting K, Ma L, Shannon MF. The transcription repressor, ZEB1, cooperates with CtBP2 and HDAC1 to suppress IL-2 gene activation in T cells. Int Immunol. (2009) 21:227–35. doi: 10.1093/intimm/dxn1143
49. Kitamura F, Kitamura N, Mori A, Tatsumi H, Nemoto S, Miyoshi H, et al. Selective down-regulation of Th2 cytokines by C-terminal binding protein 2 in human T cells. Int Arch Allergy Immunol. (2010) 152:18–21. doi: 10.1159/000312121
50. Lv C, Stewart WJ, Akanyeti O, Frederick C, Zhu J, Santos-Sacchi J, et al. Synaptic ribbons require ribeye for electron density, proper synaptic localization, and recruitment of calcium channels. Cell Rep. (2016) 15:2784–95. doi: 10.1016/j.celrep.2016.2705.2045
51. He Q, Wang F, O’donnell ME, Li H. Cryo-EM reveals a nearly complete PCNA loading process and unique features of the human alternative clamp loader CTF18-RFC. Proc Natl Acad Sci U S A. (2024) 121:e2319727121. doi: 10.2319721073/pnas.2319727121
52. Merkle CJ, Karnitz LM, Henry-Sánchez JT, Chen J. Cloning and characterization of hCTF18, hCTF8, and hDCC1. Human homologs of a Saccharomyces cerevisiae complex involved in sister chromatid cohesion establishment. J Biol Chem. (2003) 278:30051–6. doi: 10.1074/jbc.M211591200
53. Stokes K, Winczura A, Song B, Piccoli G, Grabarczyk DB. Ctf18-RFC and DNA Pol ∈ form a stabl leading strand polymerase/clamp loader complex required for normal and perturbed DNA replication. Nucleic Acids Res. (2020) 48:8128–45. doi: 10.1093/nar/gkaa541
54. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
55. Gore AV, Pillay LM, Venero Galanternik M, Weinstein BM. The zebrafish: A fintastic model for hematopoietic development and disease. Wiley Interdiscip Rev Dev Biol. (2018) 7:e312. doi: 10.1002/wdev.1312
56. Grabarczyk DB, Silkenat S, Kisker C. Structural basis for the recruitment of ctf18-RFC to the replisome. Structure. (2018) 26:137–144.e133. doi: 10.1016/j.str.2017.1011.1004
57. Surh CD, Sprent J. Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J Exp Med. (2000) 192:F9–F14. doi: 10.1084/jem.1192.1084.f1089
58. Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, et al. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell. (2012) 11:302–17. doi: 10.1016/j.stem.2012.1008.1006
59. Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. (2004) 18:2747–63. doi: 10.1101/gad.313104
60. Manesia JK, Xu Z, Broekaert D, Boon R, Van Vliet A, Eelen G, et al. Highly proliferative primitive fetal liver hematopoietic stem cells are fueled by oxidative metabolic pathways. Stem Cell Res. (2015) 15:715–21. doi: 10.1016/j.scr.2015.1011.1001
61. Karras GI, Yi S, Sahni N, Fischer M, Xie J, Vidal M, et al. HSP90 shapes the consequences of human genetic variation. Cell. (2017) 168:856–866.e812. doi: 10.1016/j.cell.2017.1001.1023
62. Landelouci K, Sinha S, Pépin G. Type-I interferon signaling in fanconi anemia. Front Cell Infect Microbiol. (2022) 12:820273. doi: 10.3389/fcimb.2022.820273
63. Washif M, Ahmad T, Hosen MB, Rahman MR, Taniguchi T, Okubo H, et al. CTF18-RFC contributes to cellular tolerance against chain-terminating nucleoside analogs (CTNAs) in cooperation with proofreading exonuclease activity of DNA polymerase ϵ. DNA Repair (Amst). (2023) 127:103503. doi: 10.1016/j.dnarep.2023.103503
64. Cancelas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Y, Williams DA. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nat Med. (2005) 11:886–91. doi: 10.1038/nm1274
65. Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A. (2007) 104:5091–6. doi: 10.1073/pnas.0610819104
66. Zeng Y, Broxmeyer HE, Staser K, Chitteti BR, Park SJ, Hahn S, et al. Pak2 regulates hematopoietic progenitor cell proliferation, survival, and differentiation. Stem Cells. (2015) 33:1630–41. doi: 10.1002/stem.1951
67. Ugale A, Shunmugam D, Pimpale LG, Rebhan E, Baccarini M. Signaling proteins in HSC fate determination are unequally segregated during asymmetric cell division. J Cell Biol. (2024) 223:e202310137. doi: 10.202311083/jcb.202310137
68. Rokade S, Damani AM, Oft M, Emmerich J. IL-2 based cancer immunotherapies: an evolving paradigm. Front Immunol. (2024) 15:1433989. doi: 10.3389/fimmu.2024.1433989
69. Shiomi Y, Masutani C, Hanaoka F, Kimura H, Tsurimoto T. A second proliferating cell nuclear antigen loader complex, Ctf18-replication factor C, stimulates DNA polymerase eta activity. J Biol Chem. (2007) 282:20906–14. doi: 10.1074/jbc.M610102200
70. Zakaria ZZ, Eisa-Beygi S, Benslimane FM, Ramchandran R, Yalcin HC. Design and Microinjection of Morpholino Antisense Oligonucleotides and mRNA into Zebrafish Embryos to Elucidate Specific Gene Function in Heart Development. J Vis Exp. (2022) 10:3791/63324. doi: 10.63791/63324
71. Kuijpers TW, Tool ATJ, van der Bijl I, De Boer M, Van Houdt M, De Cuyper IM, et al. Combined immunodeficiency with severe inflammation and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol. (2017) 140:273–277.e210. doi: 10.1016/j.jaci.2016.09.061
72. Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet. (1998) 20:394–7. doi: 10.1038/3877
73. Iwanami N, Mateos F, Hess I, Riffel N, Soza-Ried C, Schorpp M, et al. Genetic evidence for an evolutionarily conserved role of IL-7 signaling in T cell development of zebrafish. J Immunol. (2011) 186:7060–6. doi: 10.4049/jimmunol.1003907
74. Lawir DF, Hess I, Sikora K, Iwanami N, Siamishi I, Schorpp M, et al. Evolutionary transition from degenerate to nonredundant cytokine signaling networks supporting intrathymic T cell development. Proc Natl Acad Sci U.S.A. (2019) 116:26759–67. doi: 10.1073/pnas.1915223116
75. Maki K, Sunaga S, Komagata Y, Kodaira Y, Mabuchi A, Karasuyama H, et al. Interleukin 7 receptor-deficient mice lack gammadelta T cells. Proc Natl Acad Sci U.S.A. (1996) 93:7172–7. doi: 10.1073/pnas.93.14.7172
76. Van Der Slot AJ, Zuurmond AM, Bardoel AF, Wijmenga C, Pruijs HE, Sillence DO, et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J Biol Chem. (2003) 278:40967–72. doi: 10.1074/jbc.M307380200
77. Gistelinck C, Witten PE, Huysseune A, Symoens S, Malfait F, Larionova D, et al. Loss of type I collagen telopeptide lysyl hydroxylation causes musculoskeletal abnormalities in a zebrafish model of bruck syndrome. J Bone Miner Res. (2016) 31:1930–42. doi: 10.1002/jbmr.2977
78. Kasamatsu A, Uzawa K, Hayashi F, Kita A, Okubo Y, Saito T, et al. Deficiency of lysyl hydroxylase 2 in mice causes systemic endoplasmic reticulum stress leading to early embryonic lethality. Biochem Biophys Res Commun. (2019) 512:486–91. doi: 10.1016/j.bbrc.2019.03.091
79. Berkowitz KM, Sowash AR, Koenig LR, Urcuyo D, Khan F, Yang F, et al. Disruption of CHTF18 causes defective meiotic recombination in male mice. PloS Genet. (2012) 8:e1002996. doi: 10.1371/journal.pgen.1002996
80. Anderson LA, Perkins ND. Regulation of RelA (p65) function by the large subunit of replication factor C. Mol Cell Biol. (2003) 23:721–32. doi: 10.1128/MCB.23.2.721-732.2003
81. Gali VK, Balint E, Serbyn N, Frittmann O, Stutz F, Unk I. Translesion synthesis DNA polymerase η exhibits a specific RNA extension activity and a transcription-associated function. Sci Rep. (2017) 7:13055. doi: 10.1038/s41598-017-12915-1
82. Kawasumi R, Abe T, Psakhye I, Miyata K, Hirota K, Branzei D. Vertebrate CTF18 and DDX11 essential function in cohesion is bypassed by preventing WAPL-mediated cohesin release. Genes Dev. (2021) 35:1368–82. doi: 10.1101/gad.348581.348121
83. Psakhye I, Kawasumi R, Abe T, Hirota K, Branzei D. PCNA recruits cohesin loader Scc2 to ensure sister chromatid cohesion. Nat Struct Mol Biol. (2023) 30:1286–94. doi: 10.1038/s41594-41023-01064-x
84. Choudhry SK, Neal ML, Li S, Navare AT, Van Eeuwen T, Wozniak RW, et al. Nuclear pore complexes mediate subtelomeric gene silencing by regulating PCNA levels on chromatin. J Cell Biol. (2023) 222:e202207060. doi: 10.202201083/jcb.202207060
85. Comazzetto S, Murphy MM, Berto S, Jeffery E, Zhao Z, Morrison SJ. Restricted hematopoietic progenitors and erythropoiesis require SCF from leptin receptor+ Niche cells in the bone marrow. Cell Stem Cell. (2019) 24:477–486.e476. doi: 10.1016/j.stem.2018.1011.1022
86. Comazzetto S, Shen B, Morrison SJ. Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev Cell. (2021) 56:1848–60. doi: 10.1016/j.devcel.2021.1805.1018
87. Kara N, Xue Y, Zhao Z, Murphy MM, Comazzetto S, Lesser A, et al. Endothelial and Leptin Receptor(+) cells promote the maintenance of stem cells and hematopoiesis in early postnatal murine bone marrow. Dev Cell. (2023) 58:348–360.e346. doi: 10.1016/j.devcel.2023.1002.1003
88. Petrie HT. Cell migration and the control of post-natal T-cell lymphopoiesis in the thymus. Nat Rev Immunol. (2003) 3:859–66. doi: 10.1038/nri1223
89. Misslitz A, Pabst O, Hintzen G, Ohl L, Kremmer E, Petrie HT, et al. Thymic T cell development and progenitor localization depend on CCR7. J Exp Med. (2004) 200:481–91. doi: 10.1084/jem.20040383
Keywords: T lymphocyte, thymus, immunodeficiency, zebrafish, CHTF18
Citation: Sertori R, Truong B, Singh MK, Shinton S, Price R, Sharo A, Shultes P, Sunderam U, Rana S, Srinivasan R, Datta S, Font-Burgada J, Brenner SE, Puck JM and Wiest DL (2025) Disruption of the moonlighting function of CTF18 in a patient with T-lymphopenia. Front. Immunol. 16:1539848. doi: 10.3389/fimmu.2025.1539848
Received: 04 December 2024; Accepted: 22 January 2025;
Published: 14 February 2025.
Edited by:
Andrew R Gennery, Newcastle University, United KingdomCopyright © 2025 Sertori, Truong, Singh, Shinton, Price, Sharo, Shultes, Sunderam, Rana, Srinivasan, Datta, Font-Burgada, Brenner, Puck and Wiest. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David L. Wiest, RGF2aWQuV2llc3RAZmNjYy5lZHU=
†Present address: Paulameena Shultes, Case Western Reserve University School of medicine, Cleveland, OH, United States