 Manuel Alejandro Mosso-Pani
Manuel Alejandro Mosso-Pani Dante Barreda
Dante Barreda Ma. Isabel Salazar
Ma. Isabel Salazar- 1Departamento de Microbiología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Ciudad de Mexico, Mexico
- 2Laboratory of Genomics, ODIN Bioscience, Miami, FL, United States
- 3Aging and Metabolism Research Program, The Oklahoma Medical Research Foundation, Oklahoma City, OK, United States
- 4Laboratorio Nacional de Vacunología y Virus Tropicales (LNVyVT), Escuela Nacional de Ciencias Biológicas Instituto Politécnico Nacional, Ciudad de México, Mexico
Platelets, traditionally known for their roles in hemostasis and thrombosis, have emerged as key regulators of immune responses, particularly through their dynamic interactions with neutrophils. This review explores how platelets influence neutrophil functions by forming platelet-neutrophil aggregates, releasing extracellular vesicles, and secreting metabolites. These processes govern critical immune activities, including cell recruitment, activation, endothelium interactions and the resolution or exacerbation of inflammation. Additionally, platelets induce metabolic reprogramming in neutrophils, affecting glycolysis and mitochondrial pathways, while also shaping the immune microenvironment by modulating other immune cells, such as T and B cells. Understanding this complex crosstalk between platelets and neutrophils—two of the most abundant cell types in the bloodstream—might reveal new therapeutic opportunities to regulate immune responses in inflammatory and immune-mediated diseases.
1 Introduction
Platelets, derived from megakaryocytes in the bone marrow, are released into the bloodstream (1). Despite lacking nuclei, they play critical roles in hemostasis, maintaining endothelial integrity (2), and immunity. Beyond their ‘so called’ classical functions, growing experimental evidence identifies platelets as key contributors to immunity, highlighting their ability to modulate both acute and chronic inflammation (3, 4).
Neutrophils, the most abundant innate immune cells, are rapidly mobilized to sites of injury, guided by chemokine gradients secreted by different cellular types, including damaged endothelial cells (5), tissue macrophages, and platelets (6). Platelets bound to neutrophils enhance their mobilization to these sites. Once at the inflammation site, activated neutrophils contribute to pathogen clearance through specialized mechanisms such as phagocytosis, cytokine release, oxidative burst, and the secretion of neutrophil extracellular traps (NETs) (5). The emerging field of immunometabolism examines how cellular metabolism shapes immune responses. The basics of immunometabolism suggests that pro-inflammatory responses are typically driven by glycolysis, whereas anti-inflammatory responses are primarily fueled by oxidative phosphorylation (OxPhos) (7, 8). For example, accumulation of Krebs cycle intermediates, such as succinate and citrate, along with mitochondrial reactive oxygen species (ROS), trigger the activation of the NLRP3 inflammasome which in turn processes interleukin-1β (IL-1β), promoting a pro-inflammatory state. In contrast, IL-10 activates mitochondrial metabolism via AMP-activated protein kinase (AMPK), which stimulates both fatty acid oxidation (FAO) and OxPhos. This metabolic shift leads to the consumption of Krebs cycle intermediates and a reduction in mitochondrial ROS production, inhibiting NLRP3 inflammasome assembly, and preventing IL-1β processing, thereby fostering an anti-inflammatory response (9). This observation has raised the idea that repolarizing immune cells towards a non-inflammatory phenotype by modulating cellular metabolism using metabolic intermediated molecules is possible.
Although neutrophil metabolism has traditionally been viewed as largely dependent on glycolysis, recent evidence suggests that mitochondrial metabolism also plays a role in regulating their effector functions (10). Furthermore, the impact of these metabolic pathways on inflammatory and anti-inflammatory responses remains an important area for further investigation. Emerging evidence suggests that platelets contribute to this metabolic reprogramming of neutrophils, enhancing their immune responses due to their close anatomical proximity through two primary mechanisms: direct interactions via receptor-ligand binding and indirect interactions through the release of soluble molecules. These platelet-neutrophil interactions are essential for the metabolic and functional modulation of neutrophils, amplifying their immune activity (11, 12).
This review highlights the current understanding of the crosstalk between platelets and neutrophils, focusing on how platelets regulate neutrophil immune responses. Additionally, we discuss the role of platelet-derived metabolites in driving the metabolic reprogramming of neutrophils and how this reprogramming impacts their effector functions.
2 Glycolysis and the pentose phosphate pathway in neutrophil immunometabolism
Glycolysis is the predominant metabolic pathway during neutrophil immune responses, given the short lifespan of neutrophils (13, 14). Glycolysis begins when neutrophils, take up glucose via GLUT transporters, primarily GLUT1 and GLUT3, and convert it into pyruvate through a series of enzymatic reactions. The conversion yields two molecules of ATP, one molecule of NADH, and intermediates for the pentose phosphate pathway (PPP). In following metabolic reactions, the resulting products can proceed under two different conditions depending on oxygen availability. Under aerobic conditions, pyruvate is oxidized to acetyl coenzyme A (acetyl-CoA) through the Krebs cycle, producing 32 molecules of ATP along with other reduced intermediates. Under hypoxic conditions, pyruvate is reduced to lactate by lactate dehydrogenase, a process that not only sustains glycolysis but also serves as a metabolic foundation for immune cells to produce and secrete soluble molecules. These molecules actively contribute to establishing a pro-inflammatory microenvironment, linking metabolic adaptation to immune function.
Neutrophils harness glycolysis to support their effector functions. Upon activation by stimuli such as phorbol 12-myristate 13-acetate (PMA), neutrophils increase glucose uptake in proportion to the surface expression of GLUT-1. This metabolic shift triggers critical processes, including the production and release of NETs (15). NET formation is a highly regulated process that involves chromatin decondensation, histone citrullination, and the preparation of nuclear and mitochondrial DNA for expulsion (16, 17). These web-like structures play a pivotal role in trapping and neutralizing pathogens. Additionally, the metabolic activity in neutrophils provides the energy and intermediates required for such immune processes, highlighting the integral connection between cellular metabolism and the ability of neutrophils to combat infections.
Although the abrogation of neutrophil effector functions by glucose depletion has been reported (15, 18), under conditions of inflammation or hypoxia, neutrophils rely on intracellular glycogen stores accumulated through gluconeogenesis (12). These glycogen stores are essential for neutrophil survival and efficient microbial killing via the production and release of NETs. Impaired glycogenolysis is associated with reduced ability of neutrophils to produce NETs (12, 19). In COVID-19 patients, for example, increased glycogenolysis is linked to the release of large amounts of NETs into the bloodstream, contributing to the exacerbation of the inflammatory state (19).
Similar to glycolysis, the PPP operates in the cytosol and plays a crucial role in neutrophil metabolism by supporting both biosynthetic and antimicrobial functions. It produces ribose-5-phosphate for DNA and RNA synthesis, glycerol-3-phosphate for phospholipid synthesis, and NADPH molecules essential for neutrophil oxidative burst and antimicrobial activity. Glucose-6-phosphate dehydrogenase (G6PDH), the key enzyme in the PPP, facilitates the conversion of glucose-6-phosphate to ribulose-5-phosphate while generating two molecules of NADPH. This NADPH powers NADPH oxidases (NOX2), which catalyze the reduction of molecular oxygen to superoxide, leading to the production of ROS. These ROS are crucial for the oxidative burst that enables pathogen clearance during phagocytosis (12).
A shift toward increased PPP activity ensures sufficient NADPH supply for ROS production while supporting neutrophil survival and function (20, 21). This includes de novo protein synthesis, cytokine secretion, and sustained antimicrobial capacity in inflammatory microenvironments (20).
3 Mitochondrial regulation of neutrophil immunometabolism
Emerging experimental evidence in neutrophil immunometabolism suggests that mitochondrial metabolism is also active in these cells and plays crucial roles in development, survival, and effector functions (22), despite the relatively low abundance of mitochondria in neutrophils (23). Mitochondrial metabolism encompasses OxPhos, wherein electron transport chain (ETC) complexes (complex I to IV) couple with ATP synthesis, generating mitochondrial transmembrane potential and relying on metabolic intermediates supplied by the Krebs cycle. Disruption of mitochondrial function impairs energy metabolism, significantly affecting not only the ontogeny (24) and differentiation (25) of neutrophils but also their immune functions. Immature neutrophils, which exhibit lower glucose uptake, rely on mitochondrial metabolism for ROS generation (26). Moreover, inhibition of ETC complex III significantly reduces antimicrobial activity (27) and oxygen consumption, correlating with a marked decrease in ROS production (28, 29).
Furthermore, mitochondria house cytotoxic proteins and monitor metabolic and redox states, accumulating and storing Ca2+ to facilitate apoptotic processes without triggering release of toxic contents into the extracellular milieu (21). These mitochondrial processes promote neutrophil polarization, chemotaxis (30), and rely on maintenance of mitochondrial membrane potential and ATP synthase function. Consequently, inhibition of mitochondrial activity hampers neutrophil transmigration to tissues (31).
If mitochondrial dysfunction occurs, a premature apoptosis process is activated in neutrophils. Conversely, increased mitochondrial activity has been linked to heightened mitochondrial ROS production during colitis (32). Thus, tight regulation of neutrophil mitochondrial function is pivotal to maintaining homeostasis. There is experimental evidence that mitochondrial morphology/physiology can be molded by platelet factors (30, 33). In the future it would be interesting to explore how to manipulate an immune response mediated by neutrophils by regulating their mitochondrial function using platelets. When platelets are absent, neutrophils exhibit increased surveillance, which may contribute to the development of chronic inflammation and fibrosis (34).
3.1 Oxidative burst
One key mechanism of neutrophils to eliminate infectious agents is the activation of the oxidative burst, a process tightly regulated by mitochondrial activity. Upon stimulation, NADPH oxidase assembles at the cell membrane and becomes activated, catalyzing the NADPH-dependent reduction of O2 to form superoxide anions (O2−) and derivatives such as H2O2, hydroxyl radicals (OH•), and hypochlorous acid (HOCl) (35). Mitochondrial ROS production is induced by N-formyl peptides (fMLP), which are exclusively expressed on bacteria (36). Notably, inhibiting mitochondrial function reduces neutrophil oxidative burst and ROS production in response to fMLP (37). Additionally, mitochondria-targeted antioxidants such as SkQ1 can also inhibit fMLP-induced neutrophil oxidative burst and degranulation (36).
NADPH production is not limited to the PPP. In neutrophils, alternative pathways and enzymes also contribute to meeting the high demand for ROS generation (38, 39). For instance, the malic enzyme catalyzes the conversion of malate, a Krebs cycle intermediate, into NADPH (40). Additionally, isocitrate dehydrogenases 1 and 2, as well as pathways such as the folate cycle, may enhance NADPH availability, further supporting ROS production (41). In conditions like acute destructive pancreatitis, both the PPP and the Krebs cycle are significantly upregulated in neutrophils, highlighting the critical role of these metabolic pathways in sustaining the oxidative burst.
Mitochondria also contribute to ROS production by linking NADPH oxidase (NOX2) activity to metabolic processes (26, 27, 42). This connection relies on an ETC Complex III-dependent mechanism (27), coupled with increased FAO (26). During FAO, free fatty acids are metabolized to generate acetyl-CoA, which fuels the Krebs cycle and sustains NADPH production (26). Notably, neutrophils adapt to metabolic stress—such as hyperglycemia or limited glucose and glutamine availability—by relying on mitochondrial FAO to maintain ROS generation (26, 40, 43) and NETs production (44). This metabolic flexibility highlights the critical interplay between mitochondrial and cytosolic pathways in regulating neutrophil bactericidal activity under various physiological and pathological conditions.
3.2 Neutrophil extracellular traps production
The activity of the ETC complexes I and III drives the production and secretion of NETs neutrophil activation by platelet-activated factor (PAF) (45) intrinsically or by lipopolysaccharide (LPS) through a Toll-like receptor 4 (TLR-4) dependent manner during infection (27).
PAF, a key pro-inflammatory mediator, enhances neutrophil activation and ROS production by mitochondria, modulating Ca2+ signaling (46, 47). NETs production and secretion are also increased by high mobility group box 1 (HMGB1), a key regulator of cell bioenergetics that modulates the balance between apoptosis and autophagy (48). In neutrophils, HMGB1 regulates recruitment, activation, and survival by preventing mitochondrial potential reduction and inducing autophagosome formation (49). Blocking autophagic flux prevents HMGB1-induced NETs production and secretion (49). Furthermore, in obese mice, NETs formation shifts from a sole reliance on glycolysis to ATP production through mitochondrial FAO and the PPP (50, 51). This process also depends on the catabolism of glutamate and proline (52, 53), underscoring the metabolic adaptability of neutrophils in supporting NETs formation.
Our central hypothesis is that mitochondrial function plays a critical role in regulating neutrophil activity and its dysregulation has significant pathological implications. When mitochondrial dysfunction occurs, it triggers premature apoptosis in neutrophils, impairing their immune response. In contrast, increased mitochondrial activity has been associated with elevated mitochondrial ROS production, as observed in conditions such as colitis (32). External factors, including platelet-mediated regulation, may modulate neutrophil mitochondrial function, driving inflammatory states in neutrophils (30, 33). Notably, in the absence of platelets, neutrophils exhibit heightened surveillance activity, which could exacerbate chronic inflammation and fibrosis (34). This highlights the intricate interplay between platelets, neutrophils, and mitochondrial dynamics in immune regulation, as discussed further in the review.
4 Platelet roles in immune response and metabolic regulation
Platelets are recognized for their immune regulatory functions (54). Following injury, platelets facilitate vascular repair post-hemorrhage and recruit immune cells to the injury site, where infectious pathogens may enter (55). It is well-established that platelets interact with neutrophils through P-selectin, enhancing transendotelial migration and potentiating their immune functions (56). For example, during sepsis, platelets form clots in small vessels to prevent the hematogenous spread of infection, trap bacteria to facilitate their clearance, induce cellular differentiation, and present antigens, among other functions (57–59).
Beyond their mechanical roles in immune defense, platelets play a crucial role in inflammatory processes. Once recruited as single cells to the vascular wall, platelets act as pathfinders, guiding leukocytes out of the microvasculature to sites of inflammation (60). Several studies have shown that leukocytes rely on scanning activated platelets to pinpoint the exact locations for exiting circulation, highlighting the essential role of platelets in orchestrating leukocyte trafficking during inflammation (61, 62). While this review cannot cover the full range of platelet-mediated inflammatory mechanisms, others have thoroughly explored how platelets influence both inflammation and its resolution, as well as their impact on immune-mediated diseases (59, 63, 64). By discussing these selected mechanisms, we aim to illustrate the diverse ways in which platelets interact with other cells and processes, challenging the notion of platelets as simple or purely mechanical components of the immune system.
Platelets influence inflammation by secreting HMGB1, miR-15b-5p, and miR-378a-3p by means of exosomal release. These exosomal factors activate the Akt/mTOR autophagy pathway, leading to excessive release of NETs by neutrophils, which promotes inflammation and tissue damage (65). The role of platelets in driving NETs formation has been further demonstrated during dengue virus infections. Activated platelets numbers in infected patients are correlated positively with the presence of NETs, plasma leakage and worse prognosis (66). The proinflammatory functions of platelets appear to be regulated through multiple layers of control. For instance, platelet-derived factors have been shown to regulate IL-1β production by activating the inflammasome in various leukocyte populations (67). Additionally, activated platelets stimulate leukocytes to produce proinflammatory cytokines. In the case of SARS-CoV-2, the Spike protein induces the expression of P-selectin and CD40L on platelets, which bind to PSGL-1 and CD40 on monocytes, further promoting IL-1β production (68). This highlights the intricate role of platelets in amplifying inflammatory responses. Conversely, several key mechanisms have been identified that position platelets as gatekeepers of inflammation within the microvasculature, highlighting their cellular plasticity. On inflamed vasculature, platelets actively scan for fibrinogen in an Arp2/3-dependent manner to direct their spread and preserve vascular integrity, thereby preventing further inflammation (61). Furthermore, platelets opsonized with IgG can directly interact with circulating monocytes, inducing the production of anti-inflammatory IL-10 while simultaneously reducing the production of proinflammatory cytokines (69).
The tight regulated immune functions that are just begun to envision for platelets, blurs the line that separate them from simplicity. To fulfill these diverse functions, platelets must be highly metabolically active (70, 71). Upon activation, their energy demands increase, prompting them to switch between metabolic pathways depending on the stimulus and available substrates (72). In a steady state, platelets primarily rely on glycolysis, with a lesser dependence on mitochondrial OxPhos (73). Upon activation, they increase glucose uptake through GLUT3, which is translocated from α-granule membranes to the plasma membrane. Notably, genetic deletion of GLUT3 impairs platelet activation and reduces degranulation, spreading, and clot retraction (74, 75). However, there is no clear consensus on which metabolic pathway primarily fuels platelet activation. Some studies report that aerobic glycolysis is the main pathway following platelet stimulation (70, 72, 76), while others suggest that oxidative metabolism is the key driver (77, 78). Corona-de-la-Peña et al. (2017) and Ravera et al. (2023) delve into these discrepancies, which may stem from variations in experimental approaches and readouts (71, 79). Additionally, platelets’ rapid and flexible switching between metabolic pathways contributes to the observed differences. Despite these variations, the importance of ATP as the ultimate energy source for platelet functions is undisputed. ATP is essential for processes such as maintaining calcium homeostasis, which activates key signaling pathways. Unsurprisingly, about 50% of mitochondrial activity in platelets is dedicated to ATP production (80). When OxPhos is pharmacologically inhibited, granule release and thrombus formation—two of the most energy-intensive platelet functions—are impaired (78, 79). Given the constantly changing conditions platelets face in circulation; their physiological functions should be viewed in this context. Rapidly shifting environmental factors may force platelets to rely on glucose through simultaneous use of glycolysis and OxPhos, occurring outside and inside the mitochondria, respectively. Supporting this idea, research indicates that platelets utilize glucose from two different sources with distinct metabolic fates (81). Metabolic differences in resting versus activated platelets are depicted in Figure 1.
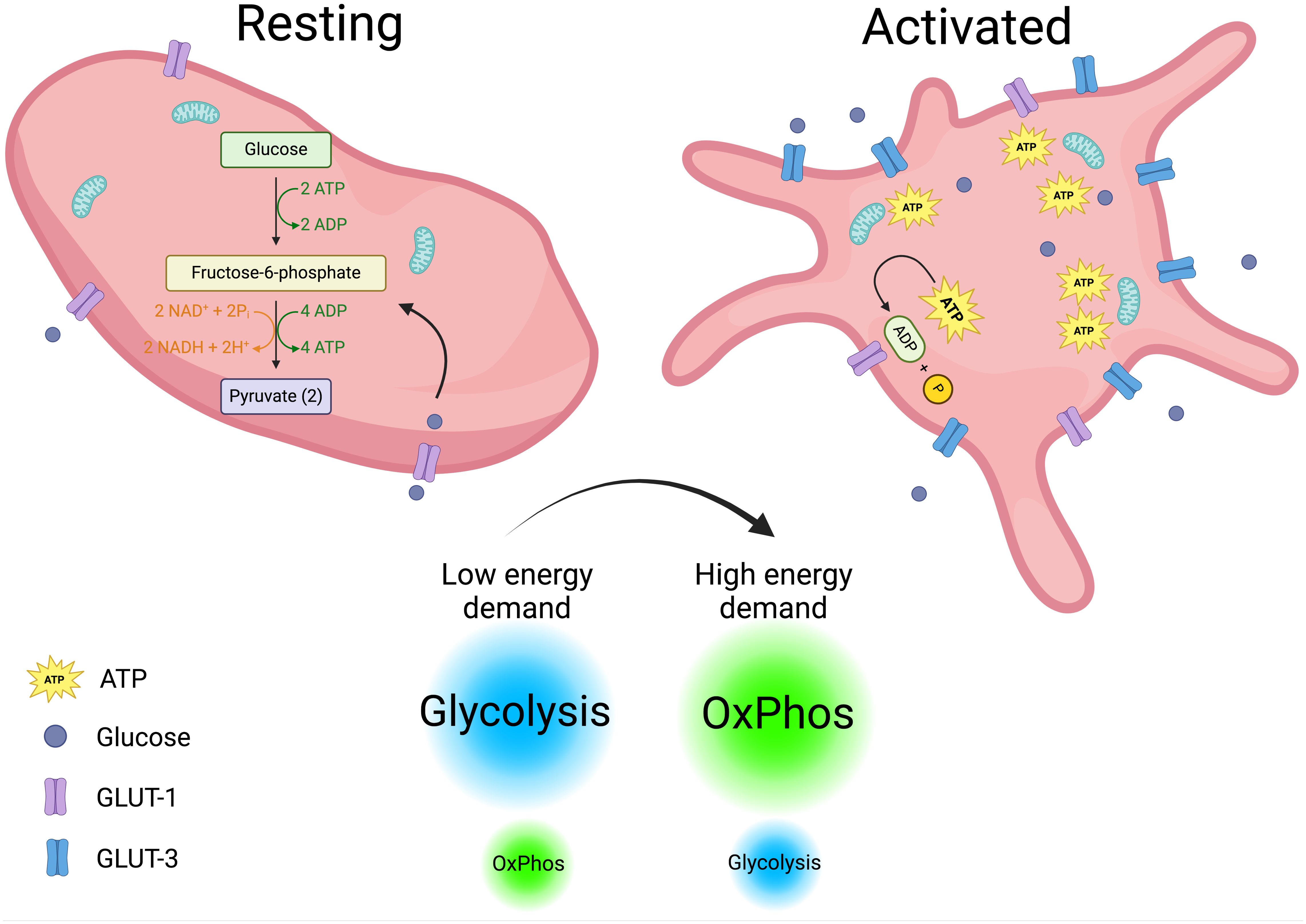
Figure 1. Platelets metabolism. Metabolic differences in resting and activated platelets are depicted. Resting platelets with a low energy demand rely mainly on glycolysis to fuel their functions. In contrast, activated platelets increase their energy demands and glucose intake, shifting from glycolysis to OxPhos (oxidative phosphorylation) to meet their energetic requirements. Created in https://BioRender.com.
5 Regulating neutrophil immunometabolism during inflammation
Experimental evidence demonstrates that direct interaction between platelets and neutrophils, forming platelet-neutrophil aggregates (PNAs), enhances neutrophil function. This interaction bridges hemostasis and inflammation (55, 82–84), regulating neutrophil immune responses by modulating their metabolic pathways. Platelet-neutrophil receptor-ligand interactions are summarized in Figure 2, illustrating bidirectional activation mechanisms.
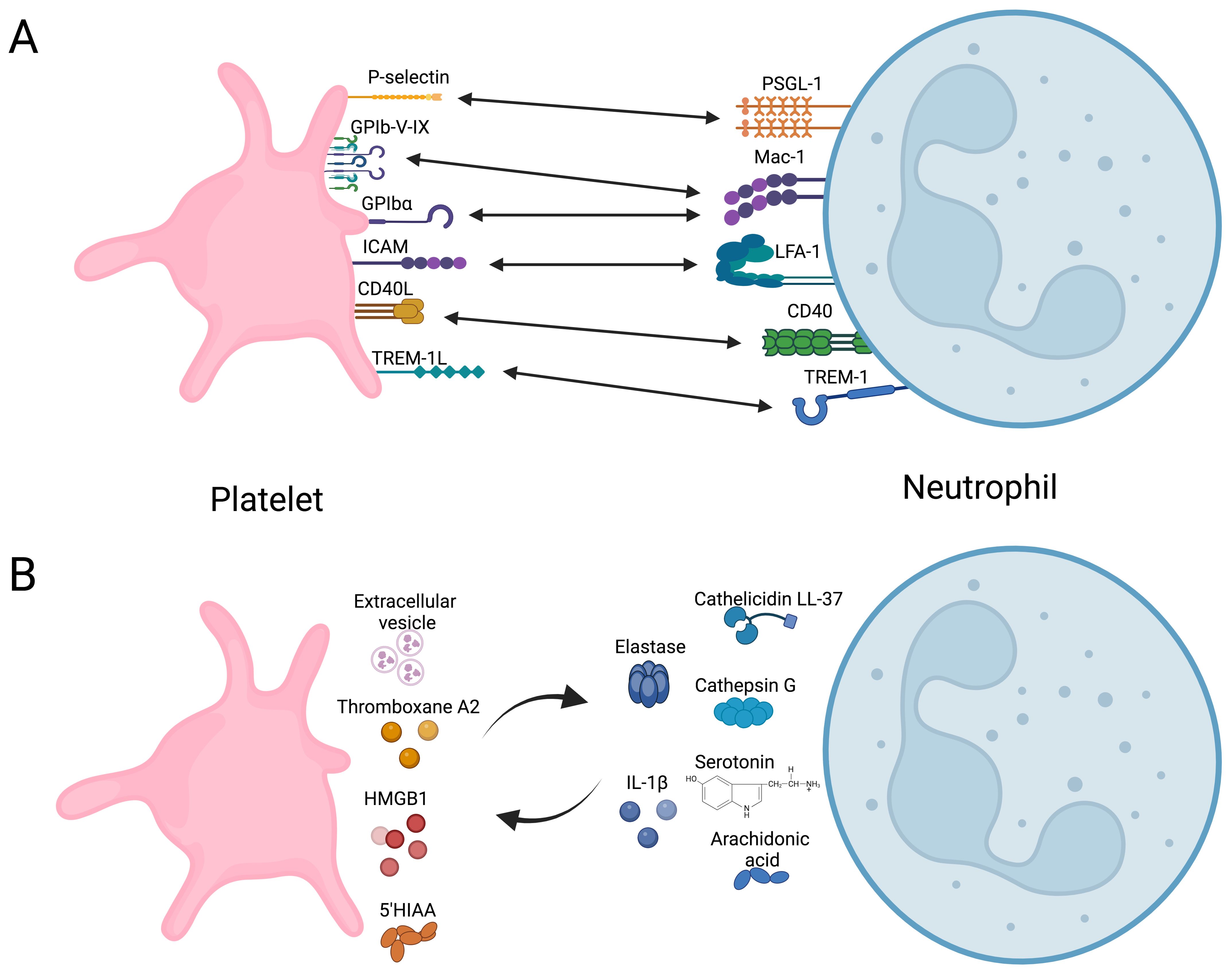
Figure 2. Overview of direct and indirect platelet-neutrophil interactions. (A) Receptors and ligand coupling that orchestrate bidirectional cellular activation. (B) Soluble molecules secreted by platelets that modulate neutrophils immune response and vice versa. HMGB1, High mobility group box 1; 5-HIAA, 5-Hydroxyindoleacetic acid. Created in https://BioRender.com.
Platelet activation via ligands detected by TLR2, TLR4, or TLR9 induces the formation of PNAs (85, 86). This enhances the adhesion of neutrophils to vascular endothelium by increasing integrin affinity, promoting their migration into the tissue (84), and contributing to the resolution of infections caused by Brucella abortus (87), Mycobacterium tuberculosis (88), Staphylococcus aureus, and Escherichia coli (88, 89). These interactions trigger oxidative burst and the production and secretion of NETs. Platelet glycoprotein Ibα, P-selectin, and integrin αIIbβ3 are recognized by Mac1, PSGL-1, and SLC44A2 on neutrophils, respectively (56, 86, 90). However, triggering receptor expressed in myeloid (TREM)-like transcript-1 (TLT-1) and TREM-1 in platelets also play roles in this process (91, 92). TLT-1 serves as a specific marker for platelet activation, stored in α-granule and expressed on the surface after activation (93). TLT-1 regulates the transmigration of neutrophils during inflammatory events (91) by dissociating platelet-neutrophil aggregates before neutrophils transmigrate into the interstitial space. Additionally, TLT-1 promotes platelet adhesion to fibrinogen and contributes to fibrinogen accumulation (91). TREM-1 plays a significant role in neutrophil immunity (92). Interaction between PNAs and TREM-1 platelet-induced neutrophil activation, ROS production, and phagocytosis (92). During PNAs formation, platelets develop an activation phenotype characterized by increased expression of P-selectin and TLT-1. However, neutrophils regulate this activation phenotype on platelets to prevent procoagulant events (94).
Neutrophil immunometabolism is orchestrated by platelets to support membrane reorganization and cellular architecture, facilitating the production and secretion of NETs following PNAs formation. This metabolic shift involves a transition to ATP production via mitochondrial FAO (50, 51) rather than glycolysis, occurring in a NADPH-dependent manner (86). In obese mice, platelet dysfunction adversely affects the production and secretion of NETs. Restoring platelet functionality also reinstates the ability of neutrophils to produce and secrete NETs. In this murine model, the formation of PNAs modulates neutrophil influx, a process dependent on leptin, IL-33, and CXCR2 signaling driven by platelet secretory intermediates. Moreover, this process is inhibited by blocking P-selectin (50, 51). While intravital microscopy has proven valuable for examining neutrophil activity in vivo in mice (95), studying platelet-neutrophil interactions has required advanced tools, such as microfluidic assays (96), to better understand these complex mechanisms.
On the other hand, once PNAs are formed, a two-way communication begins. PAF activates phospholipase C in neutrophils, which hydrolyzes phosphoinositide substrate to produce inositol trisphosphate (IP3). IP3 binds to receptors on the endoplasmic reticulum, triggering the release of intracellular Ca²+. This increase in Ca²+ activates Ca²+-dependent phospholipase A2, which cleaves the sn-2 fatty acid from membrane phospholipids, releasing arachidonic acid (97). Neutrophils secrete arachidonic acid-rich extracellular vesicles (EVs), which are internalized by platelets in a Mac1-dependent manner. Subsequently, thromboxane A2 (TxA2) and other bioactive lipid mediators, including prostaglandins, and leukotrienes are synthesized by cyclooxygenase or lipoxygenase enzymes (97) and secreted, inducing endothelial expression of intercellular adhesion molecule-1 (ICAM-1) enhancing neutrophil transmigration (90), and oxidative burst through the increase of NADPH oxidase activity, and ROS generation (98). Moreover, histamine secreted by neutrophils regulates platelet activation by modulating Akt phosphorylation (99). Conversely, platelet EVs carrying transcription factors, nucleic acids, and containing mitochondria are internalized by activated neutrophils, thereby promoting inflammation (100). Furthermore, platelet recruitment of immune cells has been observed and is crucial for both initiating and resolving inflammation (34). Following neutrophil recruitment at the onset of inflammation, platelets recruit regulatory T cells (Tregs) during the resolution phase. Each mechanism relies on differential expression of P-selectin and activation by soluble CD40L. These aggregates of platelets and Tregs are vital for modulating their transcriptome and instructing Tregs to release anti-inflammatory mediators, such as IL-10 and transforming growth factor beta (TGF-β). As a result, macrophages undergo transcriptional reprogramming and polarization towards an anti-inflammatory phenotype, leading to effective resolution of inflammation mediated by TLT-1 expressed by platelets (34). Moreover, platelet-specific deletion of CLEC-2, but not GPVI, results in enhanced systemic inflammation and accelerated organ injury in two mouse sepsis models. This deficiency is linked to a reduction in podoplanin-expressing macrophages, despite elevated cytokine and chemokine levels in the infected peritoneum, suggesting that podoplanin activation mediates the anti-inflammatory effect of CLEC-2 on platelets regulating immune cell infiltration and inflammation during sepsis (101). Finally, the phosphorylation of dynamin-related protein 1 (Drp1) at Ser616, a key event in mitochondrial fission, regulates neutrophil polarization and chemotaxis. Platelet-derived factors may influence Drp1 phosphorylation, enhancing mitochondrial fragmentation in neutrophils. This process is linked to increased neutrophil activation and the formation of NETs, contributing to immune defense (30).
6 Platelet-derived metabolites and their influence on neutrophil responses
Metabolism is central to cellular function, and platelets are no exception. While the series of events during platelet activation seems to dictate which metabolic pathways are active, emerging evidence suggests that targeting specific metabolic pathways could modulate platelet functions (76, 78, 102, 103). Recent research has mapped the global metabolism of platelets following stimulation, revealing the intricate interplay between various metabolic pathways. In response to activation, 202 metabolites are upregulated, with lipids accounting for 50% of this increase (70). This section explores how platelet-derived metabolites impact neutrophil functions by modulating oxidative burst, survival, and metabolism (85, 86, 104).
In the intestinal epithelium, low levels of IL-33 induce the release of serotonin by enterochromaffin cells, which is subsequently taken up by platelets. In conditions like chronic inflammatory bowel diseases (IBDs), where IL-33 concentrations are elevated, the heightened uptake of serotonin by platelets leads to increased clotting and neutrophil recruitment (105, 106). Platelets then metabolize serotonin into 5-HIAA, a key mediator in neutrophil recruitment through the GPR35 receptor (Figure 3). This mechanism has been validated in several murine inflammatory models. Additionally, 5-HIAA derived from platelets also aids in fungal clearance by promoting eosinophil recruitment (107, 108). Chemokines such as PF4, RANTES, CXCL4, and serotonin influence oxidative burst (104, 109), with PF4 specifically enhancing monocyte phagocytosis, prolonging ROS production, preventing apoptosis, and promoting cell differentiation (110). Another key mediator, HMGB1, supports neutrophil recruitment and activation by preserving mitochondrial potential, inducing autophagosome formation, and increasing NET production (49). Additionally, platelet-derived IDO1, a key enzyme in tryptophan metabolism, contributes to immune suppression by depleting tryptophan and generating kynurenine (111). Elevated IDO1 expression during Plasmodium yoelii infection alters plasma tryptophan and kynurenine levels, shaping immune responses of T cells, macrophages, and dendritic cells (112–114), while in severe malaria, excessive neutrophil activation exacerbates inflammation (115). Platelet-Derived Growth Factor (PDGF) induces mitochondrial fission in various cell types, including vascular smooth muscle cells (33). We hypothesize that PDGF may similarly promote mitochondrial fragmentation in neutrophils, a process linked to enhanced cellular responses such as oxidative burst and NET formation. Ahead of metabolic reprogramming, platelets can directly transfer functional mitochondria to neutrophils, influencing calcium mobilization, EVs release, and gene expression, thereby shaping immune responses (116).
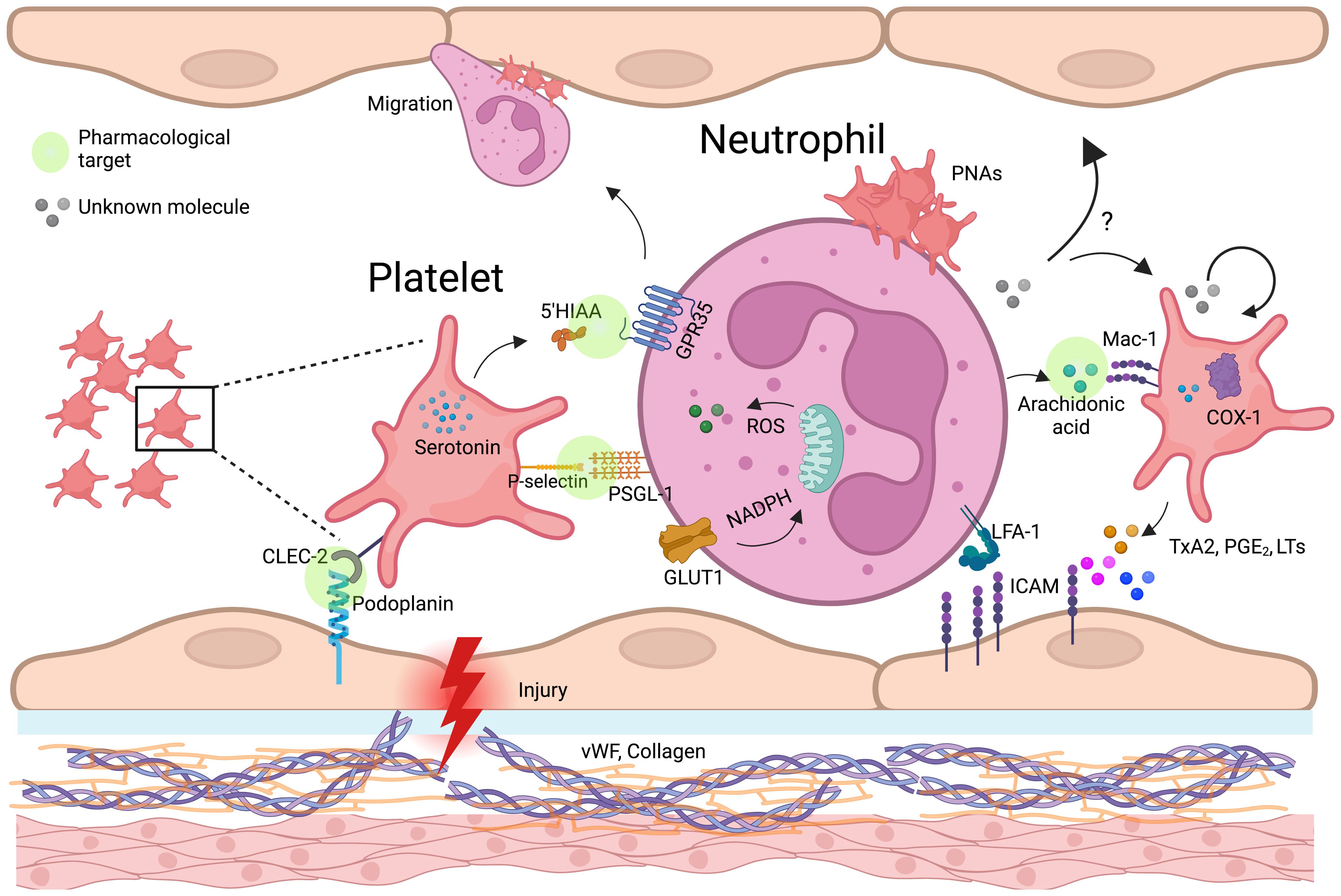
Figure 3. Platelet-neutrophil-endothelium crosstalk in vascular inflammation and immune regulation. Schematic representation of platelet-neutrophil-endothelial crosstalk in inflammation and vascular injury. Upon endothelial damage, platelets adhere to expose von Willebrand factor (vWF) and collagen, becoming activated and releasing key mediators such as serotonin, thromboxane A2 (TxA2), and arachidonic acid. Platelet P-selectin binds to neutrophil PSGL-1, facilitating platelet-neutrophil aggregates (PNAs). Neutrophils internalize platelet-derived arachidonic acid via Mac-1, leading to the production of lipid mediators (prostaglandins, leukotrienes) and reinforcing inflammatory responses. TxA2, produced by platelet COX-1, enhances endothelial ICAM-1 expression, promoting neutrophil adhesion and transmigration. Additionally, platelets transfer serotonin, which may modulate neutrophil oxidative burst through GPR35, and entry the mitochondria, potentially affecting neutrophil metabolism and immune function. Possible mechanisms, indicated by question marks (?), highlight critical knowledge gaps, such as the impact of neutrophil-derived metabolites in platelets (beyond arachidonic acid) and the endothelium. Green circle icons highlight the platelet-neutrophil-endothelium interaction points where pharmacological approaches can be developed to modulate the immune response during inflammatory diseases. Created in https://BioRender.com.
Platelet-derived EVs modulate neutrophil function through multiple pathways, influencing both immune activation and metabolic reprogramming. EVs rich in soluble CD40L strongly enhance neutrophil oxidative burst via CD40-dependent activation of PI3K and NF-κB signaling (117–120). During dengue virus infection, platelets activated through CLEC2 release EVs that stimulate neutrophils via CLEC5A and TLR2, driving NET formation (121). Blocking these pathways significantly reduces the inflammatory response (121), suggesting a role for platelet-derived CD40L in regulating neutrophil immunometabolism by promoting oxidative burst and NET release (117, 118, 122). Additionally, EVs enriched in ceramides contribute to immune regulation by enhancing neutrophil migration, phagocytosis, and cytokine production while inhibiting the ETC, leading to ROS generation and apoptosis (123). Beyond neutrophils, platelet-derived lipid metabolites such as L-carnitine and acyl-carnitine influence immune cell metabolism. L-carnitine facilitates FAO and OXPHOS, increasing ROS production and activating FoxP3 signaling to enhance Treg suppressive function (123). Meanwhile, acyl-carnitine boosts mitochondrial ETC complex activity and OXPHOS in B cells by promoting H3K27 acetylation, thereby strengthening immunity (124) (Figure 4).
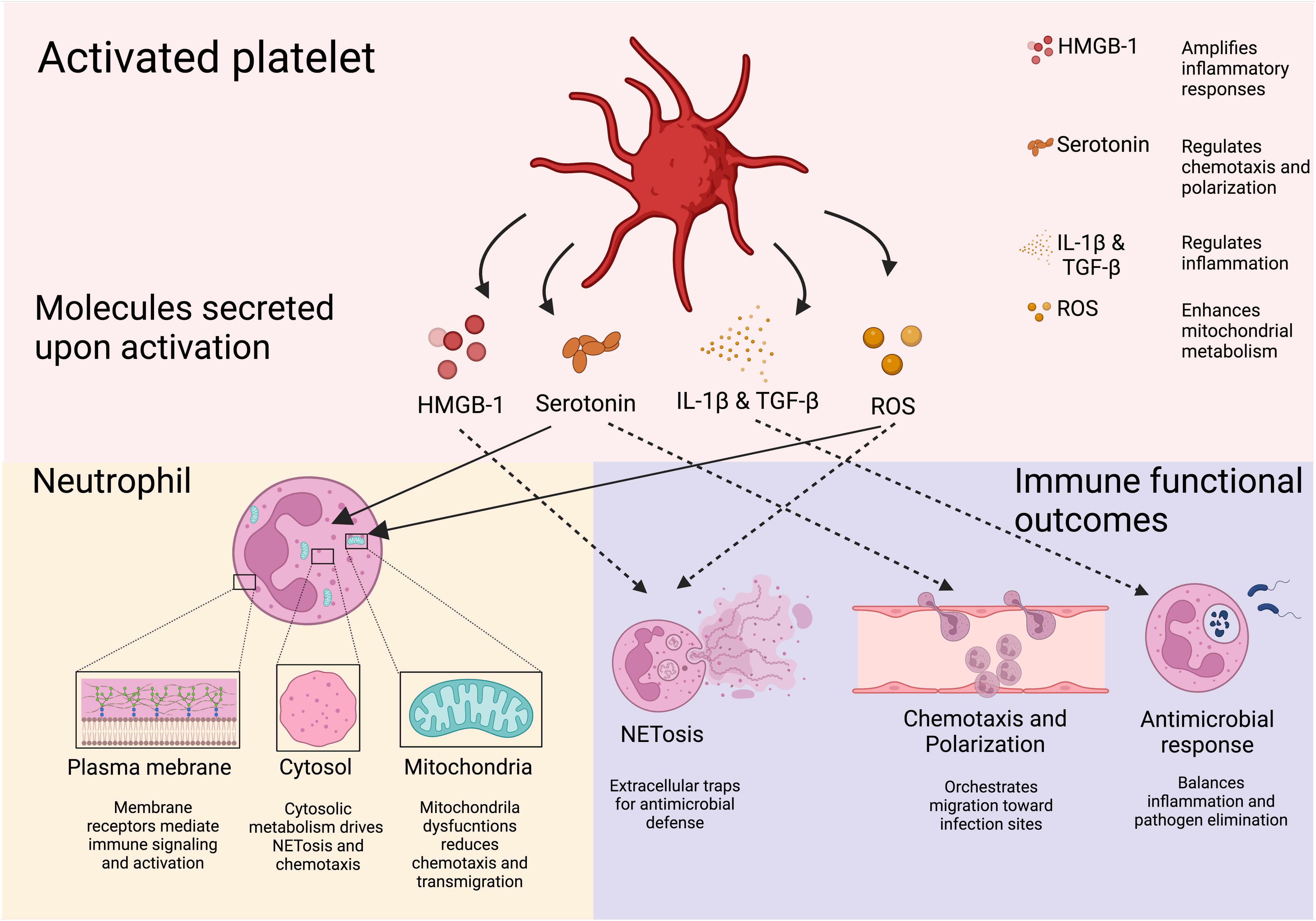
Figure 4. Soluble platelet-derive molecules regulate neutrophil immune response. Molecules derived from platelets and their effects on neutrophils are shown. Through their soluble mediators, platelets are able to induce neutrophil responses pivotal for the host defense. Downstream effects of platelet-derived molecules include NETosis, transmigration, and neutrophil recruitment. Created in https://BioRender.com.
7 Impending challenges
Platelet transfusion is a lifesaving procedure implemented worldwide. Yet the clinical interest in platelets often remains limited to their role in hemostasis overlooking the broader physiological and pathological significance of these cells. However, cumulative evidence associates thrombocytopenia with increased mortality in critically ill patients (125, 126). While preclinical studies consistently highlight the regulatory roles of platelets during conditions such as sepsis, clinical studies have yet to confirm these findings conclusively (127). Notably, the immunomodulatory interactions between platelets and neutrophils offer a promising foundation for developing novel therapeutic strategies. Exploring the immunomodulatory potential of platelets on neutrophils for broader clinical applications is an emerging but challenging field (128). This attempt raises several interesting questions and presents key areas for future investigation. The first challenge involves elucidating the specific signaling pathways and molecular mechanisms underlying platelet-neutrophil interactions across various proinflammatory contexts. An interesting approach to tackle this matter would be the use of surfaceomics to define the molecule expression patterns on platelets and neutrophils from patients under specific scenarios, such as sepsis or dengue virus infection, where unregulated platelet-neutrophils interactions accounts for the progression of the disease (121, 129). The cell surface expression profile can be used as biomarker and would help to predict the outcome of a cell-cell interaction. The best characterized receptor-ligand interaction between platelets and neutrophils is P-selectin/PSGL-1. Notably, in vitro and clinical studies have demonstrated that human anti-P-selectin antibody can inhibit cell adhesion in a safety manner in individuals suffering from sickle cell disease (130, 131). These findings stablish a proof of principle on the importance of dissecting platelet-neutrophil interactions with a perspective of blocking their downstream effects to alleviate pathological conditions. The dynamic nature of platelet-neutrophil interactions, which may vary depending on disease stage, patient heterogeneity, and underlying metabolic conditions makes of this endeavor a hard to accomplish quest. In that context, to add a layer of complexity, is important to draw attention to the role of the endothelial cells (Figure 3). Critically, in vascular dysfunctions and systemic lupus erythematosus (SLE) (132), endothelial cells make important contributions to the onset and maintenance of the disease by interacting with platelet-neutrophil complexes. The second challenge is defining the roles of soluble platelet-derived molecules in neutrophil modulation. Recent studies revealed the metabolic complexity of platelets (70), highlighting a limited known number of metabolites that influence neutrophil immune responses. Given the heightened metabolic activity of activated platelets, it is reasonable to hypothesize that they produce a broader spectrum of metabolites capable of shaping the immune microenvironment beyond neutrophil modulation. Additionally, the reciprocal impact of neutrophil-derived metabolites on platelet functions and their contributions to immune regulation remain largely unexplored and warrant further investigation. From the short list of known platelet-derived metabolites, we highlighted the role of 5-HIAA on recruiting neutrophiles. 5-HIAA is a potent chemoattractan for all GPR35+ cells not just neutrophils which make it a promising candidate to exploit pharmacological immunomodulation during proinflammatory disorders. In recent years, significant advances in chemical compounds synthesis have allow the development of “peptidomimetics”, modified peptide sequences with improved biological properties (133, 134). The design of a peptide capable to block the action of 5-HIAA is appealing as treatment of viral respiratory infections including respiratory syncytial (RSV) and Influenza virus (135, 136), where, according to clinical studies, neutrophiles contribute to the lung damage during the disease. The third challenge emphasizes the need for comparative studies to analyze human and murine platelet-neutrophil interactions under similar conditions. These studies should aim to delineate species-specific differences in receptor functionality, metabolic pathways, and immune responses. Although, different in size and number, genetically human and mouse platelets share high levels of identity. Nevertheless, the differences exist. In silico analysis reveals that human and mouse secretomes are only 75% identical (137). Variations in receptor expression, metabolic dependencies, and signaling pathway sensitivities highlight the importance of cautious interpretation when translating murine model findings to humans. For example, variations in CLEC-2 expression or arachidonic acid metabolism between mice and humans could lead to differing immune outcomes. Validation using humanized models, organoids or primary human cells is imperative. While murine models remain indispensable for exploring complex in vivo interactions—particularly in inflammatory and thrombotic contexts—their genetic tractability must be balanced with efforts to ensure relevance to human biology. Comprehensively defining the impact of platelet-neutrophil interactions on immune responses holds significant relevance for human hyperinflammatory diseases such as sepsis and cardiovascular disorders. A clearer understanding of how platelet- and neutrophil-derived metabolites influence their crosstalk could have profound implications for basic physiology, immune regulation, and responses to infection.
Author contributions
MM-P: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. DB: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. MS: Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Polytechnic Institute under grant number IPN SIP20241440.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. This manuscript incorporates the use of generative AI, specifically OpenAI’s ChatGPT, to assist with tasks such as refining the abstract, reviewing and revising titles and subtitles, and summarizing the manuscript’s scope. All content generated by AI was carefully reviewed and edited by the authors to ensure accuracy, adherence to journal guidelines, and alignment with the scientific context of the manuscript. The authors affirm that the AI tools were used as an aid in writing and formatting only, without compromising the originality, intellectual content, or scientific validity of the work.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Machlus KR, Italiano JE. The incredible journey: From megakaryocyte development to platelet formation. J Cell Biol. (2013) 201:785–96. doi: 10.1083/jcb.201304054
2. Ho-Tin-Noé B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost. (2011) 9:56–65. doi: 10.1111/j.1538-7836.2011.04317.x
3. Wagenhäuser MU, Mulorz J, Krott KJ, Bosbach A, Feige T, Rhee YH, et al. Crosstalk of platelets with macrophages and fibroblasts aggravates inflammation, aortic wall stiffening, and osteopontin release in abdominal aortic aneurysm. Cardiovasc Res. (2024) 120:417–32. doi: 10.1093/cvr/cvad168
4. Anjum A, Mader M, Mahameed S, Muraly A, Denorme F, Kliem FP, et al. Aging platelets shift their hemostatic properties to inflammatory functions. Blood. (2025) 135:1234–45. doi: 10.1182/blood.2024024901
5. Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. (2018) 371:531–9. doi: 10.1007/s00441-017-2785-7
6. Kornerup KN, Salmon GP, Pitchford SC, Liu WL, Page CP. Circulating platelet-neutrophil complexes are important for subsequent neutrophil activation and migration. J Appl Physiol. (2010) 109:758–67. doi: 10.1152/japplphysiol.01086.2009
7. O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
8. O’Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
9. Kelly B, O’Neill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. (2015) 25:771–84. doi: 10.1038/cr.2015.68
10. Jeon J-H, Hong C-W, Kim EY, Lee JM. Current understanding on the metabolism of neutrophils. Immune Netw. (2020) 20. doi: 10.4110/in.2020.20.e46
11. Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil metabolic shift during their lifecycle: impact on their survival and activation. Int J Mol Sci. (2020) 21:287. doi: 10.3390/ijms21010287
12. Sadiku P, Willson JA, Ryan EM, Sammut D, Coelho P, Watts ER, et al. Neutrophils fuel effective immune responses through gluconeogenesis and glycogenesis. Cell Metab. (2021) 33:411–423.e4. doi: 10.1016/j.cmet.2020.11.016
13. McCracken JM, Allen LAH. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death. (2014) 7:15. https://pmc.ncbi.nlm.nih.gov/articles/PMC4167320/.
14. Koenderman L, Tesselaar K, Vrisekoop N. Human neutrophil kinetics: a call to revisit old evidence. Trends Immunol. (2022) 43:868–76. doi: 10.1016/j.it.2022.09.008
15. Rodríguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MMB, López-Villegas EO, Sánchez-García FJ. Metabolic requirements for neutrophil extracellular traps formation. Immunology. (2015) 145:213–24. doi: 10.1111/imm.2015.145.issue-2
16. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Sci (80-). (2004) 303:1532–5. doi: 10.1126/science.1092385
17. McIlroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, et al. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J Crit Care. (2014) 29:1133.e1–1133.e5. doi: 10.1016/j.jcrc.2014.07.013
18. Azevedo EP, Rochael NC, Guimarães-Costa AB, de-Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J Biol Chem. (2015) 290:22174–83. doi: 10.1074/jbc.M115.640094
19. Borella R, De Biasi S, Paolini A, Boraldi F, Lo Tartaro D, Mattioli M, et al. Metabolic reprograming shapes neutrophil functions in severe COVID-19. Eur J Immunol. (2022) 52:484–502. doi: 10.1002/eji.202149481
20. Piccolo EB, Thorp EB, Sumagin R. Functional implications of neutrophil metabolism during ischemic tissue repair. Curr Opin Pharmacol. (2022) 63:102191. doi: 10.1016/j.coph.2022.102191
21. van Raam B, Verhoeven A, Kuijpers T. Mitochondria in neutrophil apoptosis. Int J Hematol. (2006) 84:199–204. doi: 10.1532/IJH97.06131
22. Kumar S, Dikshit M. Metabolic insight of neutrophils in health and disease. Front Immunol. (2019) 10:2099. doi: 10.3389/fimmu.2019.02099
23. Cao Z, Zhao M, Sun H, Hu L, Chen Y, Fan Z. Roles of mitochondria in neutrophils. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.934444
24. Six E, Lagresle-Peyrou C, Susini S, De Chappedelaine C, Sigrist N, Sadek H, et al. AK2 deficiency compromises the mitochondrial energy metabolism required for differentiation of human neutrophil and lymphoid lineages. Cell Death Dis. (2015) 6:e1856–6. doi: 10.1038/cddis.2015.211
25. Tanimura A, Miyoshi K, Horiguchi T, Hagita H, Fujisawa K, Noma T. Mitochondrial activity and unfolded protein response are required for neutrophil differentiation. Cell Physiol Biochem. (2018) 47:1936–50. doi: 10.1159/000491464
26. Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun. (2018) 9:5099. doi: 10.1038/s41467-018-07505-2
27. Dunham-Snary KJ, Surewaard BG, Mewburn JD, Bentley RE, Martin AY, Jones O, et al. Mitochondria in human neutrophils mediate killing of Staphylococcus aureus. Redox Biol. (2022) 49:102225. doi: 10.1016/j.redox.2021.102225
28. Gamara J, Davis L, Leong AZ, Pagé N, Rollet-Labelle E, Zhao C, et al. Arf6 regulates energy metabolism in neutrophils. Free Radic Biol Med. (2021) 172:550–61. doi: 10.1016/j.freeradbiomed.2021.07.001
29. Lehrer RI, Ganz T, Szklarek D, Selsted ME. Modulation of the in vitro candidacidal activity of human neutrophil defensins by target cell metabolism and divalent cations. J Clin Invest. (1988) 81:1829–35. doi: 10.1172/JCI113527
30. Zheng X, Chen M, Meng X, Chu X, Cai C, Zou F. Phosphorylation of dynamin-related protein 1 at Ser616 regulates mitochondrial fission and is involved in mitochondrial calcium uniporter-mediated neutrophil polarization and chemotaxis. Mol Immunol. (2017) 87:23–32. doi: 10.1016/j.molimm.2017.03.019
31. Zhou W, Cao L, Jeffries J, Zhu X, Staiger CJ, Deng Q. Neutrophil-specific knockout demonstrates a role for mitochondria in regulating neutrophil motility in zebrafish. Dis Model Mech. (2018) 11. doi: 10.1242/dmm.033027
32. Riffelmacher T, Giles DA, Zahner S, Dicker M, Andreyev AY, McArdle S, et al. Metabolic activation and colitis pathogenesis is prevented by lymphotoxin β receptor expression in neutrophils. Mucosal Immunol. (2021) 14:679–90. doi: 10.1038/s41385-021-00378-7
33. Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. (2013) 1:542–51. doi: 10.1016/j.redox.2013.10.011
34. Rossaint J, Thomas K, Mersmann S, Skupski J, Margraf A, Tekath T, et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J Exp Med. (2021) 218. doi: 10.1084/jem.20201353
35. Chen Y, Junger WG. Measurement of oxidative burst in neutrophils. Methods Mol Biol. (2012) 844:115–24. doi: 10.1007/978-1-61779-527-5_8
36. Vorobjeva N, Prikhodko A, Galkin I, Pletjushkina O, Zinovkin R, Sud’ina G, et al. Mitochondrial reactive oxygen species are involved in chemoattractant-induced oxidative burst and degranulation of human neutrophils in vitro. Eur J Cell Biol. (2017) 96:254–65. doi: 10.1016/j.ejcb.2017.03.003
37. Bao Y, Ledderose C, Seier T, Graf AF, Brix B, Chong E, et al. Mitochondria regulate neutrophil activation by generating ATP for autocrine purinergic signaling. J Biol Chem. (2014) 289:26794–803. doi: 10.1074/jbc.M114.572495
38. Paclet MH, Laurans S, Dupré-Crochet S. Regulation of neutrophil NADPH oxidase, NOX2: A crucial effector in neutrophil phenotype and function. Front Cell Dev Biol. (2022) 10:945749. https://pmc.ncbi.nlm.nih.gov/articles/PMC9329797/.
39. Zeng MY, Miralda I, Armstrong CL, Uriarte SM, Bagaitkar J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol Oral Microbiol. (2019) 34:27. https://pmc.ncbi.nlm.nih.gov/articles/PMC6935359/.
40. Savchenko AA, Borisov AG, Zdzitovetskiy DE, Medvedev AY, Gvozdev II. Dependence of neutrophil respiratory burst on their metabolic state in the patients with acute destructive pancreatitis of different severity. Med Immunol. (2019) 21:77–88. doi: 10.15789/1563-0625-2019-1-77-88
41. Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab. (2019) 1:404–15. doi: 10.1038/s42255-019-0043-x
42. Monteith AJ, Miller JM, Williams JM, Voss K, Rathmell JC, Crofford LJ, et al. Altered Mitochondrial Homeostasis during Systemic Lupus Erythematosus Impairs Neutrophil Extracellular Trap Formation Rendering Neutrophils Ineffective at Combating Staphylococcus aureus. J Immunol. (2022) 208:454–63. doi: 10.4049/jimmunol.2100752
43. Hatanaka E, Levada-Pires AC, Pithon-Curi TC, Curi R. Systematic study on ROS production induced by oleic, linoleic, and γ-linolenic acids in human and rat neutrophils. Free Radic Biol Med. (2006) 41:1124–32. doi: 10.1016/j.freeradbiomed.2006.06.014
44. Hellenthal KEM, Thomas K, Ludwig N, Cappenberg A, Schemmelmann L, Tekath T, et al. Glutamine modulates neutrophil recruitment and effector functions during sterile inflammation. J Leukoc Biol. (2024) 00:1–14. doi: 10.1093/jleuko/qiae243
45. Quiroga J, Alarcón P, Manosalva C, Taubert A, Hermosilla C, Hidalgo MA, et al. Mitochondria-derived ATP participates in the formation of neutrophil extracellular traps induced by platelet-activating factor through purinergic signaling in cows. Dev Comp Immunol. (2020) 113:103768. doi: 10.1016/j.dci.2020.103768
46. Quiroga J, Alarcón P, Manosalva C, Taubert A, Hermosilla C, Hidalgo MA, et al. Glycolysis and mitochondrial function regulate the radical oxygen species production induced by platelet-activating factor in bovine polymorphonuclear leukocytes. Vet Immunol Immunopathol. (2020) 226:110074. doi: 10.1016/j.vetimm.2020.110074
47. Okabayashi K, Kanai S, Katakura F, Takeuchi R, Yamauchi T, Nakayama S, et al. Activation of canine neutrophils by platelet-activating factor. Vet Immunol Immunopathol. (2021) 241:110336. doi: 10.1016/j.vetimm.2021.110336
48. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. (2011) 18:581–8. doi: 10.1038/cdd.2011.1
49. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. (2014) 12:2074–88. doi: 10.1111/jth.12710
50. Cichon I, Ortmann W, Kolaczkowska E. Metabolic pathways involved in formation of spontaneous and lipopolysaccharide-induced neutrophil extracellular traps (NETs) differ in obesity and systemic inflammation. Int J Mol Sci. (2021) 22:7718. doi: 10.3390/ijms22147718
51. Cichon I, Ortmann W, Santocki M, Opydo-Chanek M, Kolaczkowska E. Scrutinizing mechanisms of the ‘Obesity paradox in sepsis’: obesity is accompanied by diminished formation of neutrophil extracellular traps (NETs) due to restricted neutrophil–platelet interactions. Cells. (2021) 10:384. doi: 10.3390/cells10020384
52. Garcia C, de Oliveira MCX, Verlengia R, Curi R, Pithon-Curi TC. Effect of dexamethasone on neutrophil metabolism. Cell Biochem Funct. (2003) 21:105–11. doi: 10.1002/cbf.v21:2
53. Garcia C, Tc P-C, De Lourdes Firmano M, Pires De Melo M, Newsholme P, Curi R. Effects of adrenaline on glucose and glutamine metabolism and superoxide production by rat neutrophils. Clin Sci. (1999) 96:549–55. doi: 10.1042/cs0960549
54. Ali RA, Wuescher LM, Worth RG. Platelets: essential components of the immune system. Curr Trends Immunol. (2015) 16:65–78.
55. Middleton EA, Weyrich AS, Zimmerman GA. Platelets in pulmonary immune responses and inflammatory lung diseases. Physiol Rev. (2016) 96:1211–59. doi: 10.1152/physrev.00038.2015
56. Lam FW, Burns AR, Smith CW, Rumbaut RE. Platelets enhance neutrophil transendothelial migration via P-selectin glycoprotein ligand-1. Am J Physiol Circ Physiol. (2010) 300:H468–75. doi: 10.1152/ajpheart.00491.2010
57. Nicolai L, Pekayvaz K, Massberg S. Platelets: Orchestrators of immunity in host defense and beyond. Immunity. (2024) 57:957–72. doi: 10.1016/j.immuni.2024.04.008
58. Yan C, Wu H, Fang X, He J, Zhu F. Platelet, a key regulator of innate and adaptive immunity. Front Med. (2023) 10:1074878. doi: 10.3389/fmed.2023.1074878
59. Scherlinger M, Richez C, Tsokos GC, Boilard E, Blanco P. The role of platelets in immune-mediated inflammatory diseases. Nat Rev Immunol. (2023) 23:495–510. doi: 10.1038/s41577-023-00834-4
60. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8. doi: 10.1126/science.1256478
61. Nicolai L, Schiefelbein K, Lipsky S, Leunig A, Hoffknecht M, Pekayvaz K, et al. Vascular surveillance by haptotactic blood platelets in inflammation and infection. Nat Commun. (2020) 11:1–16. doi: 10.1038/s41467-020-19515-0
62. Zuchtriegel G, Uhl B, Puhr-Westerheide D, Pörnbacher M, Lauber K, Krombach F, et al. Platelets guide leukocytes to their sites of extravasation. PloS Biol. (2016) 14. doi: 10.1371/journal.pbio.1002459
63. Müller KAL, Chatterjee M, Rath D, Geisler T. Platelets, inflammation and anti-inflammatory effects of antiplatelet drugs in ACS and CAD. Thromb Haemost. (2015) 114:498–518. doi: 10.1160/TH14-11-0947
64. Margraf A, Zarbock A. Platelets in inflammation and resolution. J Immunol. (2019) 203:2357–67. doi: 10.4049/jimmunol.1900899
65. Zhao J, Zhen N, Zhou Q, Lou J, Cui W, Zhang G, et al. NETs promote inflammatory injury by activating cGAS-STING pathway in acute lung injury. Int J Mol Sci. (2023) 24(6):5125. doi: 10.3390/ijms24065125
66. Garishah FM, Rother N, Riswari SF, Alisjahbana B, Overheul GJ, van Rij RP, et al. Neutrophil extracellular traps in dengue are mainly generated NOX-independently. Front Immunol. (2021) 12:1–10. doi: 10.3389/fimmu.2021.629167
67. Rolfes V, Ribeiro LS, Hawwari I, Böttcher L, Rosero N, Maasewerd S, et al. Platelets fuel the inflammasome activation of innate immune cells. Cell Rep. (2020) 31(6).
68. Li T, Yang Y, Li Y, Wang Z, Ma F, Luo R, et al. Platelets mediate inflammatory monocyte activation by SARS-CoV-2 spike protein. J Clin Invest. (2022) 132(4):e150101. doi: 10.1172/JCI150101
69. Inui M, Tazawa K, Kishi Y, Takai T. Platelets convert peripheral blood circulating monocytes to regulatory cells via immunoglobulin G and activating-type Fcγ receptors. BMC Immunol. (2015) 16:20. doi: 10.1186/s12865-015-0086-z
70. Ghatge M, Flora GD, Nayak MK, Chauhan AK. Platelet metabolic profiling reveals glycolytic and 1-carbon metabolites are essential for GP VI-stimulated human platelets-brief report. Arterioscler Thromb Vasc Biol. (2024) 44:409–16. doi: 10.1161/ATVBAHA.123.319821
71. Ravera S, Signorello MG, Panfoli I. Platelet metabolic flexibility: A matter of substrate and location. Cells. (2023) 12:1–12. doi: 10.3390/cells12131802
72. Aibibula M, Naseem KM, Sturmey RG. Glucose metabolism and metabolic flexibility in blood platelets. J Thromb Haemost. (2018) 16:2300–14. doi: 10.1111/jth.14274
73. Wang L, Wu Q, Fan Z, Xie R, Wang Z, Lu Y. Platelet mitochondrial dysfunction and the correlation with human diseases. Biochem Soc Trans. (2017) 45:1213–23. doi: 10.1042/BST20170291
74. Heijnen HFG, Oorschot V, Sixma JJ, Slot JW, James DE. Thrombin stimulates glucose transport in human platelets via the translocation of the glucose transporter GLUT-3 from alpha-granules to the cell surface. J Cell Biol. (1997) 138:323–30. doi: 10.1083/jcb.138.2.323
75. Fidler TP, Campbell RA, Funari T, Dunne N, Balderas Angeles E, Middleton EA, et al. Deletion of GLUT1 and GLUT3 reveal multiple roles for glucose metabolism in platelet and megakaryocyte function. Cell Rep. (2017) 20:881. doi: 10.1016/j.celrep.2017.06.083
76. Kulkarni PP, Tiwari A, Singh N, Gautam D, Sonkar VK, Agarwal V, et al. Aerobic glycolysis fuels platelet activation: small-molecule modulators of platelet metabolism as anti-thrombotic agents. Haematologica. (2019) 104:806–18. doi: 10.3324/haematol.2018.205724
77. Ravi S, Chacko B, Sawada H, Kramer PA, Johnson MS, Benavides GA, et al. Metabolic plasticity in resting and thrombin activated platelets. PloS One. (2015) 10(4):e0123597. doi: 10.1371/journal.pone.0123597
78. Kulkarni PP, Ekhlak M, Singh V, Kailashiya V, Singh N, Dash D. Fatty acid oxidation fuels agonist-induced platelet activation and thrombus formation: Targeting β-oxidation of fatty acids as an effective anti-platelet strategy. FASEB J. (2023) 37(2):e22768. doi: 10.1096/fj.202201321RR
79. De La Peña NC, Gutiérrez-Aguilar M, Hernández-Reséndiz I, Marín-Hernández Á, Rodríguez-Enríquez S. Glycoprotein Ib activation by thrombin stimulates the energy metabolism in human platelets. PloS One. (2017) 12:e0182374. https://pmc.ncbi.nlm.nih.gov/articles/PMC5560607/.
80. Kramer PA, Ravi S, Chacko B, Johnson MS, Darley-Usmar VM. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biol. (2014) 2:206. doi: 10.1016/j.redox.2013.12.026
81. Prakhya KS, Vekaria H, Coenen DM, Omali L, Lykins J, Joshi S, et al. Platelet glycogenolysis is important for energy production and function. Platelets. (2023) 34(1):2222184. doi: 10.1080/09537104.2023.2222184
82. Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. (2007) 21:99–111. doi: 10.1016/j.blre.2006.06.001
83. Bauer HM. In-vitro platelet–neutrophil adherence. Am J Clin Pathol. (1975) 63:824–7. doi: 10.1093/ajcp/63.6.824
84. Page C, Pitchford S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int Immunopharmacol. (2013) 17:1176–84. doi: 10.1016/j.intimp.2013.06.004
85. Matsumoto K, Yasuoka H, Yoshimoto K, Suzuki K, Takeuchi T. Platelet CXCL4 mediates neutrophil extracellular traps formation in ANCA-associated vasculitis. Sci Rep. (2021) 11:222. doi: 10.1038/s41598-020-80685-4
86. Constantinescu-Bercu A, Grassi L, Frontini M, Salles-Crawley II, Woollard K, Crawley JT. Activated αIIbβ3 on platelets mediates flow-dependent NETosis via SLC44A2. Elife. (2020) 9:e53353. doi: 10.7554/eLife.53353
87. Trotta A, Milillo MA, Serafino A, Castillo LA, Birnberg Weiss F, Delpino MV, et al. Brucella abortus –infected platelets modulate the activation of neutrophils. Immunol Cell Biol. (2020) 98:743–56. doi: 10.1111/imcb.12373
88. Scheuermann L, Pei G, Domaszewska T, Zyla J, Oberbeck-Müller D, Bandermann S, et al. Platelets restrict the oxidative burst in phagocytes and facilitate primary progressive tuberculosis. Am J Respir Crit Care Med. (2020) 202:730–44. doi: 10.1164/rccm.201910-2063OC
89. Miedzobrodzki J, Panz T, Płonka PM, Zajac K, Dracz J, Pytel K, et al. Platelets augment respiratory burst in neutrophils activated by selected species of gram-positive or gram-negative bacteria. Folia. Histochem Cytobiol. (2008) 46(3):383–8. doi: 10.2478/v10042-008-0052-1
90. Rossaint J, Kühne K, Skupski J, Van Aken H, Looney MR, Hidalgo A, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun. (2016) 7:13464. doi: 10.1038/ncomms13464
91. Vázquez-Otero I, Rodríguez-Navedo Y, Vilá-Rivera K, Nieves-Plaza M, Morales-Ortiz J, Washington V, et al. Association of soluble TREM-like transcript-1 with clinical features and patient reported outcomes in systemic lupus erythematosus. Eur J Rheumatol. (2018) 5:244–8. doi: 10.5152/eurjrheum.2018.18074
92. Haselmayer P, Grosse-Hovest L, von Landenberg P, Schild H, Radsak MP. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood. (2007) 110:1029–35. doi: 10.1182/blood-2007-01-069195
93. Washington AV, Gibot S, Acevedo I, Gattis J, Quigley L, Feltz R, et al. TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J Clin Invest. (2009) 119:1489–501. doi: 10.1172/JCI36175
94. Etulain J, Negrotto S, Carestia A, Pozner R, Romaniuk M, D’Atri L, et al. Acidosis downregulates platelet haemostatic functions and promotes neutrophil proinflammatory responses mediated by platelets. Thromb Haemost. (2012) 107:99–110. doi: 10.1160/TH11-06-0443
95. De Filippo K, Rankin SM. The secretive life of neutrophils revealed by intravital microscopy. Front Cell Dev Biol. (2020) 8. doi: 10.3389/fcell.2020.603230
96. Frydman GH, Le A, Ellett F, Jorgensen J, Fox JG, Tompkins RG, et al. Technical Advance: Changes in neutrophil migration patterns upon contact with platelets in a microfluidic assay. J Leukoc Biol. (2017) 101:797–806. doi: 10.1189/jlb.1TA1115-517RR
97. Nakashima S, Suganuma A, Sato M, Tohmatsu T, Nozawa Y. Mechanism of arachidonic acid liberation in platelet-activating factor-stimulated human polymorphonuclear neutrophils. J Immunol. (1989) 143:295–302.
98. Herbertsson H, Bengtsson T. Role of platelets and the arachidonic acid pathway in the regulation of neutrophil oxidase activity. Scand J Clin Lab Invest. (2001) 61:641–9. doi: 10.1080/003655101753268008
99. Li H, Tang C, Zhu X, Zhang W, Abudupataer M, Ding S, et al. Histamine deficiency facilitates coronary microthrombosis after myocardial infarction by increasing neutrophil-platelet interactions. J Cell Mol Med. (2020) 24:3504–20. doi: 10.1111/jcmm.15037
100. Duchez AC, Boudreau LH, Naika GS, Bollinger J, Belleannée C, Cloutier N, et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A 2 -IIA. Proc Natl Acad Sci USA. (2015) 112(49):E6825. doi: 10.1073/pnas.1522504112
101. Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, et al. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun. (2017) 8:1–14. doi: 10.1038/s41467-017-02402-6
102. Flora GD, Nayak MK, Ghatge M, Kumskova M, Patel RB, Chauhan AK. Mitochondrial pyruvate dehydrogenase kinases contribute to platelet function and thrombosis in mice by regulating aerobic glycolysis. Blood Adv. (2023) 7:2347–59. doi: 10.1182/bloodadvances.2023010100
103. Núñez-Avellaneda D, Mosso-Pani MA, Sánchez-Torres LE, Castro-Mussot ME, Corona-De-la-Peña NA, Salazar MI. Dengue virus induces the release of sCD40l and changes in levels of membranal CD42B and CD40L molecules in human platelets. Viruses. (2018) 10(7):357. doi: 10.3390/v10070357
104. Číž M, Komrsková D, Prachařová L, Okénková K, Čížová H, Moravcová A, et al. Serotonin modulates the oxidative burst of human phagocytes via various mechanisms. Platelets. (2007) 18:583–90. doi: 10.1080/09537100701471865
105. Chen Z, Luo J, Li J, Kim G, Stewart A, Urban JF, et al. Interleukin-33 promotes serotonin release from enterochromaffin cells for intestinal homeostasis. Immunity. (2021) 54:151. doi: 10.1016/j.immuni.2020.10.014
106. Chen Z, Luo J, Li J, Kim G, Stewart A, Huang Y, et al. Intestinal IL-33 promotes platelet activity for neutrophil recruitment during acute inflammation. Blood. (2022) 139:1878–91. doi: 10.1182/blood.2021013474
107. De Giovanni M, Tam H, Valet C, Xu Y, Looney MR, Cyster JG. GPR35 promotes neutrophil recruitment in response to serotonin metabolite 5-HIAA. Cell. (2022) 185:815–830.e19. doi: 10.1016/j.cell.2022.01.010
108. De Giovanni M, Dang EV, Chen KY, An J, Madhani HD, Cyster JG. Platelets and mast cells promote pathogenic eosinophil recruitment during invasive fungal infection via the 5-HIAA-GPR35 ligand-receptor system. Immunity. (2023) 56:1548–1560.e5. doi: 10.1016/j.immuni.2023.05.006
109. Zhou H, Deng M, Liu Y, Yang C, Hoffman R, Zhou J, et al. Platelet HMGB1 is required for efficient bacterial clearance in intra-abdominal bacterial sepsis in mice. Blood Adv. (2018) M2:638–48. doi: 10.1182/bloodadvances.2017011817
110. Pervushina O, Scheuerer B, Reiling N, Behnke L, Schröder JM, Kasper B, et al. Platelet factor 4/CXCL4 induces phagocytosis and the generation of reactive oxygen metabolites in mononuclear phagocytes independently of gi protein activation or intracellular calcium transients. J Immunol. (2004) 173:2060–7. doi: 10.4049/jimmunol.173.3.2060
111. Blick-Nitko SK, Ture SK, Schafer XL, Munger JC, Livada AC, Li C, et al. Platelet Ido1 expression is induced during Plasmodium yoelii infection, altering plasma tryptophan metabolites. Blood Adv. (2024) 8:5814–25. doi: 10.1182/bloodadvances.2024013175
112. Møller SH, Wang L, Ho PC. Metabolic programming in dendritic cells tailors immune responses and homeostasis. Cell Mol Immunol. (2022) 19:370–83. doi: 10.1038/s41423-021-00753-1
113. Sudowe S, Höhn Y, Renzing A, Maxeiner J, Montermann E, Habermeier A, et al. Inhibition of antigen-specific immune responses by co-application of an indoleamine 2,3-dioxygenase (IDO)-encoding vector requires antigen transgene expression focused on dendritic cells. Amino Acids. (2020) 52:411–24. doi: 10.1007/s00726-020-02817-4
114. Fahlberg MD, Blair RV, Doyle-Meyers LA, Midkiff CC, Zenere G, Russell-Lodrigue KE, et al. Cellular events of acute, resolving or progressive COVID-19 in SARS-CoV-2 infected non-human primates. Nat Commun. (2020) 11:6078. https://pmc.ncbi.nlm.nih.gov/articles/PMC7695721/.
115. Tan AF, Sakam SSB, Piera K, Rajahram GS, William T, Barber BE, et al. Neutrophil activation, acute lung injury and disease severity in Plasmodium knowlesi malaria. PloS Negl Trop Dis. (2024) 18:e0012424. https://pmc.ncbi.nlm.nih.gov/articles/PMC11357107/.
116. Léger JL, Soucy MN, Veilleux V, Foulem RD, Robichaud GA, Surette ME, et al. Functional platelet-derived mitochondria induce the release of human neutrophil microvesicles. EMBO Rep. (2022) 23(11):e54910. doi: 10.15252/embr.202254910
117. Støy S, Patel VC, Sturgeon JP, Manakkat Vijay GK, Lisman T, Bernal W, et al. Platelet-leucocyte aggregation is augmented in cirrhosis and further increased by platelet transfusion. Aliment Pharmacol Ther. (2018) 47:1375–86. doi: 10.1111/apt.14600
118. Perazzio SF, Soeiro-Pereira PV, dos Santos VC, de Brito MV, Salu B, Oliva MLV, et al. Soluble CD40L is associated with increased oxidative burst and neutrophil extracellular trap release in Behçet’s disease. Arthritis Res Ther. (2017) 19:235. doi: 10.1186/s13075-017-1443-5
119. Jin R, Yu S, Song Z, Zhu X, Wang C, Yan J, et al. Soluble CD40 ligand stimulates CD40-dependent activation of the β2 integrin mac-1 and protein kinase C zeda (PKCζ) in neutrophils: implications for neutrophil-platelet interactions and neutrophil oxidative burst. PloS One. (2013) 8(6):e64631. doi: 10.1371/journal.pone.0064631
120. Xie RF, Hu P, Li W, Ren YN, Yang J, Yang YM, et al. The effect of platelet-derived microparticles in stored apheresis platelet concentrates on polymorphonuclear leucocyte respiratory burst. Vox Sang. (2014) 106:234–41. doi: 10.1111/vox.2014.106.issue-3
121. Sung P-S, Huang T-F, Hsieh S-L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. (2019) 10:2402. doi: 10.1038/s41467-019-10360-4
122. Xie RF, Hu P, Wang ZC, Yang J, Yang YM, Gao L, et al. Platelet-derived microparticles induce polymorphonuclear leukocyte-mediated damage of human pulmonary microvascular endothelial cells. Transfusion. (2015) 55:1051–7. doi: 10.1111/trf.12952
123. Couto-Rodriguez A, Villaseñor A, Pablo-Torres C, Obeso D, Rey-Stolle MF, Peinado H, et al. Platelet-derived extracellular vesicles as lipid carriers in severe allergic inflammation. Int J Mol Sci. (2023) 24(16):12714. doi: 10.3390/ijms241612714
124. Zhang Z, Xu X, Zhang D, Zhao S, Wang C, Zhang G, et al. Targeting Erbin-mitochondria axis in platelets/megakaryocytes promotes B cell-mediated antitumor immunity. Cell Metab. (2024) 36:541–556.e9. doi: 10.1016/j.cmet.2023.12.020
125. Garraud O, Hamzeh-Cognasse H, Chalayer E, Duchez AC, Tardy B, Oriol P, et al. Platelet transfusion in adults: An update. Transfus Clin Biol. (2023) 30:147–65. doi: 10.1016/j.tracli.2022.08.147
126. Zhou P, Xiao JH, Li Y, Zhou L, Deng Z. Platelet count has a nonlinear association with 30-day in-hospital mortality in ICU end-stage kidney disease patients: a multicenter retrospective cohort study. Sci Rep. (2024) 14:1–12. doi: 10.1038/s41598-024-73717-w
127. Brown L, Yipp BG. Inflammatory platelet death drives sepsis. Nat Cardiovasc Res. (2022) 1:689–90. doi: 10.1038/s44161-022-00111-y
128. Li S, Lu Z, Wu S, Chu T, Li B, Qi F, et al. The dynamic role of platelets in cancer progression and their therapeutic implications. Nat Rev Cancer. (2023) 24:72–87. doi: 10.1038/s41568-023-00639-6
129. Chen S, Wu A, Shen X, Kong J, Huang Y. Disrupting the dangerous alliance: Dual anti-inflammatory and anticoagulant strategy targets platelet-neutrophil crosstalk in sepsis. J Control Release. (2025) 379:814–31. doi: 10.1016/j.jconrel.2025.01.053
130. Mayer CL, Koeck K, Hottmann M, Redfern A, Davis M, Barth A, et al. A phase 1 study in healthy participants to characterize the safety and pharmacology of inclacumab, a fully human anti-P-selectin antibody, in development for treatment of sickle cell disease. Eur J Clin Pharmacol. (2023) 79:1219. doi: 10.1007/s00228-023-03514-3
131. Geng X, Mihaila R, Yuan Y, Strutt S, Benz J, Tang T, et al. Inclacumab, a fully human anti-P-selectin antibody, directly binds to PSGL-1 binding region and demonstrates robust and durable inhibition of cell adhesion. Blood. (2020) 136:10–1. doi: 10.1182/blood-2020-140530
132. Nhek S, Clancy R, Lee KA, Allen NM, Barrett TJ, Marcantoni E, et al. Activated platelets induce endothelial cell activation via an interleukin-1β pathway in systemic lupus erythematosus. Arterioscler Thromb Vasc Biol. (2017) 37:707–16. doi: 10.1161/ATVBAHA.116.308126
133. Pallara C, Cabot D, Rivas J, Brun S, Seco J, Abuasaker B, et al. Peptidomimetics designed to bind to RAS effector domain are promising cancer therapeutic compounds. Sci Rep. (2022) 12:1–15. doi: 10.1038/s41598-022-19703-6
134. Gomari MM, Abkhiz S, Pour TG, Lotfi E, Rostami N, Monfared FN, et al. Peptidomimetics in cancer targeting. Mol Med. (2022) 28:1–17. doi: 10.1186/s10020-022-00577-3
135. Kirsebom FCM, Kausar F, Nuriev R, Makris S, Johansson C. Neutrophil recruitment and activation are differentially dependent on MyD88/TRIF and MAVS signaling during RSV infection. Mucosal Immunol. (2019) 12:1244–55. doi: 10.1038/s41385-019-0190-0
136. George ST, Lai J, Ma J, Stacey HD, Miller MS, Mullarkey CE. Neutrophils and influenza: A thin line between helpful and harmful. Vaccines. (2021) 9(6):597. doi: 10.3390/vaccines9060597
Keywords: neutrophil immunometabolism, platelet-derived metabolites, mitochondrial metabolism, platelet-neutrophil aggregates, inflammation regulation
Citation: Mosso-Pani MA, Barreda D and Salazar MI (2025) Dynamic regulation of neutrophil immunometabolism by platelet-derived metabolites. Front. Immunol. 16:1542438. doi: 10.3389/fimmu.2025.1542438
Received: 09 December 2024; Accepted: 03 March 2025;
Published: 27 March 2025.
Edited by:
Jacqueline Liszeth Oliva-Ramírez, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Lee-Ann H. Allen, University of Missouri, United StatesIrina Miralda, Emory University, United States
Nonantzin Beristain-Covarrubias, University of Cambridge, United Kingdom
Copyright © 2025 Mosso-Pani, Barreda and Salazar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ma. Isabel Salazar, aXNhbGF6YXJzYW5AeWFob28uY29t
†These authors have contributed equally to this work and share first authorship