 Longyan Hong
Longyan Hong Hao Liang
Hao Liang Wenqing Man4
Wenqing Man4 Yinghui Zhao
Yinghui Zhao Pengbo Guo
Pengbo Guo- 1The First Affiliated Hospital of Shandong First Medical University, Shandong Provincial Qianfoshan Hospital, Jinan, China
- 2Department of Pathogen Biology, School of Clinical and Basic Medicine, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
- 3Department of Health Inspection and Quarantine, School of Public Health, Cheeloo College of Medicine, Shandong University, Jinan, China
- 4School of Stomatology, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
Gender differences exist in the susceptibility, incidence, progression, and prognosis of bacterial infections in males and females, influenced by various factors including lifestyle and habits. Multiple reports have indicated that estrogen plays a crucial immunomodulatory role in many pathogenic microbial infections, highlighting a complex relationship between estrogen, its receptors, and bacterial infections. Estrogen and its receptors regulate host immune responses, affecting the host’s ability to clear bacteria and thus influencing the likelihood and difficulty of infection eradication. Variations in estrogen levels may lead to differences in the occurrence and progression of bacterial infections, with estrogen playing varied roles in diseases caused by the same bacterial pathogens. The interaction between estrogen and bacterial infections represents a complex and crucial aspect of human physiology and clinical medicine. Understanding this interaction is essential for advancing infection prevention and treatment strategies. This article reviews the correlation and mechanisms between estrogen and bacterial infections, emphasizing the importance of further research in this field.
1 Introduction
Bacterial infections pose a common yet significant health challenge, continuously impacting global human health (1). Gender differences play a crucial role in the incidence, severity, and prognosis of bacterial infections, thus attracting widespread attention to the role of gender factors in infectious pathology (2). Over the past few decades, researchers have identified numerous physiological and immunological differences related to gender (3, 4). However, in recent years, there has been a growing recognition of the role of estrogen in modulating bacterial infections (5). Estrogen not only plays a pivotal role in the female reproductive system but also exerts diverse physiological functions in other organs and systems (6–8). Increasing evidence has suggested that estrogen and its receptors not only influence the host’s susceptibility to bacterial infection and clearance capabilities but also directly or indirectly impact bacterial growth, survival, and virulence (4, 9). Understanding the mechanisms of interaction between estrogen and bacterial infections is crucial for developing more effective prevention and treatment strategies. Therefore, this paper aims to review the latest research progress on the role of estrogen in modulating bacterial infections, with the hope of providing new insights and directions for future studies.
2 Estrogen
Estrogen, a growth-inducing sex hormone, is expressed in both males and females and exerts its effects through binding to estrogen receptors (ER) α, β (ERα/β), as well as G protein-coupled estrogen receptor (GPER) (10, 11). The interaction between estrogen and its receptors is integral to immune regulation and disease development. They influence the host’s susceptibility to various pathogens, regulate inflammatory responses, influence the differentiation and function of immune cells, and contribute to the initiation and progression of autoimmune diseases (12–15). In bacterial infections, estrogen and its receptors could impact the recognition and clearance of bacteria by immune cells, as well as influence the development and progression of such infections by affecting inflammatory cytokines and signaling pathways. (Effects of Estrogen on Bacteria as Seen in Supplementary Materials Tables 1, 2).
3 Estrogen and bacteria
3.1 Chlamydia trachomatis
C. trachomatis, a Gram-negative bacterium, primarily parasitizes within eukaryotic cells (16). This bacterium is the main pathogen causing trachoma and is a common pathogen of sexually transmitted diseases globally. Infections of C. trachomatis in the urogenital tract could lead to various diseases, including urethritis, cervicitis, and pelvic inflammatory disease (17). Furthermore, it can result in severe complications such as tubal infertility and ectopic pregnancy, posing serious health issues to patients (18). Estrogen plays a crucial role in the female reproductive system, and variations in estrogen levels may affect the structure and immune function of the reproductive tract, thereby increasing the risk of C. trachomatis infection (19). Studies have shown an association between estrogen and its related receptors with an increased incidence of C. trachomatis infection (20–23).
Membrane estrogen receptors (mERs) facilitate the entry of C. trachomatis into host cells, and mER signaling promotes the development of inclusions in C. trachomatis infection. Hormones that enhance the C. trachomatis infection also involve stromal signals and direct stimulation of uterine epithelial cells by estrogen (21). Activation of mERs leads to the activation of the phosphoinositide 3-kinase (PI3K) signaling pathway, which is involved in cell proliferation. The C. trachomatis III effector molecule TARP interacts with PI3K to promote infection. Estradiol also affects the expression of TLR4 and downstream signaling molecules (IRAK4 and Nuclear factor κB), reducing the gene expression of Th1-related cytokines IL-12, IL-6, TNFα, and IFNγ, resulting in incorrect recognition of C. trachomatis by dendritic cells and thus increasing susceptibility to C. trachomatis infection (22) (Figure 1).
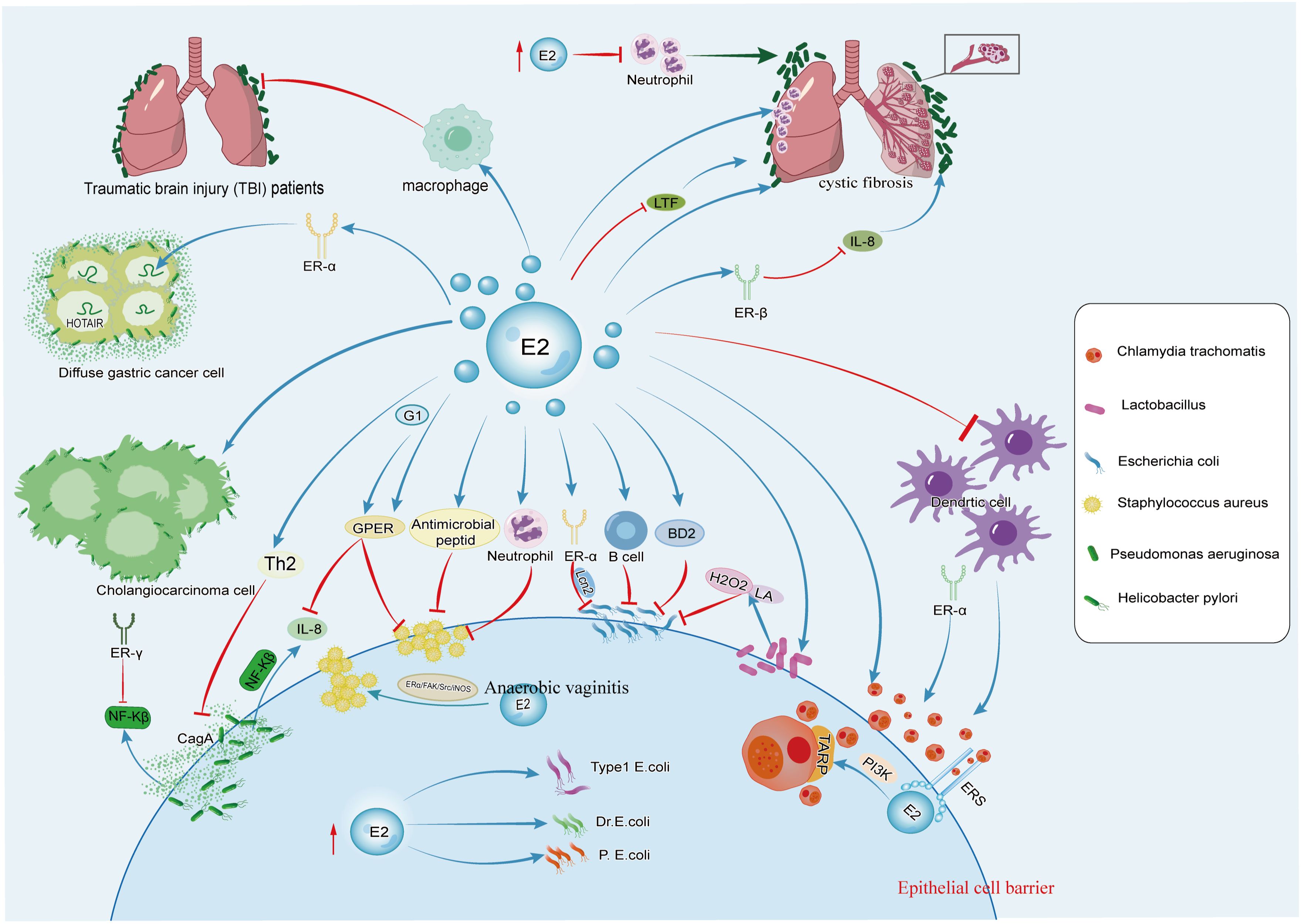
Figure 1. The figure illustrates the effects of estrogen on C. trachomatis, E. coli, Lactobacillus, S. aureus, P. aeruginosa, and H. pylori. Estrogen promotes the growth of C. trachomatis and Lactobacillus and can also synergize with P. aeruginosa to accelerate the occurrence and development of pulmonary cystic fibrosis. It has dual effects on S. aureus, H. pylori, and E. coli.
After C. trachomatis infection, inhibitors of estrogen receptor signaling reduces epithelial ER signaling exposure, affecting the MAPK signaling pathway, including ERK phosphorylation, regulation of downstream effector cPLA2, and the PI3K/AKT signaling pathway and calcium mobilization, all crucial for C. trachomatis inclusion development (19, 21).
During C. trachomatis infection, the lack of ERα alters the shedding of C. trachomatis in the vagina. Compared to WT animals expressing both ERs or only ERα, the progression of C. trachomatis infection is faster, and the clearance from the reproductive tract is more rapid in mice with only ERβ (ERαKO) (20) (Figure 1). ERα regulates T cell responses to C. trachomatis infection in mice. In ERαKO mice infected with C. trachomatis, the expression of T cells, FOXP3, and IFNϵ significantly increase. The defect of ERα expression increases stimulation of CD4+ T cells in mice’s macrophages. FOXP3, a transcription factor, is a critical marker of regulatory T cells (Tregs), indicating that Tregs are also involved in the clearance of C. trachomatis in condition of ERα deficiency (20).
C. trachomatis infection is also considered as a major cause of recurrent spontaneous abortion (24). Infection of the trophoblast layer by C. trachomatis impairs cellular cholesterol biosynthesis, depleting substrates for estrogen and progesterone synthesis, affecting functions such as implantation and placental formation in the trophoblast layer, thereby influencing pregnancy outcomes (25, 26). Studies have found that decreased progesterone levels lead to increased expression of pro-inflammatory cytokines in recurrent spontaneous abortion associated with infection, resulting in miscarriage by disrupting maternal tolerance of the embryo/fetus (27). Higher susceptibility has been observed during the proliferative phase of the menstrual cycle when estrogen levels are elevated, while progesterone enhances the innate immune response to C. trachomatis infection, thereby reducing susceptibility to C. trachomatis infection (28, 29).
3.2 Lactobacillus
Lactobacillus, representative microorganisms of normal vaginal flora, are characterized by being Gram-stain positive, microaerophilic, acid-resistant, non-spore-forming, and capable of producing lactic acid. In the vaginal microbiota of healthy women, Lactobacillus dominates, accounting for over 90% of the total vaginal flora (30, 31). Maintaining an acidic microenvironment of the vagina inhibited the growth of pathogens, including C. trachomatis, Neisseria gonorrhoeae (N. gonorrhoeae), and Escherichia coli (E. coli) (32).
During puberty, the increase in circulating estrogen promotes the proliferation of lactobacilli, leading to the production of lactic acid and hydrogen peroxide, which inhibited the growth of pathogens (33). Previous studies have shown that some postmenopausal patients who take estradiol could maintain vaginal pH around 4.5 and normal serum estradiol levels (34). The hormonal imbalance during the female reproductive cycle leads to bacterial vaginosis (BV), with the imbalance of vaginal microbiota playing a role.
The incidence of urinary tract infections begin to rise during menopause, and recurrent urinary tract infections are considered a feature of genitourinary syndrome of menopause, characterized by thinning of the vaginal epithelium, various symptoms associated with vaginal atrophy, and a relative loss of lactobacilli in the vaginal microbiota (35). The loss of estrogen during menopause lead to changes in the vaginal microbiota of women, with a decrease in the relative abundance of lactobacilli (36). Studies have shown that colonization of E. coli is more frequent in women who have not received estrogen replacement therapy, and it is negatively correlated with the presence of lactobacilli (37, 38) (Figure 1). Estrogen stimulates the proliferation of lactobacilli in the vaginal epithelium, reduces the pH value, prevents excessive colonization of intestinal bacteria in the vagina, and thus prevents urinary tract infection (39).
The use of local estrogen preparations to treat menopause-related estrogen deficiency can reduce the incidence of recurrent urinary tract infection in most women and restore lactobacilli in the vagina (35). Estrogen also lowers the pH value by stimulating the vaginal mucosa to produce acid to maintain vaginal flora balance. In postmenopausal women with recurrent cystitis, low-dose vaginal estrogen therapy controls the growth of pathogens in the vagina (40). Additionally, higher estrogen levels produced by adipose tissue increases the glycogen content in vaginal epithelial cells. The increase in glycogen promotes the colonization of lactobacilli and the production of lactic acid, thus supporting a more ideal vaginal environment (41).
Research has also found that progesterone and dehydroepiandrosterone (DHEA) have effects on lactobacilli. Progesterone increases the population of lactobacilli, affecting brain function by modulating gut microbiota and upregulating the expression of brain-derived neurotrophic factor (BDNF) genes, thereby alleviating depression and anxiety during menopause (42). Additionally, DHEA lowers vaginal pH by stimulating lactobacilli, thus relieving symptoms of atrophic vaginitis during menopause (43).
3.3 E. coli
E. coli is a Gram-negative rod bacterium commonly colonized in the intestines of animals and humans. Most strains are harmless and play important physiological roles in the intestines (44). However, certain strains may be pathogenic, causing various diseases, including sepsis, diarrhea, and urinary tract infections (UTIs) (45).
Among common urinary tract pathogens associated with the development of UTIs, uropathogenic E. coli (UPEC) is the main cause (23, 46). The incidence of UTIs significantly increases in postmenopausal women. Decreased estrogen levels may promote the colonization of UPEC in the urinary genital tract and enhance its ability for persistent infection (47–50). UPEC infection is included in a multi-stage infection pathway, including adhesion, invasion, replication, and persistent infection. Studies have shown that estrogen increases the thickness of extracellular glycosaminoglycan (GAG) layer on the bladder surface, affecting the composition and sulfation status of GAG, thereby preventing urinary tract infections caused by E. coli (51). However, some studies have found that high-dose estrogen therapy lead to a higher bacterial infection rate of three clinical isolates, type 1, P, and Dr fimbriae of UPEC, in the kidneys (52) (Figure 1).
The differences in research results may be related to the “dosage” or pathway of estrogen administration. Additionally, studies have found that under the influence of androgens, both males and females have exhibited increased susceptibility to urinary tract infections caused by UPEC, and activation of the androgen receptor (AR) have increased susceptibility to the formation of pyelonephritis and renal abscesses resulting from E. coli infection (53).
The recurrence rate of E. coli infection in the urethra is low among women receiving E2 treatment, which is related to the production of antimicrobial peptides (such as human β-defensins HBD-1, HBD-2, ribonuclease (RNase) 7) and the stability of the common microbiota (54). Among postmenopausal women receiving estrogen therapy, an increase in mRNA encoding HBD-1 and HBD-3, psoriasin, and cAMP are observed, thereby reducing the risk of bacterial infection (55).
E2 enhances epithelial integrity by increasing the production of antimicrobial peptides and strengthening intercellular connections, thus promoting epithelial barrier function (55, 56). BD2, also known as β-defensin 2, is a host defense peptide produced by host cells with bactericidal activity against various bacteria, including UPEC. Studies have shown that the concentration of BD2 in the vagina increased in patients receiving estrogen therapy, enhancing bladder innate responses and strengthening urethral epithelial integrity to prevent invasion by pathogenic E. coli (57) (Figure 1).
E2 also inhibits the production of IL-1β and lipopolysaccharide (LPS)-induced inflammatory effects by E. coli. During acute E. coli infection, estrogen restricts its proliferation and reduces residual bacteria (55, 56). Lipocalin-2 (Lcn2) is an important molecule that prevents bacterial infection by sequestering iron ions, and E. coli induces the expression of Lcn2 in the endometrium. Further research have shown that estrogen induces the expression of Lcn2 in the endometrial epithelium through ERα, thereby enhancing resistance to E. coli infection during early pregnancy (58) (Figure 1).
E. coli may also cause bacteremia, which may lead to sepsis, organ failure, and death when bacteria accumulate and persist in the body (59). Women have lower incidence and mortality rates of E. coli bacteremia (4). Studies have shown that colonic pathogenic E. coli in the bloodstream is rapidly phagocytosed by Kupffer cells in female mice, while bacterial uptake is delayed in male mice (60). There is a class of natural antibodies against colonic pathogenic E. coli in female mouse serum but not in male mouse serum. These antibodies are mainly composed of IgM and IgG3 subtypes, mainly produced by B1 cells, and estrogen indirectly promotes the production of natural antibodies by B1 cells through its effect on peritoneal macrophages (61) (Figure 1).
Antibodies against LPS-O127 reduce inflammatory tissue damage and provide life-saving protection in infants. Several research have found that estrogen-mediated interactions between peritoneal macrophages and B1 cells regulate the production of innate antibodies against LPS-O127 (60). Therefore, estrogen-driven natural antibodies against E. coli infection play a dual protective role in women and their offspring.
3.4 Pseudomonas aeruginosa (P. aeruginosa)
P. aeruginosa, is a ubiquitous Gram-negative bacillus existed in soil, water bodies, plant surfaces, as well as on the surfaces of humans and animals (62). It is a highly drug-resistant pathogen, particularly common in hospital-acquired infections and immunocompromised patients, leading to various infections including respiratory tract infections, urinary tract infections, and wound infections (63).
P. aeruginosa is one of the common pathogens causing pulmonary infections in cystic fibrosis (CF) patients, seriously impacting the health of CF patients, resulting in deteriorating lung function, respiratory distress, acute exacerbations, chronic inflammation, and potentially leading to pulmonary fibrosis and airway damage, exacerbating the condition of CF patients (64, 65). Studies have indicated a significant gender difference in cystic fibrosis (CF). Compared to male, the survival rates of female patients are lower and the prognosis is worse (66). Female mice are more susceptible to P. aeruginosa infection than male mice, as estrogen and its receptors exacerbate lung inflammation and P. aeruginosa infection in CF patients (67). Supplementation of estrogen in male and female mice infected with P. aeruginosa reduce bacterial clearance ability (66).
17β-estradiol mediates neutrophil response in the environment of P. aeruginosa infection. In ovariectomized mice treated with E2 after P. aeruginosa infection, TNF-α, KC, IL-10, and IL-6 significantly increase, along with chemoattractants including granulocyte-macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1α (MIP-1α), and MIP-1β (66, 68). Neutrophil granule proteins myeloperoxidase (MPO) and neutrophil elastase (NE) are significantly enriched in lung tissues of mice treated with E2 (66). Furthermore, E2 enhances the production of pro-inflammatory Th17 mediators (IL-23/IL-17 pathway molecules), increases granulocyte colony-stimulating factor (G-CSF) to stimulate the neutrophil production, induces IL-17-mediated chemoattractant factor MIP-2 levels, recruits neutrophils to the site of inflammation, exacerbates the severity of P. aeruginosa pneumonia, and promotes lung tissue damage (4, 68). Under gradually increasing E2 doses, the killing ability of neutrophils against P. aeruginosa is inhibited (66) (Figure 1).
In female CF patients, increased levels of estradiol mediates upregulation of secretory leukocyte protease inhibitor (SLPI) expression via ERβ, thereby inhibiting TLR-dependent IL-8 release in cystic fibrosis bronchial epithelial cells, making female with cystic fibrosis more susceptible to infection and colonization by P. aeruginosa (4). E2 also modulates the formation of P. aeruginosa biofilms by affecting antimicrobial peptide lactoferrin (LTF), a component of innate immunity that interferes with bacterial biofilm development (68) (Figure 1). E2 treatment significantly reduces mRNA levels of antimicrobial peptide LTF in lung tissues and PIP in trachea, with LTF mRNA levels almost completely eliminated after exposure to E2.
Excessive production of alginate in CF patients exacerbates respiratory symptoms and increased the risk of respiratory infections (69). P. aeruginosa actively metabolizes steroid hormones and utilized estradiol as a carbon source to promote the production of alginate (70, 71). In menstruating CF females, circulating estradiol levels positively correlates with exacerbations, and increased proportions of the mucoid transition of P. aeruginosa are isolates during the period of higher estradiol levels (72). Short-term exposure of P. aeruginosa to estradiol results in increased hydrogen peroxide levels, and inhibits hydrogen peroxide detoxifying enzyme activity. Estradiol promotes mucoid transition of P. aeruginosa, increases alginate production and selectively induces mucA gene mutations (a negative regulator of alginate synthesis), which is due to impaired catalase activity and increased hydrogen peroxide levels (72).Therefore, female CF patients are infected with P. aeruginosa earlier than males, transitioning to mucoid strains prematurely, exacerbating respiratory symptoms, and making infections more difficult to clear (72).
The incidence of bacterial pneumonia is high in patients with severe traumatic brain injury (TBI) (73), with P. aeruginosa being one of the most common pathogens (74). Studies have found that P. aeruginosa pneumonia after TBI lead to higher mortality and reduced bacterial clearance, and gender differences are observed. Compared to female mice, male mice have higher mortality rates (75). There are differences between male and female macrophages, with female macrophages secreting higher levels of TNF-α compared to male macrophages (75). Severe TBI patients are at higher risk of bacterial pneumonia due to immune suppression. The administration of estradiol reduces mortality in male or ovariectomized wild-type female mice, and increases lung bacterial clearance, possibly mediated by differences in alveolar macrophage function (Figure 1).
The role of estrogen in P. aeruginosa infection varies depending on the type of disease, possibly related to estrogen dose, route of administration, and mediated signaling pathways.
3.5 Staphylococcus aureus
S. aureus is a prevalent Gram-positive bacterium found on the human skin and mucous membranes. Although it is usually harmless, S. aureus could also cause various infections, including skin infections, food poisoning, respiratory tract infections, and sepsis (76).
As a major pathogen of global skin and soft tissue infections (SSTIs), the toxin alpha-hemolysin (Hlα) produced by S. aureus is a key virulence factor causing skin necrosis and inflammation, promoting invasive infections by destroying cell junctions (77–79). Studies have shown that males are more susceptible to SSTIs caused by S. aureus compared to females (80) (Figure 1). Furthermore, research have found that estrogen plays an important role in regulating the innate immune response of females to S. aureus pathogens (14, 80). Female mice exhibit lower levels of pro-inflammatory cytokines such as IL-1β, TNFα, IL-6, and CXCL1 at the site of S. aureus infection, along with stronger neutrophil bactericidal activity (80). Female mice show estrogen-dependent reduction in skin necrosis, lower levels of local inflammatory cytokines, and a significantly reduced bacterial burden after infection. Additionally, in the rat uterus, E2 induces antimicrobial peptides to inhibit the invasion of S. aureus (55) (Figure 1).
G-1, a highly selective ligand of GPER, has been shown to reduce the expression of the Hlα receptor ADAM10 on the surface of keratinocytes by modulating GPER, thereby limiting the disruption of epithelial barrier integrity mediated by Hlα, further alleviating the severity of S. aureus-induced SSTIs. In the mouse infection model, G-1 reduces the production of pro-inflammatory cytokines and enhances the host innate immune defense against major bacterial toxins (81) (Figure 1).
Nasal carriage of S. aureus is the most important reservoir of this pathogen, with significant implications for pathogen transmission and infection (82). Research has found that women taking hormonal contraceptives are more likely to become continuous carriers of S. aureus in the nose, and reproductive hormone intake is correlated with the persistence of S. aureus nasal colonization (83). However, this study does not specifically differentiate between estrogen and progesterone contraceptives for an in-depth investigation. While physiological concentrations of progesterone has been found to inhibit the growth of Staphylococcus aureus, the specific role of estrogen in nasal colonization by S. aureus remains unclear (84).
As an opportunistic bacterium, S. aureus may also cause anaerobic vaginitis (85). Studies suggest that ERα may be a key factor in S. aureus -induced vaginal infections. A certain dose of E2 may promote the adhesion of S. aureus to human vaginal epithelial cells through the ERα/FAK/Src/iNOS axis, thereby accelerating S. aureus infection of the vagina (86) (Figure 1).
Previous studies have shown that estrogen has a dual effect on diseases caused by S. aureus infection, and the specific role of estrogen should be determined based on the type of disease. Therefore, estrogen can be used as an intervention point to take corresponding preventive and therapeutic measures to prevent and slow down disease progression.
3.6 Helicobacter pylori
H. pylori is a Gram-negative spiral-shaped bacterium commonly found on the mucous membranes of the human stomach (87), which is considered to be the major causative factor for gastric ulcer disease and gastric cancer (88).According to epidemiological data, the incidence of gastric cancer in humans is predominantly higher in males, with a male-to-female ratio of 2:1 (89).
In H. pylori-induced gastric injury, pro-inflammatory Th1 cytokines including IFN-γ, IL-1β, and TNF-α, along with Th17 cells, participates in the pathogenesis (90, 91). Foxp3-positive regulatory T cells and pro-inflammatory Th17 cells play crucial roles in the development of H. pylori-induced gastric inflammation in mice. Studies have indicated that E2 treatment in male INS-GAS mice infected with H. pylori can reduce mRNA levels of IFN-γ and IL-1β by upregulating gastric IL-10 activity and increase mRNA expression of Foxp3 (92). Furthermore, E2 supplementation also reduces gastric epithelial cell proliferation in both H. pylori-infected and uninfected mice (92, 93). Treatment with E2 after H. pylori infection trigger a Th2-skewed cytokine profile in the gastric mucosa while alleviating gastric pathology (93) (Figure 1). Studies have also shown that the female sex hormone progesterone can mediate anti-inflammatory responses, inhibiting the growth of H. pylori and exerting a protective effect (94, 95).
GPER, a membrane estrogen receptor, mediates rapid non-genomic estrogen actions by binding with estrogen on the cell membrane (96). Cytotoxin-associated gene A (CagA) is one of the major virulent factors produced by H. pylori (97). IL-8 is a typical inflammatory cytokine upregulated in H. pylori infection and correlates with the histological severity of damaged gastric mucosa (98, 99).The activation of the CagA-mediated signaling pathway promotes the expression of the inflammatory cytokine IL-8 via the NF-κB pathway, leading to chronic gastritis and peptic ulcers (100) (Figure 1). Studies have shown that GPER can downregulate IL-8 expression in gastric cell lines by inhibiting NF-κB promoter activation induced by CagA expression, thereby mitigating inflammation and gastric mucosal damage (100) (Figure 1).
Estrogen-related receptor gamma (ER-γ) is a nuclear receptor identified as a tumor suppressor gene in several cancers, particularly sex-related tumors (101). In gastric cancer, ER-γ binds to the Trefoil factor 1(TFF1) promoter and induces TFF1 gene expression (102, 103). TFF1, a tumor suppressor, is a downstream target of ER-γ, and H. pylori infection downregulates ER-γ expression. NF-κB is an inflammatory transcription regulator, and the enhanced phosphorylation of NF-κB/p65 occurs in H. pylori infection and its related CagA pathogenic protein (104). Research has found that ER-γ protects gastric cells from H. pylori infection and inhibits gastric cancer cell growth by regulating TFF1 and NF-κB (105) (Figure 1).
H. pylori infection is also a risk factor for diffuse gastric cancer (DGC), with higher mortality rates observed in young female DGC patients (106, 107). Studies have found that estrogen affects DGC organoids and directly regulates the oncogenic HOTAIR, promoting tumorigenesis in the presence of ERα (108, 109) (Figure 1). Estrogen-dependent activation of HOTAIR can occur in various forms, and the Mixed Lineage Leukemia factor (MLL) family is a known ER co-regulator. MLL1 and MLL3 bind to the promoter of the HOTAIR gene in the presence of 17β-estradiol (110). In ERα-positive Diffuse Gastric Cancer (DGC), H. pylori secretes CagA, which recruits the co-regulatory factor MLL3 to promote HOTAIR transcription. Consequently, estrogen binds to the Estrogen Response Element on the HOTAIR promoter, thereby enhancing the expression of oncogenic HOTAIR. This leads to the induction of epithelial-mesenchymal transition (EMT) and stem cell phenotypes in both diffuse gastric cancer cells and organoids, thereby accelerating the progression of diffuse gastric cancer (109).
H. pylori also promotes the occurrence and progression of cholangiocarcinoma. In vitro observation of human intrahepatic biliary epithelial cells (HIBECs) reveals that H. pylori and 17β-estradiol, either alone or in combination, promote HIBEC proliferation, inhibit apoptosis, and induce a certain degree of invasiveness (111) (Figure 1). In general, estrogen plays different roles in diseases caused by H. pylori infection.
3.7 Non-tuberculous Mycobacteria and Mycobacterium tuberculosis
NTM are a group of Gram-positive rods widely distributed in soil, natural water, and artificial aquatic systems. Humans are often exposed to NTM, the majority of which are non-pathogenic, but some strains could cause human diseases (112, 113).
NTM infections lead to chronic pulmonary diseases, with NTM lung diseases becoming increasingly common among postmenopausal women with slender body shapes (114). Studies have suggested that postmenopausal women with NTM lung diseases show estrogen imbalance. Additionally, experimental evidence suggests that estradiol can inhibit the activation of the transcription factor NF-κB in mouse macrophages (115) (Figure 2). NF-κB inhibition results in apoptosis of macrophages infected with M. tuberculosis, which is a well-known mechanism of M. tuberculosis skilling. Thus, estrogen may enhance its killing effect by promoting the intracellular mycobacterial apoptosis (114, 116).
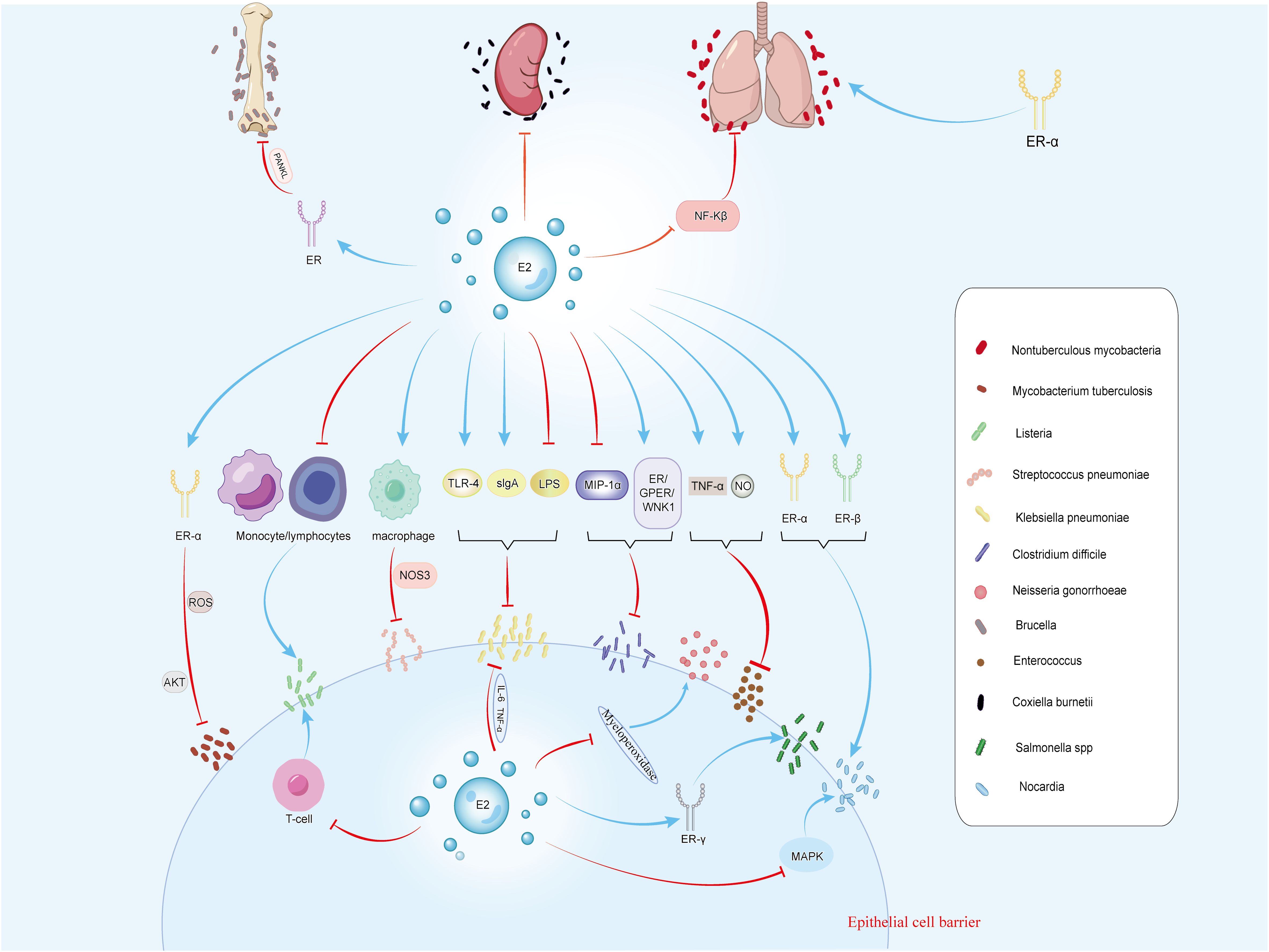
Figure 2. The figure illustrates the effects of estrogen on 12 types of bacteria, including M. tuberculosis, Listeria, S. pneumoniae, Klebsiella, and C. difficile. Estrogen can regulate bacterial infections through receptors and related signaling pathways. It inhibits infections caused by Klebsiella, C. difficile, S. pneumoniae, Enterococcus, Brucella, M. tuberculosis, and C. burnetii. Conversely, it promotes infections by Listeria, N. gonorrhoeae, Salmonella, and Nocardia. For non-tuberculous mycobacterial infections, estrogen has dual effects.
Lady Windermere syndrome, characterized by low body mass index (BMI), tall stature, and a high prevalence of kyphoscoliosis, pectus excavatum, and mitral valve prolapse, is associated with NTM pulmonary infections (117). It is believed that estrogen deficiency leads to immune response disorders and other aspects of NTM, resulting in this syndrome (118). Moreover, the relative leptin deficiency in lean individuals can lead to decreased estrogen levels, which is another potential risk factor for NTM infections (114).
The prevalence and burden of nontuberculous mycobacterial pulmonary disease (NTM-PD) are increasing globally (119). The duration of hormone replacement therapy is positively associated with the risk of NTM-PD in postmenopausal women (120). Furthermore, research has found the regulatory effect of estrogen receptors. In M. tuberculosis infection, ER-α-deficient mice produce more infection-controlling cytokines, namely IL-18 and IFN-γ (121).
M. tuberculosis is the causative agent of tuberculosis (122), which can enter the human body through droplet transmission, causing pulmonary tuberculosis, or invading other organs, leading to other forms of tuberculosis, such as lymph node tuberculosis and skeletal tuberculosis (123). After being engulfed by host cells, M. tuberculosis replicates within the infected cells and prevents the elimination of target bacteria by inhibiting the maturation of phagosomes. Host cells control this evasion mechanism by inducing autophagy, a complex cellular process that gradually eliminated bacteria and reduce the bacterial load within infected cells (124). Studies have shown that ER-α is a transcriptional activator of autophagy genes, promoting the initiation and execution of autophagy by regulating the expression of autophagy-related genes, thereby enhancing the host’s defense against intracellular pathogens (125). In the context of tracheobronchial tuberculosis infection, estradiol primarily acts through ERα binding, affecting ROS generation and the AKT signaling pathway, thereby inhibiting autophagy in M. tuberculosis-infected cells and controlling the proliferation of intracellular M. tuberculosis (126) (Figure 2).
3.8 Sepsis
Sepsis is a severe systemic inflammatory response, typically caused by bacterial infection (127). When the body responds to the infection, the immune system releases a large amount of chemicals into the bloodstream, initiating an inflammatory response which leads to systemic symptoms such as fever, accelerated heart rate, shortness of breath, and lower blood pressure (128, 129).
Clinical studies have shown a higher incidence of sepsis in males compared to age-matched female populations (130). Research has indicated that agonists of ER-β provide significant survival advantages in experimental models of bacterial sepsis, while reducing tissue damage and decreasing levels of various pro-inflammatory proteins (131). ER-β is present in intestinal epithelial cells, and the gastrointestinal mucosa is a major source of microbiota-derived mediators released systemically during sepsis (132). ER-β agonists can reduce the level of circulating endotoxin and peritonitis cytokines, decrease the number of intestinal bacteria in the bloodstream after cecal ligation and puncture in mice, thereby improving the results of experimental systemic infections, and maintaining the function and structure of the gastrointestinal mucosa (132, 133).
Heat shock protein 70 (HSP70) is a molecular chaperone involved in heat stress, promoting protein folding and preventing the secretion of inflammatory mediators, thereby reducing the mortality of sepsis (134, 135). Modulation of the heme oxygenase-1 (HO-1) pathway can offset oxidative stress, improve myocardial function in endotoxemia, and increase survival rates in septic mice (136). Studies have proved that estrogen receptor modulators upregulated HSP70 and HO-1 by mediating ER-α activation, exerting antioxidant and anti-inflammatory effects, and alleviating the severity of sepsis (137).
In septic infections, serum haptoglobin (Hp) plays a balanced role in maintaining the cascade of pro-inflammatory and anti-inflammatory responses. Estrogen can regulate the increase in Hp levels, while Hp maintains endotoxin tolerance by reducing TNFα levels (the main mediator of cytokine storm), thereby slowing the progression of bacterial sepsis (138).
In addition to the protective effects of estrogen in sepsis, progesterone can also improve the sepsis syndrome by reducing the levels of inflammatory factors IL-6 and TNF-α, and by restoring antioxidant enzyme activity in some tissues (139).
3.9 Other bacterial infections
Listeria: Listeria monocytogenes (L. monocytogenes) is a Gram-positive bacterium commonly found in soil, water, and animal feces. It can cause foodborne infections, and for pregnant women, infection with L. monocytogenes may result in miscarriage, stillbirth, or neonatal infection (140). Studies have shown a direct correlation between estrogen activity and increased susceptibility to L. monocytogenes infection. Estrogen exposure inhibits the accumulation of lymphocytes and monocytes in the peritoneal cavity of mice, possibly mediates through the estrogen receptor mechanism, partly via thymus regulation, and ultimately suppresses the activation of T-cell-dependent defense mechanisms, thereby exacerbating the infection (141) (Figure 2). Additionally, estrogen may reduce the proliferation of antigen-sensitive T lymphocytes by inhibiting IL-2 production, thereby weakening the host’s resistance to L. monocytogenes (142).
Streptococcus pneumoniae: Streptococcus pneumoniae (S. pneumoniae) is a Gram-positive bacterium commonly present in the human upper respiratory tract and oropharynx, capable of causing various diseases including pneumonia, otitis media, meningitis, and sepsis (143).Female mice exhibit stronger resistance to S. pneumoniae, with research indicating that estrogen mediates this enhanced host resistance by affecting NOS3 in pulmonary macrophages of female mice (144) (Figure 2). In pneumococcal meningitis caused by S. pneumoniae infection, ER-β influences microglial cell status by downregulating the NF-κB signaling pathway, promoting neuronal recovery through wound healing mechanisms (145–147). The activation of ER-β also induces the expression of brain-derived neurotrophic factor, a neuroprotective factor that promotes the survival of local neurons after injury (146).
Klebsiella pneumoniae: Klebsiella pneumoniae (K. pneumoniae) is a Gram-negative bacillus commonly found in the respiratory tract and can cause pneumonia (148). Research suggest that E2 enhances the transport of sIgA to the respiratory tract and increases TLR-4 expression, thereby helping to prevent K. pneumoniae-infected pneumonia (149) (Figure 2).
K. pneumoniae is also known as a pathogen colonizing on the skin (150). LPS delays several aspects of wound healing, inducing excessive cell death, stalling keratinocyte activation, enhancing local inflammation, and reducing collagen deposition (151). Studies suggest that estrogen weakens LPS-induced inflammatory signals through IL6 and TNFα, thereby reducing macrophage infiltration and improving wound closure and delays epidermal regeneration (151) (Figure 2).
Clostridium difficile: Clostridium difficile (C. difficile) is a Gram-positive bacterium commonly found in soil, water, and feces. It forms heat-resistant spores, which can cause C. difficile infection (CDI), leading to gastrointestinal inflammation and symptoms such as diarrhea, abdominal pain, and fever (152, 153). Macrophage inflammatory protein 1α (MIP-1α) mediates CDI, which typically results in epithelial damage and neutrophil infiltration in the colonic mucosa of infected mice (154). Studies have shown that soy isoflavones, acting as estrogen-like compounds, reduces mortality in infected hamsters by inducing an anti-apoptotic effect in epithelial cells mediated by ER/GPER/WNK1 and inhibiting MIP-1α expression, thereby improving cecal damage and CDI disease activity and providing protective effects against C. difficile toxins (154, 155) (Figure 2).
N. gonorrhoeae: N. gonorrhoeae, a Gram-negative bacterium, is the pathogen of gonorrhea, which is most commonly manifested by male urethritis and female cervicitis or urethritis (143). Animal studies have indicated that female mice are most susceptible to N. gonorrhoeae infection in the early stage of estrus. The treatment of estradiol affected the bactericidal activity of polymorphonuclear leukocytes mediates by myeloperoxidase, enhancing the susceptibility of mice to disseminated gonococcal infection (23, 156) (Figure 2).
Gonococcal vaginitis is the most common form of N. gonorrhoeae infection in children after the neonatal period. Compared to adolescents and adults, due to the low level of estrogen, the vaginal environment of prepubertal children is relatively normal to alkaline pH, with a thinner layer of vaginal mucosal cells, making it easier for N. gonorrhoeae to infect and colonize the vagina (157).
Brucella: Brucella is a genus of Gram-negative bacteria that can cause brucellosis, which is a zoonotic disease. The bacteria are primarily present in mammals such as cattle, sheep, and pigs, and are transmitted to humans through contact with infected animals (158).
Osteoarticular brucellosis is the most common presentation of active brucellosis in humans, where Brucella spp. directly or indirectly impaired osteoblast function and induced osteoblast apoptosis. Furthermore, Brucella infection lead to the occurrence of pathological osteoclasts, resulting in damage to the bones and joints of patients with osteoarticular brucellosis (159). Studies have shown that Brucella infection inhibited extracellular matrix deposition by osteoblasts, while DHEA treatment can reverse this response via estrogen receptor signaling (160). Additionally, Brucella infection of human synovial cells induces increased expression of RANKL. Overexpression of RANKL in pathological conditions may lead to osteoporosis, fractures, and other skeletal diseases. Research suggests that ER regulates osteoclast formation in Brucella-infected synovial cells by modulating the key molecule RANKL (161) (Figure 2). Furthermore, low estradiol levels are also an important risk factor for osteoporosis (162).
Enterococcus: Enterococcus is a genus of Gram-positive cocci and is commonly found in the intestines of both animals and humans. Enterococcus causes healthcare-associated infections, especially in patients who are already debilitated or immunocompromised (163). Estradiol administration prevents bacteremia following intraperitoneal inoculation of Enterococcus in ovariectomized rats by increasing TNF-α and NO levels (4) (Figure 2).
Coxiella burnetii: Coxiella burnetii (C. burnetii), the pathogen of Q fever, is a Gram-negative bacterium (164). Q fever is a zoonotic infectious disease caused by C. burnetii, typically transmitted to humans via respiratory tract or contact with infected sources. Symptoms of Q fever usually include high fever, headache, muscle pain, cough, and complications such as pneumonia, hepatitis, and pericarditis may occur in severe cases (4) (Figure 2).
Studies have shown that in female mice infected with C. burnetii, the number of granulomas in the spleen is lower compared to that of male mice infected with C. burnetii, and treatment with 17β-estradiol can reduce C. burnetii load and prevent the upregulation of granuloma formation (165).
Salmonella spp.: Salmonella is a genus of Gram-negative rods and is a common pathogen, causing various diseases such as food poisoning and gastrointestinal infections (166).
Research indicates a significant association between estrogen therapy and Salmonella infection. Estradiol-treated mice are more susceptible to typhoid Salmonella than the control group, with a higher mortality rate (167). Additionally, estrogen exposure weakens the antibacterial effect of Salmonella in the abdominal cavity (168).
Studies have found that S. typhimurium induces hepcidin expression, hypo-ferraemia, and ERRγ expression in mice. ERRγ acts on the downstream of IL-6 and negatively impacts host defense against infection (169) (Figure 2). Furthermore, overexpression of ERRγ induces hepcidin production and hypoferraemia, while ERRγ inverse agonists can reduce its activity. These inverse agonists improve Salmonella-induced hepcidin expression, hypo-ferraemia, and hepatic and splenic iron accumulation, while reducing bacterial load, macrophage iron content, and pro-inflammatory cytokine production, thereby enhancing host survival rates (169, 170).
Nocardia: Nocardia is a type of Gram-positive bacteria that is typically found in soil, water, and decaying organic matter (171). In medicine, Nocardia infections are often associated with respiratory infections, skin infections, and infections in other organs, often requiring long-term antibiotic treatment (172). Studies have found that female mice infected with Nocardia have a higher mortality rate. E2 is found to impair the ability of mice to clear Nocardia, as it can bind to ER-α and ER-β to facilitate the entry of Nocardia into host cells, leading to severe cellular damage. Additionally, E2 is observed to promote bacterial survival by inhibiting the inflammatory response mediated by the MAPK pathway (173) (Figure 2).
Disseminated odontogenic infections results from a large number of bacteria entering surrounding tissues from the root canal system, evading and disrupting local immune mechanisms. Bacterial dissemination lead to serious complications such as sinusitis, airway obstruction, cavernous sinus thrombosis, brain abscess, and even death (174). Studies suggest that females may have higher resistance to the spread of odontogenic infections compared to males. IL-1 promotes the activity of neutrophils and monocytes, enhancing their antimicrobial activity. Estradiol stimulates and activates neutrophils by increasing IL-1 expression, constituting a critical host defense mechanism against the spread of odontogenic infections (175).
4 Conclusion
The difference in estrogen levels between men and women may be a key factor in determining different outcomes after bacterial infections. Estrogen influences the regulatory role of the immune system against bacteria. In some bacterial infections, such as SSTI induced by S. aureus, gastric cancer caused by H. pylori infection, and bacterial infections caused by lung diseases induced by NTM, males tend to exhibit higher susceptibility. Additionally, estrogen can mediate the progression of diseases through estrogen receptor signaling pathways. For example, in C. trachomatis infection, antibodies against ERα and/or ERβ can reduce the infectivity of Chlamydia. During early pregnancy, ERα enhances resistance to E. coli infections. Furthermore, ERβ agonists lowers circulating endotoxin levels and pro-inflammatory cytokines, reducing the number of intestinal bacteria entering the bloodstream. Modulating GPER alleviates the severity of skin and soft tissue infections caused by S. aureus and mitigates the inflammatory response and gastric mucosal damage caused by H. pylori.
Extensive clinical studies have demonstrated the therapeutic potential of exogenous estrogen treatment. Administering exogenous estrogen or estrogen receptor agonists is necessary for diseases in which estrogen plays a beneficial role in bacterial infections. The availability of these treatment modalities provides important avenues for improving patient prognosis. Conversely, estrogen receptor antagonists, which mediated pro-inflammatory responses, may have beneficial therapeutic effects.
For different types of bacterial infections, individualized treatment plans for different gender and estrogen levels may be required. By comprehensively considering the biological effects of estrogen and clinical research findings, we can better understand the role of estrogen in bacterial infections and develop more effective prevention and treatment strategies to improve the prognosis and quality of life for patients.
Author contributions
LH: Writing – original draft, Writing – review & editing. HL: Conceptualization, Writing – original draft. WM: Writing – review & editing. YZ: Writing – review & editing. PG: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Natural Science Youth Foundation of Shandong Province [No. ZR2022QH039], National Natural Science Youth Foundation [No. 82402627] and Youth Innovation Science and Technology Support Plan for Universities in Shandong Province [No. 2024KJJ009].
Acknowledgments
We thank Zhujun Shao from the State Key Laboratory for Infectious Disease Prevention and Control, National Institute for Communicable Disease Control for kindly and helpful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1556683/full#supplementary-material
References
1. McCulloch TR, Wells TJ, Souza-Fonseca-Guimaraes F. Towards efficient immunotherapy for bacterial infection. Trends Microbiol. (2022) 30:158–69. doi: 10.1016/j.tim.2021.05.005
2. Dias SP, Brouwer MC, van de Beek D. Sex and gender differences in bacterial infections. Infection Immun. (2022) 90:e0028322. doi: 10.1128/iai.00283-22
3. Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front neuroendocrinol. (2014) 35:347–69. doi: 10.1016/j.yfrne.2014.04.004
4. Vázquez-Martínez ER, García-Gómez E, Camacho-Arroyo I, González-Pedrajo B. Sexual dimorphism in bacterial infections. Biol Sex Differ. (2018) 9:27. doi: 10.1186/s13293-018-0187-5
5. Caretto M, Giannini A, Russo E, Simoncini T. Preventing Urinary Tract Infections after Menopause without Antibiotics. Maturitas. (2017) 99:43–6. doi: 10.1016/j.maturitas.2017.02.004
6. Alderman MH 3rd, Taylor HS. Molecular mechanisms of estrogen action in female genital tract development. Differentiation; Res Biol Diversity. (2021) 118:34–40. doi: 10.1016/j.diff.2021.01.002
7. Chen GG, Zeng Q, Tse GM. Estrogen and its receptors in cancer. Medicinal Res Rev. (2008) 28:954–74. doi: 10.1002/med.20131
8. Lu CL, Herndon C. New roles for neuronal estrogen receptors. Neurogastroenterol Motil. (2017) 29. doi: 10.1111/nmo.13121
9. Shepherd R, Cheung AS, Pang K, Saffery R, Novakovic B. Sexual dimorphism in innate immunity: the role of sex hormones and epigenetics. Front Immunol. (2020) 11:604000. doi: 10.3389/fimmu.2020.604000
10. Jones BG, Penkert RR, Surman SL, Sealy RE, Pelletier S, Xu B, et al. Matters of life and death: how estrogen and estrogen receptor binding to the immunoglobulin heavy chain locus may influence outcomes of infection, allergy, and autoimmune disease. Cell Immunol. (2019) 346:103996. doi: 10.1016/j.cellimm.2019.103996
11. Eyster KM. The estrogen receptors: an overview from different perspectives. Methods Mol Biol (Clifton NJ). (2016) 1366:1–10. doi: 10.1007/978-1-4939-3127-9_1
12. Soucy G, Boivin G, Labrie F, Rivest S. Estradiol is required for a proper immune response to bacterial and viral pathogens in the female brain. J Immunol. (2005) 174:6391–8. doi: 10.4049/jimmunol.174.10.6391
13. Grimes DS, Hindle E, Dyer T. Respiratory infection and coronary heart disease: progression of a paradigm. QJM: monthly J Assoc Physicians. (2000) 93:375–83. doi: 10.1093/qjmed/93.6.375
14. Fahey JV, Rossoll RM, Wira CR. Sex hormone regulation of anti-bacterial activity in rat uterine secretions and apical release of anti-bacterial factor(S) by uterine epithelial cells in culture. J Steroid Biochem Mol Biol. (2005) 93:59–66. doi: 10.1016/j.jsbmb.2004.11.002
15. Chakraborty B, Byemerwa J, Krebs T, Lim F, Chang CY, McDonnell DP. Estrogen receptor signaling in the immune system. Endocrine Rev. (2023) 44:117–41. doi: 10.1210/endrev/bnac017
16. Cevenini R, Donati M, Sambri V. Chlamydia trachomatis - the agent. Best Pract Res Clin obstetrics gynaecol. (2002) 16:761–73. doi: 10.1053/beog.2002.0323
17. Guseva NV, Dessus-Babus SC, Whittimore JD, Moore CG, Wyrick PB. Characterization of estrogen-responsive epithelial cell lines and their infectivity by genital chlamydia trachomatis. Microbes Infect. (2005) 7:1469–81. doi: 10.1016/j.micinf.2005.05.004
18. den Heijer CDJ, Hoebe C, Driessen JHM, Wolffs P, van den Broek IVF, Hoenderboom BM, et al. Chlamydia trachomatis and the risk of pelvic inflammatory disease, ectopic pregnancy, and female infertility: A retrospective cohort study among primary care patients. Clin Infect Dis. (2019) 69:1517–25. doi: 10.1093/cid/ciz429
19. Kintner J, Schoborg RV, Wyrick PB, Hall JV. Progesterone antagonizes the positive influence of estrogen on chlamydia trachomatis serovar E in an ishikawa/sht-290 co-culture model. Pathog Dis. (2015) 73. doi: 10.1093/femspd/ftv015
20. Gravitte A, Kintner J, Brown S, Cobble A, Kennard B, Hall JV. The hormonal environment and estrogen receptor signaling alters chlamydia muridarum infection in vivo. Front Cell Infection Microbiol. (2022) 12:939944. doi: 10.3389/fcimb.2022.939944
21. Hall JV, Schell M, Dessus-Babus S, Moore CG, Whittimore JD, Sal M, et al. The multifaceted role of oestrogen in enhancing chlamydia trachomatis infection in polarized human endometrial epithelial cells. Cell Microbiol. (2011) 13:1183–99. doi: 10.1111/j.1462-5822.2011.01608.x
22. Agrawal T, Bhengraj AR, Vats V, Mittal A. Chlamydia trachomatis: tlr4-mediated recognition by human dendritic cells is impaired following oestradiol treatment. Br J BioMed Sci. (2013) 70:51–7. doi: 10.1080/09674845.2013.11669935
23. Sonnex C. Influence of ovarian hormones on urogenital infection. Sexually transmitted infections. (1998) 74:11–9. doi: 10.1136/sti.74.1.11
24. Ray A, Bhati T, Arora R, Pradhan D, Parvez S, Rastogi S. Differential expression of urine-circulating micro-rnas in chlamydia trachomatis-induced recurrent spontaneous aborters. Microbial pathogenesis. (2021) 160:105156. doi: 10.1016/j.micpath.2021.105156
25. Azenabor AA, Kennedy P, Balistreri S. Chlamydia trachomatis infection of human trophoblast alters estrogen and progesterone biosynthesis: an insight into role of infection in pregnancy sequelae. Int J Med Sci. (2007) 4:223–31. doi: 10.7150/ijms.4.223
26. Ray A, Pradhan D, Arora R, Siraj F, Rastogi S. Determination of chlamydial load in recurrent miscarriage in relation to some female sex hormones: A case-control study. J Infect Dev Ctries. (2023) 17:999–1006. doi: 10.3855/jidc.17677
27. Ray A, Bhati T, Arora R, Rastogi S. Progesterone-mediated immunoregulation of cytokine signaling by mirna-133a and 101-3p in chlamydia trachomatis-associated recurrent spontaneous abortion. Mol Immunol. (2023) 164:47–57. doi: 10.1016/j.molimm.2023.10.012
28. Kaushic C, Zhou F, Murdin AD, Wira CR. Effects of estradiol and progesterone on susceptibility and early immune responses to chlamydia trachomatis infection in the female reproductive tract. Infection Immun. (2000) 68:4207–16. doi: 10.1128/iai.68.7.4207-4216.2000
29. Wan C, Latter JL, Amirshahi A, Symonds I, Finnie J, Bowden N, et al. Progesterone activates multiple innate immune pathways in chlamydia trachomatis-infected endocervical cells. Am J Reprod Immunol (New York NY: 1989). (2014) 71:165–77. doi: 10.1111/aji.12168
30. Coppedge N, Garza J, Gandhi K, Sanchez A, Galloway J, Ventolini G. Lactobacillus microbiota of the female genital tract in vaginal lactobacillosis. Arch gynecol obstetrics. (2023) 307:1319–22. doi: 10.1007/s00404-023-06988-9
31. Ansari A, Son D, Hur YM, Park S, You YA, Kim SM, et al. Lactobacillus probiotics improve vaginal dysbiosis in asymptomatic women. Nutrients. (2023) 15. doi: 10.3390/nu15081862
32. Kwon MS, Lee HK. Host and microbiome interplay shapes the vaginal microenvironment. Front Immunol. (2022) 13:919728. doi: 10.3389/fimmu.2022.919728
33. Schaeffer AJ. Aetiopathology and pathogenesis of urogenital infections. Andrologia. (1998) 30 Suppl 1:3–6. doi: 10.1111/j.1439-0272.1998.tb02819.x
34. Caillouette JC, Sharp CF Jr., Zimmerman GJ, Roy S. Vaginal ph as a marker for bacterial pathogens and menopausal status. Am J Obstet Gynecol. (1997) 176:1270–5. doi: 10.1016/s0002-9378(97)70345-4
35. Stapleton AE. The vaginal microbiota and urinary tract infection. Microbiol Spectr. (2016) 4. doi: 10.1128/microbiolspec.UTI-0025-2016
36. Muhleisen AL, Herbst-Kralovetz MM. Menopause and the vaginal microbiome. Maturitas. (2016) 91:42–50. doi: 10.1016/j.maturitas.2016.05.015
37. Hudson PL, Hung KJ, Bergerat A, Mitchell C. Effect of vaginal lactobacillus species on escherichia coli growth. Female Pelvic Med Reconstr Surg. (2020) 26:146–51. doi: 10.1097/spv.0000000000000827
38. Franco AV. Recurrent urinary tract infections. Best Pract Res Clin obstetrics gynaecol. (2005) 19:861–73. doi: 10.1016/j.bpobgyn.2005.08.003
39. Raz R. Postmenopausal women with recurrent uti. Int J antimicrobial Agents. (2001) 17:269–71. doi: 10.1016/s0924-8579(00)00355-1
40. Parsons CL, Schmidt JD. Control of recurrent lower urinary tract infection in the postmenopausal woman. J Urol. (1982) 128:1224–6. doi: 10.1016/s0022-5347(17)53434-5
41. Lokken EM, Richardson BA, Kinuthia J, Mwinyikai K, Abdalla A, Jaoko W, et al. A prospective cohort study of the association between body mass index and incident bacterial vaginosis. Sex Transm Dis. (2019) 46:31–6. doi: 10.1097/OLQ.0000000000000905
42. Sovijit WN, Sovijit WE, Pu S, Usuda K, Inoue R, Watanabe G, et al. Ovarian progesterone suppresses depression and anxiety-like behaviors by increasing the lactobacillus population of gut microbiota in ovariectomized mice. Neurosci Res. (2021) 168:76–82. doi: 10.1016/j.neures.2019.04.005
43. Wang J, Wang L. The therapeutic effect of dehydroepiandrosterone (Dhea) on vulvovaginal atrophy. Pharmacol Res. (2021) 166:105509. doi: 10.1016/j.phrs.2021.105509
44. Fegan N, Gobius KS. Pathogenic escherichia coli and one health implications. Curr topics Microbiol Immunol. (2013) 366:49–62. doi: 10.1007/82_2012_261
45. Nesta B, Pizza M. Vaccines against escherichia coli. Curr topics Microbiol Immunol. (2018) 416:213–42. doi: 10.1007/82_2018_111
46. Terlizzi ME, Gribaudo G, Maffei ME. Uropathogenic escherichia coli (Upec) infections: virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front Microbiol. (2017) 8:1566. doi: 10.3389/fmicb.2017.01566
47. Wang C, Symington JW, Ma E, Cao B, Mysorekar IU. Estrogenic modulation of uropathogenic escherichia coli infection pathogenesis in a murine menopause model. Infection Immun. (2013) 81:733–9. doi: 10.1128/IAI.01234-12
48. Ferrante KL, Wasenda EJ, Jung CE, Adams-Piper ER, Lukacz ES. Vaginal estrogen for the prevention of recurrent urinary tract infection in postmenopausal women: A randomized clinical trial. Female Pelvic Med Reconstr Surg. (2021) 27:112–7. doi: 10.1097/spv.0000000000000749
49. Bi S, Li M, Liang Z, Li G, Yu G, Zhang J, et al. Self-assembled aluminum oxyhydroxide nanorices with superior suspension stability for vaccine adjuvant. J colloid Interface Sci. (2022) 627:238–46. doi: 10.1016/j.jcis.2022.07.022
50. Freiesleben H, Sack K, Woermann K, Henkel W. Humoral and cellular factors of immunological defense against infection exemplified by estrogen-facilitated pyelonephritis in rats. Zentralblatt fur Bakteriologie Parasitenkunde Infektionskrankheiten und Hygiene Erste Abteilung Originale Reihe A: Medizinische Mikrobiologie und Parasitologie. (1974) 227:314–20.
51. Anand M, Wang C, French J, Isaacson-Schmid M, Wall LL, Mysorekar IU. Estrogen affects the glycosaminoglycan layer of the murine bladder. Female Pelvic Med Reconstr Surg. (2012) 18:148–52. doi: 10.1097/SPV.0b013e31824b76bd
52. Curran EM, Tassell AH, Judy BM, Nowicki B, Montgomery-Rice V, Estes DM, et al. Estrogen increases menopausal host susceptibility to experimental ascending urinary-tract infection. J Infect Dis. (2007) 195:680–3. doi: 10.1086/511275
53. Olson PD, McLellan LK, Hreha TN, Liu A, Briden KE, Hruska KA, et al. Androgen exposure potentiates formation of intratubular communities and renal abscesses by escherichia coli. Kidney Int. (2018) 94:502–13. doi: 10.1016/j.kint.2018.04.023
54. Patel MV, Fahey JV, Rossoll RM, Wira CR. Innate immunity in the vagina (Part I): estradiol inhibits hbd2 and elafin secretion by human vaginal epithelial cells. Am J Reprod Immunol (New York NY: 1989). (2013) 69:463–74. doi: 10.1111/aji.12078
55. Medina-Estrada I, Alva-Murillo N, Lopez-Meza JE, Ochoa-Zarzosa A. Immunomodulatory effects of 17beta-estradiol on epithelial cells during bacterial infections. J Immunol Res. (2018) 2018:6098961. doi: 10.1155/2018/6098961
56. Luthje P, Brauner H, Ramos NL, Ovregaard A, Glaser R, Hirschberg AL, et al. Estrogen supports urothelial defense mechanisms. Sci Transl Med. (2013) 5:190ra80. doi: 10.1126/scitranslmed.3005574
57. Ali ASM, Mowbray C, Lanz M, Stanton A, Bowen S, Varley CL, et al. Targeting deficiencies in the tlr5 mediated vaginal response to treat female recurrent urinary tract infection. Sci Rep. (2017) 7:11039. doi: 10.1038/s41598-017-10445-4
58. Liu YF, Li MY, Yan YP, Wei W, Li B, Pan HY, et al. Erα-dependent stimulation of lcn2 in uterine epithelium during mouse early pregnancy. Reprod (Cambridge England). (2020) 159:493–501. doi: 10.1530/rep-19-0616
59. Laupland KB. Incidence of bloodstream infection: A review of population-based studies. Clin Microbiol infection. (2013) 19:492–500. doi: 10.1111/1469-0691.12144
60. Zeng Z, Surewaard BGJ, Wong CHY, Guettler C, Petri B, Burkhard R, et al. Sex-hormone-driven innate antibodies protect females and infants against epec infection. Nat Immunol. (2018) 19:1100–11. doi: 10.1038/s41590-018-0211-2
61. Koch MA. Sex bias in sepsis. Cell Host Microbe. (2018) 24:613–5. doi: 10.1016/j.chom.2018.10.014
62. Stratton CW. Pseudomonas aeruginosa. Infection control: IC. (1983) 4:36–40. doi: 10.1017/s0195941700057647
63. Mulcahy LR, Isabella VM, Lewis K. Pseudomonas aeruginosa biofilms in disease. Microbial Ecol. (2014) 68:1–12. doi: 10.1007/s00248-013-0297-x
64. Hogardt M, Heesemann J. Microevolution of pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung. Curr topics Microbiol Immunol. (2013) 358:91–118. doi: 10.1007/82_2011_199
65. Jurado-Martín I, Sainz-Mejías M, McClean S. Pseudomonas aeruginosa: an audacious pathogen with an adaptable arsenal of virulence factors. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22063128
66. Abid S, Xie S, Bose M, Shaul PW, Terada LS, Brody SL, et al. 17β-estradiol dysregulates innate immune responses to pseudomonas aeruginosa respiratory infection and is modulated by estrogen receptor antagonism. Infection Immun. (2017) 85. doi: 10.1128/iai.00422-17
67. Tyrrell J, Harvey BJ. Sexual dimorphism in the microbiology of the cf ‘Gender gap’: estrogen modulation of pseudomonas aeruginosa virulence. Steroids. (2020) 156:108575. doi: 10.1016/j.steroids.2019.108575
68. Wang Y, Cela E, Gagnon S, Sweezey NB. Estrogen aggravates inflammation in pseudomonas aeruginosa pneumonia in cystic fibrosis mice. Respir Res. (2010) 11:166. doi: 10.1186/1465-9921-11-166
69. Pedersen SS, Kharazmi A, Espersen F, Høiby N. Pseudomonas aeruginosa alginate in cystic fibrosis sputum and the inflammatory response. Infection Immun. (1990) 58:3363–8. doi: 10.1128/iai.58.10.3363-3368.1990
70. Fishman J, Bradlow HL, Gallagher TF. Oxidative metabolism of estradiol. J Biol Chem. (1960) 235:3104–7. doi: 10.1016/S0021-9258(20)81317-9
71. Benisek WF, Ogez JR. Proton nuclear magnetic resonance studies of pseudomonas testosteroni 3-oxo-delta 5-steroid isomerase and its interaction with 17 beta-estradiol. Biochemistry. (1982) 21:5816–25. doi: 10.1021/bi00266a015
72. Chotirmall SH, Smith SG, Gunaratnam C, Cosgrove S, Dimitrov BD, O’Neill SJ, et al. Effect of estrogen on pseudomonas mucoidy and exacerbations in cystic fibrosis. New Engl J Med. (2012) 366:1978–86. doi: 10.1056/NEJMoa1106126
73. Sharma R, Shultz SR, Robinson MJ, Belli A, Hibbs ML, O’Brien TJ, et al. Infections after a traumatic brain injury: the complex interplay between the immune and neurological systems. Brain behavior Immun. (2019) 79:63–74. doi: 10.1016/j.bbi.2019.04.034
74. Mulani MS, Kamble EE, Kumkar SN, Tawre MS, Pardesi KR. Emerging strategies to combat eskape pathogens in the era of antimicrobial resistance: A review. Front Microbiol. (2019) 10:539. doi: 10.3389/fmicb.2019.00539
75. Pittet JF, Hu PJ, Honavar J, Brandon AP, Evans CA, Muthalaly R, et al. Estrogen alleviates sex-dependent differences in lung bacterial clearance and mortality secondary to bacterial pneumonia after traumatic brain injury. J Neurotrauma. (2021) 38:989–99. doi: 10.1089/neu.2020.7327
76. Lowy FD. Staphylococcus aureus infections. New Engl J Med. (1998) 339:520–32. doi: 10.1056/nejm199808203390806
77. Talan DA, Krishnadasan A, Gorwitz RJ, Fosheim GE, Limbago B, Albrecht V, et al. Comparison of staphylococcus aureus from skin and soft-tissue infections in us emergency department patients, 2004 and 2008. Clin Infect Dis. (2011) 53:144–9. doi: 10.1093/cid/cir308
78. Lin S, Cheng H, Yang G, Wang C, Leung CK, Zhang S, et al. Nrf2 antagonizes hiv-1 tat and methamphetamine-induced bv2 cell ferroptosis by regulating slc7a11. Neurotoxicity Res. (2023) 41:398–407. doi: 10.1007/s12640-023-00645-4
79. Li M, Diep BA, Villaruz AE, Braughton KR, Jiang X, DeLeo FR, et al. Evolution of virulence in epidemic community-associated methicillin-resistant staphylococcus aureus. Proc Natl Acad Sci United States America. (2009) 106:5883–8. doi: 10.1073/pnas.0900743106
80. Castleman MJ, Pokhrel S, Triplett KD, Kusewitt DF, Elmore BO, Joyner JA, et al. Innate sex bias of staphylococcus aureus skin infection is driven by alpha-hemolysin. J Immunol. (2018) 200:657–68. doi: 10.4049/jimmunol.1700810
81. Triplett KD, Pokhrel S, Castleman MJ, Daly SM, Elmore BO, Joyner JA, et al. Gper activation protects against epithelial barrier disruption by staphylococcus aureus α-toxin. Sci Rep. (2019) 9. doi: 10.1038/s41598-018-37951-3
82. Winkler J, Block C, Leibovici L, Faktor J, Pitlik SD. Nasal carriage of staphylococcus aureus: correlation with hormonal status in women. J Infect Dis. (1990) 162:1400–2. doi: 10.1093/infdis/162.6.1400
83. Zanger P, Nurjadi D, Gaile M, Gabrysch S, Kremsner PG. Hormonal contraceptive use and persistent staphylococcus aureus nasal carriage. Clin Infect Dis. (2012) 55:1625–32. doi: 10.1093/cid/cis778
84. Yotis W, Stanke R. Bacteriostatic action of progesterone on staphylococci and other microorganisms. J bacteriol. (1966) 92:1285–9. doi: 10.1128/jb.92.5.1285-1289.1966
85. Bertuccini L, Russo R, Iosi F, Superti F. Effects of lactobacillus rhamnosus and lactobacillus acidophilus on bacterial vaginal pathogens. Int J immunopathol Pharmacol. (2017) 30:163–7. doi: 10.1177/0394632017697987
86. Yan L, Rui C, Zhuang B, Liu X, Luan T, Jiang L, et al. 17beta-estradiol mediates staphylococcus aureus adhesion in vaginal epithelial cells via estrogen receptor alpha-associated signaling pathway. Curr Microbiol. (2023) 80:391. doi: 10.1007/s00284-023-03488-6
87. Smith SI, Seriki A, Ndip R, Pellicano R. Helicobacter pylori infection in africa: 2018 literature update. Minerva gastroenterologica e dietologica. (2018) 64:222–34. doi: 10.23736/s1121-421x.18.02464-9
88. Fischbach W, Malfertheiner P. Helicobacter pylori infection. Deutsches Arzteblatt Int. (2018) 115:429–36. doi: 10.3238/arztebl.2018.0429
89. Lindblad M, Ye W, Rubio C, Lagergren J. Estrogen and risk of gastric cancer: A protective effect in a nationwide cohort study of patients with prostate cancer in Sweden. Cancer epidemiol Biomarkers prevention: A Publ Am Assoc Cancer Res cosponsored by Am Soc Preventive Oncol. (2004) 13:2203–7. doi: 10.1158/1055-9965.2203.13.12
90. Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced treg skewing and th17 suppression in mice. Gastroenterology. (2010) 138:1046–54. doi: 10.1053/j.gastro.2009.11.043
91. Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. (2007) 117:60–9. doi: 10.1172/jci30111
92. Ohtani M, Ge Z, Garcia A, Rogers AB, Muthupalani S, Taylor NS, et al. 17 beta-estradiol suppresses helicobacter pylori-induced gastric pathology in male hypergastrinemic ins-gas mice. Carcinogenesis. (2011) 32:1244–50. doi: 10.1093/carcin/bgr072
93. Ohtani M, Garcia A, Rogers AB, Ge Z, Taylor NS, Xu S, et al. Protective role of 17 beta -estradiol against the development of helicobacter pylori-induced gastric cancer in ins-gas mice. Carcinogenesis. (2007) 28:2597–604. doi: 10.1093/carcin/bgm150
94. Saqui-Salces M, Rocha-Gutiérrez BL, Barrios-Payán JA, Ruiz-Palacios G, Camacho-Arroyo I, Gamboa-Dominguez A. Effects of estradiol and progesterone on gastric mucosal response to early helicobacter pylori infection in female gerbils. Helicobacter. (2006) 11:123–30. doi: 10.1111/j.1523-5378.2006.00386.x
95. Hosoda K, Shimomura H, Hayashi S, Yokota K, Hirai Y. Steroid hormones as bactericidal agents to helicobacter pylori. FEMS Microbiol Lett. (2011) 318:68–75. doi: 10.1111/j.1574-6968.2011.02239.x
96. Mizukami Y. In vivo functions of gpr30/gper-1, a membrane receptor for estrogen: from discovery to functions in vivo. Endocrine J. (2010) 57:101–7. doi: 10.1507/endocrj.k09e-332
97. Takahashi-Kanemitsu A, Knight CT, Hatakeyama M. Molecular anatomy and pathogenic actions of helicobacter pylori caga that underpin gastric carcinogenesis. Cell Mol Immunol. (2020) 17:50–63. doi: 10.1038/s41423-019-0339-5
98. Crabtree JE, Wyatt JI, Trejdosiewicz LK, Peichl P, Nichols PH, Ramsay N, et al. Interleukin-8 expression in helicobacter pylori infected, normal, and neoplastic gastroduodenal mucosa. J Clin Pathol. (1994) 47:61–6. doi: 10.1136/jcp.47.1.61
99. Nozawa Y, Nishihara K, Peek RM, Nakano M, Uji T, Ajioka H, et al. Identification of a signaling cascade for interleukin-8 production by helicobacter pylori in human gastric epithelial cells. Biochem Pharmacol. (2002) 64:21–30. doi: 10.1016/s0006-2952(02)01030-4
100. Okamoto M, Miura A, Ito R, Kamada T, Mizukami Y, Kawamoto K. G-protein-coupled estrogen receptor prevents nuclear factor-kappa B promoter activation by helicobacter pylori cytotoxin-associated gene a in gastric cancer cells. J veterinary Med Sci. (2023) 85:1348–54. doi: 10.1292/jvms.23-0054
101. Shen Z, Hu Y, Zhou C, Yuan J, Xu J, Hao W, et al. Esrrg promoter hypermethylation as a diagnostic and prognostic biomarker in laryngeal squamous cell carcinoma. J Clin Lab Anal. (2019) 33:e22899. doi: 10.1002/jcla.22899
102. Resende C, Thiel A, MaChado JC, Ristimäki A. Gastric cancer: basic aspects. Helicobacter. (2011) 16 Suppl 1:38–44. doi: 10.1111/j.1523-5378.2011.00879.x
103. Lefebvre O, Chenard MP, Masson R, Linares J, Dierich A, LeMeur M, et al. Gastric mucosa abnormalities and tumorigenesis in mice lacking the ps2 trefoil protein. Sci (New York NY). (1996) 274:259–62. doi: 10.1126/science.274.5285.259
104. Takabayashi H, Shinohara M, Mao M, Phaosawasdi P, El-Zaatari M, Zhang M, et al. Anti-inflammatory activity of bone morphogenetic protein signaling pathways in stomachs of mice. Gastroenterology. (2014) 147:396–406.e7. doi: 10.1053/j.gastro.2014.04.015
105. Kang M-H, S-i E, Park Y-Y. Estrogen-related receptor-gamma influences helicobacter pylori infection by regulating tff1 in gastric cancer. Biochem Biophys Res Commun. (2021) 563:15–22. doi: 10.1016/j.bbrc.2021.05.076
106. Ansari S, Gantuya B, Tuan VP, Yamaoka Y. Diffuse gastric cancer: A summary of analogous contributing factors for its molecular pathogenicity. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19082424
107. De B, Rhome R, Jairam V, Özbek U, Holcombe RF, Buckstein M, et al. Gastric adenocarcinoma in young adult patients: patterns of care and survival in the United States. Gastric Cancer. (2018) 21:889–99. doi: 10.1007/s10120-018-0826-x
108. Takei Y, Hara T, Suzuki A, Mihara K, Yanagihara K. Long noncoding rna hotair promotes epithelial-mesenchymal transition and is a suitable target to inhibit peritoneal dissemination in human scirrhous gastric cancers. Pathobiol: J immunopathol Mol Cell Biol. (2020) 87:277–90. doi: 10.1159/000508350
109. Kang S, Park M, Cho JY, Ahn SJ, Yoon C, Kim SG, et al. Tumorigenic mechanisms of estrogen and helicobacter pylori cytotoxin-associated gene a in estrogen receptor alpha-positive diffuse-type gastric adenocarcinoma. Gastric Cancer. (2022) 25:678–96. doi: 10.1007/s10120-022-01290-0
110. Bhan A, Mandal SS. Estradiol-induced transcriptional regulation of long non-coding rna, hotair. Methods Mol Biol (Clifton NJ). (2016) 1366:395–412. doi: 10.1007/978-1-4939-3127-9_31
111. Ma F, Yang Y, Wang JD, Quan ZW, Zhou D. Helicobacter pylori and 17beta-estradiol induce human intrahepatic biliary epithelial cell abnormal proliferation and oxidative DNA damage. Hepatobiliary Pancreat Dis Int. (2017) 16:519–27. doi: 10.1016/S1499-3872(17)60038-9
112. Wassilew N, Hoffmann H, Andrejak C, Lange C. Pulmonary disease caused by non-tuberculous mycobacteria. Respiration; Int Rev thoracic Dis. (2016) 91:386–402. doi: 10.1159/000445906
113. Porvaznik I, Solovič I, Mokrý J. Non-tuberculous mycobacteria: classification, diagnostics, and therapy. Adv Exp Med Biol. (2017) 944:19–25. doi: 10.1007/5584_2016_45
114. Chan ED, Iseman MD. Slender, older women appear to be more susceptible to nontuberculous mycobacterial lung disease. Gend Med. (2010) 7:5–18. doi: 10.1016/j.genm.2010.01.005
115. Deshpande R, Khalili H, Pergolizzi RG, Michael SD, Chang MD. Estradiol down-regulates lps-induced cytokine production and nfkb activation in murine macrophages. Am J Reprod Immunol (New York NY: 1989). (1997) 38:46–54. doi: 10.1111/j.1600-0897.1997.tb00275.x
116. Bai X, Feldman NE, Chmura K, Ovrutsky AR, Su WL, Griffin L, et al. Inhibition of nuclear factor-kappa B activation decreases survival of mycobacterium tuberculosis in human macrophages. PloS One. (2013) 8:e61925. doi: 10.1371/journal.pone.0061925
117. Cataño JC, Porras Mancilla JP. Lady windermere syndrome. Postgraduate Med J. (2022) 98:e29. doi: 10.1136/postgradmedj-2021-140906
118. Holt MR, Miles JJ, Inder WJ, Thomson RM. Exploring immunomodulation by endocrine changes in lady windermere syndrome. Clin Exp Immunol. (2019) 196:28–38. doi: 10.1111/cei.13265
119. Ratnatunga CN, Lutzky VP, Kupz A, Doolan DL, Reid DW, Field M, et al. The rise of non-tuberculosis mycobacterial lung disease. Front Immunol. (2020) 11:303. doi: 10.3389/fimmu.2020.00303
120. Choi H, Han K, Yang B, Shin DW, Sohn JW, Lee H. Female reproductive factors and incidence of nontuberculous mycobacterial pulmonary disease among postmenopausal women in korea. Clin Infect Dis. (2022) 75:1397–404. doi: 10.1093/cid/ciac134
121. Curran EM, Judy BM, Newton LG, Lubahn DB, Rottinghaus GE, Macdonald RS, et al. Dietary soy phytoestrogens and eralpha signalling modulate interferon gamma production in response to bacterial infection. Clin Exp Immunol. (2004) 135:219–25. doi: 10.1111/j.1365-2249.2003.02368.x
122. Boom WH, Schaible UE, Achkar JM. The knowns and unknowns of latent mycobacterium tuberculosis infection. J Clin Invest. (2021) 131. doi: 10.1172/jci136222
123. Ketata W, Rekik WK, Ayadi H, Kammoun S. Extrapulmonary tuberculosis. Rev pneumologie clinique. (2015) 71:83–92. doi: 10.1016/j.pneumo.2014.04.001
124. Bento CF, Empadinhas N, Mendes V. Autophagy in the fight against tuberculosis. DNA Cell Biol. (2015) 34:228–42. doi: 10.1089/dna.2014.2745
125. Kim SY, Yang CS, Lee HM, Kim JK, Kim YS, Kim YR, et al. Esrra (Estrogen-related receptor alpha) is a key coordinator of transcriptional and post-translational activation of autophagy to promote innate host defense. Autophagy. (2018) 14:152–68. doi: 10.1080/15548627.2017.1339001
126. Gan Y, Hu Q, Li A, Gu L, Guo S. Estradiol inhibits autophagy of mycobacterium tuberculosis−Infected 16hbe cells and controls the proliferation of intracellular mycobacterium tuberculosis. Mol Med Rep. (2022) 25. doi: 10.3892/mmr.2022.12712
127. Lin GL, McGinley JP, Drysdale SB, Pollard AJ. Epidemiology and immune pathogenesis of viral sepsis. Front Immunol. (2018) 9:2147. doi: 10.3389/fimmu.2018.02147
128. Nagalingam K. Understanding sepsis. Br J Nurs (Mark Allen Publishing). (2018) 27:1168–70. doi: 10.12968/bjon.2018.27.20.1168
129. Teplick R, Rubin R. Therapy of sepsis: why have we made such little progress? Crit Care Med. (1999) 27:1682–3. doi: 10.1097/00003246-199908000-00066
130. Angele MK, Pratschke S, Hubbard WJ, Chaudry IH. Gender differences in sepsis: cardiovascular and immunological aspects. Virulence. (2014) 5:12–9. doi: 10.4161/viru.26982
131. Christaki E, Opal SM, Keith JC Jr., Kessinian N, Palardy JE, Parejo NA, et al. Estrogen receptor beta agonism increases survival in experimentally induced sepsis and ameliorates the genomic sepsis signature: A pharmacogenomic study. J Infect Dis. (2010) 201:1250–7. doi: 10.1086/651276
132. Opal SM, Palardy JE, Cristofaro P, Parejo N, Jhung JW, Keith JC Jr., et al. The activity of pathway-selective estrogen receptor ligands in experimental septic shock. Shock. (2005) 24:535–40. doi: 10.1097/01.shk.0000183388.90895.cb
133. Cristofaro PA, Opal SM, Palardy JE, Parejo NA, Jhung J, Keith JC Jr., et al. Way-202196, a selective estrogen receptor-beta agonist, protects against death in experimental septic shock. Crit Care Med. (2006) 34:2188–93. doi: 10.1097/01.CCM.0000227173.13497.56
134. Bruemmer-Smith S, Stüber F, Schroeder S. Protective functions of intracellular heat-shock protein (Hsp) 70-expression in patients with severe sepsis. Intensive Care Med. (2001) 27:1835–41. doi: 10.1007/s00134-001-1131-3
135. McConnell KW, Fox AC, Clark AT, Chang NY, Dominguez JA, Farris AB, et al. The role of heat shock protein 70 in mediating age-dependent mortality in sepsis. J Immunol. (2011) 186:3718–25. doi: 10.4049/jimmunol.1003652
136. Wang YL, Lam KK, Cheng PY, Lee YM. Celastrol prevents circulatory failure via induction of heme oxygenase-1 and heat shock protein 70 in endotoxemic rats. J ethnopharmacol. (2015) 162:168–75. doi: 10.1016/j.jep.2014.12.062
137. Shen HH, Huang SY, Cheng PY, Chu YJ, Chen SY, Lam KK, et al. Involvement of hsp70 and ho-1 in the protective effects of raloxifene on multiple organ dysfunction syndrome by endotoxemia in ovariectomized rats. Menopause. (2017) 24:959–69. doi: 10.1097/GME.0000000000000864
138. Raju SM, Kumar AP, Yadav AN, Rajkumar K, Mvs S, Burgula S. Haptoglobin improves acute phase response and endotoxin tolerance in response to bacterial lps. Immunol Lett. (2019) 207:17–27. doi: 10.1016/j.imlet.2019.01.002
139. Aksoy AN, Toker A, Celık M, Aksoy M, Halıcı Z, Aksoy H. The effect of progesterone on systemic inflammation and oxidative stress in the rat model of sepsis. Indian J Pharmacol. (2014) 46:622–6. doi: 10.4103/0253-7613.144922
140. Vázquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Domínguez-Bernal G, Goebel W, et al. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. (2001) 14:584–640. doi: 10.1128/cmr.14.3.584-640.2001
141. Pung OJ, Luster MI, Hayes HT, Rader J. Influence of steroidal and nonsteroidal sex hormones on host resistance in mice: increased susceptibility to listeria monocytogenes after exposure to estrogenic hormones. Infection Immun. (1984) 46:301–7. doi: 10.1128/iai.46.2.301-307.1984
142. Pung OJ, Tucker AN, Vore SJ, Luster MI. Influence of estrogen on host resistance: increased susceptibility of mice to listeria monocytogenes correlates with depressed production of interleukin 2. Infection Immun. (1985) 50:91–6. doi: 10.1128/iai.50.1.91-96.1985
143. Mitchell AM, Mitchell TJ. Streptococcus pneumoniae: virulence factors and variation. Clin Microbiol infection. (2010) 16:411–8. doi: 10.1111/j.1469-0691.2010.03183.x
144. Yang Z, Huang YC, Koziel H, de Crom R, Ruetten H, Wohlfart P, et al. Female resistance to pneumonia identifies lung macrophage nitric oxide synthase-3 as a therapeutic target. Elife. (2014) 3. doi: 10.7554/eLife.03711
145. Lee S, Lee SO, Kim GL, Rhee DK. Estrogen receptor-beta of microglia underlies sexual differentiation of neuronal protection via ginsenosides in mice brain. CNS Neurosci Ther. (2018) 24:930–9. doi: 10.1111/cns.12842
146. Meltser I, Tahera Y, Simpson E, Hultcrantz M, Charitidi K, Gustafsson JA, et al. Estrogen receptor beta protects against acoustic trauma in mice. J Clin Invest. (2008) 118:1563–70. doi: 10.1172/jci32796
147. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
148. Wang G, Zhao G, Chao X, Xie L, Wang H. The characteristic of virulence, biofilm and antibiotic resistance of klebsiella pneumoniae. Int J Environ Res Public Health. (2020) 17. doi: 10.3390/ijerph17176278
149. Ali AA, Diebel LN, Liberati DM. Estrogen modulation of pneumonia? An immunoglobulin a effect. J Trauma Acute Care Surg. (2012) 72:908–15. doi: 10.1097/TA.0b013e3182468989
150. Chhibber S, Gondil VS, Singla L, Kumar M, Chhibber T, Sharma G, et al. Effective topical delivery of H-agnps for eradication of klebsiella pneumoniae-induced burn wound infection. AAPS PharmSciTech. (2019) 20:169. doi: 10.1208/s12249-019-1350-y
151. Crompton R, Williams H, Ansell D, Campbell L, Holden K, Cruickshank S, et al. Oestrogen promotes healing in a bacterial lps model of delayed cutaneous wound repair. Lab Invest. (2016) 96:439–49. doi: 10.1038/labinvest.2015.160
152. Sandhu BK. McBride SM.Clostridioides difficile. Trends Microbiol. (2018) 26:1049–50. doi: 10.1016/j.tim.2018.09.004
153. Shen A. Clostridioides difficile spore formation and germination: new insights and opportunities for intervention. Annu Rev Microbiol. (2020) 74:545–66. doi: 10.1146/annurev-micro-011320-011321
154. Xie Y, Fontenot L, Estrada AC, Nelson B, Bullock A, Faull KF, et al. Genistein inhibits clostridioides difficile infection via estrogen receptors and lysine-deficient protein kinase 1. J Infect Dis. (2023) 227:806–19. doi: 10.1093/infdis/jiad008
155. Zhou T, Meng C, He P. Soy isoflavones and their effects on xenobiotic metabolism. Curr Drug Metab. (2019) 20:46–53. doi: 10.2174/1389200219666180427170213
156. Kita E, Takahashi S, Yasui K, Kashiba S. Effect of estrogen (17 beta-estradiol) on the susceptibility of mice to disseminated gonococcal infection. Infection Immun. (1985) 49:238–43. doi: 10.1128/iai.49.1.238-243.1985
157. Bambang AW, Idrus I, Amin S, Iswanty M. Gonorrhea vaginitis in a pediatric patient: A case report. Pan Afr Med J. (2021) 38:358. doi: 10.11604/pamj.2021.38.358.28390
158. Głowacka P, Żakowska D, Naylor K, Niemcewicz M, Bielawska-Drózd A. Brucella - virulence factors, pathogenesis and treatment. Polish J Microbiol. (2018) 67:151–61. doi: 10.21307/pjm-2018-029
159. Scian R, Barrionuevo P, Fossati CA, Giambartolomei GH, Delpino MV. Brucella abortus invasion of osteoblasts inhibits bone formation. Infection Immun. (2012) 80:2333–45. doi: 10.1128/iai.00208-12
160. Gentilini MV, Pesce Viglietti AI, Arriola Benitez PC, Iglesias Molli AE, Cerrone GE, Giambartolomei GH, et al. Inhibition of osteoblast function by brucella abortus is reversed by dehydroepiandrosterone and involves erk1/2 and estrogen receptor. Front Immunol. (2018) 9:88. doi: 10.3389/fimmu.2018.00088
161. Gentilini MV, Giambartolomei GH, Delpino MV. Adrenal steroids modulate fibroblast-like synoviocytes response during B. Abortus Infection Front Endocrinol (Lausanne). (2019) 10:722. doi: 10.3389/fendo.2019.00722
162. Chinda D, Shimoyama T, Iino C, Matsuzaka M, Nakaji S, Fukuda S. Decrease of estradiol and several lifestyle factors, but not helicobacter pylori infection, are significant risks for osteopenia in Japanese females. Digestion. (2017) 96:103–9. doi: 10.1159/000479317
163. Goh HMS, Yong MHA, Chong KKL, Kline KA. Model systems for the study of enterococcal colonization and infection. Virulence. (2017) 8:1525–62. doi: 10.1080/21505594.2017.1279766
164. España PP, Uranga A, Cillóniz C, Torres A. Q fever (Coxiella burnetii). Semin Respir Crit Care Med. (2020) 41:509–21. doi: 10.1055/s-0040-1710594
165. Leone M, Honstettre A, Lepidi H, Capo C, Bayard F, Raoult D, et al. Effect of sex on coxiella burnetii infection: protective role of 17beta-estradiol. J Infect Dis. (2004) 189:339–45. doi: 10.1086/380798
166. Kurtz JR, Goggins JA, McLachlan JB. Salmonella infection: interplay between the bacteria and host immune system. Immunol Lett. (2017) 190:42–50. doi: 10.1016/j.imlet.2017.07.006
167. Kass PH, Farver TB, Beaumont JJ, Genigeorgis C, Stevens F. Disease determinants of sporadic salmonellosis in four northern california counties. A case-control study of older children and adults. Ann Epidemiol. (1992) 2:683–96. doi: 10.1016/1047-2797(92)90013-g
168. Kita E, Yagyu Y, Nishikawa F, Hamuro A, Oku D, Emoto M, et al. Alterations of host resistance to mouse typhoid infection by sex hormones. J leukocyte Biol. (1989) 46:538–46. doi: 10.1002/jlb.46.6.538
169. Kim DK, Jeong JH, Lee JM, Kim KS, Park SH, Kim YD, et al. Inverse agonist of estrogen-related receptor Γ Controls salmonella typhimurium infection by modulating host iron homeostasis. Nat Med. (2014) 20:419–24. doi: 10.1038/nm.3483
170. Fang FC, Weiss G. Iron errs with salmonella. Cell Host Microbe. (2014) 15:515–6. doi: 10.1016/j.chom.2014.04.012
171. Wilson JW. Nocardiosis: updates and clinical overview. Mayo Clinic Proc. (2012) 87:403–7. doi: 10.1016/j.mayocp.2011.11.016
172. Margalit I, Lebeaux D, Tishler O, Goldberg E, Bishara J, Yahav D, et al. How do I manage nocardiosis? Clin Microbiol infection. (2021) 27:550–8. doi: 10.1016/j.cmi.2020.12.019
173. Han L, Ji X, Liu X, Xu S, Li F, Che Y, et al. Estradiol aggravate nocardia farcinica infections in mice. Front Immunol. (2022) 13:858609. doi: 10.3389/fimmu.2022.858609
174. Ogle OE. Odontogenic infections. Dental Clinics North America. (2017) 61:235–52. doi: 10.1016/j.cden.2016.11.004
Keywords: estrogen, bacterial infection, sex difference, immune response, molecular mechanism
Citation: Hong L, Liang H, Man W, Zhao Y and Guo P (2025) Estrogen and bacterial infection. Front. Immunol. 16:1556683. doi: 10.3389/fimmu.2025.1556683
Received: 07 January 2025; Accepted: 11 April 2025;
Published: 29 April 2025.
Edited by:
Xiaohui Sem, Women in Global Health Singapore, SingaporeReviewed by:
Irina Miralda, Emory University, United StatesCopyright © 2025 Hong, Liang, Man, Zhao and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengbo Guo, ODc3MjczNjIxQHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship