 Jichun Yang1,2†
Jichun Yang1,2† Meng Shi
Meng Shi- 1Department of Thoracic and Cardiovascular Surgery, Hua Shan Hospital, Affiliated with Fudan University, Shanghai, China
- 2School of Integrative Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
Lung diseases, including acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), are associated with various etiological factors and are characterized by high mortality rates. Current treatment strategies primarily focus on lung-protective ventilation and careful fluid management. Despite over 50 years of basic and clinical research, effective treatment options remain limited, and the search for novel strategies continues. Traditionally, platelets have been viewed primarily as contributors to blood coagulation; however, recent research has revealed their significant role in inflammation and immune regulation. While the relationship between platelet count and ALI/ARDS has remained unclear, emerging studies highlight the “dual role” of platelets in these conditions. On one hand, platelets interact with neutrophils to form neutrophil extracellular traps (NETs), promoting immune thrombosis and exacerbating lung inflammation. On the other hand, platelets also play a protective role by modulating inflammation, promoting regulatory T cell (Treg) activity, and assisting in alveolar macrophage reprogramming. This dual functionality of platelets has important implications for the pathogenesis and resolution of ALI/ARDS. This review examines the multifaceted roles of platelets in ALI/ARDS, focusing on their immunomodulatory effects, the platelet-neutrophil interaction, and the critical involvement of platelet-Treg cell complexes in shaping the inflammatory environment in ALI.
1 Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) present clinically as rapid-onset respiratory failure, with a mortality rate reaching approximately 40% in severe cases (1, 2). Recent epidemiological studies estimate the global incidence of ARDS to range from 10 to 86 cases per 100,000 people per year, depending on population demographics and diagnostic criteria (3, 4). Despite advances in intensive care, ARDS remains associated with substantial morbidity and mortality, with hospital mortality rates ranging from 30% to over 40%, particularly in moderate-to-severe cases (5). Even with optimal supportive care, a major clinical trial reported a 42.8% mortality rate by day 90 in patients with moderate-to-severe ARDS, underscoring the urgent need for improved therapeutic strategies (6). A variety of precipitating factors, including severe infections, inhalational lung injuries, ischemia-reperfusion events, multi-trauma, extensive blood transfusions, and acute pancreatitis, can directly or indirectly damage lung tissue or associated vasculature, often serving as precursors to the rapid onset of respiratory failure (7). A marked increase in ARDS cases was observed during the coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), underscoring ARDS as a critical global health challenge (8).
The concept of ALI and ARDS was first introduced by Ashbaugh et al. in 1967 (9). In 1994, a collaboration between European and American experts established diagnostic criteria for ALI and ARDS, specifying the presence of acute bilateral lung infiltrates on chest imaging, no evidence of elevated left atrial pressure, and a ratio of arterial oxygen partial pressure to fractional inspired oxygen (PaO2/FiO2) ≤ 300 mmHg for ALI or < 200 mmHg for ARDS (10, 11). The Berlin definition, introduced in 2012, further refined these parameters by standardizing the grading of ARDS severity based on oxygenation levels: mild (200 < PaO2/FiO2 ≤ 300 mmHg), moderate (100 < PaO2/FiO2 ≤ 200 mmHg), and severe (PaO2/FiO2 ≤ 100 mmHg). This updated framework also provided objective diagnostic tools and emphasized clinical markers, addressing limitations in the earlier US-European Consensus Conference criteria (12). Despite these advancements, the clinical mortality rate for ALI and ARDS remains alarmingly high, with treatment options largely limited to supportive care. Current approaches focus primarily on protective mechanical ventilation, corticosteroid-assisted fluid management, and interventions targeting the underlying cause (13, 14). However, even with optimal supportive care, a major clinical trial reported a 42.8% mortality rate by day 90 in patients with moderate-to-severe ARDS (15). Although mechanical ventilation is indispensable, it is associated with risks such as ventilator-induced ALI (VILI), which exacerbates inflammation and may increase the likelihood of pulmonary fibrosis (16, 17). These challenges highlight the need for a deeper understanding of the complex pathogenesis of ALI and ARDS, as well as for developing novel therapeutic strategies.
Recent studies suggest that ARDS is not a single disease entity but rather a syndrome composed of distinct phenotypic subgroups that exhibit different inflammatory responses, clinical outcomes, and treatment responses (18, 19). While traditional clinical trials have treated ARDS as a homogeneous condition, emerging evidence supports the need for a phenotype-driven approach to improve patient selection and therapeutic efficacy. Genome-wide association studies (GWAS) have identified platelet count as a key intermediate phenotype in ARDS, linking platelet activation to disease severity and genetic susceptibility (20). Moreover, phenotypic variability has significantly influenced the success rates of clinical trials evaluating novel ARDS treatments, highlighting the necessity of patient enrichment strategies to enhance study design and improve therapeutic targeting (21). Integrating genetic and phenotypic stratification into ARDS research and clinical management may lead to more precise and effective interventions, ultimately improving patient outcomes.
Excessive inflammatory responses are central to the pathogenesis of ALI and ARDS, leading to a massive influx of neutrophils, macrophages, and other inflammatory cells that cause extensive damage to pulmonary vascular endothelial and alveolar epithelial barriers (22, 23). Platelets, long recognized for their role in coagulation, have recently emerged as key mediators of inflammation and immune regulation (24). Increasing evidence suggests that platelets interact closely with neutrophils to form platelet-neutrophil complexes, which amplify inflammatory cascades and contribute to tissue damage (25). For instance, Semaphorin 7A (Sema7A), a glycosylphosphatidylinositol-anchored protein, interacts with PlexinC1 receptors on neutrophils, enhancing neutrophil activation, chemotaxis, and adhesion (26). This interaction facilitates the formation of platelet-neutrophil aggregates, which promote the release of neutrophil extracellular traps (NETs), a process closely linked to endothelial cell injury and alveolar-capillary barrier disruption (26, 27). NETs, though initially protective by capturing pathogens, release cytotoxic histones and proteases that exacerbate pulmonary inflammation, edema, and tissue damage (28). The Sema7A-PlexinC1 axis has also been shown to enhance neutrophil transmigration across the endothelial barrier, further reinforcing the role of platelet-neutrophil interactions in ALI and ARDS (26).
Platelets store and release a wide range of inflammatory mediators, including cytokines and chemokines, which propagate the immune response upon activation (29). For example, activated platelets release factors such as P-selectin, platelet factor 4 (CXCL4), and interleukin-1β (IL-1β), which recruit neutrophils to sites of injury and amplify leukocyte aggregation (29). Platelet-endothelial interactions also exacerbate inflammation by increasing vascular permeability and disrupting the alveolar-capillary barrier, further contributing to pulmonary edema and impaired gas exchange (30). Consequently, platelets are often regarded as amplifiers of ALI and ARDS, driving hyperreactive inflammatory cascades (31). However, in addition to their well-characterized pro-inflammatory role, platelets are increasingly recognized as key regulators of immune balance through their interactions with various immune cells, including regulatory T cells (Tregs) and alveolar macrophages (30, 32). These interactions contribute not only to modulating inflammatory responses but also to promoting tissue repair and resolution of ALI. While the immunoregulatory functions of platelets have gained attention in recent years, the precise molecular mechanisms governing these processes remain incompletely understood, highlighting the need for further investigation. Earlier reviews, such as the work by Middleton et al. (2018) (33), have provided a detailed discussion of platelet involvement in ALI/ARDS, particularly emphasizing their role in inflammation and thrombosis. Our review extends this discussion by incorporating recent findings that shed light on platelet-driven immune modulation. Specifically, we examine how platelet interactions with Tregs and macrophages shape the inflammatory milieu and influence lung recovery. Furthermore, our work takes a more translational perspective by exploring a broader range of antiplatelet and immunomodulatory therapies, assessing their potential use across different ARDS phenotypes. In addition, we discuss the emerging significance of lung-resident megakaryocytes and their potential contributions beyond platelet production, offering new insights into their role in both local and systemic immune regulation within the context of ALI/ARDS. Emerging evidence suggests that targeting platelets may provide therapeutic benefits. For instance, antiplatelet therapies such as aspirin have demonstrated the ability to attenuate hyperoxia-induced ALI by modulating platelet-driven inflammatory pathways (34, 35). Additionally, platelet glycoprotein VI (GPVI) has been identified as a key mediator of neutrophil recruitment, migration, and NETosis in early ALI and ARDS (36). Novel therapeutic approaches, such as tea polyphenol-loaded nanoparticles coated with platelet membranes, have shown promise in ameliorating lipopolysaccharide (LPS)-induced ALI, further highlighting platelets as a potential therapeutic target (37).
Interestingly, platelets exhibit a dual role in ALI and ARDS (38). In the early stages, platelets amplify inflammation by releasing pro-inflammatory mediators, while in the later stages, they contribute to the resolution of inflammation (39). In infectious pneumonia, platelets maintain the alveolar-capillary barrier and reduce the virulence of pathogens, thereby protecting against severe pulmonary complications (40, 41). During the resolution phase, platelets promote macrophage polarization from the pro-inflammatory M1 phenotype to the anti-inflammatory M2 phenotype, facilitating tissue repair and recovery (42, 43). Furthermore, platelets interact with regulatory T cells (Tregs), enhancing their activation and secretion of anti-inflammatory cytokines such as transforming growth factor-beta (TGF-β) and interleukin-10 (IL-10) (44). These interactions are crucial for suppressing excessive inflammation and promoting the clearance of apoptotic neutrophils (41, 42). This dual functionality raises critical questions: What molecular signals govern the transition of platelets from pro-inflammatory to anti-inflammatory states? How do platelets interact with other immune cells to balance inflammation and tissue repair? Addressing these questions may provide insights into novel therapeutic strategies for ALI and ARDS.
2 Activated platelets are essential inflammatory and immune effector cells in ALI/ARDS
Platelets, anucleate cells derived from megakaryocytes, are second only to red blood cells in abundance within circulation. Historically, platelets were thought to originate exclusively from bone marrow megakaryocytes. Recent studies have revealed that lung-resident megakaryocytes also actively produce circulating platelets, adding an important dimension to their biology (45, 46). Platelets are crucial for physiological hemostasis and pathological thrombosis, as they rapidly adhere to damaged vessel walls, form aggregates, and initiate clot formation (47, 48). Damage to the vascular wall triggers platelets to release procoagulant factors, recruiting leukocytes and red blood cells to form a thrombotic barrier at the injury site, preventing further bleeding and microbial invasion (32, 49).
Beyond hemostasis, platelets are now recognized as key players in immune responses. They interact with various immune cells, such as neutrophils and macrophages, releasing bioactive molecules from intracellular granules (50, 51). These molecules act to bridge innate and adaptive immunity, positioning platelets as critical drivers of inflammation, particularly in the pathogenesis of ALI/ARDS (51–53). Platelets fine-tune immune responses by modulating the phenotype and activity of immune cells, thereby influencing the progression and resolution of ALI (54).
2.1 Platelet surface adhesion molecules and receptors
Platelet activation is a multistep process triggered by vascular injury or inflammation. Once activated, platelets adhere to neutrophils, endothelial cells, and other immune cells, forming platelet-leukocyte aggregates that amplify the inflammatory response (55, 56). Platelets recognize von Willebrand factor (VWF) and collagen at the site of injury through specific receptors, such as glycoproteins (GP) Ib-IX-V, GPVI, and GPIIb/IIIa (57–59). These interactions not only promote adhesion to the endothelium but also initiate rapid intracellular signaling that stabilizes thrombus formation and amplifies inflammation (60, 61).
A key mediator of platelet-neutrophil interactions is P-selectin, which binds to P-selectin glycoprotein ligand-1 (PSGL-1) on neutrophils. This interaction triggers signaling cascades, such as extracellular signal-regulated kinase (ERK)1/2- mitogen-activated protein kinase (MAPK), activating neutrophil integrins like Mac-1 and LFA-1 (30, 62). These integrins facilitate neutrophil adhesion and migration across the endothelium, further amplifying the inflammatory response in ALI/ARDS (63, 64). The formation of platelet-neutrophil aggregates (PNAs) enhances neutrophil activation, leading to ROS production and NETs release, which exacerbate endothelial dysfunction and alveolar injury (65, 66), as described earlier.
Recent studies have highlighted the role of mitochondrial dynamics in platelets, showing that mitofusin-2 (Mfn2) regulates platelet-neutrophil interactions by influencing mitochondrial ROS production (67, 68). Dysregulation of this pathway exacerbates platelet-neutrophil aggregate formation, worsening inflammation and ALI (67). Moreover, platelet GPIIb/IIIa binds to soluble fibrinogen, creating a bridge with neutrophil Mac-1, further stabilizing platelet-neutrophil complexes during inflammation (64, 69). Additionally, P-selectin stored in platelet alpha granules and endothelial Weibel-Palade bodies mediates platelet-leukocyte adhesion, making it a key player in ALI (70).
Therapeutic strategies targeting these interactions, such as P-selectin inhibitors and GPVI signaling blockers, have shown promise in preclinical models (71, 72). These approaches reduce platelet-neutrophil aggregates, mitigate NET formation, and alleviate inflammation, offering potential avenues for ARDS treatment (63).
2.2 Platelets are rich in a variety of immune mediators
Platelets are small anucleate cells that store and release various thrombosis and immune regulation-associated bioactive substances (Table 1). These molecules, packaged in alpha granules, dense granules, and lysosomal granules, are released upon platelet activation in response to vascular injury or inflammatory stimuli (83).
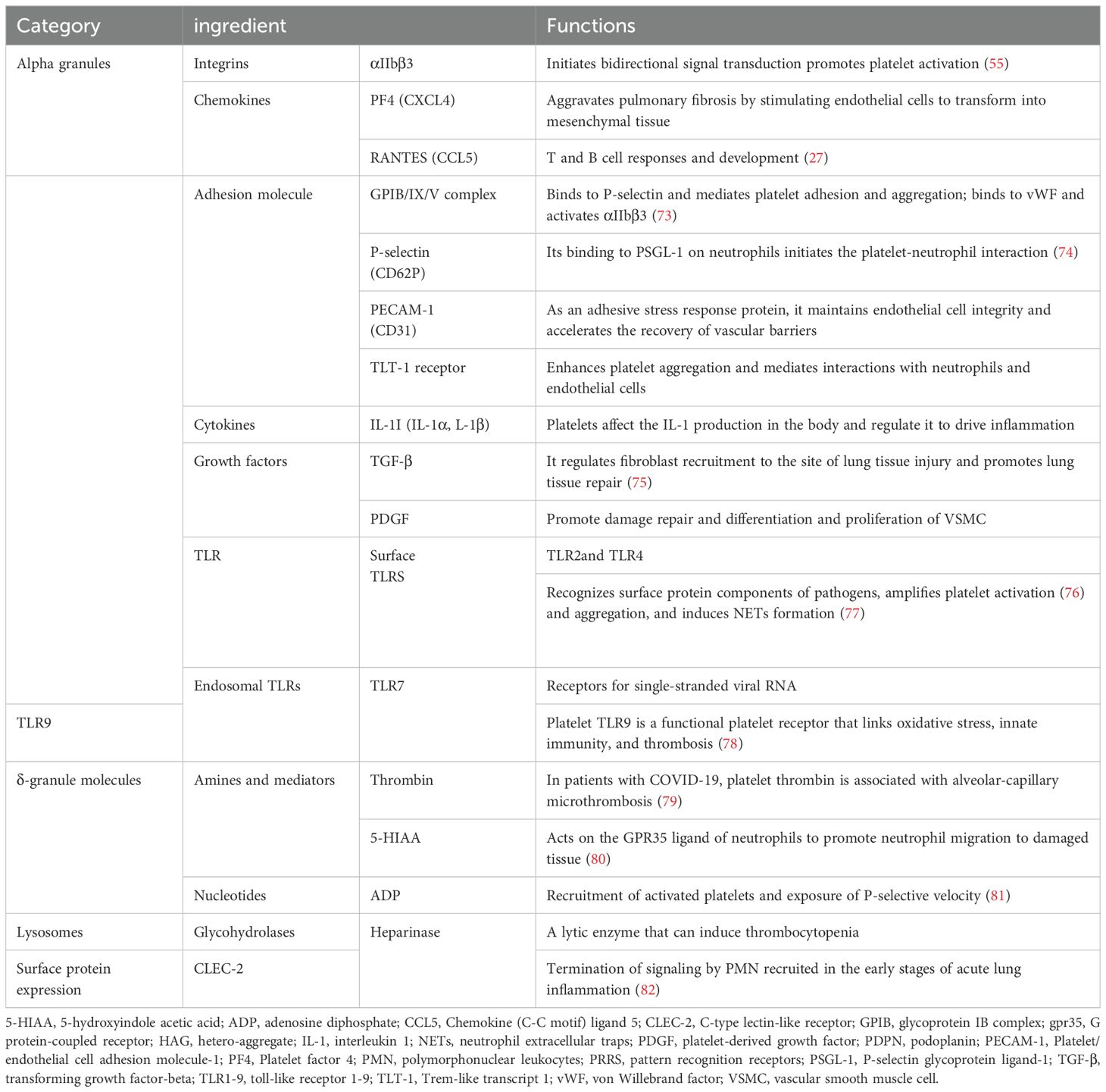
Table 1. Important platelet-derived immune mediators.
Alpha granules are particularly important for inflammation and immune regulation, as they contain mediators such as platelet factor 4 (PF4), RANTES, and interleukin-8 (IL-8). These mediators play key roles in recruiting neutrophils and other leukocytes to sites of injury (84–86). PF4, a member of the CXC chemokine family, exhibits dual roles in promoting neutrophil recruitment and modulating immune responses. Elevated PF4 levels have been correlated with disease severity in ARDS, underscoring its clinical relevance (84, 87).
Dense granules store small molecules, including ADP, ATP, and serotonin (5-HT), which amplify platelet activation through feedback mechanisms involving the P2Y12 receptor (88). Recent research has identified 5-hydroxyindoleacetic acid (5-HIAA), a serotonin metabolite released by activated platelets, as a ligand for G protein-coupled receptor 35 (GPR35) (89, 90). This signaling axis promotes neutrophil migration and adhesion, highlighting the intricate role of platelet-derived mediators in inflammation (80). Lysosomes, though less studied, contain acid hydrolases and glycohydrolases that dissolve platelet aggregates, helping regulate thrombosis and inflammation (91).
Although platelets lack nuclei, they retain functional mRNA and splicing machinery, allowing them to synthesize proteins such as interleukin-1 beta (IL-1β) in response to stimuli (92, 93). This dynamic capability enables platelets to adapt to changing inflammatory environments. Alterations in platelet transcriptomes, observed in COVID-19 patients, have revealed upregulation of inflammatory pathways, such as MAPK signaling, linked to severe ALI (94).
Platelet-derived extracellular vesicles (EVs), including exosomes and microvesicles, are another important mechanism by which platelets influence inflammation (95). These vesicles carry cytokines, chemokines, and nucleic acids, modulating endothelial barrier function and reducing inflammation in preclinical ALI models. Recent advancements in biomimetic nanoparticle technology have demonstrated the potential of platelet-derived vesicles as targeted drug delivery systems, offering new therapeutic opportunities (94, 96–100).
3 Platelets promote neutrophil recruitment, pathogen elimination, and induction of neutrophil extracellular traps release in ALI/ARDS
The activation and recruitment of neutrophils are essential processes in the pathogenesis of ALI (ALI) and acute respiratory distress syndrome (ARDS) (101). These processes are closely associated with platelet-neutrophil interactions during the inflammatory surge in ALI/ARDS. Platelet activation enhances neutrophil recruitment through P-selectin-PSGL-1 interactions and promotes the formation of platelet-neutrophil aggregates (PNAs). This interaction amplifies neutrophil activation and inflammatory mediator release, contributing to endothelial damage and alveolar injury (Figure 1) (102, 103). Evidence from lipopolysaccharide (LPS)-induced pneumonia models suggests that platelet depletion leads to a significant reduction in neutrophil recruitment, highlighting the platelet’s indispensable role in coordinating the inflammatory response (104). Furthermore, studies have demonstrated that inhibiting platelet-derived chemokines such as CCL5 and CXCL4 effectively prevents the progression of ALI (105). In addition, platelet integrin-mediated signaling inhibition reduces NET release, thereby mitigating lung tissue damage in ALI VILI models (106).
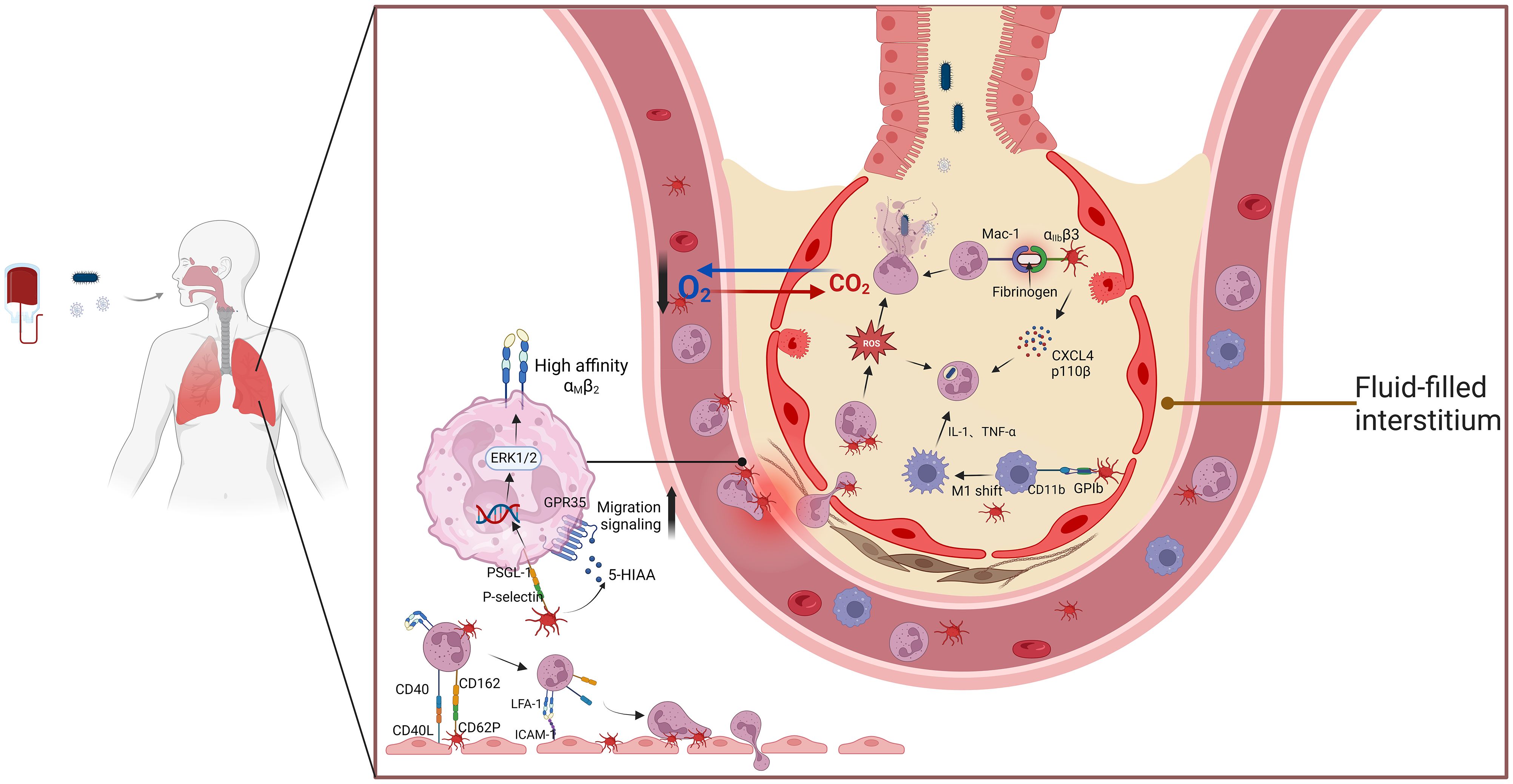
Figure 1. The interaction between platelets and neutrophils aggravates ALI/ARDs. This schematic illustration highlights the critical role of platelet-neutrophil interactions in the pathogenesis of ALI and ARDS. Upon activation, platelets release inflammatory mediators such as PF4, P-selectin, and CCL5, which facilitate neutrophil recruitment and adhesion. The formation of platelet-neutrophil aggregates enhances NET release, a process that contributes to endothelial damage, increased vascular permeability, and alveolar inflammation. Additionally, interactions between PSGL-1 on neutrophils and P-selectin on platelets promote immune cell crosstalk, amplifying inflammatory responses. Excessive NET formation releases cytotoxic histones and proteases that disrupt the alveolar-capillary barrier, exacerbating lung injury. These mechanisms collectively contribute to the progression of ARDS, underscoring the potential of targeting platelet-neutrophil interactions as a therapeutic strategy. Abbreviations: ALI, acute lung injury; ARDS, acute respiratory distress syndrome; PF4, platelet factor 4; NETs, neutrophil extracellular traps; PSGL-1, P-selectin glycoprotein ligand-1; CCL5, chemokine (C-C motif) ligand 5.
Platelet P-selectin stored in alpha granules plays a key role in mediating platelet-neutrophil interactions. Blocking P-selectin has been shown to diminish platelet-neutrophil aggregates and slow ALI progression in acid-induced ALI models (65). Protease-activated receptor 2 blockade has also been reported to inhibit carbamoyl-platelet-activating factor (PAF)-mediated neutrophil recruitment and inflammation in mouse lung tissue (107). Furthermore, cigarette smoke-induced severe influenza has been shown to worsen due to platelet-driven pulmonary microvascular occlusion, underscoring the role of platelet-neutrophil aggregation in exacerbating ALI (108).
3.1 Platelets and neutrophil recruitment in ALI/ARDS
Physiologically, red and white blood cells are primarily located in the central vascular region, while platelets are concentrated near the vascular endothelium, positioning them to interact with leukocytes under both physiological and pathological conditions (109). Even in the absence of inflammation, transient interactions occur between platelets and neutrophils near the vascular endothelium (110, 111). Once activated, platelets regulate neutrophil rolling and adhesion, which are critical for neutrophil recruitment during inflammation, as demonstrated in various pneumonia models (65).
In inflammatory environments, platelets act as navigators, bridging neutrophils and endothelial cells. P-selectin on platelets interacts with neutrophil P-selectin glycoprotein ligand-1 (PSGL-1), mediating high-affinity activation of neutrophil integrin β2 via ERK1/2 MAPK signaling. This activation enhances neutrophil adhesion and transmigration across endothelial barrier (112). Integrins such as LFA-1 and Mac-1 further stabilize platelet-neutrophil complexes and promote neutrophil extravasation (113, 114).
Besides direct interactions, platelets also maintain vascular endothelial integrity indirectly through mechanisms involving phosphoinositide 3-kinase (PI3K) isoforms, particularly p110β. In pneumococcal pneumonia-induced mouse ALI models, p110β promotes platelet activation, neutrophil extravasation, and bacterial clearance (115). These findings underscore the indispensable role of platelets in coordinating neutrophil recruitment and regulating vascular inflammation.
3.2 Neutrophil functions: reactive oxygen species and phagocytosis
Neutrophils play critical roles in pathogen elimination and the regulation of pulmonary injury (116, 117). ROS generated by neutrophils are essential for microbial killing but can exacerbate tissue injury when excessively produced (118). Platelet-neutrophil aggregates amplify ROS production. Specifically, studies have shown that when neutrophils bind to platelets on immunoglobulin G (IgG)-coated surfaces, ROS release is significantly increased (119). ROS also influences specific pathways involved in NET formation, further enhancing pathogen clearance capacity (120). Platelets further enhance neutrophil phagocytosis through signaling pathways such as TLR2/PI3K/AKT (121). These pathways facilitate bacterial engulfment by neutrophils, though the detailed molecular mechanisms remain a promising area for future investigation (122).
3.3 NET formation and its dual role in ALI/ARDS pathogenesis
NETs, composed of DNA scaffolds modified by histones, myeloperoxidase (MPO), and neutrophil elastase (NE), serve as robust antimicrobial barriers (123). Dysregulated NET formation contributes to aseptic inflammation, thrombosis inflammation, and tissue injury in ALI/ARDS (Figure 2) (124, 125). NET production is primarily driven by ROS generated through NADPH oxidase, which stimulates MPO and NE activity, leading to chromatin decondensation (126). Pore-forming proteins like gasdermin D facilitate the extracellular release of NETs in the NADPH oxidase 2 (NOX2)-ROS-dependent pathway (127).
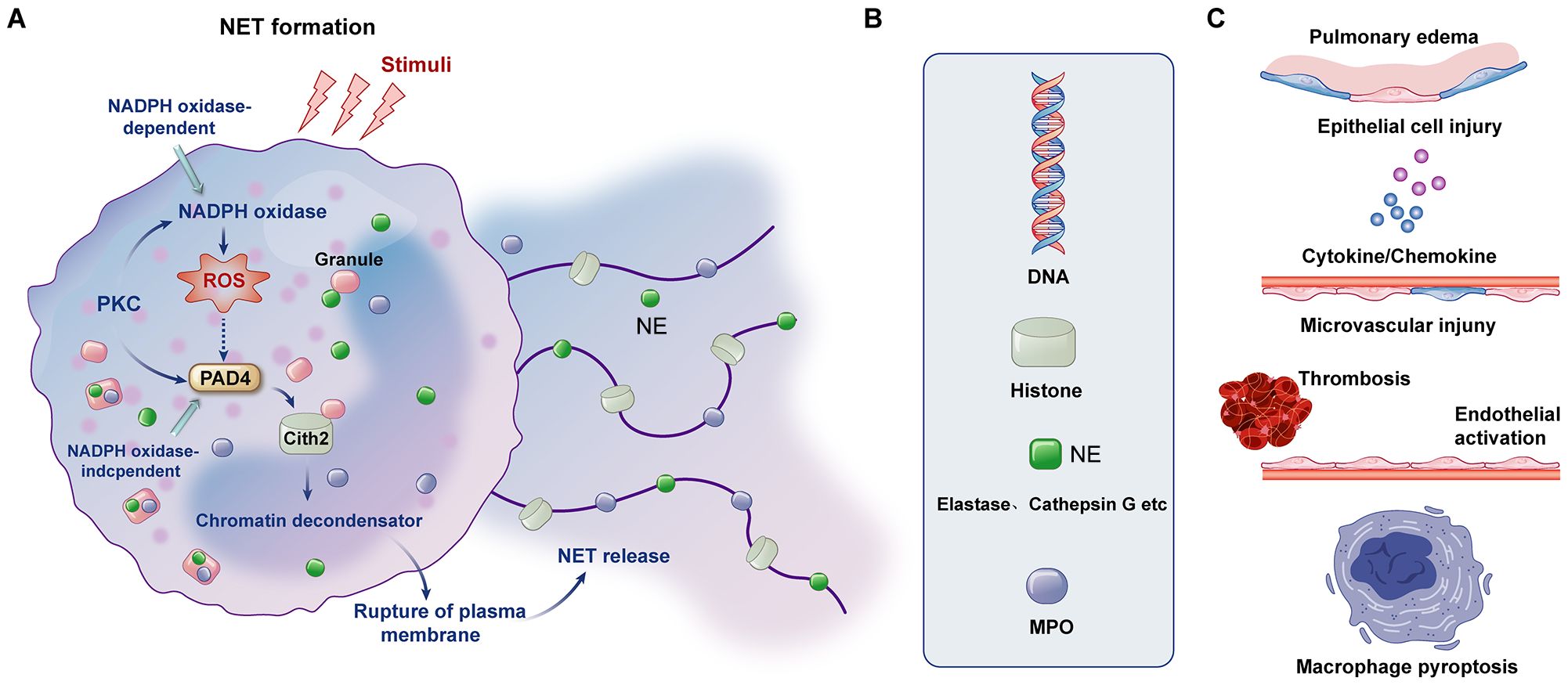
Figure 2. Formation of NETs, components of NETs and mechanisms affecting ARDS. (A) The formation of NETs can be divided into NADPH oxidase-dependent and independent formations. (B) The major components of NETs include DNA, histones, NE and MPO. (C) NETs and their components can cause pulmonary tissue edema, damage alveolar epithelial cells and microvessels, cause the formation of immune thrombosis and activation of endothelial cells, and mediate the pyroptosis of macrophages and the release of downstream cytokines.
During pulmonary infection, platelet-neutrophil complexes critically modulate NET release through multiple mechanisms. These include PSGL-1 signaling, platelet glycoprotein VI (GPVI) signaling, and high mobility group box 1 (HMGB1)-mediated autophagy (64, 128). Platelet P-selectin also promotes NET release by binding to PSGL-1, while fibrinogen-mediated interactions between platelet integrin β3 and neutrophil Mac-1 further enhance NET formation (129). Inhibition of GPVI signaling has shown promise in experimental LPS-induced ALI models by reducing NET release and mitigating lung inflammation (37).
Therapeutic strategies targeting platelet-mediated NET formation represent a potential avenue for ALI/ARDS treatment. For example, GPVI signaling inhibitors and HMGB1-targeting therapies have demonstrated efficacy in reducing NET-mediated tissue damage in experimental models (130).
3.4 Concluding remarks on platelet-neutrophil interactions in ALI/ARDS
Severe infection, inhalation injury, and massive blood transfusion are significant risk factors for ALI and ARDS. In these contexts, platelets play pivotal roles in neutrophil recruitment and NET formation, processes that are central to the inflammatory and immune responses in ALI/ARDS. For instance, platelet P-selectin-PSGL-1 interactions promote neutrophil adhesion and migration to injured alveoli. Platelet-derived chemokines, including CXCL4 and p110β, enhance neutrophil phagocytic capacity, aiding in bacterial clearance (106). Moreover, platelet-neutrophil complexes amplify ROS production and NET release, further influencing the disease’s progression.
While NETs serve protective roles by limiting pathogen spread, their dysregulated release can exacerbate tissue damage and promote immunothrombosis. Therefore, therapeutic strategies that modulate platelet-neutrophil interactions, particularly NET formation, hold promise for mitigating inflammation and improving clinical outcomes in ALI/ARDS. Future research should aim to elucidate the precise molecular mechanisms underlying these interactions and identify novel therapeutic targets.
4 Activated platelets, endothelial cells, and neutrophils mixed results in the formation of immunothrombosis aggravate ALI/ARDS
Innate immunity serves as the first line of defense against pathogens. Platelets play a crucial role not only in directly eliminating microorganisms but also in mediating the formation of immune thrombosis, a process referred to as “immunothrombosis” (131). Unlike classical thrombosis, activated platelets can interact with neutrophils and complement proteins to trigger the coagulation cascade, forming thrombi within microvessels. This disruption of the balance between coagulation and inflammation leads to severe inflammatory responses, resulting in widespread damage to pulmonary capillaries and alveolar edema (132). The complex formation of immunothrombosis involves the activation of platelets, endothelial cells, neutrophils, NETs, and microparticles, all of which are recognized as key contributors to the pathogenesis of ALI/ARDS [119]. In the case of COVID-19 caused by SARS-CoV-2, nearly 25% of patients with severe disease exhibit pronounced hypercoagulability (133). Postmortem lung tissue analyses from COVID-19 patients have consistently revealed the presence of disseminated microthrombi (134–136).
4.1 Role of endothelial cells in ALI/ARDS-related inflammation
ALI is characterized by intricate interactions among immune cells, inflammatory mediators, and tissue components (137). The progression from acute inflammation to healing involves a series of coordinated cellular pathways (138). Initially, during ALI, alveolar epithelial cells are activated by pro-inflammatory cytokines, which leads to an increase in vascular permeability and allows immune cells, especially neutrophils, to infiltrate the lung tissue (139). This inflammatory cascade is regulated by signaling pathways such as NF-κB and MAPK, with key inflammatory mediators like TNF-α and IL-1β playing crucial roles (140, 141).
An intact endothelial barrier is crucial for maintaining vascular permeability and ensuring the diffusion of nutrients, oxygen, and metabolic waste products (142). In the context of ALI/ARDS, endothelial cells regulate vascular permeability, and platelet activation and aggregation can lead to the formation of microthrombi (38). Endothelial cells are covered by a glycocalyx, a multi-layered structure that limits direct contact between endothelial cells and blood components, thereby inhibiting leukocyte and platelet adhesion, coagulation, and microthrombosis (136, 143). However, this protective barrier is compromised in the inflammatory environment of ARDS (144).
ALI/ARDS is a life-threatening lung condition characterized by the disruption of the alveolar-capillary barrier, leading to pulmonary edema and impaired gas exchange (145). Endothelial cell injury and inflammation are central to the development of ALI/ARDS (146). As a highly dynamic and metabolically active tissue, the endothelium plays a critical role in maintaining organ homeostasis (147). During inflammation, endothelial cells are among the first to respond to inflammatory stimuli (148). In ALI/ARDS, the activation of inflammatory cells (e.g., neutrophils) and the release of inflammatory mediators (e.g., TNF-α, interleukins) exacerbate endothelial cell injury (149). This endothelial damage results in the increased release of inflammatory mediators, which further attract additional immune cells, creating a vicious cycle of sustained inflammation. Consequently, endothelial injury increases the permeability of the blood vessel wall, promoting the development of pulmonary edema and impairing gas exchange, ultimately leading to symptoms such as dyspnea and hypoxemia (150). Recent studies suggest that lung endothelial cells are key regulators of both innate and adaptive immunity, playing an essential role in the pathogenesis of ARDS (142). Additionally, endothelial cell vesicles (ECVs) have been shown to exacerbate ALI/ARDS by transmitting inflammatory signals. These vesicles carry FSTL1, which activates inflammatory pathways like the TLR4/JAK3/STAT3/IRF-1 pathway (151). Moreover, TRIM47 has been implicated in enhancing the inflammatory response and promoting endothelial activation in ALI/ARDS (152).
4.2 Interaction between activated platelets and endothelial cells
Activated platelets interact with endothelial cells to induce immune responses that promote immunothrombosis. Specifically, IL-1β released by activated platelets stimulates endothelial cells, increasing their permeability and accelerating the extravasation of fluid and proteins (153). Additionally, activated platelets bind to endothelial cell PSGL1 via P-selectin, while glycoprotein GPIb interacts with von Willebrand factor on endothelial cells (154). This interaction facilitates the migration of inflammatory cells, thereby promoting the formation of immunothrombosis (155, 156). Clinical manifestations of ARDS, such as endothelial dysfunction and immune-thrombosis-related complications, are attributed to this complex interplay between platelets and endothelial cells (142, 157, 158). Flow chamber analyses have shown that platelet-endothelial interactions are critical for maintaining vascular integrity and regulating blood flow (159).
4.3 Activated platelets mediate the formation of NETs, promoting immunothrombosis
Neutrophils are central to the early stages of ALI, as they release ROS and proteolytic enzymes that damage both the endothelial and epithelial barriers (101). However, the unchecked activation of neutrophils can worsen the condition by sustaining the inflammatory response and causing further tissue damage (138). Beyond their traditional role in clot formation, platelets also contribute significantly to the regulation of inflammation (160). The interaction between platelets and neutrophils, mediated by P-selectin, amplifies the inflammatory response by enhancing neutrophil recruitment and promoting the formation of NETs (161).
As ALI progresses, activated platelets express P-selectin, which binds to neutrophil PSGL-1 to promote the formation of NETs, structures that trap pathogens (162). However, in this process, the presence of tissue factor within NETs stimulates the release of thrombin, which in turn activates platelets, initiating a vicious cycle of immune thrombosis and potentially leading to disseminated intravascular coagulation (DIC) (163, 164). Activated platelets also secrete various proinflammatory molecules, including soluble P-selectin, platelet factor 4 (PF4), platelet-activating factor, and neutrophil-activating peptides, which promote neutrophil recruitment to sites of thrombosis (131, 165). Furthermore, high mobility group box 1 (HMGB1) released from activated platelets contributes to NET formation. HMGB1 interacts with the receptor for advanced glycation end products (RAGE) on neutrophils, promoting autophagy and NET generation, independent of NADPH oxidase-mediated ROS production (130, 166, 167). Platelet TLR4 signaling plays a pivotal role in this process. When LPS binds to TLR4, activated platelets interact with neutrophils through GP1b and neutrophil integrin β2, facilitating NET formation (66, 168). In experimental models of transfusion-associated ALI, the inhibition of platelet activation via aspirin or GPIIb/IIIa blockers has been shown to reduce NET formation and alleviate ALI (169, 170). The release of tissue factor (TF) from NETs further amplifies the imbalance between inflammation and coagulation, exacerbating immunothrombosis and leading to poor ALI prognosis (164). This process is heavily influenced by the activation of the STING (stimulator of interferon genes) pathway and TLR2 on endothelial cells (171). In summary, activated platelets not only induce NET formation but also recruit platelets to vascular sites under high shear conditions, triggering further platelet activation and perpetuating a cycle of immunothrombosis (172).
5 Investigation of the role of platelets in regulating the resolution of inflammation in patients with ALI/ARDS
Damage to tissues or microbial invasion triggers an acute response to protect the host. However, excessive and prolonged acute inflammation can cause tissue damage and impair organ function, ultimately leading to disease. To limit inflammation and prevent collateral damage to healthy tissue, lung tissue orchestrates the formation of specific pro-resolving mediators, such as lipoxins, protectins, and maresins (173). These mediators act at specific nodes to prevent further leukocyte recruitment, promote apoptosis of neutrophils, eliminate apoptotic cells, convert macrophages from a pro-inflammatory to a pro-resolving phenotype, and inhibit pro-inflammatory mediators, ultimately restoring homeostasis (174, 175). Platelets play a crucial role in controlling the resolution of inflammation in ALI/ARDS by influencing macrophage activity, T cells, and the secretion of anti-inflammatory mediators (176, 177). Consequently, research on platelet immune function is increasingly focusing on their role in pro-resolution rather than in promoting inflammation (178).
5.1 Role of platelets in macrophage polarization
Macrophages and T regulatory (Treg) cells are essential in the resolution of inflammation in ALI (179). To restore balance, the effector function of macrophages shifts from a pro-inflammatory (M1) to an anti-inflammatory (M2) phenotype (180–182). During ALI, M1 macrophages initially take the lead in guiding neutrophils to eliminate pathogens (183). Platelets, in association with GPIb-CD11b interactions, promote monocyte-mediated M1 macrophage polarization (184). As ALI progresses into the resolution phase, M2 macrophages dominate, regulating the proliferation and differentiation of alveolar type 2 (AEC2) cells, thus promoting lung tissue repair (185, 186). These M2 macrophages secrete prostanoids like PGE2, which regulate the production of anti-inflammatory lipoxin A4, downregulate CXCR2 expression, reduce ROS levels, inhibit polymorphonuclear neutrophil (PMN) migration, and prevent the release of NETs (187).
The two types of pulmonary alveolar macrophages (AMs) are monocyte-derived alveolar macrophages (Mo-AMs) and tissue-resident alveolar macrophages (TR-AMs) (188). Compared to TR-AMs, Mo-AMs are more plastic and derived from monocytes that enter the alveoli after ALI (189, 190). The properties and functions of Mo-AMs depend on the regulation of the lung microenvironment. Early inflammatory Mo-AMs exhibit the M1 phenotype, exerting significant pro-inflammatory effects that can exacerbate tissue injury (191). During the resolution phase, Mo-AMs adopt a transcriptional profile favoring tissue repair (192–194). TR-AMs, which are the “guardians” of the alveoli, effectively recognize and absorb inhaled pathogens (195). TR-AMs also support the termination and resolution of ALI inflammation through mechanisms potentially driven by β-catenin-hypoxia inducible factor-1α signaling (196). TR-AMs also release anti-inflammatory mediators like transforming growth factor beta (TGF-β) and IL-10, facilitating tissue repair (197, 198).
Recent studies have shown that ALI can trigger antigen-specific CD4+ T cell activation, amplifying Treg regulatory function during acute tissue injury (199). CD4+ T cells differentiate based on the signals they receive (200). For example, IL-2 and Janus kinase (JAK) tyrosine kinases initiate T-cell receptor (TCR) signaling, engaging signal transducer and activator of transcription 5 (STAT5) and Foxp3, leading to Treg differentiation (201, 202). Tregs regulate neutrophil apoptosis, prevent neutrophil migration, and promote lung tissue repair by releasing TGF-β (203, 204). Additionally, Tregs stimulate the proliferation and differentiation of AEC2 and directly aid in the regeneration of damaged alveolar epithelial cells (205–207). In contrast, IL-6 promotes Th17 differentiation through the JAK/STAT3/RORγt pathway, exacerbating ALI by releasing the pro-inflammatory cytokine IL-17A (208–210). The synergistic interaction between macrophages and Tregs promotes ALI resolution. Macrophages regulate the Th17/Treg balance in ALI, increasing the number of anti-inflammatory cells during the resolution phase, while Tregs guide macrophages toward the M2 phenotype (179, 211). Tregs also enhance macrophage phagocytic capacity by secreting IL-13 and stimulating macrophages to release anti-inflammatory cytokines such as TGF-β and IL-10 (211, 212).
5.2 Platelet-Treg cell interactions in ALI resolution
Platelets are instrumental in the resolution of inflammation (30). As the inflammatory response shifts toward healing, platelets release anti-inflammatory mediators like TGF-β, IL-10, and PGE2 (213). These factors help promote the polarization of macrophages to an M2 phenotype, which is associated with tissue repair and the resolution of inflammation (214). Moreover, platelets engage with regulatory T cells (Treg cells), fostering their differentiation and enhancing their anti-inflammatory effects (30). This interaction is thought to be critical for transitioning the immune response from a pro-inflammatory state to a reparative phase, thus aiding in the resolution of ALI. Additionally, platelet-Treg cell aggregation supports the shift of macrophages to an anti-inflammatory state, optimizing alveolar macrophage phagocytosis (215).
Recent studies highlight that platelets play a role in the resolution of lung inflammation through their interactions with Tregs and macrophages (216) (Figure 3). Specifically, platelet P-selectin binds to PSGL-1 on T cells, forming platelet-Treg aggregates that promote CD4+ T cell differentiation into Tregs. Additionally, platelets are the primary source of soluble CD40L (sCD40L), which is released upon activation and further enhances platelet aggregation and activation. This interaction increases the number of Tregs in the lungs and facilitates the secretion of anti-inflammatory agents, contributing to inflammation resolution (215, 217). In vitro studies of platelet-T cell cocultures showed a marked increase in the proliferation and differentiation of the FoxP3+ Treg subset by day 3 (218). Platelet-derived PH4 promotes IL-10 secretion, a potent anti-inflammatory cytokine (218, 219). Platelets regulate T cell effector responses in a context-dependent manner through PF4-TGFβ interactions, with platelet coculture enhancing sTGFβRIII release, amplifying TGFβ signaling, and promoting CD4+ T cell effector functions (54). During the resolution phase of inflammation, Tregs in the lungs enhance macrophage internalization of apoptotic neutrophils through the Vav1-Rac1 pathway (211, 220). This process accelerates macrophage phagocytosis of apoptotic neutrophils and aids in the transition of macrophages from a pro-inflammatory to a reparative phenotype.
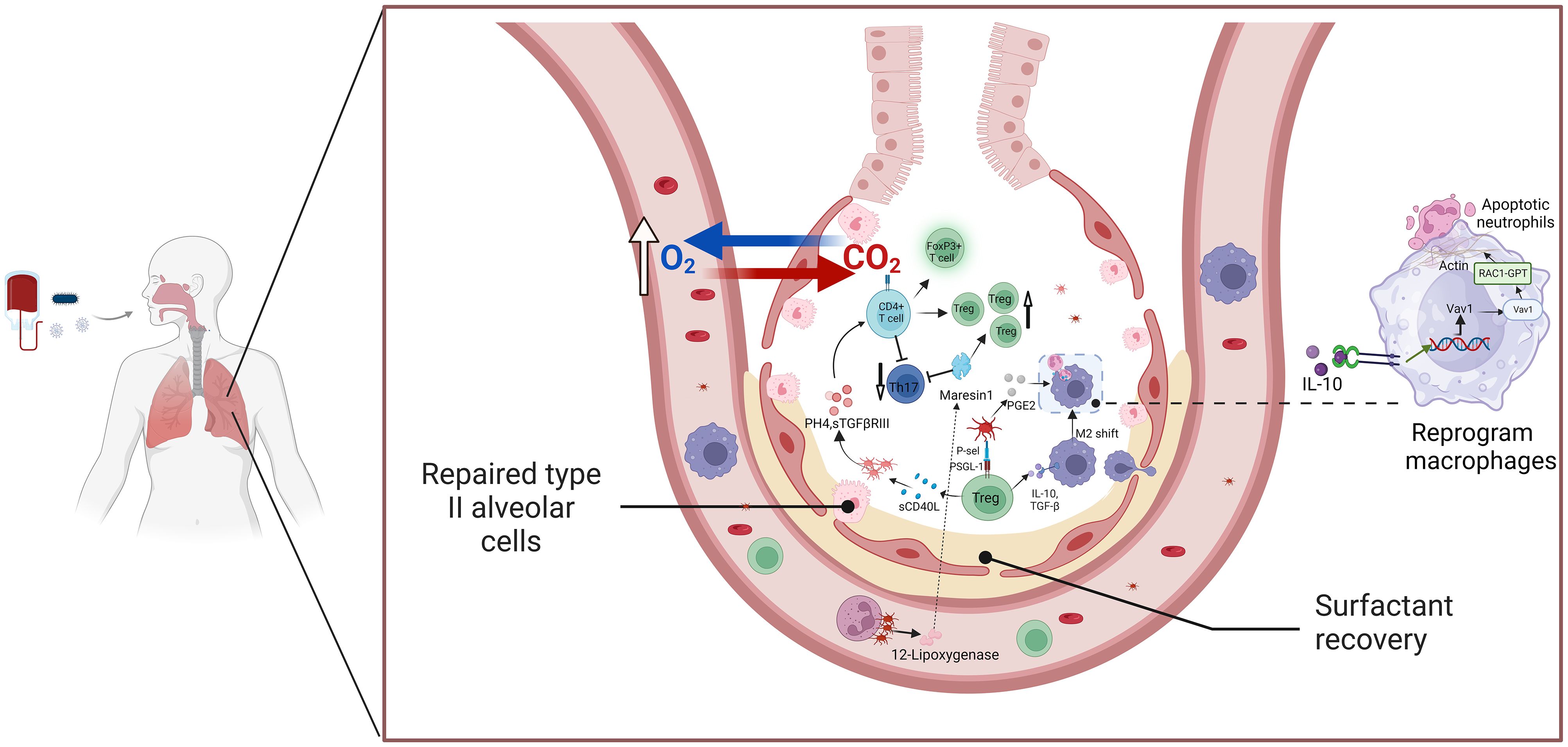
Figure 3. The platelets and Treg cells crosstalk mediate the resolution of ALI/ARD inflammation. This schematic illustration highlights the interaction between platelets and Tregs in facilitating the resolution of inflammation in ALI and ARDS. Activated platelets release key mediators such as TGF-β and PF4, which enhance Treg activation and function. In response, Tregs secrete anti-inflammatory cytokines, including IL-10 and TGF-β, which contribute to immunosuppression and the resolution of inflammation. These cytokines also promote macrophage polarization toward the M2 phenotype and assist in neutrophil apoptosis, aiding in the restoration of immune balance and tissue repair. The platelet-Treg crosstalk plays a crucial role in limiting excessive inflammation and promoting recovery in ARDS, highlighting its potential as a therapeutic target. Abbreviations: ALI, acute lung injury; ARDS, acute respiratory distress syndrome; TGF-β, transforming growth factor-beta; PF4, platelet factor 4; IL-10, interleukin-10; Treg, regulatory T cell.
5.3 Platelet-derived mediators and their role in ALI recovery
Platelets can act directly or through mediators to regulate macrophage responses, enhancing bacterial clearance and reducing inflammation. Activated platelets release PGE2, prompting mononuclear macrophages to reduce TNF-α secretion while promoting the release of anti-inflammatory mediators and facilitating inflammation resolution (211, 221). A study by Tang et al. on acute liver injury repair provides an intriguing new perspective. Typically, neutrophils use ROS production to increase the release of NETs, which exacerbates inflammation (222). However, this study hypothesized that ROS activates AMP-activated protein kinase (AMPK), which shifts macrophages toward the repair phenotype, promoting liver repair (223). It remains an exciting question whether this mechanism also plays a role in ALI recovery. The balance between Th17 and Tregs is intricately linked to the resolution of pneumonia in ALI (224). In ALI/ARDS, neutrophil-platelet aggregates enhance the synthesis of MAResin1, a pro-resolving mediator released by platelets. MAResin1 regulates the balance between Th17 and Tregs, improving lung function during acute inflammation (225–227). However, the precise mechanism by which MAResin1 affects Th17/Treg differentiation remains unclear, and further studies are needed to unravel the complex mechanisms involved in ALI resolution.
In the resolution phase of ALI/ARDS, alveolar edema is significantly reduced, and lung function improves dramatically (228). During this period, platelets coordinate the resolution of pulmonary inflammation through interactions with Tregs and macrophages (216). P-selectin binds to PSGL-1 on Tregs, forming stable platelet-Treg aggregates, which release inflammatory mediators like TGF-β and IL-10 (229). sCD40L produced by Tregs mediates platelet activation, leading to the release of factors such as PH4 and sTGFβRIII, which enhance the number of Tregs in the lung (229). Moreover, platelet-Treg aggregates promote macrophage internalization of apoptotic neutrophils through the Vav1-Rac1 pathway, enhancing the phagocytic capacity of macrophages (211, 220). The balance between Th17 and Treg differentiation plays a crucial role in alleviating lung inflammation (224). In ALI/ARDS, neutrophil-platelet aggregates promote MAResin1 synthesis, which regulates Th17/Treg signaling and improves lung function during acute inflammation (225, 226, 230). Future studies should further elucidate the mechanisms by which platelets mediate MAResin1 signaling to regulate the Th17/Treg balance in ALI resolution.
6 Antiplatelet agents in the prevention and treatment of ALI/ARDS
6.1 Clinical relevance of antiplatelet therapy in ALI/ARDS
Acute critical illnesses, such as ALI/ARDS, pose significant clinical challenges and are associated with high morbidity and mortality rates. According to the Berlin definition, a study by Hendrickson reported mortality rates of 34.9%, 40.3%, and 46.1% for mild, moderate, and severe ARDS cases, respectively (231). Notably, the 28-day mortality rates for mild, moderate, and severe cases were 29.6%, 35.2%, and 40.9%, respectively (231). The pathological features of ALI/ARDS include increased pulmonary microvascular permeability, alveolar exudates rich in proteins, and pulmonary edema (232). Given the limited treatment options, such as mechanical ventilation and restrictive fluid management, there is an urgent need for safer and more effective pharmacological interventions (233).
6.2 Efficacy of antiplatelet agents in ALI management
Animal model studies have suggested that antiplatelet agents, including aspirin (ASA) and lipid compounds, could help alleviate ALI symptoms (234–236). Retrospective clinical observations have further supported the potential of antiplatelet agents in improving ARDS symptom management (234, 237, 238). Clinical data from Philip Toner et al. have also demonstrated the potential of ASA in the treatment of ALI/ARDS (239). However, a study assessing early ASA administration for ARDS found no significant difference in morbidity between the ASA and placebo groups by day 7 (240, 241). This suggests that ASA alone may not significantly alter the progression of ALI/ARDS. It has been hypothesized that ASA does not effectively reduce platelet-driven neutrophil recruitment or prevent neutrophil rolling on endothelial cells through P-selectin, which could contribute to suboptimal therapeutic outcomes (242, 243). Additionally, the efficacy of ASA in ALI/ARDS treatment may be influenced by factors such as dosage and timing of administration (244, 245). The relatively small number of positive control patients in the ARDS trial at day 7 may have limited the study’s power to detect the full potential of ASA (240, 246). Thus, the results of the LIPS-A study could have been improved with a larger sample size.
It is important to emphasize that platelets play a crucial role in maintaining the integrity of the alveolar-capillary barrier and in promoting alveolar recovery (240, 246). Recent research has revealed that, in addition to their well-established production in the bone marrow, platelets can also originate from megakaryocytes (MKs) residing in the lungs (247). These lung-derived MKs serve as an extramedullary site of thrombopoiesis, contributing to platelet homeostasis under both physiological and pathological conditions (248). Unlike bone marrow-derived platelets, those produced within the pulmonary microenvironment may possess specialized functional properties, particularly in immune regulation (249). Studies suggest that lung MKs not only replenish platelet levels but also actively influence endothelial integrity, neutrophil trafficking, and macrophage polarization (250). These findings indicate that lung-derived platelets may play a unique role in ARDS pathogenesis, with potential contributions to both inflammatory responses and tissue repair processes. As lung-resident platelets exhibit distinct immune-regulatory roles, systemic antiplatelet therapies may differentially impact pulmonary and circulating platelet populations (251). In particular, lung-derived platelets may interact directly with the alveolar microenvironment, influencing local immune responses and endothelial stability, which could have critical implications for ARDS progression (31, 252). Understanding how these locally generated platelets contribute to immune homeostasis and tissue repair could lead to new therapeutic strategies that specifically target lung MKs and their platelet output. A more refined approach that considers the local effects of lung megakaryocytes and their platelets could improve the efficacy of pharmacological interventions in ARDS.
This emerging understanding has significant implications for antiplatelet therapies in ARDS, as systemic platelet inhibition may affect lung-resident platelet populations differently from circulating platelets. Tailoring therapeutic strategies to account for both systemic and lung-specific platelet activity could be crucial for optimizing treatment efficacy. Further investigation is needed to clarify the precise role of pulmonary MKs in ARDS and explore whether targeting lung-specific platelet production could offer novel therapeutic avenues for managing platelet-driven inflammation in ALI. In the later stages of tissue repair, once inflammation begins to resolve, continued use of antiplatelet agents could suppress these beneficial responses. While ASA and other antiplatelet agents have shown promise in ALI/ARDS treatment, the multifaceted role of platelets must be carefully considered, particularly the potential effects of pharmacological interventions at different stages of the disease.
The role of platelet activation in ARDS pathogenesis varies across distinct ARDS phenotypes, emphasizing the need for a precision medicine approach (253). Recent studies have classified ARDS into hyperinflammatory and hypoinflammatory subtypes, with the hyperinflammatory phenotype exhibiting heightened platelet activation and an increased risk of thromboinflammatory complications (254, 255). This suggests that antiplatelet strategies may be particularly effective in hyperinflammatory ARDS patients, whereas their benefit in hypoinflammatory cases remains uncertain (256). Furthermore, biomarker-based classification has enabled better patient selection in clinical trials, improving the efficacy of pharmacologic interventions in ARDS subpopulations (257). Emerging research also suggests that platelet count may serve as a biological marker for ARDS severity, providing a valuable tool for stratifying patients in future clinical studies (20). Incorporating ARDS subphenotyping into clinical practice could help optimize treatment outcomes and guide individualized therapies.
Beyond aspirin, recent investigations have identified additional antiplatelet strategies for ARDS, aiming to curb platelet-driven inflammation while maintaining essential hemostatic functions. P2Y12 receptor inhibitors, such as ticagrelor, have shown promise in attenuating platelet-neutrophil interactions and reducing immune thrombosis, thereby mitigating vascular injury associated with ARDS (258). Likewise, GP IIb/IIIa inhibitors may help regulate platelet aggregation, decreasing microvascular occlusion and improving pulmonary perfusion in severe cases (259). Moreover, pro-resolving lipid mediators, including aspirin-triggered lipoxins and resolvins, are being actively studied for their dual role in modulating platelet activity and facilitating inflammation resolution (260). Unlike traditional antiplatelet agents, which broadly suppress platelet function, these bioactive lipids appear to fine-tune platelet responses, reducing excessive inflammation while preserving their beneficial contributions to tissue repair (261). This selective mechanism makes them particularly compelling as potential therapeutic options for ARDS. Given the complexity of ARDS pathophysiology, a more structured approach to antiplatelet therapy is needed. Recent evidence suggests that the effectiveness of antiplatelet drugs may vary depending on the phase of ARDS progression (262). In the early inflammatory phase, targeting platelet activation could mitigate endothelial dysfunction, immune thrombosis, and neutrophil-driven tissue damage. However, during the resolution phase, an overly aggressive inhibition of platelet function might hinder tissue repair.
Beyond antiplatelet therapy, additional immunomodulatory and regenerative treatments are being explored for ARDS. Cytokine inhibitors, including IL-6 and TNF-α blockers, have shown potential in dampening excessive inflammation and improving oxygenation in severe cases (21). Mesenchymal stem cell (MSC) therapy has also emerged as a promising approach due to its ability to modulate immune responses, promote alveolar epithelial repair, and reduce fibrosis (256). Additionally, biologics targeting immune checkpoints and metabolic pathways involved in platelet-leukocyte interactions may present new therapeutic opportunities (21). While these treatments remain under investigation, their integration into ARDS management could enhance current strategies by complementing antiplatelet interventions, offering a more comprehensive approach across different disease stages.
6.3 Future directions for antiplatelet strategies
Emerging research suggests that combination therapy strategies, integrating multiple classes of antiplatelet and anti-inflammatory agents, may enhance therapeutic efficacy. By combining classical antiplatelet agents (e.g., P2Y12 inhibitors) with pro-resolving lipid mediators, it may be possible to suppress excessive inflammation while preserving the platelet-mediated resolution of ALI (263, 264). Moreover, precision medicine approaches that tailor antiplatelet interventions based on ARDS phenotypes and biomarkers may improve patient outcomes by optimizing drug selection and timing. These strategies warrant further investigation to determine their clinical feasibility and long-term benefits.
As research on platelet involvement in ARDS advances, it has become evident that antiplatelet interventions may need to be tailored to different stages of the disease. In the early inflammatory phase, dampening platelet-driven immune activation may help prevent endothelial dysfunction and excessive thrombosis, whereas in the later phase, a more controlled approach that preserves platelet-mediated tissue repair may be preferable (31, 176). Integrating a precision medicine approach-wherein biomarkers and ARDS phenotypes guide therapeutic decisions-could help identify patient subgroups most likely to benefit from antiplatelet interventions. Future studies should also focus on elucidating the potential role of lung-resident platelets in modulating the immune microenvironment of the alveoli and whether distinct platelet subtypes exist with differential contributions to ARDS progression. Future should explore how combining different antiplatelet agents or adjusting their administration timing can optimize their efficacy while minimizing potential adverse effects. Also should be focused on refining these therapeutic strategies, identifying optimal treatment windows, and determining which patient populations stand to benefit most from targeted antiplatelet interventions. In addition, to explore the potential synergies between antiplatelet agents and novel ARDS treatments, including immunotherapy and regenerative medicine, to optimize clinical outcomes.
7 Future directions for ALI/ARDS research and treatment
7.1 Platelet involvement in ARDS and its implications
The pathological hallmark of ALI/ARDS is the exacerbation of inflammation and disruption of the alveolar vascular endothelial barrier. Recent studies have underscored the critical role of platelets and their immunomodulatory functions in lung diseases, which can be compared to their role in thrombosis. Given that pulmonary megakaryocytes are the source of platelets, a key question arises: do platelets derived from these megakaryocytes have a distinct role in ALI/ARDS? Identifying functional and pathway differences between pulmonary megakaryocyte-derived platelets and myeloid platelets may be crucial in identifying novel markers for intervention and prognosis in ALI/ARDS, potentially offering new therapeutic strategies for lung diseases. Emerging data indicate that lung-resident megakaryocytes may give rise to platelets with distinct phenotypic and functional traits adapted to the pulmonary immune landscape. Investigating these differences could uncover localized regulatory mechanisms and aid in identifying platelet-influenced subtypes of ALI/ARDS.
Platelet-neutrophil aggregation plays a critical role in the inflammatory process of ALI/ARDS. Platelets recruit neutrophils to inflamed areas of the lung via the P-selectin-PSGL-1 axis. This interaction amplifies neutrophil-derived ROS and enhances the body’s inflammatory defense. Platelet activation plays a role in modulating NET formation, a process that, while beneficial in pathogen defense, may also drive immunothrombosis and tissue damage. Investigating how platelets influence NET dynamics could provide novel therapeutic approaches for ALI/ARDS, as previously described.
7.2 Advancing precision medicine in ARDS
Given the heterogeneity of ARDS, future studies should focus on identifying patient subgroups most likely to benefit from platelet-targeted interventions. Advances in molecular profiling and biomarker discovery could help refine precision medicine strategies, allowing for more individualized and effective therapeutic approaches (257). Additionally, ARDS subphenotyping has been shown to enhance clinical trial design, improving patient selection and minimizing variability in treatment responses (256). GWAS studies have further highlighted platelet-related pathways in ARDS, suggesting their potential as novel therapeutic targets (20). Integrating ARDS phenotyping into future clinical trials will be crucial for advancing precision medicine and improving patient outcomes.
7.3 Emerging therapeutic approaches for ARDS
The clinical management of ALI and ARDS has primarily focused on supportive care, including mechanical ventilation, fluid management, and nutritional support (265). However, there is a clear need for more targeted therapeutic approaches. While corticosteroids have been considered for their anti-inflammatory effects, their clinical use remains debated due to concerns over adverse effects such as delayed wound healing and increased infection risk.
Recent interest has emerged in using antiplatelet agents such as aspirin (ASA) to modulate platelet activity and neutrophil recruitment in ARDS. Clinical trials suggest that ASA may help alleviate ARDS symptoms by disrupting platelet-leukocyte interactions, though further studies are needed to clarify its full clinical benefit. Additionally, biological therapies targeting inflammatory cytokines, such as IL-6 inhibitors and TNF-α blockers, have demonstrated potential in reducing ARDS severity. Stem cell therapies represent another promising avenue, as they facilitate tissue repair and immune regulation, potentially accelerating inflammation resolution in ARDS (261, 266, 267).
Platelet-targeted therapies, designed to modulate platelet function and enhance their tissue-repairing roles, also hold significant promise (31, 252). Given the dual role of platelets in both fostering and resolving inflammation, optimizing the timing of platelet modulation in ALI/ARDS remains crucial. Some studies suggest that in later stages, platelet activity may support tissue repair, whereas early inhibition of platelet aggregation could help prevent further injury. Thus, a deeper understanding of the temporal dynamics of platelet activity is crucial to prevent inadvertent disruption of reparative immune processes. Future research should focus on stage-specific platelet behavior and determine optimal windows for intervention to enhance clinical outcomes while limiting potential adverse effects.
8 Conclusion
ALI/ARDS remains a challenging clinical syndrome with significant mortality and a lack of effective pharmacological treatments. Recent research has drawn attention to the multifaceted role of platelets- not only as contributors to inflammatory damage but also as regulators of immune resolution. Their interactions with neutrophils, endothelial cells, macrophages, and Tregs reveal a complex network that governs the progression and potential recovery of lung injury.
Recognizing the phase-specific functions of platelets, and how these differ among patient subgroups, may inform the design of more nuanced therapeutic approaches. The integration of platelet-related markers into ARDS subphenotyping frameworks holds promise for advancing individualized treatment. Further studies aimed at linking platelet biology to clinical phenotypes will be critical in translating these insights into practical strategies for care.
Author contributions
JY: Formal Analysis, Writing – original draft, Writing – review & editing. XZ: Resources, Writing – review & editing. XQ: Data curation, Writing – review & editing. MS: Conceptualization, Resources, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by grants from the Shanghai Science and Technology Committee (18140903400), and School of Integrative Medicine, Tianjin University of Traditional Chinese Medicine 2022 Faculty Research Initiation Fund (ZXYQDLX202201).
Acknowledgments
We thank all our laboratory members for their helpful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Li W, Li D, Chen Y, Abudou H, Wang H, Cai J, et al. Classic signaling pathways in alveolar injury and repair involved in sepsis-induced ALI/ARDS: new research progress and prospect. Dis Markers. (2022) 2022:6362344. doi: 10.1155/2022/6362344
2. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. (2016) 315:788–800. doi: 10.1001/jama.2016.0291
3. Dominguez G, Muralidharan O, Lee Him R, Harrison L, Vaivada T, Bhutta ZA. The care of preterm and term newborns with respiratory conditions: A systematic synthesis of evidence from low- and middle-income countries. Neonatology. (2025) 122:152–72. doi: 10.1159/000542482
4. Pirracchio R, Venkatesh B, Legrand M. Low-dose corticosteroids for critically ill adults with severe pulmonary infections: A review. Jama. (2024) 332:318–28. doi: 10.1001/jama.2024.6096
5. Zambon M, Vincent J-L. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest. (2008) 133:1120–7. doi: 10.1378/chest.07-2134
6. Bhadade R, De Souza R, Harde M, Khot A. Clinical characteristics and outcomes of patients with acute lung injury and ARDS. J postgraduate medicine. (2011) 57:286–90. doi: 10.4103/0022-3859.90077
7. Kaku S, Nguyen CD, Htet NN, Tutera D, Barr J, Paintal HS, et al. Acute respiratory distress syndrome: etiology, pathogenesis, and summary on management. J Intensive Care Med. (2020) 35:723–37. doi: 10.1177/0885066619855021
8. Lamers MM, Haagmans BL. SARS-coV-2 pathogenesis. Nat Rev microbiology. (2022) 20:270–84. doi: 10.1038/s41579-022-00713-0
9. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. (1967) 2:319–23. doi: 10.1016/S0140-6736(67)90168-7
10. Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. (1994) 149:818–24. doi: 10.1164/ajrccm.149.3.7509706
11. Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. Report of the American-European consensus conference on ARDS: definitions, mechanisms, relevant outcomes and clinical trial coordination. Consensus Committee. Intensive Care Med. (1994) 20:225–32. doi: 10.1007/BF01704707
12. Matthay MA, Arabi Y, Arroliga AC, Bernard G, Bersten AD, Brochard LJ, et al. A new global definition of acute respiratory distress syndrome. Am J Respiratory Crit Care Med. (2023) 209:37–47. doi: 10.1164/ajrccm-conference.2023.D16
13. Grasselli G, Calfee CS, Camporota L, Poole D, Amato MBP, Antonelli M, et al. ESICM guidelines on acute respiratory distress syndrome: definition, phenotyping and respiratory support strategies. Intensive Care Medicine. (2023) 49:727–59. doi: 10.1007/s00134-023-07050-7
14. Fernando SM, Ferreyro BL, Urner M, Munshi L, Fan E. Diagnosis and management of acute respiratory distress syndrome. CMAJ. (2021) 193:E761–E8. doi: 10.1503/cmaj.202661
15. Moss M, Huang DT, Brower RG, Ferguson ND, Ginde AA, Gong MN, et al. Early neuromuscular blockade in the acute respiratory distress syndrome. N Engl J Med. (2019) 380:1997–2008. doi: 10.1056/NEJMoa1901686
16. Albert RK. Constant vt ventilation and surfactant dysfunction: an overlooked cause of ventilator-induced lung injury. Am J Respiratory Crit Care Medicine. (2022) 205:152–60. doi: 10.1164/rccm.202107-1690CP
17. Smith BJ, Roy GS, Cleveland A, Mattson C, Okamura K, Charlebois CM, et al. Three alveolar phenotypes govern lung function in murine ventilator-induced lung injury. Front Physiol. (2020) 11:660. doi: 10.3389/fphys.2020.00660
18. Ma W, Tang S, Yao P, Zhou T, Niu Q, Liu P, et al. Advances in acute respiratory distress syndrome: focusing on heterogeneity, pathophysiology, and therapeutic strategies. Signal transduction targeted Ther. (2025) 10:75. doi: 10.1038/s41392-025-02127-9
19. Cai Y, Shang L, Zhou F, Zhang M, Li J, Wang S, et al. Macrophage pyroptosis and its crucial role in ALI/ARDS. Front Immunol. (2025) 16:1530849. doi: 10.3389/fimmu.2025.1530849
20. Tejera P, Christiani DC. Deconstructing ARDS variability: platelet count, an ARDS intermediate phenotype and novel mediator of genetic effects in ARDS. Semin Respir Crit Care Med. (2019) 40:12–8. doi: 10.1055/s-0039-1683891
21. Shankar-Hari M, Rubenfeld GD. Population enrichment for critical care trials: phenotypes and differential outcomes. Curr Opin Crit Care. (2019) 25:489–97. doi: 10.1097/MCC.0000000000000641
22. Bos LDJ, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet. (2022) 400:1145–56. doi: 10.1016/S0140-6736(22)01485-4
23. Zhou H, Fan EK, Fan J. Cell-cell interaction mechanisms in acute lung injury. Shock (Augusta Ga). (2021) 55:167–76. doi: 10.1097/SHK.0000000000001598
24. Repsold L, Joubert AM. Platelet function, role in thrombosis, inflammation, and consequences in chronic myeloproliferative disorders. Cells. (2021) 10:3034. doi: 10.3390/cells10113034
25. Denorme F, Campbell RA. Platelets net neutrophils during ALI. Blood. (2023) 142:1409–10. doi: 10.1182/blood.2023021641
26. Granja T, Köhler D, Tang L, Burkard P, Eggstein C, Hemmen K, et al. Semaphorin 7A coordinates neutrophil response during pulmonary inflammation and sepsis. Blood Adv. (2024) 8:2660–74. doi: 10.1182/bloodadvances.2023011778
27. Morrell CN, Pariser DN, Hilt ZT, Vega Ocasio D. The platelet napoleon complex-small cells, but big immune regulatory functions. Annu Rev Immunol. (2019) 37:125–44. doi: 10.1146/annurev-immunol-042718-041607
28. Xin Y, Peng J, Hong YY, Chao QC, Na S, Pan S, et al. Advances in research on the effects of platelet activation in acute lung injury (Review). BioMed Rep. (2022) 16:17. doi: 10.3892/br.2022.1500
29. Sharma S, Tyagi T, Antoniak S. Platelet in thrombo-inflammation: Unraveling new therapeutic targets. Front Immunol. (2022) 13:1039843. doi: 10.3389/fimmu.2022.1039843
30. Ludwig N, Hilger A, Zarbock A, Rossaint J. Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation. Cells. (2022) 11:1957. doi: 10.3390/cells11121957
31. Hua T, Zhang G, Yao Y, Jia H, Liu W. Research progress of megakaryocytes and platelets in lung injury. Ann medicine. (2024) 56:2362871. doi: 10.1080/07853890.2024.2362871
32. Polidoro RB, Hagan RS, de Santis Santiago R, Schmidt NW. Overview: systemic inflammatory response derived from lung injury caused by SARS-coV-2 infection explains severe outcomes in COVID-19. Front Immunol. (2020) 11:1626. doi: 10.3389/fimmu.2020.01626
33. Middleton EA, Rondina MT, Schwertz H, Zimmerman GA. Amicus or adversary revisited: platelets in acute lung injury and acute respiratory distress syndrome. Am J Respir Cell Mol Biol. (2018) 59:18–35. doi: 10.1165/rcmb.2017-0420TR
34. Sandeep B, Xiao Z, Zhao F, Feng Q, Gao K. Role of platelets in acute lung injury after extracorporeal circulation in cardiac surgery patients: A systemic review. Curr Probl Cardiol. (2022) 47:101088. doi: 10.1016/j.cpcardiol.2021.101088
35. Suresh MV, Francis S, Aktay S, Kralovich G, Raghavendran K. Therapeutic potential of curcumin in ARDS and COVID-19. Clin Exp Pharmacol Physiol. (2023) 50:267–76. doi: 10.1111/1440-1681.13744
36. Brannon ER, Kelley WJ, Newstead MW, Banka AL, Uhrich KE, O’Connor CE, et al. Polysalicylic acid polymer microparticle decoys therapeutically treat acute respiratory distress syndrome. Adv Healthc Mater. (2022) 11:e2101534. doi: 10.1002/adhm.202101534
37. Burkard P, Schonhart C, Vögtle T, Köhler D, Tang L, Johnson D, et al. A key role for platelet GPVI in neutrophil recruitment, migration, and NETosis in the early stages of acute lung injury. Blood. (2023) 142:1463–77. doi: 10.1182/blood.2023019940
38. Zoulikha M, Xiao Q, Boafo GF, Sallam MA, Chen Z, He W. Pulmonary delivery of siRNA against acute lung injury/acute respiratory distress syndrome. Acta Pharm Sin B. (2022) 12:600–20. doi: 10.1016/j.apsb.2021.08.009
39. Zarbock A, Ley K. The role of platelets in acute lung injury (ALI). Front bioscience: A J virtual library. (2009) 14:150. doi: 10.2741/3236
40. Jin H, Zhao Y, Yao Y, Zhao J, Luo R, Fan S, et al. Therapeutic effects of tea polyphenol-loaded nanoparticles coated with platelet membranes on LPS-induced lung injury. Biomater Sci. (2023) 11:6223–35. doi: 10.1039/D3BM00802A
41. Holinstat M. Normal platelet function. Cancer Metastasis Rev. (2017) 36:195–8. doi: 10.1007/s10555-017-9677-x
42. Bain W, Olonisakin T, Yu M, Qu Y, Hulver M, Xiong Z, et al. Platelets inhibit apoptotic lung epithelial cell death and protect mice against infection-induced lung injury. Blood Advances. (2019) 3:432–45. doi: 10.1182/bloodadvances.2018026286
43. Liu C, Xiao K, Xie L. Advances in the regulation of macrophage polarization by mesenchymal stem cells and implications for ALI/ARDS treatment. Front Immunol. (2022) 13:928134. doi: 10.3389/fimmu.2022.928134
44. Hotta E, Tamagawa-Mineoka R, Katoh N. Platelets are important for the development of immune tolerance: Possible involvement of TGF-β in the mechanism. Exp Dermatol. (2019) 28:801–8. doi: 10.1111/exd.2019.28.issue-7
45. Stone AP, Nascimento TF, BarraChina MN. The bone marrow niche from the inside out: how megakaryocytes are shaped by and shape hematopoiesis. Blood. (2022) 139:483–91. doi: 10.1182/blood.2021012827
46. Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. (2017) 544:105–9. doi: 10.1038/nature21706
47. Sun S, Qiao B, Han Y, Wang B, Wei S, Chen Y. Posttranslational modifications of platelet adhesion receptors. Pharmacol Res. (2022) 183:106413. doi: 10.1016/j.phrs.2022.106413
48. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: an update. Eur Heart J. (2017) 38:785–91. doi: 10.1093/eurheartj/ehw550
49. Grüner S, Prostredna M, Schulte V, Krieg T, Eckes B, Brakebusch C, et al. Multiple integrin-ligand interactions synergize in shear-resistant platelet adhesion at sites of arterial injury in vivo. Blood. (2003) 102:4021–7. doi: 10.1182/blood-2003-05-1391
50. Zhang Z, Xiyuan F, Zhou X, Zhou X, Cheng Z, Hu Y. Platelet disorders and medication strategies. Thromb haemostasis. (2025) 142:1413–25. doi: 10.1055/a-2561-8818
51. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. (2018) 122:337–51. doi: 10.1161/CIRCRESAHA.117.310795
52. Tokarz-Deptuła B, Palma J, Baraniecki Ł, Stosik M, Kołacz R, Deptuła W. What function do platelets play in inflammation and bacterial and viral infections? Front In Immunol. (2021) 12:770436. doi: 10.3389/fimmu.2021.770436
53. Mandel J, Casari M, Stepanyan M, Martyanov A, Deppermann C. Beyond hemostasis: platelet innate immune interactions and thromboinflammation. Int J Mol Sci. (2022) 23:3868. doi: 10.3390/ijms23073868
54. Min Y, Hao L, Liu X, Tan S, Song H, Ni H, et al. Platelets fine-tune effector responses of naïve CD4+ T cells via platelet factor 4-regulated transforming growth factor β signaling. Cell Mol Life Sci. (2022) 79:247. doi: 10.1007/s00018-022-04279-1
55. Estevez B, Du X. New concepts and mechanisms of platelet activation signaling. Physiol (Bethesda Md). (2017) 32:162–77. doi: 10.1152/physiol.00020.2016
56. Gremmel T, Frelinger AL 3rd, Michelson AD. Platelet Physiology. Seminars Thrombosis Hemostasis. (2024) 50:1173–86. doi: 10.1055/s-0044-1786387
57. Perrella G, Huang J, Provenzale I, Swieringa F, Heubel-Moenen FCJI, Farndale RW, et al. Nonredundant roles of platelet glycoprotein VI and integrin αIIbβ3 in fibrin-mediated microthrombus formation. Arterioscler Thromb Vasc Biol. (2021) 41:e97-e111. doi: 10.1161/ATVBAHA.120.314641
58. Quach ME. GPIb-IX-V and platelet clearance. Platelets. (2022) 33:817–22. doi: 10.1080/09537104.2021.1942815
59. Saadalla A, Seheult J, Pruthi RK, Chen D. Von willebrand factor multimer analysis and classification: A comprehensive review and updates. Semin Thromb Hemost. (2023) 49:580–91. doi: 10.1055/s-0042-1757183
60. Marin Oyarzún CP, Heller PG. Platelets as mediators of thromboinflammation in chronic myeloproliferative neoplasms. Front Immunol. (2019) 10:1373. doi: 10.3389/fimmu.2019.01373
61. Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit Care medicine. (2002) 30:S294–301. doi: 10.1097/00003246-200205001-00020
62. Kim HN, Ruan Y, Ogana H, Kim Y-M. Cadherins, selectins, and integrins in CAM-DR in leukemia. Front Oncology. (2020) 10:592733. doi: 10.3389/fonc.2020.592733
63. Dehghani T, Panitch A. Endothelial cells, neutrophils and platelets: getting to the bottom of an inflammatory triangle. Open Biol. (2020) 10:200161. doi: 10.1098/rsob.200161
64. Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets. (2021) 32:314–24. doi: 10.1080/09537104.2020.1817360
65. Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Investigation. (2006) 116:3211–9. doi: 10.1172/JCI29499
66. Niklaus M, Klingler P, Weber K, Koessler A, Boeck M, Kobsar A, et al. The involvement of toll-like receptors 2 and 4 in human platelet signalling pathways. Cell Signal. (2020) 76:109817. doi: 10.1016/j.cellsig.2020.109817
67. Jacob S, Kosaka Y, Bhatlekar S, Denorme F, Benzon H, Moody A, et al. Mitofusin-2 regulates platelet mitochondria and function. Circ Res. (2024) 134:143–61. doi: 10.1161/CIRCRESAHA.123.322914
68. Ma Y, Jiang Q, Yang B, Hu X, Shen G, Shen W, et al. Platelet mitochondria, a potent immune mediator in neurological diseases. Front Physiol. (2023) 14:1210509. doi: 10.3389/fphys.2023.1210509
69. Quach ME, Li R. Structure-function of platelet glycoprotein Ib-IX. J Thromb Haemostasis: JTH. (2020) 18:3131–41. doi: 10.1111/jth.15035
70. Jurk K, Kehrel BE. Platelets: physiology and biochemistry. Semin Thromb Hemost. (2024) 50:794–803. doi: 10.1055/s-0043-1777305
71. Perrella G, Nagy M, Watson SP, Heemskerk JW. Platelet GPVI (glycoprotein VI) and thrombotic complications in the venous system. Arteriosclerosis thrombosis Vasc Biol. (2021) 41:2681–92. doi: 10.1161/ATVBAHA.121.316108
72. Alenazy FO, Thomas MR. Novel antiplatelet targets in the treatment of acute coronary syndromes. Platelets. (2021) 32:15–28. doi: 10.1080/09537104.2020.1763731
73. Cruz MA, Diacovo TG, Emsley J, Liddington R, Handin RI. Mapping the glycoprotein Ib-binding site in the von willebrand factor A1 domain. J Biol Chem. (2000) 275:19098–105. doi: 10.1074/jbc.M002292200
74. Cleary SJ, Hobbs C, Amison RT, Arnold S, O’Shaughnessy BG, Lefrançais E, et al. LPS-induced lung platelet recruitment occurs independently from neutrophils, PSGL-1, and P-selectin. Am J Respiratory Cell Mol Biol. (2019) 61:232–43. doi: 10.1165/rcmb.2018-0182OC
75. Kim KK, Sheppard D, Chapman HA. TGF-β1 signaling and tissue fibrosis. Cold Spring Harbor Perspect In Biol. (2018) 10:a022293. doi: 10.1101/cshperspect.a022293
76. Tang X, Xu Q, Yang S, Huang X, Wang L, Huang F, et al. Toll-like receptors and thrombopoiesis. Int J Mol Sci. (2023) 24:1010. doi: 10.3390/ijms24021010
77. Maouia A, Rebetz J, Kapur R, Semple JW. The immune nature of platelets revisited. Transfusion Med Rev. (2020) 34:209–20. doi: 10.1016/j.tmrv.2020.09.005
78. Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, et al. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ Res. (2013) 112:103–12. doi: 10.1161/CIRCRESAHA.112.274241
79. Zaid Y, Guessous F, Puhm F, Elhamdani W, Chentoufi L, Morris AC, et al. Platelet reactivity to thrombin differs between patients with COVID-19 and those with ARDS unrelated to COVID-19. Blood Advances. (2021) 5:635–9. doi: 10.1182/bloodadvances.2020003513
80. Bordon Y. Platelet-derived 5-HIAA helps neutrophils enter tissue. Nat Rev Immunol. (2022) 22:206–7. doi: 10.1038/s41577-022-00699-z
81. van der Meijden PEJ, Feijge MAH, Giesen PLA, Huijberts M, van Raak LPM, Heemskerk JWM. Platelet P2Y12 receptors enhance signalling towards procoagulant activity and thrombin generation. A study with healthy subjects and patients at thrombotic risk. Thromb Haemostasis. (2005) 93:1128–36. doi: 10.1160/TH04-09-0597
82. Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Investigation. (2013) 123:908–16. doi: 10.1172/JCI65154
83. Heijnen H, van der Sluijs P. Platelet secretory behaviour: as diverse as the granules … or not? J Thromb Haemostasis: JTH. (2015) 13:2141–51. doi: 10.1111/jth.13147
84. Gray AL, Karlsson R, Roberts ARE, Ridley AJL, Pun N, Khan B, et al. Chemokine CXCL4 interactions with extracellular matrix proteoglycans mediate widespread immune cell recruitment independent of chemokine receptors. Cell Rep. (2023) 42:111930. doi: 10.1016/j.celrep.2022.111930
85. Cambier S, Gouwy M, Proost P. The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell Mol Immunol. (2023) 20:217–51. doi: 10.1038/s41423-023-00974-6
86. Ferrero MR, Tavares LP, Garcia CC. The dual role of CCR5 in the course of influenza infection: exploring treatment opportunities. Front In Immunol. (2021) 12:826621. doi: 10.3389/fimmu.2021.826621
87. Nguyen SN, Le SH, Ivanov DG, Ivetic N, Nazy I, Kaltashov IA. Structural characterization of a pathogenic antibody underlying vaccine-induced immune thrombotic thrombocytopenia (VITT). Anal Chem. (2024) 96:6209–17. doi: 10.1021/acs.analchem.3c05253
88. Golebiewska EM, Poole AW. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. (2015) 29:153–62. doi: 10.1016/j.blre.2014.10.003
89. Yang Z, Chen Z, Chen L, Xiong G. 5-Hydroxyindoleacetic acid, a new ligand for GPR35, plays an important role in inflammatory disease: 5-Hydroxyindoleacetic acid in inflammatory disease. Acta Biochim Biophys Sinica. (2022) 55:169. doi: 10.3724/abbs.2022198
90. De Giovanni M, Chen H, Li X, Cyster JG. GPR35 and mediators from platelets and mast cells in neutrophil migration and inflammation. Immunological Rev. (2023) 317:187–202. doi: 10.1111/imr.13194
91. Yadav S, Storrie B. The cellular basis of platelet secretion: Emerging structure/function relationships. Platelets. (2017) 28:108–18. doi: 10.1080/09537104.2016.1257786
92. Macaulay IC, Carr P, Gusnanto A, Ouwehand WH, Fitzgerald D, Watkins NA. Platelet genomics and proteomics in human health and disease. J Clin Investigation. (2005) 115:3370–7. doi: 10.1172/JCI26885
93. Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. (2005) 122:379–91. doi: 10.1016/j.cell.2005.06.015
94. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood. (2020) 136:1317–29. doi: 10.1182/blood.2020007214
95. French SL, Butov KR, Allaeys I, Canas J, Morad G, Davenport P, et al. Platelet-derived extracellular vesicles infiltrate and modify the bone marrow during inflammation. Blood Advances. (2020) 4:3011–23. doi: 10.1182/bloodadvances.2020001758
96. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. (2018) 7:1535750. doi: 10.1080/20013078.2018.1535750
97. Aatonen M, Grönholm M, Siljander PRM. Platelet-derived microvesicles: multitalented participants in intercellular communication. Semin Thromb Hemost. (2012) 38:102–13. doi: 10.1055/s-0031-1300956
98. Ratajczak MZ. Megakaryocyte-derived microvesicles, please stand up! Blood. (2009) 113:981–2. doi: 10.1182/blood-2008-10-182964
99. Boilard E. Extracellular vesicles and their content in bioactive lipid mediators: more than a sack of microRNA. J Lipid Res. (2018) 59:2037–46. doi: 10.1194/jlr.R084640
100. Lopez E, Srivastava AK, Pati S, Holcomb JB, Wade CE. Platelet-derived microvesicles: A potential therapy for trauma-induced coagulopathy. Shock (Augusta Ga). (2018) 49:243–8. doi: 10.1097/SHK.0000000000000974
101. Scozzi D, Liao F, Krupnick AS, Kreisel D, Gelman AE. The role of neutrophil extracellular traps in acute lung injury. Front Immunol. (2022) 13:953195. doi: 10.3389/fimmu.2022.953195
102. Wienkamp A-K, Erpenbeck L, Rossaint J. Platelets in the NETworks interweaving inflammation and thrombosis. Front In Immunol. (2022) 13:953129. doi: 10.3389/fimmu.2022.953129
103. Kim S-J, Carestia A, McDonald B, Zucoloto AZ, Grosjean H, Davis RP, et al. Platelet-mediated NET release amplifies coagulopathy and drives lung pathology during severe influenza infection. Front In Immunol. (2021) 12:772859. doi: 10.3389/fimmu.2021.772859
104. Kornerup KN, Salmon GP, Pitchford SC, Liu WL, Page CP. Circulating platelet-neutrophil complexes are important for subsequent neutrophil activation and migration. J Appl Physiol (Bethesda Md: 1985). (2010) 109:758–67. doi: 10.1152/japplphysiol.01086.2009
105. Grommes J, Alard JE, Drechsler M, Wantha S, Mörgelin M, Kuebler WM, et al. Expression of concern: disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respiratory Crit Care Medicine. (2020) 202:1199. doi: 10.1164/rccm.v202eoc1
106. Rossaint J, Herter JM, Van Aken H, Napirei M, Döring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood. (2014) 123:2573–84. doi: 10.1182/blood-2013-07-516484
107. Silva IS, Almeida AD, Lima Filho ACM, Fernandes-Braga W, Barra A, Oliveira HMC, et al. Platelet-activating factor and protease-activated receptor 2 cooperate to promote neutrophil recruitment and lung inflammation through nuclear factor-kappa B transactivation. Sci Rep. (2023) 13:21637. doi: 10.1038/s41598-023-48365-1
108. Kaminski TW, Brzoska T, Li X, Vats R, Katoch O, Dubey RK, et al. Lung microvascular occlusion by platelet-rich neutrophil-platelet aggregates promotes cigarette smoke-induced severe flu. JCI Insight. (2024) 9:e167299. doi: 10.1172/jci.insight.167299
109. Goldsmith HL, Spain S. Radial distribution of white cells in tube flow. Kroc Found Ser. (1984) 16:131–46.
110. Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Dynamics of leukocyte-platelet adhesion in whole blood. Blood. (1991) 78:1730–7. doi: 10.1182/blood.V78.7.1730.1730
111. Peters MJ, Heyderman RS, Hatch DJ, Klein NJ. Investigation of platelet-neutrophil interactions in whole blood by flow cytometry. J Immunol Methods. (1997) 209:125–35. doi: 10.1016/S0022-1759(97)00139-7
112. Zuchtriegel G, Uhl B, Puhr-Westerheide D, Pörnbacher M, Lauber K, Krombach F, et al. Platelets guide leukocytes to their sites of extravasation. PloS Biol. (2016) 14:e1002459. doi: 10.1371/journal.pbio.1002459
113. Sun W, Wang S, Zhang J, Arias K, Griffith BP, Wu ZJ. Neutrophil injury and function alterations induced by high mechanical shear stress with short exposure time. Artif organs. (2021) 45:577–86. doi: 10.1111/aor.13874
114. Lam FW, Burns AR, Smith CW, Rumbaut RE. Platelets enhance neutrophil transendothelial migration via P-selectin glycoprotein ligand-1. Am J Physiology-Heart Circulatory Physiol. (2011) 300:H468–H75. doi: 10.1152/ajpheart.00491.2010
115. Schrottmaier WC, Kral-Pointner JB, Salzmann M, Mussbacher M, Schmuckenschlager A, Pirabe A, et al. Platelet p110β mediates platelet-leukocyte interaction and curtails bacterial dissemination in pneumococcal pneumonia. Cell Rep. (2022) 41:111614. doi: 10.1016/j.celrep.2022.111614
116. Bordon J, Aliberti S, Fernandez-Botran R, Uriarte SM, Rane MJ, Duvvuri P, et al. Understanding the roles of cytokines and neutrophil activity and neutrophil apoptosis in the protective versus deleterious inflammatory response in pneumonia. Int J Infect diseases: IJID. (2013) 17:e76–83. doi: 10.1016/j.ijid.2012.06.006
117. Ye S, Ma L, Chi Y, Liu N, Liu Y, Wei W, et al. Targeting neutrophil dysfunction in acute lung injury: Insights from active components of Chinese medicine. Phytomedicine. (2025) 141:156664. doi: 10.1016/j.phymed.2025.156664
118. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxidants Redox Signaling. (2014) 20:1126–67. doi: 10.1089/ars.2012.5149
119. Wetterö J, Tengvall P, Bengtsson T. Platelets stimulated by IgG-coated surfaces bind and activate neutrophils through a selectin-dependent pathway. Biomaterials. (2003) 24:1559–73. doi: 10.1016/S0142-9612(02)00543-4
120. Ravindran M, Khan MA, Palaniyar N. Neutrophil extracellular trap formation: physiology, pathology, and pharmacology. Biomolecules. (2019) 9:365. doi: 10.3390/biom9080365
121. Assinger A, Laky M, Schabbauer G, Hirschl A, Buchberger E, Binder B, et al. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J Thromb Haemostasis. (2011) 9:799–809. doi: 10.1111/j.1538-7836.2011.04193.x
122. Seki R, Nishizawa K. Inflammatory vs. thrombotic states of platelets: a mini-review with a focus on functional dichotomy. Ann BioMed Res. (2018) 1:107. doi: 10.61545/ABR-1-107
123. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. (2021) 61:194–211. doi: 10.1007/s12016-020-08804-7
124. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
125. Sørensen OE, Borregaard N. Neutrophil extracellular traps - the dark side of neutrophils. J Clin Investigation. (2016) 126:1612–20. doi: 10.1172/JCI84538
126. Parker HA, Jones HM, Kaldor CD, Hampton MB, Winterbourn CC. Neutrophil NET formation with microbial stimuli requires late stage NADPH oxidase activity. Antioxidants. (2021) 10:1791. doi: 10.3390/antiox10111791
127. Stojkov D, Claus MJ, Kozlowski E, Oberson K, Schären OP, Benarafa C, et al. NET formation is independent of gasdermin D and pyroptotic cell death. Sci Signaling. (2023) 16:eabm0517. doi: 10.1126/scisignal.abm0517
128. Kaiser R, Escaig R, Erber J, Nicolai L. Neutrophil-platelet interactions as novel treatment targets in cardiovascular disease. Front Cardiovasc medicine. (2022) 8:824112. doi: 10.3389/fcvm.2021.824112
129. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
130. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemostasis: JTH. (2014) 12:2074–88. doi: 10.1111/jth.12710
131. Nicolai L, Pekayvaz K, Massberg S. Platelets: Orchestrators of immunity in host defense and beyond. Immunity. (2024) 57:957–72. doi: 10.1016/j.immuni.2024.04.008
132. Markin SS, Lapshin RD, Baskina OS, Korotchenko SA, Mukhina IV, Ivanov SV, et al. Nebulized non-immunogenic staphylokinase in the mice acute lung injury model. Int J Mol Sci. (2022) 23:9307. doi: 10.3390/ijms23169307
133. Akpınar S, Oran M, Doğan M, Çelikkol A, Erdem I, Turgut B. The role of oxidized phospholipids in COVID-19-associated hypercoagulopathy. Eur Rev Med Pharmacol Sci. (2021) 25:5304–9. doi: 10.26355/eurrev_202108_26551
134. Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemostasis: JTH. (2020) 18:1421–4. doi: 10.1111/jth.14830
135. Zhao J, Xu X, Gao Y, Yu Y, Li C. Crosstalk between platelets and SARS-coV-2: implications in thrombo-inflammatory complications in COVID-19. Int J Mol Sci. (2023) 24:14133. doi: 10.3390/ijms241814133
136. Wadowski PP, Panzer B, Józkowicz A, Kopp CW, Gremmel T, Panzer S, et al. Microvascular thrombosis as a critical factor in severe COVID-19. Int J Mol Sci. (2023) 24:2492. doi: 10.3390/ijms24032492
137. Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. (2020) 11:1722. doi: 10.3389/fimmu.2020.01722
138. Mowery NT, Terzian WH, Nelson AC. Acute lung injury. Curr problems surgery. (2020) 57:100777. doi: 10.1016/j.cpsurg.2020.100777
139. Bissonnette EY, Lauzon-Joset J-F, Debley JS, Ziegler SF. Cross-talk between alveolar macrophages and lung epithelial cells is essential to maintain lung homeostasis. Front Immunol. (2020) 11:583042. doi: 10.3389/fimmu.2020.583042
140. Alharbi KS, Fuloria NK, Fuloria S, Rahman SB, Al-Malki WH, Shaikh MAJ, et al. Nuclear factor-kappa B and its role in inflammatory lung disease. Chemico-biological interactions. (2021) 345:109568. doi: 10.1016/j.cbi.2021.109568
141. Chi G, Wei M, Xie X, Soromou L, Liu F, Zhao S. Suppression of MAPK and NF-κB pathways by limonene contributes to attenuation of lipopolysaccharide-induced inflammatory responses in acute lung injury. Inflammation. (2013) 36:501–11. doi: 10.1007/s10753-012-9571-1
142. Price DR, Garcia JGN. A razor’s edge: vascular responses to acute inflammatory lung injury/acute respiratory distress syndrome. Annu Rev Physiol. (2024) 86:505–29. doi: 10.1146/annurev-physiol-042222-030731
143. Qiao X, Yin J, Zheng Z, Li L, Feng X. Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: pathogenesis and therapeutic implications. Cell Commun Signal. (2024) 22:241. doi: 10.1186/s12964-024-01620-y
144. Rizzo AN, Schmidt EP. The role of the alveolar epithelial glycocalyx in acute respiratory distress syndrome. Am J Physiol Cell Physiol. (2023) 324:C799–806. doi: 10.1152/ajpcell.00555.2022
145. Dong J, Liu W, Liu W, Wen Y, Liu Q, Wang H, et al. Acute lung injury: a view from the perspective of necroptosis. Inflammation Res. (2024) 73:997–1018. doi: 10.1007/s00011-024-01879-4
146. Ren J, Deng G, Li R, Jin X, Liu J, Li J, et al. Possible pharmacological targets and mechanisms of sivelestat in protecting acute lung injury. Comput Biol Med. (2024) 170:108080. doi: 10.1016/j.compbiomed.2024.108080
147. Tuckermann J, Adams RH. The endothelium-bone axis in development, homeostasis and bone and joint disease. Nat Rev Rheumatol. (2021) 17:608–20. doi: 10.1038/s41584-021-00682-3
148. Lallo V, Bracaglia LG. Influencing endothelial cells’ Roles in inflammation and wound healing through nucleic acid delivery. Tissue Eng Part A. (2024) 30:272–86. doi: 10.1089/ten.tea.2023.0296
149. Osorio-Valencia S, Zhou B. Roles of macrophages and endothelial cells and their crosstalk in acute lung injury. Biomedicines. (2024) 12:632. doi: 10.3390/biomedicines12030632
150. Eltayeb A, Al-Sarraj F, Alharbi M, Albiheyri R, Mattar EH, Abu Zeid IM, et al. Intrinsic factors behind long COVID: IV. Hypothetical roles of the SARS-CoV-2 nucleocapsid protein and its liquid-liquid phase separation. J Cell Biochem. (2024) 125:e30530. doi: 10.1002/jcb.30530
151. Yuan HX, Chen YT, Li YQ, Wang YS, Ou ZJ, Li Y, et al. Endothelial extracellular vesicles induce acute lung injury via follistatin-like protein 1. Sci China Life Sci. (2024) 67:475–87. doi: 10.1007/s11427-022-2328-x
152. Qian Y, Wang Z, Lin H, Lei T, Zhou Z, Huang W, et al. TRIM47 is a novel endothelial activation factor that aggravates lipopolysaccharide-induced acute lung injury in mice via K63-linked ubiquitination of TRAF2. Signal Transduct Target Ther. (2022) 7:148. doi: 10.1038/s41392-022-00953-9
153. Camacho M, López-Belmonte J, Vila L. Rate of vasoconstrictor prostanoids released by endothelial cells depends on cyclooxygenase-2 expression and prostaglandin I synthase activity. Circ Res. (1998) 83:353–65. doi: 10.1161/01.RES.83.4.353
154. Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. (2005) 106:2334–9. doi: 10.1182/blood-2005-04-1530
155. Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood. (2010) 115:3632–9. doi: 10.1182/blood-2009-11-252643
156. Coenen DM, Mastenbroek TG, Cosemans JMEM. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood. (2017) 130:2819–28. doi: 10.1182/blood-2017-04-780825
157. Borek I, Birnhuber A, Voelkel NF, Marsh LM, Kwapiszewska G. The vascular perspective on acute and chronic lung disease. J Clin Investigation. (2023) 133:e170502. doi: 10.1172/JCI170502
158. Maneta E, Aivalioti E, Tual-Chalot S, Emini Veseli B, Gatsiou A, Stamatelopoulos K, et al. Endothelial dysfunction and immunothrombosis in sepsis. Front In Immunol. (2023) 14:1144229. doi: 10.3389/fimmu.2023.1144229
159. Krott KJ, Feige T, Elvers M. Flow chamber analyses in cardiovascular research: impact of platelets and the intercellular crosstalk with endothelial cells, leukocytes, and red blood cells. Hamostaseologie. (2023) 43:338–47. doi: 10.1055/a-2113-1134
160. Kaiser R, Escaig R, Nicolai L. Hemostasis without clot formation–how platelets guard the vasculature in inflammation, infection, and Malignancy. Blood. (2023). doi: 10.1182/blood.2023020535
161. Page C, Pitchford S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int immunopharmacology. (2013) 17:1176–84. doi: 10.1016/j.intimp.2013.06.004
162. Hook JS, Cao M, Potera RM, Alsmadi NZ, Schmidtke DW, Moreland JG. Nox2 regulates platelet activation and NET formation in the lung. Front Immunol. (2019) 10:1472. doi: 10.3389/fimmu.2019.01472
163. NaveenKumar SK, Hemshekhar M, Sharathbabu BN, Kemparaju K, Mugesh G, Girish KS. Platelet activation and ferroptosis mediated NETosis drives heme induced pulmonary thrombosis. Biochim Biophys Acta Mol Basis Dis. (2023) 1869:166688. doi: 10.1016/j.bbadis.2023.166688
164. Zhang H, Zhou Y, Qu M, Yu Y, Chen Z, Zhu S, et al. Tissue factor-enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis-induced lung injury. Front Cell Infect Microbiol. (2021) 11:677902. doi: 10.3389/fcimb.2021.677902
165. Scherlinger M, Richez C, Tsokos GC, Boilard E, Blanco P. Author Correction: The role of platelets in immune-mediated inflammatory diseases. Nat Rev Immunol. (2023) 23:409. doi: 10.1038/s41577-023-00869-7
166. Carestia A, Kaufman T, Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front In Immunol. (2016) 7:271. doi: 10.3389/fimmu.2016.00271
167. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep. (2018) 8:2068. doi: 10.1038/s41598-018-20479-x
168. Zha C, Zhang W, Gao F, Xu J, Jia R, Cai J, et al. Anti-β2GPI/β2GPI induces neutrophil extracellular traps formation to promote thrombogenesis via the TLR4/MyD88/MAPKs axis activation. Neuropharmacology. (2018) 138:140–50. doi: 10.1016/j.neuropharm.2018.06.001
169. Liu Y, Wang R, Song C, Ding S, Zuo Y, Yi K, et al. Crosstalk between neutrophil extracellular traps and immune regulation: insights into pathobiology and therapeutic implications of transfusion-related acute lung injury. Front In Immunol. (2023) 14:1324021. doi: 10.3389/fimmu.2023.1324021
170. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Investigation. (2012) 122:2661–71. doi: 10.1172/JCI61303
171. Zhu S, Yu Y, Qu M, Qiu Z, Zhang H, Miao C, et al. Neutrophil extracellular traps contribute to immunothrombosis formation via the STING pathway in sepsis-associated lung injury. Cell Death Discov. (2023) 9:315. doi: 10.1038/s41420-023-01614-8
172. Colicchia M, Perrella G, Gant P, Rayes J. Novel mechanisms of thrombo-inflammation during infection: spotlight on neutrophil extracellular trap-mediated platelet activation. Res Pract Thromb Haemost. (2023) 7:100116. doi: 10.1016/j.rpth.2023.100116
173. Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol. (2014) 76:467–92. doi: 10.1146/annurev-physiol-021113-170408
174. Kourtzelis I, Hajishengallis G, Chavakis T. Phagocytosis of apoptotic cells in resolution of inflammation. Front In Immunol. (2020) 11:553. doi: 10.3389/fimmu.2020.00553
175. Panigrahy D, Gilligan MM, Serhan CN, Kashfi K. Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol Ther. (2021) 227:107879. doi: 10.1016/j.pharmthera.2021.107879
176. Leung G, Middleton EA. The role of platelets and megakaryocytes in sepsis and ARDS. J Physiol. (2024) 602:6047–63. doi: 10.1113/tjp.v602.22
177. Yadav H, Kor DJ. Platelets in the pathogenesis of acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L915–23. doi: 10.1152/ajplung.00266.2015
178. Cleary SJ, Conrad C. Investigating and imaging platelets in inflammation. Int J Biochem Cell Biol. (2023) 157:106373. doi: 10.1016/j.biocel.2023.106373
179. Guan T, Zhou X, Zhou W, Lin H. Regulatory T cell and macrophage crosstalk in acute lung injury: future perspectives. Cell Death Discov. (2023) 9:9. doi: 10.1038/s41420-023-01310-7
180. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
181. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
182. Austermann J, Roth J, Barczyk-Kahlert K. The good and the bad: monocytes’ and macrophages’ Diverse functions in inflammation. Cells. (2022) 11:1979. doi: 10.3390/cells11121979
183. Song C, Li H, Li Y, Dai M, Zhang L, Liu S, et al. NETs promote ALI/ARDS inflammation by regulating alveolar macrophage polarization. Exp Cell Res. (2019) 382:111486. doi: 10.1016/j.yexcr.2019.06.031
184. Carestia A, Mena HA, Olexen CM, Wilczynski JMO, Negrotto S, Errasti AE, et al. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell reports. (2019) 28:896–908.e5. doi: 10.1016/j.celrep.2019.06.062
185. Lechner AJ, Driver IH, Lee J, Conroy CM, Nagle A, Locksley RM, et al. Recruited monocytes and type 2 immunity promote lung regeneration following pneumonectomy. Cell Stem Cell. (2017) 21:120–34.e7. doi: 10.1016/j.stem.2017.03.024
186. Noone PM, Reddy SP. Recent advances in dead cell clearance during acute lung injury and repair. Fac Rev. (2021) 10:33. doi: 10.12703/r/10-33
187. Jiao Y, Zhang T, Liu M, Zhou L, Qi M, Xie X, et al. Exosomal PGE2 from M2 macrophages inhibits neutrophil recruitment and NET formation through lipid mediator class switching in sepsis. J BioMed Sci. (2023) 30:62. doi: 10.1186/s12929-023-00957-9
188. Hou F, Xiao K, Tang L, Xie L. Diversity of macrophages in lung homeostasis and diseases. Front In Immunol. (2021) 12:753940. doi: 10.3389/fimmu.2021.753940
189. Kulikauskaite J, Wack A. Teaching old dogs new tricks? The plasticity of lung alveolar macrophage subsets. Trends Immunol. (2020) 41:864–77. doi: 10.1016/j.it.2020.08.008
190. Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, et al. Cell Origin Dictates Programming of Resident versus Recruited Macrophages during Acute Lung Injury. Am J Respiratory Cell Mol Biol. (2017) 57:294–306. doi: 10.1165/rcmb.2017-0061OC
191. Yang G, Yang Y, Liu Y, Liu X. Regulation of alveolar macrophage death in pulmonary fibrosis: a review. Apoptosis: an Int J programmed Cell Death. (2023) 28:1505–19. doi: 10.1007/s10495-023-01888-4
192. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Investigation. (2019) 129:2619–28. doi: 10.1172/JCI124615
193. Mould KJ, Jackson ND, Henson PM, Seibold M, Janssen WJ. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.126556
194. Hou F, Wang H, Zheng K, Yang W, Xiao K, Rong Z, et al. Distinct transcriptional and functional differences of lung resident and monocyte-derived alveolar macrophages during the recovery period of acute lung injury. Immune Netw. (2023) 23:e24. doi: 10.4110/in.2023.23.e24
195. Neupane AS, Willson M, Chojnacki AK, Vargas E Silva Castanheira F, Morehouse C, Carestia A, et al. Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell. (2020) 183:110–25.e11. doi: 10.1016/j.cell.2020.08.020
196. Zhu B, Wu Y, Huang S, Zhang R, Son YM, Li C, et al. Uncoupling of macrophage inflammation from self-renewal modulates host recovery from respiratory viral infection. Immunity. (2021) 54:1200–18.e9. doi: 10.1016/j.immuni.2021.04.001
197. Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front In Immunol. (2011) 2:65. doi: 10.3389/fimmu.2011.00065
198. Herold S, Tabar TS, Janssen H, Hoegner K, Cabanski M, Lewe-Schlosser P, et al. Exudate macrophages attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia. Am J Respiratory Crit Care Medicine. (2011) 183:1380–90. doi: 10.1164/rccm.201009-1431OC
199. Shin DS, Ratnapriya S, Cashin CN, Kuhn LF, Rahimi RA, Anthony RM, et al. Lung injury induces a polarized immune response by self-antigen-specific CD4+ Foxp3+ regulatory T cells. Cell Rep. (2023) 42:112839. doi: 10.1016/j.celrep.2023.112839
200. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+T cells: differentiation and functions. Clin Dev Immunol. (2012) 2012:925135. doi: 10.1155/2012/925135
201. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
202. Ono M. Control of regulatory T-cell differentiation and function by T-cell receptor signalling and Foxp3 transcription factor complexes. Immunology. (2020) 160:24–37. doi: 10.1111/imm.v160.1
203. Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, Li Z. Transforming growth factor-β1 in regulatory T cell biology. Sci Immunol. (2022) 7:eabi4613. doi: 10.1126/sciimmunol.abi4613
204. Jovisic M, Mambetsariev N, Singer BD, Morales-Nebreda L. Differential roles of regulatory T cells in acute respiratory infections. J Clin Investigation. (2023) 133:e170505. doi: 10.1172/JCI170505
205. Dial CF, Tune MK, Doerschuk CM, Mock JR. Foxp3+ Regulatory T cell expression of keratinocyte growth factor enhances lung epithelial proliferation. Am J Respiratory Cell Mol Biol. (2017) 57:162–73. doi: 10.1165/rcmb.2017-0019OC
206. Mock JR, Dial CF, Tune MK, Gilmore RC, O’Neal WK, Dang H, et al. Impact of regulatory T cells on type 2 alveolar epithelial cell transcriptomes during resolution of acute lung injury and contributions of IFN-γ. Am J Respiratory Cell Mol Biol. (2020) 63:464–77. doi: 10.1165/rcmb.2019-0399OC
207. Mock JR, Dial CF, Tune MK, Norton DL, Martin JR, Gomez JC, et al. Transcriptional analysis of Foxp3+ Tregs and functions of two identified molecules during resolution of ALI. JCI Insight. (2019) 4:e124958. doi: 10.1172/jci.insight.124958
208. Aliyu M, Zohora FT, Anka AU, Ali K, Maleknia S, Saffarioun M, et al. Interleukin-6 cytokine: An overview of the immune regulation, immune dysregulation, and therapeutic approach. Int Immunopharmacology. (2022) 111:109130. doi: 10.1016/j.intimp.2022.109130
209. Sutherland TE, Logan N, Rückerl D, Humbles AA, Allan SM, Papayannopoulos V, et al. Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol. (2014) 15:1116–25. doi: 10.1038/ni.3023
210. Raucci F, Saviano A, Casillo GM, Guerra-Rodriguez M, Mansour AA, Piccolo M, et al. IL-17-induced inflammation modulates the mPGES-1/PPAR-γ pathway in monocytes/macrophages. Br J Pharmacol. (2022) 179:1857–73. doi: 10.1111/bph.v179.9
211. Proto JD, Doran AC, Gusarova G, Yurdagul A, Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. (2018) 49:666–77.e6. doi: 10.1016/j.immuni.2018.07.015
212. Gerlach BD, Ampomah PB, Yurdagul A, Liu C, Lauring MC, Wang X, et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab. (2021) 33:2445–63.e8. doi: 10.1016/j.cmet.2021.10.015
213. Scopelliti F, Cattani C, Dimartino V, Mirisola C, Cavani A. Platelet derivatives and the immunomodulation of wound healing. Int J Mol Sci. (2022) 23:8370. doi: 10.3390/ijms23158370
214. Anitua E, Troya M, Alkhraisat MH. Immunoregulatory role of platelet derivatives in the macrophage-mediated immune response. Front Immunol. (2024) 15:1399130. doi: 10.3389/fimmu.2024.1399130
215. Rossaint J, Thomas K, Mersmann S, Skupski J, Margraf A, Tekath T, et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J Exp Med. (2021) 218:e20201353. doi: 10.1084/jem.20201353
216. Kapur R, Semple JW. Platelets instruct T reg cells and macrophages in the resolution of lung inflammation. J Exp medicine. (2021) 218. doi: 10.1084/jem.20210754
217. Scherlinger M, Richez C, Tsokos GC, Boilard E, Blanco P. The role of platelets in immune-mediated inflammatory diseases. Nat Rev Immunol. (2023) 23:495–510. doi: 10.1038/s41577-023-00834-4
218. Zhu L, Huang Z, Stålesen R, Hansson GK, Li N. Platelets provoke distinct dynamics of immune responses by differentially regulating CD4+ T-cell proliferation. J Thromb haemostasis: JTH. (2014) 12:1156–65. doi: 10.1111/jth.12612
219. Gerdes N, Zhu L, Ersoy M, Hermansson A, Hjemdahl P, Hu H, et al. Platelets regulate CD4+ T-cell differentiation via multiple chemokines in humans. Thromb Haemostasis. (2011) 106:353–62. doi: 10.1160/TH11-01-0020
220. Doran AC, Yurdagul A, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. (2020) 20:254–67. doi: 10.1038/s41577-019-0240-6
221. Linke B, Schreiber Y, Picard-Willems B, Slattery P, Nüsing RM, Harder S, et al. Activated platelets induce an anti-inflammatory response of monocytes/macrophages through cross-regulation of PGE2 and cytokines. Mediators Inflammation. (2017) 2017:1463216. doi: 10.1155/2017/1463216
222. Tang S, Zhang J, Zhang L, Zhao Y, Xiao L, Zhang F, et al. Knockdown of CXCL1 improves ACLF by reducing neutrophil recruitment to attenuate ROS production and hepatocyte apoptosis. Hepatology Commun. (2023) 7. doi: 10.1097/HC9.0000000000000257
223. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Communications. (2019) 10:1076. doi: 10.1097/HC9.0000000000000257
224. Thomas R, Qiao S, Yang X. Th17/treg imbalance: implications in lung inflammatory diseases. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24054865
225. Abdulnour R-EE, Dalli J, Colby JK, Krishnamoorthy N, Timmons JY, Tan SH, et al. Maresin 1 biosynthesis during platelet-neutrophil interactions is organ-protective. Proc Natl Acad Sci USA. (2014) 111:16526–31. doi: 10.1073/pnas.1407123111
226. Serhan CN, Dalli J, Karamnov S, Choi A, Park C-K, Xu Z-Z, et al. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. (2012) 26:1755–65. doi: 10.1096/fj.11-201442
227. Saito-Sasaki N, Sawada Y, Nakamura M. Maresin-1 and inflammatory disease. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23031367
228. Matthay MA, Ware LB. Resolution of alveolar edema in acute respiratory distress syndrome. Physiology and biology. Am J respiratory Crit Care medicine. (2015) 192:124–5. doi: 10.1164/rccm.201505-0938ED
229. Stolla M, Refaai MA, Heal JM, Spinelli SL, Garraud O, Phipps RP, et al. Platelet transfusion - the new immunology of an old therapy. Front Immunol. (2015) 6:28. doi: 10.3389/fimmu.2015.00028
230. Xia H, Wang F, Wang M, Wang J, Sun S, Chen M, et al. Maresin1 ameliorates acute lung injury induced by sepsis through regulating Th17/Treg balance. Life Sci. (2020) 254:117773. doi: 10.1016/j.lfs.2020.117773
231. Hendrickson KW, Peltan ID, Brown SM. The epidemiology of acute respiratory distress syndrome before and after coronavirus disease 2019. Crit Care Clin. (2021) 37:703–16. doi: 10.1016/j.ccc.2021.05.001
232. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. (2019) 5:18. doi: 10.1038/s41572-019-0069-0
233. Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. (2013) 369:2126–36. doi: 10.1056/NEJMra1208707
234. Hamid U, Krasnodembskaya A, Fitzgerald M, Shyamsundar M, Kissenpfennig A, Scott C, et al. Aspirin reduces lipopolysaccharide-induced pulmonary inflammation in human models of ARDS. Thorax. (2017) 72:971–80. doi: 10.1136/thoraxjnl-2016-208571
235. Xia L, Zhang C, Lv N, Liang Z, Ma T, Cheng H, et al. AdMSC-derived exosomes alleviate acute lung injury via transferring mitochondrial component to improve homeostasis of alveolar macrophages. Theranostics. (2022) 12:2928–47. doi: 10.7150/thno.69533
236. Tung Y-T, Wei C-H, Yen C-C, Lee P-Y, Ware LB, Huang H-E, et al. Aspirin attenuates hyperoxia-induced acute respiratory distress syndrome (ARDS) by suppressing pulmonary inflammation via the NF-κB signaling pathway. Front Pharmacol. (2021) 12:793107. doi: 10.3389/fphar.2021.793107
237. Dixon B, Smith RJ, Campbell DJ, Moran JL, Doig GS, Rechnitzer T, et al. Nebulised heparin for patients with or at risk of acute respiratory distress syndrome: a multicentre, randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. (2021) 9:360–72. doi: 10.1016/S2213-2600(20)30470-7
238. Erlich JM, Talmor DS, Cartin-Ceba R, Gajic O, Kor DJ. Prehospitalization antiplatelet therapy is associated with a reduced incidence of acute lung injury: a population-based cohort study. Chest. (2011) 139:289–95. doi: 10.1378/chest.10-0891
239. Toner P, McAuley DF, Shyamsundar M. Aspirin as a potential treatment in sepsis or acute respiratory distress syndrome. Crit Care (London England). (2015) 19:374. doi: 10.1186/s13054-015-1091-6
240. Kor DJ, Carter RE, Park PK, Festic E, Banner-Goodspeed VM, Hinds R, et al. Effect of aspirin on development of ARDS in at-risk patients presenting to the emergency department: the LIPS-A randomized clinical trial. JAMA. (2016) 315:2406–14. doi: 10.1001/jama.2016.6330
241. Toner P, Boyle AJ, McNamee JJ, Callaghan K, Nutt C, Johnston P, et al. Aspirin as a treatment for ARDS: A randomized, placebo-controlled clinical trial. Chest. (2022) 161:1275–84. doi: 10.1016/j.chest.2021.11.006
242. Li N, Hu H, Hjemdahl P. Aspirin treatment does not attenuate platelet or leukocyte activation as monitored by whole blood flow cytometry. Thromb Res. (2003) 111:165–70. doi: 10.1016/j.thromres.2003.08.026
243. Rinder CS, Student LA, Bonan JL, Rinder HM, Smith BR. Aspirin does not inhibit adenosine diphosphate-induced platelet alpha-granule release. Blood. (1993) 82:505–12. doi: 10.1182/blood.V82.2.505.505
244. Yadav H, Thompson BT, Gajic O. Fifty years of research in ARDS. Is acute respiratory distress syndrome a preventable disease? Am J Respiratory Crit Care Medicine. (2017) 195:725–36. doi: 10.1164/rccm.201609-1767CI
245. Reilly JP, Christie JD. Is it possible to prevent ARDS? JAMA. (2016) 315:2403–5. doi: 10.1001/jama.2016.5988
246. Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med. (2017) 5:524–34. doi: 10.1016/S2213-2600(17)30188-1
247. Zhang Z, Xu X, Zhang D, Zhao S, Wang C, Zhang G, et al. Targeting Erbin-mitochondria axis in platelets/megakaryocytes promotes B cell-mediated antitumor immunity. Cell Metab. (2024) 36:541–56.e9. doi: 10.1016/j.cmet.2023.12.020
248. Livada AC. Lung megakaryocytes are long-lived, arise from flt3-negative bone marrow cells and contribute to platelet recovery in thrombocytopenia: university of rochester. (2024). doi: 10.1101/2024.04.11.589077
249. Fox KA, Kirwan DE, Whittington AM, Krishnan N, Robertson BD, Gilman RH, et al. Platelets regulate pulmonary inflammation and tissue destruction in tuberculosis. Am J respiratory Crit Care medicine. (2018) 198:245–55. doi: 10.1164/rccm.201710-2102OC
250. Gelon L, Fromont L, Lefrançais E. Occurrence and role of lung megakaryocytes in infection and inflammation. Front Immunol. (2022) 13:1029223. doi: 10.3389/fimmu.2022.1029223
251. Pariser DN, Hilt ZT, Ture SK, Blick-Nitko SK, Looney MR, Cleary SJ, et al. Lung megakaryocytes are immune modulatory cells. J Clin Invest. (2021) 131. doi: 10.1172/JCI137377
252. Washington AV, Esponda O, Gibson A. Platelet biology of the rapidly failing lung. Br J haematology. (2020) 188:641–51. doi: 10.1111/bjh.16315
253. Xu W, Xia SH, Wang H, Chen H, Wang YL. Role of platelet activating factor in pathogenesis of acute respiratory distress syndrome. Chin Med J. (2007) 120:1840–4. doi: 10.1097/00029330-200710020-00022
254. Maddali MV, Churpek M, Pham T, Rezoagli E, Zhuo H, Zhao W, et al. Validation and utility of ARDS subphenotypes identified by machine-learning models using clinical data: an observational, multicohort, retrospective analysis. Lancet Respiratory medicine. (2022) 10:367–77. doi: 10.1016/S2213-2600(21)00461-6
255. van Amstel RBE, Bartek B, Vlaar APJ, Gay E, van Vught LA, Cremer OL, et al. Temporal transitions of the hyperinflammatory and hypoinflammatory phenotypes in critical illness. Am J respiratory Crit Care medicine. (2025) 211:347–56. doi: 10.1164/rccm.202406-1241OC
256. Torbic H, Bulgarelli L, Deliberato RO, Duggal A. Potential impact of subphenotyping in pharmacologic management of acute respiratory distress syndrome. J Pharm Pract. (2024) 37:955–66. doi: 10.1177/08971900231185392
257. Alipanah N, Calfee CS. Phenotyping in acute respiratory distress syndrome: state of the art and clinical implications. Curr Opin Crit Care. (2022) 28:1–8. doi: 10.1097/MCC.0000000000000903
258. Mansour A, Bachelot-Loza C, Nesseler N, Gaussem P, Gouin-Thibault I. P2Y(12) inhibition beyond thrombosis: effects on inflammation. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21041391
259. Sharifi-Rad J, Sharopov F, Ezzat SM, Zam W, Ademiluyi AO, Oyeniran OH, et al. An updated review on glycoprotein IIb/IIIa inhibitors as antiplatelet agents: basic and clinical perspectives. High Blood Press Cardiovasc Prev. (2023) 30:93–107. doi: 10.1007/s40292-023-00562-9
260. Romano M, Cianci E, Simiele F, Recchiuti A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur J Pharmacol. (2015) 760:49–63. doi: 10.1016/j.ejphar.2015.03.083
261. Xu H, Sheng S, Luo W, Xu X, Zhang Z. Acute respiratory distress syndrome heterogeneity and the septic ARDS subgroup. Front Immunol. (2023) 14:1277161. doi: 10.3389/fimmu.2023.1277161
262. Chen C-M, Lu H-C, Tung Y-T, Chen W. Antiplatelet therapy for acute respiratory distress syndrome. Biomedicines. (2020) 8:230. doi: 10.3390/biomedicines8070230
263. Devaney J, Contreras M, Laffey JG. Clinical review: gene-based therapies for ALI/ARDS: where are we now? Crit Care (London England). (2011) 15:224. doi: 10.1186/cc10216
264. Wang J, Peng X, Yuan N, Wang B, Chen S, Wang B, et al. Interplay between pulmonary epithelial stem cells and innate immune cells contribute to the repair and regeneration of ALI/ARDS. Trans research: J Lab Clin medicine. (2024) 272:111–25. doi: 10.1016/j.trsl.2024.05.012
265. Al-Husinat L, Azzam S, Al Sharie S, Araydah M, Battaglini D, Abushehab S, et al. A narrative review on the future of ARDS: evolving definitions, pathophysiology, and tailored management. Crit Care (London England). (2025) 29:88. doi: 10.1186/s13054-025-05291-0
266. Xie R, Tan D, Liu B, Xiao G, Gong F, Zhang Q, et al. Acute respiratory distress syndrome (ARDS): from mechanistic insights to therapeutic strategies. MedComm. (2025) 6:e70074. doi: 10.1002/mco2.70074
Keywords: ALI/ARDS, platelets, inflammation, immune regulation, neutrophil, regulatory T cell
Citation: Yang J, Zhou X, Qiao X and Shi M (2025) Friend or foe: the role of platelets in acute lung injury. Front. Immunol. 16:1556923. doi: 10.3389/fimmu.2025.1556923
Received: 07 January 2025; Accepted: 24 April 2025;
Published: 14 May 2025.
Edited by:
Simon C. Robson, Harvard University, United StatesReviewed by:
Rikesh Kumar Dubey, University of Pittsburgh, United StatesJohn-Paul Tung, Australian Red Cross Lifeblood, Australia
Copyright © 2025 Yang, Zhou, Qiao and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng Shi, bWVuZ3NoaUBmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work