 Zhen Qin
Zhen Qin Tao Xu*
Tao Xu*- Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, China
T cell receptor (TCR) signaling, also known as signal 1, plays a crucial role in the activation and proliferation of T cells. The question of whether TCR signaling exerts a deterministic role in T cell fate determination is an area of active investigation. It has been particularly challenging to address this question due to the complexities associated with genetic manipulation of TCR signaling components, which often disrupts thymic T cell development or impairs T cell activation upon TCR engagement. Recent study demonstrates that the TCR-Lck/Fyn axis directly induces STAT3 phosphorylation and synergizes with pro-inflammatory cytokines to optimize STAT3 phosphorylation during Th17 cell differentiation. Additionally, the TCR-Lck/Fyn-AKT/mTOR axis negatively regulates Treg cell differentiation. In CD8+ T cells, persistent high-affinity antigen stimulation drives differentiation along the exhaustion pathway, while acute infection or intermediate antigen levels promote differentiation into effector and memory T cells, although the underlying mechanism remains to be fully elucidated. Collectively, these studies provide compelling evidence that TCR signaling has a deterministic impact on T cell fate. This review summarizes recent advances in understanding how TCR signaling shapes T cell fate determination.
Introduction
The activation and differentiation of T cells need 3 signals: signal 1 provided by the interaction between T cell receptor (TCR) and peptide-MHC complex presented by APCs; signal 2 mediated by the interaction between CD28 expressed on T cells and CD80/86 on APC cells; and signal 3 provided by cytokines in the microenvironment (1). TCR specifically recognizes short peptide antigens (8–17 amino acids) presented by major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs) (2–4). TCR engagement with pMHC triggers a cascade of intracellular signaling leading to the activation of transcription factors such as NFATs, Jun/AP-1, and NF-κBs which translocate into the nucleus to drive the expression of many genes, such as IL-2, IL-2rα, generally essential for the activation and proliferation of all the T cells (5). The signal 2, is also called “co-stimulation signal” provided primarily through CD28 binding to CD80 or CD86 on APCs, are indispensable for enhancing T cell survival and proliferation. Other co-stimulatory receptors, such as ICOS, 4-1BB, and OX40, also contribute to fine-tuning the T cell activation (6, 7). Conversely, co-inhibitory receptors like CTLA-4 and PD-1 counterbalance these signals, regulating the magnitude of T cell activation and preventing immune overactivation (8). The signal 3, mediated by cytokines, directs the differentiation of activated T cells into different lineages (9). For CD4+ T cells, cytokines such as IL-12 and IL-4 promote Th1 and Th2 differentiation via the transcription factors T-bet and GATA-3, respectively, while TGF-β and inflammatory cytokines like IL-6 and IL-23 drive Th17 polarization through RORγt (10). In CD8+ T cells, cytokines like IL-12 and type I interferons enhance effector differentiation, while IL-15 is essential for memory formation (11). The coordinated interplay among these three signals ensures that T cells differentiate into specialized effector and memory populations tailored to the specific antigenic challenge, providing both immediate immune defense and long-term protection.
However, recent studies suggest that TCR signaling may not merely serve as an on/off switch for T cell activation; rather, it appears to provide nuanced signals that influence the specific lineage commitment of T cells (12). For instance, the strength and duration of TCR signaling can dictate whether CD4+ T cells differentiate into Th1, Th2, Th17, or regulatory T cell (Treg) subsets, each of which plays a distinct role in the immune response (13). Attenuation of TCR signaling favors the differentiation of activated CD4+ T cells into Treg cells (14–16). Similarly, CD8+ T cell differentiation into effector or memory subsets is also modulated by TCR signaling strength and duration (17). All these studies suggest that TCR signaling may play a deterministic role in T cell fate determination. Here in this review, we will summarize the recent evidence that TCR signaling itself plays a key deterministic role in T cell fate determination, as well as the underlying mechanism. Deep understanding of how TCR signaling influence T cell fate determination is crucial, particularly in the development of therapeutic strategies aimed at manipulating T cell responses in diseases such as cancer and autoimmune diseases. By deciphering the complex interplay between TCR signals, co-stimulatory signals, and the cytokine environment, researchers can potentially identify novel targets for immunotherapy. Such strategies could enhance T cell responses against tumors, improve vaccine efficacy, or even restore tolerance in autoimmune diseases.
Overview of TCR signaling
TCR is a heterodimer composed of TCRα and TCRβ chains, each containing variable and constant regions (18). TCR is associated with the CD3 subunits consisting of invariant γ, δ, ϵ, and ζ subunits responsible for transducing intracellular signals following antigen recognition, which together are called as TCR-CD3 complex (19). Upon recognizing specific peptide antigens presented by major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs), the TCR undergoes conformational changes that lead to exposure of the immunoreceptor tyrosine-based activation motifs (ITAMs, YXXM motifs) located in the cytoplasmic tails of the CD3 subunits (20), facilitating phosphorylation of the tyrosines within these motifs by Src-family protein tyrosine kinases, predominantly Lck and Fyn (21, 22). Phosphorylated ITAM motifs recruit ZAP-70 kinases, which facilitates its phosphorylation by Lck/Fyn kinases (23). Phosphorylated ZAP-70 then phosphorylates key adaptor proteins, including Linker for Activation of T Cells (LAT) and SH2 domain-containing leukocyte protein of 76 kDa (SLP-76), which form signaling complexes (24). LAT and SLP-76 provide docking sites for multiple effector proteins, enabling the activation of downstream pathways such as the MAPK/ERK pathway, the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR pathway, and the NF-κB pathway, which trigger a series of intracellular signaling cascades that lead to T cell activation, and proliferation (25, 26).
These pathways play critical roles in metabolic reprogramming and cell survival. For instance, the mTOR pathway integrates signals from TCR engagement, costimulatory receptors, and nutrient availability to regulate T cell fate decisions, including effector and memory differentiation (27). While mTOR is often activated via the canonical PI3K-AKT pathway, it can also be triggered independently of AKT through mechanisms such as amino acid sensing mediated by Rag GTPases (28, 29). PI3K activation leads to the phosphorylation of AKT, which further amplifies mTOR activity, supporting T cell growth, proliferation, and metabolic adaptation (30, 31). Meanwhile, LAT and SLP-76 also facilitate the recruitment of phospholipase C-γ1 (PLC-γ1), which catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3) (32). DAG activates protein kinase Cθ (PKCθ) and RasGRP1, which promote the activation of the NF-κB and MAPK pathways, respectively (33). IP3, on the other hand, induces the release of intracellular calcium from the endoplasmic reticulum, leading to the activation of the phosphatase calcineurin and the subsequent nuclear translocation of the transcription factor NFAT (nuclear factor of activated T cells) (34).
These signaling pathways collectively activate key transcription factors, including NFAT, AP-1 (activator protein 1), and NF-κB. These transcription factors together orchestrate the transcriptional programs necessary for T cell activation, proliferation, differentiation, and effector function (35, 36). The role of LAT in assembling these signaling complexes is critical, as mutations in LAT disrupt T cell development and cytokine production, thereby impairing immune responses (37–41).
While the role of TCR signaling in T cell activation is well established, increasing evidence supports its substantial influence on T cell fate decisions. Rather than being merely permissive, TCR signaling contributes instructively to lineage commitment by modulating transcriptional and metabolic programs. Although many studies have demonstrated causal links between TCR signal strength and specific differentiation outcomes—particularly in CD4+ and CD8+ subsets—important questions remain regarding how these signals are integrated across different cellular and environmental contexts. Continued investigation is necessary to fully decipher the precise mechanisms and to determine how TCR signaling interacts with co-stimulatory, cytokine, and metabolic cues to guide long-term fate decisions. Deepening our understanding of these pathways holds significant implications for the development of targeted immunotherapies in cancer, chronic infection, and autoimmune diseases (33).
TCR signaling and CD4+ T cell fate determination
CD4+ T cells are pivotal components of the adaptive immune system, capable of differentiating into specialized functional subsets—such as Th1, Th2, Th17, T follicular helper (Tfh), and Tregs—depending on the signals they encounter. These lineages are defined by transcription factors and unique cytokine profiles, and adopt distinct roles. Th1 cells drive cellular immunity against intracellular pathogens, Th2 cells promote humoral immunity to combat extracellular parasites, Th17 cells defend against extracellular bacteria and fungi, and Tregs maintain immune tolerance (42). The differentiation of CD4+ T cell lineages is highly context-dependent, requiring the integration of three critical signals: TCR signaling (signal 1), co-stimulatory signals (signal 2), and cytokine signals (signal 3). The strength, duration, and quality of TCR signaling are crucial factors influencing lineage commitment and functional specialization.
TCR signal strength and lineage commitment
The strength and duration of TCR signaling are critical determinants in T cell fate decisions, influencing the differentiation of naive CD4+ T cells into distinct effector or regulatory subsets (13, 43, 44). Recent studies find that the intensity of TCR signaling plays a pivotal role in directing lineage commitment, supporting a deterministic model where stronger signals drive effector T cell differentiation while weaker signals promote the development of regulatory T cells (45).
Strong TCR signaling typically favors the differentiation of Th1, Th17 and Tfh cells through the activation of lineage-defining transcription factors (10, 45–50). For instance, robust and sustained TCR signals promote Th1 differentiation by enhancing the expression of T-bet, the master transcription factor for Th1 cells (51), which drives the production of IFN-γ, a key cytokine involved in cellular immunity against intracellular pathogens (52). Likewise, the TCR-Lck/Fyn axis facilitates STAT3 activation, supporting Th17 differentiation (12). Similarly, strong TCR signals, in conjunction with costimulatory molecules and IL-21 signaling, promote the expression of Bcl-6, the transcription factor essential for Tfh cell differentiation, which supports the generation of high-affinity antibodies in germinal centers (53–55).
Expanding on the role of TCR signal quality, a recent study dissected the distinct contributions of antigen affinity and antigen dose in shaping CD4+ T cell differentiation during infection (56). The results demonstrated that high-affinity peptide-MHC interactions preferentially promote Th1 differentiation, independent of antigen dose, whereas Tfh cells can arise across a broader range of affinities but require sustained antigen availability to persist. Notably, increasing the antigen dose could not compensate for the suboptimal Th1 differentiation induced by low-affinity peptides. Furthermore, memory CD4+ T cells retained recall potential shaped by the strength of the initial TCR signal, emphasizing how early TCR engagement imprints long-term functional bias. These findings highlight antigen affinity as a critical determinant in effector subset specification and memory imprinting, with important implications for vaccine design and T cell-based immunotherapies.
Strong TCR signals have been shown to inhibit default Th2 differentiation programs by preventing early IL-4 expression and autocrine signaling through GATA3, thereby promoting Th1 over Th2 differentiation (57, 58). Studies suggest that this process is mediated by the nuclear translocation of NFATp and alterations in the DNA binding activity of AP-1 under strong TCR signaling (59, 60). This mechanism prevents IL-4-mediated feedback loops that would otherwise promote Th2 polarization, effectively guiding the cell toward a Th1 phenotype under conditions of robust antigen engagement.
A key component in this regulatory process is the adaptor protein LAT (Linker for Activation of T cells), which functions as a scaffold, facilitating the assembly of the “LAT signalosome,” a multiprotein complex that organizes and links TCR signals to intracellular pathways such as MAPK and NF-κB, thereby influencing lineage commitment (41). While LAT has been primarily understood as a positive regulator of TCR signaling, recent findings reveal its dual role. Studies of LAT mutations in mouse models, where the COOH-terminal tyrosine residues of LAT are altered, have shown that defective LAT signaling can lead to lymphoproliferative disorders characterized by polyclonal T cells with increased Th2 cytokine production (61). This unexpected finding underscores LAT’s role as a negative regulator of excessive TCR signaling, helping maintain T cell homeostasis and limiting unwarranted Th2 differentiation (Figure 1).
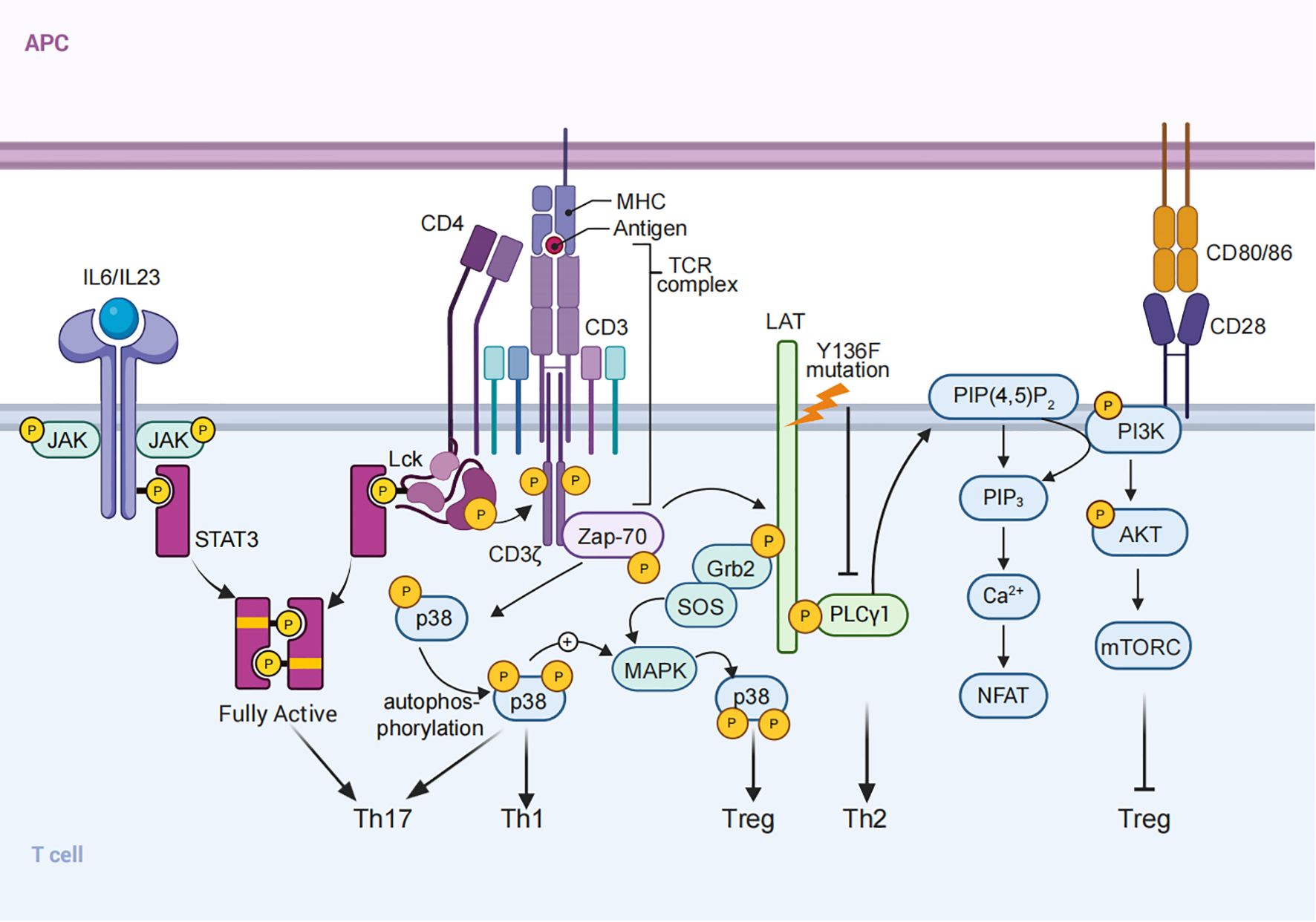
Figure 1. TCR Signaling and CD4+ T Cell Differentiation. Schematic representation of TCR signaling in CD4+ T cell activation and differentiation. Signal 1 (TCR signaling), Signal 2 (co-stimulation), and Signal 3 (cytokine signals) integrate to regulate T cell fate. Examples include: (1) the TCR-Lck/Fyn axis synergizing with cytokines to optimize STAT3 phosphorylation, promoting Th17 differentiation; (2) LAT mutations that alter COOH-terminal tyrosine residues lead to defective signaling, resulting in Th2-skewed cytokine responses; and (3) PI3K/AKT/mTOR signaling, activated by TCR stimulation, inhibits FOXP3 expression and suppresses Treg differentiation. (4) p38 MAPK signaling, activated via ZAP70 and LAT-SOS, promotes Th1 and Th17 differentiation, while also modulating Treg development.
In addition, recent studies have identified two distinct pathways for p38 MAPK activation in T cells (1): a classical MAPK cascade mediated by LAT-SOS, and (2) an alternative pathway involving direct phosphorylation by ZAP70 (62–64). Basal ZAP70 activation primes the classical p38 pathway, lowering the activation threshold and facilitating full p38 activation upon TCR engagement. This dual activation mechanism plays a critical role in maintaining immune balance, as evidenced by impaired IL-2 production and reduced Treg differentiation in the absence of Sos1 and Sos2. Furthermore, p38 MAPK signaling influences CD4+ T cell differentiation, with its inhibition leading to decreased Th1, Th17, and inducible Treg (iTreg) differentiation (Figure 1). These findings underscore the complex interplay between TCR signaling strength, adaptor proteins, and downstream kinase pathways in shaping T cell fate (64, 65).
TCR signal strength has differing implications for thymus-derived (tTreg) and peripherally induced (pTreg) subsets (14, 66, 67). Tregs, essential for maintaining immune tolerance and preventing autoimmune responses, are characterized by the expression of the transcription factor FOXP3 (68–70). tTregs are typically generated in response to moderate-to-high affinity TCR interactions with self-antigens during T cell development in the thymus (71–73). This engagement drives FOXP3 expression and establishes a stable epigenetic program, reinforced by histone modifications such as H3K4me2 and H3K4me3 at the Foxp3 loci, which prevents lineage deviation into effector T cells (66, 74–77). In contrast, the generation of pTregs in peripheral tissues involves relatively weak TCR signaling, often in response to environmental antigens or commensal microbes, rather than high-affinity self-antigens (14). Weaker TCR signals, especially when combined with anti-inflammatory cytokines like TGF-β and IL-2, promote FOXP3 induction in peripheral T cells (78, 79). This allows pTregs to modulate immune responses in peripheral tissues, providing a flexible mechanism for maintaining tolerance to non-self antigens encountered outside the thymus (15, 66, 80, 81).
Distinct intracellular pathways underscore these differences. In pTregs, weak TCR stimulation is thought to favor FOXP3 induction partly by reducing PI3K/AKT/mTOR signaling, which would otherwise antagonize FOXP3 expression (15, 82). For instance, reduced activity of the PIP3 phosphatase PTEN—a regulator of AKT signaling—is associated with suboptimal TCR stimulation, promoting FOXP3 expression in pTregs (80, 83) (Figure 1). Additionally, adaptor molecules like ITK, which are activated downstream of TCR engagement, modulate PTEN activity; ITK deficiency enhances pTreg differentiation, indicating that reduced signaling through this pathway may support FOXP3 stability in pTregs (80). In tTregs, continuous TCR signaling is generally unnecessary for maintaining FOXP3 expression once cells have fully differentiated, as evidenced by studies where TCRα deletion in mature Tregs does not lead to loss of identity of Treg cells in a resting state, although its deletion may cause functional impairment (84, 85).
Recent findings on TCR signaling complexity provide additional insights into how signal strength and duration affect Treg differentiation. The TCR-CD3 complex contains a total of 10 ITAMs, with each CD3ζ chain contributing three ITAMs (six in total from the CD3ζ homodimer), and the remaining four derived from the CD3γ, CD3δ, and two CD3ϵ subunits (20). ITAM multiplicity amplifies TCR signaling, critical for certain specialized T cell functions requiring strong or sustained TCR–ligand interactions. However, studies using knock-in mice expressing non-signaling CD3ζ chains (6Y within ITAM motifs were mutated into 6F) suggest that ITAM multiplicity is not essential for general T-cell functions like cytokine production or the development of a diverse antigen-reactive TCR repertoire (48). However, the knock-in mice exhibited greatly increased cell number of Treg cells. These findings imply that while strong ITAM-mediated signals may be vital for tTreg differentiation, weaker ITAM signaling could suffice for pTreg induction, particularly in the presence of supportive cytokine environments.
Furthermore, pharmacological interventions such as the use of rapamycin analogs (e.g., everolimus) and Srci1 (highly selective inhibitor of Lck/Fyn) enhance pTreg generation by inhibiting AKT/mTOR pathways and stabilizing FOXP3 expression through reduced DNA methylation at the Foxp3 promoter and CpG island (86, 87). Nutrient-sensing pathways, such as Rag GTPase-dependent mTORC1 activation, have also been implicated in the functional programming of Tregs, especially activated Tregs in peripheral tissues and tumors (88). These pathways link metabolic cues to Treg expansion and suppressive capacity, further emphasizing the interplay between TCR signaling, cytokines, and metabolism in Treg biology.
Under Th17-polarizing conditions, strong and sustained TCR signaling, characterized by high antigen dose and persistent stimulation, has been shown to promote IL-17 expression, the hallmark cytokine of Th17 cells (12, 89, 90), while moderate TCR stimulation, typically involving lower-affinity antigens or reduced antigen availability, tends to favor Treg development over Th17 polarization, even in the presence of similar cytokine environment (14, 80). However, the precise role of TCR signal strength in determining the balance between Th17 and Treg differentiation remains complex and the underlying mechanism is still to be determined. Our recent studies demonstrates that the TCR-Lck/Fyn axis directly phosphorylates STAT3 at Y705, synergistically with proinflammatory cytokines like IL-6 and IL-23, to achieve the optimal STAT3 phosphorylation needed for Th17 lineage commitment. Pharmacological inhibition of Lck/Fyn kinase activity, or disrupting its interaction with STAT3, significantly reduces STAT3 phosphorylation, skewing differentiation away from the Th17 pathway and toward a Treg phenotype (91). Mechanistically, Lck and Fyn interact with STAT3 as evidenced by results from co-immunoprecipitation assays. AlphaFold Multimer analysis indicates that the MAS-motif within STAT3 initiates the interaction of between STAT3 and Lck/Fyn, enhanced their kinase activity, and is essential for its phosphorylation by Lck/Fyn, which is further supported by in vitro kinase assay showing that the peptide containing WT MAS motif significantly enhanced STAT3 phosphorylation by Lck kinases. This critical interaction demonstrates the deterministic role of TCR signaling in directing Th17 differentiation. Notably, disruption of the interaction between Lck/Fyn ad STAT3 by disease causing STAT3 mutation selectively inhibits TCR stimulation induced STAT3 phosphorylation, but not proinflammatory cytokines induced STAT3 phosphorylation, and inhibits Th17 cell differentiation, which further demonstrates the significance of TCR-Lck/Fyn-STAT3 axis in Th17 cell differentiation. Administration of the Src inhibitor Srci1 or disruption of the Lck/Fyn-STAT3 interaction significantly ameliorated experimental autoimmune encephalomyelitis (EAE), a Th17-mediated autoimmune disease, by reducing Th17 differentiation and enhancing Treg polarization. These findings not only demonstrate modulation of the TCR-Lck/Fyn-STAT3 axis holds promise for therapeutic intervention, but also uncover the critical synergy between TCR signaling and cytokine networks (Signal 3) in regulating CD4+ T cell differentiation, with the TCR-Lck/Fyn axis serving as a key determinant of Th17 lineage fate (Figure 1).
Integration of TCR signaling with cytokine networks
The integration of TCR signaling with cytokine networks is critical for directing CD4+ T cell lineage commitment and plasticity. CD4+T cells differentiation is orchestrated by a dynamic interplay between intrinsic TCR signals and extrinsic cytokine inputs, which collectively shape T cell fate and function. Strong TCR signals combined with cytokines like IL-12 and IFN-γ promote Th1 differentiation by activating STAT1 and inducing T-bet expression, whereas IL-4 signaling favors Th2 lineage commitment through STAT6 and GATA3 activation (87, 92–95). In the context of Th17 differentiation, IL-6 and TGF-β act in concert with TCR signaling to activate STAT3, driving the differentiation process, which engage STAT3 and RORγt, the master transcription factor for Th17 cells, thereby reinforcing lineage-specific gene expression (12, 90, 96–99). Notably, TCR-Lck/Fyn axis directly induces STAT3 phosphorylation, establishing a critical link between TCR strength and cytokine-driven lineage determination during Th17 cell differentiation (12).
The plasticity of CD4+ T cells enables functional adaptation in response to changing cytokine environments, disruptions in the cytokine-TCR signaling axis can lead to immune dysregulation. For example, Th1 cells reactivated in Th2-polarizing conditions can express Th2 cytokines, demonstrating the dynamic nature of T cell responses (100, 101). Similarly, Tregs and Th17 cells exhibit reciprocal plasticity influenced by the balance of TGF-β and pro-inflammatory cytokines such as IL-6 and IL-23 (102). This plasticity highlights the importance of a finely tuned cytokine milieu in shaping T cell fate even after initial differentiation. These cytokines and TCR signaling pathways converge to influence CD4+ T cell fate, underscoring the coordination required between extracellular signals and intracellular transcriptional regulation.
In summary, TCR signaling plays a critical and multifaceted role in the differentiation of CD4+ T cells into distinct functional subsets. The context in which TCR signaling occurs—particularly the cytokine environment and the strength of the signal—dictates whether CD4+ T cells differentiate into Th1, Th2, Th17, or Treg subsets. Recent advances in understanding the molecular mechanisms underlying TCR signaling, such as the role of the TCR-Lck/Fyn-STAT3 axis and the MALT1 cleavage pathway (12, 103), have provided deeper insights into how TCR signaling influences immune function and tolerance. Understanding these mechanisms is crucial for developing targeted immunotherapies aimed at modulating T cell responses in autoimmune diseases, infections, and cancer.
Integration of TCR signaling with metabolic networks
Metabolic reprogramming occurs upon T cell activation, with metabolism and nutrient cues reciprocally influencing TCR signaling strength and T cell differentiation. Upon TCR engagement, T cells shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, a phenomenon known as the Warburg effect (108, 109). This metabolic switch facilitates the rapid generation of ATP and metabolic intermediates essential for proliferation and effector functions (110). The mechanistic mTOR pathway, a central regulator of metabolism, links TCR signaling to metabolic reprogramming, controlling glycolysis, lipid synthesis, and amino acid metabolism to promote effector T cell differentiation (88, 104). Intrinsic cellular energy demands are synchronized with extracellular environmental signals, such as nutrient availability and pH, to ensure proper T cell activation, proliferation, and differentiation (105, 106). Given the growing recognition of metabolism’s pivotal role in T cell differentiation, it is now often referred to as “signal 4.”
Distinct CD4+ T cell subsets exhibit unique metabolic dependencies. Effector T cells, such as Th1 and Th17 cells, rely on glycolysis, and amino acid metabolism (105), whereas Tregs favor oxidative phosphorylation and fatty acid oxidation (FAO) to support their suppressive function (106–108). This metabolic divergence is crucial for lineage specification, reinforcing how TCR signaling and metabolic programs coordinate to shape T cell fate.
Beyond glycolysis and amino acid metabolism, sterol metabolism has emerged as a key regulator of TCR signaling and CD4+ T cell differentiation (109). Upon TCR engagement, activated T cells upregulate sterol biosynthesis and uptake pathways to support membrane expansion during clonal proliferation (110). The liver-X receptors (LXRs), which serve as metabolic sensors, regulate this process (111, 112). Notably, LXRβ deficiency enhances both CD4+ and CD8+ T cell proliferation, leading to increased IFN-γ production, highlighting the metabolic influence on effector function (111).
Metabolically, sterol metabolism directly influences lineage decisions. Activation of LXRs suppresses Th17 differentiation via sterol regulatory element-binding protein-1 (SREBP-1), which competes with the aryl hydrocarbon receptor (AhR) for binding at the Il17a locus, thereby repressing Th17 lineage commitment (113, 114). In addition, a cholesterol biosynthetic intermediate has been demonstrated as endogenous Rorγt ligand to direct Th17 differentiation (115). These findings suggest that sterol metabolism not only supports proliferation but also modulates differentiation by regulating lineage-defining transcriptional programs.
TCR signaling in CD8+ T cell fate determination
TCR signaling plays a pivotal role in orchestrating the differentiation and functional specialization of CD8+ T cells, influencing their development, differentiation, and functional responses, also determining the long-term behavior of CD8+ T cells. CD8+ T cells are essential for immune defense against viral infections and tumors, and their function is closely linked to the nature and strength of TCR signals received during antigen recognition. A growing body of evidence has illuminated how TCR signaling intricately modulates CD8+ T cell differentiation, particularly the balance between short-lived effector cells and long-lived memory cells, as well as the risk of T cell exhaustion (116, 117).
TCR Signal Strength and Duration Shape CD8⁺ T Cell Fate
High-affinity TCR interactions are essential for the differentiation of cytotoxic effector T cells (CTLs), which directly target infected or malignant cells (118). Strong, sustained TCR signals—often amplified by co-stimulatory molecules like CD28—activate transcription factors such as T-bet and Eomesodermin (Eomes) (119, 120). These factors orchestrate the expression of cytotoxic machinery, including perforin and granzymes, which is crucial for immediate pathogen elimination. Studies indicate that high-affinity TCR engagement is a primary driver of robust CTL differentiation, where prolonged TCR engagement has been shown to enhance terminal effector differentiation, equipping CD8+ T cells with rapid and potent immune capabilities.
In contrast to the differentiation of effector cells, intermediate TCR signals tend to favor the formation of memory CD8+ T cells, which provide long-term protection by rapidly responding to subsequent antigen exposure (116, 121, 122). Memory T cells exhibit distinct metabolic and functional profiles, enabling their prolonged survival and capacity for robust recall responses (123). This balance between persistence and responsiveness ensures effective immunity against reinfections (124). In addition to promoting effector functions, strong TCR signals have also been shown to modulate the mode of cell division in activated CD8+ T cells (125). Under strong stimulation, asymmetric cell division (ACD) safeguards memory potential by enabling fate bifurcation within progeny, whereas symmetric divisions favor terminal effector differentiation. Inhibiting ACD under high TCR signaling impairs memory generation, suggesting that ACD acts as a regulatory mechanism that preserves long-term immunity under conditions of intense stimulation.
Moreover, the duration of TCR signaling has also been shown to directly influence memory T cell formation (123). Shorter TCR signaling durations lead to the generation of memory precursors, which maintain the ability to rapidly proliferate and acquire effector functions upon secondary antigen encounters. This dichotomy in TCR signaling strength provides a finely tuned mechanism by which CD8+ T cells balance their immediate effector functions with long-term memory formation, ensuring both rapid pathogen clearance and durable immunity (126).
In addition to governing the balance between effector and memory T cell differentiation, TCR signal strength also plays a decisive role in the development of exhausted CD8+ T cells (Tex) and progenitor exhausted T cells (Tpex) (127, 128). Factors such as peptide-MHC affinity, contact time with dendritic cells (DCs), persistent antigen load, and the number of antigen-presenting DCs determine TCR signal strength during priming (129). High TCR signal strength has been shown to increase the expression of inhibitory receptors such as PD-1 and LAG-3, driving terminal exhaustion and reduced cytotoxic function in chronic infections and tumors (130). Conversely, lower TCR signal strength favors the generation of Tpex cells, a less differentiated subset that retains proliferative capacity and responds better to immune checkpoint blockade (ICB) therapy (131–134). This highlights the need to optimize TCR signaling thresholds to balance protective immunity, persistence, and responsiveness to immunotherapy.
Regulatory mechanisms and external modulation of TCR signaling
One of the critical aspects of TCR signaling is its ability to dictate the expression of transcription factors that govern CD8+ T cell differentiation. For instance, the transcription factor interferon regulatory factor 4 (IRF4) is essential for the expansion and differentiation of CD8+ T cell. Recent studies demonstrated that the expression of IRF4 in CD8+ T cells is contingent upon the strength of TCR signaling, which is partially mediated by mTOR signaling pathways (135). This finding corroborated another study which further elucidated that graded levels of IRF4 regulate CD8+ T cell differentiation and expansion in response to acute viral infections (136), highlighting the importance of TCR signaling in this context. Additionally, the role of IL-2 inducible T-cell kinase (ITK) in regulating IRF4 expression underscores the complexity of TCR signaling pathways and their downstream effects on CD8+ T cell fate (119).
CD45, a critical phosphatase, plays a significant role in modulating TCR sensitivity in naive and memory CD8+ T cells (137). Continuous interaction with self-MHC ligands is crucial for the survival of naive T cells but not for memory cells, indicating distinct TCR sensitivity between these subsets (138). High CD45 expression in memory CD8+ T cells is associated with reduced TCR sensitivity compared to naive cells. This reduced sensitivity, linked to decreased activation of LCK and short-term TCR signaling, protects CD8+ T cells from excessive auto-MHC reactivity while preserving robust responses to foreign antigens (139). This differential regulation highlights how TCR signaling mechanisms adapt to the distinct functional requirements of naive and memory T cell populations.
The role of cytokines in modulating TCR signaling cannot be overlooked. Pathogen-specific inflammatory environments can enhance TCR signaling in CD8+ T cells, thereby tuning their antigen sensitivity and functional responses (140). This interplay between cytokine signaling and TCR engagement is crucial for effective differentiation, as transient enhanced IL-2 receptor signaling during priming amplifies the development of functional effector-memory cells (141). Furthermore, both TCR and IL-2 signaling strength control memory CD8+ T cell functional fitness via chromatin remodeling, highlighting the integration of external cues with intrinsic signaling (142).
Beyond antigen recognition, co-stimulation, and cytokine signaling, emerging evidence suggests that metabolic cues act as signal 4 in T cell activation, influencing TCR signaling strength, CD8+ T cell differentiation, and functional persistence (143–145). TCR engagement induces metabolic reprogramming, shifting from oxidative phosphorylation to glycolysis to meet the bioenergetic and biosynthetic demands of rapid expansion and effector differentiation (146). CD28 co-stimulation further enhances glycolysis, amplifying metabolic flux to support proliferation and cytokine production (147). Notably, high glycolytic activity favors effector differentiation but impairs the long-term survival of memory CD8+ T cells (148, 149). Thus, balancing glycolysis with fatty acid oxidation (FAO) is critical for sustaining effective immune responses (150).
Metabolic constraints in the tumor microenvironment (TME) further modulate TCR signaling strength and CD8+ T cell functionality (151). In nutrient-deprived conditions, such as low glucose and amino acid availability, CD8+ T cells upregulate alternative metabolic pathways to sustain their function (152). However, chronic metabolic stress can impair TCR signaling, driving metabolic exhaustion and dysfunction (153). Notably, glycolysis is directly linked to TCR-dependent IFN-γ production, as reducing glycolytic activity dampens cytokine output and cytotoxicity (154). Conversely, selectively enhancing glycolysis restores effector functions and may serve as a strategy to reinvigorate exhausted CD8+ T cells in immunotherapy settings (155).
Additionally, the mTOR pathway plays a critical role in linking TCR signaling to metabolic adaptation (104). Increased glycolytic activity activates mTOR, promoting effector differentiation but potentially contributing to metabolic dysregulation and partial T cell dysfunction (28). Interestingly, mTOR-dependent metabolic shifts may adversely affect IFN-γ production in some contexts (156), further emphasizing the necessity of fine-tuned metabolic control. Furthermore, metabolic competition between glycolysis and FAO influences the long-term survival and recall capacity of memory CD8+ T cells (157, 158). While effector cells rely on glycolysis for rapid cytotoxic responses, memory cells preferentially utilize FAO, allowing for extended persistence and robust recall responses upon reinfection (159).
TCR signaling is further modulated by the acidic conditions of the TME, where extracellular acidification suppresses T cell function (160). Acidosis directly impacts T cell metabolism by restricting one-carbon metabolism, limiting nucleotide biosynthesis and impairing activation potential. Additionally, low pH inhibits TCR signal transduction via the STS1-Cbl-b complex, a pH-sensitive phosphatase that actively suppresses T cell function. This regulatory mechanism constrains CD8+ T cell effector responses in tumors, dampening anti-tumor immunity. Notably, the deficiency of either STS1 or Cbl-b desensitizes T cells to acidic pH, leading to enhanced anti-tumor reactivity and improved T cell survival in hostile microenvironments. These findings underscore the metabolic constraints imposed by acidic niches and their impact on TCR-driven fate decisions.
Metabolic adaptations under nutrient-deprived conditions also modulate TCR signaling strength and CD8+ T cell differentiation (154). In the TME, where glucose and amino acid availability are limited, CD8+ T cells upregulate nutrient transporters and alternative metabolic pathways to sustain functionality (152, 155). However, chronic metabolic stress can impair TCR signaling and drive T cell dysfunction, ultimately promoting T cell exhaustion, a hallmark of dysfunctional tumor-infiltrating lymphocytes (153). Understanding these adaptations provides therapeutic opportunities to enhance anti-tumor immunity by modulating metabolic pathways to optimize TCR function in tumors.
In addition to biochemical and metabolic inputs, mechanical forces have emerged as a novel regulatory dimension of TCR signaling (161–163). T cells apply cytoskeletal tension at the immunological synapse to interrogate antigen-presenting cells, and TCR-pMHC interactions can behave as catch bonds that prolong signal duration under force (162). These mechanosensing mechanisms enhance antigen discrimination and influence downstream transcriptional programs, adding an underexplored but important layer of control to T cell activation and fate decisions.
Taken together, TCR signaling and metabolism are highly interdependent. Targeting metabolic pathways in conjunction with TCR signaling holds promise for enhancing T cell-based immunotherapies, improving T cell persistence, cytotoxicity, and survival in cancer and chronic infections (158, 159).
Developmental dynamics and long-term outcomes
The dynamics of TCR signaling play a pivotal role in CD8+ T cell differentiation (164, 165). These findings underscore the importance of TCR signaling not only in immediate immune responses but also in shaping long-term immune potential.
A seminal study challenged the traditional paradigm that only strong TCR ligation is sufficient to initiate T cell responses (166). Their findings demonstrated that even very low-affinity TCR–peptide-MHC interactions can activate naïve CD8+ T cells, induce rapid proliferation, and generate both effector and memory populations. Notably, while low-affinity interactions were sufficient to trigger early activation and migration—marked by faster CCR7 downregulation and earlier tissue egress—sustained T cell expansion required higher-affinity stimulation. This differential expansion contributes to a maturation of the T cell pool over time, favoring the prolonged persistence of high-affinity clones. Interestingly, despite limited expansion, low-affinity ligands were still capable of generating functional memory T cells, indicating that memory formation is not strictly dependent on strong TCR signals (Figure 2). The clonal composition of responding CD8+ T cells is shaped by TCR avidity thresholds, which govern recruitment and expansion (167). High-avidity T cells dominate protective responses, but low-avidity clones also contribute functional flexibility, especially during heterologous re-infections. This layered recruitment enhances the adaptability of T cell responses, with clonal diversity tuned by the affinity landscape of peptide-MHC interactions during priming. TCR signal strength influences both the extent of effector responses and the qualitative diversity of memory and clonal selection.
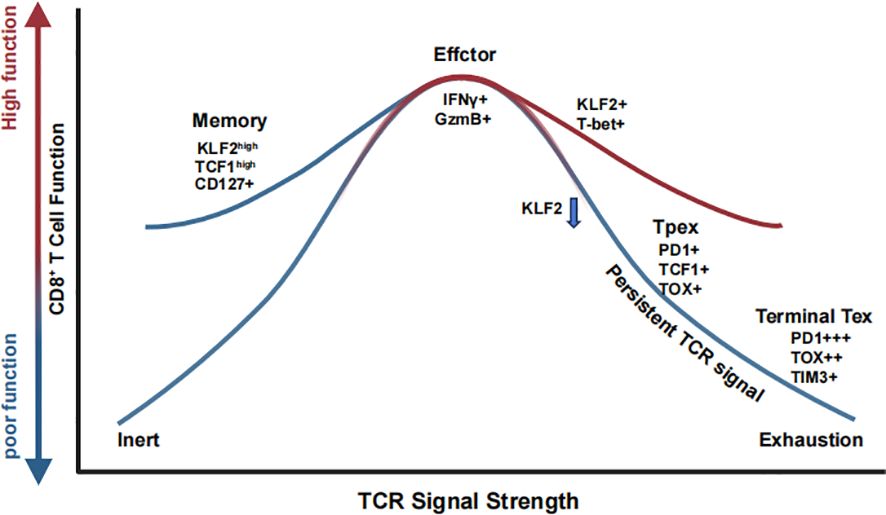
Figure 2. TCR signal strength shapes CD8+ T cell fate through coordinated transcriptional and functional programs. CD8+ T cells interpret graded TCR signals to adopt distinct functional states. Low TCR stimulation promotes memory precursor formation (KLF2+ TCF1+ CD127+), supporting long-term persistence. Intermediate TCR strength induces optimal effector differentiation (IFN-γ+ GzmB+) with sustained KLF2 and T-bet expression. Prolonged or excessive TCR signaling drives progressive loss of KLF2 and acquisition of exhaustion-associated markers (PD-1, TOX), leading to a stepwise transition from progenitor exhausted T cells (Tpex) to terminally exhausted T cells (Tex).
Recent innovative studies have further elucidated the role of TCR signaling in determining CD8+ T cell fate. Evidence now suggests that precursor exhausted T (Tpex) cells arise early in both acute and chronic infections, positioning them at a critical bifurcation point where they can either differentiate into memory precursors following pathogen clearance or progress toward terminal exhaustion under conditions of persistent antigen exposure (165). This finding emphasizes that the pMHC-TCR signal itself is a decisive factor in dictating whether CD8+ T cells differentiate into memory T cells or exhausted T cells, irrespective of infection type (168). Moreover, a groundbreaking Perturb-seq study identified Klf2 as a key transcription factor governing CD8+ T cell fate determination (169) (Figure 2). The study demonstrated that Klf2 knockout drives aberrant differentiation toward an exhaustion-like state even in acute infections, suggesting that Klf2 plays an essential role in suppressing the exhaustion-promoting transcription factor TOX while enabling T-bet to drive effector differentiation. Additionally, strong TCR stimulation was found to silence Klf2 at protein level. These findings reinforce the concept that TCR signaling is the primary determinant of CD8+ T cell differentiation, directly influencing exhaustion versus effector/memory lineage commitment.
Chronic antigen stimulation during persistent infections or cancer can lead to T cell exhaustion, characterized by diminished effector function and upregulation of inhibitory receptors such as PD-1, Tim-3, and LAG-3 (170–172). Recent studies suggest that TCR signal strength is a key determinant of exhaustion dynamics in CD8+ T cells (130, 173) (Figure 2). High-affinity, persistent TCR signaling promotes terminal exhaustion, whereas lower TCR signal intensity favors the differentiation of Tpex cells, which retain proliferative potential and responsiveness to immune checkpoint blockade therapy (127, 174, 175). Recent findings further elucidate that the formation of Tpex cells is initiated early during acute responses and is driven by strong TCR signaling and high-affinity peptide-MHC interactions. This developmental trajectory is counterbalanced by PD-1 signaling, which restricts precursor expansion (165). Together, these findings reinforce the idea that TCR signal strength not only governs early activation but imprints long-term lineage potential across both effector and memory trajectories.
Interestingly, both low- and high-affinity antigen-expressing tumors exhibit resistance to immune control, albeit through different mechanisms (176). High-affinity tumors induce deep exhaustion by promoting prolonged antigen stimulation and inhibitory receptor expression, leading to dysfunctional T cells. Conversely, low-affinity tumors fail to elicit strong TCR engagement, resulting in poor CD8+ T cell priming and weak immune responses. These findings suggest that fine-tuning TCR signaling—both during early priming in lymphoid tissues and within the tumor microenvironment—could optimize antitumor immunity (Figure 2). This principle also applies to adoptive T cell therapies, where selecting optimal TCRs or chimeric antigen receptors (CARs) based on antigen affinity and signal strength may improve T cell persistence, functionality, and therapeutic efficacy (177–180).
In addition to its role in memory formation and exhaustion, TCR signaling strength has also been implicated in tissue-resident memory T cell (TRM) differentiation (181). TRM cells are non-circulating CD8+ T cells that provide long-term localized immunity across various tissues (182). Recent evidence suggests that lower TCR signal strength favors TRM differentiation, while high-affinity TCR interactions preferentially drive circulating memory T cell formation (183). In models of influenza virus infection, weaker TCR stimulation correlated with enhanced TRM formation in the lung, whereas stronger TCR signaling biased cells toward a circulating effector memory phenotype (184).
Beyond antigen affinity, DC subsets play a crucial role in shaping TRM differentiation by influencing TCR signaling strength during priming (185). Recent studies have shown that mice lacking DNGR1 or Batf3—key markers of cross-presenting DCs—exhibited impaired TRM formation after viral infection, while circulating memory T cells were unaffected (186). These findings highlight the interplay between TCR strength, antigen-presenting cell specialization, and tissue-specific cues in determining whether CD8+ T cells persist in circulation or take up long-term residence within tissues.
Moreover, TCR strength influences the chemotactic properties of CD8+ T cells, regulating their ability to establish tissue residency (183). Weaker TCR signaling is associated with reduced expression of KLF2 and S1PR1 (187, 188), which promotes TRM retention by limiting their egress from tissues. These observations suggest that TCR signaling not only governs differentiation into effector versus memory states but also fine-tunes tissue-specific localization and persistence. TCR signal strength also governs chemotactic behavior, including the upregulation of CXCR6 and suppression of S1PR1 expression, promoting tissue residency. This chemotactic switch is dependent on Blimp1 induction by TCR re-stimulation and is essential for efficient TRM differentiation across non-lymphoid tissues (183). These findings emphasize how TCR signal strength not only instructs fate commitment but also spatial positioning of effector progeny during immune responses.
Sustained TCR signaling is also known to drive this exhausted phenotype by inducing the expression of transcription factors such as TOX and NR4A, which suppress effector functions and promote inhibitory receptor expression (189). Exhausted CD8+ T cells exhibit reduced cytotoxic capacity, impairing their ability to clear chronic infections or tumors effectively. Importantly, recent therapeutic strategies have focused on reversing T cell exhaustion by blocking inhibitory receptors with immune checkpoint inhibitors, thus restoring the effector functions of exhausted CD8+ T cells in the context of cancer immunotherapy. These approaches highlight the critical importance of understanding TCR signaling dynamics in addressing chronic diseases and cancer.
TCR signaling is a fundamental determinant of CD8+ T cell fate, shaping their development, differentiation, and functional potential. The strength, duration, and context of TCR signals, coupled with co-stimulatory inputs and metabolic programming, govern the balance between effector differentiation, memory formation, and the risk of exhaustion. These signals influence whether CD8+ T cells adopt an effector or memory phenotype or become exhausted under chronic antigen exposure. A deeper understanding of these mechanisms is essential for developing targeted therapies to enhance CD8+ T cell responses in infections, cancer, and chronic diseases. Future research into the nuances of TCR signal modulation will be critical for advancing immunotherapeutic strategies, ultimately improving outcomes across diverse clinical applications.
In conclusion, TCR signaling plays a critical role not only in T cell activation but also in fate determination. As we unravel the complex signaling networks involved in T cell differentiation, we move closer to developing therapies that can precisely modulate T cell responses in diverse disease contexts. Continued exploration of TCR signaling nuances will undoubtedly yield innovative therapeutic strategies, transforming the landscape of T cell based immunotherapy.
Author contributions
ZQ: Conceptualization, Writing – original draft, Writing – review & editing. TX: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. TX was supported by the National Natural Science Foundation of China (Grants 82371814 and 32170775), and by the Guangdong Province “Pearl River Talent Plan” Innovation and Entrepreneurship Team Project (Grant 2019ZT08Y464).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Curtsinger JM and Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. (2010) 22:333–40. doi: 10.1016/j.coi.2010.02.013
2. Hennecke J and Wiley DC. T cell receptor-MHC interactions up close. Cell. (2001) 104:1–4. doi: 10.1016/S0092-8674(01)00185-4
3. Attaf M, Legut M, Cole DK, and Sewell AK. The T cell antigen receptor: the Swiss army knife of the immune system. Clin Exp Immunol. (2015) 181:1–18. doi: 10.1111/cei.12622
4. Szeto C, Lobos CA, Nguyen AT, and Gras S. TCR recognition of peptide-MHC-I: rule makers and breakers. Int J Mol Sci. (2020) 22(1):68. doi: 10.3390/ijms22010068
5. Liao W, Lin JX, and Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. (2013) 38:13–25. doi: 10.1016/j.immuni.2013.01.004
6. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz LJ, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J Exp Med. (2015) 212:217–33. doi: 10.1084/jem.20141432
7. Roselli E, Boucher JC, Li G, Kotani H, Spitler K, Reid K, et al. 4-1BB and optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J Immunother Cancer. (2021) 9(10):e003354. doi: 10.1136/jitc-2021-003354
8. Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. (2011) 3:581–92. doi: 10.1002/emmm.201100165
9. Pham NL, Badovinac VP, and Harty JT. Differential role of “Signal 3” inflammatory cytokines in regulating CD8 T cell expansion and differentiation in vivo. Front Immunol. (2011) 2:4. doi: 10.3389/fimmu.2011.00004
10. Yamane H and Paul WE. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol Rev. (2013) 252:12–23. doi: 10.1111/imr.2013.252.issue-1
11. Kohlhapp FJ, Zloza A, O’Sullivan JA, Moore TV, Lacek AT, Jagoda MC, et al. CD8(+) T cells sabotage their own memory potential through IFN-gamma-dependent modification of the IL-12/IL-15 receptor alpha axis on dendritic cells. J Immunol. (2012) 188:3639–47. doi: 10.4049/jimmunol.1101580
12. Qin Z, Wang R, Hou P, Zhang Y, Yuan Q, Wang Y, et al. TCR signaling induces STAT3 phosphorylation to promote TH17 cell differentiation. J Exp Med. (2024) 221(3):e20230683. doi: 10.1084/jem.20230683
13. Bhattacharyya ND and Feng CG. Regulation of T helper cell fate by TCR signal strength. Front Immunol. (2020) 11:624. doi: 10.3389/fimmu.2020.00624
14. Gottschalk RA, Corse E, and Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med. (2010) 207:1701–11. doi: 10.1084/jem.20091999
15. Haxhinasto S, Mathis D, and Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. (2008) 205:565–74. doi: 10.1084/jem.20071477
16. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, and von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. (2005) 6:1219–27. doi: 10.1038/ni1265
17. Kaech SM, Wherry EJ, and Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. (2002) 2:251–62. doi: 10.1038/nri778
18. van der Merwe PA and Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. (2011) 11:47–55. doi: 10.1038/nri2887
19. Call ME and Wucherpfennig KW. The T cell receptor: critical role of the membrane environment in receptor assembly and function. Annu Rev Immunol. (2005) 23:101–25. doi: 10.1146/annurev.immunol.23.021704.115625
20. Love PE and Hayes SM. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect Biol. (2010) 2:a002485. doi: 10.1101/cshperspect.a002485
21. Sugie K, Jeon MS, and Grey HM. Activation of naive CD4 T cells by anti-CD3 reveals an important role for Fyn in Lck-mediated signaling. Proc Natl Acad Sci U S A. (2004) 101:14859–64. doi: 10.1073/pnas.0406168101
22. Smith-Garvin JE, Koretzky GA, and Jordan MS. T cell activation. Annu Rev Immunol. (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
23. Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freedman TS, et al. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol. (2010) 2:a002279. doi: 10.1101/cshperspect.a002279
24. Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol. (2002) 20:371–94. doi: 10.1146/annurev.immunol.20.092601.111357
25. Hwang JR, Byeon Y, Kim D, and Park SG. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med. (2020) 52:750–61. doi: 10.1038/s12276-020-0435-8
26. Courtney AH, Lo WL, and Weiss A. TCR signaling: mechanisms of initiation and propagation. Trends Biochem Sci. (2018) 43:108–23. doi: 10.1016/j.tibs.2017.11.008
27. Finlay D and Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. (2011) 11:109–17. doi: 10.1038/nri2888
28. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. (2009) 460:108–12. doi: 10.1038/nature08155
29. Powell JD and Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. (2010) 33:301–11. doi: 10.1016/j.immuni.2010.09.002
30. Kim EH and Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol. (2013) 4:20. doi: 10.3389/fimmu.2013.00020
31. Chen Y, Xu Z, Sun H, Ouyang X, Han Y, Yu H, et al. Regulation of CD8(+) T memory and exhaustion by the mTOR signals. Cell Mol Immunol. (2023) 20:1023–39. doi: 10.1038/s41423-023-01064-3
32. Fu G, Chen Y, Yu M, Podd A, Schuman J, He Y, et al. Phospholipase Cgamma1 is essential for T cell development, activation, and tolerance. J Exp Med. (2010) 207:309–18. doi: 10.1084/jem.20090880
33. Shah K, Al-Haidari A, Sun J, and Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther. (2021) 6:412. doi: 10.1038/s41392-021-00823-w
34. Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, and Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. (2002) 109:719–31. doi: 10.1016/S0092-8674(02)00767-5
35. Oh-hora M and Rao A. Calcium signaling in lymphocytes. Curr Opin Immunol. (2008) 20:250–8. doi: 10.1016/j.coi.2008.04.004
36. Malissen B, Gregoire C, Malissen M, and Roncagalli R. Integrative biology of T cell activation. Nat Immunol. (2014) 15:790–7. doi: 10.1038/ni.2959
37. Shen S, Chuck MI, Zhu M, Fuller DM, Yang CW, and Zhang W. The importance of LAT in the activation, homeostasis, and regulatory function of T cells. J Biol Chem. (2010) 285:35393–405. doi: 10.1074/jbc.M110.145052
38. Aguado E, Richelme S, Nunez-Cruz S, Miazek A, Mura AM, Richelme M, et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science. (2002) 296:2036–40. doi: 10.1126/science.1069057
39. Mingueneau M, Roncagalli R, Gregoire C, Kissenpfennig A, Miazek A, Archambaud C, et al. Loss of the LAT adaptor converts antigen-responsive T cells into pathogenic effectors that function independently of the T cell receptor. Immunity. (2009) 31:197–208. doi: 10.1016/j.immuni.2009.05.013
40. Brownlie R and Zamoyska R. LAT polices T cell activation. Immunity. (2009) 31:174–6. doi: 10.1016/j.immuni.2009.08.002
41. Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, and Samelson LE. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell. (1998) 92:83–92. doi: 10.1016/S0092-8674(00)80901-0
42. Zhu J, Yamane H, and Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
43. Gascoigne NR, Rybakin V, Acuto O, Brzostek J, and Signal Strength TCR. and T cell development. Annu Rev Cell Dev Biol. (2016) 32:327–48. doi: 10.1146/annurev-cellbio-111315-125324
44. Kim C, Wilson T, Fischer KF, and Williams MA. Sustained interactions between T cell receptors and antigens promote the differentiation of CD4(+) memory T cells. Immunity. (2013) 39:508–20. doi: 10.1016/j.immuni.2013.08.033
45. van Panhuys N, Klauschen F, and Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization In Vivo. Immunity. (2014) 41(1):63-74. doi: 10.1016/j.immuni.2014.06.003
46. Fazilleau N, McHeyzer-Williams LJ, Rosen H, and McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. (2009) 10:375–84. doi: 10.1038/ni.1704
47. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell. (2013) 153:785–96. doi: 10.1016/j.cell.2013.04.007
48. Hwang S, Palin AC, Li L, Song KD, Lee J, Herz J, et al. TCR ITAM multiplicity is required for the generation of follicular helper T-cells. Nat Commun. (2015) 6:6982. doi: 10.1038/ncomms7982
49. Snook JP, Kim C, and Williams MA. TCR signal strength controls the differentiation of CD4(+) effector and memory T cells. Sci Immunol. (2018) 3(25): eaas9103. doi: 10.1126/sciimmunol.aas9103
50. DiToro D, Winstead CJ, Pham D, Witte S, Andargachew R, Singer JR, et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science. (2018) 361(6407): eaao2933. doi: 10.1126/science.aao2933
51. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
52. Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, and Glimcher LH. T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci U S A. (2003) 100:7749–54. doi: 10.1073/pnas.1332767100
53. Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PloS One. (2011) 6:e17739. doi: 10.1371/journal.pone.0017739
54. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. (2013) 38:596–605. doi: 10.1016/j.immuni.2012.11.020
55. Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J Exp Med. (2015) 212:539–53. doi: 10.1084/jem.20141380
56. Keck S, Schmaler M, Ganter S, Wyss L, Oberle S, Huseby ES, et al. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc Natl Acad Sci U S A. (2014) 111:14852–7. doi: 10.1073/pnas.1403271111
57. Yamane H, Zhu J, and Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med. (2005) 202:793–804. doi: 10.1084/jem.20051304
58. Zhu J, Yamane H, Cote-Sierra J, Guo L, and Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. (2006) 16:3–10. doi: 10.1038/sj.cr.7310002
59. Brogdon JL, Leitenberg D, and Bottomly K. The potency of TCR signaling differentially regulates NFATc/p activity and early IL-4 transcription in naive CD4+ T cells. J Immunol. (2002) 168:3825–32. doi: 10.4049/jimmunol.168.8.3825
60. Jorritsma PJ, Brogdon JL, and Bottomly K. Role of TCR-induced extracellular signal-regulated kinase activation in the regulation of early IL-4 expression in naive CD4+ T cells. J Immunol. (2003) 170:2427–34. doi: 10.4049/jimmunol.170.5.2427
61. Malissen B, Aguado E, and Malissen M. Role of the LAT adaptor in T-cell development and Th2 differentiation. Adv Immunol. (2005) 87:1–25. doi: 10.1016/S0065-2776(05)87001-4
62. Dodeller F and Schulze-Koops H. The p38 mitogen-activated protein kinase signaling cascade in CD4 T cells. Arthritis Res Ther. (2006) 8:205. doi: 10.1186/ar1905
63. Ashwell JD. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol. (2006) 6:532–40. doi: 10.1038/nri1865
64. Jun JE, Kulhanek KR, Chen H, Chakraborty A, and Roose JP. Alternative ZAP70-p38 signals prime a classical p38 pathway through LAT and SOS to support regulatory T cell differentiation. Sci Signal. (2019) 12(591):eaa0736. doi: 10.1126/scisignal.aao0736
65. Carbone F, Russo C, Colamatteo A, La Rocca C, Fusco C, Matarese A, et al. Cellular and molecular signaling towards T cell immunological self-tolerance. J Biol Chem. (2024) 300:107134. doi: 10.1016/j.jbc.2024.107134
66. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. (2008) 105:7797–802. doi: 10.1073/pnas.0800928105
67. Li MO and Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol. (2016) 16:220–33. doi: 10.1038/nri.2016.26
68. Hori S, Nomura T, and Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
69. Lu L, Barbi J, and Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol. (2017) 17:703–17. doi: 10.1038/nri.2017.75
70. Zheng Y and Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. (2007) 8:457–62. doi: 10.1038/ni1455
71. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. (2011) 208:1279–89. doi: 10.1084/jem.20110308
72. Hsieh CS, Zheng Y, Liang Y, Fontenot JD, and Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. (2006) 7:401–10. doi: 10.1038/ni1318
73. Cozzo Picca C, Simons DM, Oh S, Aitken M, Perng OA, Mergenthaler C, et al. CD4(+)CD25(+)Foxp3(+) regulatory T cell formation requires more specific recognition of a self-peptide than thymocyte deletion. Proc Natl Acad Sci U S A. (2011) 108:14890–5. doi: 10.1073/pnas.1103810108
74. Feng Y, van der Veeken J, Shugay M, Putintseva EV, Osmanbeyoglu HU, Dikiy S, et al. A mechanism for expansion of regulatory T-cell repertoire and its role in self-tolerance. Nature. (2015) 528:132–6. doi: 10.1038/nature16141
75. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PloS Biol. (2007) 5:e38. doi: 10.1371/journal.pbio.0050038
76. Kim HP and Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. (2007) 204:1543–51. doi: 10.1084/jem.20070109
77. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. (2012) 37:785–99. doi: 10.1016/j.immuni.2012.09.010
78. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
79. Marie JC, Letterio JJ, Gavin M, and Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. (2005) 201:1061–7. doi: 10.1084/jem.20042276
80. Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, et al. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med. (2014) 211:529–43. doi: 10.1084/jem.20131459
81. Schmierer B and Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. (2007) 8:970–82. doi: 10.1038/nrm2297
82. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. (2015) 16:188–96. doi: 10.1038/ni.3077
83. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
84. Levine AG, Arvey A, Jin W, and Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. (2014) 15:1070–8. doi: 10.1038/ni.3004
85. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. (2014) 41:722–36. doi: 10.1016/j.immuni.2014.10.012
86. Daniel C, Wennhold K, Kim HJ, and von Boehmer H. Enhancement of antigen-specific Treg vaccination in vivo. Proc Natl Acad Sci U S A. (2010) 107:16246–51. doi: 10.1073/pnas.1007422107
87. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. (2002) 3:549–57. doi: 10.1038/ni794
88. Do MH, Wang X, Zhang X, Chou C, Nixon BG, Capistrano KJ, et al. Nutrient mTORC1 signaling underpins regulatory T cell control of immune tolerance. J Exp Med. (2020) 217(1): e20190848. doi: 10.1084/jem.20190848
89. Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, and Kopf M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci U S A. (2009) 106:876–81. doi: 10.1073/pnas.0810769106
90. Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, et al. Differential expression of interleukin-17A and -17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity. (2009) 31:587–97. doi: 10.1016/j.immuni.2009.07.009
91. Qin Z, Hou P, Lin H, Chen M, Wang R, and Xu T. Inhibition of Lck/Fyn kinase activity promotes the differentiation of induced Treg cells through AKT/mTOR pathway. Int Immunopharmacol. (2024) 134:112237. doi: 10.1016/j.intimp.2024.112237
92. Kaplan MH, Schindler U, Smiley ST, and Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. (1996) 4:313–9. doi: 10.1016/S1074-7613(00)80439-2
93. Zhu J, Guo L, Watson CJ, Hu-Li J, and Paul WE. Stat6 is necessary and sufficient for IL-4’s role in Th2 differentiation and cell expansion. J Immunol. (2001) 166:7276–81. doi: 10.4049/jimmunol.166.12.7276
94. Usui T, Nishikomori R, Kitani A, and Strober W. GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL-12Rbeta2 chain or T-bet. Immunity. (2003) 18:415–28. doi: 10.1016/S1074-7613(03)00057-8
95. Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, et al. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. (2008) 29:679–90. doi: 10.1016/j.immuni.2008.08.017
96. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell. (2012) 151:289–303. doi: 10.1016/j.cell.2012.09.016
97. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
98. Lee JY, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, et al. Serum amyloid A proteins induce pathogenic th17 cells and promote inflammatory disease. Cell. (2020) 180:79–91 e16. doi: 10.1016/j.cell.2019.11.026
99. Zhou L, Ivanov II, R. Spolski R, Shenderov K, Egawa T, Levy DE, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
100. Eizenberg-Magar I, Rimer J, Zaretsky I, Lara-Astiaso D, Reich-Zeliger S, and Friedman N. Diverse continuum of CD4(+) T-cell states is determined by hierarchical additive integration of cytokine signals. Proc Natl Acad Sci U S A. (2017) 114:E6447–56. doi: 10.1073/pnas.1615590114
101. Hegazy AN, Peine C, Niesen D, Panse I, Vainshtein Y, Kommer C, et al. Plasticity and lineage commitment of individual T(H)1 cells are determined by stable T-bet expression quantities. Sci Adv. (2024) 10:eadk2693. doi: 10.1126/sciadv.adk2693
102. Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, et al. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol. (2015) 16:286–95. doi: 10.1038/ni.3099
103. Jeltsch KM, Hu D, Brenner S, Zoller J, Heinz GA, Nagel D, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol. (2014) 15:1079–89. doi: 10.1038/ni.3008
104. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. (2012) 12:325–38. doi: 10.1038/nri3198
105. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic control of T(H)17 and induced T(reg) cell balance by an epigenetic mechanism. Nature. (2017) 548:228–33. doi: 10.1038/nature23475
106. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
107. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. (2014) 20:1327–33. doi: 10.1038/nm.3704
108. Haghikia A, Jorg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. (2015) 43:817–29. doi: 10.1016/j.immuni.2015.09.007
109. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. (2013) 14:489–99. doi: 10.1038/ni.2570
110. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. (2016) 531:651–5. doi: 10.1038/nature17412
111. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. (2008) 134:97–111. doi: 10.1016/j.cell.2008.04.052
112. Repa JJ and Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. (2000) 16:459–81. doi: 10.1146/annurev.cellbio.16.1.459
113. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science. (2014) 343:432–7. doi: 10.1126/science.1247606
114. Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest. (2011) 121:658–70. doi: 10.1172/JCI42974
115. Santori FR, Huang P, van de Pavert SA, Douglass EF Jr., Leaver DJ, Haubrich BA, et al. Identification of natural RORgamma ligands that regulate the development of lymphoid cells. Cell Metab. (2015) 21:286–98. doi: 10.1016/j.cmet.2015.01.004
116. Daniels MA and Teixeiro E. TCR signaling in T cell memory. Front Immunol. (2015) 6:617. doi: 10.3389/fimmu.2015.00617
117. Arens R and Schoenberger SP. Plasticity in programming of effector and memory CD8 T-cell formation. Immunol Rev. (2010) 235:190–205. doi: 10.1111/j.0105-2896.2010.00899.x
118. Riquelme E, Carreno LJ, Gonzalez PA, and Kalergis AM. The duration of TCR/pMHC interactions regulates CTL effector function and tumor-killing capacity. Eur J Immunol. (2009) 39:2259–69. doi: 10.1002/eji.200939341
119. Nayar R, Enos M, Prince A, Shin H, Hemmers S, Jiang JK, et al. TCR signaling via Tec kinase ITK and interferon regulatory factor 4 (IRF4) regulates CD8+ T-cell differentiation. Proc Natl Acad Sci U S A. (2012) 109:E2794–802. doi: 10.1073/pnas.1205742109
120. Rao RR, Li Q, Odunsi K, and Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. (2010) 32:67–78. doi: 10.1016/j.immuni.2009.10.010
121. Solouki S, Huang W, Elmore J, Limper C, Huang F, and August A. TCR signal strength and antigen affinity regulate CD8(+) memory T cells. J Immunol. (2020) 205:1217–27. doi: 10.4049/jimmunol.1901167
122. Pearce EL and Shen H. Making sense of inflammation, epigenetics, and memory CD8+ T-cell differentiation in the context of infection. Immunol Rev. (2006) 211:197–202. doi: 10.1111/j.0105-2896.2006.00399.x
123. Kaech SM and Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
124. Roberts AD, Ely KH, and Woodland DL. Differential contributions of central and effector memory T cells to recall responses. J Exp Med. (2005) 202:123–33. doi: 10.1084/jem.20050137
125. Grabnitz F, Stark D, Shlesinger D, Petkidis A, Borsa M, Yermanos A, et al. Asymmetric cell division safeguards memory CD8 T cell development. Cell Rep. (2023) 42:112468. doi: 10.1016/j.celrep.2023.112468
126. Kavazovic I, Polic B, and Wensveen FM. Cheating the hunger games; mechanisms controlling clonal diversity of CD8 effector and memory populations. Front Immunol. (2018) 9:2831. doi: 10.3389/fimmu.2018.02831
127. Burger ML, Cruz AM, Crossland GE, Gaglia G, Ritch CC, Blatt SE, et al. Antigen dominance hierarchies shape TCF1(+) progenitor CD8 T cell phenotypes in tumors. Cell. (2021) 184:4996–5014 e26. doi: 10.1016/j.cell.2021.08.020
128. Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, et al. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol. (2020) 21:1256–66. doi: 10.1038/s41590-020-0760-z
129. Bhandarkar V, Dinter T, and Spranger S. Architects of immunity: How dendritic cells shape CD8(+) T cell fate in cancer. Sci Immunol. (2025) 10:eadf4726. doi: 10.1126/sciimmunol.adf4726
130. Shakiba M, Zumbo P, Espinosa-Carrasco G, Menocal L, Dundar F, Carson SE, et al. TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J Exp Med. (2022) 219(2): e20201966. doi: 10.1084/jem.20201966
131. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
132. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. (2019) 50:195–211 e10. doi: 10.1016/j.immuni.2018.12.021
133. Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. (2019) 576:465–70. doi: 10.1038/s41586-019-1836-5
134. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(-)CD8(+) tumor-infiltrating T cells. Immunity. (2019) 50:181–194 e6. doi: 10.1016/j.immuni.2018.11.014
135. Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity. (2013) 39:833–45. doi: 10.1016/j.immuni.2013.10.007
136. Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, et al. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol. (2014) 192:5881–93. doi: 10.4049/jimmunol.1303187
137. Cho JH, Kim HO, Ju YJ, Kye YC, Lee GW, Lee SW, et al. CD45-mediated control of TCR tuning in naive and memory CD8(+) T cells. Nat Commun. (2016) 7:13373. doi: 10.1038/ncomms13373
138. Duong MN, Erdes E, Hebeisen M, and Rufer N. Chronic TCR-MHC (self)-interactions limit the functional potential of TCR affinity-increased CD8 T lymphocytes. J Immunother Cancer. (2019) 7:284. doi: 10.1186/s40425-019-0773-z
139. Tewari K, Walent J, Svaren J, Zamoyska R, and Suresh M. Differential requirement for Lck during primary and memory CD8+ T cell responses. Proc Natl Acad Sci U S A. (2006) 103:16388–93. doi: 10.1073/pnas.0602565103
140. Richer MJ, Nolz JC, and Harty JT. Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity. (2013) 38:140–52. doi: 10.1016/j.immuni.2012.09.017
141. Castro I, Dee MJ, and Malek TR. Transient enhanced IL-2R signaling early during priming rapidly amplifies development of functional CD8+ T effector-memory cells. J Immunol. (2012) 189:4321–30. doi: 10.4049/jimmunol.1202067
142. Chin SS, Guillen E, Chorro L, Achar S, Ng K, Oberle S, et al. T cell receptor and IL-2 signaling strength control memory CD8(+) T cell functional fitness via chromatin remodeling. Nat Commun. (2022) 13:2240. doi: 10.1038/s41467-022-29718-2
143. Balmer ML, Ma EH, Bantug GR, Grählert J, Pfister S, Glatter T, et al. Memory CD8 T cells require increased concentrations of acetate induced by stress for optimal function. Immunity. (2016) 44:1312–24. doi: 10.1016/j.immuni.2016.03.016
144. Geltink RIK, Kyle RL, and Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol. (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
145. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
146. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. (2013) 123:4479–88. doi: 10.1172/JCI69589
147. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
148. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
149. Verbist KC, Guy CS, Milasta S, Liedmann S, Kaminski MM, Wang R, et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature. (2016) 532:389–93. doi: 10.1038/nature17442
150. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
151. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
152. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
153. Wherry EJ and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
154. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
155. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
156. Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest. (2015) 125:2090–108. doi: 10.1172/JCI77746
157. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
158. Raud B, McGuire PJ, Jones RG, Sparwasser T, and Berod L. Fatty acid metabolism in CD8(+) T cell memory: Challenging current concepts. Immunol Rev. (2018) 283:213–31. doi: 10.1111/imr.2018.283.issue-1
159. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. (2017) 32:377–391 e9. doi: 10.1016/j.ccell.2017.08.004
160. Tsai YL, Arias-Badia M, Kadlecek TA, Lwin YM, Srinath A, Shah NH, et al. TCR signaling promotes formation of an STS1-Cbl-b complex with pH-sensitive phosphatase activity that suppresses T cell function in acidic environments. Immunity. (2023) 56:2682–2698 e9. doi: 10.1016/j.immuni.2023.11.010
161. Meng KP, Majedi FS, Thauland TJ, and Butte MJ. Mechanosensing through YAP controls T cell activation and metabolism. J Exp Med. (2020) 217(8): e20200053. doi: 10.1084/jem.20200053
162. Jin W, Tamzalit F, Chaudhuri PK, Black CT, Huse M, and Kam LC. T cell activation and immune synapse organization respond to the microscale mechanics of structured surfaces. Proc Natl Acad Sci U S A. (2019) 116:19835–40. doi: 10.1073/pnas.1906986116
163. Choi HK, Cong P, Ge C, Natarajan A, Liu B, Zhang Y, et al. Catch bond models may explain how force amplifies TCR signaling and antigen discrimination. Nat Commun. (2023) 14:2616. doi: 10.1038/s41467-023-38267-1
164. Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, and Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci U S A. (2010) 107:9777–82. doi: 10.1073/pnas.0914127107
165. Chu T, Wu M, Hoellbacher B, de Almeida GP, Wurmser C, Berner J, et al. Precursors of exhausted T cells are preemptively formed in acute infection. Nature. (2025) 640(8059):782-792. doi: 10.1038/s41586-024-08451-4
166. Zehn D, Lee SY, and Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. (2009) 458:211–4. doi: 10.1038/nature07657
167. Straub A, Grassmann S, Jarosch S, Richter L, Hilgendorf P, Hammel M, et al. Recruitment of epitope-specific T cell clones with a low-avidity threshold supports efficacy against mutational escape upon re-infection. Immunity. (2023) 56:1269–1284 e6. doi: 10.1016/j.immuni.2023.04.010
168. McManus DT, Valanparambil RM, Medina CB, Scharer CD, McGuire DJ, Sobierajska E, et al. An early precursor CD8 T cell that adapts to acute or chronic viral infection. Nature. (2025) 640(8059):772-781. doi: 10.1038/s41586-024-08562-y
169. Fagerberg E, Attanasio J, Dien C, Singh J, Kessler EA, Abdullah L, et al. KLF2 maintains lineage fidelity and suppresses CD8 T cell exhaustion during acute LCMV infection. Science. (2025) 387:eadn2337. doi: 10.1126/science.adn2337
170. Okoye IS, Houghton M, Tyrrell L, Barakat K, and Elahi S. Coinhibitory receptor expression and immune checkpoint blockade: maintaining a balance in CD8(+) T cell responses to chronic viral infections and cancer. Front Immunol. (2017) 8:1215. doi: 10.3389/fimmu.2017.01215
171. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. (2010) 107:14733–8. doi: 10.1073/pnas.1009731107
172. Speiser DE, Utzschneider DT, Oberle SG, Munz C, Romero P, and Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat Rev Immunol. (2014) 14:768–74. doi: 10.1038/nri3740
173. Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A. (2018) 115:4749–54. doi: 10.1073/pnas.1718217115
174. Sandu I, Cerletti D, Claassen M, and Oxenius A. Exhausted CD8(+) T cells exhibit low and strongly inhibited TCR signaling during chronic LCMV infection. Nat Commun. (2020) 11:4454. doi: 10.1038/s41467-020-18256-4
175. Zhang J, Lei F, and Tan H. The development of CD8 T-cell exhaustion heterogeneity and the therapeutic potentials in cancer. Front Immunol. (2023) 14:1166128. doi: 10.3389/fimmu.2023.1166128
176. Presotto D, Erdes E, Duong MN, Allard M, Regamey PO, Quadroni M, et al. Fine-tuning of optimal TCR signaling in tumor-redirected CD8 T cells by distinct TCR affinity-mediated mechanisms. Front Immunol. (2017) 8:1564. doi: 10.3389/fimmu.2017.01564
177. Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. (2019) 25:82–8. doi: 10.1038/s41591-018-0290-5
178. Harris DT and Kranz DM. Adoptive T cell therapies: A comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci. (2016) 37:220–30. doi: 10.1016/j.tips.2015.11.004
179. Hoffmann MM and Slansky JE. T-cell receptor affinity in the age of cancer immunotherapy. Mol Carcinog. (2020) 59:862–70. doi: 10.1002/mc.23212
180. Schlenker R, Olguin-Contreras LF, Leisegang M, Schnappinger J, Disovic A, Ruhland S, et al. Chimeric PD-1:28 receptor upgrades low-avidity T cells and restores effector function of tumor-infiltrating lymphocytes for adoptive cell therapy. Cancer Res. (2017) 77:3577–90. doi: 10.1158/0008-5472.CAN-16-1922
181. Schenkel JM and Masopust D. Tissue-resident memory T cells. Immunity. (2014) 41:886–97. doi: 10.1016/j.immuni.2014.12.007
182. Mueller SN and Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. (2016) 16:79–89. doi: 10.1038/nri.2015.3
183. Abdelbary M, Hobbs SJ, Gibbs JS, Yewdell JW, and Nolz JC. T cell receptor signaling strength establishes the chemotactic properties of effector CD8(+) T cells that control tissue-residency. Nat Commun. (2023) 14:3928. doi: 10.1038/s41467-023-39592-1
184. Fiege JK, Stone IA, Fay EJ, Markman MW, Wijeyesinghe S, Macchietto MG, et al. The impact of TCR signal strength on resident memory T cell formation during influenza virus infection. J Immunol. (2019) 203:936–45. doi: 10.4049/jimmunol.1900093
185. Enamorado M, Khouili SC, Iborra S, and Sancho D. Genealogy, dendritic cell priming, and differentiation of tissue-resident memory CD8(+) T cells. Front Immunol. (2018) 9:1751. doi: 10.3389/fimmu.2018.01751
186. Iborra S, Martinez-Lopez M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, et al. Optimal Generation of Tissue-Resident but Not Circulating Memory T Cells during Viral Infection Requires Crosspriming by DNGR-1(+) Dendritic Cells. Immunity. (2016) 45:847–60. doi: 10.1016/j.immuni.2016.08.019
187. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, and Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. (2013) 14:1285–93. doi: 10.1038/ni.2745
188. Dijkgraaf FE, Kok L, and Schumacher TNM. Formation of tissue-resident CD8(+) T-cell memory. Cold Spring Harb Perspect Biol. (2021) 13(8):a038117. doi: 10.1101/cshperspect.a038117
Keywords: TCR signaling, T cell fate, T cell differentiation, TCR signal strength, immunometabolism, T cell exhaustion, Tpex, memory T cells
Citation: Qin Z and Xu T (2025) Deciphering the deterministic role of TCR signaling in T cell fate determination. Front. Immunol. 16:1562248. doi: 10.3389/fimmu.2025.1562248
Received: 17 January 2025; Accepted: 29 April 2025;
Published: 21 May 2025.
Edited by:
Ewoud Bernardus Compeer, University of Oxford, United KingdomReviewed by:
Noa B. Martin-Cofreces, Princess University Hospital, SpainMaria N. Navarro, Spanish National Research Council (CSIC), Spain
Copyright © 2025 Qin and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Xu, eHV0YW8yNUBtYWlsLnN5c3UuZWR1LmNu