 Mingjun Yao1†
Mingjun Yao1† Wei Zhao
Wei Zhao Siyuan Song
Siyuan Song Yi Wang
Yi Wang- 1School of Medicine, University of Electronic Science and Technology of China, Chengdu, China
- 2Pathology Department, University of Texas, MD Anderson Cancer Center, Houston, TX, United States
- 3Department of Neuroscience, Baylor College of Medicine, Houston, TX, United States
- 4Department of Critical Care Medicine, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, Sichuan, China
- 5Translational Clinical Immunology Medicine Key Laboratory of Sichuan Province, Center of Organ Transplantation, Sichuan Academy of Medical Sciences and Sichuan Provincial People's Hospital, Chengdu, Sichuan, China
Idiopathic pulmonary fibrosis (IPF) is a fatal interstitial lung disease characterized by progressive scarring, alveolar destruction, and limited therapeutic options. Although the exact etiology of IPF remains unclear, emerging evidence suggests that ferroptosis, an iron-dependent form of regulated cell death driven by lipid peroxidation and oxidative stress, plays a significant role in its pathogenesis. Ferroptotic stress not only compromises alveolar epithelial cell integrity, but also triggers inflammatory responses and profibrotic signaling cascades that activate and sustain fibroblast dysfunction. This review delineates the core regulatory pathways of ferroptosis, iron metabolism, lipid peroxidation, antioxidant defenses, mitochondrial remodeling, and RNA editing, with an emphasis on their relevance in IPF. We explore how epithelial injury and macrophage-derived signals initiate ferroptosis, and how fibroblast subsets, shaped by scRNA-seq-defined heterogeneity and plasticity, respond to these cues by reinforcing ECM deposition and oxidative stress. Therapeutic avenues targeting ferroptosis, including antioxidant supplementation, iron chelation, and modulation of lipid metabolism, are discussed alongside cell-specific interventions and nanodelivery strategies. By integrating recent advances in molecular profiling and ferroptosis biology, this review provides a framework for leveraging ferroptosis as a tractable target in IPF and identifies novel directions for precision antifibrotic therapy.
1 Introduction
1.1 Overview of pulmonary fibrosis
Pulmonary fibrosis (PF) encompasses a group of chronic, progressive lung diseases characterized by excessive extracellular matrix (ECM) deposition, tissue remodeling, and loss of lung elasticity. These pathological changes impair gas exchange and ultimately lead to respiratory failure, with advanced cases often necessitating lung transplantation (1). The development of PF involves a complex interplay of genetic predisposition, environmental exposures, and dysregulated tissue repair. Repeated alveolar epithelial injury, unresolved inflammation, and aberrant fibroblast activation drive progressive scarring, as activated fibroblasts secrete ECM components that perpetuate fibrosis (2). Despite significant advances in understanding the molecular mechanisms underlying PF, effective treatment options remain limited, underscoring the need for continued research.
1.2 Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF), the most severe and common subtype of PF, affects approximately 25,000 new individuals annually in the United States. Histologically, IPF is defined by the usual interstitial pneumonia (UIP) pattern, characterized by patchy fibrosis, fibroblastic foci, and honeycombing (3). Unlike other interstitial lung diseases, IPF follows a particularly aggressive clinical course, with a median survival of only 3–5 years after diagnosis (4).Current antifibrotic therapies, including pirfenidone and nintedanib, may slow disease progression but fail to halt or reverse fibrosis, highlighting the urgent need for more effective treatments (5).
1.3 Ferroptosis and IPF
Ferroptosis, a unique form of cell death derived by iron overload and lipid peroxidation, has emerged as a critical contributor to oxidative stress and chronic inflammation in IPF. This process disrupts alveolar epithelial integrity, activating profibrotic signaling pathways, including fibroblasts proliferation, and ECM deposition—hallmarks of IPF pathogenesis (6). Unlike apoptosis, ferroptosis is non-apoptotic and pro-inflammatory, amplifying tissue injury and perpetuating fibrotic remodeling. As such, targeting ferroptosis represents a promising therapeutic strategy (7).
This review aims to synthesize emerging insights into the mechanistic role of ferroptosis in IPF, with a particular focus on how ferroptotic stress influences fibroblast responses, epithelial-fibroblast crosstalk, and fibrotic progression. We highlight recent findings on the cellular and molecular mechanisms driving ferroptosis, its impact on tissue remodeling, and potential therapeutic opportunities. By examining both epithelial and stromal contributions to ferroptosis-driven pathology, we aim to provide a framework for identifying novel targets for disease interception and treatment.
2 Ferroptosis: core mechanisms and regulatory pathways
Ferroptosis is a distinct form of regulated cell death that differs fundamentally from apoptosis, necroptosis, and pyroptosis. Unlike these pathways, which involve caspase activation, membrane pore formation, or DNA fragmentation, ferroptosis is driven by iron-catalyzed lipid peroxidation and oxidative damage to cellular membranes. Its regulation centers on four interconnected processes: iron metabolism, polyunsaturated fatty acids (PUFA) peroxidation, antioxidant defense failure, and mitochondrial dysfunction (8).
2.1 Iron metabolism and ferroptosis
Iron is central to ferroptosis, as its redox activity catalyzes the formation of reactive oxygen species (ROS) via Fenton reactions, triggering cellular membranes damage and cell death (9). Cellular iron homeostasis is maintained by tightly regulated pathways involving iron import (e.g., transferrin receptor 1 [TfR1]), export (e.g., ferroportin), and storage (e.g., ferritin) (10–12). Disruption of this balance leads to labile iron accumulation, sensitizing cells to ferroptosis (13).
A critical regulator of intracellular iron levels is ferritinophagy, the selective autophagic degradation of ferritin mediated by nuclear receptor coactivator 4 (NCOA4). This process releases stored iron into the cytosol, increasing susceptibility to ferroptosis (14). Recent studies have shown that enhanced ferritinophagy in stressed alveolar epithelial cells contributes to iron overload and ferroptotic injury in pulmonary fibrosis models (7).
In the context of IPF, elevated pulmonary iron levels and altered expression of iron-handling proteins have been observed in patient lungs, particularly in fibrotic regions (6). Experimental inhibition of iron accumulation or ferritinophagy using deferoxamine or NCOA4 knockdown attenuates epithelial cell death and collagen deposition in bleomycin-induced fibrosis models, supporting the pathogenic role of iron dysregulation in IPF (15).
Together, these findings underscore aberrant ion metabolism, notably excessive import, ferritin degradation, and impaired export, as a key initiator of ferroptosis in IPF and related fibrotic lung diseases. Targeting these dynamics may attenuate the epithelial damage and profibrotic microenvironment characteristic of IPF.
2.2 Lipid peroxidation and PUFA susceptibility
Lipid peroxidation of polyunsaturated fatty acids (PUFAs), particularly within phospholipid membranes, is a defining feature of ferroptosis (16). This process is driven by the enzymatic and non-enzymatic oxidation of PUFA-containing phosphatidylethanolamines (PUFA-PEs), such as those incorporating arachidonic acid (AA) and adrenic acid (AdA). Among these, AdA-phosphatidylethanolamine (AdA-PE) has emerged as a critical pro-ferroptotic lipid species, whose oxidation initiates membrane damage and cell death.
Key enzymes regulate the synthesis and incorporation of PUFAs into membrane phospholipids. Acyl-CoA synthetase long-chain family member 4 (ACSL4) activates arachidonic acid (AA) and adrenic acid (AdA) to form PUFA-CoAs, which are subsequently esterified into phospholipids by Lysophosphatidylcholine acyltransferase 3 (LPCAT3), yielding PUFA-PEs such as AdA-PE (17, 18). These PUFA-PEs are highly prone to peroxidation, especially under oxidative stress. Lipoxygenases (LOXs), such as ALOX15, enzymatically oxidize these substrates, producing lipid hydroperoxides (PE-OOH) that execute ferroptosis (19, 20).
In the setting of IPF, elevated ACSL4 and LOXs expression has been documented in fibrotic lung tissue and alveolar epithelial cell, correlating with enhanced lipid peroxidation and fibrotic progression (21–23). Genetic or pharmacological inhibition of ACSL4 or LOXs reduces lipid peroxidation, preserves epithelial cell integrity, and mitigates fibrotic remodeling (21, 24, 25). Furthermore, LPCAT3 deficiency confers partial resistance to ferroptosis by reducing PUFA incorporation into membranes, highlighting its potential as a regulatory checkpoint and therapeutic target (26).
These findings underscore the significance of PUFA metabolism and lipid peroxidation in ferroptosis execution and IPF pathogenesis. Targeting these upstream processes may attenuate ferroptotic epithelial damage and downstream fibroblast activation.
2.3 Antioxidant defense
Oxidative lipid damage is a hallmark of ferroptosis, counteracted by a network of antioxidant systems that preserve membrane integrity and cell survival. Among these, the glutathione peroxidase 4 (GPX4) pathway serves as the central defense mechanism. GPX4 utilizes reduced glutathione (GSH) to neutralize lipid hydroperoxides (PUFA-PE-OOH) into non-toxic lipid alcohols, thereby preventing ferroptotic cell death (Figure 1) (27). In IPF, both GPX4 activity and intracellular GSH levels are significantly diminished in injured alveolar epithelial cells, linking ferroptosis to epithelial dysfunction and fibrosis (28).
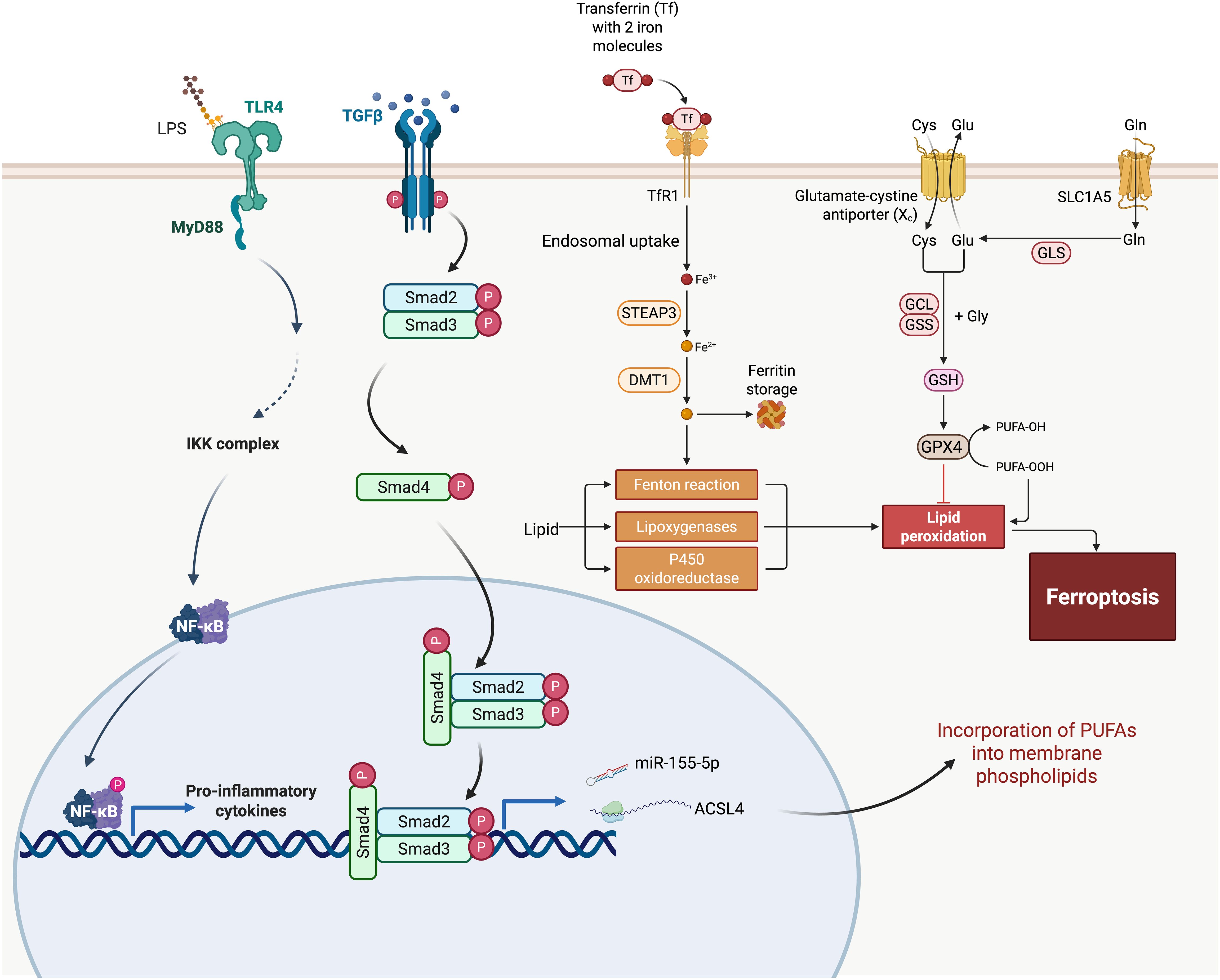
Figure 1. Integration of ferroptosis pathways with fibrogenic signaling in idiopathic pulmonary fibrosis (IPF). This schematic outlines the molecular crosstalk between ferroptosis and pro-fibrotic pathways in IPF. Iron uptake through transferrin receptor 1 (TfR1) and reduction by STEAP3 and DMT1 leads to intracellular Fe²⁺ accumulation, which catalyzes ROS production via the Fenton reaction. Oxidative lipid metabolism mediated by lipoxygenases and P450 oxidoreductase contributes to lipid peroxidation, a key step in ferroptosis. Glutathione (GSH) synthesis and GPX4 activity act as antioxidant defenses against lipid peroxides, while ACSL4 facilitates the incorporation of polyunsaturated fatty acids (PUFAs) into membrane phospholipids, increasing vulnerability to peroxidation. The figure also illustrates how ferroptosis intersects with the TGF-β/Smad and TLR4/NF-κB pathways—major drivers of fibrosis. TGF-β signaling activates Smad2/3/4 complexes, leading to transcriptional induction of fibrosis-associated genes. Concurrently, TLR4 signaling via MyD88 activates NF-κB, promoting the expression of pro-inflammatory cytokines. These pathways may be further amplified by ferroptotic cell death and ROS, exacerbating tissue damage and fibrotic remodeling. miR-155-5p and other regulators are implicated in enhancing ACSL4 expression, linking immune signaling to ferroptotic susceptibility. Together, this figure illustrates a feedforward loop between ferroptosis and fibrogenesis in IPF.
Beyond GPX4, alternative systems also protect cells from lipid peroxidation. Ferroptosis suppressor protein 1 (FSP1) functions independently of GSH/GPX4 by reducing coenzyme Q10 (CoQ10) to its antioxidant form, CoQH2, which directly quenches lipid radicals in the plasma membrane (29). Activation of FSP1 has shown protective effects against ferroptosis in non-pulmonary systems, and emerging data suggest that this axis may also safeguard lung epithelium, though its role in IPF warrants further exploration (30–35).
Another key layer of defense is governed by the transcription factor nuclear factor erythroid 2–related factor 2 (NRF2). Upon oxidative stress, NRF2 translocates to the nucleus and induces expression of genes involved in GSH synthesis (e.g., GCLC, GCLM) (36), iron sequestration (e.g., FTH1), and detoxification enzymes (37). In pulmonary fibrosis models, NRF2 deficiency exacerbates lung injury and inflammation (38), while pharmacological activation of NRF2 restores redox balance, limits ferroptotic damage, and ameliorates fibrotic remodeling (37, 37).
Additionally, the GTP cyclohydrolase-1 (GCH1)/tetrahydrobiopterin (BH4) axis has recently been identified as a novel ferroptosis defense mechanism. GCH1 catalyzes the synthesis of BH4, a potent radical-trapping antioxidant that also preserves CoQ10 levels and stabilizes lipid membranes (39, 40). BH4 supplementation or GCH1 overexpression has been shown to suppress ferroptosis in oxidative injury models and may represent a therapeutic option for fibrotic lung conditions (39, 41).
Collectively, these antioxidant mechanisms—GPX4/GSH, FSP1/CoQ10, NRF2 signaling, and GCH1/BH4—interact to mitigate ferroptotic damage in epithelial cells. In the setting of IPF, where these defenses are often compromised, restoring or enhancing antioxidant capacity may not only protect alveolar integrity but also interrupt the feedback loop of oxidative stress and fibroblast activation that drives fibrosis.
2.4 Mitochondrial involvement and metabolic rewriting
Mitochondria serve as key modulators of ferroptosis sensitivity through their roles in reactive oxygen species (ROS) production, metabolic flux, and the maintenance of iron–sulfur (Fe–S) clusters (42). Under physiological conditions, mitochondrial respiration tightly regulates ROS levels. However, in the fibrotic lung microenvironment, persistent epithelial stress and mitochondrial dysfunction promote excessive mitochondrial ROS (mtROS), which synergize with cytosolic lipid peroxidation to drive ferroptotic cell death (43).
In IPF, alveolar epithelial cells exhibit disrupted mitochondrial dynamics and defective oxidative phosphorylation, which contribute to elevated mtROS production (44). These ROS not only damage mitochondrial membranes but also amplify lipid peroxidation cascades initiated in the cytoplasm, sensitizing cells to ferroptosis (45). Targeting mtROS with agents such as MitoQ have been shown to attenuate ferroptosis and fibrosis in preclinical models by restoring mitochondrial function and reducing lipid ROS (46).
Additionally, mitochondria are crucial for the synthesis and maintenance of Fe–S clusters, which are essential cofactors for numerous redox enzymes. Loss of Fe–S cluster stability, often observed under oxidative stress, leads to the release of free iron into the mitochondrial matrix, exacerbating Fenton chemistry and fueling ferroptosis (42). This mitochondrial iron dysregulation creates a feed-forward loop of oxidative injury that accelerates epithelial dysfunction and fibrosis.
These findings highlight the pivotal role of mitochondrial dysfunction in ferroptosis-driven fibrosis. Interventions aimed at restoring mitochondrial health represent a promising strategy to break the cycle of epithelial injury and fibrogenesis.
2.5 Potential biomarkers of ferroptosis
Translating ferroptosis from a mechanistic concept into a clinical tool for IPF will require reliable, minimally invasive biomarkers that reflect iron overload, lipid peroxidation, and antioxidant failure in the lung.
2.5.1 Lipid−peroxidation byproducts
Quantification of malondialdehyde (MDA) and 4−hydroxynonenal (4−HNE) in bronchoalveolar lavage fluid (BALF) or exhaled breath condensate provides a direct readout of membrane lipid damage (47). Elevated MDA and 4−HNE levels correlate with disease severity and have been reported in fibrotic lung models (48).
2.5.2 Enzymatic and protein markers
GPX4 activity or expression can be measured by ELISA or immunohistochemistry on BALF cells or transbronchial biopsies; reduced GPX4 is a hallmark of ferroptotic susceptibility (49).
ACSL4 and LOX family members are often upregulated in IPF lung tissue; their mRNA or protein levels may serve as positive indicators of ongoing lipid remodeling and peroxidation (50).
2.5.3 Iron−handling molecules
Ferritin, transferrin saturation, and labile iron pool measurements in serum or BALF reflect systemic and alveolar iron loading (51).
NCOA4 expression in airway epithelial cells—assessed by qPCR—may indicate enhanced ferritinophagy and intracellular iron release (52).
2.5.4 Ferroptosis−related gene signatures
Transcriptomic analyses of IPF BALF have identified an eight−gene ferroptosis signature (NRAS, EMP1, SLC40A1, MYC, ANGPTL4, PRKCA, MUC1, GABARAPL1) that distinguishes IPF patients from healthy controls and associates with prognosis. In murine bleomycin models, 20 additional ferroptosis−linked genes (e.g., ALOX15, CDO1, JUN, SLC2A1, GPX2) were differentially expressed in fibrotic versus normal lung (53).
Taken together, these biomarkers capture complementary facets of ferroptosis—upstream iron dysregulation, core lipid peroxidation events, failed antioxidant defenses, and downstream gene expression changes. Future efforts should standardize assay techniques, validate candidate markers in large IPF cohorts, and develop multi−analyte panels that maximize sensitivity and specificity for early detection, patient stratification, and monitoring of ferroptosis−targeted therapies.
3 Ferroptosis in the development of IPF
3.1 Environmental and intrinsic triggers
Alveolar epithelial cells (AECs), particularly type II alveolar epithelial cells (AECIIs), are central to the initiation and progression of IPF due to their roles in maintaining alveolar integrity, surfactant production, and epithelial regeneration (54–56). A growing body of evidence indicates that these cells exhibit heightened susceptibility to ferroptosis under stress conditions, rendering them a critical cellular trigger for fibrotic remodeling in the lung (57). In lungs of IPF patients, AECIIs often show impaired antioxidant capacity and evidence of accumulated oxidative damage, correlating with increased cell death and tissue injury (58). Unlike fibroblasts or immune cells, AECIIs reside at the frontline of the alveolar surface and are continually exposed to environmental, mechanical, and metabolic stressors, which may predispose them to lipid peroxidation and iron-dependent cell death (56).
Multiple environmental and intrinsic factors converge to induce ferroptosis in AECIIs, contributing to the initiation and progression of IPF. Environmental insults such as airborne pollutants, cigarette smoke, and occupational exposures (e.g., asbestos, silica) are well-documented oxidative stress in the lung. These insults generate ROS that overwhelm cellular antioxidant defenses, promote lipid peroxidation, and drive ferroptotic cell death in AECs (56, 58). Additionally chronic or recurrent respiratory infections, such as those caused by herpesviruses and bacterial colonization, can act as exogenous triggers of epithelial stress and ferroptosis (59, 60).
Intrinsic factors, including aging and genetic predisposition, further sensitize lung epithelial cells to ferroptosis (61). Age-associated mitochondrial dysfunction, telomere shortening, and impaired antioxidant responses all contribute to a ferroptosis-permissive environment (62). Genetic mutations in surfactant protein C (SFTPC) or telomerase components (TERT, TERC) compromise proteostasis and telomere maintenance, increasing epithelial susceptibility to oxidative damage and Ferroptosis (63, 64).
These environmental and intrinsic triggers for ferroptosis are also reported causes of IPF (65–68). Additionally, they may act in concert with key fibrotic signaling pathways—notably the TGF-β/Smad and TLR4/NF-κB axes—to drive chronic epithelial injury and fibrotic remodeling.
3.2 TGF-β signaling pathway
TGF-β is a central mediator of fibrotic responses and remains persistently activated in the lungs of patients with IPF (69). Its activation is closely linked to both environmental and intrinsic stimuli that disrupt cellular homeostasis and provoke oxidative stress. Once activated, TGF-β binds to its receptor complex, initiating phosphorylation of Smad2 and Smad3, which then form a complex with Smad4 and translocate to the nucleus and regulate gene transcription (70). In AECs, this cascade not only promotes epithelial–mesenchymal transition (EMT) and cell cycle arrest, but also contributes to a pro-ferroptotic intracellular environment. Specifically, it upregulate pro-ferroptotic enzymes, such as ACSL4, increases ROS production, and suppresses antioxidant defense mechanisms—thereby facilitating lipid peroxidation and ferroptotic cell death (Figure 1) (7, 71).
Beyond transcriptional regulation, TGF-β signaling modulates ferroptosis through its interplay with microRNAs (miRNAs)—key post-transcriptional regulators implicated in IPF pathogenesis (72). TGF-β can both induce and be modulated by specific miRNAs, forming a feedback network that shapes epithelial stress responses (72, 73). For example, Kong et al. demonstrated that TGF-β/Smad4 signaling upregulates miR-155, which in turn promotes TGF-β-induced epithelial–mesenchymal transition (EMT), tight junction dissolution, and cell migration (74). In bleomycin-induced pulmonary fibrosis models, inhibition of miR-155-5p reduced expression of TGF-β1, IL-1β, and TNF-α, attenuating inflammatory and fibrotic progression (Figure 1) (75). Given miR-155’s reported role in enhancing oxidative stress and mitochondrial dysfunction, its upregulation may also sensitize epithelial cells to ferroptosis. More broadly, TGF-β-responsive miRNAs may act as intermediaries that promote ferroptotic cell death by suppressing ferroptosis-inhibitory targets (e.g., GPX4, SLC7A11) or amplifying pro-oxidative signaling (76–78). Thus, miRNA dysregulation represents a layer of ferroptosis control embedded within canonical TGF-β signaling, contributing to the chronic epithelial injury and maladaptive repair that characterize IPF.
Moreover, TGF-β has been shown to epigenetically repress ferroptosis-protective genes (79). In models of pulmonary fibrosis, TGF-β signaling increases the expression of methylation regulators (e.g., UHRF1), which silence GPX4 and FSP1, two key suppressors of ferroptosis (30). As a result, AECs become highly susceptible to lipid ROS accumulation and ferroptotic cell death.
The ferroptotic loss of AECs is a critical pathological event that precedes and perpetuates fibrotic remodeling. Dead or dying epithelial cells release damage-associated molecular patterns (DAMPs), stimulate the recruitment of profibrotic macrophages, and enhance TGF-β production in a self-amplifying loop (80). Additionally, the loss of epithelial-derived niche factors (e.g., WNT ligands, BMPs) destabilizes the epithelial-mesenchymal balance, promoting fibroblast activation and extracellular matrix (ECM) deposition (81). In this context, TGF-β/Smad signaling serves as both a driver and amplifier of ferroptosis-induced epithelial damage, establishing a mechanistic link between chronic injury, ferroptotic stress, and irreversible fibrotic progression in IPF (82, 83).
3.3 NF-κB signaling pathway
The NF-κB signaling pathway plays a pivotal role in connecting innate immune activation to ferroptotic cell death and fibrotic remodeling in the lung (84). Environmental insults such as cigarette smoke or bacterial endotoxins (e.g., LPS), as well as intrinsic triggers including DAMPs released from stressed or dying epithelial cells, can engage TLR4 receptors on AECs (85). Upon activation, TLR4 signals through MyD88-dependent pathways to induce NF-κB nuclear translocation, leading to transcription of pro-inflammatory cytokines (e.g., IL-6, TNF-α) and enzymes involved in iron metabolism and lipid peroxidation, both key components of ferroptosis (Figure 1) (86). In LPS-induced acute lung injury models, inhibition of TLR4/NF-κB signaling has been shown to alleviate ferroptosis and reduce pulmonary damage (84).
In AECs, TLR4/NF-κB activation contributes to a pro-ferroptotic intracellular environment by upregulating genes such as NOX1 and NOX4 (promoting ROS production) and ACSL4 (increasing PUFA-containing phospholipid substrates), while suppressing ferroptosis-protective genes like GPX4 through redox imbalance and inflammatory stress (86–88). Sustained NF-κB signaling also impairs the cellular antioxidant response by disrupting Nrf2 activity, further sensitizing AECs to ferroptosis (89).
Notably, there is growing evidence of functional crosstalk between TLR4/NF-κB and TGF-β/Smad signaling in fibrotic lung disease (89). For instance, NF-κB activation has been shown to enhance TGF-β expression and facilitate its autocrine and paracrine amplification, while TGF-β signaling can stabilize TLR4 expression and amplify NF-κB-driven transcriptional responses, creating a positive feedback loop that amplifies inflammatory and fibrotic responses (86, 90, 91). Together, these pathways synergistically promote AEC ferroptosis, inflammation, and the activation of fibroblasts and myofibroblasts, ultimately accelerating ECM deposition and lung architecture distortion.
Through this integrative mechanism, TLR4/NF-κB acts not only as a sensor of epithelial injury but as an amplifier of ferroptosis-associated inflammation, bridging environmental triggers with immune activation and fibrotic remodeling—hallmarks of IPF.
4 Ferroptosis and IPF progression
4.1 Inflammatory amplification
Ferroptosis contributes not only to the initiation of IPF, but also to its chronic progression through sustained inflammatory amplification by perpetuating a cycle of epithelial injury, immune activation, and fibrotic remodeling (92).
One critical consequence of ferroptosis in AECs is the release of DAMPs, including high-mobility group box 1 (HMGB1), extracellular ATP, and oxidized phospholipids (oxPLs), which act as endogenous triggers of innate immune responses (93, 94). These DAMPs activate pattern recognition receptors (PRRs), notably TLR4 and RAGE (receptor for advanced glycation end products) on neighboring epithelial cells, infiltrating neutrophils, macrophages, and resident fibroblasts (95, 96). This activation triggers the NF-κB signaling cascade, leading to a transcriptional upregulation of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6), chemokines, and adhesion molecules, which in turn recruit more immune cells to the injury site, and driving chronic inflammation and tissue damage (85).
Additionally, ferroptosis-derived oxidized lipid mediators, such as 4-HNE and MDA, can further potentiate inflammation by inducing oxidative stress and enhancing NF-κB signaling in a feed-forward loop (97–99). These lipid peroxidation products not only propagate tissue injury but also impair anti-inflammatory resolution processes (100).
Moreover, the influx of neutrophils and pro-inflammatory macrophages contributes to the formation of a self-amplifying inflammatory circuit. Neutrophils release myeloperoxidase (MPO) and ROS, exacerbating oxidative stress (101), while M1-like macrophages secrete further pro-inflammatory cytokines, establishing a chronic inflammatory niche (102). This chronicity hinders epithelial repair and sets the stage for fibrogenic signaling cascades.
Taken together, ferroptosis-induced DAMP release, cytokine production, and ROS generation converge to establish a vicious cycle of oxidative stress and inflammation, which not only amplifies immune activation but also primes the lung microenvironment for fibrotic transformation.
4.2 Macrophage polarization
Ferroptosis profoundly influences the immune landscape in the fibrotic lung, particularly by modulating macrophage recruitment and polarization—key processes that drive the transition from inflammation to fibrosis in IPF (86, 102). Ferroptotic AECs release bioactive molecules, including DAMPs, oxPLs, and lipid aldehydes (e.g.,4-HNE, MDA), promote monocyte infiltration by triggering the release of chemokines (like CCL2) and cytokines. These signals guide the infiltration of circulating monocytes into the injured alveolar niche and promoting their differentiation into macrophages (103). Among the heterogenous macrophage populations in the lung, monocytes-induced alveolar macrophage (Mo-AMs) have emerged as key fibrogenic players. Unlike tissue-resident alveolar macrophages, Mo-AMs expand during injury and persist long-term in the fibrotic lung. Genetic ablation studies have shown that depletion of Mo-AMs—but not embryonically derived resident alveolar macrophages—significantly ameliorates asbestos-induced pulmonary fibrosis (104). This persistence and functional specialization of Mo-AMs underscores their central role in driving sustained inflammation, secreting profibrotic mediators like TGF-β, IL-10, and CCL18, and orchestrating fibroblast activation and ECM deposition (103, 105).
Once recruited into the injured alveolar niche, infiltrating monocytes differentiate into macrophages under the influence of the ferroptotic microenvironment. This niche is enriched with DAMPs, oxPLs, and lipid peroxidation products, which collectively shape the phenotypic fate of these newly differentiated macrophages. Initially, exposure to DAMPs and ROS may transiently promote M1-like polarization characterized by pro-inflammatory responses. However, as the injury becomes chronic, persistent exposure to immunomodulatory signals—including IL-10, TGF-β, oxidized phosphatidylethanolamines (oxPEs), and lipid aldehydes like 4-HNE—progressively skews macrophages toward a profibrotic M2-like phenotype (106). Moreover, certain ferroptotic signaling products actively reinforce this polarization: oxidized phospholipids serve as ligands for PPARγ, a nuclear receptor that stabilizes M2 polarization while suppressing M1-like inflammatory gene expression (107, 108).
These M2-like macrophages become central orchestrators of fibrosis progression. They secrete a spectrum of profibrotic mediators, including TGF-β1, IL-10, arginase-1, and CCL18, which promote fibroblast proliferation, myofibroblast differentiation, and excessive ECM deposition (109). Among these, CCL18 is particularly notable for its strong correlation with disease severity in IPF patients (110).
In sum, ferroptosis not only recruits macrophages to sites of injury but also reprograms their phenotype, transforming them from inflammatory responders into active participants in fibrotic remodeling. This dual role makes macrophage polarization a crucial node in the ferroptosis-IPF axis and a promising target for therapeutic intervention.
4.3 Fibroblast activation and differentiation
Ferroptosis-induced loss of AECs disrupts the epithelial–mesenchymal equilibrium that is critical for maintaining pulmonary homeostasis. The depletion eliminates key antifibrotic signals—such as bone morphogenetic proteins (BMPs) and WNT pathway inhibitors—leading to the release of fibroblasts from their quiescent state and driving their proliferation and excessive collagen deposition (111, 112). The resulting imbalance forsters the expansion of fibroblastic foci, the pathological hallmark of progressive IPF, which are associated with poor clinical outcomes (113).
Fibroblasts generally exhibit resistant to ferroptosis, owning to their elevated antioxidant defenses (114). However, they are highly sensitive to ferroptotic signals released from adjacent epithelial cells. DAMPs, pro-inflammatory cytokines (e.g., IL-6, IL-1β), and lipid peroxidation products released during ferroptosis can activate fibroblasts and induce their differentiation into α-smooth muscle actin (α-SMA)-expressing myofibroblasts (115). Notably, oxPLs can directly engage TLR4 on fibroblasts, initiating downstream TGF-β and NF-κB signaling pathways that reinforce fibroblast activation and survival (116).
Differentiation into α-SMA+ myofibroblasts is a pivotal event in IPF progression. α-SMA expression confers contractile properties that enable myofibroblasts to remodel the ECM, contributing to increased tissue stiffness, impaired gas exchange, and further activation of latent TGF-β, thus perpetuating the fibrotic loop (117, 118). These myofibroblasts are the primary producers of ECM proteins, secreting abundant type I/III collagen, fibronectin, and tenascin-C, leading to progressive matrix deposition and scarring (119).
Crucially, myofibroblasts exhibit resistance to apoptosis, allowing them to persist even after the initial injury has resolved, sustaining the chronicity of IPF (120). They also secrete profibrotic cytokines such as TGF-β and IL-6, which promote further fibroblast differentiation and recruit additional inflammatory and fibrogenic cells, thereby amplifying a positive feedback loop that sustains fibrosis (121–123). Ferroptotic AECs further exacerbate this process by releasing matricellular proteins such as periostin, which stimulate fibroblast proliferation and ECM deposition within fibrotic niches (124).
Recent studies highlight that fibroblast activation is not a uniform process. Single-cell transcriptomic profiling has revealed marked heterogeneity among alveolar fibroblast subpopulations, including age-dependent subsets with distinct sensitivity to oxidative stress and ferroptotic signals (125). These subpopulations follow different differentiation trajectories—some favoring fibrosis progression while others may contribute to resolution—thus influencing the spatial and temporal heterogeneity of fibrotic remodeling. Ferroptosis likely exerts differential effects on these subsets, selectively promoting the expansion of profibrotic myofibroblast lineages while sparing or even suppressing anti-fibrotic populations (125).
This heterogeneity is further reflected in the characteristic patchy architecture of IPF. Areas with intensive ferroptotic activity exhibit high lipid peroxidation and iron accumulation, often colocalizing with fibroblastic foci, suggesting spatial coupling between ferroptosis-induced epithelial injury and fibroblast activation (126).
Importantly, these findings imply that ferroptosis inhibitors such as liproxstatin-1 may not exert uniform effects across fibroblast subsets. Instead, they may preferentially suppress the expansion of profibrotic fibroblast lineages, offering a potential avenue for targeted antifibrotic therapy tailored to fibroblast heterogeneity.
4.4 ECM production and fibrotic niche reinforcement
Ferroptosis-induced oxidative stress significantly influences ECM remodeling, a hallmark of IPF. The oxidative milleu enhances ECM stability and stiffness through the upregulation of enzymes like lysyl oxidases (LOX and LOXL2), which catalyzes the crosslinking of collagen and elastin fibers, increasing EMC tensile strength (127, 128). This stiffened ECM not only impairs lung compliance but also serves as a mechanical stimulus that activates fibroblasts through mechanotransduction pathways (117).
One consequence of increased ECM stiffness is suppression of miR-29, a key negative regulator the expression of stromal genes, resulting to increased translation of ECM components and further matrix deposition. Simultaneously, mechanical cues activates the Hippo pathway effector Yes-associated protein 1 (YAP), which translocates to the nucleus, where it promotes the transcription of profibrotic genes, reinforcing ECM deposition and stiffness (129). Additionally, mesenchymal progenitor cells acquire a mechanical memory mediated by miR-21, enabling them to maintain a fibrogenic phenotype even after the initial stiffening stimulus has subsided (130).
Ferroptosis of AECs exacerbates these effects by generating localized oxidative stress, which contributes to ECM stiffening and creates a fibrotic niche that fosters myofibroblast persistence. This self-reinforcing loop—linking ferroptosis, oxidative ECM remodeling, and fibroblast activation—underscores the central role of ferroptosis in sustaining chronic fibrotic remodeling in IPF.
Given the heterogeneity of fibroblast populations, such ECM-driven reinforcement of fibrosis may disproportionately affect specific fibroblast subsets that are more prone to profibrotic activation, further emphasizing the importance of context-dependent therapeutic strategies that consider fibroblast diversity and their variable sensitivity to ferroptotic stress.
5 Ferroptosis as therapeutic target for IPF
Given its pivotal role in alveolar epithelial injury and its contribution to inflammation and fibrotic remodeling, ferroptosis has emerged as a compelling therapeutic target in IPF. Multiple strategies have been proposed to modulate ferroptosis, targeting different components of its regulatory network and downstream signaling cascades. These approaches can be broadly categorized according to their target, including regulators of oxidative stress, lipid peroxidation, iron dysregulation, DAMP-mediated inflammation, profibrotic cytokines, and ECM remodeling (Table 1).
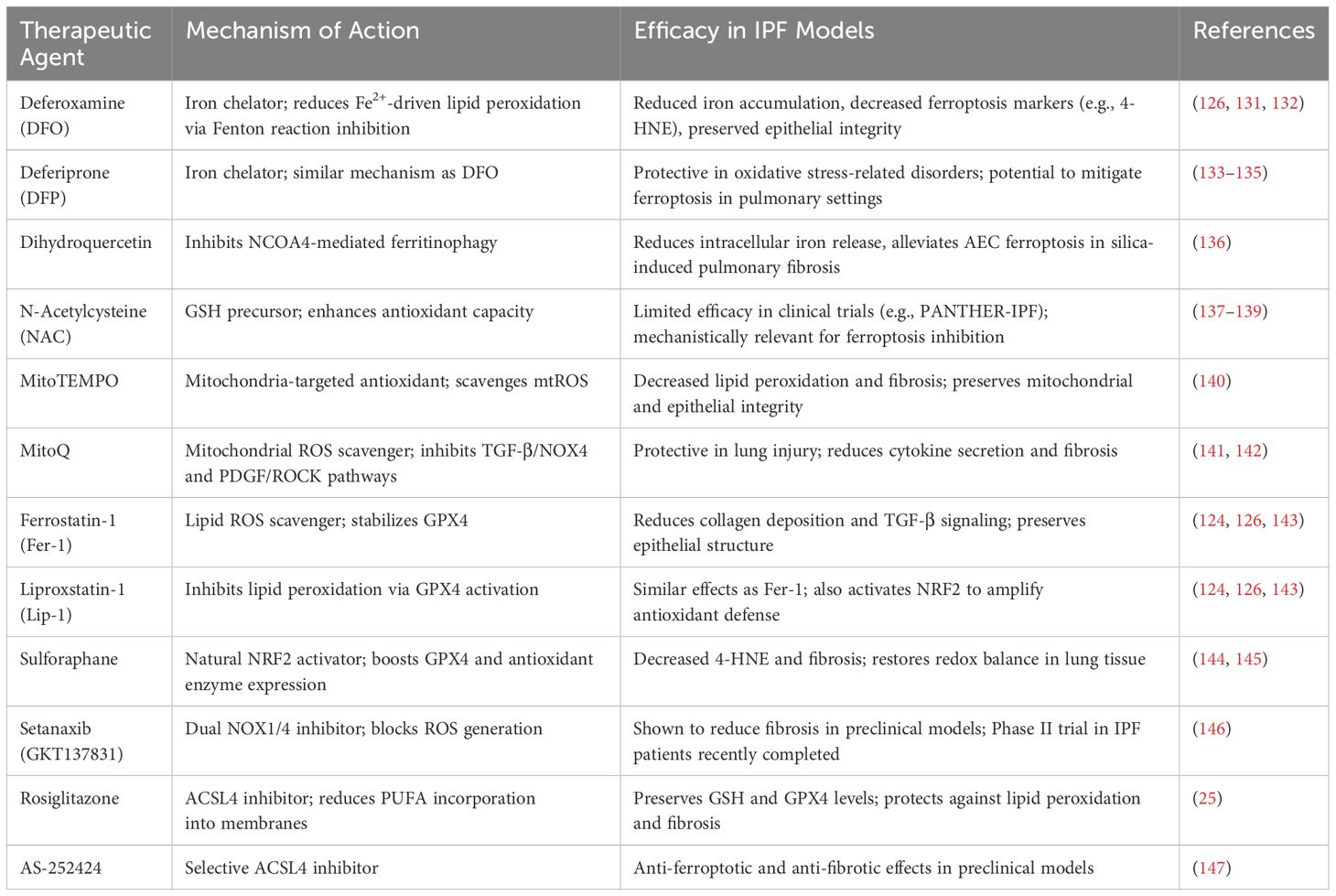
Table 1. Summary of ferroptosis inhibitors and their efficacy in IPF models.
5.1 Regulators of ferroptosis core machinery
5.1.1 Iron chelation and ROS management
Ferroptosis is an iron-dependent, regulated form of cell death characterized by iron-driven lipid peroxidation. In IPF, iron overload in alveolar space exacerbates oxidative stress and sensitizes AECs to ferroptosis by promoting Fenton chemistry and lipid peroxidation (148, 149). This process is especially damaging in the context of chronic lung injury, where sustained epithelial death and release of DAMPs propagate inflammation and fibrotic remodeling.
Supporting the translational relevance of these mechanisms, a clinical study by Puxeddu et al. (131) demonstrated the presence of iron-laden macrophages and elevated ferritin expression in lung tissue from IPF patients (131). These findings provide direct evidence of local iron accumulation and oxidative stress in the human fibrotic lung, reinforcing the pathophysiological rationale for targeting iron in therapeutic strategies.
Accordingly, strategies that reduce the bioavailable iron pool within the lung microenvironment have shown promise in mitigating ferroptosis and preserving epithelial integrity. Clinically approved iron chelators such as deferoxamine (DFO) and deferiprone (DFP), which are used to treat systemic iron overload, have demonstrated potential antifibrotic effects through inhibition of ferroptosis. In preclinical model of lung injury, DFO reduced iron accumulation, decreased ferroptosis markers such as 4-HNE, and preserve epithelial integrity, ultimately leading to attenuated fibrotic remodeling (126, 132, 133). Similarly, DFP has shown protective effects in models of neurodegenerative and retinal disorders by alleviating oxidative stress caused by excess iron (134–136). These chelators act by binding free Fe2+, thereby inhibiting the Fenton reactions and thus blocking a key driver of ferroptotic cell death.
Beyond chelation, modulation of ferritinophagy—a process that release stored iron from ferritin—offer another promising approach to reduce intracellular iron level (137). NCOA4—mediated ferritinophagy increases the available iron pool by releasing stored iron from ferritin complexes, thereby promotes ferroptosis under oxidative conditions. As discussed in Section 2.1, pharmacologic inhibition of ferritinophage, such as with dihydroquercetin, has been shown to ameliorate silica-induced pulmonary fibrosis by protecting AECs from ferroptosis (137). Thus, targeting NCOA4 could provide a more complementary, cell-intrinsic strategy for limiting ferroptotic injury. When combined with iron chelation, NCOA4 inhibition could yield a more robust and sustained suppression of ferroptosis, reinforcing epithelial resilience in the fibrotic lung.
5.1.2 Antioxidant defense modulators
Oxidative stress is a key upstream driver of ferroptosis in IPF, making antioxidant defense modulators as attractive therapeutic candidates. By replenish glutathione (GSH), scavenging ROS, or enhancing endogenous antioxidant enzymes, these agents can interrupt the lipid peroxidation cascade and protect AECs from ferroptotic death.
N−Acetylcysteine (NAC), a precursor for GSH synthesis, has long been explored for oxidative stress mitigation. However, large randomized controlled trials, including the PANTHER−IPF study, revealed limited clinical efficacy of NAC alone therapy in slowing disease progression or improving survival in unselected IPF cohort (138, 139, 150). Although NAC’s ability to restore intracellular GSH and modulate ferroptotic markers remains mechanistically relevant, its limited benefit in clinical practice suggests a need to prioritize more targeted and potent antioxidants in future therapeutic design.
A growing body of evidence highlights the pivotal role of mitochondrial ROS (mtROS) in amplifying lipid peroxidation and sensitizing cells to ferroptosis (140). Accordingly, mitochondria-targeted antioxidants have gained traction. MitoTEMPO, a superoxide dismutase mimetic designed to localize within mitochondria, effectively scavenges mtROS and preserves mitochondrial integrity, significantly reducing ferroptosis and fibrosis exacerbation in experimental models (141). Similarly, MitoQ, a mitochondria-directed ubiquinone derivative, demonstrates protective effects in lung injury models by suppressing mtROS, decreasing proinflammatory cytokine secretion, and inhibiting TGF-β/NOX4 and PDGF/ROCK signaling pathways (142, 143). These agents exemplify organelle-specific antioxidant defense strategies that not only block ferroptosis but also interfere with pro-fibrotic signaling cascades.
Ferroptosis-specific inhibitors offer another line of therapeutic intervention. Small molecules such as ferrostatin-1 (Fer-1) and liproxstatin-1 (Lip-1) stabilize the activity of GPX4, the core enzyme that detoxifies lipid peroxides and suppresses ferroptotic cell death. In bleomycin-induced lung fibrosis models, Fer-1 and Lip-1 preserve epithelial structure, reduce collagen deposition, and attenuate TGF-β signaling. These agents also activate the NRF2 pathway, further amplifying antioxidant gene expression (124, 126, 144). While these inhibitors remain at the preclinical stage, they continue to inform the mechanistic underpinnings of ferroptosis inhibition and serve as a proof of concept for targeting lipid peroxidation in IPF.
The NRF2 transcription factor orchestrates expression of GPX4, SLC7A11, and a suite of antioxidant enzymes.
Central to the antioxidant response is the NRF2–GPX4 signaling axis. NRF2 transcriptionally activates genes encoding antioxidant enzymes such as GPX4, SLC7A11, and others involved in GSH metabolism (7, 145). Pharmacological NRF2 activators have demonstrated robust protective effects in preclinical models by reducing 4-HNE, restoring redox homeostasis, and limiting AEC ferroptosis (146). Sulforaphane, a natural NRF2 agonist, has shown antifibrotic activity in lung models and may be a candidate for further development (145). Newer synthetic NRF2 activators with improved bioavailability are currently under preclinical investigation, with some poised for translational studies.
Translating antioxidant modulation into clinical application has proven challenging, but emerging agents are bridging this gap. Setanaxib (GKT137831) is a first-in-class dual inhibitor of NOX1/4, two isoforms of NADPH oxidase that generate ROS and contribute to ferroptosis-associated tissue injury. Setanaxib has shown antifibrotic efficacy in preclinical models of liver, kidney, and lung fibrosis (151). It is the first NADPH oxidase inhibitor to enter clinical trials for IPF. A phase II randomized, placebo-controlled, multicenter trial (NCT03865927) was recently completed to evaluate Setanaxib in ambulatory patients with IPF. Although results have not yet been posted, preliminary investigator updates suggest that Setanaxib may significantly reduce pulmonary injury and disease progression by limiting NOX-derived ROS and interfering with ferroptosis-associated pathways. This agent represents one of the most clinically advanced antioxidant-based therapies aimed at IPF.
Together, these antioxidant modulators offer multiple points of intervention along the ferroptotic cascade. While clinical translation has been challenging, ongoing trials of NRF2 activators and novel formulations may yet unlock significant benefits for IPF patients.
5.1.3 Targeting lipid metabolism
Lipid peroxidation is a hallmark of ferroptosis and a major contributor to alveolar epithelial injury in IPF (57). Therapeutic strategies that disrupt the enzymatic or non-enzymatic formation of lipid hydroperoxides may directly suppress ferroptotic cell death and attenuate the downstream fibrotic cascade. Among these, ACSL4 plays a key role by facilitating the incorporation of PUFAs into membrane phospholipids, thereby sensitizing cells to peroxidation and ferroptosis (147).
Inhibition of ACSL4 has shown therapeutic promise in preclinical models of fibrosis. Rosiglitazone, a thiazolidinedione initially developed as a PPARγ agonist, was found to attenuate ischemia–reperfusion–induced lung injury by suppressing ACSL4 activity. This intervention was associated with reduced lipid peroxidation and preserved intracellular levels of glutathione (GSH) and glutathione peroxidase 4 (GPX4), two essential components of cellular defense against ferroptosis (25). More recently, a selective small-molecule ACSL4 inhibitor, AS-252424, exhibited both anti-ferroptotic and anti-fibrotic effects in experimental settings, further underscoring the therapeutic potential of directly modulating lipid metabolism in the context of IPF (152).
Although the clinical translation of ACSL4 inhibitors remains in its early stages, these preclinical findings highlight the importance role of lipid remodeling in ferroptosis-driven fibrotic progression and suggest that future therapies strategies may benefit from targeting this metabolic axis.
5.2 Inflammatory signaling and immune activation
Regulated cell death pathways, including non−lytic apoptosis and lytic forms such as necroptosis, pyroptosis, and ferroptosis, release DAMPs that trigger sterile inflammation in the lung (153, 154). Key DAMPs such as HMGB1 and ATP activate PRRs, including TLR4 and receptor for advanced glycation end products (RAGE), on macrophages, neutrophils, and fibroblasts. This engagement leads to NF−κB–dependent upregulation of pro−inflammatory cytokines (TNF−α, IL−1β, IL−6) and chemokines, perpetuating tissue injury and fibrogenesis (155, 156).
Small−molecule inhibitors of TLR4 signaling have shown preclinical efficacy in lung injury models. TAK−242 (Resatorvid), a selective TLR4 inhibitor, suppresses NF−κB activation, reduces cytokine releases, and mitigates alveolar damage in models of acute lung injury (157). Eritoran (E5564), an MD2–TLR4 antagonist, was well tolerated in a Phase II sepsis trial (NCT00046072) and demonstrated trends toward lower mortality, suggesting translational potential in IPF (158).
Targeting RAGE signaling may also offer therapeutic benefit. FPS−ZM1, a blood−brain−barrier−permeable RAGE inhibitor, significantly attenuates HMGB1-mediated pulmonary inflammation and tissue injury in murine emphysema models, illustrating proof of concept for its use in fibrotic lung disease (159).
Direct inhibition of NF−κB signaling can dampen cytokine storms and fibrotic remodeling. BAY 11−7082 irreversibly inhibits IκB kinase, suppressing TNF−α–induced IκBα phosphorylation and reducing pro−coagulant and inflammatory markers in alveolar epithelial cells (160). Dimethyl fumarate (DMF), an FDA−approved NRF2 agonist for multiple sclerosis, indirectly inhibits NF−κB and has demonstrated antifibrotic effects by restoring redox homeostasis in aged IPF models (161).
The chemokine CCL2 (MCP−1) drives monocyte recruitment and differentiation into profibrotic monocyte−derived alveolar macrophages (Mo−AMs). Carlumab, an anti−CCL2 monoclonal antibody, demonstrated safety in Phase 1b oncology trials, though with limited monotherapy activity; its repurposing for IPF is under investigation (162). MLN1202, an anti−CCR2 antibody, reduced macrophage infiltration in Phase II rheumatoid arthritis studies, highlighting its potential to disrupt monocyte−macrophage-mediated fibrosis (163).
5.3 Cytokines and macrophage polarization
Ferroptosis-driven inflammation promotes the polarization of macrophages toward an M2-like, pro-fibrotic phenotype, largely mediated by cytokines such as TGF-β and IL-10 (164). Targeting these cytokines or their downstream signaling pathways offers a strategy to disrupt the self-perpetuating loop linking ferroptotic cell death to fibroblast activation and ECM deposition.
TGF−β and IL−10 are central to M2 macrophage polarization in the fibrotic lung. TGF−β, elevated downstream of ferroptosis, not only stimulates collagen production by fibroblasts but also induces the expression of M2 markers including arginase−1 and the mannose receptor (CD26) in macrophages, cementing their M2 identity. Therapeutic approaches targeting TGF-β signaling have shown partial success in clinical trials for IPF. Fresolimumab (GC1008), a pan−TGF−β neutralizing monoclonal antibody, completed a Phase I trial (NCT00125385) in IPF patients, demonstrating good tolerability and biomarker reductions suggestive of anti−fibrotic activity (165). Pirfenidone, an approved IPF antifibrotic, indirectly inhibits TGF−β1–mediated epithelial-mesenchymal transitions by suppressing MUC1−CT phosphorylation and β−catenin signaling, reducing α−SMA and collagen expression in preclinical models (166). Nintedanib, another approved IPF therapy, targets multiple tyrosine kinases downstream of PDGF and FGF, which intersect with TGF−β signaling to limit myofibroblast proliferation. In a Phase II trial, nintedanib effectively slowed lung function decline and reduced the frequence of acute exacerbation in patients with IPF (167).
IL−10, another key cytokine released by ferroptotic AECs, reinforces M2 polarization and tissue−repair functions in macrophages. Although IL−10 blockade remains preclinical in fibrosis, studies in murine models demonstrate that anti−IL−10 antibodies or genetic deletion of IL−10 signaling shifts macrophages toward a less fibrogenic profile and reduces collagen accumulation (168).
IL−6 lies at the interaction of inflammation and fibrosis. By promoting STAT3 activation, IL−6 fosters macrophage survival and M2 marker expression, while also driving fibroblast–myofibroblast transition. Tocilizumab, an IL−6 receptor antagonist approved for systemic sclerosis–associated interstitial lung disease (SSc−ILD), has shown promising results in preserving lung function and reducing M2−associated biomarkers in clinical studies (169, 170). While its role in IPF remains under investigation, the findings from SSc−ILD suggest that IL−6 blockade may help rebalance macrophage phenotypes and limit fibrotic progression.
Collectively, targeting cytokine and chemokine involved in ferroptosis-induced macrophage polarization presents a compelling strategy to reprogram the fibrotic immune environment in IPF. Future trials should explore these interventions—ideally in combination with ferroptosis inhibitors—to achieve synergistic suppression of epithelial injury and pro−fibrotic immune activation.
5.4 Fibroblast activation and ECM remodeling
Ferroptosis−associated inflammation activates myofibroblasts, which secrete matricellular proteins such as periostin and fibronectin. These proteins accumulate in the lung interstitium, and drive matrix stiffening and epithelial stress (129). This remodeled, rigid extracellular matrix drives nuclear translocation of YAP and TAZ, the Hippo pathway effectors, which in turn induce glycolytic and glutaminolytic genes that support myofibroblast survival and ECM production (171). Simultaneously, oxidative modifications of chromatin—such as histone H3K27 demethylation—enable YAP/TAZ to access fibrogenic promoters, sustaining an activated fibroblast phenotype (172). Therapeutic strategies targeting this axis, such as verteporfin to agents that modulate redox-sensitive epigenetic marks, hold promise for reversing fibroblast activation in IPF.
Several ECM-targeted therapies have advanced to clinical evaluation. Pamrevlumab (FG−3019), a fully human monoclonal antibody against connective tissue growth factor (CTGF/CCN2), was tested in the Phase II PRAISE trial (NCT01890265). In 103 IPF patients, pamrevlumab reduced the decline in forced vital capacity (FVC) by 60.3% compared to placebo over 48 weeks (–2.9% vs –7.2%; between−group difference of 4.3%, p=0.033) and halved the rate of disease progression (10.0% vs 31.4%), with a favorable safety profile (173).
PRM−151, a recombinant form of human pentraxin−2, also showed potential in IPF. In a Phase II randomized, placebo−controlled study (NCT02550873) of 111 IPF patients, PRM−151 slowed FVC decline over 28 weeks and preserved 6−minute walk distance, with good tolerability (174). A 76-week open−label extension confirmed sustained effects on lung function, consistent with its role in inhibiting fibrocyte differentiation and ECM production (175). However, the subsequent Phase III STARSCAPE trial (NCT04552899) was terminated early for lack of efficacy, though prior data still validate pentraxin−2 as a regulator of fibroblast activity (175).
Admilparant (BMS−986278), an oral lysophosphatidic acid (LPA) receptor antagonist, has been evaluated in a Phase II randomized trial (NCT04308681) in IPF and progressive pulmonary fibrosis. A 26-week course of 60 mg twice daily led to a 69% relative reduction in the rate of percent predicted FVC decline versus placebo (treatment difference 2.9%; 95% CI 0.4-5.5), with a favorable safety profile (176). These encouraging results support its progression to Phase III evaluation.
Emerging evidence indicates that ferroptosis−associated lipid peroxidation and iron overload not only contribute to ECM stiffening but also reprogram fibroblast metabolism and epigenetics to keep cells into an activated state. Disrupting key steps in ECM remodeling, including matricellular protein deposition, collagen crosslinking, and mechanotransduction, represents a promising strategy to halt fibrosis progression. When combined with upstream ferroptosis inhibitors, this multipronged strategy holds promise for preserving lung architecture and halting IPF progression.
6 Conclusion and perspective
Ferroptosis, an iron−dependent, lipid peroxidation–driven form of regulated cell death, has emerged as a key pathophysiological mechanism in IPF, linking alveolar epithelial injury to persistent inflammation, macrophage activation, and fibrotic remodeling of the lung parenchyma (7). This review integrates core mechanisms—dysregulated iron metabolism, compromised antioxidant defenses, enzymatic and non−enzymatic lipid peroxidation, and mechanotransduction via YAP/TAZ—with cellular crosstalk among epithelial cells, immune populations, and fibroblasts. We also discuss therapeutic strategies ranging from iron chelators to inhibitors of mechanosensitive pathway (6). Together, these insights offer a systems-level view of ferroptosis as both a driver and a modifiable target in IPF pathogenesis (177).
However, several major challenges must be addressed to enable clinical translation of ferroptosis-targeted therapies. Iron chelators such as deferoxamine and deferiprone, while effective in mitigating epithelial ferroptotic injury in preclinical models, carry the risk of systemic iron depletion and may exacerbate anemia-a condition already prevalent among IPF patients due to chronic inflammation and reduced erythropoiesis (178). Similarly, antioxidant therapies like NAC, while aim to restore intracellular glutathione and reduce ROS-dedicated lipid peroxidation, have shown inconsistent efficacy in IPF. Notably, PANTHER−IPF study reported no improvement in lung function and even raised safety concerns when NAC was combined with immunosuppressants (139, 150). These findings highlight the complex and context-dependent role of ROS in IPF pathogenesis, where both insufficient and excessive ROS can be detrimental. Direct ferroptosis inhibitors, including ferrostatin−1 and liproxstatin−1, exhibit potent in vitro protection but suffer from poor oral bioavailability, uncertain pharmacokinetics, and potential off−target toxicity, complicating their clinical development (177). Enzyme−targeted approaches, such as ACSL4 inhibition, remain confined to early preclinical stages, with no candidates yet advancing to human trials.
Beyond these individual drug challenges, broader translational barriers exist: achieving sufficient distribution in fibrotic lung tissue, avoiding disruption of physiological redox signaling, and minimizing toxicity during long-term administration (177). Emerging delivery platforms such as nanoparticle−based delivery systems and inhalable formulations offer promising avenues to enhance lung targeting and reduce systemic exposure, but their efficacy and safety in IPF-specific contexts remain to be rigorously validated (179).
Future research efforts should prioritize the identification of ferroptosis-specific biomarker to improve clinical translation. These may include genetic, proteomic, or metabolomic indicators that not only confirm the presence of ferroptotic activity but also help stratify patients likely to benefit from targeted interventions. Such biomarkers would be invaluable for selecting appropriate candidates for clinical trials and for monitoring treatment responses in real time (180).
Another important direction is the development of combination therapies that simultaneously target ferroptosis and other pro-fibrotic mechanisms. For instance, co-targeting ferroptotic pathways along with TGF-β/Smad signaling or YAP/TAZ-driven mechanotransduction may produce synergistic effects that surpass those of monotherapies. Achieving this will require a deeper mechanistic understanding of how ferroptosis interacts with these parallel pathways in different lung cell types and microenvironments (7).
Advances in spatial multi-omics technologies hold promise for resolving the spatial heterogeneity of ferroptotic activity within the fibrotic lung. These approaches can identify distinct cell populations and regions with heightened ferroptotic stress, enabling the design of more precise and localized therapies. By integrating spatial transcriptomics, proteomics, and metabolomics, researchers can develop targeted interventions based on the cellular context and disease stage (181).
Recent discoveries also highlight the role of RNA editing and epigenetic regulation in ferroptosis. In particular, ADAR1−mediated adenosine−to−inosine RNA editing has been shown to modulate ferroptosis-regulated microRNAs such as let−7d, which intersect with both TGF−β signaling and oxidative stress pathways; restoring normal ADAR1 function could mitigate ferroptosis−driven miRNA dysregulation and stabilize both epithelial and fibroblast phenotypes (182).
In conclusion, ferroptosis represents both a mechanistic cornerstone and a therapeutic target in IPF. By addressing these key knowledge gaps—ranging from biomarker discovery to epigenetic control—future studies can establish the foundation for ferroptosis-targeted IPF therapies (183). Such advances hold the potential to transform how we diagnose, monitor, and treat this devastating and currently incurable disease.
Author contributions
MY: Writing – original draft. ZL: Writing – original draft. WZ: Writing – original draft. SS: Conceptualization, Investigation, Supervision, Writing – review & editing. XH: Conceptualization, Funding acquisition, Investigation, Supervision, Validation, Writing – review & editing. YW: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the National Natural Science Foundation of China This study was supported by the National Natural Science Foundation of China (81802504), the Sichuan Science and Technology Program (2025YFHZ0123), Chengdu Science and Technology Program (2024-YF05-01315-SN), and a grant from Shenzhen Weixin (2024HX0008) for Dr. Yi Wang.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Noble PW, Barkauskas CE, and Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. (2012) 122:2756–62. doi: 10.1172/JCI60323
2. Wilson MS and Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. (2009) 2:103–21. doi: 10.1038/mi.2008.85
3. Meltzer EB and Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. (2008) 3:8. doi: 10.1186/1750-1172-3-8
4. Kaunisto J, Salomaa ER, Hodgson U, Kaarteenaho R, Kankaanranta H, Koli K, et al. Demographics and survival of patients with idiopathic pulmonary fibrosis in the FinnishIPF registry. ERJ Open Res. (2019) 5:00170–2018. doi: 10.1183/23120541.00170-2018
5. Lancaster L, Crestani B, Hernandez P, Inoue Y, Wachtlin D, Loaiza L, et al. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: pooled data from six clinical trials. BMJ Open Respir Res. (2019) 6:e000397. doi: 10.1136/bmjresp-2018-000397
6. Wang C, Hua S, and Song L. Ferroptosis in pulmonary fibrosis: an emerging therapeutic target. Front Physiol. (2023) 14:1205771. doi: 10.3389/fphys.2023.1205771
7. Hu Y, Huang Y, Zong L, Lin J, Liu X, and Ning S. Emerging roles of ferroptosis in pulmonary fibrosis: current perspectives, opportunities and challenges. Cell Death Discov. (2024) 10:301. doi: 10.1038/s41420-024-02078-0
8. Kist M and Vucic D. Cell death pathways: intricate connections and disease implications. EMBO J. (2021) 40:e106700. doi: 10.15252/embj.2020106700
9. Endale HT, Tesfaye W, and Mengstie TA. ROS induced lipid peroxidation and their role in ferroptosis. Front Cell Dev Biol. (2023) 11:1226044. doi: 10.3389/fcell.2023.1226044
10. Wang S, He X, Wu Q, Jiang L, Chen L, Yu Y, et al. Transferrin receptor 1-mediated iron uptake plays an essential role in hematopoiesis. Haematologica. (2020) 105:2071–82. doi: 10.3324/haematol.2019.224899
11. Galy B, Conrad M, and Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. (2024) 25:133–55. doi: 10.1038/s41580-023-00648-1
12. Ghio AJ, Turi JL, Yang F, Garrick LM, and Garrick MD. Iron homeostasis in the lung. Biol Res. (2006) 39:67–77. doi: 10.4067/S0716-97602006000100008
13. Sun Y, Ren Y, Song Ly, Wang Yy, Li Tg, Wu Y, et al. Targeting iron-metabolism:a potential therapeutic strategy for pulmonary fibrosis. Biomed Pharmacother. (2024) 172:116270. doi: 10.1016/j.biopha.2024.116270
14. Flórez A and Alborzinia H eds. Ferroptosis: mechanism and diseases. Cham: Springer (2021). p. 1. (Advances in experimental medicine and biology).
15. Zhai X, Zhu J, Li J, Wang Z, Zhang G, and Nie Y. Fraxetin alleviates BLM-induced idiopathic pulmonary fibrosis by inhibiting NCOA4-mediated epithelial cell ferroptosis. Inflamm Res. (2023) 72:1999–2012. doi: 10.1007/s00011-023-01800-5
16. Xu C, Liu Z, and Xiao J. Ferroptosis: A double-edged sword in gastrointestinal disease. IJMS. (2021) 22:12403. doi: 10.3390/ijms222212403
17. Das UN. Saturated fatty acids, MUFAs and PUFAs regulate ferroptosis. Cell Chem Biol. (2019) 26:309–11. doi: 10.1016/j.chembiol.2019.03.001
18. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. (2019) 51:575–586.e4. doi: 10.1016/j.devcel.2019.10.007
19. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. (2022) 185:2401–21. doi: 10.1016/j.cell.2022.06.003
20. Mortensen MS, Ruiz J, and Watts JL. Polyunsaturated fatty acids drive lipid peroxidation during ferroptosis. Cells. (2023) 12:804. doi: 10.3390/cells12050804
21. Tomitsuka Y, Imaeda H, Ito H, Asou I, Ohbayashi M, Ishikawa F, et al. Gene deletion of long-chain acyl-CoA synthetase 4 attenuates xenobiotic chemical-induced lung injury via the suppression of lipid peroxidation. Redox Biol. (2023) 66:102850. doi: 10.1016/j.redox.2023.102850
22. Wilborn J, Bailie M, Coffey M, Burdick M, Strieter R, and Peters-Golden M. Constitutive activation of 5-lipoxygenase in the lungs of patients with idiopathic pulmonary fibrosis. J Clin Invest. (1996) 97:1827–36. doi: 10.1172/JCI118612
23. Cheng T, Liu Q, Zhang R, Zhang Y, Chen J, Yu R, et al. Lysyl oxidase promotes bleomycin-induced lung fibrosis through modulating inflammation. J Mol Cell Biol. (2014) 6:506–15. doi: 10.1093/jmcb/mju039
24. Zhang L, Luo YL, Xiang Y, Bai XY, Qiang RR, Zhang X, et al. Ferroptosis inhibitors: past, present and future. Front Pharmacol. (2024) 15:1407335. doi: 10.3389/fphar.2024.1407335
25. Xu Y, Li X, Cheng Y, Yang M, and Wang R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. (2020) 34:16262–75. doi: 10.1096/fj.202001758R
26. Reed A, Ichu TA, Milosevich N, Melillo B, Schafroth MA, Otsuka Y, et al. LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem Biol. (2022) 17:1607–18. doi: 10.1021/acschembio.2c00317
27. Forcina GC and Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. (2019) 19:1800311. doi: 10.1002/pmic.201800311
28. Zhang W, Liu Y, Liao Y, Zhu C, and Zou Z. GPX4, ferroptosis, and diseases. Biomed Pharmacother. (2024) 174:116512. doi: 10.1016/j.biopha.2024.116512
29. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
30. Liu Y, Cheng D, Wang Y, Xi S, Wang T, Sun W, et al. UHRF1-mediated ferroptosis promotes pulmonary fibrosis via epigenetic repression of GPX4 and FSP1 genes. Cell Death Dis. (2022) 13:1070. doi: 10.1038/s41419-022-05515-z
31. Du G, Zhang Q, Huang X, and Wang Y. Molecular mechanism of ferroptosis and its role in the occurrence and treatment of diabetes. Front Genet. (2022) 13:1018829. doi: 10.3389/fgene.2022.1018829
32. Zeng F, Lan Y, Wang N, Huang X, Zhou Q, and Wang Y. Ferroptosis: A new therapeutic target for bladder cancer. Front Pharmacol. (2022) 13:1043283. doi: 10.3389/fphar.2022.1043283
33. Zhou Q, Yang L, Li T, Wang K, Huang X, Shi J, et al. Mechanisms and inhibitors of ferroptosis in psoriasis. Front Mol Biosci. (2022) 9:1019447. doi: 10.3389/fmolb.2022.1019447
34. Long L, Guo H, Chen X, Liu Y, Wang R, Zheng X, et al. Advancement in understanding the role of ferroptosis in rheumatoid arthritis. Front Physiol. (2022) 13:1036515. doi: 10.3389/fphys.2022.1036515
35. Zhou Q, Li T, Qin Q, Huang X, and Wang Y. Ferroptosis in lymphoma: Emerging mechanisms and a novel therapeutic approach. Front Genet. (2022) 13:1039951. doi: 10.3389/fgene.2022.1039951
36. Silva MM, Rocha CRR, Kinker GS, Pelegrini AL, and Menck CFM. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci Rep. (2019) 9:17639. doi: 10.1038/s41598-019-54065-6
37. Kerins MJ and Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. (2018) 29:1756–73. doi: 10.1089/ars.2017.7176
38. Zhao C, Pu W, Wazir J, Jin X, Wei L, Song S, et al. Long-term exposure to PM2.5 aggravates pulmonary fibrosis and acute lung injury by disrupting Nrf2-mediated antioxidant function. Environ Pollut. (2022) 313:120017. doi: 10.1016/j.envpol.2022.120017
39. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
40. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. (2020) 16:1351–60. doi: 10.1038/s41589-020-0613-y
41. Hu Q, Wei W, Wu D, Huang F, Li M, Li W, et al. Blockade of GCH1/BH4 axis activates ferritinophagy to mitigate the resistance of colorectal cancer to erastin-induced ferroptosis. Front Cell Dev Biol. (2022) 10:810327. doi: 10.3389/fcell.2022.810327
42. Liu Y, Lu S, lei W, Yang L, Yang L, and Wang J. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. (2023) 14:519. doi: 10.1038/s41419-023-06045-y
43. Cala-Garcia JD, Medina-Rincon GJ, Sierra-Salas PA, Rojano J, and Romero F. The role of mitochondrial dysfunction in idiopathic pulmonary fibrosis: new perspectives for a challenging disease. Biol (Basel). (2023) 12:1237. doi: 10.3390/biology12091237
44. Moss BJ, Ryter SW, and Rosas IO. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev Pathol. (2022) 17:515–46. doi: 10.1146/annurev-pathol-042320-030240
45. Fu C, Cao N, Zeng S, Zhu W, Fu X, Liu W, et al. Role of mitochondria in the regulation of ferroptosis and disease. Front Med (Lausanne). (2023) 10:1301822. doi: 10.3389/fmed.2023.1301822
46. Sulaimon LA, Afolabi LO, Adisa RA, Ayankojo AG, Afolabi MO, Adewolu AM, et al. Pharmacological significance of MitoQ in ameliorating mitochondria-related diseases. Adv Redox Res. (2022) 5:100037. doi: 10.1016/j.arres.2022.100037
47. Bhargava M, Viken K, Wang Q, Jagtap P, Bitterman P, Ingbar D, et al. Bronchoalveolar lavage fluid protein expression in acute respiratory distress syndrome provides insights into pathways activated in subjects with different outcomes. Sci Rep. (2017) 7:7464. doi: 10.1038/s41598-017-07791-8
48. Zhang Y, Zhou L, Cheng G, Zhou Y, Guo Q, Wu J, et al. Cordyceps sinensis ameliorates idiopathic pulmonary fibrosis in mice via inhibiting mitochondrion-mediated oxidative stress. MedComm &8211. Future Med. (2024) 3:e91. doi: 10.1002/mef2.v3.3
49. Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. (2019) 10:3145. doi: 10.1038/s41467-019-10991-7
50. Hayek H, Rehbini O, Kosmider B, Brandt T, Chatila W, Marchetti N, et al. The Regulation of Fatty Acid Synthase by Exosomal miR-143-5p and miR-342-5p in Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol. (2024) 70:259–82. doi: 10.1165/rcmb.2023-0232OC
51. He X, Ji J, Chen X, Luo Z, Fang S, Yan H, et al. Serum ferritin as a significant biomarker for patients with idiopathic inflammatory myopathy-associated interstitial lung disease: A systematic review and meta-analysis. Semin Arthritis Rheumat. (2024) 64:152350. doi: 10.1016/j.semarthrit.2023.152350
52. Wang J, Wu N, Peng M, Oyang L, Jiang X, Peng Q, et al. Ferritinophagy: research advance and clinical significance in cancers. Cell Death Discov. (2023) 9:463. doi: 10.1038/s41420-023-01753-y
53. He Y, Shang Y, Li Y, Wang M, Yu D, Yang Y, et al. An 8-ferroptosis-related genes signature from Bronchoalveolar Lavage Fluid for prognosis in patients with idiopathic pulmonary fibrosis. BMC Pulm Med. (2022) 22:15. doi: 10.1186/s12890-021-01799-7
54. Günther A, Korfei M, Mahavadi P, von der Beck D, Ruppert C, and Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev. (2012) 21:152–60. doi: 10.1183/09059180.00001012
55. Di Bonaventura G, Lupetti V, De Fabritiis S, Piccirilli A, Porreca A, Di Nicola M, et al. Giving Drugs a Second Chance: Antibacterial and Antibiofilm Effects of Ciclopirox and Ribavirin against Cystic Fibrosis Pseudomonas aeruginosa Strains. Int J Mol Sci. (2022) 23:5029. doi: 10.3390/ijms23095029
56. Parimon T, Yao C, Stripp BR, Noble PW, and Chen P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci. (2020) 21:2269. doi: 10.3390/ijms21072269
57. Zhang F, Xiang Y, Ma Q, Guo E, and Zeng X. A deep insight into ferroptosis in lung disease: facts and perspectives. Front Oncol. (2024) 14:1354859. doi: 10.3389/fonc.2024.1354859
58. Selman M, King TE, Pardo A, American Thoracic Society, European Respiratory Society, and American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. (2001) 134:136–51. doi: 10.7326/0003-4819-134-2-200101160-00015
59. Tang YW, Johnson JE, Browning PJ, Cruz-Gervis RA, Davis A, Graham BS, et al. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. (2003) 41:2633–40. doi: 10.1128/JCM.41.6.2633-2640.2003
60. Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiology-Lung Cell Mol Physiol. (2008) 294:L1119–26. doi: 10.1152/ajplung.00382.2007
61. Spagnolo P and Semenzato U. Revealing the pathogenic and ageing-related mechanisms of the enigmatic idiopathic pulmonary fibrosis (and chronic obstructive pulmonary disease). Curr Opin Pulm Med. (2022) 28:296–302. doi: 10.1097/MCP.0000000000000876
62. Zeidan RS, Han SM, Leeuwenburgh C, and Xiao R. Iron homeostasis and organismal aging. Ageing Res Rev. (2021) 72:101510. doi: 10.1016/j.arr.2021.101510
63. Blackwell TS. Lung injury and fibrosis induced by a mutant form of surfactant protein C. J Clin Investig. (2018) 128:3745–6. doi: 10.1172/JCI122727
64. Liu HJ, Hu HM, Li GZ, Zhang Y, Wu F, Liu X, et al. Ferroptosis-related gene signature predicts glioma cell death and glioma patient progression. Front Cell Dev Biol. (2020) 8:538. doi: 10.3389/fcell.2020.00538
65. Gandhi S, Tonelli R, Murray M, Samarelli AV, and Spagnolo P. Environmental causes of idiopathic pulmonary fibrosis. Int J Mol Sci. (2023) 24:16481. doi: 10.3390/ijms242216481
66. Collard HR. The age of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2010) 181:771–2. doi: 10.1164/rccm.201001-0049ED
67. Markart P, Ruppert C, Wygrecka M, Schmidt R, Korfei M, Harbach H, et al. Surfactant protein C mutations in sporadic forms of idiopathic interstitial pneumonias. Eur Respir J. (2007) 29:134–7. doi: 10.1183/09031936.00034406
68. Armanios MY, Chen JJL, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. (2007) 356:1317–26. doi: 10.1056/NEJMoa066157
69. Khalil N, O’Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, et al. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. (1991) 5:155–62. doi: 10.1165/ajrcmb/5.2.155
70. Deng Z, Fan T, Xiao C, Tian H, Zheng Y, Li C, et al. TGF-β signaling in health, disease and therapeutics. Sig Transduct Target Ther. (2024) 9:61. doi: 10.1038/s41392-024-01764-w
71. Meyer KC. Pulmonary fibrosis, part I: epidemiology, pathogenesis, and diagnosis. Expert Rev Respir Med. (2017) 11:343–59. doi: 10.1080/17476348.2017.1312346
72. Wang S, Yu H, Liu S, Liu Y, and Gu X. Regulation of idiopathic pulmonary fibrosis: a cross-talk between TGF-β signaling and MicroRNAs. Front Med (Lausanne). (2024) 11:1415278. doi: 10.3389/fmed.2024.1415278
73. Xu X, Hong P, Wang Z, Tang Z, and Li K. MicroRNAs in transforming growth factor-beta signaling pathway associated with fibrosis involving different systems of the human body. Front Mol Biosci. (2021) 8:707461. doi: 10.3389/fmolb.2021.707461
74. Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, et al. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. (2008) 28:6773–84. doi: 10.1128/MCB.00941-08
75. Adel RM, Helal H, Ahmed Fouad M, and Sobhy Abd-Elhalem S. Regulation of miRNA-155-5p ameliorates NETosis in pulmonary fibrosis rat model via inhibiting its target cytokines IL-1β, TNF-α and TGF-β1. Int Immunopharmacol. (2024) 127:111456. doi: 10.1016/j.intimp.2023.111456
76. Xu P, Wang Y, Deng Z, Tan Z, and Pei X. MicroRNA-15a promotes prostate cancer cell ferroptosis by inhibiting GPX4 expression. Oncol Lett. (2022) 23:67. doi: 10.3892/ol.2022.13186
77. Li X, Wang J, Wu C, Lu X, and Huang J. MicroRNAs involved in the TGF-β signaling pathway in atherosclerosis. Biomed Pharmacother. (2022) 146:112499. doi: 10.1016/j.biopha.2021.112499
78. Saha S. Role of microRNA in oxidative stress. Stresses. (2024) 4:269–81. doi: 10.3390/stresses4020016
79. Kim DH, Kim WD, Kim SK, Moon DH, and Lee SJ. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. (2020) 11:406. doi: 10.1038/s41419-020-2618-6
80. Yin J, Xu X, Guo Y, Sun C, Yang Y, Liu H, et al. Repair and regeneration: ferroptosis in the process of remodeling and fibrosis in impaired organs. Cell Death Discov. (2024) 10:424. doi: 10.1038/s41420-024-02181-2
81. Volckaert T and De Langhe SP. Wnt and FGF mediated epithelial-mesenchymal crosstalk during lung development. Dev Dynam. (2015) 244:342–66. doi: 10.1002/dvdy.v244.3
82. Chen Y, Dai Y, Huang Y, Zhang L, Zhang C, Gao H, et al. Inhibition of tubular epithelial cells ferroptosis alleviates renal interstitial fibrosis by reducing lipid hydroperoxides and TGF-β/Smad signaling. Cell Commun Signal. (2025) 23:81. doi: 10.1186/s12964-025-02068-4
83. Bao R, Wang Q, Yu M, Zeng Y, Wen S, Liu T, et al. AAV9-HGF cooperating with TGF-beta/Smad inhibitor attenuates silicosis fibrosis via inhibiting ferroptosis. BioMed Pharmacother. (2023) 161:114537. doi: 10.1016/j.biopha.2023.114537
84. Li Y, Huang L, Li J, Li S, Lv J, Zhong G, et al. Targeting TLR4 and regulating the Keap1/Nrf2 pathway with andrographolide to suppress inflammation and ferroptosis in LPS-induced acute lung injury. Chin J Natural Med. (2024) 22:914–28. doi: 10.1016/S1875-5364(24)60727-2
85. Bolourani S, Brenner M, and Wang P. The interplay of DAMPs, TLR4, and proinflammatory cytokines in pulmonary fibrosis. J Mol Med (Berl). (2021) 99:1373–84. doi: 10.1007/s00109-021-02113-y
86. Chen X, Kang R, Kroemer G, and Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med. (2021) 218:e20210518. doi: 10.1084/jem.20210518
87. Maloney E, Sweet IR, Hockenbery DM, Pham M, Rizzo NO, Tateya S, et al. Activation of NF-kappaB by palmitate in endothelial cells: a key role for NADPH oxidase-derived superoxide in response to TLR4 activation. Arterioscler Thromb Vasc Biol. (2009) 29:1370–5. doi: 10.1161/ATVBAHA.109.188813
88. Echizen K, Horiuchi K, Aoki Y, Yamada Y, Minamoto T, Oshima H, et al. NF-κB-induced NOX1 activation promotes gastric tumorigenesis through the expansion of SOX2-positive epithelial cells. Oncogene. (2019) 38:4250–63. doi: 10.1038/s41388-019-0702-0
89. Gao W, Guo L, Yang Y, Wang Y, Xia S, Gong H, et al. Dissecting the crosstalk between nrf2 and NF-κB response pathways in drug-induced toxicity. Front Cell Dev Biol. (2022) 9:809952. doi: 10.3389/fcell.2021.809952
90. Zhang C, Zhu X, Hua Y, Zhao Q, Wang K, Zhen L, et al. YY1 mediates TGF-β1-induced EMT and pro-fibrogenesis in alveolar epithelial cells. Respir Res. (2019) 20:249. doi: 10.1186/s12931-019-1223-7
91. Chen Y, Fang ZM, Yi X, Wei X, and Jiang DS. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. (2023) 14:205. doi: 10.1038/s41419-023-05716-0
92. Ling H, Xiao H, Luo T, Lin H, and Deng J. Role of ferroptosis in regulating the epithelial–mesenchymal transition in pulmonary fibrosis. Biomedicines. (2023) 11:163. doi: 10.3390/biomedicines11010163
93. Ma TL, Zhou Y, Wang C, Wang L, Chen JX, Yang HH, et al. Targeting ferroptosis for lung diseases: exploring novel strategies in ferroptosis-associated mechanisms. Bravo L, editor. Oxid Med Cell Longev. (2021) 2021:1098970. doi: 10.1155/2021/1098970
94. Wen Q, Liu J, Kang R, Zhou B, and Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. (2019) 510:278–83. doi: 10.1016/j.bbrc.2019.01.090
95. Hudson BI and Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. (2018) 69:349–64. doi: 10.1146/annurev-med-041316-085215
96. Friedmann Angeli JP, Krysko DV, and Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. (2019) 19:405–14. doi: 10.1038/s41568-019-0149-1
97. Zhang H and Forman HJ. 4-hydroxynonenal-mediated signaling and aging. Free Radic Biol Med. (2017) 111:219–25. doi: 10.1016/j.freeradbiomed.2016.11.032
98. Breitzig M, Bhimineni C, Lockey R, and Kolliputi N. 4-Hydroxy-2-nonenal: a critical target in oxidative stress? Am J Physiol Cell Physiol. (2016) 311:C537–43. doi: 10.1152/ajpcell.00101.2016
99. Liu X, Lin R, Zhao B, Guan R, Li T, and Jin R. Correlation between oxidative stress and the NF-κB signaling pathway in the pulmonary tissues of obese asthmatic mice. Mol Med Rep. (2016) 13:1127–34. doi: 10.3892/mmr.2015.4663
100. Dalleau S, Baradat M, Guéraud F, and Huc L. Cell death and diseases related to oxidative stress:4-hydroxynonenal (HNE) in the balance. Cell Death Differ. (2013) 20:1615–30. doi: 10.1038/cdd.2013.138
101. Warheit-Niemi HI, Huizinga GP, Edwards SJ, Wang Y, Murray SK, O’Dwyer DN, et al. Fibrotic lung disease alters neutrophil trafficking and promotes neutrophil elastase and extracellular trap release. ImmunoHorizons. (2023) 6:817–34. doi: 10.4049/immunohorizons.2200083
102. Pérez S and Rius-Pérez S. Macrophage polarization and reprogramming in acute inflammation: A redox perspective. Antioxid (Basel). (2022) 11:1394. doi: 10.3390/antiox11071394
103. Dash SP, Gupta S, and Sarangi PP. Monocytes and macrophages: Origin, homing, differentiation, and functionality during inflammation. Heliyon. (2024) 10:e29686. doi: 10.1016/j.heliyon.2024.e29686
104. Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P, et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur Respir J. (2020) 55:1900646. doi: 10.1183/13993003.00646-2019
105. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. (2017) 214:2387–404. doi: 10.1084/jem.20162152
106. Covarrubias A, Byles V, and Horng T. ROS sets the stage for macrophage differentiation. Cell Res. (2013) 23:984–5. doi: 10.1038/cr.2013.88
107. Van Ginderachter JA, Movahedi K, Van den Bossche J, and De Baetselier P. Macrophages, PPARs, and cancer. PPAR Res. (2008) 2008:169414. doi: 10.1155/ppar.v2008.1
108. Penas F, Mirkin GA, Vera M, Cevey Á, González CD, Gómez MI, et al. Treatment in vitro with PPARα and PPARγ ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim Biophys Acta. (2015) 1852:893–904. doi: 10.1016/j.bbadis.2014.12.019
109. Ge Z, Chen Y, Ma L, Hu F, and Xie L. Macrophage polarization and its impact on idiopathic pulmonary fibrosis. Front Immunol. (2024) 15:1444964. doi: 10.3389/fimmu.2024.1444964
110. Liu S, Liu C, Wang Q, Liu S, and Min J. CC chemokines in idiopathic pulmonary fibrosis: pathogenic role and therapeutic potential. Biomolecules. (2023) 13:333. doi: 10.3390/biom13020333
111. Wu DH and Hatzopoulos AK. Bone morphogenetic protein signaling in inflammation. Exp Biol Med (Maywood). (2019) 244:147–56. doi: 10.1177/1535370219828694
112. Morrisey EE. Wnt signaling and pulmonary fibrosis. Am J Pathol. (2003) 162:1393–7. doi: 10.1016/S0002-9440(10)64271-X
113. Yamaguchi M, Hirai S, Tanaka Y, Sumi T, Miyajima M, Mishina T, et al. Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial–mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab Investig. (2017) 97:232–42. doi: 10.1038/labinvest.2016.135
114. Zhang F, Xiao Y, Huang Z, Wang Y, Wan W, Zou H, et al. Upregulation of GPX4 drives ferroptosis resistance in scleroderma skin fibroblasts. Free Radical Biol Med. (2024) 221:23–30. doi: 10.1016/j.freeradbiomed.2024.05.013
115. Qiu X, Bi Q, Wu J, Sun Z, and Wang W. Role of ferroptosis in fibrosis: From mechanism to potential therapy. Chin Med J (Engl). (2024) 137:806–17. doi: 10.1097/CM9.0000000000002784
116. Erridge C, Kennedy S, Spickett CM, and Webb DJ. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J Biol Chem. (2008) 283:24748–59. doi: 10.1074/jbc.M800352200
117. Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. (2012) 47:340–8. doi: 10.1165/rcmb.2012-0050OC
118. Wang Z, Castro N, Bernstein AM, and Wolosin JM. TGFβ1-driven SMAD2/3 phosphorylation and myofibroblast emergence are fully dependent on the TGFβ1 pre-activation of MAPKs and controlled by maternal leucine zipper kinase. Cell Signal. (2024) 113:110963. doi: 10.1016/j.cellsig.2023.110963
119. Klingberg F, Hinz B, and White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. (2013) 229:298–309. doi: 10.1002/path.2013.229.issue-2
120. Hinz B and Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol. (2020) 16:11–31. doi: 10.1038/s41584-019-0324-5
121. Porte J, Jenkins G, and Tatler AL. Myofibroblast TGF-β Activation measurement in vitro. Methods Mol Biol. (2021) 2299:99–108. doi: 10.1007/978-1-0716-1382-5_6
122. Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. (2010) 107:20009–14. doi: 10.1073/pnas.1013805107
123. Li Y, Zhao J, Yin Y, Li K, Zhang C, and Zheng Y. The role of IL-6 in fibrotic diseases: molecular and cellular mechanisms. Int J Biol Sci. (2022) 18:5405–14. doi: 10.7150/ijbs.75876
124. Yang L, Li X, and Wang Y. Ferrostatin-1 inhibits fibroblast fibrosis in keloid by inhibiting ferroptosis. PeerJ. (2024) 12:e17551. doi: 10.7717/peerj.17551
125. Lingampally A, Truchi M, Shi X, Zhou Y, Vasquez-Pacheco E, Panagiotidis GD, et al. Unraveling alveolar fibroblast and activated myofibroblast heterogeneity and differentiation trajectories during lung fibrosis development and resolution in young and old mice. Aging Cell. (2025), e14503. doi: 10.1111/acel.14503
126. Pei Z, Qin Y, Fu X, Yang F, Huo F, Liang X, et al. Inhibition of ferroptosis and iron accumulation alleviates pulmonary fibrosis in a bleomycin model. Redox Biol. (2022) 57:102509. doi: 10.1016/j.redox.2022.102509
127. Bellaye PS, Shimbori C, Upagupta C, Sato S, Shi W, Gauldie J, et al. Lysyl oxidase-like 1 protein deficiency protects mice from adenoviral transforming growth factor-β1-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. (2018) 58:461–70. doi: 10.1165/rcmb.2017-0252OC
128. Cox TR, Bird D, Baker AM, Barker HE, Ho MWY, Lang G, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. (2013) 73:1721–32. doi: 10.1158/0008-5472.CAN-12-2233
129. Herrera J, Henke CA, and Bitterman PB. Extracellular matrix as a driver of progressive fibrosis. J Clin Investig. (2018) 128:45–53. doi: 10.1172/JCI93557
130. Li CX, Talele NP, Boo S, Koehler A, Knee-Walden E, Balestrini JL, et al. MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat Mater. (2017) 16:379–89. doi: 10.1038/nmat4780
131. Puxeddu E, Comandini A, Cavalli F, Pezzuto G, D’Ambrosio C, Senis L, et al. Iron laden macrophages in idiopathic pulmonary fibrosis: The telltale of occult alveolar hemorrhage? Pulmon Pharmacol Ther. (2014) 28:35–40. doi: 10.1016/j.pupt.2013.12.002
132. Liang Y, Wang Y, Sun C, Xiang Y, and Deng Y. Deferoxamine reduces endothelial ferroptosis and protects cerebrovascular function after experimental traumatic brain injury. Brain Res Bull. (2024) 207:110878. doi: 10.1016/j.brainresbull.2024.110878
133. Zhu W, Huang Y, Ye Y, and Wang Y. Deferoxamine preconditioning ameliorates mechanical ventilation-induced lung injury in rat model via ROS in alveolar macrophages: a randomized controlled study. BMC Anesthesiol. (2018) 18:116. doi: 10.1186/s12871-018-0576-7
134. Rayatpour A, Foolad F, Heibatollahi M, Khajeh K, and Javan M. Ferroptosis inhibition by deferiprone, attenuates myelin damage and promotes neuroprotection in demyelinated optic nerve. Sci Rep. (2022) 12:19630. doi: 10.1038/s41598-022-24152-2
135. Hadziahmetovic M, Song Y, Wolkow N, Iacovelli J, Grieco S, Lee J, et al. The oral iron chelator deferiprone protects against iron overload-induced retinal degeneration. Invest Ophthalmol Vis Sci. (2011) 52:959–68. doi: 10.1167/iovs.10-6207
136. Ye Z, Yan Y, Jin F, Jiang J, Deng C, Wang L, et al. Deferiprone protects photoreceptors by inhibiting ferroptosis after experimental retinal detachment. Exp Eye Res. (2025) 250:110156. doi: 10.1016/j.exer.2024.110156
137. Du J, Yu L, Yang X, Shao F, Xia J, Jin W, et al. Regulation of NCOA4-mediated iron recycling ameliorates paraquat-induced lung injury by inhibiting ferroptosis. Cell Commun Signal. (2024) 22:146. doi: 10.1186/s12964-024-01520-1
138. The Idiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, azathioprine, and N -acetylcysteine for pulmonary fibrosis. N Engl J Med. (2012) 366:1968–77. doi: 10.1056/NEJMoa1113354
139. Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA, Anstrom KJ, King TE, and Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2093–101. doi: 10.1056/NEJMoa1401739
140. Lyamzaev KG, Panteleeva AA, Simonyan RA, Avetisyan AV, and Chernyak BV. Mitochondrial lipid peroxidation is responsible for ferroptosis. Cells. (2023) 12:611. doi: 10.3390/cells12040611
141. Yue D, Zhang Q, Zhang J, Liu W, Chen L, Wang M, et al. Diesel exhaust PM2.5 greatly deteriorates fibrosis process in pre-existing pulmonary fibrosis via ferroptosis. Environ Int. (2023) 171:107706. doi: 10.1016/j.envint.2022.107706
142. Chen L, Lan C, Xiao H, Zhang X, Qi X, Ouyang L, et al. Mechanism of yifei decoction combined with mitoQ on inhibition of TGFβ1/NOX4 and PDGF/ROCK signal pathway in idiopathic pulmonary fibrosis. Evid Based Complement Alternat Med. (2021) 2021:6615615. doi: 10.1155/2021/6615615
143. Li Y, Du Z, Li T, Ren X, Yu Y, Duan J, et al. MitoQ ameliorates PM2.5-induced pulmonary fibrosis through regulating the mitochondria DNA homeostasis. Chemosphere. (2023) 330:138745. doi: 10.1016/j.chemosphere.2023.138745
144. Li X, Duan L, Yuan S, Zhuang X, Qiao T, and He J. Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. J Inflamm. (2019) 16:11. doi: 10.1186/s12950-019-0216-0
145. Liu P, Luo G, Dodson M, Schmidlin CJ, Wei Y, Kerimoglu B, et al. The NRF2-LOC344887 signaling axis suppresses pulmonary fibrosis. Redox Biol. (2021) 38:101766. doi: 10.1016/j.redox.2020.101766
146. Wang Y, Wei J, Deng H, Zheng L, Yang H, and Lv X. The role of nrf2 in pulmonary fibrosis: molecular mechanisms and treatment approaches. Antioxid (Basel). (2022) 11:1685. doi: 10.3390/antiox11091685
147. Chen J, Yang L, Geng L, He J, Chen L, Sun Q, et al. Inhibition of acyl-coA synthetase long-chain family member 4 facilitates neurological recovery after stroke by regulation ferroptosis. Front Cell Neurosci. (2021) 15:632354. doi: 10.3389/fncel.2021.632354
148. Vani Raju M, Kaniyur Chandrasekaran M, Muthaiyan Ahalliya R, and Velliyur Kanniappan G. Reconnoitering the role of lipid metabolites in ferroptosis. Adv Redox Res. (2025) 14:100117. doi: 10.1016/j.arres.2024.100117
149. Yan H, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Sig Transduct Target Ther. (2021) 6:49. doi: 10.1038/s41392-020-00428-9
150. Kam M, Caliez J, Nunes H, and Gille T. The road to hell is paved with good intentions: a look back at the PANTHER-IPF trial. Breathe. (2022) 18:220074. doi: 10.1183/20734735.0074-2022
151. Thannickal VJ, Jandeleit-Dahm K, Szyndralewiez C, and Török NJ. Pre-clinical evidence of a dual NADPH oxidase 1/4 inhibitor (setanaxib) in liver, kidney and lung fibrosis. J Cell Mol Med. (2023) 27:471–81. doi: 10.1111/jcmm.17649
152. Huang Q, Ru Y, Luo Y, Luo X, Liu D, Ma Y, et al. Identification of a targeted ACSL4 inhibitor to treat ferroptosis-related diseases. Sci Adv. (2025) 10:eadk1200. doi: 10.1126/sciadv.adk1200
153. Choudhury SM, Sarkar R, Karki R, and Kanneganti TD. A comparative study of apoptosis, pyroptosis, necroptosis, and PANoptosis components in mouse and human cells. PLoS One. (2024) 19:e0299577. doi: 10.1371/journal.pone.0299577
154. Chen R, Kang R, and Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. (2022) 54:91–102. doi: 10.1038/s12276-022-00736-w
155. Shirey KA, Blanco JCG, and Vogel SN. Targeting TLR4 signaling to blunt viral-mediated acute lung injury. Front Immunol. (2021) 12:705080. doi: 10.3389/fimmu.2021.705080
156. Liu X, Lu F, and Chen X. Examination of the role of necroptotic damage-associated molecular patterns in tissue fibrosis. Front Immunol. (2022) 13:886374. doi: 10.3389/fimmu.2022.886374
157. Bhattacharyya S, Wang W, Tamaki Z, Shi B, Yeldandi A, Tsukimi Y, et al. Pharmacological inhibition of toll-like receptor-4 signaling by TAK242 prevents and induces regression of experimental organ fibrosis. Front Immunol. (2018) 9:2434. doi: 10.3389/fimmu.2018.02434
158. Tidswell M, Tillis W, Larosa SP, Lynn M, Wittek AE, Kao R, et al. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med. (2010) 38:72–83. doi: 10.1097/CCM.0b013e3181b07b78
159. Lee H, Park JR, Kim WJ, Sundar IK, Rahman I, Park SM, et al. Blockade of RAGE ameliorates elastase-induced emphysema development and progression via RAGE-DAMP signaling. FASEB J. (2017) 31:2076–89. doi: 10.1096/fj.201601155R
160. Sieber P, Schäfer A, Lieberherr R, Caimi SL, Lüthi U, Ryge J, et al. NF-κB drives epithelial-mesenchymal mechanisms of lung fibrosis in a translational lung cell model. JCI Insight. (2023) 8:e154719. doi: 10.1172/jci.insight.154719
161. Kato K, Papageorgiou I, Shin YJ, Kleinhenz JM, Palumbo S, Hahn S, et al. Lung-targeted delivery of dimethyl fumarate promotes the reversal of age-dependent established lung fibrosis. Antioxid (Basel). (2022) 11:492. 10.3390/antiox11030492
162. Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. (2015) 10:111–23. doi: 10.1007/s11523-014-0320-2
163. Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, Van Den Bosch F, et al. Modulation of CCR2 in rheumatoid arthritis: A double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheumat. (2008) 58:1931–9. doi: 10.1002/art.23591
164. Yang Y, Wang Y, Guo L, Gao W, Tang TL, and Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis. (2022) 13:355. doi: 10.1038/s41419-022-04775-z
165. Shenderov K, Collins SL, Powell JD, and Horton MR. Immune dysregulation as a driver of idiopathic pulmonary fibrosis. J Clin Invest. (2021) 131:e143226. doi: 10.1172/JCI143226
166. Ballester B, Milara J, and Cortijo J. Pirfenidone anti-fibrotic effects are partially mediated by the inhibition of MUC1 bioactivation. Oncotarget. (2020) 11:1306–20. doi: 10.18632/oncotarget.v11i15
167. Richeldi L, Du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2071–82. doi: 10.1056/NEJMoa1402584
168. Steen EH, Wang X, Balaji S, Butte MJ, Bollyky PL, and Keswani SG. The role of the anti-inflammatory cytokine interleukin-10 in tissue fibrosis. Adv Wound Care (New Rochelle). (2020) 9:184–98. doi: 10.1089/wound.2019.1032
169. Roofeh D, Lin CJF, Goldin J, Kim GH, Furst DE, Denton CP, et al. Tocilizumab prevents progression of early systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol. (2021) 73:1301–10. doi: 10.1002/art.41668
170. Sheng XR, Gao X, Schiffman C, Jiang J, Ramalingam TR, Lin CJF, et al. Biomarkers of fibrosis, inflammation, and extracellular matrix in the phase 3 trial of tocilizumab in systemic sclerosis. Clin Immunol. (2023) 254:109695. doi: 10.1016/j.clim.2023.109695
171. Cabrera-Fuentes HA, Barreto G, Al-Suhaimi EA, and Liehn EA. Fibroblast plasticity in pulmonary and cardiovascular fibrosis: Mechanistic insights and emerging therapeutic targets. Arch Med Res. (2025) 56:103173. doi: 10.1016/j.arcmed.2024.103173
172. Wei Y, Hui VLZ, Chen Y, Han R, Han X, and Guo Y. YAP/TAZ: Molecular pathway and disease therapy. MedComm (2020). (2023) 4:e340. doi: 10.1002/mco2.v4.4
173. Richeldi L, Fernández Pérez ER, Costabel U, Albera C, Lederer DJ, Flaherty KR, et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. (2020) 8:25–33. doi: 10.1016/S2213-2600(19)30262-0
174. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: A randomized clinical trial. JAMA. (2018) 319:2299–307. doi: 10.1001/jama.2018.6129
175. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: an open-label extension study. Lancet Respir Med. (2019) 7:657–64. doi: 10.1016/S2213-2600(19)30172-9
176. Corte TJ, Behr J, Cottin V, Glassberg MK, Kreuter M, Martinez FJ, et al. Efficacy and safety of admilparant, an LPA1 antagonist, in pulmonary fibrosis: A phase 2 randomized clinical trial. Am J Respir Crit Care Med. (2025) 211:230–8. doi: 10.1164/rccm.202405-0977OC
177. von Samson-Himmelstjerna FA, Kolbrink B, Riebeling T, Kunzendorf U, and Krautwald S. Progress and setbacks in translating a decade of ferroptosis research into clinical practice. Cells. (2022) 11:2134. doi: 10.3390/cells11142134
178. Velasquez J and Wray AA. Deferoxamine. In: StatPearls. StatPearls Publishing, Treasure Island (FL (2025). Available at: http://www.ncbi.nlm.nih.gov/books/NBK557654/.
179. Han S, Zou J, Xiao F, Xian J, Liu Z, Li M, et al. Nanobiotechnology boosts ferroptosis: opportunities and challenges. J Nanobiotechnol. (2024) 22:606. doi: 10.1186/s12951-024-02842-5
180. Chen X, Comish PB, Tang D, and Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. (2021) 9:637162. doi: 10.3389/fcell.2021.637162
181. Liu X, Peng T, Xu M, Lin S, Hu B, Chu T, et al. Spatial multi-omics: deciphering technological landscape of integration of multi-omics and its applications. J Hematol Oncol. (2024) 17:72. doi: 10.1186/s13045-024-01596-9
182. Díaz-Piña G, Rubio K, Ordoñez-Razo RM, Barreto G, Montes E, Becerril C, et al. ADAR1 isoforms regulate let-7d processing in idiopathic pulmonary fibrosis. Int J Mol Sci. (2022) 23:9028. doi: 10.3390/ijms23169028
Keywords: ferroptosis, immune homeostasis, idiopathic pulmonary fibrosis (IPF), type I interferon, interleukin, tumor necrosis factor, transforming growth factor, toll-like receptor
Citation: Yao M, Liu Z, Zhao W, Song S, Huang X and Wang Y (2025) Ferroptosis in idiopathic pulmonary fibrosis: mechanisms, impact, and therapeutic opportunities. Front. Immunol. 16:1567994. doi: 10.3389/fimmu.2025.1567994
Received: 28 January 2025; Accepted: 05 May 2025;
Published: 21 May 2025.
Edited by:
William Boisvert, University of Hawaii at Manoa, United StatesReviewed by:
Hector A. Cabrera-Fuentes, Imam Abdulrahman bin Faisal University, Saudi ArabiaTingting Wei, Fudan University, China
Copyright © 2025 Yao, Liu, Zhao, Song, Huang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Wang, d195aTIwMjJAMTYzLmNvbQ==; Xiaobo Huang, ZHJodWFuZ3hiQDE2My5jb20=; Siyuan Song, c2kteXVhbi5zb25nQGJjbS5lZHU=
†These authors have contributed equally to this work