Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is a series of obesity-related metabolic liver diseases, ranging from relatively benign hepatic steatosis to metabolic-associated steatohepatitis (MASH). With the changes in lifestyle, its incidence and prevalence have risen to epidemic proportions globally. In recent years, an increasing amount of evidence has indicated that the hepatic microenvironment is involved in the pathophysiological processes of MASH-induced liver fibrosis and the formation of hepatocellular carcinoma (HCC). The hepatic microenvironment is composed of various parenchymal and non-parenchymal cells, which communicate with each other through various factors. In this review, we focus on the changes in hepatocytes, cholangiocytes, liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), Kupffer cells (KC), dendritic cells (DC), neutrophils, monocytes, T and B lymphocytes, natural killer cells (NK), natural killer T cells (NKT), mucosal-associated invariant T cells (MAIT), γδT cells, and gut microbiota during the progression of MASLD. Furthermore, we discuss promising therapeutic strategies targeting the microenvironment of MASLD-MASH-HCC.
Introduction
Current findings reveal a global prevalence of metabolic dysfunction-associated steatotic liver disease (MASLD) at around 25% (1), with the Middle East exhibiting the highest prevalence at 32%, followed by South America at 31%, and Africa at the lowest at 14% (2). Over the past three decades, there has been a significant increase in MASLD prevalence, rising from 25.3% in 1990–2006 to 38.2% in 2016-2019 (3). Metabolic disorders such as hyperglycemia, obesity, dyslipidemia, and hypertension are known contributors to the development of MASLD (4). MASLD is characterized by hepatic steatosis, defined as >5% accumulation of triglycerides in hepatocytes. In contrast, metabolically associated steatohepatitis (MASH) is identified by >5% hepatic steatosis accompanied by hepatocytes damage, inflammation, and potentially fibrosis (5). Approximately 20% of MASLD cases progress to MASH, with a subset of MASH patients (about 2%) advancing to MASH-associated hepatocellular carcinoma (MASH-HCC) (see Figure 1 (5). Following the rebranding of the American Association for the Study of Liver Diseases (AASLD) from non-alcoholic fatty liver disease (NAFLD) to MASLD and from non-alcoholic steatohepatitis (NASH) to MASH, the new nomenclature has garnered significant recognition among scholars for its enhanced applicability in both clinical and academic settings.
Figure 1

The development of MASLD, MASH and HCC.
Approximately 10% (range 1-38%) of liver cancer cases are linked to MASLD, with higher prevalence rates (>20%) observed in studies conducted in the US, UK, India, Germany, and the Middle East, and lower rates (1-2%) reported in China and Japan (6). Utilizing mathematical models to forecast the incidence of MASH-HCC, projections indicate a substantial increase between 2016 and 2030, with a predicted rise of 47% in Japan, 82% in China, 88% in the UK, 117% in France, and 130% in the USA (7). Despite MASH-HCC having a lower incidence compared to virus-induced hepatocellular carcinoma (virus-HCC), the escalating prevalence of MASLD in conjunction with advancements in viral hepatitis treatment is anticipated to elevate its relative proportion and incidence (5).
Novel insights propose that the liver microenvironment could represent a pivotal determinant in the evolution of MASLD, MASH, and MASH-related HCC, comprising parenchymal cells, non-parenchymal cells, and the extracellular matrix (see Figure 2). Parenchymal cells, such as hepatocytes and cholangiocytes, are integral components of the liver microenvironment. Non-parenchymal cells, including liver sinusoidal endothelial cells (LSECs), hepatic satellite cells (HSCs), Kupffer cells (KCs), dendritic cells (DCs), neutrophils, monocytes, T and B lymphocytes, natural killer cells (NK), natural killer T cells (NKT), mucosal-associated invariant T cells (MAITs), and γδT cells, are believed to have significant implications in the initiation and progression of HCC (8).
Figure 2
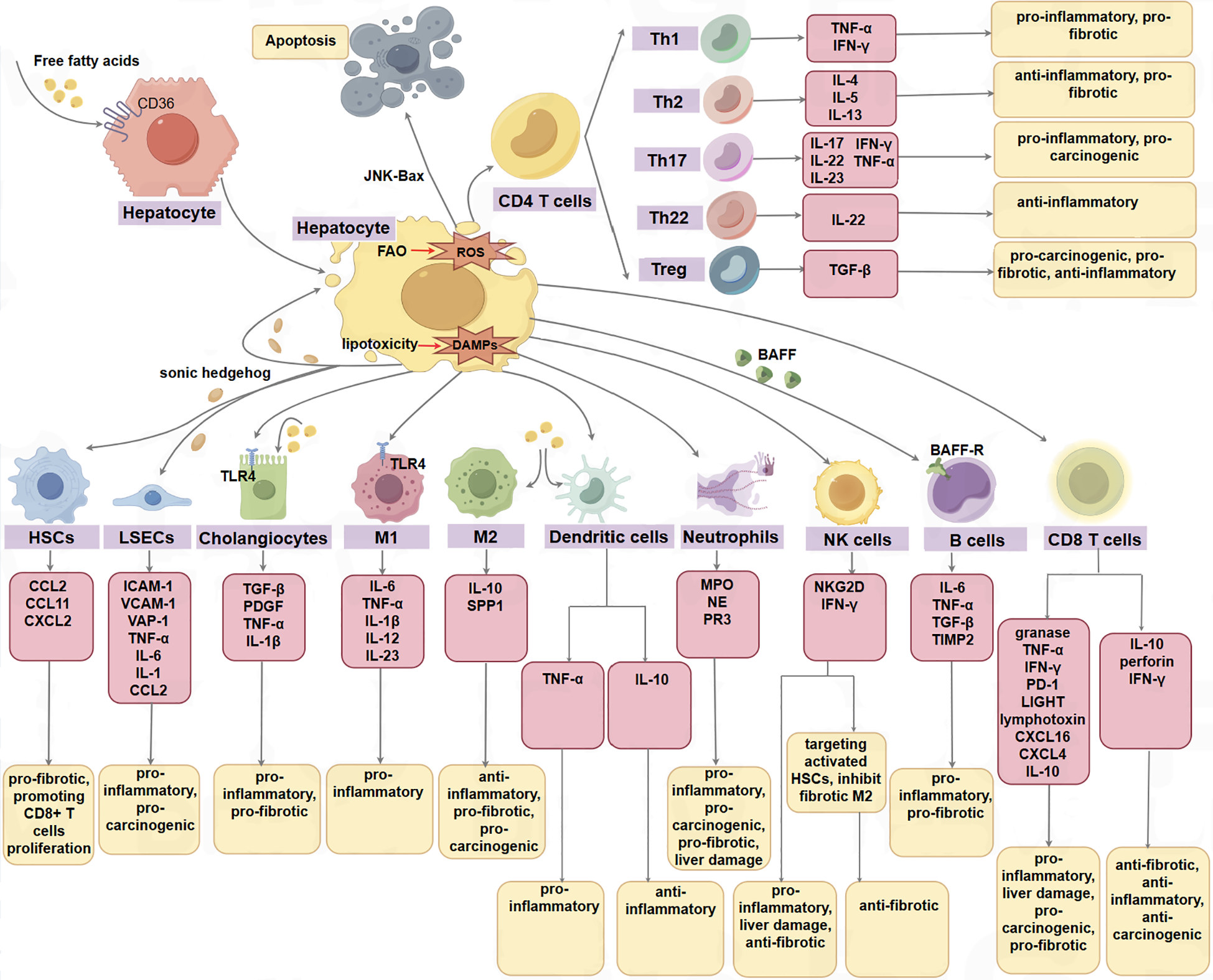
The parenchymal cells, non-parenchymal cells, and the extracellular matrix in the hepatic microenvironment function in the progression of MASLD, MASH, and MASH-HCC. Some cells have opposite effects in different phenotypes or disease stages, such as cx3cr1-expressing myeloid DCs can produce TNF-α to promote inflammation, while CD103+ dc plays a protective role in steatohepatitis by producing IL10. In addition, CD8T cells can not only promote the progression of NALFD by producing a series of inflammatory factors such as granase, TNF-α, IFN-γ, but also play an anti-inflammatory and anti-tumor role by producing IL-10 and perforin. M1 M1 macrophage, M2 M2 macrophage.
The current therapeutic strategies for MASH-HCC mirror those employed for other etiologies of HCC, encompassing transplantation, resection, or locoregional therapies for early or intermediate-stage disease (9). However, due to the distinct pathological characteristics of MASH-HCC, its tumor microenvironment differs substantially from virus-induced HCC. Consequently, while the prognosis following local treatment for early or intermediate-stage MASH-HCC and virus-induced HCC is generally comparable, advanced MASH-HCC exhibits limited efficacy and survival rates in immune checkpoint blockade (ICB) therapy (10, 11). Therefore, further investigations into the intricate interplay of the liver microenvironment in the pathogenesis of MASLD, MASH, and MASH-HCC are imperative for advancing our understanding of these conditions.
Hepatocytes
The accumulation of lipids within hepatocytes is associated with lipotoxicity, leading to cellular stress and eventual cell death. This process triggers the release of damage-associated molecular patterns (DAMPs), which encompass a variety of molecules such as metabolites, microRNAs, mitochondrial double-stranded RNA, mitochondrial DNA, purine nucleotides, cytoplasmic proteins, and mitochondrial compounds (12). Metabolites considered as DAMPs mainly include fatty acid, uric acid, ceramide, cardiolipin, cholesterol, citrate, succinate, ATP, etc. All of these can induce the activation of the NLRP3 inflammasome (13). Subsequently, these DAMPs are recognized by members of the Toll-like receptor (TLR) family, such as TLR2, TLR4, TLR5, and TLR9 (14), leading to the activation of Kupffer cells and the recruitment of monocyte-derived macrophages (MDMs) and other immune cells to orchestrate a sterile inflammatory response aimed at restoring tissue homeostasis. For example, DAMPs can recruit KCs through TLR on the cell surface and activate them to release tumor necrosis factor-α (TNF-α), IL-1β, CCL2, etc. DAMPs can also recruit mast cells through TLR on the cell surface and activate them to release TNF-α, IL-1β, TGFβ, etc. Additionally, DAMPs can recruit DCs through TLR on the cell surface and activate them to release TNF-α and IL-1s (14). However, in cases where the proinflammatory stimulus persists, the inflammatory response may become chronic, leading to tissue remodeling, fibrosis, and sustained inflammation.
Furthermore, hepatocytes apoptosis is considered a pivotal mechanism contributing to liver fibrosis in the pathogenesis of MASH. Macrophages and apoptotic hepatocytes are significant sources of reactive oxygen species (ROS) production (15). The entry of long-chain fatty acids (LCFAs) into hepatocytes mitochondria via FAT/CD36 facilitates fatty acid oxidation (FAO), consequently increasing ROS generation (16). Elevated levels of ROS have the potential to activate c-Jun N-terminal kinase (JNK), also known as stress-activated protein kinase. The activation of JNK by ROS promotes the lipoapoptosis of hepatocytes by upregulating the pro-apoptotic Bcl-2 protein Bax, thereby initiating the mitochondrial apoptosis pathway (17). Conversely, ROS can also stimulate AMP-activated protein kinase (AMPK), which acts to suppress ROS production, thereby preserving metabolic equilibrium and cell survival. Notably, AMPK activity is diminished in MASLD and MASH, with AMPK demonstrating the ability to alleviate liver fat accumulation and MASH-associated hepatocytes apoptosis (18).
Ballooned hepatocytes represent a characteristic feature of MASH and are implicated in the pathogenesis of the disease. Hedgehog (Hh), a ligand involved in the developmental hedgehog signaling pathway (19), is released from distended hepatocytes, leading to the activation of LSECs and HSCs (20). However, dysregulated activation of the Hedgehog signaling pathway during MASH and fibrogenesis may ultimately contribute to the development of liver cancer (21).
Cholangiocytes
Elevated levels of fatty acids (FAs) have been demonstrated to induce cholangiocytes lipoapoptosis in a FoxO3/miR-34a-dependent manner. MASLD patients present heightened levels of Ductular reaction (DR) and fibrosis, serving as markers of cholangiocytes damage. DR and biliary senescence are recognized as characteristic features of cholangiopathies, with their prevalence heightened in individuals with MASLD and MASH, suggesting a pivotal role in the progression of MASLD (22).
During the development and progression of MASLD, hepatocyte-derived DAMPs have been observed to interact with TLR4 expressed by biliary epithelial cells, leading to the manifestation of duct reactions and biliary senescence (23). Senescent cholangiocytes exhibit an augmented secretion of senescence-associated secretory phenotype (SASP) factors, including transforming growth factor-beta (TGF-β), platelet-derived growth factor (PDGF), TNF-α, and IL-1β (24). This increased secretion plays a role in the attraction and infiltration of immune cells, thereby worsening microvesicular steatosis and fibrosis as MASLD progresses.
NKT cells have been shown to become activated upon recognition of lipid antigens through cluster differentiation 1 molecules on bile duct cells, leading to the release of inflammatory cytokines and active participation in the inflammatory response (25). Furthermore, both secretin (SCT) and its receptor (SCTR) exhibit upregulation in MASLD/MASH patients compared to healthy controls. Notably, mice lacking SCT or SCTR demonstrated reduced levels of DR and biliary cell senescence, concomitant with an upregulation of miR-125b, a specific inhibitor targeting various enzymes involved in lipid biosynthesis, such as Elovl1 (26).
LSECs
Under normal physiological circumstances, LSECs orchestrate a highly immunosuppressive microenvironment that acts as a protective shield against intestinal antigen-induced inflammation (27). In the initial phases of MASLD, LSECs demonstrate a downregulation of pro-inflammatory chemokines via a mitogen-activated protein kinase (MAPK)-dependent pathway, including CCL2, potentially serving as a compensatory mechanism to impede disease progression (28). The above conclusion is contrary to that CCL2 is upregulated in other cells in MASLD. The downregulation of CCL2 in LSECs may be related to the enhanced autophagy of LSECs in the early stage of MASLD, which aims to maintain cellular homeostasis (29). However, as MASH advances, LSECs transition to a pro-inflammatory state characterized by the heightened expression of adhesion molecules and pro-inflammatory mediators, such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), vascular adhesion protein-1 (VAP-1), TNF-α, IL-6, IL-1, and CCL2 (30, 31). Consequently, dysfunctional LSECs contribute to the inflammatory cascade and activation of KCs (32), thereby accelerating the progression of MASLD towards MASH and HCC (33). Furthermore, lipotoxicity has been shown to induce the generation of ROS by LSECs, thereby fostering a pro-carcinogenic milieu (31).
In addition to alterations in phenotype and functionality, the morphology of LSECs undergoes changes during the progression from MASLD to MASH. A prominent morphological change observed in LSECs is capillarization, which manifests early in the development of MASLD (34). The capillarization of LSECs hampers the exchange of substances between sinusoids and blood, obstructs the removal of hepatocyte-derived very low-density lipoprotein (VLDL), and leads to the accumulation of cholesterol and triglycerides within the liver (35), exacerbating hepatocellular steatosis (36). Moreover, capillarization reduces the transfer of chylomicrons to hepatocytes, stimulates new adipogenesis, and triggers a compensatory rise in cholesterol and triglyceride synthesis in hepatocytes (37). Notably, primary human LSECs exposed to oxidized low-density lipoprotein (LDL) exhibited reduced fenestrae diameter and porosity (38), while a 48-hour fasting period in rats led to an enlargement in the diameter of LSEC fenestrae (39). Additionally, consumption of a high-fat diet has been linked to a reduction in both the size and number of fenestrae in LSECs (40).
HSCs
The potential dual role of HSCs in MASH has been postulated, suggesting a beneficial impact in the early stages and a deleterious effect in the later stages of the disease progression (41). In the context of MASLD, HSC activation is triggered by hepatocellular-derived Hh ligands and osteopontin (OPN), leading to the upregulation of transcription factors ETS1 and RUNX1 in HSCs (42, 43). Utilizing single-cell RNA sequencing (scRNA-seq) analysis in murine MASH models, it was observed that activated HSCs play a role in modulating macrophage functions through the secretion of stellakines, including CCL2, CCL11, and CXCL2 (44). Furthermore, HSC activation is induced by the secretion of IL-10 from CD8+ T cells, subsequently promoting CD8+ T cells proliferation and exacerbating MASH progression (45). HSCs are pivotal in the development of liver fibrosis (46), with quiescent HSCs capable of differentiating into myofibroblasts in response to hepatocyte-derived lipid mediators and pro-fibrotic cytokines such as TGF-β in MASH (47). The therapeutic targeting of both apoptosis and senescence of activated HSCs represents crucial strategies in mitigating hepatic fibrosis (47).
However, significant upregulation of key signaling molecules, including SHh, β-catenin, and Wnt10b, involved in the Hedgehog and Wnt signaling pathways, was observed in senescent HSCs. These findings suggest a potential role for these molecules in the process of hepatocellular malignant transformation within a microenvironment characterized by steatosis, inflammation, and fibrosis (48).
Gut microbiota
The composition of gut microbiota has been shown to influence the immunological landscape of the liver, impacting both adaptive immunity (Tregs, Th1, Th2, Th17, NKT, and B cells) and innate immunity (Neutrophils, KCs, dendritic cells, and NK cells) (49). Dysbiosis of the gut microbiota is frequently observed in MASLD (50), with MASH patients exhibiting increased levels of Actinomyces, Ruminococcus, Blautia, and Dorea, and decreased levels of Bacteroides compared to healthy individuals (51). Conversely, alternative studies have reported lower levels of Faecalibacterium and Anaerosporobacter but higher levels of Parabacteroides and Allisonella in MASH patients (52). Furthermore, MASH fibrosis has been associated with higher levels of Ruminococcus, Proteobacteria, and Escherichia coli, and lower levels of Firmicutes (53).
In addition, the diameter and quantity of LSECs may exhibit a negative correlation with the presence of Bacteroides and a positive correlation with the abundance of Firmicutes (40). Furthermore, an elevated prevalence of alcohol-producing bacteria has been noted in individuals with MASH, suggesting a potential involvement of these bacterial strains in the pathogenesis of MASH (54). Recent research involving animal models has demonstrated that Faecalibacterium prausnitzii can mitigate inflammation in adipose tissue and enhance liver function in MASH mice (55).
The migration of bacterial byproducts from the gut microbiota, such as lipopolysaccharide (LPS), peptidoglycan, and bacterial DNA, through the portal vein can activate TLRs on KCs, triggering an inflammatory response and exacerbating MASH (56). Notably, individuals with MASLD often experience bacterial overgrowth, leading to heightened levels of bacterial products like LPS (57). Activation of KCs occurs upon binding of LPS to TLR4 (58). Oral administration of Lactobacillus casei to mice has been shown to suppress methionine-choline-deficient (MCD) diet-induced MASH by inhibiting TLR-4 signaling, resulting in reduced serum LPS levels, decreased liver inflammation, and fibrosis (59).
Recent findings suggest that the gut microbiota may play a role in the development of MASH-HCC by virtue of the immunomodulatory properties of their metabolites (50). Studies have linked HCC with alterations in gut microbiota composition and inflammation in MASLD patients, characterized by decreased levels of Bifidobacterium but increased abundance of Bacteroides and Ruminococcaceae (60). Furthermore, a recent investigation revealed an enrichment of short-chain fatty acid (SCFA)-producing bacteria in patients with MASLD-HCC, leading to an immunomodulatory shift towards immunosuppression marked by elevated IL-10+ Tregs and reduced CD8+ T cells (61). Notably, interventions such as antibiotic administration and intestinal sterilization have been shown to decrease the incidence of HCC in obese mice, underscoring the significant role of microbiota dysbiosis in the pathogenesis of HCC (62).
Macrophages
In the conventional understanding, liver macrophages are primarily composed of liver-resident KCs and circulating MDMs (63). In patients with MASLD, the infiltration of portal macrophages is detected at an early stage, preceding the onset of inflammation, and their activation plays a role in the initiation and advancement of the disease (64). The primary mechanism underlying this process appears to involve lipid accumulation leading to the depletion of KCs in the liver. In MASH, hepatocytes damage induces the recruitment of CCR2+/CCR3+ MDMs by motivating KCs to release proinflammatory chemokines such as CCL2, CCL5, and CXCL10. As MASH progresses, KCs are gradually replaced by Ly-6C+ monocytes (65). These MDMs demonstrate reduced efficacy in promoting liver triglyceride storage and exacerbate liver damage in the context of MASH. This phenomenon is supported by the significant decrease in liver inflammation observed in CCR2-/- and CXCR3-/- MASH mice (66). Apart from MDMs, KCs have the capacity to recruit other cell types that contribute to the acceleration of MASH progression (67).
Furthermore, liver macrophages have the capacity to secrete a diverse array of cytokines that play a role in driving the progression of MASH. The balance between M1 and M2 phenotypes within hepatic macrophages is disrupted by hepatic steatosis, influenced by factors such as cytokines, lipid metabolites, and various signaling molecules (68). In the context of MASLD, KCs are activated upon recognition of DAMPs via TLR4 receptors (69). Upon activation, KCs release cytokines such as IL-6, TNF-α, and IL-1β, which in turn enhance the infiltration of NKT and CD8+ T cells into the liver, thereby promoting the progression of MASH (70).
Additionally, the unsaturated fatty acid oleic acid (OA) has been found to induce M2 macrophage polarization in the context of MASLD -MASH (71). The polarization of macrophages towards the M2 phenotype has been implicated in the promotion of MASH-associated liver cancer by creating an immunosuppressive microenvironment (72). Consequently, it has been postulated that the activity of HSCs may exhibit a beneficial effect in the early stages of MASH but a detrimental effect in the later stages (63). This suggests a potential transition from a pro-inflammatory M1 macrophage phenotype to an anti-inflammatory M2 macrophage phenotype during the progression of MASLD-MASH, mainly attributed to the secretion of IL10 by M2 macrophage, which activates arginase in M1 macrophage and promotes M1 apoptosis (73).
In the context of MASH, M2-type KCs exhibit the ability to mitigate hepatic inflammation by suppressing the activation of M1-type KCs, yet they possess the potential to promote fibrosis (74). Various investigations have delineated a distinct subset of macrophages characterized by the expression of myeloid cells 2 (TREM2), CD9, GPNMB, and SPP1 within the liver affected by MASH (75). These TREM2-expressing macrophages demonstrate elevated levels of CCR2 and CX3CR1 and are recruited to hepatic crown-like structures (CLSs) in the steatotic liver in a CCR2-dependent manner (76). Correspondingly, a separate scRNA-seq analysis has demonstrated a pathogenic subset of TREM2+CD9+ macrophages within the fibrotic microenvironment of human livers affected by MASH, exhibiting a positive correlation with the degree of MASH-induced liver fibrosis (77). Conversely, studies have revealed that the absence of TREM2 accelerates the progression of MASLD in murine models, potentially due to its anti-inflammatory properties. Mechanistically, TREM2 interacts with and signals through DAP12, leading to the downregulation of inflammatory gene transcription, such as TNF-α, IL-1β, and NOS2 (78).
Consequently, investigations have demonstrated that heightened recruitment of TREM2+ macrophages within the hepatic milieu and elevated levels of soluble TREM2 in the systemic circulation are linked to favorable prognoses in individuals afflicted with fibrotic MASH (79). Furthermore, prolonged exposure to excessive nutrition in the context of fatty liver conditions results in the compromise of TREM2-dependent macrophage efferocytic function, consequently exacerbating hepatic inflammation and the advancement of MASH (80). Noteworthy is the observation that the uptake and accumulation of fatty acids prompt macrophage polarization towards an immunosuppressive phenotype in HCC (81).
Dendritic cells
Alongside macrophages, the hepatic environment harbors a population of DCs comprising distinct subsets of classical and plasmacytoid DCs (pDCs) that are situated in the subcapsular region, interstitial spaces between hepatocytes, and within the vasculature (82). Investigations have revealed that DCs with lower lipid content exhibit a more tolerogenic phenotype, capable of promoting Tregs induction, whereas lipid-rich hepatic DCs possess immunogenic properties, stimulating the activation of T cells, NK cells, and NKT cells (83). Recent findings have indicated an increase in CX3CR1-expressing myeloid DCs in mice subjected to a MCD diet, which led to heightened TNF-α production; conversely, blockade of CX3CR1 resulted in amelioration of steatohepatitis in mice, implicating a disease-promoting role for myeloid DCs in MASLD (84). Utilizing scRNA-seq and transcriptional network analyses, it was demonstrated that conventional type 1 dendritic cells (cDC1s) are more prevalent and exhibit enhanced production of proinflammatory cytokines and chemokines in both human and murine MASH (85). These XCR1+ cDC1s were found to drive inflammatory T cell activation and exacerbate MASH liver pathologies in murine models (86).
Conversely, a reduction in the frequency of pDCs is observed in MASH patients, with the decline in pDCs correlating inversely with the extent of liver damage as indicated by serum alanine transferase levels (87). A study utilizing the MCD diet reveals an increase in DCs within the liver of mice; however, following diphtheria toxin-induced depletion of CD11c+ cells, the severity of steatohepatitis worsens, and DC ablation leads to the proliferation of CD8+ T cells and reduction in Tregs, indicating a potential regulatory function (88). Conversely, during the transition from MASH to HCC, hepatic lipid accumulation may exacerbate DC impairment by enhancing triglyceride uptake via scavenger receptor A, thereby compromising tumor-specific antigen presentation to CD8+ T cells and anti-tumor immune responses (89). A targeted investigation has demonstrated that mice lacking CD103+ DCs exhibited more pronounced steatohepatitis, underscoring a protective role for CD103+ conventional dendritic cells (cDCs) (86). The divergent outcomes regarding DC involvement in MASH could be attributed to variations in distinct DC subtypes.
Neutrophils
In MASH, there is an upregulation of key chemokines that attract neutrophils from hepatocytes (90). The extent of neutrophil infiltration is positively associated with the severity of MASLD (64). The involvement of neutrophils in MASLD is widely supported by the observation of elevated levels of Myeloperoxidase (MPO) (91), neutrophil elastase (NE) (64), and proteinase 3 (PR3) (92), as well as neutrophil extracellular traps (NETs) (93) in MASLD patients, all of which have been implicated in promoting liver injury and the progression of MASH. Apart from their role in activating HSCs and driving liver fibrosis through the induction of M2-macrophage polarization (90), MPO has been implicated in the formation of CLS in MASH (94). NE has been shown to modulate insulin signaling, with its deletion leading to improved insulin sensitivity in an obesity mouse model (95). Modulating the expression or activity of MPO, NE, or PR3 through genetic manipulation or pharmacological approaches presents a promising therapeutic strategy for ameliorating pathological changes in MASH (64, 96). NETs represent a novel mechanism for neutrophil elimination, with elevated free fatty acids (FFAs) triggering NETs formation in neutrophils, and inhibition of NETs resulting in reduced severity of steatohepatitis (93). Indeed, NETs formation appears to foster an immune-suppressive milieu through programmed death ligand 1 (PD-L1) signaling, contributing to T-cell exhaustion in murine models of MASH-induced HCC. Specifically, NETs promote Treg differentiation by binding and activating TLR4 in immature CD4+T cells to enhance mitochondrial OXPHOS, while Treg can inhibit Teffs’ inhibitory effect on tumors (97). Consequently, beyond their traditional role in immune response, neutrophils are now recognized as potential mediators of MASH-HCC progression.
NK cells
In individuals with MASH, elevated levels of hepatic CXCL9, CXCL10, CXCL11, CCL3, CCL4, and CCL5 were observed compared to healthy controls, with circulating CXCL10 levels correlating with the severity of lobular inflammation (66). Following MCD treatment, increased CXCL10 expression facilitated the recruitment of NK cells to the liver (98). The upregulation of chemokine expression, including CXCL10, CCL2, and CCL5, was found to be reliant on IL-15 signaling in hepatocytes in response to a high-fat diet (HFD), with IL-15-deficient mice exhibiting reduced MASH severity and diminished NK cell presence in the liver (99). Similarly, NK cells are activated by cytokines such as IL-12, IL-15, and IL-18 to produce interferon-gamma (IFN-γ), thereby driving the progression from MASLD to MASH (99).
Studies have revealed a notable increase in the population of NK cells expressing nature killer group 2D (NKG2D) in MASH compared to healthy individuals (100). Additionally, elevated levels of the NKG2D ligand major histocompatibility complex (MHC) class I associated chains A and B (MICA/B) in the liver have been observed in MASH patients, suggesting a potential involvement of NK cells in MASH progression through interactions with MICA/B (101). This interaction may trigger JAK-STAT1/3 and nuclear factor κB (NF-κB) signaling pathways in the liver, leading to hepatocytes damage (100). Interestingly, our investigations have demonstrated that NK cells exhibit cytotoxic activity against MICA-expressing cancer cells via the NKG2D receptor during HCC progression (102). Conversely, NK cells may play a protective role in the development of MASH-related fibrosis. Notably, NK cells have been shown to regulate liver fibrosis by directly targeting activated hepatic stellate cells through receptors such as NKG2D and NKp46, as well as the p38/PI3K/AKT pathway (103).
A recent investigation has reported a negative correlation between liver stiffness measurement and the total number of NK cells (104). In a murine model utilizing the MCD diet, an increase in CD49b+ NKp46+ conventional NK (cNK) cells is observed to inhibit fibrotic M2 phenotypic differentiation by skewing macrophages towards M1-like phenotypes. This effect is dependent on the cNK cells’ secretion of IFN-γ rather than granzyme-mediated cytotoxicity (105).
B cells
B lymphocytes constitute approximately 6% of intrahepatic cells and represent around 50% of intrahepatic lymphocytes in mice (106). Focal clusters of B cells have been identified in the livers of both MASH patients and mice (107), which secrete pro-inflammatory cytokines such as TNF-α and IL-6 (108). While it is commonly believed that B cells produce fewer inflammatory cytokines compared to macrophages and neutrophils, a subset of B cells expressing high levels of BAFF-R has been detected during MASH. Upregulated BAFF in the liver may serve as one of the cytokines that facilitate the maturation and activation of these B cells (109). Treatment with BAFF depletion or induction of B-cell deficiency has been shown to ameliorate steatohepatitis in mice (110).
In MASH, B cells exhibit not only the capacity to generate pro-inflammatory mediators but also to participate in antigen presentation. The activation of B cells during MASH is mechanistically linked to signaling pathways involving the innate adaptor protein myeloid differentiation primary response protein 88 (MyD88). Specifically, the deletion of MyD88 in B cells has been shown to diminish hepatic T cell-mediated inflammation and fibrosis (109). Furthermore, RNA-sequencing analyses have unveiled that intrahepatic B cells play a role in MASH by expressing genes associated with fibrosis, such as TGF-β and TIMP2 (111). The pro-fibrotic actions of B cells are mediated through the production of pro-inflammatory molecules that stimulate HSCs and macrophages. Activated HSCs, in turn, promote the survival and maturation of liver B cells by releasing retinoic acid. Notably, the presence of circulating IgA has been identified as an independent predictor of advanced fibrosis and HCC development in MASH (111). Collectively, these findings underscore the dual role of B cells in driving inflammation and fibrosis progression in MASH.
CD8+ T cells
The presence of ROS and its byproduct derived from lipid peroxidation, malondialdehyde (MDA), has been shown to trigger the recruitment of CD8+ T cells to the liver in both human subjects and murine models of MASLD (10, 11, 67, 112). In response to metabolic triggers like acetate and extracellular ATP, CXCR6+CD8+ T cells demonstrate self-destructive tendencies regardless of MHC-class-I molecules. This behavior is characterized by increased expression of granase, TNF-α, IFN-γ, and programmed death 1 (PD-1), which is facilitated by IL-15-induced downregulation of the transcription factor FOXO1. Consequently, this cascade of events promotes hepatocytes injury and contributes to the development of MASH and HCC (10).
Furthermore, a study has demonstrated that the administration of anti-PD-1 immunotherapy to mice fed a choline-deficient high-fat diet (CD-HFD) led to an increase in hepatic resident-like (CXCR6+) and effector (GZMK+ and GZMA+) CD8+ T cells expressing PD-1. These cells are found to secrete tumor necrosis factor superfamily member 14 (TNFSF14/LIGHT) and lymphotoxin, which respectively activate the lymphotoxin β receptor (LTβR) and NF-κB signaling pathways in hepatocytes. This activation serves as a crucial intrinsic regulatory mechanism in hepatocytes, influencing the transition from MASH to HCC (67). Moreover, B2 microglobulin knockout (b2m-/-) mice, which exhibit a severe deficiency in CD8+ T cells, are found to be protected from liver damage and tumorigenesis induced by a CD-HFD diet (67).
Additionally, in the murine model of MASH with cirrhosis, hepatic fibrosis has been linked to an elevated CD8+/CD4+ T cell ratio (113). Within the MASH mouse model, hepatic CD8+ T cells expressing CXCR1 and CXCR6 have been identified to effectively activate HSCs through the secretion of IL-10, CXCL4, and CXCL16. The reduction of CD8+ T cells within the MASH liver has been associated with decreased mRNA levels of fibrotic genes, including α-smooth muscle actin, TGF-β, collagen type 1 alpha 1, and collagen type 1 alpha 2 (112).
While the current evidence supports the pathogenic role of total CD8+ T cells in MASH progression, the application of single-cell genomics has unveiled functionally distinct subsets within the CD8+ T cell population. Through single-cell RNA analysis, researchers have characterized a subset of hepatic resident memory T cells marked by the expression of CD44+CD62L− CD69+CD8+ T cells, which also exhibit CXCR6. These resident T cells have been found to play a crucial role in resolving the fibrotic process. Specifically, these cells interact with HSCs through CCR5-driven chemotaxis and induce apoptosis in HSCs via Fas-FasL contact (114).
Concurrently, enhanced intrahepatic retention of activated NKT cells and CD8+ T cells has been observed to mitigate glucose intolerance, liver steatosis, the release of inflammatory factors (such as IL-10), and tumor growth (via the IRF1/CXCL10/CXCR3 axis) in MASH, consequently suppressing the development of HCC (115–117). Furthermore, CD8+ T cells have been identified to secrete perforin, which triggers apoptosis in M1 macrophages, thereby inhibiting the synthesis of pro-inflammatory cytokines in MCD-fed mice (118). Hence, it is evident that specific subsets of CD8+ T cells also exhibit a protective role in the context of MASH.
CD4+ T cells
Linoleic acid has been shown to disrupt mitochondrial function in CD4+ T cells, culminating in elevated ROS generation, activation of caspases, and subsequent cell death, leading to an overall reduction in CD4+ T cell numbers (119). Despite the decline in the total count of CD4+ T cells observed in MASH and MASH-HCC, a specific subset of T helper (Th) cells has been found to increase (120). Based on their functional characteristics and cytokine secretion profiles, CD4+ T cells are further categorized into distinct subtypes including Th1, Th2, Th17, Th22, Tregs, and other subsets (121).
Th1 cells
In MASLD, Th1 cells differentiate from naïve CD4+ T cells and secrete cytokines such as IFN-γ and TNF-α, exerting pro-inflammatory effects (122). Clinical evidence further substantiates these findings, demonstrating an increased proportion of liver and circulating Th1 cells producing IFN-γ in both pediatric and adult patients with MASH (123, 124). Additionally, elevated plasma levels of IFN-γ have been positively correlated with the abundance and size of hepatic lymphocyte clusters, as well as the severity of fibrosis in MASH (107). IFN-γ can activate KCs, leading to the progression of MASH and MASH-associated HCC (125). Furthermore, the IFN-γ-induced chemokine CXCL10 is capable of attracting T cells expressing the CXCR3 receptor, and the absence of CXCR3 or deletion of CXCL10 has been shown to alleviate liver inflammation, injury, and fibrosis (66). Studies have revealed an upregulation of the TNF family co-stimulatory molecule OX40 in CD4+ T cells of mice fed a high-fat diet, with OX40 deficiency resulting in reduced infiltration of CD4+ T cells and diminished Th1 cell differentiation, thereby ameliorating steatohepatitis (126).
Th2 cells
Currently, limited research exists on the role of Th2 cells in MASLD and MASH, which predominantly secrete IL-4, IL-5, and IL-13 and activate signal transducer and activator of transcription (STAT) proteins 5 and 6 (124). While investigations have indicated an elevated Th1/Th2 ratio in mesenteric lymph nodes of MASLD-afflicted mice (127), a contrasting scenario is observed in peripheral blood of MASLD patients, where Th2 cell levels are heightened compared to healthy individuals (128). Notable differences in Th2 cell populations in peripheral blood or liver have not been observed among patients with MASH, MASLD, or healthy controls (124). Despite the potential anti-inflammatory role of Th2 cells in MASLD (129), their involvement in liver fibrosis progression, particularly in the presence of IL-13, is evident. Elevated levels of circulating IL-13 and increased expression of IL-13 receptor alpha 2 (IL-13Rα2) in the liver have been reported in individuals with MASH, with liver fibrosis mitigation observed upon targeting IL-13Rα2+ cells, including HSCs (130).
Th17 cells
IL-17 secreted by Th17 cells exacerbates inflammation in MASLD by inducing ROS and enhancing neutrophil infiltration, thereby hastening disease progression (131). Both in MASLD patients and murine models, hepatic Th17 cell populations exhibit a continuous increase throughout disease advancement, peaking at the onset of steatohepatitis and later stages of the condition (132, 133). Steatotic hepatocytes display heightened sensitivity to IL-17A, leading to upregulation of its receptor IL-17RA, augmented cytokine release encompassing IL-6, TNF-α, CXCL1, and increased lipid synthesis (134). Recent investigations have unveiled that MASLD triggers a rise in circulating and liver-resident CXCR3+ Th17 cells through glycolysis induction, enhancing their capacity to produce IL-17A, IFN-γ, and TNF-α (134). Mice subjected to glycolytic inhibition or lacking IL-17A or its receptor, IL-17RA, exhibited milder steatohepatitis symptoms (131). Furthermore, Th17 cells prompt macrophages to release pro-inflammatory cytokines such as IL-6, TGF-β, TNF-α, and IFN-γ through IL-17, thereby amplifying the inflammatory cascade (135).
Studies have documented that the expansion of Th17 cells, along with their secretion of IL-17, IL-22, and IL-23, contributes to the promotion of hepatocarcinogenesis in MASH (136). In mice fed a CD-HFD diet, IL-17A released by hepatic Th17 cells induces insulin resistance in the adjacent adipose tissue, leading to enhanced influx of fatty acids into the liver, thereby triggering MASH-related HCC. Inhibition of IL-17A signaling mitigates liver damage and prevents the development of HCC (137).
Th22 cells
Within the context of MASH, CD4+ T cells producing IL-22 (Th22) are notably abundant during the initial and subsequent expansions of Th17 cells (129). IL-22 has been implicated in exerting inhibitory and protective effects on the advancement of MASLD. Administration of recombinant IL-22 has shown significant improvement in the liver damage and steatohepatitis in animal models of MASH, potentially mediated by STAT3 pathway (138). IL-22 also demonstrates the ability to mitigate lipotoxicity induced by palmitate by inhibiting JNK in a phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)-dependent manner (129). Notably, the beneficial effects of IL-22 are observed in the absence of IL-17, as IL-17 can upregulate phosphatase and tensin homolog (PTEN), an antagonist of PI3K-AKT signaling (129). Furthermore, IL-22 can upregulate the expression of anti-apoptotic genes such as bcl2 and suppress the expression of lipid biosynthesis genes such as scd1 (139).
Tregs
In the MASH context, the generation of NETs has been demonstrated to promote the differentiation of Tregs from naive CD4+ T cells (93). Notably, the population of liver-resident Tregs (CD4+Foxp3+) expands during MASH, with FOXP3+ Tregs demonstrating the ability to suppress the proliferation and activity of CD8+ T cells and Th1 cells, which play a crucial role in immune surveillance against cancer. Notably, the depletion of Tregs (CD4+Foxp3+) has been found to significantly impede the progression of HCC in a MASH model induced by choline deficiency, high-fat diet consumption, and diethylnitrosamine administration (97). Conversely, the transfer of Tregs in MASH exacerbates the severity of the disease (140). Furthermore, Tregs are known to secrete TGF-β, which possesses pro-fibrotic properties (141). However, a decline in hepatic Treg numbers has been observed in MASLD, potentially attributed to the induction of Treg apoptosis by KCs and DCs through the generation of oxidative stress, TNF-α, and interferon I (142, 143).
Additionally, the upregulation of leptin production in obesity has been demonstrated to impede the differentiation of Tregs while concurrently promoting the activation of dendritic cells and the polarization of CD4+ T cells towards the Th1 and Th17 subsets (144). Remarkably, a recent investigation revealed that the reduction in Treg numbers in mice achieved through the combined deficiency of the co-stimulatory molecules CD80 and CD86 exacerbated adipose tissue inflammation and steatohepatitis induced by a high-fat diet (145). Moreover, the transfer of Tregs has been shown to mitigate TNF-α signaling induced by a high-fat diet and LPS-triggered hepatotoxicity (142). These seemingly contradictory findings may be ascribed to the dual role of Tregs in MASH, acting protectively during the inflammatory phase and promoting tumor development in later stages.
Innate-like T lymphocytes
Hepatic innate-like T lymphocytes, such as γδT cells, NKT cells, and MAITs, have been implicated in the pathogenesis of MASH in humans and mice.
γδ T cells
γδ T cells have been implicated in the advancement of MASLD. In murine models of MASH, there is a notable increase in the population of γδ T cells within the liver, contributing to the development of steatohepatitis, hepatic injury, and perturbed glucose metabolism via the secretion of IL-17A (146). The recruitment of γδ T cells, particularly IL-17high Ly6C− CD44+ γδ T cells, is dependent on the chemokine receptors CCR2 and CCR5 in the liver afflicted with steatohepatitis, facilitating the infiltration of inflammatory monocytes while dampening IFN-γ production in CD4+ T cells through the release of vascular endothelial growth factor (VEGF) and IL-15 (147). Conversely, the cytokines VEGF and IL-15 derived from γδ T cells in a CD1d-dependent manner have been shown to suppress IFN-γ, which exerts protective effects in certain cases of MASH (147). Additionally, γδ T cells can promote the progression of fibrosis by activating HSCs and KCs through the production of IL-17 (148). Notably, MASH mice lacking γδ T cells exhibits milder liver injury and steatohepatitis (149).
NKT cells
An increase of 10% in NKT cells has been observed in the livers of individuals with cirrhosis unrelated to MASH, while a 20% increase is noted in those with MASH-related cirrhosis (150). In the context of MASH, NKT cells not only instigate inflammation and steatosis by releasing LIGHT, IFN-γ, TNF-α, and IL-17A upon recognition of lipid antigens but also motivate HSCs to promote fibrosis via the secretion of OPN and Hh ligands in response to IL-15 stimulation and Hh pathway activation (67, 150, 151). Mice lacking IL-15 or IL-15Rα exhibit reduced intrahepatic CD4+, CD8+, NKT cells, and HSCs and, when subjected to a high-fat diet, display less severe steatosis and lobular inflammation compared to their wild-type counterparts (152). While the aforementioned findings support the role of NKT cells in driving the progression of MASH, their impact on the development or inhibition of MASH-related liver cancer remains uncertain. Research has indicated that liver NKT cells secrete LIGHT, a ligand for the lymphotoxin beta receptor (LTβR), which activates NF-kB signaling by binding to LTβR on hepatocytes and collaborates with CD8+ T cells to facilitate malignant transformation (67). Notably, targeted deletion of LTβR on hepatocytes or depletion of NKT cells has been shown to mitigate high-fat diet-induced HCC (67).
The involvement of distinct subpopulations of NKT cells in the regulation of disease progression presents a complex scenario. Several investigations have documented a decrease in the abundance of NKT cells in both human and murine MASLD (11, 153). This reduction in NKT cell numbers has been linked to heightened apoptosis triggered by IL-12 produced by KCs (154) and intensified Tim-3/Gal-9 signaling pathways (155). Mice deficient in Ja18 and CD1d, which lack NKT cells, exhibit elevated rates of weight gain and liver steatosis in the context of MASH (10). Conversely, leptin-deficient mice demonstrate diminished hepatic steatosis following the transfer of NKT cells, potentially associated with the down-regulation of IL-10 (116). Moreover, the accumulation of hepatic cholesterol selectively impedes the anti-tumor immune surveillance function of NKT cells through the buildup of lipid peroxidation and SREBP2-dependent cytotoxic impairments in diet-induced mouse models of MASH-HCC (156).
MAITs
CCR5 facilitates the migration of MAITs to sites of hepatic steatosis through its interaction with CCL5 (157). In individuals with MASLD-related cirrhosis, MALTs secrete IL-17 and TNF-α to stimulate the activation of hepatic fibrotic cells (158). Concurrently, the functions of circulating MALT cells undergo alterations, characterized by an increase in IL-4 secretion and a decrease in IFN-γ production (157). IL-4, known for its anti-inflammatory properties, may induce a shift KCs towards an M2 phenotype in the context of MASLD (73). This protective role is underscored by studies in animal models, where mice lacking MAITs exhibit pronounced hepatic steatosis (157) or exacerbated diabetes (159). In essence, MAITs appear to exert an anti-inflammatory protective function in MASLD while simultaneously promoting the progression of fibrosis.
Therapeutic targeting the microenvironment of MASLD-MASH-HCC
For the treatment of MASH, the goal should be to improve clinical outcomes, that is, to reduce the MASH-related mortality rate and decrease the incidence of progression to cirrhosis and HCC. The only treatment that has been strongly proven to improve liver injury in patients with MASLD without severe hepatic fibrosis is weight loss through diet (160). In addition, some studies have shown that in some patients, after lifestyle intervention and Roux-en-Y gastric bypass, MASH has been improved. In these improved patients, the inflammatory-related pathways, such as the complement, TNF-α, and IL-6 signaling pathways, have all been inhibited, mainly manifested by the downregulation of CXCL9, CXCL10 and lysozyme (161).
With the in-depth research on the mechanisms of metabolic damage, as well as the activation of inflammatory and fibrotic pathways in MASH, some drugs targeting several cellular components or molecular pathways of MASH have been preliminarily explored. Resmetirom is a selective agonist of the thyroid hormone receptor β (THR-β) and is the first treatment approved by the US Food and Drug Administration (FDA) for MASH (162–164). In MASH, the function of THR-β in the liver is impaired, leading to reduced mitochondrial function and fatty acid β-oxidation, which in turn increases fibrosis. Stimulating THR-β can promote the regulation of hepatic lipid metabolism (165). THR increases cholesterol metabolism through the hepatic enzyme CYP7A1 and reduces de novo lipogenesis (165). In contrast, the thyroid hormone activity outside the liver, including in the heart and bones, is mainly mediated by thyroid hormone receptor α (THR-α) (166).
In a mouse model of MASH with fibrosis, Resmetirom treatment significantly improved the MASLD activity score and reduced hepatic fibrosis (167). Data from phase II/III clinical trials reflect the safety and potential efficacy of Resmetirom in adult patients with MASH (162, 163). The activation of THR-β in the liver by Resmetirom has been shown to reduce triglycerides by 30.8%, low-density lipoprotein cholesterol (LDLC) by 22.3%, and lipoprotein (a) by 37.9% (162). Both 80 mg and 100 mg of Resmetirom can not only significantly improve the progression of steatohepatitis (24.2% for 80 mg, 25.9% for 100 mg, compared with 14.2% in the placebo group), but also reverse the hepatic fibrosis in MASH (25.9% for 80 mg, 29.9% for 100 mg, compared with 9.7% in the placebo group) (168). Compared with the placebo, among every 1,000 patients treated with Resmetirom, the number of decompensated cirrhosis events decreases by 87, the number of HCC events decreases by 59, and the number of liver transplants decreases by 30 (169). However, for patients with decompensated cirrhosis (Child-Pugh class B or C), Resmetirom should be avoided because as the dosage of Resmetirom increases, the risk of adverse events also increases (170).
Since the immune response seems to play a key role in MASH and its progression to HCC, methods to address this issue need to be explored. Cenicriviroc, a CCR2/CCR5 antagonist targeting pro-inflammatory monocytes, has been shown in a mouse model. In a phase II clinical trial, it was confirmed that Cenicriviroc improved the fibrosis in patients with MASLD (171). However, in a larger-scale phase III (AURORA) clinical trial, after one year of treatment, Cenicriviroc did not show sustained antifibrotic efficacy, leading to the termination of the study on its monotherapy for MASH. In another phase II clinical trial, Cenicriviroc was combined with the farnesoid X receptor (FXR) agonist Tropifexor to improve the hepatic fibrosis in patients with MASH, but the result analysis is still ongoing (172). FXR is mainly expressed in the liver, intestine, kidney, adipose tissue, and immune cells, participates in lipid metabolism, and exerts anti-inflammatory and antifibrotic effects (173).
In another phase IIa clinical trial, patients with MASH and type 2 diabetes mellitus received the treatment of CD3 monoclonal antibody (OKT3) to induce Tregs, and the transaminase levels and insulin resistance were improved (174). Currently, a phase II clinical trial (NCT03291249) is ongoing to determine the safety and effectiveness of Foralumab, a new CD3 monoclonal antibody, in patients with MASH and type 2 diabetes mellitus (T2DM) (175). However, despite a large number of explorations in regulating the immune system to control the progression of MASLD, conclusive evidence is still lacking.
In the clinical trials of drug development for MASH, the improvement of hepatic fibrosis is often used as the primary or secondary endpoint. Given the central role of HSCs in hepatic fibrosis, inhibiting HSCs activation has long been proposed as a therapeutic strategy to prevent the progression of MASH-related fibrosis. In MASH, the activation of the Notch signaling pathway in hepatocytes induces an increase in the secretion of OPN, which in turn activates HSCs (42). Therefore, Nicastrin antisense oligonucleotide (Ncst ASO), an inhibitor of Notch, can reduce the fibrosis in MASH diet-fed mice by inhibiting the activation of HSCs through the secretion of OPN by hepatocytes (42).
In addition, in MASH, the nuclear factor of activated T cells 4 (NFATc4) in hepatocytes translocates from the cytoplasm to the nucleus, which can inhibit the transcriptional activity of peroxisome proliferator-activated receptor α (PPARα) and induce the expression of OPN. Inhibiting the activation of NFATc4 can alleviate the lipid deposition and the progression of fibrosis in MASH mice (176). Widjaja et al. developed a neutralizing anti-IL-11 antibody and a neutralizing anti-IL-11 receptor α (IL-11RA) antibody, and found that they significantly alleviated the hepatic steatosis, liver inflammation, and hepatic fibrosis in MASH mice by inhibiting the activation of HSCs by IL-11 (177). Protease-activated receptor-2 (PAR2) is an emerging new target expressed on hepatic stellate cells and hepatocytes, which can regulate liver injury, inflammation, and fibrosis (178). Pepducin PZ-235 is a complete antagonist of PAR2. Pharmacological inhibition of PAR2 can not only prevent the activation of HSCs and fibrosis, but also reduce the production of ROS mediated by hepatocytes through inhibiting PAR2 (178).
The therapeutic efficacy of MASH-related HCC is akin to virus-related HCC in surgical, liver transplantation, radiofrequency ablation (RFA), transarterial chemoembolization (TACE) interventions, and molecular targeted therapy (see Table 1) (179–185), but variations in the immune milieu result in divergent responses to ICB therapy. Although favorable outcomes are observed across different etiologies, a meta-analysis indicates that patients with MASLD etiology may derive comparatively lesser benefits from anti-PD-1 treatment. Notably, a combinatorial approach utilizing both anti-PD-L1 and anti-VEGF therapies has demonstrated enhanced survival rates (186).
Table 1
| Treatment | Date | n(MASLD/non- MASLD) | Outcome |
|---|---|---|---|
| Liver resection (179) | 2022.11.29 | 11477(2470/9007) | Slightly higher overall survival (HR 0.87; 95% CI: 0.75–1.02) and recurrence-free survival (HR 0.93; 95% CI: 0.84–1.02) than those with HCC of other aetiologies. |
| All curative therapy (resection, transplantation, ablation) (180) | 2021.01.13 | 5579(NA) | Liver resection: improved disease-free survival (HR 0.85; 95% CI: 0.74–0.98, p = 0.03) and overall survival (HR 0.87; 95% CI: 0.81–0.93; p < 0.0001) than those with HCC of other aetiologies. All curative therapy: improved overall survival (HR 0.96; 95% CI: 0.86–1.06; p = 0.40) and disease-free survival (HR 0.85; 95% CI: 0.74–0.98; p = 0.03) than those with HCC of other aetiologies. |
| Radiofrequency ablation (181) | 2022.01.05 | 520(62/458) | There were no differences in morbidity, tumour recurrence and overall survival among patients with MASLD-HCC vs other aetiologies as well as no prognostic impact of metabolic components (p = 0.3168). |
| Transarterial chemoembolization (182) | 2019.11.04 | 220(30/190) | There was a non-significant increased overall survival in the non-MASH [median 1078 days (95% CI: 668–1594)] as compared to the MASH cohort [median 706 days (95% CI: 314–not reached)] (p = 0.08) |
| ICIs, TKI and anti-VEGF (183) | 2021.06.12 | 22113(NA) | ICIs: improved overall survival in patients with viral-related HCC (HR=0.64; 95% CI: 0.5–0.83) compared with nonviral-related HCC (HR=0.92, 95% CI 0.77–1.11) (p = 0.0259). TKI and anti-VEGF: no impact of etiology in outcome was observed with TKI/anti-VEGF therapies. The overall survival in patients with viral-related HCC (HR=0.81; 95% CI: 0.71–0.92) compared with nonviral-related HCC (HR=0.82; 95% CI: 0.67–1.01) (p = 0.8828). |
| Sorafenib (184) | 2021.10.24 | 180(37/143) | There was no significant differences in overall survival between the Virus/alcohol group and the MASLD/MASH group in patients who received sequential therapy (median survival times was 23.4 and 27.0 months. p = 0.173, respectively). |
| Lenvatinib (185) | 2021.11.27 | 1232(236/996) | MASH-HCC was associated with longer overall survival (22.2 versus 15.1 months; HR 0.69; 95% CI: 0.56-0.85; p = 0.0006). MASH-HCC was associated with longer disease-free survival (7.5 versus 6.5 months; HR 0.84; 95% CI: 0.71-0.99; p = 0.0436). |
Therapeutic variance between MASH-HCC and virus-induced HCC.
ICI, immune checkpoint inhibitor; TKI, tyrosine kinase inhibitor.
In a murine model of MASH-HCC, treatment with anti-PD-1 immunotherapy results in an escalation in the occurrence of MASH-HCC and the quantity and dimensions of tumor nodules. This augmentation is linked to heightened levels of hepatic CD8+ PD1+ T cells and TNF+ T cells, and is averted by the depletion of CD8+ T cells or TNF+ neutralization (10). The primary rationale behind this phenomenon may stem from the fact that liver tumors in mice treated with anti-PD1 exhibited an elevated presence of CD8+/PD1+ T cells, which, in contrast to resident hepatic CD8+ T cells, display amplified expression of effector and exhaustion markers alongside impaired proliferative capacity. These CD8+/PD1+ T cells not only exhibit compromised immune surveillance capabilities but also possess tissue-damaging effects (11). This mechanistic insight could elucidate the observed acceleration in tumor progression in around 13% of advanced HCC patients undergoing anti-PD-1 therapy, a phenomenon recognized as hyperprogressive syndrome (11). Consequently, the imperative for improved stratification of HCC patients undergoing immunotherapy based on the etiology of liver cancer is underscored, necessitating the development of tailored immunotherapeutic strategies for MASH-HCC.
Conclusion
MASH represents a significant global health challenge and is anticipated to emerge as a prominent contributor to HCC incidence with the rise in metabolic disorders (5). The transition from MASH to HCC is governed by a multitude of factors encompassing molecular modifications, germline variability, fibrotic conditions, immune milieu, and the microbiome. All cellular constituents implicated in MASLD, MASH, and MASH-related HCC, such as T lymphocytes, macrophages, hepatocytes, and hepatic stellate cells, exhibit immunomodulatory characteristics. Numerous investigations have underscored the presence of a distinct immune cell subset in MASH-related HCC in contrast to virus-induced HCC, partially elucidating the dissimilarities in immune checkpoint inhibitor responsiveness between the two conditions. Nonetheless, the intricate interplay among tumor cells, immune cells, and stromal cells remains largely unexplored. Consequently, further exploration into the variances in the microenvironment between MASH-related HCC and virus-induced HCC is imperative to unravel the distinctions in treatment outcomes, facilitating the implementation of more efficacious, personalized therapeutic strategies in the foreseeable future.
Statements
Author contributions
QW: Writing – original draft. YY: Writing – original draft. SL: Writing – original draft. DG: Conceptualization, Project administration, Writing – review & editing. YHY: Conceptualization, Funding acquisition, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by The National Natural Science Foundation of China (82360526, Y.H.Y.), The Joint Project on Regional High-Incidence Diseases Research of Guangxi Natural Science Foundation (2023GXNSFDA026011, Y.H.Y.), NIH 5R24DK139775-02 (D.A.G.), NIH P30DK120531 (D.A.G.), The Victor and Anna Mae Beghini Charitable Foundation (D.A.G.), and Mr. John Mills generous support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
- AASLD
American Association for the Study of Liver Diseases
- AMPK
AMP-activated protein kinase
- ASH
Alcoholic steatohepatitis
- cDC1s
Conventional type 1 dendritic cells
- CD-HFD
Choline-deficient high-fat diet
- CTLA-4
Cytotoxic T-lymphocyte-associated protein 4
- CHB
Chronic hepatitis B
- DCs
Dendritic cells
- DAMPs
Damage-associated molecular patterns
- DR
Ductular reaction
- FABP4
Fatty acid-binding protein 4
- FAO
Fatty acid oxidation
- FDA
Food and Drug Administration
- FFAs
Free fatty acids
- HBV
Hepatitis B virus
- HCV
Hepatitis C virus
- HCC
Hepatocellular carcinoma
- Hh
Hedgehog
- HFD
High-fat diet
- ICB
Immune checkpoint blockade
- ICAM-1
Intercellular adhesion molecule-1
- IFN-γ
interferon-gamma
- IL-13Rα2
IL-13 receptor alpha 2
- JNK
C-Jun N-terminal kinase
- KCs
Kupffer cells
- LSECs
Liver sinusoidal endothelial cells
- LCFAs
Long-chain fatty acids
- LDLC
low-density lipoprotein cholesterol
- LDL
Low-density lipoprotein
- LPS
Lipopolysaccharide
- LTβR
Lymphotoxin β
- MAPK
Mitogen-activated protein kinase
- MASLD
Metabolic dysfunction-associated steatotic liver disease
- MASH
Metabolically associated steatohepatitis
- MASH-HCC
MASH-associated hepatocellular carcinoma
- MAITs
Mucosal-associated invariant T cells
- MDMs
Monocyte-derived macrophages
- MCD
Methionine-choline-deficient
- MPO
Myeloperoxidase
- MICA/B
Major histocompatibility complex (MHC) class I associated chains A and B
- MyD88
Myeloid differentiation primary response protein 88
- NAFLD
Non-alcoholic fatty liver disease
- NASH
Non-alcoholic steatohepatitis
- NETs
Neutrophil extracellular traps
- NKG2D
Nature killer group 2D
- NF-κB
Nuclear factor κB
- NE
Neutrophil elastase
- NKT
Natural killer T cells
- NK
Natural killer cells
- OPN
Osteopontin
- PD-L1
Programmed death ligand 1
- PR3
Proteinase 3
- PD-1
Programmed death 1
- PTEN
Phosphatase and tensin homolog
- RFA
Radiofrequency ablation
- ROS
Reactive oxygen species
- SASP
Senescence-associated secretory phenotype
- SCFA
Short-chain fatty acid
- STAT
Signal transducer and activator of transcription
- TACE
Transarterial chemoembolization
- TLR
Toll-like receptor
- TGF-β
Transforming growth factor-beta
- TNF-α
Tumor necrosis factor-alpha
- THR-β
thyroid hormone receptor β
- THR-α
thyroid hormone receptor α
- TKI
Tyrosine kinase inhibitor
- TNFSF14/LIGHT
Tumor necrosis factor superfamily member 14
- VEGF
Vascular endothelial growth factor
- Virus-HCC
Viral associated hepatocellular Carcinoma
- VCAM-1
Vascular cell adhesion molecule-1
- VAP-1
Vascular adhesion protein-1
- VLDL
Very low-density lipoprotein.
References
1
Sheka AC Adeyi O Thompson J Hameed B Crawford PA Ikramuddin S . Nonalcoholic steatohepatitis: A review. JAMA. (2020) 323:1175–83. doi: 10.1001/jama.2020.2298
2
Younossi ZM Koenig AB Abdelatif D Fazel Y Henry L Wymer M . Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64:73–84. doi: 10.1002/hep.28431
3
Li M Gong W Wang S Li Z . Trends in body mass index, overweight and obesity among adults in the USA, the NHANES from 2003 to 2018: a repeat cross-sectional survey. BMJ Open. (2022) 12:e065425. doi: 10.1136/bmjopen-2022-065425
4
Lazarus JV Mark HE Anstee QM Arab JP Batterham RL Castera L et al . Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol. (2022) 19:60–78. doi: 10.1038/s41575-021-00523-4
5
Huang DQ El-Serag HB Loomba R . Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2021) 18:223–38. doi: 10.1038/s41575-020-00381-6
6
Llovet JM Willoughby CE Singal AG Greten TF Heikenwälder M El-Serag HB et al . Nonalcoholic steatohepatitis-related hepatocellular carcinoma: Pathogenesis and treatment. Nat Rev Gastroenterol Hepatol. (2023) 20:487–503. doi: 10.1038/s41575-023-00754-7
7
Estes C Anstee QM Arias-Loste MT Bantel H Bellentani S Caballeria J et al . Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol. (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
8
Loomba R Friedman SL Shulman GI . Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. (2021) 184:2537–64. doi: 10.1016/j.cell.2021.04.015
9
Vogel A Meyer T Sapisochin G Salem R Saborowski A . Hepatocellular carcinoma. Lancet. (2022) 400:1345–62. doi: 10.1016/S0140-6736(22)01200-4
10
Dudek M Pfister D Donakonda S Filpe P Schneider A Laschinger M et al . Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature. (2021) 592:444–9. doi: 10.1038/s41586-021-03233-8
11
Pfister D Núñez NG Pinyol R Govaere O Pinter M Szydlowska M et al . NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. (2021) 592:450–6. doi: 10.1038/s41586-021-03362-0
12
Yahoo N Dudek M Knolle P Heikenwälder M . Role of immune responses in the development of NAFLD-associated liver cancer and prospects for therapeutic modulation. J Hepatol. (2023) 79:538–51. doi: 10.1016/j.jhep.2023.02.033
13
Ma M Jiang W Zhou R . DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity. (2024) 57:752–71. doi: 10.1016/j.immuni.2024.03.002
14
Cai J Zhang XJ Li H . The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology. (2019) 70:1026–37. doi: 10.1002/hep.30506
15
van Son KC Verschuren L Hanemaaijer R Reeves H Takkenberg RB Drenth J et al . Non-parenchymal cells and the extracellular matrix in hepatocellular carcinoma in non-alcoholic fatty liver disease. Cancers (Basel). (2023) 15:1308. doi: 10.3390/cancers15041308
16
Zeng S Wu F Chen M Li Y You M Zhang Y et al . Inhibition of fatty acid translocase (FAT/CD36) palmitoylation enhances hepatic fatty acid β-oxidation by increasing its localization to mitochondria and interaction with long-chain acyl-coA synthetase 1. Antioxid Redox Signal. (2022) 36:1081–100. doi: 10.1089/ars.2021.0157
17
Lu Z Sun GF Pan XA Qu XH Yang P Chen ZP et al . BCATc inhibitor 2 ameliorated mitochondrial dysfunction and apoptosis in oleic acid-induced non-alcoholic fatty liver disease model. Front Pharmacol. (2022) 13:1025551. doi: 10.3389/fphar.2022.1025551
18
Rabinovitch RC Samborska B Faubert B Ma EH Gravel SP Andrzejewski S et al . AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. (2017) 21:1–9. doi: 10.1016/j.celrep.2017.09.026
19
Della Corte CM Viscardi G Papaccio F Esposito G Martini G Ciardiello D et al . Implication of the Hedgehog pathway in hepatocellular carcinoma. World J Gastroenterol. (2017) 23:4330–40. doi: 10.3748/wjg.v23.i24.4330
20
Wu X Shu L Zhang Z Li J Zong J Cheong LY et al . Adipocyte fatty acid binding protein promotes the onset and progression of liver fibrosis via mediating the crosstalk between liver sinusoidal endothelial cells and hepatic stellate cells. Adv Sci (Weinh). (2021) 8:e2003721. doi: 10.1002/advs.202003721
21
Anstee QM Reeves HL Kotsiliti E Govaere O Heikenwalder M . From NASH to HCC: Current concepts and future challenges. Nat Rev Gastroenterol Hepatol. (2019) 16:411–28. doi: 10.1038/s41575-019-0145-7
22
Sato K Marzioni M Meng F Francis H Glaser S Alpini G . Ductular reaction in liver diseases: Pathological mechanisms and translational significances. Hepatology. (2019) 69:420–30. doi: 10.1002/hep.30150
23
Fabris L Spirli C Cadamuro M Fiorotto R Strazzabosco M . Emerging concepts in biliary repair and fibrosis. Am J Physiol Gastrointest Liver Physiol. (2017) 313:G102–102G116. doi: 10.1152/ajpgi.00452.2016
24
Wu N Meng F Invernizzi P Bernuzzi F Venter J Standeford H et al . The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-β1 biliary secretion in mice. Hepatology. (2016) 64:865–79. doi: 10.1002/hep.28622
25
Schrumpf E Tan C Karlsen TH Sponheim J Björkström NK Sundnes O et et al . The biliary epithelium presents antigens to and activates natural killer T cells. Hepatology. (2015) 62:1249–59. doi: 10.1002/hep.27840
26
Chen L Wu N Kennedy L Francis H Ceci L Zhou T et al . Inhibition of secretin/secretin receptor axis ameliorates NAFLD phenotypes. Hepatology. (2021) 74:1845–63. doi: 10.1002/hep.31871
27
Diehl L Schurich A Grochtmann R Hegenbarth S Chen L Knolle PA . Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology. (2008) 47:296–305. doi: 10.1002/hep.21965
28
McMahan RH Porsche CE Edwards MG Rosen HR . Free fatty acids differentially downregulate chemokines in liver sinusoidal endothelial cells: Insights into non-alcoholic fatty liver disease. PloS One. (2016) 11:e0159217. doi: 10.1371/journal.pone.0159217
29
Hammoutene A Biquard L Lasselin J Kheloufi M Tanguy M Vion AC et al . A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol. (2020) 72:528–38. doi: 10.1016/j.jhep.2019.10.028
30
Weston CJ Shepherd EL Claridge LC Rantakari P Curbishley SM Tomlinson JW et al . Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest. (2015) 125:501–20. doi: 10.1172/JCI73722
31
Hammoutene A Rautou PE . Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. (2019) 70:1278–91. doi: 10.1016/j.jhep.2019.02.012
32
Tateya S Rizzo NO Handa P Cheng AM Morgan-Stevenson V Daum G et al . Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes. (2011) 60:2792–801. doi: 10.2337/db11-0255
33
Velliou RI Legaki AI Nikolakopoulou P Vlachogiannis NI Chatzigeorgiou A . Liver endothelial cells in NAFLD and transition to NASH and HCC. Cell Mol Life Sci. (2023) 80:314. doi: 10.1007/s00018-023-04966-7
34
Pasarín M La Mura V Gracia-Sancho J García-Calderó H Rodríguez-Vilarrupla A García-Pagán JC et al . Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PloS One. (2012) 7:e32785. doi: 10.1371/journal.pone.0032785
35
Tanaka M Iwakiri Y . The hepatic lymphatic vascular system: Structure, function, markers, and lymphangiogenesis. Cell Mol Gastroenterol Hepatol. (2016) 2:733–49. doi: 10.1016/j.jcmgh.2016.09.002
36
Kawashita E Ozaki T Ishihara K Kashiwada C Akiba S . Endothelial group IVA phospholipase A(2) promotes hepatic fibrosis with sinusoidal capillarization in the early stage of non-alcoholic steatohepatitis in mice. Life Sci. (2022) 294:120355. doi: 10.1016/j.lfs.2022.120355
37
Herrnberger L Hennig R Kremer W Hellerbrand C Goepferich A Kalbitzer HR et al . Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle-associated protein and is required for lipoprotein passage. PloS One. (2014) 9:e115005. doi: 10.1371/journal.pone.0115005
38
Zhang Q Liu J Liu J Huang W Tian L Quan J et al . oxLDL induces injury and defenestration of human liver sinusoidal endothelial cells via LOX1. J Mol Endocrinol. (2014) 53:281–93. doi: 10.1530/JME-14-0049
39
O’Reilly JN Cogger VC Fraser R Le Couteur DG . The effect of feeding and fasting on fenestrations in the liver sinusoidal endothelial cell. Pathology. (2010) 42:255–8. doi: 10.3109/00313021003636469
40
Cogger VC Mohamad M Solon-Biet SM Senior AM Warren A O'Reilly JN et al . Dietary macronutrients and the aging liver sinusoidal endothelial cell. Am J Physiol Heart Circ Physiol. (2016) 310:H1064–70. doi: 10.1152/ajpheart.00949.2015
41
Schwabe RF Tabas I Pajvani UB . Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology. (2020) 158:1913–28. doi: 10.1053/j.gastro.2019.11.311
42
Zhu C Kim K Wang X Bartolome A Salomao M Dongiovanni P et al . Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med. (2018) 10:eaat0344. doi: 10.1126/scitranslmed.aat0344
43
Marcher AB Bendixen SM Terkelsen MK Hohmann SS Hansen MH Larsen BD et al . Transcriptional regulation of Hepatic Stellate Cell activation in NASH. Sci Rep. (2019) 9:2324. doi: 10.1038/s41598-019-39112-6
44
Xiong X Kuang H Ansari S Liu T Gong J Wang S et al . Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell. (2019) 75:644–60.e5. doi: 10.1016/j.molcel.2019.07.028
45
Breuer DA Pacheco MC Washington MK Montgomery SA Hasty AH Kennedy AJ . CD8(+) T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. (2020) 318:G211–211G224. doi: 10.1152/ajpgi.00040.2019
46
Hammerich L Tacke F . Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. (2023) 20:633–46. doi: 10.1038/s41575-023-00807-x
47
Bates J Vijayakumar A Ghoshal S Marchand B Yi S Kornyeyev D et al . Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J Hepatol. (2020) 73:896–905. doi: 10.1016/j.jhep.2020.04.037
48
Zhou Y Zhang L Ma Y Xie L Yang YY Jin C et al . Secretome of senescent hepatic stellate cells favors Malignant transformation from nonalcoholic steatohepatitis-fibrotic progression to hepatocellular carcinoma. Theranostics. (2023) 13:4430–48. doi: 10.7150/thno.85369
49
Ma C Han M Heinrich B Fu Q Zhang Q Sandhu M et al . Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. (2018) 360:eaan5931. doi: 10.1126/science.aan5931
50
Zhang X Coker OO Chu ES Fu K Lau H Wang YX et al . Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. (2021) 70:761–74. doi: 10.1136/gutjnl-2019-319664
51
Del Chierico F Nobili V Vernocchi P Russo A De Stefanis C Gnani D et al . Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology. (2017) 65:451–64. doi: 10.1002/hep.28572
52
Wong VW Tse CH Lam TT Wong GL Chim AM Chu WC et al . Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis–a longitudinal study. PloS One. (2013) 8:e62885. doi: 10.1371/journal.pone.0062885
53
Loomba R Seguritan V Li W Long T Klitgord N Bhatt A et al . Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. (2019) 30:607. doi: 10.1016/j.cmet.2019.08.002
54
Zhu L Baker SS Gill C Liu W Alkhouri R Baker RD et al . Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. (2013) 57:601–9. doi: 10.1002/hep.26093
55
Sokol H Pigneur B Watterlot L Lakhdari O Bermúdez-Humarán LG Gratadoux JJ et al . Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. (2008) 105:16731–6. doi: 10.1073/pnas.0804812105
56
Bäckhed F Ding H Wang T Hooper LV Koh GY Nagy A et al . The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. (2004) 101:15718–23. doi: 10.1073/pnas.0407076101
57
Wigg AJ Roberts-Thomson IC Dymock RB McCarthy PJ Grose RH Cummins AG . The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. (2001) 48:206–11. doi: 10.1136/gut.48.2.206
58
Miele L Valenza V La Torre G Montalto M Cammarota G Ricci R et al . Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. (2009) 49:1877–87. doi: 10.1002/hep.22848
59
Wagnerberger S Spruss A Kanuri G Stahl C Schröder M Vetter W et al . Lactobacillus casei Shirota protects from fructose-induced liver steatosis: a mouse model. J Nutr Biochem. (2013) 24:531–8. doi: 10.1016/j.jnutbio.2012.01.014
60
Ponziani FR Bhoori S Castelli C Putignani L Rivoltini L Del Chierico F et al . Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. (2019) 69:107–20. doi: 10.1002/hep.30036
61
Behary J Amorim N Jiang XT Raposo A Gong L McGovern E et al . Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun. (2021) 12:187. doi: 10.1038/s41467-020-20422-7
62
Yoshimoto S Loo TM Atarashi K Kanda H Sato S Oyadomari S et al . Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. (2013) 499:97–101. doi: 10.1038/nature12347
63
Tacke F Zimmermann HW . Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
64
Gadd VL Skoien R Powell EE Fagan KJ Winterford C Horsfall L et al . The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. (2014) 59:1393–405. doi: 10.1002/hep.26937
65
Tran S Baba I Poupel L Dussaud S Moreau M Gélineau A et al . Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity. (2020) 53:627–40.e5. doi: 10.1016/j.immuni.2020.06.003
66
Zhang X Shen J Man K Chu ES Yau TO Sung JC et al . CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J Hepatol. (2014) 61:1365–75. doi: 10.1016/j.jhep.2014.07.006
67
Wolf MJ Adili A Piotrowitz K Abdullah Z Boege Y Stemmer K et al . Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. (2014) 26:549–64. doi: 10.1016/j.ccell.2014.09.003
68
Lefere S Tacke F . Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. (2019) 1:30–43. doi: 10.1016/j.jhepr.2019.02.004
69
Takimoto Y Chu PS Nakamoto N Hagihara Y Mikami Y Miyamoto K et al . Myeloid TLR4 signaling promotes post-injury withdrawal resolution of murine liver fibrosis. iScience. (2023) 26:106220. doi: 10.1016/j.isci.2023.106220
70
Van Herck MA Weyler J Kwanten WJ Dirinck EL De Winter BY Francque SM et al . The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Front Immunol. (2019) 10:82. doi: 10.3389/fimmu.2019.00082
71
Luo W Xu Q Wang Q Wu H Hua J . Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep. (2017) 7:44612. doi: 10.1038/srep44612
72
Liu Y Chen H Yan X Zhang J Deng Z Huang M et al . MyD88 in myofibroblasts enhances nonalcoholic fatty liver disease-related hepatocarcinogenesis via promoting macrophage M2 polarization. Cell Commun Signal. (2024) 22:86. doi: 10.1186/s12964-024-01489-x
73
Wan J Benkdane M Teixeira-Clerc F Bonnafous S Louvet A Lafdil F et al . M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. (2014) 59:130–42. doi: 10.1002/hep.26607
74
Lee JL Wang YC Hsu YA Chen CS Weng RC Lu YP et al . Galectin-12 modulates Kupffer cell polarization to alter the progression of nonalcoholic fatty liver disease. Glycobiology. (2023) 33:673–82. doi: 10.1093/glycob/cwad062
75
Jaitin DA Adlung L Thaiss CA Weiner A Li B Descamps H et al . Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell. (2019) 178:686–98.e14. doi: 10.1016/j.cell.2019.05.054
76
Daemen S Gainullina A Kalugotla G He L Chan MM Beals JW et al . Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep. (2021) 34:108626. doi: 10.1016/j.celrep.2020.108626
77
Ramachandran P Dobie R Wilson-Kanamori JR Dora EF Henderson B Luu NT et al . Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
78
Takahashi K Prinz M Stagi M Chechneva O Neumann H . TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PloS Med. (2007) 4:e124. doi: 10.1371/journal.pmed.0040124
79
Hendrikx T Porsch F Kiss MG Rajcic D Papac-Miličević N Hoebinger et al . Soluble TREM2 levels reflect the recruitment and expansion of TREM2(+) macrophages that localize to fibrotic areas and limit NASH. J Hepatol. (2022) 77:1373–85. doi: 10.1016/j.jhep.2022.06.004
80
Wang X He Q Zhou C Xu Y Liu D Fujiwara N et al . Prolonged hypernutrition impairs TREM2-dependent efferocytosis to license chronic liver inflammation and NASH development. Immunity. (2023) 56:58–77.e11. doi: 10.1016/j.immuni.2022.11.013
81
Huang J Wu Q Geller DA Yan Y . Macrophage metabolism, phenotype, function, and therapy in hepatocellular carcinoma (HCC). J Transl Med. (2023) 21:815. doi: 10.1186/s12967-023-04716-0
82
David BA Rezende RM Antunes MM Santos MM Freitas Lopes MA Diniz AB et al . Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology. (2016) 151:1176–91. doi: 10.1053/j.gastro.2016.08.024
83
Ibrahim J Nguyen AH Rehman A Ochi A Jamal M Graffeo CS et al . Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology. (2012) 143:1061–72. doi: 10.1053/j.gastro.2012.06.003
84
Sutti S Locatelli I Bruzzì S Jindal A Vacchiano M Bozzola C et al . CX3CR1-expressing inflammatory dendritic cells contribute to the progression of steatohepatitis. Clin Sci (Lond). (2015) 129:797–808. doi: 10.1042/CS20150053
85
Deczkowska A David E Ramadori P Pfister D Safran M Li B et al . XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med. (2021) 27:1043–54. doi: 10.1038/s41591-021-01344-3
86
Heier EC Meier A Julich-Haertel H Djudjaj S Rau M Tschernig T et al . Murine CD103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol. (2017) 66:1241–50. doi: 10.1016/j.jhep.2017.01.008
87
Stiglund N Strand K Cornillet M Stål P Thorell A Zimmer CL et al . Retained NK cell phenotype and functionality in non-alcoholic fatty liver disease. Front Immunol. (2019) 10:1255. doi: 10.3389/fimmu.2019.01255
88
Henning JR Graffeo CS Rehman A Fallon NC Zambirinis CP Ochi A et al . Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology. (2013) 58:589–602. doi: 10.1002/hep.26267
89
Herber DL Cao W Nefedova Y Novitskiy SV Nagaraj S Tyurin VA et al . Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. (2010) 16:880–6. doi: 10.1038/nm.2172
90
Liu K Wang FS Xu R . Neutrophils in liver diseases: Pathogenesis and therapeutic targets. Cell Mol Immunol. (2021) 18:38–44. doi: 10.1038/s41423-020-00560-0
91
Miyagi T Takehara T Uemura A Nishio K Shimizu S Kodama T et al . Absence of invariant natural killer T cells deteriorates liver inflammation and fibrosis in mice fed high-fat diet. J Gastroenterol. (2010) 45:1247–54. doi: 10.1007/s00535-010-0272-y
92
Mirea AM Toonen E van den Munckhof I Munsterman ID Tjwa E Jaeger M et al . Increased proteinase 3 and neutrophil elastase plasma concentrations are associated with non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes. Mol Med. (2019) 25:16. doi: 10.1186/s10020-019-0084-3
93
van der Windt DJ Sud V Zhang H Varley PR Goswami J Yazdani HO et al . Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. (2018) 68:1347–60. doi: 10.1002/hep.29914
94
Rensen SS Bieghs V Xanthoulea S Arfianti E Bakker JA Shiri-Sverdlov R et al . Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PloS One. (2012) 7:e52411. doi: 10.1371/journal.pone.0052411
95
Talukdar S Oh DY Bandyopadhyay G Li D Xu J McNelis J et al . Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. (2012) 18:1407–12. doi: 10.1038/nm.2885
96
Chen J Liang B Bian D Luo Y Yang J Li Z et al . Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem Biophys Res Commun. (2019) 518:691–7. doi: 10.1016/j.bbrc.2019.08.111
97
Wang H Zhang H Wang Y Brown ZJ Xia Y Huang Z et al . Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. (2021) 75:1271–83. doi: 10.1016/j.jhep.2021.07.032
98
Fan Y Zhang W Wei H Sun R Tian Z Chen Y . Hepatic NK cells attenuate fibrosis progression of non-alcoholic steatohepatitis in dependent of CXCL10-mediated recruitment. Liver Int. (2020) 40:598–608. doi: 10.1111/liv.14307
99
Cepero-Donates Y Lacraz G Ghobadi F Rakotoarivelo V Orkhis S Mayhue M et al . Interleukin-15-mediated inflammation promotes non-alcoholic fatty liver disease. Cytokine. (2016) 82:102–11. doi: 10.1016/j.cyto.2016.01.020
100
Wang F Zhang X Liu W Zhou Y Wei W Liu D et al . Activated natural killer cell promotes nonalcoholic steatohepatitis through mediating JAK/STAT pathway. Cell Mol Gastroenterol Hepatol. (2022) 13:257–74. doi: 10.1016/j.jcmgh.2021.08.019
101
Kahraman A Schlattjan M Kocabayoglu P Meziletoglu S Schlensak M Fingas CD et al . Major histocompatibility complex class I-related chains A and B (MIC A/B): a novel role in nonalcoholic steatohepatitis. Hepatology. (2010) 51:92–102. doi: 10.1002/hep.23253
102
Li X Huang J Wu Q Du Q Wang Y Huang Y et al . Inhibition of checkpoint kinase 1 (CHK1) upregulates interferon regulatory factor 1 (IRF1) to promote apoptosis and activate anti-tumor immunity via MICA in hepatocellular carcinoma (HCC). Cancers (Basel). (2023) 15:850. doi: 10.3390/cancers15030850
103
Gur C Doron S Kfir-Erenfeld S Horwitz E Abu-Tair L Safadi R et al . NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut. (2012) 61:885–93. doi: 10.1136/gutjnl-2011-301400
104
Diedrich T Kummer S Galante A Drolz A Schlicker V Lohse AW et al . Characterization of the immune cell landscape of patients with NAFLD. PloS One. (2020) 15:e0230307. doi: 10.1371/journal.pone.0230307
105
Tosello-Trampont AC Krueger P Narayanan S Landes SG Leitinger N Hahn YS . NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology. (2016) 63:799–812.
106
Novobrantseva TI Majeau GR Amatucci A Kogan S Brenner I Casola S et al . Attenuated liver fibrosis in the absence of B cells. J Clin Invest. (2005) 115:3072–82. doi: 10.1172/JCI24798
107
Bruzzì S Sutti S Giudici G Burlone ME Ramavath NN Toscani A et al . B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic Biol Med. (2018) 124:249–59. doi: 10.1016/j.freeradbiomed.2018.06.015
108
Zhang F Jiang WW Li X Qiu XY Wu Z Chi YJ et al . Role of intrahepatic B cells in non-alcoholic fatty liver disease by secreting pro-inflammatory cytokines and regulating intrahepatic T cells. J Dig Dis. (2016) 17:464–74. doi: 10.1111/cdd.2016.17.issue-7
109
Barrow F Khan S Fredrickson G Wang H Dietsche K Parthiban P et al . Microbiota-driven activation of intrahepatic B cells aggravates NASH through innate and adaptive signaling. Hepatology. (2021) 74:704–22. doi: 10.1002/hep.31755
110
Nakamura Y Abe M Kawasaki K Miyake T Watanabe T Yoshida O et al . Depletion of B cell-activating factor attenuates hepatic fat accumulation in a murine model of nonalcoholic fatty liver disease. Sci Rep. (2019) 9:977. doi: 10.1038/s41598-018-37403-y
111
Shalapour S Lin XJ Bastian IN Brain J Burt AD Aksenov AA et al . Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. (2017) 551:340–5. doi: 10.1038/nature24302
112
Bhattacharjee J Kirby M Softic S Miles L Salazar-Gonzalez RM Shivakumar P et al . Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun. (2017) 1:299–310. doi: 10.1002/hep4.1041
113
Locatelli I Sutti S Vacchiano M Bozzola C Albano E . NF-κB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin Sci (Lond). (2013) 124:279–87. doi: 10.1042/CS20120289
114
Koda Y Teratani T Chu PS Hagihara Y Mikami Y Harada Y et al . CD8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun. (2021) 12:4474. doi: 10.1038/s41467-021-24734-0
115
Zigmond E Zangen SW Pappo O Sklair-Levy M Lalazar G Zolotaryova L et al . Beta-glycosphingolipids improve glucose intolerance and hepatic steatosis of the Cohen diabetic rat. Am J Physiol Endocrinol Metab. (2009) 296:E72–8. doi: 10.1152/ajpendo.90634.2008
116
Elinav E Pappo O Sklair-Levy M Margalit M Shibolet O Gomori M et al . Adoptive transfer of regulatory NKT lymphocytes ameliorates non-alcoholic steatohepatitis and glucose intolerance in ob/ob mice and is associated with intrahepatic CD8 trapping. J Pathol. (2006) 209:121–8. doi: 10.1002/(ISSN)1096-9896
117
Yan Y Zheng L Du Q Yazdani H Dong K Guo Y et al . Interferon regulatory factor 1(IRF-1) activates anti-tumor immunity via CXCL10/CXCR3 axis in hepatocellular carcinoma (HCC). Cancer Lett. (2021) 506:95–106. doi: 10.1016/j.canlet.2021.03.002
118
Wang T Sun G Wang Y Li S Zhao X Zhang C et al . The immunoregulatory effects of CD8 T-cell-derived perforin on diet-induced nonalcoholic steatohepatitis. FASEB J. (2019) 33:8490–503. doi: 10.1096/fj.201802534RR
119
Ma C Kesarwala AH Eggert T Medina-Echeverz J Kleiner DE Jin P et al . NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. (2016) 531:253–7. doi: 10.1038/nature16969
120
Ramadori P Kam S Heikenwalder M . T cells: Friends and foes in NASH pathogenesis and hepatocarcinogenesis. Hepatology. (2022) 75:1038–49. doi: 10.1002/hep.32336
121
Zhou Y Zhang H Yao Y Zhang X Guan Y Zheng F . CD4(+) T cell activation and inflammation in NASH-related fibrosis. Front Immunol. (2022) 13:967410. doi: 10.3389/fimmu.2022.967410
122
Li Z Soloski MJ Diehl AM . Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. (2005) 42:880–5. doi: 10.1002/hep.20826
123
Inzaugarat ME Ferreyra Solari NE Billordo LA Abecasis R Gadano AC Cherñavsky AC . Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J Clin Immunol. (2011) 31:1120–30. doi: 10.1007/s10875-011-9571-1
124
Ferreyra Solari NE Inzaugarat ME Baz P De Matteo E Lezama C Galoppo M et al . The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J Clin Immunol. (2012) 32:611–21. doi: 10.1007/s10875-011-9635-2
125
Fu JT Liu J Wu WB Chen YT Lu GD Cao Q et al . Targeting EFHD2 inhibits interferon-γ signaling and ameliorates non-alcoholic steatohepatitis. J Hepatol. (2024) 81:389–403. doi: 10.1016/j.jhep.2024.04.009
126
Sun G Jin H Zhang C Meng H Zhao X Wei D et al . OX40 regulates both innate and adaptive immunity and promotes nonalcoholic steatohepatitis. Cell Rep. (2018) 25:3786–99.e4. doi: 10.1016/j.celrep.2018.12.006
127
Su L Wu Z Chi Y Song Y Xu J Tan J et al . Mesenteric lymph node CD4(+) T lymphocytes migrate to liver and contribute to non-alcoholic fatty liver disease. Cell Immunol. (2019) 337:33–41. doi: 10.1016/j.cellimm.2019.01.005
128
Rau M Schilling AK Meertens J Hering I Weiss J Jurowich C et al . Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of th17 cells in the liver and an increased th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol. (2016) 196:97–105. doi: 10.4049/jimmunol.1501175
129
Rolla S Alchera E Imarisio C Bardina V Valente G Cappello P et al . The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci (Lond). (2016) 130:193–203. doi: 10.1042/CS20150405
130
Shimamura T Fujisawa T Husain SR Kioi M Nakajima A Puri RK . Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J Immunol. (2008) 181:4656–65. doi: 10.4049/jimmunol.181.7.4656
131
Harley IT Stankiewicz TE Giles DA Softic S Flick LM Cappelletti M et al . IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology. (2014) 59:1830–9. doi: 10.1002/hep.26746
132
Van Herck MA Vonghia L Kwanten WJ Julé Y Vanwolleghem T Ebo DG et al . Diet reversal and immune modulation show key role for liver and adipose tissue T cells in murine nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. (2020) 10:467–90. doi: 10.1016/j.jcmgh.2020.04.010
133
Rajoriya N Fergusson JR Leithead JA Klenerman P . Gamma delta T-lymphocytes in hepatitis C and chronic liver disease. Front Immunol. (2014) 5:400. doi: 10.3389/fimmu.2014.00400
134
Moreno-Fernandez ME Giles DA Oates JR Chan CC Damen M Doll JR et al . PKM2-dependent metabolic skewing of hepatic Th17 cells regulates pathogenesis of non-alcoholic fatty liver disease. Cell Metab. (2021) 33:1187–204.e9. doi: 10.1016/j.cmet.2021.04.018
135
Meng F Wang K Aoyama T Grivennikov SI Paik Y Scholten D et al . Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. (2012) 143:765–76.e3. doi: 10.1053/j.gastro.2012.05.049
136
Kuang DM Peng C Zhao Q Wu Y Chen MS Zheng L . Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. (2010) 51:154–64. doi: 10.1002/hep.23291
137
Gomes AL Teijeiro A Burén S Tummala KS Yilmaz M Waisman A et al . Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell. (2016) 30:161–75. doi: 10.1016/j.ccell.2016.05.020
138
Yang L Zhang Y Wang L Fan F Zhu L Li Z et al . Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J Hepatol. (2010) 53:339–47. doi: 10.1016/j.jhep.2010.03.004
139
Hamaguchi M Okamura T Fukuda T Nishida K Yoshimura Y Hashimoto Y et al . Group 3 innate lymphoid cells protect steatohepatitis from high-fat diet induced toxicity. Front Immunol. (2021) 12:648754. doi: 10.3389/fimmu.2021.648754
140
Dywicki J Buitrago-Molina LE Noyan F Davalos-Misslitz AC Hupa-Breier KL Lieber M et al . The detrimental role of regulatory T cells in nonalcoholic steatohepatitis. Hepatol Commun. (2022) 6:320–33. doi: 10.1002/hep4.1807
141
Yadav H Quijano C Kamaraju AK Gavrilova O Malek R Chen W et al . Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab. (2011) 14:67–79. doi: 10.1016/j.cmet.2011.04.013
142
Ma X Hua J Mohamood AR Hamad AR Ravi R Li Z . A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology. (2007) 46:1519–29. doi: 10.1002/hep.21823
143
Roh YS Kim JW Park S Shon C Kim S Eo SK et al . Toll-like receptor-7 signaling promotes nonalcoholic steatohepatitis by inhibiting regulatory T cells in mice. Am J Pathol. (2018) 188:2574–88. doi: 10.1016/j.ajpath.2018.07.011
144
Francisco V Pino J Campos-Cabaleiro V Ruiz-Fernández C Mera A Gonzalez-Gay MA et al . Obesity, fat mass and immune system: Role for leptin. Front Physiol. (2018) 9:640. doi: 10.3389/fphys.2018.00640
145
Chatzigeorgiou A Chung KJ Garcia-Martin R Alexaki VI Klotzsche-von Ameln A Phieler J et al . Dual role of B7 costimulation in obesity-related nonalcoholic steatohepatitis and metabolic dysregulation. Hepatology. (2014) 60:1196–210. doi: 10.1002/hep.27233
146
Li F Hao X Chen Y Alexaki VI Klotzsche-von Ameln A Phieler J et al . The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat Commun. (2017) 7:13839. doi: 10.1038/ncomms13839
147
Torres-Hernandez A Wang W Nikiforov Y Tejada K Torres L Kalabin A et al . γδ T cells promote steatohepatitis by orchestrating innate and adaptive immune programming. Hepatology. (2020) 71:477–94. doi: 10.1002/hep.30952
148
Tan Z Qian X Jiang R Liu Q Wang Y Chen C et al . IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol. (2013) 191:1835–44. doi: 10.4049/jimmunol.1203013
149
Chen Y Tian Z . Roles of hepatic innate and innate-like lymphocytes in nonalcoholic steatohepatitis. Front Immunol. (2020) 11:1500. doi: 10.3389/fimmu.2020.01500
150
Syn WK Oo YH Pereira TA Karaca GF Jung Y Omenetti A et al . Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. (2010) 51:1998–2007. doi: 10.1002/hep.23599
151
Syn WK Agboola KM Swiderska M Michelotti GA Liaskou E Pang H et al . NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. (2012) 61:1323–9. doi: 10.1136/gutjnl-2011-301857
152
Maricic I Marrero I Eguchi A Nakamura R Johnson CD Dasgupta S et al . Differential activation of hepatic invariant NKT cell subsets plays a key role in progression of nonalcoholic steatohepatitis. J Immunol. (2018) 201:3017–35. doi: 10.4049/jimmunol.1800614
153
Baranek T Lebrigand K de Amat Herbozo C Gonzalez L Bogard G Dietrich C et al . High dimensional single-cell analysis reveals iNKT cell developmental trajectories and effector fate decision. Cell Rep. (2020) 32:108116. doi: 10.1016/j.celrep.2020.108116
154
Kremer M Thomas E Milton RJ Perry AW van Rooijen N Wheeler MD et al . Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis. Hepatology. (2010) 51:130–41. doi: 10.1002/hep.23292
155
Tang ZH Liang S Potter J Jiang X Mao HQ Li Z . Tim-3/galectin-9 regulate the homeostasis of hepatic NKT cells in a murine model of nonalcoholic fatty liver disease. J Immunol. (2013) 190:1788–96. doi: 10.4049/jimmunol.1202814
156
Tang W Zhou J Yang W Feng Y Wu H Mok M et al . Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol Immunol. (2022) 19:834–47. doi: 10.1038/s41423-022-00872-3
157
Li Y Huang B Jiang X Chen W Zhang J Wei Y et al . Mucosal-associated invariant T cells improve nonalcoholic fatty liver disease through regulating macrophage polarization. Front Immunol. (2018) 9:1994. doi: 10.3389/fimmu.2018.01994
158
Hegde P Weiss E Paradis V Wan J Mabire M Sukriti S et al . Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun. (2018) 9:2146. doi: 10.1038/s41467-018-04450-y
159
Rouxel O Da Silva J Beaudoin L Nel I Tard C Cagninacci L et al . Cytotoxic and regulatory roles of mucosal-associated invariant T cells in type 1 diabetes. Nat Immunol. (2017) 18:1321–31. doi: 10.1038/ni.3854
160
Pugliese N Plaz Torres MC Petta S Valenti L Giannini EG Aghemo A . Is there an ‘ideal’ diet for patients with NAFLD. Eur J Clin Invest. (2022) 52:e13659. doi: 10.1111/eci.13659
161
Haas JT Vonghia L Mogilenko DA Verrijken A Molendi-Coste O Fleury S et al . Author Correction: Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat Metab. (2019) 1:744. doi: 10.1038/s42255-019-0093-0
162
Harrison SA Bashir MR Guy CD Zhou R Moylan CA Frias JP et al . Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. (2019) 394:2012–24. doi: 10.1016/S0140-6736(19)32517-6
163
Harrison SA Taub R Neff GW Lucas KJ Labriola D Moussa SE et al . Resmetirom for nonalcoholic fatty liver disease: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. (2023) 29:2919–28. doi: 10.1038/s41591-023-02603-1
164
Harrison SA Bedossa P Guy CD Schattenberg JM Loomba R Taub R et al . A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med. (2024) 390:497–509. doi: 10.1056/NEJMoa2309000
165
Karim G Bansal MB . Resmetirom: an orally administered, smallmolecule, liver-directed, β-selective THR agonist for the treatment of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. touchREV Endocrinol. (2023) 19:60–70. doi: 10.17925/EE.2023.19.1.60
166
Hönes GS Sivakumar RG Hoppe C König J Führer D Moeller LC . Cell-specific transport and thyroid hormone receptor isoform selectivity account for hepatocyte-targeted thyromimetic action of MGL-3196. Int J Mol Sci. (2022) 23:13714. doi: 10.3390/ijms232213714
167
Kannt A Wohlfart P Madsen AN Veidal SS Feigh M Schmoll D . Activation of thyroid hormone receptor-β improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br J Pharmacol. (2021) 178:2412–23. doi: 10.1111/bph.15427
168
Guirguis E Dougherty J Thornby K Grace Y Mack K . Resmetirom: the first food and drug administration-approved medication for nonalcoholic steatohepatitis (NASH). Ann Pharmacother. (2025) 59:162–73. doi: 10.1177/10600280241259528
169
Javanbakht M Fishman J Moloney E Rydqvist P Ansaripour A . Early cost-effectiveness and price threshold analyses of resmetirom: an investigational treatment for management of nonalcoholic steatohepatitis. Pharmacoecon Open. (2023) 7:93–110. doi: 10.1007/s41669-022-00370-2
170
Keam SJ . Resmetirom: First approval. Drugs. (2024) 84:729–35. doi: 10.1007/s40265-024-02045-0
171
Ratziu V Sanyal A Harrison SA Wong VW Francque S Goodman Z et al . Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: Final analysis of the phase 2b CENTAUR study. Hepatology. (2020) 72:892–905. doi: 10.1002/hep.31108
172
Pedrosa M Seyedkazemi S Francque S Sanyal A Rinella M Charlton M et al . A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp Clin Trials. (2020) 88:105889. doi: 10.1016/j.cct.2019.105889
173
Lefebvre P Cariou B Lien F Kuipers F Staels B . Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. (2009) 89:147–91. doi: 10.1152/physrev.00010.2008
174
Lalazar G Mizrahi M Turgeman I Adar T Ben Ya’acov A Shabat Y et al . Oral administration of OKT3 MAb to patients with NASH, promotes regulatory T-cell induction, and alleviates insulin resistance: Results of a phase IIa blinded placebo-controlled trial. J Clin Immunol. (2015) 35:399–407. doi: 10.1007/s10875-015-0160-6
175
Ilan Y Shailubhai K Sanyal A . Immunotherapy with oral administration of humanized anti-CD3 monoclonal antibody: a novel gut-immune system-based therapy for metaflammation and NASH. Clin Exp Immunol. (2018) 193:275–83. doi: 10.1111/cei.13159
176
Du M Wang X Yuan L Liu B Mao X Huang D et al . Targeting NFATc4 attenuates non-alcoholic steatohepatitis in mice. J Hepatol. (2020) 73:1333–46. doi: 10.1016/j.jhep.2020.07.030
177
Widjaja AA Singh BK Adami E Viswanathan S Dong J D’Agostino GA et al . Inhibiting interleukin 11 signaling reduces hepatocyte death and liver fibrosis, inflammation, and steatosis in mouse models of nonalcoholic steatohepatitis. Gastroenterology. (2019) 157:777–92.e14. doi: 10.1053/j.gastro.2019.05.002
178
Shearer AM Rana R Austin K Baleja JD Nguyen N Bohm A et al . Targeting liver fibrosis with a cell-penetrating protease-activated receptor-2 (PAR2) pepducin. J Biol Chem. (2016) 291:23188–98. doi: 10.1074/jbc.M116.732743
179
Su JY Deng ZJ Teng YX Koh YX Zhang WG Zheng MH et al . Prognosis after hepatic resection of patients with hepatocellular carcinoma related to non-alcoholic fatty liver disease: Meta-analysis. BJS Open. (2023) 7:zrac167. doi: 10.1093/bjsopen/zrac167
180
Chin KM Prieto M Cheong CK Di Martino M Ielpo B Goh B et al . Outcomes after curative therapy for hepatocellular carcinoma in patients with non-alcoholic fatty liver disease: a meta-analysis and review of current literature. HPB (Oxford). (2021) 23:1164–74. doi: 10.1016/j.hpb.2021.01.009
181
Nguyen N Rode A Trillaud H Aubé C Manichon AF Hocquelet A et al . Percutaneous radiofrequency ablation for hepatocellular carcinoma developed on non-alcoholic fatty liver disease. Liver Int. (2022) 42:905–17. doi: 10.1111/liv.15129
182
Young S Sanghvi T Rubin N Hall D Roller L Charaf Y et al . Transarterial chemoembolization of hepatocellular carcinoma: Propensity score matching study comparing survival and complications in patients with nonalcoholic steatohepatitis versus other causes cirrhosis. Cardiovasc Intervent Radiol. (2020) 43:65–75. doi: 10.1007/s00270-019-02363-x
183
Haber PK Puigvehí M Castet F Lourdusamy V Montal R Tabrizian P et al . Evidence-based management of hepatocellular carcinoma: Systematic review and meta-analysis of randomized controlled trials (2002-2020). Gastroenterology. (2021) 161:879–98. doi: 10.1053/j.gastro.2021.06.008
184
Shimose S Hiraoka A Nakano M Iwamoto H Tanaka M Tanaka T et al . First-line sorafenib sequential therapy and liver disease etiology for unresectable hepatocellular carcinoma using inverse probability weighting: A multicenter retrospective study. Cancer Med. (2021) 10:8530–41. doi: 10.1002/cam4.4367
185
Rimini M Kudo M Tada T Shigeo S Kang W Suda G et al . Nonalcoholic steatohepatitis in hepatocarcinoma: New insights about its prognostic role in patients treated with lenvatinib. ESMO Open. (2021) 6:100330. doi: 10.1016/j.esmoop.2021.100330
186
Finn RS Qin S Ikeda M Galle PR Ducreux M Kim TY et al . Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. (2020) 382:1894–905. doi: 10.1056/NEJMoa1915745
Summary
Keywords
microenvironment, MASLD, MASH, HCC, therapeutics
Citation
Wu Q, Yang Y, Lin S, Geller DA and Yan Y (2025) The microenvironment in the development of MASLD-MASH-HCC and associated therapeutic in MASH-HCC. Front. Immunol. 16:1569915. doi: 10.3389/fimmu.2025.1569915
Received
07 February 2025
Accepted
08 April 2025
Published
30 April 2025
Volume
16 - 2025
Edited by
Lisardo Bosca, Autonomous University of Madrid, Spain
Reviewed by
Helena Solleiro-Villavicencio, Universidad Autónoma de la Ciudad de México, Mexico
Angela M Valverde, Spanish National Research Council (CSIC), Spain
Updates
Copyright
© 2025 Wu, Yang, Lin, Geller and Yan.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yihe Yan, yanyihe12@sr.gxmu.edu.cn; David A. Geller, gellerda@upmc.edu
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.